

Abstract
Cancer-secreted, extracellular vesicle (EV)-encapsulated miRNAs enable cancer cells to communicate with each other and with non-cancerous cells in tumor pathogenesis and response to therapies. Here we show that treatment with a sub-lethal dose of chemotherapeutic agents induces breast cancer cells to secrete EV with the capacity to stimulate a cancer stem-like cell (CSC) phenotype, rendering cancer cells resistance to therapy. Chemotherapy induced breast cancer cells to secrete multiple EV miRNAs including miR-9–5p, miR-195–5p, and miR-203a-3p, which simultaneously targeted the transcription factor One Cut Homeobox 2 (ONECUT2), leading to induction of CSC traits and expression of stemness-associated genes including NOTCH1, SOX9, NANOG, OCT4, and SOX2. Inhibition of these miRNAs or restoration of ONECUT2 expression abolished the CSC-stimulating effect of EV from chemotherapy-treated cancer cells. In mice bearing xenograft mammary tumors, docetaxel treatment caused elevations of miR-9–5p, miR-195–5p, and miR-203a-3p in circulating EV and decreased ONECUT2 expression and increased levels of stemness-associated genes. These effects following chemotherapy were diminished in tumors deficient in exosome secretion. In human breast tumors, neoadjuvant chemotherapy decreased ONECUT2 expression in tumor cells. Our results indicate a mechanism by which cancer cells communicate with each other and self-adapt to survive in response to cytotoxic treatment. Targeting these adaptation mechanisms along with chemotherapy, such as by blocking the EV miRNA-ONECUT2 axis, represents a potential strategy to maximize the anti-cancer effect of chemotherapy and reduce chemoresistance in cancer management.
Keywords: Chemotherapy, Cancer stem-like cells, Breast cancer, Extracellular vesicles, miRNA, ONECUT2
Introduction
Extracellular vesicles (EVs), including exosomes that have an endosomal origin and a typical diameter of 30–150 nm, are released into the interstitial spaces and blood by many cell types including cancer cells (1,2). Serving as vehicles to transfer a variety of cellular materials including RNA, DNA, proteins, and lipids between adjacent or distant cells, EVs have been recognized for their unique role in intercellular communication (3). Many reported functions of EVs are mediated by microRNAs (miRNAs) through post-transcriptionally regulating gene expression in recipient cells (4–9). Cancer-secreted EVs and their cargo have been implicated in the adaptations of cancer and niche cells to promote multiple aspects of tumor progression including tissue invasion, angiogenesis, immune evasion, and metastasis (1,2).
Solid tumors exhibit remarkable intratumoral heterogeneity in cellular composition, tissue architecture (e.g., vasculature), and abundance of matrix proteins, cytokines, metabolites, and therapeutic agents. Cargo of cancer-secreted EVs may mediate the dynamic crosstalk between different cancer cell populations in response to environmental changes, thereby contributing to cancer evolution. We set out to examine the hypothesis that under chemotherapy stress, a subset of cancer cells may alter their secreted miRNAome to reprogram other cancer cells that would survive and remain in the tumor after therapy (e.g., those not exposed to a lethal dose of drug or exhibiting intrinsic resistance). These dynamic intercellular events would best represent the scenario of preoperative or neoadjuvant therapy (NT), which is increasingly used in patients with locally advanced or inflammatory breast cancer (BC). Although NT has been shown to significantly improve clinical outcomes in BC and other human cancers (10,11), a significant subset of patients, including up to 50% of triple-negative (TN) or HER2+ BC and >70% of hormone receptor-positive (HR+) BC, do not exhibit pathologic complete response (pCR). In addition, some patients with initial pCR relapse with more advanced disease (12,13). These observations suggest potential chemotherapy-induced cancer cell behaviors contributing to adaptation to the treatment and tumor cell evolution. In this study, we focused on the effect of chemotherapy-induced, BC-secreted EV miRNAs for the dynamic regulation of a cancer stem-like cell (CSC) phenotype, which has been associated with tumor refractoriness and progression (14).
Materials and Methods
Cells and constructs
Human BC cell lines MDA-MB-231 (MDA231), MCF-7, and BT474 were obtained from the American Type Culture Collection (Manassas, VA) and cultured in the recommended media. The non-cancerous mammary epithelial cell line MCF10A was cultured as reported (15). MDA231 cells with stable Rab27a knockdown (MDA231/Rab27aKD) were generated by transducing cells with lentivirus carrying MISSION pLKO.1-puro-RAB27A shRNA (#SHCLNG-NM_004580, TRCN0000380306) followed by puromycin selection, whereas control cells were generated using pLKO.1-puro empty vector (Sigma-Aldrich; St. Louis, MO). The ONECUT2 overexpression plasmid (#RC211951) was purchased from OriGene (Rockville, MD); stable transfection of MDA231 cells was achieved by G418 selection to generate the MDA231/ONECUT2 cells. All cells used herein were tested to be free of mycoplasma and authenticated by short tandem repeat profiling at the beginning and end of study. The miRNA mimics and inhibitors and their corresponding negative controls were purchased from Dharmacon (Lafayette, CO). ONECUT2 siRNAs (FlexiTube GeneSolution/GS including equal mixture of four preselected siRNAs SI04346384 (#1), SI04340049 (#2), SI04181709, and SI00665504, or individual siRNA #1 and #2), negative control siRNAs, and miRNA miScript primers were purchased from Qiagen (Venlo, Netherlands). Transient transfection with RNA or DNA was performed using Lipofectamine RNAiMAX (Invitrogen; Carlsbad, CA) or Lipofectamine 2000 (Invitrogen), respectively, following the manufacturer’s protocols. Cells were seeded on 6-well plates at 2×105 per well one day prior to transfection, and a total of 25 pmol siRNA/miRNA mimics/miRNA inhibitors or 2 µg plasmid DNA with 7.5 µL Lipofectamine were used. MG132 was purchased from Selleckchem (Houston, TX) and used at 10 µM.
EV purification and characterization
EVs secreted by cultured cells were prepared as previously reported (5,7). Conditioned medium was first prepared by incubating cells grown at sub-confluence in growth media containing EV-depleted fetal bovine or horse serum (prepared by overnight ultracentrifugation of medium-diluted sera at 100,000 ×g at 4 °C) for 48 h, and pre-cleared by centrifugation at 500 ×g for 15 min and then at 10,000 ×g for 20 min. EVs were pelleted by ultracentrifugation at 110,000 ×g for 70 min, and washed in PBS under the same ultracentrifugation conditions. Pelleted EVs were resuspended in PBS and characterized by nanoparticle tracking analysis using a NanoSight NS300 system (Malvern; Westborough, MA) and by Western blot (Fig. S1A,B). For cell treatment, 2 μg of EVs (equivalent to those collected from ~5×106 producer cells) based on protein measurement using Pierce™ BCA protein assay kit (Thermo Fisher Scientific; Waltham, MA) were added to 2×105 recipient cells. To fluorescently label EVs, resuspended EVs were incubated with 20 µM CFSE (5(6)-carboxyfluorescein diacetate N-succinimidyl ester; Sigma-Aldrich) at 37 °C for 2 h before the PBS wash. When indicated, EVs were treated with RNase A (100 μg/mL; Thermo Fisher Scientific) and Proteinase K (200 μg/mL; Thermo Fisher Scientific) at 37 °C for 2 h before the PBS wash, and then used to treat cells.
RNA extraction and quantitative reverse transcription PCR (RT-qPCR)
RNA extraction by TRIZOL or TRIZOL LS (Thermo Fisher Scientific) and RT-qPCR were performed as described previously (5,7). Sequences of primers used in RT-qPCR are indicated in Table S1. An annealing temperature of 55 °C was used for all primers. For detection of intracellular miRNAs, U6 small nuclear RNA was used as an internal control. For detection of EV miRNAs, 20 fmol of synthetic cel-miR-39–3p was added to EVs from an equal number of cells during RNA extraction, and the levels of this spike-in control were used for data normalization following miScript miRNA RT-qPCR assays (Qiagen).
Western blot analysis
Cell lysates were resolved by electrophoresis on a 10% SDS polyacrylamide gel. Detection of selected proteins was performed using antibodies listed in Table S1. Scanned images were quantified using ImageJ (Fiji Software) with normalization to the GAPDH blot.
Luciferase reporter assay
PCR-amplified fragments of human ONECUT2 3’UTR were digested with XhoI and NotI, and inserted into the same sites of psiCHECK-2 reporter vector (Promega; Madison, WI) downstream to the Renilla luciferase gene. Sequences of the PCR primers are indicated in Table S1. All plasmid constructs were verified by sequencing. Luciferase activities were measured as described previously (7).
Sphere formation assay
Mammosphere formation assay was performed as previously described (16). Cells were seeded in ultralow attachment 6-well plates (Corning; Corning, NY). The number of spheres (defined as diameter ≥ 70μm) was counted on day 28 (for MDA231), day 15 (for MCF-7), or day 12 (for BT474). Sphere-forming efficiency was calculated based on the number of initially seeded cells.
ALDEFLUOR assay
Cells were analyzed by an ALDEFLUOR assay kit (Stemcell Technologies; Vancouver, BC, Canada) following the manufacturer’s protocol. Flow cytometry was performed on a CyAn ADP flow cytometer (Dako; Carpinteria, CA) and analyzed by FlowJo software (TreeStar; Ashland, OR).
