

Abstract
Post-traumatic epilepsy (PTE) is one of the most devastating long-term, network consequences of traumatic brain injury (TBI). There is currently no approved treatment that can prevent onset of spontaneous seizures associated with brain injury, and many cases of PTE are refractory to antiseizure medications. Post-traumatic epileptogenesis is an enduring process by which a normal brain exhibits hypersynchronous excitability after a head injury incident. Understanding the neural networks and molecular pathologies involved in epileptogenesis are key to preventing its development or modifying disease progression. In this article, we describe a critical appraisal of the current state of PTE research with an emphasis on experimental models, molecular mechanisms of post-traumatic epileptogenesis, potential biomarkers, and the burden of PTE-associated comorbidities. The goal of epilepsy research is to identify new therapeutic strategies that can prevent PTE development or interrupt the epileptogenic process and relieve associated neuropsychiatric comorbidities. Therefore, we also describe current preclinical and clinical data on the treatment of PTE sequelae. Differences in injury patterns, latency period, and biomarkers are outlined in the context of animal model validation, pathophysiology, seizure frequency, and behavior. Improving TBI recovery and preventing seizure onset are complex and challenging tasks; however, much progress has been made within this decade demonstrating disease modifying, anti-inflammatory, and neuroprotective strategies, suggesting this goal is pragmatic. Our understanding of PTE is continuously evolving, and improved preclinical models allow for accelerated testing of critically needed novel therapeutic interventions in military and civilian persons at high risk for PTE and its devastating comorbidities.
Significance Statement
Post-traumatic epilepsy is a chronic seizure condition after brain injury. With few models and limited understanding of the underlying progression of epileptogenesis, progress is extremely slow to find a preventative treatment for PTE. This study reviews the current state of modeling, pathology, biomarkers, and potential interventions for PTE and comorbidities. There’s new optimism in finding a drug therapy for preventing PTE in people at risk, such as after traumatic brain injury, concussion, and serious brain injuries, especially in military persons.
I. Introduction to Brain Injury and Post-Traumatic Epilepsy
Traumatic brain injury (TBI) remains a significant source of death and permanent disability, contributing to nearly one-third of all injury-related deaths in the United States and exacting a profound personal and economic toll. TBI is defined as a disruption in the normal function of the brain that can be caused by a bump, blow, or jolt to the head, or a penetrating head injury. Common causes of TBI include sports-related injuries, falls, car accidents, and military incidents. About 2.87 million Americans experience a TBI each year, with more than 56,000 deaths and 280,000 individuals requiring hospitalization (Taylor et al., 2017). The number of emergency department visits related to TBI increased over 50% between 2007 and 2013; this rise is attributed to an ever-aging population and increased number of fall-related TBIs (DeGrauw et al., 2018).
The extent of damage varies widely based on age, sex, and severity of injury (Christian et al., 2020). An individual’s physical burden stretches beyond the initial damage, as TBI is often accompanied by a collection of secondary health consequences that negatively affect daily life. These complications include headache, vision impairment, tinnitus, difficulty focusing, imbalance, loss of hand-eye coordination, cognitive impairment, and affective disorders (Malec et al., 2019). The impact of TBI on close family members and caregivers is also extraordinary, ranging from physical strain and emotional stress of living with a person whose abilities, behavior, and personality have been altered, to additional demands on the caregivers for ongoing monitoring and assistance with daily tasks (Malec et al., 2017). Furthermore, the burden of TBI may unevenly fall on lower-income households (Tropeano et al., 2019), shedding light on the inequity of access to critical health care both in the United States and abroad. Moreover, TBI puts patients at higher risk of sleep disturbances and post-traumatic seizures (Gilbert et al., 2015). By some estimates, seizures occur in one out of every 10 hospitalized persons with moderate or severe TBI. Identified risk factors for post-traumatic seizures include loss of consciousness, intracranial hemorrhage, chronic alcoholism, depressed skull fractures, and cerebral contusions (Englander et al., 2003). Despite the increased resources that have been generated to improve our understanding of TBI and its comorbidities, the development of new diagnostic approaches has been disappointingly slow.
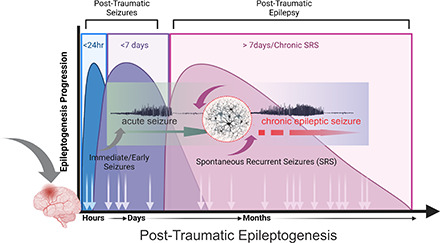
Post-traumatic epilepsy (PTE) is characterized by spontaneous recurrent seizures (SRSs) occurring as a result of TBI. Seizures have been temporally categorized into immediate (within 24 hour), early (1–7 days postinjury), and late seizures (>7 days postinjury) (Christensen, 2015). Immediate and early seizures are not considered to be “epileptic” and are thought to be provoked from the injury itself, rather than arising from a multitude of cellular and molecular changes. Thus, PTE represents a complex and chronic network disorder after head trauma, which induces epileptogenicity in the brain.
The cumulative risk of developing PTE ranges from 2%–50% depending on the location and severity of injury (Annegers et al., 1998; Chen et al., 2009; DVBIC, 2019; Tubi et al., 2019). It is well-established that the incidence of PTE increases with severity of initial TBI; however, the mechanism by which these seizures develop is still unclear. Many mechanisms have been identified through studies of status epilepticus and temporal lobe epilepsy (Pitkanen et al., 2015; Clossen and Reddy, 2017; Reddy et al., 2021). Once a patient with TBI experiences a single late seizure, their chance of experiencing a subsequent event increases by 80%, and seizure reoccurrence is most likely within 2 years of the first spontaneous late seizure (Haltiner et al., 1997; Englander et al., 2015). Acute or immediate seizures after TBI are treated with symptomatic antiseizure medications (ASMs), but these drugs are ineffective at preventing long-term or epileptic seizure occurrence (Marion, 1999). Although there are more than 20 ASMs in clinical use, approximately 30% of patients with epilepsy still experience drug-refractory epileptic seizures (Kobau et al., 2008; Reddy, 2020).
In addition to seizures, neuropsychiatric comorbidities are a significant source of burden after TBI. The mortality rate is nearly three times higher in patients with TBI who have seizures compared with TBI alone (Englander et al., 2003). When injury severity and location was accounted for, the most significant difference between patients who have PTE and those who don’t was the presence of focal cortical contusions. Neuropathology associated with TBI and PTE has recently been identified as a risk factor for developing serious neurologic disorders later in life, including Parkinson’s disease and dementia (Gardner et al., 2015; Fann et al., 2018). Moderate to severe head injury triggers amyloid plaque buildup in some patients, suggesting a possible link to Alzheimer’s disease as well (Barnes et al., 2014). Furthermore, a variety of comorbidities, including difficulty focusing, anxiety, learning and memory impairment, motor dysfunction, and sleep disturbances reduce the quality of life for many patients with PTE (Hammond et al., 2019).
Prevention of epilepsy and its progression is one of the major U.S. National Institutes of Health National Institute of Neurologic Disorders and Stroke research benchmarks. Experimental studies have demonstrated great understanding of neuropathology and PTE-associated comorbidities, including seizures, psychologic changes, and motor dysfunction. However, clinical translation of therapeutic strategies is lacking or has been unsuccessful in preventing post-traumatic seizures (Temkin, 2009). Therefore, our current animal models need to be further refined to discover novel biomarkers for PTE and better capture the mechanisms involved in epileptogenesis as related to the human condition. This article describes the current state of animal models used in experimental PTE studies, briefly examines mechanisms and biomarkers of post-traumatic epileptogenesis, then discusses the current progress in prophylactic and preventative therapeutics for PTE. Critical differences in injury patterns, variable latency period, and biomarkers are outlined in the context of model validation and correlation of pathophysiology, seizure frequency, and behavior. It also covers preclinical and clinical trials of new candidate treatments for PTE sequalae and associated comorbidities.
II. Animal Models of Post-Traumatic Epilepsy
PTE is a condition characterized by at least two SRSs after a head injury. Since spontaneous seizures often do not emerge until months or years after the initial TBI, it is extremely challenging to study the epileptogenic process in the clinical setting. The required studies that would provide insight from immediate injury through to diagnosis of epilepsy are invasive, time consuming, and perhaps not always ethically feasible. Furthermore, longitudinal studies using less invasive procedures, such as magnetic resonance imaging (MRI) and surface electroencephalogram (EEG), could take decades of compliance to complete and assume these patients do eventually develop seizures.
Rodent models of PTE allow for a more extensive investigation into the causal relationship between brain injury and seizures. Here, we can begin to understand the broad and cellular pathophysiology of epileptogenesis, discover and validate new biomarkers for PTE, and assess the efficacy of therapeutics for PTE. Rodent disease models are cost-efficient, more tightly controlled, and consume far less time to complete long-term reports. However, it is important to distinguish models of TBI from those of PTE. PTE requires the occurrence of at least two seizures after brain injury. Therefore, it is important as researchers, we strive to include seizure detection methods within the models. Without the presence of seizures or ability to detect epileptiform abnormalities, the focus of the model becomes brain injury and its comorbidities, rather than post-traumatic epileptogenesis. Presently, a handful of studies report seizure incidences from 0%–50% after mild to moderate TBI, which can slow progress on therapeutic developments (Kochanek et al., 2006; Hunt et al., 2009; Hunt et al., 2010; Rodgers et al., 2015; Szu et al., 2020). Researchers have attempted to remedy this challenge by including additional electrical or chemical convulsion approaches after injury, to ensure they observe seizures (Kharatishvili et al., 2006; Chrzaszcz et al., 2010; Bolkvadze and Pitkänen, 2012). Although a reduction in seizure threshold to these electrical or chemical stimulations has been observed, these models do not truly depict PTE since the seizures are not naturally generated. Table 1 summarizes the PTE models with in vivo recordings of neuronal hyperexcitability. Similarly, in vitro preparations of slices or cell cultures are an inadequate replacement for the invaluable data that spontaneously progressing in vivo models can provide. Lastly, a crucial benefit of rodent models over the clinical setting is the ability to explore novel targets and investigate efficacy of therapeutics. Even when biomarkers are discovered in humans, it is unethical to test pharmacological agents in patients until the safety and efficacy has been critically evaluated and confirmed.
TABLE 1.
Summarized PTE models with in vivo recordings of neuronal hyperexcitability
| Model | Species/ Preparation | Seizure Susceptibility | EEG Recording | Animals with Epilepsy (%) | Latency to SRS | Seizure Frequency | Seizure Duration | Epileptiform Spiking, Discharges, HFOs | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Parasagittal FPI | |||||||||
| Rat: Sprague Dawley, young male; 3.75–4 atm | N/A | 14 wks continuous recording beginning 2 wks postinjury | Probability of 92% epilepsy | 2–4 weeks | N/A | N/A | N/A | D'Ambrosio et al., 2004 | |
| Rat: Sprague Dawley, young male; 3.75–4 atm | N/A | 8 hr/rat/week for 30 wks beginning 2 wks postinjury | Probability of 100% epilepsy | 3–5 weeks | 4.1 ±1 seizures/week | 4.2–11.8 s | N/A | D'Ambrosio et al., 2005 | |
| Rat; Sprague Dawley, young male; 1.8–2.2 atm | Increased susceptibility to PTZ-induced seizures at 12 wks postinjury | N/A | N/A | N/A | N/A | N/A | N/A | Atkins et al., 2010 | |
| Rat: Sprague Dawley, adult young male; 1.9–2.1 atm | Increased susceptibility to PTZ-induced seizures at 2 weeks postinjury | N/A | N/A | N/A | N/A | N/A | N/A | Bao et al., 2011 | |
| Rat: Sprague Dawley, young male; 2.0 or 3.4 atm | N/A | Up to 17 weeks continuous recording, beginning 2 wks postinjury | Probability of 100% epilepsy | 2–18 weeks | 0.6 –3.0/hr | 1–144 s | N/A | Curia et al., 2011 | |
| Rat: Sprague Dawley, young male; 3.4–3.7 atm | N/A | 1–2 24 hr recordings/rat/week for up to 2.5 months postinjury | N/A | 2–5 weeks | 0.25–1.75 seizures/hr | 1 s–4.5 min | N/A | D'Ambrosio et al., 2013 | |
| Rat: Wistar, adult male | N/A | 2 wks continuous recording beginning 1wk postinjury | 30% | N/A | 6.3 seizures in 2 wks | 52.9 s | 11.5 discharges in 2 wks | Shultz et al., 2013 | |
| Rat: Sprague Dawley, adult male; 2.0 or 3.4 atm | N/A | Up to 12 months continuous recording, beginning 2 wks postinjury | 0% | N/A | N/A | N/A | Spike/wave discharges observed in both sham and injured rats. No epileptiform discharges/spikes observed in injured or sham rats. | Rodgers et al., 2015 | |
| Rat: Sprague Dawley, young male; 1.8 atm | N/A | Random increments of 12 hrs on/12 hrs off for 1–14 wks postinjury | 25% | 4–7 weeks | 2.83 seizures/week | Mean = 47.5 s | Interictal spikes observed in 58.3% of rats | Nemes et al., 2016 | |
| Lateral FPI | |||||||||
| Rat: Sprague Dawley, adult male; 0.5–1.2 atm, 1.5–1.6 atm, or 2.0–2.2 atm | Increased granule cell hyperexcitability at 1 wk postinjury | N/A | N/A | N/A | N/A | N/A | N/A | Lowenstein et al., 1992 | |
| Rat: Sprague Dawley, adult male; 2.0 atm | Increased inhibition in dentate gyrus at 15 days postinjury | N/A | N/A | N/A | N/A | N/A | N/A | Reeves et al., 1997 | |
| Rat: Sprague Dawley, adult male; 2.6–3.3 atm | Increased susceptibility to PTZ-induced seizures at 12 months postinjury | 4–7 wks continuous recording intervals for up to 12 months postinjury | 46% | 4–11 weeks | 0.3 seizures/day | Mean = 137 ± 57 s | Interictal spikes observed in epileptic rats | Kharatishvili et al., 2006 | |
| Rat: Sprague Dawley, adult male; 2.3–3.2 atm | Increased susceptibility to PTZ-induced seizures at 12 months postinjury | 3 wks continuous recording at 11 months postinjury | 0% | N/A | N/A | N/A | Isolated spiking observed in 80% of TBI rats | Kharatishvili et al., 2007 | |
| Rat: Wistar, P21; 2.0–2.2 atm | Increased susceptibility to kainate-induced seizures at 6 wks postinjury | N/A | N/A | N/A | N/A | N/A | N/A | Echegoyen et al., 2009 | |
| Rat: Sprague Dawley, P21 male; 2.2 atm | No change in PTZ susceptibility at 20 wks postinjury | Behavioral observation | 0% | N/A | N/A | N/A | N/A | Gurkoff et al., 2009 | |
| Rat: Sprague Dawley, adult male; 1.8–2.2 atm | Increased susceptibility to PTZ-induced seizures at 12 wks post-TBI | N/A | N/A | N/A | N/A | N/A | N/A | Atkins et al., 2010 | |
| Rat: Sprague Dawley, adult male; 2.2 atm | No change in fluorothyl-induced seizures at 3 and 6 wks postinjury | N/A | N/A | N/A | N/A | N/A | N/A | Schwartzkroin et al., 2010 | |
| Mouse: C57BL/6S, adult male; 2.8–3.0 atm | Increased susceptibility to PTZ-induced seizures at 6 months postinjury | Three 2-wk continuous recordings at 6 m and 9 m post-TBI | 3% (1/30) | N/A | 1 total seizure recorded during observation period | 91 s | Observed epileptiform spiking in 71% injured mice | Bolkvadze and Pitkänen, 2012 | |
| Rat: Long-Evans, adult male; 2.2–2.4 atm | N/A | Continuous recording for 12 wks at 3 months postinjury | 100% | N/A | 151 ±44 seizures/hr | 10–60 s | N/A | Goodrich et al., 2013 | |
| Mouse: C57BL/6S, adult male; 1.5–1.7 atm | Increased susceptibility to PTZ-induced seizures at 1month postinjury | N/A | N/A | N/A | N/A | N/A | N/A | Mukherjee et al., 2013 | |
| Rat: Wistar, adult male; 3.2–3.5 atm | N/A | 2 wks continuous recording at 1 wk postinjury | 30% | N/A | Mean = 6.3 seizures/rat over 2 wks | Mean = 52.9 s | Observed epileptic discharges in 22% of injured rats | Shultz et al., 2013 | |
| Rat: Sprague Dawley, adult male; 2.4–2.8 atm | N/A | Up to 50 total hrs of continuous recording per rat at 5–33 wks post-TBI | 94% (16/17) incidence of nonconvulsive and 20% (1/5) convulsive seizures | N/A | 7/hrs nonconvulsive seizures; 0.05/hr convulsive seizures | 78–110 s | N/A | Campbell et al., 2014 | |
| Rat: Sprague Dawley, adult male; 3.2–3.5 atm | N/A | Continuous recording up to 26 weeks post-TBI, divided into early, intermediate, and late recording groups | 50% at 1 year follow-up | N/A | N/A | 14–30 s | HFOs observed in 71% epileptic rats | Reid et al., 2016 | |
| Rat: Sprague Sawley, adult male; 2.8–3.3 atm | Increased susceptibility to PTZ-induced seizures at 6 wks postinjury | N/A | N/A | N/A | N/A | N/A | N/A | Wang et al., 2016b | |
| Rat: Sprague Dawley, adult male; 2.8–3.3 atm | N/A | Intermittent recording of 24 hrs every 2 wks, up to 12 months postinjury | 55% | 2 weeks–10 months | Mean = 6 seizures/rat | N/A | N/A | Wang et al., 2016d | |
| Rat: Sprague Dawley, young adult; mild: 1.2–1.5 atm, moderate: 1.8–2.2 atm, severe: 2.4–3.0 atm | N/A | 1 year after injury, rats were recorded in 1 hr ECoG increments | N/A | N/A | N/A | N/A | Greater spike/wave discharges observed in TBI rats | Sick et al., 2017 | |
| Rat: Wistar, adult male; 2.8–3.2 atm | Increased susceptibility to PTZ-induced seizures at 46 days post-TBI | Intermittent recordings of 48 hrs every wk at 2–5 wks post-TBI | 62.50% | N/A | Mean = 7.8 seizures/rat | N/A | Higher frequency and power of spike-wave discharges and spiking observed in TBI versus sham rats | Smith et al., 2018 | |
| Central FPI | |||||||||
| Rat: Sprague Dawley, adult male; 2.1 atm + PTZ kindling | Sham and injured rats developed an equivalent convulsant response to PTZ. | N/A | N/A | N/A | N/A | N/A | N/A | Hamm et al., 1995 | |
| CCI | |||||||||
| Rat: Sprague Dawley, P17; 2.0 mm depth | No change in threshold for tonic hind limb extension or minimal clonic seizures at P34–40; reduced threshold for minimal clonic seizures at P60–63 | N/A | N/A | N/A | N/A | N/A | N/A | Statler et al., 2008 | |
| Mouse: CD-1, adult; 0.5 or 1.0 mm depth | N/A | Random 1–2 hr intervals of recording, up to 18 hrs total per mouse, beginning 42 days post-TBI | 20% (0.5 mm); 36% (1.0 mm) | N/A | N/A | N/A | N/A | Hunt et al., 2009 | |
| Rat: Sprague Dawley, P17; 2.0 mm depth | N/A | Continuous recording in 2-8 wk epochs for up to 3 months, beginning 4–8 months post-TBI | 13% (1/8) | N/A | N/A | 45-60 s | Observed EEG spiking in 87.5% injured rats | Statler et al., 2009 | |
| Mouse: CD-1, adult; 1.0 mm depth | N/A | Random 1–2 hr intervals totaling 4–6hr/wks up to 10 wks post-TBI | 40% | 6.5 ±1.3 weeks | N/A | N/A | N/A | Hunt et al., 2010 | |
| Mouse: C57BL/6S, adult male; 0.5 mm depth | Increased susceptibility to PTZ-induced seizures at 6 months postinjury | Three 2-wk continous recordings at 6m and 9m postTBI | 9% | N/A | 0.25/day | 50 ±14 s | Observed epileptiform spiking in 81% injured mice | Bolkvadze and Pitkänen, 2012 | |
| Mouse: CD-1, adult male; 2.0 mm depth | N/A | Continuous recording for 16 wks post-TBI | 50% of untreated TBI mice | 82.3 10.2 days | 0.55 ±0.16 seizures/day | 35.5 ±2.8 s | Interictal spike discharges were seen in some TBI mice | Guo et al., 2013 | |
| Mouse: Plau deficient on C57BL/6J background, adult male; 0.5 mm depth | Both wild-type and Plau mice demonstrated increased susceptibiity to PTZ-induced seizures compared with shams | 2 wks continuous recording at 6 or 8 months post-TBI | 0% of wild-type or Plau mice | N/A | N/A | N/A | N/A | Bolkvadze et al., 2015 | |
| Mouse: CD-1, adult male; 1.0 mm depth | N/A | Random 1–2hr intervals totaling 6hrs/wk at 6–10 wks post-TBI | 40% | N/A | N/A | >30 s | N/A | Butler et al., 2015 | |
| Rat: Sprague Dawley, adult male; 2.8 mm depth | N/A | Week-long recordings up to 619 days post-TBI in some rats | 20% | N/A | N/A | 32 ±3 s (nonconvulsive); 91 ±12 (convulsive) | N/A | Kelly et al., 2015 | |
| Mouse: PlauR deficient on C57BL6/J background, adult male; 0.5 mm depth | Sham and injured mice developed an equivalent convulsant response to PTZ | 2 wks continuous recording at 12 wks post-TBI | 14% (1/6) wild-type mice | N/A | 1 observed seizure in 2 wks | 33 s | N/A | Bolkvadze et al., 2016 | |
| Rat: Wistar, adult male; 2.0 mm depth + PTZ kindling | Accelerated PTZ kindling compared with sham | N/A | N/A | N/A | N/A | N/A | N/A | Eslami et al., 2016 | |
| Mouse: APP/PS1 on C57BL/6 background, adult male; 0.5 mm depth | N/A | 2 wks continuous recording at 6 or 14 wks post-TBI | 11% wild-type TBI mice; 88% APP/PS TBI mice | N/A | 0.47/day | 35 s | N/A | Miszczuk et al., 2016 | |
| Mouse: C57BL/6, adult male; 1.0 mm depth | Increased firing of LV pyramidal cells within 1-2 mm of lesion core at 14 days post-TBI | N/A | N/A | N/A | N/A | N/A | N/A | Ping and Jin, 2016b | |
| Mouse: C57BL/6, adult male; 1.0 mm depth | Increased susceptibility to PTZ-induced seizures at 3 days postinjury | Random 30 min recordings at 0, 3, or 24 hrs after TBI | 74% mice experienced immediate post-traumatic seizures | <24 hr | N/A | N/A | N/A | Wang et al., 2017 | |
| Mouse: C57BL/6S, adult male; 0.5 mm depth | Increased susceptibility to PTZ-induced seizures at 14 wks postinjury | 2 wks continuous recording at 10 wks postinjury | 10% | N/A | N/A | N/A | N/A | Pijet et al., 2018 | |
| Rat: Sprague Dawley, adult male; 2.5–3.0 mm depth | N/A | Undefined intermittent recording parameters, up to 17 months post-TBI | 20.3% | N/A | N/A | N/A | N/A | Sun et al., 2018 | |
| Mouse: CD1, adult male; 1.0 mm depth | N/A | 1 wk continuous recording at 7, 14, 30, 60, or 90 days post-TBI | 27.80% | N/A | 0.43–0.71 seizures/day | N/A | N/A | Szu et al., 2020 | |
| Mouse: C57BL/6, adult male; 1.0 or 2.0 mm depth | N/A | Continuous recording for up to 4 months post-TBI, starting 10 days post-TBI | 33% (1.0 mm); 87% (2.0 mm) | 19.5 ±3 days (1.0 mm); 52.9 ±8 days (2.0 mm) | 0–2.74 seizures/day (1.0 mm); 0–4.17 seizures/day (2.0 mm) | 10–90 s | Epileptiform discharges observed in 42% (1.0 mm) and 87% (2.0 mm) TBI mice | Golub and Reddy, 2022 | |
| Weight Drop | |||||||||
| Rat: Sprague Dawley, adult male; 20 g weight at 20 cm | Increased susceptibility to PTZ-induced seizures at 15 days or 7 weeks | N/A | N/A | N/A | N/A | N/A | N/A | Golarai et al., 2001 | |
| Adult CD1 mice, male: closed skull injury + electroconvulsive shock 1 wk post-TBI | Increased susceptibility to PTZ-induced seizures at 15 days or 7 weeks | N/A | N/A | N/A | N/A | N/A | N/A | Chrzaszcz et al., 2010 | |
| Rat: Sprague Dawley, adult male; 50g weight at 60cm + PTZ administration on days 7 and 21 post-TBI | Animals subjected to both TBI and PTZ showed higher sensitivity to PTZ-induced seizures than noninjured PTZ-only rats | 1.5 hr recordings on days 7 and 21 post-TBI during PTZ testing | N/A | N/A | N/A | N/A | N/A | Efendioglu et al., 2020 | |
| Mouse: C57BL/6, adult; 100 g weight at 50 cm | N/A | Continuous recording up to 107 days post-TBI, starting 4 days post-TBI | 50% | >3 weeks | 1–11 seizures/mouse during observation period | 12–15 s | N/A | Shandra and Robel, 2020 | |
| Mouse: C57BL/6, adult male; closed skull, 50g weight at 80cm | Increased susceptibility to pilocarpine-induced seizures and status epilepticus | N/A; observational scoring | N/A | N/A | N/A | N/A | N/A | Ben Shimon et al., 2020 | |
| Penetrating and Blast Injury | |||||||||
| Rat: Sprague Dawley, adult; 0.48, 0.63, or 0.7 cm balloon diameter | N/A | 72 hrs continuous recording at time of injury and 2 hrs recording on days 7 and 14 post-TBI | 13% (mild); 39% (moderate); 59% (severe) | Immediate seizures; N/A | 3.8 ±0.8 seizures/rat in 72 hrs (moderate); 10.5 ±2.6 seizures/rat in 72 hrs (severe) | 31.4 ±9.6 s (moderate); 33.6 ±5.0 s (severe) | Epileptiform discharges observed in 30% (moderate) and 65% (severe) TBI animals | Lu et al., 2011 | |
| Rat: Sprague Dawley, adult male; 0.63 cm balloon diameter | N/A | 72 hrs continuous recording at time of injury | 71% immediate seizures | Immediate seizures; N/A | 9.5 ±2.5 seizures/rat in 72 hrs | 34.5 ±4 s | N/A | Mountney et al., 2013 | |
| Rat: Sprague Dawley, adult male; hippocampal lesion with either copper or stainless steel wire insertion | N/A | Random 2-wk continuous recording intervals for up to 11 months postinjury, beginning 6 wks postinjury | 96% copper; 15% stainless steel | N/A | 3/day copper; 0.2/day stainless steel | 33.8 ±2.6 s copper; 25.7 ±0.7 s stainless steel | N/A | Kendirli et al., 2014 | |
| Mouse: C57BL/6, adult male; peak overpressure of 14.6 psi, up to 3 blasts | N/A | 72 hrs continuous recording at 2 days post-TBI, and intermittent 48–72 hr recordings at monthly intervals for up to 10 months | 0% (single blast); 33% (double); 50% (triple) | N/A | 2.2 ±0.4 seizures/mouse (double) and 15 ±6.6 seizures/mouse (triple) during observation period | 39 ±4 s (>30 days post-TBI) | N/A | Bugay et al., 2020 | |
| Cortical Undercut | |||||||||
| Cat: adult both sexes; 3–4 mm depth and 13–15 mm distance | N/A | Not described | 40% | 2–3 hrs after undercut | N/A | 5–50 s | Observed interictal spikes, sharp waves, spike-wave, and polyspike-wave complexes in 60% of animals | Topolnik et al., 2003a; 2003b | |
| Cat: adult both sexes; 3–4 mm depth and 13–15 mm distance | N/A | Intermittent weekly recording under anesthesia | 92% | N/A | N/A | N/A | Low-frequency spike-waves and polyspike-wave complexes observed intermingled with fast 10–20 Hz runs in all injured cats | Nita et al., 2006 | |
| Mouse - C57BL/6, adult male; 1.2 mm depth and 1-2 mm distance | Increased susceptibility to PTZ-induced seizures at 15 days post-TBI | Continuous recording for 50 or 90 days post-TBI | 50% | 17-34 days | 0.17 ± 0.17 seizures/day | 8-18 s | Epileptic spikes observed in 71.4% of mice | Ping and Jin, 2016a | |
ECoG = Electrocorticography.
Four TBI models that have been adapted to induce PTE include the fluid percussion injury, controlled cortical impact, impact-acceleration, and the blast injury models. A recent study confirmed neuronal excitability and seizure activity after a repetitive blast model of TBI (Bugay et al., 2020). Additional models such as cortical undercut and penetration injury have also been used to model PTE. However, these models are less widely used, and therefore, less data on the translational properties are available. Table 2 compares the advantages and disadvantages of various animal models of PTE.
TABLE 2.
Advantages and disadvantages of animal models of PTE
| Model | Advantages | Disadvantages |
|---|---|---|
| CCI | Produces similar histopathology to the human condition, including concussion, acute subdural hematoma, loss of cortical tissue, axonal injury, inflammation, loss of gray matter, and blood-brain barrier dysfunction | Varied incidence of epilepsy between research groups and cohorts |
| Increased seizure susceptibility to chemical convulsants | Prolonged time-course for the development of SRS and some comorbidities | |
| Decreased seizure threshold to electrical stimulation | Mechanical variation | |
| Demonstrates persistent sensori-functional and cognitive deficits | Requires complex technical device to produce injury | |
| Produces epileptiform EEG activity, including discharges, high-frequency oscillations, and seizures in some animals | ||
| Highly studied and reproducible | ||
| Can produce varying degrees of severity | ||
| Has been used in rodent (mouse/rat), swine, canine, and primate models | ||
| FPI | Produces similar histopathology to the human condition, including diffuse or focal injury, contusion, edema, progressive loss of gray matter, inflammation, and blood-brain barrier dysfunction | Varied incidence of epilepsy between research groups and cohorts |
| Increased seizure susceptibility to chemical convulsants | Prolonged time-course for the development of SRS and some comorbidities | |
| Decreased seizure threshold to electrical stimulation | Lacks some translation to therapeutic validity | |
| Demonstrates persistent sensori-functional and cognitive deficits | Requires complex technical device to produce injury | |
| Produces epileptiform EEG activity, including discharges, high-frequency oscillations, and seizures in some animals | Variation in injury can occur because pressure wave is highly sensitive to operational factors | |
| Highly studied and reproducible | ||
| Can produce varying degrees of severity | ||
| Cortical Undercut | Simple surgical procedure that produces hyperexcitability | Not widely studied |
| Inexpensive and relatively simple protocol | Failure to consistently produce spontaneous recurrent seizures | |
| Impact-Acceleration/Weight Drop Model | Inexpensive and relatively simple protocol | Not widely studied |
| Can produce varying degrees of severity | Difficulty with reliable reproducibility | |
| Focal injury with axonal injury and hemorrhage | Failure to consistently produce spontaneous recurrent seizures | |
| Blast Injury Model | Produces similar histopathology to the human condition, including diffuse or focal injury, intracranial hemorrhage, inflammation, and blood-brain barrier dysfunction | Not widely studied |
| Injury mechanism similar to military TBI | Failure to consistently produce spontaneous recurrent seizures | |
| Penetrating Ballisticlike Brain Injury Model | Injury mechanism close to human missile or bullet wound injury | Not widely studied |
| Inexpensive and relatively simple protocol | Needs standardization | |
| Canine Model | Conducted in a species that naturally develops epilepsy disorders | Not widely studied |
| Needs standardization | ||
| Large Animal Models (Pig, Etc.) | Species is an intermediate state between rodent and primates | Not widely studied |
| Needs standardization | ||
| Requires additional resources and time for surgeries and care |
A. Fluid Percussion Injury Model
Fluid percussion injury (FPI) is perhaps the most extensively used and studied model of PTE, largely due to its ability to easily modify the severity of injury, impact site, and species used. FPI can be applied centrally over the sagittal suture between bregma and lambda (midline FPI) or laterally over the parietal cortex (lateral FPI) and has been adapted for mouse, rat, rabbit, cat, and pig models. FPI produces a fluid pulse injury directly onto the surface of the dura after a craniectomy. The fluid percussion device consists of an adjustable pendulum that strikes the piston end of a fluid-filled cylinder, generating a fluid pulse that is transmitted along the horizontal axis of the cylinder onto the brain.
