

Abstract
Oncogenic mutations in the small GTPase RAS contribute to ~30% of human cancers. In a Drosophila genetic screen, we identified novel and evolutionary conserved cancer genes that affect Ras-driven tumorigenesis and metastasis in Drosophila including confirmation of the tetraspanin Tsp29Fb. However, it was not known whether the mammalian Tsp29Fb orthologue, TSPAN6, has any role in RAS-driven human epithelial tumors. Here we show that TSPAN6 suppressed tumor growth and metastatic dissemination of human RAS activating mutant pancreatic cancer xenografts. Whole-body knockout as well as tumor cell autonomous inactivation using floxed alleles of Tspan6 in mice enhanced KrasG12D-driven lung tumor initiation and malignant progression. Mechanistically, TSPAN6 binds to the EGFR and blocks EGFR-induced RAS activation. Moreover, we show that inactivation of TSPAN6 induces an epithelial-to-mesenchymal transition and inhibits cell migration in vitro and in vivo. Finally, low TSPAN6 expression correlates with poor prognosis of patients with lung and pancreatic cancers with mesenchymal morphology. Our results uncover TSPAN6 as a novel tumor suppressor receptor that controls epithelial cell identify and restrains RAS-driven epithelial cancer.
Subject terms: Cell polarity, Growth factor signalling, Cancer genetics
Introduction
The 3 RAS proteins (H-RAS, N-RAS, and K-RAS) are small GTPase binary molecular switches, that switch between active (GTP bound) and inactive (GDP bound) states [1]. Members of the RAS family are central players in many signaling networks and are amongst the most commonly identified oncogenes in human cancer [2, 3]. Gain of function mutations of RAS, such as RASV12, that increase the levels of the GTP-bound version of the protein were first observed in cancers approximately 40 years ago [4]. Activating mutation in RAS leads to a cascade of molecular events affecting cell proliferation, cell survival, and, thus, lead to carcinogenesis [5, 6]. Although activating RAS mutations are common, this event is rarely sufficient to promote tumorigenesis, as it promotes cellular senescence [7–10]. As such, additional mutations or silencing of tumor suppressors is frequently needed to promote tumorigenesis and metastasis together with activation of the RAS signaling pathway [11–13].
In order to identify novel tumor suppressors that cooperate with activated Ras (RasV12) to promote tumorigenesis, we undertook a genome-wide genetic screen using transgenic RNAi lines in the vinegar fly, Drosophila melanogaster, model organism [14]. This screen identified many novel potential tumor suppressor genes, and we verified that 80 out of the top 100 genes whose orthologs were downregulated in human cancer were able to cooperate with RasV12 to promote epithelial tissue overgrowth in Drosophila. One of these conserved tumor suppressor genes was Drosophila Tsp29Fb, the mammalian ortholog of which is TSPAN6 (TM4SF6) [14]. Tetraspanins are small transmembrane proteins expressed in all multicellular organisms, which have been implicated in multiple biological functions [15]. They are known to interact with one another, other transmembrane molecules such as integrins and other adhesion receptors as well as growth factor and cytokine receptors to modulate cellular signaling [16, 17]. Tetraspanin mammalian family members, such as CD9, CD63, CD81, CD82, CD151, or Net2, have previously been implicated in tumorigenesis [18, 19]. In contrast to many human Tetraspanin family members with broad expression [20–22], TSPAN6 mRNA and protein are primarily expressed in epithelial cells. Further to this, we found that the Drosophila TSPAN6 ortholog, Tsp29Fb, regulates epithelial architecture and EGFR-Ras signaling [14]. Intriguingly, Tsp29Fb was also found to affect the junctional localization of the Dlg1 cell polarity protein and genetically interact with the Scribble (Scrib)/Dlg1 polarity module [14]. In Drosophila, loss of function mutations of the cell polarity genes dlg1 and scrib lead to uncontrolled proliferation, loss of epithelial polarity and invasive phenotypes, and they cooperate with oncogenic Ras in tumorigenesis in both Drosophila and mouse models [23–27].
As the function of TSPAN6 in mammalian systems was unknown, in this current study we investigate the role of TSPAN6 in human cell lines and mouse cancer models. We show that the TSPAN6 knockdown mirrors the loss of either human SCRIB or DLG1 in the presence of H-RASV12, leading to an increase in invasive spiking in MCF10A normal human breast cells grown in 3D conditions and in 2D cultures TSPAN6 knockdown reduces epithelial sheet migration, similar to SCRIB and DLG1 knockdowns. Furthermore, we show that human TSPAN6 acts as a tumor suppressor in a mouse orthotopic model of pancreatic cancer, and that full body as well as tissue specific knockout of Tspan6 in mice enhances lung cancer driven by activated Kras. Mechanistically, TSPAN6 binds to the EGFR and suppresses EGFR-RAS-ERK signaling and promotes an epithelial cell morphology in vitro and in vivo. Furthermore, we show in lung and pancreatic cancer cohorts that low TSPAN6 expression correlates with poor survival in cancers with mesenchymal features. Altogether, our results reveal TSPAN6 as a novel tumor suppressor gene that cooperates with activated RAS.
Results
Human TSPAN6 knockdown phenocopies SCRIB and DLG1 knockdowns in affecting cell migration and invasive phenotypes in vitro
Given our findings in Drosophila that Tsp29Fb genetically interacts with scrib [14], we first sought to test whether TSPAN6 functions similarly to Scrib and Dlg1 in mammalian cells. In mammalian systems, both Dlg1 and Scrib have also been implicated in wound healing and the coordinated movement of epithelial cells [28, 29] (Galea and Humbert, unpublished data), and SRIB or DLG1 knockdown cooperates with H-RASV12 to drive cell invasion in 3D epithelial cell cultures [11, 24, 30] (Galea and Humbert, unpublished data). To determine whether human TSPAN6 also regulates cell migration and invasion similarly to SCRIB and DLG1, we generated MCF10A breast epithelial cell lines where SCRIB, DLG1 or TSPAN6 were stably knocked down (using 2 independent hairpins) in the presence of absence of oncogenic H-RASV12. 2D culture scratch wound assays, which measure epithelial sheet migration, a directed cell migration, revealed that knockdown of TSPAN6 phenocopied the knockdown of SCRIB and DLG1 in inhibiting wound closure, with a delay of ~6 h relative to the control (Fig. 1a). This result indicates that, similar to the function of SCRIB and DLG1, epithelial sheet migration is positively regulated by TSPAN6.
Fig. 1. TSPAN6 controls epithelial cell migration and cooperates with H-RASV12 to induce cell invasion in normal human mammary epithelial cells.

a The knockdown of SCRIB, DLG1, and TSPAN6 leads to decreased epithelial sheet migrative potential. Representative phase contrast microscope images of Scrib, Dlg1, or TSPAN6 knockdown MCF10A epithelial cell monolayers at 0, 12, and 18 h after wounding, and quantification of wound closure from 0–24 h. In each case, the corresponding shRNA constructs had a slower wound closing time as compared to the Scramble shRNA control (SCRIB sh vs Scram P < 0.0001; DLG1 sh vs Scram P < 0.0001; TSPAN6 sh vs Scram P < 0.0001; two-way ANOVA, 3 independent experiments). Scale bar = 100 μM. b SCRIB, DLG1 and TSPAN6 knockdown promote H-RASV12-induced invasion in 3D cultures. In combination with low or high levels of H-RASV12, knockdown of SCRIB, DLG1, and TSPAN6 led to increased invasive acini in 3D culture. (*P < 0.0001, ** P < 0.01, ***P < 0.05; one-way ANOVA, three independent experiments). See Supplementary data for microscope images.