Small RNA-seq and bioinformatics
Illumina sequencing was performed by City of Hope Integrative Genomics Core using RNA extracted from EVs of MDA231 cells previously treated with docetaxel (DTX; 4 nM; Sigma-Aldrich), doxorubicin (DOXO; 125 nM; Sigma-Aldrich), or PBS for 48 h. All small RNAs of 15–52 nts were selected and sequenced using the Hiseq 2500 system, following the manufacturer’s protocol (Illumina; San Diego, CA) as previously described (5). Raw counts were normalized by trimmed mean of M value method and differentially expressed miRNAs between different treatments were identified using Bioconductor package “edgeR”. The small RNA-seq data are deposited into NCBI Gene Expression Omnibus (accession code GSE126419).
Cell viability (MTS assay)
Cell viability was assessed using CellTiter 96 AQueous MTS assay (Promega). In brief, cells were seeded at 5,000 cells/well in flat-bottomed 96-well culture plates and treated with DTX (10 nM) or DOXO (500 nM). At indicated time points, 20 μl MTS/PMS solution was added to each well followed by an 1.5-h incubation at 37 °C. Optical density was read at 490 nm using a Varioskan LUX reader (Thermo Fisher Scientific). Each individual experiment was repeated at least three times.
Animals
All animal experiments were approved by the institutional animal care and use committee at the University of California, San Diego. Eight-week-old female NOD/SCID/IL2Rγ-null (NSG) mice were used. For limiting-dilution transplantation, pre-treated MDA231 cells were injected into the mammary fat pads at the indicated numbers. Tumor incidence after 25 days was shown. The tumor-initiating cell (TIC) frequency was estimated by extreme limiting dilution analysis (ELDA) (17). For in vivo drug treatment, xenograft tumors were established by injecting 2×105 MDA231, MDA231/Rab27aKD, or MDA231/ONECUT2 cells into the #4 mammary fat pad. When tumor size reached approximately 300 mm3, mice were treated weekly with DTX (15 mg/kg) for 3 weeks. Serum was collected before and after the entire chemotherapy via retro-orbital bleeding. Tumor volume was determined by caliper measurements. Tumors were collected before the first DTX treatment (pre-DTX) or 3 days after the last DTX treatment (post-DTX). Each tumor was cut into three pieces for Western blot, RT-qPCR, and IHC analyses.
Clinical specimens
Human tumor specimens were obtained from BC patients who had received NT at the City of Hope National Medical Center (Duarte, CA) or the Tianjin Medical University Cancer Institute and Hospital (Tianjin, China). Written informed consents were obtained from all patients. The studies were conducted in accordance with recognized ethical guidelines, and were approved by an institutional review board. Clinical information, including age, tumor stage and pathology, as well as NT starting time, regimen, and response, is summarized in Table S1.
Immunohistochemistry (IHC)
IHC of formaldehyde-fixed, paraffin-embedded tumor tissues was performed using a Ventana Discovery Ultra system (Roche; Basel, Switzerland) with antibodies listed in Table S1. Stained slides were scored according to intensity of staining (-: 0; +: 1; ++: 2; and +++: 3) and percentage of tumor cells staining positive for the antigen (0%: 0; 1~29%: 1; 30~69%: 2; and ≥ 70%: 3). The intensity score was multiplied by the percentage score to obtain a final score, which was used in the statistical analyses.
Statistics and reproducibility
All quantitative data are presented as mean ± standard deviation. Statistical tests were performed using GraphPad Prism 7.01. Two-sided Student’s t-tests were used for comparisons of the means of data between two groups. For multiple independent groups, one-way ANOVA was used. Nonparametric Wilcoxon test was used for comparison of paired pre- and post-NT patient samples. Values of P<0.05 were considered significant. All Western blots were repeated independently three times with similar results, and representative images are shown.
Data and material availability
All materials, data, and protocols described in the article are available from the corresponding author on reasonable request.
Results
EVs and their cargo RNA secreted by chemotherapy-treated BC cells induce a CSC phenotype
EVs were isolated from the conditioned medium of MDA231 or MCF10A cells that had been treated with PBS (as a control) or a sub-lethal dose of chemotherapeutic agent DTX or DOXO (herein referred to as “chemo-EVs”). MDA231 BC cells that had never been treated with drugs exhibited efficient EV uptake (Fig. S1C), and were assessed for CSC-associated properties following EV treatment. We observed significant inductions of sphere-forming efficiency (Fig. 1A) and ALDEFLUORbright population known to be enriched in CSCs (18) (Fig. 1B) in cells that had received chemo-EVs from MDA231 but not from untreated BC cells or non-cancerous MCF10A cells. In addition, when varying numbers of MDA231 cells pre-treated with chemo-EVs or control EVs were injected into the mammary fat pads of NSG mice in a limiting-dilution transplantation assay, EVs derived from DTX- or DOXO-treated MDA231 cells showed the ability to stimulate tumorigenicity (Fig. 1C). The CSC-promoting effect was accompanied by a remarkable induction of stemness-associated genes, including NOTCH1, SOX9, NANOG, and POU5F1/OCT4, at both RNA and protein levels in chemo-EV-treated MDA231 (Fig. 1D,E). Some stemness-associated genes were also induced in MCF-7 and BT474 BC cells treated with their corresponding chemo-EVs (Fig. 1E), indicating induced stemness gene expression is a general effect of chemo-EVs from BC cells. Chemo-EVs’ effect on CSC phenotype was not affected by pre-treating EVs with RNase A in combination with Proteinase K to remove any free RNA and proteins outside of EV membranes (Fig. 1F), and the effect could be recapitulated by transfecting recipient BC cells with RNA isolated from the EVs (Fig. 1G,H), indicating this function is mediated by the cargo RNA of chemo-EVs.
Fig. 1. EVs and their cargo RNA secreted by chemotherapy-treated BC cells induce a CSC phenotype.
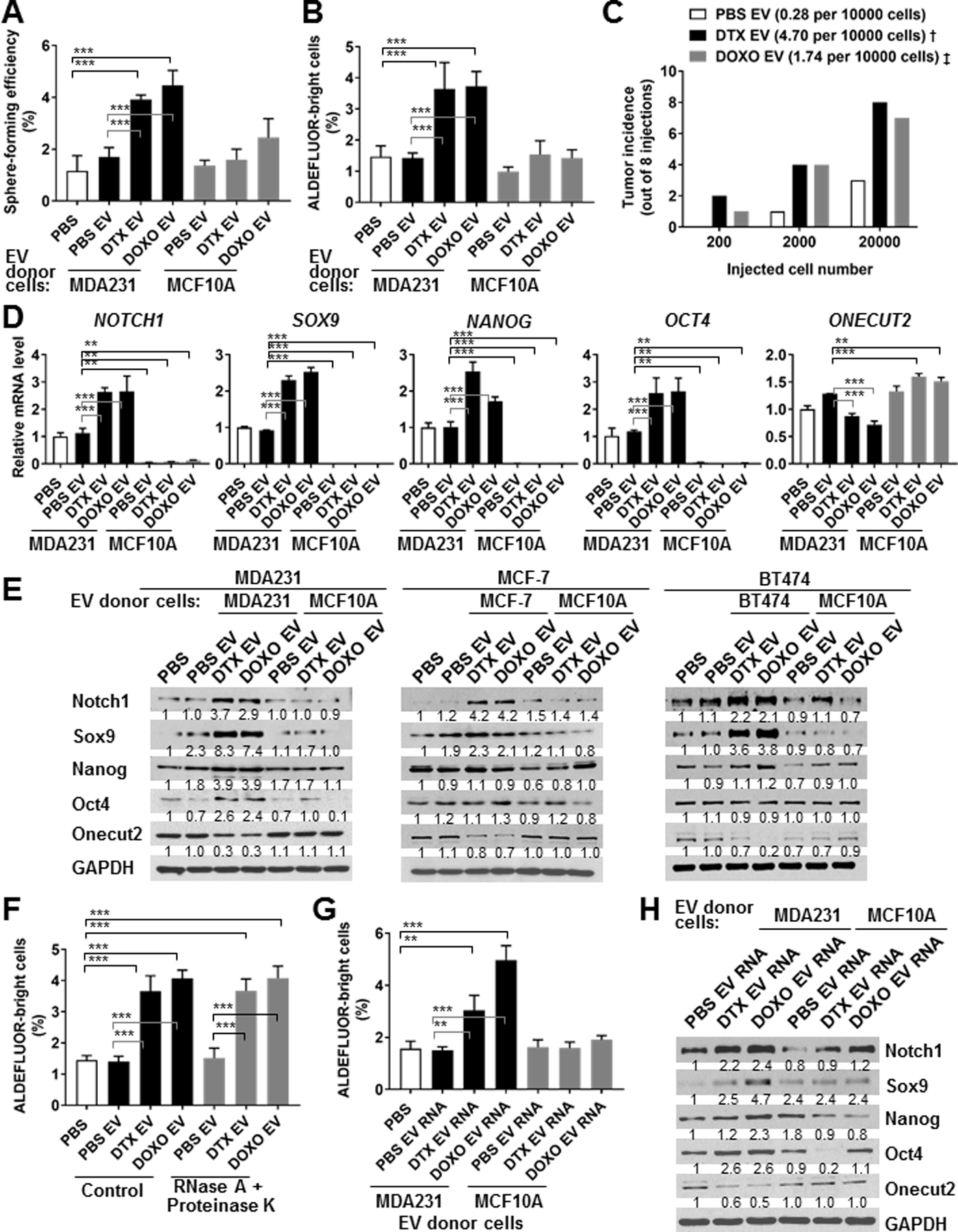
(A,B) MDA231 cells were treated with PBS or an equal amount of EVs from either MDA231 or MCF10A cells that had been exposed to PBS, docetaxel (DTX; 4 nM), or doxorubicin (DOXO; 125 nM) for 48 h before EV collection. EV-treated MDA231 cells were analyzed at 48 h by sphere formation assay (A) and at 72 h by ALDEFLUOR assay (B). (C) MDA231 cells pre-treated with indicated EVs from either PBS- or drug-treated MDA231 cells for 48 h were injected into the mammary fat pads of female NSG mice at the indicated numbers for limiting-dilution transplantation. Tumor incidence (out of 8 injections) after 25 days was shown. The estimated TIC frequency per 10,000 cells was determined by ELDA at the confidence level of 0.95 and indicated in brackets. Pairwise test for difference: †Pr(>ChiSq)<0.001, ‡Pr(>ChiSq)<0.01 compared to the PBS EV group. (D) Total RNA was extracted from MDA231 cells treated as in A, and subjected to RT-qPCR for levels of indicated genes using 18S rRNA level for normalization. (E) MDA231, MCF-7, and BT474 cells were treated with PBS or EVs from the same BC cell line or MCF10A cells that had been exposed to PBS, DTX, or DOXO as in A-C. EV-treated BC cells were analyzed at 48 h by Western blots for indicated protein levels. (F) EVs collected from MDA231 cells were treated with RNase A (100 μg/mL) and Proteinase K (200 μg/mL) for 2 h or left untreated (as control), washed with PBS, and then used to treat MDA231 cells for ALDEFLUOR assay analyzed at 72 h. (G,H) Total RNA extracted from the MDA231- and MCF10A-derived EVs examined in A-C were transfected into MDA231 cells (200 ng RNA added to 106 cells), which were subsequently analyzed at 72 h by ALDEFLUOR assay (G) and at 48 h by Western blots (H). **P<0.01, ***P<0.001. Numbers below Western images indicate quantification after normalization to GAPDH with the first lane set as 1.