The model was initially described by McIntosh et al. (1989) in rats with a 4.8 mm craniectomy mid-distance between bregma and lambda and centered between the sagittal suture and lateral ridge. An injury cap is positioned over the craniectomy and secured with dental cement or glue to ensure the fluid bolus remained within the cranial cavity. The injury is produced by a pressure pulse lasting 20 milliseconds, and the severity of injury can be altered depending on the location and magnitude of pressure used (low 1.5 atm to high 3.5 atm in rats). Parasagittal and lateral FPI are common models for studying PTE. After FPI, electrodes can be implanted either immediately or after a short recovery period.
The FPI model can reproduce neuropathology associated with both diffuse or focal injuries as well as other aspects of human TBI, such as acute hypertension, bradycardia, increased plasma glucose levels, hemorrhages, inflammation, and cognitive deficits (Eakin et al., 2015). The damage of FPI is most severe in the ipsilateral cortex, hippocampus, and thalamus, although mild lesions and cell loss has also been detected contralaterally. Additionally, many laboratories have demonstrated the development of epileptogenesis, including a reduction in seizure threshold, presence of epileptiform discharges, and subgroups with SRS (Kharatishvili et al., 2006; Mukherjee et al., 2013; D’Ambrosio et al., 2004).
Seizure incidence after FPI has been inconsistent, with ranges from 0% to a cumulative probability of 100% epilepsy. A major limitation in many of the earlier reports is the lack of continuous long-term EEG recording. Random or intermittent epochs of recording do not allow researchers to determine latency to seizure onset, and in many cases, seizure occurrence will be underestimated or over-reported due to missing data. In one of the most cited reports of FPI-induced seizures, Kharatishvili et al. (2006) found approximately 50% of rats developed PTE over a period of 12 months when monitored with 24/7 video-EEG. Seizures were described as partial or secondarily generalized with a duration between 67–59 seconds; however, seizure frequency was quite low, averaging 0.3 ±0.2 seizures/d/animal. Neuronal loss was observed within the ipsilateral hippocampus, and aberrant mossy fiber sprouting was evident within the dentate gyrus. Similar data have now been collected from several laboratories (Zhang et al., 2008; Andrade et al., 2017; Mukherjee et al., 2013; Shultz et al., 2013; Carver et al., 2021).
B. Controlled Cortical Impact Model
Controlled cortical impact (CCI) has also been widely characterized since its first description by Lighthall in 1988 (Lighthall, 1988). Originating as a model of TBI, CCI has been adapted for studying PTE with the addition of EEG-recording electrodes and measurements of electrophysiological changes. To date, CCI techniques have been replicated in mice, rats, swine, monkeys, and ferrets (Xiong et al., 2013). This method requires general anesthesia of the subject as well as a craniectomy—similar to the FPI model. A computer-controlled impactor is used to deliver a unilateral strike to the intact dura directly at a velocity and depth specified by the researcher. This model can be customized further by choosing the diameter of the impact tip as well as whether the tip is blunt or rounded. Due to the levels of precision involved, CCI can mimic focal injury or widespread degeneration and can remove a potential source of error regarding the position and depth of the impact site. CCI studies have mimicked acute subdural hematoma, axonal injury, cell and tissue loss, blood-brain barrier disruption, and inflammation (Osier and Dixon, 2016). Additionally, many studies have described psychologic, functional, and cognitive changes associated with TBI (Kochanek et al., 2002; Watanabe et a., 2013; Yen et al., 2018).
Over time, long-term changes in the organization of neural circuitry due to trauma can lead to significant cell loss as well as an imbalance of excitatory and inhibitory neurotransmission. Seizures after CCI and FPI have been described in similar terms, both behaviorally and electrographically; however, CCI-induced spontaneous seizures appear to have a swifter onset compared with lateral FPI in rats (Kharatishvili et al., 2006). Previous studies have suggested limbic involvement may not appear for several months after FPI, resulting in a longer latency between initial injury and seizure occurrence (D’Ambrosio et al., 2004; 2005). This shorter, yet clinically relevant timeline, allows for greater flexibility in pharmacological testing.
A full spectrum of interictal activity including early (within the first week postinjury) or late seizures (after first week postinjury), isolated spikes, epileptiform discharges, absencelike spike wave discharges, and high-frequency oscillations (HFOs) have been successfully reproduced after moderate and severe CCI (Hunt et al., 2009; 2010; Statler et al., 2009; Golub and Reddy, 2022; Konduru et al., 2021). Sham mice do not show evidence of seizure activity, but high-frequency oscillations, interictal spike runs or “absencelike” spike-wave discharges have been observed in both injured and sham mice (Konduru et al., 2021). However, this study also pointed out that sham-injured rodents are not always appropriate controls for EEG studies since sham rodents have sometimes demonstrated interictal spiking or discharge activity. Computed tomography (CT) imaging revealed mild lesions compared with noncraniectomy mice, which could have contributed to abnormal activity.
Similar to FPI studies, seizure incidence after CCI is largely dependent on the severity of impact, ranging between 10% and 85% of mice exhibiting increased epileptic discharge spiking and seizures in the weeks after CCI (Hunt et al., 2010; Bolkvadze and Pitkänen, 2012; Golub and Reddy, 2022). Additionally, CCI studies have demonstrated progressive hyperexcitability in neocortical circuits within the first two weeks after TBI (Yang et al., 2010). These changes in electroencephalography are often accompanied with mossy fiber sprouting, hippocampal lesions, and changes in neurogenesis—all of which are defined as hallmarks of temporal lobe epilepsy (Hall et al., 2008; Hånell et al., 2010). Like FPI studies, CCI injuries provide consistency, reproducibility, and an overall accepted construct validity. This model also has the added benefit of easily altering injury severity, thereby changing the course of epileptogenesis and hippocampal pathology.
C. Weight-Drop or Impact-Acceleration Model
The impact-acceleration model, also known as the weight-drop model, simulates a diffuse injury to the brain and was first described by Mararmou and colleagues (Mararmou et al., 1994). After general anesthesia, rats or mice are placed below the weight-drop device on a foam block or platform intended to provide a consistent placement of the animal’s head and body relative to the apparatus. Skull fracture can be prevented by securing a small impact surface onto the location where the force will be applied, using cement or another adhesive. The impact-acceleration model can produce graded injuries, based on the heaviness and height at which the weight is dropped. This model can also be used as a form of repetitive TBI, producing a number of cumulative, but lesser injuries (Bailes et al., 2014).
The clear advantage to this model is the simplicity to induce trauma. The weight-drop apparatus itself can be constructed using inexpensive supplies and does not require a craniotomy within a stereotaxic rig, allowing for low cost and high throughput efficiency. Although this model leads to many known sequelae of human TBI, a critical downside is that weight-drop only produces PTE at extreme intensities, at which most animals do not survive. Furthermore, to identify and analyze abnormalities in EEG activity, such as post-traumatic seizures, typically an electrode is implanted at various focal sites, thereby still requiring access of a stereotaxic apparatus. Although this model is rarely used to identify seizure activity, it has been recently adapted to identify subtle changes in seizure threshold to chemical convulsant compounds, such as pilocarpine, without the need of recording electrode placement (Ben Shimon et al., 2020).
D. Blast Injury Model
Blast-related injuries are a frequent outcome of military exposure to explosive detonation. A leading hypothesis for the mechanism by which an explosive blast causes TBI is through the transmission of shock waves across the target tissue, causing rapid acceleration and deformation of the brain (Magnuson et al., 2012). These oscillating blast waves have sometimes been referred to as the “bobblehead effect” (Rosenfeld et al., 2013). Indirect transmission of kinetic energy from blast shock can travel through vasculature, playing a large role in TBI. Several models of blast TBI have been developed, although open-field blast, blast tubes, and shock tubes are the most frequently used (Kovacs et al., 2014).
Open-field blast occurs when an explosive detonation occurs within an outdoor open area, either by overhead suspension or placement on the ground. This is one of the most accurate representations of the human blast injury condition since subjects are located a standoff distance away from the blast. However, since debris and clouds from the primary explosion can contribute to the injury, blast tubes provide another method of blast injury in which a combined shock wave and blast wind is initiated by an explosion. In this case, animals are placed at the end of a pressurized blast tube, and the head-on explosion occurs at the opposite end of the tube. The torso and abdomen of the mouse are protected from exposure to prevent confounding injuries of the lungs, heart, and gastrointestinal tract. This method allows the rodent to be subjected to a “clean” blast without the reflection of shock fronts from the ground or other surfaces. Another advantage of blast tubes versus open-field blasts is that an equivalent blast intensity can be achieved with a much smaller explosive charge. Lastly, shock tubes use compressed gasses, such as helium, rather than explosives to achieve injury. Shock tubes are generally safer for both the subject and researcher to perform, more cost effective, and can be performed indoors—thereby not affected by weather conditions. However, the physics of shock tubes differs from that of explosive shock waves, which may not be as comparable to the human condition.
Blast injuries re-create several pathophysiological processes that likely play a role in the development of PTE, including intracranial hemorrhages, vasospasm, neuronal damage and degeneration, focal or diffuse axonal injury, and inflammatory reactions (Nakagawa et al., 2011). Although blast models have revealed neurofunctional changes, only one study has reported post-traumatic seizures and reduction in seizure threshold. Bugay et al. (2020) observed 46% incidence of spontaneous seizures in mice within a long-term study after up to three consecutive days of repetitive blast injury. Most seizures were electrographic, with little to no behavioral component observed. They also reported a shortened latency to spiking and hyperpolarization of action potential threshold in patch clamp recording of the hippocampus. These results produced a graded response to the number of blasts each mouse received (i.e., one, two, or three consecutive blasts). Although this is the first study to investigate TBI effects on neuronal excitability, the data clearly demonstrate increased risk of post-traumatic seizures as a measure of severity and repetitive injury.
E. Penetrating Ballistic-Like Brain Injury Model
The penetrating ballistic-like brain injury (PBBI) was designed to model two aspects of high-energy bullet wounds to the head: a large temporary cavity produced from energy dissipation and a permanent injury tract created by the path of the bullet (Williams et al., 2005). The PBBI itself is generated by inserting a custom probe into the brain at the desired location, creating the permanent injury tract, followed by the sudden inflation of an attached balloon to mimic the temporary cavity. The rodent is first placed into a stereotaxic device under anesthesia, scalp incised along the midline, and a small cranial window is created to allow the insertion of the PBBI probe. The probe can be constructed from a thin, 20-gauge stainless steel tube with spatially fixed perforations at one end. The perforations are sealed with airtight elastic tubing, which forms an inflatable balloon when an air pulse is delivered through the steel tube. The probe is then retracted, and the craniectomy is sealed with sterile bone wax or dental cement. Screw or depth electrodes can also be placed either before or after PBBI to obtain EEG recording. Since the relationship between the bullet’s impact velocity and the diameter of the cavity is linear, the parameters of this model can be altered to generate varied injury types or severities. However, this model of TBI mimics a specific and severe form of injury generally seen only in military populations.
One of the most notable consequences of PBBI is intracerebral hemorrhage, which is most common along the route of the probe entry. Additionally, PBBI re-creates pathologies found in the human condition of missile injury, such as extensive zones of radiating neurodegeneration, inflammation, neurologic impairments, edema, and post-traumatic seizures (Wei et al., 2010).
F. Cortical Undercut Model
The cortical undercut model of PTE was developed to mimic penetrating cortical injuries and has been used in both in vivo and ex vivo studies. Cortical undercut has been performed on rats, mice, and cats of varying age (Graber and Prince, 1999; 2006; Chauvette et al., 2016). Typically, rats are anesthetized and placed into a stereotaxic frame before unilateral exposure of the area of interest, typically the sensorimotor cortex. The dura mater and blood supply should be left intact after the craniotomy. A custom-made surgical knife or thin gauge needle, bent at a 90° angle 2mm from the tip is lowered to the white matter below cortical layer VI and rotated 180°, raised, then rotated back to its original position before being withdrawn to transect the underlying white matter. Attention should be paid to avoid any damage to major blood supplies. When a more complete undercut is needed, a second transcortical cut can be made with the needle without rotation (Graber and Prince, 1999; 2006). After a maturation period, animals are sacrificed, and coronal slices are obtained for electrophysiologic recordings and histology.
When preparing for an in vivo EEG study, depth electrodes can be placed in the surrounding or contralateral cortex, hippocampus, or other regions of interest. Array placement of electrodes is common in larger species, such as cats (Nita et al., 2006; Timofeev et al., 2013). Previous experiments demonstrate an immediate reaction to partial cortical deafferentation, resulting in significant reduction of local field potential amplitudes in regions above the transected white matter. Furthermore, 70% of animals experienced a shift in slow oscillatory activity to paroxysmal discharges (Topolnik et al., 2003a; 2003b). Seizure onset in this model evolves from slow oscillation and is characterized by the shortening of silence periods and increased amplitude of depolarization during active periods. Within a month after cortical undercut, electrographic paroxysmal activity spreads to regions distal to the initial transection and can be detected from corresponding electrodes. In mice, generalized seizures can be detected from the leading electrode, located just proximal to the undercut, within hours or days postinjury (Chauvette et al., 2016).
Cortical undercut is a valuable model for investigating the details of functional and structural alteration of neocortical GABAergic interneurons and pyramidal neurons occurring at the site of a focal injury without the spread of widespread damage and inflammation. However, this model is rarely used for pharmacological research due to its lack of translatability to the human condition. Clean transection of the white matter is rarely seen in human TBI without more extensive injury. Furthermore, there are few behavioral studies with this model, although available data indicate no significant motor deficits or cognitive impairment (Graber and Prince, 1999).
G. Repetitive Traumatic Brain Injury and Concussion Model
The majority of TBIs are mild in severity and are often underreported and, therefore, undertreated. Close-head impact injuries are a common cause of concussion and TBI. The consequences of repetitive traumatic brain injury (rTBI) and concussions have gained increasing attention with emerging reports of altered mood, behavior, and neurologic function. Concussions are extremely common in sports-related injuries, especially in contact-collision sports, such as boxing or American football, putting athletes at a higher risk for neurologic injuries, such as chronic traumatic encephalopathy and PTE (Mez et al., 2017).
The rTBI model mimics cellular and molecular changes induced by diffuse TBI, representing mild, concussive TBIs. Shandra and Robel (2020) recently published a detailed protocol for reproducing rTBI using a modified weight-drop model in mice. Briefly, an anesthetized mouse is placed in an induction chamber with its head positioned under the weight-drop tube on a foam pad. The pin on the weight-drop tube is released so that a 100 g weight is dropped from a height of 50 cm directly onto the scalp. A total of 2–4 weight drops are induced, with a 45-minute recovery period in between. EEG electrode implants can be placed via stereotaxic surgery either on the same day or the next day.
The rTBI model is characterized by a lack of focal lesion to the brain, loss of consciousness, high survivability, and late seizure onset (Shandra and Robel, 2020). Progressive tauopathy has also been observed in both experimental rTBI models and after repeated sports-related injuries (McKee et al., 2009; Tagge et al., 2018). Contrary to FPI or CCI models, the rTBI model does not require a scalp or cranial opening, reducing the risk of increased inflammation or infection.
H. Large Animal Models of Post-Traumatic Epilespy
Large animal species, such as pigs, are used in translational research because of their gyrencephalic neuroanatomy and significant white matter composition. However, one limitation of using these animals is that the laminar structure of the pig hippocampus has not been well characterized compared with that of a rat or mouse. Nevertheless, the Wolf group has described a porcine model of brain injury-related hyperexcitability and PTE and has been working toward elucidating the structure and characterization of the pig hippocampus (Ulyanova et al., 2018).
In the pig model of TBI, the primary neuropathological finding is diffuse axonal injury; however, hippocampal axonal and synaptic dysfunction as well as regional hyperexcitability have been observed, suggesting this model can be adapted for PTE (Meaney et al., 1995; Johnson et al., 2016; Wolf et al., 2017). Closed-head rotation induces a diffuse brain injury using a HYGE pneumatic actuator at controlled rotational acceleration levels to obtain the intended injury severity (Cullen et al., 2016). Briefly, the animal’s head is secured to a custom large-animal stereotaxic rig equipped with a padded snout clamp. The HYGE pneumatic actuator is mounted to the surgical rig using a custom linkage assembly that converts the linear motion to an angular motion. Rapid head rotation is performed within the coronal plane at velocities between 131 and 195 radians/s. Pathologies observed in the pig model of TBI include axonal shearing, tau accumulation, inflammation, and increased network excitability in the hippocampus (Smith et al., 1999; Johnson et al., 2016; Wolf et al., 2017; Grovola et al., 2020). Furthermore, motor and cognitive dysfunction has been documented in pigs after TBI (Friess et al., 2007).
A major downfall of larger animal models is the additional resources and time required to perform the appropriate surgical procedures and care. Therefore, pig studies have been inconsistently used due to their difficulty to implement and increased cost. Furthermore, induction of TBI in cats and dogs have been established but have not been widely used for studying PTE (Morganti-Kossmann et al., 2010). More recently, however, naturally occurring canine epilepsy has been proposed as a translational platform for novel therapeutics for epilepsy disorders (Davis et al., 2011). The prevalence and pathology of naturally occurring canine epilepsy are similar to the human condition (Löscher et al., 1985). In a study evaluating risk of seizures in dogs after head injury, patient records from 1343 diverse breeds were reviewed for previous head injury and recurrent seizures (Steinmetz et al., 2013). Of the 236 dogs with previous head injury, 18.6% exhibited early and/or late post-traumatic seizures. Observed seizure types included convulsive status epilepticus, partial and generalized tonic-clonic seizures, and cluster seizures. Although these data seem promising, the study has a few shortcomings, including difficulty obtaining enough questionnaires or telephone interviews to obtain statistical power. Retrospective studies such as these are not as feasible for the testing of therapeutic interventions.
The broad etiology after TBI presents a challenge for a singular paradigm to re-create all pathologies associated with PTE. The most common causes of human brain injury are car accidents, falls, recreational or sports injuries, and military incidents—all of which present differently within the clinic. Furthermore, acceleration-deceleration injuries differ from blunt force closed-head injuries or penetrating trauma in tearing, scar formation, and contusion (Dixon, 2017). Desirable features in animal models include a high frequency of epilepsy with an absence of extreme seizure clustering, low intersubject variability in seizure presentation, and a rapid and defined evolution of epileptogenesis. Ideally, this model would also be high throughput and low cost. Although the current animal models of PTE have provided much needed insight, no current model can fully recapitulate the full experience of human TBI. Therefore, it is important to understand the strengths and shortcomings of each model to determine which has the optimum conditions to evaluate specific research questions.
I. Translational Relevancy of Animal Models for Post-Traumatic Epilepsy
There has been much debate about the translational relevancy of animal models to the human condition for brain trauma research. As outlined above, numerous animal models have been developed to replicate various aspects of TBI and used for testing potential treatments. Although larger animals are closer in size and physiology to humans, rodents, such as rats and mice, are most commonly used in PTE research due to convenience research operations. The most widely used models include the CCI model, the FPI model, the weight drop-impact model, and the blast injury model. However, these models show intense negative effects, such as skull fracture, intracerebral hemorrhage, axonal injury, neuronal cell, and tissue death. Figure 1 outlines important considerations of translational relevancy between using small rodents to model the human condition. Like poststatus epilepticus models (Reddy and Kuruba, 2013), small animal models are most frequently used in preclinical post-TBI studies, with the aim to improve and develop better understanding of the recovery mechanisms and discover new biomarkers or clinical therapeutics. However, there are differences between small animals and humans as well as limitations to consider. Humans have 23 pairs of chromosomes, whereas rats and mice have 21 and 20, respectively. Although the known human genes associated with disease pathologies have corresponding orthologs in the rat and mice genome, their rates of synonymous substitution are different in the remaining genes. Furthermore, there are many differences in brain anatomy and complexity between small animals and humans. There are also some analogies, i.e., cerebrovascular parameters (Cernak, 2006), but the ratio of white:gray matter differs tremendously, making interpretation of behavioral alterations more challenging (Cordeiro and Horn, 2015). Furthermore, human TBI tends to be much more heterogenous in both injury location and cause (e.g., fall, car accident, sports injury, military events, etc.) than controlled laboratory experiments. However, there are many well-established neurologic and functional tests that can identify sensitive changes in recovery, cognition, or psychiatric function. These functional assessments are critical in comparing injury severity and outcomes not only between cohorts, but also other laboratory groups, similar to how the Glasgow coma scale is the gold standard for identifying injury severity upon clinic arrival. We discuss some of these neurologic and functional assessments in detail in section V of this manuscript, “Comorbidities of PTE.”
Fig. 1.

Translational relevance of rodent to human TBI. Despite significant investment in advancing technology and basic science to increase knowledge of human TBI pathology, translation from bench-to-bedside into therapeutic advances has been slower than expected. One of the factors limiting the translation of scientific knowledge from preclinical studies into the clinic is the limitation of small rodent in vivo disease models. Although these models have been developed to simulate and mimic the human condition, there are innate differences between rodents and humans, which can limit the impact of these studies. Likewise, there are many important similarities as well as practical hints that can be used to overcome these limitations. This figure discusses important considerations of rodent to human translational relevance.
TBI is also associated with greater risk and frequency of neurodegenerative diseases, such as Alzheimer’s disease, but also enhanced risk of epileptic seizures. Temporally, seizures after brain injury have been categorized into immediate, early, and late seizures. Due to anesthesia protocols and difficulty in recording EEG directly after impact, immediate seizures in rodents are rarely identified; however, studies have reported early and late seizures after FPI or CCI-induced trauma (see sections II and VI). Animal models allow for experimental strategies in determining cellular and molecular interactions within the latency period and epileptic onset, thus provide a reasonable platform to develop new therapeutic interventions for PTE.
As trauma is frequently associated with damage of skin and soft tissues, differences of wound healing between rodents and humans should also be considered. The epidermis and dermis of small animals is thinner than in humans, which creates a challenge for wound suturing. However, wound clips provide a suitable alternative to classic suturing techniques, which is both time and cost effective. Moreover, rats and mice are at a lower risk for infection compared with humans due to faster wound healing processes and their ability to convert L-gluconogammalactone to vitamin C (Abdullahi et al., 2014).
TBI-induced coagulopathy manifests as disseminated intracranial hemorrhage, systemic bleeding, or intracerebral hematoma and is closely correlated to poor clinical outcomes and early onset seizures (Abdelmalik et al., 2016). An animal model using Sprague Dawley rats mimics this specific clinical scenario, and the study completed by Gangloff et al. (2018) suggests great similarity to human acute traumatic coagulopathy in terms of temporality, type of injury, compensatory mechanisms, and impairments in the coagulation systems. However, quantitative results in blood coagulation are not entirely transferable due to species-specific differences in clotting factors in the serum, such as coagulation factors (F)V, FII, FXII, and FXIII, which were all elevated in rats compared with healthy human donors (Karges et al., 1994). Additionally, coagulation factors FVIII, XI, X, and XI were all reduced in rats when compared with pooled human plasma. Other studies have identified decreased platelet count and reduced responsiveness to thrombin in small animals compared with humans (Derian et al., 1995). These factors may affect identification of biomarkers for TBI-induced pathologies.
Another caveat of rodent models is that animals are anesthetized at the time of injury, in comparison with humans, who are typically awake upon injury impact. Ethical reasons prevent a study in which the injury is given without anesthesia, and therefore the impact of anesthesia at time of injury on subsequent neuropathology and behavioral manifestations is unknown. Perhaps future studies can be designed to administer the trauma event in an unanesthetized animal to truly understand the impact of TBI, but this requires working with Animal Care and Use Committees locally or nationally. When planning and performing trauma-related experiments, the “3 R’s” of Reduction, Replacement, and Refinement, published by Russell and Burch in 1959, should always be considered and reflect both the complex pathophysiology and the immunologic alterations induced by the trauma event. Although there appear to be several differences between humans and small animals, there are also many processes in basic trauma and regeneration that have been conserved, making these models suitable for translational research.
III. Neuropathological Mechanisms of Post-Traumatic Epilepsy
TBI sets into motion a multifaceted cascade of temporally overlapping cellular and molecular events, ultimately leading to PTE. Primary injury refers to the immediate trauma and tissue deformation that occurs within seconds to minutes after insult. Within this timeline, a flood of neurotransmitters is released, followed by ion channel activation and calcium influx. Immediate and early seizures are thought to occur as a direct result of the excitotoxic environment, mitochondrial damage, inflammation, and tissue injury. Although these seizures are not considered to be “epileptic,” they can exacerbate initial damage (Temkin, 2009). Secondary injury involves several physiologic mechanisms associated with progressive damage (Pitkanen et al., 2002). Chronic activation of inflammatory cascades, oxidative stress, and edema causes buildup of free radicals and reactive oxygen species. These factors become compounded by neurodegeneration, mitochondrial dysfunction, and the extended disruption of homeostasis. Furthermore, self-repair mechanisms occur concurrently and include plastic processes, such as structural axonal remodeling, neurogenesis, gliosis, and angiogenesis (Lucke-Wold et al., 2015).
Classically, epileptogenesis is defined as the period of time in which a normal brain is functionally altered, resulting in increased seizure susceptibility and risk of SRS. Within the framework of acquired epilepsy, researchers relied on the context of a “latent period” in which an epileptogenic insult (mechanical, chemical, or otherwise) triggered a series of changes and ultimately ended with occurrence of seizure output. However, certain processes, such as molecular and cellular plasticity, inflammatory cascades, and neurodegeneration, can continue indefinitely beyond the occurrence of the first seizure (Pitkanen et al., 2002; Dudek and Staley, 2012). Recently, the International League Against Epilepsy revised the definition of epileptogenesis to include disease modification and the concept of continuous epilepsy progression. Thus, the term “disease modification” has two main components: (i) alleviation or prevention of seizure development, termed “antiepileptogenesis,” and (ii) modification of PTE-associated comorbidities. In this next section, we discuss some of the major mechanisms associated with the progression of epileptogenesis, including changes in neuroinflammation, blood-brain barrier (BBB) breakdown, alteration of the epigenetic landscape, and reorganization of neural circuitry (Fig. 2).
Fig. 2.

Acute pathologies of post-traumatic epileptogenesis. Brain injury triggers several acute pathologies. Direct insult compromises the blood-brain barrier, allowing infiltration of peripherally circulating immune cells, such as leukocytes, macrophages, and neutrophils. NF-κB translocates to the nuclei of microglia, transforming them to an activated phenotype. This induces cellular proliferation and the release of inflammatory amplifiers, such as chemokines, cytokines, reactive oxygen species (ROS), and nitric oxide synthase (NOS). Macrophages participate in the cleanup of damaged cells and debris, but based on their functional activation state, may either exacerbate damage or initiate repair mechanisms. Lactate release from astrocytes contributes to water retention and edema. Excess iron from a leaky BBB can contribute to hyperexcitability. Excessive accumulation of glutamate and aspartate neurotransmitters due to spillage from damaged neurons or impaired reuptake by astrocytes activates NMDA and AMPA receptors located on postsynaptic membranes, allowing for influx of calcium ions. Together with the release of Ca2+ stores from the endoplasmic reticulum, increases in Ca2+ leads to production of ROS and activation of calpains. Damaged or dysfunctional mitochondria create a deficit of available ATP, leading to Na+/K+ pump failure, activation of Ca2+ channels, and further production of ROS/NOS. Cytochrome C released into the cytosol activates cell death pathways via caspase proteins. Epigenetic modifications, in the form of increased HDAC activity and altered DNA/histone methylation, changes transcriptionally active sites, including many genes associated with hyperexcitability and serotonin-to-melatonin conversion. Furthermore, DNA damage leads to apoptosis and cell loss. Progressive axonal damage and tau tangles lead to impaired axonal transport and results in both neurodegeneration and hyperexcitability. Together, these acute pathologies are both adaptive and maladaptive. The former contributes to functional and beneficial recovery, whereas the latter exacerbates epileptogenesis and the progression of abnormal electrographic activity.
A. Neuroinflammation
Local inflammation is intended as a beneficial protective measure after tissue insult; however, aberrant inflammatory responses can alter neuronal function and lead to serious consequences, such as BBB disruption and seizure development (Vezzani et al., 2013). Activated microglia and astrocytes play a large role in inflammation by releasing proinflammatory cytokines into the neuronal environment and promoting scar formation around tissue injury. Cytokine cascades in the brain regulate important pathways, such as neuroendocrine function, synaptic plasticity, metabolism of neurotransmitters, neurogenesis, and the kynurenine pathway (Paudel et al., 2018). These innate processes play significant roles in cell excitability and survival, thereby promoting network hyperexcitability. In particular, the interleukin (IL)-1/toll-like receptor (TLR) signaling pathway is disrupted and the associated receptors IL-1R1, TLR2, TLR3, and TLR4 are rapidly upregulated after both cell injury and seizures (Ravizza and Vezzani, 2006). The excitatory effects of IL-1β have been reported in several brain regions (Vezzani et al., 2011). IL-1β reduces GABA inhibition within the Cornu Ammonis area 3 (CA3) of the hippocampus and increases neuronal excitability in the CA1 by reducing N-methyl D-aspartate (NMDA) and voltage-gated calcium channel efflux (Zhang et al., 2010). Furthermore, lipopolysaccharide-induced inflammation is associated with reduction in seizure threshold in both postnatal and adult rodent models (Galic et al., 2009). This effect on seizure threshold can be reversed by blocking cytokine induction in activated microglia (Galic et al., 2009).
Induction of cyclooxygenase-2 (COX-2) has also been shown to promote epileptogenesis and contribute to neuronal damage in several animal models of epilepsy (Kulkarni and Dhir, 2009). Overexpression of COX-2 intensifies kainic-acid–induced seizures and mortality in mice (Kelley et al., 1999). Wei et al. (2018) confirmed COX-2 mRNA expression was significantly elevated after maximal electroshock. Although the modulation of the COX-2/prostaglandin E2 (PGE2) pathway has been pursued as an alternative therapeutic strategy for controlling seizures, careful study of COX-2 inhibitors could not fully prevent the appearance and development of spontaneous seizures in a rat model of status epilepticus (Holtman et al., 2010). Furthermore, inhibition of COX-2 has been found to either exacerbate or attenuate epilepsy-induced neurodegeneration, depending on the strategies used to interfere with the COX-2 pathway (Baik et al., 1999; Polascheck et al., 2010). These data highlight the ways in which the COX-2 pathway affects epileptogenesis, but mediation of this pathway alone is not sufficient for preventing seizure development.
An important consideration of inflammation in PTE is its contribution to progressive cell loss after injury. Free radicals and proteases accumulate during periods of inflammation, supporting lipid and protein peroxidation, DNA damage, mitochondrial dysfunction, and induction of apoptosis (Vezzani et al., 2013). Tissue damage, stress, and their subsequent cytokine release also adversely interfere with neurogenesis and neuroplasticity through their interactions with brain-derived neurotrophic factor (BDNF) and tropomyosin receptor kinase B (TrkB) receptor signaling (Goshen and Yirmiya, 2007; Ibrahim et al., 2016; Reddy et al., 2020). In a healthy brain, BDNF plays a crucial role in neuron maturation by regulating chloride levels and modifying inhibitory GABAergic signaling from depolarizing to hyperpolarizing (Rivera et al., 2002). However, within the context of injury, the upregulation of BDNF and its receptor TrkB are believed to promote aberrant mossy fiber sprouting (Dinocourt et al., 2006). Furthermore, brain injury causes a selective cluster of differentiation-74 (CD74)-dependent peripheral lymphocyte activation that may exacerbate neurodegeneration (Tobin et al., 2014).
Prolonged neuroinflammation also greatly affects quality of life and complicates comorbidities, giving cause for identifying therapeutics that explore the mechanistic association between PTE and neurobehavior dysfunction (Paudel et al., 2018). interferon-α can decrease BDNF levels, thereby slowing the rate of cell proliferation in the hippocampus and negatively affecting learning and memory consolidation (Lotrich et al., 2013). Additionally, increased cytokine production causes an imbalance of neurotransmitters, such as serotonin and dopamine, by deregulating the kynurenine pathway and disrupting neurotransmitter transport function (De la Garza and Asnis, 2003). Meta-analyses of existing research have concluded the most reliable biomarkers of inflammation in patients with depression are heightened levels of IL-6, tumor necrosis factor (TNF)-α, IL-1β, and C-reactive protein—all of which are significantly increased with TBI (Miller et al., 2009). Together, these inflammatory processes work in concert to promote depression, anxiety, cognitive impairment, and disturb sleep (Dantzer et al., 2008; Mukherjee et al., 2020).
