

Abstract
Mitochondria can function as signaling organelles, and part of this output leads to epigenetic remodeling. The full extent of this far-reaching interplay remains undefined. Here, we show that MYC transcriptionally activates IDH2 and increases alpha-ketoglutarate (αKG) levels. This regulatory step induces the activity of αKG-dependent DNA hydroxylases and RNA demethylases, thus reducing global DNA and RNA methylation. MYC, in a IDH2-dependent manner, also promotes the nuclear accumulation of TET1-TET2-TET3, FTO and ALKBH5. Notably, this subcellular movement correlated with the ability of MYC, in an IDH2-dependent manner, and, unexpectedly, of αKG to directly induce O-GlcNAcylation. Concordantly, modulation of the activity of OGT and OGA, enzymes that control the cycling of this non-canonical mono-glycosylation, largely recapitulated the effects of the MYC-IDH2-αKG axis on the subcellular movement of DNA and RNA demethylases. Together, we uncovered a hitherto unsuspected crosstalk between MYC, αKG and O-GlcNAcylation which could influence the epigenome and epitranscriptome homeostasis.
Introduction
Alpha-ketoglutarate (αKG) is an essential intermediate metabolite. The main physiologic source of αKG is the oxidative decarboxylation of isocitrate executed by three isocitrate dehydrogenases (IDH), the NADP-dependent cytoplasmic IDH1 and mitochondrial IDH2, and the evolutionary distinct NAD-dependent mitochondrial IDH31. In addition, in normal and malignant proliferating cells, anaplerotic reactions can also contribute to the αKG pools. In this instance, glutamine is first deaminated to glutamate by glutaminase (GLS), and glutamate, through the action of glutamate dehydrogenase (GDH) or transaminases (glutamate pyruvate transaminases, GPTs, and glutamate oxaloacetate transaminases, GOT) is converted into αKG1, 2. Furthermore, the metabolic repair enzymes D2HGDH and L2HGDH promote the oxidation of the D-2-HG and L-2-HG into αKG3, 4. In physiologic conditions these reactions are likely to be only minor contributors to the cellular pool of αKG, given the low physiologic levels of the precursor metabolite 2-HG. However, multiple metabolic disturbances associate with aberrant accumulation of D-2-HG and L-2-HG, and in those instances their oxidation to αKG may be quantitatively relevant5–7.
Once present, αKG activities are pleotropic. As a TCA cycle intermediate, it participates in the generation of reducing equivalents, contributes to energy production, and it is a source of precursors for biosynthesis of macromolecules8. In addition, αKG is an important nitrogen scavenger in multiple metabolic pathways, and given the reversibility of GDH activity, αKG also contributes to the glutamate pool1, 2, 8. Importantly, αKG is an obligatory co-substrate for a large class of dioxygenases, including multiple enzymes that control epigenetics, including histone demethylases, DNA hydroxylases, RNA demethylases, and prolyl-hydroxylases9. Further, in a variety of model systems, αKG supplementation was recently shown to be clinically beneficial, positively impacting longevity in C elegans and mammals and decreasing frailty in mice10, 11.
The role of αKG as a rate-limiting co-substrate for multiple dioxygenases came to the fore with the discovery of mutant neomorphic IDH1 and IDH2 isoforms in a variety of cancer types12. These enzymes aberrantly reduce αKG to D-2-HG, instead of to isocitrate, resulting in D-2-HG accumulation. The structural similarity between αKG and D-2-HG results in competitive inhibition of αKG-dependent dioxygenases, and a phenotype of DNA, RNA and histone hypermethylation emerges, which contributes to the pathogenesis of cancers harboring IDH1/2 mutations9, 13. These observations link αKG homeostasis to cancer biology14.
Remarkably, despite the importance of αKG generation by IDHs, and the presence of IDH1 and IDH2 mutations in cancer, very little is known about the transcriptional regulation of these genes, except for an association between SREBP (sterol regulatory element binding protein) and C/EBPβ with IDH1, and PCAF (p300/CBP-associated factor) with IDH215–17. Conspicuously, no regulatory relationship has been established between IDH1 and IDH2 and transcription factors that play a significant role in cancer metabolism, a putative interplay that deserves close examination.