BC-derived, EV-encapsulated miR-9–5p, miR-203a-3p, and miR-195–5p are induced by chemotherapy and required for CSC regulation
We performed small RNA-seq to characterize the miRNAs present in chemo-EVs secreted by MDA231 cells. Compared to PBS treatment, DTX and DOXO respectively induced 35 and 26 miRNAs in the EVs by >2 folds (only miRNAs with >40 counts were selected). Among these, 19 miRNAs were induced by both drugs (Table S2; Fig. S2A). We further selected the miRNAs broadly conserved across vertebrates with the exclusion of let-7 miRNAs, due to the presence of multiple let-7 family members sharing the same seed sequence and thereby with potential functional redundancy, and due to their very high endogenous expression (5) making the intracellular level unlikely to be significantly altered by EV transfer. The four selected miRNAs were then transfected into MDA231 cells to examine their individual effect on sphere formation; miR-9–5p, miR-203a-3p, and miR-195–5p were able to stimulate this CSC phenotype (Fig. S2B). RT-qPCR confirmed that these three miRNAs were dramatically induced by both DTX and DOXO in a dose-dependent manner in MDA231-derived EVs, and were also induced by at least one drug in the EVs from MCF-7 and BT474 BC cells, but not from MCF10A cells (Fig. 2A; Fig. S3). The intracellular levels of these miRNAs, however, did not always exhibit consistent changes compared to their levels in the EVs (Fig. 2B; Fig. S3), suggesting that chemotherapy may regulate the selective EV packing and secretion of these miRNAs. The putative target genes of these three miRNAs were determined by miRDB, and we initially focused on genes simultaneously targeted by all (Table S2). Among these, ONECUT2 stood out as a master transcription factor in cell fate specification, and its expression was indeed inhibited by chemo-EVs from BC cells (Fig. 1D,E,H). In contrast, expression of ONECUT1 and ONECUT3 did not show significant regulation (Fig. S4). Previously reported ONECUT2-regulated genes KLF4, LMO2, and POU6F2 (19) showed increased expression following chemo-EV treatment (Fig. S4). To determine if the three selected miRNAs indeed mediate the CSC-promoting effect of chemo-EVs, we individually blocked each miRNA in BC cells using anti-miRNA inhibitors and found that all three miRNAs were required to achieve maximal suppression of ONECUT2 and induction of stemness-associated genes (Fig. 2C,D). Direct chemotherapy treatment failed to show a significant and consistent effect on the endogenous expression of ONECUT family members and ONECUT2 targets, regardless of the presence of a proteasome inhibitor (Fig. S5). This further suggests that the herein studied regulation of ONECUT2 and CSC phenotype requires cargo transfer of chemo-EVs from chemo-stressed to treatment-naïve BC cells.
Fig. 2. BC-derived, EV-encapsulated miR-9–5p, miR-203a-3p, and miR-195–5p are induced by chemotherapy and required for CSC regulation.
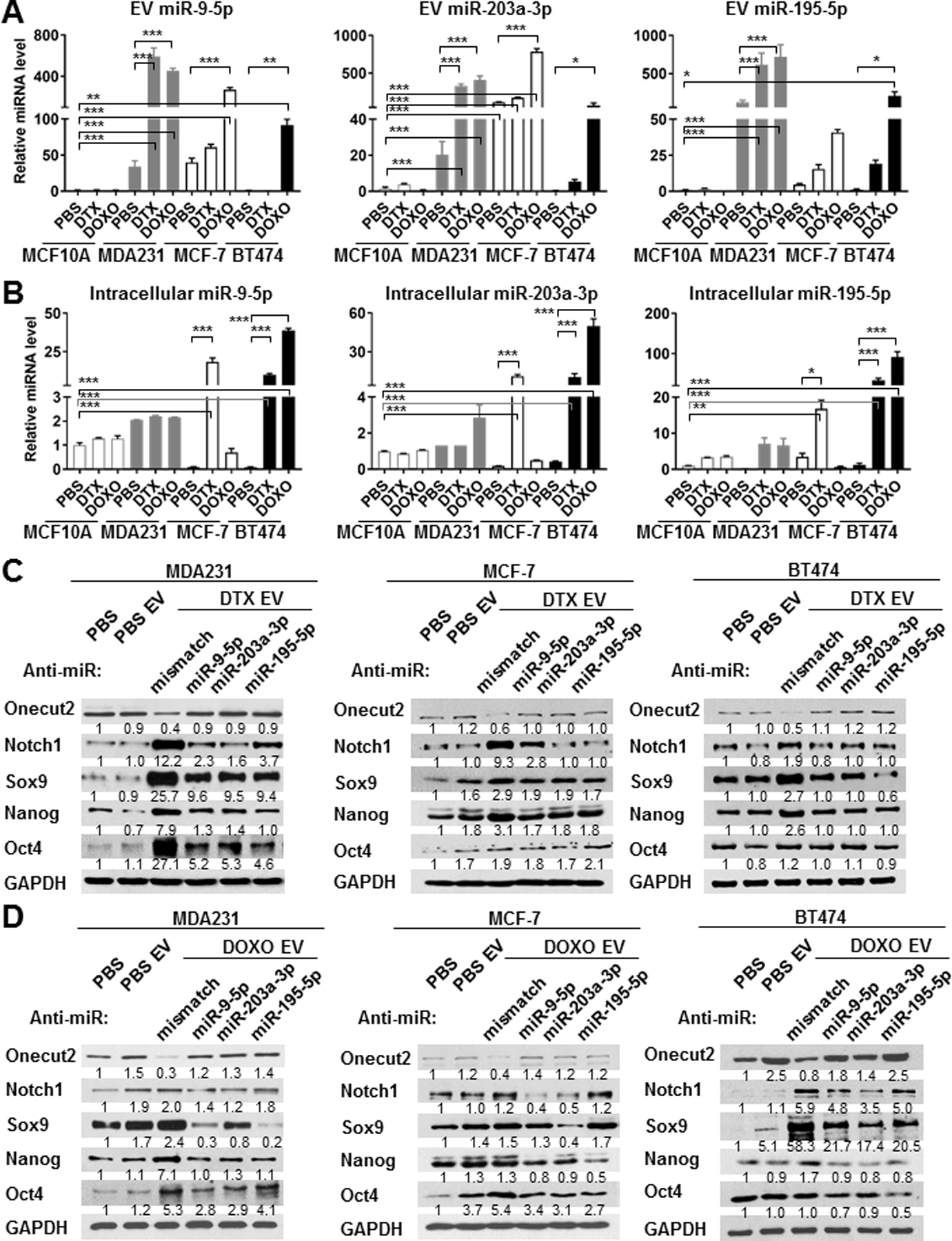
(A,B) Total RNA was extracted from an equal amount of EVs (A) or cell lysates (B) of PBS- or chemotherapy-treated MCF10A, MDA231, MCF-7, and BT474 cells. RT-qPCR was performed to determine levels of miR-9–5p, miR-203a-3p, and miR-195–5p using a cel-miR-39–3p spike-in control (for EV miRNA levels) or U6 internal control (for intracellular miRNA levels) for normalization. (C,D) MDA231, MCF-7, and BT474 cells were transfected with anti-miR-9–5p, anti-miR-203a-3p, anti-miR-195–5p oligonucleotides or mismatch control (25 pmol per 2×105 cells), and treated with EVs from the indicated producer cells for 48 h, before being analyzed by Western blots. *P<0.05, **P<0.01, ***P<0.001. Numbers below Western images indicate quantification after normalization to GAPDH with the first lane set as 1.
miR-9–5p, miR-203a-3p, and miR-195–5p regulate CSC-associated phenotype through directly targeting ONECUT2
We determined if the three selected miRNAs were sufficient to induce CSC properties by transfecting BC cells with miRNA mimics either individually or in combination. In MDA231 and BT474 cells, each miRNA mimic stimulated sphere-forming efficiency to a certain degree, whereas in MCF-7 cells only the combination of all three was sufficient to induce sphere formation (Fig. 3A). miR-203a-3p was also able to induce the ALDEFLUORbright population in MDA231 cells (Fig. 3B). In various BC cell lines, mimics of the three miRNAs decreased ONECUT2 expression and induced a full or partial set of examined stemness-associated genes as indicated by changes at the RNA and protein levels (Fig. 3C,D).
