

Abstract
Calreticulin (CALR) is recurrently mutated in myelofibrosis via a frameshift that removes an endoplasmic reticulum retention signal, creating a neoepitope potentially targetable by immunotherapeutic approaches. We developed a specific rat monoclonal IgG2α antibody, 4D7, directed against the common sequence encoded by both insertion and deletion mutations. 4D7 selectively bound to cells co‐expressing mutant CALR and thrombopoietin receptor (TpoR) and blocked JAK‐STAT signalling, TPO‐independent proliferation and megakaryocyte differentiation of mutant CALR myelofibrosis progenitors by disrupting the binding of CALR dimers to TpoR. Importantly, 4D7 inhibited proliferation of patient samples with both insertion and deletion CALR mutations but not JAK2 V617F and prolonged survival in xenografted bone marrow models of mutant CALR‐dependent myeloproliferation. Together, our data demonstrate a novel therapeutic approach to target a problematic disease driven by a recurrent somatic mutation that would normally be considered undruggable.
Keywords: calreticulin, monoclonal antibody, myelofibrosis, myeloproliferative neoplasm, stem cell progenitor
Subject Categories: Cancer, Immunology, Signal Transduction
CALR is recurrently mutated in myelofibrosis, creating a surface‐exposed neoepitope that activates the thrombopoietin receptor. A monoclonal antibody (4D7) with mutation‐specific efficacy in CALR mutant progenitors disrupts the receptor‐CALR complex and inhibits proliferation.
Introduction
Myeloproliferative neoplasms (MPNs) are haematological stem‐cell‐derived proliferation disorders, commonly characterized by an expansion of the myeloid lineage of cells and constitutive activation of the signalling pathways involved in haematopoiesis. MPNs can be categorized into three main subtypes: primary myelofibrosis (PMF), polycythaemia vera (PV) and essential thrombocytopaenia (ET), with both ET and PV having the potential to deteriorate to PMF (Tefferi & Pardanani, 2015). Common mutually exclusive driver mutations within Janus Kinase 2 (JAK2), Calreticulin (CALR) and myeloproliferative leukaemia virus oncogene (MPL) are often identified in MPN patients. While JAK2 mutations are exclusive to PV, the mutational status of these genes is not specifically associated with a particular MPN (Tefferi & Vainchenker, 2011).
Primary myelofibrosis is the most severe Philadelphia‐negative MPN and is characterized by marrow fibrosis and chronic inflammatory symptoms with a 5‐year survival of less than 50% (Baade et al, 2019). Although rare, PMF affects both young and older adults and can evolve into acute leukaemia in > 15% of cases (Passamonti et al, 2012). Mutations within CALR, the second most common genetic aberration associated with PMF, are observed in 70% of non‐JAK2V617F and non‐MPL cases (Klampfl et al, 2013; Nangalia et al, 2013) and are found in 20–30% of ET (Tefferi et al, 2014). Importantly, patients with CALR mutations do not effectively respond to JAK inhibitor therapy and no CALR‐specific therapy has been developed (Ross et al, 2021).
Virtually, all CALR mutations identified in PMF are small insertions or deletions clustered within exon 9. The two most common mutations identified include a 52 bp deletion (type 1) or a 5 bp insertion (type 2) (Klampfl et al, 2013; Nangalia et al, 2013). These frameshift mutations lead to a neoepitope peptide sequence which is thought to directly or indirectly activate the thrombopoietin receptor (TpoR) by a poorly defined mechanism that is dependent on glycan‐binding sites, N‐terminal chaperone domain and the novel C‐terminal tail of the mutant protein (Araki et al, 2016; Chachoua et al, 2016; Elf et al, 2016; Marty et al, 2016). All somatic CALR mutations observed in MPNs result in a +1 frameshift, leading to a common altered peptide sequence with loss of the negative‐charged C‐terminal calcium‐binding domain, gain of a lysine/arginine‐rich segment followed by a stop codon and loss of the KDEL sequence that constitutes an endoplasmic reticulum retention signal (Klampfl et al, 2013; Nangalia et al, 2013). Recently, it has been demonstrated that without this localization signal, mutant CALR protein is passively secreted from cells, and is detectable in cultured cell supernatants (Han et al, 2016; Liu et al, 2020; Masubuchi et al, 2020), with evidence that TpoR activation occurs after cell surface exposure, implying it may be accessible to an extracellularly acting therapeutic (How et al, 2019). Interestingly, recent data suggest that multimerization of mutant CALR monomers is absolutely required for mutant CALR TPO‐independent proliferation (Araki et al, 2019). There are subtle differences in prognosis and biochemistry between type 1 and type 2 CALR mutations (How et al, 2019), which are classified by the extent of elimination of negatively charged residues in the mutant protein compared to wild type. Ideally, a therapeutic would have activity against both type 1 and type 2 CALR mutations with minimal to no effect on normal haematopoiesis.
Here, we demonstrate a novel therapeutic strategy for MPNs by developing a monoclonal antibody with specificity for the mutant CALR peptide that inhibits TpoR activation through a distinct mechanism. Treatment of mutant CALR cells with the 4D7 monoclonal antibody inhibits binding of mutant CALR to the TpoR, blocking TpoR tyrosine phosphorylation and constitutive STAT and ERK phosphorylation. Biologically, 4D7 blocks TPO‐independent megakaryocyte differentiation from patients with both type 1 and type 2 frameshift mutations and prolong survival in cell‐line xenograft models of mutant CALR‐driven proliferation. Together, our data demonstrate a novel approach to target frameshift mutations in cancer that were previously considered undruggable.
Results and Discussion
Both type 1 and type 2 CALR mutations in myelofibrosis result in a loss of the endoplasmic reticulum retention signal KDEL producing aberrant localization of mutant protein (Klampfl et al, 2013; Nangalia et al, 2013; How et al, 2019) and the acquisition of a 31 amino‐acid‐long sequence towards the C‐terminus (Fig 1A). We reasoned that this neoepitope would be an ideal target for precision medicine approaches and set out to develop a panel of rat monoclonal antibodies to a chemically synthesized peptide corresponding to the C‐terminal mutant CALR neoepitope sequence (Fig 1A). Of three hybridoma screens, several antibodies showed positive binding to the peptide and to the CALR‐mutant protein in immunoblotting with clone 4D7 showing the strongest reactivity (Fig EV1A). By using peptides covering different regions of the immunizing neopeptide, we mapped the binding of 4D7 to the 11 amino acids of the N‐terminus with no binding of 4D7 observed to a scrambled peptide or the C‐terminal portion of the immunogen (Fig 1B). Clone 4D7 mAb showed a dissociation constant (K d) of 1.53 nM to the full‐length peptide as measured by 125I‐Scatchard analysis (Figs 1C and EV1B). To investigate the specific binding of 4D7 on CALR mutant‐bearing cells, we generated cytokine‐independent TF‐1 TpoR CALRdel61 and TF‐1 TpoR CALRdel52 (Fig EV1C). Using these cells in a horizontal co‐culturing system, we found no evidence of a paracrine effect by the mutant CALR protein (Fig EV1D). Specific binding of phycoerythrin (PE)‐conjugated 4D7 to the cell surface of mutant CALR‐expressing cells, but not TF‐1 TpoR CALRWT‐expressing cells, was observed in comparison to unstained or isotype‐PE control (Figs 1D and EV1E).
Figure 1. Anti‐mutant calreticulin antibody binds to cell surface and inhibits TPO‐independent proliferation.
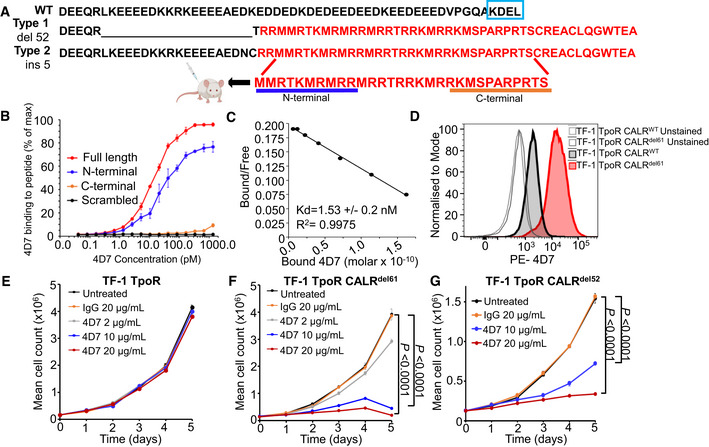
- Schematic showing wild‐type C‐terminal calreticulin protein sequence and neoepitope sequences for 52 bp deletion or 5 bp insertion and peptide sequence used for immunization.
- Diluting concentrations of 4D7 to bound full‐length 30 amino acid peptide compared to scrambled, C‐terminal and N‐terminal 11 amino acid peptides as shown in A (n = 3 biological replicates).
- Scatchard analysis showing dissociation constant of 4D7 using 125I‐labelled and unlabelled 4D7 bound to full‐length 30 amino acid peptide (n = 3 biological replicates).
- Histogram showing fluorescence intensity of 4D7 conjugated to phycoerythrin at a 4 µg/ml TF‐1 TpoR CALRdel61 vs. TF‐1 TpoR CALRWT compared to unstained.
- TF‐1 TpoR cells with endogenous wild‐type CALR cultured in the presence of TPO and 2, 10 or 20 µg/ml 4D7 anti‐mutant CALR antibody or 20 µg/ml control IgG antibody (n = 3 biological replicates).
- Proliferation curves of factor‐independent TF‐1 TpoR CALRdel61 cells cultured with 2, 10 or 20 µg/ml 4D7 or 20 µg/ml of control IgG antibody (n = 3 biological replicates).
- Proliferation curves of factor‐independent TF‐1 TpoR CALRdel52 cells cultured with 10 or 20 µg/ml 4D7 or 20 µg/ml of control IgG antibody (n = 3 biological replicates).
Data information: For panels (E–G), bars represent standard error of the mean summarizing three biological replicates performed in triplicate, with a one‐way ANOVA with Bonferroni correction used to determine statistical significance.
Figure EV1. Characterization of 4D7 antibody as a potential therapeutic tool.