We then examined the cooperation of TSPAN6 knockdown with H-RASV12 in 3D MCF10A cultures, where SCRIB or DLG1 knockdown have been shown to cooperate with H-RASV12 in inducing an invasive phenotype [11]. Knockdown of TSPAN6, SCRIB, or DLG1 alone did not alter acini morphology, although occasional invasive spikes were observed in TSPAN6 knockdown cells (Fig. 1b, Supplementary Fig. 1). Whereas low or high expression of H-RASV12 in cells resulted in a small number of invasive spikes in the acini, we found that knockdown of TSPAN6, greatly increased the number of invasive spikes in acini, similar to the knockdown of SCRIB or DLG1 (Fig. 1b and Supplementary Fig. 1) [11]. Thus, TSPAN6 acts similarly to SCRIB and DLG1 in preventing cell invasion in H-RASV12-transformed epithelial cells.
Tspan6 controls growth and metastases of Ras-transformed epithelial cells in vivo
To address the functional role of Tspan6 in Ras-mutant cancers, we first examined the effect of reduced Tspan6 expression in mouse mammary epithelial cells (Eph4) stably transfected with oncogenic H-Ras, termed EpRas cells [31]. Stable knockdown of Tspan6 in Ras-transformed EpRas cells (Fig. 2a) increased proliferation in vitro (Fig. 2b). EpRas cells exhibit a stable epithelial phenotype and normally undergo an epithelial-to-mesenchymal transition (EMT) only upon TGFβ treatment [31]. Intriguingly, RNAi-mediated knockdown of Tspan6 in EpRas cells by itself was sufficient to induce an EMT-like phenotype, with cells invading the collagen 3D matrix showing reduction in ZO-1 and gain of Vimentin expression (Fig. 2c).
Fig. 2. Tspan6 knockdown in EpRas mouse mammary epithelial cells affects cell polarity, proliferation, and invasion.

a Efficacy of shRNA mediated Tspan6 knockdown in murine mammary epithelial EpRas cells. Data for two different shRNA knockdown lines, termed shRNAm1 and shRNAm2, are shown relative to the scrambled shRNA control. Cells were transfected using a lentiviral approach. Data were obtained by qPCR. b Proliferation of EpRas mammary cancer cells infected with lentiviral vectors encoding two distinct shRNAs targeting Tspan6, or scrambled shRNA as a control. Proliferation was determined in triplicate at 8 h using tritiated thymidine incorporation (mean ± SEM). Experiments were conducted twice. c Knockdown of Tspan6 results in EMT in H-RasV12 transformed epithelial EpRas cells. Top panels show representative cell morphologies of EpRas cells transfected with scrambled shRNA (control) or two distinct shRNAs targeting Tspan6 5 days after seeding of 5 × 103 cells in collagen gels. Experiments were conducted twice. Yellow arrow indicates typical tubular structures. Black arrows indicate spindle-like cell morphologies indicative of EMT. Lower panel shows immunostaining for the epithelial marker ZO-1 (green) and the mesenchymal marker vimentin (red). DAPI (blue) is shown to label nuclei. Images were taken by confocal microscopy. Scale bars, 80μM top panels, 50μM bottom panels. d Left: Representative macroscopic appearances of tumors from control EpRas and Tspan6 knockdown EpRas cells 4 weeks after inoculation. EpRas cells transfected with lentiviruses targeting Tspan6 or scrambled shRNA (control) were inoculated in mammary fat pads of female nu/nu hosts (2.5 × 105 cells each). Experiments were conducted twice. Right: Mean tumor volume 4 weeks after inoculation (n = 8 per group). *P < 0.05 (Student’s t test). e In vivo tumor growth, and effect on Fra1 and E-cadherin expression. Top panel, Hematoxylin and Eosin (H&E) staining of cross tumor sections from designated xenografts. Black arrows indicate large necrotic areas formed by EpRas Tspan6 shRNA cells. Middle panel, Fra1 immunostaining in tumors from control EpRas and Tspan6 knockdown EpRas cells showing that the Ras pathway target, Fra1 is upregulated in Tspan6 knockdown tumors. Bottom panel, E-cadherin immunostaining in tumors from control EpRas and Tspan6 knockdown EpRas cells indicate dissolution of adherens junction upon Tspan6 knockdown. Representative images are shown at 4 weeks after injection into female nu/nu hosts. Scale bars = 1000 μM. f Lung metastases of control (scrambled shRNA) and Tspan6 knockdown murine EpRas epithelial cells 30 days after tail vein injection of 5 × 105 cells/mouse. Arrows indicate metastases. Scale bar is 1000 µm. For quantification (right panel), at least ten different planes from each lung were H&E stained and analyzed in a blinded fashion. Data are from at least 4 mice per genotype, and experiments were conducted twice. Mean foci numbers ± s.e.m. per lung section are shown. *p < 0.05 (Student’s t test).
To study the in vivo tumorigenic potential, we performed orthotopic injection of EpRas ± Tspan6-shRNA cells into the mammary fat pad of female nu/nu mice. Stable knockdown of Tspan6 in Ras-transformed EpRas cells indeed enhanced tumor growth in vivo following orthotopic injection (Fig. 2d). Immunohistochemistry of the in vivo tumors formed by EpRas cells with stable Tspan6 knockdown revealed upregulation of the Ras pathway target, Fra1, and strongly reduced E-cadherin expression (Fig. 2e), indicative of an EMT [32]. Since we did not observe metastases in this orthotopic injection model, we performed tail vein injections and assayed for lung metastases. Stable depletion of Tspan6 in EpRas cells significantly increased numbers and sizes of lung metastases as compared to cells transduced with scrambled control RNAi (Fig. 2f). Taken together, our experiments using stable knockdown of endogenous Tspan6 in H-Ras-transformed mouse epithelial cells identify Tspan6 as a regulator of RAS-oncogene dependent epithelial identity, tumor growth, and invasion/metastasis.
TSPAN6 controls growth and metastases of human K-RAS mutant pancreatic cancer by interacting with the EGFR
Next, to determine the role of elevated expression of TSPAN6 in suppressing RAS-mutant human cancers in vivo, we used a doxycyclin (Dox)-inducible Tet ON/OFF switch to regulate TSPAN6 expression in two human K-RAS activating mutant pancreatic cancer cell lines, MIA PaCa2 and PANC1 (Fig. 3a and Supplementary Fig. 2a). We used these two pancreatic adenocarcinoma cell lines because they carry constitutively activated endogenous K-RAS mutations (K-RASG12C and K-RASG12D respectively) and in an initial pre-screen of a panel of pancreatic cancer cell lines, we observed low expression of TSPAN6 in these lines compared to others examined (data not shown). Overexpression of TSPAN6 in both cell lines reduced RAS activation at baseline and, importantly, following EGFR stimulation (Fig. 3b and Supplementary Fig. 2b). Using phospho-kinase arrays we observed that induction of TSPAN6 led to reduced phosphorylation of EGFR, ERK1/2, and P38 at basal levels and following EGFR stimulation (Supplementary Fig. 2c). Interestingly, the Phospho-3-Inositol Kinase (PI3K) arm of EGFR-RAS signaling (indicated by pAKT1/2/3 and pTOR) was not similarly repressed upon TSPAN6 expression in EGF-stimulated cells (Supplementary Fig. 2c), and in fact the phosphorylation of AKT1/2/3 and TOR was increased, suggesting that TSPAN6 inhibits EGFR-RAS-ERK signaling, but has the opposite effect upon PI3K-AKT-TOR signaling. Reduced EGF-induced phosphorylation in TSPAN6 expressing PANC1 cells was confirmed using an antibody that detects phosphorylated tyrosine 1173 of the activated EGFR (Supplementary Fig. 2d), indicating that TSPAN6 affects proximal EGFR signaling. Thus, similar to the function of Drosophila Tsp29Fb, mammalian TSPAN6 represses RAS activation and EGFR signaling.
