

Abstract
Age‐associated obesity and muscle atrophy (sarcopenia) are intimately connected and are reciprocally regulated by adipose tissue and skeletal muscle dysfunction. During ageing, adipose inflammation leads to the redistribution of fat to the intra‐abdominal area (visceral fat) and fatty infiltrations in skeletal muscles, resulting in decreased overall strength and functionality. Lipids and their derivatives accumulate both within and between muscle cells, inducing mitochondrial dysfunction, disturbing β‐oxidation of fatty acids, and enhancing reactive oxygen species (ROS) production, leading to lipotoxicity and insulin resistance, as well as enhanced secretion of some pro‐inflammatory cytokines. In turn, these muscle‐secreted cytokines may exacerbate adipose tissue atrophy, support chronic low‐grade inflammation, and establish a vicious cycle of local hyperlipidaemia, insulin resistance, and inflammation that spreads systemically, thus promoting the development of sarcopenic obesity (SO). We call this the metabaging cycle. Patients with SO show an increased risk of systemic insulin resistance, systemic inflammation, associated chronic diseases, and the subsequent progression to full‐blown sarcopenia and even cachexia. Meanwhile in many cardiometabolic diseases, the ostensibly protective effect of obesity in extremely elderly subjects, also known as the ‘obesity paradox’, could possibly be explained by our theory that many elderly subjects with normal body mass index might actually harbour SO to various degrees, before it progresses to full‐blown severe sarcopenia. Our review outlines current knowledge concerning the possible chain of causation between sarcopenia and obesity, proposes a solution to the obesity paradox, and the role of fat mass in ageing.
Keywords: Sarcopenia, Obesity, Proto‐sarcopenia, Myosteatosis, Insulin resistance, Inflammation
Introduction
The animal body's remarkable ability to rapidly adapt to ever‐changing environmental and internal signals is vital for organismal health and survival. Modern human ageing is possibly a maladaptive programmed response to modern changes in food intake, sanitation and environmental pollution after several millennia of human civilization. Most notable in human ageing are the changes in physiology and body composition, such as the changes or redistribution in muscle and adipose mass, even if the total body weight remains unchanged.[ 1 ] Across populations today, muscle mass remains relatively stable during early life, but after age ~30 years, muscle mass declines at a rate of ~0.5–1.0% per year.[ 2 ] With ageing, the impaired balance between protein synthesis and proteolysis in skeletal muscle results in a progressive decline in skeletal muscle mass, strength, and function and is defined as sarcopenia.[ 3 ] As the strength of limb muscles and respiratory muscles gradually decrease, normal physical functions and activities such as breathing, standing, walking, and running will also decline.[ 2 ] On average, the peak strength decreases by 20–40% between 30 and 80 years old. Loss of muscle has a serious consequence on many chronic diseases, and on ageing itself, because it leads to weakness, loss of independence, and increased risk of death.[ 4 ] Various myokines released from active muscles act as signalling mediators between skeletal muscle and other vital organs, such as the liver, fat, and brain.[ 5 ] This signalling influences the progression of chronic disease such as diabetes, cardiovascular disease (CVD), osteoporosis, osteoarthritis, cancer, and many other ageing‐related diseases. Thus, sarcopenia represents a major health problem and an important burden for healthcare systems across the globe.[ 6 ]
Sarcopenic muscles show reduced numbers of stem cells (called satellite cells) and terminally differentiated myofibres. Moreover, type II glycolytic myofibres become hypotrophic with reduced diameter, and the muscles become infiltrated with adipose tissue and, at later stages, fibrotic tissue.[ 7 ] Fatty infiltration into both muscle and bone, and the redistribution of subcutaneous fat to the intra‐abdominal area (visceral fat), all result in decreased overall strength and functionality, increasing the risk of falls and fractures, and a potential increase in morbidity.[ 8 ] The proportions of visceral fat and intramuscular fat also increase with age, reaching a peak between 60 and 75 years old, as the proportion of subcutaneous fat decreases.[ 9 ] Interestingly, a major geriatric study of nonagenarians indicated that, muscle fat infiltration is still positively correlated with the area of pericardial and visceral adipose tissue in these oldest old individuals, although visceral fat mass strangely become protective against frailty in this age group (part of a phenomenon known as the obesity paradox, which we will address in a later section).[ 10 ] It is noteworthy that while the redistribution of fat does not necessarily manifest as obesity, intramuscular fat infiltration generally increases the chances of progression to obesity across adults.[ 11 ] In obesity, muscle progenitor cells could also differentiate to an adipocyte‐like phenotype as a result of paracrine signals from cytokines, leading to reduced muscular renewal, increased fatty infiltration, and thus a vicious cycle.[ 12 ] This synergy between muscle loss and fatty infiltration might trigger and aggravate the pathogenesis of sarcopenic obesity (SO).[ 13 ] SO refers to an obese disease condition accompanied by low skeletal muscle quality, strength and/or function, more common in the elderly, and seriously affects their quality of life by directly causing unstable walking, balance disorders, falls, and fractures.[ 14 ] Deposition of intramyocellular lipids further promotes lipotoxicity, which induces and aggravates mitochondrial dysfunction, oxidative stress, insulin resistance (IR), and inflammation.[ 15 ] These molecular changes interact with each other, leading to a vicious cycle that impairs the regeneration of muscle and blocks the recovery of physical function.[ 16 ] Hormone dysfunction is another explanation for the ageing‐induced increase in adipogenesis and the accelerated loss of bone and muscle mass after menopause in women and andropause in men.[ 17 ] Studies have increasingly recognized the importance of muscular fatty infiltrations for the age‐mediated loss of skeletal muscle function, and are beginning to suggest that this new important factor is closely linked to physical inactivity.[ 18 ]
In general, two major problems have plagued the field for many years. Firstly, sarcopenia and obesity are both considered multifactorial syndromes sharing various overlapping causes and feedback mechanisms. However, different studies have presented confusing views on the pathogenic relationship between sarcopenia and obesity, with no clear answer. Secondly, inflammation and insulin resistance both play important roles in sarcopenia and obesity, but the origins of local inflammation and insulin resistance, and how they cause systemic inflammation, systemic insulin resistance, and changes in body composition, had remained unclear.
Our review posits that assuming adipocyte dysfunction is the first of many drivers of local inflammation and insulin resistance (IR), followed by a vicious cycle (the metabaging cycle) that spreads systemically, may be the best unifying explanation for the metabolic syndrome that causes both obesity and sarcopenia during ageing, and further explains the obesity paradox in certain age groups. While previous reviews had focused on other aspects of sarcopenia and SO,[ 19 , 20 , 21 , 22 ] our review re‐analyses the current knowledge from new perspectives based on the following biomedical puzzles: (1) Does ectopic fatty infiltration into skeletal muscles lead to muscle atrophy even before sarcopenia is diagnosed? (2) Does lipid‐induced inflammaging and IR unify the chronic conditions of obesity and sarcopenia mechanistically, suggesting a vicious cycle causes SO? (3) If the mortality risk of sarcopenia patients is higher than SO patients, is fat mass really protective, as suggested in the obesity paradox? This review examines the possibility of a pathogenic mechanism with multiple drivers and triggers, proceeding from local hyperlipidaemia to fat redistribution and local inflammation to sarcopenia, suggesting that a metabaging cycle that spreads systemically may be the root cause for obesity, sarcopenia and the metabolic syndrome during ageing.
Methods
A comprehensive literature search was performed on the PubMed, EMBASE, OVID, and Cochrane library databases, using data from 1 January 2015 to 31 June 2021. The following search terms were used: sarcopenia or muscle wasting or muscle atrophy or sarcopenic or muscle attenuation, and obesity or obese or adiposity or fat mass. Primary studies were included if they met the following criteria: (1) study type: animal research, clinical research and cell experiments; (2) subjects: elderly persons and aged animals with sarcopenia or sarcopenic obesity; (3) primary indexes: muscle atrophy, ectopic fat infiltration, fat wasting, and decline of physical function. Studies were excluded if (1) study topic: non‐sarcopenia‐relevant basic research, neuromuscular junction mechanism and gut microbiota mechanisms; (2) subjects: young adults.
Hyperlipidaemia initially leads to local adipose inflammation and fat redistribution
During transient hyperlipidaemia after food intake, adipose tissues are driven to expand. The expansion of adipose depots can be driven either by the increase in adipocyte size (hypertrophy), or by the formation of new adipocytes from perivascular preadipocytes and pericytes (hyperplasia).[ 23 ] Under conditions of over‐nutrition, chronic stress, and/or long periods of physical inactivity, maladaptive adipose tissue expansion causes an increase in hypoxia in these cells, because of their expanded size.[ 24 ] Adipose tissue hypoxia either increases the expression of pro‐fibrotic genes and leads to adipose fibrosis, or directly leads to adipose necrosis, inducing infiltration by immune cells and adipose inflammation.[ 25 ]
The metabolic and redox states of adipocytes also dictate the adipose tissue responses.[ 26 ] During ageing, the extant adipocytes gradually develop resistance to insulin, leptin, fibroblast growth factor‐21 (FGF21), and other secreted factors, leading to further elevation in local glucose and lipid concentrations.[ 27 ] Furthermore, the extant adipocytes' impaired insulin signalling permits lipolysis, further promoting local hyperlipidaemia.[ 28 ]
Thus, even before obesity becomes obvious, persistent hyperlipidaemia contributes to fat redistribution, for example, subcutaneous adipose inflammation and lipolysis, fuelling visceral adipose tissue (VAT) expansion.[ 23 ] Many other extracellular signals have also been reported to influence adipogenesis.[ 23 ] Besides lipids themselves, local levels of insulin, glucocorticoids, sex steroids, and bone morphogenetic proteins (BMPs) also directly regulate peroxisome proliferator‐activated receptor‐γ (PPARγ) and CCAAT/enhancer‐binding protein‐α (CEBPα), to facilitate preadipocyte differentiation.[ 29 ] Thus, hyperlipidaemia, hyperinsulinaemia, and other metabolic signalling cues drive the creation and expansion of new lipid storage depots during fat redistribution.[ 29 ]
Separately, systemic physiological states, such as those associated with inflammation, excessive oxidative stress, and the production of reactive oxygen species (ROS), have complex and context‐dependent effects on adipose tissue homeostasis and fibrosis.[ 30 ] ROS overproduction in myoblasts causes accumulation of S100B, which promote the myoblast‐to‐adipocyte transition, and myoblasts from sarcopenic subjects show properties of (pre)adipocytes.[ 31 ] In turn, S100B stimulates NF‐κB activity with resultant upregulation of YY1 and YY1‐dependent inhibition of microRNA‐133, a promyogenic and anti‐adipogenic microRNA,[ 32 ] ultimately leading to the transition of myoblasts into adipocytes.[ 31 ] Chronic inflammation in extant adipose depots and other tissues also contribute to the pathophysiology of adipose tissue dysfunction, including the gradual impairment of adipogenesis that contributes to IR and metabolic disease.[ 23 ] Pro‐inflammatory cytokines such as interleukin (IL)‐1β inhibit adipogenesis, or the ability of pre‐adipocytes to mature into functional lipid‐enriched adipocytes.[ 33 ] In a chronic inflammation microenvironment, preadipocytes can transdifferentiate into macrophage‐like cells, with increased expression of pro‐inflammatory cytokines and decreased adipogenic capacity in response to inflammatory stimuli.[ 34 ] This ultimately contributes to a vicious cycle of adipose tissue inflammation, lipolysis and fibrosis as well.