Fig. 3. Mimics of miR-9–5p, miR-203a-3p, and miR-195–5p promote CSC-associated properties and downregulate ONECUT2.
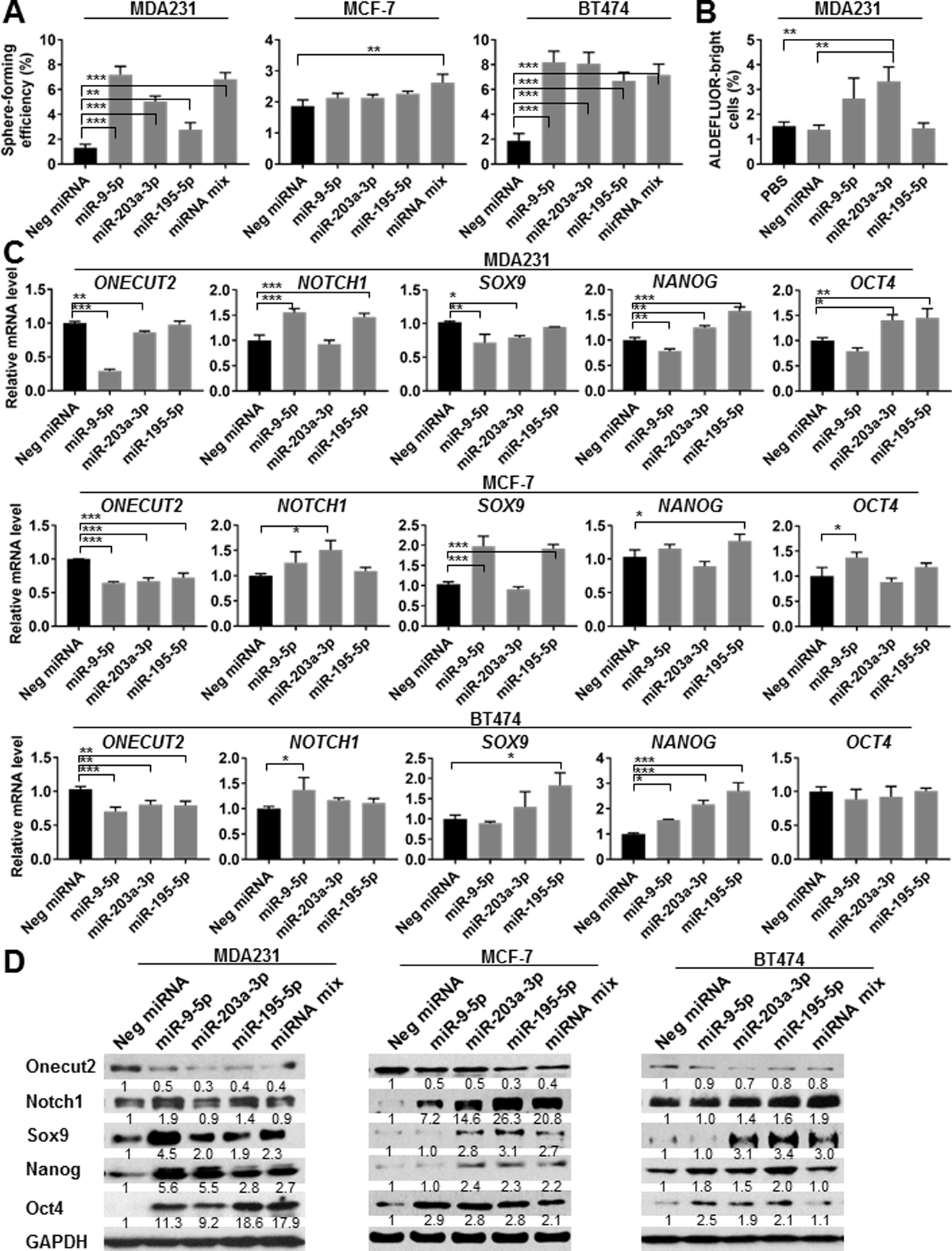
(A) MDA231, MCF-7, and BT474 cells were transfected with indicated miRNA mimics or a 1:1:1 mixture of miR-9–5p, miR-203a-3p, and miR-195–5p mimics, or with a negative control mimic. A total of 25 pmol miRNA mimics were used to transfect 2×105 cells seeded on 6-well plates one day prior to transfection. After 48 h, cells were collected for sphere formation assay. (B) ALDEFLUOR assay of MDA231 cells transfected with indicated miRNA mimics for 72 h. (C) RT-qPCR-determined RNA levels of indicated genes in various BC cells at 48 h following transfection with indicated miRNA mimics. (D) Western blots showing the expression levels of indicated proteins in BC cells at 48 h following transfection of indicated miRNA mimics. *P<0.05, **P<0.01, ***P<0.001. Numbers below Western images indicate quantification after normalization to GAPDH with the first lane set as 1.
Knockdown of ONECUT2 with siRNAs significantly increased sphere-forming efficiency and the ALDEFLUORbright population in BC cells (Fig. 4A,B), and enhanced the expression of stemness-associated genes (Fig. 4C,D). CSCs characteristically express high levels of ATP-binding cassette (ABC) transporters on their cell surface as a mechanism to efflux chemotherapeutic drugs. Among the large family of ABC transporters, ABCB1 (multidrug resistance 1 or P-glycoprotein), ABCG2 (breast cancer resistance protein), ABCB5, and ABCC1 are the most well-characterized (20). In MDA231 cells with ONECUT2 knockdown, we detected increased partial resistance to DTX and DOXO as well as the expression of selected ABC transporters (Fig. 4E,F). To further establish that chemo-EVs induce CSC properties through downregulating ONECUT2, we transfected MDA231 cells with a ONECUT2 cDNA construct lacking 3’UTR and thus resistant to miRNA regulation. This significantly blocked the effect of chemo-EVs on inducing sphere-forming efficiency and stemness gene expression (Fig. 5A,B). In contrast to the effect of ONECUT2 knockdown, overexpression of ONECUT2 suppressed the expression of stemness-associated genes (Fig. 5C,D), and enhanced sensitivity to chemotherapy drugs (Fig. 5E,F).
Fig. 4. ONECUT2 is a negative regulator of CSC-associated properties.