Fig. 3. TSPAN6 inhibits growth and metastasis of activating RAS mutant human epithelial tumor cells.

a Schematic vector map of doxycycline (Dox) inducible mCherry-IRES-TSPAN6. Bottom panel shows doxycycline-inducible TSPAN6 protein expression in stably transfected MIA PaCa2 and PANC1 cells. b Ras activation assay measured upon EGFα stimulation in PANC1 in the absence of or upon induced TSPAN6 expression. Experiments were performed in six repeats. c Transwell migration of TSPAN6-PANC1 cells in response to treatment with EGFα and/or growing FBS gradient. For each condition, experiments were carried out in triplicate. d Representative bioluminescence images of nu/nu recipient mice orthotopically transplanted with the Dox-inducible TSPAN6 human pancreatic cancer cell lines MIA PaCa2 (upper panels) and PANC1 (lower panels) in the presence or absence of Dox. e Representative H&E staining of liver and diaphragm sections of TSPAN6 ON and TSPAN6 OFF PANC1 cohorts 12 weeks after orthotopic grafting, showing reduction of metastatic foci (arrows) upon TSPAN6 expression. Scale bars = 200 μM. f Representative intestinal sections (H&E staining) of mice following orthotopic injection of TSPAN6 ON and TSPAN6 OFF PANC1 cells. Scale bars = 500 μM.
Functionally, we found that Dox-induced TSPAN6 expression in both MIA PaCa2 and PANC1 pancreas cancer cells decreased proliferation at baseline (Supplementary Fig. 2e). Moreover, TSPAN6 overexpression reduced EGF-dependent invasion using transwell migration assays (Fig. 3c); in vitro scratch assays further confirmed that TSPAN6 overexpression in these K-RAS activating mutant pancreatic cancer cells leads to impaired epithelial sheet migration (Supplementary Fig. 2f). EGFR stimulation had no apparent effect on the ability of TSPAN6 to inhibit proliferation (Supplementary Fig. 2e), but markedly increased invasion, which was abrogated by TSPAN6 expression (Fig. 3c). Furthermore, Dox-induced TSPAN6 expression in EGFα-stimulated PANC1 cells reduced the expression of the EMT markers, N-Cadherin and Vimentin (Supplementary Fig. 3a), and also reduced the expression of the EMT transcription factor, Slug, in both EGFα-stimulated PANC1 and MIA PaCa2 cells (Supplementary Fig. 3b), indicating that TSPAN6 expression dictates an epithelial cell state.
Importantly, when we implanted control (no Dox) and Dox-induced MIA PaCa2 and PANC1 cells orthotopically into the pancreas of immunodeficient nu/nu mice, TSPAN6 overexpression markedly reduced tumor growth within the primary pancreatic injection site and significantly decreased metastatic spread to distant sites (Fig. 3d–f and Supplementary Fig. 4a–e). Thus, overexpression of TSPAN6 in the two K-RAS-activating mutant human pancreatic cancer cell lines, PANC1 and MIA PaCa2, impaired proliferation and migration in vitro and significantly reduced tumor growth and metastasis in vivo.
To further explore how Tspan6 was regulating EGFR-RAS signaling we examined whether TSPAN6 interacts with EGFR in the PANC1 and MIA PaCa2 cells. Endogenous TSPAN6 was co-immunoprecipitated with EGFR under mild stringency conditions (1% Brij 98) (Fig. 4a), but not under stringent conditions (0.5% Triton X100) (not shown). Additionally, using the converse IP approach, an interaction was also observed between TSPAN6 and EGFR when extracts from cells expressing a Flag tagged-TSPAN6 were immunoprecipitated with an anti-FLAG antibody (Fig. 4b). The interaction observed between TSPAN6 and EGFR only under mild stringency conditions is consistent with previous observations that tetraspanins form a multimolecular transmembrane complex called the “tetraspanins web” that is maintained in nonionic detergent [21, 33]. We also investigated the interaction between TSPAN6 and DLG1-SCRIB complex and did not observe any interactions (Fig. 4b). Altogether the results showing that TSPAN6 and the EGFR physically interact suggests that TSPAN6 directly regulates the EGFR. Moreover, since TSPAN6 does not interact with DLG1 or SCRIB it suggests that TSPAN6 and DLG1/SCRIB regulate EGFR-RAS signaling by parallel pathways.
Fig. 4. TSPAN6 Interacts with the EGFR but not with SCRIB or DLG1.
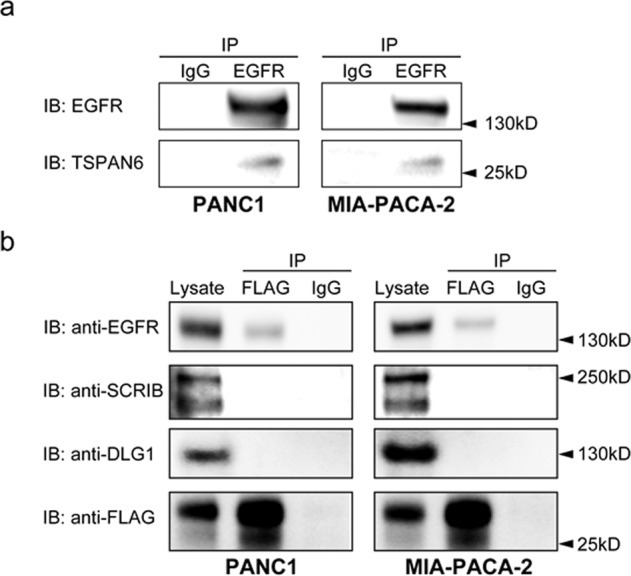
a Co-immunoprecipitation between TSPAN6 and the EGFR. PANC1 or MIA-PaCa2 cells were lysed in 1% Brij 98 protein extraction buffer. After immunoprecipitation (IP) with anti-EGFR or IgG control antibodies, proteins were separated by SDS-PAGE and detected by immunoblotting with anti-TSPAN6 or anti-EGFR antibodies (IB). b Co-immunoprecipitation between FLAG-TSPAN6 overexpressing pancreatic cell lines and the EGFR. PANC1 or MIA-PaCa2 cells were lysed in 1% Brij 98 protein extraction buffer. After immunoprecipitation (IP) with anti-FLAG or IgG control antibodies, proteins were separated by SDS-PAGE and detected by immunoblotting with anti-EGFR, anti-SCRIB, anti-DLG1, or anti-FLAG antibodies (IB) The Lysate is also shown. Experiments were conducted once.
Tspan6 controls KrasG12D driven lung cancer in vivo
To obtain definitive proof that Tspan6 plays a role in Ras-driven tumorigenesis, we generated whole-body Tspan6 mutant mice using gene targeting via homologous recombination (Supplementary Fig. 5a, b). Homozygous mutant mice were born at the expected Mendelian frequency and exhibited normal fertility with no obvious anatomical or histological abnormalities. Tspan6 mutant mice were then crossed to mice carrying the Lox-Stop-Lox-KrasG12D transgene. Lox-Stop-Lox-KrasG12D mice develop non-small-cell lung carcinomas (NSCLCs) upon Cre-mediated deletion and clonal induction of the mutant KrasG12D allele in a well-described stepwise process that leads from epithelial hyperplasia and benign adenomas to adenocarcinomas [34]. We used male Tspan6 mutant offspring (Tspan6−/y KrasG12D) and their Tspan6 expressing littermates (Tspan6+/y KrasG12D) as controls since Tspan6 is located on the X-chromosome. Clonal activation of KrasG12D-expression by adeno-viral Cre delivery (Ad-Cre) [34] significantly reduced survival in the Tspan6-knockout background compared to their respective littermate controls (P < 0.01, Fig. 5a). Quantification of overall tumor burden revealed a significant increase of the tumor areas in the lungs of Tspan6−/y KrasG12D mice compared to Tspan6+/y KrasG12D littermates (Fig. 5b and Supplementary Fig. 5c). We next assessed tumor initiation and staged the malignant progression of the lung cancers. At 4 weeks after Ad-Cre inhalation, Tspan6−/y KrasG12D mice harbored significantly more hyperplastic lesions than their Tspan6 expressing littermates (Fig. 5c). Eight weeks after Ad-Cre inhalation, we again observed significantly more hyperplastic regions as well as increased numbers of adenomas in Tspan6−/y KrasG12D mice (Fig. 5c). At 16 weeks after Ad-Cre infection, knockout of Tspan6 resulted in markedly increased progression to adenocarcinomas (Fig. 5c). These data show that genetic inactivation of Tspan6 results in enhanced KrasG12D-driven tumor initiation and malignant lung cancer progression.
