

Abstract
Redox biology is at the core of life sciences, accompanied by the close correlation of redox processes with biological activities. Redox homeostasis is a prerequisite for human health, in which the physiological levels of nonradical reactive oxygen species (ROS) function as the primary second messengers to modulate physiological redox signaling by orchestrating multiple redox sensors. However, excessive ROS accumulation, termed oxidative stress (OS), leads to biomolecule damage and subsequent occurrence of various diseases such as type 2 diabetes, atherosclerosis, and cancer. Herein, starting with the evolution of redox biology, we reveal the roles of ROS as multifaceted physiological modulators to mediate redox signaling and sustain redox homeostasis. In addition, we also emphasize the detailed OS mechanisms involved in the initiation and development of several important diseases. ROS as a double‐edged sword in disease progression suggest two different therapeutic strategies to treat redox‐relevant diseases, in which targeting ROS sources and redox‐related effectors to manipulate redox homeostasis will largely promote precision medicine. Therefore, a comprehensive understanding of the redox signaling networks under physiological and pathological conditions will facilitate the development of redox medicine and benefit patients with redox‐relevant diseases.
Keywords: hydrogen peroxide, oxidative stress, reactive oxygen species, redox‐relevant disease, redox signaling, redox therapy
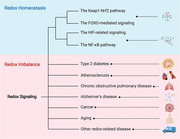
Redox homeostasis is essential for human health, supported by multiple functional signaling pathways including the Keap1‐Nrf2, FOXO, HIF and NF‐κB pathways. However, redox imbalance causes multiple serious diseases such as type 2 diabetes, atherosclerosis, chronic obstructive pulmonary disease, Alzheimer's disease, cancer, and aging. Our review emphasizes the significance of redox manipulation in clinical therapeutics and points out the existing challenges involved in this field.
1. INTRODUCTION
Electron flow is one of the most fundamental and common perspectives in understanding biology. 1 As Nobel prize‐winning biochemist Albert Szent‐Györgyi said, “Life is nothing but an electron looking for a place to rest.” Although life seems to be much more complex than electrons, the electron transfer process, which refers to the transformation of energy from an excited state (high‐energy electrons) to a ground state (low‐energy resting electrons) is fundamental. The rules of quantum mechanics restrain the electron transfer process whereby high‐energy electrons drop spontaneously to low‐energy states. Instead, they need to be offered feasible pathways, which tightly regulate the rearrangement of electrons along the energy scale. These electron transfer and transformation of energy processes make life possible. 1
The delivery of electrons from the donor molecules to the terminal acceptor molecules triggers oxidative (for donor molecules) and reductive (for acceptor molecules) processes, defined as redox reactions. Redox reactions are the sources of intracellular energy, harvested from electron flow during the transfer from one reductant (oxidation) to another oxidant (reduction). High‐energy electrons must move to a new resting acceptor, usually molecular oxygen for aerobes, ultimately yielding H2O. For humans, intracellular enzymes reduce oxygen, which enters the cell and captures energy to accelerate ATP production. However, in some specific conditions (i.e., redox control), the reduction of oxygen is blocked at an intermediate state, such as hydrogen peroxide (H2O2), which functions as an active oxidant to modulate multiple physiological and pathological redox signaling pathways on the basis of the level of oxidants. 2 , 3
The delivery of electrons on the basis of energy gradients prompts the rearrangements of chemical bonds and induces cellular responses at all levels of regulation, triggering redox chemistry and redox biology simultaneously. However, how does electron flow arouse resting cells and instruct them to determine cell fate, from proliferation and differentiation to death? Many essential intermediate proteins are involved between electron flow and cellular responses, whose activities are dynamically modulated by the electron transfer process through disulfide bridges. These functional proteins act as the main executors in regulating cellular biological processes and making cell fate decisions. In the broadest sense, identifying the coordination and crosstalk between electron gradients and cellular responses is the main purpose of redox biology. Nevertheless, understanding the redox modification of these functional intermediate proteins seems to be equally important due to the close correlation of multiple diseases with aberrant redox modulation, which will largely promote the development of redox medicine. Hence, redox reactions are intimately linked to human health, in which the concept of redox homeostasis is emphasized. Constant intracellular surveillance is vital for maintaining redox homeostasis, termed “homeodynamics” due to its dynamic property. 4
Recent studies on redox biology have indicated a relatively complete redox architecture that is closely linked to physiological function, 5 with a set of intricate mechanisms denoted as the “redox code.” 6 H2O2 as a key second messenger is central to the redox code, contributing to cell fate decisions. 6 In addition, the redox modifications of cysteines, such as intra‐ or intermolecular disulfide bond formation, S‐sulfonation, S‐glutathionylation, and S‐nitrosylation, have also been reported to reset the function of proteins, thus, mediating the process of various cellular biological events.
Redox imbalance between oxidants and antioxidants, especially for oxidative stress (OS), accounts for many diseases including neurological disorders, immune system disorders, cardiovascular diseases, and skeletal diseases. 7 Nonetheless, not all redox‐related disorders are caused by excessive reactive oxygen species (ROS) production. Several pathological conditions may also result from other reactive species, such as reactive nitrogen species and reactive sulfur species, or other small signaling molecules, such as H2, NH3, and CO, which are also capable of engendering redox imbalance.
In this review, we look back at the evolution of redox biology and summarize the cellular redox landscape with a particular focus on the core redox signaling metabolite–H2O2. 2 , 8 , 9 Considering the central role redox plays in determining cell fates and overall organization of living organisms, we also discuss the impacts of redox imbalance on several human diseases with an emphasis on the enormous potential of developing redox medicine. It is hoped that this systematic perspective will facilitate a better understanding of redox biology and attract more interest in the field.
2. AN OVERVIEW OF REDOX BIOLOGY FROM A HISTORICAL PERSPECTIVE
Oxygen originates from the photosynthetic activity of cyanobacteria which first entered the earth's atmosphere approximately 2.3 billion years ago. 10 , 11 The dramatic increase of O2 killed most anaerobic living creatures present at that period, with only a few aerobic life forms emerging that gradually occupied a major part in all organisms. The existence of enough O2 and its significance in producing energy for aerobes prompted the coining of the term “redox biology.”
Flohe and de Villiers et al. have reviewed several important events at the early stages of redox biology, 11 , 12 which we have addressed and added other representative findings along this timeline (Figure 1). Generally, oxygen is highly active and can be transformed into multiple ROS, such as H2O2, singlet molecular oxygen(1O2), ozone (O3), superoxide anion radicals (O2 −•), and hydroxyl radicals (OH . ), by endogenous and exogenous factors. 13 , 14 Among the major members of ROS family, H2O2 is the most frequently explored and first discovered in redox chemistry by Louis Jaques Thénard in 1818, 12 though the underlying roles in redox biology were not defined until 1954. 15 In 1900, catalase, which may be the first discovered antioxidant, was defined as a catalyst of H2O2. 16 Intriguingly, selenium was first found and identified as a toxic chemical catalyst in 1818 and was later proven to be an essential component of the glutathione peroxidase (GPX) family. 12 GPX was claimed to be a novel peroxidase that was independent of typical heme peroxidase in 1957, which was initially widely debated, but has gradually been accepted as GPX1 in the GPX family. 17 In addition to the GPX family, thioredoxin reductases (TrxR), as catalysts of Trx, are well‐studied selenoproteins that are classified as important oxidoreductases. Trx and peroxiredoxin (Prx) were first discovered in 1964 and 1968, respectively. 18 , 19 They function as two essential antioxidant systems that determine cell fates and pathophysiological changes in response to various stresses. 20 , 21 , 22 , 23 , 24 , 25
FIGURE 1.
Key events in the history of redox biology
Intracellular H2O2 is endogenously derived from NADPH oxidase (NOX), mitochondrial oxidative phosphorylation (mtOXPHOS ) (i.e., the electron transport chain [ETC] of mitochondria), and other cell compartments, including peroxisomes and the endoplasmic reticulum (ER), 2 , 26 or exogenously arises from stress such as ultraviolet radiation, ionizing radiation, and toxic compounds. Early in 1961, Iyer et al. found that H2O2 was released from phagocytosis of guinea pig polymorphonuclear leukocytes. 27 Later, in 1964, Rossi and colleagues confirmed that NOX was the upstream event in H2O2 production, answering how phagocytes generated H2O2 by respiratory burst. 28 However, NOX was first disclosed to generate superoxide radicals and H2O2 by Sbarra and Karnowski in 1959. 29 In 1973, a study by Babior et al. confirmed that in the process of respiratory burst, O2 −• but not H2O2 was the direct product, meaning that H2O2 originated from O2 −• during metabolic activity. 30 In fact, before this finding, in 1969 McCord and Fridovich reported that O2 −• can be converted into two new forms–oxygen and H2O2 by bovine erythrocyte‐derived superoxide dismutase (SOD) 31. ETC of mitochondria was first proven to be the source of H2O2 in 1967, 32 and in 1974, Loschen et al. similarly elucidated that O2 −• was also the precursor of H2O2 in mitochondria. 33
For a long time, ROS were identified as toxic byproducts of redox reactions, damaging biomacromolecules (e.g., DNA, 34 , 35 , 36 RNA, 37 , 38 proteins, 39 and lipids 40 , 41 , 42 ) and causing cell death or malignant transformation. In 1954 and 1956, Gershman and Harman described that oxidant burden was closely correlated with tissue injury and aging, which greatly promoted the theory of ROS as poisonous byproducts. 43 , 44 However, when in‐depth studies were carried out, the roles of ROS in not only damaging but also regulating physiological signaling pathways depending on ROS levels were defined. 2 , 7 Thus, for H2O2, under the tight control of cellular physiological signaling and antioxidant systems, the concentration of intracellular H2O2 is maintained at a physiological level (ranging from 1–100 nM) and modulates cell proliferation, differentiation and death, whereas a supraphysiological H2O2 level (above 100 nM) destroys biomacromolecules that control cell fates. In between these levels, there is a small window of the adjustable interval, where intracellular adaptive antioxidant systems are activated through Kelch‐like ECH‐associated protein 1‐NF‐E2‐related factor 2 (Nrf2/Keap1) and nuclear factor kappa B (NF‐κB), which partially reverse the detrimental impacts. Nrf2, which can induce the expression of phase II detoxifying enzyme genes through antioxidant response elements (AREs), was defined as a master regulator of antioxidant systems in 1997. 45 A typical example of ROS acting as an essential physiological signaling agent is reflected in immune defense, 13 in which intracellular H2O2 triggers the migration of leukocytes to lesions through several possible mechanisms. 46 , 47 , 48 , 49
In summary, research in the last century has found multiple pivotal redox regulators and disclosed their roles in redox modulation. In addition, the main sources of ROS were also partially illustrated. Additionally, ROS was found to have more functions including physiological and pathological significance. Currently, the role of redox biology is continually being extended. For instance, researchers have found more NOX subunits in recent years. 50 , 51
3. REDOX SIGNALING NETWORKS IN CELLULAR HOMEOSTASIS
3.1. H2O2 as the main messenger in redox signaling
The most physiologically relevant ROS include O2 −• and H2O2, which are generally derived from NOX and ETC. Cellular compartmentalization of H2O2 facilitates new levels of regulation and confines certain signaling pathways to specific compartments. 52 Therefore, H2O2 is considered as one of the main types of ROS involved in redox regulation of intracellular biological processes, 2 , 53 the generation of which is tightly controlled through various growth factors, chemokines, and physical stress. 26 Intracellular physiological levels of H2O2 (1–100 nM) act as major activators to modulate specific targets through reversible oxidative modification and further influence the activity, localization, and interactions of protein targets and cellular behaviors in response to moderate environmental stresses. The regulation of biological activities under physiological levels of H2O2 is also called “oxidative eustress.” However, supraphysiological intracellular H2O2 levels (above 100 nM) cause cell and tissue dysfunction, attributed to indiscriminate attacks on biomacromolecules. Excessive and irreversible oxidation directly leads to cell senescence, death, and even malignant transformation (“oxidative distress” compared to “oxidative eustress”), a term defining redox imbalance in favor of oxidant burden. In the next section, we mainly focus on H2O2 functioning as a physiological modulator in the redox regulation of four transcription factors (TFs)—Keap1‐Nrf2, forkhead box class O (FOXO), hypoxia‐inducible factors (HIFs), and NF‐κB (Figure 2).
FIGURE 2.
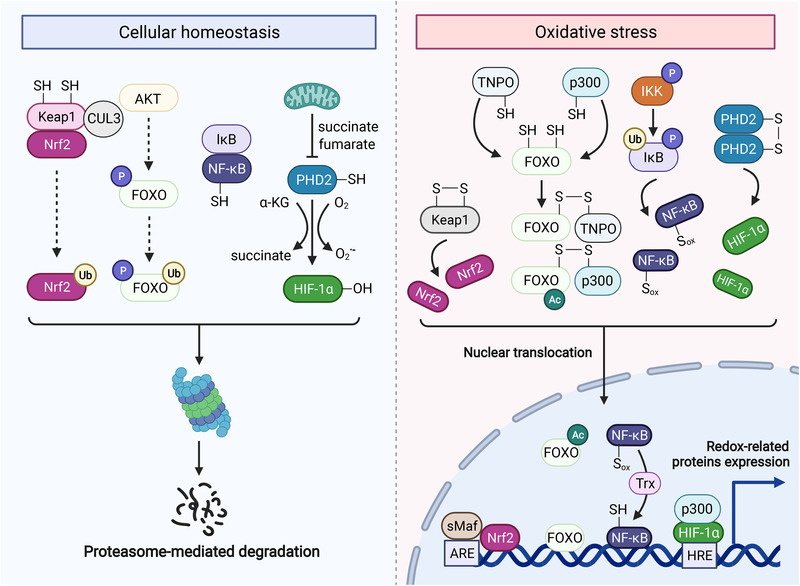
Redox regulation of Keap1‐Nrf2, FOXO, NF‐κB, and HIF. Keap1‐Nrf2: Keap1 binds to Nrf2 and CUL3, inducing the proteasomal degradation of Nrf2. Keap1 as a redox sensor is tightly regulated by H2O2, which helps to form an intramolecular disulfide and releases NRF2. FOXO: Under moderate levels of H2O2, FOXO engages proteasomal degradation through AKT signaling. However, pathological levels of H2O2 facilitate the formation of an intermolecular disulfide between TNPO and FOXO, promoting the nuclear translocation of FOXO. Intriguingly, FOXO can also form an intermolecular disulfide with acetyltransferase p300, enhancing the activity of FOXO. NF‐κB: Normally, NF‐κB remains inactive in the cytoplasm by interacting with IκB that can be phosphorylated and inhibited by IKK. Under OS, H2O2‐mediated IKK activation drives the proteasomal degradation of IκB and subsequent nuclear translocation of NF‐κB. Notably, NF‐κB is also oxidized by H2O2, which needs to be reversed by Trx in the nucleus. HIF: The redox sensor PHD2 inactivates HIF‐1α via hydroxylation, which can be prevented by forming intermolecular disulfide between PHD2 under H2O2 regulation. As a result, HIF‐1α enters the nucleus and prompts the transcription of target genes
3.2. ROS scavenging: The Keap1‐Nrf2 pathway
The Keap1‐Nrf2 pathway is a thiol‐based redox switch that responds to various oxidant stresses and, thus, plays an essential role in maintaining redox homeostasis in eukaryotes. 54 , 55 Keap1 functions as a redox sensor, inhibiting Nrf2 activity by targeting Nrf2 for ubiquitination and consequent degradation under quiescent circumstances. But under oxidative insults, Nrf2 is released from Keap1 and acts as a master TF to induce the expression of proteins involved in the cellular antioxidant responses. 56 , 57 , 58
Nrf2, along with Nrf1, Nrf3, and p45 NF‐E2, is a member of the Cap “n” Collar family. 59 , 60 There are seven functional domains (Neh1‐7) that regulate the stability and transcriptional activity of Nrf2. 61 The N‐terminal Neh2 domain is involved in the Keap1‐Nrf2 interaction, 62 as well as Nrf2 stability and ubiquitination. 63 The Neh4 and Neh5 domains are transcription activation domains that interact with the CREB binding protein (CBP) to promote Nrf2 transcription, 64 leading to enhanced expression of Nrf2‐targeted ARE genes. It has been reported that retinoid X receptor alpha (RXR‐α) suppresses the function of Nrf2 by interacting with the Neh7 domain (also known as the RXR‐α interaction domain). 65 The Neh6 domain regulates Nrf2 degradation in the absence of Keap1. 66 The Neh1 domain enables Nrf2 to connect to the ARE sequence and activate transcription through its basic leucine zipper motif. 67 Additionally, the Neh1 domain is important for the stability of the Nrf2 protein by interacting with the ubiquitin‐conjugating enzyme UbcM2. 68 Following its release from Keap1, the Neh1 domain mediates the nuclear translocation of Nrf2. 69 The C‐terminal Neh3 domain is required for interaction with the chromo‐ATPase/helicase DNA‐binding protein family member CHD6, 70 which is required for the transcription of ARE‐dependent genes.
Keap1, a repressor of Nrf2, is composed of four domains: broad complex‐Tramtrack‐Bric‐a‐brac (BTB), intervening region (IVR), double‐glycine repeat (DGR; also known as Kelch domain), and C‐terminal region (CTR). 71 The BTB domain has been reported to be associated with dimer formation. 72 The IVR domain contains a nuclear export sequence (NES), which is important for the cytoplasmic localization and reactivity of Keap1 in response to oxidative stimuli. This is mediated by two reactive cysteine residues in the IVR: cysteine 273 (C273) and cysteine 288 (C288). 71 , 73 The two DGR/Kelch domains recruit Neh2 by differentially binding to the ETGE and DLG motifs of the Nrf2 molecule. 74 , 75 When oxidants are present, the DLG motif is freed from Keap1, preventing Nrf2 ubiquitination and subsequent destruction. 76 , 77 The interaction between Nrf2 and the DGR domain in Keap1 is competitively inhibited by proteins with specific motifs such as p62 and partner and localizer of BRCA2 (PALB2). 78 The CTR of Keap1 has also been reported to be essential for interacting with Nrf2 and subsequent suppression. 79 , 80
Functioned as a physiological thiol‐based sensor–effector mechanism, the Keap1‐Nrf2 system plays a central role in responding to oxidant stress and maintaining cellular redox homeostasis. 7 , 81 Normally, Nrf2 binds to the E3 ligase Keap1, continually targeting Nrf2 for proteasomal destruction through Cul3‐mediated ubiquitination. 82 , 83 Nrf2 disassociates from the Keap1‐Nrf2 complex and translocates to the nucleus under OS circumstances. More than 500 antioxidant genes involved in redox balance and stress response are induced by Nrf2. 84 , 85 Keap1 harbors several cysteine sensors that can sense Nrf2‐activating chemicals, with oxidation of critical cysteine residues and induction of conformational changes in Keap1. 86 , 87 As a result, the Nrf2‐Keap1 interaction is partially disrupted, preventing Nrf2 ubiquitination and subsequent destruction, as mentioned above. However, a study has also shown that, in some cases, Nrf2 may not be released from Keap1. 88 Additionally, it was found that de novo synthesis of Nrf2 occurs extremely rapid in response to low concentrations of H2O2 at a pace that surpasses the rate of Nrf2 nuclear translocation. 88 This may be another indication that cells may have other powerful, but as yet unidentified, redox sensors associated with Nrf2 activation in addition to Keap1.
3.3. ROS scavenging: The FOXOs‐mediated signaling
FOXO TFs are critical regulators of the physiological stress response and have been reported to be activated in response to OS. 89 , 90 , 91 The term “fork head” was first described in Drosophila as a potential transcriptional regulator, 92 and it was discovered to contain a so‐called winged‐helix DNA binding domain that exists in other transcriptional regulators. 93 , 94 , 95 Members of the FOXO family (or FOXO orthologues) have been found in a variety of species. For instance, FOXO is also known as daf‐16 in worms 96 and dFOXO in flies. 97 FOXO1a, FOXO3a, FOXO4, and FOXO6 are the four FOXO proteins found in humans and are widely expressed in a range of organs. 91 , 98 FOXOs have a conserved nuclear localization signal domain, a NES domain, a DNA‐binding domain, and a C‐terminal transactivation domain (TAD) that regulate their transactivation activities. 99 The activity of FOXO proteins is mainly controlled by post‐transcriptional modifications and cytoplasmic or nuclear distribution. AKT functions as a primary regulator of FOXO phosphorylation through IKK and IκB kinases. The phosphorylated FOXOs bind to the 14‐3‐3 protein, resulting in the ubiquitination and degradation of FOXO proteins in the cytoplasm. 100 Under OS, arginine methyltransferase 1 (PRMT1) can bind to and methylate FOXOs, which blocks AKT‐mediated phosphorylation of FOXOs, and promotes subsequent nuclear translocation. 101
Various cysteine residues have been identified in FOXO proteins: FOXO1 has seven cysteine residues, 102 FOXO3a and FOXO4 have five, 103 and FOXO6 has ten cysteine residues. 89 Oxidative modifications on these cysteines and consequent contribution to redox signaling play crucial roles in the regulation of OS. 104 , 105 , 106 The oxidation of cysteines in FOXOs participates in stabilizing protein–protein interactions by promoting the formation of disulfide bridges with transportin (TNPO) in an oxidative state, which is required for the activation of FOXOs. 107 , 108 Furthermore, the FOXO target genes that encode antioxidant proteins are involved in suppressing the production of oxygen. For instance, Mn‐SOD (SOD2) is a FOXO‐regulated antioxidant that catalyzes the dismutation of superoxide to oxygen and H2O2 109. H2O2 is further dismutated into water and oxygen by catalase, which is regulated by FOXO3a. 110 In addition, FOXO3a has been demonstrated to alter the expression of mitochondrial TrxR2 and Trx2, which might contribute to the reduction of mitochondrial Prx3. 111 Taken together, this evidence demonstrates that FOXOs function as regulators in response to OS and maintain redox homeostasis.
