

Abstract
Oncolytic virotherapies have shown excellent promise in a variety of cancers by promoting antitumor immunity. However, the effects of oncolytic virus-mediated type I interferon (IFN-I) production on antitumor immunity remain unclear. Recent reports have highlighted immunosuppressive functions of IFN-I in the context of checkpoint inhibitor and cell-based therapies. In this study, we demonstrate that oncolytic virus-induced IFN-I promotes the expression of PD-L1 in tumor cells and leukocytes in a IFN receptor (IFNAR)-dependent manner. Inhibition of IFN-I signaling using a monoclonal IFNAR antibody decreased IFN-I-induced PD-L1 expression and promoted tumor-specific T cell effector responses when combined with oncolytic virotherapy. Furthermore, IFNAR blockade improved therapeutic response to oncolytic virotherapy in a manner comparable with PD-L1 blockade. Our study highlights a critical immunosuppressive role of IFN-I on antitumor immunity and uses a combination strategy that improves the response to oncolytic virotherapy.
Keywords: oncolytic virus, PD-L1, IFN, IFNAR, immunotherapy, immune checkpoint
Graphical abstract
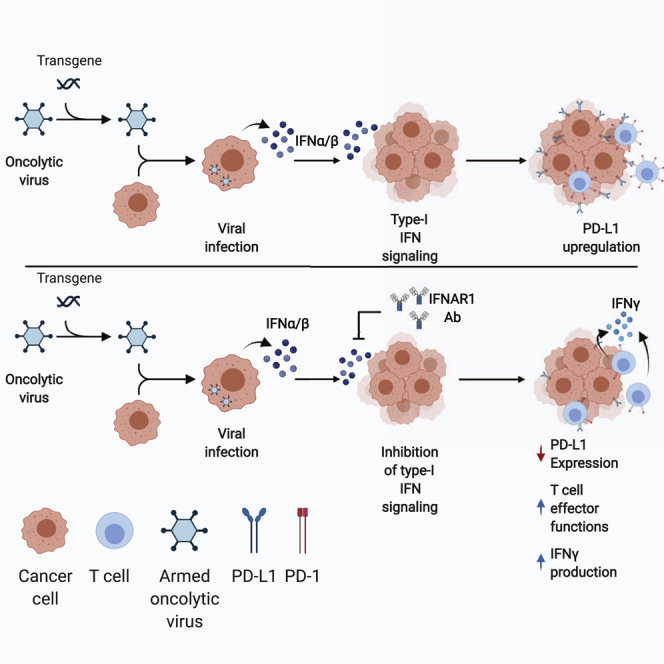
Oncolytic virotherapies have shown promise in cancer by promoting antitumor immunity. In this study, we demonstrate that oncolytic virus-induced IFNα/β promotes the expression of PD-L1 in tumor cells and leukocytes. Inhibition of IFNα/β signaling decreased promoted tumor-specific T cell effector responses and improved therapeutic response to oncolytic virotherapy.
Introduction
Oncolytic viruses (OVs) are a growing class of biotherapeutics that have demonstrated remarkable potential in the treatment of solid tumors.1 OVs are multimodal agents that induce cancer cell death through a variety of mechanisms. Although the most direct mechanism of OV-mediated cytotoxicity is oncolysis of cancer cells, OVs can also generate a robust antitumor immune response by inducing localized inflammation in the tumor microenvironment (TME).2, 3, 4 OVs have been shown to sensitize otherwise immune “cold” tumors to immune checkpoint inhibitor (ICI) therapy, a strategy that has seen some success in both pre-clinical and clinical studies.5, 6, 7 Furthermore, OVs are attractive candidates for use as vectors for tumor-associated antigens (TAAs) and chemoattractants because of their selective replication at the tumor site, with active replication changing the dynamics of the TME. Most notably, the presence of a virus will highly upregulate the production of type I interferons (IFN-I). The role of IFN-I in cancer immunotherapy has become highly controversial, as they have been shown to have both protumor and antitumor properties.8 IFN-I can enhance immunogenicity of the tumor by upregulating the surface expression of major histocompatibility complex-I (MHC-I) and TAAs.9, 10, 11 However, IFN-I signaling can also potentiate resistance through upregulation of T cell inhibitory receptors and their respective ligands, including programmed death-ligand 1 (PD-L1) and galectin-9.12, 13, 14, 15, 16 There is also accumulating evidence that IFN-I can potentiate resistance to ICI therapy via PD-L1-independent mechanisms and that blocking IFN-I signaling can restore the function of exhausted T cell subsets.17,18 Furthermore, IFN-I potentiates autoimmune side effects of antigen-targeted adoptive cell therapy, and modulation of IFN-I signaling ameliorates side effects without compromising antitumor efficacy.19 Finally, the antiviral functions of IFN-I can be detrimental to OV therapy by preventing infection of tumor cells and subsequent expression of encoded tumor antigens from the viral vector. Indeed, several groups have seen improved outcomes in pre-clinical models by using small-molecule inhibitors of type I IFN signaling in combination with OV therapy.20, 21, 22
In this study, we show that OVs can induce upregulation of PD-L1 in tumors in an IFN-I-dependent manner. Differences in PD-L1 upregulation induced by oncolytic vesicular stomatitis virus (VSV) and Vaccinia virus are directly correlated with the level of IFN-I induction. Additionally, IFNα/β receptor (IFNAR)-knockout (KO) cells demonstrate vastly reduced OV-mediated expression of PD-L1, a phenomenon that was also observed in vivo. Finally, IFNAR blockade prevented OV-mediated upregulation of PD-L1 in circulating leukocytes and vastly improved antitumor CD8+ T cell activity resulting in improved therapeutic efficacy. Our strategy of combining OVs with IFNAR blockade can prevent the onset of IFN-I-mediated immunosuppression in the TME. These findings will be applicable for improving OV therapy in multiple solid tumor types.
Results
Virus-mediated PD-L1 upregulation corresponds with IFN-I production
To assess the role of OVs in regulating the expression of PD-L1, we decided to compare two commonly used OVs: VSVΔ51-GFP (VSV) and Vaccinia virus-YFP (VacV). B16F10 cells were infected with VSV or VacV at a multiplicity of infection (MOI) of 1, and the relative expression of PD-L1 mRNA was assessed using RT-PCR 24 h post-infection. GFP or YFP expression was used to confirm initiation of replication (Figure S1). We found that VSV, but not VacV, significantly induced mRNA expression of PD-L1 (Figure 1A). VSV also upregulated PD-L1 in MC38 cells (Figure S2), which express higher basal levels of PD-L1 and are responsive to PD-L1 blockade therapy.23 To see how PD-L1 data compare with other virus-induced genes, we assessed the mRNA expression of the interferon-stimulated gene IFIT1. Like PD-L1, IFIT1 mRNA was highly expressed in cells infected with VSV (Figure 1B). As IFIT1 expression can be independent of IFN-I signaling,24 we assessed virus-induced expression of IFN-I. Here we also found that VSV induced much higher expression of IFNα/β mRNA (Figures 1C and 1D) and protein (Figures 1E and 1F) compared with VacV.