Fig. 5. Tspan6 is a tumor suppressor in KRasG12D-driven lung cancer.

a Kaplan–Meier survival plots for whole-body Tspan6-/y KrasG12D mice (n = 8) and TSPAN6+/y KrasG12D (n = 9) littermate controls. Log-rank test was used for statistical analysis. **P < 0.01. b Representative histological H&E sections of Tspan6-/y KrasG12D and Tspan6+/y KrasG12D lungs 12 weeks after Ad-Cre infection. Magnifications x5. c Quantification of hyperplasic regions, adenomas, and adenocarcinomas in Tspan6-/y KrasG12D and Tspan6+/y KrasG12D lungs at the indicated time points following Ad-Cre inhalation. n = 5 for each cohort and time point analyzed. Data are shown as means ± s.e.m. * P < 0.05; ** P < 0.01; N.S. = not significant (Student’s t test). d Representative in situ immunostaining for phosphorylated EGFR (pEGFR) 8 weeks post Ad-Cre delivery to lungs of Tspan6-/y KrasG12D (n = 5) and Tspan6+/y KrasG12D mice (n = 5). Right panel shows quantification of % pEGFR+ cells in hyperplasic regions, adenomas, and adenocarcinomas. Data are shown as mean ± s.e.m. * P < 0.05; ***P < 0.001; N.S. = not significant (Student’s t test). Scale bars = 200 μM. e Representative immunostaining for the mesenchymal markers, vimentin, and N-cadherin, and the epithelial marker E-cadherin in lung tumors 8 weeks after Ad-Cre inhalation. Sections were counterstained with haematoxylin. Scale bars, top left panel = 100 μM, bottom left panel = 200 μM, middle panels = 200 μM, right panels = 500 μM.
Proliferation, as detected by Ki67 staining, was significantly enhanced in hyperplastic regions, as well as in early (4 weeks after Ad-Cre inhalation) adenomas and adenocarcinomas from Tspan6−/y KrasG12D mice compared to their Tspan6+/y KrasG12D littermates (Supplementary Fig. 5d, e). We detected only a few apoptotic cells in KrasG12D-induced lung tumors from both Tspan6 mutant and wild-type mice and we did not find significant differences in the percentages of apoptotic cells among the different cohorts (not shown). Immunohistochemistry for phospho-EGFR revealed markedly increased phospho-EGFR staining in Tspan6 mutant lung tumors, in particular in the hyperplastic regions and adenomas (Fig. 5d). We also detected enhanced expression of vimentin and N-cadherin, as well as reduced E-cadherin expression in the Tspan6−/y KrasG12D lung tumors (Fig. 5e), further supporting the notion that loss of Tspan6 induces an EMT.
Profiling Tspan6 mRNA expression in the lung revealed transcripts in alveolar pneumocytes bronchial epithelium, endothelial cells, as well as resident macrophages (not shown). To therefore test whether the effects of Tspan6 on KrasG12D-induced lung cancer are cell autonomous to the transformed lung epithelium, we generated mice that carry a Tspan6floxed allele. Cre-mediated recombination results in excision of Exons 2–6 and a shift in the reading frame (Supplementary Fig. 6a, b). Female Tspan6flox/flox mice were then crossed to male Lox-Stop-Lox-KrasG12D animals to generate male Tspan6fl/y KrasG12D and Tspan6+/y KrasG12D mice. In this model system, Ad-Cre inhalation results in concurrent deletion of Tspan6 in the cells that express oncogenic KrasG12D in the lung (Supplementary Fig. 6c). Selective loss of Tspan6 in the KrasG12D-expressing lung epithelial cells again resulted in significantly reduced survival and increased tumor burden as compared to Tspan6-expressing KrasG12D littermates (Fig. 6a–c). Tspan6fl/y KrasG12D mice also harbored significantly more hyperplastic lesions and increased numbers of adenomas and adenocarcinomas as compared to their Tspan6-expressing littermates (Fig. 6d). Western blotting of tumor tissue confirmed efficient loss of Tspan6 in Tspan6fl/y KrasG12D mice (Supplementary Fig. 5d). Similar to the whole-body mutant mice, cell proliferation was significantly enhanced in hyperplastic regions and adenomas from the conditional Tspan6fl/y KrasG12D mice (Fig. 6e). There were no significant differences in numbers of apoptotic cells (detected by cleaved caspase 3), intratumoral CD3+ T cells, intratumoral FoxP3+ regulatory T cells, and tumor angiogenesis as detected by CD31 immunostaining (Supplementary Fig. 7). In conclusion, our genetic data show that Tspan6 exerts a cell autonomous function in suppressing KRasG12D-driven initiation and progression of lung cancer.
Fig. 6. TSPAN6 has tumor cell-intrinsic functions.

a Conditional Tspan6 deletion significantly decreases survival (Kaplan Meier blot) in lung cancer-bearing Tspan6fl/y KrasG12D mice as compared to Tspan6+/y KrasG12D control littermates. n = 10 per group. *P < 0.05 (Log-rank test). b Representative H&E-stained histological sections of both cohorts 6 weeks after Ad-Cre inhalation. Magnifications ×5. c Representative microCT images to detect lung tumors in Tspan6fl/y KrasG12D and Tspan6+/y KrasG12D littermates 8 weeks after Ad-Cre inhalation. d Quantification of hyperplasic regions, adenomas and adenocarcinomas in lungs from Tspan6fl/y KrasG12D (n = 5) and control Tspan6+/y KrasG12D (n = 5) littermates at the indicated time points following Ad-Cre inhalation. Data are shown as means ± s.e.m. *P < 0.05; **P < 0.01; N.S. = not significant (Student’s t test). e Quantification of % Ki67+ cells in hyperplasic regions, adenomas, and adenocarcinomas 4 and 8 weeks post Ad-Cre infection. Data are shown as mean ± s.e.m. *P < 0.05; **P < 0.01; N.S. = not significant (Student’s t test). f p53 ablation in KrasG12D lung tumors abrogates the survival advantage conferred by Tspan6 expression. Kaplan Meier survival plots are shown for whole-body Tspan6-/y p53fl/fl KrasG12D mice (n = 4) and Tspan6+/y p53fl/fl KrasG12D (n = 6) littermates, and conditional Tspan6fl/y p53fl/fl KrasG12D mice (n = 5) and their respective Tspan6+/y p53fl/fl KrasG12D (n = 8) littermates. A Log-rank test was used for statistical analysis.
