

Abstract
Immunohistochemical staining of mouse brains is a routine technique commonly used in neuroscience to investigate central mechanisms underlying the regulation of energy metabolism and other neurobiological processes. However, the quality, reliability, and reproducibility of brain histology results may vary among laboratories. For each staining experiment, it is necessary to optimize the key procedures based on differences in species, tissues, targeted proteins, and the working conditions of the reagents. This paper demonstrates a reliable workflow in detail, including intra-aortic perfusion, brain sectioning, free-floating immunostaining, tissue mounting, and imaging, which can be followed easily by researchers in this field.
Also discussed are how to modify these procedures to satisfy the individual needs of researchers. To illustrate the reliability and efficiency of this protocol, perineuronal nets were stained with biotin-labeled Wisteria florbunda agglutinin (WFA) and arginine vasopressin (AVP) with an anti-AVP antibody in the mouse brain. Finally, the critical details for the entire procedure have been addressed, and the advantages of this protocol compared to those of other protocols. Taken together, this paper presents an optimized protocol for free-floating immunostaining of mouse brain tissue. Following this protocol makes this process easier for both junior and senior scientists to improve the quality, reliability, and reproducibility of immunostaining studies.
Introduction
The prevalence of obesity and associated comorbidities has reached epidemic levels, causing a tremendous socioeconomic burden1,2. Various mouse models have been developed to better understand the biological processes responsible for obesity3,4. Because central mechanisms are important for the regulation of energy homeostasis in these animal models, neuroanatomical studies of mouse brains have become a necessary technique in this field. However, the quality, reliability, and reproducibility of brain histology techniques vary considerably among laboratories and even researchers within the same laboratory for various reasons (e.g., antibodies, tissues, treatments, species, research objectives). Therefore, it is necessary to establish a general protocol for histological studies of the mouse brain, including perfusion, brain sectioning, free-floating immunostaining, tissue mounting, and imaging. Meanwhile, beginners can quickly learn, master, and adjust this protocol to satisfy their individual needs.
Immunohistochemical staining is an established method that has been used extensively to visualize specific cell types, mRNAs, and proteins in a variety of tissues (e.g., brain and peripheral tissues)5,6. More specifically, an antigen of interest can be labeled by a specific primary antibody and a corresponding secondary antibody linked to an enzyme (e.g., chromogenic immunohistochemistry) or a fluorescent dye (fluorescein isothiocyanate)6. As an example of the utility of these techniques, β-endorphin [one peptide encoded by pro-opiomelanocortin (POMC)] and c-fos (a marker of neuronal activity) were stained in the arcuate nucleus. Deletion of tryptophan hydroxylase 2 (an enzyme integral to serotonin synthesis) in the dorsal raphe nucleus was shown to decrease c-fos expression in POMC neurons in the arcuate nucleus7. In addition, the distribution of vitamin D receptor mRNA was mapped in the mouse brain via in situ hybridization (RNAscope)8. This paper presents a reliable and efficient method with a step-by-step workflow for free-floating immunostaining, aiming to improve the quality and reproducibility of histological studies of the mouse brain.
Protocol
C57BL/6J mice of both sexes (8–16 weeks of age) were used in the present study. Care of all animals and all procedures were approved by Baylor College of Medicine’s Institutional Animal Care and Use Committees.
1. Perfusion
NOTE: Steps 1.1 – 1.6 are performed in a fume hood.
1. Anesthesia
-
Pour 5 mL of isoflurane (see the Table of Materials) onto a paper towel placed at the bottom of a desiccator. Introduce the mouse onto the holed barrier inside the desiccator and wait until signs of respiration have disappeared.
NOTE: Without respiration, the mouse still has heartbeat for a short period of time, and it will be perfused before the disappearance of the heartbeat.
Before proceeding, confirm there is no reflex to a toe-pinch.
Fix the mouse to a foam board by driving a pin through each foot. Ensure that the foam board is placed in a tray to collect liquid spillover.