Figure 1.

Virus-induced expression of PD-L1 correlates with type I IFN production
B16F10 cells were infected with VSVΔ51-GFP (VSV) or vaccinia virus-YFP (VacV) at an MOI of 1. RNA was harvested 24 h post-infection, and RT-PCR was used to assess mRNA expression of (A) PD-L1, (B) IFIT1, (C) IFNα, and (D) IFNβ. Supernatants were also collected and used to measure the concentration of (E) IFNα and (F) IFNβ secreted into the supernatant. Statistical significance was calculated using ordinary one-way ANOVA for (A)–(D) and two-way ANOVA with multiple comparisons for (E) and (D). p values are displayed for relevant comparisons.
While IFNγ is a known and potent inducer of PD-L1 expression in the TME,25, 26, 27 several emerging studies suggest that IFN-I also regulates PD-L1 expression.14, 15, 16 As we see a correlation between virus-induced PD-L1 expression and IFN-I production, we tested the effects of IFN-I on PD-L1 expression in B16F10 and MC38 cells. Treatment with either IFNα or IFNβ induced expression of PD-L1 mRNA in both cell lines (Figures 2A and 2B). Furthermore, IFNα/β receptor knockout cells showed no increase in PD-L1 expression when treated with either IFNα or IFNβ (Figures 2A and 2B). These data were consistent with the IFN-induced expression of IFIT1 (Figures S3A and S3B). Finally, these results were validated at the protein level by measuring the mean fluorescence intensity (MFI) of PD-L1 surface expression using flow cytometry analysis, in which we saw a similar expression profile (Figures 2C–2F).
Figure 2.

Type I IFN induces PD-L1 upregulation in murine cancer cell lines
MC38/MC38 IFNAR-KO cells and B16F10/B16F10 IFNAR-KO cells were treated with 100 U/mL of IFNα or IFNβ. (A and B) RNA was harvested and used to assess mRNA expression of PD-L1 via RT-PCR. (C and D) Cells were stained with anti-PD-L1-BV711 antibody, and MFI was measured using flow cytometry. (E and F) Representative histograms of PD-L1-BV711 fluorescence intensity. Statistical significance was calculated using two-way ANOVAs with multiple comparisons. p values are displayed for relevant comparisons.
Virus-induced PD-L1 cell surface expression is dependent on IFN-I signaling
Although type I IFNs induce PD-L1 expression through IFNAR, several reports have also demonstrated that PD-L1 can be induced via IFN-independent inflammatory pathways that generally converge on IRF1, a transcription factor involved in PD-L1 regulation.28, 29, 30, 31 We thus used IFNAR-KO cells to determine the dependence of VSV-induced PD-L1 upregulation on IFN-I signaling. Although both MC38 and B16F10 IFNAR-KO cells showed upregulation of PD-L1 when infected with VSV, the expression of PD-L1 mRNA in IFNAR-KO cells was significantly lower (Figures 3A and 3B). Despite a small increase in PD-L1 expression at the mRNA level, flow cytometry analysis of B16F10 cells failed to detect an increase in PD-L1 surface expression in VSV-infected IFNAR-KO cells, while parental B16F10 cells demonstrated a significant increase in PD-L1 surface expression when infected with VSV (Figures 3C and 3D). GFP expression analysis confirmed similar initiation of infection in parental and KO cells (Figures S4A–S4C). Although data were consistent at the protein level in B16F10 cells, monitoring surface expression of PD-L1 on MC38 cells was not feasible, because of the sensitivity of this cell line to VSV cytotoxicity (Figure S4D). As a result, we used polyinosinic:polycytidylic acid (poly I:C) as a replacement of viral infection to initiate antiviral signaling while maintaining cell viability. Indeed, poly I:C transfection also induced an upregulation of PD-L1 at both the mRNA and protein levels on MC38 and B16F10 cells. Although there was a small increase in PD-L1 mRNA expression in IFNAR-KO cells stimulated with poly I:C, there was no significant change in surface expression of PD-L1 (Figures 3E–3J). Altogether these data strongly suggest that the virus-induced expression of PD-L1 is largely dependent on IFN-I signaling.
Figure 3.

Virus-induced PD-L1 expression is dependent on type I IFN signaling
Wild-type and knockout MC38 and B16F10 cells were infected with VSVΔ51-GFP (VSV) at an MOI of 1. (A and B) RNA was harvested and used to assess mRNA expression of PD-L1 via RT-PCR. (C) Representative histogram and (D) MFIs of PD-L1 surface expression on B16F10 and B16 IFNAR-KO were measured using flow cytometry. (E) MC38/MC38 IFNAR-KO cells and (F) B16F10/B16F10 IFNAR-KO cells were transfected with 1 μg poly I:C. RNA was harvested and used to assess mRNA expression of PD-L1 using RT-PCR. (G and H) Representative histograms and (I and J) MFIs of PD-L1 surface expression were measured using flow cytometry. Statistical significance was calculated using two-way ANOVAs with multiple comparisons. p values are displayed for relevant comparisons.
Oncolytic VSV-induced PD-L1 upregulation in murine tumors is partially dependent on type I IFN signaling
Although our data strongly suggest that virus-induced PD-L1 expression is dependent on IFN-I signaling in vitro, the TME in vivo contains a complex network of additional factors that regulate the expression of PD-L1. This includes the presence of other cytokines and chemokines that can regulate PD-L1 expression.29,30 To this end, we assessed the effect of VSV on PD-L1 expression in vivo and characterized the role of IFN-I signaling. MC38 or B16F10 tumors were implanted subcutaneously and intradermally, respectively, into C57BL/6 mice. One week later, tumors were treated with anti-IFNAR or isotype control antibody by intraperitoneal (i.p.) injection followed by intravenous administration of VSV. Tumors were harvested and PD-L1 surface expression was assess via immunohistochemistry 24 h after treatment. In both MC38 and B16F10 tumors, treatment with VSV induced much higher PD-L1 expression relative to untreated mice (Figure 4A). Pre-treatment with anti-IFNAR antibody decreased, but did not eliminate, virus-induced PD-L1 expression. Next, we validated these data by comparing virus-induced PD-L1 expression in MC38 IFNAR-KO tumors. MC38 and MC38 IFNAR-KO cells were implanted subcutaneously, and mice were treated with VSV intravenously 1 week later. Consistent with anti-IFNAR antibody treatment, the loss of IFNAR in MC38 tumors reduced but did not eliminate PD-L1 expression following VSV treatment (Figure 4B). These data suggest that early virus-induced expression of PD-L1 in the TME is partially dependent on IFN-I signaling.