Finally, we asked the question of whether the survival advantage conferred by Tspan6 expression in KrasG12D transformed cells is dependent on p53 using p53floxed mice. Using either whole-body Tspan6 knockouts or Tspan6floxed alleles (Fig. 6f), tumor-specific deletion of p53 abolished the protective effects of Tspan6 expression on KrasG12D transformed cells. In conclusion, our data indicate that Tspan6 exerts a cell autonomous function in suppressing KrasG12D-driven initiation and progression of lung cancer, and Tspan6’s tumor suppressor function is p53 dependent.
TSPAN6 expression stratifies survival in human lung and pancreatic cancer
Subgroups of Non-Small Cell Lung Cancer (NSCLC) were identified based on EMT and epithelial-expression signatures with differing therapy responses [35, 36]. As our results showed that depletion of TSPAN6 in epithelial cells leads to an EMT and tumor progression, we asked whether TSPAN6 expression could be used to differentiate between EMT and epithelial subgroups of NSCLC tumors. TSPAN6 expression indeed differentiated between the EMT and epithelial NSCLC tumor subgroups, with high TSPAN6 expression being associated with an epithelial signature (Fig. 7a), which correlates with better therapy response and overall patient survival [36]. We then asked whether TSPAN6 expression in NSCLC tumors predicts patient survival. Survival analysis conducted for 1926 NSCLC patients revealed a significant correlation (p = 0.0008) between low TSPAN6 tumor expression and reduced patient survival (Fig. 7b).
Fig. 7. Low TSPAN6 expression is correlated with poor survival in human lung and pancreatic cancer patient survival with mesenchymal signatures.

a TSPAN6 expression is significantly reduced in lung cancer (NSCLC) cancers from the GSE10245 cohort with an EMT signature ((NLCLC-EMT) versus an epithelial (NLCLCepith) signature (**p < 0.01)). The TSPAN6 probes used are indicated. b Low TSPAN6 expression levels (Affymetrix probes) predict poor overall survival of lung cancer patients from this cohort. Survival analysis was calculated using a log-rank test. Data presented were obtained using KM plotter. P values (log-rank test) and total numbers of patients with either low (black) or high (red) TSPAN6 expression are indicated. c TSPAN6 expression is significantly reduced in pancreatic adenocarcinoma (PDA) cancers with an EMT signature (PDA-exoc and PDA-QM) relative to those with an epithelial signature (PDA-epith) in the GSE17891 and GSE11838 cohorts. d Low TSPAN6 expression is correlated with poor survival in pancreatic ductal carcinoma patients from these cohorts (data presented as in b). Green bar-TSPAN6 low expression identifies PDA patients with advanced disease and early relapse with lethal outcome (18–24 months). NSCLCepith Non-Small Cell Lung Cancer epithelial subclass, NSCLCmes Non-Small Cell Lung Cancer mesenchymal subclass [36]; PDAepith Pancreatic Ductual Adenocarcinoma epithelial subclass, PDAexocrine Pancreatic Ductual Adenocarcinoma exocrine subclass, PDA-QM Pancreatic Ductual Adenocarcinoma quasi-mesenchymal subclass [37].
Similarly, pancreatic adenocarcinomas have been classified based on their EMT and epithelial-expression signatures [37]. We found that low expression of TSPAN6 correlated with pancreatic adenocarcinomas with an EMT signature (Panc-exoc and Panc-Qm) relative to those with an epithelial signature (Panc-epith) in two pancreatic cancer cohorts [37, 38] (Fig. 7c). Importantly, low TSPAN6 expression also significantly correlated with poor survival in these pancreatic adenocarcinoma cohorts (p = 0.028 for GSE17891 and p = 0.001 for GSE11838) (Fig. 7d). Together these data indicate that low TSPAN6 expression levels correlate with lung and pancreatic cancer EMT signatures and also with poor survival in these cancer patients.
Discussion
Our study has revealed a novel role for the Tetraspanin TSPAN6 as a tumor suppressor in Ras-driven cancer (Fig. 8). We show that in normal human mammary epithelial cells, knockdown of TSPAN6 cooperates with H-RASV12 to drive invasive properties, similarly to knockdown of the cell polarity regulators SCRIB and DLG1. In mouse orthotopic transplantation experiments, Tspan6 knockdown promotes tumorigenesis of H-RasV12-transformed mouse mammary epithelial (EpRas) cells, and overexpression of TSPAN6 abrogates the tumorigenic potential of K-Ras activating mutant human pancreatic cancer cells. Moreover, whole-body and lung-specific knockout of Tspan6 cooperates with KrasG12D to promote lung cell tumors, by promoting cell proliferation, an EMT and invasive/metastatic properties. Moreover, Tspan6’s tumor suppressor function in KrasG12D-driven lung cancer in the mouse model is p53 dependent. Importantly, low TSPAN6 expression was correlated with an EMT signature and poor survival of human patients with non-small cell lung and pancreatic cancers, indicating that TSPAN6 functions as a tumor suppressor in human epithelial cancers. Mechanistically, TSPAN6 binds to the EGFR to inhibit EGFR-RAS-ERK signaling.
Fig. 8. Model for the role of TSPAN6 in the regulation of signaling pathways in tumorigenesis.
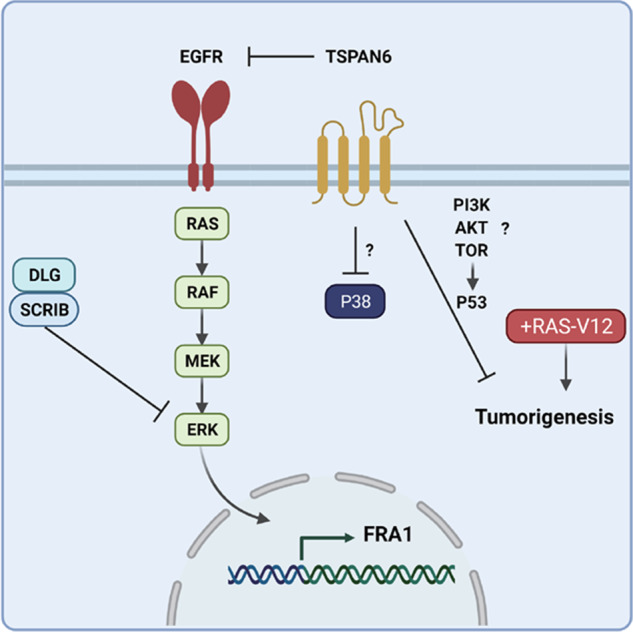
We show in this study that depletion of TSPAN6 cooperates with oncogenic RAS in tumorigenesis and that the tumor suppressor role of TSPAN6 requires P53. We show that TSPAN6 binds to and inhibits EGFR activation and thereby inhibits RAS activation. TSPAN6 does not bind to SCRIB or DLG1, suggesting that they act in parallel to each other in the regulation of the EGFR-RAS-ERK pathway, with TSPAN6 regulating EGFR and SCRIB/DLG1 regulating ERK. TSPAN6 also inhibits P38 activation by an unknown mechanism. TSPAN6 may be required for the activation of the tumor suppressor P53 by a pathway involving the activation of PI3K-AKT-TOR. Additionally, TSPAN6 alters epithelial cell morphology with its depletion leading to an EMT, which may further activate RAS-ERK signaling through SCRIB/DLG1 impairment. “?”, indicate that the potential involvement of TSPAN6 in these pathways needs to be confirmed.