Lipid derivatives accumulate in muscle cells to cause myosteatosis
Inflammation‐induced lipolysis, hyperlipidaemia, and the progressive redistribution of lipids to newly formed visceral fat depots and nearby muscles lead to ectopic fat infiltration in many organs. Fat redistribution is a hallmark of modern ageing that has been validated amongst nonagenarians as well.[ 10 ] Excess lipids are thought to ‘spill over’ to other tissues, especially the skeletal muscles,[ 35 ] which accumulate the lipids as intermuscular adipose tissue (IMAT), intramuscular adipose tissue and intramyocellular lipid droplets (IMCLs) containing triacylglycerol or fatty acid derivatives such as diacylglycerols (DAG), long chain acyl CoAs, and ceramides.[ 36 ] Skeletal muscle ectopic fat infiltration, or myosteatosis, is an abnormal phenomenon that increases with ageing and is recognized to negatively correlate with muscle mass, strength, mobility and normal metabolism.[ 37 ] Evidence shows that skeletal muscle fat infiltration accelerates with increasing age amongst middle aged and older men.[ 38 ] Furthermore, changing rates in myosteatosis are likely more related to ageing than any particular disease.[ 39 ] Clinical data showed that skeletal muscle fat infiltration occurred in both lean and obese individuals, although the infiltrated muscle volume was positively associated with body mass index (BMI).[ 11 ] Skeletal muscle fat infiltration appears to be independent of muscle mass, however, suggesting it precedes muscle atrophy to cause sarcopenia.[ 37 ]
While the biological mechanisms underlying increased myosteatosis or skeletal muscle fat infiltration are currently unclear, recent evidence suggest the importance of leptin signalling,[ 40 ] fibro‐adipogenic precursor (FAP) cells,[ 18 ] and mitochondrial dysfunction.[ 14 ] The muscular environment contains various populations of tissue‐resident progenitors, including the muscular satellite cells and FAPs.[ 18 ] FAPs appear to be the main stem cell population implicated in sarcopenia‐related IMAT development.[ 18 ] In muscle disuse or pathological conditions, such as Duchenne muscular dystrophy, FAPs proliferate and differentiate into adipose and fibrous tissue.[ 41 ] Inflammatory deregulation and the possible overlap between tumour necrosis factor‐α (TNF‐α) and transforming growth factor β1 (TGF‐β1) signalling may promote survival of FAPs and adipocytic differentiation leading to IMAT development and fat accumulation during sarcopenia.[ 42 ] FAPs are highly influenced by environmental changes during ageing. The Notch signalling pathway may also be involved in FAP‐driven IMAT development during ageing.[ 43 ] This pathway is a major mechanism implicated in the maintenance of satellite cell quiescence as well as promotion of their proliferation after injury. During ageing, an attenuated Notch signalling response results in impaired proliferation of satellite cells and a failure in muscle regeneration.[ 44 ] Furthermore, TGF‐β is a Notch inhibitor upregulated during ageing and may explain the spontaneous entry of satellite cells into the cell cycle during ageing.[ 45 ] During ageing, altered Wnt10b signalling increases expression of key adipogenic genes to promote IMAT accumulation.[ 46 ] The Wnt signalling pathway appears to be particularly complex, especially when related to ageing because expression levels of Wnt proteins will not all vary in the same direction. Ageing is also linked to altered synthesis of nitric oxide (NO) in skeletal muscle,[ 47 ] which regulates FAP fate through inhibition of their differentiation into adipocytes.
Ectopic lipid deposition has also been linked to cellular stress responses and apoptosis via increased lipid metabolism.[ 48 ] IMCLs are mostly found in lipid droplets (LDs) containing perilipins (PLINs), which are believed to be involved in important aspects of muscle physiology such as the breakdown of stored lipids.[ 49 ] In skeletal muscle, lipolysis in the IMCL stores is mediated by lipoprotein lipases, including the adipose triglyceride lipase (ATGL) and hormone‐sensitive lipase (HSL).[ 50 ] HSL and ATGL need to work in concert to regulate lipolysis in skeletal muscle, and their discordant activity are thought to be responsible for dysregulated lipolysis and IR in the skeletal muscle of obese and type II diabetes mellitus (T2DM) individuals.[ 51 ] One of the major mechanisms that cause muscle IR is thought to be the myocytes' accumulation of secondary products of lipid metabolism such as DAG, ceramides, and other lipids in IMCLs.[ 13 ] Ceramide levels were found to be higher in obese and insulin‐resistant individuals compared to insulin‐sensitive muscle. Ceramides can directly induce cellular IR by blocking the downstream effects of insulin signalling, such as glucose transporter‐4 (GLUT4) translocation.[ 52 ]
Besides hyperlipidaemia‐induced ectopic lipid deposition, dysregulated lipid oxidation in mitochondria during ageing also predisposes individuals to muscle IR.[ 53 ] If not for the cumulative ROS damage to mitochondria with ageing, ectopic lipid deposition per se might not induce muscle IR, as is often seen in young endurance athletes, a phenomenon known as the Athletes' Paradox. But as a result of mitochondrial dysfunction with ageing, the lipid oxidation‐induced ROS and local inflammation lead to the activation of stress signalling pathways, such as c‐Jun N‐terminal kinase (JNK), IκB kinase (IKK), protein kinase C (PKC) and p38‐mitogen‐activated protein kinase (p38‐MAPK), all of which also suppress insulin‐PI3K‐mTOR signalling.[ 54 ] In addition, the impaired insulin‐PI3K‐mTOR signalling further inhibits muscle mitochondrial function, leading to further lipid accumulation and lipid oxidation‐induced ROS, thus creating a vicious cycle of myosteatosis, lipotoxicity and muscle IR with ageing.[ 55 ]
Myosteatosis leads to systemic inflammaging, systemic insulin resistance, and sarcopenic obesity
The vicious cycle of local myosteatosis and muscle IR can now complete a larger vicious cycle with local adipose inflammation to create a storm of inflammation to induce systemic IR (Figure 1 ). One important reason is that the local muscle IR leads to reduced lipid uptake and increases local free fatty acid (FFA) concentrations, thereby worsening the local hyperlipidaemia and local IMAT inflammation, as explained in the earlier section, and boosting the secretion of inflammatory factors by both adipose and muscle tissues.[ 56 ] Inflammaging, defined as the increase in senescence‐associated secretory phenotype (SASP) inflammatory cytokines with ageing, can spread this metabolic dysfunction systemically as this low‐grade chronic inflammatory state spreads inflammation‐induced IR, lipid dysfunction, and senescence across the adipose depots, muscles and other tissues.[ 57 ] Senescent cells accumulate in a range of tissues with age, secreting numerous pro‐inflammatory cytokines, chemokines, growth factors, and extracellular matrix (ECM) degrading proteins.[ 58 ] SASP is thought to have evolved as an adaptive signal to acutely and rapidly recruit immune cells to the vicinity of senescent cells in order to mediate their clearance during tissue development, wound healing or carcinogenesis. In the setting of chronic metabolic dysfunction, however, SASP becomes a maladaptive signal to progressively spread inflammation‐induced IR.
Figure 1.
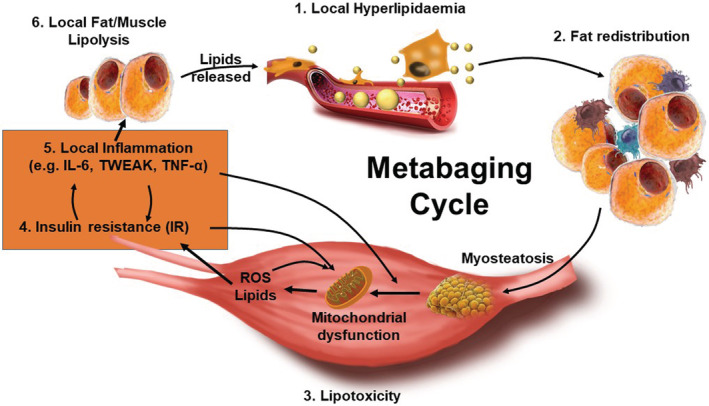
The metabaging cycle. (1) during transient hyperlipidaemia after food intake or inactivity, or local hyperlipidaemia due to lipolysis, nearby adipose tissues are driven to expand. (2) Excess lipids are thought to ‘spill over’ and redistribute to other tissues, especially the skeletal muscles. (3) Lipids and their derivatives accumulate both within and between muscle cells (myosteatosis), inducing mitochondrial dysfunction, disturbing β‐oxidation of fatty acids, and enhancing reactive oxygen species (ROS) production, leading to lipotoxicity, which also induces (4) insulin resistance and (5) inflammation. The pro‐inflammatory factors further induce infiltration of macrophages and other immune cells into adipose and muscle, which further secrete a large amount of pro‐inflammatory cytokines and chemokines, thus broadening the local chronic inflammation into a low‐grade systemic inflammaging state in adipose depots and muscles, which further spreads inflammation‐induced insulin resistance and lipid dysfunction. (6) The vicious cycle of local myosteatosis and muscle insulin resistance can now complete a larger vicious cycle leading to reducing lipid uptake and increasing local free fatty acid concentrations, and thus local lipolysis. The resultant local hyperlipidaemia, lipotoxicity, and insulin resistance that triggered the local inflammaging, further worsens the lipid dysfunction and insulin resistance in a spreading vicious cycle that results in sarcopenic obesity. We call this the metabaging cycle.