Figure 4.

IFNAR blockade reduces virus-induced PD-L1 expression in the tumor
(A) MC38 and B16F10 cells were implanted into C57BL/6 mice subcutaneously and intradermally, respectively. Mice were treated with 1 mg anti-IFNAR antibody by i.p. followed by intravenous (i.v.) injection of 2 × 108 pfu VSVΔ51 (VSV) 2 h later. (B) MC38 and MC38 IFNAR-KO cells were implanted into C57BL/6 mice subcutaneously. Mice were treated with VSV by i.v. injection. Twenty-four hours after VSV administration, tumors were harvested and stained for PD-L1 expression using immunohistochemistry.
Oncolytic VSV induces PD-L1 upregulation in circulating leukocytes
Expression of PD-L1 on T cells, B cells, macrophages, and other leukocytes can enhance immunosuppression and promote tumor tolerance.32, 33, 34 We investigated whether PD-L1 expression on circulating leukocytes is upregulated following OV treatment and if early expression of PD-L1 is dependent on IFN-I signaling. We used tumors and virus expressing gp33, an immunodominant antigen of lymphocytic choriomeningitis virus, as a surrogate antigen. Mice harboring B16F10-gp33 tumors were treated with anti-IFNAR antibody followed by intravenous delivery of VSVΔ51-gp33 (VSV-gp33). Peripheral blood mononuclear cells (PBMCs) were isolated, and PD-L1 surface expression was assessed using flow cytometry 24 h later. We found that there was a substantial increase in the percentage of PD-L1-positive T cells and B cells after treatment with VSV-gp33. Moreover, pre-treatment with IFNAR blockade partially abrogated virus-mediated upregulation of PD-L1 on these cells (Figures 5A and 5B). Interestingly, this trend was not entirely consistent among different cell types. Although monocytes and macrophages also demonstrated VSV-mediated PD-L1 expression, IFNAR blockade only mildly prevented virus-mediated expression (Figures 5C and 5D). The percentage of PD-L1-positive neutrophils was substantially increased following VSV-gp33 treatment, but unlike the other immune cell types, neutrophils demonstrated a further increase in percentage PD-L1-positive cells after IFNAR blockade (Figure 5E). Finally, circulating natural killer (NK) cells demonstrated a mild reduction in percentage PD-L1-positive cells following treatment with VSV-gp33 (Figure 5F). Collectively, these data suggest that the level of dependence of VSV-induced PD-L1 expression on IFN-I signaling varies among cell types. Virus-mediated upregulation of PD-L1 on T and B lymphocytes is moderately dependent on IFN-I signaling, while similar PD-L1 upregulation on monocytes and macrophages seems to be largely independent of IFN-I signaling, or even dampened by IFN-I signaling.
Figure 5.

Blocking IFNAR signaling alters virus-mediated PD-L1 expression on circulating leukocytes
B16F10-gp33 cells (105) were implanted intradermally in C57BL/6 mice. Mice were treated with 1 mg anti-IFNAR by i.p. followed by i.v. injection of 2 × 108 pfu VSVΔ51-gp33 2 h later. Expression of PD-L1 on circulating mononuclear cells was assessed 24 h after VSV treatment using flow cytometry. (A) Representative flow plots of PD-L1 expression on circulating T cells (top) and B cells (bottom). (B) Percentage of circulating T cells (top) and B cells (bottom) expressing PD-L1. (C–F) Percentage of circulating monocytes, macrophages, neutrophils, and NK cells expressing PD-L1. Statistical significance was calculated using ordinary one-way ANOVAs. p values are displayed for relevant comparisons.
IFNAR blockade potentiates activation of antitumor CD8+ T cells and prevents upregulation of exhaustion markers
IFN-I signaling is required for priming antitumor CD8+ T cells and can promote effector functions in CD8+ T cells.35,36 Therefore, we decided to investigate the effect of IFNAR blockade on the activity of antitumor CD8+ T cells and determine if IFNAR blockade dampens the generation of effector CD8+ T cells. We used tumors and virus expressing gp33 as a surrogate antigen to assess the priming and magnitude of antitumor T cell responses. C57BL/6 mice harboring B16-gp33 tumors were treated with anti-IFNAR or anti-PD-L1 antibodies followed by vaccination with VSV-gp33. PBMCs were isolated 7 days later and stimulated with gp33 peptide. The magnitude of the gp33-specific T cell response was measured by assessing IFNγ production in response to gp33 antigenic stimulation by intracellular cytokine staining. As expected, vaccination with VSV-gp33 increased the number of IFNγ+ gp33-specific CD8+ T cells, which was improved further by the addition of anti-PD-L1 therapy. Interestingly, IFNAR blockade was substantially more effective than PD-L1 blockade at increasing the number of IFNγ+ gp33-specific T cells when combined with VSV-gp33 (Figures 6A and 6B). Next, we decided to further characterize the phenotype of T cells by assessing the expression of CD44 and CD62L, which are surface markers used to differentiate between naive (CD44−, CD62L+), central memory (CM; CD44+, CD62L+), effector memory (EM; CD44+, CD62L−) and double-negative (DN; CD44−, CD62L−) cells (Figure 6C). We found that vaccination with VSV-gp33 resulted in a higher fraction of EM CD8+ T cells (TEM cells) in circulation. Addition of either PD-L1 or IFNAR blockade further increased the percentage of TEM cells (Figures 6C and 6D). This trend was consistent when assessing the absolute number of TEM cells in circulation, with PD-L1 and IFNAR blockade substantially increasing the number of TEM cells when combined with VSV-gp33 (Figure 6E). Finally, we assessed the level of T cell exhaustion characterized by the overexpression of programmed death-1 (PD-1) and T cell immunoglobulin and mucin domain-containing-3 (TIM-3). These markers can be upregulated following T cell effector functions and chronic antigen stimulation, leading to exhaustion and dysfunctional T cell activity.37 Although vaccination with VSV-gp33 marginally increased the absolute number of PD-1+ and TIM-3+ CD8+ T cells in circulation, the addition of PD-L1 blockade significantly increased the number of CD8+ T cells expressing exhaustion markers (Figures 6F–6H). Impressively, the combination of IFNAR blockade with VSV-gp33 resulted in a negligible increase in PD-1+ and TIM-3+ T cells compared with VSV-gp33 monotherapy. Taken together, these data demonstrate that IFNAR blockade can promote tumor-specific CD8+ T cell activation and TEM cell proliferation in a manner similar to that of PD-L1 blockade, while maintaining lower expression of exhaustion markers such as PD-1 and TIM-3.