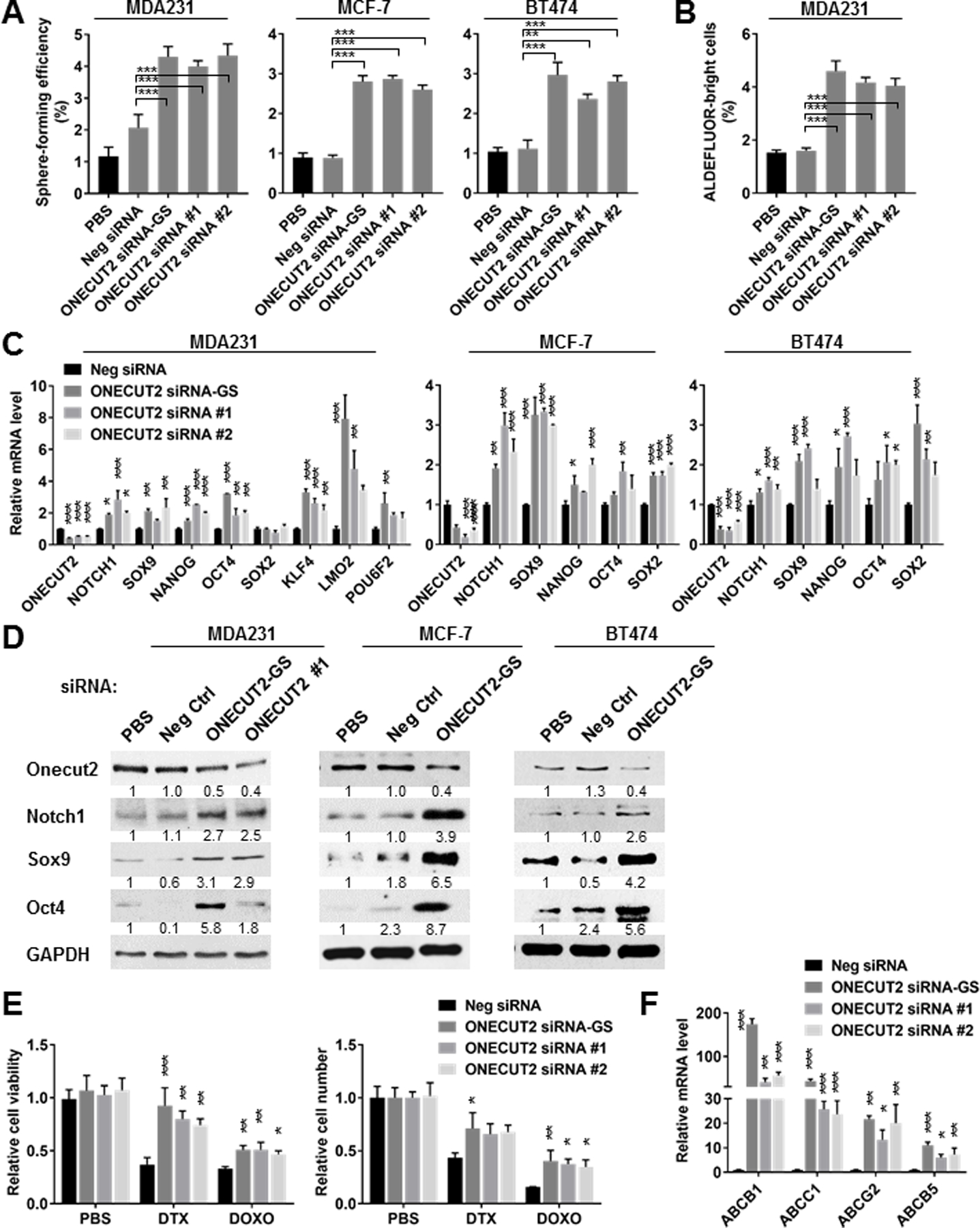
(A) MDA231, MCF-7, and BT474 cells were transfected with siRNA against ONECUT2 (GeneSolution/GS including equal mixture of 4 preselected siRNAs, or individual siRNA #1 and #2), or a control siRNA, or with PBS. After 48 h, cells were collected for sphere formation assay. (B) ALDEFLUOR assay of MDA231 cells transfected with siRNA as indicated for 72 h. (C) RT-qPCR-determined RNA levels of indicated genes in various BC cells at 48 h following transfection with indicated siRNA. (D) Western blots showing the expression levels of indicated proteins in BC cells at 48 h following transfection of ONECUT2 siRNA-GS or control siRNA. (E) MDA231 cells were transfected with indicated siRNA and seeded at an equal number on day 0. DTX (10 nM) or DOXO (500 nM) was added on day 1, and replenished every 24 h. On day 3, cell viability (left) and cell number (right) were determined by MTS assay and cell counting, respectively, and compared to the PBS treatment group. (F) RT-qPCR of indicated genes in MDA231 cells at 72 h after transfection with indicated siRNA. *P<0.05, **P<0.01, ***P<0.001 compared to control siRNA or as indicated. Numbers below Western images indicate quantification after normalization to GAPDH with the first lane set as 1.
Fig. 5. Downregulation of ONECUT2 is required for chemo-EV-induced CSC properties.
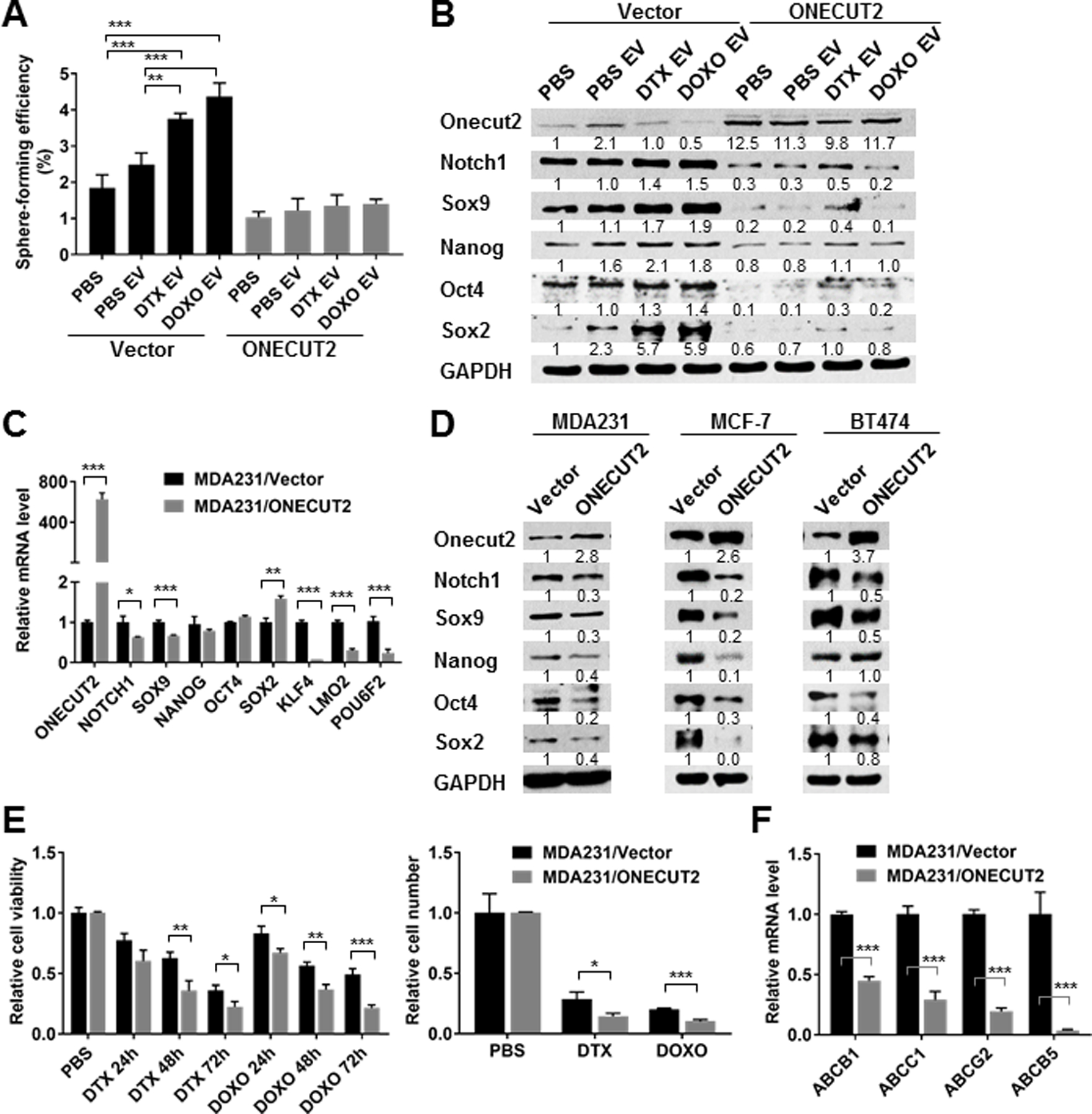
(A,B) MDA231 cells stably expressing a mammalian expression plasmid of human ONECUT2 cDNA or the empty vector were exposed to PBS or EVs from PBS/DTX/DOXO-treated MDA231 cells for 48 h before being analyzed by sphere formation assay (A) and Western blots (B). (C) RT-qPCR-determined RNA levels of indicated genes in MDA231 cells stably expressing ONECUT2 or vector. (D) MDA231, MCF-7, and BT474 cells transfected with the ONECUT2 expression plasmid or empty vector were analyzed by Western blots. (E) MDA231 cells stably expressing ONECUT2 or vector were seeded at an equal number on day 0. DTX (10 nM) or DOXO (500 nM) was added after 6 h, and replenished every 24 h. Cell viability (left) was measured by MTS assay every 24 h and cell number (right) was determined by cell counting on day 3. Data were normalized to the PBS control group. (F) RT-qPCR of indicated genes in MDA231 cells stably expressing ONECUT2 or vector. *P<0.05, **P<0.01, ***P<0.001. Numbers below Western images indicate quantification after normalization to GAPDH with the first lane set as 1.
Due to the length of ONECUT2 3’UTR which is >14,000 bps, we constructed luciferase reporters containing various fragments of ONECUT2 3’UTR that encompass most of the putative binding sites of the three selected miRNAs to confirm the miRNA-mediated regulation. Fragments 1 and 3 contain one or more binding sites for all three miRNAs, whereas Fragment 2 only contains binding sites for miR-9–5p and miR-203a-3p (Fig. 6A). When MDA231 cells transfected with the 3’UTR reporters were treated with chemo-EVs, Fragments 1 and 3 showed significant responses by reducing the expression of luciferase reporter, whereas Fragment 2 showed less significant responses (Fig. 6B). When co-transfected with miRNA mimics, Fragment 1 responded to miR-9–5p and modestly to miR-203a-3p, Fragment 2 responded to miR-203a-3p and slightly to miR-9–5p, and Fragment 3 responded to miR-9–5p and modestly to the other two miRNAs when compared to control mimic (Fig. 6C). This result thus confirmed that ONECUT2 is a shared target of miR-9–5p, miR-203a-3p, and miR-195–5p, which recognize various regions of the gene’s 3’UTR.
Fig. 6. miR-9–5p, miR-203a-3p, and miR-195–5p target the 3’UTR of ONECUT2.