The pro‐inflammatory factors further induce infiltration of macrophages and other immune cells into adipose, muscle and other tissues, which further secrete a large amount of pro‐inflammatory cytokines and chemokines, thus broadening the local chronic inflammation into a systemic inflammaging condition in adipose depots, muscles and other tissues.[ 57 ] Notably, it was local hyperlipidaemia, lipotoxicity, and IR that triggered the local inflammaging, which worsened the lipid metabolism dysfunction in a spreading vicious cycle. We call this the metabaging cycle (Figure 1 ). As a result, this lipotoxic vicious cycle becomes the major mechanism that drives systemic IR and sarcopenic obesity.[ 57 ]
Sarcopenic obesity (SO), a combination of sarcopenia and obesity, is the concurrence of muscle loss and excessive body fat accrual.[ 59 ] The definition of SO has not been universally established, and that is partly due to the existence of various diagnostic criteria for both sarcopenia and obesity. Korea's recommended SO diagnostic criteria are defined as subjects fulfilling both the criteria for obesity (men body fat ≥27%, women body fat ≥38%) and the European Working Group on Sarcopenia in Older People 2 (EWGSOP2) criteria for sarcopenia.[ 60 ] However, current diagnostic criteria are likely inaccurate, because the concurrent loss of muscle and gain in fat could bring little or zero net change in body weight or BMI. This means many ‘normal’ subjects in current studies could actually be SO subjects that were not suspected nor tested for (pre‐)sarcopenia, and we might be grossly underestimating both the prevalence and the impact of SO. Nevertheless, in the longitudinal Korean Sarcopenic Obesity Study, the extent of visceral obesity at the start of the study was shown to correlate with the extent of appendicular muscle loss over ~2 years of follow‐up, indicating that fat redistribution may play a causal role in this association.[ 61 ] Compared with obesity alone, SO is associated with a heightened risk for adverse health outcomes, such as disability or impairment, CVD, metabolic diseases, morbidity, and mortality.[ 62 ]
In both animal models and human patients, obesity‐induced hyperlipidaemia and lipotoxicity‐induced inflammation is thought to be one of the drivers for SO.[ 63 ] As mentioned before at the local level, but now at the systemic level, the main effect of lipotoxicity is the marked impairment of mitochondria, characterized by enhanced ROS production, disturbed mitophagy and reduced mitochondrial biogenesis. This is accompanied by significant functional disturbances, such as reduced lipid β‐oxidation of fatty acids and lipolysis,[ 64 ] resulting in systemic IR.
Enhanced release of fatty acids and dysregulated production of adipokines can activate and recruit macrophages via binding chemokine (C‐C motif) receptor 2 (CCR2), CD36 and/or toll‐like receptor 4 (TLR4),[ 65 ] as well as other immune cells via activation of adhesion molecules and triggering chemotaxis, worsening the pro‐inflammatory vicious cycle. For example, ectopic lipids induce Nod‐like receptors protein 3 (NLRP3) inflammasome formation to trigger caspase‐1, which subsequently activate IL‐1β and IL‐18, thus activating the pro‐inflammatory M1 macrophages resident in the skeletal muscle.[ 66 ] These macrophages release TNFα, IL‐1β, IL‐6, monocyte‐chemoattractant protein‐1 (MCP‐1), and TGFβ, which can also stimulate trans‐differentiation of fibroblasts into myofibroblasts to cause fibrosis and further tissue damage, worsening the systemic inflammaging.
Macrophages have emerged as an important player in tissue inflammaging. Macrophages undergo polarization from an anti‐inflammatory (M2) to a pro‐inflammatory (M1) phenotype, apparently under the influence of accumulating Th1 and Th17 CD4+ cells, and CD8+ cells.[ 67 ] M1 macrophages activated by Th1 mediators, such as interferon γ (IFNγ), go on to secrete pro‐inflammatory molecules like TNFα, IL1‐β, IL‐6, and MCP‐1/CCL2. Such a M2‐to‐M1 switch is linked to the emergence of systemic IR.[ 67 ] Adipose tissue‐resident B cells, which are localized around macrophage clusters, have also been shown to have a predominant pathogenic role in obesity‐related IR in mice.[ 68 ] These pathogenic B cells activate IFNγ‐producing CD4+ and CD8 + T cells and/or IL‐17‐secreting Th1 cells within the visceral adipose tissues, presumably through LTB4/leukotriene‐B4 receptor 1 (LTB4R1) signalling. All these pro‐inflammatory immune cells secrete a complex mixture of cytokines and chemokines to induce, support, and exacerbate tissue inflammation.
While adipose tissue responds to pro‐inflammatory cytokines with fibrosis, lipolysis, fat redistribution and central obesity, as explained in the earlier section: skeletal muscle tissue responds to pro‐inflammatory cytokines with atrophy and sarcopenia. Cytokines such as TNF‐α, TNF‐like weak promoter of apoptosis (TWEAK), IL‐18, and IL‐6, are thought to induce muscle atrophy during sarcopenia.[ 69 ] Increased levels of TNF‐α and TWEAK have been shown to inhibit the expression of anabolism hormones such as insulin and insulin growth factor 1 (IGF‐1).[ 70 ] Additionally, the local expression of IGF‐1 is blunted by pro‐inflammatory cytokines TNFα and IL‐1β, leading to impairment of glucose disposal into the muscle and the resistance of mTORC1 activation from insulin receptor substrate 1 (IRS1) phosphorylation and its downstream cascade, an example of the synergy between IR and anabolic resistance.[ 70 ] High TNF‐α concentrations also result in the activation of the caspase cascade via ROS production and TNF receptor 1 (TNFR1) activation. TNFR1 activation increases muscle cell apoptosis, leading to the release of more TNF‐α to exacerbate the vicious cycle.[ 71 ] Besides its role in programmed cell death, TNF‐α also upregulates the expression of the muscle ubiquitin ligase muscle ring finger1 (MuRF1) via the NF‐κB pathway, to promote muscle proteolysis and muscle atrophy.[ 71 ] In addition, TNF‐α also upregulates ROS via increased lipolysis and lipid oxidation, activating NF‐κB either directly or indirectly.[ 72 ] In turn, the increased amount of NF‐κB may contribute to an increased accumulation of ceramide in sarcopenic muscle and the attenuation of anabolic signalling by resistance exercise.[ 73 ] Indeed, ceramide, a fatty acid‐derived second messenger in TNF‐α and IL‐1β signalling pathways, was a key element in insulin/IGF‐1 resistance and thus anabolic resistance in muscle cells.[ 74 ] Under pathological conditions, TWEAK also becomes overexpressed and activates the NF‐κB pathway via the FGF‐inducible 14 receptors, leading to increased expression of MuRF1 and activation of the ubiquitin‐proteasome (UPS) pathway as well.[ 69 ] Thus, systemic inflammaging induces a complex variety of mechanisms to cause muscle atrophy during SO.
Cross‐talk in inflammaging and insulin resistance leads to full‐blown sarcopenia
Sarcopenic obesity patients may appear as ‘normal’ subjects due to the mutual masking effect of sarcopenia and obesity. But after a long period of systemic inflammaging, chronic inflammation in adipose depots would lead to the systemic atrophy and wasting observed in cachexia and full‐blown sarcopenia.[ 23 ] White adipose tissue (WAT) lipolysis and WAT browning are examples of how chronic inflammation can cause adipose tissue wasting.[ 75 ] In cancer cachexia, lipolysis and (reversible) adipose wasting may occur to an extent before (irreversible) muscle wasting is detected.[ 76 ] This suggests that SO may progress to full‐blown sarcopenia in the same way that cachexia develops: systemic inflammation‐induced adipose atrophy, which unmasks and exacerbates the muscle atrophy (Figure 2 ).
Figure 2.