Our data showing knockdown of TSPAN6 phenocopies the knockdown of SCRIB and DLG1 in cooperation with H-RasV12 is consistent with our observations that TSPAN6 knockdown induces cell morphology changes leading to an EMT in cooperation with oncogenic RAS. Our previous studies in Drosophila have revealed that the TSPAN6 ortholog, Tsp29Fb, regulates Dlg1 protein localization and genetically interacts with scrib in epithelial development [14], and whether TSPAN6 also affects DLG1/SCRIB localization/function in mammalian cells is of interest to determine. In Drosophila, various Tetraspanins have been linked to the assembly or function of the Septate Junction [39, 40], which functions similarly to mammalian Tight Junctions in epithelial barrier formation [41]. Interestingly, the Drosophila Tetraspanin protein, Tsp2A, regulates the stability of the cell polarity protein, atypical protein kinase C (aPKC), which is a regulator of Hippo-Yki and JAK-STAT signaling in intestinal epithelial cells, thereby limiting cell proliferation [40]. Whether Tsp29Fb/TSPAN6 also regulates aPKC to mediate its tumor suppressor effects remains to be determined.
Mechanistically, our genetic and biochemical experiments show that TSPAN6 modulates EGFR-RAS-ERK signaling (Fig. 8). Moreover, TSPAN6 can bind to the EGFR suggesting that it directly negatively regulates EGFR phosphorylation and signaling. Indeed a recent study has shown that TSPAN6 controls the production of the extracellular vesicles containing the transmembrane form of TGF-alpha, a EGFR ligand, and thereby downregulates EGFR signaling [42]. In this newly described mechanism, TSPAN6 is physically linked to TNF-alpha via the adapter protein syntenin-1, and upon TSPAN6 downregulation increased production of extracellular tm-TGF-alpha occurs leading to increased EGFR signaling [42]. Although our findings show that TSPAN6 knockdown phenocopies DLG1 and SCRIB knockdown, and our analysis in Drosophila has linked Tsp29Fb to Scrib/Dlg1 regulation/function [14], and that Scrib/Dlg1 are known to regulate Ras signaling in Drosophila and mammalian cells [11, 30, 43, 44], since TSPAN6 does not bind to DLG1 or SCRIB, it is likely that these proteins regulate EGFR-RAS signaling by parallel pathways, with TSPAN6 functioning at the level of the EGFR by binding to it and inhibiting its activation, and SCRIB/DLG1 directly binding to and negatively regulating ERK activation [43]. However, it is possible that knockdown of Tsp29Fb/TSPAN6 may also deregulate RAS-ERK signaling indirectly through inducing an EMT and impairing SRIB/DLG1 function.
Whether Tsp29Fb/TSPAN6 regulates other signaling pathways needs to be further explored; however the decrease in phospho-P38 observed upon TSPAN6 overexpression, suggests that it also negatively regulates the P38-stress response pathway (Fig. 8), a JUN-kinase-related pathway that cooperates with oncogenic Ras in tumorigenesis in Drosophila and mammalian systems [45, 46]. Furthermore, how the tumor suppressor role of TSPAN6 in RAS-driven tumorigenesis is linked to P53 function requires further analysis. P53 is a key tumor suppressor that is mutated in ~50% of human cancer, which functions by inhibiting cell cycle progression, promoting senescence and inducing cell death [47–49]. Since high levels of RAS signaling can also induce cell cycle arrest and senescence [7, 8], it is possible that P53 contributes to these tumor-suppression mechanisms and TSPAN6 might be involved in P53 activation. Interestingly, another tetraspanin, CD9, induces senescence by inducing P53 via the PI3K-AKT-TOR pathway [50]. Intriguingly, TSPAN6 overexpression led to an increase in phospho-AKT and phospho-TOR suggesting that it may also function similarly to CD9 in inducing P53 activity through the PI3K-AKT-TOR pathway (Fig. 8). Further analysis is required to confirm and further elucidate the potential regulation of P38 and PI3K-AKT-TOR signaling and P53 activity by TSPAN6.
In Drosophila, apical-basal cell polarity mutants or deregulation of cytoskeletal proteins (such as Src, Rac1, RhoGEF2), result in epithelial cells undergoing cell morphology changes and becoming migratory, resembling an EMT in mammalian cells [25], and knockdown of the TSPAN6 ortholog, Tsp29Fb, in Drosophila affects Dlg1 localization, enhances scrib mutant cell polarity-impaired phenotypes and promotes invasive overgrowth together with oncogenic Ras [14]. Importantly, knockdown of Tspan6 was sufficient to trigger an EMT in Ras-transformed mouse mammary epithelial cells and in in vivo tumor experiments we also observed an EMT, as defined by altered Vimentin and E-Cadherin expression, in Tspan6 knockdown cells. Moreover, induction of TSPAN6 in human pancreatic cancer cells inhibits EGFR-induced expression of the EMT markers Vimentin and N-Cadherin, as well as the critical EMT transcription factor Slug, thereby linking TSPAN6 to the inhibition of EGFR-mediated EMT in human tumor cells. Together, these observations in Drosophila [14] and mammalian cells (this study) suggest that TSPAN6 plays a role in regulating apico-basal cell polarity and controlling cellular architecture. Whether these effects on epithelial architecture modulate EGFR-RAS-ERK activation (perhaps through altering SCRIB/DLG1 function) or affect other signaling pathways to contribute to the tumor suppressor function of Tsp29Fb/TSPAN6 needs to be examined. Considering our results that TSPAN6 had no apparent effect on EGFR-induced proliferation but markedly altered invasive behavior in response to EGF, TSPAN6-regulated cell polarity and an EMT could conceivably explain why the modulation of TSPAN6 expression affects metastatic tumor spread.
In summary, we have shown that TSPAN6 binds to the EGFR and inhibits EGFR-RAS-ERK signaling. Furthermore, our results show that overexpression of TSPAN6 impairs cell proliferation and invasion of pancreatic tumor cells in vitro and abrogates tumor growth and metastatic spread in orthotopic tumor implants in vivo. Conversely, knockdown of human TSPAN6 in human mammary epithelial cells enhances the oncogenic RAS-driven invasive phenotype, and knockdown of mouse Tspan6 enhances cell proliferation and in vivo tumor growth and metastases of oncogenic Ras-transformed mouse epithelial cells. Importantly, by generating Tspan6 whole-body mutant mice and a conditional Tspan6floxed allele, we provide definitive evidence that loss of Tspan6 markedly enhances the initiation as well as malignant progression of oncogenic Kras-driven lung cancer in a tumor cell autonomous manner. Additionally, our results indicate that loss of Tspan6 results in enhanced epithelial proliferation and altered epithelial architecture implicated in cell morphology, migration and tumor invasion/metastasis. Thus, TSPAN6 constitutes an evolutionary conserved bona fide suppressor of oncogenic RAS-driven tumor growth and invasion/metastasis in human pancreatic cancer cells, human and mouse mammary epithelial cells and mouse lung tumors. Moreover, our analysis of human lung and pancreatic cancer cohorts shows a correlation of low TSPAN6 expression with cancers with an EMT signature and with poor patient survival. In addition to lung and pancreatic cancer, mining of various databases, including TCGA, revealed mutations, deletions, and copy number variations of TSPAN6 in multiple human cancers (not shown). A recent study has revealed that TSPAN6 also plays a tumor suppressor role in colorectal cancer [42], but whether TSPAN6 has a tumor suppressor role in additional cancer types, apart from pancreatic, lung and colorectal, needs to be explored in future experiments.
Materials and methods
Mammary and pancreatic cell line analysis
Generation of Tspan6 mutant mice
Whole-body Tspan6 mutant mice were generated by DeltaGene. Briefly, a targeting vector was inserted into exon 2 of the murine Tspan6 locus. The linearized construct was electroporated into embryonic stem (ES) cells derived from the 129/OlaHsd mouse sub-strain. Correctly targeted ES cell clones were confirmed by Southern blotting and used to generate chimeric mice. Germline transmitted F1 mice were backcrossed to C57BL/6 females. The following primers were used for genotyping giving a mutant band of 405 bp and a 260 bp wild type band: endogenous forward primer TGTGATCAAGGACTCAAGCTTGTAC; the Neo primer GGGTGGGATTAGATAAATGCCTGCTCT; and the endogenous reverse primer CTTACTCACCAGTTTCAGCATCCAG.
