

Abstract
Background
Nicotinamide phosphoribosyltransferase (Nampt), a key enzyme in NAD salvage pathway is decreased in metabolic diseases, and its precise role in skeletal muscle function is not known. We tested the hypothesis, Nampt activation by P7C3 (3,6‐dibromo‐α‐[(phenylamino)methyl]‐9H‐carbazol‐9‐ethanol) ameliorates diabetes and muscle function.
Methods
We assessed the functional, morphometric, biochemical, and molecular effects of P7C3 treatment in skeletal muscle of type 2 diabetic (db/db) mice. Nampt+/− mice were utilized to test the specificity of P7C3.
Results
Insulin resistance increased 1.6‐fold in diabetic mice compared with wild‐type mice and after 4 weeks treatment with P7C3 rescued diabetes (P < 0.05). In the db‐P7C3 mice fasting blood glucose levels decreased to 0.96‐fold compared with C57Bl/6J wild‐type naïve control mice. The insulin and glucose tolerance tests blood glucose levels were decreased to 0.6‐fold and 0.54‐folds, respectively, at 120 min along with an increase in insulin secretion (1.76‐fold) and pancreatic β‐cells (3.92‐fold) in db‐P7C3 mice. The fore‐limb and hind‐limb grip strengths were increased to 1.13‐fold and 1.17‐fold, respectively, together with a 14.2‐fold increase in voluntary running wheel distance in db‐P7C3 mice. P7C3 treatment resulted in a 1.4‐fold and 7.1‐fold increase in medium‐sized and larger‐sized myofibres cross‐sectional area, with a concomitant 0.5‐fold decrease in smaller‐sized myofibres of tibialis anterior (TA) muscle. The transmission electron microscopy images also displayed a 1.67‐fold increase in myofibre diameter of extensor digitorum longus muscle along with 2.9‐fold decrease in mitochondrial area in db‐P7C3 mice compared with db‐Veh mice. The number of SDH positive myofibres were increased to 1.74‐fold in db‐P7C3 TA muscles. The gastrocnemius and TA muscles displayed a decrease in slow oxidative myosin heavy chain type1 (MyHC1) myofibres expression (0.46‐fold) and immunostaining (6.4‐fold), respectively. qPCR analysis displayed a 2.9‐fold and 1.3‐fold increase in Pdk4 and Cpt1, and 0.55‐fold and 0.59‐fold decrease in Fgf21 and 16S in db‐P7C3 mice. There was also a 3.3‐fold and 1.9‐fold increase in Fabp1 and CD36 in db‐Veh mice. RNA‐seq differential gene expression volcano plot displayed 1415 genes to be up‐regulated and 1726 genes down‐regulated (P < 0.05) in db‐P7C3 mice. There was 1.02‐fold increase in serum HDL, and 0.9‐fold decrease in low‐density lipoprotein/very low‐density lipoprotein ratio in db‐P7C3 mice. Lipid profiling of gastrocnemius muscle displayed a decrease in inflammatory lipid mediators n‐6; AA (0.83‐fold), and n‐3; DHA (0.69‐fold) and EPA (0.81‐fold), and a 0.66‐fold decrease in endocannabinoid 2‐AG and 2.0‐fold increase in AEA in db‐P7C3 mice.
Conclusions
Overall, we demonstrate that P7C3 activates Nampt, improves type 2 diabetes and skeletal muscle function in db/db mice.
Keywords: P7C3, Nampt, Skeletal muscle, Insulin sensitivity, Physical performance, Pathophysiology
Introduction
Type 2 diabetes mellitus is one of the most common and chronic metabolic disease worldwide. 1 Type 2 diabetes has a debilitating impact on the whole body with major changes affecting the musculoskeletal system. It is mainly characterized by insulin resistance with the inability of the metabolic tissues such as skeletal muscle and liver to utilize insulin for maintaining glucose homeostasis, resulting in elevated blood glucose levels leading to hyperglycaemia. 2 , 3 The occurrence of type 2 diabetes has increased rapidly in the last few decades and is the 7th major cause of mortality in the USA (CDC, USA). There is increased prevalence of type 2 diabetes in younger population due to obesity, which is a predisposing factor to the metabolic disease. 4 Due to an increase in the number of older populations, the incidence of type 2 diabetes has also increased worldwide. 5 Type 2 diabetes is a major cause of cardiovascular diseases and neurodegenerative disorders. 6 , 7 Diabetes affects skeletal muscle mass, strength and function through impaired energy metabolism, decreased blood flow, mitochondrial dysfunction and cell death. 8
Skeletal muscle is a major metabolic organ that not only utilizes glucose to produce the energy needed for contraction but also plays a key role in insulin‐stimulated glucose uptake in the postprandial state, and overall energy expenditure in mammals. 9 , 10 Thus, skeletal muscle insulin resistance plays an important role in the pathophysiology of type 2 diabetes even before the pancreatic β cells dysfunction and the development of overt hyperglycaemia. 11 Disruption of key insulin signalling pathways prevents the normal utilization of circulating blood glucose and storage of excess glucose as glycogen in the muscle and liver leading to hyperglycaemia and insulin resistance. 12 Further, the secretion of pro‐inflammatory cytokines by immune cells and the increase in lipolysis and free fatty acids, and the accumulation of intermediary lipid metabolites from other tissues such as adipose tissue can induce myocyte inflammation in obesity and negatively regulate the muscle cell metabolism resulting in insulin resistance. 3 The skeletal muscle inflammation is also caused by the ectopic fat deposition, that is, inter‐muscular and intra‐muscular lipid accumulation and can add to the degree of insulin resistance through paracrine effects. 13 In addition, mitochondrial dysfunction has also been attributed in the development of insulin resistance and type 2 diabetes. 14
Nampt is a key rate‐limiting enzyme in the NAD salvage pathway, which is dysregulated in the metabolic disease such as diabetes, and during ageing. 15 , 16 , 17 , 18 , 19 Nampt is ubiquitously expressed, and the homozygous‐null genotype is embryonically lethal in mice. 20 Muscle‐specific knockdown of Nampt in mice reduces the intramuscular NAD+ levels and causes myofibre degeneration and loss of physical functions like strength and endurance capacity mimicking type 2 diabetes in humans. 21 Furthermore, knockdown of Nampt in mouse C2C12 myoblasts also reduces the NAD+ levels and results in decreased mitochondrial biogenesis. 22 The Nampt activator, P7C3 was initially identified as a neuroprotective aminopropyl carbazole agent, which binds to and enhances the Nampt activity. 23 , 24 However, the precise role of Nampt in skeletal muscle function and pathophysiology of metabolic disease such as type 2 diabetes is not known. In the present study, we hypothesized that P7C3‐mediated Nampt activation in the db/db mouse model of type 2 diabetes ameliorates the skeletal muscle diabetic phenotype and improves insulin sensitivity and function.
Methods
Animals
The C57Bl/6J wild‐type (WT), and the type 2 diabetic B6.BKS(D)‐Leprdb/J (db/db) male mice were purchased from Jackson laboratories. The Nampt heterozygous (Nampt+/−) mice were provided by Dr. Junichi Sadoshima (Rutgers Medical School, NJ, USA), and the colonies were bred and maintained at the Morsani College of Medicine, University of South Florida, USA. The 16‐weeks old male db/db mice were treated with Nampt activator, P7C3 (db‐P7C3) (10 mg/kg body weight for 4 weeks, daily, i/p). The vehicle control db/db (db‐Veh) mice were treated with an equivalent dose of DMSO dissolved in 10% Kolliphor oil (Sigma‐Aldrich, MO, USA) in phosphate buffered saline (PBS). The age‐matched C57Bl/6J WT male mice were used as naïve control. In addition, 20–24 weeks old Nampt heterozygous (Nampt+/−) and their littermate WT male mice were also treated with Nampt activator, 10 mg/kg body weight P7C3 (P7C3) once, i/p. The vehicle control Nampt+/− (vehicle) male mice were treated with an equivalent dose of DMSO dissolved in 10% Kolliphor oil in phosphate buffered saline (PBS). A total of 156 mice were used in this study, that is, 50 db‐Veh and 51 db‐P7C3 treated mice, and 35 C57Bl/6J WT and 10 each of the Nampt+/− and littermate WT mice. All mice used in this study were fed with food and water ad libitum. All animal work was approved before start of the study by the Institutional Animal Care and Use Committee at the University of South Florida, Tampa, FL, USA.
Fasting blood glucose and serum lipids
Mice were fasted 6 h, and tail vein blood was used for glucose measurement with ACCU‐CHECK blood glucose meter (Roche Diagnostics, Mannheim, Germany) on a weekly basis for 4 weeks. At the end of 4 week treatment, blood was collected by retro orbital plexus technique for the assessment of serum lipids content using the Rat/Mouse Insulin ELISA kit (Millipore, MA, USA) following manufacturer's instructions. The serum high‐density lipoprotein (HDL) and low‐density lipoprotein/very low‐density lipoprotein (LDL/VLDL) were quantified using HDL and LDL/VLDL quantification kit (Sigma‐Aldrich, MO, USA USA).
Insulin tolerance test
Both db‐Veh and db‐P7C3 mice were fasted for 6 hours and were injected with a single dose of 1 U/kg body weight Novolin R regular human Insulin (Novo Nordisk, NJ, USA), i/p for the insulin tolerance test (ITT). The blood glucose levels were measured at 0, 30, 60, 90 and 120 min after the administration of human insulin with ACCU‐CHEK glucose analyser (Roche Diagnostics, Mannheim, Germany).
Glucose tolerance test
The glucose tolerance test (GTT) was performed in the Lepr−/− (db/db) and Nampt+/− mice. After 4 weeks of treatment, both the db‐Veh and db‐P7C3 treated mice were fasted overnight for GTT assay. GTT was performed by a single dose of intraperitoneal injection of 2 g/kg body weight of D‐(+)‐glucose (Sigma‐Aldrich, MO, USA). Blood samples were obtained by the retro orbital plexus technique. Serum glucose levels were determined prior to glucose administration (0 min) and after 30, 60, 90, and 120 min of glucose injection, using glucose oxidase peroxidase kit (Pointe Scientific Inc., MI, USA) and following manufacturer's instructions. Similarly, the Nampt+/− and their littermate WT mice were fasted overnight and given a single dose of vehicle control or P7C3 30 min prior to the administration of 2 g/kg body weight D‐(+)‐glucose, i/p. The circulating blood glucose levels were measured with ACCU‐CHECK blood glucose meter at 0, 15, 30, 60, 120 minutes of glucose administration. The index of the total glucose shift was calculated as the area under the curve (AUC) using trapezium method.
Immunostaining
Immunostaining of the pancreas was performed for the measurement of insulin secreting β cells area, as previously described. 25 Briefly, the frozen pancreatic tissues were fixed into tissue freezing media (TFM). Pancreas were cut into 10 μm thick sections and kept at −80°C. For immunostaining, the slides were first air dried and washed with phosphate buffer saline (PBS). The sections were then fixed in 4% paraformaldehyde for 30 min and blocked with SuperBlock™ T20 (TBS) blocking buffer (Thermo scientific, MA, USA) for 30 min, at room temperature (RT). Slides were then incubated with insulin primary antibody (sc‐9168, Santa Cruz, TX, USA) diluted in blocking buffer (1:200), overnight at 4°C. Slides were then washed in PBS and incubated with Alexa Fluor®488 secondary antibody (Abcam, MA, USA) diluted in blocking buffer for 1 h at RT. Following washes, the slides were then mounted with ProLong® Gold antifade mountant containing DAPI (Molecular Probe, OR, USA). Nuclei were labelled by DAPI. The images obtained with confocal multiphoton laser scanning microscope (Olympus FV1000 MPE, Olympus Inc., USA) were quantified by using ImageJ software (NIH, USA). The cross‐sectional areas of pancreas and β cells were determined from multiple sections (n = 3 mice per group), separated by at least 50 μm from each section. Pancreatic β cell area was calculated by using the following formula: islet β cell area (%) = (area stained by insulin antibody/total islets area) × 100.