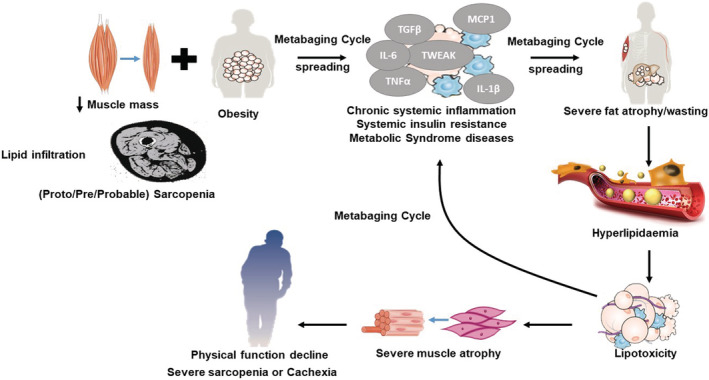
Resolving the obesity paradox. During ageing, adipose inflammation leads to the redistribution of fat to the intra‐abdominal area (visceral fat) and fatty infiltration into muscle. Intramuscular fat infiltration increases the chances of progression to obesity, which reduced muscular renewal, progressing to pre‐sarcopenia. This synergy between muscle loss (pre‐sarcopenia) and fatty infiltration (myosteatosis) might trigger and aggravate the pathogenesis of sarcopenic obesity (SO), low‐grade inflammation (inflammaging) and systemic insulin resistance. SO patients may appear as ‘normal’ subjects due to the mutual masking effect of sarcopenia and obesity. The vicious cycle of local myosteatosis and muscle insulin resistance can complete a larger vicious cycle leading to increasing lipolysis and local free fatty acid concentrations (the metabaging cycle), thereby worsening and spreading the local hyperlipidaemia. But after a long period of systemic inflammaging, chronic inflammation would exhaust both extant and newly formed adipose depots, leading to the systemic atrophy and wasting observed in cachexia and full‐blown sarcopenia. This suggests that SO may progress to full‐blown sarcopenia in the same way that cachexia develops: systemic inflammation‐induced adipose atrophy, which unmasks and exacerbates the muscle atrophy.
Increased lipolysis and lipid oxidation, decreased lipogenesis, lipid deposition, and adipogenesis, as well as WAT browning, are known to underlie adipose atrophy.[ 77 ] Inflammatory TNF‐α, or cachectin, is a primary cytokine in the repertoire of pro‐inflammatory factors and a strong promoter of the hydrolysis of intracellular TGs in fat cells.[ 71 ] It can stimulate lipolysis by at least three separate mechanisms. One is by inhibiting insulin receptor signalling, thereby counteracting the anti‐lipolytic effect of the insulin hormone. Another is by inhibiting signalling through the Gi‐protein‐coupled adenosine receptor to counteract the anti‐lipolytic effect of adenosine. The third way is via direct stimulation of basal (non‐hormonal) lipolysis through interactions with the lipid‐binding protein perilipin. In human adipocytes, TNF‐α‐mediated lipolysis is dependent on down‐regulation of perilipin expression via mitogen activated protein kinases (MAPKs) p44/42 and JNK, which could be of etiological importance for the development of IR in both cachexia and obesity.[ 78 ] Correlation analyses indicated a significantly positive association between serum TNF‐α and serum FFA in early‐stage cachexia. In fact, serum IL‐6, another pro‐inflammatory cytokine, is also positively correlated with serum FFA in both early‐stage and late‐stage cachexia.[ 75 ] Inflammatory cytokines like IL‐6 and tumour‐derived parathyroid hormone‐related protein (PTHrP) can also mediate WAT browning by inducing expression of thermogenic genes.[ 79 ] WAT browning was associated with increased expression of brown fat markers including UCP‐1, peroxisome proliferator‐activated receptor‐γ co‐activator‐1α (PGC‐1α), PPARγ and cell death‐inducing DNA fragmentation factor‐like effector A (CIDEA) in cachectic mice.[ 80 ] Thus, inflammatory factors can directly cause lipolysis and WAT browning to induce adipose atrophy (Figure 1 and 2 ).
As mentioned in the earlier section, inflammation can also cause IR. In humans, insulin and catecholamines are the most important hormones regulating adipocyte lipolysis under normal conditions.[ 81 ] Catecholamines such as adrenaline stimulate human adipocyte lipolysis through an increase in cyclic adenosine monophosphate (cAMP) levels, resulting in protein kinase A (PKA)‐mediated phosphorylation and activation of HSL.[ 82 ] Insulin usually inhibits catecholamine‐induced lipolysis through the disruption of PKA scaffolding induced by β‐adrenoceptor signalling.[ 83 ] In IR subjects, however, the lipolytic effect of catecholamines is increased in VAT. Lipolysis in VAT enables direct delivery of FFAs to the liver, which may lead to elevated hepatic triglyceride (TG) production, increased very low‐density lipoprotein secretion, and higher plasma TGs which exacerbates an already dysregulated hyperlipidaemic state.[ 79 ]
In the presence of chronic inflammation and the resultant IR, muscle atrophy is exacerbated as well. Besides the direct inflammation‐induced mechanisms explained in earlier section, the muscle protein synthesis response to amino acids is also impaired, in part via the suppression of insulin signalling, which is required to drive mTORC1 signalling for protein synthesis.[ 84 ] Meanwhile, increased mitochondrial ROS production and insulin resistance can activate the forkhead box O family of transcription factors (FoxO) pathway. FoxOs co‐ordinate a variety of stress‐response genes, including autophagy and ROS detoxification, during catabolic conditions.[ 85 ] Yet a ROS‐FoxO cross‐talk occurs in cells whereby ROS and FoxO reciprocally regulate their levels,[ 86 ] and old sedentary rat muscles show elevated levels of the atrophy genes (atrogenes), atrogin‐1 and MuRF‐1, both of which are FoxO targets.[ 87 ] In turn, the atrogenes trigger the UPS and the lysosome‐autophagy system that significantly contribute to disturbed mitochondrial fission and muscle remodelling, causing severe muscle atrophy.
Sarcopenic obesity often correlates with chronic disease incidence, while sarcopenia often correlates with chronic disease severity
As a pathological characteristic for several diseases, local IMAT could have a feedback effect on whole‐body metabolism and influence other chronic diseases. Cross‐sectional studies have reported a higher incidence of cardiovascular risk factors and metabolic syndrome in SO subjects.[ 88 ] Furthermore, the effect of muscle fat infiltration on vascular alterations may constitute a significant mechanism that augments cardiovascular risk in lean patients with chronic obstructive pulmonary disease (COPD).[ 89 ] SO was associated with a higher risk of hypertension, dyslipidaemia and IR, and up to an 8‐fold increased risk for the metabolic syndrome compared with non‐sarcopenic and non‐obese subjects.[ 90 ] Similar findings were reported in a large cross‐sectional analysis of over 14 000 adults from the National Health and Nutrition Examination Survey III, in which the SO group (defined by BIA‐measured muscle mass and BMI) had the highest risk of IR and dysglycaemia.[ 91 ] Nearly 20% of treatment‐seeking overweight and obese adult women displayed SO and showed a significantly higher prevalence of T2D and hypertension than those without SO.[ 92 ] Logistic regression analysis showed that SO increases the odds of having T2D and hypertension by nearly 550% [odds ratio (OR) = 5.42, 95% confidence interval (95% CI): 1.37–21.40, P < 0.05].[ 92 ] Amongst 239 SO patients, 43.5% subjects had chronic kidney disease, 12.1% subjects had ischaemic heart disease, and 5.9% subjects had stroke.[ 93 ] SO was significantly associated with reduced index scores for immediate memory and language in fully adjusted models with corresponding βs −2.71 (95% CI: −5.06 to −0.36; P = 0.024) and −2.48 (95% CI: −4.87 to −0.08; P = 0.043).[ 94 ] Obesity itself leads to hepatic steatosis or non‐alcoholic fatty liver disease, which can proceed to non‐alcoholic steatohepatitis,[ 95 ] as well as obesity‐related cancers such as colorectal and breast cancer.[ 96 ]
Full‐blown sarcopenia, on the other hand, often leads to loss of respiratory function, and increased risk of death.[ 4 ] Sarcopenic individuals also have a significantly higher risk of life‐threatening falls (cross‐sectional studies: OR = 1.60, 95% CI: 1.37–1.86, P < 0.001, I2 = 34%; prospective studies: OR = 1.89, 95% CI: 1.33–2.68, P < 0.001, I2 = 37%) and fractures (cross‐sectional studies: OR = 1.84, 95% CI 1.30–2.62, P = 0.001, I2 = 91%; prospective studies: OR = 1.71, 95% CI 1.44–2.03, P = 0.011, I2 = 0%), compared with non‐sarcopenic individuals.[ 97 ] Early studies have shown that 15.7% patients with late‐stage diabetes had full‐blown sarcopenia, and skeletal muscle mass index values were significantly decreased in patients with diabetes.[ 98 ] A cross‐sectional analysis from KNHANES 2008–2011 reported that 12.2% of non‐alcoholic fatty liver disease subjects had sarcopenia, and full‐blown sarcopenia was significantly associated with severe liver fibrosis.[ 99 ] Moreover, in both liver cirrhosis and chronic heart failure patients, skeletal muscle loss seemed to reflect a steady decline in health, rather than acute severity of disease,[ 98 , 99 ] with both sarcopenia and the chronic diseases concurrently worsening each other.
Thus, it seems that SO is frequently associated with increased risk of chronic disease incidence, while full‐blown sarcopenia is frequently associated with increased chronic disease severity and risk of death. On one hand, this suggests that as muscle wasting progresses from SO to sarcopenia, it is associated with a worsened prognosis for several chronic diseases. On the other hand, in the context of sarcopenia, this might also suggest that obesity might be ‘protective’ against chronic disease severity and death, a phenomenon similar to the ‘obesity paradox’.