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| Alexa Flour 594 donkey anti-rabbit IgG (H+L) | Invitrogen | A21207 | |
| 30% Sucrose | VWR | 470302 | 30 g Sucrose dissoved into 100 mL of PBS |
| Neutral Buffered Formalin | VWR | 16004–128 | 10%, 25 °C, pH 6.8–7.2 |
| 1 mL Sub-Q Syringe | BD | 309597 | |
| 48 Well Cell Culture Plate | Corning | 3548 | |
| 6 Well Cell Culture Plate | Corning | 3516 | |
| Antifading mounting media with DAPI | Vector Laboratories | H-1200 | |
| Autoclavable plastic desiccator | Thermo Scientific Nalgene | 5315–0150 | |
| AVP antibody | Phoenix Pharmaceuticals | H-065–07 | |
| Cell Strainer | Corning | 431752 | |
| Cryoprotectant buffer | User preference | Not applicable | 20% glycerol, 30% ethylene glycol, and 50% PBS |
| Isoflurane | Covetrus | 11695–6777-2 | |
| Leica DFC310FX microscope | Leica | Not applicable | |
| Microscope Slide Boxes (50-place) | VWR | Not applicable | |
| PBT | User preference | Not applicable | 2.5 mL of Triton X-100 dissolved into 1000 mL of PBS |
| Perfusion two automated Perfusion System | Leica | 39471005 | |
| Phosphate-buffered saline (PBS) 20x | VWR | VWRVE703–1L | 25 °C, pH 7.3–7.5, 1x composition:137 mM NaCl, 2.7 mM KCl, 9.8 mM Phosphate buffer |
| Slideing Microtome Microm HM450 | ThermoFisher | Microm HM450 | |
| Sodium Chloride | RICCA Chemical | 7220–32 | 0.9%, 25 °C, pH 7.4 |
| Streptavidin Protein, DyLight 488 | ThermoFisher | #21832 | |
| Triton X-100 | Sigma-Aldrich | 089k01921 | |
| WFA antibody | Sigma-Aldrich | L1516 | |
| Zeiss Axio Z1 Scanner | Zeiss | Not applicable | |
| Zen 3.1 software | scanner software |
2. Exposing the heart
Make a longitudinal superficial incision along the midline over the thorax and abdomen, then move the skin aside to expose the muscle wall of the thorax and abdomen. Next, make an incision in the muscle layer to expose the liver and the intestine. Finally, cut the rib cage with scissors to open the thorax and expose the heart and lungs. Use hemostatic forceps to pull the ribcage aside to widen the work area.
3. Collection of terminal blood (optional)
Insert a 1 mL syringe (see the Table of Materials) carefully into the right atrium of the heart until the tip is completely embedded. Hold the syringe steady and draw blood slowly until the desired volume is reached.</p>NOTE: Take care not to penetrate past the right atrium; Collection of 300–400 µL of blood is achievable per adult mouse. Additives such as a clot accelerator or anticoagulant may be used, depending on the purpose of blood collection.
4. Placement of the perfusion cannula
For beginners, cut a small hole (<1 mm) in the left ventricle of the heart with scissors and insert a blunt cannula (18 G needle for young mice and 21 G needle for aged mice) through the left ventricle into the ascending aorta. Alternatively, for experienced experimenters, penetrate the left heart ventricle with a blunt cannula directly and insert it into the ascending aorta carefully.
-
Place pins around the conjunction of the cannula and coupled tubing on the foam board to prevent movement during the perfusion. Alternatively, use hemostatic forceps to fix the cannula in place. Proceed to cut the right atrium to allow the outflow of blood from circulation.
NOTE: The perfusion cannula and coupled tubing are connected to the perfusion pump.
5. Perfusion with saline
-
Turn on the saline pressure switch, and perfuse the mouse transcardially with 40–60 mL of saline (0.9% NaCl: 25 °C, pH 7.4). Observe the outflow from the right atrium and the color of the liver closely.
NOTE: An automated perfusion pump (see the Table of Materials) is used to flush the blood within 1–2 min. As the pressure range of the pump is 1–300 mmHg, the speed can be adjusted accordingly. If saline is leaking from the nose and/or the lung is inflated, decrease the pressure and adjust the cannula. Once the outflow no longer contains blood, the brain is sufficiently flushed with saline. Meanwhile, the liver is devoid of blood and becomes brownish-gray in color.
6. Perfusion with formalin
-
Turn off the saline pressure switch and turn on the formalin pressure switch to perfuse the mouse with 40 mL of 10% neutral buffered formalin (10% NBF: 25 °C, pH 6.8–7.2, see the Table of Materials). Observe the animal’s limbs for evidence of tremors.
NOTE: Tail movement may also be observed after infusion of 10% NBF.CAUTION: As NBF is hazardous, it is suggested that researchers wear personal protective equipment (e.g., appropriate respirator, face shield) throughout the procedure.
7. Brain isolation9
Use scissors to remove the head. Make a middle line incision along the integument to expose the skull. Trim off the skin and muscle attachment with scissors.
