

In this study, Hu et al. investigated the specific functions of the two main PPARγ isoforms by generating mouse lines in which endogenous PPARγ1 and PPARγ2 were epitope-tagged to interrogate isoform-specific genomic binding, and mice deficient in either PPARγ1 or PPARγ2 to assess isoform-specific gene regulation. They show that PPARγ isoforms have specific and separable metabolic functions that may be targeted to improve therapy for insulin resistance and diabetes.
Keywords: PPARγ, adipocyte, diabetes, gene regulation, nuclear receptor, obesity
Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear receptor that is a vital regulator of adipogenesis, insulin sensitivity, and lipid metabolism. Activation of PPARγ by antidiabetic thiazolidinediones (TZD) reverses insulin resistance but also leads to weight gain that limits the use of these drugs. There are two main PPARγ isoforms, but the specific functions of each are not established. Here we generated mouse lines in which endogenous PPARγ1 and PPARγ2 were epitope-tagged to interrogate isoform-specific genomic binding, and mice deficient in either PPARγ1 or PPARγ2 to assess isoform-specific gene regulation. Strikingly, although PPARγ1 and PPARγ2 contain identical DNA binding domains, we uncovered isoform-specific genomic binding sites in addition to shared sites. Moreover, PPARγ1 and PPARγ2 regulated a different set of genes in adipose tissue depots, suggesting distinct roles in adipocyte biology. Indeed, mice with selective deficiency of PPARγ1 maintained body temperature better than wild-type or PPARγ2-deficient mice. Most remarkably, although TZD treatment improved glucose tolerance in mice lacking either PPARγ1 or PPARγ2, the PPARγ1-deficient mice were protected from TZD-induced body weight gain compared with PPARγ2-deficient mice. Thus, PPARγ isoforms have specific and separable metabolic functions that may be targeted to improve therapy for insulin resistance and diabetes.
The obesity epidemic is a major public health issue, as it is highly associated with type 2 diabetes (T2D), cardiovascular diseases, and other metabolic syndromes (Caballero 2007). Adipose tissue, a nutrient-storing and fuel-burning organ, is increased in obesity and likely plays a role in T2D progression (Rosen and Spiegelman 2006; Iozzo 2009). The nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) is required for adipocyte differentiation and metabolic functions (Chawla and Lazar 1994; Tontonoz et al. 1994). Whole-body deficiency of PPARγ causes embryonic lethality, whereas mice lacking a single PPARγ allele were protected from the development of insulin resistance in the setting of diet-induced obesity (DIO) (Kubota et al. 1999), suggesting a dose-dependent effect of PPARγ in adipose metabolism. Adipocyte-specific PPARγ knockout mice exhibit lipoatrophy and severe insulin resistance (He et al. 2003; Wang et al. 2013), while activation of PPARγ in adipocytes improves insulin sensitivity (Sugii et al. 2009). Correspondingly, in humans, dominant-negative mutations of PPARγ cause lipodystrophy and insulin resistance (Barroso et al. 1999). Other rare variants impair adipocyte differentiation and predispose individuals to T2D (Majithia et al. 2014), while a common coding region variant (P12A) improves adiposity, plasma lipids, and insulin sensitivity (Altshuler et al. 2000; Majithia et al. 2016).
There are two main PPARγ isoforms, γ1 and γ2, derived from separate transcriptional start sites. γ1 and γ2 are identical except for an additional 30 amino acids at the N terminus of PPARγ2 (Werman et al. 1997; Ricote et al. 1998). Both γ1 and γ2 are predominantly expressed in adipocytes, with PPARγ1 also expressed at low levels in other tissues, such as macrophages, liver, brain, and muscle (Vidal-Puig et al. 1996). Besides the critical role of PPARγ in adipose tissue, several studies have shown that knockout of PPARγ in other nonadipose tissues, such as macrophages (Odegaard et al. 2007), brain (Lu et al. 2011), muscle (Hevener et al. 2003; Norris et al. 2003), liver (Matsusue et al. 2003), and T cells (Cipolletta et al. 2012), impaired the glucose homeostasis or the response to TZDs. However, the isoform-specific regulation and function of γ1 and γ2 remain unclear. It was shown that γ1 and γ2 respond differently to developmental signal and nutrition signals (Vidal-Puig et al. 1996; Secco et al. 2017; Soccio et al. 2017), and γ1 expression level is negatively correlated with adiposity in human subcutaneous and visceral fat, while γ2 expression level positively correlates with adiposity in human fat mass (Li et al. 2016). The deficiency of γ2 in mice caused controversial metabolic phenotypes (Zhang et al. 2004; Medina-Gomez et al. 2005, 2007), largely because of the different genetic backgrounds of the mice used. However, there is no prior model of γ1-deficient mice, which are very challenging to generate, to study its isoform-specific function.
PPARγ is related to nuclear receptors for hormones and metabolites (Chawla et al. 2001) and is activated by fatty acids, although a dominant endogenous ligand has remained elusive (Tontonoz and Spiegelman 2008). However, PPARγ binds with high affinity to TZD drugs that can effectively reverse the insulin resistance central to the pathophysiology of T2D (Lehmann et al. 1995; Soccio et al. 2014). There is strong evidence that TZDs function via PPARγ to enhance insulin action. TZDs activate the PPARγ/RXR heterodimer by recruiting coactivators to the promoters/enhancers of PPARγ target genes (Step et al. 2014). Moreover, deletion of PPARγ abrogates the ability of TZDs to regulate gene expression (Nelson et al. 2018).
The relative abundance of PPARγ in adipocytes suggests that adipose tissue is the major site of action of TZDs. TZDs promote insulin sensitivity by enhancing fat storage in adipocytes, which serves to reduce lipotoxicity in other metabolic organs, as well as altering adipokine expression and release (Soccio et al. 2014). Indeed, TZDs are ineffective at lowering blood glucose in mice with severe lipodystrophy (Fiorenza et al. 2011; Soccio et al. 2014), although there is evidence for a role for PPARγ in nonadipocytes, such as T cells, macrophages, muscle, and brain in response to TZDs (Hevener et al. 2003; Odegaard et al. 2007; Lu et al. 2011; Cipolletta et al. 2012). TZDs also have notable side effects that limit their clinic use, including weight gain, edema, and bone loss (Soccio et al. 2014; Hu et al. 2019). At present, the mechanisms by which TZDs uniquely promote insulin sensitivity and cause the adverse metabolic effects remain uncertain, and determining specific functions of PPARγ isoforms could provide important clues leading to more targeted approaches.
Here, we generated unique mouse models to dissect the isoform-specific functions of γ1 and γ2. Although γ1 and γ2 contain identical DNA binding domains, we identified numerous isoform-specific PPARγ genomic binding sites in addition to shared binding sites. Moreover, γ1 and γ2 regulated a different set of genes in adipose tissue depots. In brown adipose tissue (BAT), mice with selective deficiency of γ1 exhibited increased thermogenic gene expression and maintained body temperature better than wild-type or γ2-deficient mice. Most strikingly, TZDs retained their antidiabetic effects in either γ1- or γ2-deficient mice, but the γ1-deficient mice were uniquely protected from TZD-induced body weight gain. These data demonstrate PPARγ isoform-specific molecular and physiological functions that discriminate between salutary and adverse effects of TZD drugs.