3.4. ROS generation: The hypoxia‐inducible factor‐related signaling
HIF is a TF that binds to particular nuclear cofactors and transactivates a wide range of genes in response to low oxygen levels. 112 , 113 HIF is a heterodimer made up of two subunits: an oxygen‐labile subunit (HIF‐α, including HIF‐1α, HIF‐2α, and HIF‐3α) and a common stable β‐subunit (HIF‐1β/ARNT). 114 , 115 Both subunits are members of the basic helix‐loop‐helix (bHLH‐PAS) TF family. 116 There are three genes that encode different HIF‐α isoforms: HIF1A, which encodes HIF‐1α; EPAS1, which encodes HIF‐2α; and HIF3A, which is alternatively spliced to generate various HIF‐3α variants in humans. 117 , 118 The N‐terminal TAD and oxygen‐dependent degradation domain (ODD) exist in HIF‐1α, HIF‐2α, and variants 1–3 of HIF‐3α, while HIF‐1α and HIF‐2α have an additional C‐terminal TAD. 119
HIF interacts with and binds to the von Hippel‐Lindau (VHL) protein, activating the ubiquitin ligase system and causing the degradation of HIF by the proteasome under normoxic conditions. 120 , 121 Hydroxylation of proline residues in HIFs is required for VHL binding and is mediated by HIF prolyl hydroxylases (also known as prolyl hydroxylase domain, PHD), ketoglutarate‐dependent dioxygenases, and asparaginyl hydroxylase. 122 , 123 , 124 The oxygen sensor proteins PHD and factor inhibiting HIF‐1 become inactive during hypoxia, which causes stabilization of HIF‐α and subsequent dimerization with HIF‐1β and coactivator P300. 125 HIF dimerizes in the nucleus and binds to E‐box‐like hypoxia response elements, leading to the activation of genes involved in cellular oxygen homeostasis. 126 Hypoxic cells respond to stress via transcriptional and post‐transcriptional processes, which are primarily controlled by HIF. 127 , 128 These molecular modifications enable cells to respond to stress by decreasing oxygen consumption. It has been reported that hypoxia is associated with the production of H2O2 as a result of mitochondrial ETC inhibition. 129 , 130 Chronic intermittent hypoxia, a potentially fatal condition that occurs in several breathing disorders, has also been shown to activate redox signaling, resulting in a variety of systemic and cellular responses. 131 , 132 , 133
3.5. ROS generation: The NF‐κB pathway
The NF‐κB family comprises five TFs, including p65/RELA, RELB, c‐REL, p50 (its progenitor p105), and p52 (its precursor p100), 134 which play essential roles in inflammation, 135 immunology, 136 cell proliferation, 137 and differentiation. 138 The mature proteins p65/RELA, RELB, and c‐REL possess a C‐terminal TAD within their respective C‐terminal portions. 139 The TAD area endows p65/RELA, RELB, and c‐REL with the potential to boost the initiation of gene transcription. 140 , 141 p105 and p100 lack TAD but possess a CTR containing ankyrin repeats that are cleaved post‐translationally to generate p50 and p52, respectively. 142 , 143
NF‐κB activity is tightly controlled due to its potential to regulate the expression of numerous genes. The NF‐κB pathway is primarily regulated by inhibitors of NF‐κB (IκB) and IκB kinase (known as IKK, a kinase that phosphorylates IκB). 144 , 145 NF‐κB is maintained inactive in the cytosol during physiological resting states by interacting with an inhibitor of IκB. 146 IKK phosphorylates IκB, resulting in the ubiquitination and degradation of IκB in response to numerous stimuli, including OS, after which NF‐κB is released and translocates into the nucleus to activate downstream target genes. 147 , 148
Since NF‐κB was discovered as a regulator of B‐cell development in 1986, 149 this family of TFs has been extensively investigated, and aberrant activation of NF‐κB has been found in a wide variety of human disease states. 136 , 150 , 151 In eukaryotic cells, NF‐κB was the first TF to be classified as a redox‐sensitive factor. Staal and colleagues demonstrated for the first time in 1990 that TNF (tumor necrosis factor)‐induced NF‐κB activation is reliant on the intracellular thiol redox state. 152 One year later in 1991, Schreck and colleagues demonstrated a favorable association between intracellular levels of H2O2 and NF‐κB activation. 153 Nonetheless, contradictory evidence about the association between H2O2 and NF‐κB activation has also been published. Currently, depending on the circumstances, H2O2 is regarded as either a stimulatory or inhibitory factor for NF‐κB activity. Cytosolic H2O2 may stimulate the NF‐κB pathway through oxidation and activation of IKK, which functions as a negative regulator of IκB stability. 60 , 154 Due to the oxidizable cysteines in NF‐κB, H2O2 may also directly influence NF‐κB. 155 Enhanced nuclear H2O2 accumulation hinders DNA binding, decreasing the transcriptional activity of NF‐κB. 156 Moreover, increased levels of nuclear Prx1 and Trx have also been reported to promote NF‐κB transcriptional activity. 157
Although H2O2 best fulfills the requirements of being a main messenger in redox regulation, other ROS have also been elucidated to function as physiological mediators. For example, O2 −• activates the ras/rac‐Raf1‐ MAPK and ERK signaling pathways, thus, serving as a growth signal in different cells. 158 Organic hydroperoxides (ROOH) were reported to act as an essential regulator in cell signaling and determining cell fates. 159 , 160 In addition, similar to H2O2, 1 O2 regulates cell fate in a location and dose‐dependent manner. A lower or physiological level of 1 O2 can mediate multiple signaling pathways and promote programmed cell death. 161 , 162 However, limited by the detection techniques, the underlying mechanisms of these ROS mediating cell signaling are still ambiguous. Further exploration on how diverse ROS regulate intracellular physiological signaling will extend our knowledge of redox biology and help in developing precise redox medicine.
4. REDOX IMBALANCE AND OXIDATIVE DAMAGE IN HUMAN DISEASE
Considering the significance of redox signaling in physiological conditions, redox imbalance occurring in different tissues, cells, or organelles may be intimately linked to multiple pathophysiological events. In this section, we focus on several common redox‐relevant diseases, highlighting how precision medicine is now possible by modulating aberrant ROS accumulation and leaving physiological signaling intact.
4.1. Type 2 diabetes
The increasing prevalence of diabetes and some related end‐stage organ damage has become a major cause of death and disability around the world. 163 , 164 Unfortunately, the underlying mechanisms are still not fully understood, which is a major obstacle in developing targetable antidiabetic therapies. Thus, a deeper understanding of the detailed mechanisms of the initiation and progression of diabetes is an unmet clinical need of great importance for public health.
Mounting evidence has indicated that redox imbalance plays an essential role in diabetes, in which ROS are found to widely influence insulin signaling (Figure 3). Superficially, markers of OS have been largely found at elevated levels in the body fluids of patients with type 2 diabetes, including plasma 8‐hydroxydeoxyguanosine (8‐OHdG, a marker of DNA oxidative damage), 165 oxidized low‐density lipoprotein (oxLDL) to LDL ratio, 166 GSH conjugation to haemoglobin, 167 8‐iso‐PGF2‐α, 168 protein carbonyls, 169 urine 8‐OHdG, and 8‐iso‐PGF2‐α. 170 However, to date small‐molecule antioxidant drugs have exhibited poor outcomes, suggesting that a different redox‐based perspective should be urgently sought. In recent years, important conceptual breakthroughs have attracted much interest, turning “general redox biology” into “precise redox biology” on the basis of increased knowledge of the disease‐relevant ROS sources and physiological roles of ROS. The increased ROS levels mainly arise from dysfunctional mitochondria 171 and NOX1, 172 which are accompanied by the common symptoms of diabetes—hyperglycemia and dyslipidemia. Therefore, more targetable therapeutic strategies are now available for exploitation.
FIGURE 3.
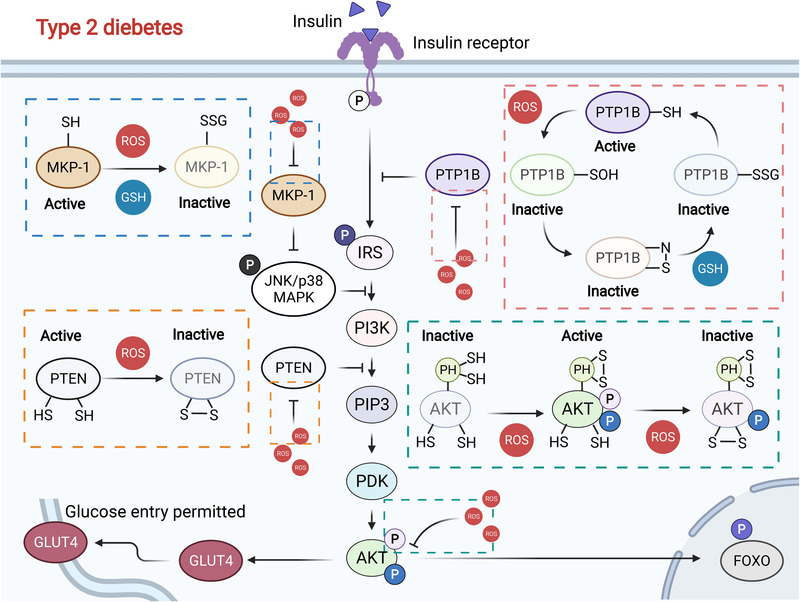
Redox regulation of the insulin signaling pathway. Normally, moderate ROS levels inactivate PTPB1 and PTEN and activate AKT, prompting glucose uptake through GLUT4. However, excessive ROS accumulation leads to glutathionylation modification and proteasomal degradation of MKP‐1, which prevents insulin signaling. Simultaneously, ROS burden also inactivates AKT, inhibiting GLUT4 and subsequent glucose uptake, which eventually causes type 2 diabetes
Multiple key regulators and targets of insulin receptor (IR) signaling have been revealed to be redox‐sensitive. Their aberrant forms are extensively found in type 2 diabetes, 173 , 174 including AKT, protein tyrosine phosphatase 1B (PTP1B), phosphatase and tensin homologue (PTEN), JUN amino‐terminal kinase (JNK), and FOXO. Insulin binds to the IR, phosphorylating IR substrate proteins (e.g., IRS1 and IRS2) and activating the phosphatidylinositol 3‐kinase (PI3K)‐AKT signaling pathway, which eventually promotes the translocation and activation of glucose transporter 4 (GLUT4) and glucose uptake. 175 , 176 PTP1B, a negative regulator of IR, dephosphorylates important tyrosine residues of IR and is tightly regulated by its in vivo redox status. 175 Physiologically, a low level of ROS was found to increase insulin sensitivity by modulating stress‐response kinases and further dephosphorylation and decreased activity of PTP1B and PTEN, which dephosphorylated IR and downregulated the PI3K signaling pathway, respectively. 175 , 176 Simultaneously, NOX4‐mediated ROS activated MAP kinase phosphatase‐1 (MKP‐1), which indirectly led to reduced IRS‐1 phosphorylation on serine residues, thereby increasing IRS‐1 tyrosine phosphorylation by attenuating ERK1/2 signaling. 177 Nevertheless, high levels of ROS potently promote insulin resistance and are associated with several abnormalities in type 2 diabetes including hyperglycemia, increased nonenzymatic glycosylation, inflammation, and activation of ETC production. 178
As a major insulin resistance mechanism, high ROS levels prompted the activation of JNK, which largely outweighed PTP1B inactivation. 179 ROS activated JNK and p38 by modulating their regulatory proteins. A representative example is that MKP‐1 inactivated JNK and p38 MAPK by dephosphorylation. However, the process can be manipulated by glutathionylation modification, which targeted MKP‐1 for proteasomal degradation. 180 The FOXO family of TFs has four main members—FOXO1, FOXO3, and FOXO4, which share similar properties, and FOXO6, which occupies a different expression form. FOXOs have several cysteine residues that are highly sensitive to redox stress and are conducive to producing antioxidants and relieving ROS overload. 181 However, hyperactivation of FOXOs significantly induces hypertriglyceridemia, hyperglycemia, and insulin resistance that eventually cause incurable diabetes. 182 , 183 There is a compensatory process of the insulin signaling pathway to target FOXOs. Phosphorylating and blocking them in the cytoplasm with ROS‐modulated PI3K‐AKT decreased DNA binding to its consensus response elements and increased nuclear exclusion. 174
4.2. Atherosclerosis
In pathological conditions of atherosclerosis, plaque appears in the intimal layer of arteries, which persistently accumulates and eventually causes stroke and infarction. 184 Redox disease—diabetes is an important inducer of the initiation of atherosclerosis, and accumulating evidence also reveals that oxidant burden plays an important role in atherosclerosis. 185
Similar to type 2 diabetes, multiple markers of OS were shown to have elevated levels in patients suffering from atherosclerosis. For instance, lipid hydroperoxides were first identified in human atherosclerotic aortae in 1952. 186 Following that, increasing research has shown that oxidized lipids and other oxidant markers aggregate in atherosclerotic lesions. In freshly isolated human atherosclerotic plaques, 20% of cholesteryl linoleate was oxidized, in contrast to undetectable levels in normal arteries. 187 OS was reported to function in the conversion of LDL into oxLDL, and modified LDL was considered a pivotal marker of the initiation and development of atherosclerosis. 188 For example, 4‐hydroxy‐2‐nonenal (HNE)‐modified LDL was elevated by 50% in the plasma of patients with atherosclerosis compared with healthy volunteers. 189 Malondialdehyde (MDA), the product of lipid peroxide that originates from prostanoid metabolism, is the other modification pattern of LDL. 188 Circulating MDA‐LDL levels have also been identified as a marker of ROS overload in atherosclerosis and are elucidated to be closely related to the prognosis of coronary artery disease. 190 , 191 , 192 In addition, isoprostanes, as peroxidation products of arachidonic acid, were proven to be fivefold higher in atherosclerotic lesions than in umbilical veins. 193 All of these oxidant markers confirm that atherosclerosis may also be a redox‐relevant disease.
How ROS contribute to the initiation and development of atherosclerosis is a vital question worthy of exploration. Multiple underlying mechanisms have been proposed on the basis of its sources—NOXs and mitochondria (Figure 4). 194 , 195 Among NOXs, NOX4 is abundantly found in the plagues of atherosclerosis, which was reported to correlate with atherosclerosis in 2002. 196 NOX4 is a double‐edged sword in atherosclerosis that can both relieve and exacerbate the disease. In mouse models of atherosclerosis, NOX4‐derived ROS were essential for vessel homeostasis. Two NOX4‐knockout models—NOX4–/–/Ldlr–/– and NOX4–/–/ApoE–/– mice, suffer remarkable endothelial damage and plaque burden. 197 , 198 In contrast, global NOX4 deletion can both protect against and aggravate diabetes‐induced atherosclerosis, depending on the time frame. 199 Evidence has shown that NOX4 deletion promoted diabetes‐induced plaque formation in the early stage, but in the progressive stage, NOX4 deletion helped to repress inflammation. 200 Besides, NOX4 can increase plaque burden with less T‐cell activation and infiltration in a 10‐week model of diabetes, but conversely, in the 20‐week model of diabetes, NOX4 can decrease collagen deposition and proliferation, blocking advanced lesions. 199 , 201 In addition to NOX4, other NOXs have also been found to be aberrantly upregulated in atherosclerosis. For example, NOX1 was considered an important risk factor in OS‐mediated inflammation and atherosclerosis. 172 NOX2 was found to have a particular function, which enabled the recruitment of macrophages through ROS‐vascular cell adhesion molecule‐1 signaling and the activation of endothelial cells. 202 Chen et al. revealed that NOX2 could also upregulate peroxisome proliferator‐activated receptor activity and CD36 expression by augmenting ROS levels, accelerating the formation of macrophage‐foam cells, and development of atherosclerosis. 203 Calcium‐dependent NOX5 was also a source of ROS, reflecting a marked increase and possible application potential in atherosclerosis. 204 Intriguingly, global inhibition of NOXs transcription via histone deacetylase (HDAC) blockade dramatically relieved OS and inflammation, suggesting that HDAC was upstream of NOXs and could become a good target for atherosclerosis. 205 Moreover, NOXs increased ROS levels, helping internalize oxLDL into macrophages through CD36 and generating foam cells, which may be a universal mechanism underlying NOX‐mediated progression of atherosclerosis judging from available reports. 203 , 206
FIGURE 4.
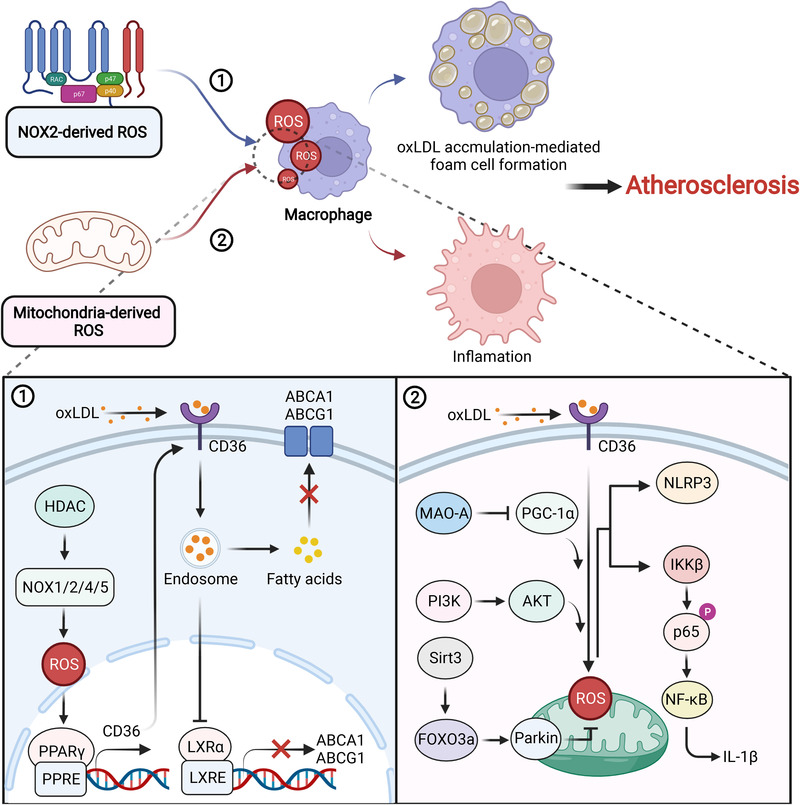
The roles of NOX and mitochondria‐derived ROS in atherosclerosis. NOX‐derived ROS promote CD36 expression, which facilitates the transportation of oxLDL and inhibition of ABCA1 and ABCG1 transcription. Under these conditions, fatty acids accumulate in the cytoplasm that engenders macrophage transforming into foam cells. On the other hand, mitochondria‐derived ROS elicit the production of NLRP3 and IL‐1β through multiple signaling pathways, accounting for an inflammatory phenotype in atherosclerosis pathogenesis. Both of them are important inducers of atherosclerosis
Mitochondrial ROS (mitoROS)‐mediated aberrant signaling has been largely elucidated to accelerate atherosclerosis and causes associated vascular complications, which are frequently accompanied by plaque accumulation. 207 , 208 , 209 Elimination or genetic inhibition of mitoROS prevented the development of atherosclerosis and associated complications. Melatonin was capable of efficiently scavenging mitoROS through the Sirtuin3 (Sirt3)/FOXO3a/Parkin signaling pathway, preventing the production of the nucleotide‐binding domain and leucine‐rich repeat pyrin domain containing 3 (NLRP3), thus, largely ameliorating atherosclerosis. 210 Besides, renal denervation, an agent that inactivates mitochondrial monoamine oxidase A and subsequently peroxisome‐proliferator‐activated receptor‐γ coactivator‐1α (PGC‐1α), was able to attenuate mitoROS‐mediated inflammation and atherosclerosis. 211 In wild‐type (WT) macrophages, LPS and oxLDL‐induced mitoROS remarkably strengthened IKK‐β and downstream p65 phosphorylation which could be inhibited by overexpressing mitoROS‐targeted catalase that efficiently repressed attenuated lesion progression and immune cell infiltration. 212 Another publication described PI3K/AKT was upstream of mitoROS, and blocking it may be an efficient method for treating atherosclerosis. 213 The ROS scavenging system comprises many regulatory proteins, among which the proteins of paraoxanase (PON) family were able to resist mitoROS in the development of atherosclerosis due to their capacity to hydrolyze lipid peroxides. 214 , 215 , 216 Overexpression of the PON1/2/3 cluster facilitated collagen synthesis, narrowed the necrotic core area, and decreased oxLDL and inflammatory markers, inhibiting mitochondrial dysfunction and stimulating plaque stability to alleviate atherosclerosis. 217 , 218 PON1 can also inhibit monocyte‐to‐macrophage differentiation and help macrophages resist oxLDL‐induced foam cell formation, 219 which prevented the formation of an inflammatory phenotype. 220 In contrast, PON1 deficiency was frequently observed in lesions of atherosclerosis, which resulted in vascular OS and leukocyte adhesion. Therefore, downregulation of PON expression may be a prerequisite for atherosclerosis. 221
4.3. Chronic obstructive pulmonary disease
Incurable chronic obstructive pulmonary disease (COPD) is characterized by progressive dyspnea and functional loss of the lung, and is a major public health issue. 222 COPD includes progressive chronic bronchitis and emphysema, often eliciting associated complications, including lung cancer, cardiovascular disease, skeletal muscle wasting, and osteoporosis. 223 , 224 , 225 , 226 Induction of COPD has many causes, including OS, inflammation, protease antiprotease imbalance, and apoptosis, 227 , 228 , 229 among which OS is probably the fundamental mechanism due to its central roles in other processes. Likewise, several oxidized products or markers of OS have been discovered in patients with COPD. Increasing ROS generation was found in the airways of patients with COPD, with superoxide and MDA being detected in the blood, sputum, airspaces, and lungs. 230 , 231 8‐Isoprostane was also consistently detectable in the exhaled breath condensate of COPD patients compared with healthy controls. 232 In addition, HNE was abundantly found in the airways and alveolar epithelial cells, endothelial cells, and neutrophils, with levels being elevated by at least 50% in patients with COPD. 233 Simultaneously, in urinary samples from COPD patients, the level of 8‐OHdG was also dramatically increased. 234 Furthermore, ROS levels were significantly elevated with COPD exacerbation, and the level of OS was inversely associated with the lung function of patients with COPD, hinting that oxidant burden may be an essential risk factor in the initiation and progression of the disease. 233 , 235
Sources of oxidants in patients with COPD originate from both endogenous and exogenous elements (Figure 5). For exogenous sources, cigarette smoke (CS), biomass smoke, and air pollution are the main causes of COPD, which can generate various ROS, including O2 −•, ONOO−, H2O2, and OH•. 222 , 236 Airway and pulmonary vascular remodeling are two characteristics of COPD. Zhu et al. disclosed that CS‐induced ROS‐activated calpain, which led to COPD exacerbation through the airway and pulmonary vascular remodeling. 237 OS‐mediated mitochondrial dysfunction was also a cause of airway remodeling and inflammation in COPD. 233 Additionally, ROS stimulated the overgrowth of airway smooth muscle through ASK1/JNK/P38 MAPK pathway. 238 ROS impaired the expression and function of CTFR, which was another contributor to CS in COPD. 239 Notably, CTFR dysfunction could be reversed by S‐nitrosoglutathione, which restored autophagy impairment and alleviated chronic inflammatory‐OS in CS‐induced COPD. 240 Consistently, insufficient mitophagy resulted in PTEN‐induced putative protein kinase 1 (PINK1) accumulation and parkin RBR E3 ubiquitin protein ligase reduction via PINK1‐mediated proteasomal degradation, which further accelerated insufficient mitophagy and ROS release, becoming a significant inducer of COPD pathogenesis. 241 Additionally, a similar study revealed that CS impaired PARK2 function and mitophagy, causing ROS‐mediated cell senescence. Conversely, severe CS exposure induced excessive mitophagy, contributing to apoptosis and necrosis of primary human bronchial epithelial cells. 242 Intriguingly, CS induced the accumulation of iron in mitochondria and cytosol, following which mitoROS and lipid peroxidation occurred that further engendered necrosis and ferroptosis. 243 Exposure to cigarette smoking evoked inflammatory responses and related chain reactions, such as cell death and fibrosis, which are important to the development of COPD. 244 Additionally, CS‐induced OS significantly reduced phagocytosis of macrophages from COPD, which was linked to enhanced inflammation. 245
FIGURE 5.