Figure 6.

IFNAR blockade promotes tumor-specific CD8 T cell activation and the generation of TEM cells while maintaining lower expression of exhaustion markers
(A) Schematic representation of the treatment regimen used. B16F10-gp33 cells (105) were implanted intradermally in C57BL/6 mice. Mice were treated with 1 mg anti-IFNAR or 250 μg anti-PD-L1 antibodies by i.p. followed by i.v. injection of 2 × 108 pfu VSVΔ51-gp33 2 h later. PBMCs were isolated 7 days post-treatment and re-stimulated with gp33 peptide, and IFNγ production was assessed using ICS. (B) Representative contour plots showing the percentage of CD8 T cells that are IFNγ+. (C) Percentages of IFNγ+ CD8 T cells were graphed; lines represent the means. PBMCs were isolated 7 days post-treatment, and expression of T cell markers was assessed using flow cytometry. (D) Representative contour plots showing the percentage of naive, central memory (CM), effector memory (EM), and double-negative (DN) CD8 T cells. (E) Quantification of the T cell populations for each group. (F) Boxplot showing the absolute number of EM CD8 T cells. Boxplots showing absolute numbers of (G) PD-1+ and (H) TIM-3+ CD8 T cells. (I) Representative contour plots showing percentage of PD-1+ and TIM-3+ CD8 T cells. Statistical significance was calculated using ordinary one-way ANOVAs. p values are displayed for relevant comparisons.
IFNAR blockade synergizes with oncolytic VSV to improve therapeutic outcomes in murine melanoma model
Our data show that IFNAR blockade can limit early virus-induced expression of PD-L1 in the tumor and promote tumor-specific T cell activity. To further establish the relevance of these observations in the context of therapeutic efficacy, we assessed the therapeutic potential of IFNAR blockade when combined with VSV-gp33 in tumor-bearing mice. We chose to use B16F10 tumors because of their documented resistance to ICI therapy.38 C57BL/6 mice were implanted intradermally with B16F10-gp33 cells and treated with anti-IFNAR and/or anti-PD-L1 antibodies 10 days later. After 2 h, the mice were then treated with VSV-gp33. Tumor volumes were monitored and anti-PD-L1 treatments were administered every 3 days according to a previously established therapeutic regimen (Figure 7A).39 VSV-gp33 monotherapy significantly delayed tumor progression relative to the control, but the combination of VSV-gp33 and anti-PD-L1 resulted in a significant delay in tumor progression and prolonged survival (Figures 7B–7D). Of interest, the combination of VSV-gp33 and a single dose of anti-IFNAR antibody was comparable with VSV-gp33 plus continual anti-PD-L1 administration (Figures 7B–7D). Finally, the addition of anti-PD-L1 to VSV-gp33 plus anti-IFNAR treatment did not improve therapeutic outcomes further (Figures 7B–7D). These demonstrate comparable therapeutic efficacy of IFNAR blockade and PD-L1 blockade when used in combination with VSV-gp33, suggesting that PD-L1 blockade holds no additional benefit to IFNAR blockade in this context.
Figure 7.

IFNAR blockade synergizes with oncolytic VSV to improve therapeutic outcomes in tumor-bearing mice
(A) Schematic representation of the treatment regimen used. B16F10-gp33 cells (105) were implanted intradermally in C57BL/6 mice. Mice were treated with 1 mg anti-IFNAR and/or 250 μg anti-PD-L1 (checkpoint) antibody by i.p. followed by i.v. injection of 2 × 108 pfu VSVΔ51-gp33 2 h later. Tumor volumes were monitored every 2–3 days until endpoint (1,000 mm3). (B) Tumor volumes graphed for each mouse over time. (C) Average tumor volumes over time. (D) Kaplan-Meier survival curves. Statistical significance of survival was calculated using Gehan-Bresloq-Wilcoxon test. p values are displayed for relevant comparisons.
Discussion
Oncolytic viruses have gained traction as a potent cancer immunotherapy in the past decade. The presence of an actively replicating virus in the TME can cause substantial upregulation of IFN-I production; however, the role of IFN-I signaling in immunotherapy has become a topic of controversy. In this study, we show that IFNAR blockade is an effective therapeutic strategy when combined with oncolytic VSV. Efficacy of this combination in tumor-bearing mice is comparable with that of PD-L1 blockade and oncolytic VSV. Furthermore, addition of PD-L1 blockade does not improve therapeutic efficacy any further. This observation suggests that the efficacy of IFNAR blockade could be dependent on inhibition of PD-L1. Furthermore, we use IFNAR-knockout cells in vitro to show that OV-mediated upregulation of PD-L1 is highly dependent on IFN-I signaling. Importantly, this observation is consistent in vivo, where a variety of other factors can be involved in the expression of PD-L1. Namely, IFNγ is a key player for cancer immunotherapy and is known as a potent inducer of PD-L1.25,26 In the context of OV therapy, however, systemic IFNα/β is induced as early as 5 h after OV treatment,40 while IFNγ is only detected in circulation a few days post-infection. Our data suggest that early induction of PD-L1 expression in the TME following OV therapy is at least partially dependent on IFN-I signaling. Our results match similar findings which show that sustained IFN-I and IFN-II signaling confer resistance to immunotherapy and induce expression of inhibitory ligands.41 However, Benci et al.41 demonstrated that IFN-mediated resistance is only partially dependent on PD-L1 upregulation, and that upregulation of other inhibitory ligands, including galectin-9, HVEM and MHCII, contributes to IFN-mediated resistance. This observation means that IFNAR blockade could potentially function as a multimodal inhibitor of several immune checkpoints, which could have widespread clinical implications.