Sarcopenic obesity progresses to full‐blown sarcopenia: solving the obesity paradox
In many cardiometabolic diseases, the so‐called ‘obesity paradox’ could perhaps be partly explained by our theory that SO progresses to full‐blown sarcopenia in a continuum during ageing and ageing‐related chronic diseases (Figure 2 ). In 2002, Gruberg et al. observed better health outcomes in overweight and obese patients with coronary heart disease, compared with their normal‐weight counterparts. This unexpected phenomenon, which contradicted the commonly held view that obesity worsens metabolic and health outcomes, was described as the ‘obesity paradox’.[ 102 ] A systematic review of 40 cohort studies with 250 152 patients confirmed that overweight patients had significantly lower risks for total mortality (risk ratio = 0.87) and cardiovascular mortality (risk ratio = 0.88).[ 103 ] This ‘obesity paradox’ phenomenon was also confirmed in patients with hypertension, chronic heart failure, peripheral arterial disease, stroke, thromboembolism, T2D, COPD, and osteoporosis.[ 104 ] Similarly, obese sarcopenic elderly show significantly lower mortality than non‐obese sarcopenic elderly. A cross‐section study indicated that robust nonagenerians presented significantly greater pericardial and abdominal adipose volume than the frail nonagenarians, who are at higher risk of death.[ 10 ] In another elderly cohort where the mean age was 77.22 ± 7.22 years, sarcopenic subjects (HR = 9.26, P < 0.001) had nearly twice as high a risk of mortality as SO subjects (HR = 5.23, P < 0.001), relative to control subjects.[ 22 ] While many explanations have been offered to explain the obesity paradox,[ 105 ] including confounding variables like smoking status, collider stratification bias, and the imperfections of BMI as an obesity measure, none have been definitive. Many of these studies neglected the metabolic effects of inconspicuous fat tissue, like visceral adiposity, and obesity masked by sarcopenia.[ 106 ] In fact, a systematic review of older adults aged ≥65 years found that BMI in the overweight range is not associated with a significantly increased mortality risk, and BMI in the moderately obese range is only associated with a modest increase in mortality risk.[ 107 ] Similarly, a more recent large meta‐analysis of nearly 200 000 individuals aged 65 or older showed a U‐shaped relationship between BMI and mortality, with the lowest risk seen in those with a BMI between 24.0 and 30.0 kg/m2 and risk only began to increase when BMI exceeded 33 kg/m2 in severe obesity.[ 108 ] A larger elderly cohort study showed the association between mortality and combined measurements of BMI and waist‐to‐hip ratio, even after adjusting for various factors. They found that central adiposity was associated with mortality, even amongst subjects with a normal BMI,[ 109 ] suggesting that the obesity paradox may at least partly result from failing to accurately account for central adiposity in some patients with ‘normal BMI’.
Our metabaging theory that SO progresses to sarcopenia, and the extensive evidence presented in sections, further suggests that the higher mortality in the ‘normal BMI’ elderly groups might also be partly because the underlying chronic disease has actually progressed to the point of being associated with full‐blown sarcopenia, instead of SO, in some patients. In other words, fat loss is not an independent causative factor but a tightly associated feature of chronic disease (including sarcopenia) progression, severity, and death in the oldest old. First, we know unintentional weight loss (>5% reduction in both lean and fat mass within 12 months) occurs in ~20% of older adults, and this trend is associated with increased mortality and morbidities, such as malignancy, non‐malignant gastrointestinal disease, and psychiatric conditions.[ 110 ] Second, this is likely an underestimate since ageing‐related lean mass decreases tend to be masked and offset by gains in fat mass or obesity in the early stages of chronic disease,[ 109 , 110 ] due to metabolic imbalance, such that lean mass loss only manifests upon severe inflammation and then fat loss in the later stages.[ 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 ] Third, in cancer, fat loss is often a result of cancer progression, which advances regardless of a patient's body weight. For example, a cross‐sectional investigation for gastric cancer, the mean subcutaneous fat area was significantly lower in deceased gastric cancer patients [108.0 (±70.0) cm2], than in patients who were still alive over 6 months [155.5 (±74.3) cm2](P < 0.001).[ 113 ] Thus, fat mass is merely another effect of cancer progression, the ultimate cause of mortality in these cases. The metabaging cycle can explain this too, by accounting for the severe inflammation induced by advanced cancers and the resultant lipolysis in cancer cachexia. In a final thought experiment to illustrate this point, given that obesity and hyperinsulinaemia also increases the risk of many ageing‐related cancers (and other chronic diseases), if one were to compare early‐stage (obesity‐enriched) cancer patients with late‐stage cancer (cachexia‐enriched) patients, our theory predicts and explains why one would observe obesity ‘protects’ against cancer mortality as well, that is, the obesity paradox.
Conclusion: importance of diagnostic criteria and methods
Besides supporting our theory of sarcopenia progression with metabaging and the role of fat loss in the obesity paradox (Figure 2 ), all these findings also suggest that we should search for indices that are better than BMI to measure body fat redistribution and visceral obesity more accurately. Unfortunately, the correct assessment of SO and full‐blown sarcopenia still represents a challenge for clinicians. Various techniques have emerged for assessing muscle loss, amongst which, computed tomography (CT) and magnetic resonance imaging are deemed as the gold standard for distinguishing fat and other soft tissues from muscle mass. However, the high costs of magnetic resonance imaging and the risks of CT radiation make them difficult to recommend for sarcopenia diagnosis in general.[ 114 ] Another low radiation method called dual‐energy‐X‐ray absorptiometry is recommended to estimate the lean mass and fat mass of the whole body or certain regions of the body. However, the high costs and scarce availability of this technique have led to the search for more convenient alternatives. As an affordable and readily available tool, bioelectrical impedance analysis (BIA) can be used to estimate total muscle mass as well. However, current BIA devices cannot accurately quantify muscle mass, for it indirectly reflects whole body composition, which is influenced by fluid and food intake, internal body fat, and disease‐related conditions.[ 115 ] Thus, BIA is mainly used as a method for screening sarcopenia while dual‐energy‐X‐ray absorptiometry is better for distinguishing muscle from fat and other tissues in elderly persons. One possible area of improvement is to use portable BIA devices that can accurately measure the muscle mass of locally defined body parts, such as the forearm, thigh or calf.
In oncology, CT images are a routine part of treatment and are available from patient records as a chart review.[ 77 ] CT image analysis has emerged as the gold standard for body composition assessment in cancer patients due to its ability to discriminate and quantify muscle, adipose tissue and other organ masses. Inconsistent methods of assessing fat and reporting values as cross‐sectional area (cm2) or volume (cm3), total fat mass (kg or %), change in area or the rate of changes limit the ability to interpret and compare studies, however.[ 77 ] The recent development of the D3‐creatine dilution method[ 116 ] with high reproducibility and minimized invasiveness has accomplished promising results in the estimation of skeletal muscle mass, but its adoption in the clinical setting as a routine method remains difficult. A robust panel of body fluid biomarkers to detect the first signs of muscular loss has not been established either. Another frequently seen comorbidity in these patients is cachexia, which is also in itself often accompanied by reduced hand grip strength and/or low walking speed, as well as worse performance in the short physical performance battery test.[ 117 ] Recently, in a study based on 469 830 UK Biobank participants, associations of sarcopenia with adverse outcomes (all‐cause mortality, incidence and mortality from CVD, respiratory disease, and COPD) were strongest when sarcopenia was defined as slow gait speed plus low muscle mass, followed by severe sarcopenia, strongly suggesting that this combination of physical capability markers should still be considered in the diagnosis of sarcopenia.[ 118 ] However, recent data indicated that gait speed lacked strong association with physical function, while hand grip strength could robustly and conveniently predict sarcopenia.[ 119 ]
Serum homocysteine (OR = 1.9, 95% CI: 1.0–3.6) and highly sensitive C‐reactive protein (hsCRP) (OR = 3.9, 95% CI: 2.2–6.9) were also independent predictors of sarcopenia in 1582 participants, with stronger correlations seen in women.[ 120 ] More innovative efforts are needed to establish a globally standardized definition, assessment method and prediction method for sarcopenia.
Part of this involves recognizing that significant changes in body composition do take place during ageing, even if the BMI appears unchanged, for example during fat redistribution, myosteatosis, and muscle weakness/atrophy. In modern ageing, adipose tissue undergoes expansion, inflammation, fibrosis and atrophy, due to the sedentary lifestyle, chronic stress, and excess nutritional intake. The resultant transfer of lipids from the subcutaneous adipose depot to the VAT, intermuscular and intramuscular spaces lead to myosteatosis, lipotoxicity, IR, and a chronic inflammatory environment. This lipotoxic vicious cycle spreads systemically in what we term the metabaging cycle (Figure 1 ). It is worth noting that myosteatosis does not imply obesity, but a special state of fat redistribution accompanied by skeletal muscle atrophy and the persistent secretion of proinflammatory factors, although a study on nonagenerians suggested that myosteatosis is well‐correlated with visceral adiposity.[ 10 ] New diagnosis methodologies are needed to capture this state early, for example by using reliable predictive serum or urine biomarkers for inflammaging, or by measuring limb muscle mass via portable BIA, or perhaps by taking CT scans of muscle vs. fat distribution. Given that myosteatosis is likely to cause local muscle IR, inflammation and atrophy, that could rapidly spread systemically via the metabaging cycle to cause SO and full‐blown sarcopenia, the field may need to study and define another state of ‘proto‐sarcopenia’, defined by a certain threshold of myosteatosis, for timely diagnosis and early metabolic interventions. It might also be worthwhile to expand current SO studies to include definitions of pre‐sarcopenia and probable sarcopenia according to muscle weakness, and definitions of obesity according to abdominal circumference or waist‐to‐hip ratios, to correct for current BMI‐based underestimations of SO prevalence and clinical impact.
For many years, an outstanding unresolved problem is whether a ‘healthy’ obesity phenotype exists, and which is the best level of BMI to maintain, given the obesity paradox? This has been very controversial, especially regarding the effect of obesity on the elderly. Certainly, deciphering the underlying mechanisms is important not only for achieving scientific progress in the field of metabolism, but also for creating new prophylactic and therapeutic interventions in the clinic. Based on the latest evidence on adipose and skeletal muscle pathology, as reviewed above, we believe there exists a complex cross‐talk between lipotoxicity, adipose and skeletal muscle atrophy, two of the largest tissues in the human body by mass, via the establishment and spreading of a metabaging cycle, in which lipid‐induced chronic inflammation and IR leads to sarcopenia and obesity concurrently, and heightens the risk for metabolic syndrome diseases. It should be mentioned that several key questions concerning the process of fat redistribution and skeletal muscle atrophy still remain unanswered. For example, what are the dominant molecular, nutritional, and genetic factors that drive ectopic fat redistribution and/or myosteatosis? What are the key lipids that mediate lipotoxicity? Is severe sarcopenia equivalent to cachexia? What interventions can halt the metabaging cycle to reverse SO and the metabolic syndrome? Additional mechanistic, longitudinal, prospective, and well‐controlled studies will go a long way towards fighting the current epidemic of metabolic syndrome diseases.