Gomori aldehyde fuchsin staining
The Gomori aldehyde fuchsin staining of the pancreatic β cells was performed as per previously published protocol. 26 Briefly, the frozen pancreas was TFM embedded and cut into 6 μm thick transverse serial cryosections using Microm cryostat machine. The slides were air dried at 60°C and dewaxed using xylene and then hydrated through decreasing concentrations of ethyl alcohol. The slides were then rinsed in cold tap water followed by a single rinse in 50% ethanol and immersed in aldehyde fuchsin (Electron Microscopy Services, PA, USA) for 1 h. The excess stain was removed by rinsing in 70% ethanol and tap water. The slides were then counter‐stained with Light Green (American Master Tech Scientific, CA, USA) through immersion for 60 s and dehydrated with increasing concentration of ethanol, and cleared with xylene. The sections were then mounted with disterene phthalate xylene (DPX) mountant (Sigma‐Aldrich, MO, USA). Images were obtained at ×20 magnification with the Olympus IX73 inverted microscope using CellSens Standard software (Olympus Soft Imaging Solution, USA) and quantified with ImageJ software (NIH, USA).
Grip strength and voluntary running wheel
The fore‐limb and hind‐limb grip strengths were measured with the use of Chatillon force measurement DFE II grip strength meter (Ametek, Columbus Instruments, OH, USA). The average of five measurements per mice is represented as KGF/kg body weight for both the fore‐limb and hind‐limb grip strengths measured. The overnight voluntary running wheel experiments were carried out with the Columbus Instruments 0297, the CI Multi‐Device Interface (Columbus Instruments, OH, USA) per manufacturer's recommendations.
Ex vivo myofibre contractility and force‐frequency relationship
Animals were euthanized in compliance with University of South Florida IACUC guidelines. The extensor digitorum longus (EDL) muscles of each animal were tied with surgical silk at the proximal and distal tendons and dissected free. Muscles were immediately transferred to a customized vertical organ bath system (Aurora Scientific Inc., Canada) with the suture on the distal tendon clamped in place and that on the proximal tendon tied to a force transducer such that the muscles were positioned between two platinum wire stimulating electrodes. The buffer (137 mM NaCl, 0.4 mM NaH2PO4, 5.1 mM KCl, 1.05 mM MgCl2, 2.0 mM CaCl2 and 10 mM glucose; pH 7.4) in the bath was maintained at 22°C and continuously aerated with 95%/5% O2/CO2. Muscles were equilibrated for 10–15 min after which, the optimal muscle length (l 0 ) for each muscle was calculated by adjusting the length until a maximal force response to a single twitch was attained. Following determination of l 0 , the muscles were subjected to stimulation trains (500 ms duration) of frequencies ranging from 1–125 Hz, 1 per minute to determine the force‐frequency relationship (FFR). Stimulation was delivered with a S8800 dual pulse digital stimulator (Grass Products, West Warwick, RI, USA), and all pulses were 1 ms in duration. The peak forces of each contraction at the different stimulation frequencies were used to plot the FFR. Muscle force was expressed in both absolute (mN) and normalized terms (mN/mg muscle mass) of myofibre FFR. n = 5–6 EDL muscles from three mice per group.
Haematoxylin and eosin staining
The tibialis anterior (TA) muscle was dissected from the mice, and embedded in optimal cutting temperature (OCT), frozen in isopentane chilled in liquid nitrogen and stored at −80°C. The 10 μm thick transverse serial cryosections were cut at the mid‐belly region using Microm cryostat machine, and stained with haematoxylin and eosin 27 for the analysis of myofibre cross‐sectional area of the WT, db‐Veh and db‐P7C3 treated mice. Images were obtained at ×10 and ×20 magnifications with the Olympus IX73 inverted microscope and were quantified using CellSens Standard software (Olympus Soft Imaging Solution, USA).
Transmission electron microscopy
The EDL muscles were fixed with 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer for 2 h at RT, and overnight at 4°C, followed by washing in buffer and fixation with 1% osmium tetroxide in buffer for 2 h at 4°C. After rinsing in buffer, the samples were then dehydrated in ethanol and acetone at RT and infiltrated in acetone: pure resin (Embed812) mix in vacuum with desiccant for 1 h and in pure resin for 4 h and embedded in the embedding mix. Polymerization of the embedding mix was carried out overnight at 70°C in the embedding moulds. Semithin (1 μm) and ultrathin (70 nm) sections were cut using a Reichert Ultracut microtome machine. The sections were then stained with 2% uranyl acetate and contrasted with lead citrate as per standard protocol. Images were taken at ×15 000, ×30 000, and ×50 000 magnifications with a JOEL1400 transmission electron microscope for the analysis of myofibre diameter and the mitochondrial area.
Mitochondrial DNA (mtDNA) copy number
Total DNA was extracted from gastrocnemius (GAS) muscle using DNeasy Blood and Tissue kit (Qiagen, MD, USA) according to the manufacturer's protocol. The quantitative real‐time PCR (qPCR) analysis of the mtDNA/nDNA was performed using iTaq Universal SYBR green supermix (Bio‐Rad Laboratories, CA, USA). The mouse 16S was used as the mitochondrial gene and HK2 was used as the nuclear gene to obtain the mitochondrial copy number.
Succinate dehydrogenase staining
The 10 μm thick transverse cryosections of TA muscles were cut at the mid‐belly region using Leica CM 1860 AgProtect cryostat machine. Briefly, the tissue sections were air‐dried for 45 minutes, and incubated in 50 mM sodium succinate dibasic hexahydrate and 1.25 mM nitroblue tetrazolium in 0.1 M phosphate buffer (Sigma‐Aldrich, MO, USA) for 30 min. The reaction was stopped washing in deionized water, and the sections were dehydrated in a series of graded ethanol and cleared using xylene. The sections were then mounted with DPX mountant, and the images were captured at ×20 magnification of the Olympus IX73 inverted microscope using CellSens Standard software (Olympus Soft Imaging Solution, USA) and the succinate dehydrogenase (SDH) positive myofibres were quantified with ImageJ software.
Quantitative real‐time PCR
Total RNA was isolated from GAS muscle using the Exiqon miRCURY RNA Isolation kit (Exiqon, MA, USA) according to the manufacturer's protocol. Complimentary DNA from total RNA was synthesized with iScript cDNA synthesis kit (Bio‐Rad Laboratories, CA, USA) and quantitative real‐time PCR (qPCR) analysis was performed on key transcriptome targets using iTaq Universal SYBR Green Supermix (Bio‐Rad Laboratories, CA, USA). The cDNA synthesis and qPCR procedures were performed as described previously. 28 The mouse HPRT gene was used as an internal control.
Luminex Magpix IP‐10 (C‐X‐C motif chemokine ligand 10) immunoassay
The serum inflammatory markers were analysed using a mouse cytokine/chemokine antibody‐immobilized premixed 32‐plex Luminex magnetic beads based 96‐Well plate immunoassay kit (Milliplex Map kit—MCYTMAG‐70K‐PX32, EMD Millipore Corporation, MA, USA) according to the manufacturer's instruction. Briefly, the serum samples were diluted at 1:2 in the assay buffer and measured in duplicates along with the standards and quality controls. The serum samples were incubated overnight at 4°C with the magnetic beads. The wells were then washed with wash buffer and incubated with the detection antibodies for 1 h at RT, followed by a streptavidin‐phycoerythrin incubation for 30 min at RT. After washing, the plate was run on MAGPIX with xPONENT software. The mean fluorescent intensity data was analysed using a five‐parameter logistic curve‐fitting Multi Analyst software for calculating the cytokine/chemokine concentrations of the samples (n = 6 mice per group).
Whole transcriptome sequencing (RNA Seq)
The RNA Seq of GAS muscle was carried out using Novogene Next Generation Sequencing platform NovaSeq 6000 PE150 (Novogene, Sacramento, CA, USA). Total RNA was extracted from GAS muscle and the RNA quality and integrity were determined. RNA Seq library was then prepared and the cDNA library quality were determined before sequencing with Illumina PE150 platform. The data generated were analysed between groups (n = 3 mice/group) for bioinformatics analysis.
Bioinformatic analysis
The Heatmap, Volcano Plot and Venn diagram were generated using R 4.1.1 software for differentially expressed genes. The gene network and pathways were analysed using DAVID v6.8 for GO enrichment analysis with Benjamini significant P < 0.05 for both up‐regulated and down‐regulated genes. The Gene interaction network construction and visualization was performed using Cytoscape v3.8.2. 29 , 30 The identified treatment responsive differentially expressed genes were created with the Search Tool for the Retrieval of Interacting Genes and proteins (STRING) database. Default settings of confidence score and maximum additional interactors were used for visualization. The Molecular Complex Deletion (MCODE) plugin for Cytoscape was used to identify the network modules. 31 Densely connected regions/clusters in the co‐expression network were identified with the following parameters; degree cut‐off = 2, haircut, k‐core = 2, node score cut off = 0.2 and max. depth = 100.
Myofibre staining
The 10 μm thick transverse serial cryosections of TA muscles were cut at the mid‐belly region using Microm cryostat machine. Briefly, the tissue sections were blocked with 0.2% bovine serum albumin (BSA) and 10% normal sheep serum (NSS) in PBS‐Triton X100 (PBST) for 1 h at RT. All washes were carried out with PBS. Following washing, the sections were incubated in 5 μg/mL of mouse monoclonal primary antibody against myosin heavy chain type‐1 (A4.840s DSHB, IA, USA) in 0.2% BSA and 5% NSS antibody dilution buffer in PBST overnight at 4°C. After washing, the sections were then fixed with 10% neutral buffered formalin for 5 min at RT, washed and incubated in 1:300 dilution of biotinylated sheep anti‐mouse IgG (ab6807, Abcam, MA, USA) and 1:1000 dilution of rabbit anti‐laminin (L9393, Sigma‐Aldrich, MO, USA) in the antibody dilution buffer for 1 h at RT. Following washes, the tissue sections were then incubated in 1:400 dilution of streptavidin AF488 conjugate and goat anti‐rabbit AF594 (Invitrogen, CA, USA) and then washed and mounted with Prolong gold anti‐fade mounting medium with DAPI (Invitrogen, CA, USA). Images were obtained at ×10 and ×20 magnifications with the Olympus IX73 inverted microscope using CellSens Standard software (Olympus Soft Imaging Solution, USA) and the MyHC1 positive myofibres were quantified with ImageJ software. 28
Lipidomics
The lipidomic profiling of GAS muscle was performed using a liquid chromatography–tandem mass spectrometry (LC–MS/MS) method as described previously. 32 , 33 Briefly, the GAS muscle samples were collected, and flash frozen in liquid nitrogen. Around 50–100 mg of the muscle tissue was homogenized using a bead‐beating method, then the obtained supernatant was further cleaned and concentrated by solid phase extraction. The extracted LMs samples were dried under vacuum and stored at −80°C for future LC–MS/MS analysis. The dried extracts were reconstituted with 50 μL of methanol, and 10 μL was injected into LC–MS/MS to observe lipid profiles in GAS muscles. The LC–MS/MS system components were from Shimadzu Scientific Instruments Inc. (Columbia, MD, USA). All analyses and data processing were completed with the use of Shimadzu Lab Solutions V5.91 software (Shimadzu Scientific, Tokyo, Japan).
Nampt enzymatic activity
The Nampt enzymatic activity was measured from the GAS muscles of WT naïve control, db‐Veh and db‐P7C3 treated mice using the commercially available CycLex Nampt Colorimetric Assay Kit (MBL international, MA, USA) as per manufacturer's instructions. The OD values were obtained at 450 nm with the Synergy Hybrid H4 96‐Well Plate Reader (Biotek, VT, USA) using Gen5 software after the sample incubation at 30°C, and the Nampt enzymatic activity was assessed.