Make a cut at the orbital ridge, and then place the sharp end of iris scissors into the foramen magnum. Advance the scissors along the inner surface of the skull, maintaining upward pressure to avoid damage to the brain. Remove the parietal/frontal bones and meninges carefully. Finally, remove the brain from the opened skull gently.
8. Post-fixation
-
Place the brain in a 15 mL tube filled with 10 mL of 5% NBF and 15% sucrose for overnight fixation at 4 °C.
NOTE: To prepare 10 mL of 5% NBF and 15% sucrose, combine 5 mL of 10% NBF and 5 mL of 30% sucrose (w/v, dissolved into phosphate-buffered saline (PBS), see the Table of Materials). Make sure the volume of the fixative buffer is at least 10x larger than the sample itself. Initially, the brain will float in the buffer and will sink to the bottom after overnight fixation.
9. Dehydration
Transfer the brain sample to a 15 mL tube filled with 10 mL of 30% sucrose for dehydration at 4 °C until it sinks.
2. Cryosectioning (coronal sections)
1. Preparation
-
Place dry ice on top of the height adjustment plate of a sliding microtome, and wait until white frost is visible. Carefully spread 5 mL of 30% sucrose on top of the plate to form a layer of a solid base after the sucrose solution is fully frozen. Place all brain samples (up to 5 brain samples in one batch) horizontally in a line on top of the sucrose, and then add 0.5 mL of 30% sucrose to the bottom of each brain.
NOTE: The brain will immediately stick to the frozen sucrose base and gradually freeze from bottom to top. Place additional sucrose around the mounted portion of the brain to form a sturdier base for cutting.
2. Sectioning
-
After 5–10 min of freezing, when the brain has become hard and white, trim the brain until the desired layer/region is reached.
NOTE: Adjust the amount of dry ice on the plate to control the temperature. The temperature is too low when ice crystals appear on the surface of the brain. The temperature is too high when the brain becomes soft and is no longer white.
Switch from the Trim mode to the Feed mode and section brain tissue to 25 µm thickness. Prepare a 48-well plate filled with 1x PBS, and mark five wells for one mouse brain. Use a paintbrush to collect each section from one mouse and place it into one well. Collect the subsequent section of the same mouse and place it into the 2nd well.
-
Repeat 2.2.2 until the 5th well is reached, placing sections 6–10 in the 1st −5th well and so on. Repeat the same procedure for the rest of the brains simultaneously. Repeat until all the sections from one mouse are collected into the 5 wells in anatomical order.
NOTE: Following this procedure, the brain sections from each mouse are aliquoted into 5 series, which can be used for up to 5 different histological studies.
3. Storage
-
Use a paintbrush to transfer the sections from each well to a 1.5 mL microtube filled with cryoprotectant buffer (20% glycerol, 30% ethylene glycol, and 50% PBS), and store the samples at −20 °C.
NOTE: These sections stored in small tubes (1.5 mL) can save physical space in the freezer, and the integrity of the sections can be preserved for several months.
3. Free-floating WFA staining and anti-AVP immunostaining
-
Place a cell strainer into a well of a 6-well cell culture plate filled with PBS, and use a paintbrush to transfer one series of brain sections into the cell strainer. Rinse the sections in PBS by transferring the cell strainer to another well filled with PBS. Rinse for 6 × 10 min on a shaker for sections stored in cryoprotectant buffer and 3 × 10 min for freshly cut sections.
NOTE: All the washing procedures are performed in a 6-well cell culture plate with a cell strainer inside each well (see the Table of Materials). Each strainer holds the brain sections of interest from each mouse. To save reagents, all the incubation procedures described below are performed in a 1.5 mL microcentrifuge tube. Between washing and incubation procedures, use a paintbrush to transfer the brain sections between each cell strainer and each tube. For washing with PBS, 12 mL is adequate for each well.
Prepare 1 mL of biotin-labeled WFA (1:1,000) solution in PBT (2.5 mL of Triton X-100 dissolved in 1,000 mL of PBS) buffer in 1.5 mL tube. Transfer brain sections from the cell strainer to the tube and incubate at room temperature on a rocking platform ~50 rpm overnight.
Rinse the brain sections with PBS for 3 × 10 min as described in 3.1.
-
Prepare 1 mL of streptavidin-Dylight 488 (1:500) solution in PBT in a 1.5 mL tube. Transfer brain sections into the tube and incubate at room temperature for 2 h on a rocking platform at ~50 rpm.
NOTE: Cover the plate with foil to avoid light exposure from this step forward.