Conflict of interest
Chun‐wei Li, Kang Yu, Ng Shyh‐Chang, Zongmin Jiang, Taoyan Liu, Shilin Ma, Lanfang Luo, Lu Guang, Kun Liang, Wenwu Ma, Hefan Miao, Wenhua Cao, Ruirui Liu, Ling‐juan Jiang, Song‐lin Yu, Chao Li, Hui‐jun Liu, Long‐yu Xu, Rong‐ji Liu, Xin‐yuan Zhang, and Gao‐shan Liu declare that they have no conflicts of interest.
Acknowledgements
This material is based upon work supported by; National Natural Science Foundation of China: 1191957202, 81900782; Strategic Priority Research Program of the CAS: XDA16010109; National Key R&D Program of China: 2018YFE0201103, 2018YFC1004102, 2019YFA0801701, 2021YFE0111800; Whole People Nutrition Research Fund: CNSNNSRG2021‐129; Key Research of Program of the CAS: KJZD‐SW‐L04;CAS Project for Young Scientists in Basic Research: YSBR‐012; Strategic Collaborative Research Program of the Ferring Institute of Reproductive Medicine: FIRMA180301, FIRMA200507; Nutrition Scientific Research Foundation of BY‐HEALTH:TY0171102; Foundation of Tianjin Union Medical Center (2017YJ023). The authors' responsibilities were as follows—KY, NS‐C, and C‐WL designed the program; C‐WL, Z‐MJ, T‐YL, S‐LM, L‐FL, LG, KL, W‐WM, H‐FM, R‐RL, L‐JJ, S‐LY, CL, H‐JL, L‐YX, R‐JL, X‐YZ, and G‐SL wrote the paper; KY and NS‐C guided the whole programme and revising the manuscript. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.
Li C.‐w., Yu K., Shyh‐Chang N., Jiang Z., Liu T., Ma S., Luo L., Guang L., Liang K., Ma W., Miao H., Cao W., Liu R., Jiang L.‐j., Yu S.‐l., Li C., Liu H.‐j., Xu L.‐y., Liu R.‐j., Zhang X.‐y., and Liu G.‐s. (2022) Pathogenesis of sarcopenia and the relationship with fat mass: descriptive review, Journal of Cachexia, Sarcopenia and Muscle, 13, 781–794, 10.1002/jcsm.12901
Contributor Information
Kang Yu, Email: yuk1997@sina.com.
Ng Shyh‐Chang, Email: huangsq@ioz.ac.cn.
References
- 1. Zamboni M, Mazzali G, Fantin F, Rossi A, Francesco VD. Sarcopenic obesity: a new category of obesity in the elderly. Nutr Metab Cardiovasc Dis Actions. 2008;18:388–395. [DOI] [PubMed] [Google Scholar]
- 2. Kemmler W, Stengel SV, Schoene D. Longitudinal changes in muscle mass and function in older men at increased risk for sarcopenia—the FrOST‐Study. J Frailty Aging. 2019;8:57–61. [DOI] [PubMed] [Google Scholar]
- 3. Murton AJ, Marimuthu K, Mallinson JE, Selby AL, Smith K, Rennie MJ, et al. Obesity appears to be associated with altered muscle protein synthetic and breakdown responses to increased nutrient delivery in older men, but not reduced muscle mass or contractile function. Diabetes. 2015;64:3160–3171. [DOI] [PubMed] [Google Scholar]
- 4. Kob R, Fellner C, Bertsch T, Wittmann A, Mishura D, Sieber CC, et al. Gender‐specific differences in the development of sarcopenia in the rodent model of the ageing high‐fat rat. J Cachexia Sarcopenia Muscle. 2015;6:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hong SH, Choi KM. Sarcopenic obesity, insulin resistance, and their implications in cardiovascular and metabolic consequences. Int J Mol Sci. 2020;21:494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakuma K, Aoi W, Yamaguchi A. Current understanding of sarcopenia: possible candidates modulating muscle mass. Pflugers Arch. 2015;467:213–229. [DOI] [PubMed] [Google Scholar]
- 7. Verdijk LB, Snijders T, Drost M, Delhaas T, Kadi F, Loon LJCV. Satellite cells in human skeletal muscle; from birth to old age. Age (Dordr). 2014;36:545–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cruz‐Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European consensus on definition and diagnosis: report of the European Working Group on Sarcopenia in Older People. Age Ageing. 2010;39:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stenholm S, Harris TB, Rantanen T, Visser M, Kritchevsky SB, Ferrucci L. Sarcopenic obesity: definition, cause and consequences. Curr Opin Clin Nutr Metab Care. 2008;11:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Idoate F, Cadore EL, Casas‐Herrero A, Zambom‐Ferraresi F, Marcellan T, Gordoa AR, et al. Adipose tissue compartments, muscle mass, muscle fat infiltration, and coronary calcium in institutionalized frail nonagenarians. Eur Radiol. 2015;25:2163–2175. [DOI] [PubMed] [Google Scholar]
- 11. Vivodtzev I, Moncharmont L, Tamisier R, Borel JC, Arbib F, Wuyam B, et al. Quadriceps muscle fat infiltration is associated with cardiometabolic risk in COPD. Clin Physiol Funct Imaging. 2018;38:788–797. [DOI] [PubMed] [Google Scholar]
- 12. Kob R, Bollheimer LC, Bertsch T, Fellner C, Djukic M, Sieber CC, et al. Sarcopenic obesity: molecular clues to a better understanding of its pathogenesis? Biogerontology. 2015;16:15–29. [DOI] [PubMed] [Google Scholar]
- 13. Tardif N, Salles J, Guillet C, Tordjman J, Reggio S, Landrier JF, et al. Muscle ectopic fat deposition contributes to anabolic resistance in obese sarcopenic old rats through eIF2α activation. Aging Cell. 2014;13:1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee DC, Shook RP, Drenowatz C, Blair SN. Physical activity and sarcopenic obesity: definition, assessment, prevalence and mechanism. Future Sci OA. 2016;2:FSO127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. D'Souza K, Mercer A, Mawhinney H, Pulinilkunnil T, Udenigwe CC, Kienesberger PC. Whey peptides stimulate differentiation and lipid metabolism in adipocytes and ameliorate lipotoxicity‐induced insulin resistance in muscle cells. Nutrients. 2020;12:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lipina C, Hundal HS. Lipid modulation of skeletal muscle mass and function. J Cachexia Sarcopenia Muscle. 2017;8:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abildgaard J, Ploug T, Al‐Saoudi E, Wagner T, Thomsen C, Ewertsen C , et al. Changes in abdominal subcutaneous adipose tissue phenotype following menopause is associated with increased visceral fat mass. Sci Rep. 2021;11:14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brioche T, Pagano AF, Py G, Chopard A. Muscle wasting and aging: experimental models, fatty infiltrations, and prevention. Mol Asp Med. 2016;50:56–87. [DOI] [PubMed] [Google Scholar]
- 19. Ham DJ, Borsch A, Lin S, Thurkauf M, Weihrauch M, Reinhard JR, et al. The neuromuscular junction is a focal point of mTORC1 signaling in sarcopenia. Nat Commun. 2020;11:4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He LX, Khanal P, Morse CI, Williams A, Thomis M. Associations of combined genetic and epigenetic scores with muscle size and muscle strength: a pilot study in older women. J Cachexia Sarcopenia Muscle. 2020;11:1548–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Picca A, Ponziani FR, Calvani R, Marini F, Biancolillo A, Coelho‐Junior HJ, et al. Gut microbial, inflammatory and metabolic signatures in older people with physical frailty and sarcopenia: results from the BIOSPHERE Study. Nutrients. 2019;12:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Atmis V, Yalcin A, Silay K, Ulutas S, Bahsi R, Turgut T, et al. The relationship between all‐cause mortality sarcopenia and sarcopenic obesity among hospitalized older people. Aging Clin Exp Res. 2019;31:1563–1572. [DOI] [PubMed] [Google Scholar]
- 23. Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20:242–258. [DOI] [PubMed] [Google Scholar]
- 24. Halberg N, Khan T, Trujillo ME, Wernstedt‐Asterholm I, Attie AD, Sherwani S, et al. Hypoxia‐inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29:4467–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khan T, Muise ES, Iyengar P, Wang ZC, Chandalia M, Abate N, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29:1575–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao H. Adipocytokines in obesity and metabolic disease. J Endocrinol. 2014;220:T47–T59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stern JH, Rutkowski JM, Scherer PE. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016;23:770–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scherer PE. The multifaceted roles of adipose tissue‐therapeutic targets for diabetes and beyond: the 2015 Banting lecture. Diabetes. 2016;65:1452–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Back K, Arnqvist HJ. Changes in insulin and IGF‐I receptor expression during differentiation of human preadipocytes. Growth Hormon IGF Res. 2009;19:101–111. [DOI] [PubMed] [Google Scholar]
- 30. Aggarwal A, Costa MJ, Rivero‐GutiErrez B, Ji L, Morgan SL, Feldman BJ. The circadian clock regulates adipogenesis by a Per3 crosstalk pathway to Klf15. Cell Rep. 2017;21:2367–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morozzi G, Beccafico S, Bianchi R, Riuzzi F, Bellezza I, Giambanco I, et al. Oxidative stress‐induced S100B accumulation converts myoblasts into brown adipocytes via an NF‐κB/YY1/miR‐133 axis and NF‐κB/YY1/BMP‐7 axis. Cell Death Differ. 2017;24:2077–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yin H, Pasut A, Soleimani VD, Bentzinger CF, Antoun G, Thorn S, et al. MicroRNA‐133 controls brown adipose determination in skeletal muscle satellite cells by targeting Prdm16. Cell Metab. 2013;17:210–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McGillicuddy FC, Reynolds CM, Finucane O, Coleman E, Harford KA, Grant C, et al. Long‐term exposure to a high‐fat diet results in the development of glucose intolerance and insulin resistance in interleukin‐1 receptor I‐deficient mice. Am J Physiol Endocrinol Metab. 2013;305:E834–E844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Isakson P, Hammarstedt A, Gustafson B, Smith U. Impaired preadipocyte differentiation in human abdominal obesity: role of Wnt, tumor necrosis factor‐alpha, and inflammation. Diabetes. 2009;58:1550–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pienkowska J, Brzeska B, Kaszubowski M, Kozak O, Jankowska A, Szurowska E. MRI assessment of ectopic fat accumulation in pancreas, liver and skeletal muscle in patients with obesity, overweight and normal BMI in correlation with the presence of central obesity and metabolic syndrome. Diabetes Metab Syndr Obes. 2019;12:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ritter O, Jelenik T, Roden M. Lipid‐mediated muscle insulin resistance: different fat, different pathways? J Mol Med (Berl). 2015;93:831–843. [DOI] [PubMed] [Google Scholar]