Serum pyruvate enzymatic activity
Serum pyruvate levels were measured using the pyruvate assay kit (Sigma‐Aldrich, MO, USA) as per manufacturer's protocol. Briefly, 5 μL of the serum samples were added to each well along with the pyruvate assay buffer, probe solution and the enzyme mix, and incubated for 30 min at RT. The absorbance was measured at 570 nm in the Synergy H4 Hybrid 96‐Well Plate Reader (Biotek, VT, USA) using Gen5 software, and the concentration of the serum pyruvate levels were assessed.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 9.0.2. Two‐way ANOVA with Tukey's multiple comparison post‐hoc test was utilized for all groups. An unpaired Student's t test was utilized when comparing only two groups. For the myofibre force‐frequency data, two‐way repeated‐measures ANOVA (treatment × frequency), with frequency as the repeated factor was used to test for the main effect of treatment and the treatment × frequency interaction. Unless otherwise specified each black dot or square, and the red triangle represent the data obtained from a mouse. Data are expressed as mean ± SEM. The comparison‐wise error rate, α, was set at 0.05 for statistical analysis of all data analysed using GraphPad Prism 9.0.2 software (La Jolla, CA, USA), where asterisk (*) denotes significant difference from the post‐hoc test.
Results
P7C3 treatment alleviates the fasting blood glucose, insulin resistance and glucose intolerance in db/db mice
The db‐P7C3 treated mice showed a significant decrease in the fasting blood glucose levels compared with the vehicle‐treated db/db mice, while the db‐Veh mice exhibited an increase compared with WT mice (Figure 1A). The C57Bl/6J WT mice displayed the same amount of circulating blood glucose levels during the weekly assessment for the entire 4 weeks. These data suggest that P7C3 improves the insulin sensitivity or glucose utilization in the type 2 diabetic db/db mice. Circulating blood glucose levels in response to the ITT were found to be significantly reduced at all‐time points analysed in db‐P7C3 vs. db‐Veh mice (Figure 1B) confirming the insulin sensitivity of the db‐P7C3 treated mice. A similar response to treatment was observed for the intraperitoneal GTT (Figure 1C). The index of the total glucose shift calculated as per cent AUC using trapezium method also displayed a significant decrease in the circulating blood glucose levels in the db‐P7C3 mice compared with db‐Veh mice (Figure 1D). This validates that the db/db mice treated with P7C3 has increased glucose uptake, possibly through insulin‐dependent mechanism. Subsequently, we also found that the circulating insulin levels were also increased in the db‐P7C3 treated mice with decreased homeostatic model of insulin resistance (HOMA‐IR) and increased β cells function (HOMA‐B) compared with db‐Veh mice (Figure 1E–G). Together, this clearly demonstrates that the intraperitoneal administration of P7C3 in the type 2 diabetic, db/db mice increases the glucose uptake, and promotes pancreatic β cells function.
Figure 1.
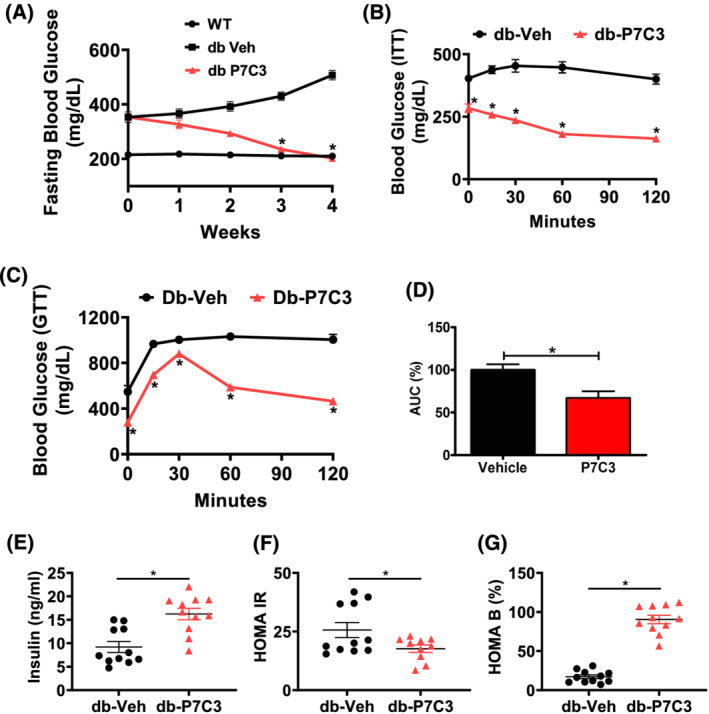
P7C3 reverses hyperinsulinaemia and hyperglycaemia of the diabetic mice. The 16‐week‐old type 2 diabetic (db/db) mice were administered daily with intraperitoneal dose of P7C3 (10 mg/kg/day) or vehicle for 4 weeks. (A) Weekly 6 h fasting blood glucose levels of the db/db mice treated with P7C3 or vehicle (n = 11 each), and the C57Bl/6J WT (n = 6) naïve control mice. (B) Insulin tolerance test (ITT) of 6 h fasted db/db mice injected with 1 U/kg body weight human insulin intraperitoneally (n = 4–5 per group). (C) Glucose tolerance test (GTT) of overnight fasted db/db mice injected with 2 g/kg body weight D‐(+)‐glucose intraperitoneally and measured with GOD‐POD method (n = 5 mice per group). (D) The area under the curve of GTT was obtained using the trapezium method and expressed in percentage. (E) The circulating insulin levels in blood serum and (F) homeostatic model of insulin resistance (HOMA IR) and (G) homoeostatic model of pancreatic β‐cells (HOMA B) expressed in percentage. Data are expressed as mean ± SEM; *P < 0.05; P7C3 vs. vehicle‐treated db/db mice. The age‐matched WT mice were used as naïve control to determine the baseline weekly fasting blood glucose levels.
P7C3 treatment increases the number of pancreatic β cells in db/db mice
To assess whether db‐P7C3 mice improve pancreatic β cells function of the islets of Langerhans, we utilized the db/db mice treated with vehicle or P7C3 for 4 weeks. Immunohistochemical analysis of the pancreatic islets of Langerhans displayed a significant increase in the insulin positive β cells area per islet of Langerhans area (Figure 2A, B). In addition, histological analysis of the pancreatic tissue cryosections stained with Gomori trichrome also displayed a significant increase in the number of β cells and islet of Langerhans per tissue section along with increased number of β cells per islets of Langerhans in the db‐P7C3 mice compared with db‐Veh treated mice (Figure 2C–F). Taken together, this clearly suggests that there is increased number of pancreatic β cells in the P7C3 treated type 2 diabetic db/db mice, which contributed to the increased glucose uptake in the db‐P7C3 mice.
Figure 2.
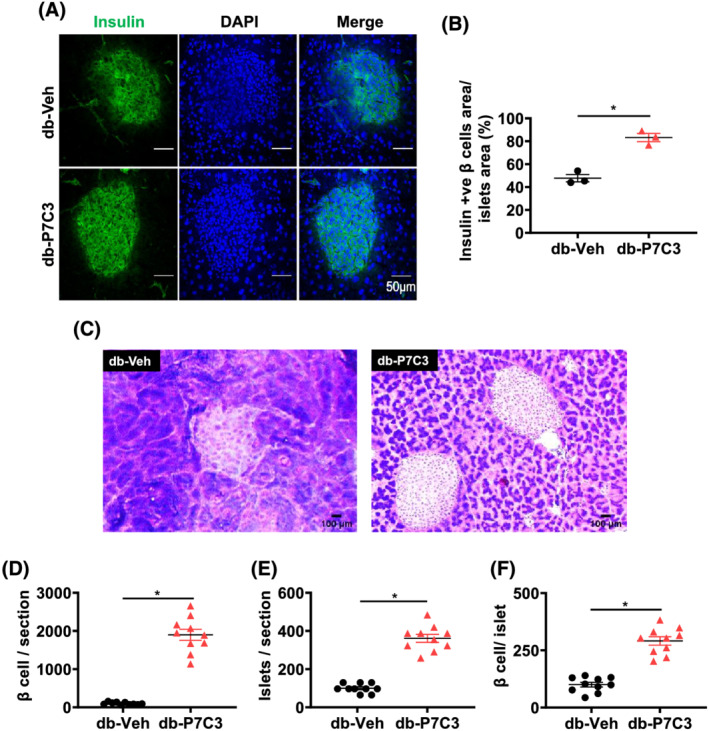
P7C3 increases the pancreatic β cells number and function of the db/db mice. (A) Immunohistochemical staining of insulin secreting pancreatic β cells (green). (B) The insulin secreting pancreatic β cells area per islet of Langerhans expressed as percentage. (C) Gomori aldehyde fuchsin staining of pancreas. (D) The number of pancreatic β cells per section, and (E) number of islet of Langerhans per section and (F) number of pancreatic β cells per islet of Langerhans. Data are expressed as mean ± SEM; *P < 0.05; P7C3 vs. vehicle‐treated db/db mice.
P7C3 treatment increases the physical performance of db/db mice
To determine whether P7C3 treatment modulates the physical performance in db/db mice, we compared fore‐limb and hind‐limb grip strength and voluntary free running wheel performance, along with the ex vivo force frequency of the extensor digitorum longus muscle. The db‐P7C3 treated mice displayed a significant increase in the fore‐limb and hind‐limb grip strengths compared with db‐Veh treated mice (Figure 3A,B). The db‐Veh mice also displayed a significant decrease in the fore‐limb and hind‐limb grip strengths compared with WT mice (supporting information, Figure S3A,B). There was significant group × frequency for both the absolute and normalized force frequency relationship data (P = 0.05 and 0.04, respectively) when comparing the WT, db‐Veh, and db‐P7C3 mice, although no main effect of treatment was observed for either measure of the ex vivo extensor digitorum longus muscle contractility. These data indicate a trend for reduced force in the db‐Veh group at higher frequencies, which is mitigated in the db‐P7C3 group (Figure 3C,D). However, analysis of the area under curve through the trapezium method displayed a significant increase in the absolute myofibre force frequency in db‐P7C3 mice compared with db‐Veh treated mice (Figure 3E). The db‐Veh mice also displayed a significant decrease in the absolute myofibre force frequency area under curve compared with WT mice (Figure S3C). The normalized myofibre force frequency also showed a similar increase in the db‐P7C3 treated mice (Figure 3F). Even though the db‐Veh mice displayed a decrease in the normalized myofibre force frequency area under curve compared with WT mice, it was found to be non‐significant (Figure S3D). In addition, the voluntary running wheel performance of the db‐P7C3 treated mice also displayed a significant increase in the distance covered compared with db‐Veh treated mice (Figure 3G). Together, these data suggest that P7C3 treatment increases the physical performance of the type 2 diabetic db/db mice.
Figure 3.
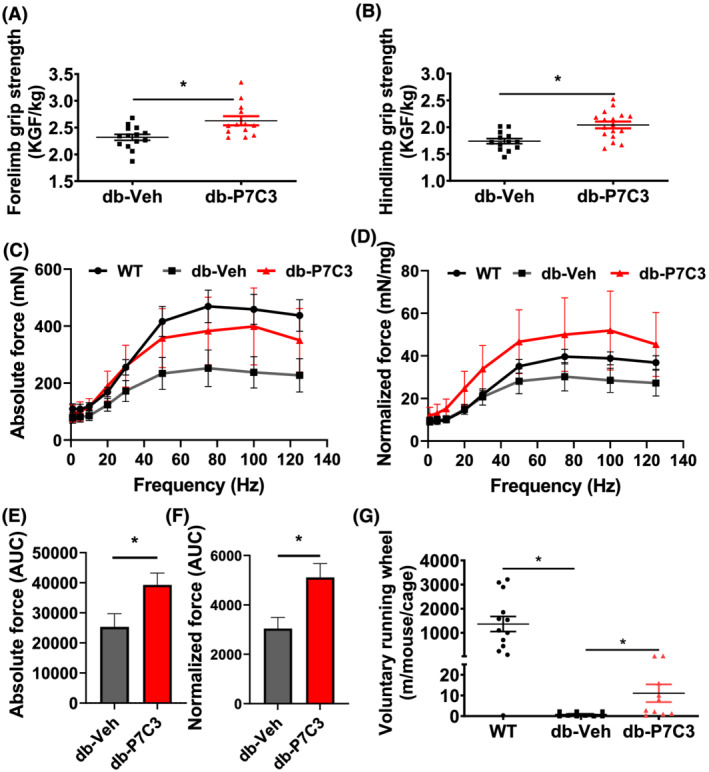
P7C3 enhances physical performance of db/db mice. (A) Fore‐limb grip strength and (B) hind‐limb grip strength measured and expressed as KGF/kg body weight. (C) The absolute force frequency and (D) normalized force frequency of the extensor digitorum longus muscles. (E) The area under the curve of the absolute muscle force frequency and (F) the normalized muscle force frequency. (G) The voluntary running wheel performance of the mice expressed in metres. Data are expressed as mean ± SEM; *P < 0.05; P7C3 vs. vehicle‐treated db/db mice. The age‐matched WT mice were used as naïve control to determine the baseline muscle force frequencies of the extensor digitorum longus muscle.