Rinse the brain sections with PBS for 3 × 10 min. Incubate the brain sections in blocking buffer (3% normal donkey serum diluted in PBT) for 2 h at room temperature.</p>NOTE: The choice of blocking serum is determined by the species from which the secondary antibody is generated. e.g., if the secondary antibody is from goat, normal goat serum should be used.
-
Incubate the brain sections in primary antibody (rabbit anti-AVP, 1:500) at room temperature on a rocking platform at ~50 rpm overnight.
NOTE: Prepare both primary and secondary antibodies in blocking buffer. The concentration, duration of incubation, and temperature of incubation need to be optimized in pilot studies.
Rinse the brain sections with PBS for 3 × 10 min. Incubate the brain sections in secondary antibody (Alexa Flour 594 donkey anti-rabbit IgG (H+L), 1:500) at room temperature for 2 h on a rocking platform at ~50 rpm. Rinse the brain sections with PBS for 3 × 10 min.
4. Mounting
-
Fill two Petri dishes (a diameter of 150 mm) with 100 mL of 1x PBS each. Transfer all the brain sections from one strainer into the first dish, and align the brain sections in neuroanatomical order from caudal to rostral.
NOTE: To avoid mixing sections from different animals, mount one series of brain sections from one animal at a time.
After all the brain sections are aligned in the first dish, submerge one slide into the second dish with one end slightly tilted with a stand (see Figure 1 and Figure 2). Use a fine paintbrush to gently place a brain section just below the air-buffer interface onto the tilted slide. Repeat the same procedure with another brain section and mount it side by side with the first section.
-
Use a transfer pipette to slowly and gently remove the buffer to lower its level until both brain sections are entirely above the air-buffer interface.
NOTE: Take care to minimize the disturbance of the buffer surface, or the brain sections may float away. When dry, this row of brain sections will adhere to the slide firmly. If necessary, gently adjust the sections on the slide to ensure there are no wrinkles or folds in the tissue when the sections are still wet.
Repeat this process until the bottom of the slide is reached. Continue to repeat until all sections are mounted onto the slide(s).
Figure 1: Flow chart of fluorescence immunohistochemistry with mouse brains.
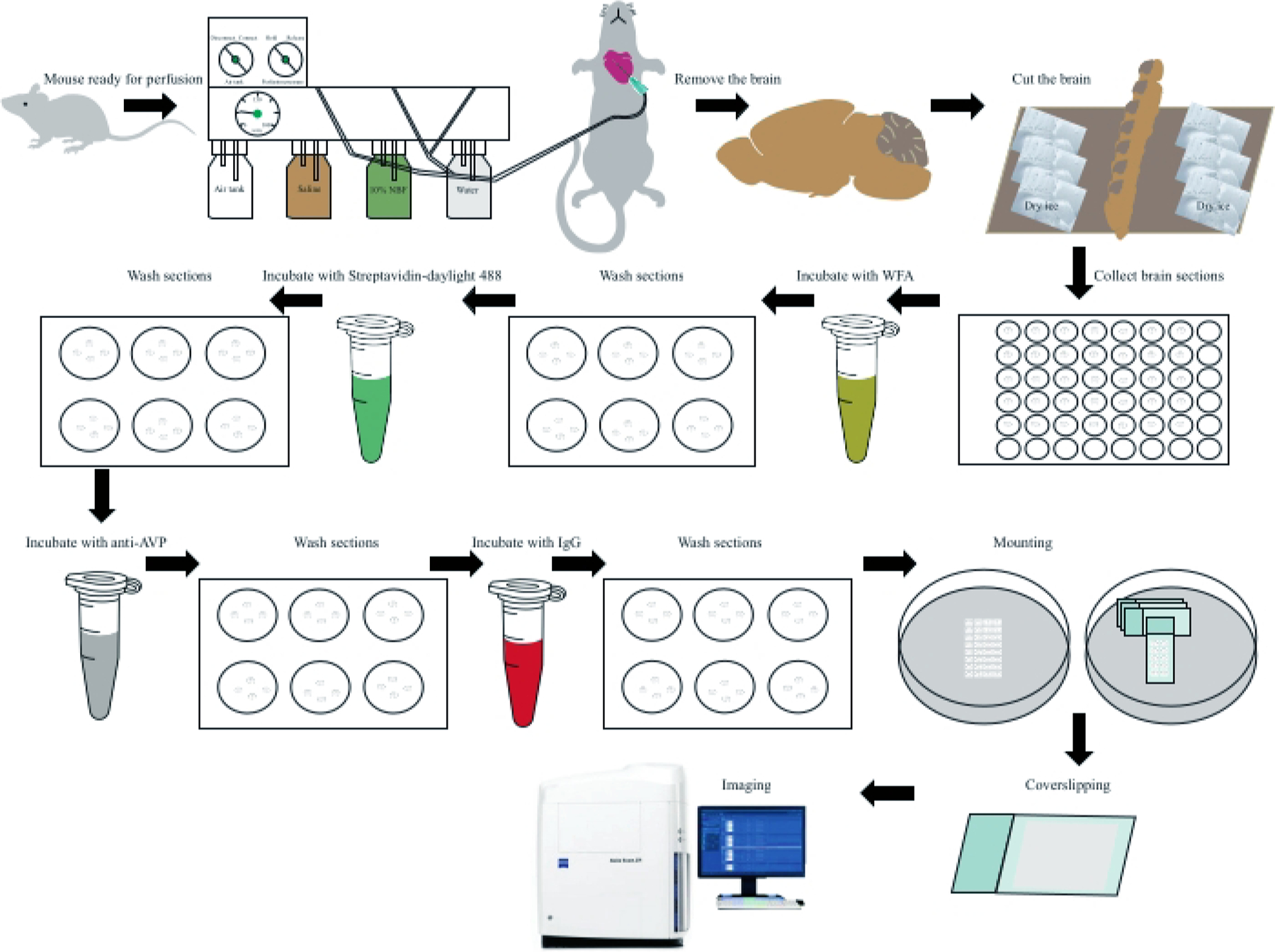