B. Breakdown of the Blood-Brain Barrier
The blood-brain barrier (BBB) is a particularly important structure for central nervous system (CNS) homeostasis. There is increasing evidence demonstrating the BBB as a multifactorial pathophysiologic process involving faulty angiogenesis, neuroinflammation, altered glial physiology, leukocyte-endothelial interactions, and hemodynamic changes resulting in hyperexcitability (Marchi et al., 2012). Epilepsy disorders and TBI manifest with variable extent of BBB dysfunction; however, the link between BBB permeability and seizures has been posed as “the puzzle of the chicken and the egg” (Friedman, 2011). Acute vascular failure with BBB damage is sufficient to cause seizures in the absence of CNS pathologies or concomitant chemical convulsants (Marchi et al., 2007). Additionally, focal chronic seizures are frequent in patients with vascular malformations, such as cavernous angiomas (Kraemer and Awad, 1994). Magnetic resonance images of cavernous angiomas often present with BBB dysfunction, intracerebral deposits of iron, and albumin accumulation—all three of these factors have been identified as common features of TBI and temporal lobe epilepsy (van Vliet et al., 2007; Raabe et al., 2012).
BBB damage has been demonstrated to both trigger and sustain seizures in animal models and the human experience (Marchi et al., 2007; van Vliet et al., 2007; Raabe et al., 2012). Tomkins et al. (2008) observed greater association of BBB pathology in patients with PTE compared to patients with seizure-free TBI, suggesting a correlation between BBB breakdown and hyperexcitability. Areas of BBB disruption were linked to decreased brain glucose uptake, hypometabolism, and abnormal neuronal activity. After exposure of the cerebral cortex in rats, hypersynchronous epileptiform activity involving glutamatergic and GABAergic neurotransmission as well as significant endothelial tight junction impairment was observed (Seiffert et al., 2004). Accumulated albumin within the parenchyma is associated with downregulation of inward-rectifying potassium channels in astrocytes, affecting buffering capacity and contributing to hyperexcitability (Ivens et al., 2007). Moreover, loss of aquaporins expressed in the end feet of astrocytes affects water flux and potassium regulation, further disrupting the homeostatic environment of the brain (Binder and Steinhauser, 2006). Additionally, BBB damage could allow circulating levels of zinc to gain entry into the brain with devastating consequences, including excessive hyperexcitability and seizures (Carver et al., 2016; Chuang and Reddy, 2019). Zinc is an important neuromodulator, and its ability to persistently block extrasynaptic GABA-A receptors in the brain have dramatic consequences on epileptogenesis.
Neuroinflammation also plays a critical role in BBB permeability. Elevated levels of IL-1β, IL-6, and TNF-α can increase the permeability of the BBB and facilitate the movement of peripherally located cytokines into the CNS. These cytokines bind to receptors in the brain vasculature, producing secondary messengers and toxic by-products that further compromise its integrity (Fabene et al., 2010; Yarlagadda et al., 2009). Furthermore, these factors can trigger the activation of astrocytes and resident microglia, contributing to their dysfunction of neurotransmitter clearance and subsequent secretion of immunoregulatory markers. Systemic injection of lipopolysaccharide has been shown to lower seizure threshold to pentylenetetrazol, suggesting peripheral inflammation leads to a leaky BBB and possible infiltration of peripherally circulating leukocytes (Marchi et al., 2012). BBB dysfunction represents a convergence of pathogenic aspects that often create positive-feedback loops for further exacerbation of inflammation, functional impairment, and BBB permeability. For full review on how the breakdown of BBB affects PTE development, see Dadas and Janigro, 2019.
C. Epigenetic Modifications
Epigenetics refers to the plastic changes in gene expression that occur without alteration of the DNA sequence itself. Under normal conditions, epigenetic modifications are essential for growth, development, learning and memory, and the immune response (Hwang et al., 2017). Epigenetic modifications, such as DNA/Histone methylation, acetylation, and phosphorylation etc., have been implicated in a vast number of diseases, most notably cancer (Weber, 2010). Evidence suggests that epigenetic regulation of gene expression may play a critical role in the physiology of both epilepsy and TBI (Younus and Reddy, 2017; Nagalakshmi et al., 2018). Reddy et al. (2018a) demonstrated the histone deacetylase (HDAC) inhibitor, sodium butyrate, significantly slowed the kindling process in a mouse model of temporal lobe epilepsy when administered prior to electrical stimulation. This study suggests HDAC inhibitor compounds may possess antiseizure effects with an ability to curtail the process of epileptogenesis. Moreover, valproate has been administered as an antiseizure medication for decades, although its inhibitory effect on HDACs was unknown until 2001 (Göttlicher et al., 2001).
Histone modification is perhaps the most widely studied epigenetic modification in both epilepsy and TBI. Reduced H4 acetylation has been observed after pilocarpine administration at GluR/Gria2 promoter loci, a region that encodes for AMPA receptor subunits and limits calcium permeability (Huang et al., 2002). Downregulation of GluR/Gria2 is associated with hyperexcitability and initiating epileptogenesis. The same study also noted H4 acetylation at the BDNF promoter, increasing after seizure activity. H3 phosphorylation is thought to promote acetylation of histone proteins, and multiple studies have found striking evidence of H3 phosphorylation after pilocarpine and kainic-acid induced seizures (Crosio et al., 2003; Sng et al., 2006). Furthermore, hyperactivity of HDAC proteins occurs at early timepoints after lateral FPI (Zhang et al., 2008). This increased H3/H4 acetylation can be found throughout the hippocampus but is particularly visible in the CA3 (Gao et al., 2006). Increased HDAC activity leads to seizure susceptibility and post-traumatic epilepsy in both experimental models and in the clinical setting (Huang et al., 2012; Dash et al., 2009).
Changes in cell-specific global DNA/histone methylation have been shown to persist for up to 8 months post-TBI (Haghighi et al., 2015). Many of the affected genes have been associated with hyperexcitability, disruption of the sleep cycle, and neuropsychiatric disorders, such as Nos1, Il1r1, Homer1, Per3, and the Aanat gene, which encodes the enzyme responsible for catalyzing the serotonin to melatonin conversion (Haghighi et al., 2015). DNA methylation also plays a role in the inflammatory response to injury. Within 24 hours post-TBI, hypomethylation of microglia promotes active gene transcription in areas of widespread necrosis (Zhang et al., 2007). Furthermore, a study of patients with intractable temporal lobe epilepsy found expression of Dnmt1 and Dnmt3a were significantly higher in epileptic versus healthy controls, suggesting aberrant DNA methyltransferases may contribute to the pathogenesis of seizures (Zhu et al., 2012). DNA methyltransferase inhibitors have shown some promise for suppressing neuronal excitability in hippocampal neurons (Nelson et al., 2008; Levenson et al., 2006).
D. Reorganization of Neural Circuitry
The culmination of neuroinflammatory cascades, weakened BBB integrity, and epigenetic modification leads to consequent reorganization of neural circuitry through progressive cell loss, aberrant axonal sprouting, and neurogenesis. Several experimental models have highlighted the loss of inhibitory interneurons coupled with recurrent excitatory circuits as a basis for hypersynchronous epileptiform activity (Dudek and Spitz, 1997; McCormick and Contreras, 2001; Golub and Reddy, 2022). The hippocampus is a model system to study circuitry changes since it is particularly susceptible to injury and undergoes structural reorganization after TBI and in epilepsy disorders (Kharatishvili et al., 2006; Hunt et al., 2009;).
GABAA receptors are responsible for the majority of inhibitory signaling in the brain. GABAergic interneurons form robust local synaptic connections with excitatory principal cells to control activity in two primary ways: phasic (synaptic) and tonic (extrasynaptic) inhibition (Farrant and Nusser, 2005; Chuang and Reddy, 2018). Phasic inhibition refers to the rapid transmission of information and activation of receptors at the synaptic junction after exposure to high concentrations of GABA released from presynaptic vesicles. Tonic inhibition, on the other hand, is mediated by extrasynaptic GABAA receptors, persistently activated by low concentrations of ambient GABA. A common histopathologic feature of PTE is the drastic loss of inhibitory interneurons in the dentate gyrus and hilar regions (Lowenstein et al., 1992). Loss of these cells is correlated to an increase in tonic current amplitude in the dentate gyrus contralateral to TBI (Mtchedlishvili et al., 2010). Additional studies have reported changes in the subunit configuration of GABAA receptors after CCI injury, which may also affect inhibitory control (Gupta et al., 2012; Raible et al., 2012).
Neurodegeneration after TBI affects both principal neurons and interneurons, although it was unclear whether one population is preferentially lost. Carron et al. (2020) examined how TBI affects different populations of interneurons, observing heterogenous changes of calbindin, parvalbumin, calretinin, neuropeptide Y, and somatostatin expressing interneurons in the hippocampus. Their findings suggest a differential vulnerability of interneurons across various brain regions and function after TBI. In a recent study, we performed a time-course of unbiased stereological quantification in the hippocampus contralateral to CCI at days 1, 3, 7, 30, 60, and 120. Populations of principal neurons and inhibitory PV+ GABAergic interneurons had decreased by roughly 30% and 45%, respectively, at 4 months postinjury; however, the degeneration in interneurons was accelerated to that of excitatory cells. Furthermore, the steep decline in interneurons coincided with the onset of spontaneous seizures (Golub and Reddy, 2022). Figure 3 highlights the linear regressions of cell loss during epileptogenesis and their temporal association to seizure onset. Our data agree with a previous study by the Hunt group that found a dramatic shift of interneuron diversity and loss after contusion injury (Frankowski et al., 2019). As previous studies have pointed to, the dentate gyrus and hilar regions showed the greatest loss of inhibitory interneurons (Hunt et al., 2011; Gupta et al., 2012). Loss of cells, whether excitatory or inhibitory, forces reorganization of these neural circuits, contributing the ongoing pathology of epileptogenesis. Restoration of the excitatory and inhibitory balance may be possible with transplantation of neuronal stem cells (Ngwenya et al., 2018). These animals also exhibited improved recovery and novel object recognition compared with nontransplant FPI animals.
Fig. 3.

Relationship of long-term neurodegeneration and spontaneous seizures. TBI induces a state of immediate inflammation and hyperexcitation in the brain, which exacerbate cell loss both ipsilateral and contralateral to the lesion. PTE was induced via a severe 2.0 mm depth CCI model of TBI. After injury, mice were tethered to 24/7-videoEEG for up to 4 months and seizures were identified by a customized MATLAB script and validated by unbiased researchers. Stereological quantification of two cell populations in the contralateral hippocampus was performed at days 1, 3, 7, 30, 60, and 120 post-TBI in subsets of these recorded mice. (A) Linear fit of remaining NeuN+ principal neurons overlayed the linear regression of average seizure output from responding mice to highlight the temporal relationship between cell loss and seizure occurrence. (B) Linear fit of remaining PV+ GABAergic interneurons overlayed the linear regression of average seizure output from responding mice to highlight the temporal relationship between cell loss and seizure occurrence.
Excitatory dentate granule cells are not typically connected to each other. However, several laboratories have demonstrated the reactive plasticity of these circuits in TBI and epilepsy models (Kharatishvili et al., 2006; Hunt et al., 2009; 2010; Bolkvadze and Pitkänen, 2012). Aberrant mossy fiber sprouting refers to the germination of axon collaterals from dentate granule cells into the inner molecular layer, forming functionally recurrent excitatory circuits. These local circuit changes are easy to detect with Timm’s immunohistochemistry and have been consistently reproduced in human and rodent tissues (Sutula et al., 1989; Hunt et al., 2010). Mossy fiber sprouting is generally more robust after severe versus mild TBI, suggesting degree of sprouting is correlated to both severity of injury and seizure risk (Hunt et al., 2012). Sprouting is most often noted in the hippocampus ipsilateral to TBI; however, damage is not constrained to the injured hemisphere and may influence circuitry reorganization with increased mossy fiber density (Pischiutta et al., 2018). Aberrant sprouting may provide a means for regional network synchronization, which is particularly vulnerable if basal inhibition is lost or impaired by cell loss or dysfunction.
Neural precursors proliferate in areas both proximal and distal to TBI impact. Much of this proliferation makes up the astrogliotic scar that forms around the injury site (Kernie et al., 2001). Changes in the rate of neurogenesis have also been found after TBI, and ectopic migration of these newborn cells may affect the excitability of the neural circuitry. TBI-induced newborn cells have increased dendritic branching proximal to the soma and wider dendritic reach that persists through cell maturity (Villasana et al., 2015). Neurogenesis has been a point of controversy in epileptogenesis, with some reports suggesting increased cell proliferation after TBI (Dash et al., 2001; Gao et al., 2009), whereas others observe reduced neurogenesis (Rola et al., 2006). However, differences in the rate of neurogenesis may be in part due to proximity to injury, timepoint of tissue sampling after TBI, or even the possibility of selective death of vulnerable newborn cells (Gao et al., 2008). Yu et al. (2008) observed both upregulation of type-1 quiescent progenitor cell activation in the injured hippocampus as well as progressive elimination of type-2 doublecortin-expressing progenitors. At the same time, the contralateral hippocampus also saw upregulation of type-1 progenitors, suggesting TBI may differentially impact damage versus compensatory signaling. Furthermore, severity of injury affects neurogenesis at different stages (Wang et al., 2016a). Regardless of location, severity, or timing after injury, fluctuation of cells born into the hilar and molecular layers of the dentate gyrus have been suggested to play a role in epileptogenesis (Danzer, 2019). Villasana et al. (2015) found TBI-induced newborn granule cells receive a normal balance of excitatory and inhibitory inputs and are involved in information processing, but suggested TBI-induced anatomic changes and dendritic projection patterns may be the root cause of maladaptive neurogenesis network properties. Insulinlike growth factor-1 overexpression has been found to increase the survival of newly born granule cells while inhibiting ectopic migration, the main implication of neurogenesis-associated circuitry changes (Carlson et al., 2014; Littlejohn et al., 2020). Therefore, conditional expression of astrocytic IGF-1 may be beneficial in reducing reactivity of astrocytes and preserving cognition after TBI.
E. Mammalian Target of Rapamycin Pathway Hyperactivity
The mammalian target of rapamycin (mTOR) pathway regulates several physiologic functions, and, in the brain, it is involved in cell proliferation and survival, neuronal morphology, and protein synthesis (Bockaert and Marin, 2015). Dysregulation of this pathway has been implicated in several brain disorders, including tuberous sclerosis complex, ganglioglioma, and focal cortical dysplasia—all of which may potentially or certainly lead to epilepsy (Liu et al., 2014a). Moreover, a role of mTOR signaling has been identified in brain trauma, although it is shrouded in controversy (Chen et al., 2007a). Some studies suggest the inhibition of mTOR, via administration of the mTOR inhibitor rapamycin, prevents neuronal injury and cell death after TBI (Erlich et al., 2007; Nikolaeva et al., 2016), whereas others suggest increasing mTOR signaling promotes greater recovery of function and regeneration, and this transient increase in mTOR signaling after TBI may be critical in stimulating neural stem cell proliferation (Wang et al., 2016b).
Hyperactivation of mTOR seems to play a critical role in the pathogenesis of acquired epilepsy, such as PTE, and rapamycin administration has prevented epileptogenic mechanisms and reduced seizure burden in certain models. In a rat hippocampal organotypic slice culture model of PTE, inhibition of Akt, PI3K, or mTOR reduced both ictal activity and cell death (Berdichevsky et al., 2013). In the clinical setting, rapamycin and its derivatives have been tested mainly on severe, refractory epilepsy disorders, such as tuberous sclerosis complex. Rapamycin and everolimus treatments improved seizure control in phase I/II studies (Krueger et al., 2013), and, in some cases, patients experienced complete cessation from previously intractable seizures (Perek-Polnik et al., 2012). The mechanisms by which mTOR inhibition reduces seizure activity in experimental models is still largely unclear but seems to point toward neurocircuitry reorganization. Inactivation of phosphatase and tensin homolog induces aberrant mossy fiber sprouting in animal and human hippocampal granule cells. Sutula and Dudek demonstrated that phosphatase and tensin homolog deletion was sufficient to trigger spontaneous seizures and that mTOR hyperactivation played a central role in this process (Sutula and Dudek, 2007). Furthermore, rapamycin inhibition of mTOR signaling reduces abnormal axonal sprouting and other pathologies associated with epileptogenesis, including neuronal excitability (Zeng et al., 2009). Although these data are positive, other studies have found rapamycin treatment reduces mossy fiber sprouting but has little effect on decreasing seizure frequency or duration (Buckmaster et al., 2009). Inhibitors of mTOR possess low efficacy in halting seizures within the preclinical models since multiple days of treatment are needed to achieve an antiseizure impact and beneficial effects typically cease after drug discontinuation.
When considering the mechanisms of epileptogenesis for prophylactic approaches, it is important to consider that both adaptive and maladaptive processes are activated by brain injury. The former can contribute to functional and beneficial recovery, whereas the latter may contribute to epileptogenesis. Questions emerge to determine whether these processes are separate or concurrent, if they are distinctly different forms of plasticity involved in either functional recovery or epileptogenesis, and if these processes are not distinct, do they differ quantitatively in their timing or intensity? Preventative or curative treatments may interfere with both epileptogenesis and development of comorbidities. It is likely that multiple agents will be needed to provide full spectrum symptomatic relief to patients. The answers to these questions are critical to pharmacological progress in epilepsy and head trauma.
IV. Emerging Biomarkers of Post-Traumatic Epilepsy
In 2015, the U.S. Food and Drug Administration (FDA) developed the Biomarkers, EndpointS, and other Tools effort to promote consistent use of biomarker terms and concepts. The use of the Biomarkers, EndpointS, and other Tools resource has evolved the term “biomarker” to be labeled as “a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions.” This definition includes characteristics such as changes in a patient’s molecular, radiographic, genetic, or genomic, electrographic, physiologic, and histologic traits. Biomarkers are objectively measured and quantifiable and should ideally be cost-effective and noninvasive for both the patient and healthcare provider. Furthermore, an effective biomarker should have limited variability among the general population. Within the context of disease or environmental exposure, the presence of biomarkers can indicate change in a biologic condition, predict risk or development of comorbidities, and measure progression of a disease state. In this section, we review the current state of PTE biomarker discovery and use, both in the clinical state as well as experimental animal model research.
A. Imaging Biomarkers
Imaging within the context of TBI has thus far focused largely on primary lesion formation and the evolution of gliosis; however, imaging techniques have the potential to capture progression of pathologies of PTE. Neuroimaging biomarkers are incredibly appealing not only due to their noninvasive procedures but also because they are routinely performed as part of a patient’s routine medical care. Furthermore, imaging can detect patterns across specific structures or within the whole brain (Reddy et al., 2019).
CT scans have been used for decades to assess global structural damage after TBI (D’Alessandro et al., 1988). Characterization of early CT scans have identified an increased risk of PTE development in patients with depressed skull fracture, dural penetration, and intraparenchymal, subdural, or epidural hemorrhage. Additionally, patients with cortical/subcortical contusions or large lesions in the temporal lobe have shown higher rates of PTE incidence, regardless of injury severity (Englander et al., 2003; Tubi et al., 2019).
Positron emission tomography (PET) allows researchers to visualize inflammatory responses and the metabolic impact of neuronal injury after TBI. 18F-Fluorodeoxyglucose (FDG) is a radioactive tracer of cerebral metabolism often used in PET experiments. Hypometabolism has been observed within the first 24 hours after kainic acid-induced status epilepticus, suggesting abnormal metabolism may play a role in epileptogenesis (Jupp et al., 2012). Conversely, FDG-PET scans of patients with severe TBI demonstrate hyperglycolysis occurs up to 2 weeks after initial damage (Bergsneider et al., 1997). This increase in glucose consumption may be due to the rise in inflammatory cell populations surrounding the impact site.
Gliosis appears bright on T2-weighted MRI and fluid attenuated inversion recovery techniques. Inflammation and glial scarring are extremely common around the impact site after TBI in both rodent models and in the clinical setting (Dixon, 2017). Activation of astrocytes and microglia through maladaptive inflammatory cascades have been correlated to excitotoxicity, mitochondria dysfunction, and cell loss—all of which have also been indicated as epileptogenic factors (Alyu and Dikmen, 2017; Shandra et al., 2019). Magnetic resonance spectroscopy is also a useful tool for providing quantitative data on altered metabolite profiles related to inflammation and hyperexcitability. Reductions in glucose and GABA neurotransmitter, as well as increased glutamate have been observed after TBI using magnetic resonance spectroscopy (Friedman et al., 1999). To further assess metabolite profiles as a function of epileptogenesis, Filibian et al. (2012) investigated glutathione levels, an antioxidant produced by activated astrocytes, in rats with pilocarpine-induced status epilepticus. Glutathione was negatively correlated with seizure frequency, providing strong evidence of astrocytic involvement in seizure generation.
MRI has excellent resolution and tissue classification, using strong magnetic fields to form images of the anatomic and physiologic processes of given organs (Reddy et al., 2019). Structural MRI studies have revealed minor, but significant, changes in the ipsilateral hippocampus relative to baseline in rats experiencing post-traumatic seizures versus nonepileptic rats one week after lateral FPI (Shultz et al., 2014). The same study boasted high predictability of PTE incidence using a multivariate logistic regression model of serial FDG-PET parameters among all injured and noninjured cohorts. Similarly, hyperexcitability in the hippocampus has been correlated to early 3-hour postinjury decreases and later 1- to 12-month postinjury increases in hippocampus diffusion (Kharatishvili et al., 2007). Furthermore, mossy fiber sprouting scores, which have been long considered a hallmark of epilepsy, were correlated with diffusion values after lateral FPI, providing greater evidence that quantitative diffusion MRI is a potential tool for facilitating the prediction of increased seizure risk after TBI. In a follow-up study, the same group accurately predicted seizure susceptibility to pentylenetetrazol (PTZ)-administration using similar MRI diffusion patterns, with the greatest accuracy to be found with a combinational biomarker calculated from diffusion in the ipsilateral somatosensory cortex and thalamic regions at 2 months post-TBI (Immonen et al., 2013).
Functional MRI can be used to analyze connectivity, plasticity, and remodeling within the brain after injury. There is extensive reporting on functional changes after TBI and clinical outcome, but studies focusing on epileptogenesis and PTE are scarce and inconsistent. Evidence of both hypoconnectivity and hyperconnectivity have been observed in several networks after TBI, and these abnormalities have been linked to changes in behavior, cognitive impairment, and motor control (Hillary et al., 2011; Tang et al., 2011; Stevens et al., 2012; Palacios et al., 2013). For example, abnormal frontoparietal network connectivity after mild TBI affected performance in working memory tasks, suggesting that interhemispheric connectivity between left and right inferior frontal gyri may contribute to learning and memory impairments seen in the clinical setting (Kasahara et al., 2010). A single experimental study has investigated functional MRI biomarkers in relation to epileptogenesis using lateral FPI and PTZ susceptibility as a model. Mishra et al. (2014) observed decreased connectivity between the ipsilateral and contralateral parietal cortex and between the hippocampus and parietal cortex in the injured hemisphere compared with sham-operated rats. However, no significant relationship was found between functional connectivity and seizure susceptibility during the PTZ test.
Blood-brain barrier (BBB) dysfunction has long been suggested to play a key role in seizure susceptibility after TBI and in epilepsy disorders (Cornford and Oldendorf, 1986). Recent studies demonstrate that abnormalities in the BBB can be visualized using dynamic contrast-enhanced MRI and fluid attenuated inversion recovery techniques. Disrupted BBB function has been found with increased frequency and to a larger extent in patients with post-traumatic seizures versus nonepileptic patients with TBI (Tomkins et al., 2011). Furthermore, reduced BBB integrity within the cortical regions surrounding the impact site or within the piriform network have been suggested as sensitive predictors of epilepsy (Bar-Klein et al., 2017). There are several hypotheses that associate cerebrovascular permeability with epileptogenesis, including imbalance of ion and molecule distribution and disturbance of neuronal homeostasis (Dadas and Janigro, 2019).
Two of the biggest challenges regarding imaging biomarkers is the heterogeneity of PTE-related injuries and the rarity of longitudinal studies. Thus, a consistent and validated imaging biomarker for PTE has yet to be discovered. For a full review of imaging biomarkers in PTE, see Garner et al. (2019) and Immonen et al. (2019).
B. Electrographic Biomarkers
Electrographic biomarkers may predict the onset of seizures and epileptogenesis, allowing for development of targeted preventative therapies. Currently, there are no validated electrophysiological biomarkers for PTE; however, experimental EEG studies using lateral FPI and CCI in rodents have identified potential candidates, including pathologic high-frequency oscillations (HFOs), reduction in sleep spindle duration, changes in theta oscillations, dominant frequency at the stage III to rapid eye movement sleep, and epileptiform spiking/discharges preceding seizure onset.
HFOs are commonly classified into ripples (80–250 Hz) and fast ripples (250–500 Hz) and are believed to be a naturally occurring phenomena involved in both physiologic and pathologic processes (Zijlmans et al., 2012). Although physiologic and pathologic HFOs cannot be differentiated on spectral frequency alone, increased rhythmic patterns and power of HFOs have been correlated to epileptic foci (Jirsch et al., 2006; Staba, 2012). Early investigators of HFOs questioned whether HFOs could be evaluated by standard invasive macroelectrodes, but recent reports have shown that HFOs can be detected for long periods of time by a wide variety of methods, including the standardized scalp EEG (Andrade-Valenca et al., 2011).
The premise that HFOs may play a role in epileptogenesis was first hypothesized by Bragin et al. (2000) who observed fast ripple occurrence preceded spontaneous seizures by weeks in a kainic acid model of status epilepticus. A follow-up study found that 19 out of 26 rats developed pathologic HFOs in the first month postkainic acid injection and that all 19 rats later developed epilepsy, whereas the remaining seven rats did not exhibit pathologic HFOs or seizures (Bragin et al., 2004). In a lateral FPI study, repetitive HFOs occurred at a significantly higher rate after moderate or severe injury, compared with mild or sham-injured animals, providing additional evidence that injury severity correlates to epilepsy (Reid et al., 2016). Among these FPI rats, HFOs tended to occur more frequently in the early stages of the study before seizure onset. These studies suggest a pattern of intense and increasing abnormal epileptic activity during the epileptogenic period, starting with increased HFOs and ending the latency period with spontaneous seizures (Fig. 4). Nevertheless, alterations in characteristics of HFOs in people at risk for epilepsy or epileptogenesis may serve as valuable noninvasive biomarker of epilepsy or risk prediction. For full review of HFOs as biomarkers for epilepsy, see Perucca et al. (2019) and Jacobs and Zijlmans (2020).
Fig. 4.

Evolution of TBI-induced hyperexcitability and seizure activity. Electrographic biomarkers may predict onset of seizures, as hyperactivity in the brain progresses over time to the culmination of spontaneous recurrent seizures. TBI induces a state of heavy inflammation, disrupting both neurotransmitter and metabolic homeostasis. The emergence of these abnormal electrographic activities may reflect different stages of the epileptogenic process postinjury. Pathologic HFOs often precede seizures by weeks, followed by reduced frequency and duration of sleep spindles during the transition between stage 3 and REM sleep. Disruption of normal sleep spindles contributes to several sleep-wake disorders reported by patients with TBI. EEG spiking and discharges represent an advanced hyperactive disturbance that has been described as epileptiform abnormalities in animal brain slices and in vivo at various time-points postinjury. The final stage is the end of latency indicated by the occurrence of spontaneous seizures; however, epileptogenesis can continue to progress even after the first seizure.
Sleep spindles occur at a frequency between 10–20 Hz (typically 12–14 Hz) with a duration of 500–2000 milliseconds. Spindle generation involves interaction between inhibitory neurons in the thalamic regions to function as pacemakers. An increase in spindle frequency is known to occur immediately preceding REM sleep in both rodents and humans (Vyazovskiy et al., 2004; Purcell et al., 2017). Sleep disturbances are well documented after TBI, and disruption of normal spindle activity may contribute to epileptogenesis (Duclos et al., 2014). A study by Andrade and colleagues discovered 92% of detected seizures occurred during the transition period between stage III and REM sleep after lateral FPI in rats (Andrade et al., 2017). Sleep spindles in these epileptic rats were significantly shorter and slower during the transition from slow-wave N3 to REM sleep, compared with nonepileptic rats. These changes were identified at 9 weeks postinjury but were not observed within the first 2–3 weeks (Bragin et al., 2016), providing greater evidence of the progression of abnormal EEG over time. However, these results somewhat contradict those of a more recent study in which Konduru et al. (2021) confirm shorter spindle duration and lower band frequency in injured versus noncraniectomy mice but did not find significant differences in these features between TBI mice with and without a seizure response. Furthermore, sham-injured animals have been known to exhibit abnormal EEG activity compared with noncraniectomy groups, suggesting sham cohorts may not always be the best control for all parameters.
EEG spikes, sometimes called interictal spiking, have been observed in both rats and mice after brain injury (Statler et al., 2009; Bolkvadze and Pitkänen, 2012). These spikes can take the form of isolated spikes, runs of spike-wave discharges, or absence-like spike-wave discharges and represent abnormal fluctuations in brain waves but have yet to be correlated with the onset of SRS. Furthermore, stimulation-evoked hyperexcitability in the hippocampus has been demonstrated after weight drop and in neocortical brain slices after CCI (Golarai et al., 2001; Yang et al., 2010). It is unclear whether these electrophysiological disturbances are associated directly with the injury state or with epileptogenesis. It should be noted that not all studies show a correlation between animals with spiking activity and documented seizures, although recordings in this case were short 1-week recordings (Konduru et al., 2021). Recently, Golub and Reddy (2022) observed short bursts of high-energy activity lasting between 3 and 9 seconds after CCI injury in mice. Although this definition of epileptiform discharges is intermediary to interictal spikes and seizures, it does provide further evidence of the evolution of abnormal EEG activity to seizure development.
Although there are currently no validated electrophysiological biomarkers for PTE, experimental studies continue to identify potential candidates that may be instrumental in predicting and, therefore, preventing epilepsy with targeted therapeutic approach. These EEG abnormalities include pathologic or repetitive HFOs, sleep spindle disturbances, and abnormal interictal spiking. Further experimental studies using a controlled means of EEG analytics are needed to determine which features may be reliable biomarkers of PTE in the clinical setting. Machine learning, which has been recently instituted for seizure detection (Abbasi and Goldenholz, 2019), may also help to classify these abnormal EEG patterns in more meaningful ways. Furthermore, standardization of detection protocols, analysis algorithms, and sampling of EEG recordings will aid the progress of neurophysiologic biomarker discovery.
C. Molecular Biomarkers
Biofluid markers are useful for determining TBI severity and play a critical role in monitoring disease progression and clinical prognosis (Sharma and Laskowitz, 2012). Molecular biomarkers refer to nonimaging factors that have biophysical properties allowing for measurement in biologic samples, such as blood, plasma, cerebrospinal fluid (CSF), saliva, or biopsy. Circulating biofluid markers, such as microRNAs (miRNAs), proteins, extracellular vesicles, and cytokines have been widely studied in both TBI and epilepsy conditions (see reviews Engel et al., 2013; Agoston et al., 2017), but few studies have combined their efforts to identify biomarkers for epileptogenesis and PTE. Furthermore, there is little evidence to suggest biomarkers for TBI overlap with those for epileptogenesis; therefore, this subsection will briefly cover molecular biomarkers of TBI that have been indicated as potential risk factors for seizures.
Severe and penetrating TBI represent the highest risk of PTE development due to the extent of tissue damage, bleeding, inflammation, and bone fracture. These processes produce a graded increase in circulating levels of inflammatory chemo- and cytokines, regulators, and bone morphogenic proteins, providing a molecular basis for classifying injury severity (Heggeness et al., 2017). Deposition of bone particles and other foreign bodies in the brain parenchyma are among the most important risk factors for developing seizures after TBI (Salazar and Grafman, 2015). Likewise, increased serum and/or CSF levels of claudin-5, VEGF, occludin, aquaporin-4, and von Willebrand factor may indicate a breakdown of the BBB and/or vascular injury (Croll et al., 2004; De Oliveira et al., 2007; Jiao et al., 2011; Thrane et al., 2011; Ahmed et al., 2015).
Iron levels, due to excessive bleeding, increase risk of PTE (Ding et al., 2016). Accumulation of iron in the blood can be cytotoxic, resulting in mitochondrial dysfunction and oxidative stress through the generation of free radical particles. In the clinical setting, patients with TBI and low ceruloplasmin, an important protein involved in iron metabolism and injury repair, develop increased intracranial pressure that can lead to post-traumatic seizures (Dash et al., 2010a). Neuroinflammation around the impact site also contributes to the rise in intracranial pressure. Increased cytokines and inflammatory proteins, such as IL-1, IL-6, TNFα, CD53, fibrinogen, and MIP1α, have been implemented in prolonged inflammation in human and animal studies (McManus et al., 1998; Katayama et al., 2009; Woodcock and Morganti-Kossmann, 2013). A genetic biomarker study demonstrated higher CSF:serum IL-1β ratios were associated with increased PTE incidence (Diamond et al., 2015a). Furthermore, due to the astrocytic roles in both inflammation and glucose metabolism, CSF and/or serum levels of glial fibrillary acidic protein (GFAP) can provide insight into more than one pathology associated with PTE. It is likely that a combination of disrupted neural connectivity due to cell loss, metabolic dysregulation, and inflammation are critical components of immediate and early seizure onset after TBI (Tubi et al., 2019).