Results
Generation and validation of PPARγ1 and PPARγ2 endogenous epitope-tagged mice
To date, due to the lack of reagents and mouse models, it is not known whether the two PPARγ protein isoforms, PPARγ1 (γ1) and PPARγ2 (γ2), have distinct and separable functions. Commercial γ2-specific antibodies are of poor affinity and specificity, and it has not been possible to generate a γ1-specific antibody since all of γ1 is contained in γ2 (Zhu et al. 1995). To test the hypothesis that γ1 and γ2 are functionally distinct and demonstrate isoform-specific roles in adipocytes, we used CRISPR–Cas9 technology to generate strains of mice with knock-in of an epitope tag to either γ1 or γ2. Specifically, we inserted a 6xHis-HA tag into the N-terminal of the γ2 locus of C57Bl/6J mice as well as a tag containing three copies of the HA sequence (3xHA) at the PPARγ1-b exon of the PPARγ locus in C57Bl6/J mice (Fig. 1A). The PPARγ1-b exon is normally untranslated (Pap et al. 2016), so we introduced a Kozak sequence and AUG start site “ATG” to force its translation from the γ1-b exon (Kozak 1989). The epitope-tagged γ1 and γ2 proteins were specifically detected by Western blotting of epididymal adipose tissue from 12-wk-old male mice at levels similar to the endogenous proteins (Fig. 1B,C). mRNA expression levels of Pparγ1 and Pparγ2 showed little difference between epitope-tagged mice and their littermate controls except for modestly higher Pparγ2 level in HHA-PPARγ2 mice (Supplemental Fig. S1A,B).
Figure 1.

Generation and validation of PPARγ1 and PPARγ2 epitope-tagged mice and knockout mice. (A) Schematic representation of the insertion of the 3xHA and His-HA (HHA) epitope tag in PPARγ1 and PPARγ2 loci, respectively. (B–E) Western blot of PPARγ1 and PPARγ2 using endogenous PPARγ antibody or HA antibody in iWAT of 3HA-PPARγ1 (B), HHA-PPARγ2 (C), PPARγ1 KO (D), and PPARγ2 KO (E) mice. n = 3. (F,G) Western blot of PPARγ1 and PPARγ2 across many tissues in 3HA-PPARγ1 (F) and HHA-PPARγ2 (G) mice. The bottom blot for each panel is from HA immunoprecipitated lysates. (H,I) Representative images of HA staining of iWAT from 3HA-PPARγ1 (H) and HHA-PPARγ2 (I) mice. Scale bar, 100 μm. (J) Representative images of coimmunostaining of HA with perilipin, PDGFRα, or CD31 from iWAT of 3HA-PPARγ1 and HHA-PPARγ2 mice. Scale bar, 50 μm. (K) The percentage of HA-positive cells in perilipin-labeled mature adipocytes, PDGFRα-labeled preadipocytes, or CD31-labeled endothelial cells. n = 3. Three fields per mice.
Generation and validation of selective PPARγ1- and PPARγ2-deficient mice
While generating the epitope tag knock-ins, we found some gene-edited mouse lines with premature stop codons near the gRNA targeted region, suggesting these mice could be specific knockouts (KOs) for γ1 and γ2. Indeed, γ2 protein was not detected in one mouse strain that we refer to as γ2 KO mice (Fig. 1E). Pparγ2 mRNA was increased in γ2 KO mice, possibly due to a compensatory effect (Supplemental Fig. S1D). Interestingly, while γ1 mRNA was undetectable in the γ1 KO mice (Supplemental Fig. S1C), we noted a small amount of γ1 protein (Fig. 1D), which we suspect is derived from internal translation of γ2 mRNA and could explain why γ2 mRNA is more abundant than the protein (Soccio et al. 2017). Nevertheless, by densitometry, the γ1 KO expressed only ∼25% of the normal amount of endogenous γ1, with no changes at γ2 mRNA and protein levels. Previous studies have demonstrated the expression of γ1 in adipose tissue, macrophage, brain, liver, and muscle, whereas γ2 expression is highly restricted to adipose depots (Vidal-Puig et al. 1996), which we confirmed by RT-qPCR in numerous mouse tissues (Supplemental Fig. S1E–G). However, the expression patterns of γ1 and γ2 across multiple mouse tissues have been difficult to discern by Western blot with a commercial antibody (Fig. 1F,G). In contrast, Western blotting of tissues from the epitope-tagged mouse models using the HA antibody clearly demonstrated the adipose-restricted expression of γ2 as well as the nonadipose expression of γ1 (Fig. 1F,G). Moreover, we did not observe significant changes of γ1 and γ2 in response to various temperatures (Supplemental Fig. S1H,I), although PPARγ is critical for thermogenesis (Lasar et al. 2018). We further explored the protein location of γ1 and γ2 in inguinal white adipose tissue (iWAT). As expected, both γ1 and γ2 were located in the nucleus. Strikingly, the data also revealed that γ1 is expressed in the majority of cells in iWAT, while γ2 is expressed in considerably fewer cells (Fig. 1H,I). Moreover, γ1 was dominantly expressed in perilipin-labeled mature adipocytes and PDGFRα-labeled preadipocytes, in comparison with the expression of γ2 (Fig. 1J,K). In addition, γ1 was modestly expressed in CD31-labeled endothelial cells, but we did not detect any expression of γ2 in endothelial cells (Fig. 1J,K), consistent with its adipocyte-specific expression. These data validated the epitope knock-in mice as a tool to probe for isoform-specific functions of PPARγ.
PPARγ1 and PPARγ2 bind to both common and isoform-specific genomic binding sites
We performed HA-ChIP-seq in iWAT, epididymal WAT (eWAT), and BAT from 12-wk-old male mice. PPARγ binding sites on the genome in each mouse strain were highly reproducible across three to four biological replicates (Supplemental Fig. S2A–F). Importantly, these sites overlapped well with antibody-based PPARγ cistromes from mouse adipose depots (Fig. 2A; Soccio et al. 2015). Of the γ1 and γ2 binding sites, 33.4% and 42.4%, respectively, contained the canonical PPAR motif and were also enriched for the motifs of classical PPARγ cooperation factors, CEBPA and NFI (Fig. 2B,C; Supplemental Fig. S2G; Soccio et al. 2015; Hiraike et al. 2017). The majority of γ1 and γ2 binding sites were shared in all three adipose depots (Fig. 2D–F; Supplemental Fig. S2H), and the genes near these common PPARγ binding sites were enriched for PPAR signaling and fatty acid metabolic pathways, as would be expected (Supplemental Fig. S3A–C). We also detected hundreds to thousands of binding sites preferred by γ1 or γ2, suggesting isoform-specific genomic functions (Fig. 2G–I; Supplemental Fig. S3D,E). KEGG analysis revealed that the γ1-specific peaks were enriched for pathways that include the PPAR signaling pathway and regulation of lipolysis in adipocytes, while γ2-specific peaks were enriched for the cAMP signaling pathway and vascular smooth muscle contraction pathway (Fig. 2J,K).
Figure 2.

PPARγ1 and PPARγ2 exhibit isoform-specific genomic occupancy. (A) Venn diagram showing the overlap of the cistromes of HA-PPARγ1 and HA-PPARγ2 with previous endogenous PPARγ cistromes in iWAT and eWAT. (B,C) Top motifs enriched in PPARγ1 (B) and PPARγ2 (C) binding sites using HOMER de novo motif analysis. (D–F) Scatter plots showing the isoform-specific PPARγ binding sites in iWAT (D), eWAT (E), and BAT (F). Fold change > 2 and P value < 0.05 (Student's t-test) were used for identifying isoform-specific binding sites. (G–I) Heat map showing PPARγ1- or PPARγ2-selective sites in three or four biological replicates from iWAT (G), eWAT (H), and BAT (I). (J,K). KEGG analysis for the nearest genes of PPARγ1-specific (J) or PPARγ2-specific (K) sites in iWAT.