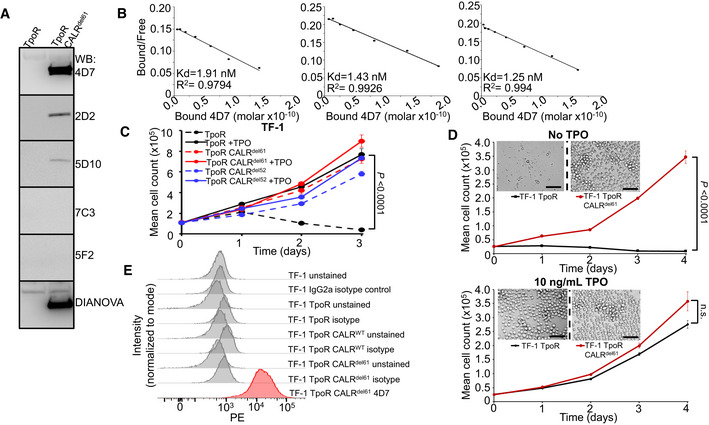
- Western blot screening of various mutant CALR antibody clones produced from hybridomas in TF‐1 TpoR and TF‐1 TpoR CALRdel61 cells. Varying levels of intensity can be observed compared to commercial Dianova monoclonal mutCALR antibody. 4D7 is able to detect mutant CALR protein in CALRdel61 cells but not in CALR wild‐type TF‐1 cells.
- Biological replicate Scatchard analyses of 4D7 using 125I‐labelled and unlabelled 4D7 bound to full‐length peptide (n = 3 technical replicates).
- TF‐1 cells expressing TpoR and CALRdel61 or CALRdel52 demonstrate factor independence in absence of TPO. Cells were cultured in the presence or absence of 10 ng/ml TPO (n = 3 biological replicates).
- Paracrine CALR‐mutant protein is not sufficient to maintain TPO‐sensitive cells in culture. TF‐1 TpoR and TF‐1 TpoR CALRdel61 cells were seeded at the same density and cultured in the presence or absence of TPO. Cells were separated by semi‐permeable membrane in a horizontal co‐culture system. Cell populations on either side of the membrane were counted every 24 h over 4 days in triplicate and representative images were taken on day 4. Exogenous CALR secreted by TF‐1 TpoR CALRdel61 was unable to assist growth of factor‐dependent TF‐1 TpoR cells. Scale bar indicates 100 µm (n = 3 biological replicates).
- Histogram overlays showing fluorescence intensity of unstained and PE‐conjugated IgG2a isotype control in TF‐1, TF‐1 TpoR, TF‐1 TpoR CALRWT and TF‐1 TpoR CALRdel61 compared to 4D7 conjugated to PE.
Source data are available online for this figure.
Monoclonal antibody 4D7 blocks TPO‐independent signalling
We then tested cytokine‐independent TF‐1 TpoR CALRdel61 and TF‐1 TpoR CALRdel52 cells for their ability to proliferate after treatment with increasing concentrations of 4D7 antibody or isotype control. Firstly, we noted no inhibition by 4D7 of TF‐1 cells expressing TpoR alone (with endogenous wild‐type CALR; Fig 1E). In contrast, we observed blockade of TPO‐independent cell growth over 5 days using 2, 10 and 20 µg/ml 4D7 in cytokine‐independent cells (Fig 1F and G) with evidence of concentration‐dependent inhibition observed after 48 h. No inhibition was observed with IgG isotype control antibody at 20 µg/ml. No inhibition by 4D7 was observed in TF‐1 cells with an overexpression of wild‐type CALR (Fig EV2A). Additionally, TF‐1 cells harbouring either the CALR del52 or CALR del61 mutations but lacking TpoR were unaffected when treated with 4D7 (Fig EV2B and C). Interestingly, MARIMO cells also did not show inhibition with 4D7, consistent with CALR no longer acting as a driver mutation in this NRAS mutant cell line (Han et al, 2018) (Fig EV2D). In addition, no inhibition was observed in factor‐independent JAK2 V617F SET2 cells (Fig EV2E) or factor‐independent TF‐1 expressing the PTPN11 E76K mutation (Fig EV2F) indicating a mutation‐specific effect.
Figure EV2. Biological specificity of 4D7.
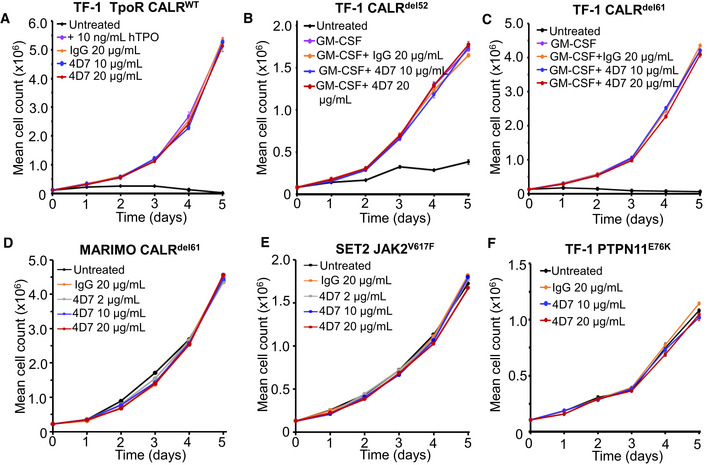
- Cytokine‐dependent TF‐1 TpoR cells with an overexpression of WT CALR cultured in the absence of TPO, 10 ng/ml hTPO and 10 or 20 µg/ml 4D7 or 20 µg/ml control IgG antibody for 5 days and the number of trypan blue‐negative cells were counted every 24 h (n = 3 biological replicates with three technical replicates).
- Cytokine‐dependent TF‐1 CALRdel52 cells lacking TpoR were cultured in the presence of 2 ng/ml GM‐CSF and 10 or 20 µg/ml 4D7 or 20 µg/ml control IgG antibody (n = 3 biological replicates with three technical replicates).
- Cytokine‐dependent TF‐1 CALRdel61 cells lacking TpoR were cultured in the presence of 2 ng/ml GM‐CSF and 10 or 20 µg/ml 4D7 or 20 µg/ml control IgG antibody (n = 3 biological replicates with three technical replicates).
- MARIMO cells from which the CALRdel61 mutation was originally amplified were cultured in the presence of 2, 10 or 20 µg/ml 4D7 or 20 µg/ml control IgG antibody (n = 3 biological replicates with three technical replicates).
- Cytokine‐independent SET2 cells which harbour the pathogenic JAK2V617F mutation were cultured in the presence of 2, 10 or 20 µg/ml 4D7 or 20 µg/ml control IgG antibody (n = 3 biological replicates with three technical replicates).
- Cytokine‐independent TF‐1 PTPN11E76K cells were cultured in the presence of 10 or 20 µg/ml 4D7 or 20 µg/ml control IgG antibody (n = 3 biological replicates with three technical replicates).
Data information: For all panels, bars represent standard error of the mean.
To understand the mechanism of action of 4D7, we performed signalling experiments in TF‐1 cells. Firstly, we observed no inhibition of signalling in cytokine‐dependent TF‐1 cells expressing TpoR alone, which showed the expected down‐regulation of phospho‐STAT1,3,5 and phospho‐ERK only after TPO withdrawal (Figs 2A and EV3A). In contrast, we observed blockade of constitutive factor‐independent phospho‐STAT1/3/5 and phospho‐ERK after incubation with 10 or 20 µg/ml 4D7, but not with IgG control in both CALRdel61 and CALRdel52 cells (Figs 2B and C, EV3B and C). Importantly, this blockade was also observed in peripheral blood mononuclear cells (PBMNCs) from a primary CALRdel52 PMF (Fig 2D) but not in JAK2V617F PMF (Fig EV3D). Treatment with 4D7 resulted in a significant reduction in phospho‐signalling, whereas ruxolitinib resulted in complete suppression. This was expected as ruxolitinib is known to inhibit all JAK‐dependent signalling, lacking mutation specificity (Tvorogov et al, 2018). A significant increase in the sub‐G0 fraction was observed by 4D7 compared to IgG control (P = 0.001; Fig 2E) in the absence of a major effect on G2‐M on cell cycle analysis (Fig EV3E) consistent with induction of an apoptotic response. This was confirmed by caspase 3 cleavage that occurred within 24 h and peaked at 48 h after 4D7 treatment (Fig 2F).
Figure 2. 4D7 disrupts interaction with TpoR and downstream signalling.
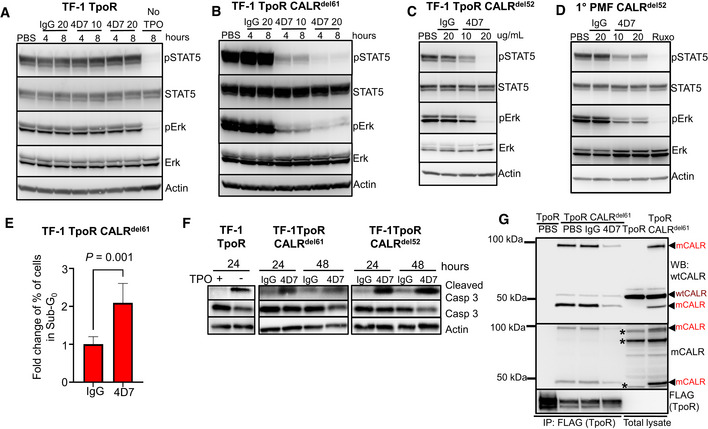
- Cell extracts blotted for phospho‐STAT5, total STAT5, phospho‐ERK, total ERK and actin from TF‐1 TpoR cells after incubation with 10 or 20 µg/ml 4D7 or IgG for 4 or 8 h, in presence of TPO. The last line indicates TPO withdrawal.
- Similar experiment using TPO‐independent TF‐1 TpoR CALRdel61 cells.
- Similar experiment using TPO‐independent TF‐1 TpoR CALRdel52 cells at 8 h.
- Similar experiment using PBMNCs from CALRdel52 PMF primary cells at 8 h. Additionally, cells were treated with 280 nM of ruxolitinib as a positive control.