Expression of PD-L1 in the TME is not restricted to cancer cells. Indeed, studies have demonstrated that PD-L1-knockout tumor models can still benefit from anti-PD-L1 therapy.42,43 Furthermore, expression of PD-L1 on immune cells can also suppress antitumor immune responses. Indeed, one study by Diskin et al.34 demonstrated that PD-L1 expression on T cells can promote tumor tolerance and suppression of effector T cells. Expression of PD-L1 on antigen-presenting cells can also attenuate tumor antigen presentation and priming CD8+ T cells.43, 44, 45 In this study we show that OV-mediated PD-L1 expression on T cells, B cells, and monocytes is partially dependent on IFN-I signaling. We also demonstrated that IFNAR blockade can promote the generation of TEM cells and increase the number of IFNγ+ tumor-specific T cells to similar levels as with PD-L1 blockade. This finding suggests that transient blocking IFN-I signaling promotes priming and/or boosting of tumor-specific T cells, but the relevance of inhibiting PD-L1 expression on other immune cells has yet to be determined. Interestingly, the dependance of PD-L1 expression on IFN-I signaling varies by cell type. Whereas OV-mediated PD-L1 expression on T cells, B cells, and monocytes is partially dependent on IFN-I signaling, PD-L1 upregulation on macrophages is mostly IFN-I independent. Furthermore, IFNAR blockade caused an upregulation of PD-L1 on circulating neutrophils, suggesting that IFN-I signaling negatively regulates PD-L1 expression on neutrophils. Although we demonstrate that IFNAR blockade can enhance antitumor T cell activity, it has been well established that IFN-I signaling is important for promoting effector functions in CD8+ T cells.35,36 Interestingly, existing reports have demonstrated that specifically prolonged IFN-I signaling is detrimental to antitumor immunity.41 Another study shows that IFN-I can still confer therapeutic benefit when combined with PD-L1 blockade to counteract the IFN-mediated upregulation of PD-L1.46 Future studies should continue to focus on the kinetics of IFN-I signaling in the TME to establish the effects of acute versus sustained signaling on resistance to immunotherapy. The timing of anti-IFNAR administration must be considered to allow priming of tumor-specific T cell responses while preventing IFN-I-mediated exhaustion.
Finally, IFNAR blockade as a therapeutic strategy has unique implications for OV therapy. Several OVs induce expression of PD-L1 in the tumor, so OVs are often combined with anti-PD-1/PD-L1 therapy to improve therapeutic outcomes.47, 48, 49, 50 Our study demonstrates that IFNAR blockade can prevent the onset of IFN-mediated PD-L1 expression and synergize with oncolytic VSV in a similar manner to PD-L1 blockade. Future studies are required to determine if IFNAR blockade will synergize with OVs that induce lower levels of IFN-I expression, such as oncolytic VacV and HSV. On the basis of the data in this study, VacV induces lower levels of PD-L1 compared with VSV, but other studies have demonstrated synergistic tumor control by combining oncolytic VacV with anti-PD-L1 therapy.51,52 Unlike anti-PD-L1 therapy, however, IFNAR blockade has the added benefit of potentially enhancing OV replication in the TME. Indeed, several groups have shown that inhibition of IFN-I signaling can enhance OV replication and promote therapeutic efficacy in several OV platforms without compromising the safety profile of OVs.21,22 We observed no additional adverse effects from the combination of VSV and IFNAR blockade. The transient nature of a single dose of anti-IFNAR antibody seems to be enough to enhance tumor control while maintaining VSV’s selectivity for malignant cells. Furthermore, previous studies have shown that IFNAR blockade can ameliorate adverse effects caused by other immunotherapies.19 However, additional experiments will be required to fully elucidate the safety implications of this combination strategy. Furthermore, OVs can function as effective vectors for transgenes, including pro-inflammatory chemokines and tumor antigens that can further boost the generation of a robust antitumor response. Promoting OV replication by inhibiting IFN-I signaling will, in turn, increase the production of encoded transgenes. Therefore, our development of an IFNAR blockade strategy has broad clinical implications for the future of OV therapy.
Materials and methods
Cell lines
MC38 cells (American Type Culture Collection [ATCC]) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS; 30-2020; ATCC), 2 mmol/L L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). B16F10 (ATCC) and B16-gp33 cells (B16F10 cells stably transfected with a minigene corresponding to the gp33 peptide)53 were maintained in minimum essential medium with Earle’s salts (MEM-Earle’s) supplemented with 1× MEM vitamin solution (11120052; Thermo Fisher Scientific), 10% FBS, 2 mmol/L L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. MC38 and B16F10 IFNAR knockouts were obtained from the lab of Dr. John Bell (Ottawa Hospital Research Institute [OHRI], Ottawa, ON, Canada) and cultured under the same conditions as their respective wild-type cell lines.
Virus propagation and peptides
VSVΔ51 is an oncolytic attenuated variant of the VSV Indiana strain. VSV was propagated, purified and quantified on Vero cells as described previously.54 Briefly, virus stocks were purified from cell culture supernatants by filtration through a 0.22 μm Steritop filter (Millipore) and centrifugation at 30,000 × g before resuspension in PBS. VSV-gp33 is a recombinant VSVΔ51 expressing the dominant epitopes of the lymphocytic choriomeningitis virus glycoprotein (LCMV-gp33-41 and LCMV-gp61-80, respectively).55 The VacV used in this study is the wild-type Copenhagen strain and was produced in HeLa cells and quantified in U2OS cells. For VacV purification, virus was collected by repeated freeze-thaw cycles. Further purification was done by centrifugation at 20,700 × g through a 36% sucrose cushion before resuspension in 1 mM Tris (pH 9). The H-2Db-restricted peptide of LCMV-GP33 (GP33-41; KAVYNFATM) was synthesized by Biomer Technology (Pleasanton, CA).
In vivo experiments
Mice were maintained at the McMaster University Central Animal Facility, and all procedures were performed in full compliance with the Canadian Council on Animal Care and approved by the Animal Research Ethics Board of McMaster University. MC38 tumors: 2 × 105 cells were implanted subcutaneously into the left flank of 6- to 8-week-old female C57BL/6 mice (Charles River Laboratories, Wilmington, MA). B16F10 tumors: 1 × 105 cells were implanted intradermally into C57BL/6 mice. Mice were treated 7 days after B16F10 tumor challenge or 10 days after MC38 tumor challenge. αIFNAR (1 mg, one dose; clone MAR1-5A3; InVivoMab) and/or 250 μg αPD-L1 (clone 10F.9G2; InVivoMab) antibodies were administered by i.p. injection. Experimental groups receiving αPD-L1 followed a dosing schedule of 250 μg treatments every 3 days for a total of 8 doses. Two hours after αPD-L1 and/or αIFNAR treatment, 2 × 108 pfu of VSVΔ51 was administered via tail vein injection. About 150 μL blood was collected via retro-orbital bleed 1 and 7 days following treatment for immune analysis (described in another section). Tumor volumes were monitored and measured every 2–3 days until they reached their endpoint volume (1,000 mm3).