PPARγ1-specific genomic binding is associated with ETS factor GABPα
Given their identical DNA binding domain (DBD), we hypothesized that other transcription factors may mediate the isoform-specific genomic binding of PPARγ, potentially due to positive or negative effects of the additional N-terminal amino acids in γ2 (Suzuki et al. 2010). De novo motif analysis revealed that γ1-specific binding sites from all three adipose depots were enriched for the ETS motif (Fig. 3A–C), while the common PPARγ binding sites were enriched classical motifs for PPARγ, CEBP, and NFI (Supplemental Fig. S4A–C). Intriguingly, ETS family member GABPα is highly expressed in iWAT (Supplemental Fig. S4D) and was recently shown to promote formation of a subset of beige adipocytes (Chen et al. 2019). To test whether GABPα was indeed cobound selectively with γ1, we performed GABPα ChIP-seq in iWAT. As expected, the ETS motif was the top enriched motif in the 8776 GABPα peaks identified (Fig. 3D). Remarkably, 731 of the γ1-specific binding peaks were shared with GABPα (Fig. 3E,G), whereas we observed almost no overlap between GABPα binding sites and γ2-specific sites (Fig. 3F). These data suggest a role for GABPα in mediating γ1-specific functions in adipose tissue.
Figure 3.

GABPα contributes to PPARγ1-specific genomic binding. (A–C) De novo motif analysis reveals the top enriched motifs for PPARγ1- or PPARγ2-specific sites in iWAT (A), eWAT (B), and BAT (C). (D) Top hit from HOMER de novo motif search at all GABPα binding sites. (E,F) Venn diagram showing the overlap of the PPARγ1-specific (E) and PPARγ2-specific (F) binding sites with GABPα binding sites in iWAT. (G) The percentage of GABPα binding sites shared with PPARγ1- and PPARγ2-specific binding sites.
PPARγ1 and PPARγ2 regulate differential sets of genes in WAT
To determine the transcriptomic functions of γ1 and γ2 in adipose, we performed RNA-seq in γ1 KO and γ2 KO mice, and compared them with their littermate controls using three to five biological replicates for each adipose depot. This analysis revealed that γ1 and γ2 regulate distinct groups of genes in eWAT, iWAT, and BAT. While many genes were specifically regulated by γ1 or γ2 in iWAT (Fig. 4A,B), relatively few genes were affected by loss of either γ1 or γ2 (Fig. 4C), suggesting that regulation of these genes, many bound by both isoforms, was redundant, as is common for transcription factors (Mechta-Grigoriou et al. 2001; Kuntz et al. 2012). Intriguingly, gene ontology analysis implicated the down-regulated genes in γ1 KO mice in lipid metabolic and fat cell differentiation pathways, while the PPAR signaling pathway and glucose metabolism pathway were enriched in the down-regulated genes in γ2 KO mice (Fig. 4D,E). Similar findings were observed in eWAT, in which γ1 and γ2 also regulated a different set of genes (Supplemental Fig. S5A–C) that played different roles in adipose metabolism (Supplemental Fig. S5D,E).
Figure 4.

PPARγ1 and PPARγ2 regulate differential set of genes in adipose tissue. (A,B) Heat map of the genes differentially expressed in PPARγ1 KO (A) and PPARγ2 KO (B) mice in iWAT. Three biological replicates; DE cutoff: |FC| > 1.5, FDR < 0.01. (C) Venn diagram showing the comparison of the PPARγ1- and PPARγ2-regulated genes in iWAT. (D,E) Gene ontology analysis of genes differentially expressed in PPARγ1 KO (D) and PPARγ2 KO (E) mice. (F,G) Heat map of the genes differentially expressed in PPARγ1 KO (F) and PPARγ2 KO (G) mice in BAT. Three biological replicates; DE cutoff: |FC| > 1.5, FDR < 0.01. (H) Venn diagram showing the comparison of the PPARγ1- and PPARγ2-regulated genes in BAT. (I,J) GSEA showing the enrichment of cold-induced thermogenesis pathways for PPARγ1-regulated genes (I), but not for PPARγ2-regulated genes (J). Genes were ranked by average fold change in KO versus WT. (K) Heat map of the cold-induced thermogenesis genes in PPARγ1 KO and PPARγ2 KO mice in BAT. The color bar indicates log2(fold change) in KO versus WT. (L) mRNA expression of Ucp1 in PPARγ1 KO and PPARγ2 KO mice in BAT, normalized to Arbp; WT was set to 1, as measured by qRT-PCR. Data are expressed as mean ± SEM. (*) P < 0.05; Student's t-test. n = 5–6 per group.
PPARγ1 and PPARγ2 differentially regulate gene expression in thermogenic BAT
In BAT, a thermogenic tissue, deficiency of γ1 or γ2 also affected the expression levels of hundreds of genes (Fig. 4F–H). Interestingly, the cold-induced thermogenesis pathway was specifically up-regulated in BAT of γ1 KO mice, but not γ2 KO mice (Fig. 4I,J). In contrast, other PPARγ-regulated gene programs, such as adipose tissue development and inflammatory response, were similarly regulated in γ1 KO and γ2 KO mice (Supplemental Fig. S5F,G). For example, Ucp1, Elovl3, and Dio2 mRNAs were highly up-regulated in γ1 KO but not significantly altered (FC > 1.5, FDR < 0.01) in γ2 KO BAT (Fig. 4K). Up-regulation of Ucp1 was validated in a different cohort of mice using RT-qPCR (Fig. 4L). The isoform-specific gene regulation was highly associated with isoform-specific genomic binding, suggesting a direct mechanism of the differential functions of γ1 and γ2 (Supplemental Fig. S6A,B). For example, the Slc1a1 gene was specifically down-regulated in γ1 KO BAT but unchanged in γ2 KO BAT. Consistent with this, γ1 but not γ2 binding was detected at the Slc1a1 locus (Supplemental Fig. S6C). In contrast, greater γ2 binding was noted at the Slc6a13 locus, and this was associated with γ2-specific regulation of the Slc6a13 gene (Supplemental Fig. S6D). Brown adipocyte determination factor EBF2 regulates thermogenic genes through cooperation with PPARγ (Rajakumari et al. 2013), and a higher percentage of γ1-specific binding peaks was located near EBF2 binding sites relative to γ2-specific binding peaks (Supplemental Fig. S6E). Of note, the loss of either γ1 or γ2 was associated with both up-regulation and down-regulation of nearby gene expression in adipose tissue. The dependence of gene expression on PPARγ is expected based on its role as a master regulator of adipogenic gene expression (Lefterova et al. 2014). However it is also known that PPARγ can repress basal gene expression on some genes by recruiting the nuclear receptor corepressor (NCoR) complex (Guan et al. 2005). Indeed, both NCoR and its stoichiometric partner histone deacetylase 3 (HDAC3) were bound more strongly at γ1-specific sites near genes that were up-regulated in the γ1 KO than near down-regulated genes (Supplemental Fig. S6F,G), consistent with basal repression of these specific genes. Furthermore, genes basally repressed genes by γ1 that had nearby NCoR and/or HDAC3 binding sites were up-regulated with TZD treatment (Supplemental Fig. S6H). The lower number of γ2-specific sites precluded a similar analysis for that isoform.