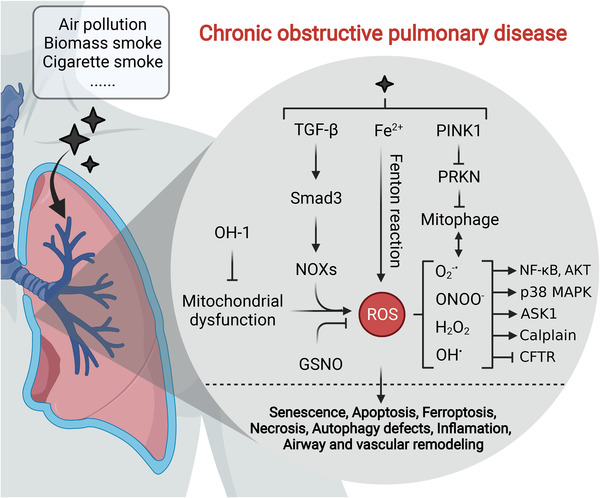
Exogenous and endogenous factor‐induced ROS mediate COPD pathogenesis. Exogenous sources (i.e., air pollution, biomass smoke, and CS) and endogenous sources (i.e., mitochondria and NOX) are important inducers of COPD, which accelerate various ROS accumulation in the lung through multiple pathways including TGF‐β signaling, Fenton reaction and mitochondrial dysfunction, stimulating cell senescence and death, autophagy defects, inflammation and airway, and vascular remodeling
Endogenous sources—mitochondria, NOXs, myeloperoxidase (MPO), and inducible nitric oxide synthase (iNOS)—also play pivotal roles in COPD. 244 The levels of mitoROS were elevated in patients and animal models with COPD, which was proven to be an inducer of inflammation and airway remodeling. 246 Heme oxygenase protected mitochondria from mitochondrial dysfunction, decreasing OS‐mediated senescence of lung fibroblasts that was closely linked with COPD. 247 In COPD, a study was conducted to explore the specific sites of mitoROS generation, in which complex III was identified as a main site of mitoROS production, and seemed to closely correlate with muscle oxidative damage. 248 NOXs are major intracellular sources of abundant ROS and are composed of multiple subtypes, as discussed earlier. Accumulation of NOX‐derived ROS has been largely found in diverse chronic diseases of the respiratory system, which ultimately progress to COPD. 249 , 250 For example, the NOX‐ROS‐NF‐κB transduction pathway was observed to be involved in the pathogenesis of patients with COPD. 251 NOX4 was a contributor to multiple lung diseases, including acute respiratory distress syndrome, pulmonary fibrosis, and pulmonary vascular disease, leading to COPD and cancer. 252 Airway and alveolar epithelial cells aberrantly express NOX1/4, resulting in the progression of acute lung injury, neutrophilic asthma, and pulmonary fibrosis. 253 , 254 , 255 Besides, Smad3‐NOX4‐derived ROS‐mediated p38 MAPK/AKT signaling was involved in TGF‐β (transforming growth factor)‐induced airway remodeling, which may be a general mechanism underlying COPD pathogenesis. 256
4.4. Alzheimer's disease
Among the many widely studied neurological disorders, neurodegenerative diseases, such as Alzheimer's disease (AD), 257 Parkinson's disease (PD), 258 Huntington's disease (HD), 259 and multiple sclerosis 260 are closely related to redox imbalance. Intracellular mitochondrial dysfunction and excitotoxicity of the brain and spinal cord frequently occur in these diseases, directly resulting in apoptosis and functional loss of nerve cells and increased risk of these diseases.
AD is characterized by the accumulation of extracellular amyloid β‐peptide (Aβ) plaques and intracellular neurofibrillary tangles (NFTs). 257 , 261 Multiple risk factors, such as genetics, 262 , 263 environment, 264 , 265 diet, 266 , 267 age, 268 , 269 sex, 270 , 271 and race, 272 have been confirmed in AD, but the underlying mechanisms remain largely unknown. Many studies have illustrated that increased OS occurs in different parts of brains in patients with AD, including elevated levels of F2‐isoprostane‐α in cerebrospinal fluid and frontal and temporal poles, 273 , 274 acrolein in the amygdala and hippocampus/parahippocampal gyrus, 275 and HNE in ventricular fluid, hippocampus, inferior parietal lobule, and cortex. 276 , 277 , 278 Additionally, nuclear and mitochondrial DNA oxidation were also observed in the frontal, parietal, and temporal lobes of the brain in AD patients. 279 Hence, we can justifiably conclude that OS may play a crucial role in AD through the involved signaling pathways. 257
Oxidant burden is derived from multiple causes in the brains of patients with AD, such as Aβ, activated microglia, iron accumulation, and dysfunctional mitochondria. 280 , 281 , 282 , 283 To relieve ROS overload, Nrf2 was considered as a “dark horse” in AD treatments, as a decline in Nrf2 function was frequently observed in patients with AD, and Nrf2 activation exhibited potent therapeutic potential in several AD models. 284 Nrf2 deficiency in an AD mouse model exacerbates spatial learning and memory disorders. 285 Notably, Nrf2 knockout mice also displayed a similar mRNA pattern as patients with AD. 286 Eukaryotic elongation factor‐2 kinase was upregulated in patients with AD, which was found to negatively regulate Nrf2 and lead to Aβ generation. 287 The interaction between inhibitor of apoptosis‐stimulating protein of p53 (iASPP) and Keap1 can stabilize Nrf2 and restore cellular redox balance. 288 Mounting evidence has revealed that glycogen synthase kinase (GSK)‐3β activity is involved in phosphorylation‐mediated Nrf2 degradation. 289 Increased expression of GSK‐3β has been observed in different studies on AD. 290 , 291 GSK‐3β phosphorylation and inhibition via AMPK or PI3K/AKT favored an increase in Nrf2 expression, which significantly relieved Aβ‐induced oxidative damage. 292 , 293 , 294 β‐secretase enzyme (BACE1) is responsible for the generation of Aβ, and a slight increase in BACE1 expression contributes to remarkable Aβ accumulation. In patients with AD, BACE1 is aberrantly upregulated and positively correlates with the levels of Aβ. A noteworthy example showed that Nrf2 exerted an essential role in the BACE1‐Aβ axis. 284 Nrf2 bound to the ARE in the promoter of BACE1, suppressing its expression in an animal model of AD and reducing subsequent Aβ generation. 295 As a result, Nrf2 activation ameliorated cognitive deficits. By contrast, Nrf2 deletion significantly elevated BACE1 and Aβ levels, exacerbating cognitive deficits. Nrf2 stabilization increased SOD1 synthesis and inhibited the NF‐κB‐NOX2‐ROS axis, which prevented BACE1 expression and Aβ production. 296
Misfolding of the tau protein causes tauopathies and accelerates the production of β‐sheet fibrils and NFTs, which has been proven to be an important risk factor for AD. 297 , 298 , 299 Overexpression of GSK‐3β caused neuronal loss and memory disorders, which may be the result of tau hyperphosphorylation. 300 , 301 PINK1 was capable of restoring the activity of PI3K/AKT/GSK3β signaling, stabilizing Nrf2, and alleviating tau hyperphosphorylation. 302 Similarly, direct inhibition using antisense oligonucleotide of GSK‐3β also activated Nrf2, reducing the phosphorylated tau protein and leading to improved learning and memory in animal models. 303 Intriguingly, an inhibitor—dimethyl fumarate, can simultaneously prevent both GSK‐3β and Keap1 activity in a preclinical model. 304 As expected, dual inhibition potently activated Nrf2, which provided a promising method for reducing tau phosphorylation and treating neurodegenerative diseases including AD. Interestingly, Aβ‐induced ROS were an important cause of tau activation and AD exacerbation, which was mediated by the regulator of calcineurin gene (RCAN1) synthesis and subsequent calcineurin inactivation and GSK‐3β activation. 305 A schematic outlining Aβ accumulation and tau phosphorylation has been presented in this text (Figure 6).
FIGURE 6.
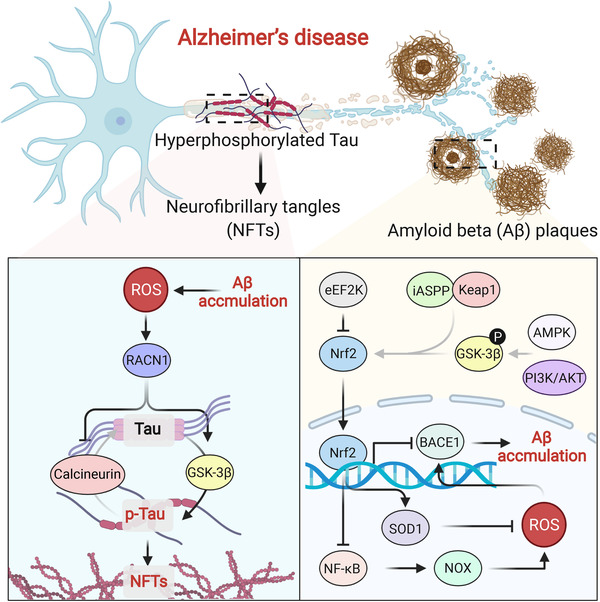
ROS accelerates Aβ accumulation and p‐tau formation in AD. Phosphorylated tau protein (p‐tau) and Aβ are essential risk factors in AD, which originate from the downregulated Nrf2 and increased ROS burden. Dysregulated Nrf2 is controlled by different proteins, promoting ROS overproduction and BACE1 expression which elicits Aβ accumulation. Aβ accumulation further causes ROS generation and activates RACN1, phosphorylating tau protein through calcineurin and GSK‐3β
Autophagy has been extensively reported to be involved in the clearance of tau protein. p62 is a pleiotropic protein involved in multiple intracellular activities, in which the autophagic degradation of aggregated proteins is mostly reported. 284 p62‐knockout mice exhibited neurodegenerative features, indicating that p62 may be involved in the initiation and progression of AD. Using an animal model of AD, Zheng and colleagues revealed that p62‐Keap1‐Nrf2 signaling was involved in this process, with p62 degradation downregulating Nrf2 expression. 306 These data suggest that p62 may be a pivotal upstream protein that regulates tau hyperphosphorylation and largely influences the development of AD. Gu et al. also reported that activation of p62 could overcome Aβ‐induced cell death, which was mediated by the activation of Nrf2 and autophagy. 307 In addition, Xu and colleagues demonstrated that p62 controlled autophagic clearance of pathogenic microtubule‐associated proteins tau, which dramatically reduced neurofibrillary tangle accumulation and pathological spreading. 308 Nuclear dot protein 52 (NDP52), an autophagy adaptor protein, is capable of efficiently promoting autophagic degradation of p‐tau. Jo et al. found that Nrf2 decreased p‐tau levels and alleviated AD by inducing NDP52 expression. 309 Mechanistically, Nrf2 bound to AREs in the promoter of NDP52 and enhanced its transcription. Another study also suggested a similar function of Nrf2 in clearing p‐tau and relieving AD. Nrf2, as a key regulator, modulated selective autophagy that helped to eliminate tau species, which was mediated by the expression of p62, NDP52, NBR1, and BAG3. 310 Additionally, Kim and colleagues illustrated the detailed mechanisms of Nrf2, TFEB, p62, and NDP52 in autophagic elimination of tau protein. 311 In support of the large application potential of Nrf2 in AD, various Nrf2 activators like benfotiamine and sulforaphane have shown excellent therapeutic effects on the basis of inhibiting the tau protein. 312 , 313
4.5. Cancer
The hallmarks of cancer involve redox alterations, which are complex based on distinct stages in cancer progression. 314 , 315 , 316 ROS are involved in each stage of tumorigenesis including the malignant transformation of normal cells, metastasis, and resistance to therapy. 317 , 318 , 319 , 320 The underlying mechanisms mainly result from the disruption of physiological redox signaling, producing pro‐oncogenic and antiapoptotic signals in cancer initiation and progression. Increasing ROS levels in cancer cells greatly strengthen proliferation and metastasis by regulating multiple signaling pathways and TFs, 321 and under these conditions, metabolic reprogramming also occurs in response to nutrition deficiency and hypoxia. 322 , 323 , 324 Similar to other redox‐relevant diseases, various oxidants have been widely detected in the body fluids of cancer patients including elevated H2O2 levels in patients with non‐small‐cell lung cancer and increased levels of 8‐OHdG in patients with prostate cancer and lung cancer. 325 , 326 , 327
ROS‐modulated expression of oncogenes and alterations of involved signaling pathways have been extensively reported to be associated with cancer initiation. As a molecule downstream of WNT signaling, RAC1 was found to potently trigger ROS production and activate the ROS‐NF‐κB pathway, leading to the proliferation of intestinal stem cells and colorectal cancer initiation. 328 Myeloid‐derived H2O2 induced genome mutations in intestinal epithelial cells, initiating the malignant transformation of normal epithelium. Furthermore, H2O2 also drove additional mutations in transformed epithelial cells, evoking cancer metastasis through the NF‐κB‐AKT‐TNFα‐H2O2 feedback loop. 329 The accumulation of self‐renewing tumor‐initiating cells is a primary driving force for thyroid cancer. An inspiring study found that in CD133+ self‐renewing tumor‐initiating cells, NOX1 was phosphorylated and activated by STAT3, and subsequent ROS overload stimulated the PI3K/AKT pathway, which increased the self‐renewal activity and tumorigenicity of CD133+ thyroid cells. 330 Notably, reducing ROS levels may also be a cause of cancer initiation. Cheung et al. reported that ROS could be dynamically regulated by TIGAR, an antioxidant protein, in different phases of tumorigenesis. 318 In premalignant lesions, a higher level of TIGAR resulted in a lower ROS level, which was conducive to cancer initiation. However, in metastasizing tumors, TIGAR was downregulated and ROS were abundantly generated, decreasing dual‐specificity phosphatase 6 expression to activate ERK signaling and drive metastasis.
Metastasis, the spread of primary cancer cells to distant organs and coupled with drug resistance, has become the main cause of cancer patient death. In solid cancers, metastasis generally begins with epithelial‐mesenchymal transition (EMT), 331 leading to the detachment and infiltration of primary tumor cells from the basement membrane into the local vasculature and/or lymphatics. Circulating tumor cells (CTCs), which enter the systemic circulation and evade anoikis and immune surveillance, is the other prerequisite for colonization of distal organs. These processes are largely regulated by altered redox status (Figure 7). ROS‐mediated TGF‐β signaling has been extensively reported to be involved in EMT, leading to cancer metastasis. 332 ROS dynamically interact with TGF‐β, modulating EMT through multiple signaling pathways. 332 Canonical TGF‐β signaling was mediated by phosphorylated Smad2/3 proteins that entered the nucleus and activated Smad4, which activated the EMT process. In addition, noncanonical pathways mediated by RAC, 333 RAS, MAPK, 334 and TGF‐β‐activated kinase 1 335 signaling also play essential roles in ROS‐mediated EMT. The fate of CTCs followed by the detachment of primary cancer cells from the basement membrane is also determined by ROS. CTCs are capable of resisting anoikis, which largely depends on redox regulation. An elegant study demonstrated that tumor‐derived angiopoietin‐like 4 protein (ANGPTL4) specifically bound to integrins and subsequently activated FAK and Rac1, which further prompted NOX‐dependent ROS generation. Increased ROS oxidized the redox sensor c‐Src, stimulating downstream PI3K and ERK‐mediated survival and preventing Bad‐mediated apoptosis. 336 However, excessive ROS burden facilitated CTC death without KLF4‐mediated induction of β‐globin (HBB). 337 In the tumor microenvironment, myeloid‐derived suppressor cells (MDSCs) inhibit the toxic effects of T cells and further accelerate cancer progression. Intriguingly, MDSCs were also found to promote cancer metastasis by regulating CTCs. Mechanistically, MDSCs interacted with CTCs, which aided MDSC‐derived ROS to elevate Notch1 receptor expression in CTCs via the ROS‐Nrf2‐ARE axis. Jagged1‐expressing MDSCs contributed to the activation of Notch signaling in CTCs by engaging the Notch1 receptor, leading to the dissemination and metastasis of CTCs. 338
FIGURE 7.