- Fraction of apoptotic sub‐G0 population of TF‐1 TpoR CALRdel61 cells after 48 h of 4D7 or IgG treatment (n = 3 biological replicates, bars represent standard deviation for three replicates, normalized to IgG, with a Student's unpaired t‐test used to determine statistical significance).
- Western blot showing caspase 3 cleavage occurring within 24 h of TPO withdrawal in TPO‐dependent TF‐1 TpoR cells. An increase in cleaved caspase 3 is observed after 48 h of treatment with 20 µg/ml 4D7 in TF‐1 TpoR CALRdel61 and TF‐1 TpoR CALRdel52 cells.
- Western blot of TpoR immunoprecipitation under non‐reducing conditions showing associated CALR 50 kDa monomers and 100 kDa dimers (red arrowheads) present only in TF‐1 TpoR CALRdel61 disrupted by 8‐h treatment with 20 µg/ml 4D7 but not PBS or 20 µg/ml IgG. CALR monomers and dimers are detectable by polyclonal anti‐wild‐type CALR or anti‐mutant CALR monoclonal antibodies. Red arrowheads, detected mutant CALR protein; brown arrowheads, detected wild‐type CALR protein; asterisk, non‐specific bands.
Source data are available online for this figure.
Figure EV3. Monoclonal antibody blocks STAT1, 3 signalling and TpoR phosphorylation.
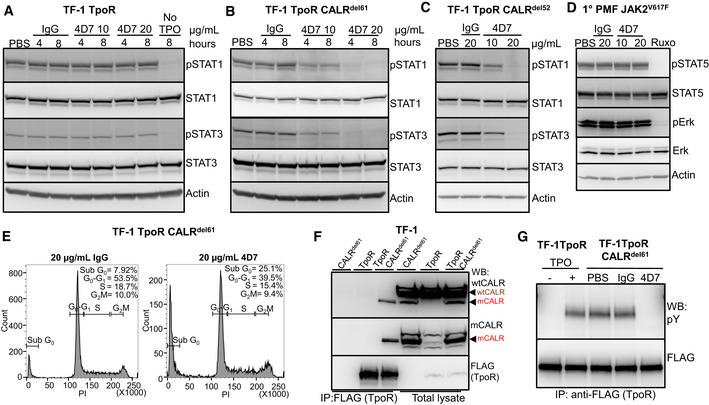
- Cell extracts blotted for phospho‐STAT1, total STAT1, phospho‐STAT3, total STAT3 and actin from TF‐1 TpoR cells after incubation with 10 or 20 µg/ml 4D7 or IgG for 4 or 8 h as indicated.
- Similar experiment using TPO‐independent TF‐1 TpoR CALRdel61 cells at 4 and 8 h.
- Similar experiment shown using TPO‐independent TF‐1 TpoR CALRdel52 cells, 8 h.
- Similar experiment using PBMNCs from JAK2V617F PMF primary cells at 8 h. Additionally, cells were treated with 280 nM of ruxolitinib as a positive control.
- Flow cytometry analysis for cell cycle distribution of TF‐1 TpoR CALRdel61 cells exposed to 20 µg/ml IgG or 4D7 for 48 h. Cells were harvested and fixed and stained with propidium iodide and their DNA contents were analysed. Results from one representative experiment shown. Percentages of cells in Sub G0, G1, S and G2/M cycle indicated.
- Western blot showing specific co‐immunoprecipitation of mutant CALRdel61 cells with TpoR anti‐FLAG antibody under reducing conditions detected by WT and mutant‐specific CALR antibodies. Red arrowheads, detected mutant CALR protein; brown arrowheads, detected wild‐type CALR protein.
- Western blot showing decreased TpoR phosphorylation in a TpoR immunoprecipitated after 8 h of 20 µg/ml 4D7 treatment compared to PBS or IgG control in TF‐1 TpoR CALRdel61.
Source data are available online for this figure.
4D7 disrupts the mutCALR/TpoR complex
To investigate the mechanism of action of 4D7, we examined how it affects mutant CALR interactions with TpoR. Analysis of TF‐1 TpoR CALRdel61 cells revealed that CALR mutants constitutively existed in complex with the TpoR (Fig EV3F) consistent with previous findings (Araki et al, 2019). Importantly, pre‐treatment with 4D7 diminished the presence of CALR monomers (slightly < 50 kDa) and dimers (~ 100 kDa) bound to TpoR (Fig 2G), supporting the notion that 4D7 interferes with the mutCALR/TpoR complexes at the cell surface by disrupting the constitutive association of mutCALR to TpoR. Similar results were obtained with TF‐1 TpoR CALRdel52 cells (Fig EV4A). 4D7 also prevented TpoR activation in the presence of CALR‐mutant protein as demonstrated by its inhibition of TpoR tyrosine phosphorylation (Fig EV3G) and downstream signalling (Fig 2B–D), consistent with inhibition of receptor activation.
Figure EV4. 4D7 blocks CALR interaction with TpoR in CALRdel52 cells.
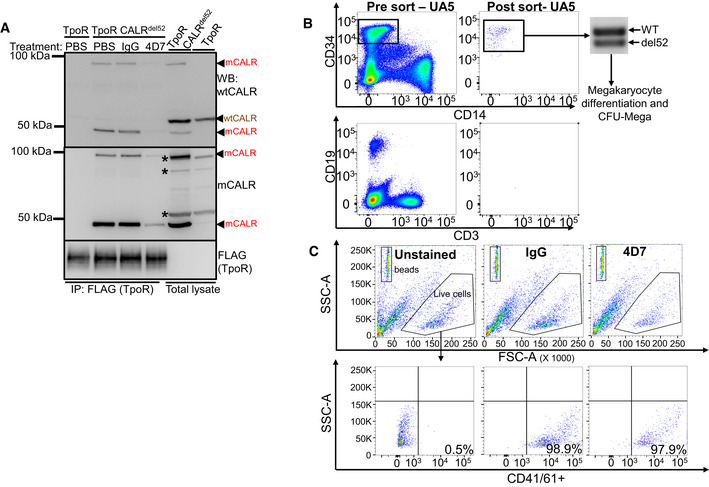
- Western blot of TpoR immunoprecipitation under non‐reducing conditions showing associated CALR 50 kDa monomers and 100 kDa dimers (red arrowheads) present only in TF‐1 TpoR CALRdel52 disrupted by 8‐h treatment with 20 µg/ml 4D7 but not PBS or 20 µg/ml IgG. CALR monomers and dimers are detectable by polyclonal anti‐wild‐type CALR or anti‐mutant CALR monoclonal antibodies. Red arrowheads, detected mutant CALR protein; brown arrowheads, detected wild‐type CALR protein; asterisk, non‐specific bands.
- Peripheral blood mononuclear cells from PMF samples were thawed and stained for CD34, CD14, CD19 and CD3 prior to FACS sorting. Each population was collected and purity was verified prior to proceeding with any further analysis. PCR amplification of CALR exon 9 was carried out to confirm mutational status of CD34+ cells which were utilized in megakaryocyte differentiation assays and colony forming assays in the presence of 4D7 or IgG.
- Representative flow cytometry plots for determination of CD41+/61+ populations from liquid culture assay from one PMF patient. Beads shown in the upper left panel with high SSC‐A. Live cell population shown in hexagon gate (top panel). CD41/61+ population gates shown in lower panel with % CD41/61+ cells indicated for unstained, IgG‐ and 4D7‐treated cells over 12 days. The number of CD41/61+ cells‐to‐bead ratio used to enumerate effect of 4D7 on megakaryopoiesis.
Source data are available online for this figure.
Monoclonal antibody 4D7 inhibits TPO‐independent megakaryocyte formation
Primary myelofibrosis is characterized by abnormal proliferation and morphology of clonal megakaryocytes. To examine whether 4D7 suppressed CALR‐mutant megakaryocytes, we tested its activity on purified primary CD34+ cells obtained from patients with CALR‐mutant myelofibrosis using two orthogonal assays: (i) TPO‐independent megakaryocyte differentiation in liquid culture, and (ii) TPO‐independent megakaryocyte colony formation on a collagen‐based medium. Five of eight patients had the most common type 1 (52 base pair deletion) CALR mutation which was heterozygous in the bulk of the CD34+ fraction (Fig 3A). The sorting strategy for myelofibrosis stem cells is shown in Fig EV4B and clinical details for all patients are listed in Table EV1. Strikingly, four of five mutant CALR MF patient samples that displayed robust TPO‐independent growth of CD41+CD61+ megakaryocyte progenitors showed inhibition by 4D7 of at least 50% (Figs 3B and EV4C). Interestingly, the patient sample exhibiting less inhibition harboured a 34 base pair deletion. No significant inhibition was observed when JAK2 V617F was present in all three myelofibrosis samples (black bars; Fig 3B). An additional two patient samples obtained from the University of Graz also showed evidence of inhibition compared to IgG control, including a type 2 CALR patient (five base pair insertion; Fig 3C). Overall, a mean decrease of 55% was observed across CALR‐mutant samples after 4D7 treatment (P < 0.0001; Fig 3D). Similarly, we saw a dramatic reduction in the absolute numbers of primary TPO‐independent megakaryocyte colonies cultured on collagen (colony‐forming unit‐mega) treated with 4D7 in multiple patient samples (decrease of 62%, P < 0.0001; Fig 3E–G). Strikingly, the size of 4D7‐treated megakaryocyte colonies was substantially smaller as enumerated in a patient with a type 2 mutation (Fig 3H).
Figure 3. 4D7 monoclonal antibody specifically inhibits primary megakaryocyte differentiation of mutated CALR myelofibrosis samples.