Distinct thermogenic functions of PPARγ1 and PPARγ2 in organismal metabolism
PPARγ plays an important role in the development and function of BAT (Nedergaard et al. 2005), and the differential genomic binding and gene regulation suggested that γ1 and γ2 may play distinct roles. To interrogate the functions of γ1 and γ2 in thermogenesis, isoform-deficient and control mice were subjected to cold temperature (4°C) challenge after acclimatization to thermoneutrality for 2 wk. Remarkably, γ1 KO mice maintained their body temperatures better than control littermates (Fig. 5A,B), whereas this was not the case for γ2-deficient mice (Fig. 5D). We then determined the requirement for each isoform in regulating BAT thermogenic capacity by measuring norepinephrine (NE)-induced whole-body oxygen consumption in anaesthetized mice. Twelve-week-old male γ1 KO mice exhibited a more rapid and robust increase in oxygen consumption following NE treatment compared with that observed in control littermates (Fig. 5C), whereas γ2 KO mice showed no significant difference (Fig. 5E). Consistent with this, upon exposure to cold temperature (4°C) for 5 d, the browning of iWAT was greater in γ1 KO mice than in their littermates, but such was not the case for γ2 KO mice (Supplemental Fig. S7A,B). These data suggest that the γ1 isoform normally restricts cold tolerance, consistent with the up-regulation of thermogenic gene expression observed in BAT of mice lacking γ1.
Figure 5.
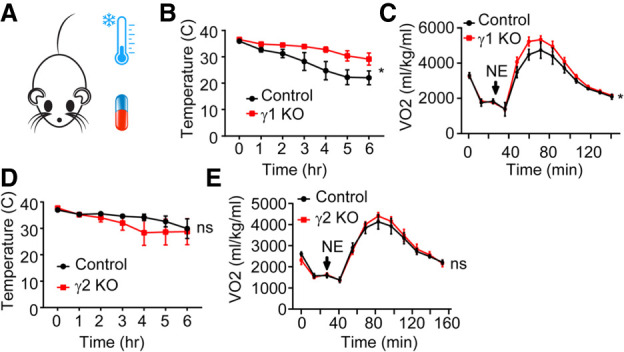
Distinct function of PPARγ1 and PPARγ2 on thermogenesis. (A) Scheme of experiment design of mice receiving norepinephrine or acute cold challenge. (B,D) Effect of acute cold exposure from housing at 29°C–4°C on PPARγ1 KO (B) and PPARγ2 KO (D) mice versus control littermates. n = 6–9 per group. (C,E) Oxygen consumption rates of anaesthetized PPARγ1 KO (C) and PPARγ2 KO (E) mice versus control littermates after injection of 1 mg/kg (body weight) norepinephrine (NE). n = 4–5 per group. Data are expressed as mean ± SEM. (*) P < 0.05, (ns) not significant; two-way ANOVA.
Deficiency of PPARγ1 or PPARγ2 protects against diet-induced obesity
PPARγ is required for adipocyte differentiation (Rosen et al. 1999), and mutations in PPARγ cause lipodystrophy in mice and humans (Barroso et al. 1999; Auclair et al. 2013). As adults, γ1 KO mice had normal glucose tolerance, body weights, and fat pad weights compared with their littermate controls under normal chow diets (NCDs) (Supplemental Fig. S7C–E). Similarly, we also did not observe significant changes of glucose tolerance, body weights, and fat pad weights in γ2 KO mice under normal chow diets (Supplemental Fig. S7F–H). These data are consistent with the GSEA indicating no enrichment of the adipose tissue development pathway for genes specifically regulated by either γ1 or γ2, likely because this is a fundamental and redundant property of both PPARγ isoforms. Moreover, the energy expenditure, respiratory exchange ratio, and physical activity were not affected in γ1 KO or γ2 KO mice (Supplemental Fig. S8A–L). We also did not detect significant changes in food intake in these mice under NCDs. However, when the mice were fed an obesogenic high-fat diet (HFD) for 12 wk, weight gain was lessened in both the γ1 KO and γ2-deficient mice (Fig. 6A,D). The γ2 KO was apparently effective in alleviating obesity, as their fat pads weighed less than those of their littermate controls, whereas no significant differences were observed between γ1 KO mice and control mice (Fig. 6B,E). These data suggest that PPARγ1 and PPARγ2 deletion protects against HFD-induced obesity. Both γ1 KO and γ2 KO mice showed improved glucose tolerance (Fig. 6C,F), which was not surprising given the reduced weight gain, as well as a previous study showing that mice with mutation of one PPARγ allele also exhibited increased glucose tolerance (Kubota et al. 1999).
Figure 6.

Metabolic phenotypes of PPARγ1 KO and PPARγ2 KO mice under HFD treatment. (A,D) Body weight gain of PPARγ1 KO (A) and PPARγ2 KO (D) mice and their control littermates on HFD. n = 5–13 per group. (B,E) iWAT, eWAT, and BAT weights from PPARγ1 KO (B) and PPARγ2 KO (E) mice and their control littermates after 12 wk of HFD. n = 5–13 per group. (C,F) Intraperitoneal glucose tolerance test of PPARγ1 KO (C) and PPARγ2 KO (F) mice and their control littermates under 12-wk HFD treatment. n = 5–13 per group. Data are expressed as mean ± SEM. (*) P < 0.05, (**) P < 0.01, (ns) not significant; Student's t-test.
Antidiabetic TZD rosiglitazone has different effects on PPARγ1 KO and PPARγ2 KO mice
TZDs such as rosiglitazone (rosi) function via PPARγ to enhance insulin sensitivity but lead to the adverse effect of weight gain (Soccio et al. 2014). To ascertain whether γ1 and γ2 play differential roles in mediating the actions of TZDs, male γ1 and γ2 KO mice were given a HFD for 10 wk starting at 8 wk of age, with the HFD continued with or without rosi (36 mg/kg diet) for an additional 6 wk (Fig. 7A). Rosi improved glucose tolerance in γ1 KO mice (Fig. 7B), suggesting that γ2 can mediate the antidiabetic effects of rosi. Rosi was even more effective in γ1 KO mice than in control littermates (Fig. 7C). Rosi also improved glucose tolerance in γ2 KO mice (Fig. 7I), suggesting some redundancy of γ1 and γ2 in mediating this therapeutic effect, although the effect was similar to that in wild-type littermates (Fig. 7J), suggesting that the γ1 KO (and hence endogenous γ2) may be a more powerful mediator of the antidiabetic effect of rosi. Rosi also causes adverse metabolic effects that reduce its clinical utility, especially weight gain (Soccio et al. 2014). Strikingly, γ1 KO mice were protected from body weight gain after rosi treatment (Fig. 7D,E) whereas γ2 KO mice gained amounts of weight similar to control mice (Fig. 7K,L). In accordance with this, fat pads of γ1 KO mice weighed less than those of control mice after rosi treatment (Fig. 7F–H), but were similar between γ2 KO mice and control mice (Fig. 7M–O). Glycerol kinase, a PPARγ target gene that controls BAT inducibility (Guan et al. 2002; Lasar et al. 2018), was induced by rosi in both γ1 KO and γ2 KO mice (Supplemental Fig. S8M,N). Together, these data suggest that γ1 and γ2 are largely redundant for the therapeutic effect of rosi. However, rosi-induced weight gain was likely attributable to γ1, since it was ameliorated in the γ1-deficient mice.
Figure 7.

Differential response of PPARγ1 KO and PPARγ2 KO mice to antidiabetic drug treatment. (A) Scheme of experiment design of HFD and rosiglitazone treatment. (B,I) Intraperitoneal glucose tolerance test of PPARγ1 KO (B) and PPARγ2 KO (I) mice after 6 wk of rosiglitazone treatment or vehicle treatment. n = 5 per group. (C,J) Intraperitoneal glucose tolerance test of PPARγ1 KO (C) and PPARγ2 KO (J) mice and their control littermates after 6 wk of rosiglitazone treatment. n = 5–7 per group. (D,K) Body weights from PPARγ1 KO (D) and PPARγ2 KO (K) mice and their control littermates after 6 wk of rosiglitazone treatment. n = 5–7 per group. (E,L) Body weight gain of PPARγ1 KO (E) and PPARγ2 KO (L) mice and their control littermates after 6 wk of rosiglitazone treatment. n = 5–7 per group. (F–H) iWAT (F), eWAT (G), and BAT (H) weights from PPARγ1 KO mice and their control littermates after 6 wk of rosiglitazone treatment. n = 5–6 per group. (M–O) iWAT (M), eWAT (N), and BAT (O) weights from PPARγ2 KO mice and their control littermates after 6 wk of rosiglitazone treatment. n = 5–7 per group. Data are expressed as mean ± SEM. (*) P < 0.05, (ns) not significant; Student's t-test for D–H and K–O. (*) P < 0.05, (**) P < 0.01, (****) P < 0.0001; two-way ANOVA for B, C, I, and J.