Following complete anesthesia, a mouse is perfused with saline and then 10% NBF. The brain is carefully removed and cut into sections after fixation and dehydration. The sections were incubated with WFA followed by Streptavidin-Dylight 488 after three washes with PBS. The brain sections were blocked and then incubated with a primary anti-AVP antibody. Then, the sections were washed 3 times with PBS followed by incubation with the secondary antibody, Alexa Flour 594 donkey anti-rabbit IgG (H+L). The brain sections were mounted on slides and coverslips placed on the slides with antifading mounting medium with DAPI before imaging. Abbreviations: NBF = neutral buffered formalin; WFA = Wisteria florbunda agglutinin; PBS = phosphate-buffered saline; AVP = arginine vasopressin; DAPI = 4′,6-diamidino-2-phenylindole.
Figure 2: Photos illustrating practical cryosectioning and mounting in the laboratory.

(A) Build up a base on top of the plate with 30% sucrose to hold all samples horizontally, cut the tissues into sections (25 µm/section) and collect the sections into a 48-well cell culture plate filled with phosphate-buffered saline. (B) Submerge a slide in the dish with one end slightly tilted with a stand. Place each row of sections under the air-buffer interface and lower the buffer to bring the sections out of the buffer until the bottom of the slide. The white line indicates the interface of buffer and air. (C) A fully mounted slide with brain sections.
5. Coverslipping
-
After all sections have dried (1–24 h at room temperature), place 80–100 µL of anti-fading mounting medium with DAPI (see the Table of Materials) on the slide, and gently apply a glass coverslip to cover the samples.
NOTE: Apply the glass coverslip carefully and slowly to avoid bubbles.
-
Place the slides in a microscope slide box (see the Table of Materials) and store them at 4 °C.
NOTE: Image all sections within 1–3 days to avoid fading of fluorescence and the development of auto fluorescence.
6. Imaging
-
Turn on the scanner and the computer (see Table of Materials). Position the slides in the slide holder with the loading device and insert the holder in the scanner.
NOTE: The scanner enables scanning of multiple channels (e.g., 4′,6-diamidino-2-phenylindole (DAPI), green, and red channels) simultaneously. The scanner scans slides only with a 20x magnification, and a traditional fluorescent microscope (see Table of Materials) can be used when a higher magnification (e.g., 40x) is needed.
Open the software for the scanner (see the Table of Materials). Choose the appropriate storage location and scanning profile.
Start the preview scan by clicking on the Start Preview Scan button. After the preview scan, open the tissue detection wizard and circle the regions of interest for imaging.
-
Click on the Start Scan button after choosing the regions for imaging. Wait for the machine to finish scanning. Check the result file and export the images.
NOTE: If the profile is unsuitable for the slide, adjust by refocusing and changing the exposure time or light strength to adapt the profile to the tissues. Refer to the instructions for the scanner for more technical details (Table of Materials).