MYC is a prototypical oncogene, and its “driver” role in multiple hematological and epithelial malignancies is unquestionable18, 19. MYC oncogenicity has many facets, including a role on cancer-associated metabolic reprograming20. MYC transcriptionally regulates various enzymes that positively influence anapleorotic mitochondrial metabolism (e.g., GLS and GDH), glycolysis (LDHA), and the transport of glucose and glutamine across the cell membrane (GLUT1, SLC1A5)20. In addition, more recently, we showed that MYC transcriptionally induces D2HGDH and L2HGDH, adding a new layer to MYC’s regulation of intermediary metabolism5. These recent data, together with the known role of MYC in glutaminolysis, suggest that the activities of this transcriptional factor may be an important hub for the control of αKG homeostasis. Still, it remains to be defined if MYC also transcriptionally regulates IDHs, the main contributors to the cellular pool of αKG.
Here, focusing on the IDH isoforms that play a role in cancer, we show that MYC binds to the promoter and transcriptionally activates IDH2, but not IDH1. The regulation of IDH2 by MYC has a significant impact on the cellular levels of αKG, and on the function of the dioxygenases that control DNA and RNA methylation. Importantly, we found that in addition to increasing the activity of these enzymes, MYC, in an IDH2-dependent manner, also influences their sub-cellular localization. In this regard, we discovered that the MYC-IDH2-αKG axis promotes nuclear accumulation of the TET DNA hydroxylases and the RNA demethylases in association with O-GlcNAcylation, a post-translational modification characterized by the attachment of single O-linked N-acetylglucosamine (O-GlcNAc) moiety to serine or threonine residues in hundreds of cellular proteins21. In addition, we showed that D2HGDH and L2HGDH are also intermediaries in the MYC-mediated regulation of O-GlcNAcylation. These data suggest that MYC functions as a control hub for αKG homeostasis and uncover a hitherto unrecognized participation of O-GlcNAcylation in the MYC-driven, and αKG-executed, activation and sub-cellular movement of TET enzymes and RNA demethylases. These findings also suggest that pharmacological targeting of O-GlcNAc transferase (OGT) or of O-GlcNAcase (OGA), enzymes that control the cycling of O-GlcNAcylation, may be a valid approach to modulate the epigenome and epitranscriptome.
Materials and Methods:
Cell lines.
The Burkitt’s lymphoma cell line HS-Sultan, and P493-6, a human B cell line carrying a conditional MYC gene22, were cultured at 37°C in 5%CO2 in RPMI-1640 medium (Invitrogen) containing 10% (vol/vol) fetal bovine serum (FBS), as we described23. HEK-293T were maintained in Dulbecco’s modified Eagle media (DMEM; Mediatech) with 10% FBS, as we described24, 25. The cell lines were preexistent in the investigator’s laboratory and earlier purchased from Invitrogen or ATTC. MYC suppression with tetracycline, cell line authenticity, Mycoplasma testing were done as we described5.
Mice and isolation of murine mature splenic B lymphocytes.
Spleens from Eμ-Myc transgenic (B6.Cg-Tg[IgHMyc]22Bri/J) or WT mice, 2 to 3-months old, male and female, were harvested and analyzed, as we recently described5. Mature B lymphocytes were and purity, validate by FACS and RNA isolated as we reported26–28. The investigators were not blind to the mice genotype during the experiment; the mice were not randomized. All animal procedures were approved by the Institute Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio, San Antonio, TX, USA.
Compounds.
Exposure of cell lines to dimethyl 2-oxoglutarate (DMαKG; Sigma-Aldrich, #349631) octyl-D-2-HG, octyl-L-2-HG (Toronto Research Chemicals, #H942595 and #H942596), dimethyloxalylglycine (DMOG; Frontier Scientific, D1070) was performed as we described5. Cells were also exposed to Thiamet G (5μM, OGA inhibitor, #HY-12588, MedChemExpress) and to OSMI-1 (20μM, OGT inhibitor, #SML1621, Sigma-Aldrich) for 16h.
Luciferase Reporter assays.
Two DNA fragments, 1013bp and 1147bp, surrounding the transcriptional start site (TSS) of the genes IDH1 and IDH2, respectively, were cloned into pGL3-basic vector (Promega). The reporter assays in HEK-293T cells were performed as we recently described5, 29. The primers sequences are listed in Supplementary Table S1.
Chromatin Immune Precipitation (ChIP)-PCR assay:
HS-Sultan cells were used in these assays performed as we recently described in details5, 30, and included as positive control for MYC binding the well-characterized E-box in the LIN28B promoter (CATGTG)31.
Generation of genetic models.