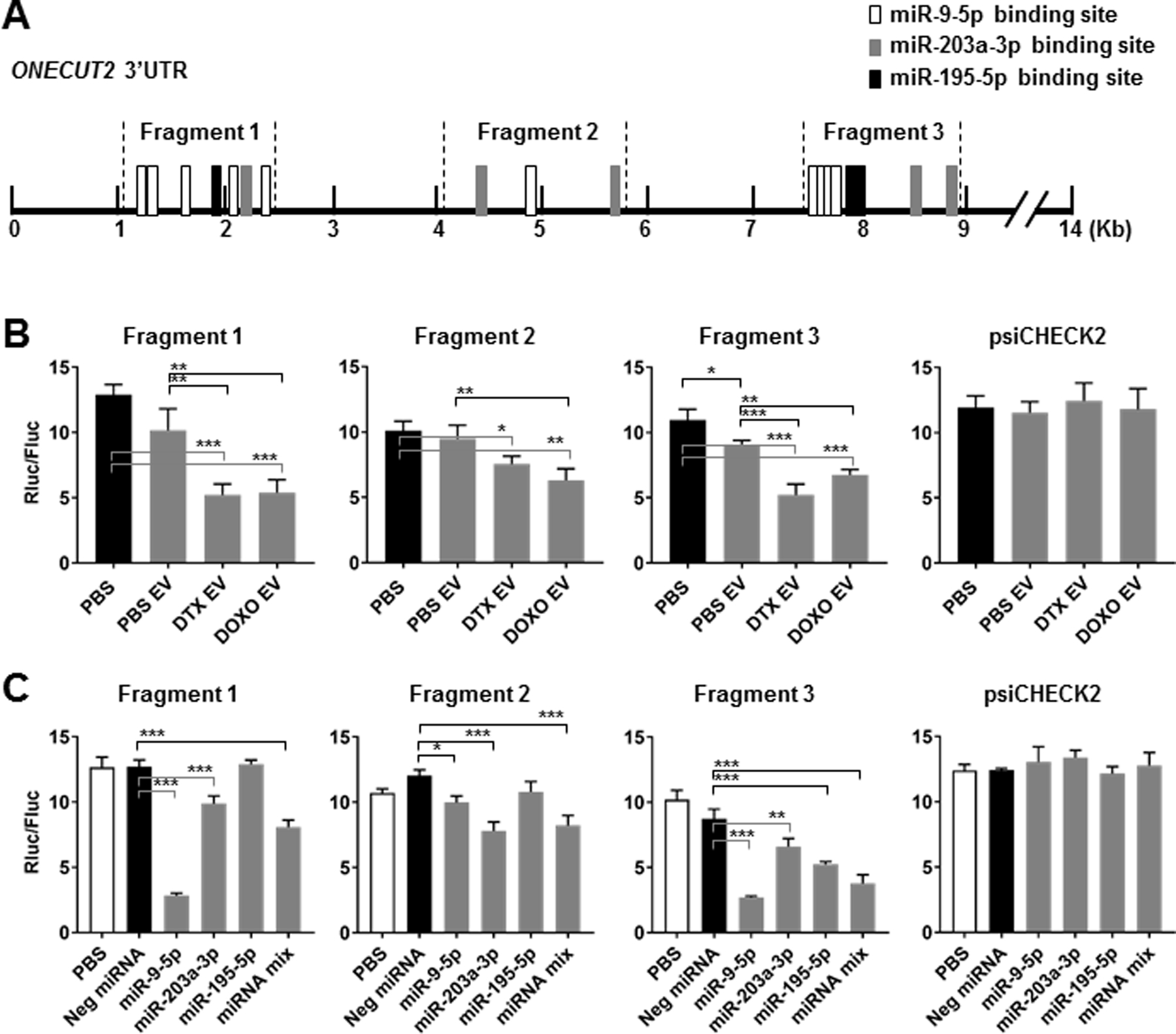
(A) A schematic representative showing putative binding sites of miR-9–5p, miR-203a-3p, and miR-195–5p in the 3’UTR of human ONECUT2 and the regions cloned into the luciferase reporter plasmid constructs. (B) MDA231 cells were transfected with psiCHECK2 reporter plasmids containing indicated ONECUT2 3’UTR region or with the psiCHECK2 vector (2 µg DNA per 2×105 cells). After 12 h, transfected cells were exposed to PBS or EVs from PBS/DTX (4 nM)/DOXO (125 nM)-treated MDA231 cells for 48 h before luciferase activities were measured. Ratio between Renilla luciferase and firefly luciferase activities (Rluc/Fluc) is shown. (C) MDA231 cells were co-transfected with indicated psiCHECK2 reporter plasmids (2 µg DNA per 2×105 cells) and miRNA mimics (individually or with a 1:1:1 mixture of miR-9–5p, miR-203a-3p, and miR-195–5p mimics for a total of 25 pmol). Luciferase activities were analyzed at 48 h. *P<0.05, **P<0.01, ***P<0.001.
ONECUT2 level in tumor is regulated by chemotherapy through EV secretion and affects therapeutic response
To examine ONECUT2 regulation by chemotherapy in vivo and the role of cancer-derived EVs in this process, we generated MDA231 cells with stable knockdown of RAB27A (MDA231/Rab27aKD), a gene required for exosome secretion (21), and confirmed significant reduction of EV secretion by this line (Fig. 7A). In addition, MDA231 cells stably overexpressing ONECUT2 were also generated (Fig. 7A). These modified cells as well as the control MDA231 cells were injected into the #4 mammary fat pad of NSG mice. When tumor size reached ~300 mm3, mice were treated with DTX for 3 weeks. Tumors with Rab27a knockdown grew slower than the control MDA231 tumors from the beginning, and exhibited enhanced response to DTX (Fig. 7B; Fig. S6A). Tumors with ONECUT2 overexpression grew faster than controls, also exhibiting better response to DTX-induced tumor regression (Fig. 7B; Fig. S6A). DTX treatment decreased ONECUT2 but increased CSC-associated gene expression in MDA231 tumors, but these effects were diminished in tumors with Rab27aKD or ONECUT2 overexpression (Fig. 7C,D; Fig. S6A). Following DTX treatment, levels of miR-9–5p, miR-203a-3p, and miR-195–5p were significantly elevated in the EVs isolated from the sera of mice bearing MDA231, but not MDA231/Rab27aKD tumors (Fig. 7E), which is consistent with the defective EV secretion capacity of the latter. Our data from the MDA231 tumor model collectively suggest that although ONECUT2 enhances tumor growth, its downregulation by chemo-EVs can promote stemness-associated gene expression, contributing to therapeutic resistance. Lastly, we compared ONECUT2 levels in 12 primary human breast tumors before and after NT, and detected decreased tumor cell expression of ONECUT2 following chemotherapy treatment (Fig. 7F; Fig. S6B).
Fig. 7. ONECUT2 level in tumor is regulated by chemotherapy through EV secretion and affects therapeutic response.