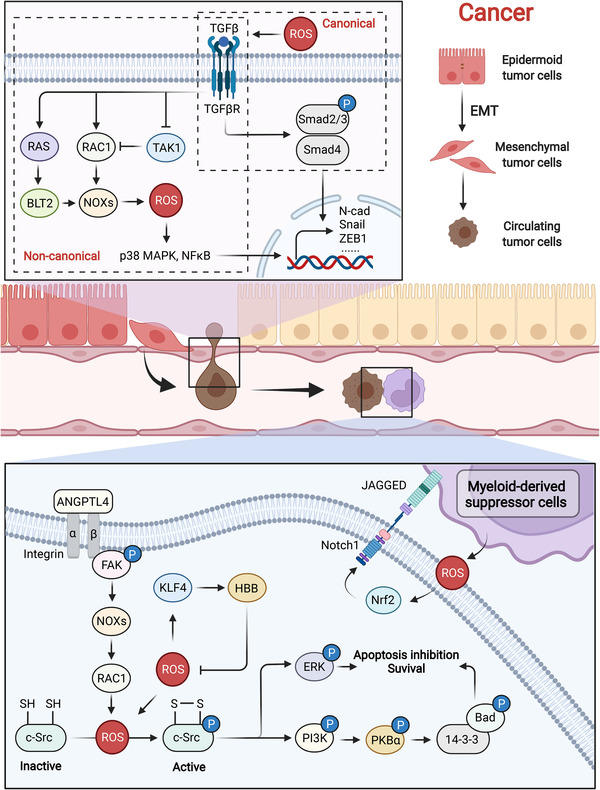
ROS promote the EMT of epithelial cells and survival of CTCs during cancer metastasis. During cancer metastasis, extracellular and intracellular ROS activate TGF‐β signaling and trigger an EMT phenotype through canonical and noncanonical pathways. CTCs entering the blood stream also face ROS stress, which protects them from apoptosis and immune surveillance
Cancer therapeutic resistance is the other main reason for treatment failure and patient death. Accumulating research has found that the metabolic shift from glycolysis toward mtOXPHOS is an important cause of cancer chemoradiotherapy resistance. 339 In chemotherapy‐resistant breast cancer stem cells (CSCs), MYC and MCL1 were upregulated and cooperated to promote mtOXPHOS, exerting a resistant role to chemotherapy via HIF‐1α. 340 OXPHOS is essential for maintaining the stemness of cancer cells, which can be targeted to exploit anticancer drugs. The antidiabetic drug metformin, a well‐established agent for mitochondrial inhibition, has been proposed to benefit cancer therapy. 341 However, drug resistance frequently occurs, which may be due to the MYC/PGC‐1α balance. Moderate levels of MYC/PGC‐1α prompted a plastic phenotype, an intermediate state between differentiated cancer cells and CSCs. MYC prevention through a BET inhibitor augmented PGC‐1α expression, engendering the stemness of cancer cells that can be further killed by metformin. 342 Venetoclax, an FDA‐approved BCL2 inhibitor, has achieved some success in managing lymphoid malignancies, but resistance to this drug is emerging. A study revealed the underlying mechanisms that involved apoptotic and metabolic pathways simultaneously, implying multiple possible strategies for reversing drug resistance. 343 Likewise, under venetoclax treatment, monocytic subclones were shown to embrace a specific transcriptome profile, which downregulated BCL2 and triggered MCL1‐mediated OXPHOS and survival. 344 In addition, venetoclax is also an inhibitor of amino acid metabolism, exerting a killing effect on leukemia stem cells. Treatment failure of venetoclax targeting amino acid metabolism may partially be attributed to nicotinamide metabolism. The nicotinamide phosphoribosyltransferase inhibitor is a potent agent for reversing venetoclax resistance. 345
4.6. Aging/lifespan
As reviewed above, dysregulated redox homeostasis is intimately associated with aging‐related diseases such as AD and cancer. Aging itself involves altered redox status, whose hallmarks are tightly regulated by excessive ROS. 346 Reviews of the numerous available reports suggest that each hallmark of aging can be linked to the damaging roles of oxidants. 347 As discussed above, physiological redox signaling is at the core of adaptive homeostasis mechanisms, but aging is always followed by an increasing level of ROS. 348 Under this condition, an imbalanced redox status prompts ROS‐mediated pathophysiological redox signaling including the classical Nrf2, NF‐κB, and AMPK signaling pathways. 349 , 350 , 351 From the perspective of a single cell, redox imbalance in aging accounts for mitochondrial dysfunction, 352 protein misfolding and aggregate formation, 353 abnormal cell membranes and intercellular communication, 354 and cell death and senescence 355 by aberrantly modulating multiple signaling pathways, among which mitochondrial dysfunction is strongly associated with aging and aging‐related disorders through ROS control. For example, aging‐induced ROS increased DNA damage and activated DNA‐dependent protein kinase, phosphorylating and counteracting the chaperone function of HSP90‐α for clients that aggravated mitochondrial dysfunction. 356 Hughes et al. found that the spatial compartmentalization of amino acids by vacuoles was essential for mitochondrial homeostasis in normal cells. 357 However, aging triggered the breakdown of vacuoles and cysteine accumulation, which remarkably inhibited iron bioavailability and mitochondrial function via ROS. Elevated ROS levels were observed in oocytes during postovulatory aging, which blocked the Sirt1‐FOXO3a‐SOD2 pathway and further increased ROS generation, preventing AKT and ERK1/2 activation and leading to mitochondrial apoptosis. 358 Wang et al. confirmed that decreased Sirt1 may break mitochondrial biogenesis by increasing PGC‐1‐α acetylation. 359 On the other hand, mitochondrial dysfunction is also a pivotal inducer of aging. ROS overload was widely found during mitochondrial dysfunction, mediating multiple signaling pathways, such as JNK, to engender cell senescence. 360 For example, mitochondrial DNA double strand break‐mediated ROS generation accelerated the aging of certain tissues, which can be partially attributed to the activation of cell cycle arrest proteins (p21/p53 pathway). 361 Thus, there is a positive feedback loop of the aging‐ROS‐mitochondrial dysfunction‐ROS axis, indicating a great potential of modulating ROS levels for attenuating aging (Figure 8).
FIGURE 8.
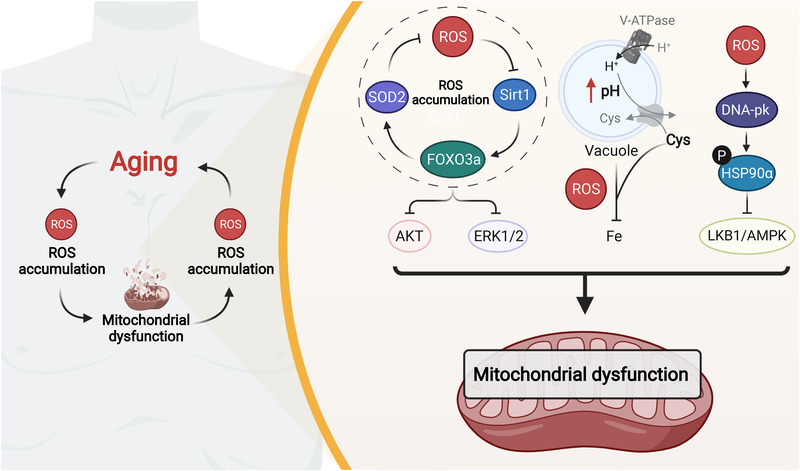
The positive feedback loop of the aging‐ROS‐mitochondrial dysfunction‐ROS axis. Aging generally accompanies ROS overproduction, which contributes to mitochondrial dysfunction via different mechanisms. Notably, mitochondrial dysfunction further induces ROS generation and accelerates aging, suggesting that scavenging ROS could efficiently attenuate aging
Dysregulated redox signaling influences lifespan, for which the levels, types, and sites of ROS generation have been identified as important elements. 362 Different kinds of ROS can have distinct impacts on lifespan. Mounting evidence has indicated the contribution of oxidants to the extension of lifespan. The analysis of genome sequencing of multiple species indicated that the frequency of mitochondria‐encoded cysteine residues negatively correlated with lifespan, 363 implying that redox biology plays an important part in lifespan and possesses a large therapeutic and commercial potential. Notably, a study conducted by Xiao et al. exploited a new tool to profile the mouse cysteine redox proteome in vivo, which has greatly promoted our knowledge of the underlying mechanisms of redox regulation in aging science. 364 mitoROS levels seemed to correlate inversely with lifespan, with the overexpression of mitochondrial catalase downregulating H2O2 levels and extending the lifespan of aged mice. 365 , 366 Suhm et al. described how mitochondrial protein translation influenced protein homeostasis and lifespan. 367 Hypoaccurate translation caused ROS accumulation, limiting lifespan, and proteostasis. However, ROS also seem to function in prolongevity responses through unique pathways. For example, under stress conditions, the intrinsic apoptosis pathway augmented protective mechanisms by sensing mitoROS, which greatly helped control harmful factors. 368 Independent of DNA damage, cells can also sense mitoROS through epigenetic silencing, which mediates mitochondrial stress‐induced longevity. 369 AMPK activation strengthened mitochondrial homeostasis and fatty‐acid oxidation, which was essential for the increased lifespan of Caenorhabditis elegans. 352 Another study revealed that ROS generation functioned as an important signal in sustaining mitochondrial homeostasis and extending lifespan in Drosophila by inducing reverse electron transport. 370 Additionally, mitoROS signaling was also found to act on mitohormesis, becoming a significant prolongevity mechanism in response to caloric restriction, hypoxia, body temperature, and physical activity. 371 , 372 Intriguingly, a finding by Bazopoulou et al. implied that early‐life H2O2 exposure benefited later‐life stress resistance and lifespan extension, which may benefit from ROS‐sensitive epigenetic changes in C. elegans. 373
4.7. Other diseases
In addition to the above‐mentioned common and serious pathophysiological conditions, other diseases are also intimately correlated with redox dysregulation and oxidative damage. For example, systemic inflammatory response syndrome is an exaggerated host defense response to exogenous factors that generally derive from infectious and noninfectious stimuli, such as sepsis, surgery, and pregnancy, recruiting immune cells to the sites of infection and trauma. 374 In this process, neutrophils exert essential functions, whose hyperactivation releases a ROS burst and causes subsequent tissue damage. Though the detailed mechanisms still remain to be resolved, the elimination of neutrophils and ROS has been proven to be effective in relieving systemic inflammatory response syndrome. 375 Fatty liver disease, known as a common liver disorder with excessive accumulation of lipids in the liver, is a redox‐centered disease due to the pivotal function of ROS in hepatic metabolism. 376 , 377 In patients with fatty liver diseases, dysregulated lipid metabolism increases the mitochondrial damage and causes ROS overproduction, which facilitates the activation of NACHT, LRR, and PYD domains‐containing protein 3 (NLRP3) inflammasome by damaged hepatic cells, resulting in metabolic inflammation of liver tissues. 378 Though the underlying mechanisms are still unknown, several studies have suggested that multiple nuclear receptors act as redox sensors and they are promising therapeutic targets in treating fatty liver diseases. 377 Ischemia‐reperfusion injury (IRI) is accompanied by several major diseases, like heart disease and stroke, which occur under excessive inflammatory responses and OS. 184 In the reperfusion phase, NO, ONOO−, O2 −• and other oxidants are markedly increased, 379 , 380 , 381 which is followed by elevated levels of OS markers in patients with IRI. 382 Intriguingly, dysregulation of the circadian rhythm is highly redox‐relevant and contributes to the cardiovascular disease and other pathological conditions. 383 , 384 Therefore, several exogenous ROS sources—air pollution and exposure of traffic noise are important risk factors for cardiovascular diseases. The circadian rhythm is tightly controlled by redox status, which is mediated by the redox regulation on cryptochrome 1 (CRY1) and period 2 (PER2) and subsequent activation or repression of circadian locomotor output cycles protein kaput/brain and muscle arnt‐like protein‐1 complex. 383 OS is a direct cause of acute poisoning, like the chemical herbicide ‐ paraquat. The intake of paraquat leads to rapid and constant accumulation of O2 −•, causing pneumonitis, progressive lung fibrosis, and even death. 385
5. THERAPEUTIC IMPLICATIONS OF REWIRING THE REDOX STATE
Current redox‐based treatment regimens have been widely applied to medical practice. Mainly these are therapeutic pro‐oxidants such as ionizing radiation, oxidant‐generating chemotherapeutics, and photodynamic therapy (PDT). Therapeutic antioxidants are also attracting much interest, some of which have entered clinical trials. However, clinical observations have frequently found only limited clinical benefits and even adverse outcomes for conventional redox‐based therapeutic interventions 386 , 387 , 388 , 389 (e.g., severe side‐effects and ineffective treatments), which may primarily be attributed to the disruption of ROS signaling and combined metabolic functions. In light of the extensive effects of redox reactions in physiological and pathological conditions, a deeper understanding of the underlying mechanisms of ROS function with a focus on appropriate sources and targets of disease‐relevant ROS will facilitate the development of new‐generation redox and precision medicine. Notably, these concepts are now coming true with some precise therapeutic interventions targeting ROS entering clinical trials. 7 , 390
5.1. Manipulating redox homeostasis by exogenous pro‐oxidants or antioxidants
Although indiscriminately manipulating redox status to treat diseases has achieved some successes, typically patients suffer from severe side‐effects. On the basis of host defense against invading pathogens that directly release a ROS burst, ionizing radiation has been extensively applied to medical practice, especially for cancer patients. Radiotherapy is one of the earliest paradigms addressing redox medicine and is characterized as the generation of powerful and toxic OH•. The adverse effects of radiotherapy on normal tissues vary greatly and include hair loss, anemia, inflammation, epithelial damage, and lung injury. 391 Although some strategies aimed at precisely identifying the risk of radiation injury, modulating an appropriate radiotherapy dose and improving symptomatic management have been proposed. 391 , 392 However, further studies are required in related research fields to further advance treatment potential. Exploiting next‐generation radiotherapy technologies and applying radioprotective drugs could be an option for relieving adverse effects in cancer radiotherapy. 393 , 394 , 395 Traditional oxidant‐generating chemotherapeutics, mostly described as cytotoxic anticancer chemotherapy, target DNA and cause the direct or indirect OS to induce cancer cell death. 396 Increasing evidence is indicating that the efficacy of conventional anticancer agents not only depends on nonspecific cytotoxic effects but also reactivation of the immune responses of cancer cells, and there is frequent drug resistance and considerable side‐effects (e.g., alopecia, fatigue, nausea, anemia, inflammation, and immunosuppression) which largely restrain their clinical applications. 397 , 398 Some studies are trying to resolve the underlying molecular networks behind drug resistance, 398 but a barrier still remains due to a lack of clinical trials and the intricate tumor heterogeneity.
Photodynamic therapy is another clinically approved and ROS‐elevated treatment regimen, accumulating cytotoxic ROS levels in cancer cells. 399 PDT utilizes the property of cancer cells preferentially taking up a sensitive photosensitizer (PS), a particular chemical activated by a specific wavelength that releases much ROS ( 1 O2). 400 It seems that cancer cells can be targeted and eliminated by PDT, but normal cells can equally absorb PS, leading to the occurrence of unavoidable deleterious side‐effects. In addition, 1 O2 only has a short lifetime, which greatly limits its diffusion and narrows the range of action of PDT. 401 Therefore, although showing some clinical promise, the potential adverse effects and insufficient therapeutic efficacy constrain the further development of PDT.
OS and subsequent disruption of redox signaling are the direct or indirect causes of multiple physiological disorders, which suggest its pharmacological role in choosing therapeutic antioxidants as treatment strategies. However, although many small molecules have been employed as therapeutic antioxidants, clinical observations frequently exhibit poor outcomes. 184 A clinical trial was conducted to study the effects of several antioxidants (e.g., vitamin C, vitamin E, selenium, I‐carnitine, zinc, folic acid lycopene, and placebo) on semen parameters or DNA integrity among men with infertility. While preclinical studies are promising, the final results suggest that there are no significant improvements in pregnancy or live‐birth rates (NCT02421887). 402 Another clinical trial was conducted to evaluate the effects of vitamin C and E (initiated between the 9th to 16th weeks) on the risk of pregnancy‐associated hypertension, which also eventually indicated a negative outcome (NCT00135707). 403 However, systematic antioxidant supplements do help relieve patients with cystic fibrosis, which may be attributed to the specific disease itself or increased systemic antioxidant levels (NCT01018303). 404 In addition, some antioxidant enzyme mimics also revealed poor prognosis. N‐Acetylcysteine (NAC), a well‐established antioxidant agent, has been proven to work in multiple diseases. 405 , 406 , 407 , 408 However, clinical observation of NAC has been disappointing in many patients including those with hypertrophic cardiomyopathy and type 2 diabetes (NCT01537926, NCT01394510). 409 , 410 Therefore, revisiting redox‐modulated treatments from a novel perspective is urgently required to develop new treatments.
5.2. Directly targeting ROS sources and redox‐related effectors
Recent ROS‐based drug development emphasizes the importance of modulating the sources and targets of disease‐relevant ROS compared with conventional systematic pro‐oxidant or antioxidant therapy. 411 Accumulating evidence confirms its feasibility, and some drugs have entered the clinic. NOXs, which are the only enzyme family that are responsible for ROS production, have been shown to be closely related to multiple pathological conditions, such as aging, COPD, diabetes, cardiac dysfunction, and cancer. 250 , 412 , 413 , 414 Small‐molecule NOX inhibitors based on disease‐related ROS sources have exhibited significant treatment effects. For example, during liver fibrosis, Aoyama and colleagues revealed that the excessive activation of NOX1 in hepatic stellate cells upregulated NOX4 expression, which greatly induced ROS generation and suggested that NOX1/4 were promising therapeutic targets for liver fibrosis. 415 In vivo experiments confirmed that dual‐targeting treatment with GKT137831, a well‐established NOX1/4 inhibitor, 416 remarkably attenuated liver fibrosis. Similarly, a dual NOX1/4 inhibitor was also applied to neuroglial cell inflammation and achieved the expected outcomes. 417 In light of the potent therapeutic effects in preclinical studies, several ongoing or completed clinical trials have been conducted to evaluate the safety and efficacy of GKT137831 in patients with idiopathic pulmonary fibrosis, diabetes, and primary biliary cirrhosis, although the results have not yet been reported (NCT03865927, NCT02010242, NCT03226067). MPO is regarded as an essential promotor in chemoresistant acute myeloid leukemia (AML) cells, preventing increased mitochondrial and cytosolic ROS levels and sensitizing AML cells to cytarabine treatment. 418 Furthermore, inhibiting MPO activity using the specific inhibitor 4‐aminobenzoic acid hydrazide (ABAH) also dramatically relieved inflammation, increased neurogenesis, and suppressed atherogenesis based on specific mechanisms. 419 , 420 , 421 As well as ABAH, several other MPO inhibitors have also entered clinical studies such as AZD3241 in patients with PD (NCT01603069, NCT01527695) and AZD4831 in patients with heart failure (NCT04232345).
With respect to targets of disease‐relevant ROS, mounting evidence has reported many common and independent downstream molecules in different disease situations. Nrf2, a well‐recognized redox sensor, is activated by elevated ROS levels and evades degradation. Nevertheless, Nrf2 activity is frequently blocked, leading to ROS overload and systematic disorders such as atherogenesis, diabetes, and diabetic nephropathy. 422 , 423 , 424 Nrf2 is capable of stimulating endogenous ROS elimination, which assists the modulation of physiological ROS signaling. Hence, selectively activating Nrf2 is a promising therapeutic strategy for redox‐relevant diseases. Nrf2 activators have been extensively applied in clinical research, among which sulforaphane, synthetic triterpenoids, and dimethyl fumarate are the most successful examples. 184 , 411 In preclinical studies, these three compounds were widely reported to function in many disease situations including type 2 diabetes, 425 epilepsy, 426 pachyonychia congenita, 427 cancers, 428 , 429 , 430 autoimmunity, 431 AD, 432 and infections. 433 A large number of clinical trials are currently being carried out in patients with a range of disorders, some of which have undergone preliminary tests and some have even been approved (NCT01335971, NCT03517995, NCT00322140, NCT02683863).
In summary, treatment avenues aimed at inhibiting the sources and targets of disease‐relevant ROS have achieved encouraging preclinical and clinical benefits with excellent safety profiles, which will help facilitate the discovery of more accessible targets and exploitation of more targeted drugs in redox‐relevant diseases. Sophisticated redox control is now gradually replacing conventional systematic redox interventions. Nonetheless, some pivotal questions remain to be resolved. First, identifying the functions of ROS in diseases is the basis, which requires numerous preclinical studies to ensure the reliability. The pathophysiological roles of ROS sources and targets should be complemented under different conditions. Besides, we would better determine the comprehensive landscape of ROS sources and targets in diverse diseases and status of disease progression, achieving which may be conducive to more effective, safe, and precise medical interventions. Various ROS from different sources have been proven to take an important part in disease progression through certain targets and signaling pathways. For monitoring different ROS levels to timely adjust and change therapeutic strategies, a general, accurate, and convenient device needs to be developed. Resolving these problems, the agents directly targeting ROS sources and redox‐related effectors can better serve as next‐generation redox medicine.
6. CONCLUSIONS AND PERSPECTIVES
Redox reactions are central to the production of energy, which have given rise to the terms “redox chemistry” and “redox biology.” Generally, redox chemistry involves the transformation of energy and electron transfer processes, which is the basis of human activity. With accumulating knowledge of redox reactions in physiological and pathophysiological conditions, the term “redox biology” has been proposed. Redox homeostasis is mediated by multiple reactive species, in which ROS have been mostly identified as the pivotal mediator. Redox imbalance in favor of oxidant burden frequently emerges, causing various diseases. To overcome this, therapeutic strategies targeting excessive ROS have been developed for many diseases. Unfortunately, while antioxidant therapy has achieved promising effects in many preclinical studies, subsequent clinical trials have often been unsuccessful. Hence, a deep understanding of redox status in pathophysiological conditions is urgently needed to develop new‐generation redox‐relevant agents.
ROS are double‐edged swords. Mounting evidence has indicated a dual role of ROS in human health, which is intricate because of the many elements influencing the final ROS outcome. For example, a moderate level of H2O2 stimulates multiple physiological signaling pathways through reversible oxidation of specific protein targets (oxidation of sulfur in target proteins), which contributes to the orchestration of multiple cellular biological activities including proliferation, differentiation, and migration. In contrast, excessive H2O2 generation causes physiological disorders, which result from unspecific oxidation of proteins and irreversible damage to biomacromolecules that are directly linked to growth arrest and cell death. Finding detectable markers of OS in patients with redox‐relevant diseases should be a priority. Balancing the beneficial and toxic effects of ROS will confer great application potential to redox medicine.