CRISPR-Cas9 technology was used to knockout (KO) the IDH2, D2HGDH and L2HGDH genes in P493-6 cells, as we reported5. For the KO of IDH2, independent transductions were performed to create unique cell models termed IDH2-KO-1 (RNA guide#2 cloned into lentiCRISPR v2-puromycin) and IDH2-KO-2 (combination of RNA-guides #3 into lentiCRISPR v2-puromycin, and #4 cloned into pL-CRISPR.EFS.GFP)32. For double D2HGDH/L2HGDH KO, a combination of lentiCRISPR v2 puromycin (expressing L2HGDH guide#1) and pL-CRISPR.EFS.GFP (expressing D2HGDH guide#2)5, 33 was used. Virus generation was performed as we previously described34, 35. IDH2, D2HGDH and L2HGDH knockout was confirmed by western blotting. For each gene, multiple unique guide RNAs were used (Table S1).
Protein isolation, subcellular fractionation and western blots.
Whole-cell lysates (WCL) were examined as we described36. Nuclear and cytoplasmic proteins were extracted and analyzed as we recently reported5. The following antibodies were used: c-MYC (clone 9E10; #sc-40, Santa Cruz Biotechnology), IDH2 (#ab55271, Abcam), OGT (#24083, Cell Signaling Technology), OGA (Cat #14711–1-AP, Proteintech), O-GlcNAc (clone RL2, #MA1–072, Thermo-Fisher Scientific), D2HGDH (#13895–1-AP, Proteintech), L2HGDH (#15707–1-AP, Proteintech), ALKBH5 (# ab69325, Abcam), FTO (c-3, #sc-271713, Santa Cruz Biotechnology), TET1 (#SAB2700730, Sigma-Aldrich), TET2 (#18950, Cell Signaling Technology), TET3 (#: ABS463, EMD Millipore Corporation), METTL3, METTL14, WTAP (# 69391, # 51104 and #56501, all from Cell Signaling Technology), β-actin (# A2228, Sigma-Aldrich), Lamin A (#A303–433A-M, Bethyl Laboratories), Tubulin (#62204 Invitrogen). PVDF membranes were stripped and re-probed with relevant antibodies for loading control, as reported37.
Quantification of m6A in RNA and 5hmC/5mC in DNA.
m6A levels and 5hmC/5mC marks were quantified using ELISA-based EpiQuik™ m6A RNA Methylation Quantification Kit (Epigentek, #P-9005), and with the Hydroxymethylated DNA (5hmC) Quantification Kit and Methylated DNA (5mC) Quantification Kit (Epigentek, #P-1034 and #P-1036), respectively, as we described5, 13. All assays were performed in triplicate and multiple biological replicates completed.
Demethylases activity.
The activity of m6A RNA demethylases and DNA hydroxylases (TET enzymes) was quantified in nuclear protein of the relevant models, as before5, using the m6A Demethylase Activity Kit and the 5mC-Hydroxylase TET Activity Assay Kit, respectively (Epigentek, #P-9013 and #P-3086).
Metabolite quantification by mass spectrometry.
The measurements of the D-2-HG, L-2-HG and αKG were performed by liquid chromatography-tandem mass spectrometry (LC-MS/MS), as we described earlier3, 5.
Quantification and Statistical Analysis.
Analyses were performed using a one-way or two-way ANOVA, with Bonferroni’s multiple comparison post-hoc test, and two-tailed Student’s t-test. P<0.05 was considered significant. When appropriate, equal variance was calculated with an F-test (t-test) or with a Bartlett’s statistics for equal variances (ANOVA). All data involving statistics are presented as mean ± SD or ± SEM. The number of replicates and the statistical test used are described in the figure legends. Data analyses were performed in the GraphPad Prism 8 software (version 8.3.1, GraphPad Software Inc).
Results:
IDH2 is a transcriptional target of MYC.
Using reporter assays, we identified DNA sequences near the annotated transcription start sites for IDH1 and IDH2 containing basal promoter activity (Figure 1A and Supplementary Figure S1A). We used the MatInspector software and the “regulation” function of the UCSC genome browser to identify putative transcription factor binding sites common to both promoter regions; two putative E-boxes were found in the IDH2 promoter and one in the IDH1 promoter (Supplementary Table S2). To test the ability of MYC to transactivate these promoters, we performed reporter assays in HEK-293T cells co-transfected with MYC or control vectors and identified an MYC-dependent reporter activity in IDH2 but not IDH1 (Figure 1B and Supplementary Figure S1B). We then performed chromatin immunoprecipitation of MYC in the Burkitt’s lymphoma cell line HS-Sultan followed by real-time qPCR (ChIP-qPCR) and showed that MYC binds to E-box #2 in the IDH2 locus (Figure 1C). To confirm the relevance of these interactions, we used the human B cell line P493-6, which has MYC expression under the control of a tetracycline responsive element; turning MYC OFF in these cells decreased IDH2 expression, while restoring MYC expression progressively increased its levels (Figure 1D). This tight regulation was not detected in respect to the IDH1 transcript (Supplementary Figure S1C). Finally, we isolated splenic mature B cells from Eμ-Myc mice and littermate wild-type (WT) controls (n = 4), and using qRT-PCR, we showed that the expression of Idh2, but not Idh1 was significantly higher in mature B cells from Eμ-Myc mice than from WT controls (Figure 1E and Supplementary Figure S1D). We concluded that IDH2 is a transcriptional target of MYC.