P7C3 treatment ameliorates the diabetic skeletal muscle phenotype of the db/db mice
We then sought to measure the myofibre size of the type 2 diabetic db/db mice treated with vehicle or P7C3. Histological analysis of the haematoxylin and eosin (H&E) stained tibialis anterior muscle cross‐sectional area around the mid‐belly region displayed muscle atrophy with increased number of smaller (0–1500 μm2) myofibres cross‐sectional areas in db‐Veh treated mice compared with WT and db‐P7C3 treated mice (Figure 4A–C). The C57Bl/6J WT mice showed decreased number of small (0–1500 μm2) myofibres and increased number of medium (1500–3000 μm2) and larger (>3000μm2) myofibres cross‐sectional areas (Figure 4C). Noticeably, the db‐P7C3 treated mice displayed a significant decrease in the small myofibres cross‐sectional areas compared with the db‐Veh treated mice, with a shift towards an increase in medium and larger sized myofibres cross‐sectional areas (Figure 4A–C). There was also a significant increase in the medium‐sized myofibres and decrease in larger sized myofibres in the db‐Veh treated mice compared with the WT mice (Figure 4C). Subsequent transmission electron microscopy (TEM) image analysis of the longitudinal sections of the extensor digitorum longus muscles also displayed a significant decrease in the myofibre diameter in the db‐Veh treated mice compared with the WT mice (Figure 4D,E). Similar to the H&E‐stained tibialis anterior muscle cryosections, the TEM images of the db‐P7C3 treated mice also displayed an increase in the myofibre diameter of the extensor digitorum longus muscle compared with the db‐Veh treated EDL muscle. However, it was found to be significantly lower than the WT mice (Figure 4E). Interestingly, the TEM images also displayed increased mitochondrial area in the db‐Veh treated mice compared with WT and db‐P7C3 treated mice (Figure 4D,F). To assess the metabolic status and mitochondrial function in the skeletal muscle the SDH staining was performed. Figure 4G,H shows the decrease in the SDH activity in db‐Veh mice compared with WT, whereas treatment with P7C3 for 4 weeks significantly improved the SDH positive myofibres in db‐P7C3 mice skeletal muscle. To gain further insight into the mitochondrial DNA (mtDNA) content, 16S expression was measured. Figure 4I shows the increased expression of mtDNA in db‐Veh compared with WT (P = 0.06), whereas, the 16S expression was significantly decreased in db‐P7C3 group compared with db‐Veh mice. Together, this suggests that P7C3 treatment rescues the type 2 diabetic skeletal muscle phenotype of the db/db mice with an increase in the myofibre size and SDH positive myofibres, along with reduced mitochondrial copy number likely due to decreased stress resulting in improved mitochondrial structure and function.
Figure 4.
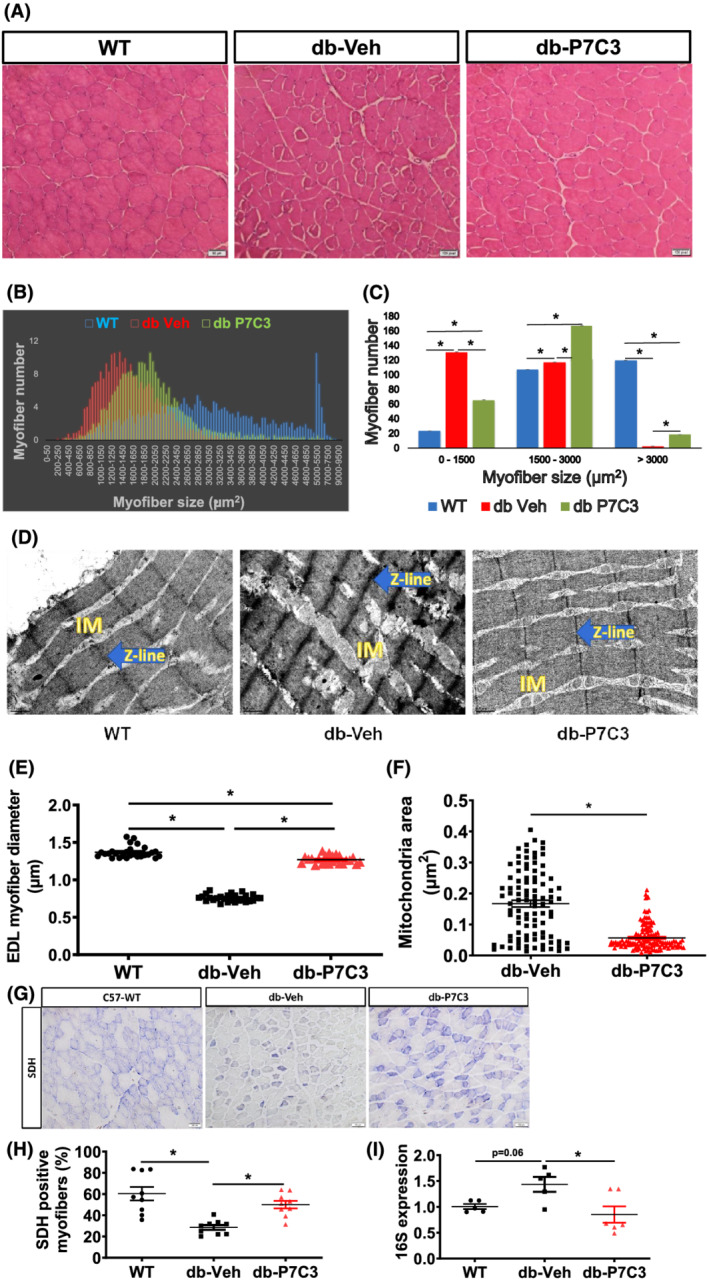
P7C3 ameliorates the diabetic skeletal muscle phenotype of the db/db mice. (A) Haematoxylin and eosin stained ×20 magnification images of the tibialis anterior muscles of the diabetic mice treated with vehicle or P7C3 and the wild‐type naïve control mice. (B,C) analysis of 250 random myofibres cross‐sectional area of the haematoxylin and eosin‐stained images per section and 3 such section was counted per mice, where n = 3 mice per group displaying a shift towards medium‐sized myofibres in the db‐P7C3 treated mice (green) compared to db‐Veh treated mice (red). The age‐matched WT naïve control mice myofibres display an increased larger sized myofibres (blue). (D) Transmission electron microscopy images of the extensor digitorum longus muscle of the WT naïve control, db‐Veh and db‐P7C3 treated mice. The intermyofibrillar mitochondria (IM), and the Z‐lines (blue arrows) are distinguished in the TEM images taken at ×30 000 magnification. (E) Analysis of the extensor digitorum longus muscles myofibre diameter of TEM images at ×30 000 magnifications (n = 8–9 myofibres per image, and 3 such images were analysed per group). Data are expressed as mean ± SEM; *P < 0.05. (F) Mitochondrial area measured from db‐Veh or db‐P7C3 expressed as μm2. (G) Succinate dehydrogenase (SDH) staining of TA muscle, (H) quantification of SDH stained myofibres, and (I) relative mRNA expression of 16S in wild‐type (WT), db‐Veh, and db‐P7C3. Data are expressed as mean ± SEM, P < 0.05.
P7C3 treatment decreases MyHC1 expression and number in db/db mice
We next sought to determine the myofibre types in the db‐P7C3 treated mice due to the observed shift in myofibre size. The db‐P7C3 treated mice displayed a significant decrease in the MyHC type 1 (MyHC1) expression levels in the gastrocnemius muscle compared with the db‐Veh mice (Figure 5A). Moreover, the MyHC1 expression levels were found to be non‐significant between the WT and db‐P7C3 treated mice. In agreement with other finding, we found a many‐fold increase in the expression levels of MyHC1 in the db‐Veh treated mice compared with WT mice, suggestive of an alternative increased slow oxidative metabolism in the diabetic mice. 34 Further, qPCR analysis of the db‐P7C3 mice gastrocnemius muscle for myosin heavy chain fibre types, that is, MyHC‐2a, MyHC‐2b, and MyHC‐2x displayed no significant change in their expression levels compared with db‐Veh treated mice and the WT mice (Figure 5B–D). Subsequent analysis of the immunostained tibialis anterior muscle cryosections displayed a significant decrease in the number of MyHC1 positive myofibres in the db‐P7C3 treated mice compared with db‐Veh treated mice, which was found to be non‐significant compared with the WT mice (Figure 5E,F). These data suggest that the diabetic skeletal muscle phenotype of db/db mice is rescued by P7C3 treatment with a decrease in the slow oxidative myofibres.
Figure 5.
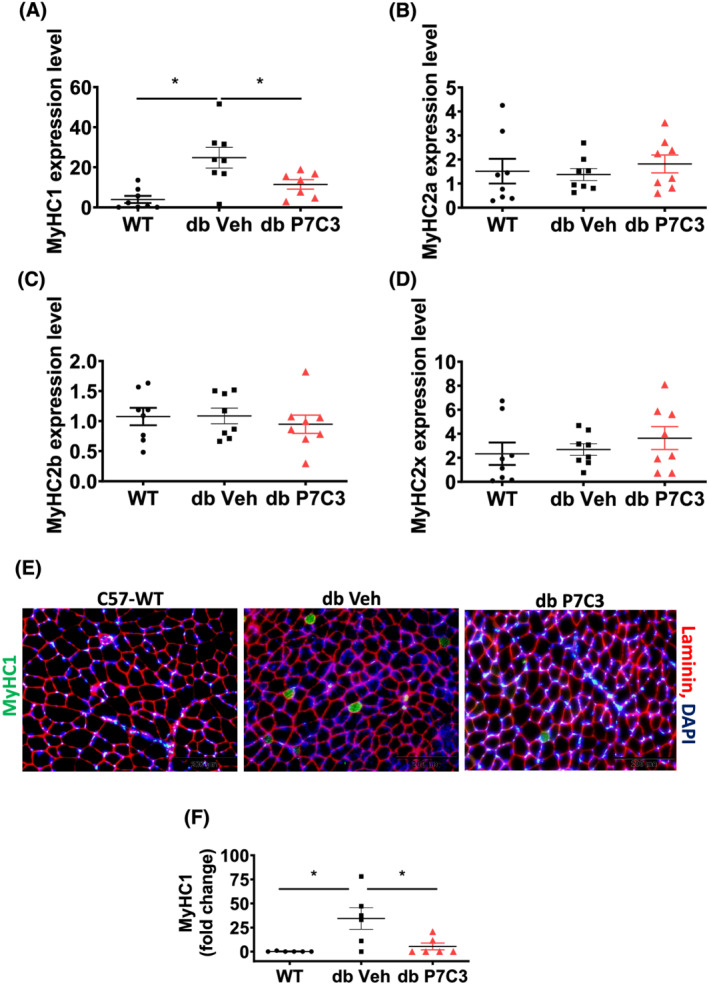
P7C3 decreases MyHC1 expression levels and immunostained myofibres numbers in gastrocnemius muscle. Relative gene expression levels of myosin heavy chain fibre types: (A) MyHC1, (B) MyHC2a, (C) MyHC2b, and (D) MyHC2x. (E) Immunolabelling of tibialis anterior muscle cryosections of WT, db‐Veh, and db‐P7C3 treated mice. MyHC1 positive myofibres are immunostained in green, laminin in red and DAPI for nuclei in blue. (F) Quantification of MyHC1 depicting the fold change in green myofibre immunostaining. Data are expressed as mean ± SEM; *P < 0.05. The age‐matched WT mice was used as naïve control to determine the baseline expression levels of the myosin heavy chain fibre types in gastrocnemius muscle and immunostaining of MyHC1 in tibialis anterior muscle.