In light of the significance of ROS in physiological and pathological conditions, approaches targeting ROS detoxification and production have been widely applied in medical practice. Radiotherapy, a first‐line therapy, can release a ROS burst to kill cancer cells, although this frequently exhibits unwanted and inescapable side‐effects. On the other hand, although ROS detoxification seems to have a huge potential in redox‐relevant diseases, clinical studies have often been associated with poor or even harmful outcomes. Thus, revisiting the role of intervention targeting ROS from a novel perspective will further facilitate the development of redox medicine.
Accumulating reports show that the switch from general to sophisticated redox manipulation on the basis of an understanding of the regulatory mechanisms and sources of disease‐relevant ROS in treating physiological disorders is attracting much interest. This will lead to the discovery of more novel ROS‐related targets and the exploitation of targeted therapies. These methods have prompted new therapeutic strategies on the basis of ROS, some of which are being evaluated in the clinic. In addition, the emerging field of ROSbased nanomedicine also holds great promise for improved clinical applications. The rise of network medicine, integrating phenotypic analysis and intracellular and intercellular connectivity, will link the different omics approaches to mitochondrial function and find more rationale targets on the basis of ROS regulation.
In summary, redox homeostasis is essential for human health, whose imbalance involves nearly all major diseases. Redox regulation as a promising treatment avenue has achieved some success, but the limited efficacy often observed requires the intrinsic mechanisms involved to be further investigated. With new emerging technological advancements in associated fields, we believe that redox regulation will play a more important role in disease management and advancing precision redox medicine.
CONFLICT OF INTEREST
Canhua Huang is an editorial board member of MedComm. Author Canhua Huang was not involved in the journal's review of, or decisions related to, this manuscript. The other authors have no conflicts of interest to declare.
ETHICS STATEMENT
Not applicable.
AUTHOR’S CONTRIBUTIONS
CHH, WZ, and CW conceived the structure of the manuscript. JZ, ZZ, MCL, and LZ drafted initial manuscript. ECN prepared and revised the manuscript. ZZ and JZ prepared the figures. All authors read and approved the final manuscript.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Key Research and Development Project (2020YFA0509400), Guangdong Basic and Applied Basic Research Foundation (2019B030302012), 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC21004) and the National Natural Science Foundation of China (81821002, 82130082, 81790251 and 82171527). These figures were created by Biorender.
Zuo J, Zhang Z, Luo M, et al. Redox signaling at the crossroads of human health and disease. MedComm. 2022;3:e127. 10.1002/mco2.127
Jing Zuo, Zhe Zhang, and Maochao Luo contributed equally to this work.
Contributor Information
Wei Zhang, Email: weizhang27@scu.edu.cn.
Chuang Wang, Email: wangchuang@nbu.edu.cn.
Canhua Huang, Email: hcanhua@scu.edu.cn.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Herrmann JM, Dick TP. Redox biology on the rise. Biol Chem. 2012;393(9):999‐1004. [DOI] [PubMed] [Google Scholar]
- 2. Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 2017;11:613‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mantzaris MD, Bellou S, Skiada V, Kitsati N, Fotsis T, Galaris D. Intracellular labile iron determines H2O2‐induced apoptotic signaling via sustained activation of ASK1/JNK‐p38 axis. Free Radic Biol Med. 2016;97:454‐465. [DOI] [PubMed] [Google Scholar]
- 4. Lloyd D, Aon MA, Cortassa S. Why homeodynamics, not homeostasis? Scientific World J. 2001;1:133‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Santolini J, Wootton SA, Jackson AA, Feelisch M. The redox architecture of physiological function. Curr Opin Physiol. 2019;9:34‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones DP, Sies H. The redox code. Antioxid Redox Signl. 2015;23(9):734‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363‐383. [DOI] [PubMed] [Google Scholar]
- 8. Li X, Liang M, Jiang J, et al. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib‐induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int J Biol Sci. 2018;14(10):1291‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H(2)O(2), and protein chaperones. Antioxid Redox Signal. 2011;15(3):781‐794. [DOI] [PubMed] [Google Scholar]
- 10. Lyons TW, Reinhard CT, Planavsky NJ. The rise of oxygen in earth's early ocean and atmosphere. Nature. 2014;506(7488):307‐315. [DOI] [PubMed] [Google Scholar]
- 11. de Villiers D, Potgieter M, Ambele MA, Adam L, Durandt C, Pepper MS. The role of reactive oxygen species in adipogenic differentiation. Adv Exp Med Biol. 2018;1083:125‐144. [DOI] [PubMed] [Google Scholar]
- 12. Flohe L. Looking back at the early stages of redox biology. Antioxidants (Basel). 2020;9(12):1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nathan C, Cunningham‐Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat Rev Immunol. 2013;13(5):349‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zuo J, Zhang Z, Li M, et al. The crosstalk between reactive oxygen species and noncoding RNAs: from cancer code to drug role. Mol Cancer. 2022;21(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Commoner B, Townsend J, Pake GE. Free radicals in biological materials. Nature. 1954;174(4432):689‐691. [DOI] [PubMed] [Google Scholar]
- 16. Loew O. A new enzyme of general occurrence in organisms. Science. 1900;11(279):701‐702. [DOI] [PubMed] [Google Scholar]
- 17. Mills GC. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J Biol Chem. 1957;229(1):189‐197. [PubMed] [Google Scholar]
- 18. Laurent TC, Moore EC, Reichard P. Enzymatic synthesis of deoxyribonucleotides. Iv. Isolation and characterization of thioredoxin, the hydrogen donor from Escherichia coli B. J Biol Chem. 1964;239:3436‐3444. [PubMed] [Google Scholar]
- 19. Harris JR. Release of a macromolecular protein component from human erythrocyte ghosts. Biochim Biophys Acta. 1968;150(3):534‐537. [DOI] [PubMed] [Google Scholar]
- 20. Detienne G, De Haes W, Mergan L, Edwards SL, Temmerman L, Van Bael S. Beyond ROS clearance: peroxiredoxins in stress signaling and aging. Ageing Res Rev. 2018;44:33‐48. [DOI] [PubMed] [Google Scholar]
- 21. Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem Sci. 2015;40(8):435‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park MH, Jo M, Kim YR, Lee CK, Hong JT. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol Ther. 2016;163:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muri J, Kopf M. Redox regulation of immunometabolism. Nat Rev Immunol. 2021;21(6):363‐381. [DOI] [PubMed] [Google Scholar]
- 24. Zhang J, Li X, Han X, Liu R, Fang J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol Sci. 2017;38(9):794‐808. [DOI] [PubMed] [Google Scholar]
- 25. Balsera M, Buchanan BB. Evolution of the thioredoxin system as a step enabling adaptation to oxidative stress. Free Radic Biol Med. 2019;140:28‐35. [DOI] [PubMed] [Google Scholar]
- 26. Parvez S, Long MJC, Poganik JR, Aye Y. Redox signaling by reactive electrophiles and oxidants. Chem Rev. 2018;118(18):8798‐8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iyer GY, Islam MF, Quastel JH. Biochemical aspects of phagocytosis. Nature. 1961;192(480):535‐541. [Google Scholar]
- 28. Rossi F, Zatti M. Biochemical aspects of phagocytosis in polymorphonuclear leucocytes. NADH and NADPH oxidation by the granules of resting and phagocytizing cells. Experientia. 1964;20(1):21‐23. [DOI] [PubMed] [Google Scholar]
- 29. Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. 1959;234(6):1355‐1362. [PubMed] [Google Scholar]
- 30. Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52(3):741‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244(22):6049‐6055. [PubMed] [Google Scholar]
- 32. Hinkle PC, Butow RA, Racker E, Chance B. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin‐cytochrome beta region of the respiratory chain of beef heart submitochondrial particles. J Biol Chem. 1967;242(22):5169‐5173. [PubMed] [Google Scholar]
- 33. Loschen G, Azzi A, Richter C, Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42(1):68‐72. [DOI] [PubMed] [Google Scholar]
- 34. Tan J, Duan M, Yadav T, et al. An R‐loop‐initiated CSB‐RAD52‐POLD3 pathway suppresses ROS‐induced telomeric DNA breaks. Nucleic Acids Res. 2020;48(3):1285‐1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khan S, Zafar A, Naseem I. Redox cycling of copper by coumarin‐di(2‐picolyl)amine hybrid molecule leads to ROS‐mediated modulation of redox scavengers, DNA damage and cell death in diethylnitrosamine induced hepatocellular carcinoma. Bioorg Chem. 2020;99:103818. [DOI] [PubMed] [Google Scholar]
- 36. Liu N, Wang KS, Qi M, et al. Vitexin compound 1, a novel extraction from a Chinese herb, suppresses melanoma cell growth through DNA damage by increasing ROS levels. J Exp Clin Cancer Res. 2018;37(1):269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fimognari C. Role of oxidative RNA damage in chronic‐degenerative diseases. Oxid Med Cell Longev. 2015;2015:358713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Willi J, Kupfer P, Evequoz D, et al. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 2018;46(4):1945‐1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Veskoukis AS, Tsatsakis AM, Kouretas D. Dietary oxidative stress and antioxidant defense with an emphasis on plant extract administration. Cell Stress Chaperones. 2012;17(1):11‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stepniak J, Karbownik‐Lewinska M. 17beta‐estradiol prevents experimentally‐induced oxidative damage to membrane lipids and nuclear DNA in porcine ovary. Syst Biol Reprod Med. 2016;62(1):17‐21. [DOI] [PubMed] [Google Scholar]
- 41. Felix R, Valentao P, Andrade PB, Felix C, Novais SC, Lemos MFL. Evaluating the in vitro potential of natural extracts to protect lipids from oxidative damage. Antioxidants (Basel). 2020;9(3):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li X, Rommelaere S, Kondo S, Lemaitre B. Renal purge of hemolymphatic lipids prevents the accumulation of ROS‐induced inflammatory oxidized lipids and protects Drosophila from tissue damage. Immunity. 2020;52(2):374‐387 e6. [DOI] [PubMed] [Google Scholar]
- 43. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298‐300. [DOI] [PubMed] [Google Scholar]
- 44. Gerschman R, Gilbert DL, Nye SW, Dwyer P, Fenn WO. Oxygen poisoning and x‐irradiation: a mechanism in common. Science. 1954;119(3097):623‐626. [DOI] [PubMed] [Google Scholar]
- 45. Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313‐322. [DOI] [PubMed] [Google Scholar]
- 46. Kuiper JW, Sun C, Magalhaes MA, Glogauer M. Rac regulates PtdInsP(3) signaling and the chemotactic compass through a redox‐mediated feedback loop. Blood. 2011;118(23):6164‐6171. [DOI] [PubMed] [Google Scholar]
- 47. Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue‐scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459(7249):996‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yoo SK, Starnes TW, Deng Q, Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature. 2011;480(7375):109‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11(5):411‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245‐313. [DOI] [PubMed] [Google Scholar]
- 51. Jiang F, Zhang Y, Dusting GJ. NADPH oxidase‐mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63(1):218‐242. [DOI] [PubMed] [Google Scholar]
- 52. Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014;2:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Winterbourn CC. Biological production, detection, and fate of hydrogen peroxide. Antioxid Redox Signal. 2018;29(6):541‐551. [DOI] [PubMed] [Google Scholar]
- 54. Yamamoto M, Kensler TW, Motohashi H. The KEAP1‐NRF2 system: a thiol‐based sensor‐effector apparatus for maintaining redox homeostasis. Physiol Rev. 2018;98(3):1169‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burgener AV, Bantug GR, Meyer BJ, et al. SDHA gain‐of‐function engages inflammatory mitochondrial retrograde signaling via KEAP1‐Nrf2. Nat Immunol. 2019;20(10):1311‐1321. [DOI] [PubMed] [Google Scholar]
- 56. Cuadrado A. Brain‐protective mechanisms of transcription factor NRF2: toward a common strategy for neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 2022;62:255‐277. [DOI] [PubMed] [Google Scholar]
- 57.Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. LeBoeuf SE, Wu WL, Karakousi TR, et al. Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non‐essential amino acids. Cell Metab. 2020;31(2):339‐350 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu P, Kerins MJ, Tian W, Neupane D, Zhang DD, Ooi A. Differential and overlapping targets of the transcriptional regulators NRF1, NRF2, and NRF3 in human cells. J Biol Chem. 2019;294(48):18131‐18149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29(17):1727‐1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen QM. Nrf2 for cardiac protection: pharmacological options against oxidative stress. Trends Pharmacol Sci. 2021;42(9):729‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mou Y, Wen S, Li YX, Gao XX, Zhang X, Jiang ZY. Recent progress in Keap1‐Nrf2 protein‐protein interaction inhibitors. Eur J Med Chem. 2020;202:112532. [DOI] [PubMed] [Google Scholar]
- 63. Siswanto FM, Oguro A, Imaoka S. Sp1 is a substrate of Keap1 and regulates the activity of CRL4A(WDR23) ubiquitin ligase toward Nrf2. J Biol Chem. 2021;296:100704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chang M, Wilson CJ, Karunatilleke NC, Moselhy MH, Karttunen M, Choy WY. Exploring the conformational landscape of the Neh4 and Neh5 domains of Nrf2 using two different force fields and circular dichroism. J Chem Theory Comput. 2021;17(5):3145‐3156. [DOI] [PubMed] [Google Scholar]
- 65. Wang H, Liu K, Geng M, et al. RXRalpha inhibits the NRF2‐ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013;73(10):3097‐3108. [DOI] [PubMed] [Google Scholar]
- 66. Cloer EW, Goldfarb D, Schrank TP, Weissman BE, Major MB. NRF2 activation in cancer: from DNA to protein. Cancer Res. 2019;79(5):889‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ulasov AV, Rosenkranz AA, Georgiev GP, Sobolev AS. Nrf2/Keap1/ARE signaling: towards specific regulation. Life Sci. 2022;291:120111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu T, Lv YF, Zhao JL, You QD, Jiang ZY. Regulation of Nrf2 by phosphorylation: consequences for biological function and therapeutic implications. Free Radic Biol Med. 2021;168:129‐141. [DOI] [PubMed] [Google Scholar]
- 69. Sivandzade F, Prasad S, Bhalerao A, Cucullo L. NRF2 and NF‐B interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019;21:101059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mohan S, Gupta D. Crosstalk of toll‐like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed Pharmacother. 2018;108:1866‐1878. [DOI] [PubMed] [Google Scholar]
- 71. Camina N, Penning TM. Genetic and epigenetic regulation of the NRF2‐KEAP1 pathway in human lung cancer. Br J Cancer. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mena EL, Kjolby RAS, Saxton RA, et al. Dimerization quality control ensures neuronal development and survival. Science. 2018;362(6411):eaap8236. [DOI] [PubMed] [Google Scholar]
- 73. Ogura T, Tong KI, Mio K, et al. Keap1 is a forked‐stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C‐terminal domains. Proc Natl Acad Sci USA. 2010;107(7):2842‐2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015;88(Pt B):101‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213‐223. [DOI] [PubMed] [Google Scholar]
- 76. Baird L, Lleres D, Swift S, Dinkova‐Kostova AT. Regulatory flexibility in the Nrf2‐mediated stress response is conferred by conformational cycling of the Keap1‐Nrf2 protein complex. Proc Natl Acad Sci USA. 2013;110(38):15259‐15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu S, Pi J, Zhang Q. Mathematical modeling reveals quantitative properties of KEAP1‐NRF2 signaling. Redox Biol. 2021;47:102139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tu W, Wang H, Li S, Liu Q, Sha H. The anti‐inflammatory and anti‐oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019;10(3):637‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Unni S, Deshmukh P, Krishnappa G, Kommu P, Padmanabhan B. Structural insights into the multiple binding modes of dimethyl fumarate (DMF) and its analogs to the Kelch domain of Keap1. FEBS J. 2021;288(5):1599‐1613. [DOI] [PubMed] [Google Scholar]
- 80.Dayalan Naidu S, Dinkova‐Kostova AT. KEAP1, a cysteine‐based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020;10(6):200105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cvetko F, Caldwell ST, Higgins M, et al. Nrf2 is activated by disruption of mitochondrial thiol homeostasis but not by enhanced mitochondrial superoxide production. J Biol Chem. 2021;296:100169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cebula M, Schmidt EE, Arner ES. TrxR1 as a potent regulator of the Nrf2‐Keap1 response system. Antioxid Redox Signal. 2015;23(10):823‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jerabkova K, Cullin Sumara I. 3, a cellular scripter of the non‐proteolytic ubiquitin code. Semin Cell Dev Biol. 2019;93:100‐110. [DOI] [PubMed] [Google Scholar]
- 84. Shrishrimal S, Chatterjee A, Kosmacek EA, Davis PJ, McDonald JT, Oberley‐Deegan RE. Manganese porphyrin, MnTE‐2‐PyP, treatment protects the prostate from radiation‐induced fibrosis (RIF) by activating the NRF2 signaling pathway and enhancing SOD2 and sirtuin activity. Free Radic Biol Med. 2020;152:255‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hirotsu Y, Katsuoka F, Funayama R, et al. Nrf2‐MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012;40(20):10228‐10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cuadrado A, Rojo AI, Wells G, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov. 2019;18(4):295‐317. [DOI] [PubMed] [Google Scholar]
- 87. Horie Y, Suzuki T, Inoue J, et al. Molecular basis for the disruption of Keap1‐Nrf2 interaction via Hinge & Latch mechanism. Commun Biol. 2021;4(1):576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Baird L, Dinkova‐Kostova AT. Diffusion dynamics of the Keap1‐Cullin3 interaction in single live cells. Biochem Biophys Res Commun. 2013;433(1):58‐65. [DOI] [PubMed] [Google Scholar]
- 89. Klotz LO, Sanchez‐Ramos C, Prieto‐Arroyo I, Urbanek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Barnes PJ. Oxidative stress‐based therapeutics in COPD. Redox Biol. 2020;33:101544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Calissi G, Lam EW, Link W. Therapeutic strategies targeting FOXO transcription factors. Nat Rev Drug Discov. 2021;20(1):21‐38. [DOI] [PubMed] [Google Scholar]
- 92. Weigel D, Jurgens G, Kuttner F, Seifert E, Jackle H. The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell. 1989;57(4):645‐658. [DOI] [PubMed] [Google Scholar]
- 93. Gajiwala KS, Chen H, Cornille F, et al. Structure of the winged‐helix protein hRFX1 reveals a new mode of DNA binding. Nature. 2000;403(6772):916‐921. [DOI] [PubMed] [Google Scholar]
- 94. Mukhopadhyay S, Das T, Bose M, et al. Residues at the interface between zinc binding and winged helix domains of human RECQ1 play a significant role in DNA strand annealing activity. Nucleic Acids Res. 2021;49(20):11834‐11854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li J, Dai S, Chen X, et al. Mechanism of forkhead transcription factors binding to a novel palindromic DNA site. Nucleic Acids Res. 2021;49(6):3573‐3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Senchuk MM, Dues DJ, Schaar CE, et al. Activation of DAF‐16/FOXO by reactive oxygen species contributes to longevity in long‐lived mitochondrial mutants in Caenorhabditis elegans . PLoS Genet. 2018;14(3):e1007268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dobson AJ, Ezcurra M, Flanagan CE, et al. Nutritional programming of lifespan by FOXO inhibition on sugar‐rich diets. Cell Rep. 2017;18(2):299‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Matsuzaki T, Alvarez‐Garcia O, Mokuda S, et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci Transl Med. 2018;10(428):eaan0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ma J, Matkar S, He X, Hua X. FOXO family in regulating cancer and metabolism. Semin Cancer Biol. 2018;50:32‐41. [DOI] [PubMed] [Google Scholar]
- 100. Maiese K, Chong ZZ, Shang YC. OutFOXOing disease and disability: the therapeutic potential of targeting FoxO proteins. Trends Mol Med. 2008;14(5):219‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang XS, Wang T, Lin XW, Denlinger DL, Xu WH. Reactive oxygen species extend insect life span using components of the insulin‐signaling pathway. Proc Natl Acad Sci USA. 2017;114(37):E7832‐E7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tsitsipatis D, Gopal K, Steinbrenner H, Klotz LO. FOXO1 cysteine‐612 mediates stimulatory effects of the coregulators CBP and PGC1alpha on FOXO1 basal transcriptional activity. Free Radic Biol Med. 2018;118:98‐107. [DOI] [PubMed] [Google Scholar]