Figure 1. IDH2 is a transcriptional target of MYC.

A) Luciferase activity of the putative IDH2 promoter region – arrows indicate transcriptional start sites; gray and red squares are putative E-boxes (n=3). B) Luciferase activity of the IDH2 promoter in HEK-293T cells co-transfected with MYC. C) ChIP-qPCR of MYC binding to E-boxes in IDH2 promoter; +ctrl is the well-characterized E-box of the LIN28B promoter, -ctrl is a promoter region of IDH2 lacking a predicted E-box (n=3). D) left panel: q-RT-PCR of IDH2 mRNA in P493-6 cells (n=3); Right panel: representative immunoblot of one of the assays with tetracycline-regulated MYC expression in P493-6 cells. E) q-RT-PCR of Idh2 mRNA in mature B-cells from Eμ-Myc mice and WT mice (n=4). Data shown are mean −/+ SD; p values in b) and e) are from two-tailed Student’s t-test.
IDH2 contributes to the MYC-driven elevation of αKG levels and modulation of DNA and RNA methylation.
We found that MYC transcriptionally activates IDH2 (Figure 1). Earlier, we reported that MYC regulates the mitochondrial enzymes D2HGDH and L2HGDH, and through them the activity of TET DNA hydroxylases and RNA demethylases5. Here, we sought to determine the contribution of the newly found MYC-IDH2 interplay to these processes. We speculated that MYC, via modulation of IDH2 transcription, increases αKG levels and the activity of the αKG-dependent TET DNA hydroxylases and RNA demethylases.
To test this concept, we first generated CRISPR-based two IDH2 knockout (KO) models (using distinct guideRNAs) in the MYC-inducible P493-6 cells. Thereafter, we quantified αKG levels in MYC-inducible IDH2 WT or KO P493-6 cells. At baseline (MYC ON), WT cells expectedly displayed significantly higher αKG levels than IDH2 KO isogenic models (Figure 2A). To investigate the kinetics of the MYC/IDH2-dependent αKG production, we suppressed and restored MYC expression in IDH2 WT or KO cells and measured αKG. In absence of MYC and early after it was re-expressed, the levels of αKG were similar irrespectively of the genetic integrity of IDH2. However, 24h post-MYC re-expression the “baseline” status re-emerged and αKG levels were again higher in IDH2 WT cells (Figure 2A), which corresponds to the transcriptional activation of IDH2 shown in Figure 1D. To examine the potential relationship between MYC, IDH2 and αKG-dependent dioxygenases, we quantified 5hmC/5mC and m6A levels in the MYC-inducible IDH2 WT or KO P493-6 cells; as an additional relevant control, we also created and tested new double D2HGDH/L2HGDH KO P493-6 cells, since in our earlier study we showed that these genes are also directly regulated by MYC, but examined exclusively individual KO models5. In WT P493-6 cells, turning MYC expression ON, resulted in a ~ 50% increase in the abundance of 5hmC DNA marks. In contrast, in IDH2 KO cells, MYC-mediated increase in the deposition of 5hmC was limited to ~15% (ranging from 11% to 19%, Figure 2B); in agreement with our earlier data in single D2HGDH or L2HGDH KO cells, double genetic removal of these enzymes also significantly blocked MYC-mediated increase in 5hmC abundance (Figure 2B). Corresponding changes were detected with 5mC quantification, with KO of IDH2 and of D2HGDH/L2HGDH limiting MYC’s ability to reduce 5mC levels (Figure 2B). Correlating with the 5hmC/5mC data, we found that MYC-mediated induction of TET activity was significantly reduced in IDH2 KO, as well as in the double D2HGDH/L2HGDH KO cells (Figure 2C). Notably, these genetic and metabolic perturbations did not modify the TET1–3 levels (Figure 2C). We next examined the role of IDH2, downstream to MYC, in controlling RNA m6A levels and RNA demethylase activity. MYC induction in WT P493-6 cells resulted in a significantly more pronounced decrease in m6A levels than in the IDH2 KO counterpart cells. In addition, in cells lacking IDH2, or D2HGDH/L2HGDH, MYC-mediated activation of RNA demethylases was significantly reduced (Figure 2D); importantly, in these genetic models the expression of RNA demethylases FTO and ALKBH5, or of components of the RNA methyltransferase complex, METTL3, METTL14 and WTAP was unchanged (Figure 2D). Of note, while the effects of D2HGDH/L2HGDH deficiency on DNA and RNA methylation result from a combination of abnormal accumulation 2-HG and limited αKG generation, as we showed recently5, in respect to IDH2 only the latter applies, since D-2HG and L-2-HG levels were, expectedly, unmodified in IDH2 KO cells (Supplementary Figure S2). We concluded that MYC-mediated transcriptional activation of IDH2, as well as of other mitochondrial dehydrogenases, influence the epigenome and epitranscriptome.