P7C3 treatment enhances mitochondrial fatty acid oxidation and decreases the myofibre stress in the db/db mice
Given that P7C3 treatment rescued the diabetic skeletal muscle phenotype of the db/db mice we next examined a panel of metabolic markers through qPCR gene expression analysis of the gastrocnemius muscle. We found that the key genes involved in fatty acid uptake (Fabp1 and CD36) to be significantly down‐regulated in the db‐P7C3 treated mice compared with db‐Veh treated mice, while there was no significant difference between the db‐P7C3 and WT mice (Figure 6A,B). Whereas, the expression levels of key genes involved in fatty acid oxidation (Pdk4 and Cpt1) was found to significantly up‐regulated in db‐P7C3 treated mice compared to both db‐Veh treated mice and the C57Bl/6J naïve control mice (Figure 6C,D). Noticeably, the db‐P7C3 treated mice also resulted in a significant reduction in the expression level of Fgf21, the stress‐induced myokine compared with the db‐Veh treated mice, while it was non‐significant compared with the WT mice (Figure 6E). To evaluate the treatment responsive genes in diabetic skeletal muscle, RNA‐seq analysis was performed. Figure 6G shows heat map with significant key differentially expressed genes in the db‐P7C3 group compared with db‐Veh. The key genes include increased Ppargc1a (PGC1a), decreased inflammation or stress responses along with improved myogenic genes with P7C3 treatment. As shown in the Venn diagram and Volcano plot Figure 6H,I, respectively, the RNA‐seq analysis showed high number of treatment responsive genes with 772/1415 genes up‐regulated along with 1213/1726 genes down‐regulated. We identified significant pathways that are up‐regulated due to P7C3 treatment, which include muscle structure development, metabolic pathways involved in energy metabolism (NADH, pyruvate, and ATP), calcium signalling, and glycolysis and gluconeogenesis. The pathways that are significantly down‐regulated include inflammation (NFkB, IL‐6,8, TNF, and cytokine production), TLR signalling, TGF‐β, apoptotic and cell cycle processes, fatty acid elongation, and cytokine receptor and NADPH oxidase activity (Figure 6J). Taken together, this suggest that P7C3 treatment of the type 2 diabetic db/db mice increases mitochondrial fatty acid β‐oxidation and thereby enhancing insulin sensitivity and energy metabolism of diabetic skeletal muscle phenotype with decreased oxidative stress and inflammation (Figure 6F,G).
Figure 6.
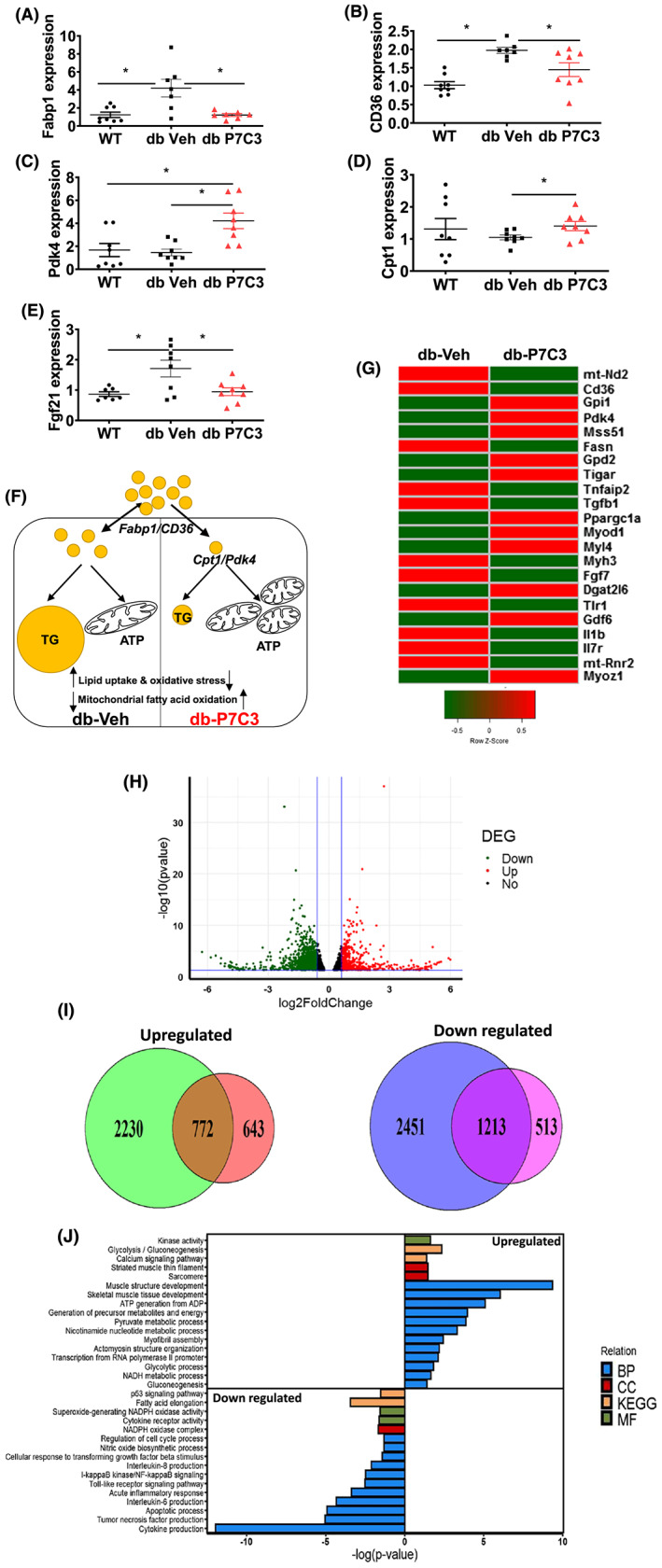
P7C3 treatment increases fatty acid oxidation and decrease myofibre stress in db/db mice. Relative mRNA expression levels of (A) Fabp1, (B) CD36, (C) Pdk4, (D) Cpt1, (E) Fgf21, and (F) schematic depiction of differential gene responses in db‐P7C3 and db‐Veh showing increased mitochondrial fatty acid oxidation and decreased fatty acid uptake and oxidative stress with P7C3 treatment in diabetic mice (G) RNA‐seq analysis of skeletal muscle from db‐Veh and db‐P7C3 (H) volcano plot showing differentially expressed genes (DEG) in db_P7C3‐db_Veh. The up‐regulated genes (red), and the down‐regulated genes (green) with a fold change <0.6, and with P < 0.05 (I) Venn diagram showing comparison of the differentially up‐regulated genes in db_P7C3‐db_Veh and down‐regulated genes in db_Veh‐WT_Veh, and provides the P7C3 treatment responsive genes with 772 genes up‐regulated and 1213 genes down‐regulated, respectively (J) the top 17 up‐regulated and 16 down‐regulated pathways in the biological processes (BPs), chemical component (CC), Kyoto encyclopaedia of genes and genomes (KEGG) and molecular function (MF) pathways. Data expressed is mean ± SEM, *P < 0.05. The age‐matched WT mice was used as naïve control to determine the baseline expression levels of the key genes involved in fatty acid uptake, oxidation, and oxidative stress in gastrocnemius muscle.
P7C3 treatment increases the circulating HDL levels and decreases the inflammatory lipid mediators of the db/db mice
We next sought to determine the circulating levels of HDL and LDL/VLDL lipoproteins in blood plasma and the LC–MS/MS‐based skeletal muscle lipid mediators. The db‐P7C3 treated mice displayed a significant increase in the HDL lipoproteins level compared with the db‐Veh treated mice (Figure 7A). Furthermore, the db‐P7C3 treated mice also displayed a significant decrease in the LDL/VLDL lipoproteins level compared with the db‐Veh treated mice (Figure 7B). Based on the increase in lipids in the obesogenic diabetic model, we further evaluated the lipids signalling in the skeletal muscle. The omega‐6 polyunsaturated fatty acid (n‐6 PUFA) arachidonic acid (AA) and its derivatives 11‐HETE, 15‐HETE, were significantly increased in db‐Veh treated group, while P7C3 treatment of the db/db mice significantly attenuated the pro‐inflammatory LMs in the gastrocnemius muscle. Additionally, P7C3 treatment of the diabetic mice also lowered the levels of the anti‐inflammatory LMs, (n‐3 PUFA) docosahexaenoic acid (DHA) and its derivatives, 7‐HDoHE, 13‐HDoHE, 16‐HDoHE, 17‐HDoHE, 20‐HDoHE, and the eicosapentaenoic acid (EPA) and its derivative 5‐HEPE and allowed the muscle to recover and bring the levels closer to that of the WT mice (Figure 7C–F). Interestingly, the levels of endocannabinoid, 2‐arachidonoyl glycerol (2‐AG) was found to be significantly decreased in the db‐P7C3 mice compared with the db‐Veh treated mice, while it was more similar to the WT mice (Figure 7G). However, anandamide (AEA) was found to be significantly higher in the db‐P7C3 treated mice compared with the db‐Veh and the WT mice (Figure 7H). To verify the inflammatory cytokines/chemokines, we assessed the interferon γ‐induced protein 10 (IP‐10/C‐X‐C motif chemokine ligand 10), a key pro‐inflammatory target in serum using Luminex‐Magpix magnetic bead immunoassay. As shown in Figure 7I, we detected increased IP‐10 levels in db‐Veh group compared with WT, whereas treatment of diabetic mice with P7C3 led to significant decrease in IP‐10 levels compared with db‐Veh, while it was still high compared with WT mice. To provide an in‐depth analysis and overview of the gene network and pathways that are activated with P7C3 treatment in diabetic skeletal muscle the STRING network analysis was performed (Figures S1 and S2). The data show that the glycolytic, myogenesis, and NADH pathways were up‐regulated, whereas the inflammation, apoptosis and stress responsive genes are down‐regulated. Furthermore, TNFα expression was detected at high levels in db‐Veh compared with db‐P7C3 skeletal muscle (Figure S3E). Overall, these data suggest that P7C3 treatment of the diabetic mice improves the circulating good lipoproteins and lipid mediators of the lipoxygenase (LOX) pathway (Figures S1–S5) and contribute towards skeletal muscle recovery from the chronic low‐grade inflammation associated with obesity and diabetes.
Figure 7.