Discussion
PPARγ1 and PPARγ2 differ in tissue distribution and gene expression, and their expression levels change differently in response to HFD, fasting, and developmental signals. Here, using novel knock-in and knockout mouse models, including what we believe to be the first γ1-specific-deficient mouse, we have identified isoform-specific roles of γ1 and γ2 in adipose gene expression, metabolism, and the response to antidiabetic TZD drugs.
We found that, in addition to binding at a majority of common sites, γ1 and γ2 each bound uniquely at hundreds of specific genomic binding sites. De novo motif analysis revealed enrichment of the ETS motif at γ1-specific binding sites in three adipose depots, suggesting that an ETS factor might cooperate with γ1 but not γ2 to drive binding at specific sites despite the identical DNA binding domains of γ1 and γ2. A previous study suggested that an ETS factor can modulate DNA binding activity of another nuclear receptor, androgen receptor, through direct interaction (Wasmuth et al. 2020). Here, we observed that the highly expressed ETS factor in iWAT, GABPα, bound at γ1-specific binding sites but not at γ2-specific sites. Given that γ2 contains all the amino acids in γ1, these results suggest that the additional 30 amino acids at the unique N terminus of γ2 play an inhibitory role in the cooperation and cobinding with GABPα, although the precise nature of this effect remains to be discovered. Nevertheless, these findings demonstrate that a specific GABPα can bind near a subset of PPARγ sites in an isoform-specific way. Intriguingly, GABPα has been shown to play a key role in glycolytic beige adipocyte differentiation (Chen et al. 2019). Further studies will also need to examine the role of Sox family members, which are also enriched in γ1-specific binding sites.
Since PPARγ1 and PPARγ2 are largely identical, it is not surprising that the majority of their binding sites are identical, and thus the lack of one but not both isoforms has little effect on the majority of genes that are PPARγ targets. For example, the classical PPARγ target genes (Lefterova et al. 2014) Fabp4 and Adipoq, which have similar γ1 and γ2 genomic binding, were not altered in γ1 KO or γ2 KO adipose tissues due to the redundant functions of γ1 and γ2 in the regulation of these genes. In contrast, a small number of genes, including thermogenic genes, were differentially regulated in γ1 KO and γ2 KO mice. The underlying mechanism may involve the role of distinct cooperating factors, such as GABPα for γ1, but this will require further experimentation. In addition, the γ1 protein is expressed in nonadipocyte cells within adipose tissue, whereas the γ2 protein is restricted to adipocytes, which may also contribute to the differential gene regulation.
Of note, our study demonstrates that the deficiency of either PPARγ1 or PPARγ2 may cause genes to be either up-regulated or down-regulated. Genes that are up-regulated are thus normally repressed by that specific isoform, presumably by interaction with nuclear receptor corepressor complexes known to be recruited by unliganded PPARγ in adipocytes (Guan et al. 2005). Down-regulation of gene expression upon deletion of PPARγ implies that the gene was normally activated, which could be due to the binding of an endogenous ligand to PPARγ, or basal activation through the N terminus, which can function as a ligand-independent activation domain in a subset of nuclear receptors (Mangelsdorf et al. 1995). In this regard, the N terminus of the γ2 isoform has been reported to be a stronger activator domain (Bugge et al. 2009). Consistent with this, we found that a higher percentage of γ2-responsive genes was down-regulated in γ2 KO eWAT and BAT compared with the percentage of genes that was down-regulated in the γ1 KO mice.
Deficiency of PPARγ1 and γ2 did not cause lipoatrophy and glucose intolerance in mice fed normal chow. However, after 12 wk of HFD, both mouse strains were protected from diet-induced obesity and improved glucose tolerance. This is similar to previous observations of heterozygous PPARγ KO mice (Kubota et al. 1999), suggesting a dose-dependent effect of PPARγ in adipose metabolism. More interestingly, γ1 KO mice were protected from rosi-induced body weight gain while retaining the therapeutic effect of rosi on glucose tolerance. In contrast, rosi treatment still increased the body weight of γ2 KO mice. Although γ1 KO mice maintained their body temperature better upon an acute cold challenge and had higher energy expenditure after norepinephrine treatment, no significant differences in food intake or energy expenditure were noted between γ1- and γ2-deficient mice on a HFD. Thus, the mechanism underlying the difference in TZD-induced weight gain related to the absence of γ1 versus γ2 will require further study. It has been reported that PPARγ in preadipocytes regulated metabolic homeostasis independent of weight changes through visceral WAT remodeling (Shao et al. 2018). In our models, deletion of γ1 and γ2 in the germline affects not only mature adipocytes but also preadipocytes, endothelial cells, macrophages, and other cell types, which could also contribute to the effects on body weight in the context of HFD and rosi treatment. Moreover, isoform-specific functions of γ1 and γ2 in nonadipose tissues such as brain, liver, and muscle should be elucidated in the future. Nevertheless, these data suggest that the γ1 isoform is not necessarily for insulin sensitization yet is largely responsible for rosi-induced weight gain, pointing to the γ2 isoform as a more specific and safer target for future therapies.
Materials and methods
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania. Mice were housed in a temperature-controlled specific pathogen-free facility under 12-h light/dark cycles, with free access to water and standard chow (LabDiet 5010) or high-fat diet composed of 60:20:20 kcal percentage of fat/carbohydrate/protein (Research Diets D12492i) starting at 8 wk old. Unless otherwise specified in the figure legends, all experiments were carried out on 12- to16-wk-old male mice. 3HA-PPARγ1, HHA-PPARγ2, PPARγ1 KO, and PPARγ2 KO mice were generated as described below and maintained on a C57BL/6J genetic background. For drug treatment, rosiglitazone (Selleckchem) was incorporated into the diets by Research Diets at 36 mg/kg of diet, such that a 30-g mouse eating 3 g of diet per day received a rosiglitazone dose of 3.6 mg/kg/d. PPARγ1 KO and PPARγ2 KO mice and their littermates were fed a HFD for 10 wk and the rosiglitazone-containing HFD or the control HFD for the final 6 wk.