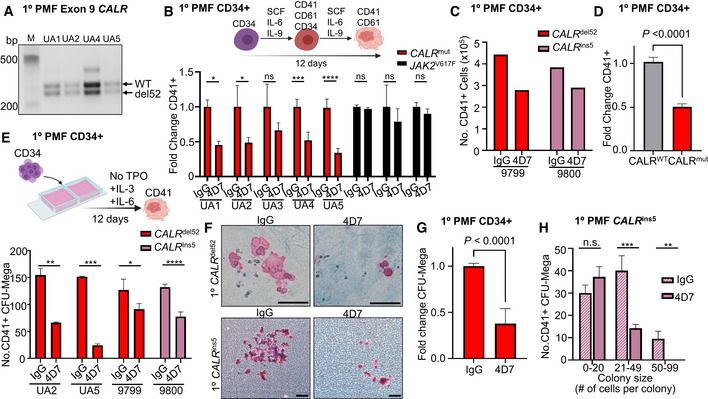
- PCR amplification of CALR exon 9 from patients confirming heterozygous del52 mutation in sorted CD34+ cells obtained from CALR‐mutated myelofibrosis samples.
- Graph showing the decreased fold change of CD41+CD61+ megakaryocytes cultured in 4D7 compared to IgG. FACS‐sorted CD34+ from myelofibrosis patients with CALR or JAK2 mutation was cultured over 12 days without TPO in the presence of SCF, IL‐6 and IL‐9. Black columns show JAK2 V617F mutation‐positive samples (n = 4 technical replicates of samples from different patients).
- Number of CD41+ megakaryocytes derived from isolated CD34+ progenitors from myelofibrosis patients. Number of CD41/CD61+ cells counted on day 12 using trypan blue exclusion (n = 1 technical replicate).
- Summary of fold change reduction of CD41/CD61+ megakaryocytes by 4D7 in all tested CALR‐mutated patient samples compared to CALR wild type, normalized to IgG (n = 11 samples from different patients, with four technical replicates per sample).
- Number of CD41+ megakaryocyte colonies from patient samples after 4D7 treatment. CD34+ from patients with myelofibrosis with CALR del52 or CALR ins5 was plated on collagen‐based matrix in presence of 20 µg/ml 4D7 or IgG control (n = 3 biological replicates).
- Representative micrographs showing CD41+ colonies in pink and CD41‐ colonies in blue after treatment with 4D7 or IgG at 100× or 40× magnification. Scale bar indicates 100 µm.
- Summary of fold change reduction in CD41+ megakaryocytes in CALR‐mutated samples cultured MegaCult treated with 4D7 compared to IgG (n = 4 biological replicates).
- Number of megakaryocyte colony forming units grouped according to colony cell number after 4D7 treatment in a mutant CALR ins5 sample (n = 3 biological replicates).
Data information: Bars represent standard error of means in (D) and (G). Unpaired Student's t‐test used to determine statistical significance in (B, D, E, G and H). Bars represent standard deviations in (B, E and H). *P = 0.05–0.01, **P = 0.01–0.001, ***P = 0.001–0.0001, ****P < 0.0001, n.s, not significant.
Monoclonal antibody 4D7 inhibits the growth of ruxolitinib‐persistent cells
Ongoing follow‐up of COMFORT‐1 and COMFORT‐2 studies (Harrison et al, 2012; Verstovsek et al, 2012) suggests patients with CALR mutations are less responsive to the JAK inhibitor ruxolitinib than patients with JAK2 mutations, but cytopenia limits the use of higher treatment doses (Ross et al, 2021). We therefore tested whether an immunotherapeutic approach would have efficacy in cells with ruxolitinib “persistence/resistance” (Meyer et al, 2015). We first showed that ruxolitinib had inhibitory effects on megakaryocyte formation from healthy cord blood with > 30% inhibition after 12 days at 50 or 100 nM (P = 0.008 and 0.07, respectively; Fig 4A). In contrast, 4D7 had no effect on normal haematopoiesis. At the highest concentration, 4D7 did not inhibit physiological megakaryopoiesis in liquid culture (Fig 4B), haematopoietic granulocyte‐macrophage colony formation and erythroid colony formation (Fig 4C), or collagen‐plated megakaryocyte colony numbers (CFU‐Mega; Fig 4D and E) compared to IgG control.
Figure 4. 4D7 has no effect on normal progenitor cells and shows activity against ruxolitinib‐resistant cells without haematological toxicity.
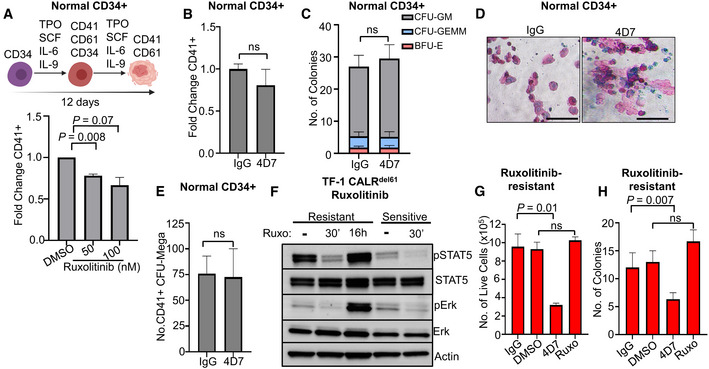
- Graph showing effect of ruxolitinib on megakaryocyte differentiation at 50 and 100 nM in healthy CD34+ cells (n = 4 different cords collected with three technical replicates).
- Effect of 4D7 on TPO‐dependent megakaryocyte differentiation of healthy CD34+ cells cultured with 20 µg/ml 4D7 or IgG control (n = 2 biological replicates, with two technical replicates).
- Total numbers of haematopoietic colonies plated in MethoCult from healthy cord blood after treatment with 4D7 or IgG. CFU‐GM, colony forming unit–granulocyte macrophage; BFU‐E, blast forming unit–erythroid; CFU‐GEMM, colony forming unit–granulocyte, erythroid, monocyte and megakaryocyte (n = 3 biological replicates).
- CD41+ megakaryocyte colonies from healthy CD34+ cells observed on collagen matrix after 4D7 treatment. Colonies were cultured in TPO, SCF, IL‐9 and IL‐3. Scale bar indicates 100 µm.
- Number of megakaryocyte colony forming units observed in MegaCult assay after treatment of healthy cord blood CD34+ cells with either IgG or 4D7 (n = 3 biological replicates).
- Western blot showing signalling in ruxolitinib‐resistant TF‐1 TpoR CALRdel61 compared to ruxolitinib‐sensitive TF‐1 TpoR CALRdel61 cells after treatment with 100 nM ruxolitinib for 16 h or 30 min and blotted for phospho‐STAT5, total STAT5, phospho‐ERK, total ERK and actin as indicated. Ruxolitinib‐sensitive cells were non‐viable after 16 h of treatment.
- Comparison of cell growth after DMSO, 20 µg/ml 4D7, IgG or 100 nM ruxolitinib treatment over 4 days of ruxolitinib‐resistant TF‐1 TpoR CALRdel61. Cells counted using trypan blue exclusion (n = 3 biological replicates with three technical replicates).
- Number of colonies observed from cells plated in MethoCult following 72 h of treatment with DMSO, 20 µg/ml 4D7, IgG or 100 nM ruxolitinib performed in ruxolitinib‐resistant TF‐1 TpoR CALRdel61 cells (n = 3 biological replicates with three technical replicates).
Data information: Error bars on (A, B, C and E) indicate standard error of mean. Error bars on (G and H) represent standard deviation. All P‐values are unpaired Student's unpaired t‐test.
Source data are available online for this figure.
To generate ruxolitinib‐resistant cells, we cultured TF‐1 TpoR CALRdel61 cells with increasing concentrations of ruxolitinib over 4 weeks, up to 100 nM. Ruxolitinib‐resistant cells showed enhanced phospho‐STAT5 and phospho‐ERK phosphorylation in the presence of ruxolitinib but still showed some inhibition after withdrawal and re‐stimulation (Fig 4F). Importantly, 4D7 had strong inhibitory activity on cells that were resistant to ruxolitinib, in both liquid culture at 96 h (Fig 4G) and in colony formation (Fig 4H). Together, these results suggest that an immunotherapeutic approach with 4D7 may have clinical utility in CALR‐mutant patients who develop resistance/persistence of myelofibrosis during ruxolitinib treatment.
A recent report suggests TpoR co‐expressed with mutant CALR may have a distinct conformation that is less responsive to physiological TPO compared to normal TpoR (Basso‐Valentina et al, 2021). We therefore performed rescue experiments with 10 ng/ml TPO in the presence of 4D7. We found in the presence of 4D7, TPO could only partially rescue the proliferation defect in both TF‐1 TpoR CALRdel61 cell line and patient‐derived megakaryocyte colonies (Fig EV5A–C), suggesting endogenous TPO may not be sufficient to overcome 4D7 effect in vivo.
Figure EV5. Effect of 4D7 antibody on TPO receptor biology.
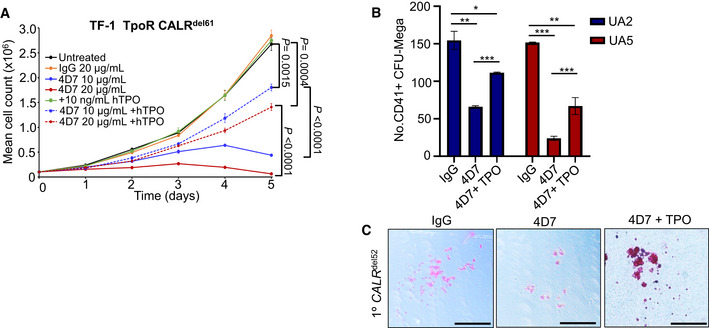
- Proliferation curves of factor‐independent TF‐1 TpoR CALRdel61 cells cultured with 10 or 20 µg/ml 4D7 or 20 µg/ml of control IgG antibody in presence or absence of 10 ng/ml TPO (n = 3 biological replicates with three technical replicates).
- Number of CD41+ megakaryocyte colonies CALRdel52 patient after 4D7 treatment in the presence of TPO. Samples were seeded in a collagen‐based matrix in presence of 20 µg/ml 4D7 or IgG control with 50 ng/ml TPO (n = 2 patient samples with two technical replicates).
- Representative micrographs showing CD41+ colonies in pink and CD41‐ colonies in blue after treatment with IgG or 4D7 in absence or presence of 50 ng/ml TPO. Scale bar indicates 100 µm.