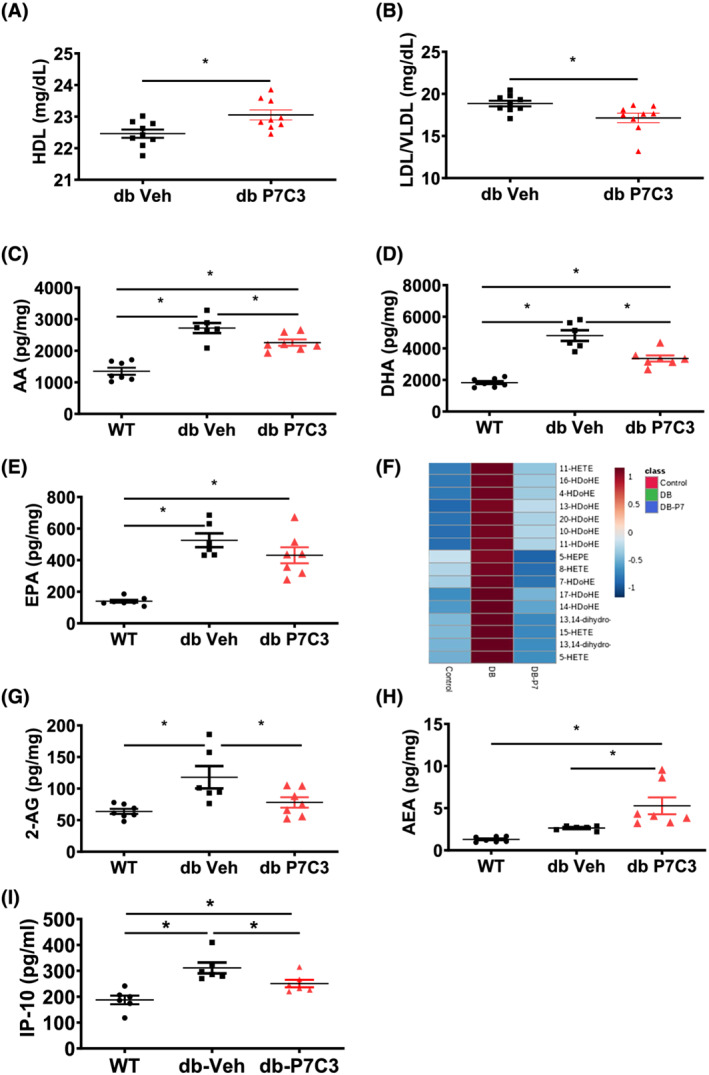
Cholesterol and lipid mediators are differentially altered in P7C3 treated db/db mice. (A) high‐density lipoprotein, (B) low‐density lipoprotein/very low‐density lipoprotein ratio. Panels (C–H) lipid mediators measured in wild‐type (WT), db‐Veh and db‐P7C3 treated mice, where (C) arachidonic acid; AA, (D) docosahexaenoic acid; DHA, (E) Eicosapentaenoic acid; EPA, (F) heat map of the pro‐inflammatory AA derivatives HETEs, and the anti‐inflammatory DHA derivatives HDoHEs and the EPA derivative HEPE (G) endocannabinoids, 2‐arachidonoylglycerol; 2‐AG and (H) anandamide; AEA. (I) Levels of serum IP‐10 (C‐X‐C motif chemokine ligand 10) measured by Luminex‐MagPix magnetic bead immunoassay. Data are expressed as mean ± SEM; *P < 0.05. The age‐matched WT mice were used as naïve control to determine the baseline lipid mediators levels in gastrocnemius muscle.
P7C3 treatment enhances the Nampt activity both in vitro and in vivo
Recent report show that administration of P7C3 in animal models and tissue cultures enhance the enzymatic activity of Nampt, the key rate‐limiting enzyme in the NAD salvage pathway from nicotinamide. 35 Therefore, we tested the P7C3 specificity of Nampt activity using enzyme assay with or without P7C (Figure 8A). As shown in Figure 8A, the Nampt activity was found to be significantly decreased with the Nampt inhibitor (FK866) and increased with the Nampt activator (P7C3). The Nampt enzymatic activity was at its basal level and was found to be non‐significant between the negative control DMSO, and the inhibitor and activator alone (Figure 8A). Furthermore, db‐P7C3 treated mice displayed a significant increase in the Nampt activity in gastrocnemius muscle compared with the db‐Veh treated mice (Figure 8B). In addition to Nampt activity, the levels of pyruvate in serum were assessed from diabetic mice treated with Nampt activator P7C3. As shown in Figure 8C, the serum pyruvate levels were significantly high in db‐P7C3 treated mice compared with db‐Veh. Together, this shows the specificity of P7C3 as the Nampt activator, and these data suggest that P7C3 administration increased Nampt activity in the type 2 diabetic db/db mice.
Figure 8.
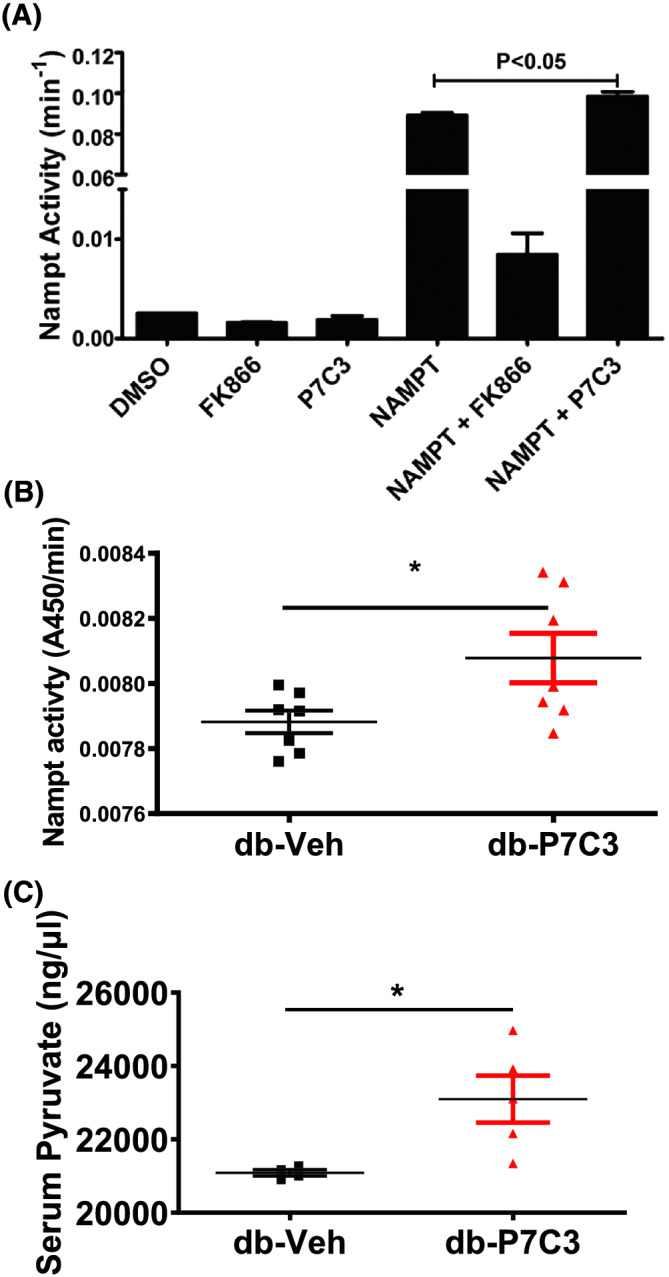
P7C3 treatment improves the Nampt enzymatic activity. (A) Enzymatic activity of recombinant Nampt was measured every 5 min for 1 h. Negative control (DMSO), positive control (Nampt enzyme), Nampt inhibitor (FK866), and Nampt activator (P7C3) were all utilized to assess the enzymatic activity. (B) The gastrocnemius muscle Nampt enzymatic activity was measured every 5 min for a total duration of 40 min. Data are expressed as mean ± SEM; *P < 0.05 of the absorbance measured at 20 min during the linear range. (C) Serum pyruvate activity of db‐Veh and db‐P7C3 treated mice. Data expressed as mean ± SEM with P < 0.05.
P7C3‐mediated effects are Nampt‐dependent
Next, to test whether the observed P7C3‐mediated effects are Nampt‐dependent, we utilized the Nampt heterozygous‐null (Nampt+/−) and their littermate WT mice that were administered with a single dose of 10 mg/kg body weight P7C3, i/p. The tail snip PCR analysis clearly identified the Nampt+/− and their littermate WT mice, with a double (300 bp and 500 bp) and a single (300 bp) intensity bands, respectively (Figure S6A). The fore‐limb grip strength of the Nampt+/− mice was found to be significantly decreased compared with the littermate WT mice (Figure S6B). Subsequent, GTT clearly showed a significant decrease in the circulating blood glucose levels of the Nampt littermate WT mice treated with P7C3 compared with the vehicle‐treated littermate WT mice. The Nampt+/− mice treated with P7C3 did not show any significant effect on the blood glucose levels tested at all‐time points analysed compared with the vehicle‐treated Nampt+/− mice (Figure S6C,D). The index of the total glucose shift was calculated as per cent AUC also displayed a significant decrease in the circulating blood glucose levels in the P7C3 treated Nampt littermate WT mice compared with vehicle‐treated Nampt littermate WT mice (Figure S6E), with no significant differences between the P7C3 and vehicle‐treated Nampt+/− mice (Figure S6F). These data demonstrate that P7C3 modulates its anti‐glycaemic effect through the activation of Nampt and suggest that the amelioration of the hyperglycaemic effect and its associated skeletal muscle phenotype observed in the type 2 diabetic db/db mice treated with P7C3 is Nampt‐dependent.
Discussion
The P7C3 is an aminopropyl carbazole that was reported as a neuroprotective compound in animal models of neurodegenerative diseases or nerve cell injury. 23 Recently, it has been reported that P7C3‐A20 treatment of C57Bl/6J mice on a high‐fat diet rescued the fatty liver syndrome with improved insulin sensitivity and hepatic inflammation. 36 P7C3 increases NAD synthesis from nicotinamide by activating Nampt, a key rate limiting enzyme in the NAD salvage pathway, 35 and analogues of P7C3 have been shown to bind to Nampt. 24 The enzymatic activity of Nampt and the bioavailability of NAD have been reported to be decreased in metabolic diseases and during ageing. 24 Currently, a multidrug approach is aimed for the treatment of diabetes, which includes skeletal muscle and other tissues in the body. 37 We hypothesized that P7C3‐mediated activation of Nampt in the type 2 diabetic db/db mice ameliorates the diabetic skeletal muscle phenotype and improves the insulin sensitivity. In the present study, we demonstrate P7C3 as a novel compound in the amelioration of type 2 diabetic skeletal muscle phenotype and function in the Leprdb homozygous‐null (db/db) mice. Furthermore, our data provide insights into the P7C3‐mediated insulin sensitivity through Nampt activation.
Based on the study, we demonstrate that the blood glucose levels of fasting blood glucose, insulin resistance, and GTTs were reduced in the db‐P7C3 treated mice. Furthermore, there was increased number of pancreatic β cells and islets of Langerhans, and β cells function in the db‐P7C3 treated mice. There was also increased physical performance in the db‐P7C3 treated mice as measured with the fore‐limb and hind‐limb grip strengths, ex vivo myofibre force frequency and voluntary running wheel performance. Previous report identifies that doxorubicin induced decrease in NAD can be compensated by P7C3 via Nampt activity. 24 This study also shows direct cross‐linking of P7C3 with recombinant Nampt enzyme. However, previous studies did not report if the observed decrease in the blood glucose levels is through direct P7C3‐mediated Nampt activation, involved in NAD biosynthetic salvage pathway or due to an indirect effect. To address this question, we utilized the recombinant Nampt enzyme and assessed the Nampt enzymatic activity with P7C3 or the Nampt inhibitor FK866 in vitro. In addition, we also utilized the type 2 diabetic, db/db mice treated with P7C3 or vehicle for assessing the gastrocnemius muscle Nampt and serum pyruvate activities. We found that the Nampt enzymatic activity was increased with P7C3 and inhibited by FK866 in the in vitro assay, and it was also increased in the db‐P7C3 treated mice. Similarly, the serum pyruvate levels were found to be increased in the db‐P7C3 treated mice. To address whether the observed increase in insulin sensitivity and glucose uptake with the administration of P7C3 in the type 2 diabetic db/db mice is directly through Nampt activation, we utilized the Nampt+/− and their littermate WT mice treated once with P7C3 or vehicle for assessing the blood glucose levels of the intraperitoneal GTT. The glucose tolerance was improved in the Nampt littermate WT mice treated with P7C3 but did not improve in the Nampt+/− mice treated with P7C3. This clearly indicates that the P7C3‐mediated effect on glucose uptake in the type 2 diabetic db/db mice is via Nampt activation.