For the generation of conditional Tspan6floxed mice we performed recombineering to introduce loxP sites, and positive and negative selection markers, into BAC DNA by homologous recombination. BAC clones were ordered from bacpac.chori.org/vectorsdet.htm containing the region of interest of the Tspan6 gene. The final targeting construct was electroporated into A9 ES cells which were screened for correct recombination events via PCR. For PCR screening the following primers were used:
Neo/Short arm primers—product size 998:
Forward 5′ TGGACGTAAACTCCTCTTCAGACCT 3′ (Tm = 61C)
-
Reverse 5′ CCCTGTCTTAGTTGGCCCTAACAGTA 3′ (Tm = 61C)
Short arm/genomic region primers - product size 1052
Forward 5′ TCCTTATCGCCTCCAAGGAAAGGAT 3′ (Tm = 61C)
Reverse 5′CAGAGGCAGGTAGATCTTCAGATCTTC 3′ (Tm = 60C)
Conditional Tspan6fl/fl and whole-body Tspan6 mutant mice were backcrossed for at least ten generations onto a C57BL/6J background and then crossed to LSL-K-RasG12D mice to generate Tspan6fl/y LSL-K-RasG12D and Tspan6+/y LSL-K-RasG12D mice and their respective Tspan6 expressing littermate control cohorts. Mouse genotypes were determined by PCR and, if required for confirmation, by DNA blot analysis. In all experiments, only age and sex-matched littermate mice were used. All mice were maintained according to the ethical animal license protocol complying with the Austrian and European legislation.
Induction of lung cancer
Inhalation of mice with Ad-Cre viruses was performed as previously reported [51]. In brief, experimental animals were anesthetized with 10% Ketasol/Xylasol and placed on a heated pad. An Ad-Cre-CaCl2 precipitate was produced by mixing 60 μl MEM, 2.5 μl Ad-Cre (1010 pfu/ml; University of Iowa, Gene Transfer Vector Core Iowa, USA) and 0.6 μl CaCl2 (1 M) for each mouse and incubated for 20 min at room temperature (21–22 °C).
Histology, immunohistochemistry, and lung/tumor ratio quantification
Histological and immunohistochemical analysis of lung tumors was performed as previously described [51]. Briefly, 2 µm sections from at least 3 different planes of the lung were cut and stained with H&E. Sections were scanned using a Mirax slide scanner and lung/tumor areas automatically scored by an algorithm programmed and executed using the Definiens software suite and visually controlled in a blinded way. Immunohistochemistry staining was done using an automatic staining machine (Leica Bond3) or manually processed. Sections were dehydrated and antigenic epitopes retrieved using a 10-mM citrate buffer and microwaving for 10 min Specimens were then incubated with anti-Foxp3 (eBioscience, 13-5773), anti-CD3ε (Santa Cruz, 101442), anti-Ki67 (Novocastra), anti-CD31 (Abcam, ab28364), and anti-cleaved Caspase 3 (Cell signaling, 9661). Primary antibody staining was detected by peroxidase-conjugated anti-rabbit IgG. Positive cells were counted on 20 randomly chosen tumor areas at ×400 magnifications in a double-blinded fashion. Images were captured with a Zeiss AxioImager Z1. Quantitative analysis was performed using HistoQuestTm software (TissueGnostics GmbH, Vienna, Austria, www.tissuegnostics.com). These experiments were independently replicated, with biological and technical replicates as detailed in the legend of the corresponding figure, with similar results.
MicroCT scanning
Formaldehyde perfused and fixed lungs were stained in a solution of 1% elemental iodine and 2% potassium iodide in distilled water for three days. After staining, specimens were rinsed mounted in plastic tubes for microCT scanning. Lungs were scanned using a SCANCO µCT 35 (SCANCO Medical AG, Brüttisellen, Switzerland) with a source energy of 70keV and an intensity of 114 µA using a 0.2 mm copper filter. Projection images were recorded with an angular increment of 0.36°. Reconstructed microCT slices measured 1024 × 1024 pixels (voxel size = 20 µm). Image stacks were imported into Amira® 5.3 (Visualization Sciences Group, Mérignac Cedex, France) and filtered with a 3D median filter (3 × 3 × 3 kernel). For segmentation of lung and tumor tissue, specific attenuation thresholds were used. For discriminating background from lung tissue an X-ray attenuation value of µ = 0.3987 and for discriminating lung tissue from tumor tissue an attenuation value of μ = 1.2776 was used, respectively. Based on segmentation, lung tissue and tumor volumes were calculated. These experiments were independently replicated, with biological and technical replicates as detailed in the figure legend, with similar results.
Human cancer data and statistical analysis
Supplementary information
Acknowledgements
We thank all members of our laboratories for helpful discussions as well as VDRC and IMP/IMBA core facilities for excellent technical support. We also acknowledge Life Science Editors for editorial support (www.lifescienceeditors.com). JMP was supported by IMBA, ERC Advanced Investigator grant, an Innovator Award from Era of Hope, the T. von Zastrow foundation, and a Canada 150 Chair in Functional Genetics. TZP was supported by a Marie Curie Excellence grant. RK was supported by Infla-Care and Infect-ERA grants. RdMS was supported by a grant from Invest NI RD0412515. AF was supported from funds from the Cancer Council Victoria (Australia) to HER, POH, and JMP. HER and POH were supported by fellowships from the National Health & Medical Research Council (NHMRC), Australia, and by funds from La Trobe University.
Author contributions
JMP, HER, AF, KF wrote the paper, JMP, TFM, POH, HER coordinated the study, TZP, BP, AF, KF, RK, IK, GGN, AH conducted experiments, JMP, POH, HER analyzed data and finalized figures, RdMS, SJFC, conducted bioinformatic analysis, POH provided editorial advice.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Tamara Zoranovic Pryjda, Blanka Pranjic.
Change history
3/15/2022
A Correction to this paper has been published: 10.1038/s41388-022-02265-2
Contributor Information
Patrick O. Humbert, Email: P.Humbert@latrobe.edu.au
Helena E. Richardson, Email: H.Richardson@latrobe.edu.au
Josef M. Penninger, Email: josef.penninger@ubc.ca
Supplementary information
The online version contains supplementary material available at 10.1038/s41388-022-02223-y.