- 103. Putker M, Vos HR, van Dorenmalen K, et al. Evolutionary acquisition of cysteines determines FOXO paralog‐specific redox signaling. Antioxid Redox Signal. 2015;22(1):15‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hopkins BL, Neumann CA. Redoxins as gatekeepers of the transcriptional oxidative stress response. Redox Biol. 2019;21:101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tothova Z, Kollipara R, Huntly BJ, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325‐339. [DOI] [PubMed] [Google Scholar]
- 106. Putker M, Madl T, Vos HR, et al. Redox‐dependent control of FOXO/DAF‐16 by transportin‐1. Mol Cell. 2013;49(4):730‐742. [DOI] [PubMed] [Google Scholar]
- 107. Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35(9):505‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Soh R, Hardy A, Zur Nieden NI. The FOXO signaling axis displays conjoined functions in redox homeostasis and stemness. Free Radic Biol Med. 2021;169:224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Klotz LO, Steinbrenner H. Cellular adaptation to xenobiotics: interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017;13:646‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Young D, Pedre B, Ezerina D, et al. Protein promiscuity in H2O2 signaling. Antioxid Redox Signal. 2019;30(10):1285‐1324. [DOI] [PubMed] [Google Scholar]
- 111. Olmos Y, Sanchez‐Gomez FJ, Wild B, et al. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC‐1alpha complex. Antioxid Redox Signal. 2013;19(13):1507‐1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Meng X, Grotsch B, Luo Y, et al. Hypoxia‐inducible factor‐1alpha is a critical transcription factor for IL‐10‐producing B cells in autoimmune disease. Nat Commun. 2018;9(1):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Choudhry H, Harris AL. Advances in hypoxia‐inducible factor biology. Cell Metab. 2018;27(2):281‐298. [DOI] [PubMed] [Google Scholar]
- 114. Choueiri TK, Bauer TM, Papadopoulos KP, et al. Inhibition of hypoxia‐inducible factor‐2alpha in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med. 2021;27(5):802‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Burrows N, Maxwell PH. Hypoxia‐inducible factor 2 inhibitors show promise in advanced kidney cancer. Nat Rev Urol. 2021;18(9):516‐517. [DOI] [PubMed] [Google Scholar]
- 116. Peek CB. Metabolic implications of circadian‐HIF crosstalk. Trends Endocrinol Metab. 2020;31(6):459‐468. [DOI] [PubMed] [Google Scholar]
- 117. Schodel J, Ratcliffe PJ. Mechanisms of hypoxia signalling: new implications for nephrology. Nat Rev Nephrol. 2019;15(10):641‐659. [DOI] [PubMed] [Google Scholar]
- 118. Ivan M, Kaelin WG Jr., The EGLN‐HIF O2‐sensing system: multiple inputs and feedbacks. Mol Cell. 2017;66(6):772‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Semenza GL. The genomics and genetics of oxygen homeostasis. Annu Rev Genomics Hum Genet. 2020;21:183‐204. [DOI] [PubMed] [Google Scholar]
- 120. Tarade D, Ohh M. The HIF and other quandaries in VHL disease. Oncogene. 2018;37(2):139‐147. [DOI] [PubMed] [Google Scholar]
- 121. Maniaci C, Hughes SJ, Testa A, et al. Homo‐PROTACs: bivalent small‐molecule dimerizers of the VHL E3 ubiquitin ligase to induce self‐degradation. Nat Commun. 2017;8(1):830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Semenza GL. Pharmacologic targeting of hypoxia‐inducible factors. Annu Rev Pharmacol Toxicol. 2019;59:379‐403. [DOI] [PubMed] [Google Scholar]
- 123. Samanta D, Semenza GL. Metabolic adaptation of cancer and immune cells mediated by hypoxia‐inducible factors. Biochim Biophys Acta Rev Cancer. 2018;1870(1):15‐22. [DOI] [PubMed] [Google Scholar]
- 124. Rodriguez J, Haydinger CD, Peet DJ, Nguyen LK, von Kriegsheim A. Asparagine hydroxylation is a reversible post‐translational modification. Mol Cell Proteomics. 2020;19(11):1777‐1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hayashi Y, Yokota A, Harada H, Huang G. Hypoxia/pseudohypoxia‐mediated activation of hypoxia‐inducible factor‐1alpha in cancer. Cancer Sci. 2019;110(5):1510‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Peek CB, Levine DC, Cedernaes J, et al. Circadian clock interaction with HIF1alpha mediates oxygenic metabolism and anaerobic glycolysis in skeletal muscle. Cell Metab. 2017;25(1):86‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ma CP, Liu H, Yi‐Feng Chang I, et al. ADAR1 promotes robust hypoxia signaling via distinct regulation of multiple HIF‐1alpha‐inhibiting factors. EMBO Rep. 2019;20(5):e47107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. 2020;21(5):268‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Dunham‐Snary KJ, Wu D, Potus F, et al. Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen‐sensing and hypoxic pulmonary vasoconstriction. Circ Res. 2019;124(12):1727‐1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Balogh E, Toth A, Mehes G, Trencsenyi G, Paragh G, Jeney V. Hypoxia triggers osteochondrogenic differentiation of vascular smooth muscle cells in an HIF‐1 (hypoxia‐inducible factor 1)‐dependent and reactive oxygen species‐dependent manner. Arterioscler Thromb Vasc Biol. 2019;39(6):1088‐1099. [DOI] [PubMed] [Google Scholar]
- 131. Lucking EF, O'Connor KM, Strain CR, et al. Chronic intermittent hypoxia disrupts cardiorespiratory homeostasis and gut microbiota composition in adult male guinea‐pigs. EBioMedicine. 2018;38:191‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Guan P, Sun ZM, Wang N, et al. Resveratrol prevents chronic intermittent hypoxia‐induced cardiac hypertrophy by targeting the PI3K/AKT/mTOR pathway. Life Sci. 2019;233:116748. [DOI] [PubMed] [Google Scholar]
- 133. Song JQ, Jiang LY, Fu CP, et al. Heterozygous SOD2 deletion deteriorated chronic intermittent hypoxia‐induced lung inflammation and vascular remodeling through mtROS‐NLRP3 signaling pathway. Acta Pharmacol Sin. 2020;41(9):1197‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. De Donatis GM, Le Pape E, Pierron A, et al. NF‐kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene. 2016;35(21):2813. [DOI] [PubMed] [Google Scholar]
- 135. Sun SC. The non‐canonical NF‐kappaB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Taniguchi K, Karin M. NF‐kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18(5):309‐324. [DOI] [PubMed] [Google Scholar]
- 137. Fomicheva M, Macara IG. Genome‐wide CRISPR screen identifies noncanonical NF‐kappaB signaling as a regulator of density‐dependent proliferation. Elife. 2020;9:e63603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Mishra R, Sehring I, Cederlund M, Mulaw M, Weidinger G. NF‐kappaB signaling negatively regulates osteoblast dedifferentiation during zebrafish bone regeneration. Dev Cell. 2020;52(2):167‐182 e7. [DOI] [PubMed] [Google Scholar]
- 139. Mulero MC, Wang VY, Huxford T, Ghosh G. Genome reading by the NF‐kappaB transcription factors. Nucleic Acids Res. 2019;47(19):9967‐9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ngo KA, Kishimoto K, Davis‐Turak J, et al. Dissecting the regulatory strategies of NF‐kappaB RelA target genes in the inflammatory response reveals differential transactivation logics. Cell Rep. 2020;30(8):2758‐2775 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Moser BA, Steinhardt RC, Escalante‐Buendia Y, et al. Increased vaccine tolerability and protection via NF‐kappaB modulation. Sci Adv. 2020;6(37):eaaz8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF‐kappaB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1‐2):37‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rius‐Perez S, Perez S, Marti‐Andres P, Monsalve M, Sastre J. Nuclear factor kappa B signaling complexes in acute inflammation. Antioxid Redox Signal. 2020;33(3):145‐165. [DOI] [PubMed] [Google Scholar]
- 144. Singh S, Singh TG. Role of nuclear factor kappa B (NF‐kappaB) signalling in neurodegenerative diseases: an mechanistic approach. Curr Neuropharmacol. 2020;18(10):918‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kang C, Shin WS, Yeo D, Lim W, Zhang T, Ji LL. Anti‐inflammatory effect of avenanthramides via NF‐kappaB pathways in C2C12 skeletal muscle cells. Free Radic Biol Med. 2018;117:30‐36. [DOI] [PubMed] [Google Scholar]
- 146. Wang T, Wang Y, Liu L, et al. Research progress on sirtuins family members and cell senescence. Eur J Med Chem. 2020;193:112207. [DOI] [PubMed] [Google Scholar]
- 147. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF‐kappaB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5(1):209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lu MC, Zhao J, Liu YT, et al. CPUY192018, a potent inhibitor of the Keap1‐Nrf2 protein‐protein interaction, alleviates renal inflammation in mice by restricting oxidative stress and NF‐kappaB activation. Redox Biol. 2019;26:101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer‐binding protein Nf‐kappa B by a posttranslational mechanism. Cell. 1986;47(6):921‐928. [DOI] [PubMed] [Google Scholar]
- 150. Zannas AS, Jia M, Hafner K, et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF‐kappaB‐driven inflammation and cardiovascular risk. Proc Natl Acad Sci USA. 2019;116(23):11370‐11379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ke W, Hong E, Saito RF, et al. RNA‐DNA fibers and polygons with controlled immunorecognition activate RNAi, FRET and transcriptional regulation of NF‐kappaB in human cells. Nucleic Acids Res. 2019;47(3):1350‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Staal FJ, Roederer M, Herzenberg LA, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci USA. 1990;87(24):9943‐9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF‐kappa B transcription factor and HIV‐1. EMBO J. 1991;10(8):2247‐2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF‐kappaB signaling. Cell Res. 2011;21(1):103‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Halvey PJ, Hansen JM, Johnson JM, Go YM, Samali A, Jones DP. Selective oxidative stress in cell nuclei by nuclear‐targeted d‐amino acid oxidase. Antioxid Redox Signal. 2007;9(7):807‐816. [DOI] [PubMed] [Google Scholar]
- 156. Fong CC, Zhang Y, Zhang Q, et al. Dexamethasone protects RAW264.7 macrophages from growth arrest and apoptosis induced by H2O2 through alteration of gene expression patterns and inhibition of nuclear factor‐kappa B (NF‐kappaB) activity. Toxicology. 2007;236(1‐2):16‐28. [DOI] [PubMed] [Google Scholar]
- 157. Kim SU, Park YH, Min JS, et al. Peroxiredoxin I is a ROS/p38 MAPK‐dependent inducible antioxidant that regulates NF‐kappaB‐mediated iNOS induction and microglial activation. J Neuroimmunol. 2013;259(1‐2):26‐36. [DOI] [PubMed] [Google Scholar]
- 158. Buetler TM, Krauskopf A, Ruegg UT. Role of superoxide as a signaling molecule. News Physiol Sci. 2004;19:120‐123. [DOI] [PubMed] [Google Scholar]
- 159. Flohe L, Toppo S, Cozza G, Ursini F. A comparison of thiol peroxidase mechanisms. Antioxid Redox Signal. 2011;15(3):763‐780. [DOI] [PubMed] [Google Scholar]
- 160. Yoshida Y, Ito N, Shimakawa S, Niki E. Susceptibility of plasma lipids to peroxidation. Biochem Biophys Res Commun. 2003;305(3):747‐753. [DOI] [PubMed] [Google Scholar]
- 161. Wagner D, Przybyla D, Op den Camp R, et al. The genetic basis of singlet oxygen‐induced stress responses of Arabidopsis thaliana . Science. 2004;306(5699):1183‐1185. [DOI] [PubMed] [Google Scholar]
- 162. Koh E, Fluhr R. Singlet oxygen detection in biological systems: uses and limitations. Plant Signal Behav. 2016;11(7):e1192742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88‐98. [DOI] [PubMed] [Google Scholar]
- 164. Rodriguez‐Gutierrez R, Gonzalez‐Gonzalez JG, Zuniga‐Hernandez JA, McCoy RG. Benefits and harms of intensive glycemic control in patients with type 2 diabetes. BMJ. 2019;367:l5887. [DOI] [PubMed] [Google Scholar]
- 165. Al‐Aubaidy HA, Jelinek HF. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur J Endocrinol. 2011;164(6):899‐904. [DOI] [PubMed] [Google Scholar]
- 166. Girona J, Manzanares JM, Marimon F, et al. Oxidized to non‐oxidized lipoprotein ratios are associated with arteriosclerosis and the metabolic syndrome in diabetic patients. Nutr Metab Cardiovasc Dis. 2008;18(5):380‐387. [DOI] [PubMed] [Google Scholar]
- 167. Niwa T, Naito C, Mawjood AH, Imai K. Increased glutathionyl hemoglobin in diabetes mellitus and hyperlipidemia demonstrated by liquid chromatography/electrospray ionization‐mass spectrometry. Clin Chem. 2000;46(1):82‐88. [PubMed] [Google Scholar]
- 168. Gopaul NK, Anggard EE, Mallet AI, Betteridge DJ, Wolff SP, Nourooz‐Zadeh J. Plasma 8‐epi‐PGF2 alpha levels are elevated in individuals with non‐insulin dependent diabetes mellitus. FEBS Lett. 1995;368(2):225‐229. [DOI] [PubMed] [Google Scholar]
- 169. Pandey KB, Mishra N, Rizvi SI. Protein oxidation biomarkers in plasma of type 2 diabetic patients. Clin Biochem. 2010;43(4‐5):508‐511. [DOI] [PubMed] [Google Scholar]
- 170. Davi G, Ciabattoni G, Consoli A, et al. In vivo formation of 8‐iso‐prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99(2):224‐229. [DOI] [PubMed] [Google Scholar]
- 171. Nishikawa T, Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2007;9(3):343‐353. [DOI] [PubMed] [Google Scholar]
- 172. Gray SP, Di Marco E, Okabe J, et al. NADPH oxidase 1 plays a key role in diabetes mellitus‐accelerated atherosclerosis. Circulation. 2013;127(18):1888‐1902. [DOI] [PubMed] [Google Scholar]
- 173. Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol. 2018;19(1):31‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lennicke C, Cocheme HM. Redox regulation of the insulin signalling pathway. Redox Biol. 2021;42:101964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85‐96. [DOI] [PubMed] [Google Scholar]
- 176. Mahadev K, Motoshima H, Wu X, et al. The NAD(P)H oxidase homolog Nox4 modulates insulin‐stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24(5):1844‐1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Schroder K, Wandzioch K, Helmcke I, Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol. 2009;29(2):239‐245. [DOI] [PubMed] [Google Scholar]
- 178. Kaneto H, Katakami N, Matsuhisa M, Matsuoka TA. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. 2010;2010:453892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Onyango AN. Cellular stresses and stress responses in the pathogenesis of insulin resistance. Oxid Med Cell Longev. 2018;2018:4321714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, HO Pae. Mitogen‐activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Klotz L, Sánchez‐Ramos C, Prieto‐Arroyo I, Urbánek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox biology. 2015;6:51‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27(16):2320‐2336. [DOI] [PubMed] [Google Scholar]
- 183. Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005;16(4):183‐189. [DOI] [PubMed] [Google Scholar]
- 184. Forman HJ, Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov. 2021;20(9):689‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Yuan T, Yang T, Chen H, et al. New insights into oxidative stress and inflammation during diabetes mellitus‐accelerated atherosclerosis. Redox Biol. 2019;20:247‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Glavind J, Hartmann S, Clemmesen J, Jessen KE, Dam H. Studies on the role of lipoperoxides in human pathology. II. The presence of peroxidized lipids in the atherosclerotic aorta. Acta Pathol Microbiol Scand. 1952;30(1):1‐6. [DOI] [PubMed] [Google Scholar]
- 187. Suarna C, Dean RT, May J, Stocker R. Human atherosclerotic plaque contains both oxidized lipids and relatively large amounts of alpha‐tocopherol and ascorbate. Arterioscler Thromb Vasc Biol. 1995;15(10):1616‐1624. [DOI] [PubMed] [Google Scholar]
- 188. Martin‐Ventura JL, Rodrigues‐Diez R, Martinez‐Lopez D, Salaices M, Blanco‐Colio LM, Briones AM. Oxidative stress in human atherothrombosis: sources, markers and therapeutic targets. Int J Mol Sci. 2017;18(11):2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Salomon RG, Kaur K, Podrez E, Hoff HF, Krushinsky AV, Sayre LM. HNE‐derived 2‐pentylpyrroles are generated during oxidation of LDL, are more prevalent in blood plasma from patients with renal disease or atherosclerosis, and are present in atherosclerotic plaques. Chem Res Toxicol. 2000;13(7):557‐564. [DOI] [PubMed] [Google Scholar]
- 190. Toshima S, Hasegawa A, Kurabayashi M, et al. Circulating oxidized low density lipoprotein levels. A biochemical risk marker for coronary heart disease. Arterioscler Thromb Vasc Biol. 2000;20(10):2243‐2247. [DOI] [PubMed] [Google Scholar]
- 191. Tanaga K, Bujo H, Inoue M, et al. Increased circulating malondialdehyde‐modified LDL levels in patients with coronary artery diseases and their association with peak sizes of LDL particles. Arterioscler Thromb Vasc Biol. 2002;22(4):662‐666. [DOI] [PubMed] [Google Scholar]
- 192. Kotani K, Tashiro J, Yamazaki K, et al. Investigation of MDA‐LDL (malondialdehyde‐modified low‐density lipoprotein) as a prognostic marker for coronary artery disease in patients with type 2 diabetes mellitus. Clin Chim Acta. 2015;450:145‐150. [DOI] [PubMed] [Google Scholar]
- 193. Gniwotta C, Morrow JD, Roberts LJ, 2nd, Kuhn H. Prostaglandin F2‐like compounds, F2‐isoprostanes, are present in increased amounts in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1997;17(11):3236‐3241. [DOI] [PubMed] [Google Scholar]
- 194. Zhang Y, Murugesan P, Huang K, Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol. 2020;17(3):170‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Wei Y, Corbalan‐Campos J, Gurung R, et al. Dicer in macrophages prevents atherosclerosis by promoting mitochondrial oxidative metabolism. Circulation. 2018;138(18):2007‐2020. [DOI] [PubMed] [Google Scholar]
- 196. Di Minno A, Turnu L, Porro B, et al. 8‐Hydroxy‐2‐deoxyguanosine levels and cardiovascular disease: a systematic review and meta‐analysis of the literature. Antioxid Redox Signal. 2016;24(10):548‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Langbein H, Brunssen C, Hofmann A, et al. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J. 2016;37(22):1753‐1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Schurmann C, Rezende F, Kruse C, et al. The NADPH oxidase Nox4 has anti‐atherosclerotic functions. Eur Heart J. 2015;36(48):3447‐3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Di Marco E, Gray SP, Chew P, et al. Differential effects of NOX4 and NOX1 on immune cell‐mediated inflammation in the aortic sinus of diabetic ApoE‐/‐ mice. Clin Sci (Lond). 2016;130(15):1363‐1374. [DOI] [PubMed] [Google Scholar]
- 200. Gray SP, Di Marco E, Kennedy K, et al. Reactive oxygen species can provide atheroprotection via NOX4‐dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol. 2016;36(2):295‐307. [DOI] [PubMed] [Google Scholar]
- 201. Di Marco E, Gray SP, Kennedy K, et al. NOX4‐derived reactive oxygen species limit fibrosis and inhibit proliferation of vascular smooth muscle cells in diabetic atherosclerosis. Free Radic Biol Med. 2016;97:556‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Douglas G, Bendall JK, Crabtree MJ, et al. Endothelial‐specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE(‐)/(‐) mice. Cardiovasc Res. 2012;94(1):20‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Chen CH, Leu SJ, Hsu CP, Pan CC, Shyue SK, Lee TS. Atypical antipsychotic drugs deregulate the cholesterol metabolism of macrophage‐foam cells by activating NOX‐ROS‐PPARgamma‐CD36 signaling pathway. Metabolism. 2021;123:154847. [DOI] [PubMed] [Google Scholar]
- 204. Guzik TJ, Chen W, Gongora MC, et al. Calcium‐dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol. 2008;52(22):1803‐1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Manea SA, Vlad ML, Fenyo IM, et al. Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E‐deficient mice; potential implications for human atherosclerosis. Redox Biol. 2020;28:101338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Violi F, Pignatelli P. Clinical application of NOX activity and other oxidative biomarkers in cardiovascular disease: a critical review. Antioxid Redox Signal. 2015;23(5):514‐532. [DOI] [PubMed] [Google Scholar]
- 207. Martinez‐Reyes I, Diebold LP, Kong H, et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol Cell. 2016;61(2):199‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Vendrov AE, Vendrov KC, Smith A, et al. NOX4 NADPH oxidase‐dependent mitochondrial oxidative stress in aging‐associated cardiovascular disease. Antioxid Redox Signal. 2015;23(18):1389‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Mercer JR, Yu E, Figg N, et al. The mitochondria‐targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/‐/ApoE‐/‐ mice. Free Radic Biol Med. 2012;52(5):841‐849. [DOI] [PubMed] [Google Scholar]
- 210. Ma S, Chen J, Feng J, et al. Melatonin ameliorates the progression of atherosclerosis via mitophagy activation and NLRP3 inflammasome inhibition. Oxid Med Cell Longev. 2018;2018:9286458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Li Z, Li Q, Wang L, et al. Targeting mitochondria‐inflammation circle by renal denervation reduces atheroprone endothelial phenotypes and atherosclerosis. Redox Biol. 2021;47:102156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor‐kappaB‐mediated inflammation in macrophages. Circ Res. 2014;114(3):421‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Liu Z, Li J, Lin S, Wu Y, He D, Qu P. PI3K regulates the activation of NLRP3 inflammasome in atherosclerosis through part‐dependent AKT signaling pathway. Exp Anim. 2021;70(4):488‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Durrington PN, Mackness B, Mackness MI. Paraoxonase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21(4):473‐480. [DOI] [PubMed] [Google Scholar]