Representative Results
The flow chart of this protocol is briefly illustrated in Figure 1. This laboratory’s cryosectioning procedure is demonstrated in Figure 2A, in which 5 brain samples were sectioned simultaneously. The mounting of brain sections is shown in Figure 2B, and a fully mounted slide with brain sections is illustrated in Figure 2C. In Figure 3, representative fluorescence immunohistochemistry images of a mouse brain section with co-staining of WFA and AVP at lower and higher magnification at Bregma −0.82 mm. AVP signals were observed in the paraventricular nucleus and the supraoptic nucleus. WFA signals were observed in the perifornical area of the anterior hypothalamus and the reticular nucleus.
Figure 3: An example of double immunofluorescence staining.
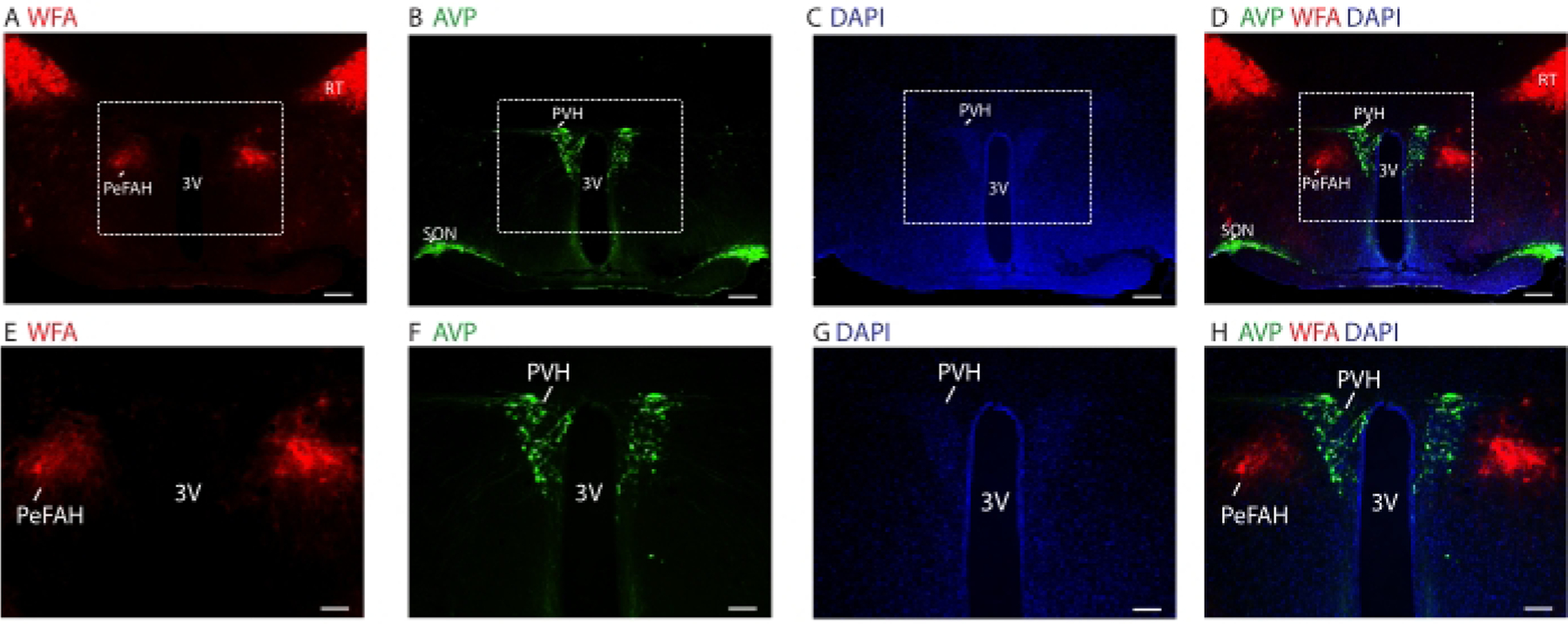
(A-D) Microscopic images showing the distribution of WFA (red, A), AVP (green, B), DAPI (blue, C), and merged (D) in coronal mouse brain sections at Bregma −0.82 mm. Scale bars = 200 µm. (E-H) Higher magnification microscopic images of the white boxes in A-D, respectively. Scale bars = 100 µm. Abbreviations: 3V = third ventricle; PeFAH = perifornical area of the anterior hypothalamus; PVH = paraventricular nucleus; RT = reticular nucleus; SON = supraoptic nucleus.
Discussion
This protocol provides an established method for neuroanatomical studies of the mouse brain, including perfusion, tissue sectioning, free-floating immunostaining, tissue mounting, and imaging. However, a few key details essential for consistent and reliable results must be optimized.