Immune analysis and flow cytometry
Following blood collection, red blood cells were lysed with ACK buffer, and peripheral blood mononuclear cells were transferred to a round-bottom 96-well plate. For in vitro cell lines, confluent cells were treated with TrypLE Express Enzyme (Thermo Fisher Scientific), resuspended in PBS, and then transferred to a round-bottom 96-well plate. The cell suspensions were stained with fixable viability stain 510 (catalog no. 564406; BD Biosciences) for 30 min at room temperature, then treated with anti-CD16/CD32 (Fc block; catalog no. 553141; BD Biosciences) for 15 min at 4°C. Cell surface staining was done for 30 min at 4 degrees. Intracellular staining was done using Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences). For analysis of gp33-specific T cells, PBMCs were treated with 1 μg/mL gp33 peptide (KAVYNFATM; Biomer Technology, CA) and incubated for 1 h at 37°, 5% CO2, followed by treatment with a protein transfer inhibitor (GolgiPlug; BD Biosciences) and incubated for another 3.5 h. The cells were then stained as described above. Data acquisition was done on the LSRFortessa (BD), and data were analyzed using FlowJo. The following antibodies were purchased from BD Biosciences: BV711 rat anti-mouse CD274 (PD-L1; clone MIH5, catalog no. 563369), BV786 rat anti-mouse CD3 (clone 17A2, catalog no. 564010), PE-Cy7 rat anti-mouse CD8α (clone 53 6.7, catalog no. 552877), PerCP-Cy5.5 rat anti-mouse CD62L (clone MEL-14, catalog no. 560513), BV711 rat anti-mouse CD44 (clone IM7, catalog no. 563971), APC-Cy7 rat anti-mouse CD4 (clone GK1.5, catalog no. 561830), BV650 rat anti-mouse CD279 (PD-1; clone RMP1-30, catalog no. 748266), BV421 mouse anti-mouse CD366 (TIM-3; clone 5D12, catalog no. 747626), APC rat anti-mouse IFNγ (clone XMG1.2, catalog no. 554413), BV421 hamster anti-mouse CD11c (clone HL3, catalog no. 562782), BV650 rat anti-mouse F4/80 (clone T45-2342, catalog no. 743282), PerCP-Cy5.5 rat anti-mouse Ly6G (clone 1A8, catalog no. 560602), FITC rat anti-mouse Ly6C (clone AL 21, catalog no. 553104), PE-Cy7 rat anti-mouse CD19 (clone 1D3, catalog no. 552854), and BV605 rat anti-mouse CD11b (clone M1/70, catalog no. 563015).
RT-PCR
RNA extraction was performed using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s protocol. Purified RNA (500 ng) was reverse-transcribed using the iScript gDNA Clear cDNA Synthesis Kit (Bio-Rad). Real-time PCRs were performed with Ssofast EvaGreen kit (Bio-Rad), and data were acquired on a 7500 Fast Real-Time PCR system (Applied Biosystems). Relative mRNA expression was normalized to GAPDH, and fold induction was calculated relative to the untreated/uninfected controls using the Pfaffl method.56 Primer sequences are as follows:
PD-L1 F: CAGCAACTTCAGGGGGAGAG
PD-L1 R: TTTGCGGTATGGGGCATTGA
IFIT1 F: GCC TAT CGC CAA GAT TTA GAT GA
IFIT1 R: TTC TGG ATT TAA CCG GAC AGC
IFNα F: CGGAATTCTCTCCTGCCTGAAGGAC
IFNα R: AAGGGTACCACACAGTGATCCTGTGGAA
IFNβ F: AGC TCC AAG AAA GGA CGA ACA
IFNβ R: GCC CTG TAG GTG AGG TTG AT
GAPDH F: AATGGATTTGGACGCATTGGT
GAPDH R: TTTGCACTGGTACGTGTTGAT
Immunohistochemistry
Tumors were harvested and flash-frozen in liquid nitrogen. Frozen tissue sections were cut at 5 μm onto coated slides. Sections were air-dried overnight and then fixed in 10% neutral buffered formalin (NBF) for 5 min before being treated with 1% H2O2 in dH2O for 15 min at room temperature. Slides were then washed in dH2O to remove excess H2O2. Slides were rinsed in Bond Wash (Leica) and placed on the Leica Bond Automated stainer. The slides were stained with Rat primary PDL-1 (13-5982-52; EBio) 1:500 in Animal Free Diluent (SP-5035; Vector Labs). The BOND Polymer Refine Red Detection kit (Leica) was used according to the manufacturer’s protocol. The slides were then digitized according to a previously described protocol.57
ELISA
Supernatants from cells were collected 8, 16, and 24 h post-infection and treated with a protease inhibitor cocktail (Thermo Fisher Scientific) and centrifugated for 10 min at 4°C. Supernatants were transferred to a new tube and either frozen at −80°C or used immediately for ELISA. Verikine mouse IFN beta ELISA kit (PBL Assay Science) and Verikine mouse IFN alpha ELISA kit (PBL Assay Science) were used for data acquisition according to the manufacturer’s protocol. Levels of secreted cytokines (pg/mL) were interpolated from experimental standard curves.
Statistical analysis
Results are presented as mean ± SD. Log rank (Mantel-Cox) tests were used to analyze the statistical significance between treatment groups for Kaplan-Meier survival graphs. Ordinary one-way ANOVA was used to determine the statistical significance between means of treated groups according to the normality of their distributions. In all cases, the null hypothesis was rejected at p < 0.05. All statistical analysis was performed using GraphPad Prism 9.
Acknowledgments
This work was funded by the Terry Fox Research Institute (project 1073). N.E. was the recipient of the Ontario Graduate Scholarship. We would like to acknowledge the histology facility at McMaster University for processing samples for histology. MC38 and B16F10 IFNAR-knockout cell lines were received from the laboratory of Dr. John Bell (Ottawa Hospital Research Institute, Ottawa, ON) and validated using flow cytometry.
Author contributions
Conceptualization, N.E., S.W., and K.M.; Methodology, N.E., S.W., K.A., Y.W., and K.M.; Investigation, N.E., S.W., A.V., and A.R.; Writing – Original Draft, N.E.; Writing – Review & Editing, N.E., S.W., Y.W., and K.M.; Funding Acquisition, K.M.; Resources, Y.W. and K.M.; Supervision, K.M., S.W., and Y.W.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.03.006.