- 215. Shih DM, Xia YR, Wang XP, et al. Decreased obesity and atherosclerosis in human paraoxonase 3 transgenic mice. Circ Res. 2007;100(8):1200‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. She ZG, Chen HZ, Yan Y, Li H, Liu DP. The human paraoxonase gene cluster as a target in the treatment of atherosclerosis. Antioxid Redox Signal. 2012;16(6):597‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. She ZG, Zheng W, Wei YS, et al. Human paraoxonase gene cluster transgenic overexpression represses atherogenesis and promotes atherosclerotic plaque stability in ApoE‐null mice. Circ Res. 2009;104(10):1160‐1168. [DOI] [PubMed] [Google Scholar]
- 218. Devarajan A, Shih D, Reddy ST. Inflammation, infection, cancer and all that…the role of paraoxonases. Adv Exp Med Biol. 2014;824:33‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Rosenblat M, Volkova N, Ward J, Aviram M. Paraoxonase 1 (PON1) inhibits monocyte‐to‐macrophage differentiation. Atherosclerosis. 2011;219(1):49‐56. [DOI] [PubMed] [Google Scholar]
- 220. Aharoni S, Aviram M, Fuhrman B. Paraoxonase 1 (PON1) reduces macrophage inflammatory responses. Atherosclerosis. 2013;228(2):353‐361. [DOI] [PubMed] [Google Scholar]
- 221. Ng DS, Chu T, Esposito B, Hui P, Connelly PW, Gross PL. Paraoxonase‐1 deficiency in mice predisposes to vascular inflammation, oxidative stress, and thrombogenicity in the absence of hyperlipidemia. Cardiovasc Pathol. 2008;17(4):226‐232. [DOI] [PubMed] [Google Scholar]
- 222. Bernardo I, Bozinovski S, Vlahos R. Targeting oxidant‐dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacol Ther. 2015;155:60‐79. [DOI] [PubMed] [Google Scholar]
- 223. Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379(9823):1341‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Passey SL, Hansen MJ, Bozinovski S, McDonald CF, Holland AE, Vlahos R. Emerging therapies for the treatment of skeletal muscle wasting in chronic obstructive pulmonary disease. Pharmacol Ther. 2016;166:56‐70. [DOI] [PubMed] [Google Scholar]
- 225. Decramer M, Janssens W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir Med. 2013;1(1):73‐83. [DOI] [PubMed] [Google Scholar]
- 226. MacDonald MI, Shafuddin E, King PT, Chang CL, Bardin PG, Hancox RJ. Cardiac dysfunction during exacerbations of chronic obstructive pulmonary disease. Lancet Respir Med. 2016;4(2):138‐148. [DOI] [PubMed] [Google Scholar]
- 227. Barnes PJ. New anti‐inflammatory targets for chronic obstructive pulmonary disease. Nat Rev Drug Discov. 2013;12(7):543‐559. [DOI] [PubMed] [Google Scholar]
- 228. Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):71‐86. [DOI] [PubMed] [Google Scholar]
- 229. Hillas G, Nikolakopoulou S, Hussain S, Vassilakopoulos T. Antioxidants and mucolytics in COPD management: when (if ever) and in whom? Curr Drug Targets. 2013;14(2):225‐234. [DOI] [PubMed] [Google Scholar]
- 230. Rahman I. The role of oxidative stress in the pathogenesis of COPD: implications for therapy. Treat Respir Med. 2005;4(3):175‐200. [DOI] [PubMed] [Google Scholar]
- 231. Stanojkovic I, Kotur‐Stevuljevic J, Milenkovic B, et al. Pulmonary function, oxidative stress and inflammatory markers in severe COPD exacerbation. Respir Med. 2011;105(1):S31‐S37. [DOI] [PubMed] [Google Scholar]
- 232. Montuschi P, Collins JV, Ciabattoni G, et al. Exhaled 8‐isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1175‐1177. [DOI] [PubMed] [Google Scholar]
- 233. Rahman I, van Schadewijk AA, Crowther AJ, et al. 4‐Hydroxy‐2‐nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(4):490‐495. [DOI] [PubMed] [Google Scholar]
- 234. Igishi T, Hitsuda Y, Kato K, et al. Elevated urinary 8‐hydroxydeoxyguanosine, a biomarker of oxidative stress, and lack of association with antioxidant vitamins in chronic obstructive pulmonary disease. Respirology. 2003;8(4):455‐460. [DOI] [PubMed] [Google Scholar]
- 235. Antus B, Harnasi G, Drozdovszky O, Barta I. Monitoring oxidative stress during chronic obstructive pulmonary disease exacerbations using malondialdehyde. Respirology. 2014;19(1):74‐79. [DOI] [PubMed] [Google Scholar]
- 236. Taniguchi A, Tsuge M, Miyahara N, Tsukahara H. Reactive oxygen species and antioxidative defense in chronic obstructive pulmonary disease. Antioxidants (Basel). 2021;10(10):1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Zhu J, Kovacs L, Han W, et al. Reactive oxygen species‐dependent calpain activation contributes to airway and pulmonary vascular remodeling in chronic obstructive pulmonary disease. Antioxid Redox Signal. 2019;31(12):804‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Eapen MS, Kota A, Vindin H, et al. Apoptosis signal‐regulating kinase 1 inhibition attenuates human airway smooth muscle growth and migration in chronic obstructive pulmonary disease. Clin Sci (Lond). 2018;132(14):1615‐1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Shi J, Li H, Yuan C, Luo M, Wei J, Liu X. Cigarette smoke‐induced acquired dysfunction of cystic fibrosis transmembrane conductance regulator in the pathogenesis of chronic obstructive pulmonary disease. Oxid Med Cell Longev. 2018;2018:6567578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Bodas M, Silverberg D, Walworth K, Brucia K, Vij N. Augmentation of S‐nitrosoglutathione controls cigarette smoke‐induced inflammatory‐oxidative stress and chronic obstructive pulmonary disease‐emphysema pathogenesis by restoring cystic fibrosis transmembrane conductance regulator function. Antioxid Redox Signal. 2017;27(7):433‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Araya J, Tsubouchi K, Sato N, et al. PRKN‐regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy. 2019;15(3):510‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Ito S, Araya J, Kurita Y, et al. PARK2‐mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11(3):547‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Mizumura K, Gon Y. Iron‐regulated reactive oxygen species production and programmed cell death in chronic obstructive pulmonary disease. Antioxidants (Basel). 2021;10(10):1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Barnes P. Oxidative stress‐based therapeutics in COPD. Redox biology. 2020;33:101544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Belchamber KBR, Singh R, Batista CM, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J. 2019;54(4):1802244. [DOI] [PubMed] [Google Scholar]
- 246. Wiegman CH, Michaeloudes C, Haji G, et al. Oxidative stress‐induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136(3):769‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Even B, Fayad‐Kobeissi S, Gagliolo JM, et al. Heme oxygenase‐1 induction attenuates senescence in chronic obstructive pulmonary disease lung fibroblasts by protecting against mitochondria dysfunction. Aging Cell. 2018;17(6):e12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Puente‐Maestu L, Tejedor A, Lazaro A, et al. Site of mitochondrial reactive oxygen species production in skeletal muscle of chronic obstructive pulmonary disease and its relationship with exercise oxidative stress. Am J Respir Cell Mol Biol. 2012;47(3):358‐362. [DOI] [PubMed] [Google Scholar]
- 249. Griffith B, Pendyala S, Hecker L, Lee PJ, Natarajan V, Thannickal VJ. NOX enzymes and pulmonary disease. Antioxid Redox Signal. 2009;11(10):2505‐2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Schiffers C, Reynaert NL, Wouters EFM, van der Vliet A. Redox dysregulation in aging and COPD: role of NOX enzymes and implications for antioxidant strategies. Antioxidants (Basel). 2021;10(11):1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Zhuan B, Yu Y, Yang Z, Zhao X, Li P. Mechanisms of oxidative stress effects of the NADPH oxidase‐ROS‐NF‐kappaB transduction pathway and VPO1 on patients with chronic obstructive pulmonary disease combined with pulmonary hypertension. Eur Rev Med Pharmacol Sci. 2017;21(15):3459‐3464. [PubMed] [Google Scholar]
- 252. Li ZM, Xu SY, Feng YZ, et al. The role of NOX4 in pulmonary diseases. J Cell Physiol. 2021;236(3):1628‐1637. [DOI] [PubMed] [Google Scholar]
- 253. Carnesecchi S, Deffert C, Pagano A, et al. NADPH oxidase‐1 plays a crucial role in hyperoxia‐induced acute lung injury in mice. Am J Respir Crit Care Med. 2009;180(10):972‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Carnesecchi S, Deffert C, Donati Y, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. 2011;15(3):607‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Wan WY, Hollins F, Haste L, et al. NADPH oxidase‐4 overexpression is associated with epithelial ciliary dysfunction in neutrophilic asthma. Chest. 2016;149(6):1445‐1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Hao B, Sun R, Guo X, et al. NOX4‐derived ROS promotes collagen I deposition in bronchial smooth muscle cells by activating noncanonical p38MAPK/Akt‐mediated TGF‐beta signaling. Oxid Med Cell Longev. 2021;2021:6668971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20(3):148‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. [DOI] [PubMed] [Google Scholar]
- 259. Schapira AH, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet. 2014;384(9942):545‐555. [DOI] [PubMed] [Google Scholar]
- 260. Dong Y, D'Mello C, Pinsky W, et al. Oxidized phosphatidylcholines found in multiple sclerosis lesions mediate neurodegeneration and are neutralized by microglia. Nat Neurosci. 2021;24(4):489‐503. [DOI] [PubMed] [Google Scholar]
- 261. Graff‐Radford J, Yong KXX, Apostolova LG, et al. New insights into atypical Alzheimer's disease in the era of biomarkers. Lancet Neurol. 2021;20(3):222‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Tang M, Ryman DC, McDade E, et al. Neurological manifestations of autosomal dominant familial Alzheimer's disease: a comparison of the published literature with the Dominantly Inherited Alzheimer Network observational study (DIAN‐OBS). Lancet Neurol. 2016;15(13):1317‐1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Quiroz YT, Zetterberg H, Reiman EM, et al. Plasma neurofilament light chain in the presenilin 1 E280A autosomal dominant Alzheimer's disease kindred: a cross‐sectional and longitudinal cohort study. Lancet Neurol. 2020;19(6):513‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Eid A, Mhatre I, Richardson JR. Gene‐environment interactions in Alzheimer's disease: a potential path to precision medicine. Pharmacol Ther. 2019;199:173‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Dunn AR, O'Connell KMS, Kaczorowski CC. Gene‐by‐environment interactions in Alzheimer's disease and Parkinson's disease. Neurosci Biobehav Rev. 2019;103:73‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Cryan JF, O'Riordan KJ, Sandhu K, Peterson V, Dinan TG. The gut microbiome in neurological disorders. Lancet Neurol. 2020;19(2):179‐194. [DOI] [PubMed] [Google Scholar]
- 267. Fyfe I. High‐salt diet promotes Alzheimer disease‐like changes. Nat Rev Neurol. 2020;16(1):2‐3. [DOI] [PubMed] [Google Scholar]
- 268. Pan G, King A, Wu F, et al. The potential roles of genetic factors in predicting ageing‐related cognitive change and Alzheimer's disease. Ageing Res Rev. 2021;70:101402. [DOI] [PubMed] [Google Scholar]
- 269. Xia X, Jiang Q, McDermott J, Han JJ. Aging and Alzheimer's disease: comparison and associations from molecular to system level. Aging Cell. 2018;17(5):e12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Oveisgharan S, Arvanitakis Z, Yu L, Farfel J, Schneider JA, Bennett DA. Sex differences in Alzheimer's disease and common neuropathologies of aging. Acta Neuropathol. 2018;136(6):887‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Ferretti MT, Iulita MF, Cavedo E, et al. Sex differences in Alzheimer disease—the gateway to precision medicine. Nat Rev Neurol. 2018;14(8):457‐469. [DOI] [PubMed] [Google Scholar]
- 272. Kunkle BW, Schmidt M, Klein HU, et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta‐analysis. JAMA Neurol. 2021;78(1):102‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Montine TJ, Beal MF, Cudkowicz ME, et al. Increased CSF F2‐isoprostane concentration in probable AD. Neurology. 1999;52(3):562‐565. [DOI] [PubMed] [Google Scholar]
- 274. Pratico D, MYL V, Trojanowski JQ, Rokach J, Fitzgerald GA. Increased F2‐isoprostanes in Alzheimer's disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998;12(15):1777‐1783. [DOI] [PubMed] [Google Scholar]
- 275. Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer's disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging. 2001;22(2):187‐194. [DOI] [PubMed] [Google Scholar]
- 276. Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4‐hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging. 1997;18(5):457‐461. [DOI] [PubMed] [Google Scholar]
- 277. Perluigi M, Sultana R, Cenini G, et al. Redox proteomics identification of 4‐hydroxynonenal‐modified brain proteins in Alzheimer's disease: role of lipid peroxidation in Alzheimer's disease pathogenesis. Proteomics Clin Appl. 2009;3(6):682‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69(2):155‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93(4):953‐962. [DOI] [PubMed] [Google Scholar]
- 280. Butterfield DA, Hensley K, Harris M, Mattson M, Carney J. beta‐Amyloid peptide free radical fragments initiate synaptosomal lipoperoxidation in a sequence‐specific fashion: implications to Alzheimer's disease. Biochem Biophys Res Commun. 1994;200(2):710‐715. [DOI] [PubMed] [Google Scholar]
- 281. Simpson DSA, Oliver PL. ROS generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants (Basel). 2020;9(8):743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox‐generated free radicals. Proc Natl Acad Sci U S A. 1997;94(18):9866‐9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Swerdlow RH, Parks JK, Cassarino DS, et al. Cybrids in Alzheimer's disease: a cellular model of the disease? Neurology. 1997;49(4):918‐925. [DOI] [PubMed] [Google Scholar]
- 284. Osama A, Zhang J, Yao J, Yao X, Fang J. Nrf2: a dark horse in Alzheimer's disease treatment. Ageing Res Rev. 2020;64:101206. [DOI] [PubMed] [Google Scholar]
- 285. Branca C, Ferreira E, Nguyen TV, Doyle K, Caccamo A, Oddo S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer's disease. Hum Mol Genet. 2017;26(24):4823‐4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Rojo AI, Pajares M, Rada P, et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer's patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Jan A, Jansonius B, Delaidelli A, et al. eEF2K inhibition blocks Abeta42 neurotoxicity by promoting an NRF2 antioxidant response. Acta Neuropathol. 2017;133(1):101‐119. [DOI] [PubMed] [Google Scholar]
- 288. Wang CY, Zhang Q, Xun Z, et al. Increases of iASPP‐Keap1 interaction mediated by syringin enhance synaptic plasticity and rescue cognitive impairments via stabilizing Nrf2 in Alzheimer's models. Redox Biol. 2020;36:101672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Fao L, Mota SI, Rego AC. Shaping the Nrf2‐ARE‐related pathways in Alzheimer's and Parkinson's diseases. Ageing Res Rev. 2019;54:100942. [DOI] [PubMed] [Google Scholar]
- 290. Ochalek A, Mihalik B, Avci HX, et al. Neurons derived from sporadic Alzheimer's disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res Ther. 2017;9(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Talman V, Pascale A, Jantti M, Amadio M, Tuominen RK. Protein kinase C activation as a potential therapeutic strategy in Alzheimer's disease: is there a role for embryonic lethal abnormal vision‐like proteins? Basic Clin Pharmacol Toxicol. 2016;119(2):149‐160. [DOI] [PubMed] [Google Scholar]
- 292. Sotolongo K, Ghiso J, Rostagno A. Nrf2 activation through the PI3K/GSK‐3 axis protects neuronal cells from Abeta‐mediated oxidative and metabolic damage. Alzheimers Res Ther. 2020;12(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Li S, Zhao X, Lazarovici P, Zheng W. Artemether activation of AMPK/GSK3beta(ser9)/Nrf2 signaling confers neuroprotection towards beta‐amyloid‐induced neurotoxicity in 3xTg Alzheimer's mouse model. Oxid Med Cell Longev. 2019;2019:1862437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Rong H, Liang Y, Niu Y. Rosmarinic acid attenuates beta‐amyloid‐induced oxidative stress via Akt/GSK‐3beta/Fyn‐mediated Nrf2 activation in PC12 cells. Free Radic Biol Med. 2018;120:114‐123. [DOI] [PubMed] [Google Scholar]
- 295. Bahn G, Park JS, Yun UJ, et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer's models. Proc Natl Acad Sci USA. 2019;116(25):12516‐12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Kim SY, Chae CW, Lee HJ, et al. Sodium butyrate inhibits high cholesterol‐induced neuronal amyloidogenesis by modulating NRF2 stabilization‐mediated ROS levels: involvement of NOX2 and SOD1. Cell Death Dis. 2020;11(6):469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Almansoub H, Tang H, Wu Y, et al. Tau abnormalities and the potential therapy in Alzheimer's disease. J Alzheimers Dis. 2019;67(1):13‐33. [DOI] [PubMed] [Google Scholar]
- 298. Aschenbrenner AJ, Gordon BA, Benzinger TLS, Morris JC, Hassenstab JJ. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology. 2018;91(9):e859‐e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76(8):915‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Engel T, Hernandez F, Avila J, Lucas JJ. Full reversal of Alzheimer's disease‐like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase‐3. J Neurosci. 2006;26(19):5083‐5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Hernandez F, Borrell J, Guaza C, Avila J, Lucas JJ. Spatial learning deficit in transgenic mice that conditionally over‐express GSK‐3beta in the brain but do not form tau filaments. J Neurochem. 2002;83(6):1529‐1533. [DOI] [PubMed] [Google Scholar]
- 302. Wang XJ, Qi L, Cheng YF, et al. PINK1 overexpression prevents forskolin‐induced tau hyperphosphorylation and oxidative stress in a rat model of Alzheimer's disease. Acta Pharmacol Sin. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Farr SA, Ripley JL, Sultana R, et al. Antisense oligonucleotide against GSK‐3beta in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic Biol Med. 2014;67:387‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Cuadrado A, Kugler S, Lastres‐Becker I. Pharmacological targeting of GSK‐3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018;14:522‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Bonet‐Costa V, Pomatto LC, Davies KJ. The proteasome and oxidative stress in Alzheimer's disease. Antioxid Redox Signal. 2016;25(16):886‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Zheng X, Wang W, Liu R, Huang H, Zhang R, Sun L. Effect of p62 on tau hyperphosphorylation in a rat model of Alzheimer's disease. Neural Regen Res. 2012;7(17):1304‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Gu L, Yu Q, Li Q, Zhang L, Lu H, Zhang X. Andrographolide protects PC12 cells against beta‐amyloid‐induced autophagy‐associated cell death through activation of the Nrf2‐mediated p62 signaling pathway. Int J Mol Sci. 2018;19(9):2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Xu Y, Zhang S, Zheng H. The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy. 2019;15(4):583‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun. 2014;5:3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Tang M, Ji C, Pallo S, Rahman I, Johnson GVW. Nrf2 mediates the expression of BAG3 and autophagy cargo adaptor proteins and tau clearance in an age‐dependent manner. Neurobiol Aging. 2018;63:128‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Kim S, Choi KJ, Cho SJ, et al. Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors. Sci Rep. 2016;6:24933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Kim HV, Kim HY, Ehrlich HY, Choi SY, Kim DJ, Kim Y. Amelioration of Alzheimer's disease by neuroprotective effect of sulforaphane in animal model. Amyloid. 2013;20(1):7‐12. [DOI] [PubMed] [Google Scholar]
- 313. Tapias V, Jainuddin S, Ahuja M, et al. Benfotiamine treatment activates the Nrf2/ARE pathway and is neuroprotective in a transgenic mouse model of tauopathy. Hum Mol Genet. 2018;27(16):2874‐2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 315. Hornsveld M, Dansen TB. The hallmarks of cancer from a redox perspective. Antioxid Redox Signal. 2016;25(6):300‐325. [DOI] [PubMed] [Google Scholar]
- 316. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50‐64. [DOI] [PubMed] [Google Scholar]
- 317. Kudo Y, Sugimoto M, Arias E, et al. PKClambda/iota loss induces autophagy, oxidative phosphorylation, and NRF2 to promote liver cancer progression. Cancer Cell. 2020;38(2):247‐262 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Cheung EC, DeNicola GM, Nixon C, et al. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell. 2020;37(2):168‐182 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Cui Q, Wang JQ, Assaraf YG, et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1‐25. [DOI] [PubMed] [Google Scholar]
- 320. Zhang Z, Qin S, Chen Y, et al. Inhibition of NPC1L1 disrupts adaptive responses of drug‐tolerant persister cells to chemotherapy. EMBO Mol Med. 2022;14(2):e14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Kalyanaraman B, Cheng G, Hardy M, Ouari O, Bennett B, Zielonka J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018;15:347‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Kim J, Kim J, Bae JS. ROS homeostasis and metabolism: a critical liaison for cancer therapy. Exp Mol Med. 2016;48(11):e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Parascandolo A, Laukkanen MO. Carcinogenesis and reactive oxygen species signaling: interaction of the NADPH oxidase NOX1‐5 and superoxide dismutase 1–3 signal transduction pathways. Antioxid Redox Signal. 2019;30(3):443‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Chan HP, Tran V, Lewis C, Thomas PS. Elevated levels of oxidative stress markers in exhaled breath condensate. J Thorac Oncol. 2009;4(2):172‐178. [DOI] [PubMed] [Google Scholar]