The quality of perfusion is critical for successful staining. Staining results might be affected if blood remains in the brain, given that blood cells (e.g., red blood cells) can generate an artificial ‘positive’ staining10. We infer the presence of a brownish-gray liver to indicate a high quality of perfusion, which usually results in blood-free brains. The automated perfusion pump used in this protocol helps to perfuse one animal successfully within a short period. Insufficient fixation will generate soft and fragile brain sections in the subsequent procedures, while over fixation will reduce the sensitivity of antigen reactions due to the enhanced formaldehyde cross-linking of proteins. Different conditions were tested, and overnight fixation at 4 °C was sufficient for post-fixation of mouse brains. In addition to 10% NBF used in the present protocol as a fixative buffer, 4% (w/v) of freshly prepared paraformaldehyde (PFA) in PBS has also been extensively used for tissue fixation11.
Regarding cryosectioning, the thickness of sections needs to be decided depending on the specific needs. For instance, RNAscope studies require a thickness of 14 µm instead of 25 µm, commonly used in free-floating staining. Meanwhile, RNAscope studies require that all procedures are performed in RNAase-free solutions to preserve the integrity of the target mRNAs. Some researchers also use a section thickness of 30–40 µm for a variety of staining procedures. Conventional cryosectioning (i.e., optimal cutting temperature (O.C.T.) compound-embedded samples) allows for much thinner (e.g., 10 µm) brain sections that might be crucial for intracellular structures or other applications. The cryosectioning strategy presented here does not necessarily involve O.C.T. compound-embedding of brain samples and allows for 14–40 µm sections. There may be no significant difference for 3,3-diaminobenzidine (DAB) staining using 25 or 40 µm thick brain sections. However, thinner sections offer better-quality fluorescence images.
The benefit of the strategy presented here is that multiple brain samples (up to 5 brains) can be cut at one time. However, the limitation of this method is that brain samples need to be cut within 1 week after dehydration because submersion in 30% sucrose for too long is more likely to cause protein degradation and other issues. To avoid this potential issue, these brain sections can be transferred into the cryoprotectant buffer and stored at −20 °C. For free-floating staining, the duration of incubation and concentration of both primary and secondary antibodies should be optimized in pilot studies. Generally, overnight incubation at 4 °C or room temperature with mild shaking is appropriate for most primary antibodies, if not instructed otherwise by manufacturers. For secondary antibodies, incubation at room temperature for 1–3 h works well in most situations. However, these details must be optimized for various circumstances. For example, for c-fos staining, we typically incubate the brain sections with a concentration of 1:1,000 overnight at 4 °C for immunofluorescence staining. However, using the same antibody for c-fos DAB staining, we prefer to incubate brain sections with a concentration of 1:5,000 for 48 h at 4 °C.
A cocktail of primary antibodies and secondary antibodies might be used for double-staining to speed up the procedure. More specifically, two different primary antibodies from different species (e.g., one is from rabbit, the other one is from guinea pig, chicken, or mouse) are mixed before incubation, as are the corresponding secondary antibodies. The choice of secondary antibody is dependent on the primary antibody. If the primary antibody is from rabbit, the secondary antibody must be anti-rabbit, for example, donkey anti-rabbit or goat anti-rabbit. The selection of blocking serum depends on the secondary antibody, for example, normal donkey serum will be used if the secondary antibody is from donkey. Antigen retrieval is suggested if the immunostaining still does not work even if all guidelines have been followed strictly.
Mounting and coverslipping of brain sections must be performed in a very delicate manner. The whole process requires no wrinkles, folds, or air bubbles. It will take several trials to determine the optimal exposure time for imaging. We recommend the same exposure time for the same antibody across different sections, which is essential for comparing the signal intensities among different animals or groups. It is reasonable that exposure time might not be the same for different antibodies, even in the same section. For example, the exposure time for DAPI might be shorter than the c-fos signal in most cases.
A few procedures presented in the protocol are helpful to improve both reliability and efficiency throughout the whole process. 1) Using an automated perfusion pump for perfusion can considerably shorten perfusion time and significantly improve tissue quality. 2) This cryosectioning strategy enables slicing multiple brain samples simultaneously, which is much more efficient than conventional practice. This method is also easy for new researchers to learn and master. 3) For free-floating staining, as brain samples are stained in suspension, antibodies can penetrate the sections from both sides. We optimized the incubation strategy by placing all sections from one sample/series into a 1.5 mL microcentrifuge tube for primary and secondary antibodies, which saves antibodies, particularly when we need to stain brain samples in bulk. Another benefit of the free-floating approach is that it can be modified and applied to other histochemical staining methods (e.g., chromogenic IHC, hematoxylin and eosin, cresyl violet) in addition to immunofluorescence staining12.