MiRNAs are noncoding RNA molecules that have emerged as potential molecular biomarkers for a number of neurologic disorders. miRNAs are considered good biomarker candidates since they are more stable than mRNA and proteins, are present in biofluids, such as blood and CSF, and are inexpensive to assay. The human genome contains approximately 2300 miRNAs, and about 500–700 of these are also present in laboratory rodents, such as mice and rats (Alles et al., 2019). In plasma, miRNA can be either bound to argonaute2 protein or held within extracellular vesicles. Extracellular vesicles are involved in intercellular communication and can carry biomolecules such as DNA, mRNA, miRNA, proteins, and lipids. Raoof et al. (2017) conducted a study including patients with temporal lobe epilepsy and status epilepticus that identified miR-19-3p was largely argonaute2-bound in both epileptic conditions, and miR-21-5p was mostly carried within extracellular vesicles in status. A follow-up study discovered the proportion of argoaute2-bound miR-328-3p increases after a spontaneous seizure in temporal lobe epilepsy (Raoof et al., 2018). Lastly, patient levels of transfer RNA fragments 5′GluCTC, 5′AlaTGC, and 5′GlyGCC have been found to be upregulated in preseizure versus postseizure samples (Hogg et al., 2019). Since extracellular vesicles can be extracted from all biologic fluids, they have exciting potential for identifying biomarkers of post-traumatic epileptogenesis. However, most clinical research in this area have so far investigated miRNA levels in patients with pre-existing epilepsy, rather than conditions with an epileptogenic trigger.
Dysregulation of miRNAs has been observed in both patients with epilepsy and TBI; however, no study has been done to identify biofluid miRNA biomarkers of epileptogenesis in humans. There is little overlap between TBI and epilepsy in potential miRNA candidates, and previous studies have presented a high frequency of contradictory data regarding serum and plasma miRNA dysregulation and their usability in disease trajectory for PTE. Table 3 shows a list of potential miRNA biomarker candidates and whether they have been shown to be upregulated or downregulated in epilepsy, TBI, and/or PTE. Data from this table are mixed between preclinical and clinical studies, as there is limited knowledge in this field at present (Simonato et al., 2021). miRNAs, such as miR-21-5p, miR-27b-3p, miR-93, miR-135a, miR-146a, miR-155, miR-203, and miR-451, have all been shown to be upregulated in various epilepsy conditions as well as after brain injury, whereas miR-27a-3p, miR-128, and miR-221-3p are downregulated (Redell et al., 2010; Gorter et al., 2014; Liu et al., 2014b; Roncon et a., 2015; Atif and Hicks, 2019; Brennan et al., 2020). These data hold promise for PTE biomarker discovery in that there seems to be some similarity between these two neurologic conditions. However, there are many other candidates with opposing dysregulation, such as miR-23a, miR-30a-5p, miR-153, miR-182, miR-219, miR-300, miR-328-3p, and miR-574. We are currently at the beginning of the process to adequately validate the first biomarkers for epileptogenesis, and more time and effort will need to be dedicated toward discovery before their subsequent clinical use.
TABLE 3.
Putative miRNA targets with highest biomarker potential for epilepsy, TBI, and PTE
| Epilepsy | TBI | PTE | |
|---|---|---|---|
| miR-7-5p | — | ↓ | — |
| miR-9a | ↑ | ↑ | — |
| miR-10a/b-5p | ↑ | ↑ | — |
| miR-16-5p | — | ↑ | — |
| miR-21-5p | ↑ | ↑ | — |
| miR-23a | ↑ | ↓ | — |
| miR-26b-5p | — | ↑ | — |
| miR-27a-3p | ↓ | ↓ | — |
| miR-27b-3p | ↑ | ↑ | — |
| miR-29a/c | — | ↓ | — |
| miR-30a-5p | ↓ | ↑ | — |
| miR-93 | ↑ | ↑ | — |
| miR-124 | ↓ | Mixed | — |
| miR-128 | ↓ | ↓ | — |
| miR-129-2-3p | ↑ | — | — |
| miR-132 | ↑ | — | — |
| miR-134 | ↑ | — | — |
| miR-135a | ↑ | ↑ | — |
| miR-142 | ↑ | ↑ | — |
| miR-146a | ↑ | ↑ | ↑ |
| miR-151a-3p | — | ↑ | — |
| miR-153 | ↓ | ↑ | — |
| miR-155 | ↑ | ↑ | — |
| miR-181a | — | ↑ | — |
| miR-181b | ↓ | — | — |
| miR-181c | ↑ | — | — |
| miR-182 | ↑ | ↓ | — |
| miR-203 | ↑ | ↑ | — |
| miR-211 | ↑ | — | — |
| miR-219 | ↓ | ↑ | — |
| miR-221-3p | ↓ | ↓ | — |
| miR-300 | ↓ | ↑ | — |
| miR-320c | — | ↑ | — |
| miR-324-5p | ↑ | — | — |
| miR-328-3p | ↓ | ↑ | — |
| miR-423-3p | — | ↑ | — |
| miR-451 | ↑ | ↑ | ↑ |
| miR-532-5p | — | ↑ | — |
| miR-574 | ↓ | ↑ | — |
| miR-629-5p | — | ↓ | — |
| miR-1307-3p | — | ↑ | — |
↑ = upregulated; ↓ = downregulated; — = unknown
The first step in establishing plasma miRNA or protein biomarker discovery is to harmonize protocols and procedures used to collect data and perform experiments, providing an objective framework for quality control. Recently, there has been a great effort across three international sites (University of Eastern Finland, University of Melbourne/Monash University, and the University of California, Los Angeles) to standardize the protocols for miRNA biomarker validation and analysis for PTE (van Vliet et al., 2017; Kamnaksh et al., 2019). These studies highlight the need for rigorous quality assessment, as hemolysis and anesthesia presented as confounding factors. Moreover, the study suggests improved training is needed for technicians to obtain a more precise venipuncture, faster blood draws, and less coagulation in the catheter lines.
D. Genetic and Genomic Influences
A genetic biomarker is a sequence of DNA that causes or is associated with susceptibility of a disease. Genetic biomarkers typically present as a genetic variant, such as a copy number variant or single-nucleotide polymorphism in the clinical setting. Likewise, genomic biomarkers reflect the expression, function, and regulation of a gene and its interrelationships to identify a combined influence within a biologic state or within the context of disease progression (WHO, 2002). Genomic biomarkers include both DNA and RNA characteristics. DNA characteristics include single nucleotide polymorphisms, DNA modifications (e.g., methylation or acetylation), variability within short sequence repeats, insertions, deletions, copy number variations, or cytogenetic rearrangements (e.g., duplications, deletions, inversions, translocations). Characteristics of potential RNA biomarkers include changes in RNA expression levels and sequence, alteration of RNA processes, such as splicing or editing, and variation of microRNA levels. These two terms “genetic” and “genomic” biomarkers are similar in that they represent a change at the level of DNA or RNA but should not be used interchangeably. The main difference between genetic and genomic biomarkers is their respective focus on a specific gene and heredity versus an organism’s entire genetic make-up including coding and noncoding DNA and their interactions. For example, Huntington’s disease is caused by a genetic mutation in the HTT gene, representing a genetic biomarker; whereas, significantly altered mRNAs found in the peripheral blood of patients with Huntington’s serve as a genomic biomarker (Borovecki et al., 2005). Genomic biomarkers could be a useful prognostic marker for PTE by identifying individuals with a higher risk of epileptogenesis and enriching the population of antiepileptogenesis trials.
Preclinical rodent trials have included genetically modified mice and/or animals with varying genetic backgrounds to help identify genomic influence on epileptogenesis and TBI. Adenosine is known to exhibit some anticonvulsant effects within epilepsy (Knutsen and Murray, 1997; Avsar and Empson, 2004). Knockout of the Adora1 gene, affecting the adenosine receptor A1, was found to increase the incidence of acute postimpact status epilepticus (Kochanek et al., 2006). Adenosine receptor deficiency also exacerbated the microglial and neuronal damage response after TBI (Haselkorn et al., 2010). In the clinical setting, variants of adenosine kinase influenced the rate of epileptogenesis after TBI (Diamond et al., 2015b). This same study also identified the genotype rs1143634, a variation in the IL-1 proinflammatory cytokine profile, increased risk for developing seizures.
Several studies have focused on genes related to plasticity of the extracellular matrix. Pijet et al. (2018) used matrix metallopeptidase 9 (MMP-9) knockout and overexpression mice to determine the role of extracellular matrix restructuring in post-traumatic epileptogenesis. Two peaks of MMP-9 expression were found at 30 minutes and 6 hours postinjury. Their results demonstrate overexpression of MMP-9 resulted in greater seizure frequency and lowered PTZ seizure threshold after TBI. Prevalence of post-traumatic seizures was also correlated to increased lesion volume in these mice. APP/PS1 mice, which are predisposed to plaque deposition and gliosis, were found to have more pronounced epileptogenesis and robust comorbidities, such as cognitive impairment, after TBI (Miszczuk et al., 2016). A series of studies investigated the role of the extracellular matrix proteinase urokinase-type plasminogen activator and its receptor on PTE. Neither mutation of the Plua or Pluar gene, resulting in the deficiency of urokinase or its receptor, affected the progression of PTE after TBI. The authors concluded that epileptogenesis and seizure susceptibility was not worsened with urokinase-type plasminogen activator or its receptor deficiency, although comorbidities, such as cognitive impairment and motor function, were exacerbated (Bolkvadze et al., 2016). However, a recent follow-up study using a double knockout of both Plau and Plaur genes found significantly increased susceptibility to PTZ without brain injury (Kyyriäinen et al., 2019).
In the clinical setting, mutations affecting the balance of inhibitory-excitatory circuitry have been linked with an increased risk of PTE. Variations in the GAD1 gene, responsible for producing the enzyme that catalyzes the production of inhibitory GABA from glutamate, increases a patient’s risk for exhibiting post-traumatic seizures. Three high-risk genotypes have been identified for higher risk thus far: rs769391, rs3791878, and rs3828275 (Darrah et al., 2013). Genetic variation in the SLC1A1 gene responsible for neuronal glutamate transporters has also been associated with an increased risk for post-traumatic seizures and excitotoxicity (Ritter et al., 2016). Moreover, a major neuroprotective and inhibitory molecule, adenosine, has become increasingly important in identifying risk of PTE. Multivariate analysis of rs3766553 revealed a strong link between variability in the adenosine A1 receptor and increased risk of late seizures after injury (Wagner et al., 2010). Furthermore, a military-cohort study found a common variant in the methylenetetrahydrofolate enzyme C677T may predispose an individual to PTE and other epilepsy disorders (Scher et al., 2011). Lastly, the role of APOPE gene has also been studied within the context of clinical PTE. Although no statistically significant associations were found, Miller et al. (2010) found half of the individuals with the E4/E4 genotype of APOPE had exhibited chronic post-traumatic seizures, suggesting this variant may be at greater risk for delayed PTE. The lack of studies reporting the effect of genetic and genomic factors on epileptogenesis after TBI limits the progress to be made in determining targeted measures for therapeutics and clinical trials (Kumar et al., 2019). There is a great need to continue research on these mechanisms to contribute to the progress in preventative therapies for PTE.
Discovery for PTE biomarkers is at an early stage. Each potential biomarker discussed has both advantages and disadvantages; therefore, it is unreasonable to expect a single biomarker to measure the progression of a heterogenous disease, such as PTE. It is more likely that a combination of multimodal biomarkers will be needed to accurately identify epileptogenesis after brain injury. Relatively noninvasive biomarkers, such as imagining, surface EEG, and blood/plasma samples, are promising for patients with TBI in which the likelihood for seizures is uncertain. However, there is little evidence to suggest that biomarkers for epileptogenesis after brain injury overlap with those of TBI in general. These differences may be exaggerated by variance in models, detecting methods, analysis platforms used, and stage of epileptogenesis. Furthermore, translating preclinical studies into clinical biomarkers is challenging. The timeline of molecular and cellular changes that occur in the development of epilepsy is much shorter in rodents than in humans, explaining why dozens of successful experimental pharmacotherapies have failed in clinical trials (Agoston et al., 2019). Longitudinal data in both rodent and clinical studies are lacking; therefore, no standard temporal window comparing imaging pathology or sampling has been followed. A concerted effort to standardize biomarker efforts for TBI, PTE, and other epilepsies may be an optimal strategy for discovery of novel biomarkers that are translatable throughout these related disorders.
V. Comorbidities of Post-Traumatic Epilepsy
Apart from seizures, traumatic insults are well-known to be associated with an assortment of behavioral and psychiatric dysfunctions, including depressive symptoms, learning and memory deficits, personality changes, anxiety-like behavior, difficulty with social interactions, balance, motor impairment, as well as sleep disturbances. These features can have a profoundly negative effect on an individual’s quality of life, perhaps more so than even the seizures themselves (Boylan et al., 2004). A population-based cohort study found patients with PTE have 7.85 times as many medical visits per year compared with nonepileptic patients with TBI, suggesting a significantly increased medical burden (Lin et al., 2019). Preinjury behavior and functioning are also strong indicators of long-term behavioral and recovery outcomes, including development of psychiatric disorders after TBI. Children who experience high stress or significant life changes were found to be at greater risk for persistent postconcussion symptoms after brain injury (Smyth et al., 2014). The relationship between behavioral functions and seizures is extraordinarily complex, likely influenced by both recurrent seizure activity, but also the therapeutic regimens used to control seizures (Szemere and Jokeit, 2015). A retrospective clinical study on rehabilitation after TBI found patients given prophylactic antiseizure medications predicted poorer recovery, independent of whether these patients experienced post-traumatic seizures (Pingue et al., 2021). Despite the well-established overlap of comorbidities between TBI and epilepsy patients, there is little research looking into the prevalence, presentation, or mechanisms associated with these impaired recovery outcomes in PTE. Moreover, most ASMs interact with each other and with other medications. The effects of these drug-drug interactions can vary greatly and can be potentially dangerous. Therefore, it is important for future studies and practicing clinicians to consider the potential challenges and treatments of PTE comorbidities to find the best combination of symptom relief in affected patients. Table 4 briefly outlines major comorbidities associated with PTE.
TABLE 4.
Comorbidities of PTE
| Conditions/Diagnoses | Symptoms | |
|---|---|---|
| Sensorimotor Deficits | Vision disturbances | Difficulty recognizing or processing objects, blurry or lost vision, double vision, loss of depth perception |
| Tinnitus | High-pitched ringing or buzzing in one or both ears, often associated with hearing loss | |
| Neuropathic itch | Skin tingling or pain resulting from nervous system damage | |
| Vestibular agnosia | Loss of vertigo perception and imbalance | |
| Motor deficits | Imbalance, changes in ambulatory gait, loss of fine motor skills | |
| Cognitive Impairments | Mental fatigue and attention impairment | Inability to concentrate or focus, even on simple tasks |
| Short-term memory impairment | Inability to recall or remember information to which the subject was recently exposed | |
| Difficulty with critical thinking or problem solving | Difficulty processing, analyzing, evaluating, or synthesizing information to reflect, reason, communicate, or solve problems | |
| Psychiatric Disorders | Depression/affective disorders | Constant feeling of sadness, loss or interest, and irritability; often associated with fatigue, lack of motivation, difficulty with recall, or suicidal thoughts |
| Anxiety | Intense, excessive, and persistent worry or fear about everyday situations; avoidance; phobias; obsessive compulsive symptoms | |
| PTSD | Difficulty recovering after a traumatic event that triggers moments of intense emotional and physical reactions, such as headache, nightmares, pain, flashbacks, amnesia, or difficulty concentrating | |
| ADHD | Hyperactivity, impaired attention, reduced work speed, and difficulty with working or short-term memory | |
| Sleep Disorders | Insomnia | Difficulty falling or staying asleep, insufficient number of hours or sleep despite adequate opportunity |
| Parasomnia | Night terrors, sleep walking/talking, confusion arousals, REM sleep behavior disorder | |
| Idiopathic hypersomnia | Excessive daytime sleepiness | |
| Narcolepsy | Overwhelming daytime sleepiness and sudden attacks of sleep, cataplexy, sleep paralysis, sleep-related hallucinations | |
| Sleep apnea | Snoring, restlessness, apnea, open-mouth breathing, sleep fragmentation | |
| Circadian rhythm disorder | Sleep-wake cycle is not aligned with environment/schedule and interferes with daily activities; difficulty falling asleep or staying asleep |
A. Sensorimotor Abnormalities
Risk of complications increases with the severity of trauma, although mild TBI can also result in disabilities that interfere with daily life (van der Naalt, 2001). Patients may experience sensory problems, especially complications with vision (Ripley and Politzer, 2010). One of the brain’s primary functions is to integrate information from the outside environment, process it, and determine an appropriate reaction or response. Disturbances in vision, either in recognition or registration of objects, can lead to clumsiness and poor hand-eye coordination. Double vision also affects depth perception and ambulatory balance. Other sensory deficits, such as those affecting hearing, taste, smell, or touch, are less common but not unlikely. Damage to regions of the brain that process taste or smell may cause the perception of bitter and/or noxious smell. Likewise, injury to sensory pathways can trigger neuropathic itch, skin tingling, or pain (Oaklander, 2011).
The motor and somatosensory cortices are among the most vulnerable brain regions affected by TBI and diffuse axonal injury due to their superficial position. Damage to these regions is associated with impaired motor control and function. Patients with TBI often report difficulty with balance, changes in ambulatory stride, and loss of fine motor control. Recently, a first-of-its-kind study characterized the presence of a newly defined neurologic disorder called vestibular agnosia in patients with TBI (Calzolari et al., 2021). This condition results in the loss of vertigo perception and imbalance. The Seemungal group found that patients with TBI who exhibit vestibular agnosia have worse balance problems and are unlikely to experience dizziness. Therefore, these patients are at higher risk of subsequent falls or TBI.
In preclinical studies, researchers have focused on changes in motor functionality as a parameter of recovery outcomes. These changes in motor function have been measured with numerous behavioral tests, including comprehensive neurologic scoring, beam walk, and rotarod testing. Furthermore, tasks, such as open field and water maze, can also provide insight on functional recovery by evaluating walking and swim speed between injured and noninjured cohorts. Rodents with TBI and/or PTE demonstrate significantly worsened sensorimotor complications compared with sham-injured controls (Gold et al., 2013; Nissinen et al., 2017; Golub and Reddy, 2022). Insulin-like growth factor-1 overexpression attenuates post-traumatic motor dysfunction, suggesting sensorimotor recovery may be influenced by overactive inflammatory signals or reactive astrocytes (Madathil et al., 2013). Although locomotive ability progressively recovers over time, sensory and cognitive deficits often persist for months or years after injury. Therefore, it is critical to assess sensorimotor outcomes as an indicator of rehabilitation in both preclinical and clinical trials.
B. Memory and Cognitive Dysfunction
There are many forms of cognitive dysfunction, whether it be difficulty retaining or recalling information, disrupted focus, or higher order impairments, such as inability to plan, lack of motivation, or psychosocial disability. Psychometric tests found patients with PTE exhibited a significantly reduced ability to plan, showed a lack of initiative, and had a higher incidence of disinhibited behaviors compared with patients with TBI and no seizures (Mazzini et al., 2003). Furthermore, clinical studies have found evidence for impairments in attention (Fenwick and Anderson, 1999), problem solving (Cazalis et al., 2006), short-term and working memory (Vallat-Azouvi et al., 2007), and mental fatigue (Ziino and Ponsford, 2005).
Preclinical research has hypothesized altered synaptic plasticity and neurodegeneration induced by TBI may play a critical role in cognitive impairment. Mild to moderate TBI is associated with altered hippocampal bursts, with longer duration and lower interburst spike frequency (Munyon et al., 2014). Shorter interval bursts in CA1 hippocampal neurons provoke long-term potentiation and plays a role in synaptic plasticity, thus affecting information coding after TBI and resulting in hippocampal-associated cognitive impairments (Thomas et al., 1998; Ouyang et al., 2017).
TBI is known to affect both short- and long-term memory (Enomoto et al., 2005; Carron et al., 2020). Preclinical in vivo studies often use behavioral tests, such as the Morris water maze or Barnes maze, to evaluate spatial learning and memory (Barnes, 1979; Morris, 1984; Reddy, 1988). Both tests involve training rodents to use visual spatial cues to escape the arena. Over a series of trials, rodents remember the location of the hidden platform or escape box and complete the task progressively faster. Another common task is the Novel Object Recognition Test, which was initially described in 1988 (Ennaceur and Delacour, 1988). The Novel Object Recognition Test examines recognition memory by exposing rodents to two identical objects during a familiarization phase, followed by the replacement of one of those objects with a novel object. Healthy rodents recognize the new object, spending more time investigating it, using hippocampal-dependent recognition memory (Bevins and Besheer, 2006). Several research groups have evaluated short- and long-term cognitive deficits within the context of PTE, finding PTE cohorts perform poorly at these tasks compared with uninjured controls (Scheff et al., 1997; Lu et al., 2015; Nissinen et al., 2017; Golub and Reddy, 2022). These studies reflect similar cognitive deficits found after TBI (Paterno et al., 2017). Due to the extent of cognitive impairment often seen after TBI and how this relates to poorer quality of life in the human condition, it is our opinion that the success of future clinical trials of PTE depends on preclinical models that incorporate both the measurement of cellular and molecular pathologies associated with hyperexcitability as well as memory and behavior tasks.
C. Depression, Mood Disorders, and Anxiety
Cognitive deficits resulting from brain injury can also overlap with development of affective disorders. For example, emotion-recognition difficulties, such as facial affect recognition disorder, contribute to a suite of social functional impairments in patients with TBI (Babbage et al., 2011). Social dysfunction in TBI is well-documented and can negatively affect a person’s ability to form relationships, impairs empathy, and results in low social participation and stress (Hammond et al., 2004).
Among the general population, anxiety and depression have the highest prevalence of any other group of psychiatric disorders, with a lifetime occurrence reported at approximately 30% (Barlow, 2004). A growing body of research indicates that mood and anxiety disorders are even more prevalent in patients with TBI. Approximately 70% of patients with TBI experience a psychiatric illness within the first year after initial injury (Bombardier et al., 2010; Ponsford, 2017). The most common manifestations of psychiatric disorders among adolescents and adults with TBI are generalized anxiety, depression, phobias, post-traumatic stress disorder, and obsessive-compulsive disorder. A long-term neuropsychiatric study found that patients with PTE showed higher incidence of irritability and agitated behaviors, aggression, and personality disorders compared with nonepileptic patients with TBI (Mazzini et al., 2003). Aggressive behaviors may limit access to rehabilitation treatment, participation in employment and social activities, as well as contribute to drifting friendships and romantic relationships.
Neuropathological changes associated with TBI can lead to dysfunction of the lateral and dorsal prefrontal cortices and increased activation of the limbic and paralimbic structures, including the amygdala. Altered amygdala connectivity has been identified as a possible biomarker of both comorbid depression and anxiety in patients with TBI. Relative increases in amygdala connectivity were found in the left dorsomedial and right dorsolateral prefrontal cortices and thalamus as well as the brainstem with spatially dissociable patterns of correlation between this increased connectivity and symptom severity (Han et al., 2015). Another study investigating psychiatric symptoms associated with TBI found that patients with a history of major depression often exhibit comorbid anxiety (77%) and aggressive behavior (57%) (Jorge et al., 2004). These patients had significantly worse executive functionality compared with their nondepressed counterparts. Furthermore, neurotransmitters, such as norepinephrine, serotonin, dopamine, and GABA, are mediators of anxiety and depression symptoms. Disruption of these systems, by either direct impact of TBI or indirect pathologies, such as chronic stress or inflammation, can negatively influence comorbid development of psychiatric disorders.
D. Attention Deficit Hyperactivity Disorder
A meta-analysis documented a significant association between mild TBI and attention deficit hyperactivity disorder (ADHD) (Adeyemo et al., 2014). ADHD is a neurodevelopmental, childhood onset, and persistent disorder characterized by increased impulsivity and risk-taking behavior in individuals. Not only has ADHD been shown to heighten risk of experiencing a TBI, but pre-existing diagnosis of ADHD may also result in worsened recovery outcomes after injury (Bonfield et al., 2013). Compared with patients with TBI but not ADHD, individuals with both diagnoses were significantly more impaired on individual scores of working memory, planning/organization, metacognition indices, and behavioral regulation (Ponsford and Kinsella, 1992; Biederman et al., 2015).
ADHD that develops as a comorbidity of TBI is referred to as secondary ADHD and has been shown to occur in 10%–20% of patients post-TBI (Gerring et al., 1998). Many studies support the hypothesis that dysfunction in the prefrontal cortex, basal ganglia, and their related neurotransmitter systems underlie deficits in inhibitory regulation found in patients with ADHD (Dickstein et al., 2006). Therefore, damage to these areas have the potential to manifest as secondary ADHD or other psychiatric disorders in children and adults. Secondary ADHD may be more prevalent in children and adolescents due to the underdevelopment of brain regions linked to inattention and hyperactivity. Anatomic studies of youth with developmental ADHD demonstrate a loss of volume of frontal and striatal structures without lesions. One case study describes secondary ADHD development in a 10-year-old boy who suffered head trauma and early post-traumatic seizures (Ceylan and Akca, 2013). Cranial CT scans demonstrated decreased density of the right basal ganglia and loss of corticomedullar differentiation in the right frontal area. Six months after the accident, reports from the patient’s parents and teachers describe a change in the child’s behavior of overactivity, boredom at class and home, difficulty maintaining concentration, and depressive-like symptoms. Social disinhibition, hyperactivity, poor impulse control, forgetfulness, and lack of judgement/foresight are among the chronic sequelae of closed head injuries (Kaitaro et al., 1995).
E. Sleep Disorders
Sleep-wake dysfunction after brain injury is common, affecting up to 70% of patients (Viola-Saltzman and Watson, 2012). Common diagnoses after TBI include insomnia, parasomnia, idiopathic hypersomnia, narcolepsy, sleep apnea, and circadian rhythm disorder (Morse and Garner, 2018). This sleep-wake cycle is regulated by a concerted effort between circadian rhythms, sleep-wake homeostasis, metabolism, and external environmental factors, such as diet, stress, medication, and surroundings. Pre-existing sleep conditions can increase the likelihood of post-TBI sleep-wake disturbances. Additionally, comorbid sleep disorders contribute to psychologic instability, resulting in behavioral problems, mood or emotional lability, and worsened daily functioning (Shay et al., 2014; Reddy et al., 2018c).
Alertness and cortical arousal are mediated by several pathways that project from the brainstem near the junction of the midbrain and pons to innervate the thalamus, posterior hypothalamus, and forebrain (Fuller et al., 2006). These ascending pathways are populated by key cell types, including cholinergic, histaminergic, noradrenergic, dopaminergic, and serotoninergic neurons, that fire in a distinct pattern to promote wakefulness. Orexin peptides produced in the lateral hypothalamus reinforce the arousal state. However, this system is inhibited roughly every 24 hours by sleep-active GABAergic and galaninergic neurons of the ventrolateral preoptic nucleus (VLPO) to promote sleep onset (Schwartz and Roth, 2008). The VLPO is a known sleep center influenced by the suprachiasmatic nucleus and activated by endogenous sleep-promoting substances, such as adenosine and prostaglandin D2. Melatonin production from the pineal gland plays a role in REM sleep and aids in synchronizing circadian rhythms.
The pathophysiology of post-traumatic sleep disorders has not been fully explored; however, decreased levels of wake-promoting neurotransmitters, such as hypocretin (orexin) and histamine, are believed to play a contributing role. Post-mortem assessments of brains from patients with TBI demonstrate significant reductions in hypocretin neurons (Shekleton et al., 2010). Even after 6 months of recovery, many patients with TBI exhibit persistently low hypocretin-1 levels, suggesting sleep disorders as a chronic consequence of TBI (Baumann et al., 2007). Disturbed orexin signaling contributes to sleep-wake disorders by causing excessive daytime sleepiness and circadian rhythm disruption (Nishino et al., 2000; Baumann et al., 2005). Moreover, decreased CSF levels of orexin have been associated with poorer clinical outcomes and higher risk of depression and sleep disorders (Brundin et al., 2007). Low CSF histamine levels have also been reported in patients with narcolepsy and after TBI (Kanbayashi et al., 2009). Injury may cause lifestyle changes that can affect sleep and arousal, including changes to diet or medication, exercise routines, or environmental factors. Furthermore, insomnia may develop because of psychologic trauma resulting from personal assaults or accidents that resulted in TBI. Lastly, direct injury to sleep-wake brain centers, such as the VLPO or suprachiasmatic nucleus, may also explain development of circadian rhythm disorders.
Comorbid disturbances in the sleep-wake cycle have also been observed after severe TBI in mice. One critical study tracked nonrapid eye movement sleep for up to 4 months postinjury and found changes in delta power contributed to predictive seizure modeling (Konduru et al., 2021). The authors state that delta power increased in injured mice versus no craniectomy control mice; however, responding injured mice displayed lower delta power at a chronic time point compared with injured mice without seizure activity. Moreover, sleep spindle duration and dominant band frequency is lower in PTE rats compared with sham controls during the transition from stage III to REM sleep (Andrade et al., 2017). These studies suggest sleep-wake disturbances may be a potential biomarker for post-traumatic seizures.
Once the prevalence of PTE-associated comorbidities is established, the search for biomarkers for PTE and recovery may benefit from the neurocognitive network model of affective and/or sleep disorders currently applied to primary epilepsy. Since sensorimotor, cognitive, psychiatric, and sleep-wake comorbidities are so common after TBI, it is important for future work to incorporate quality of life measurements as indicators of recovery outcomes when assessing new therapeutic targets. Additionally, the potential of drug-drug interactions between antiepileptogenesis or antiseizure medications and those that reduce comorbid symptoms should also be evaluated. By examining cellular and synaptic changes in the affected neural circuitry, we can better understand how learning and memory deficits occur. Using newer technologies, such as chemogenetics or optogenetics, could substantiate the relationship between behavior and physiology in a meaningful way.
VI. Pharmacological Interventions for Post-Traumatic Epilepsy
Epilepsy and epileptogenesis are both associated with a wide range of comorbidities, ranging from mild to severe, and often originating from overlapping processes. Therefore, disease modification has two major components: antiepileptogenesis and/or the reduction of its associated comorbidities. Antiepileptogenesis includes the prospect of the prevention of epilepsy, seizure modification, or cure. Prevention can be either partial or complete. Partial prevention consists of delayed epilepsy development or reduced severity, whereas complete prevention is the termination of the development of epilepsy. Modification of seizures is considered a form of partial protection and can come in many forms: reduced frequency, milder seizure type, and shorter duration (Cross and Lagae, 2020). Antiepileptogenic treatments can be administered before or after the onset of seizures to prevent or delay the development of epilepsy. This contrasts modification of the epileptogenic insult in which a treatment is given before the onset of seizures and alters epileptogenesis by modifying the injury or insult directly. Modification of seizures, if they occur in either case, can be milder in their progression, severity, frequency, or duration (Reddy and Estes, 2016a). On the other hand, a cure refers to the absolute and permanent reversal of the epileptic state such that no seizures occur after withdrawal of treatment. Treatments that treat or modify comorbidities of PTE may alleviate or reverse the progression of somatosensory or functional impairment, cognitive decline, anxiety, depression, or any other epilepsy-associated comorbidity. Treatments of comorbidities may be singular or broad with their ability to affect a range of symptoms.
To date, the current management of clinical PTE remains as prophylactic treatment with first-line therapies, such as levetiracetam or phenytoin, for the first seven days after initial injury (Temkin et al., 2001). These ASMs have shown efficacy in focal epilepsies but have limited effectiveness in PTE. Many studies have reported evidence that there is currently no pharmacological prevention or treatment of post-traumatic seizures, and PTE is often refractory to medical management (Piccenna et al., 2017; Zimmermann et al., 2017). Novel drug discovery requires reliable animal models to elucidate the complex pathophysiology of epileptogenesis, validate targets, and test agents for efficacy and safety.