Generation of HA epitope-tagged PPARγ and PPARγ1/2 KO mice
To generate Cas9 mRNA, a plasmid containing Cas9-HA-2NLS was linearized with XbaI (a gift from Jorge Henao-Mejia, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA). Approximately 1 μg of linearized plasmid was incubated with HiScribe T7 Quick high-yield RNA synthesis kit (NEB E2050S). RNA was purified using RNeasy minicolumns (Qiagen 74106), and the capping reaction used the Vaccinia capping system (NEB M2080S). RNA was purified using RNeasy micro cleanup columns (Qiagen 74004). Capped Cas9 mRNA was then subject to polyadenylation (NEB M0276S) and purified over a RNeasy micro cleanup column and eluted in RNase-free water. Cas9 mRNA integrity was validated using an RNA Bioanalyzer. To construct 3HA-PPARγ1 mice, T7 promoter was added onto gRNA template targeting the second exon (γ1-b) by PCR amplification using specific primers (targeting guide sequence for TCTGATGTACATACCAGTAA). To construct 6His-HA-PPARγ2 mice, T7 promoter was added onto gRNA template targeting the ATG start site of γ2 by PCR amplification using specific primers (targeting guide sequence for GCTGTTATGGGTGAAACTCT). The T7-sgRNA product was purified using a PCR purification kit (Qiagen) and was used as the template for in vitro transcription using the MegaShortScript kit (Life Technologies) following the manufacturer's instructions. Subsequent sgRNA was purified using the MegaClear kit (Life Technologies) and verified by RNA Bioanalyzer before dilution for microinjection. The ssDNA homology donor (IDT) containing the 3xHA tag or 6His-HA tag was resuspended in water and prepared using DNA Clean and Concentrator (Zymo): A*T*A*TAACTGATTAATTATATTAATATAATTTATTCTGATGTACATACCAGTAGCGTAATCTGGAACGTCATAAGGATACGATCCTGCATAGTCCGGGACGTCATAGGGATAGCCCGCATAGTCAGGAACATCGTATGGATACATGGTGGCAAAGGGTAGTCTTGTTTTTAAAAATGTCCTGAATATCAGTGGTTCAC*C*G*C (3xHA tag with Kozak sequence for 3HA-PPARγ1 mice) and C*C*A*ACCAATCTTTTGCAAGACATAGACAAAACACCAGTGTGAATTACAGCAAATCTCTGTTTTATGCTGTTATGCATCATCACCATCACCACTACCCATACGATGTTCCAGATTACGCTGGTGGCGGCCGCGGTGAAACTCTGGGAGATTCTCCTGTTGACCCAGAGCATGGTGCCTTCGCTGATGCACTGCCTATG*A*G*C (6His-HA tag for HHA-PPARγ2 mice). Microinjection was performed by the Transgenic and Chimeric Mouse Facility at the University of Pennsylvania using C57BL/6J mice from JAX. Microinjection buffer consisted of 1 mM Tris (pH 8.0), 0.1 mM EDTA, 100 ng/μL Cas9 mRNA, 50 ng/μL sgRNA, and 100 ng/μL ssDNA homology donor. Correct insertion of epitope tag or generation of early stop codon was detected by PCR and confirmed by Sanger sequencing. All mice were backcrossed to the C57BL/6J genetic background for at least four to five generations and genotyped using the PCR primers in Supplemental Table S1.
Western blot and gene expression analysis
For Western blotting, fat pads were washed with cold PBS and lysed with RIPA buffer, and then tissue lysates were separated on SDS-PAGE, transferred onto nitrocellulose membrane, and blotted with the indicated primary antibodies. The membrane was detected by a secondary antibody conjugated to HRP. To examine PPARγ1/2 expression profiles, multiple organs were collected from 3HA-PPARγ1 and HHA-PPARγ2 mice, with brain samples taken from the cortex. We also performed immunoprecipitation using HA magnetic beads following the manufacturer's instructions to enrich the PPARγ1/2. For gene expression analysis, total RNA samples were collected using TRIzol (Invitrogen) followed by RNeasy kit (Qiagen) according to the manufacturer's instructions. The RNA for each reaction was reverse-transcribed to cDNA using high-capacity cDNA reverse transcription kit. Quantitative real-time PCR was subsequently conducted with specific primers and Power SYBR Green PCR master mix (Applied Biosystems). The relative expression levels were normalized against the internal control (HPRT). Primers used are listed in Supplemental Table S1.
Immunostaining
Isolated tissues were fixed in 4% PFA overnight, dehydrated, and embedded in paraffin for sectioning. Following deparaffinization, heat antigen retrieval was performed in a pressure cooker using Bulls Eye decloaking buffer (Biocare). Slides were incubated with primary antibodies (anti-HA [Cell Signaling Technology] and anti-Ucp1 [Abcam]) overnight at 4°C and then with the appropriate fluorescent probe-conjugated secondary antibodies for 1 h at room temperature. Images were captured with fluorescence microscope or Leica TCS SP8.
ChIP-seq and data processing
ChIP-seq of adipose tissue was performed as previously described (Soccio et al. 2015). HA magnetic beads (Pierce) or GABPα antibody (Santa Cruz Biotechnology sc-22810) were used to perform the immunoprecipitation. Three to four biological replicates were used for HA ChIP sequencing, and WT mice were used as negative control. Two biological replicates were used for GABPα ChIP sequencing, and IgG ChIP was used as a negative control. The library preparation for ChIP-seq followed the guide provided by Illumina. ChIP-seq libraries were sequenced single end at 50-bp or 100-bp read length on Illumina HiSeq 2000 by the Functional Genomics Core of the Penn Diabetes Research Center. ChIP-seq raw reads were trimmed using Fastp v0.19.5. Trimmed reads were then aligned to the mouse reference genome (mm10) using Bowtie2 v2.3.0 with default parameters. SAMtools v1.9 was used to extract unique mapped reads and remove duplicated reads. Tag directories were generated from alignment files using HOMER (v.4.9.1). Peaks were called using HOMER's findPeaks function with parameter -style factor –size 200.
RNA-seq and data processing
Total RNA samples were prepared with TRIzol followed by RNeasy kit (Qiagen) according to the manufacturer's instructions. More than 2 ug of RNA from three to five biological replicates was sent to Novogene for library preparation (Illumina) and sequencing at paired ends at 150-bp read length on Novaseq 6000. Raw reads were trimmed using Fastp v0.19.5 to remove reads with low quality, that were too short, or that had too many Ns. Trimmed reads were then aligned to the mouse reference genome (mm10) using Hisat2 v2.1 with default parameters. Only unique mapped reads extracted by SAMtools v1.9 were considered for downstream analyses. Quantification of genes annotated in GRCm38.99 from Ensembl database was estimated using StringTie v1.3.4. Genes with normalized expression value, fragments per kilobase of exons per million reads mapped (FPKM), >1 in at least one sample were considered. Read counts that were measured for each gene using featureCounts v1.5.1 were used as the input to DESeq2 for differential expression analysis with adjusted P-value (Benjamini–Hochberg) < 0.01 and fold change > 1.5.
Glucose tolerance test
Mice were fasted for 16 h with ad libitum access to water. Following an initial blood glucose measurement, mice were intraperitoneally injected with glucose (1.5 g/kg for mice under normal chow condition; 0.75 g/kg for mice under HFD condition) and measured the blood glucose levels over a period of 120 min using a glucometer.
Cold tolerance test
Mice were singly housed in climate-controlled rodent incubators (Powers Scientific) maintained for 2 wk at 29°C with free access to food and water. Then, mice were placed in prechilled cages at 4°C–5°C with bedding, free access to standard chow and water, and the cage lid partly open. Rectal temperatures were recorded every 60 min. Individual mice were removed from the study and euthanized if core body temperature fell ≥10°C from baseline measurement.
Whole-animal energy expenditure in response to norepinephrine
Oxygen consumption rates were measured using the CLAMS as previously described (Emmett et al. 2017). Briefly, mice were anesthetized with 75 mg/kg pentobarbital (Nembutal) and placed into CLAMS cages preacclimated to 30°C. A subcutaneous injection of 1 mg/kg L-(−)-norepinephrine (+)-bitartrate salt monohydrate (Sigma A9512) was performed in the dorsal nuchal region, and oxygen consumption rates were recorded until rates began to decline.
Statistical analysis
Data are presented as mean ± SEM. Graphing and statistical analysis were performed using Graphpad Prism. As described in the figure legends, statistical analyses were performed using a two-tailed Student's t-test for comparison between two groups, and two-way ANOVA for assessment of variables effects (time, diet, treatment, and genotype; P < 0.05 [*], P < 0.01 [**], and P < 0.001 [***]).