Data information. Error bars represent standard error of the mean in (A) and standard deviation (B). Unpaired Student's t‐test used to determine statistical significance in (B). *P = 0.05–0.01, **P = 0.01–0.001, ***P = 0.001–0.0001.
The 4D7 monoclonal antibody blocks CALR‐dependent proliferation and prolongs survival in xenograft models
Myeloproliferative neoplasms are fundamentally disorders of unregulated proliferation. To test whether 4D7 could block mutant CALR‐dependent proliferation in vivo, we developed two distinct cell‐line xenograft models. The first was a bone marrow engraftment model, which measures mutant CALR‐dependent proliferation of TF‐1 TpoR CALRdel61 cells in the bone marrow microenvironment. The second was a chloroma model created by subcutaneous injection of TF‐1 TpoR CALRdel61 cells into the flanks of NSG mice, which mimics extramedullary haematopoiesis. In both models, fibrosis is not an evaluable disease outcome. In the bone marrow engraftment model (Fig 5A), 4D7 treatment (12.5 mg/kg twice weekly via intraperitoneal injection starting day 7) showed an excellent pharmacokinetic profile, achieving a serum concentration of more than 100 µg/ml 48 h post‐injection (Fig 5B), lowered peripheral blood engraftment of human CD33+ cells at 3 weeks (0.04 vs. 19.8% CD33+ 4D7 vs. IgG, P = 0.01; Fig 5C) and significantly prolonged survival (log‐rank hazard ratio 0.24, P = 0.003; Fig 5D). In the chloroma model, 4D7 treatment significantly slowed tumour growth at 21 days post‐engraftment (353 vs. 3,317 mm3 mean tumour volume, 4D7 vs. IgG, P = 0.04; Fig 5E) and prolonged survival (hazard ratio 0.19, P = 0.02; Fig 5F). Strikingly, mutant CALR cells induced to be resistant to 100 nM ruxolitinib also showed a survival advantage after treatment with 4D7 (12.5 mg/kg twice weekly beginning at day 7) in the bone marrow engraftment model (hazard ratio 0.26, P = 0.0008; Fig 5G). Together, these results suggest an immunotherapeutic approach with antibody 4D7 may have clinical utility in CALR‐driven MPNs as well as in CALR‐mutant patients who develop resistance/persistence to ruxolitinib.
Figure 5. 4D7 blocks mutant CALR‐dependent proliferation in vivo and prolongs survival.
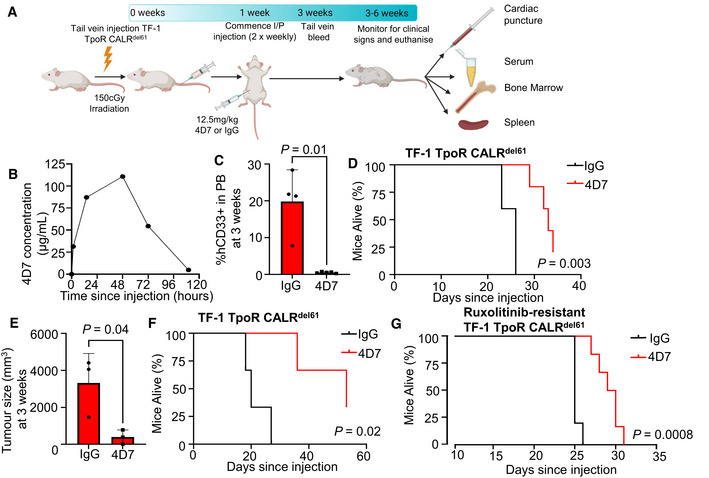
- Illustration showing bone marrow NSG engraftment model with TPO‐independent TF‐1 TpoR CALRdel61 cells treated with 4D7 or IgG control twice weekly, starting 7 days after engraftment via intraperitoneal injection and final measurements taken from euthanized mice.
- Pharmacokinetic measurements of serum levels of 4D7 in mice after intraperitoneal injection at 0, 1, 24, 48, 72 and 110 h since administration.
- Percentage of TF‐1 TpoR CALRdel61 human CD33+ cells measured in peripheral blood at 3 weeks post‐tail vein engraftment (n = 5 mice per treatment).
- Kaplan–Meier survival curve of bone marrow engraftment model of TF‐1 TpoR CALRdel61 cells showing improved survival of mice treated with 4D7 compared with IgG control commencing 1 week after tail vein injection (n = 5 mice per treatment).
- Mean tumour volume at 21 days after subcutaneous injection of TF‐1 TpoR CALRdel61 cells following treatment with 4D7 or IgG. Cells were pre‐treated with 4D7 or IgG control for 1 h prior to injection and treatment continued twice weekly at 12.5 mg/kg until euthanasia (n = 3 mice per treatment).
- Kaplan–Meier survival curve of TF‐1 TpoR CALRdel61 chloroma mice treated with 4D7 or IgG until humane killing due to tumour diameter > 30 mm or ulceration (n = 3 mice per treatment).
- Kaplan–Meier survival curve of NSG mice engrafted with ruxolitinib‐resistant TF‐1 TpoR CALRdel61 treated with 12.5 mg/kg 4D7 or IgG twice weekly (n = 5 and 6 mice for IgG and 4D7, respectively).
Data information: For (C and E), a Student's unpaired t‐test was used to determine statistical significance. For all survival curves, the log‐rank Mantel–Cox test P‐value is shown.
Primary myelofibrosis is an insidious and poorly understood disorder that encompasses features of both cancer and chronic inflammation. It is a clonal neoplasm driven by a handful of somatic mutations that activate cell signalling, presumably residing in the long‐term stem cell compartment (Wernig et al, 2006; Nangalia et al, 2013; Reinisch et al, 2016), which also has a constellation of cytokine‐mediated symptoms that are disproportionately severe. After the discovery of activating JAK2 mutations in 50–60% of patients with PMF (Baxter et al, 2005; Kralovics et al, 2005), clinicians were hopeful that myelofibrosis would respond to tyrosine kinase inhibitor therapy in a similar fashion to chronic myeloid leukaemia. However, maturing data from the COMFORT studies and other trials have shown ruxolitinib also does not cause widespread regression of fibrosis or eradicate disease clones in a mutation‐specific manner (Verstovsek et al, 2012; Cervantes et al, 2013; Harrison et al, 2016). Exceptional cases of complete haematological or molecular remission have been reported (Masarova et al, 2016), but are uncommon. Typically, there is a gradual loss of response over time, with only 27% of patients remaining on first‐line ruxolitinib treatment after 5 years in the COMFORT studies, underscoring a need for alternative approaches.
Somatic mutations in exon 9 of the endoplasmic chaperone protein CALR are found in 70–80% of patients with JAK2‐negative PMF, accounting for ~ 30% of PMF cases overall, and also present in a similar proportion of essential thrombocythemia (Klampfl et al, 2013; Nangalia et al, 2013). In a subsequent analysis of patients from the COMFORT‐2 study, the response rate to ruxolitinib was 20% (confidence interval 5.7–43.7%) in patients with a CALR mutation vs. 34% (24.8–44.1%) in CALR‐negative patients, most of whom had a JAK2 V617F mutation (Guglielmelli et al, 2016), suggesting that CALR‐mutant myelofibrosis may be less responsive to JAK inhibitor therapy. Although more than 50 CALR mutations have been described, the majority can be classified as type 1 like or type 2 like based on predicted protein structures (Eder‐Azanza et al, 2014). Our data suggest that both type 1 and type 2 CALR mutations could be amenable to an immunotherapeutic approach.
Here, we show for the first time that primary cells from patients with CALR mutations can be targeted with a mutation selective immunotherapy. Our data confirm the observations from other studies using human cells that mutant CALR protein is secreted and accessible on the cell surface and can drive growth factor independence, provided TpoR is co‐expressed. Data from independent laboratories have shown that mutant CALR protein requires TpoR for signalling and factor‐independent cell growth, and that the normal lectin domain of CALR is essential to bind glycosylated sites on TpoR (Elf et al, 2016, 2018; Pecquet et al, 2019). The relative contribution of autocrine vs. paracrine vs. endosomal signalling of mutant CALR protein has not been fully examined, with most studies to date performed in murine IL‐3‐dependent Ba/F3 cells. In our human TF‐1 model of mutant CALR‐induced proliferation, no evidence of a paracrine effect could be demonstrated in a horizontal co‐culturing assay (Fig EV1D). Remarkably, despite a demonstrated lack of autocrine/paracrine signalling, mutant CALR protein appears to be exquisitely sensitive to 4D7 binding in a dose‐dependent manner with concomitant inhibition of cell proliferation. Our results indicate that the CALR‐TpoR complex can be disrupted via a monoclonal antibody, and specifically we identified evidence for mutCALR bound constitutively to TpoR that could be inhibited by 4D7, thus blocking TpoR phosphorylation as well as downstream STAT and ERK signalling in primary samples. Future studies should determine the 3D structure of 4D7 bound to CALR and help test whether TPO analogues can overcome the effects that we describe (Basso‐Valentina et al, 2021).
Recently, it was reported that mutant CALR protein is present at high levels in the plasma of myelofibrosis patients (compared to normal individuals) (Sollazzo et al, 2016; Pecquet et al, 2018). The cell surface expression of mutant CALR is an ideal target for immunological therapies. It is present on the cell surface (Pecquet et al, 2018; Balligand et al, 2020; Masubuchi et al, 2020) and contains sequences not normally present in healthy mammalian cells or conserved across mammalian species. CALR mutations appear to be an early event in MPN ontogeny (perhaps only preceded by TET2 in some cases). This results in it being present in all of the cells of the clone, unlike JAK2 V617F which can be either an initiating or a secondary lesion, and found in only around half of patients with AML arising from an antecedent JAK2 V617F MPN (Ross et al, 2021).