Rodent studies have been particularly useful for discovering novel therapeutic targets, although there are limited studies that have specifically investigated interventions for PTE. This next section reviews research that has focused on pharmacological prevention of PTE. Table 5 details the available data on experimental and clinical trials of therapeutic interventions for PTE. Since there are not many studies that have tested therapeutic interventions for epileptogenesis after brain injury, we have also included studies that identify changes in hyperexcitability—either through chemically or electrically induced seizure threshold changes. Furthermore, as recovery factors from the TBI often interfere with the ability to record immediate (<24 hour) and early seizures (first week), most of these studies focus on late seizure (>1 week) occurrence. As previously stated, immediate and early seizures are thought to occur as a result of the impact itself and not considered classically epileptic. Moreover, immediate and early seizures do not have predictive validity for late seizure occurrence. Therefore, studies that seek to demonstrate therapeutic potential in PTE should focus on models that use long-term EEG recording and can identify late and/or early seizures after a latent period. Figure 5 depicts the timeline progression of immediate/early seizures and spontaneous epileptic seizures after TBI.
TABLE 5.
Therapeutic interventions for PTE
| System/Focus | Model | Species | Effect on Epileptiform Activity | Disease Modification | Reference |
|---|---|---|---|---|---|
| AEDs | |||||
| Carbamazepine | Human condition | Clinical | Not effective for preventing or modifying seizures | N/A | Temkin, 2009 |
| Phenytoin | Human condition | Clinical | Not effective for preventing or modifying seizures | N/A | Temkin, 2009 |
| Phenobarbital | Human condition | Clinical | Not effective for preventing or modifying seizures | N/A | Temkin, 2009 |
| Valproate | Human condition | Clinical | Not effective for preventing or modifying seizures | N/A | Temkin, 2009 |
| Carbamazepine | FPI | Rat | Not effective for preventing or modifying seizures | N/A | Eastman et al., 2010 |
| Valproate | FPI | Rat | Reduced seizure frequency; reduced cumulative seizure duration | N/A | Eastman et al., 2010 |
| Carisbamate | FPI | Rat | Not effective for preventing or modifying seizures | N/A | Eastman et al., 2011 |
| Gabapentin | Cortical undercut | Rat | Evoked epileptiform discharges in cortical slices 1d and 14 d post-gabapentin | Reduced GFAP expression | Li et al., 2012 |
| Phenytoin | Penetrating brain injury | Rat | Reduced incidence, frequency, and duration of nonconvulsive seizures in a dose-dependent manner | No effect on lesion volume or body weight with treatment | Mountney et al., 2013 |
| Ethosuximide | Penetrating brain injury | Rat | Reduced incidence, frequency, and duration of nonconvulsive seizures in a dose-dependent manner | No effect on lesion volume or body weight with treatment | Mountney et al., 2013 |
| Levetiracetam | Human condition | Clinical | Trended with lower seizure incidence in clinical patients but never reached significance | N/A | Hazama et al., 2018 |
| Retigabine | CCI | Mouse | Reduced seizure susceptibility; reduced seizure frequency | Reduced inflammation; lessened BBB breakdown; reduced neurodegeneration | Vigil et al., 2020 |
| Inhibitory/Excitatory Pathways | |||||
| Halothane (anesthetic, GABAA agonist) | FPI | Rat | No seizures occurred while subjects were under anesthesia | N/A | Eastman et al., 2010 |
| Ceftriaxone (beta-lactam antibiotic and stimulator of GLT1 expression) | FPI | Rat | Reduced cumulative seizure duration | Preserved GLT1 expression | Goodrich et al., 2013 |
| Ceftriaxone (beta-lactam antibiotic and stimulator of GLT1 expression) | FPI | Rat | Reduced cumulative seizure duration | Preserved cortical inhibitory interneuron function with continuous treatment | Hameed et al., 2019 |
| 2-deoxyglucose (glycolysis inhibitor) | CCI | Mouse | Restored excitatory and inhibitory synaptic activity; reduced epileptiform activity | Reduced neurodegeneration of PV+ interneurons | Koenig et al., 2019 |
| Imidizodiazepine KRM-II-81 (selective for alpha2/3 containing GABAAR) | CCI | Mouse | Reduced hyperactivity | N/A | Witkin et al., 2020 |
| mTOR pathway | |||||
| Rapamycin | CCI | CD1 mouse | Reduced seizure incidence (13% versus 50%); reduced seizure frequency; lessened behavioral component of seizures | Reduced neurodegeneration; attenuated mossy fiber sprouting | Guo et al., 2013 |
| Rapamycin | CCI | Mouse | Reduced seizure frequency | Reduced neurodegeneration; attenuated mossy fiber sprouting; stabilized neurogenesis | Butler et al., 2015 |
| Rapamycin | CCI | Mouse | Modified synaptic and tonic GABAAR-mediated currents. | N/A | Butler et al., 2016 |
| Inflammation | |||||
| Minozac | CCI | CD1 mice | Reduced seizure susceptibility | Reduced inflammation; improved cognitive impairments | Chrzaszcz et al., 2010 |
| Kineret (IL-1R antagonist) | CCI | Pediatric mouse | Reduced seizure susceptibility | Improved neuropathology; improved cognitive impairments | Semple et al., 2017 |
| Monophosphoryl lipid A and Pam3Cys | CCI; kindling | Rat | Reduced seizure susceptibility; slowed kindling rate | Reduced TNF-alpha brain levels | Hesam et al., 2018 |
| Glycyrrhizin (HMGB1 inhibitor) | CCI | Pediatric mouse | Not effective for preventing or modifying seizures | Reduced HMGB1 brain levels, edema, and microglial activation | Webster et al., 2019 |
| Plasticity | |||||
| BDNF blocker (TrkB-Fc) | Ex vivo Schaffer collateral lesions in organotypic hippocampal slice cultures | Pediatric mouse | Reduced hyperexcitability of CA3 neurons | Attenuate mossy fiber sprouting | Gill et al., 2013 |
| LM22A-4 (partial agonist of TrkB) | Cortical Undercut | Rat | Decreased incidence of epileptiform discharges | Increased parvalbumin immunoreactivity | Gu et al., 2018 |
| PTX BD4-3 (partial agonist of TrkB) | Cortical Undercut | Rat | Reduced PTZ susceptibility. | N/A | Gu et al., 2018 |
| Hypothermia | |||||
| Hypothermia | FPI | Rat | Reduced PTZ susceptibility | Attenuate mossy fiber sprouting; did not exhibit neuroprotective effects on cell loss | Atkins et al., 2010 |
| Hypothermia | FPI | Rat | Abolished ictal activity up to 10 weeks after hypothermia treatment | N/A | D'Ambrosio et al., 2013 |
| Electrical Stimulation | |||||
| Deep Brain Stimulation | Human Condition | Clinical | Reduced seizure frequency | N/A | Piacentino et al., 2018 |
| Hyperphosphorylation of Tau | |||||
| Sodium Selenate | FPI | Rat | Increased latency to seizures/epileptiform activity | Ameliorated enlargement of ventricles and hippocampal atrophy | Liu et al., 2016. |
| Cell Transplantation Therapy | |||||
| Cell transplantation therapy (GABAergic progenitors from medial ganglionic eminence) | CCI | Mouse | Reduced incidence of PTE | Improved cognitive impairments; | Zhu et al., 2019 |
| Other | |||||
| Rimonabant CB1R antagonist 1 or 10 mg/kg immediately or 20min after TBI | FPI | Rat | Reduced KA susceptibility | N/A | Echegoyen et al., 2009 |
| Ketogenic Diet | FPI | Rat | Not effective for preventing or modifying seizures | N/A | Schwartzkroin et al., 2010 |
| Creatine | FPI | Rat | Not effective for preventing or modifying seizures | Reduced oxidative stress markers | Saraiva et al., 2012 |
| Exercise (Treadmill) | FPI | Rat | Lengthened latency to seizures; reduced seizure duration; reduced PTZ susceptibility | N/A | Silva et al., 2013 |
| C-10068 (Dextromethorphan Derivative) | PBBI | Rat | Lengthen latency to seizures; reduced seizure frequency, reduced cumulative seizure duration | Reduced inflammation | Lu et al., 2015 |
| Atipamezole, Alpha2 Adrenergic Receptor Antagonist | FPI | Rat | Reduced PTZ susceptibility | Improved functional recovery | Nissinen et al., 2017 |
| Creatine Supplementation, May Be Needed Long-Term; 300mg/kg P.O. 4 wks | FPI | Rat | Lengthened latency to seizures; decreased duration of tonic-clonic seizures; reduced PTZ-induced epileptiform discharges | Reduced neurodegeneration | Gerbatin et al., 2019 |
| Progesterone | Weight drop | Rat | Reduced length of PTZ-induced seizures; lessened behavioral component of seizures | N/A | Ghadiri et al., 2019 |
Fig. 5.

Timeline progression of TBI-induced early seizures and late spontaneous recurrent seizures. TBI triggers acute cascades resulting in immediate or early seizure, referred to as post-traumatic seizures, and propels the process of epileptogenesis. ultimately resulting in chronic epileptic state with spontaneous recurrent seizures. The general premise about epileptogenesis can be divided into three distinct stages. The first stage occurs with an initial brain injury event. This is followed by the second latent stage that can last a varied amount of time. The third stage is the chronic period in which the patient suffers from spontaneous seizures. The time required to reach chronic stage represents a window of opportunity for testing interventions in people at high risk for epilepsy after brain injuries.
A. Antiseizure Medications
Antiepileptic drugs, recently renamed as antiseizure medications, are the mainstay for the control of early and late seizures, irrespective of the source of such seizures. More than three dozen ASMs, including first, second, and third, or recent generation, have been extensively tested against post-traumatic seizures (Reddy, 2020). Perhaps the most accessible experiments for PTE are those that evaluate the effect of clinically available ASMs for beneficial effects on disease modification or recovery outcomes. Early studies with positive but uncontrolled results led to widespread use of phenobarbital and/or phenytoin for prophylaxis against immediate and long-term post-traumatic seizures. One survey, conducted in 1973, reported over 60% of clinicians supported immediate pharmacological prophylaxis for head injuries, with 40.3% of respondents prescribing treatment of 1 or more years after injury (Rapport II and Penry, 1973). Current evidence suggests treatment of early seizures does not influence the incidence of PTE (Agrawal et al., 2006). Within the clinical setting, carbamazepine, phenytoin, phenobarbital, and valproate all failed to prevent long-term PTE after head injury (Temkin, 2009). In a recent clinical trial, Hazama et al. (2018) evaluated the benefit of levetiracetam after head trauma. The study consisted of 403 patients, 227 of whom were treated with levetiracetam for early post-traumatic seizure prophylaxis. Although patients receiving levetiracetam treatment trended with lower seizure incidence, this trend never reached statistical significance. These ASMs have shown successful seizure control in other epilepsy disorders but have little effect on preventing or modifying epileptogenesis.
Preclinical research has demonstrated a similar lack of efficacy in reducing PTE incidence with ASMs. Eastman et al. (2010) used the FPI model to systematically detect potential antiepileptic effects of carbamazepine and valproate 1-month postinjury. Carbamazepine (up to 12mg/kg/d for 4.5 days) treatment did not reduce seizure frequency or duration, nor did it reduce incidence of PTE. However, valproate (480 mg/kg/d for 7 days) reduced seizure frequency and cumulative seizure duration. Comorbidities were not evaluated as part of these investigations. In a follow-up study, carisbamate administration was also found to be ineffective at reducing seizure burden or preventing PTE (Eastman et al., 2011) Recently, retigabine, an ASM that was removed from the commercial market in 2017, has shown some promise in reducing seizure burden after CCI in mice. Not only did retigabine administration reduce inflammation, BBB breakdown, and neurodegeneration at the impact site, but also significantly reduced frequency of spontaneous seizures and enhanced susceptibility to chemoconvulsants (Vigil et al., 2020). Gabapentin administration inhibited synapse formation and decreased excitatory synaptic activity after cortical injury (Li et al., 2012). Gabapentin treatment also reduced the expression of astrocytic GFAP expression and thrombospondin-1 protein, as well as the number of fluoro-jade B+ stained cells. These results suggest gabapentin may modify pathways associated with plastic changes in brain excitability, but a controlled in vivo study with continuous EEG recording has yet to confirm an antiepileptogenic effect.
Lastly, Mountney et al. (2013) demonstrated administration of ethosuximide, or phenytoin dose-dependently attenuates in the incidence, frequency, and duration of nonconvulsive seizures after a penetrating blast-like brain injury. In this study, a loading dose of either ethosuximide or phenytoin was given 30-minute post-TBI, followed by a maintenance dose 8 hours later. Four doses of each drug were used to provide a full dose-response curve, with the two highest doses of each drug showing significance in reducing seizure burden. Although these data are promising, this study only follows the rats for up to 72 hours postinjury and identifies immediate and early nonconvulsive seizures, which are considered to be caused by direct impact rather than epileptogenesis. Furthermore, there is little evidence that suggests early seizure prophylaxis to reduce incidence of long-term spontaneous seizures.
B. Targeting Neuronal Excitability Network Pathways
The balance of inhibition and excitation in the brain is very delicate. TBI triggers changes that ultimately result in the disruption of this harmony, leading to increased risk of seizures and epileptogenesis. One approach aimed at minimizing excitotoxicity and preserving cortical inhibition after TBI is the administration of ceftriaxone, a common β-lactam antibiotic with BBB permeability. Ceftriaxone is also a known potent stimulator of glutamate transporter-1 (GLT-1) expression, a critical protein responsible for 95% of total glutamate clearance in the rat brain (Lehre and Danbolt, 1998). Ceftriaxone (200 mg/kg) rescues TBI-induced downregulation of GLT-1 expression within the first week after injury (Goodrich et al., 2013). Authors also report a reduction in regional GFAP expression relative to untreated rats. A follow-up study reported ceftriaxone significantly suppressed both the frequency and duration of post-traumatic seizures weeks after injury (Hameed et al., 2019).
Enhancing GABAergic inhibitory tone is a well-established mechanism for seizure prophylaxis, as many ASMs are approved in this class. GABAA receptors are also targets of general anesthetics, such as halothane, that can completely suspend seizure activity after FPI, although it leaves the subjects immobile (Eastman et al., 2010). Neurosteroids that modulate tonic inhibition can suppress seizures (Carver et al., 2016; Chuang and Reddy, 2020). Recent attention has been directed toward selective, rather than nonselective (e.g., diazepam), positive allosteric modulators of GABA due to a lower risk for somnolence, motor impairment, and drug abuse (Witkin et al., 2018). KRM-II-81 is an orally bioavailable compound selective for α2/3-containing GABAA receptors. Witkin et al. (2020) used several models of pharmaco-resistant epilepsy, including kainate-induced mesial temporal lobe seizures, CCI-induced focal injury, and corneal-kindling, to investigate the antiepileptogenic properties of KRM-II-81. They report complete suppression of corneal-kindling seizures in treated mice as well as reduced paroxysmal discharges after kainate injection. Furthermore, repeated injection of KRM-II-81 administration immediately reduced neural hyperactivity for weeks after CCI, suggesting drug resistance or tolerance was negligible. Many ASMs have little therapeutic impact after TBI for controlling post-traumatic seizures; therefore, the cortical excitatory reductions by KRM-II-81 encourage future preclinical and clinical studies.
Lastly, 2-deoxyglucose (2-DG), a competitive inhibitor of hexokinase, the rate-limiting enzyme in glycolysis, has also shown anticonvulsive properties both in vivo and in vitro. Recently, Koenig et al. (2019) explored the compound as a disease modifying agent to prevent epileptogenesis after TBI. Using the CCI model of brain contusion, they report acute 2-DG treatment attenuated hyperexcitability in the brain and prevented development of epileptiform activity in slices taken 3–5 weeks after injury. Additionally, 2-DG treatment reduced the loss of parvalbumin-expressing interneurons, thereby showing neuroprotection against TBI-induced cell loss.
C. Neuroinflammatory Modulation
Targeting neuroinflammation has been a therapeutic strategy in epilepsy disorders for decades, for good reason. Neuroinflammation involves both resident microglia and astrocytes, as well as peripheral immune signaling when the BBB integrity becomes compromised (Reddy et al., 2016b). Furthermore, reactive glial cells can be the drivers of abnormal neuronal activity by impairing the inhibitory action of GABA receptors, reduced neurotransmitter clearance, and disrupted homeostasis (Robel and Sontheimer, 2016). To this end, a handful of compounds have been tested as potential antiepileptogenics for PTE. Although glycyrrhizin, a HMGBI1 inhibitor, was recently found to be ineffective at preventing seizures or reducing susceptibility after TBI, its administration did reduce edema and microglial activation after CCI in pediatric mice (Webster et al., 2019). Toll-like receptor agonists, such as monophosphoryl lipid A and Pam3Cys, significantly slowed amygdala kindling after TBI, demonstrating a reduction in seizure susceptibility (Hesam et al., 2018). Treated rats also exhibited a lessened behavioral response to kindled seizures similar to nontraumatic rats. Given that monophosphoryl lipid A and Pam3Cys are safe and have clinical use as components in vaccines, they have the potential to be used in combination with other agents as a therapeutic strategy for PTE.
Minozac, a suppressor of proinflammatory cytokine upregulation, has been shown to significantly reduce electroconvulsive shock seizures after TBI in mice (Chrzaszcz et al., 2010). These data were coupled with a favorable reduction in TBI-induced metallothionein expression in the CA1, suggesting a reduction in oxidative stress with treatment. Similarly, kineret, an IL-1 receptor antagonist, reduces seizure susceptibility and improved neuropathology associated with epileptogenesis 2 weeks after CCI (Semple et al., 2017). Moreover, mice treated with kineret showed significantly lower seizure frequency compared with vehicle-treated controls at 5 months postinjury. Combined with improved neurobehavioral function, these data provide evidence of IL-1 signaling as a mediator of injury-associated epileptogenesis.
D. Disrupting the Mammalian Target of Rapamycin Pathway
As discussed above, the mTOR signaling pathway is implicated in the regulation of multiple cellular functions that contribute to epileptogenesis. The current hypothesis is such that mTOR signaling is hyperactivated after TBI and triggers multiple downstream mechanisms of PTE. Several studies of mTOR inhibition have demonstrated a beneficial effect of rapamycin, an mTOR inhibitor, in the treatment of epilepsy disorders and improving recovery outcomes after TBI.
Within PTE models, three studies have shown reduced seizure incidence, frequency, or duration after CCI in mice (Guo et al., 2013; Butler et al., 2015; Butler et al., 2016). Guo et al. (2013) observed a significant reduction in seizure incidence, dropping from 50% to 13% in treated versus untreated mice. It was also noted that when rapamycin-treated mice did exhibit seizures, the behavioral component was lessened, suggesting seizure intensity was also reduced by mTOR inhibition. Butler et al. (2015) reported a trend of reduced seizure frequency and incidence with continuous rapamycin treatment but indicated mTOR inhibition was not sufficient to prevent epileptogenesis after CCI. Furthermore, a follow-up study confirmed rapamycin treatment can modify synaptic and tonic GABAA receptor-mediated currents, hinting toward another mechanism of antiepileptogenic properties (Butler et al., 2016).
Similar to reports on status epilepticus, rapamycin administration resulted in reduced neurodegeneration and attenuated mossy fiber sprouting. Unfortunately, the beneficial effects of rapamycin appear to cease upon discontinuation of the drug in both animal and clinical trials (Buckmaster et al., 2009). Lifelong treatment with mTOR inhibitors is questionable given the known adverse effects, such as immunosuppression, risk of cognitive impairment, and negative influences on normal growth and development in children or adolescents (Bissler et al., 2008).
E. Targeting of Neuronal Plasticity
Neurotrophic factors like BDNF, nerve growth factor, and fibroblast growth factor promote cell survival, growth, and differentiation through activation of signaling pathways after TBI. Abnormalities of interneurons and cell loss play critical roles in epileptogenesis after TBI; therefore, enhancing the function of remaining parvalbumin+ interneurons may be a novel therapeutic approach to preventing PTE. BDNF is known to have positive influences on interneuron growth and function through activation of its receptor, TrkB (Marty et al., 1997). A recent study tested the hypothesis that supporting TrkB function may decrease epileptogenesis after cortical undercut (Gu et al., 2018). LM22A-4 or PTXBD4-3, both partial agonists of the TrkB receptor, were administered up to 2 weeks after injury. PTXBD4-3 reduced seizure susceptibility to PTZ but did not affect seizure duration or latency. LM22A-4, however, decreased the incidence of epileptiform discharges compared with untreated controls. Activation of the TrkB receptor also resulted in increased parvalbumin immunoreactivity, suggesting that partial activation of TrkB may be neuroprotective to interneurons, thereby preserving inhibitory circuity and reducing seizure susceptibility. A different approach explored the use of BDNF blockers in ex vivo organotypic hippocampal slice cultures with collateral lesions and observed reduced hyperexcitability of CA3 pyramidal neurons as well as a reduction in aberrant mossy fiber sprouting (Gill et al., 2013). These differing approaches provide supporting evidence for TrkB signaling modification, but further investigations are needed to confirm antiepileptogenic effects.
F. Hypothermia
Focal cooling can be broadly neuroprotective and has suppressed seizures in animal models, providing evidence that therapeutic hypothermia should be investigated within the context of PTE. Blind, randomized studies of FPI found that a graded cooling up to 2°C significantly reduced seizure frequency and duration in rats. Cessation of ictal activity lasted up to 10 weeks after hypothermia treatment ended (D’Ambrosio et al., 2013). Another study reported reduction of seizure threshold to chemical convulsants and reduced mossy fiber sprouting with hypothermia treatment but did not exhibit neuroprotective effects on cell loss (Atkins et al., 2010). Hypothermia appears to be safe with few negative consequences; however, more research is needed to optimize treatments and define its clinical value.
G. Electrical Stimulation
Deep brain stimulation (DBS) has shown to be remarkably effective, safe, and practical for the treatment of movement disorders, such as tremor, dystonia, and Parkinson’s disease (Deuschl et al., 2006). These successes inspired the application of DBS for other neurologic disorders, including epilepsy. The action of DBS is multifaceted and complex, with high-frequency stimulation (>50 Hz) mimicking the effects of ablative procedures by inducing a reversible functional lesion (Benabid et al., 2002). Lower-frequency stimulation has been associated with anticonvulsant effects, changes in adenosine receptor expression, and altered levels of neurotransmitters and hormones in cerebrospinal fluid. The basis of DBS is to improve abnormal synchrony between different brain regions. One case study was found using DBS as a potential therapeutic intervention for PTE. The patient reported a post-traumatic episode during childhood that resulted in subsequent seizures throughout adulthood. ASM treatments, such as carbamazepine, phenobarbital, and clonazepam, had all been unsuccessful in providing adequate symptomatic control of seizures. Bilateral DBS hippocampal stimulation resulted in a progressive decrease in seizure frequency over the 8 years of follow-up, although some clinicians postulate whether the surgical placement of the DBS system may have contributed to the disruption of the epileptic foci in this case (Piacentino et al., 2018). Other studies have found behavioral improvements and functionality using DBS after TBI, although seizure outcomes were not applicable in these cases (Lee et al., 2013; Shin et al., 2014; Rezai et al., 2016).
H. Tau Hyperphosphorylation
Tau is a microtubule-associated protein that has roles in maintaining neuronal health, axonal transport, and microtubule stabilization. Tau phosphorylation is a normal metabolic process critical to tau’s ability to bind to microtubules. Hyperphosphorylation of tau can cause aggregation and form insoluble fibrillar deposits in tissues. Hyperphosphorylated tau is a long-known hallmark of neurodegenerative diseases, such as Alzheimer’s disease and dementia; however, the causal role of TBI-induced tauopathy has been debated for decades (Castellani and Perry, 2019). Recent studies have confirmed moderate and severe brain injury can trigger the formation of pathologic tau aggregates, linking TBI to increased risk of Alzheimer’s disease (Edwards et al., 2020; Wu et al., 2020).
Sodium selenate mitigates hyperphosphorylated tau by antagonizing PP2A activity and can improve TBI outcomes. Liu et al. (2016) administered sodium selenate (1mg/kg/d), a less toxic form of selenium, via osmotic pumps for 12 weeks after FPI. Treatment with sodium selenate ameliorated the enlargement of ventricles and hippocampal atrophy, which often accompanies brain injury. Additionally, the latency period to spontaneous epileptiform activity, such as seizures or discharges, was extended compared with vehicle-treated rats, suggesting possible interruption of the epileptogenic process. Although this is the only study to specifically investigate the effects of sodium selenate on PTE, sodium selenate has been demonstrated to suppress seizures, improve comorbidities, and reduce seizure susceptibility in experimental models of Lafora disease and temporal lobe epilepsy (Jones et al., 2012; Sanchez-Elexpuru et al., 2017).
I. Cell Transplant Therapies
Cell transplantation, including genetically modified cell types, have been tested as a recovery-enhancing treatment after TBI. Outcome measures include the effects of treatment on lesion volume, severity of neurodegeneration, axonal injury, edema, motor ability, and cognitive function (Jackson et al., 2017). Although positive data have emerged from these studies, most reports have not measured development of spontaneous seizures as an outcome. One recent study, however, found the transplantation of GABAergic progenitors derived from the embryonic medial ganglionic eminence migrated, integrated, and restored post-traumatic decreases in synaptic inhibition (Zhu et al., 2019). Using a CCI model of PTE, mice were recorded using continuous 24/7 video-EEG for between 7 and 20 days at 4 months postinjury. Since most models of PTE find the onset of seizures to be between 20 and 90 days, this timeline was assumed to be after onset of spontaneous epileptiform activity. Incredibly, no electrographic seizures were observed in TBI mice that were implanted with medial ganglionic eminence cells, whereas five of eight untreated mice exhibited repeated ictal events. Cell transplantation therapy also resulted in improved memory precision in transplant mice using the novel object location test. Together, these results provide powerful evidence of antiepileptogenesis using cell-based therapies by restoring long-term deficits in both synaptic inhibition and neurobehavioral impairments. However, this study was limited by the relatively small cohort sizes and shortened length of EEG recording. Follow-up studies could easily address these limitations. This work establishes a promising framework for future studies to evaluate other populations of neurons for cell transplantation therapies for PTE.
J. Dietary and Phytochemical Therapeutic Strategies
Studies dating back to the 1920s have shown that diet and exercise can improve seizure control for patients with epilepsy, with a special emphasis on the ketogenic diet (Wilder, 1921). The ketogenic diet is a high-fat, adequate-protein, and low-carbohydrate diet that has been shown to reduce seizure frequency by over 50% in children and adolescents but has not demonstrated evidence of disrupting the epileptogenic process (Neal et al., 2008). Similar to clinical indications, the ketogenic diet was found to not be effective in preventing PTE after brain injury (Schwartzkroin et al., 2010). However, regular treadmill exercise not only lengthened the latency to seizure onset, but also reduced ictal duration and susceptibility to PTZ-induced seizures in rats (Silva et al., 2013). Creatine supplementation to the diet, on the other hand, has produced contradicting results. Gerbatin et al. (2019) report decreased duration of tonic-clonic seizures and reduced PTZ-induced epileptiform discharges. Creatine supplementation was also found to reduce neurodegeneration of parvalbumin+ interneurons in the hippocampus at 1-month postinjury. These results required long-term administration of daily creatine. Saraiva et al., however, found no reduction on convulsive parameters induced by PTZ in the first week after TBI, but did reduce oxidative damage at the impact site (2012). Together, these studies suggest creatine supplementation may not be a sufficient prophylactic for early post-traumatic seizures but may possess long-term downstream effects which stabilize epileptogenesis.
Cannabis and related compounds have recently broke foreground in epilepsy disorders with the USA FDA-approval of Epidiolex for Lennox-Gastaut and Dravet syndromes in 2018 (Golub and Reddy, 2020a). Cannabidiol has yet to be evaluated for post-traumatic epilepsy, but Rimonabant (SR141716A), a CB1 receptor antagonist may modify disease progression. CB1 receptors are present on both excitatory and inhibitory nerve terminals, where they inhibit glutamate and GABA release, respectively. Therefore, agents which act at this receptor may have multiple effects on neurotransmission. Interestingly, rimonabant is a proconvulsant that has been demonstrated to lower threshold to kainate-induced seizures. Blocking CB1 receptors prevented increased seizure susceptibility in an experimental model of febrile seizures (Chen et al., 2007b). Similar results were found in a rat model of PTE in which a single injection of rimonabant reduced long-term hyperexcitability and susceptibility to kainic acid (Echegoyen et al., 2009). Rimonabant administration after LFPI reversed the overexpression of mGluR5 in late-stage brain injury, thereby lengthening latency to PTZ-induced seizures (Wang et al., 2016c; 2016d). However, these studies are limited by their use of secondary convulsants, such as PTZ and kainic acid, and did not use continuous EEG recording. Moreover, a recent report found investigated the therapeutic effects of Δ9-tetrahydrocannabinol on repetitive mild traumatic brain injury and found postinjury administration reduced anxiety, depressive-like behaviors, and mitigated injury-induced deficits in short-term working memory (Bhatt et al., 2020). More research is needed in this field to fully understand how these compounds may positively or negatively affect recovery outcomes.
Atipamezole, a synthetic α2 adrenergic receptor antagonist, is used mainly in veterinary medicine as it is indicated for the reversal of sedative and analgesic effects of dexmedetomidine and medetomidine in dogs. Treatment with atipamezole reduced PTZ seizure susceptibility at 14 weeks after TBI and improved motor performance but did not prevent the development in spontaneous seizures (Nissinen et al., 2017).
Dextromethorphan, typically used as an over-the-counter cough suppressant, has multiple mechanisms of action including acting as a nonselective serotonin reuptake inhibitor, sigma-1 receptor agonist, and blocks NMDA glutamate receptors at high doses. The dextromethorphan derivative, C-10068, was found to reduce non-convulsive seizure frequency and cumulative seizure duration in a penetrating ballistic-like brain injury model of PTE (Lu et al., 2015). The most efficacious dose of C-10068 also reduced inflammation and reactive microglia accumulation around the lesion site. However, this study reported little improvement in neurobehavioral function and continuous EEG recording was only performed for up to 72 hour postinjury.
Lastly, steroid hormones have been proven to be neuroactive and protective in a variety of CNS disorders (for review see Reddy and Estes, 2016a). Neurosteroids regulate the plasticity of synaptic and extrasynaptic GABAA receptors involved in the pathophysiology of epilepsy. Progesterone is the precursor to allopregnanolone, which acts as a positive allosteric modulator and direct activator of GABAA receptors to enhance inhibition in the brain. A recent study utilizing the weight-drop model of PTE reported a reduced duration of PTZ-induced seizures after progesterone treatment (Ghadiri et al., 2019). The behavioral component of seizures was also reduced, suggesting decreased seizure intensity and neuroprotection.
K. Novel Epigenetic Interventions
Epigenetic interventions, such as histone modifiers, represent a novel therapeutic pathway that remains to be fully explored. Global and regional changes in gene expression due to epigenetic modification have been observed after TBI and in epilepsy disorders (Golub and Reddy, 2020b). TBI is known to increase HDAC activity in the brain, resulting in reduced H4 acetylation and increased seizure susceptibility (Dash et al., 2010b; Huang et al., 2012; Reddy et al., 2018b). Reversing hyperacetylation improves motor function and reduces the inflammatory response (Zhang et al., 2008). Furthermore, post-translational histone modifications, such as histone methylation and acetylation, have also strongly implicated in TBI-induced neuropsychiatric disorders (Sagarkar et al., 2017).
Epigenetic HDAC inhibitor treatments, such as valproic acid, sodium butyrate, and suberoylanilide hydroxamic acid (SAHA), have been recently identified as potential disease modifying agents. Valproate has been administered as an anticonvulsant drug for decades, yet its inhibitory effect on HDAC activity was not discovered until 2001 (Gottlicher et al., 2001). Valproate administration reduced neuronal damage, improved cognitive outcomes, and decreased BBB permeability in a CCI model of TBI (Dash et al., 2010b). Although experimental and clinical trials using valproate for PTE have not been effective, some of the observed neuroprotective properties may be linked to this mode of action (Temkin, 2009; Eastman et al., 2010).
Recently, Reddy and team discovered inhibiting HDAC hyperactivity via sodium butyrate treatment retarded the rate of hippocampal kindling in a model of temporal lobe epilepsy (Reddy et al., 2018a). Sodium butyrate treatment also reduced aberrant mossy fiber sprouting in the hilar region of the hippocampus. In addition to blocking a broad spectrum of HDAC enzymes, sodium butyrate is also a known anti-inflammatory agent and shows neuroprotection after stroke (Reddy et al., 2017; Park and Sohrabji, 2016). Stroke is a common cause of epileptogenesis in animal models and humans (Reddy et al., 2017). These data point toward the potential of sodium butyrate as a multifunctional approach to reducing several pathologies associated with epileptogenesis.