Data availability
The data sets generated during this study are available at GSE186277. NCoR and HDAC3 ChIP-seq data are from public data GSE83926. EBF2 ChIP-seq data are from public data GSE97114. RNA-seq data from public data set GSE140259 were used to determine the expression levels of γ1 and γ2 in response to various temperatures.
Supplementary Material
Acknowledgments
We thank Yuxia Guan, Jarrett R. Remsberg, and other members of the Lazar laboratory for technical support and valuable discussions. We also thank Patrick Seale for advice and helpful discussions. We thank Lan Cheng from the Penn Molecular Pathology and Imaging Core for help with histology. We also thank the Functional Genomics Core of the Penn Diabetes Research Center (P30 DK19525) for next-generation sequencing, as well as the Transgenic Mouse Genome Editing Core of the Penn Diabetes Research Center and Jorge Henao-Mejia for CRISPR/Cas9 editing. This work was supported by the Cox Medical Institute, the JPB Foundation (to M.A.L.), and National Institutes of Health grants (R01-DK049780 to M.A.L., R01-DK121801 to D.J.S., and K01-DK125602 to D.G.). W.H. was supported by American Diabetes Association training grant 1-18-PDF-132. Y.X. was supported by American Heart Association training grant 827529.
Author contributions: W.H. and M.A.L. designed research. W.H., M.K., Y.X., H.J.R., D.G., K.Z., B.M.K., A.N.R., J.M., and D.J.S. performed research. C.J. performed bioinformatics analysis. M.A.L., W.H., and C.J. analyzed data and wrote the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.349232.121.
Competing interest statement
M.A.L. is an advisory board member for Pfizer and Flare Therapeutics and a consultant to Novartis and Madrigal Pharmaceuticals, and receives research support from Pfizer for work unrelated to the present study.
References
- Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, et al. 2000. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26: 76–80. 10.1038/79216 [DOI] [PubMed] [Google Scholar]
- Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, Lascols O, Capeau J, Caron-Debarle M. 2013. Peroxisome proliferator-activated receptor-γ mutations responsible for lipodystrophy with severe hypertension activate the cellular renin–angiotensin system. Arterioscler Thromb Vasc Biol 33: 829–838. 10.1161/ATVBAHA.112.300962 [DOI] [PubMed] [Google Scholar]
- Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, et al. 1999. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402: 880–883. 10.1038/47254 [DOI] [PubMed] [Google Scholar]
- Bugge A, Grøntved L, Aagaard MM, Borup R, Mandrup S. 2009. The PPARγ2 A/B-domain plays a gene-specific role in transactivation and cofactor recruitment. Mol Endocrinol 23: 794–808. 10.1210/me.2008-0236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero B. 2007. The global epidemic of obesity: an overview. Epidemiol Rev 29: 1–5. 10.1093/epirev/mxm012 [DOI] [PubMed] [Google Scholar]
- Chawla A, Lazar MA. 1994. Peroxisome proliferator and retinoid signaling pathways co-regulate preadipocyte phenotype and survival. Proc Natl Acad Sci 91: 1786–1790. 10.1073/pnas.91.5.1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. 2001. Nuclear receptors and lipid physiology: opening the X-files. Science 294: 1866–1870. 10.1126/science.294.5548.1866 [DOI] [PubMed] [Google Scholar]
- Chen Y, Ikeda K, Yoneshiro T, Scaramozza A, Tajima K, Wang Q, Kim K, Shinoda K, Sponton CH, Brown Z, et al. 2019. Thermal stress induces glycolytic beige fat formation via a myogenic state. Nature 565: 180–185. 10.1038/s41586-018-0801-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D. 2012. PPARγ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486: 549–553. 10.1038/nature11132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmett MJ, Lim HW, Jager J, Richter HJ, Adlanmerini M, Peed LC, Briggs ER, Steger DJ, Ma T, Sims CA, et al. 2017. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature 546: 544–548. 10.1038/nature22819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorenza CG, Chou SH, Mantzoros CS. 2011. Lipodystrophy: pathophysiology and advances in treatment. Nat Rev Endocrinol 7: 137–150. 10.1038/nrendo.2010.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan HP, Li Y, Jensen MV, Newgard CB, Steppan CM, Lazar MA. 2002. A futile metabolic cycle activated in adipocytes by antidiabetic agents. Nat Med 8: 1122–1128. 10.1038/nm780 [DOI] [PubMed] [Google Scholar]
- Guan HP, Ishizuka T, Chui PC, Lehrke M, Lazar MA. 2005. Corepressors selectively control the transcriptional activity of PPARγ in adipocytes. Genes Dev 19: 453–461. 10.1101/gad.1263305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM, Evans RM. 2003. Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci 100: 15712–15717. 10.1073/pnas.2536828100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G, Olson P, Wilkes J, Evans RM, Olefsky J. 2003. Muscle-specific Pparg deletion causes insulin resistance. Nat Med 9: 1491–1497. 10.1038/nm956 [DOI] [PubMed] [Google Scholar]
- Hiraike Y, Waki H, Yu J, Nakamura M, Miyake K, Nagano G, Nakaki R, Suzuki K, Kobayashi H, Yamamoto S, et al. 2017. NFIA co-localizes with PPARγ and transcriptionally controls the brown fat gene program. Nat Cell Biol 19: 1081–1092. 10.1038/ncb3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Jiang C, Guan D, Dierickx P, Zhang R, Moscati A, Nadkarni GN, Steger DJ, Loos RJF, Hu C, et al. 2019. Patient adipose stem cell-derived adipocytes reveal genetic variation that predicts antidiabetic drug response. Cell Stem Cell 24: 299–308.e6. 10.1016/j.stem.2018.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo P. 2009. Viewpoints on the way to the consensus session: where does insulin resistance start? The adipose tissue. Diabetes Care 32 Suppl 2: S168–S173. 10.2337/dc09-S304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. 1989. The scanning model for translation: an update. J Cell Biol 108: 229–241. 10.1083/jcb.108.2.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, et al. 1999. PPARγ mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell 4: 597–609. 10.1016/S1097-2765(00)80210-5 [DOI] [PubMed] [Google Scholar]
- Kuntz SG, Williams BA, Sternberg PW, Wold BJ. 2012. Transcription factor redundancy and tissue-specific regulation: evidence from functional and physical network connectivity. Genome Res 22: 1907–1919. 10.1101/gr.133306.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasar D, Rosenwald M, Kiehlmann E, Balaz M, Tall B, Opitz L, Lidell ME, Zamboni N, Krznar P, Sun W, et al. 2018. Peroxisome proliferator activated receptor γ controls mature brown adipocyte inducibility through glycerol kinase. Cell Rep 22: 760–773. 10.1016/j.celrep.2017.12.067 [DOI] [PubMed] [Google Scholar]
- Lefterova MI, Haakonsson AK, Lazar MA, Mandrup S. 2014. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol Metab 25: 293–302. 10.1016/j.tem.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. 1995. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J Biol Chem 270: 12953–12956. 10.1074/jbc.270.22.12953 [DOI] [PubMed] [Google Scholar]