Figure 2. MYC-IDH2-αKG axis, DNA/RNA methylation and demethylase activity.
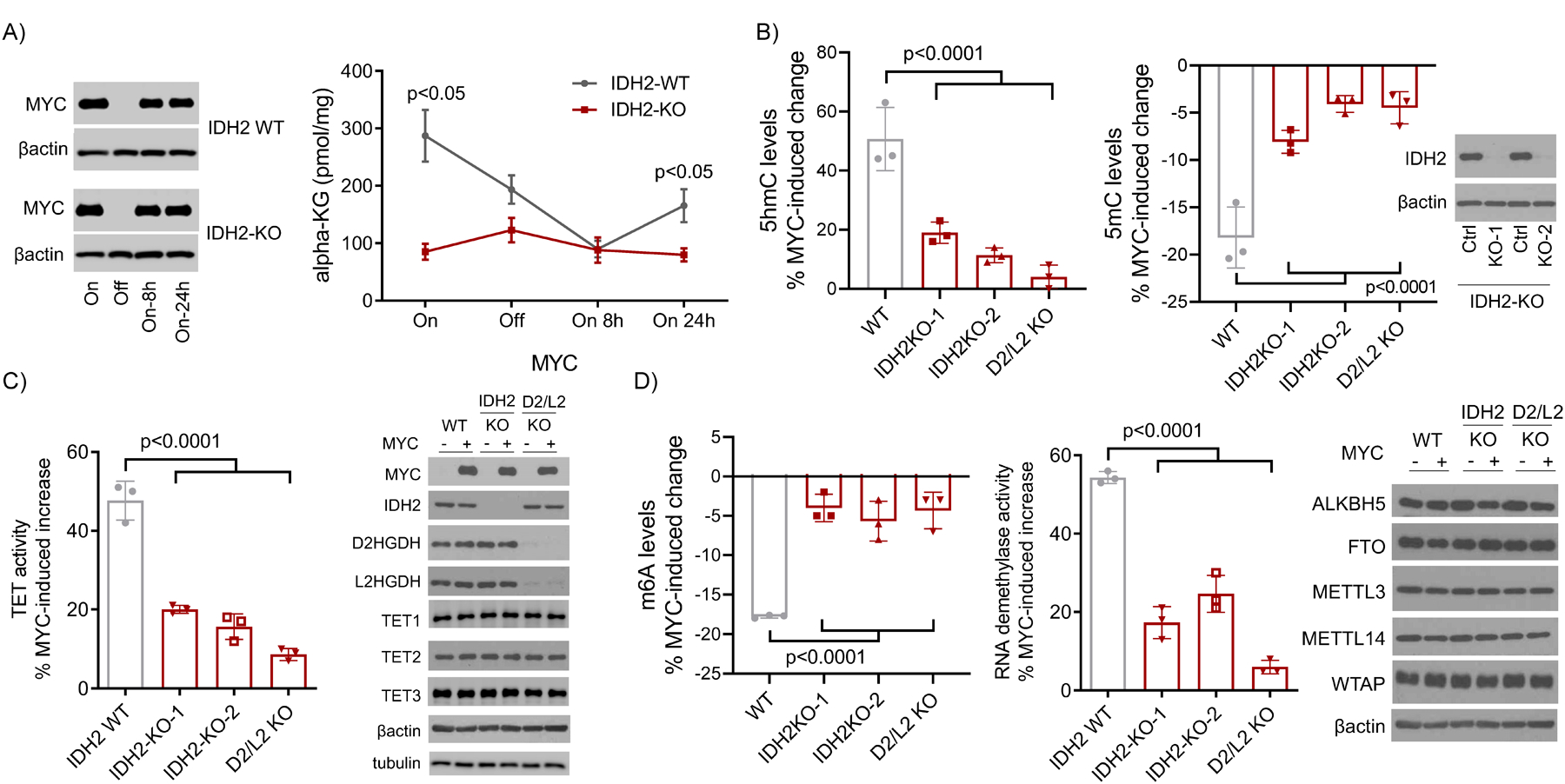
A) Quantification of αKG in P493-6 cells with WT or KO for IDH2 (MYC ON or OFF); MYC immunoblots are on the left. Data shown are mean −/+ SEM (n=3), p<0.05, two-tailed Student’s t-test. B) Left to right panels: quantification of MYC-induced increase or decrease of 5hmC and 5mC, respectively, in WT, IDH2 KOs, and double D2HGDH/L2HGDH KO P493-6 cells. Data are mean −/+ SD (n=3), p<0.0001, one-way ANOVA. Bonferroni post-test, 5hmC: p=0.0007 WT vs IDH2 KO-1, p=0.0001 WT vs. IDH2 KO-2, p<0.0001 WT vs. D2/L2 KO; 5mC: p=0.0006 WT vs. IDH2 KO-1, p<0.0001 WT vs. IDH2 KO-2, p<0.0001 WT vs. D2/L2 KO. WB of IDH2 in KO cells is at the right of the panels. C) Quantification of MYC-induced TET activity in WT, IDH2 KO, and D2HGDH/L2HGDH double KO P493-6 cells. Data are mean −/+ SD (n=3), p<0.0001, one-way ANOVA. Bonferroni post-test: p<0.0001 WT vs. IDH2 KO-1, p<0.0001 WT vs. IDH2 KO-2, p<0.0001 WT vs. D2/L2 KO; WBs of MYC, IDH2, D2HGDH, L2HGDH, TET1, TET2, TET3, and are shown to the right. D) Left to right panels: quantification of MYC-induced decrease and increase in m6A levels and RNA demethylase activity, respectively, in WT, IDH2 KOs, and double D2HGDH/L2HGDH KO P493-6 cells. Data are mean −/+ SD (n=3), p<0.0001, one-way ANOVA. Bonferroni post-test, m6A: p<0.0001 WT vs. IDH2 KO-1, p=0.0002 WT vs. IDH2 KO-2, p<0.0001 WT vs. D2/L2 KO; RNA demethylase activity: p<0.0001 WT vs. IDH2 KO-1, p<0.0001 WT vs. IDH2 KO-2, p<0.0001 WT vs. D2/L2 KO. WBs of FTO, ALKBH5, METL3, WTAP, and METTL14 in WT, IDH2 KOs, and double D2HGDH/L2HGDH KO P493-6 cells are shown to the right.
αKG rescues the genetic models of IDH2 knockout.