- 37. Nachit M, Rudder MD, Thissen JP, Schakman O, Bouzin C, Horsmans Y, et al. Myosteatosis rather than sarcopenia associates with non‐alcoholic steatohepatitis in non‐alcoholic fatty liver disease preclinical models. J Cachexia Sarcopenia Muscle. 2021;12:144–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miljkovic I, Kuipers AL, Cvejkus R, Bunker CH, Patrick AL, Gordon CL, et al. Myosteatosis increases with aging and is associated with incident diabetes in African ancestry men. Obesity (Silver Spring). 2016;24:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beattie KA, MacIntyre NJ, Ramadan K, Inglis D, Maly MR. Longitudinal changes in intermuscular fat volume and quadriceps muscle volume in the thighs of women with knee osteoarthritis. Arthritis Care Res (Hoboken). 2012;64:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koteish A, Diehl DM. Animal models of steatohepatitis. J Cell Sci. 2002;16(5):679–690. [DOI] [PubMed] [Google Scholar]
- 41. Uezumi A, Ito T, Morikawa D, Shimizu N, Yoneda T, Segawa M, et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci. 2011;124:3654–3664. [DOI] [PubMed] [Google Scholar]
- 42. Merritt EK, Stec MJ, Thalacker‐Mercer A, Windham ST, Cross JM, Shelley DP, et al. Heightened muscle inflammation susceptibility may impair regenerative capacity in aging humans. J Appl Physiol. 2013;115:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bjornson CRR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells. 2012;30:232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch‐mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–1577. [DOI] [PubMed] [Google Scholar]
- 45. Xiao ZC, Zhang J, Peng XG, Dong YJ, Jia LX, Li HH Jr, et al. The Notch γ‐secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF‐β/Smad2/3 signaling pathway activation. Int J Biochem Cell Biol. 2014;55:65–71. [DOI] [PubMed] [Google Scholar]
- 46. Vertino AM, Taylor‐Jones JM, Longo KA, Bearden ED, Lane TF, McGehee Jr RE, et al. Wnt10b deficiency promotes co‐expression of myogenic and adipogenic programs in myoblasts. Mol Biol Cell. 2005;16:2039–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samengo G, Avik A, Fedor B, Whittaker D, Myung KH, Wehling‐Henricks M, et al. Age‐related loss of nitric oxide synthase in skeletal muscle causes reductions in calpain S‐nitrosylation that increase myofibril degradation and sarcopenia. Aging Cell. 2012;11:1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, et al. Lipid‐induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid‐induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:e12481, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pourteymour S, Lee S, Langleite TM, Eckardt K, Hjorth M, Bindesboll C, et al. Perilipin 4 in human skeletal muscle: localization and effect of physical activity. Physiol Rep. 2015;3:e12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Badin PM, Langin D, Moro C, et al. Dynamics of skeletal muscle lipid pools. Trends Endocrinol Metab. 2013;24:607–615. [DOI] [PubMed] [Google Scholar]
- 51. Kase ET, Feng YZ, Badin PM, Bakke SS, Laurens C, Coue M, et al . Primary defects in lipolysis and insulin action in skeletal muscle cells from type 2 diabetic individuals. Biochim Biophys Acta. 2015;1851:1194–1201. [DOI] [PubMed] [Google Scholar]
- 52. Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin‐stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol. 1998;18:5457–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Holloway GP, Chou CJ, Lally J, Stellingwerff T, Maher AC, Gavrilova O, et al . Increasing skeletal muscle fatty acid transport protein 1 (FATP1) targets fatty acids to oxidation and does not predispose mice to diet‐induced insulin resistance. Diabetologia. 2011;54:1457–1467. [DOI] [PubMed] [Google Scholar]
- 54. Aon MA, Bhatt N, Cortassa SC. Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol. 2014;5:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim KH, Choi S, Zhou Y, Kim EY, Lee JY, Saha PK, et al. Hepatic FXR/SHP axis modulates systemic glucose and fatty acid homeostasis in aged mice. Hepatology. 2017;66:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sachs S, Zarini S, Kahn DE, Harrison KA, Perreault L, Phang T, et al . Intermuscular adipose tissue directly modulates skeletal muscle insulin sensitivity in humans. Am J Physiol Endocrinol Metab. 2019;316:E866–E879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lackey DE, Olefsky JM, et al. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. 2016;12(1):15–28. [DOI] [PubMed] [Google Scholar]
- 58. Cecco MD, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, et al. L1 drives IFN in senescent cells and promotes age‐associated inflammation. Nature. 2019;566:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hong SH, Choi KM. Sarcopenic Obesity, Insulin Resistance, and Their Implications in Cardiovascular and Metabolic Consequences. Int J Mol Sci.. 2020;21:e115407, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim TN, Yang SJ, Yoo HJ, Lim KI, Kang HJ, Song W, et al . revalence of sarcopenia and sarcopenic obesity in Korean adults: the Korean sarcopenic obesity study. Int J Obes (Lond). 2009;33:885–892. [DOI] [PubMed] [Google Scholar]
- 61. Kim TN, Park MS, Ryu JY, Choi HY, Hong HC, Yoo YJ, et al . Impact of visceral fat on skeletal muscle mass and vice versa in a prospective cohort study: the Korean Sarcopenic Obesity Study (KSOS). PLoS One. 2014;9:e115407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Scott D, Chandrasekara SD, Laslett LL, Cicuttini F, Ebeling PR, Jones G. Associations of Sarcopenic Obesity and Dynapenic Obesity with Bone Mineral Density and Incident Fractures Over 5‐10 Years in Community‐Dwelling Older Adults. Calcif Tissue Int. 2016;99:30–42. [DOI] [PubMed] [Google Scholar]
- 63. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:2237–2238. [DOI] [PubMed] [Google Scholar]
- 65. Li L, Renier G. Adipocyte‐derived lipoprotein lipase induces macrophage activation and monocyte adhesion: role of fatty acids. Obesity (Silver Spring). 2007;15:102920, 2595–2604. [DOI] [PubMed] [Google Scholar]
- 66. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NALP3/NLRP3 Inflammasome Instigates Obesity‐Induced Autoinflammation and Insulin Resistance. Nat Med. 2011;17:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu JH, Zhang L, Shi JJ, He RZ, Yang WJ, Habtezion A, et al. Macrophage phenotypic switch orchestrates the inflammation and repair/regeneration following acute pancreatitis injury. EBioMedicine. 2020;58:102920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. DeFuria J, Belkina AC, Jagannathan‐Bogdan M, Snyder‐Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T‐cell function and an inflammatory cytokine profile. Proc Natl Acad Sci USA. 2013;110:5133–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li CW, Yu K, Ng SC, Li GX, Jiang LJ, Yu SL, et al . Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J Cachexia Sarcopenia Muscle.. 2019;10:586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kawakami M, Murase T, Ogawa H, Ishibashi S, Mori N, Takaku F, et al. Human recombinant TNF suppresses lipoprotein lipase activity and stimulates lipolysis in 3T3‐L1 cells. J Biochem. 1987;101:331–338. [DOI] [PubMed] [Google Scholar]
- 71. Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S, Leeuwenburgh C, et al. Age‐related activation of mitochondrial caspase‐independent apoptotic signaling in rat gastrocnemius muscle. Mech Ageing Dev. 2008;129(9):542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kadoguchi T, Shimada K, Miyazaki T, Kitamura K, Kunimoto M, Aikawa T, et al. Promotion of oxidative stress is associated with mitochondrial dysfunction and muscle atrophy in aging mice. Geriatr Gerontol Int. 2020;20:78–84. [DOI] [PubMed] [Google Scholar]
- 73. Rivas DA, Morris EP, Haran PH, Pasha EP, Morais MS, Dolnikowski GG, et al . Increased ceramide content and NFκB signaling may contribute to the attenuation of anabolic signaling after resistance exercise in aged males. J Appl Physiol (1985). 2012;113:1727–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Strle K, Broussard SR, McCusker RH, Shen WH, Johnson RW, Freund GG, et al. Proinflammatory cytokine impairment of insulin‐like growth factor I‐induced protein synthesis in skeletal muscle myoblasts requires ceramide. Endocrinology. 2004;145:4592–4602. [DOI] [PubMed] [Google Scholar]
- 75. Jun H, Qingyang M, Lei S, Guohao W. Interleukin‐6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018;17:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Suman KD, Sandra E, Silvia S, Clemens D, Hannes T, Barbara G, et al . Adipose triglyceride lipase contributes to cancer‐associated cachexia. Science. 2001;333:233–238. [DOI] [PubMed] [Google Scholar]