- Li D, Zhang F, Zhang X, Xue C, Namwanje M, Fan L, Reilly MP, Hu F, Qiang L. 2016. Distinct functions of PPARγ isoforms in regulating adipocyte plasticity. Biochem Biophys Res Commun 481: 132–138. 10.1016/j.bbrc.2016.10.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Sarruf DA, Talukdar S, Sharma S, Li P, Bandyopadhyay G, Nalbandian S, Fan W, Gayen JR, Mahata SK, et al. 2011. Brain PPARγ promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat Med 17: 618–622. 10.1038/nm.2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majithia AR, Flannick J, Shahinian P, Guo M, Bray MA, Fontanillas P, Gabriel SB, GoT2D Consortium, NHGRI JHS/FHS Allelic Spectrum Project, SIGMA T2D Consortium, et al. 2014. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc Natl Acad Sci 111: 13127–13132. 10.1073/pnas.1410428111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majithia AR, Tsuda B, Agostini M, Gnanapradeepan K, Rice R, Peloso G, Patel KA, Zhang X, Broekema MF, Patterson N, et al. 2016. Prospective functional classification of all possible missense variants in PPARG. Nat Genet 48: 1570–1575. 10.1038/ng.3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. 1995. The nuclear receptor superfamily: the second decade. Cell 83: 835–839. 10.1016/0092-8674(95)90199-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B Jr, Reitman ML, Gonzalez FJ. 2003. Liver-specific disruption of PPARγ in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest 111: 737–747. 10.1172/JCI200317223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechta-Grigoriou F, Gerald D, Yaniv M. 2001. The mammalian Jun proteins: redundancy and specificity. Oncogene 20: 2378–2389. 10.1038/sj.onc.1204381 [DOI] [PubMed] [Google Scholar]
- Medina-Gomez G, Virtue S, Lelliott C, Boiani R, Campbell M, Christodoulides C, Perrin C, Jimenez-Linan M, Blount M, Dixon J, et al. 2005. The link between nutritional status and insulin sensitivity is dependent on the adipocyte-specific peroxisome proliferator-activated receptor-γ2 isoform. Diabetes 54: 1706–1716. 10.2337/diabetes.54.6.1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Gomez G, Gray SL, Yetukuri L, Shimomura K, Virtue S, Campbell M, Curtis RK, Jimenez-Linan M, Blount M, Yeo GS, et al. 2007. PPARγ2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet 3: e64. 10.1371/journal.pgen.0030064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, Petrovic N, Lindgren EM, Jacobsson A, Cannon B. 2005. PPARγ in the control of brown adipocyte differentiation. Biochim Biophys Acta 1740: 293–304. 10.1016/j.bbadis.2005.02.003 [DOI] [PubMed] [Google Scholar]
- Nelson VL, Nguyen HCB, Garcìa-Cañaveras JC, Briggs ER, Ho WY, DiSpirito JR, Marinis JM, Hill DA, Lazar MA. 2018. PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes Dev 32: 1035–1044. 10.1101/gad.312355.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC, Hirshman MF, Rosen ED, Goodyear LJ, Gonzalez FJ, et al. 2003. Muscle-specific PPARγ-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest 112: 608–618. 10.1172/JCI17305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, et al. 2007. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447: 1116–1120. 10.1038/nature05894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap A, Cuaranta-Monroy I, Peloquin M, Nagy L. 2016. Is the mouse a good model of human PPARγ-related metabolic diseases? Int J Mol Sci 17: 1236. 10.3390/ijms17081236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumari S, Wu J, Ishibashi J, Lim HW, Giang AH, Won KJ, Reed RR, Seale P. 2013. EBF2 determines and maintains brown adipocyte identity. Cell Metab 17: 562–574. 10.1016/j.cmet.2013.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. 1998. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 391: 79–82. 10.1038/34178 [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. 2006. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444: 847–853. 10.1038/nature05483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. 1999. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 4: 611–617. 10.1016/S1097-2765(00)80211-7 [DOI] [PubMed] [Google Scholar]
- Secco B, Camiré E, Brière MA, Caron A, Billong A, Gèlinas Y, Lemay AM, Tharp KM, Lee PL, Gobeil S, et al. 2017. Amplification of adipogenic commitment by VSTM2A. Cell Rep 18: 93–106. 10.1016/j.celrep.2016.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao M, Vishvanath L, Busbuso NC, Hepler C, Shan B, Sharma AX, Chen S, Yu X, An YA, Zhu Y, et al. 2018. De novo adipocyte differentiation from Pdgfrβ+ preadipocytes protects against pathologic visceral adipose expansion in obesity. Nat Commun 9: 890. 10.1038/s41467-018-03196-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Lazar MA. 2014. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 20: 573–591. 10.1016/j.cmet.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, Emmett MJ, Briggs ER, Fang B, Everett LJ, et al. 2015. Genetic variation determines PPARγ function and anti-diabetic drug response in vivo. Cell 162: 33–44. 10.1016/j.cell.2015.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Li Z, Chen ER, Foong YH, Benson KK, Dispirito JR, Mullican SE, Emmett MJ, Briggs ER, Peed LC, et al. 2017. Targeting PPARγ in the epigenome rescues genetic metabolic defects in mice. J Clin Invest 127: 1451–1462. 10.1172/JCI91211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Step SE, Lim HW, Marinis JM, Prokesch A, Steger DJ, You SH, Won KJ, Lazar MA. 2014. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARγ-driven enhancers. Genes Dev 28: 1018–1028. 10.1101/gad.237628.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, Hong SH, Castro GL, Yin YQ, Nelson MC, et al. 2009. PPARγ activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci 106: 22504–22509. 10.1073/pnas.0912487106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Sasaki S, Morita H, Oki Y, Turiya D, Ito T, Misawa H, Ishizuka K, Nakamura H. 2010. The role of the amino-terminal domain in the interaction of unliganded peroxisome proliferator-activated receptor γ-2 with nuclear receptor co-repressor. J Mol Endocrinol 45: 133–145. 10.1677/JME-10-0007 [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. 2008. Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem 77: 289–312. 10.1146/annurev.biochem.77.061307.091829 [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Spiegelman BM. 1994. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 79: 1147–1156. 10.1016/0092-8674(94)90006-X [DOI] [PubMed] [Google Scholar]
- Vidal-Puig A, Jimenez-Liñan M, Lowell BB, Hamann A, Hu E, Spiegelman B, Flier JS, Moller DE. 1996. Regulation of PPARγ gene expression by nutrition and obesity in rodents. J Clin Invest 97: 2553–2561. 10.1172/JCI118703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA. 2013. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. Proc Natl Acad Sci 110: 18656–18661. 10.1073/pnas.1314863110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmuth EV, Hoover EA, Antar A, Klinge S, Chen Y, Sawyers CL. 2020. Modulation of androgen receptor DNA binding activity through direct interaction with the ETS transcription factor ERG. Proc Natl Acad Sci 117: 8584–8592. 10.1073/pnas.1922159117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werman A, Hollenberg A, Solanes G, Bjørbaek C, Vidal-Puig AJ, Flier JS. 1997. Ligand-independent activation domain in the N terminus of peroxisome proliferator-activated receptor γ (PPARγ). differential activity of PPARγ1 and -2 isoforms and influence of insulin. J Biol Chem 272: 20230–20235. 10.1074/jbc.272.32.20230 [DOI] [PubMed] [Google Scholar]
- Zhang J, Fu M, Cui T, Xiong C, Xu K, Zhong W, Xiao Y, Floyd D, Liang J, Li E, et al. 2004. Selective disruption of PPARγ2 impairs the development of adipose tissue and insulin sensitivity. Proc Natl Acad Sci 101: 10703–10708. 10.1073/pnas.0403652101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Qi C, Korenberg JR, Chen XN, Noya D, Rao MS, Reddy JK. 1995. Structural organization of mouse peroxisome proliferator-activated receptor γ (mPPARγ) gene: alternative promoter use and different splicing yield two mPPARγ isoforms. Proc Natl Acad Sci 92: 7921–7925. 10.1073/pnas.92.17.7921 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated during this study are available at GSE186277. NCoR and HDAC3 ChIP-seq data are from public data GSE83926. EBF2 ChIP-seq data are from public data GSE97114. RNA-seq data from public data set GSE140259 were used to determine the expression levels of γ1 and γ2 in response to various temperatures.