In a drug screening study with 870 unique compounds, SAHA was identified as a potential antiseizure drug with selective inhibition of HDAC1 and HDAC3 proteins (Ibhazehiebo et al., 2018). Downstream activity of SAHA results in increased transcription factors crucial to expression of genes needed to induce cell differentiation. Additionally, recent study concluded that SAHA administration after TBI protected against neuronal injury by reducing oxidative stress and inflammation by inducing the inducible nitric oxide synthase/nuclear factor-erythroid factor-2related factor/antioxidant response element (iNOS/Nrf2/ARE) pathway (Xu et al., 2018). Mice treated with SAHA exhibited significantly improved grip test scores and reduced water content in the brain compared with untreated mice. SAHA (Vorinostat) is FDA-approved in patients with cutaneous T cell lymphoma; therefore, validating additional indications in the clinical setting will be a much faster and cost-effective process. Currently, an ongoing phase 2 clinical trial is evaluating the safety and efficacy of SAHA in pediatric patients with drug resistant epilepsy (NCT03894826). Results from this study are forthcoming.
As new biomarkers for PTE are identified, future therapeutic strategies could also include miRNA mimics or antisense oligonucleotides. For example, recent preclinical studies of epilepsy have attempted to control epileptogenesis by regulating expression of miR-146a and found that intranasal delivery of miRNA-146a mimics improved seizure onset and reduced hippocampal damage after pilocarpine administration (Tao et al., 2017). An additional study found similar results in immature rats after an intracerebroventricular injection of miR-146a (Wang et al., 2018). Furthermore, miRNA-146a mimics have been shown to ameliorate injury cascades after TBI (Zhang et al., 2020). Although this method has yet to be studied in PTE directly, studies involving epilepsy or TBI as a singular pathology model have been promising.
There are many studies that highlight disease-modifying effects of test treatments on TBI and comorbidities (Yun Ng and Lee, 2019), but evidence of an antiepileptogenic effect is rare. Furthermore, many studies investigating PTE have done so by measuring seizure threshold to chemical convulsant administration after injury. Although this information is valuable, it does not speak toward the agent’s effect on development of spontaneous seizures and prevalence of long-term epilepsy.
Traditional management of epilepsies has involved the evaluation of the electroclinical phenotype; however, seizures are a symptom of many different causes. Interventions for PTE require the disruption of underlying maladaptive network processes as well as protection against functional impairment. Antiseizure medications are often used for prophylaxis against early TBI-induced seizures but are ineffective at preventing long-term PTE (Wat et al., 2019). Although progress in the field continues to be made, the issue remains as to whether more effective agents will be discovered once the distinct causal processes of PTE are determined and whether these more precise therapeutics will have an improved efficacy and beneficial impact when used earlier in the epileptogenic process.
VII. Conclusions and Future Directions
Head injury is a leading cause of acquired epilepsy. Epileptogenicity that occurs after TBI is a complex chronic network disorder with hyperexcitability and neural connectivity for hypersychronization. PTE research has progressed slowly due to many complex issues. The federal funding agencies, such as the U.S. Department of Defense and National Institutes of Health, have made TBI and PTE research a top priority because of the disease burden in military and civilian people at risk for chronic disabling conditions after brain injuries. TBI is a leading cause of PTE, especially for young adult persons. About 35%–40% of people with PTE have onset within six months; 50% within one year; and nearly 80% within two years of brain injury. The PTE latency is highly variable and may occur even 15 years after head injury. The delay in SRS development presents a significant challenge for clinical and preclinical investigations. However, such latency period represents an exceptional opportunity for therapeutic interventions. Presently, there is renewed emphasis to identify both the cellular and molecular pathways through which seizures are orchestrated by TBI. The clinical prognosis via controlled studies is essential.
Our understanding of epileptogenesis is continuously evolving as animal models are improved to simulate the human condition. FPI and CCI models are the leading experimental models of PTE, and several research groups have implemented continuous EEG recording as the gold standard of epileptiform brain activity. These models are very helpful for further elucidating the mechanisms of epileptogenesis and testing novel therapeutic interventions. There are critical differences in injury patterns, genetic factors, and outcome parameters that need to be considered in the context of model validation for pathophysiology, seizure frequency, and behavior deficits reminiscent of human PTE. Furthermore, a new line of imaging and protein biomarkers is emerging, and advances in machine learning EEG analysis will aid in seizure prediction and patient diagnoses. An evolving interest in this field is finding the genetic basis of differential susceptibility to PTE by identifying factors that may contribute to variable outcomes among populations exposed to the same brain injury, even though only a fraction will later develop seizures. (Chuang and Reddy, 2018a; Reddy et al., 2021).
Treating TBI and preventing PTE is a complex and daunting challenge. Heterogenous injury categories, variances in pathologic responses, differential diagnosis of epileptic seizures versus psychogenic nonepileptic seizures, and difficulty powering both clinical and preclinical trials make this task even more complex. The critical goal of epilepsy research is to identify therapeutics that can prevent, interrupt, or reverse the epileptogenic process (Clossen and Reddy, 2017). Ideally, therapeutic strategies should also relieve PTE-associated comorbidities and thereby help improving the quality-of-life issues, including social and employment opportunities. As discussed in this review, such an intervention has yet to be identified; however, the last decade has provided promising data demonstrating disease modifying, anti-inflammatory, and neuroprotective effects of selected test compounds, suggesting this goal is not unrealistic. Overall, there is greater need to optimize preclinical and clinical research to prevent PTE after TBI. To optimize and achieve this critical goal, there are many challenges and critical gaps in knowledge that need to be addressed, including: (a) recruitment of new teams with multidisciplinary expertise to study TBI/PTE and their comorbidities; (b) optimize preclinical models and markers to reduce replication of efforts and to improve predictive value of experimental models to the clinic; (c) uncover cellular and molecular pathologic signals and network reorganization in brain regions associated with PTE; (d) identify precise latency periods and valid biomarkers for longitudinal monitoring of therapeutic strategies for prevention of PTE; and (e) identify the most promising strategies for preclinical and clinical development of treatments to prevent PTE and its comorbidities. Therefore, future research efforts should be directed toward filling these gaps to open new frontiers in the field of PTE therapeutics. The national research agencies are actively considering special programs and roadmaps to encourage multidisciplinary thematic research as per the PTE research benchmarks. Accelerated collaborative efforts are essential for uncovering key milestones in the pathophysiology and intervention of PTE.
Acknowledgments
The authors are grateful to the Reddy laboratory staff for its technical assistance in experimental studies.
Abbreviations
- ADHD
attention deficit hyperactivity disorder
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ASM
antiseizure medication
- BBB
blood-brain barrier
- BDNF
brain-derived neurotrophic factor
- CA1
Cornu Ammonis area 1
- CA3
Cornu Ammonis area 3
- CCI
controlled cortical impact
- CD
cluster of differentiation
- COX
cyclooxygenase
- CSF
cerebrospinal fluid
- CNS
central nervous system
- CT
computed tomography
- DBS
deep brain stimulation
- 2-DG
2-deoxyglucose
- EEG
electroencephalogram
- FDA
Food and Drug Administration
- FDG
18F-fluorodeoxyglucose
- FPI
fluid percussion injury
- GFAP
glial fibrillary acidic protein
- HDAC
histone deacetylase
- HFO
high-frequency oscillation
- IL
interleukin
- NOS
nitric oxide synthase
- NMDA
N-methyl D-aspartate
- MMP
matrix metallopeptidase
- MRI
magnetic resonance imaging
- miRNA
microRNA
- mTOR
mammalian target of rapamycin
- PBBI
penetrating ballistic-like brain injury
- PET
positron emission tomography
- PTE
post-traumatic epilepsy
- PTZ
pentylenetetrazol
- REM
rapid eye movement
- ROS
reactive oxygen species
- rTBI
repetitive traumatic brain injury
- SRS
spontaneous recurrent seizure
- TBI
traumatic brain injury
- TLR
toll-like receptor
- TrkB
tropomyosin receptor kinase B
- VLPO
ventrolateral preoptic nucleus
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Golub, Reddy.
Footnotes
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the FY2015 Epilepsy Research Program [Grant W81XWH-16-1-0660; Grant EP210006]; the CounterACT Program, Office of the Director, National Institutes of Health National Institute of Neurologic Disorders and Stroke [Grants R21-NS076426, U01-NS083460, R21-NS099009, U01-NS117209, U01-NS117278, R01-NS051398, and R21-NS071597]; and Texas A&M Presidential X-Grant award [X-Grant] to (D.S.R.). Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Department of Defense. The views expressed in this paper are those of the authors and do not reflect the official policy of the National Institutes of Health or the U.S. Government.
No author has an actual or perceived conflict of interest with the contents of this article.
References
- Abbasi B, Goldenholz DM (2019) Machine learning applications in epilepsy. Epilepsia 60:2037–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmalik PA, Boorman DW, Tracy J, Jallo J, Rincon F (2016) Acute traumatic coagulopathy accompanying isolated traumatic brain injury is associated with worse long-term functional and cognitive outcomes. Neurocrit Care 24:361–370. [DOI] [PubMed] [Google Scholar]
- Abdullahi A, Amini-Nik S, Jeschke MG (2014) Animal models in burn research. Cell Mol Life Sci 71:3241–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeyemo BO, Biederman J, Zafonte R, Kagan E, Spencer TJ, Uchida M, Kenworthy T, Spencer AE, Faraone SV (2014) Mild traumatic brain injury and ADHD: a systematic review of the literature and meta-analysis. J Atten Disord 18:576–584. [DOI] [PubMed] [Google Scholar]
- Agoston DV, Shutes-David A, Peskind ER (2017) Biofluid biomarkers of traumatic brain injury. Brain Inj 31:1195–1203. [DOI] [PubMed] [Google Scholar]
- Agoston DV, Vink R, Helmy A, Risling M, Nelson D, Prins M (2019) How to translate time: the temporal aspects of rodent and human pathobiological processes in traumatic brain injury. J Neurotrauma 36:1724–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal A, Timothy J, Pandit L, Manju M (2006) Post-traumatic epilepsy: an overview. Clin Neurol Neurosurg 108:433–439. [DOI] [PubMed] [Google Scholar]
- Ahmed F, Plantman S, Cernak I, Agoston DV (2015) The temporal pattern of changes in serum biomarker levels reveals complex and dynamically changing pathologies after exposure to a single low-intensity blast in mice. Front Neurol 6:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alles JFehlmann TFischer UBackes CGalata VMinet MHart MAbu-Halima MGrässer FALenhof HP, et al. (2019) An estimate of the total number of true human miRNAs. Nucleic Acids Res 47:3353–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alyu F, Dikmen M (2017) Inflammatory aspects of epileptogenesis: contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr 29:1–16. [DOI] [PubMed] [Google Scholar]
- Andrade P, Nissinen J, Pitkänen A (2017) Generalized seizures after experimental traumatic brain injury occur at the transition from slow-wave to rapid eye movement sleep. J Neurotrauma 34:1482–1487. [DOI] [PubMed] [Google Scholar]
- Andrade-Valenca LP, Dubeau F, Mari F, Zelmann R, Gotman J (2011) Interictal scalp fast oscillations as a marker of the seizure onset zone. Neurology 77:524–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, Rocca WA (1998) A population-based study of seizures after traumatic brain injuries. N Engl J Med 338:20–24. [DOI] [PubMed] [Google Scholar]
- Atif H, Hicks SD (2019) A review of MicroRNA biomarkers in traumatic brain injury. J Exp Neurosci 13:1179069519832286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Truettner JS, Lotocki G, Sanchez-Molano J, Kang Y, Alonso OF, Sick TJ, Dietrich WD, Bramlett HM (2010) Post-traumatic seizure susceptibility is attenuated by hypothermia therapy. Eur J Neurosci 32:1912–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsar E, Empson RM (2004) Adenosine acting via A1 receptors, controls the transition to status epilepticus-like behaviour in an in vitro model of epilepsy. Neuropharmacology 47:427–437. [DOI] [PubMed] [Google Scholar]
- Babbage DR, Yim J, Zupan B, Neumann D, Tomita MR, Willer B (2011) Meta-analysis of facial affect recognition difficulties after traumatic brain injury. Neuropsychology 25:277–285. [DOI] [PubMed] [Google Scholar]
- Baik EJ, Kim EJ, Lee SH, Moon C (1999) Cyclooxygenase-2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res 843:118–129. [DOI] [PubMed] [Google Scholar]
- Bailes JE, Dashnaw ML, Petraglia AL, Turner RC (2014) Cumulative effects of repetitive mild traumatic brain injury. Prog Neurol Surg 28:50–62. [DOI] [PubMed] [Google Scholar]
- Bao YH, Bramlett HM, Atkins CM, Truettner JS, Lotocki G, Alonso OF, Dietrich WD (2011) Post-traumatic seizures exacerbate histopathological damage after fluid-percussion brain injury. J Neurotrauma 28:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Klein GLublinsky SKamintsky LNoyman IVeksler RDalipaj HSenatorov VV JrSwissa ERosenbach DElazary N, et al. (2017) Imaging blood-brain barrier dysfunction as a biomarker for epileptogenesis. Brain 140:1692–1705. [DOI] [PubMed] [Google Scholar]
- Barlow DH (2004) Anxiety and Its Disorders: The Nature and Treatment of Anxiety and Panic. 2nd Edition. Guilford Press, New York. [Google Scholar]
- Barnes CA (1979) Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93:74–104. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K (2014) Traumatic brain injury and risk of dementia in older veterans. Neurology 83:312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann CR, Stocker R, Imhof HG, Trentz O, Hersberger M, Mignot E, Bassetti CL (2005) Hypocretin-1 (orexin A) deficiency in acute traumatic brain injury. Neurology 65:147–149. [DOI] [PubMed] [Google Scholar]
- Baumann CR, Werth E, Stocker R, Ludwig S, Bassetti CL (2007) Sleep-wake disturbances 6 months after traumatic brain injury: a prospective study. Brain 130:1873–1883. [DOI] [PubMed] [Google Scholar]
- Benabid AL, Benazzous A, Pollak P (2002) Mechanisms of deep brain stimulation. Mov Disord 17 (Suppl 3):S73–S74. [DOI] [PubMed] [Google Scholar]
- Ben Shimon M, Shavit-Stein E, Altman K, Pick CG, Maggio N (2020) Thrombin as key mediator of seizure development following traumatic brain injury. Front Pharmacol 10:1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky Y, Dryer AM, Saponjian Y, Mahoney MM, Pimentel CA, Lucini CA, Usenovic M, Staley KJ (2013) PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post-traumatic epilepsy. J Neurosci 33:9056–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsneider MHovda DAShalmon EKelly DFVespa PMMartin NAPhelps MEMcArthur DLCaron MJKraus JF, et al. (1997) Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg 86:241–251. [DOI] [PubMed] [Google Scholar]
- Bevins RA, Besheer J (2006) Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc 1:1306–1311. [DOI] [PubMed] [Google Scholar]
- Bhatt D, Hazari A, Yamakawa GR, Salberg S, Sgro M, Shultz SR, Mychasiuk R (2020) Investigating the cumulative effects of Δ9-tetrahydrocannabinol and repetitive mild traumatic brain injury on adolescent rats. Brain Commun 2:fcaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederman JFeinberg LChan JAdeyemo BOWoodworth KYPanis WMcGrath NBhatnagar SSpencer TJUchida M, et al. (2015) Mild traumatic brain injury and attention-deficit hyperactivity disorder in young student athletes. J Nerv Ment Dis 203:813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Steinhäuser C (2006) Functional changes in astroglial cells in epilepsy. Glia 54:358–368. [DOI] [PubMed] [Google Scholar]
- Bissler JJMcCormack FXYoung LRElwing JMChuck GLeonard JMSchmithorst VJLaor TBrody ASBean J, et al. (2008) Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358:140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockaert J, Marin P (2015) Mtor in brain physiology and pathologies. Physiol Rev 95:1157–1187. [DOI] [PubMed] [Google Scholar]
- Bolkvadze T, Pitkänen A (2012) Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J Neurotrauma 29:789–812. [DOI] [PubMed] [Google Scholar]
- Bolkvadze T, Puhakka N, Pitkänen A (2016) Epileptogenesis after traumatic brain injury in Plaur-deficient mice. Epilepsy Behav 60:187–196. [DOI] [PubMed] [Google Scholar]
- Bolkvadze T, Rantala J, Puhakka N, Andrade P, Pitkänen A (2015) Epileptogenesis after traumatic brain injury in Plau-deficient mice. Epilepsy Behav 51:19–27. [DOI] [PubMed] [Google Scholar]
- Bombardier CH, Fann JR, Temkin NR, Esselman PC, Barber J, Dikmen SS (2010) Rates of major depressive disorder and clinical outcomes following traumatic brain injury. JAMA 303:1938–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfield CM, Lam S, Lin Y, Greene S (2013) The impact of attention deficit hyperactivity disorder on recovery from mild traumatic brain injury. J Neurosurg Pediatr 12:97–102. [DOI] [PubMed] [Google Scholar]
- Borovecki FLovrecic LZhou JJeong HThen FRosas HDHersch SMHogarth PBouzou BJensen RV, et al. (2005) Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc Natl Acad Sci USA 102:11023–11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan KR, Bieling PJ, Marriott M, Begin H, Young LT, MacQueen GM (2004) Impact of comorbid anxiety disorders on outcome in a cohort of patients with bipolar disorder. J Clin Psychiatry 65:1106–1113. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Almajano J, Mody I, Engel J Jr (2004) High-frequency oscillations after status epilepticus: epileptogenesis and seizure genesis. Epilepsia 45:1017–1023. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Engel J Jr (2000) Chronic epileptogenesis requires development of a network of pathologically interconnected neuron clusters: a hypothesis. Epilepsia 41 (Suppl 6):S144–S152. [DOI] [PubMed] [Google Scholar]
- Bragin A, Li L, Almajano J, Alvardao-Rojas C, Reid AY, Staba RJ, Engel J (2016) Pathologic electrographic changes after experimental traumatic brain injury. Epilepsia 57:735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan GPBauer SEngel TJimenez-Mateos EMDel Gallo FHill TDMConnolly NMCCostard LSNeubert VSalvetti B, et al. (2020) Genome-wide microRNA profiling of plasma from three different animal models identifies biomarkers of temporal lobe epilepsy. Neurobiol Dis 144:105048. [DOI] [PubMed] [Google Scholar]
- Brundin L, Björkqvist M, Petersén A, Träskman-Bendz L (2007) Reduced orexin levels in the cerebrospinal fluid of suicidal patients with major depressive disorder. Eur Neuropsychopharmacol 17:573–579. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Ingram EA, Wen X (2009) Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci 29:8259–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugay VBozdemir EVigil FAChun SHHolstein DMElliott WRSprague CJCavazos JEZamora DORule G, et al. (2020) A mouse model of repetitive blast traumatic brain injury reveals post-trauma seizures and increased neuronal excitability. J Neurotrauma 37:248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler CR, Boychuk JA, Smith BN (2015) Effects of rapamycin treatment on neurogenesis and synaptic reorganization in the dentate gyrus after controlled cortical impact injury in mice. Front Syst Neurosci 9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler CR, Boychuk JA, Smith BN (2016) Differential effects of rapamycin treatment on tonic and phasic GABAergic inhibition in dentate granule cells after focal brain injury in mice. Exp Neurol 280:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzolari EChepisheva MSmith RMMahmud MHellyer PJTahtis VArshad QJolly AWilson MRust H, et al. (2021) Vestibular agnosia in traumatic brain injury and its link to imbalance. Brain 144:128–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JN, Gandhi A, Singh B, Churn SB (2014) Traumatic brain injury causes a tacrolimus-sensitive increase in non-convulsive seizures in a rat model of post-traumatic epilepsy. Int J Neurol Brain Disord 1:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SW, Madathil SK, Sama DM, Gao X, Chen J, Saatman KE (2014) Conditional overexpression of insulin-like growth factor-1 enhances hippocampal neurogenesis and restores immature neuron dendritic processes after traumatic brain injury. J Neuropathol Exp Neurol 73:734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carron S, Dezsi G, Ozturk E, Nithianantharajah J, Jones NC (2019) Cognitive deficits in a rat model of temporal lobe epilepsy using touchscreen-based translational tools. Epilepsia 60:1650–1660. [DOI] [PubMed] [Google Scholar]
- Carron SF, Sun M, Shultz SR, Rajan R (2020) Inhibitory neuronal changes following a mixed diffuse-focal model of traumatic brain injury. J Comp Neurol 528:175–198. [DOI] [PubMed] [Google Scholar]
- Carver CM, Chuang SH, Reddy DS (2016) Zinc Selectively Blocks Neurosteroid-Sensitive Extrasynaptic δGABAA Receptors in the Hippocampus. J Neurosci 36:8070–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CM, DeWitt HR, Stoja AP, Shapiro MS (2021) Blockade of TRPC channels limits cholinergic-driven hyperexcitability and seizure susceptibility after traumatic brain injury. Front Neurosci 15:681144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Perry G (2019) Tau biology, tauopathy, traumatic brain injury, and diagnostic challenges. J Alzheimers Dis 67:447–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalis F, Feydy A, Valabrègue R, Pélégrini-Issac M, Pierot L, Azouvi P (2006) fMRI study of problem-solving after severe traumatic brain injury. Brain Inj 20:1019–1028. [DOI] [PubMed] [Google Scholar]
- Ceylan MF, Akça ÖF (2013) Secondary attention deficit/hyperactivity disorder due to right basal ganglia injury: a case report. J Exp Clin Med 30:189–191. [Google Scholar]
- Cernak I (2006) Recent advances in neuroprotection for treating traumatic brain injury. Expert Opin Investig Drugs 15:1371–1381. [DOI] [PubMed] [Google Scholar]
- Chauvette S, Soltani S, Seigneur J, Timofeev I (2016) In vivo models of cortical acquired epilepsy. J Neurosci Methods 260:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Ruff RL, Eavey R, Wasterlain CG (2009) Posttraumatic epilepsy and treatment. J Rehabil Res Dev 46:685–696. [DOI] [PubMed] [Google Scholar]
- Chen S, Atkins CM, Liu CL, Alonso OF, Dietrich WD, Hu BR (2007a) Alterations in mammalian target of rapamycin signaling pathways after traumatic brain injury. J Cereb Blood Flow Metab 27:939–949. [DOI] [PubMed] [Google Scholar]
- Chen K, Neu A, Howard AL, Földy C, Echegoyen J, Hilgenberg L, Smith M, Mackie K, Soltesz I (2007b) Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J Neurosci 27:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Reddy DS, Maguire J, Forcelli PA (2020) Sex differences in the epilepsies and associated comorbidities: implications for use and development of pharmacotherapies. Pharmacol Rev 72:767–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzaszcz M, Venkatesan C, Dragisic T, Watterson DM, Wainwright MS (2010) Minozac treatment prevents increased seizure susceptibility in a mouse “two-hit” model of closed skull traumatic brain injury and electroconvulsive shock-induced seizures. J Neurotrauma 27:1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S-H, Reddy DS (2018a) Genetic and molecular regulation of extrasynaptic GABA-A receptors in the brain: therapeutic insights for epilepsy. J Pharmacol Exp Ther 364:180–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S-H, Reddy DS (2018b) 3β-Methyl-neurosteroid analogs are preferential positive allosteric modulators and direct activators of extrasynaptic δGABA-A receptors in the hippocampus dentate gyrus subfield. J Pharmacol Exp Therap 365:583–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S-H, Reddy DS (2019) Zinc reduces antiseizure activity of neurosteroids by selective blockade of extrasynaptic GABA-A receptor-mediated tonic inhibition in the hippocampus. Neuropharmacology 148:244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S-H, Reddy DS (2020) Isobolographic analysis of anticonvulsant activity of GABA-A receptor-modulating synthetic neurosteroids brexanolone and ganaxolone with tiagabine and midazolam. J Pharmacol Exp Therap 372:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clossen BL, Reddy DS (2017) Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim Biophys Acta Mol Basis Dis 1863:1519–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornford EM, Oldendorf WH (1986) Epilepsy and the blood-brain barrier. Adv Neurol 44:787–812. [PubMed] [Google Scholar]
- Croll SD, Goodman JH, Scharfman HE (2004) Vascular endothelial growth factor (VEGF) in seizures: a double-edged sword. Adv Exp Med Biol 548:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C, Heitz E, Allis CD, Borrelli E, Sassone-Corsi P (2003) Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci 116:4905–4914. [DOI] [PubMed] [Google Scholar]
- Cross JH, Lagae L (2020) The concept of disease modification. Eur J Paediatr Neurol 24:43–46. [DOI] [PubMed] [Google Scholar]
- Cullen DK, Harris JP, Browne KD, Wolf JA, Duda JE, Meaney DF, Margulies SS, Smith DH (2016) A porcine model of traumatic brain injury via head rotational acceleration. Methods Mol Biol 1462:289–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curia G, Levitt M, Fender JS, Miller JW, Ojemann J, D’Ambrosio R (2011) Impact of injury location and severity on posttraumatic epilepsy in the rat: role of frontal neocortex. Cereb Cortex 21:1574–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadas A, Janigro D (2019) Breakdown of blood brain barrier as a mechanism of post-traumatic epilepsy. Neurobiol Dis 123:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro R, Ferrara R, Benassi G, Lenzi PL, Sabattini L (1988) Computed tomographic scans in posttraumatic epilepsy. Arch Neurol 45:42–43. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio REastman CLDarvas FFender JSVerley DRFarin FMWilkerson HWTemkin NRMiller JWOjemann J, et al. (2013) Mild passive focal cooling prevents epileptic seizures after head injury in rats. Ann Neurol 73:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL, Miller JW (2004) Post-traumatic epilepsy following fluid percussion injury in the rat. Brain 127:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Fender JS, Fairbanks JP, Simon EA, Born DE, Doyle DL, Miller JW (2005) Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain 128:174–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer SC (2019) Adult neurogenesis in the development of epilepsy. Epilepsy Curr 19:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah SD, Miller MA, Ren D, Hoh NZ, Scanlon JM, Conley YP, Wagner AK (2013) Genetic variability in glutamic acid decarboxylase genes: associations with post-traumatic seizures after severe TBI. Epilepsy Res 103:180–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Mach SA, Moore AN (2001) Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res 63:313–319. [DOI] [PubMed] [Google Scholar]
- Dash PK, Redell JB, Hergenroeder G, Zhao J, Clifton GL, Moore A (2010a) Serum ceruloplasmin and copper are early biomarkers for traumatic brain injury-associated elevated intracranial pressure. J Neurosci Res 88:1719–1726. [DOI] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Moore AN (2009) Histone deacetylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience 163:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Zhang M, Grill RJ, Pati S, Zhao J, Moore AN (2010b) Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS One 5:e11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KA, Sturges BK, Vite CH, Ruedebusch V, Worrell G, Gardner AB, Leyde K, Sheffield WD, Litt B (2011) A novel implanted device to wirelessly record and analyze continuous intracranial canine EEG. Epilepsy Res 96:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGrauw X, Thurman D, Xu L, Kancherla V, DeGrauw T (2018) Epidemiology of traumatic brain injury-associated epilepsy and early use of anti-epilepsy drugs: an analysis of insurance claims data, 2004–2014. Epilepsy Res 146:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Garza R II, Asnis GM (2003) The non-steroidal anti-inflammatory drug diclofenac sodium attenuates IFN-alpha induced alterations to monoamine turnover in prefrontal cortex and hippocampus. Brain Res 977:70–79. [DOI] [PubMed] [Google Scholar]
- De Oliveira CO, Reimer AG, Da Rocha AB, Grivicich I, Schneider RF, Roisenberg I, Regner A, Simon D (2007) Plasma von Willebrand factor levels correlate with clinical outcome of severe traumatic brain injury. J Neurotrauma 24:1331–1338. [DOI] [PubMed] [Google Scholar]
- Derian CK, Santulli RJ, Tomko KA, Haertlein BJ, Andrade-Gordon P (1995) Species differences in platelet responses to thrombin and SFLLRN. receptor-mediated calcium mobilization and aggregation, and regulation by protein kinases. Thromb Res 78:505–519. [DOI] [PubMed] [Google Scholar]
- Deuschl GSchade-Brittinger CKrack PVolkmann JSchäfer HBötzel KDaniels CDeutschländer ADillmann UEisner W, et al. ; German Parkinson Study Group, Neurostimulation Section (2006) A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 355:896–908. [DOI] [PubMed] [Google Scholar]
- Diamond ML, Ritter AC, Failla MD, Boles JA, Conley YP, Kochanek PM, Wagner AK (2015a) IL-1β associations with posttraumatic epilepsy development: A genetics and biomarker cohort study. Epilepsia 56:991–1001. [DOI] [PubMed] [Google Scholar]
- Diamond ML, Ritter AC, Jackson EK, Conley YP, Kochanek PM, Boison D, Wagner AK (2015b) Genetic variation in the adenosine regulatory cycle is associated with posttraumatic epilepsy development. Epilepsia 56:1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein SG, Bannon K, Castellanos FX, Milham MP (2006) The neural correlates of attention deficit hyperactivity disorder: an ALE meta-analysis. J Child Psychol Psychiatry 47:1051–1062. [DOI] [PubMed] [Google Scholar]
- Ding K, Gupta PK, Diaz-Arrastia R (2016) Epilepsy after traumatic brain injury, in Translational Research in Traumatic Brain Injury (Laskowitz D, Grant G eds) CRC Press/Taylor and Francis Group, Boca Raton, FL (Chapter 14). [PubMed] [Google Scholar]
- Dinocourt C, Gallagher SE, Thompson SM (2006) Injury-induced axonal sprouting in the hippocampus is initiated by activation of trkB receptors. Eur J Neurosci 24:1857–1866. [DOI] [PubMed] [Google Scholar]
- Dixon KJ (2017) Pathophysiology of traumatic brain injury. Phys Med Rehabil Clin N Am 28:215–225. [DOI] [PubMed] [Google Scholar]
- Duclos C, Dumont M, Wiseman-Hakes C, Arbour C, Mongrain V, Gaudreault PO, Khoury S, Lavigne G, Desautels A, Gosselin N (2014) Sleep and wake disturbances following traumatic brain injury. Pathol Biol (Paris) 62:252–261. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Spitz M (1997) Hypothetical mechanisms for the cellular and neurophysiologic basis of secondary epileptogenesis: proposed role of synaptic reorganization. J Clin Neurophysiol 14:90–101. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Staley KJ (2012) The time course and circuit mechanisms of acquired epileptogenesis, in Japsper’s Basic Mechanisms of the Epilepsies (Noebels JL, et al., eds) National Center for Biotechnology Information, Bethesda, MD (Chapter 1). [PubMed] [Google Scholar]
- DVBIC (2019) DoD TBI Statistics 2000–2019 Q3, pp 1–5, Defense and Veterans Brain Injury Center, Falls Church, VA. [Google Scholar]
- Eakin K, Rowe RK, Lifshitz J (2015) Modeling fluid percussion injury, in Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects (Kobeissy FH, ed) CRC Press/Taylor & Francis, Boca Raton, FL (Chapter 19). [Google Scholar]
- Eastman CL, Verley DR, Fender JS, Stewart TH, Nov E, Curia G, D’Ambrosio R (2011) Antiepileptic and antiepileptogenic performance of carisbamate after head injury in the rat: blind and randomized studies. J Pharmacol Exp Ther 336:779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman CL, Verley DR, Fender JS, Temkin NR, D’Ambrosio R (2010) ECoG studies of valproate, carbamazepine and halothane in frontal-lobe epilepsy induced by head injury in the rat. Exp Neurol 224:369–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echegoyen J, Armstrong C, Morgan RJ, Soltesz I (2009) Single application of a CB1 receptor antagonist rapidly following head injury prevents long-term hyperexcitability in a rat model. Epilepsy Res 85:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G III, Zhao J, Dash PK, Soto C, Moreno-Gonzalez I (2020) Traumatic brain injury induces tau aggregation and spreading. J Neurotrauma 37:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efendioglu M, Basaran R, Akca M, Ceman D, Demirtas C, Yildirim M (2020) Combination therapy of gabapentin and N-acetylcysteine against posttraumatic epilepsy in rats. Neurochem Res 45:1802–1812. [DOI] [PubMed] [Google Scholar]