However, one limitation of free-floating staining is that very thin sections can be difficult to handle. An on-slide staining method might be considered if only a few sections need to be collected and stained immediately, as is frequently the case in clinical pathology. We also tested the on-slide staining method using brain sections generated from this protocol, and it worked well. To do this, mount the brain sections onto slides, wait for the sections to dry, and follow a traditional frozen section on-slide staining protocol. 4) Mounting free-floating sections on the slides can be tedious for certain researchers, especially for beginners. We use a fine paintbrush to gently coax sections onto the slide at the air-buffer interface and then use a transfer pipette to gently remove the buffer to lower the air-buffer interface as mounting advances from the top to the bottom of the slide. Although time-consuming, this strategy is friendly to beginners. Experienced experimenters can mount all the brains sections onto the slides in PBS at one time and only remove the buffer to bring the slide out after the last section is mounted. 5) Finally, we use a scanner for imaging, which is more efficient than fluorescence microscopy, especially when there are a large number of slides for imaging. The scanner enables scanning up to 12 slides in one batch with a 20x magnification. Alternatively, standard fluorescence microscopy can be employed in certain circumstances, for example, when a specific cluster of neurons in the brain must be showcased with a higher magnification (e.g., 40x or even 60x)13,14
In conclusion, this paper presents an established methodology for histological studies of mouse brains that has been proven to be reproducible, reliable, and efficient. The protocol will help generate optimal and consistent histological results among different researchers and laboratories and serve as a reference for beginners to learn this technique.
Acknowledgments
The investigators were supported by grants from the NIH (K01DK119471 to CW; P01DK113954, R01DK115761, R01DK117281, R01DK125480, and R01DK120858 to YX), USDA/CRIS (51000-064-01S to YX), and American Heart Association Postdoctoral Fellowship (#829565) to LT.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/62876.
Disclosures
The authors have no conflicts of interest to disclose.
References
- 1.Must A et al. The Disease burden associated with overweight and obesity. Journal of the American Medical Association. 282 (16), 1523–1529 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Apovian CM Obesity: definition, comorbidities, causes, and burden. The American Journal of Managed Care. 22 (7 Suppl), s176–185 (2016). [PubMed] [Google Scholar]
- 3.Wong SK, Chin KY, Suhaimi FH, Fairus A, Ima-Nirwana S Animal models of metabolic syndrome: a review. Nutrition & Metabolism. 13 (1), 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennedy AJ, Ellacott KL, King VL, Hasty AH Mouse models of the metabolic syndrome. Disease Models & Mechanisms. 3 (3–4), 156–166 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schacht V, Kern JS Basics of immunohistochemistry. The Journal of Investigative Dermatology. 135 (3), 1–4 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Mepham BL, Britten KJM Immunostaining methods for frozen and paraffin sections. in Lymphoproliferative Diseases. Jones DB, Wright DH (eds), Springer, Dordrecht, 15, 187–211 (1990). [Google Scholar]
- 7.Liu H et al. TPH2 in the dorsal raphe nuclei regulates energy balance in a sex-dependent manner. Endocrinology. 162 (1), 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu H et al. Defining vitamin D receptor expression in the brain using a novel VDR(Cre) mouse. Journal of Comparative Neurology. 529 (9), 2362–2375 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gage GJ, Kipke DR, Shain W Whole animal perfusion fixation for rodents. Journal of Visualized Experiments: JoVE. (65), e3564 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whittington NC, Wray S Suppression of red blood cell autofluorescence for immunocytochemistry on fixed embryonic mouse tissue. Current Protocols in Neuroscience. 81 (1), 2–28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeller R Fixation, embedding, and sectioning of tissues, embryos, and single cells. Current Protocols in Pharmacology. Appendix 3: 3D (2001). [DOI] [PubMed] [Google Scholar]
- 12.Potts EM, Coppotelli G, Ross JM Histological-based stainings using free-floating tissue sections. Journal of Visualized Experiments: JoVE. (162), e61622 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Y et al. A small potassium current in AgRP/NPY neurons regulates feeding behavior and energy metabolism. Cell Reports. 17 (7), 1807–1818 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu P et al. Activation of serotonin 2C receptors in dopamine neurons inhibits binge-like eating in mice. Biololgical Psychiatry. 81 (9), 737–747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]