Interestingly, in the db‐P7C3 treated mice there was an increase in myofibre size in hind‐limb tibialis anterior and the extensor digitorum longus muscles, which shows a healthy muscle phenotype in the db‐P7C3 treated mice. The type 2 diabetic db/db mice treated with P7C3 also displayed improved mitochondria morphology and function. The mitochondrial DNA was measured in the diabetic skeletal muscle and showed significant increase in 16S expression, which might be due to increased oxidative stress and previous report have shown similar increase in diabetic muscle. 38 Based on P7C3 treatment, we demonstrate that 16S expression was restored to the levels in WT muscle. More recently, P7C3 has been shown to stabilize the mitochondrial membrane potential in an in vitro dopaminergic cell culture model. 39 In our study, we found that P7C3 treatment of the type 2 diabetic, db/db mice resulted in the restoration of mitochondrial morphology and function. Furthermore, the observed shift in myofibre size from small to medium and larger myofibres was accompanied by a decrease in the MyHC1 (slow oxidative) myofibres expression levels and number, with no significant difference in the other MyHC fibre types expression levels in the db‐P7C3 treated mice. Subsequently, we found a significant increase in the succinate dehydrogenase positive oxidative myofibres and increased Pgc1α expression in the db‐P7C3 treated mice. Together this confirms that there is increased mitochondrial biogenesis in the P7C3 treated type 2 diabetic (db/db) mice that might be involved in energy metabolism.
To test this, we next looked at the expression level of the key markers in energy metabolism in gastrocnemius muscle and found decreased Fabp1 and CD36 expression levels along with an increase in Pdk4 and Cpt1 expression levels in the db‐P7C3 treated mice. Strikingly, the expression level of the stress‐induced myokine, Fgf21 was found to be decreased suggestive of improved skeletal muscle health as evident in the H&E cryosections of the tibialis anterior muscle analysed for the myofibre transverse sectional areas and the TEM images of the longitudinal section of extensor digitorum longus muscle for the diameter. Together, this suggest that there is decreased fatty acid uptake and increased mitochondrial fatty acid β‐oxidation along with decreased myofibre stress and inflammatory markers, including IP‐10 and Tnfaip2 in the type 2 diabetic db/db mice treated with P7C3 for energy metabolism and tissue homeostasis of the skeletal muscle.
Low HDL and increased VLDL levels in blood are often associated with insulin resistance and type 2 diabetes, which leads to increased risk of developing cardiovascular diseases. 40 , 41 , 42 In this study, P7C3 treatment of the type 2 diabetic (db/db) mice showed a significant increase in the circulating levels of HDL along with a decrease in LDL/VLDL levels in blood. Furthermore, excess lipid accumulation in skeletal muscle is well known to cause muscle inflammation, and their levels have been reported to be increased in type 2 diabetic and obese individuals and animal models. 43 , 44 , 45 , 46 In this study, we found an increase in the long chain poly‐unsaturated fatty acids (LC‐PUFA), especially the arachidonic acid (AA, 20:4; n‐6), docosahexaenoic acid (DHA, 22:6; n‐3) and eicosapentaenoic acid (EPA, 20:5; n‐3) and their derivatives HETE, HDoHE and HEPE of the lipoxygenase (LOX) pathway through LC–MS/MS lipidomics of gastrocnemius muscle of the type 2 diabetic (db/db) mice. Intriguingly, the db‐P7C3 treated mice significantly decreased the levels of AA, DHA and EPA, and their derivatives in gastrocnemius muscle, suggestive of decreased muscle inflammation in the db‐P7C3 treated mice. The arachidonic acid is also known to serve as a precursor for eicosanoids and endocannabinoids, and the most studied endocannabinoids are 2‐arachidonoyl glycerol (2‐AG) and anandamide (AEA). P7C3 treatment of the type 2 diabetic (db/db) mice also resulted in a decrease in 2‐AG levels consistent with the AA levels in these mice. However, there was an increase in the AEA levels in the db‐P7C3 treated mice, the significance of which is not known and could be involved in the neuromuscular junction. The increase in HDL cholesterol in the db‐P7C3 treated mice might contribute to the cardioprotective effects with increased cholesterol efflux, and anti‐oxidative and anti‐inflammatory properties. 47 Moreover, the decrease in LDL/VLDL cholesterol levels in the db‐P7C3 treated mice might also decrease atherosclerosis in heart disease, and alleviate insulin resistance and type 2 diabetes. Furthermore, the lipidomic analysis reveal that P7C3 treatment of the type 2 diabetic (db/db) mice attenuates the skeletal muscle inflammatory disease. In the present study, we utilized STRING network analysis and identified that key genes and clusters responsible for muscle development, calcium signalling, glycolysis and gluconeogenesis were up‐regulated, whereas, the inflammatory genes, cytokine production and apoptotic genes were down‐regulated (Figures S1 and S2; Data S1). In line with the present findings, a previous report shows similar increase in inflammatory markers using human myocytes from type 2 diabetic individuals with dysregulated myogenesis and gene network associations for development of diabetes. 48 Overall, based on P7C3 treatment and in‐depth RNA‐Seq and network analysis we identify beneficial molecular responses that attenuate diabetes and improves muscle function.
In summary, in the present study we demonstrate that intraperitoneal administration of P7C3 in type 2 diabetic (db/db) mice, daily for 4 weeks improved the glucose uptake by decreasing the fasting blood glucose and insulin resistance, and increasing glucose tolerance, along with increased number of pancreatic β‐cells, islet of Langerhans, and β cells function. The P7C3 treatment also improved the physical performance (fore‐limb and hind‐limb grip strengths, ex vivo myofibre force frequency and the voluntary free running wheel performance) alleviating the diabetic muscle phenotype of the db/db mice. Subsequent, histological analysis of the hind‐limb tibialis anterior and extensor digitorum longus muscles displayed an increase in the myofibre size, with a simultaneous decrease in both MyHC1 expression levels and immunostaining of the slow oxidative myofibres in the db‐P7C3 treated mice. The transmission electron microscopy also revealed an improved mitochondrial morphology in the db‐P7C3 treated mice, probably with improved mitochondrial membrane potential. The increased SDH positive myofibres and Pgc‐1α expression in the db‐P7C3 treated mice demonstrate improved mitochondrial function. Mechanistically, there was increased mitochondrial fatty acid β‐oxidation with decreased fatty acid uptake and oxidative stress in the db‐P7C3 treated mice in energy metabolism. RNA Seq analysis also displayed an increase in fatty acid oxidation, mitochondrial biogenesis, muscle function with reduced inflammation. The LC–MS/MS lipidomic analysis of gastrocnemius muscle also displayed a decrease in the inflammatory lipids, along with an increase in the circulating HDL and decrease in the VLDL/LDL lipids in the db‐P7C3 treated mice resulting in reduced muscle inflammation. The serum pro‐inflammatory marker IP‐10 levels also displayed a decrease in db‐P7C3 treated mice. The specificity of P7C3 treatment was confirmed with an increase in the Nampt activity both in vitro and in vivo. The GTT of the P7C3 treated Nampt+/− mice further validated the hyperglycaemic effects of P7C3 as Nampt‐mediated. Together, our findings highlight the importance of P7C3‐mediated Nampt activation in ameliorating the diabetic skeletal muscle phenotype of db/db mice and Nampt actvator P7C3 as a potential treatment strategy in type 2 diabetes.
Conflict of interest
The authors declare that there is no conflict of interest.
Disclosures
Patent awarded to USF: US Patent 11,007,178, 2021. USF Provisional patent filed: 19A039PR.
Supporting information
Figure S1. Gene network interaction using Cytoscape v3.8.2 for the up‐regulated genes as a STRING network in (A) MCODE (molecular complex detection) used to identify dense clusters with enrichment scores in panel (B‐D) top three clusters. Cluster 1 has enrichment score = 17.579, Cluster 2 has enrichment score = 8.500, and Cluster 3 has enrichment score = 8.286.
Figure S2. Gene network interaction using Cytoscape v3.8.2 for the down‐regulated genes as a STRING network in (A) MCODE (molecular complex detection) was used to identify dense clusters with enrichment scores in panel (B) top three clusters. Cluster 1 has enrichment score = 38.789, Cluster 2 has enrichment score = 29.760, and Cluster 3 has enrichment score = 16.833.
Figure S3. Physical performance of wild‐type and db‐Veh mice, and Tnfα expression. (A) Forelimb grip strength and (B) Hindlimb grip strength measured and expressed as KGF/kg body weight (C) The area under the curve (AUC) of the absolute muscle force frequency, and (D) the normalized muscle force frequency of the extensor digitorum longus muscles (E). The relative mRNA expression levels of Tnfα in db‐Veh and db‐P7C3 gastrocnemius muscle. Data are expressed as mean ± SEM; *P < 0.05.
Figure S4. P7C3 treatment of the diabetic db/db mice enhances the inflammatory lipid mediators. (A, B) Heat map of the key lipid mediators analysed (48 in numbers) in gastrocnemius muscle of the wild type naïve (Control) mice, db‐Veh (DB) and db‐P7C3 (db P7) treated mice.
Figure S5. P7C3 treatment of the diabetic db/db mice enhances the n‐6 and n‐3 PUFA anti‐inflammatory lipid mediators of the LOX pathway. The n‐6 PUFA AA derivatives (A) 11‐HETE, (B) 15‐HETE, and the n‐3 PUFA DHA derivatives (C) 7‐HDoHE, (D) 13‐HDoHE, (E) 16‐HDoHE, (F) 17‐HDoHE, (G) 20‐HDoHE, and the n‐3 PUFA EPA derivative (H) 5‐HEPE of the wild type naïve control (WT), db‐Veh and db‐P7C3 treated mice.
Figure S6. P7C3 reverses hyperglycaemia in a Nampt‐dependent manner. (A) Genotype of Nampt littermate wildtype (WT) with a single 300 bp nucleotide band and Nampt heterozygous mice (Nampt+/−) with a double band, i.e., 300 bp wildtype and the 500 bp heterozygous nucleotide bands. (B) Forelimb grip strength of the Nampt+/+ WT and Nampt+/− mice expressed in KGF/kg. (C) Glucose tolerance test of the Nampt+/+ wild type and (D) Nampt+/− heterozygous mice. Percentage of area under curve (AUC) for IPGTT obtained by using trapezium method for both the (E) Wildtype and (F) Nampt+/− heterozygous mice. Data are expressed as mean ± SEM; *P < 0.05 of P7C3 vs. Vehicle treated in wild type and Nampt+/− heterozygous mice.
Data S1. Supplemental Results.
Acknowledgements
The authors thank Ms Viviana Puig and Ms Jazmine Virzi for the technical assistance with mouse work, and Ms Amanda Garces for the Transmission Electron Microscopy work. This work was supported by NIH R01 DK119066 funding (to SMT, MB), Saunders Endowed Chair in Geriatric Pharmacotherapy (to SMT). The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.
Manickam R., Tur J., Badole S. L., Chapalamadugu K. C., Sinha P., Wang Z., Russ D. W., Brotto M., and Tipparaju S. M. (2022) Nampt activator P7C3 ameliorates diabetes and improves skeletal muscle function modulating cell metabolism and lipid mediators, Journal of Cachexia, Sarcopenia and Muscle, 13, 1177–1196, 10.1002/jcsm.12887
Contributor Information
Marco Brotto, Email: marco.brotto@uta.edu.
Srinivas M. Tipparaju, Email: stippara@usf.edu.