Supplemental information
References
- 1.Patel M.R., Kratzke R.A. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl. Res. 2013;161:355–364. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 2.Workenhe S.T., Mossman K.L. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014;22:251–256. doi: 10.1038/mt.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Workenhe S.T., Mossman K.L. Rewiring cancer cell death to enhance oncolytic viro-immunotherapy. Oncoimmunology. 2013;2:e27138. doi: 10.4161/onci.27138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Workenhe S.T., Simmons G., Pol J.G., Lichty B.D., Halford W.P., Mossman K.L. Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol. Ther. 2014;22:123–131. doi: 10.1038/mt.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourgeois-Daigneault M.-C., Roy D.G., Aitken A.S., El Sayes N., Martin N.T., Varette O., Falls T., St-Germain L.E., Pelin A., Lichty B.D., et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018;10:eaao1641. doi: 10.1126/scitranslmed.aao1641. [DOI] [PubMed] [Google Scholar]
- 6.Workenhe S.T., Nguyen A., Bakhshinyan D., Wei J., Hare D.N., MacNeill K.L., Wan Y., Oberst A., Bramson J.L., Nasir J.A., et al. De novo necroptosis creates an inflammatory environment mediating tumor susceptibility to immune checkpoint inhibitors. Commun. Biol. 2020;3:645. doi: 10.1038/s42003-020-01362-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun L., Funchain P., Song J.M., Rayman P., Tannenbaum C., Ko J., Mcnamara M., Marcela Diaz-Montero C., Gastman B. Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: a case series. J. Immunother. Cancer. 2018;6:36. doi: 10.1186/s40425-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musella M., Manic G., De Maria R., Vitale I., Sistigu A. Type-I-interferons in infection and cancer: unanticipated dynamics with therapeutic implications. OncoImmunology. 2017;6:e1314424. doi: 10.1080/2162402X.2017.1314424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sivinski C.L., Lindner D.J., Borden E.C., Tempero M.A. Modulation of tumor-associated antigen expression on human pancreatic and prostate carcinoma cells in vitro by α- and γ-interferons. J. Immunother. 1995;18:156–165. doi: 10.1097/00002371-199510000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Blaauboer A., Sideras K., van Eijck C.H.J., Hofland L.J. Type I interferons in pancreatic cancer and development of new therapeutic approaches. Crit. Rev. Oncol. Hematol. 2021;159:103204. doi: 10.1016/j.critrevonc.2020.103204. [DOI] [PubMed] [Google Scholar]
- 11.Liang Y., Tang H., Guo G., Qiu X., Yang Z., Ren Z., Sun Z., Bian Y., Xu L., Xu H., et al. Targeting IFNα to tumor by anti-PD-L1 creates feedforward antitumor responses to overcome checkpoint blockade resistance. Nat. Commun. 2018;9:4586. doi: 10.1038/s41467-018-06890-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Den Hoogen L.L., Van Roon J.A.G., Mertens J.S., Wienke J., Lopes A.P., De Jager W., Rossato M., Pandit A., Wichers C.G.K., Van Wijk F., et al. Galectin-9 is an easy to measure biomarker for the interferon signature in systemic lupus erythematosus and antiphospholipid syndrome. Ann. Rheum. Dis. 2018;77:1810–1814. doi: 10.1136/annrheumdis-2018-213497. [DOI] [PubMed] [Google Scholar]
- 13.Yang R., Sun L., Li C.F., Wang Y.H., Yao J., Li H., Yan M., Chang W.C., Hsu J.M., Cha J.H., et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021;12:832. doi: 10.1038/s41467-021-21099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao W., Klement J.D., Lu C., Ibrahim M.L., Liu K. IFNAR1 controls autocrine type I IFN regulation of PD-L1 expression in myeloid-derived suppressor cells. J. Immunol. 2018;201:264–277. doi: 10.4049/jimmunol.1800129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morimoto Y., Kishida T., Kotani S.I., Takayama K., Mazda O. Interferon-β signal may up-regulate PD-L1 expression through IRF9-dependent and independent pathways in lung cancer cells. Biochem. Biophys. Res. Commun. 2018;507:330–336. doi: 10.1016/j.bbrc.2018.11.035. [DOI] [PubMed] [Google Scholar]
- 16.Bazhin A.V., von Ahn K., Fritz J., Werner J., Karakhanova S. Interferon-α Up-regulates the expression of PD-L1 molecules on immune cells through STAT3 and p38 signaling. Front. Immunol. 2018;9:2129. doi: 10.3389/fimmu.2018.02129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacquelot N., Yamazaki T., Roberti M.P., Duong C.P.M., Andrews M.C., Verlingue L., Ferrere G., Becharef S., Vétizou M., Daillère R., et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019;29:846–861. doi: 10.1038/s41422-019-0224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benci J.L., Xu B., Qiu Y., Maity A., Wherry E.J., Minn Correspondence A.J., Wu T.J., Dada H., Twyman-Saint Victor C., Cucolo L., et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade article tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167:1540–1554.e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh S.R., Bastin D., Chen L., Nguyen A., Storbeck C.J., Lefebvre C., Stojdl D., Bramson J., Bell J.C., Wan Y. Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J. Clin. Invest. 2019;129:518–530. doi: 10.1172/JCI121004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebrahimi S., Ghorbani E., Khazaei M., Avan A., Ryzhikov M., Azadmanesh K., Hassanian S.M. Interferon-mediated tumor resistance to oncolytic virotherapy. J. Cell Biochem. 2017;118:1994–1999. doi: 10.1002/jcb.25917. [DOI] [PubMed] [Google Scholar]
- 21.Selman M., Ou P., Rousso C., Bergeron A., Krishnan R., Pikor L., Chen A., Keller B.A., Ilkow C., Bell J.C., et al. Dimethyl fumarate potentiates oncolytic virotherapy through NF-B inhibition. Sci. Transl. Med. 2018;10:eaao1613. doi: 10.1126/scitranslmed.aao1613. [DOI] [PubMed] [Google Scholar]
- 22.Dornan M.H., Krishnan R., Macklin A.M., Selman M., el Sayes N., Son H.H., Davis C., Chen A., Keillor K., Le P.J., et al. First-in-class small molecule potentiators of cancer virotherapy. Sci. Rep. 2016;6:26786. doi: 10.1038/srep26786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleinovink J.W., Marijt K.A., Schoonderwoerd M.J.A., van Hall T., Ossendorp F., Fransen M.F. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology. 2017;6:e1294299. doi: 10.1080/2162402X.2017.1294299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noyce R.S., Collins S.E., Mossman K.L. Identification of a novel pathway essential for the immediate-early, interferon-independent antiviral response to enveloped virions. J. Virol. 2006;80:226–235. doi: 10.1128/JVI.80.1.226-235.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coli M.L., Hill J.L.E., Marroqui L., Chaffey J., Santos R.S.D., Leete P., Paula F.M.M., Beeck A.O.de., Castela A., Marselli L., et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine. 2018;36:367–375. doi: 10.1016/j.ebiom.2018.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian J., Wang C., Wang B., Yang J., Luo F., Xu J., Zhao C., Liu R., Chu Y. The IFN-γ/PD-L1 axis between T cells and tumor microenvironment: hints for glioma anti-PD-1/PD-L1 therapy. J. neuroinflammation. 