We showed that IDH2 integrity is important for the MYC-mediated increase in αKG levels, and subsequent activation of TET DNA hydroxylases and RNA demethylases (Figure 2). Next, we tested the essentiality of αKG to this process. In brief, MYC expression was turned OFF in the P493-6 cells WT or KO for IDH2, and then restored in the presence or absence of cell-permeable dimethyl-αKG for 24h, and DNA and RNA collected. In absence of αKG we confirmed that IDH2 KO significantly blunted the MYC-driven increase in the deposition of 5hmC, and removal of m6A marks (Figure 3A). Conversely, restoring MYC expression in the presence of αKG fully rescued the IDH2 KO phenotype (Figure 3A). Notably, αKG also rescued the DNA and RNA hypermethylation associated with the double genetic deletion of D2HGDH and L2HGDH (Figure 3A). This observation supports our earlier report that in addition to the excessive accumulation of 2-HG, loss of these dehydrogenases also decreases αKG generation3, 5. In addition, these data support the concept that the competitive inhibition of αKG-dependent dioxygenases by D-2-HG and L-2-HG may be overcome by αKG, an indication that this intermediate metabolite is rate-limiting for the activity of TET hydroxylases and RNA demethylases. To cross-validate the role of αKG in the MYC-driven regulation of DNA and RNA methylation, we turned MYC expression OFF and restored it in the presence of cell-permeable octyl-D-2-HG, octyl-L-2-HG or DMOG (Dimethyloxallyl Glycine), a classical competitive inhibitor of αKG-dependent enzymes. We once more validated the ability of MYC to significantly increase 5hmC deposition and removal of m6A and showed that DMGO and 2-HG block MYC-mediated modulation of these markers (Figure 3B), even though we recognize that the effects of 2-HG on DNA and RNA methylation may be detected irrespective of the mechanistic context. We concluded that the transcriptional activation of mitochondrial dehydrogenases by MYC influences the epigenome and epitranscriptome at least in part by promoting αKG generation.