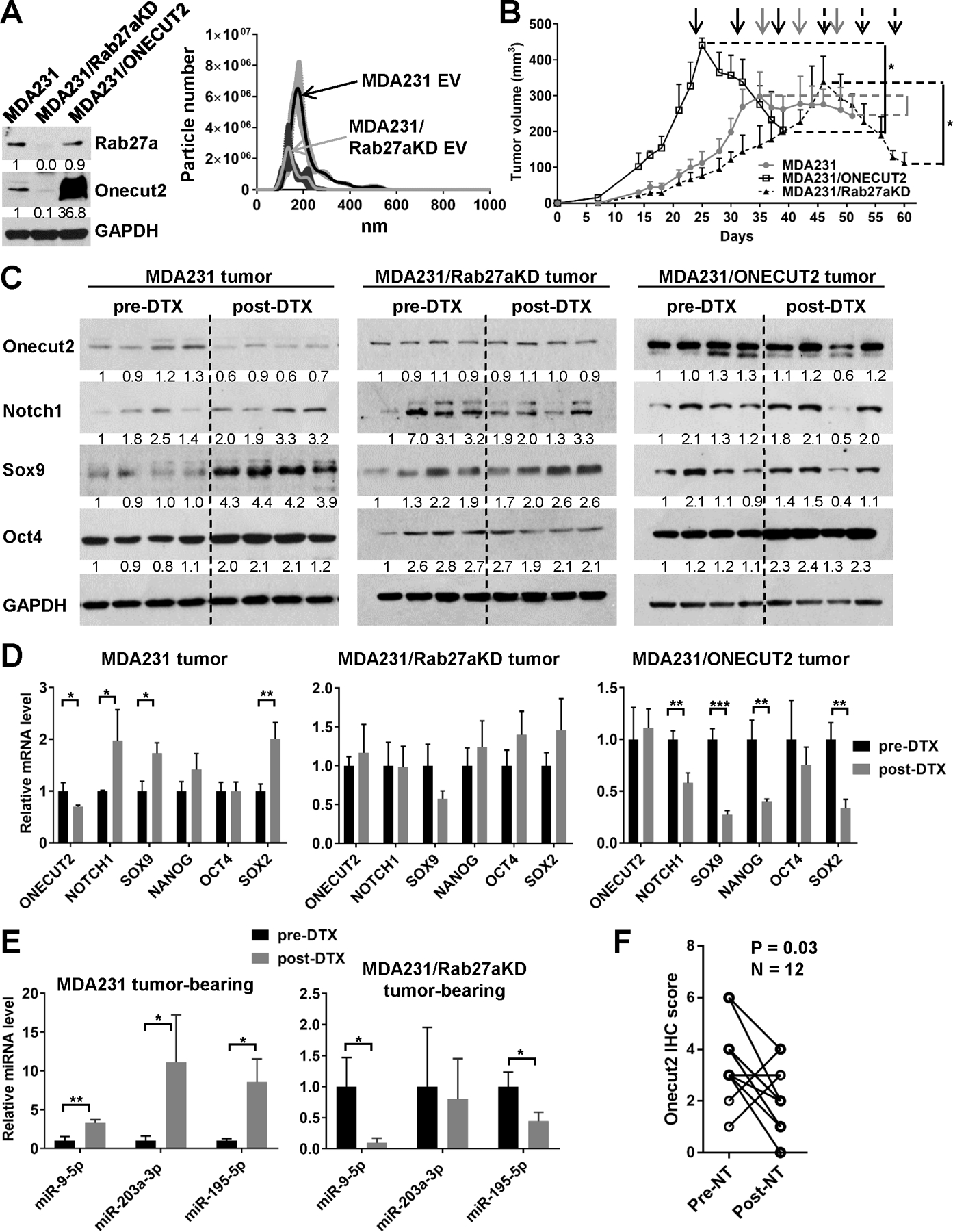
(A) Left: Western blots showing modified expression of Rab27a and Onecut2 in indicated cell lines. Right: nanoparticle tracking analysis of EVs from an equal number of producing cells showing reduced EV secretion by MDA231/Rab27aKD cells. (B-E) Xenograft tumors were established in NSG mice by injecting 2×105 of indicated cells into the #4 mammary fat pad. When tumor size reached ~300 mm3, mice were treated weekly with DTX (15 mg/kg) for 3 weeks. (B) Tumor onset and volume. The time of DTX treatments were indicated by arrows. (C) Western blots of indicated proteins using tumors collected before and after DTX treatment. Numbers below Western images indicate quantification after normalization to GAPDH with the first lane set as 1. (D) RT-qPCR analysis of indicated genes using tumors collected before and after DTX treatment. (E) EVs were prepared from the sera of indicated mice before and after the 3-week DTX treatment. Levels of miRNAs were determined by RT-qPCR using a cel-miR-39–3p spike-in control for normalization. (F) Twelve pairs of pre- and post-NT human breast tumors were analyzed by IHC to determine the ONECUT2 expression levels in tumor cells. Wilcoxon test was performed. *P<0.05, **P<0.01, ***P<0.001.
Discussion
Here we show a mechanism through which chemotherapy-treated BC cells, by secreting certain EV miRNAs, communicate with and reprogram nearby cancer cells to induce a CSC phenotype. Given the established associations between CSCs and tumor refractoriness, this may serve as a means for cancer’s self-adaptation to survive the therapy, and may contribute to chemotherapy-induced tumor progression and metastasis. Indeed, previous studies have shown that chemotherapy can induce stemness-related genes through various mechanisms independent of EV secretion, such as by directly activating hypoxia-inducible factors and Ca2+ release in cancer cells exposed to the drugs (22), and by stimulating monocytosis leading to systemic elevation of monocyte chemoattractant proteins (MCPs) (23). Our current study adds another mechanism potentially mediating therapy-driven evolution of cancer, and may represent a unique mode of EV adaptation in those cancer cells without direct exposure to chemotherapeutic drugs due to the poor bio-accessibility of these drugs to tumor tissues, as well as in cancer cells that do not express significant amounts of MCP receptors.
We identified miR-9–5p, miR-203a-3p, and miR-195–5p as the EV cargo induced by chemotherapy and in turn reprogramming BC cells through simultaneously targeting ONECUT2. These miRNAs have been previously reported to inhibit cell proliferation in BC models by targeting MTHFD2 (miR-9–5p), BIRC5 and LASP1 (miR-203a-3p), and CCNE1 (miR-195–5p) (24–26). Their seemingly tumor suppressor-like function does not conflict with the herein studied effect on inducing cancer stemness, as CSCs are known to be slowly proliferating and the induction of these miRNAs could be transient in tumor as an acute response to chemotherapy. Indeed, the function of these miRNAs during tumor growth and progression is complex, likely depending on the cellular context and stage of tumor. miR-9–5p is activated by MYC oncogene and promotes metastasis by targeting E-cadherin (27). It also promotes vascular-like structure formed by BC cells (28), and has been associated with BC local recurrence and poor survival (29,30). miR-203a-3p has also been shown to promote hepatocellular carcinoma cell proliferation and metastasis by targeting IL-24 (31). Thus, the significance of these miRNAs as chemotherapy-responsive EV cargo may be reflected by their combined effect, such as the induction of CSC properties shown in our study.
The ONECUT family of transcription factors are highly conserved and play important roles in organ development (32). Different ONECUT factors exhibit similar functions but their expression patterns are under different spatial and temporal control to allow proper cell fate specification during organogenesis. ONECUT2 expression is detected in developing liver, pancreas, nervous system, and gut endoderm, exhibiting a developmental stage-specific and tissue-restricted pattern (33). Overexpression of ONECUT2 has been reported in a variety of human cancers, including those of the prostate, ovaries, colorectum, and liver (34–37). In these previous studies, ONECUT2 plays a tumor-promoting role by stimulating cancer cell proliferation and invasion. The function of ONECUT2 in BC hasn’t been characterized. In a study identifying five upregulated miRNAs (miR-30b-5p, miR-96–5p, miR-182–5p, miR-374b-5p, and miR-942–5p) in the blood of BC patients, ONECUT2 was proposed as a common putative target of the five miRNAs, and its lower expression was associated with worse relapse-free survival of BC patients (38).
A recent study identifies ONECUT2 as a master regulator in metastatic castration-resistant prostate cancer, where ONECUT2 suppresses the expression of androgen receptor (AR) and binds to the promoters of AR target genes to suppress their expression—but only in the presence of AR ligand dihydrotestosterone (39). In this cellular context, ONECUT2 also activates genes associated with neural differentiation and cancer progression. Here in BC models, we found that ONECUT2 negatively regulated the CSC phenotype and stemness-associated genes including NOTCH1, SOX9, NANOG, OCT4, and SOX2. In addition, KLF4, LMO2, and POU6F2 have been reported to be regulated by ONECUT2 in embryonic retinas (19). These genes showed increased expression following chemo-EV treatment or ONECUT2 knockdown (Fig. S4; Fig. 4C) and decreased expression when ONECUT2 was overexpressed (Fig. 5C), and have all been implicated in the regulation of CSCs or tumor aggressiveness. KLF4 is found to be highly expressed in CSCs of mouse mammary tumors and BC cell lines, and is required for tumorigenesis through maintaining the stem/progenitor cell population and NOTCH1 expression likely through binding to the proximal NOTCH1 promoter (40,41). In embryonic stem cells, KLF2, KLF4, and KLF5 are coordinately upregulated and required for the cells’ self-renewal through regulating the promoter of NANOG gene and targets of NANOG, whereas simultaneous depletion of the KLF genes results in cell differentiation (42). Similarly, LMO2 is highly expressed in patient-derived glioma stem cells and is required for tumorigenicity through transcriptional regulation of NOTCH signaling (43). In basal-type BC, LMO2 promotes tumor cell invasion and metastasis, and its high expression is associated with lymph node metastases in patients (44). Upregulation of POU6F2 has been associated with mammary adenocarcinoma metastases to lungs (45). Thus, it is possible that ONECUT2 regulates the gene expression program associated with CSCs through these previously reported mechanisms.
It was noted that the MDA231/Rab27aKD cells, when cultured in vitro, exhibited reduced expression of ONECUT2 (Fig. 7A). Although this might be related to altered secretion of exosomal miRNAs and proteins, we did not observe a significant difference in ONECUT2 expression in pre-treatment xenograft tumors formed from these cells compared to the MDA231 tumors, possibly suggesting the presence of other mechanisms and factors in the tumor microenvironment that can regulate ONECUT2 expression. Several miRNAs, including miR-9, miR-429, and miR-218, have been previously reported to target ONECUT2 (34,35,46–48). In embryonic stem cells, the promoter of ONECUT2 is simultaneously marked with histone H3 trimethyl-lysine 4 (K4me3) and histone H3 trimethyl-lysine 27 (K27me3), a pattern known as bivalent domains, which silence developmental genes while keeping them poised for activation during differentiation (49). Bivalent domains are associated with genes coding for transcription factors critical for embryonic development and lineage specification. In differentiated cells, these domains adopt either an active or repressed state to maintain lineage-specific expression or repression (49). Chromatin regions exhibiting bivalency, including ONECUT2 promoter, constitute a major group of cancer-associated promoters in gastric adenocarcinomas (50). Thus, repression of ONECUT2 and other bivalent genes via miRNA targeting and other mechanisms would reprogram the levels of gene expression towards a pluripotent state. Future studies deciphering the spatiotemporal pattern of ONECUT2 expression and its regulation during tumor development, progression, and treatment may provide novel insights into the dynamic control of CSC-like population towards improved cancer management.
Supplementary Material
Significance:
Findings reveal a critical mechanism of resistance to chemotherapy by which breast cancer cells secrete miRNA-containing extracellular vesicles to stimulate cancer stem cell-like features.