The development of a therapeutic antibody directed against mutant CALR protein has enormous clinical potential. The mouse model utilized within this study shows safety and efficacy for a rapidly proliferating tumour that is mutant CALR dependent, however, is not a relevant disease model for MPN. Future studies should investigate efficacy in relevant heterozygous mouse models (Li et al, 2018; Balligand et al, 2020; Achyutuni et al, 2021), including evidence for fibrosis reversal. Additionally, the antibody that we have developed against the common neoepitope C‐terminus of CALR can be used in both insertion and deletion CALR mutation‐positive patients and appears to have minimal or no effects on normal cells ex vivo. The safety and tolerability of this antibody therapy could lead to its application in the earlier stages of PMF to prevent clinical progression and the acquisition of clonal heterogeneity. In parallel, an anti‐mutant CALR antibody such as 4D7 could also be combined with other treatments with the aim of augmenting their effect, as has been successfully applied in B‐cell lymphoma using anti‐CD20 antibodies (Czuczman et al, 1999). Future studies should examine a similar approach in patients with essential thrombocythemia with CALR mutations and secondary acute myeloid leukaemia that has transformed from CALR‐mutant myeloproliferative disorders.
Myelofibrosis can be associated with immune defects which are compounded by the effects of JAK inhibitors like ruxolitinib. There is, therefore, an argument in favour of immunotherapeutics that can be used in the less advanced stages of myeloproliferative neoplasms. Although immunodeficiency may limit the potential of antibody therapeutics that require intact complement‐mediated cytotoxicity, this can be overcome by the addition of toxin conjugates or the use of chimeric antigen receptor T cells. Importantly, our data suggest that antibody‐mediated inhibition of cell signalling may also contribute to suppression of CALR‐mutant cell proliferation, independent of any immune‐mediated effects. Physiologically, the SIRP alpha protein is normally present as a ligand for the “don't eat me” CD47 signal protecting cells from phagocytosis (Majeti et al, 2009; Majeti, 2011; Liu et al, 2020) but inhibition of macrophage phagocytosis does not appear to be involved in the disease progression of MPN (Daitoku et al, 2016). It will be intriguing to test anti‐CALR monoclonal antibody efficacy in the setting of macrophage activation or CAR T‐cell therapies.
Materials and Methods
Cell lines
TF‐1 cells were cultured in RPMI with 10% (v/v) foetal calf serum (FCS) supplemented with 2 ng/ml of GM‐CSF and 25 mM HEPES. HEK293T cells were cultured in DMEM with 10% FCS. TF‐1 TpoR cells were cultured in RPMI with 10% FCS supplemented with 10 ng/ml of human TPO (Peprotech). MARIMO and SET2 cells were cultured in RPMI with 10% FCS.
CALR mutation cloning
Human CALR (del61) with a 61 base pair deletion in CALR exon 9 was PCR amplified from MARIMO cDNA with oligonucleotide primers 5′‐TAT AGA ATT CGC CAC CAT GCT GCT ATC CGT GCC G‐3′ and 5′‐TAT AGA ATT CAG GCC TCA GTC CAG CCC T‐3′. The resultant PCR product was cloned into pCX4‐IRES eGFP (Neubauer et al, 2016) following digestion with EcoRI. Sequencing verified integrity and orientation of the cDNA. CALRdel61 protein was expressed in TF‐1 TpoR. This cell line was designated as TF‐1 TpoR CALRdel61 and was shown to obtain cytokine independence.
Rat monoclonal antibody production
Rats were immunized with a CALR‐mutant peptide “MMRTKMRMRRMRRT RRKMRRKMSPARPRTS” coupled to KLH, this peptide sequence is unique to CALR mutant. Serum from the immunized rats was screened by enzyme‐linked immunoassay, to verify a strong titre to the peptide immunogen, prior to performing the hybridoma fusion. Immunized rat spleen cells were harvested and a spleen single‐cell suspension was combined with Sp2/O myeloma cell line. The cell mixture was treated with polyethylene glycol plus DMEM prior to being plated in 96‐well flat bottom plates. The expanding hybridomas were screened initially by ELISA and the positive clones were further verified by western blot for reactivity to the expressed CALR‐mutant protein before subcloning the hybridomas for antibody production.
Lentiviral expression constructs
Human CALR and PTPN11 vectors were produced using a codon‐optimized coding sequence encoding CALRWT, CALRdel52 or PTPN11E76K mutations purchased as gBlocks from Integrated DNA Technologies (Iowa, USA). Mutations were cloned into pLVX‐eF1α‐IRES‐ZsGreen1 lentiviral expression construct (Takara Bio Inc., Shiga, Japan). For virus production, HEK293T cells were seeded at 3 × 106 cells per 75 cm2 flask 40 h prior to transfection. Each flask was transfected using Lipofectamine 2000 with 13.5 µg of CALR WT or ‐mutant pLVX vectors together with 13 µg envelope plasmids, VsVg and PAX2. Viral supernatants were collected 48 h after transfection. To establish cell lines expressing various CALR constructs, viral supernatants were applied to RetroNectin‐coated (Takara) tissue culture plates and spun at 2,000 g for 2 h at 37°C. Cell lines were applied to virus and spun at 200 g and cultured in the presence of virus for 24 h. ZsGreen1‐positive cells were sorted using the FACS Melody (BD Biosciences).
Antibody 125Iodine‐labelling and Scatchard analysis
4D7 was radio‐iodinated with125I (Perkin‐Elmer) using Pierce Pre‐Coated Iodination tubes (Thermo Scientific) with an estimated specific activity of 7,733 cpm/ng. For saturation‐binding assays, plates were coated overnight at 4°C with full‐length neoepitope CALR peptide (2 µg/ml) or control scrambled peptide (2 µg/ml). Wells were washed three times with phosphate‐buffered saline + 0.05% Tween20, blocked with 5% bovine serum albumin for 1 h followed by further washing.125I‐4D7 was added at concentrations ranging from 2 pM to 2.5 nM in triplicate and incubated at room temperature for 2 h. After washing, 1 M HCl was added for 30 min to elute bound antibody. Two hundred microliter aliquots from each well were counted on a PerkinElmer 2470 Wizard2 Automatic Gamma Counter. Dissociation constants were calculated using the EBDA and LIGAND programmes.
Cell proliferation to assess 4D7 effect on various cell lines
In brief, cells were seeded at 5 × 104/ml in standard growth media, with any necessary cytokines, and the addition of 20 µg/ml IgG or 2, 10 or 20 µg/ml 4D7. Cells were seeded in triplicate and each well was counted in triplicate every 24 h for 5 days using trypan blue exclusion.
Primary myelofibrosis stem cell experiments
Umbilical cord blood (UCB) was collected with written consent from full‐term deliveries at the Women's Health Unit, Lyell McEwin Hospital (Adelaide, South Australia), or the Department of Obstetrics and Gynaecology, Medical University of Graz (Austria), with institutional review board approval (IRB approval: 31‐322 ex 18/19; HREC/20/WCHN/65). Samples were processed using a Ficoll‐Paque (GE Healthcare) density gradient to isolate mononuclear cells (800 g, room temperature, 30 min, deceleration off), followed by red cell lysis (ammonium–chloride–potassium lysing buffer) to remove remaining red blood cells. CD34 enrichment was performed by magnetic cell separation using CD34 Microbead kit (Miltenyi Biotech). Alternatively, purified CD34+ cells from cord blood were purchased from Lonza. CD34+ cells with a purity above 90% were either cryopreserved or directly cultured in serum‐free stem cell retention media (StemSpan SFEMII, Stemcell Technologies, Vancouver, BC, Canada) supplemented with human recombinant SCF, TPO, FLT3, IL‐6 (all 20 ng/ml), UM729 (1.75 µM) and StemReginin 1 (SR1, 300 nM). All cytokines and SR1 were purchased from Peprotech (Rocky Hill, NJ, USA), whereas UM729 was purchased from Stemcell Technologies.
PMF patient samples
Patient samples were collected with informed consent from the South Australian Cancer Research Biobank (SACRB; Adelaide, Australia). Primary CD34+ stem/progenitor cells from peripheral blood samples from patients with JAK2V617F or CALR‐mutant myelofibrosis were purified by flow cytometry using CD34‐APC antibody, anti‐human CD45RA Brilliant Violet 605, anti‐CD90 FITC, anti‐human CD123‐PE, anti‐human CD38 PE‐Cy7, or with CD34‐APC antibody, anti‐human CD3 FITC, anti‐human CD19‐PE (clone H1B19) and anti‐human CD14 PE‐Cy7.
Mutational screening of patient samples
DNA was extracted from primary PMF samples using QuickExtractTM (Epicentre Biotechnologies) following the manufacturers protocol. PCR reactions were set up in a final concentration of 1X PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.2 µM forward and reverse primers (CALR‐F; 5′‐TAA CAA AGG TGA GGC CTG GT‐3′, CALR‐R; 5′ GCC TCT CTA CAG CTC GTC CTT‐3′), 0.5 units Taq Polymerase (Promega) and 50 ng genomic DNA in a final volume of 25 µl. Reactions were undertaken in a T100 Thermocycler (BioRad). Samples were activated/denatured at 95°C for 10 min, then were cycled at 95°C for 30 s, 58°C for 30 s and 72°C for 1 min for 40 cycles, followed by a 10‐min incubation at 72°C. Samples were visualized by electrophoresis on a 2% agarose gel containing 1 X Gel Red (Biotium) and imaged on the BioRad GelDoc XR+ (BioRad).
Haematopoietic colony formation
For megakaryocyte differentiation assays, CD34+ cells derived from cord blood or from patient peripheral blood mononuclear fractions were plated in MegaCult (MegaCult™‐C Collagen and Medium with Lipids) with added IL‐3 and IL‐6 (10 ng/ml) in the presence or absence of 50 ng/ml TPO. Cells were plated at an initial density of 1,800 cells/well and cultured for 12 days at 37°C and 5% CO2. Primary PMF CD34+ cells were cultured in StemCell Pro supplemented with human recombinant SCF (25 ng/ml), IL‐6 and IL‐9 (10 ng/ml) to allow differentiation into megakaryocytes. CD34+ cord blood cells were cultured in 50 ng/ml SCF, 50 ng/ml TPO, 20 ng/ml IL‐6 and 20 ng/ml IL‐9. Both IgG and 4D7 were added at 20 µg/ml. All cytokines are from Peprotech. Culture media were replenished every 3 days and cells were assessed for megakaryocyte differentiation on day 12 using anti‐human CD41/61 PE‐Cy7 (Clone A2A9/6) and CountBright beads (Thermo Fisher) to enumerate cell numbers. CD34+ obtained from UCB was plated in MethoCult containing cytokines (H4434). Cells were seeded at approximately 100 cells per well and were cultured in the presence of 20 µg/ml 4D7 or control IgG antibody and scored for colony and blast forming units at 12–14 days.