- 77. Maryam E. Evidence and mechanisms of fat depletion in cancer. Nutrients. 2014;6:5280–5297.–Vera. –CM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ryden M, Arvidsson E, Blomqvist L, Perbeck L, Dicker K, Arner P, et al. Targets for TNF‐alpha‐induced lipolysis in human adipocytes. Biochem Biophys Res Commun. 2004;318:168–175. [DOI] [PubMed] [Google Scholar]
- 79. Hellmer J, Marcus C, Sonnenfeld T, Arner P. Mechanisms for differences in lipolysis between human subcutaneous and omental fat cells. J Clin Endocrinol Metab. 1992;75:15–20. [DOI] [PubMed] [Google Scholar]
- 80. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VF, et al. Tumour‐derived PTH‐related protein triggers adipose tissue browning and cancer cachexia. Nature. 2014;513:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A. Inflammasome ‐driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature. 2017;550:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Inoue T, Kobayashi K, Inoguchi T, Sonoda N, Fujii M, Maeda Y, et al . Reduced expression of adipose triglyceride lipase enhances tumor necrosis factor alpha‐induced intercellular adhesion molecule‐1 expression in human aortic endothelial cells via protein kinase C‐dependent activation of nuclear factor‐kappaB. J Biol Chem. 2011;286:32045–32053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. Insulin disrupts beta‐adrenergic signalling to protein kinase A in adipocytes. Nature.. 2005;437:569–573. [DOI] [PubMed] [Google Scholar]
- 84. Bond P. Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J Int Soc Sports Nutr. 2016;13:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Klotz LO, Steinbrenner H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017;13:646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ribeiro MBT, Guzzoni V, Hord JM, Lopes GN, Marqueti RC, Andrade RV, et al. Resistance training regulates gene expression of molecules associated with intramyocellular lipids, glucose signaling and fiber size in old rats. Sci Rep. 2017;7:8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kang SY, Lim GE, Kim YK, Kim HW, Lee K, Park TJ, et al. Association between Sarcopenic Obesity and Metabolic Syndrome in Postmenopausal Women: A Cross‐sectional Study Based on the Korean National Health and Nutritional Examination Surveys from 2008 to 2011. J Bone Metab. 2017;24:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang J, Cui C, Chim YN, Yao H, Shi L, Xu J. Vibration and β‐hydroxy‐β‐methyl butyrate treatment suppresses intramuscular fat infiltration and adipogenic differentiation in sarcopenic mice. J Cachexia Sarcopenia Muscle. 2020;11:e10805, 564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kim TN, Park MS, Lim KI, Choi HY, Yang SJ, Yoo HJ, et al . Relationships between sarcopenic obesity and insulin resistance, inflammation, and vitamin D status: the Korean Sarcopenic Obesity Study. Clin Endocrinol (Oxf). 2013;78:525–532. [DOI] [PubMed] [Google Scholar]
- 91. Srikanthan P, Hevener AL, Karlamangla AS. Sarcopenia exacerbates obesity‐associated insulin resistance and dysglycemia: findings from the National Health and Nutrition Examination Survey III. PLoS One. 2010;5:e10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kreidieh D, Itani L, Masri DE, Tannir H, Citarella R, Ghoch ME, et al. Association between Sarcopenic Obesity, Type 2 Diabetes, and Hypertension in Overweight and Obese Treatment‐Seeking Adult Women. J Cardiovasc Dev Dis. 2018;5:e107265, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Low S, Goh KS, Ng TP, Ang SF, Moh A, Wang J, et al . The prevalence of sarcopenic obesity and its association with cognitive performance in type 2 diabetes in Singapore. Clin Nutr. 2020;39:2274–2281. [DOI] [PubMed] [Google Scholar]
- 94. Negrin KA, Flach RJR, DiStefano MT, Matevossian A, Friedline RH, Jung DY, et al . IL‐1 signaling in obesity‐induced hepatic lipogenesis and steatosis. PLoS One. 2014;9(9):e107265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lee YS, Olefsky J. Chronic tissue inflammation and metabolic disease. Genes Dev. 2021;35:307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Durheim MT, Slentz CA, Bateman LA, Mabe SK, Kraus WE. Relationships between exercise‐induced reductions in thigh intermuscular adipose tissue, changes in lipoprotein particle size, and visceral adiposity. Am J Physiol Endocrinol Metab. 2008;295:E407–E412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Yeung SSY, Reijnierse EM, Pham VK, Trappenburg MC, Lim WK, Meskers CGM, et al. Sarcopenia and its association with falls and fractures in older adults: A systematic review and meta‐analysis. J Cachexia Sarcopenia Muscle. 2019;10:485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kim TN, Park MS, Yang SJ, Yoo HJ, Kang HJ, Song W, et al. Prevalence and determinant factors of sarcopenia in patients with type 2 diabetes: the Korean Sarcopenic Obesity Study (KSOS). Diabetes Care. 2010;33:1497–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lee Y, Kim SU, Song K, Park JY, Kim DY, Ahn SH, et al . Sarcopenia is associated with significant liver fibrosis independently of obesity and insulin resistance in nonalcoholic fatty liver disease: Nationwide surveys (KNHANES 2008‐2011). Hepatology. 2016;63:776–786. [DOI] [PubMed] [Google Scholar]
- 100. Montano‐Loza AJ, Angulo P, Meza‐Junco J, Prad CMM, Sawyer MB, Beaumont C, et al. Sarcopenic obesity and myosteatosis are associated with higher mortality in patients with cirrhosis. J Cachexia Sarcopenia Muscle. 2016;7:126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Haehling SV, Ebner N, Santos MRD, Springer J, Anke SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol. 2017;14:323–341. [DOI] [PubMed] [Google Scholar]
- 102. Gruberg L, AWaksman R, Ajani AE, Kim HS, White RL, Pinnow EE, et al . The effect of intracoronary radiation for the treatment of recurrent in‐stent restenosis in patients with diabetes mellitus. J Am Coll Cardiol. 2002;39(12):1930–1936. [DOI] [PubMed] [Google Scholar]
- 103. Romero‐Corral A, Montori VM, Somers VK, Korinek J, Thomas RJ, Allison TJ, et al . Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: a systematic review of cohort studies. Lancet. 2006;368:666–678. [DOI] [PubMed] [Google Scholar]
- 104. Hainer V, Aldhoon‐Hainerova I. Obesity paradox does exist. Diabetes Care. 2013;36:S276–S281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Banack HR, Stokes A. The 'obesity paradox' may not be a paradox at all. Int J Obes (Lond). 2017;41:1162–1163. [DOI] [PubMed] [Google Scholar]
- 106. Bosello O. Obesity paradox and aging. Eat Weight Disord. 2021;26:27–35. [DOI] [PubMed] [Google Scholar]
- 107. Lyu SQ, Yang YM, Zhu J, Wang J, Wu S, Zhang H, et al. Gender‐specific association between body mass index and all‐cause mortality in patients with atrial fibrillation. Clin Cardiol. 2020;43:706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Winter JE, MacInnis RJ, Wattanapenpaiboon N, Nowson CA. BMI and all‐cause mortality in older adults: a meta‐analysis. Am J Clin Nutr. 2014;99:d1732, 875–890. [DOI] [PubMed] [Google Scholar]
- 109. Bowman K, Atkins JL, Delgado J, Kos K, Kuchel GA, Ble A, et al. Central adiposity and the overweight risk paradox in aging: follow‐up of 130,473 UK Biobank participants. Am J Clin Nutr. 2017;106:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. McMinn J, Steel C, Bowman A. Investigation and management of unintentional weight loss in older adults. BMJ. 2011;342:d1732. [DOI] [PubMed] [Google Scholar]
- 111. Rios‐Diaz AJ, Lin E, Williams K, Jiang W, Patel V, Shimizu N, et al. The obesity paradox in patients with severe soft tissue infections. Am J Surg. 2017;214:385–389. [DOI] [PubMed] [Google Scholar]
- 112. Park Y, Peterson LL, Colditz GA. The Plausibility of Obesity Paradox in Cancer‐Point. Cancer Res. 2018;78:1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Tegels JJW, Vugt JLA, Reisinger KW, Hulsewe KWE, Hoofwijk AGM, Derikx JPM, et al . Sarcopenia is highly prevalent in patients undergoing surgery for gastric cancer but not associated with worse outcomes. J Surg Oncol. 2015;112(4):403–407. [DOI] [PubMed] [Google Scholar]
- 114. Donini LM, Busetto L, Bauer JM, Bischoff S, Boirie Y, Cederholm T, et al . Critical appraisal of definitions and diagnostic criteria for sarcopenic obesity based on a systematic review. Clin Nutr. 2020;39:2368–2388. [DOI] [PubMed] [Google Scholar]
- 115. Wang M, Tan Y, Shi Y, Wang X, Liao Z, Wei P, et al. Diabetes and Sarcopenic Obesity: Pathogenesis, Diagnosis, and Treatments. Front Endocrinol (Lausanne). 2020;11:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Evans WJ, Hellerstein M, Orwoll E, Cummings S, Cawthon PM. D3‐Creatine dilution and the importance of accuracy in the assessment of skeletal muscle mass. J Cachexia Sarcopenia Muscle. 2019;10:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Neves T, Fett CA, Ferriolli E, Souza MGC, Filho ADDR, Lopes MBM, et al. Correlation between muscle mass, nutritional status and physical performance of elderly people. Osteoporos Sarcopenia. 2018;4:e12989, 145–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Petermann‐Rocha F, Ho FK, Welsh P, Mackay D, Brown R, Gill JMR, et al . Physical capability markers used to define sarcopenia and their association with cardiovascular and respiratory outcomes and all‐cause mortality: A prospective study from UK Biobank. Maturitas. 2020;138:110832, 69–75. [DOI] [PubMed] [Google Scholar]
- 119. Lee WJ, Peng LN, Loh CH, Chen LK. Sex‐different associations between serum homocysteine, high‐sensitivity C‐reactive protein and sarcopenia: Results from I‐Lan Longitudinal Aging Study. Exp Gerontol. 2020;132:110832. [DOI] [PubMed] [Google Scholar]