- Engel J JrPitkänen ALoeb JADudek FEBertram EH IIICole AJMoshé SLWiebe SJensen FEMody I, et al. (2013) Epilepsy biomarkers. Epilepsia 54 (Suppl 4):61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander J, Bushnik T, Duong TT, Cifu DX, Zafonte R, Wright J, Hughes R, Bergman W (2003) Analyzing risk factors for late posttraumatic seizures: a prospective, multicenter investigation. Arch Phys Med Rehabil 84:365–373. [DOI] [PubMed] [Google Scholar]
- Englander J, Cifu DX, Diaz-Arrastia R (2015) Seizures after traumatic brain injury. Arch Phys Med Rehabil 95:1223–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J (1988) A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behav Brain Res 31:47–59. [DOI] [PubMed] [Google Scholar]
- Enomoto T, Osugi T, Satoh H, McIntosh TK, Nabeshima T (2005) Pre-injury magnesium treatment prevents traumatic brain injury-induced hippocampal ERK activation, neuronal loss, and cognitive dysfunction in the radial-arm maze test. J Neurotrauma 22:783–792. [DOI] [PubMed] [Google Scholar]
- Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R (2007) Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis 26:86–93. [DOI] [PubMed] [Google Scholar]
- Eslami M, Ghanbari E, Sayyah M, Etemadi F, Choopani S, Soleimani M, Amiri Z, Hadjighassem M (2016) Traumatic brain injury accelerates kindling epileptogenesis in rats. Neurol Res 38:269–274. [DOI] [PubMed] [Google Scholar]
- Fabene PF, Bramanti P, Constantin G (2010) The emerging role for chemokines in epilepsy. J Neuroimmunol 224:22–27. [DOI] [PubMed] [Google Scholar]
- Fann JR, Ribe AR, Pedersen HS, Fenger-Grøn M, Christensen J, Benros ME, Vestergaard M (2018) Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry 5:424–431. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z (2005) Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 6:215–229. [DOI] [PubMed] [Google Scholar]
- Fenwick T, Anderson V (1999) Impairments of attention following childhood traumatic brain injury. Child Neuropsychol 5:213–223. [DOI] [PubMed] [Google Scholar]
- Filibian M, Frasca A, Maggioni D, Micotti E, Vezzani A, Ravizza T (2012) In vivo imaging of glia activation using 1H-magnetic resonance spectroscopy to detect putative biomarkers of tissue epileptogenicity. Epilepsia 53:1907–1916. [DOI] [PubMed] [Google Scholar]
- Frankowski JC, Kim YJ, Hunt RF (2019) Selective vulnerability of hippocampal interneurons to graded traumatic brain injury. Neurobiol Dis 129:208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A (2011) Blood-brain barrier dysfunction, status epilepticus, seizures, and epilepsy: a puzzle of a chicken and egg? Epilepsia 52 (Suppl 8):19–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SD, Brooks WM, Jung RE, Chiulli SJ, Sloan JH, Montoya BT, Hart BL, Yeo RA (1999) Quantitative proton MRS predicts outcome after traumatic brain injury. Neurology 52:1384–1391. [DOI] [PubMed] [Google Scholar]
- Friess SH, Ichord RN, Owens K, Ralston J, Rizol R, Overall KL, Smith C, Helfaer MA, Margulies SS (2007) Neurobehavioral functional deficits following closed head injury in the neonatal pig. Exp Neurol 204:234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller PM, Gooley JJ, Saper CB (2006) Neurobiology of the sleep-wake cycle: sleep architecture, circadian regulation, and regulatory feedback. J Biol Rhythms 21:482–493. [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ (2008) Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 28:6904–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff C, Grimault O, Theron M, Pichavant K, Galinat H, Mingant F, Ozier Y (2018) A clinically relevant and bias-controlled murine model to study acute traumatic coagulopathy. Sci Rep 8:5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WM, Chadha MS, Kline AE, Clark RS, Kochanek PM, Dixon CE, Jenkins LW (2006) Immunohistochemical analysis of histone H3 acetylation and methylation—evidence for altered epigenetic signaling following traumatic brain injury in immature rats. Brain Res 1070:31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Deng-Bryant Y, Cho W, Carrico KM, Hall ED, Chen J (2008) Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. J Neurosci Res 86:2258–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Enikolopov G, Chen J (2009) Moderate traumatic brain injury promotes proliferation of quiescent neural progenitors in the adult hippocampus. Exp Neurol 219:516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K (2015) Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol 77:987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner R, La Rocca M, Vespa P, Jones N, Monti MM, Toga AW, Duncan D (2019) Imaging biomarkers of posttraumatic epileptogenesis. Epilepsia 60:2151–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbatin RRSilva LFAHoffmann MSDella-Pace IDdo Nascimento PSKegler Ade Zorzi VNCunha JMBotelho PNeto JBT, et al. (2019) Delayed creatine supplementation counteracts reduction of GABAergic function and protects against seizures susceptibility after traumatic brain injury in rats. Prog Neuropsychopharmacol Biol Psychiatry 92:328–338. [DOI] [PubMed] [Google Scholar]
- Gerring JP, Brady KD, Chen A, Vasa R, Grados M, Bandeen-Roche KJ, Bryan RN, Denckla MB (1998) Premorbid prevalence of ADHD and development of secondary ADHD after closed head injury. J Am Acad Child Adolesc Psychiatry 37:647–654. [DOI] [PubMed] [Google Scholar]
- Ghadiri T, Vakilzadeh G, Hajali V, Khodagholi F (2019) Progesterone modulates post-traumatic epileptogenesis through regulation of BDNF-TrkB signaling and cell survival-related pathways in the rat hippocampus. Neurosci Lett 709:134384. [DOI] [PubMed] [Google Scholar]
- Gilbert KS, Kark SM, Gehrman P, Bogdanova Y (2015) Sleep disturbances, TBI and PTSD: implications for treatment and recovery. Clin Psychol Rev 40:195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R, Chang PK, Prenosil GA, Deane EC, McKinney RA (2013) Blocking brain-derived neurotrophic factor inhibits injury-induced hyperexcitability of hippocampal CA3 neurons. Eur J Neurosci 38:3554–3566. [DOI] [PubMed] [Google Scholar]
- Golarai G, Greenwood AC, Feeney DM, Connor JA (2001) Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci 21:8523–8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub V, Reddy DS (2020a) Chapter 7 - Cannabidiol Therapy for Refractory Epilepsy and Seizure Disorders. In: Cannabinoids and Neuro-psychiatric Disorders, Advances in Experimental Medicine and Biology 1264 (Editors: Monti Jaime M., Murillo-Rodríguez Eric, Pandi-Perumal S.R.), Springer Nature; Switzerland AG, pp 93–110. [DOI] [PubMed] [Google Scholar]
- Golub V, Reddy DS (2020b) Treating neurological diseases through epigenetic inhibition, in Epigenetic Inhibitors and Their Use to Treat Human Diseases (Campeau E, ed) pp 1–17, Wiley & Sons, Hoboken, NJ. [Google Scholar]
- Golub V, Reddy DS (2022) Contusion brain injury in mice for modelling of post-traumatic epilepsy with contralateral hippocampus sclerosis: a comprehensive characterization of seizures, neuropathology, and neurological comorbidities. Exp Neurol 348:113946 DOI: 10.1016/j.expneurol.2021.113946. [DOI] [PubMed] [Google Scholar]
- Gold EM, Su D, Lopez-Velazquez L, Haus DL, Perez H, Lacuesta GA, Anderson AJ, Cummings BJ (2013) Functional assessment of long-term deficits in rodent models of traumatic brain injury. Regen Med 8:483–516. [DOI] [PubMed] [Google Scholar]
- Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A (2013) Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma 30:1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, Iyer A, White I, Colzi A, van Vliet EA, Sisodiya S, Aronica E (2014) Hippocampal subregion-specific microRNA expression during epileptogenesis in experimental temporal lobe epilepsy. Neurobiol Dis 62:508–520. [DOI] [PubMed] [Google Scholar]
- Goshen I, Yirmiya R (2007) The role of pro-inflammatory cytokines in memory processes and neural plasticity, in Psychoneuroimmunology, 4th Edition (Ader R, et al., eds) Elsevier, Inc. Chapter 16; pp 337–378. [Google Scholar]
- Göttlicher MMinucci SZhu PKrämer OHSchimpf AGiavara SSleeman JPLo Coco FNervi CPelicci PG, et al. (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20:6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber KD, Prince DA (1999) Tetrodotoxin prevents posttraumatic epileptogenesis in rats. Ann Neurol 46:234–242. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA(2006) Chronic partial cortical isolation, in Animal Models of Epilepsy (Pitkanen A, Schwarzkroin PA, Moshe S, eds) pp 477–494, Elsevier, New York. [Google Scholar]
- Grovola MR, Paleologos N, Wofford KL, Harris JP, Browne KD, Johnson V, Duda JE, Wolf JA, Cullen DK (2020) Mossy cell hypertrophy and synaptic changes in the hilus following mild diffuse traumatic brain injury in pigs. J Neuroinflammation 17:44 10.1186/s12974-020-1720-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F, Parada I, Yang T, Longo FM, Prince DA (2018) Partial TrkB receptor activation suppresses cortical epileptogenesis through actions on parvalbumin interneurons. Neurobiol Dis 113:45–58. [DOI] [PubMed] [Google Scholar]
- Guo D, Zeng L, Brody DL, Wong M (2013) Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One 8:e64078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Elgammal FS, Proddutur A, Shah S, Santhakumar V (2012) Decrease in tonic inhibition contributes to increase in dentate semilunar granule cell excitability after brain injury. J Neurosci 32:2523–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi F, Ge Y, Chen S, Xin Y, Umali MU, De Gasperi R, Gama Sosa MA, Ahlers ST, Elder GA (2015) Neuronal DNA methylation profiling of blast-related traumatic brain injury. J Neurotrauma 32:1200–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurkoff GG, Giza CC, Shin D, Auvin S, Sankar R, Hovda DA (2009) Acute neuroprotection to pilocarpine-induced seizures is not sustained after traumatic brain injury in the developing rat. Neuroscience 164:862–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Bryant YD, Cho W, Sullivan PG (2008) Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma 25:235–247. [DOI] [PubMed] [Google Scholar]
- Haltiner AM, Temkin NR, Dikmen SS (1997) Risk of seizure recurrence after the first late posttraumatic seizure. Arch Phys Med Rehabil 78:835–840. [DOI] [PubMed] [Google Scholar]
- Hameed MQ, Hsieh TH, Morales-Quezada L, Lee HHC, Damar U, MacMullin PC, Hensch TK, Rotenberg A (2019) Ceftriaxone treatment preserves cortical inhibitory interneuron function via transient salvage of GLT-1 in a rat traumatic brain injury model. Cereb Cortex 29:4506–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm RJ, Pike BR, Temple MD, O’Dell DM, Lyeth BG (1995) The effect of postinjury kindled seizures on cognitive performance of traumatically brain-injured rats. Exp Neurol 136:143–148. [DOI] [PubMed] [Google Scholar]
- Hammond FM, Corrigan JD, Ketchum JM, Malec JF, Dams-O’Connor K, Hart T, Novack TA, Bogner J, Dahdah MN, Whiteneck GG (2019) Prevalence of medical and psychiatric comorbidities following traumatic brain injury. J Head Trauma Rehabil 34:E1–E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond FM, Hart T, Bushnik T, Corrigan JD, Sasser H (2004) Change and predictors of change in communication, cognition, and social function between 1 and 5 years after traumatic brain injury. J Head Trauma Rehabil 19:314–328. [DOI] [PubMed] [Google Scholar]
- Han K, Chapman SB, Krawczyk DC (2015) Altered amygdala connectivity in individuals with chronic traumatic brain injury and comorbid depressive symptoms. Front Neurol 6:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hånell AClausen FBjörk MJansson KPhilipson ONilsson LNGHillered LWeinreb PHLee DMcIntosh TK, et al. (2010) Genetic deletion and pharmacological inhibition of Nogo-66 receptor impairs cognitive outcome after traumatic brain injury in mice. J Neurotrauma 27:1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselkorn MLShellington DKJackson EKVagni VAJanesko-Feldman KDubey RKGillespie DGCheng DBell MJJenkins LW, et al. (2010) Adenosine A1 receptor activation as a brake on the microglial response after experimental traumatic brain injury in mice. J Neurotrauma 27:901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazama A, Ziechmann R, Arul M, Krishnamurthy S, Galgano M, Chin LS (2018) The effect of keppra prophylaxis on the incidence of early onset, post-traumatic brain injury seizures. Cureus 10:e2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggeness MH, Strong N, Wooley PH, Yang SY (2017) Quiescent pluripotent stem cells reside within murine peripheral nerves that can be stimulated to proliferate by recombinant human bone morphogenic protein 2 or by nerve trauma. Spine J 17:252–259. [DOI] [PubMed] [Google Scholar]
- Hesam S, Khoshkholgh-Sima B, Pourbadie HG, Babapour V, Zendedel M, Sayyah M (2018) Monophosphoryl lipid A and Pam3Cys prevent the increase in seizure susceptibility and epileptogenesis in rats undergoing traumatic brain injury. Neurochem Res 43:1978–1985. [DOI] [PubMed] [Google Scholar]
- Hillary FG, Slocomb J, Hills EC, Fitzpatrick NM, Medaglia JD, Wang J, Good DC, Wylie GR (2011) Changes in resting connectivity during recovery from severe traumatic brain injury. Int J Psychophysiol 82:115–123. [DOI] [PubMed] [Google Scholar]
- Hogg MC, Raoof R, El Naggar H, Monsefi N, Delanty N, O’Brien DF, Bauer S, Rosenow F, Henshall DC, Prehn JH (2019) Elevation in plasma tRNA fragments precede seizures in human epilepsy. J Clin Invest 129:2946–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman L, van Vliet EA, Edelbroek PM, Aronica E, Gorter JA (2010) Cox-2 inhibition can lead to adverse effects in a rat model for temporal lobe epilepsy. Epilepsy Res 91:49–56. [DOI] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R (2002) Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci 22:8422–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhao F, Wang L, Yin H, Zhou C, Wang X (2012) Increased expression of histone deacetylases 2 in temporal lobe epilepsy: a study of epileptic patients and rat models. Synapse 66:151–159. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Haselhorst LA, Schoch KM, Bach EC, Rios-Pilier J, Scheff SW, Saatman KE, Smith BN (2012) Posttraumatic mossy fiber sprouting is related to the degree of cortical damage in three mouse strains. Epilepsy Res 99:167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN (2009) Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 215:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN (2010) Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J Neurophysiol 103:1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN (2011) Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J Neurosci 31:6880–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JY, Aromolaran KA, Zukin RS (2017) The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat Rev Neurosci 18:347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibhazehiebo KGavrilovici Cde la Hoz CLMa SCRehak RKaushik GMeza Santoscoy PLScott LNath NKim DY, et al. (2018) A novel metabolism-based phenotypic drug discovery platform in zebrafish uncovers HDACs 1 and 3 as a potential combined anti-seizure drug target. Brain 141:744–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim S, Hu W, Wang X, Gao X, He C, Chen J (2016) Traumatic brain injury causes aberrant migration of adult-born neurons in the hippocampus. Sci Rep 6:21793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immonen R, Harris NG, Wright D, Johnston L, Manninen E, Smith G, Paydar A, Branch C, Grohn O (2019) Imaging biomarkers of epileptogenecity after traumatic brain injury—preclinical frontiers. Neurobiol Dis 123:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immonen R, Kharatishvili I, Gröhn O, Pitkänen A (2013) MRI biomarkers for post-traumatic epileptogenesis. J Neurotrauma 30:1305–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A (2007) TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130:535–547. [DOI] [PubMed] [Google Scholar]
- Jackson ML, Srivastava AK, Cox CS Jr (2017) Preclinical progenitor cell therapy in traumatic brain injury: a meta-analysis. J Surg Res 214:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J, Zijlmans M (2020) HFO to measure seizure propensity and improve prognostication in patients with epilepsy. Epilepsy Curr 20:338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao H, Wang Z, Liu Y, Wang P, Xue Y (2011) Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci 44:130–139. [DOI] [PubMed] [Google Scholar]
- Jirsch JD, Urrestarazu E, LeVan P, Olivier A, Dubeau F, Gotman J (2006) High-frequency oscillations during human focal seizures. Brain 129:1593–1608. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Weber MT, Cullen DK, Siman R, Smith DH (2016) SNTF immunostaining reveals previously undetected axonal pathology in traumatic brain injury. Acta Neuropathol 131:115–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NC, Nguyen T, Corcoran NM, Velakoulis D, Chen T, Grundy R, O’Brien TJ, Hovens CM (2012) Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol Dis 45:897–901. [DOI] [PubMed] [Google Scholar]
- Jorge RE, Robinson RG, Moser D, Tateno A, Crespo-Facorro B, Arndt S (2004) Major depression following traumatic brain injury. Arch Gen Psychiatry 61:42–50. [DOI] [PubMed] [Google Scholar]
- Jupp B, Williams J, Binns D, Hicks RJ, Cardamone L, Jones N, Rees S, O’Brien TJ (2012) Hypometabolism precedes limbic atrophy and spontaneous recurrent seizures in a rat model of TLE. Epilepsia 53:1233–1244. [DOI] [PubMed] [Google Scholar]
- Kaitaro T, Koskinen S, Kaipio ML (1995) Neuropsychological problems in everyday life: a 5-year follow-up study of young severely closed-head-injured patients. Brain Inj 9:713–727. [DOI] [PubMed] [Google Scholar]
- Kamnaksh APuhakka NAli ISmith GAniceto RMcCullough JDas Gupta SNdode-Ekane XEBrady RCasillas-Espinosa P, et al. (2019) Harmonization of pipeline for preclinical multicenter plasma protein and miRNA biomarker discovery in a rat model of post-traumatic epileptogenesis. Epilepsy Res 149:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanbayashi T, Kodama T, Kondo H, Satoh S, Inoue Y, Chiba S, Shimizu T, Nishino S (2009) CSF histamine contents in narcolepsy, idiopathic hypersomnia and obstructive sleep apnea syndrome. Sleep 32:181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karges HE, Funk KA, Ronneberger H (1994) Activity of coagulation and fibrinolysis parameters in animals. Arzneimittelforschung 44:793–797. [PubMed] [Google Scholar]
- Kasahara M, Menon DK, Salmond CH, Outtrim JG, Taylor Tavares JV, Carpenter TA, Pickard JD, Sahakian BJ, Stamatakis EA (2010) Altered functional connectivity in the motor network after traumatic brain injury. Neurology 75:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T, Tanaka H, Yoshida T, Uehara T, Minami M (2009) Neuronal injury induces cytokine-induced neutrophil chemoattractant-1 (CINC-1) production in astrocytes. J Pharmacol Sci 109:88–93. [DOI] [PubMed] [Google Scholar]
- Kelley KA, Ho L, Winger D, Freire-Moar J, Borelli CB, Aisen PS, Pasinetti GM (1999) Potentiation of excitotoxicity in transgenic mice overexpressing neuronal cyclooxygenase-2. Am J Pathol 155:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly KM, Miller ER, Lepsveridze E, Kharlamov EA, Mchedlishvili Z (2015) Posttraumatic seizures and epilepsy in adult rats after controlled cortical impact. Epilepsy Res 117:104–116. [DOI] [PubMed] [Google Scholar]
- Kendirli MT, Rose DT, Bertram EH (2014) A model of posttraumatic epilepsy after penetrating brain injuries: effect of lesion size and metal fragments. Epilepsia 55:1969–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernie SG, Erwin TM, Parada LF (2001) Brain remodeling due to neuronal and astrocytic proliferation after controlled cortical injury in mice. J Neurosci Res 66:317–326. [DOI] [PubMed] [Google Scholar]
- Kharatishvili I, Immonen R, Gröhn O, Pitkänen A (2007) Quantitative diffusion MRI of hippocampus as a surrogate marker for post-traumatic epileptogenesis. Brain 130:3155–3168. [DOI] [PubMed] [Google Scholar]
- Kharatishvili I, Nissinen JP, McIntosh TK, Pitkänen A (2006) A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience 140:685–697. [DOI] [PubMed] [Google Scholar]
- Knutsen LJS, Murray TF (1997) Adeosine and ATP in epilepsy, in Purinergic Approaches in Experimental Therapeutics (Jacobson KA, Jarvis MF, eds) pp 423–447, Wiley-Liss Inc., NY. [Google Scholar]
- Kobau R, Zahran H, Thurman DJ, Zack MM, Henry TR, Schachter SC, Price PH; Centers for Disease Control and Prevention (CDC) (2008) Epilepsy surveillance among adults—19 states, Behavioral Risk Factor Surveillance System, 2005. MMWR Surveill Summ 57:1–20. [PubMed] [Google Scholar]
- Kochanek PM, Hendrich KS, Dixon CE, Schiding JK, Williams DS, Ho C (2002) Cerebral blood flow at one year after controlled cortical impact in rats: assessment by magnetic resonance imaging. J Neurotrauma 19:1029–1037. [DOI] [PubMed] [Google Scholar]
- Kochanek PMVagni VAJanesko KLWashington CBCrumrine PKGarman RHJenkins LWClark RSHomanics GEDixon CE, et al. (2006) Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab 26:565–575. [DOI] [PubMed] [Google Scholar]
- Koenig JB, Cantu D, Low C, Sommer M, Noubary F, Croker D, Whalen M, Kong D, Dulla CG (2019) Glycolytic inhibitor 2-deoxyglucose prevents cortical hyperexcitability after traumatic brain injury. JCI Insight 5:e126506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konduru SS, Wallace EP, Pfammatter JA, Rodrigues PV, Jones MV, Maganti RK (2021) Sleep-wake characteristics in a mouse model of severe traumatic brain injury: relation to posttraumatic epilepsy. Epilepsia Open 6:181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs SK, Leonessa F, Grimes J, Ling GSF (2014) A neuropathology approach to understanding of explosive blast TBI seizure risk. J Neurol Disord Stroke 2:1071. [Google Scholar]
- Kraemer DL, Awad IA (1994) Vascular malformations and epilepsy: clinical considerations and basic mechanisms. Epilepsia 35 (Suppl 6):S30–S43. [DOI] [PubMed] [Google Scholar]
- Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, Mays M, Lopez CM, Kim MO, Franz DN (2013) Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol 74:679–687. [DOI] [PubMed] [Google Scholar]
- Kulkarni SK, Dhir A (2009) Cyclooxygenase in epilepsy: from perception to application. Drugs Today (Barc) 45:135–154. [DOI] [PubMed] [Google Scholar]
- Kumar RG, Breslin KB, Ritter AC, Conley YP, Wagner AK (2019) Variability with astroglial glutamate transport genetics is associated with increased risk for post-traumatic seizures. J Neurotrauma 36:230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyyriäinen J, Bolkvadze T, Koivisto H, Lipponen A, Pérez LO, Ekolle Ndode-Ekane X, Tanila H, Pitkänen A (2019) Deficiency of urokinase-type plasminogen activator and its receptor affects social behavior and increases seizure susceptibility. Epilepsy Res 151:67–74. [DOI] [PubMed] [Google Scholar]
- Lee DJ, Gurkoff GG, Izadi A, Berman RF, Ekstrom AD, Muizelaar JP, Lyeth BG, Shahlaie K (2013) Medial septal nucleus theta frequency deep brain stimulation improves spatial working memory after traumatic brain injury. J Neurotrauma 30:131–139. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC (1998) The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18:8751–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD (2006) Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 281:15763–15773. [DOI] [PubMed] [Google Scholar]
- Li H, Graber KD, Jin S, McDonald W, Barres BA, Prince DA (2012) Gabapentin decreases epileptiform discharges in a chronic model of neocortical trauma. Neurobiol Dis 48:429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lighthall JW (1988) Controlled cortical impact: a new experimental brain injury model. J Neurotrauma 5:1–15. [DOI] [PubMed] [Google Scholar]
- Lin WJ, Harnod T, Lin CL, Kao CH (2019) Mortality risk and risk factors in patients with posttraumatic epilepsy: A population-based cohort study. Int J Environ Res Public Health 16:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlejohn EL, Scott D, Saatman KE (2020) Insulin-like growth factor-1 overexpression increases long-term survival of posttrauma-born hippocampal neurons while inhibiting ectopic migration following traumatic brain injury. Acta Neuropathol Commun 8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Reeves C, Michalak Z, Coppola A, Diehl B, Sisodiya SM, Thom M (2014a) Evidence for mTOR pathway activation in a spectrum of epilepsy-associated pathologies. Acta Neuropathol Commun 2:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Sun T, Liu Z, Chen X, Zhao L, Qu G, Li Q (2014b) Traumatic brain injury dysregulates microRNAs to modulate cell signaling in rat hippocampus. PLoS One 9:e103948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJZheng PWright DKDezsi GBraine ENguyen TCorcoran NMJohnston LAHovens CMMayo JN, et al. (2016) Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 139:1919–1938. [DOI] [PubMed] [Google Scholar]
- Löscher W, Schwartz-Porsche D, Frey HH, Schmidt D (1985) Evaluation of epileptic dogs as an animal model of human epilepsy. Arzneimittelforschung 35:82–87. [PubMed] [Google Scholar]
- Lotrich FE, Albusaysi S, Ferrell RE (2013) Brain-derived neurotrophic factor serum levels and genotype: association with depression during interferon-α treatment. Neuropsychopharmacology 38:985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK (1992) Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci 12:4846–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XC, Hartings JA, Si Y, Balbir A, Cao Y, Tortella FC (2011) Electrocortical pathology in a rat model of penetrating ballistic-like brain injury. J Neurotrauma 28:71–83. [DOI] [PubMed] [Google Scholar]
- Lu XC, Shear DA, Graham PB, Bridson GW, Uttamsingh V, Chen Z, Leung LY, Tortella FC (2015) Dual therapeutic effects of C-10068, a dextromethorphan derivative, against post-traumatic nonconvulsive seizures and neuroinflammation in a rat model of penetrating ballistic-like brain injury. J Neurotrauma 32:1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BPNguyen LTurner RCLogsdon AFChen YWSmith KEHuber JDMatsumoto RRosen CLTucker ES, et al. (2015) Traumatic brain injury and epilepsy: underlying mechanisms leading to seizure. Seizure 33:13–23. [DOI] [PubMed] [Google Scholar]
- Madathil SK, Carlson SW, Brelsfoard JM, Ye P, D’Ercole AJ, Saatman KE (2013) Astrocyte-specific overexpression of insulin-like growth factor-1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in mice. PLoS One 8:e67204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson J, Leonessa F, Ling GS (2012) Neuropathology of explosive blast traumatic brain injury. Curr Neurol Neurosci Rep 12:570–579. [DOI] [PubMed] [Google Scholar]
- Malec JF, Ketchum JM, Hammond FM, Corrigan JD, Dams-O’Connor K, Hart T, Novack T, Dahdah M, Whiteneck GG, Bogner J (2019) Longitudinal effects of medical comorbidities on functional outcome and life satisfaction after traumatic brain injury: an individual growth curve analysis of NIDILRR traumatic brain injury model system data. J Head Trauma Rehabil 34:E24–E35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malec JF, Van Houtven CH, Tanielian T, Atizado A, Dorn MC (2017) Impact of TBI on caregivers of veterans with TBI: burden and interventions. Brain Inj 31:1235–1245. [DOI] [PubMed] [Google Scholar]
- Marchi NAngelov LMasaryk TFazio VGranata THernandez NHallene KDiglaw TFranic LNajm I, et al. (2007) Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 48:732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Granata T, Ghosh C, Janigro D (2012) Blood-brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia 53:1877–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion DW (1999) Management of traumatic brain injury: past, present, and future. Clin Neurosurg 45:184–191. [PubMed] [Google Scholar]
- Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K (1994) A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics. J Neurosurg 80:291–300. [DOI] [PubMed] [Google Scholar]
- Marty S, Berzaghi MdaP, Berninger B (1997) Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends Neurosci 20:198–202. [DOI] [PubMed] [Google Scholar]
- Mazzini L, Cossa FM, Angelino E, Campini R, Pastore I, Monaco F (2003) Posttraumatic epilepsy: neuroradiologic and neuropsychological assessment of long-term outcome. Epilepsia 44: 569–574. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D (2001) On the cellular and network bases of epileptic seizures. Annu Rev Physiol 63:815–846. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL (1989) Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience 28:233–244. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CM, Brosnan CF, Berman JW (1998) Cytokine induction of MIP-1 alpha and MIP-1 beta in human fetal microglia. J Immunol 160:1449–1455. [PubMed] [Google Scholar]
- Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA (1995) Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma 12:689–694. [DOI] [PubMed] [Google Scholar]
- Mez JDaneshvar DHKiernan PTAbdolmohammadi BAlvarez VEHuber BRAlosco MLSolomon TMNowinski CJMcHale L, et al. (2017) Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 318:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Conley Y, Scanlon JM, Ren D, Ilyas Kamboh M, Niyonkuru C, Wagner AK (2010) APOE genetic associations with seizure development after severe traumatic brain injury. Brain Inj 24:1468–1477. [DOI] [PubMed] [Google Scholar]
- Mishra MA, Bai X, Sanganahalli BG, Waxman SG, Shatillo O, Grohn O, Hyder F, Pitkanen, Blumenfeld H (2014) Decreased resting functional connectivity after traumatic brain injury in the rat. PLOS One 9: e105899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miszczuk D, Dębski KJ, Tanila H, Lukasiuk K, Pitkänen A (2016) Traumatic brain injury increases the expression of Nos1, Aβ clearance, and epileptogenesis in APP/PS1 mouse model of Alzheimer’s disease. Mol Neurobiol 53:7010–7027. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Yan E, Bye N (2010) Animal models of traumatic brain injury: is there an optimal model to reproduce human brain injury in the laboratory? Injury 41 (Suppl 1):S10–S13. [DOI] [PubMed] [Google Scholar]
- Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60. [DOI] [PubMed] [Google Scholar]
- Morse AM, Garner DR (2018) Traumatic brain injury, sleep disorders, and psychiatric disorders: an underrecognized relationship. Med Sci (Basel) 6:6010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountney A, Shear DA, Potter B, Marcsisin SR, Sousa J, Melendez V, Tortella FC, Lu XC (2013) Ethosuximide and phenytoin dose-dependently attenuate acute nonconvulsive seizures after traumatic brain injury in rats. J Neurotrauma 30:1973–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Lepsveridze E, Xu H, Kharlamov EA, Lu B, Kelly KM (2010) Increase of GABAA receptor-mediated tonic inhibition in dentate granule cells after traumatic brain injury. Neurobiol Dis 38:464–475. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Arisi GM, Mims K, Hollingsworth G, O'Neil K, Shapiro LA (2020) Neuroinflammatory mechanisms of post-traumatic epilepsy. J Neuroinflammation 17:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Zeitouni S, Cavarsan CF, Shapiro LA (2013) Increased seizure susceptibility in mice 30 days after fluid percussion injury. Front Neurol 4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munyon C, Eakin KC, Sweet JA, Miller JP (2014) Decreased bursting and novel object-specific cell firing in the hippocampus after mild traumatic brain injury. Brain Res 1582:220–226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi B, Sagarkar S, Sakharkar AJ (2018) Epigenetic mechanisms of traumatic brain injuries. Prog Mol Biol Transl Sci 157:263–298. [DOI] [PubMed] [Google Scholar]
- Nakagawa A, Manley GT, Gean AD, Ohtani K, Armonda R, Tsukamoto A, Yamamoto H, Takayama K, Tominaga T (2011) Mechanisms of primary blast-induced traumatic brain injury: insights from shock-wave research. J Neurotrauma 28:1101–1119. [DOI] [PubMed] [Google Scholar]
- Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH (2008) The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol 7:500–506. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM (2008) Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci 28:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes A, Najm IM, Gale JT, Ying Z, Johnson M, Gonzalez-Martinez J (2016) Underlying cortical dysplasia as risk factor for traumatic epilepsy: an animal study. J Neurotrauma 33:1883–1891. [DOI] [PubMed] [Google Scholar]
- Ngwenya LB, Mazumder S, Porter ZR, Minnema A, Oswald DJ, Farhadi HF (2018) Implantation of neuronal stem cells enhances object recognition without increasing neurogenesis after lateral fluid percussion injury in mice. Stem Cells Int 2018:4209821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaeva I, Crowell B, Valenziano J, Meaney D, D’Arcangelo G (2016) Beneficial effects of early mTORC1 inhibition after traumatic brain injury. J Neurotrauma 33:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E (2000) Hypocretin (orexin) deficiency in human narcolepsy. Lancet 355:39–40. [DOI] [PubMed] [Google Scholar]