Figure 3. Intermediate metabolites modulate MYC-IDH2 effects on DNA and RNA methylation.

A) Left to right panels - 5hmC and m6A levels in P493-6 cells (MYC OFF or ON; WT, IDH2 KO or D2/L2HGDH KO), with or without DMαKG rescue (5 mM) (n=3); p values are from two-tailed Student’s t test. B) Left to right panels - 5hmC and m6A levels in P493-6 cells (MYC OFF or ON) exposed to DMOG (1 mM), octyl-D-2-HG or L-2-HG (100 μM) (n=3); p values are from two-tailed Student’s t test.
IDH2 contributes to the MYC effects on the subcellular localization of DNA hydroxylases and RNA demethylases.
We have recently discovered that at least part of the positive effects of MYC and of αKG on the activity of TET enzymes and RNA demethylases is secondary to modulation of their sub-cellular localization, in particular nuclear accumulation5. In this earlier report, we demonstrated that single KO of D2HGDH or L2HGDH blunted MYC-driven nuclear localization of TET1/2/3 and FTO or ALKBH5 and that synthetic αKG promoted, while 2-HG and DMOG limited nuclear accumulation of these enzymes5. Here, we examined whether this interplay extends to IDH2 function. To this end, we modulated MYC expression in IDH2 WT or KO P493-6 cells, and isolated nuclear and cytosolic fractions, as well as whole cell lysates. The double D2HGDH/L2HGDH KO cells were also included in these assays for additional validation of our earlier data obtained in single D2HGDH or L2HGDH KO cells5. In WT P493-6 cells, inducing MYC expression led to a marked increase of TET1-2-3, FTO and ALKBH5 in the nucleus, in association with a decrease in the cytoplasmic fraction, even though in some instances we cannot exclude the possibility that part of the proteins that are excluded from the nucleus relocate to insoluble cellular fractions. More importantly, we found that in IDH2 KO cells the effects of MYC expression on the nuclear accumulation and cytoplasmic depletion of DNA hydroxylases and RNA demethylases were significantly blunted (Figure 4). Combined deletion of D2HGDH and L2HGDH also limited MYC effect of the nuclear accumulation of these proteins, and further validated our earlier results with single D2HGDH or L2HGDH KO cells5. In these KO models, nuclear accumulation of DNA and RNA demethylases following MYC re-expression was severely blunted but not completely abolished, suggesting residual effect of the intact genes (i.e., IDH2 in D2/L2HGDH KO cells, and D2HGDH/L2HGDH in the IDH2 KO models). A similar interpretation can be ascribed to the marked, but incomplete, block in MYC-driven 5hmC increase and m6A decrease in IDH2 and D2/L2HGDH KO models (Figure 3A). Future generation of triple KO cells should address this possibility. We concluded that the higher TET and RNA demethylase activities associated with MYC’s transcription activation of IDH2 may relate both to an increase in the cellular pool of the co-substrate αKG, and to the accumulation of the enzymes in the relevant nuclear compartment.