- 326. Ohtake S, Kawahara T, Ishiguro Y, et al. Oxidative stress marker 8‐hydroxyguanosine is more highly expressed in prostate cancer than in benign prostatic hyperplasia. Mol Clin Oncol. 2018;9(3):302‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. An AR, Kim KM, Park HS, et al. Association between expression of 8‐OHdG and cigarette smoking in non‐small cell lung cancer. J Pathol Transl Med. 2019;53(4):217‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Myant KB, Cammareri P, McGhee EJ, et al. ROS production and NF‐kappaB activation triggered by RAC1 facilitate WNT‐driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12(6):761‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Canli O, Nicolas AM, Gupta J, et al. Myeloid cell‐derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell. 2017;32(6):869‐883 e5. [DOI] [PubMed] [Google Scholar]
- 330. Wang C, Wang Z, Liu W, Ai Z. ROS‐generating oxidase NOX1 promotes the self‐renewal activity of CD133+ thyroid cancer cells through activation of the Akt signaling. Cancer Lett. 2019;447:154‐163. [DOI] [PubMed] [Google Scholar]
- 331. Yang J, Antin P, Berx G, et al. Guidelines and definitions for research on epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21(6):341‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Chang CH, Pauklin S. ROS and TGFbeta: from pancreatic tumour growth to metastasis. J Exp Clin Cancer Res. 2021;40(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Tobar N, Villar V, Santibanez JF. ROS‐NFkappaB mediates TGF‐beta1‐induced expression of urokinase‐type plasminogen activator, matrix metalloproteinase‐9 and cell invasion. Mol Cell Biochem. 2010;340(1‐2):195‐202. [DOI] [PubMed] [Google Scholar]
- 334. Lee MK, Pardoux C, Hall MC, et al. TGF‐beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26(17):3957‐3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Lam CR, Tan C, Teo Z, et al. Loss of TAK1 increases cell traction force in a ROS‐dependent manner to drive epithelial‐mesenchymal transition of cancer cells. Cell Death Dis. 2013;4(10):e848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Zhu P, Tan MJ, Huang RL, et al. Angiopoietin‐like 4 protein elevates the prosurvival intracellular O2(‐):H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell. 2011;19(3):401‐415. [DOI] [PubMed] [Google Scholar]
- 337. Zheng Y, Miyamoto DT, Wittner BS, et al. Expression of beta‐globin by cancer cells promotes cell survival during blood‐borne dissemination. Nat Commun. 2017;8:14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Sprouse ML, Welte T, Boral D, et al. PMN‐MDSCs enhance CTC metastatic properties through reciprocal interactions via ROS/Notch/Nodal signaling. Int J Mol Sci. 2019;20(8):1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Zhang L, Yao Y, Zhang S, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med. 2019;11(491):eaau1167. [DOI] [PubMed] [Google Scholar]
- 340. Lee KM, Giltnane JM, Balko JM, et al. MYC and MCL1 cooperatively promote chemotherapy‐resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017;26(4):633‐647 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143‐156. [DOI] [PubMed] [Google Scholar]
- 342. Sancho P, Burgos‐Ramos E, Tavera A, et al. MYC/PGC‐1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22(4):590‐605. [DOI] [PubMed] [Google Scholar]
- 343. Guieze R, Liu VM, Rosebrock D, et al. Mitochondrial reprogramming underlies resistance to BCL‐2 inhibition in lymphoid malignancies. Cancer Cell. 2019;36(4):369‐384 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Pei S, Pollyea DA, Gustafson A, et al. Monocytic subclones confer resistance to venetoclax‐based therapy in patients with acute myeloid leukemia. Cancer Discov. 2020;10(4):536‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Jones CL, Stevens BM, Pollyea DA, et al. Nicotinamide metabolism mediates resistance to venetoclax in relapsed acute myeloid leukemia stem cells. Cell Stem Cell. 2020;27(5):748‐764 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Guo Z, Wang G, Wu B, et al. DCAF1 regulates Treg senescence via the ROS axis during immunological aging. J Clin Invest. 2020;130(11):5893‐5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Pomatto LCD, Davies KJA. The role of declining adaptive homeostasis in ageing. J Physiol. 2017;595(24):7275‐7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Yamamoto M, Kensler T, Motohashi H. The KEAP1‐NRF2 system: a thiol‐based sensor‐effector apparatus for maintaining redox homeostasis. Physiologicl Rev. 2018;98(3):1169‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Srinivasan V, Kriete A, Sacan A, Jazwinski SM. Comparing the yeast retrograde response and NF‐kappaB stress responses: implications for aging. Aging Cell. 2010;9(6):933‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Nejabati HR, Schmeisser K, Shahnazi V, et al. N1‐methylnicotinamide: an anti‐ovarian aging hormetin? Ageing Res Rev. 2020;62:101131. [DOI] [PubMed] [Google Scholar]
- 352. Weir HJ, Yao P, Huynh FK, et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 2017;26(6):884‐896 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Hanzen S, Vielfort K, Yang J, et al. Lifespan control by redox‐dependent recruitment of chaperones to misfolded proteins. Cell. 2016;166(1):140‐151. [DOI] [PubMed] [Google Scholar]
- 354. Zhang H, Li C, Wen D, et al. Melatonin improves the quality of maternally aged oocytes by maintaining intercellular communication and antioxidant metabolite supply. Redox Biol. 2022;49:102215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Saxena S, Vekaria H, Sullivan PG, Seifert AW. Connective tissue fibroblasts from highly regenerative mammals are refractory to ROS‐induced cellular senescence. Nat Commun. 2019;10(1):4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Park SJ, Gavrilova O, Brown AL, et al. DNA‐PK promotes the mitochondrial, metabolic, and physical decline that occurs during aging. Cell Metab. 2017;25(5):1135‐1146 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Hughes CE, Coody TK, Jeong MY, Berg JA, Winge DR, Hughes AL. Cysteine toxicity drives age‐related mitochondrial decline by altering iron homeostasis. Cell. 2020;180(2):296‐310 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Xu W, Li L, Sun J, et al. Putrescine delays postovulatory aging of mouse oocytes by upregulating PDK4 expression and improving mitochondrial activity. Aging (Albany N Y). 2018;10(12):4093‐4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Wang J, Li S, Wang J, et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging (Albany N Y). 2020;12(1):650‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Joy J, Barrio L, Santos‐Tapia C, et al. Proteostasis failure and mitochondrial dysfunction leads to aneuploidy‐induced senescence. Dev Cell. 2021;56(14):2043‐2058 e7. [DOI] [PubMed] [Google Scholar]
- 361. Pinto M, Pickrell AM, Wang X, et al. Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS‐dependent but p53/p21‐independent manner. Cell Death Differ. 2017;24(2):288‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Lennicke C, Cocheme HM. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol Cell. 2021;81(18):3691‐3707. [DOI] [PubMed] [Google Scholar]
- 363. Moosmann B, Behl C. Mitochondrially encoded cysteine predicts animal lifespan. Aging Cell. 2008;7(1):32‐46. [DOI] [PubMed] [Google Scholar]
- 364. Xiao H, Jedrychowski MP, Schweppe DK, et al. A quantitative tissue‐specific landscape of protein redox regulation during aging. Cell. 2020;180(5):968‐983 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Page MM, Robb EL, Salway KD, Stuart JA. Mitochondrial redox metabolism: aging, longevity and dietary effects. Mech Ageing Dev. 2010;131(4):242‐252. [DOI] [PubMed] [Google Scholar]
- 366. Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909‐1911. [DOI] [PubMed] [Google Scholar]
- 367. Suhm T, Kaimal JM, Dawitz H, et al. Mitochondrial translation efficiency controls cytoplasmic protein homeostasis. Cell Metab. 2018;27(6):1309‐1322 e6. [DOI] [PubMed] [Google Scholar]
- 368. Yee C, Yang W, Hekimi S. The intrinsic apoptosis pathway mediates the pro‐longevity response to mitochondrial ROS in C. elegans. Cell. 2014;157(4):897‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Schroeder EA, Raimundo N, Shadel GS. Epigenetic silencing mediates mitochondria stress‐induced longevity. Cell Metab. 2013;17(6):954‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Scialo F, Sriram A, Fernandez‐Ayala D, et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016;23(4):725‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Palmeira CM, Teodoro JS, Amorim JA, Steegborn C, Sinclair DA, Rolo AP. Mitohormesis and metabolic health: the interplay between ROS, cAMP and sirtuins. Free Radic Biol Med. 2019;141:483‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol. 2010;45(6):410‐418. [DOI] [PubMed] [Google Scholar]
- 373. Bazopoulou D, Knoefler D, Zheng Y, et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature. 2019;576(7786):301‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Margraf A, Ludwig N, Zarbock A, Rossaint J. Systemic inflammatory response syndrome after surgery: mechanisms and protection. Anesth Analg. 2020;131(6):1693‐1707. [DOI] [PubMed] [Google Scholar]
- 375. Melley DD, Evans TW, Quinlan GJ. Redox regulation of neutrophil apoptosis and the systemic inflammatory response syndrome. Clin Sci (Lond). 2005;108(5):413‐424. [DOI] [PubMed] [Google Scholar]
- 376. Podszun MC, Frank J. Impact of vitamin E on redox biomarkers in non‐alcoholic fatty liver disease. Redox Biol. 2021;42:101937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Hong T, Chen Y, Li X, Lu Y. The role and mechanism of oxidative stress and nuclear receptors in the development of NAFLD. Oxid Med Cell Longev. 2021;2021:6889533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Kumar S, Duan Q, Wu R, Harris EN, Su Q. Pathophysiological communication between hepatocytes and non‐parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv Drug Deliv Rev. 2021;176:113869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Granger DN. Role of xanthine oxidase and granulocytes in ischemia‐reperfusion injury. Am J Physiol. 1988;255(6 Pt 2):H1269‐H1275. [DOI] [PubMed] [Google Scholar]
- 380. Matsushima S, Tsutsui H, Sadoshima J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia‐reperfusion. Trends Cardiovasc Med. 2014;24(5):202‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Duilio C, Ambrosio G, Kuppusamy P, DiPaula A, Becker LC, Zweier JL. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280(6):H2649‐H2657. [DOI] [PubMed] [Google Scholar]
- 382. Delanty N, Reilly MP, Pratico D, et al. 8‐epi PGF2 alpha generation during coronary reperfusion. A potential quantitative marker of oxidant stress in vivo. Circulation. 1997;95(11):2492‐2499. [DOI] [PubMed] [Google Scholar]
- 383. Daiber A, Frenis K, Kuntic M, et al. Redox regulatory changes of circadian rhythm by the environmental risk factors traffic noise and air pollution. Antioxid Redox Signal. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Wilking M, Ndiaye M, Mukhtar H, Ahmad N. Circadian rhythm connections to oxidative stress: implications for human health. Antioxid Redox Signal. 2013;19(2):192‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Bus JS, Gibson JE. Paraquat: model for oxidant‐initiated toxicity. Environ Health Perspect. 1984;55:37‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Gaziano JM, Glynn RJ, Christen WG, et al. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2009;301(1):52‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6(221):221ra15. [DOI] [PubMed] [Google Scholar]
- 388. Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med. 2014;371(2):177‐178. [DOI] [PubMed] [Google Scholar]
- 389. D'Andrea GM. Use of antioxidants during chemotherapy and radiotherapy should be avoided. CA Cancer J Clin. 2005;55(5):319‐321. [DOI] [PubMed] [Google Scholar]
- 390. Elbatreek MH, Pachado MP, Cuadrado A, Jandeleit‐Dahm K, Schmidt H. Reactive oxygen comes of age: mechanism‐based therapy of diabetic end‐organ damage. Trends Endocrinol Metab. 2019;30(5):312‐327. [DOI] [PubMed] [Google Scholar]
- 391. De Ruysscher D, Niedermann G, Burnet NG, Siva S, Lee AWM, Hegi‐Johnson F. Radiotherapy toxicity. Nat Rev Dis Primers. 2019;5(1):13. [DOI] [PubMed] [Google Scholar]
- 392. Kerns SL, Ostrer H, Rosenstein BS. Radiogenomics: using genetics to identify cancer patients at risk for development of adverse effects following radiotherapy. Cancer Discov. 2014;4(2):155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Brown PD, Pugh S, Laack NN, et al. Memantine for the prevention of cognitive dysfunction in patients receiving whole‐brain radiotherapy: a randomized, double‐blind, placebo‐controlled trial. Neuro Oncol. 2013;15(10):1429‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Schroeck FR, Jacobs BL, Bhayani SB, Nguyen PL, Penson D, Hu J. Cost of new technologies in prostate cancer treatment: systematic review of costs and cost effectiveness of robotic‐assisted laparoscopic prostatectomy, intensity‐modulated radiotherapy, and proton beam therapy. Eur Urol. 2017;72(5):712‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Guckenberger M, Allgauer M, Appold S, et al. Safety and efficacy of stereotactic body radiotherapy for stage 1 non‐small‐cell lung cancer in routine clinical practice: a patterns‐of‐care and outcome analysis. J Thorac Oncol. 2013;8(8):1050‐1058. [DOI] [PubMed] [Google Scholar]
- 396. Hayes JD, Dinkova‐Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;38(2):167‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690‐714. [DOI] [PubMed] [Google Scholar]
- 398. Wijdeven RH, Pang B, Assaraf YG, Neefjes J. Old drugs, novel ways out: drug resistance toward cytotoxic chemotherapeutics. Drug Resist Updat. 2016;28:65‐81. [DOI] [PubMed] [Google Scholar]
- 399. Agostinis P, Berg K, Cengel KA, et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61(4):250‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Manda G, Isvoranu G, Comanescu MV, Manea A, Debelec Butuner B, Korkmaz KS. The redox biology network in cancer pathophysiology and therapeutics. Redox Biol. 2015;5:347‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Dysart JS, Patterson MS. Characterization of photofrin photobleaching for singlet oxygen dose estimation during photodynamic therapy of MLL cells in vitro. Phys Med Biol. 2005;50(11):2597‐2616. [DOI] [PubMed] [Google Scholar]
- 402. Steiner AZ, Hansen KR, Barnhart KT, et al. The effect of antioxidants on male factor infertility: the males, antioxidants, and infertility (MOXI) randomized clinical trial. Fertil Steril. 2020;113(3):552‐560 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Roberts JM, Myatt L, Spong CY, et al. Vitamins C and E to prevent complications of pregnancy‐associated hypertension. N Engl J Med. 2010;362(14):1282‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Sagel SD, Sontag MK, Anthony MM, Emmett P, Papas KA. Effect of an antioxidant‐rich multivitamin supplement in cystic fibrosis. J Cyst Fibros. 2011;10(1):31‐36. [DOI] [PubMed] [Google Scholar]
- 405. Le Gal K, Ibrahim MX, Wiel C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7(308):308re8. [DOI] [PubMed] [Google Scholar]
- 406. Huang CC, Pan WY, Tseng MT, et al. Enhancement of cell adhesion, retention, and survival of HUVEC/cbMSC aggregates that are transplanted in ischemic tissues by concurrent delivery of an antioxidant for therapeutic angiogenesis. Biomaterials. 2016;74:53‐63. [DOI] [PubMed] [Google Scholar]
- 407. Hu L, Cheng H, Gao Y, et al. Antioxidant N‐acetyl‐l‐cysteine increases engraftment of human hematopoietic stem cells in immune‐deficient mice. Blood. 2014;124(20):e45‐e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Wang B, Yee Aw T, Stokes KY. N‐acetylcysteine attenuates systemic platelet activation and cerebral vessel thrombosis in diabetes. Redox Biol. 2018;14:218‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Marian AJ, Tan Y, Li L, et al. Hypertrophy regression with N‐acetylcysteine in hypertrophic cardiomyopathy (HALT‐HCM): a randomized, placebo‐controlled, double‐blind pilot study. Circ Res. 2018;122(8):1109‐1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Szkudlinska MA, von Frankenberg AD, Utzschneider KM. The antioxidant N‐acetylcysteine does not improve glucose tolerance or beta‐cell function in type 2 diabetes. J Diabetes Complications. 2016;30(4):618‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Casas AI, Nogales C, Mucke HAM, et al. On the clinical pharmacology of reactive oxygen species. Pharmacol Rev. 2020;72(4):801‐828. [DOI] [PubMed] [Google Scholar]
- 412. Yan J, Tie G, Wang S, et al. Diabetes impairs wound healing by Dnmt1‐dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat Commun. 2018;9(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Infanger DW, Cao X, Butler SD, et al. Silencing nox4 in the paraventricular nucleus improves myocardial infarction‐induced cardiac dysfunction by attenuating sympathoexcitation and periinfarct apoptosis. Circ Res. 2010;106(11):1763‐1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Patel PH, Penalva C, Kardorff M, et al. Damage sensing by a Nox‐Ask1‐MKK3‐p38 signaling pathway mediates regeneration in the adult Drosophila midgut. Nat Commun. 2019;10(1):4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Aoyama T, Paik YH, Watanabe S, et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012;56(6):2316‐2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Laleu B, Gaggini F, Orchard M, et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2010;53(21):7715‐7730. [DOI] [PubMed] [Google Scholar]
- 417. Deliyanti D, Wilkinson‐Berka JL. Inhibition of NOX1/4 with GKT137831: a potential novel treatment to attenuate neuroglial cell inflammation in the retina. J Neuroinflammation. 2015;12:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Hosseini M, Rezvani HR, Aroua N, et al. Targeting myeloperoxidase disrupts mitochondrial redox balance and overcomes cytarabine resistance in human acute myeloid leukemia. Cancer Res. 2019;79(20):5191‐5203. [DOI] [PubMed] [Google Scholar]
- 419. Kim HJ, Wei Y, Wojtkiewicz GR, Lee JY, Moskowitz MA, Chen JW. Reducing myeloperoxidase activity decreases inflammation and increases cellular protection in ischemic stroke. J Cereb Blood Flow Metab. 2019;39(9):1864‐1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Kim H, Wei Y, Lee JY, et al. Myeloperoxidase inhibition increases neurogenesis after ischemic stroke. J Pharmacol Exp Ther. 2016;359(2):262‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Tiyerili V, Camara B, Becher MU, et al. Neutrophil‐derived myeloperoxidase promotes atherogenesis and neointima formation in mice. Int J Cardiol. 2016;204:29‐36. [DOI] [PubMed] [Google Scholar]
- 422. Chen B, Lu Y, Chen Y, Cheng J. The role of Nrf2 in oxidative stress‐induced endothelial injuries. J Endocrinol. 2015;225(3):R83‐R99. [DOI] [PubMed] [Google Scholar]
- 423. Liu TS, Pei YH, Peng YP, Chen J, Jiang SS, Gong JB. Oscillating high glucose enhances oxidative stress and apoptosis in human coronary artery endothelial cells. J Endocrinol Invest. 2014;37(7):645‐651. [DOI] [PubMed] [Google Scholar]
- 424. Huang K, Huang J, Xie X, et al. Sirt1 resists advanced glycation end products‐induced expressions of fibronectin and TGF‐beta1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic Biol Med. 2013;65:528‐540. [DOI] [PubMed] [Google Scholar]
- 425. Axelsson AS, Tubbs E, Mecham B, et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci Transl Med. 2017;9(394):eaah4477. [DOI] [PubMed] [Google Scholar]
- 426. Pauletti A, Terrone G, Shekh‐Ahmad T, et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain. 2019;142(7):e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Kerns ML, Hakim JM, Lu RG, et al. Oxidative stress and dysfunctional NRF2 underlie pachyonychia congenita phenotypes. J Clin Invest. 2016;126(6):2356‐2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Choi SH, Kim BG, Robinson J, et al. Synthetic triterpenoid induces 15‐PGDH expression and suppresses inflammation‐driven colon carcinogenesis. J Clin Invest. 2014;124(6):2472‐2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Tran K, Risingsong R, Royce DB, et al. The combination of the histone deacetylase inhibitor vorinostat and synthetic triterpenoids reduces tumorigenesis in mouse models of cancer. Carcinogenesis. 2013;34(1):199‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Schmitt A, Xu W, Bucher P, et al. Dimethyl fumarate induces ferroptosis and impairs NF‐kappaB/STAT3 signaling in DLBCL. Blood. 2021;138(10):871‐884. [DOI] [PubMed] [Google Scholar]
- 431. Liebmann M, Korn L, Janoschka C, et al. Dimethyl fumarate treatment restrains the antioxidative capacity of T cells to control autoimmunity. Brain. 2021;144(10):3126‐3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Sun X, Suo X, Xia X, Yu C, Dou Y. Dimethyl fumarate is a potential therapeutic option for Alzheimer's disease. J Alzheimers Dis. 2022;85(1):443‐456. [DOI] [PubMed] [Google Scholar]
- 433. Olagnier D, Farahani E, Thyrsted J, et al. SARS‐CoV2‐mediated suppression of NRF2‐signaling reveals potent antiviral and anti‐inflammatory activity of 4‐octyl‐itaconate and dimethyl fumarate. Nat Commun. 2020;11(1):4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.