- Nissinen JAndrade PNatunen THiltunen MMalm TKanninen KSoares JIShatillo OSallinen JNdode-Ekane XE, et al. (2017) Disease-modifying effect of atipamezole in a model of post-traumatic epilepsy. Epilepsy Res 136:18–34. [DOI] [PubMed] [Google Scholar]
- Nita DA, Cissé Y, Timofeev I, Steriade M (2006) Increased propensity to seizures after chronic cortical deafferentation in vivo. J Neurophysiol 95:902–913. [DOI] [PubMed] [Google Scholar]
- Oaklander AL (2011) Neuropathic itch. Semin Cutan Med Surg 30:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osier ND, Dixon CE (2016) The controlled cortical impact model: applications, considerations for researchers, and future directions. Front Neurol 7:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Yan Q, Zhang Y, Fan Z (2017) Moderate injury in motor-sensory cortex causes behavioral deficits accompanied by electrophysiological changes in mice adulthood. PLoS One 12:e0171976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios EM, Sala-Llonch R, Junque C, Roig T, Tormos JM, Bargallo N, Vendrell P (2013) Resting-state functional magnetic resonance imaging activity and connectivity and cognitive outcome in traumatic brain injury. JAMA Neurol 70:845–851. [DOI] [PubMed] [Google Scholar]
- Park MJ, Sohrabji F (2016) The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. J Neuroinflammation 13:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterno R, Folweiler KA, Cohen AS (2017) Pathophysiology and treatment of memory dysfunction after traumatic brain injury. Curr Neurol Neurosci Rep 17:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel YN, Shaikh MF, Shah S, Kumari Y, Othman I (2018) Role of inflammation in epilepsy and neurobehavioral comorbidities: implication for therapy. Eur J Pharmacol 837:145–155. [DOI] [PubMed] [Google Scholar]
- Perek-Polnik M, Jóźwiak S, Jurkiewicz E, Perek D, Kotulska K (2012) Effective everolimus treatment of inoperable, life-threatening subependymal giant cell astrocytoma and intractable epilepsy in a patient with tuberous sclerosis complex. Eur J Paediatr Neurol 16:83–85. [DOI] [PubMed] [Google Scholar]
- Perucca P, Smith G, Santana-Gomez C, Bragin A, Staba R (2019) Electrophysiological biomarkers of epileptogenicity after traumatic brain injury. Neurobiol Dis 123:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacentino M, Beggio G, Zordan L, Bonanni P (2018) Hippocampal deep brain stimulation: persistent seizure control after bilateral extra-cranial electrode fracture. Neurol Sci 39:1431–1435. [DOI] [PubMed] [Google Scholar]
- Piccenna L, Shears G, O’Brien TJ (2017) Management of post-traumatic epilepsy: an evidence review over the last 5 years and future directions. Epilepsia Open 2:123–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijet B, Stefaniuk M, Kostrzewska-Ksiezyk A, Tsilibary PE, Tzinia A, Kaczmarek L (2018) Elevation of MMP-9 levels promotes epileptogenesis after traumatic brain injury. Mol Neurobiol 55:9294–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping X, Jin X (2016a) Chronic posttraumatic epilepsy following neocortical undercut lesion in mice. PLoS One 11:e0158231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping X, Jin X (2016b) Transition from initial hypoactivity to hyperactivity in cortical layer V pyramidal neurons after traumatic brain injury in vivo. J Neurotrauma 33:354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingue V, Mele C, Nardone A (2021) Post-traumatic seizures and antiepileptic therapy as predictors of the functional outcome in patients with traumatic brain injury. Sci Rep 11:4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pischiutta FMicotti EHay JRMarongiu ISammali ETolomeo DVegliante GStocchetti NForloni GDe Simoni MG, et al. (2018) Single severe traumatic brain injury produces progressive pathology with ongoing contralateral white matter damage one year after injury. Exp Neurol 300:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Lukasiuk K, Dudek FE, Staley KJ (2015) Epileptogenesis. Cold Spring Harb Perspect Med 5:a022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Nissinen J, Nairismägi J, Lukasiuk K, Gröhn OHJ, Miettinen R, Kauppinen R (2002) Progression of neuronal damage after status epilepticus and during spontaneous seizures in a rat model of temporal lobe epilepsy. Prog Brain Res 135:67–83. [DOI] [PubMed] [Google Scholar]
- Polascheck N, Bankstahl M, Löscher W (2010) The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp Neurol 224:219–233. [DOI] [PubMed] [Google Scholar]
- Ponsford J (2017) Anxiety and depression following TBI, in Neurobehavioural Disability and Social Handicap Following Traumatic Brain Injury (McMillian TM, Wood RL, eds) p 167, Psychology Press, London. [Google Scholar]
- Ponsford J, Kinsella G (1992) Attentional deficits following closed-head injury. J Clin Exp Neuropsychol 14:822–838. [DOI] [PubMed] [Google Scholar]
- Purcell SMManoach DSDemanuele CCade BEMariani SCox RPanagiotaropoulou GSaxena RPan JQSmoller JW, et al. (2017) Characterizing sleep spindles in 11,630 individuals from the National Sleep Research Resource. Nat Commun 8:15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe A, Schmitz AK, Pernhorst K, Grote A, von der Brelie C, Urbach H, Friedman A, Becker AJ, Elger CE, Niehusmann P (2012) Cliniconeuropathologic correlations show astroglial albumin storage as a common factor in epileptogenic vascular lesions. Epilepsia 53:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raible DJ, Frey LC, Cruz Del Angel Y, Russek SJ, Brooks-Kayal AR (2012) GABA(A) receptor regulation after experimental traumatic brain injury. J Neurotrauma 29:2548–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoof RBauer SEl Naggar HConnolly NMCBrennan GPBrindley EHill TMcArdle HSpain EForster RJ, et al. (2018) Dual-center, dual-platform microRNA profiling identifies potential plasma biomarkers of adult temporal lobe epilepsy. EBioMedicine 38:127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoof RJimenez-Mateos EMBauer STackenberg BRosenow FLang JOnugoren MDHamer HHuchtemann TKörtvélyessy P, et al. (2017) Cerebrospinal fluid microRNAs are potential biomarkers of temporal lobe epilepsy and status epilepticus. Sci Rep 7:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapport RL II, Penry JK (1973) A survey of attitudes toward the pharmacological prophylaxis of posttraumatic epilepsy. J Neurosurg 38:159–166. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Vezzani A (2006) Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience 137:301–308. [DOI] [PubMed] [Google Scholar]
- Reddy DS (1998) Preclinical and clinical behavioral paradigms for testing drugs that affect learning and memory processes. Methods Find Exp Clin Pharmacol 20:249–277. [PubMed] [Google Scholar]
- Reddy DS, Kuruba R (2013) Experimental models of status epilepticus and neuronal injury for evaluation of therapeutic interventions. Int J Mol Sci 14:18284–18318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Estes WA (2016a) Clinical potential of neurosteroids for CNS disorders. Trends Pharmacol Sci 37:543–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Clossen B, Medi H (2016b) Cellular and molecular mechanisms of neuroinflammation in epilepsy. Int J Pharm Sci Nanotech 9:3331–3336. [Google Scholar]
- Reddy DS, Bhimani A, Kuruba R, Park MJ, Sohrabji F (2017) Prospects of modeling poststroke epileptogenesis. J Neurosci Res 95:1000–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy SD, Clossen BL, Reddy DS (2018a) Epigenetic histone deacetylation inhibition prevents the development and persistence of temporal lobe epilepsy. J Pharmacol Exp Ther 364:97–109. [DOI] [PubMed] [Google Scholar]
- Reddy DS, Wu X, Golub V, Dashwood WM, Dashwood RH (2018b). Measuring histone deacetylase inhibition in the brain. Curr Protoc Pharmacol 81:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Chuang S-H, Hunn D, Crepeau AZ, Maganti R (2018c) Neuroendocrine aspects of improving sleep in epilepsy. Epilepsy Res 147:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy SD, Younus I, Sridhar V, Reddy DS (2019) Neuroimaging biomarkers of experimental epileptogenesis and refractory epilepsy. Int J Mol Sci 20:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS (2020) Clinical pharmacology and therapeutics of antiepileptic drugs for epilepsy and seizure disorders. Int J Pharm Sci Nanotechnology 13:5165–5184. [Google Scholar]
- Reddy DS, Thompson W, Calderara G (2020). Does stress trigger seizures? Evidence from experimental models, in Current Topics in Behavioral Neuroscience (Jones N, Kanner A, Damodharan A, eds) pp 1–25, Springer Nature Switzerland AG. [DOI] [PubMed] [Google Scholar]
- Reddy DS, Thompson W, Calderara G (2021) Molecular mechanisms of sex differences in epilepsy and seizure susceptibility in chemical, genetic and acquired epileptogenesis. Neurosci Lett 750:135753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, Thompson W, Calderara G (2021) Comparative evaluation of experimental models of refractory status epilepticus following exposure of cholinergic agents pilocarpine, DFP, and soman. Neuropharmacology 191:108571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redell JB, Moore AN, Ward NH III, Hergenroeder GW, Dash PK (2010) Human traumatic brain injury alters plasma microRNA levels. J Neurotrauma 27:2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Lyeth BG, Phillips LL, Hamm RJ, Povlishock JT (1997) The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res 757:119–132. [DOI] [PubMed] [Google Scholar]
- Reid AY, Bragin A, Giza CC, Staba RJ, Engel J Jr (2016) The progression of electrophysiologic abnormalities during epileptogenesis after experimental traumatic brain injury. Epilepsia 57:1558–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai AR, Sederberg PB, Bogner J, Nielson DM, Zhang J, Mysiw WJ, Knopp MV, Corrigan JD (2016) Improved function after deep brain stimulation for chronic, severe traumatic brain injury. Neurosurgery 79:204–211. [DOI] [PubMed] [Google Scholar]
- Ripley DL, Politzer T (2010) Vision disturbance after TBI. NeuroRehabilitation 27:215–216. [DOI] [PubMed] [Google Scholar]
- Ritter AC, Kammerer CM, Brooks MM, Conley YP, Wagner AK (2016) Genetic variation in neuronal glutamate transport genes and associations with posttraumatic seizure. Epilepsia 57:984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera CLi HThomas-Crusells JLahtinen HViitanen TNanobashvili AKokaia ZAiraksinen MSVoipio JKaila K, et al. (2002) BDNF-induced TrkB activation down-regulates the K+–Cl- cotransporter KCC2 and impairs neuronal Cl-extrusion. J Cell Biol 159:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Sontheimer H (2016) Glia as drivers of abnormal neuronal activity. Nat Neurosci 19:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers KM, Dudek FE, Barth DS (2015) Progressive, seizure-like, spike-wave discharges are common in both injured and uninjured Sprague-dawley rats: implications for the fluid percussion injury model of post-traumatic epilepsy. J Neurosci 35:9194–9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rola R, Mizumatsu S, Otsuka S, Morhardt DR, Noble-Haeusslein LJ, Fishman K, Potts MB, Fike JR (2006) Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp Neurol 202:189–199. [DOI] [PubMed] [Google Scholar]
- Roncon PSoukupovà MBinaschi AFalcicchia CZucchini SFerracin MLangley SRPetretto EJohnson MRMarucci G, et al. (2015) MicroRNA profiles in hippocampal granule cells and plasma of rats with pilocarpine-induced epilepsy—comparison with human epileptic samples. Sci Rep 5:14143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JV, McFarlane AC, Bragge P, Armonda RA, Grimes JB, Ling GS (2013) Blast-related traumatic brain injury. Lancet Neurol 12:882–893. [DOI] [PubMed] [Google Scholar]
- Sagarkar S, Bhamburkar T, Shelkar G, Choudhary A, Kokare DM, Sakharkar AJ (2017) Minimal traumatic brain injury causes persistent changes in DNA methylation at BDNF gene promoters in rat amygdala: a possible role in anxiety-like behaviors. Neurobiol Dis 106:101–109. [DOI] [PubMed] [Google Scholar]
- Salazar AM, Grafman J (2015) Post-traumatic epilepsy: clinical clues to pathogenesis and paths to prevention. Handb Clin Neurol 128:525–538. [DOI] [PubMed] [Google Scholar]
- Sánchez-Elexpuru G, Serratosa JM, Sánchez MP (2017) Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of Lafora disease. Epilepsia 58:467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva AL, Ferreira AP, Silva LF, Hoffmann MS, Dutra FD, Furian AF, Oliveira MS, Fighera MR, Royes LFF (2012) Creatine reduces oxidative stress markers but does not protect against seizure susceptibility after severe traumatic brain injury. Brain Res Bull 87:180–186. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Baldwin SA, Brown RW, Kraemer PJ (1997) Morris water maze deficits in rats following traumatic brain injury: lateral controlled cortical impact. J Neurotrauma 14:615–627. [DOI] [PubMed] [Google Scholar]
- Scher AI, Wu H, Tsao JW, Blom HJ, Feit P, Nevin RL, Schwab KA (2011) MTHFR C677T genotype as a risk factor for epilepsy including post-traumatic epilepsy in a representative military cohort. J Neurotrauma 28:1739–1745. [DOI] [PubMed] [Google Scholar]
- Schwartz JRL, Roth T (2008) Neurophysiology of sleep and wakefulness: basic science and clinical implications. Curr Neuropharmacol 6:367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzkroin PA, Wenzel HJ, Lyeth BG, Poon CC, Delance A, Van KC, Campos L, Nguyen DV (2010) Does ketogenic diet alter seizure sensitivity and cell loss following fluid percussion injury? Epilepsy Res 92:74–84. [DOI] [PubMed] [Google Scholar]
- Shultz SR, Tan XL, Wright DK, Liu SJ, Semple BD, Johnston L, Jones NC, Cook AD, Hamilton JA, O’Brien TJ (2014) Granulocyte-macrophage colony-stimulating factor is neuroprotective in experimental traumatic brain injury. J Neurotrauma 31:976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A (2004) Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci 24:7829–7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, O’Brien TJ, Gimlin K, Wright DK, Kim SE, Casillas-Espinosa PM, Webster KM, Petrou S, Noble-Haeusslein LJ (2017) Interleukin-1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J Neurosci 37:7864–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shandra O, Robel S (2020) Inducing post-traumatic epilepsy in a mouse model of repetitive diffuse traumatic brain injury. J Vis Exp 156:e60360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shandra OWinemiller ARHeithoff BPMunoz-Ballester CGeorge KKBenko MJZuidhoek IABesser MNCurley DEEdwards GF III, et al. (2019) Repetitive diffuse mild traumatic brain injury causes an atypical astrocyte response and spontaneous recurrent seizures. J Neurosci 39:1944–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Laskowitz DT (2012) Biomarkers in traumatic brain injury. Curr Neurol Neurosci Rep 12:560–569. [DOI] [PubMed] [Google Scholar]
- Shay N, Yeates KO, Walz NC, Stancin T, Taylor HG, Beebe DW, Caldwell CT, Krivitzky L, Cassedy A, Wade SL (2014) Sleep problems and their relationship to cognitive and behavioral outcomes in young children with traumatic brain injury. J Neurotrauma 31:1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekleton JA, Parcell DL, Redman JR, Phipps-Nelson J, Ponsford JL, Rajaratnam SM (2010) Sleep disturbance and melatonin levels following traumatic brain injury. Neurology 74:1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SS, Dixon CE, Okonkwo DO, Richardson RM (2014) Neurostimulation for traumatic brain injury. J Neurosurg 121:1219–1231. [DOI] [PubMed] [Google Scholar]
- Shultz SRCardamone LLiu YRHogan REMaccotta LWright DKZheng PKoe AGregoire MCWilliams JP, et al. (2013) Can structural or functional changes following traumatic brain injury in the rat predict epileptic outcome? Epilepsia 54:1240–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sick T, Wasserman J, Bregy A, Sick J, Dietrich WD, Bramlett HM (2017) Increased expression of epileptiform spike/wave discharges one year after mild, moderate, or severe fluid percussion brain injury in rats. J Neurotrauma 34:2467–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LFHoffmann MSGerbatin RdaRFiorin FdaSDobrachinski FMota BCWouters ATBPavarini SPSoares FAAFighera MR, et al. (2013) Treadmill exercise protects against pentylenetetrazol-induced seizures and oxidative stress after traumatic brain injury. J Neurotrauma 30:1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonato MAgoston DVBrooks-Kayal ADulla CFureman BHenshall DCPitkänen ATheodore WHTwyman REKobeissy FH, et al. (2021) Identification of clinically relevant biomarkers of epileptogenesis—a strategic roadmap. Nat Rev Neurol 17:231–242. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen X-H, Nonaka M, Trojanowski JQ, Lee VM-Y, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF (1999) Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol 58:982–992. [DOI] [PubMed] [Google Scholar]
- Smith D, Rau T, Poulsen A, MacWilliams Z, Patterson D, Kelly W, Poulsen D (2018) Convulsive seizures and EEG spikes after lateral fluid-percussion injury in the rat. Epilepsy Res 147:87–94. [DOI] [PubMed] [Google Scholar]
- Smyth K, Sandhu SS, Crawford S, Dewey D, Parboosingh J, Barlow KM (2014) The role of serotonin receptor alleles and environmental stressors in the development of post-concussive symptoms after pediatric mild traumatic brain injury. Dev Med Child Neurol 56:73–77. [DOI] [PubMed] [Google Scholar]
- Sng JC, Taniura H, Yoneda Y (2006) Histone modifications in kainate-induced status epilepticus. Eur J Neurosci 23:1269–1282. [DOI] [PubMed] [Google Scholar]
- Staba RJ (2012) Normal and pathologic high-frequency oscillations, in Jasper’s Basic Mechanisms of the Epilepsies (Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, eds) pp 202–212, Oxford University Press, New York. [Google Scholar]
- Statler KD, Scheerlinck P, Pouliot W, Hamilton M, White HS, Dudek FE (2009) A potential model of pediatric posttraumatic epilepsy. Epilepsy Res 86:221–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statler KD, Swank S, Abildskov T, Bigler ED, White HS (2008) Traumatic brain injury during development reduces minimal clonic seizure thresholds at maturity. Epilepsy Res 80:163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz S, Tipold A, Löscher W (2013) Epilepsy after head injury in dogs: a natural model of posttraumatic epilepsy. Epilepsia 54:580–588. [DOI] [PubMed] [Google Scholar]
- Stevens MC, Lovejoy D, Kim J, Oakes H, Kureshi I, Witt ST (2012) Multiple resting state network functional connectivity abnormalities in mild traumatic brain injury. Brain Imaging Behav 6:293–318. [DOI] [PubMed] [Google Scholar]
- Sun Z, Liu S, Kharlamov EA, Miller ER, Kelly KM (2018) Hippocampal neuropeptide Y protein expression following controlled cortical impact and posttraumatic epilepsy. Epilepsy Behav 87:188–194. [DOI] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L (1989) Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol 26:321–330. [DOI] [PubMed] [Google Scholar]
- Sutula TP, Dudek FE (2007) Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog Brain Res 163:541–563. [DOI] [PubMed] [Google Scholar]
- Szemere E, Jokeit H (2015) Quality of life is social—towards an improvement of social abilities in patients with epilepsy. Seizure 26:12–21. [DOI] [PubMed] [Google Scholar]
- Szu JI, Chaturvedi S, Patel DD, Binder DK (2020) Aquaporin-4 dysreguilation in a controlled cortical impact injury model of posttraumatic epilepsy. Neuroscience 428:140–153. [DOI] [PubMed] [Google Scholar]
- Tagge CAFisher AMMinaeva OVGaudreau-Balderrama AMoncaster JAZhang XLWojnarowicz MWCasey NLu HKokiko-Cochran ON, et al. (2018) Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 141:422–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Ge Y, Sodickson DK, Miles L, Zhou Y, Reaume J, Grossman RI (2011) Thalamic resting-state functional networks: disruption in patients with mild traumatic brain injury. Radiology 260:831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao HZhao JLiu TCai YZhou XXing HWang YYin MZhong WLiu Z, et al. (2017) Intranasal delivery of miRNA-146a mimics delayed seizure onset in the lithium-pilocarpine mouse model. Mediators Inflamm 2017:6512620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CA, Bell JM, Breiding MJ, Xu L (2017) Traumatic brain injury-related emergency department visits, hospitalizations, and deaths—United States, 2007 and 2013. MMWR Surveill Summ 66:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR (2009) Preventing and treating posttraumatic seizures: the human experience. Epilepsia 50 (Suppl 2):10–13. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Jarell AD, Anderson GD (2001) Antiepileptogenic agents: how close are we? Drugs 61:1045–1055. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O’Dell TJ (1998) Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci 18:7118–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrane ASRappold PMFujita TTorres ABekar LKTakano TPeng WWang FRangroo Thrane VEnger R, et al. (2011) Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci USA 108:846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Sejnowski TJ, Bazhenov M, Chauvette S, Grand LB (2013) Age dependency of trauma-induced neocortical epileptogenesis. Front Cell Neurosci 7:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin RP, Mukherjee S, Kain JM, Rogers SK, Henderson SK, Motal HL, Newell Rogers MK, Shapiro LA (2014) Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol Commun 2:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins O, Feintuch A, Benifla M, Cohen A, Friedman A, Shelef I (2011) Blood-brain barrier breakdown following traumatic brain injury: a possible role in posttraumatic epilepsy. Cardiovasc Psychiatry Neurol 2011:765923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkins O, Shelef I, Kaizerman I, Eliushin A, Afawi Z, Misk A, Gidon M, Cohen A, Zumsteg D, Friedman A (2008) Blood-brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry 79:774–777. [DOI] [PubMed] [Google Scholar]
- Topolnik L, Steriade M, Timofeev I (2003a) Hyperexcitability of intact neurons underlies acute development of trauma-related electrographic seizures in cats in vivo. Eur J Neurosci 18:486–496. [DOI] [PubMed] [Google Scholar]
- Topolnik L, Steriade M, Timofeev I (2003b) Partial cortical deafferentation promotes development of paroxysmal activity. Cereb Cortex 13:883–893. [DOI] [PubMed] [Google Scholar]
- Tropeano MP, Spaggiari R, Ileyassoff H, Park KB, Kolias AG, Hutchinson PJ, Servadei F (2019) A comparison of publication to TBI burden ratio of low- and middle-income countries versus high-income countries: how can we improve worldwide care of TBI? Neurosurg Focus 47:E5. [DOI] [PubMed] [Google Scholar]
- Tubi MALutkenhoff EBlanco MBMcArthur DVillablanca PEllingson BDiaz-Arrastia RVan Ness PReal CShrestha V, et al. (2019) Early seizures and temporal lobe trauma predict post-traumatic epilepsy: A longitudinal study. Neurobiol Dis 123:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulyanova AVKoch PFCottone CGrovola MRAdam CDBrowne KDWeber MTWeber MTRusso RJGagnon KG, et al. (2018) Electrophysiological signature reveals laminar structure of the porcine hippocampus. eNeuro 5:0102–0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat-Azouvi C, Weber T, Legrand L, Azouvi P (2007) Working memory after severe traumatic brain injury. J Int Neuropsychol Soc 13:770–780. [DOI] [PubMed] [Google Scholar]
- van der Naalt J (2001) Prediction of outcome in mild to moderate head injury: a review. J Clin Exp Neuropsychol 23:837–851. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, da Costa Araújo S, Redeker S, van Schaik R, Aronica E, Gorter JA (2007) Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130:521–534. [DOI] [PubMed] [Google Scholar]
- van Vliet EAPuhakka NMills JDSrivastava PKJohnson MRRoncon PDas Gupta SKarttunen JSimonato MLukasiuk K, et al. (2017) Standardization procedure for plasma biomarker analysis in rat models of epileptogenesis: focus on circulating microRNAs. Epilepsia 58:2013–2024. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Friedman A, Dingledine RJ (2013) The role of inflammation in epileptogenesis. Neuropharmacology 69:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T (2011) IL-1 receptor/toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun 25:1281–1289. [DOI] [PubMed] [Google Scholar]
- Vigil FABozdemir EBugay VChun SHHobbs MSanchez IHastings SDVeraza RJHolstein DMSprague SM, et al. (2020) Prevention of brain damage after traumatic brain injury by pharmacological enhancement of KCNQ (Kv7, “M-type”) K+ currents in neurons. J Cereb Blood Flow Metab 40:1256–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasana LE, Kim KN, Westbrook GL, Schnell E. (2015) Functional integration of adult-born hippocampal neurons after traumatic brain injury. eNeuro 2:ENEURO.0056-15.2015 0056-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola-Saltzman M, Watson NF (2012) Traumatic brain injury and sleep disorders. Neurol Clin 30:1299–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Achermann P, Borbély AA, Tobler I (2004) The dynamics of spindles and EEG slow-wave activity in NREM sleep in mice. Arch Ital Biol 142:511–523. [PubMed] [Google Scholar]
- Wagner AK, Miller MA, Scanlon J, Ren D, Kochanek PM, Conley YP (2010) Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res 90:259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang FWang XShapiro LACotrina MLLiu WWang EWGu SWang WHe XNedergaard M, et al. (2017) NKCC1 up-regulation contributes to early post-traumatic seizures and increased post-traumatic seizure susceptibility. Brain Struct Funct 222:1543–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gao X, Michalski S, Zhao S, Chen J (2016a) Traumatic brain injury severity affects neurogenesis in adult mouse hippocampus. J Neurotrauma 33:721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Seekaew P, Gao X, Chen J (2016b) Traumatic brain injury stimulates neural stem cell proliferation via mammalian target of rapamycin signaling pathway activation. eNeuro 3:ENEURO.0162-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhang C, Liu C, Yang HF, Hu WH, Zhang JG, Zhang K (2016c) Endogenous cannabinoid system alterations and their role in epileptogenesis after brain injury in rat. Epilepsy Res 128:35–42. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhang C, Liu C, Zhao B, Wei N, Zhang JG, Zhang K (2016d. c,) CB1 receptor antagonism prevents long-term hyperexcitability after head injury by regulation of dynorphin-KOR system and mGluR5 in rat hippocampus. Brain Res 1646:174–181. [DOI] [PubMed] [Google Scholar]
- Wang X, Yin F, Li L, Kong H, You B, Zhang W, Chen S, Peng J (2018) Intracerebroventricular injection of miR-146a relieves seizures in an immature rat model of lithium-pilocarpine induced status epilepticus. Epilepsy Res 139:14–19. [DOI] [PubMed] [Google Scholar]
- Wat RMammi MParedes JHaines JAlasmari MLiew ALu VMArnaout OSmith TRGormley WB, et al. (2019) The effectiveness of antiepileptic medications as prophylaxis of early seizure in patients with traumatic brain injury compared with placebo or no treatment: a systematic review and meta-analysis. World Neurosurg 122:433–440. [DOI] [PubMed] [Google Scholar]
- Watanabe J, Shetty AK, Hattiangady B, Kim DK, Foraker JE, Nishida H, Prockop DJ (2013) Administration of TSG-6 improves memory after traumatic brain injury in mice. Neurobiol Dis 59:86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber W (2010) Cancer epigenetics. Prog Mol Biol Transl Sci 95:299–349. [DOI] [PubMed] [Google Scholar]
- Webster KM, Shultz SR, Ozturk E, Dill LK, Sun M, Casillas-Espinosa P, Jones NC, Crack PJ, O’Brien TJ, Semple BD (2019) Targeting high-mobility group box protein 1 (HMGB1) in pediatric traumatic brain injury: Chronic neuroinflammatory, behavioral, and epileptogenic consequences. Exp Neurol 320:112979. [DOI] [PubMed] [Google Scholar]
- Wei H, Duan G, He J, Meng Q, Liu Y, Chen W, Meng Y (2018) Geniposide attenuates epilepsy symptoms in a mouse model through the PI3K/Akt/GSK-3β signaling pathway. Exp Ther Med 15:1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, Lu XC, Yang X, Tortella FC (2010) Intracranial pressure following penetrating ballistic-like brain injury in rats. J Neurotrauma 27:1635–1641. [DOI] [PubMed] [Google Scholar]
- Wilder RM (1921) High fat diets in epilepsy. May Clin Bull 2:308. [Google Scholar]
- Williams AJ, Hartings JA, Lu XC, Rolli ML, Dave JR, Tortella FC (2005) Characterization of a new rat model of penetrating ballistic brain injury. J Neurotrauma 22:313–331. [DOI] [PubMed] [Google Scholar]
- Witkin JMLi GGolani LKXiong WSmith JLPing XRashid FJahan RCerne RCook JM, et al. (2020) The positive allosteric modulator of α2/3-containing GABAA receptors, KRM-II-81, is active in pharmaco-resistant models of epilepsy and reduces hyperexcitability after traumatic brain injury. J Pharmacol Exp Ther 372:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JMSmith JLPing XGleason SDPoe MMLi GJin XHobbs JSchkeryantz JMMcDermott JS, et al. (2018) Bioisosteres of ethyl 8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo [1,5-a][1,4]diazepine-3-carboxylate (HZ-166) as novel alpha 2,3 selective potentiators of GABAA receptors: Improved bioavailability enhances anticonvulsant efficacy. Neuropharmacology 137:332–343. [DOI] [PubMed] [Google Scholar]
- Wolf JAJohnson BNJohnson VEPutt MEBrowne KDMietus CJBrown DPWofford KLSmith DHGrady MS, et al. (2017) Concussion induces hippocampal circuitry disruption in swine. J Neurotrauma 34:2303–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock T, Morganti-Kossmann MC (2013) The role of markers of inflammation in traumatic brain injury. Front Neurol 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2002) Genomics and World Health: Report of the Advisory Committee on Health Research, WHO, Geneva. [Google Scholar]
- Wu Z, Wang ZH, Liu X, Zhang Z, Gu X, Yu SP, Keene CD, Cheng L, Ye K (2020) Traumatic brain injury triggers APP and Tau cleavage by delta-secretase, mediating Alzheimer’s disease pathology. Prog Neurobiol 185:101730. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M (2013) Animal models of traumatic brain injury. Nat Rev Neurosci 14:128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shi J, Zhang J, Zhang Y (2018) Vorinostat: a histone deacetylases (HDAC) inhibitor ameliorates traumatic brain injury by inducing iNOS/Nrf2/ARE pathway. Folia Neuropathol 56:179–186. [DOI] [PubMed] [Google Scholar]
- Yang L, Afroz S, Michelson HB, Goodman JH, Valsamis HA, Ling DS (2010) Spontaneous epileptiform activity in rat neocortex after controlled cortical impact injury. J Neurotrauma 27:1541–1548. [DOI] [PubMed] [Google Scholar]
- Yarlagadda A, Alfson E, Clayton AH (2009) The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry (Edgmont) 6:18–22. [PMC free article] [PubMed] [Google Scholar]
- Yen TL, Chang CC, Chung CL, Ko WC, Yang CH, Hsieh CY (2018) Neuroprotective effects of platonin, a therapeutic immunomodulating medicine, on traumatic brain injury in mice after controlled cortical impact. Int J Mol Sci 19:1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younus I, Reddy DS (2017) Epigenetic interventions for epileptogenesis: a new frontier for curing epilepsy. Pharmacol Ther 177:108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T-S, Zhang G, Liebl DJ, Kernie SG (2008) Traumatic brain injury-induced hippocampal neurogenesis requires activation of early nestin-expressing progenitors. J Neurosci 28:12901–12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Ng S, Lee AYW (2019) Traumatic brain injuries: pathophysiology and potential therapeutic targets. Front Cell Neurosci 13:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L-H, Rensing NR, Wong M (2009) The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci 29:6964–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, West EJ, Van KC, Gurkoff GG, Zhou J, Zhang XM, Kozikowski AP, Lyeth BG (2008) HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res 1226:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhao L, Zhu W, Ding Y, Chen H, Chi N (2020) miR-146a mimics ameliorates traumatic brain injury involving JNK and NF-κB signaling pathway. Neuromolecular Med 22:484–492. [DOI] [PubMed] [Google Scholar]
- Zhang R, Sun L, Hayashi Y, Liu X, Koyama S, Wu Z, Nakanishi H (2010) Acute p38-mediated inhibition of NMDA-induced outward currents in hippocampal CA1 neurons by interleukin-1beta. Neurobiol Dis 38:68–77. [DOI] [PubMed] [Google Scholar]
- Zhang ZY, Zhang Z, Fauser U, Schluesener HJ (2007) Global hypomethylation defines a sub-population of reactive microglia/macrophages in experimental traumatic brain injury. Neurosci Lett 429:1–6. [DOI] [PubMed] [Google Scholar]
- Zhu B, Eom J, Hunt RF (2019) Transplanted interneurons improve memory precision after traumatic brain injury. Nat Commun 10:5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Wang L, Zhang Y, Zhao FH, Luo J, Xiao Z, Chen GJ, Wang XF (2012) Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy. J Mol Neurosci 46:420–426. [DOI] [PubMed] [Google Scholar]
- Ziino C, Ponsford J (2005) Measurement and prediction of subjective fatigue following traumatic brain injury. J Int Neuropsychol Soc 11:416–425. [DOI] [PubMed] [Google Scholar]
- Zijlmans M, Jiruska P, Zelmann R, Leijten FS, Jefferys JG, Gotman J (2012) High-frequency oscillations as a new biomarker in epilepsy. Ann Neurol 71:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann LL, Martin RM, Girgis F (2017) Treatment options for posttraumatic epilepsy. Curr Opin Neurol 30:580–586. [DOI] [PubMed] [Google Scholar]