Flow cytometry assessment of 4D7 binding to the cell surface
TF‐1 cells expressing TpoR, CALRWT and mutant CALR were incubated for 30 min 4D7 conjugated to phycoerythrin (PE; Abcam, #AB102918) or IgG2a PE isotype control (ThermoFisher #12‐4321‐42) at the concentrations specified after pre‐incubation with mouse serum. Cells were washed 1× before analysis on a CytoFLEX (Beckman Coulter).
Analysis of cell cycle
Cell cycle analysis was carried out by flow cytometry. Briefly, TF‐1 TpoR and TF‐1 TpoR CALRdel61 cells were seeded at a concentration of ~ 2 × 105 per well in triplicate under standard culture conditions, with the addition of 20 µg/ml IgG or 4D7. Cells were treated for 48 h and were fixed by 70% (v/v) ethanol at 4°C overnight. Fixed cells were rinsed twice with PBS and stained with 50 µg/ml propidium iodide (PI; ThermoFisher) + 100 µg/ml RNAse A (QIAGEN). Cells were incubated for 30 min at RT in the dark and analysed on the BD FACSCantoII (BD Biosciences).
Co‐culture of CALR‐secreting cells
In brief, TF‐1 TpoR and TF‐1 TpoR CALRdel61 were seeded at the same density in opposing wells of a UniWellsTM Horizontal co‐culture plate (FujiFilm, #2501‐02FW) separated by a 0.75 µm filter. Cells were seeded in the presence or absence of 10 ng/ml TPO in triplicate and counted every day for 4 days by trypan blue exclusion. Representative images of wells were taken on day 4.
Protein lysate preparation and Western blot analysis
For immunoblotting of total lysates, cells were lysed in NP40 buffer containing 150 mM NaCl, 50 mM Tris pH 7.6, 1% NP‐40, supplemented with protease inhibitors (Complete, Roche) and phosphatase inhibitor cocktails, and boiled for 5 min after addition of sample buffer (60 mM Tris pH 6.8, 5% glycerol, 1% SDS, 2% β‐mercaptoethanol, and 0.02% bromophenol blue) before SDS gel electrophoresis followed by western blotting (Tvorogov et al, 2010). Primary antibodies against pERK (#9101), pSTAT5 (#9359), ERK (#4695), actin (#4970), caspase 3 (#14220) and wild‐type CALR (#12238) were purchased from Cell Signalling. Primary antibodies against pSTAT1 (#612233), pSTAT3 (#612357), STAT1 (#610186), STAT3 (#610190), STAT5 (#610192) and anti‐phosphotyrosine 4G10 (#610012) were purchased from BD Biosciences. Mutant CALR monoclonal antibody CAL2 was purchased from Dianova (Hamburg, Germany). Immunoprecipitation of TpoR was performed in NP40 lysis buffer with additional 50 mM iodoacetamide to avoid any de novo disulphide bond formation post‐lysis. To reduce IgG background during immunoprecipitation, anti‐FLAG conjugated to magnetic beads (Sigma, #M8823) was used. For this same reason, anti‐FLAG‐HRP conjugated antibodies (Sigma, #A8592) were used in western blotting.
Ruxolitinib‐resistant cells
Ruxolitinib‐resistant TF‐1 TpoR CALRdel61 cells were established by exposing cells of untreated TF‐1 TpoR CALRdel61 cells to increasing concentrations of ruxolitinib (Selleckchem) over a 4‐week period. Cells were initially treated with 10 nM and increased to 100 nM.
Xenograft models of disease
All procedures were approved by the South Australian Health and Medical Research Animal Ethics Committee (Protocol SAM21‐018). Bone marrow engraftment model: 4‐ to 6‐week‐old NSG mice were X‐ray irradiated with 150 cGy and 5 × 105 cells (TF‐1‐TpoR CALRdel61) or 5 × 104 cells (100 nM ruxolitinib‐resistant TF‐1 TpoR CALRdel61) were intravenously injected into mice. Seven days after injection, mice were administered 12.5 mg/kg IgG or 4D7 by intraperitoneal injection twice weekly. Tail vein bleeds were taken at 3 weeks and analysed via flow cytometry to determine human leukaemia content in the peripheral blood. Leukemic cells were propidium iodide negative, mCD45.1 negative, hCD33+ and ZsGreen1+. Mice were monitored daily and euthanized once clinical symptoms were observed. At the time of euthanasia, cardiac puncture was performed to obtain both peripheral blood and serum for analysis. Additionally, bone marrow from both femurs, spine and spleens was analysed for leukaemic content as detailed above. Chloroma model: 4‐ to 6‐week‐old NSG mice were anaesthetized by isoflurane inhalation and shaved on the left back/flank and 1 × 107 cells were injected into the mice. Cells were pre‐treated with 20 µg/ml IgG or 4D7 for 1 h prior to injection. Seven days after injection, mice were administered 12.5 mg/kg IgG or 4D7 by intraperitoneal injection twice weekly. Tumour progression was assessed through calliper measurements every 2 days. Mice were euthanized once tumours began to impede movement, show signs of ulceration or if any measurement of the tumour exceeded 30 mm (length, width or depth).
Author contributions
Denis Tvorogov: Conceptualization; Formal analysis; Supervision; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Chloe A L Thompson‐Peach: Formal analysis; Supervision; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Johannes Foßelteder: Formal analysis; Investigation; Methodology; Writing—review and editing. Mara Dottore: Validation; Investigation; Methodology. Frank Stomski: Investigation; Methodology. Suraiya A Onnesha: Investigation. Kelly Lim: Investigation; Methodology. Paul A Moretti: Resources; Investigation; Methodology. Stuart M Pitson: Resources; Investigation; Methodology. David M Ross: Resources; Writing—original draft; Writing—review and editing. Andreas Reinisch: Resources; Investigation; Methodology; Writing—review and editing. Daniel Thomas: Conceptualization; Data curation; Supervision; Funding acquisition; Writing—original draft; Writing—review and editing. Angel F Lopez: Conceptualization; Funding acquisition; Writing—original draft; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
DTv, DTh and AFL conceptualized the data. DTv, CALT‐P, MD, JF, FS, SAO, KL, AR and DTh performed experiments and analysed data. DTv, FS, PABM and SMP generated reagents. AR and DMR provided patient samples. DTh, DTv, CALT‐P, DMR and AFL wrote the manuscript.
Supporting information
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Source Data for Figure 2
Source Data for Figure 4
Acknowledgements
DTv, DT and AFL are supported by the National Health and Medical Research Council Ideas Grant APP1182564 and APP 2004288, and the Australian Cancer Research Foundation (ACRF) Cancer Genomics Facility and the ACRF Discovery Accelerator. DT is supported by a Commonwealth Serum Laboratories Centenary Fellowship, a Leukemia & Lymphoma Society Translational Research Program, The Hospital Research Fund and the Australian Medical Research Future Fund for Rare Diseases, Rare Cancers and Unmet Need. AR was supported by the Austrian Science Fund (P32783 and I5021), the Austrian Society of Internal Medicine (Joseph‐Skoda Award), the Austrian Society of Haematology and Medical Oncology (Clinical Research Grant) and MEFOgraz. We thank Professor Gus Dekker and the Northern Adelaide Local Health Network for provision of fresh cord blood. We thank Richard D’Andrea for TpoR‐FLAG construct. Flow cytometry analysis and cell sorting were performed at the Adelaide Health and BioMedical Precinct Cytometry Facility (AHMS and SAHMRI). The facility is generously supported by the Detmold Hoopman Group, ACRF and Australian Government through the Zero Childhood Cancer Program. Several figures were generated with BioRender with permissions to be published. Open access publishing facilitated by The University of Adelaide, as part of the Wiley ‐ The University of Adelaide agreement via the Council of Australian University Librarians.
Disclosure and competing interests statement
DT, DTv and AR are recipients of Australian Medical Research Future Fund for Rare Diseases, Rare Cancers and Unmet Need. DR receives research support from Novartis and Bristol Myers Squib, Honoraria and consultancy from Novartis, Bristol Myers Squib and Keros Therapeutics. AR receives research support from Celgene and is a consultant for Pfizer. None of these agencies have had a role in the preparation of this manuscript. The remaining authors declare no competing interests.
EMBO reports (2022) 23: e52904.
Contributor Information
Daniel Thomas, Email: daniel.thomas@adelaide.edu.au.
Angel F Lopez, Email: angel.lopez@sa.gov.au.
Data availability
No large primary datasets have been generated and deposited.