References
- 1.Bar-Sagi D. Mechanisms of signal transduction by Ras. Semin Cell Biol. 1992;3:93–8. doi: 10.1016/s1043-4682(10)80018-2. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2:344–58. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287–92. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Li F, Xu D, Hou K, Fang W, Li Y. The function of RAS mutation in cancer and advances in its drug research. Curr Pharm Des. 2019;25:1105–14. [DOI] [PubMed]
- 6.Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeNicola GM, Tuveson DA. RAS in cellular transformation and senescence. Eur J Cancer. 2009;45(Suppl 1):211–6. doi: 10.1016/S0959-8049(09)70036-X. [DOI] [PubMed] [Google Scholar]
- 8.Dimauro T, David G. Ras-induced senescence and its physiological relevance in cancer. Curr Cancer Drug Targets. 2010;10:869–76. doi: 10.2174/156800910793357998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sacco E, Spinelli M, Vanoni M. Approaches to Ras signaling modulation and treatment of Ras-dependent disorders: a patent review (2007—present) Expert Opin Ther Pat. 2012;22:1263–87. doi: 10.1517/13543776.2012.728586. [DOI] [PubMed] [Google Scholar]
- 10.Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
- 11.Dow LE, Kauffman JS, Caddy J, Zarbalis K, Peterson AS, Jane SM, et al. Loss of human Scribble cooperates with H-Ras to promote cell invasion through deregulation of MAPK signalling. Oncogene. 2008;27:5988–6001. doi: 10.1038/onc.2008.219. [DOI] [PubMed] [Google Scholar]
- 12.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell. 2017;171:1301–15e1314. doi: 10.1016/j.cell.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet. 2015;47:996–1002. doi: 10.1038/ng.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zoranovic T, Manent J, Willoughby L, Matos de Simoes R, La Marca JE, Golenkina S, et al. A genome-wide Drosophila epithelial tumorigenesis screen identifies Tetraspanin 29Fb as an evolutionarily conserved suppressor of Ras-driven cancer. PLoS Genet. 2018;14:e1007688. doi: 10.1371/journal.pgen.1007688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubinstein E. The complexity of tetraspanins. Biochem Soc Trans. 2011;39:501–5. doi: 10.1042/BST0390501. [DOI] [PubMed] [Google Scholar]
- 16.Charrin S, Jouannet S, Boucheix C, Rubinstein E. Tetraspanins at a glance. J Cell Sci. 2014;127:3641–8. doi: 10.1242/jcs.154906. [DOI] [PubMed] [Google Scholar]
- 17.Kummer D, Steinbacher T, Schwietzer MF, Tholmann S, Ebnet K. Tetraspanins: integrating cell surface receptors to functional microdomains in homeostasis and disease. Med Microbiol Immunol. 2020;209:397–405. doi: 10.1007/s00430-020-00673-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer. 2014;14:49–60. doi: 10.1038/nrc3640. [DOI] [PubMed] [Google Scholar]
- 19.Lang T, Hochheimer N. Tetraspanins. Curr Biol. 2020;30:R204–6. doi: 10.1016/j.cub.2020.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Espana A, Chung PJ, Sarkar IN, Stiner E, Sun TT, Desalle R, et al. Appearance of new tetraspanin genes during vertebrate evolution. Genomics. 2008;91:326–34. doi: 10.1016/j.ygeno.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Yanez-Mo M, Barreiro O, Gordon-Alonso M, Sala-Valdes M, Sanchez-Madrid F. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 2009;19:434–46. doi: 10.1016/j.tcb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Zoller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer. 2009;9:40–55. doi: 10.1038/nrc2543. [DOI] [PubMed] [Google Scholar]
- 23.Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769–79. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elsum I, Yates L, Humbert PO, Richardson HE. The Scribble-Dlg-Lgl polarity module in development and cancer: from flies to man. Essays Biochem. 2012;53:141–68. doi: 10.1042/bse0530141. [DOI] [PubMed] [Google Scholar]
- 25.Humbert PO, Grzeschik NA, Brumby AM, Galea R, Elsum I, Richardson HE. Control of tumourigenesis by the Scribble/Dlg/Lgl polarity module. Oncogene. 2008;27:6888–907. doi: 10.1038/onc.2008.341. [DOI] [PubMed] [Google Scholar]
- 26.Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol. 2006;16:1139–46. doi: 10.1016/j.cub.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 27.Leong GR, Goulding KR, Amin N, Richardson HE, Brumby AM. Scribble mutants promote aPKC and JNK-dependent epithelial neoplasia independently of Crumbs. BMC Biol. 2009;7:62. doi: 10.1186/1741-7007-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caddy J, Wilanowski T, Darido C, Dworkin S, Ting SB, Zhao Q, et al. Epidermal wound repair is regulated by the planar cell polarity signaling pathway. Dev Cell. 2010;19:138–47. doi: 10.1016/j.devcel.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dow LE, Kauffman JS, Caddy J, Zarbalis K, Peterson AS, Jane SM,, et al. The tumour-suppressor Scribble dictates cell polarity during directed epithelial migration: regulation of Rho GTPase recruitment to the leading edge. Oncogene. 2007;26:2272–82. doi: 10.1038/sj.onc.1210016. [DOI] [PubMed] [Google Scholar]
- 30.Stephens R, Lim K, Portela M, Kvansakul M, Humbert PO, Richardson HE. The scribble cell polarity module in the regulation of cell signaling in tissue development and tumorigenesis. J Mol Biol. 2018;430:3585–612. [DOI] [PubMed]
- 31.Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10:2462–77. [DOI] [PubMed]
- 32.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Termini CM, Gillette JM. Tetraspanins function as regulators of cellular signaling. Front Cell Dev Biol. 2017;5:34. doi: 10.3389/fcell.2017.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuner R, Muley T, Meister M, Ruschhaupt M, Buness A, Xu EC, et al. Global gene expression analysis reveals specific patterns of cell junctions in non-small cell lung cancer subtypes. Lung Cancer. 2009;63:32–8. doi: 10.1016/j.lungcan.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 36.Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med. 2014;6:1279–93. doi: 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–3. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balagurunathan Y, Morse DL, Hostetter G, Shanmugam V, Stafford P, Shack S, et al. Gene expression profiling-based identification of cell-surface targets for developing multimeric ligands in pancreatic cancer. Mol Cancer Ther. 2008;7:3071–80. doi: 10.1158/1535-7163.MCT-08-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fox RM, Andrew DJ. Changes in organelle position and epithelial architecture associated with loss of CrebA. Biol Open. 2015;4:317–30. doi: 10.1242/bio.201411205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu C, Tang HW, Hung RJ, Hu Y, Ni X, Housden BE,, et al. The septate junction protein Tsp2A restricts intestinal stem cell activity via endocytic regulation of aPKC and hippo signaling. Cell Rep. 2019;26:670–88e676. doi: 10.1016/j.celrep.2018.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tepass U. The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu Rev Cell Dev Biol. 2012;28:655–85. doi: 10.1146/annurev-cellbio-092910-154033. [DOI] [PubMed] [Google Scholar]
- 42.Andrijes R, Hejmadi RK, Pugh M, Rajesh S, Novitskaya V, Ibrahim M, et al. Tetraspanin 6 is a regulator of carcinogenesis in colorectal cancer. Proc Natl Acad Sci USA. 2021;118:e2011411118.. doi: 10.1073/pnas.2011411118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagasaka K, Pim D, Massimi P, Thomas M, Tomaic V, Subbaiah VK, et al. The cell polarity regulator hScrib controls ERK activation through a KIM site-dependent interaction. Oncogene. 2010;29:5311–21. doi: 10.1038/onc.2010.265. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto M, Ohsawa S, Kunimasa K, Igaki T. The ligand Sas and its receptor PTP10D drive tumour-suppressive cell competition. Nature. 2017;542:246–50. doi: 10.1038/nature21033. [DOI] [PubMed] [Google Scholar]
- 45.La Marca JE, Richardson HE. Two-faced: roles of JNK signalling during tumourigenesis in the Drosophila model. Front Cell Dev Biol. 2020;8:42. doi: 10.3389/fcell.2020.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 47.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–13. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6:a026104. doi: 10.1101/cshperspect.a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rizzotto D., Englmaier L., Villunger A. At a crossroads to cancer: how p53-induced cell fate decisions secure genome integrity. Int J Mol Sci. 2021;22:10883. 10.3390/ijms221910883. [DOI] [PMC free article] [PubMed]
- 50.Cho JH., Kim E-C., Son Y., lee D-W., Park YS., Choi JH, et al. CD9 induces cellular senescence and aggravates atherosclerotic plaque formation. Cell Death Differ. 2020;27:2681–96. [DOI] [PMC free article] [PubMed]
- 51.Schramek D, Sigl V, Penninger JM. RANKL and RANK in sex hormone-induced breast cancer and breast cancer metastasis. Trends Endocrinol Metab. 2011;22:188–94. doi: 10.1016/j.tem.2011.02.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.