References
- 1. Wojciechowska J, Krajewski W, Bolanowski M, Kręcicki T, Zatoński T. Diabetes and cancer: a review of current knowledge. Exp Clin Endocrinol Diabetes 2016;124:263–275. [DOI] [PubMed] [Google Scholar]
- 2. Phielix E, Mensink M. Type 2 diabetes mellitus and skeletal muscle metabolic function. Physiol Behav 2008;94:252–258. [DOI] [PubMed] [Google Scholar]
- 3. Galicia‐Garcia U, Benito‐Vicente A, Jebari S, Larrea‐Sebal A, Siddiqi H, Uribe KB, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci 2020;21. 10.3390/ijms21176275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Twig G, Zucker I, Afek A, Cukierman‐Yaffe T, Bendor CD, Derazne E, et al. Adolescent obesity and early‐onset type 2 diabetes. Diabetes Care 2020;43:1487–1495. [DOI] [PubMed] [Google Scholar]
- 5. Mesinovic J, Zengin A, De Courten B, Ebeling PR, Scott D. Sarcopenia and type 2 diabetes mellitus: a bidirectional relationship. Diab Metab Syndr Obes 2019;12:1057–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Williams R, Van Gaal L, Lucioni C. Assessing the impact of complications on the costs of type II diabetes. Diabetologia 2002;45:S13–S17. [DOI] [PubMed] [Google Scholar]
- 7. Liu L, Simon B, Shi J, Mallhi AK, Eisen HJ. Impact of diabetes mellitus on risk of cardiovascular disease and all‐cause mortality: evidence on health outcomes and antidiabetic treatment in United States adults. World J Diabetes 2016;7:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bianchi L, Volpato S. Muscle dysfunction in type 2 diabetes: a major threat to patient's mobility and independence. Acta Diabetol 2016;53:879–889. [DOI] [PubMed] [Google Scholar]
- 9. Manickam R, Duszka K, Wahli W. PPARs and microbiota in skeletal muscle health and wasting. Int J Mol Sci 2020;21: 10.3390/ijms21218056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryder JW, Gilbert M, Zierath JR. Skeletal muscle and insulin sensitivity: pathophysiological alterations. Front Biosci 2001;6:D154–D163. [DOI] [PubMed] [Google Scholar]
- 11. Tenenbaum A, Motro M, Schwammenthal E, Fisman EZ. Macrovascular complications of metabolic syndrome: an early intervention is imperative. Int J Cardiol 2004;97:167–172. [DOI] [PubMed] [Google Scholar]
- 12. Jensen J, Rustad PI, Kolnes AJ, Lai YC. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front Physiol 2011;2:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hegarty BD, Furler SM, Ye J, Cooney GJ, Kraegen EW. The role of intramuscular lipid in insulin resistance. Acta Physiol Scand 2003;178:373–383. [DOI] [PubMed] [Google Scholar]
- 14. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387. [DOI] [PubMed] [Google Scholar]
- 15. de Guia RM, Agerholm M, Nielsen TS, Consitt LA, Søgaard D, Helge JW, et al. Aerobic and resistance exercise training reverses age‐dependent decline in NAD+ salvage capacity in human skeletal muscle. Physiol Rep 2019;7:e14139. 10.14814/phy2.14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet‐ and age‐induced diabetes in mice. Cell Metab 2011;14:528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol 2015;11:535–546. [DOI] [PubMed] [Google Scholar]
- 18. Alshahrani A, AlDubayee M, Zahra M, Alsebayel FM, Alammari N, Alsudairy F, et al. Differential expression of human N‐alpha‐acetyltransferase 40 (hNAA40), nicotinamide phosphoribosyltransferase (NAMPT) and sirtuin‐1 (SIRT‐1) pathway in obesity and T2DM: modulation by metformin and macronutrient intake. Diab Metab Syndr Obes 2019;12:2765–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hallschmid M, Randeva H, Tan BK, Kern W, Lehnert H. Relationship between cerebrospinal fluid visfatin (PBEF/Nampt) levels and adiposity in humans. Diabetes 2009;58:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Revollo JR, Körner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab 2007;6:363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frederick DW, Loro E, Liu L, Davila A Jr, Chellappa K, Silverman IM, et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab 2016;24:269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Agerholm M, Dall M, Jensen BAH, Prats C, Madsen S, Basse AL, et al. Perturbations of NAD+ salvage systems impact mitochondrial function and energy homeostasis in mouse myoblasts and intact skeletal muscle. Am J Physiol Endocrinol Metab 2018;314:E377–e95. [DOI] [PubMed] [Google Scholar]
- 23. Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, et al. Discovery of a proneurogenic, neuroprotective chemical. Cell 2010;142:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, et al. P7C3 neuroprotective chemicals function by activating the rate‐limiting enzyme in NAD salvage. Cell 2014;158:1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim W, Doyle ME, Liu Z, Lao Q, Shin YK, Carlson OD, et al. Cannabinoids inhibit insulin receptor signaling in pancreatic β‐cells. Diabetes 2011;60:1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Badole SL, Bodhankar SL. Antidiabetic activity of cycloart‐23‐ene‐3beta, 25‐diol (B2) isolated from Pongamia pinnata (L. Pierre) in streptozotocin‐nicotinamide induced diabetic mice. Eur J Pharmacol 2010;632:103–109. [DOI] [PubMed] [Google Scholar]
- 27. Chapalamadugu KC, Panguluri SK, Bennett ES, Kolliputi N, Tipparaju SM. High level of oxygen treatment causes cardiotoxicity with arrhythmias and redox modulation. Toxicol Appl Pharmacol 2015;282:100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chapalamadugu KC, Tur J, Badole SL, Kukreja RC, Brotto M, Tipparaju SM. Physiological role of Kvβ2 (AKR6) in murine skeletal muscle growth and regulation. Acta Physiol (Oxf) 2018;224:e13083, 10.1111/apha.13083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen Z, Chen Q, Ying H, Ma Z, Bi X, Li X, et al. Identification of differentially expressed genes in the endothelial precursor cells of patients with type 2 diabetes mellitus by bioinformatics analysis. Exp Ther Med 2020;19:499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cirillo E, Kutmon M, Gonzalez Hernandez M, Hooimeijer T, Adriaens ME, Eijssen LMT, et al. From SNPs to pathways: Biological interpretation of type 2 diabetes (T2DM) genome wide association study (GWAS) results. PLoS ONE 2018;13:e0193515, 10.1371/journal.pone.0193515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform 2003;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shochat C, Wang Z, Mo C, Nelson S, Donaka R, Huang J, et al. Deletion of SREBF1, a functional bone‐muscle pleiotropic gene, alters bone density and lipid signaling in zebrafish. Endocrinology 2021;162: 10.1210/endocr/bqaa189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marrelli MT, Wang Z, Huang J, Brotto M. The skeletal muscles of mice infected with Plasmodium berghei and Plasmodium chabaudi reveal a crosstalk between lipid mediators and gene expression. Malar J 2020;19:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klueber KM, Feczko JD, Schmidt G, Watkins JB 3rd. Skeletal muscle in the diabetic mouse: histochemical and morphometric analysis. Anat Rec 1989;225:41–45. [DOI] [PubMed] [Google Scholar]
- 35. Pieper AA, McKnight SL. Benefits of enhancing nicotinamide adenine dinucleotide levels in damaged or diseased nerve cells. Cold Spring Harb Symp Quant Biol 2018;83:207–217. [DOI] [PubMed] [Google Scholar]
- 36. Hua X, Sun DY, Zhang WJ, Fu JT, Tong J, Sun SJ, et al. P7C3‐A20 alleviates fatty liver by shaping gut microbiota and inducing FGF21/FGF1, via the AMP‐activated protein kinase/CREB regulated transcription coactivator 2 pathway. Br J Pharmacol 2020;178:2111–2130. [DOI] [PubMed] [Google Scholar]
- 37. Vitali F, Mulas F, Marini P, Bellazzi R. Network‐based target ranking for polypharmacological therapies. J Biomed Inform 2013;46:876–881. [DOI] [PubMed] [Google Scholar]
- 38. Antonetti DA, Reynet C, Kahn CR. Increased expression of mitochondrial‐encoded genes in skeletal muscle of humans with diabetes mellitus. J Clin Invest 1995;95:1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gu C, Zhang Y, Hu Q, Wu J, Ren H, Liu CF, et al. P7C3 inhibits GSK3β activation to protect dopaminergic neurons against neurotoxin‐induced cell death in vitro and in vivo . Cell Death Dis 2017;8:e2858, 10.1038/cddis.2017.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet 2014;384:626–635. [DOI] [PubMed] [Google Scholar]
- 41. Siddiqi HK, Kiss D, Rader D. HDL‐cholesterol and cardiovascular disease: rethinking our approach. Curr Opin Cardiol 2015;30:536–542. [DOI] [PubMed] [Google Scholar]
- 42. Fruchart JC. Insulin‐resistance and lipoprotein abnormalities. Diabete Metab 1991;17:244–248. [PubMed] [Google Scholar]
- 43. Pellegrinelli V, Rouault C, Rodriguez‐Cuenca S, Albert V, Edom‐Vovard F, Vidal‐Puig A, et al. Human adipocytes induce inflammation and atrophy in muscle cells during obesity. Diabetes 2015;64:3121–3134. [DOI] [PubMed] [Google Scholar]
- 44. Warfel JD, Bermudez EM, Mendoza TM, Ghosh S, Zhang J, Elks CM, et al. Mitochondrial fat oxidation is essential for lipid‐induced inflammation in skeletal muscle in mice. Sci Rep 2016;6:37941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Romic S, Krskova K, Olszanecki R, Balazova L, Lory V, Koricanac G, et al. Obesity‐ and age‐related alterations in FAT/CD36 translocation and lipin‐1 subcellular localization in skeletal muscle of the Zucker rats. Gen Physiol Biophys 2017;36:399–406. [DOI] [PubMed] [Google Scholar]
- 46. Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is necessary for long‐term but not short‐term high‐fat diet‐induced insulin resistance. Diabetes 2011;60:2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krauss RM. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004;27:1496–1504. [DOI] [PubMed] [Google Scholar]
- 48. Väremo L, Henriksen TI, Scheele C, Broholm C, Pedersen M, Uhlén M, et al. Type 2 diabetes and obesity induce similar transcriptional reprogramming in human myocytes. Genome Med 2017;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gene network interaction using Cytoscape v3.8.2 for the up‐regulated genes as a STRING network in (A) MCODE (molecular complex detection) used to identify dense clusters with enrichment scores in panel (B‐D) top three clusters. Cluster 1 has enrichment score = 17.579, Cluster 2 has enrichment score = 8.500, and Cluster 3 has enrichment score = 8.286.
Figure S2. Gene network interaction using Cytoscape v3.8.2 for the down‐regulated genes as a STRING network in (A) MCODE (molecular complex detection) was used to identify dense clusters with enrichment scores in panel (B) top three clusters. Cluster 1 has enrichment score = 38.789, Cluster 2 has enrichment score = 29.760, and Cluster 3 has enrichment score = 16.833.
Figure S3. Physical performance of wild‐type and db‐Veh mice, and Tnfα expression. (A) Forelimb grip strength and (B) Hindlimb grip strength measured and expressed as KGF/kg body weight (C) The area under the curve (AUC) of the absolute muscle force frequency, and (D) the normalized muscle force frequency of the extensor digitorum longus muscles (E). The relative mRNA expression levels of Tnfα in db‐Veh and db‐P7C3 gastrocnemius muscle. Data are expressed as mean ± SEM; *P < 0.05.
Figure S4. P7C3 treatment of the diabetic db/db mice enhances the inflammatory lipid mediators. (A, B) Heat map of the key lipid mediators analysed (48 in numbers) in gastrocnemius muscle of the wild type naïve (Control) mice, db‐Veh (DB) and db‐P7C3 (db P7) treated mice.
Figure S5. P7C3 treatment of the diabetic db/db mice enhances the n‐6 and n‐3 PUFA anti‐inflammatory lipid mediators of the LOX pathway. The n‐6 PUFA AA derivatives (A) 11‐HETE, (B) 15‐HETE, and the n‐3 PUFA DHA derivatives (C) 7‐HDoHE, (D) 13‐HDoHE, (E) 16‐HDoHE, (F) 17‐HDoHE, (G) 20‐HDoHE, and the n‐3 PUFA EPA derivative (H) 5‐HEPE of the wild type naïve control (WT), db‐Veh and db‐P7C3 treated mice.
Figure S6. P7C3 reverses hyperglycaemia in a Nampt‐dependent manner. (A) Genotype of Nampt littermate wildtype (WT) with a single 300 bp nucleotide band and Nampt heterozygous mice (Nampt+/−) with a double band, i.e., 300 bp wildtype and the 500 bp heterozygous nucleotide bands. (B) Forelimb grip strength of the Nampt+/+ WT and Nampt+/− mice expressed in KGF/kg. (C) Glucose tolerance test of the Nampt+/+ wild type and (D) Nampt+/− heterozygous mice. Percentage of area under curve (AUC) for IPGTT obtained by using trapezium method for both the (E) Wildtype and (F) Nampt+/− heterozygous mice. Data are expressed as mean ± SEM; *P < 0.05 of P7C3 vs. Vehicle treated in wild type and Nampt+/− heterozygous mice.
Data S1. Supplemental Results.