References
- Achyutuni S, Nivarthi H, Majoros A, Hug E, Schueller C, Jia R, Varga C, Schuster M, Senekowitsch M, Tsiantoulas D et al (2021) Hematopoietic expression of a chimeric murine‐human CALR oncoprotein allows the assessment of anti‐CALR antibody immunotherapies in vivo . Am J Hematol 96: 698–707 [DOI] [PubMed] [Google Scholar]
- Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, Mizukami Y, Kan S, Shirane S, Edahiro Y et al (2016) Activation of the thrombopoietin receptor by mutant calreticulin in CALR‐mutant myeloproliferative neoplasms. Blood 127: 1307–1316 [DOI] [PubMed] [Google Scholar]
- Araki M, Yang Y, Imai M, Mizukami Y, Kihara Y, Sunami Y, Masubuchi N, Edahiro Y, Hironaka Y, Osaga S et al (2019) Homomultimerization of mutant calreticulin is a prerequisite for MPL binding and activation. Leukemia 33: 122–131 [DOI] [PubMed] [Google Scholar]
- Baade PD, Ross DM, Anderson LA, Forsyth C, Fritschi L (2019) Changing incidence of myeloproliferative neoplasms in Australia, 2003–2014. Am J Hematol 94: E107–E109 [DOI] [PubMed] [Google Scholar]
- Balligand T, Achouri Y, Pecquet C, Gaudray G, Colau D, Hug E, Rahmani Y, Stroobant V, Plo I, Vainchenker W et al (2020) Knock‐in of murine Calr del52 induces essential thrombocythemia with slow‐rising dominance in mice and reveals key role of Calr exon 9 in cardiac development. Leukemia 34: 510–521 [DOI] [PubMed] [Google Scholar]
- Basso‐Valentina F, Levy G, Varghese LN, Oufadem M, Neven B, Boussard C, Balayn N, Marty C, Vainchenker W, Plo I et al (2021) CALR mutant protein rescues the response of MPL p. R464G variant associated with CAMT to eltrombopag. Blood 138: 480–485 [DOI] [PubMed] [Google Scholar]
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N et al (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365: 1054–1061 [DOI] [PubMed] [Google Scholar]
- Cervantes F, Vannucchi AM, Kiladjian J‐J, Al‐Ali HK, Sirulnik A, Stalbovskaya V, McQuitty M, Hunter DS, Levy RS, Passamonti F et al (2013) Three‐year efficacy, safety, and survival findings from COMFORT‐II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 122: 4047–4053 [DOI] [PubMed] [Google Scholar]
- Chachoua I, Pecquet C, El‐Khoury M, Nivarthi H, Albu R‐I, Marty C, Gryshkova V, Defour J‐P, Vertenoeil G, Ngo A et al (2016) Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127: 1325–1335 [DOI] [PubMed] [Google Scholar]
- Czuczman MS, Grillo‐López AJ, White CA, Saleh M, Gordon L, LoBuglio AF, Jonas C, Klippenstein D, Dallaire B, Varns C (1999) Treatment of patients with low‐grade B‐cell lymphoma with the combination of chimeric anti‐CD20 monoclonal antibody and CHOP chemotherapy. J Clin Oncol 17: 268–276 [DOI] [PubMed] [Google Scholar]
- Daitoku S, Takenaka K, Yamauchi T, Yurino A, Jinnouchi F, Nunomura T, Eto T, Kamimura T, Higuchi M, Harada N et al (2016) Calreticulin mutation does not contribute to disease progression in essential thrombocythemia by inhibiting phagocytosis. Exp Hematol 44: 817–825.e813 [DOI] [PubMed] [Google Scholar]
- Eder‐Azanza L, Navarro D, Aranaz P, Novo FJ, Cross NC, Vizmanos JL (2014) Bioinformatic analyses of CALR mutations in myeloproliferative neoplasms support a role in signaling. Leukemia 28: 2106–2109 [DOI] [PubMed] [Google Scholar]
- Elf S, Abdelfattah NS, Chen E, Perales‐Patón J, Rosen EA, Ko A, Peisker F, Florescu N, Giannini S, Wolach O et al (2016) Mutant calreticulin requires both its mutant C‐terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov 6: 368–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf S, Abdelfattah NS, Baral AJ, Beeson D, Rivera JF, Ko A, Florescu N, Birrane G, Chen E, Mullally A (2018) Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood 131: 782–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmelli P, Rotunno G, Bogani C, Mannarelli C, Giunti L, Provenzano A, Giglio S, Squires M, Stalbovskaya V, Gopalakrishna P et al (2016) Ruxolitinib is an effective treatment for CALR‐positive patients with myelofibrosis. Br J Haematol 173: 938–940 [DOI] [PubMed] [Google Scholar]
- Han L, Schubert C, Köhler J, Schemionek M, Isfort S, Brümmendorf TH, Koschmieder S, Chatain N (2016) Calreticulin‐mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi‐mediated secretion. J Hematol Oncol 9: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Czech J, Maurer A, Brummendorf TH, Chatain N, Koschmieder S (2018) Mutant NRAS Q61K is responsible for MAPK pathway activation in the MARIMO cell line and renders these cells independent of the CALR‐MPL‐JAK2‐STAT5 pathway. Leukemia 32: 2087–2090 [DOI] [PubMed] [Google Scholar]
- Harrison C, Kiladjian J‐J, Al‐Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L et al (2012) JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 366: 787–798 [DOI] [PubMed] [Google Scholar]
- Harrison CN, Vannucchi AM, Kiladjian J‐J, Al‐Ali HK, Gisslinger H, Knoops L, Cervantes F, Jones MM, Sun K, McQuitty M et al (2016) Long‐term findings from COMFORT‐II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 30: 1701–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- How J, Hobbs GS, Mullally A (2019) Mutant calreticulin in myeloproliferative neoplasms. Blood 134: 2242–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NCC, Berg T, Gisslinger B, Pietra D et al (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 369: 2379–2390 [DOI] [PubMed] [Google Scholar]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC (2005) A gain‐of‐function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352: 1779–1790 [DOI] [PubMed] [Google Scholar]
- Li J, Prins D, Park HJ, Grinfeld J, Gonzalez‐Arias C, Loughran S, Dovey OM, Klampfl T, Bennett C, Hamilton TL et al (2018) Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self‐renewal advantage. Blood 131: 649–661 [DOI] [PubMed] [Google Scholar]
- Liu P, Zhao L, Loos F, Marty C, Xie W, Martins I, Lachkar S, Qu BO, Waeckel‐Énée E, Plo I et al (2020) Immunosuppression by mutated calreticulin released from malignant cells. Mol Cell 77: 748–760.e749 [DOI] [PubMed] [Google Scholar]
- Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, van Rooijen N, Weissman IL (2009) CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138: 286–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeti R (2011) Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene 30: 1009–1019 [DOI] [PubMed] [Google Scholar]
- Marty C, Pecquet C, Nivarthi H, El‐Khoury M, Chachoua I, Tulliez M, Villeval J‐L, Raslova H, Kralovics R, Constantinescu SN et al (2016) Calreticulin mutants in mice induce an MPL‐dependent thrombocytosis with frequent progression to myelofibrosis. Blood 127: 1317–1324 [DOI] [PubMed] [Google Scholar]
- Masarova L, Wang W, Newberry KJ, Kantarjian H, Verstovsek S (2016) Complete remission in a patient with JAK2‐ and IDH2‐positive myelofibrosis. Blood 128: 877–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masubuchi N, Araki M, Yang Y, Hayashi E, Imai M, Edahiro Y, Hironaka Y, Mizukami Y, Kihara Y, Takei H et al (2020) Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia 34: 499–509 [DOI] [PubMed] [Google Scholar]
- Meyer SC, Keller M, Chiu S, Koppikar P, Guryanova O, Rapaport F, Xu KE, Manova K, Pankov D, O’Reilly R et al (2015) CHZ868, a type II JAK2 inhibitor, reverses type I JAK inhibitor persistence and demonstrates efficacy in myeloproliferative neoplasms. Cancer Cell 28: 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG et al (2013) Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 369: 2391–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer HA, Pham DH, Zebol JR, Moretti PAB, Peterson AL, Leclercq TM, Chan H, Powell JA, Pitman MR, Samuel MS et al (2016) An oncogenic role for sphingosine kinase 2. Oncotarget 7: 64886–64899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passamonti F, Maffioli M, Merli M, Ferrario A, Caramazza D (2012) Clinical predictors of outcome in MPN. Hematol Oncol Clin North Am 26: 1101–1116 [DOI] [PubMed] [Google Scholar]
- Pecquet C, Balligand T, Chachoua I, Roy A, Vertenoeil G, Colau D, Fertig E, Marty C, Nivarthi H, Defour J‐P et al (2018) Secreted mutant calreticulins as rogue cytokines trigger thrombopoietin receptor activation specifically in CALR mutated cells: perspectives for MPN therapy. Blood 132: 4 29976778 [Google Scholar]
- Pecquet C, Chachoua I, Roy A, Balligand T, Vertenoeil G, Leroy E, Albu R‐I, Defour J‐P, Nivarthi H, Hug E et al (2019) Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic‐defective pathogenic TpoR mutants. Blood 133: 2669–2681 [DOI] [PubMed] [Google Scholar]
- Reinisch A, Thomas D, Corces MR, Zhang X, Gratzinger D, Hong WJ, Schallmoser K, Strunk D, Majeti R (2016) A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med 22: 812–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross DM, Babon JJ, Tvorogov D, Thomas D (2021) Persistence of myelofibrosis treated with ruxolitinib: biology and clinical implications. Haematologica 106: 1244–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollazzo D, Forte D, Polverelli N, Perricone M, Romano M, Luatti S, Vianelli N, Cavo M, Palandri F, Catani L (2016) Circulating calreticulin is increased in myelofibrosis: correlation with interleukin‐6 plasma levels, bone marrow fibrosis, and splenomegaly. Mediators Inflamm 2016: 5860657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, Vainchenker W (2011) Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol 29: 573–582 [DOI] [PubMed] [Google Scholar]
- Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, Gangat N, Fjerza R, Belachew AA, Lasho TL et al (2014) Long‐term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124: 2507–2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, Pardanani A (2015) Myeloproliferative neoplasms: a contemporary review. JAMA Oncol 1: 97–105 [DOI] [PubMed] [Google Scholar]
- Tvorogov D, Anisimov A, Zheng W, Leppänen V‐M, Tammela T, Laurinavicius S, Holnthoner W, Heloterä H, Holopainen T, Jeltsch M et al (2010) Effective suppression of vascular network formation by combination of antibodies blocking VEGFR ligand binding and receptor dimerization. Cancer Cell 18: 630–640 [DOI] [PubMed] [Google Scholar]
- Tvorogov D, Thomas D, Liau NPD, Dottore M, Barry EF, Lathi M, Kan WL, Hercus TR, Stomski F, Hughes TP et al (2018) Accumulation of JAK activation loop phosphorylation is linked to type I JAK inhibitor withdrawal syndrome in myelofibrosis. Sci Adv 4: eaat3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT et al (2012) A double‐blind, placebo‐controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 366: 799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG (2006) Expression of Jak2V617F causes a polycythemia vera‐like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 107: 4274–4281 [DOI] [PMC free article] [PubMed] [Google Scholar]