2018;15:290. doi: 10.1186/s12974-018-1330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farrukh H., El-Sayes N., Mossman K. Mechanisms of PD-L1 regulation in malignant and virus-infected cells. Int. J. Mol. Sci. 2021;22:4893. doi: 10.3390/ijms22094893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loke P., Allison J.P. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. U S A. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S.J., Jang B.C., Lee S.W., Yang Y., Suh S., Park Y.M., Oh S., Shin J.G., Yao S., Chen L., et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274) FEBS Lett. 2006;580:755–762. doi: 10.1016/j.febslet.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 30.Wang X., Yang L., Huang F., Zhang Q., Liu S., Ma L., You Z. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 2017;184:7–14. doi: 10.1016/j.imlet.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamano J.B., Lamano J.B., Li Y.D., DiDomenico J.D., Choy W., Veliceasa D., Oyon D.E., Fakurnejad S., Ampie L., Kesavabhotla K., et al. Glioblastoma-Derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin. Cancer Res. 2019;25:3643–3657. doi: 10.1158/1078-0432.CCR-18-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W., Liu Y., Yan Z., Yang H., Sun W., Yao Y., Chen Y., Jiang R. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J. Immunother. Cancer. 2020;8:e000285. doi: 10.1136/jitc-2019-000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan H., Viswanadhapalli S., Chupp D., Fernandez M., Wu S., Wang J., et al. B cells produce IL-27 to upregulate PD-L1 expression and promote breast cancer development. J. Immunol. 2019;202:195–196. [Google Scholar]
- 34.Diskin B., Adam S., Cassini M.F., Sanchez G., Liria M., Aykut B., Buttar C., Li E., Sundberg B., Salas R.D., et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat. Immunol. 2020;21:442–454. doi: 10.1038/s41590-020-0620-x. [DOI] [PubMed] [Google Scholar]
- 35.Fuertes M.B., Kacha A.K., Kline J., Woo S.-R., Kranz D.M., Murphy K.M., Gajewski T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huber J.P., Farrar J.D. Regulation of effector and memory T-cell functions by type I interferon. Immunology. 2011;132:466. doi: 10.1111/j.1365-2567.2011.03412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pauken K.E., Wherry E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woods D.M., Sodré A.L., Villagra A., Sarnaik A., Sotomayor E.M., Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015;3:1375–1385. doi: 10.1158/2326-6066.CIR-15-0077-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Workenhe S.T., Nguyen A., Bakhshinyan D., Wei J., Hare D.N., MacNeill K.L., Wan Y., Oberst A., Bramson J.L., Nasir J.A., et al. De novo necroptosis creates an inflammatory environment mediating tumor susceptibility to immune checkpoint inhibitors. Commun. Biol. 2020;3:645. doi: 10.1038/s42003-020-01362-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walsh S.R., Bastin D., Chen L., Nguyen A., Storbeck C.J., Lefebvre C., Stojdl D., Bramson J.L., Bell J.C., Wan Y. Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J. Clin. Invest. 2019;129:518–530. doi: 10.1172/JCI121004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benci J.L., Xu B., Qiu Y., Wu T.J., Dada H., Twyman-Saint Victor C., Cucolo L., Lee D.S.M., Pauken K.E., Huang A.C., et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167:1540–1554.e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lau J., Cheung J., Navvaro A., Lianoglou S., Haley B., Totpal K., Sanders L., Koeppen H., Caplazi P., McBride J., et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat. Commun. 2017;8:14572. doi: 10.1038/ncomms14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noguchi T., Ward J.P., Gubin M.M., Arthur C.D., Lee S.H., Hundal J., Selby M.J., Graziano R.F., Mardis E.R., Korman A.J., et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol. Res. 2017;5:106–117. doi: 10.1158/2326-6066.CIR-16-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y., Harrison D.L., Song Y., Ji J., Huang J., Hui E. Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Rep. 2018;24:379–390.e6. doi: 10.1016/j.celrep.2018.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng Q., Qiu X., Zhang Z., Zhang S., Zhang Y., Liang Y., Guo J., Peng H., Chen M., Fu Y.-X., et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020;11:4835. doi: 10.1038/s41467-020-18570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang Y., Tang H., Guo J., Qiu X., Yang Z., Ren Z., Sun Z., Bian Y., Xu L., Xu H., et al. Targeting IFNα to tumor by anti-PD-L1 creates feedforward antitumor responses to overcome checkpoint blockade resistance. Nat. Commun. 2018;9:4586. doi: 10.1038/s41467-018-06890-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen C.-Y., Hutzen B., Wedekind M.F., Cripe T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virotherapy. 2018;7:65–77. doi: 10.2147/OV.S145532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zamarin D., Ricca J.M., Sadekova S., Oseledchyk A., Yu Y., Bluemenschein W.M., Wong J., Gigoux M., Merghoub T., Wolchok J.D. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J. Clin. Invest. 2018;128:1413–1428. doi: 10.1172/JCI98047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Z., Ravindranathan R., Kalinski P., Guo Z.S., Bartlett D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017;8:14754. doi: 10.1038/ncomms14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bourgeois-Daigneault M.-C., Roy D.G., Aitken A.S., el Sayes N., Martin N.T., Varette O., Falls T., St-Germain L.E., Pelin A., Lichty B.D., et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018;10:eaao1641. doi: 10.1126/scitranslmed.aao1641. [DOI] [PubMed] [Google Scholar]
- 51.Lou J., Dong J., Xu R., Zeng H., Fang L., Wu Y., Liu Y., Wang S. Remodeling of the tumor microenvironment using an engineered oncolytic vaccinia virus improves PD-L1 inhibition outcomes. Biosci. Rep. 2021;41 doi: 10.1042/BSR20204186. BSR20204186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z., Ravindranathan R., Kalinski P., Guo Z.S., Bartlett D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017;8:14754. doi: 10.1038/ncomms14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prévost-Blondel A., Zimmermann C., Stemmer C., Kulmburg P., Rosenthal F.M., Pircher H. Tumor-infiltrating lymphocytes exhibiting high ex vivo cytolytic activity fail to prevent murine melanoma tumor growth in vivo. J. Immunol. 1998;161:2187–2194. [PubMed] [Google Scholar]
- 54.Stojdl D.F., Lichty B.D., tenOever B.R., Paterson J.M., Power A.T., Knowles S., Marius R., Reynard J., Poliquin L., Atkins H., et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 55.Zhang L., Bridle B.W., Chen L., Pol J., Spaner D., Boudreau J.E., Rosen A., Bassett J.D., Lichty B.D., Bramson J.L., et al. Delivery of viral-vectored vaccines by B cells represents a novel strategy to accelerate CD8(+) T-cell recall responses. Blood. 2013;121:2432–2439. doi: 10.1182/blood-2012-06-438481. [DOI] [PubMed] [Google Scholar]
- 56.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mekhael O., Naiel S., Vierhout M., Hayat A.I., Revil S.D., Abed S., Inman M.D., Kolb M.R.J., Ask K. Mouse models of lung fibrosis. Methods Mol. Biol. 2021;2299:291–321. doi: 10.1007/978-1-0716-1382-5_21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.