Acknowledgements
This work was supported by the California Breast Cancer Research Program grant 20IB-0118 (SEW) and Breast Cancer Research Foundation and American Association for Cancer Research joint grant 12–60-26-WANG (SEW).
Financial support:
California Breast Cancer Research Program grant 20IB-0118 (SEW); Breast Cancer Research Foundation and American Association for Cancer Research joint grant 12–60-26-WANG (SEW)
Footnotes
Disclosure of potential conflicts of interest
The authors declare no competing financial interests.
Conflict of interest: The authors declare no potential conflicts of interest.
References
- 1.Chin AR, Wang SE. Cancer-derived extracellular vesicles: the ‘soil conditioner’ in breast cancer metastasis? Cancer Metastasis Rev 2016;35:669–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016;30:836–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tkach M, Thery C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016;164:1226–32 [DOI] [PubMed] [Google Scholar]
- 4.Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 2015;17:183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, et al. Cancer-Secreted miR-105 Destroys Vascular Endothelial Barriers to Promote Metastasis. Cancer Cell 2014;25:501–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Redzic JS, Balaj L, van der Vos KE, Breakefield XO. Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol 2014;28:14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan W, Wu X, Zhou W, Fong MY, Cao M, Liu J, et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat Cell Biol 2018;20:597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aleckovic M, Kang Y. Regulation of cancer metastasis by cell-free miRNAs. Biochim Biophys Acta 2015;1855:24–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014;26:707–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson AM, Moulder-Thompson SL. Neoadjuvant treatment of breast cancer. Ann Oncol 2012;23 Suppl 10:x231–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Group NM-aC. Preoperative chemotherapy for non-small-cell lung cancer: a systematic review and meta-analysis of individual participant data. Lancet 2014;383:1561–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cortazar P, Geyer CE Jr. Pathological complete response in neoadjuvant treatment of breast cancer. Ann Surg Oncol 2015;22:1441–6 [DOI] [PubMed] [Google Scholar]
- 13.Specht J, Gralow JR. Neoadjuvant chemotherapy for locally advanced breast cancer. Semin Radiat Oncol 2009;19:222–8 [DOI] [PubMed] [Google Scholar]
- 14.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008;8:755–68 [DOI] [PubMed] [Google Scholar]
- 15.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002;111:29–40. [DOI] [PubMed] [Google Scholar]
- 16.Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res 2012;72:2768–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009;347:70–8 [DOI] [PubMed] [Google Scholar]
- 18.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1:555–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sapkota D, Chintala H, Wu F, Fliesler SJ, Hu Z, Mu X. Onecut1 and Onecut2 redundantly regulate early retinal cell fates during development. Proc Natl Acad Sci U S A 2014;111:E4086–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 2010;12:19–30; sup pp 1–13 [DOI] [PubMed] [Google Scholar]
- 22.Lu H, Chen I, Shimoda LA, Park Y, Zhang C, Tran L, et al. Chemotherapy-Induced Ca(2+) Release Stimulates Breast Cancer Stem Cell Enrichment. Cell reports 2017;18:1946–57 [DOI] [PubMed] [Google Scholar]
- 23.Liu L, Yang L, Yan W, Zhai J, Pizzo DP, Chu P, et al. Chemotherapy Induces Breast Cancer Stemness in Association with Dysregulated Monocytosis. Clin Cancer Res 2018;24:2370–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selcuklu SD, Donoghue MT, Rehmet K, de Souza Gomes M, Fort A, Kovvuru P, et al. MicroRNA-9 inhibition of cell proliferation and identification of novel miR-9 targets by transcriptome profiling in breast cancer cells. The Journal of biological chemistry 2012;287:29516–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, Zheng X, Shen C, Shi Y. MicroRNA-203 suppresses cell proliferation and migration by targeting BIRC5 and LASP1 in human triple-negative breast cancer cells. Journal of experimental & clinical cancer research : CR 2012;31:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Q, Wei C, Li X, Li J, Chen L, Huang Y, et al. MicroRNA-195–5p is a potential diagnostic and therapeutic target for breast cancer. Oncology reports 2014;31:1096–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 2010;12:247–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Ippolito E, Plantamura I, Bongiovanni L, Casalini P, Baroni S, Piovan C, et al. miR-9 and miR-200 Regulate PDGFRbeta-Mediated Endothelial Differentiation of Tumor Cells in Triple-Negative Breast Cancer. Cancer research 2016;76:5562–72 [DOI] [PubMed] [Google Scholar]
- 29.Zhou X, Marian C, Makambi KH, Kosti O, Kallakury BV, Loffredo CA, et al. MicroRNA-9 as potential biomarker for breast cancer local recurrence and tumor estrogen receptor status. PLoS One 2012;7:e39011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gwak JM, Kim HJ, Kim EJ, Chung YR, Yun S, Seo AN, et al. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast cancer research and treatment 2014;147:39–49 [DOI] [PubMed] [Google Scholar]
- 31.Huo W, Du M, Pan X, Zhu X, Gao Y, Li Z. miR-203a-3p.1 targets IL-24 to modulate hepatocellular carcinoma cell growth and metastasis. FEBS open bio 2017;7:1085–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kropp PA, Gannon M. Onecut transcription factors in development and disease. Trends Dev Biol 2016;9:43–57 [PMC free article] [PubMed] [Google Scholar]
- 33.Jacquemin P, Pierreux CE, Fierens S, van Eyll JM, Lemaigre FP, Rousseau GG. Cloning and embryonic expression pattern of the mouse Onecut transcription factor OC-2. Gene Expr Patterns 2003;3:639–44 [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Cheng J, Zeng Z, Wang Y, Li X, Xie Q, et al. Comprehensive profiling of novel microRNA-9 targets and a tumor suppressor role of microRNA-9 via targeting IGF2BP1 in hepatocellular carcinoma. Oncotarget 2015;6:42040–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y, Shen S, Liu X, Tang H, Wang Z, Yu Z, et al. MiR-429 inhibits cells growth and invasion and regulates EMT-related marker genes by targeting Onecut2 in colorectal carcinoma. Mol Cell Biochem 2014;390:19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu T, Wu B, Yu Y, Zhu W, Zhang S, Zhang Y, et al. Blockade of ONECUT2 expression in ovarian cancer inhibited tumor cell proliferation, migration, invasion and angiogenesis. Cancer Sci 2018;109:2221–34 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Leyten GH, Hessels D, Smit FP, Jannink SA, de Jong H, Melchers WJ, et al. Identification of a Candidate Gene Panel for the Early Diagnosis of Prostate Cancer. Clin Cancer Res 2015;21:3061–70 [DOI] [PubMed] [Google Scholar]
- 38.Zhang K, Wang YW, Wang YY, Song Y, Zhu J, Si PC, et al. Identification of microRNA biomarkers in the blood of breast cancer patients based on microRNA profiling. Gene 2017;619:10–20 [DOI] [PubMed] [Google Scholar]
- 39.Rotinen M, You S, Yang J, Coetzee SG, Reis-Sobreiro M, Huang WC, et al. ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. Nat Med 2018;24:1887–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene 2011;30:2161–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Z, Teng L, Bailey SK, Frost AR, Bland KI, LoBuglio AF, et al. Epithelial transformation by KLF4 requires Notch1 but not canonical Notch1 signaling. Cancer Biol Ther 2009;8:1840–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol 2008;10:353–60 [DOI] [PubMed] [Google Scholar]
- 43.Kim SH, Kim EJ, Hitomi M, Oh SY, Jin X, Jeon HM, et al. The LIM-only transcription factor LMO2 determines tumorigenic and angiogenic traits in glioma stem cells. Cell Death Differ 2015;22:1517–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Wang Z, Huang D, Wu C, Li H, Zhang X, et al. LMO2 promotes tumor cell invasion and metastasis in basal-type breast cancer by altering actin cytoskeleton remodeling. Oncotarget 2017;8:9513–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krol M, Polanska J, Pawlowski KM, Turowski P, Skierski J, Majewska A, et al. Transcriptomic signature of cell lines isolated from canine mammary adenocarcinoma metastases to lungs. J Appl Genet 2010;51:37–50 [DOI] [PubMed] [Google Scholar]
- 46.Jafarian A, Taghikani M, Abroun S, Allahverdi A, Lamei M, Lakpour N, et al. The Generation of Insulin Producing Cells from Human Mesenchymal Stem Cells by MiR-375 and Anti-MiR-9. PLoS One 2015;10:e0128650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simion A, Laudadio I, Prevot PP, Raynaud P, Lemaigre FP, Jacquemin P. MiR-495 and miR-218 regulate the expression of the Onecut transcription factors HNF-6 and OC-2. Biochem Biophys Res Commun 2010;391:293–8 [DOI] [PubMed] [Google Scholar]
- 48.Madelaine R, Sloan SA, Huber N, Notwell JH, Leung LC, Skariah G, et al. MicroRNA-9 Couples Brain Neurogenesis and Angiogenesis. Cell reports 2017;20:1533–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006;125:315–26 [DOI] [PubMed] [Google Scholar]
- 50.Muratani M, Deng N, Ooi WF, Lin SJ, Xing M, Xu C, et al. Nanoscale chromatin profiling of gastric adenocarcinoma reveals cancer-associated cryptic promoters and somatically acquired regulatory elements. Nat Commun 2014;5:4361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.