Figure 4. Control of the subcellular localization of TET1-3, FTO and ALKBH5 by the MYC-IDH2 and MYC- D2HGDH/L2HGDH interplays.
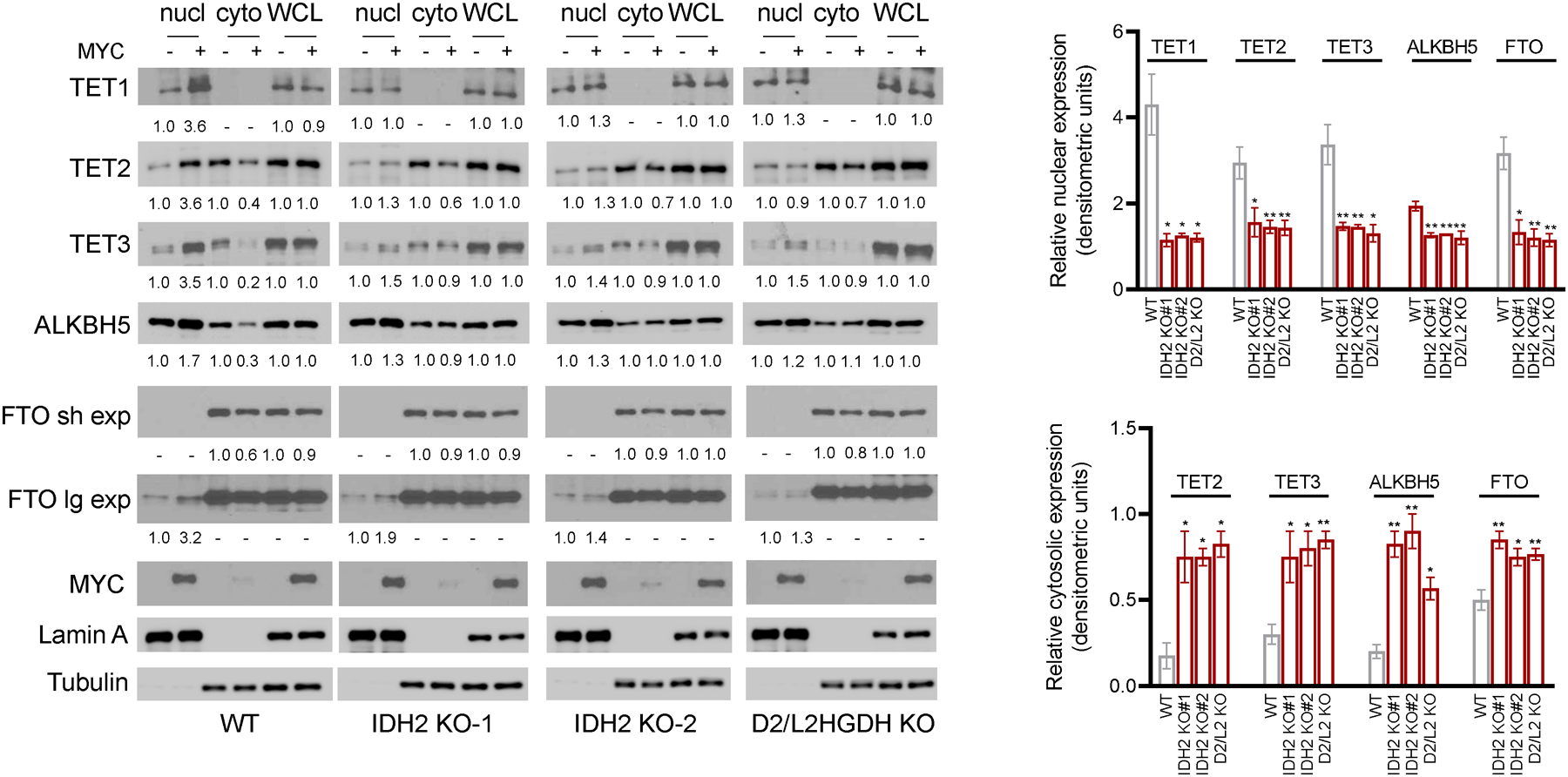
WBs of TET1-3, FTO and ALKBH5 in subcellular fractions and WCL of P493-6 cells (WT, IDH2-KO or D2HGDH/L2HGDH KO) in MYC OFF and ON status. Densitometric measurements of protein levels are shown at the bottom of the WB displays; the bar-graphs on the right represent mean ± SEM of densitometric quantifications of biological replicates for this assay (n=2 to 5). P values are from two-sided Student’s t-test (each KO vs. WT), *p≤0.05, **p≤0.01. All values are relative to controls (MYC OFF) and corrected by lamin A or tubulin; sh and lg exp = short and long exposures, respectively.
IDH2, D2HGDH/L2HGDH and αKG influence O-GlcNAcylation to modulate the sub-cellular localization of TET enzymes and RNA demethylases FTO and ALKBH5.
We showed that MYC, in part via transcription activation of IDH2, promotes the nuclear localization of TET1-2-3 and FTO and ALKBH5 (Figure 4). Earlier, we reported that D2HGDH, L2HGDH, and also supplementation of exogenous αKG could rapidly promote enrichment of these enzymes in the nucleus at the cost of cytoplasmic depletion5. These observations suggested that a covalent modification, possibly promoted by the interplay between MYC and mitochondrial intermediary metabolism, could contribute to the subcellular movement of these αKG-dependent dioxygenases. To examine this hypothesis, we considered O-GlcNAcylation as a prime candidate because this post-translational modification is nutrient responsive and has been shown to influence sub-cellular localization of proteins21. First, we showed that inducing MYC expression progressively increased the deposition of O-GlcNAc moieties to proteins (Figure 5A) – see densitometric quantification below the WB panels. Of note, MYC did not significantly influence the expression of enzymes that control the cycling of this mono-glycosylation; it did not modify the expression levels of O-GlcNAcase (OGA) and had only a small effect on O-GlcNAc transferase (OGT) (Figure 5A), as reported earlier38. Indeed, the induction of OGT expression appeared disproportionally modest when compared to the marked increase in O-GlcNAcylation, suggesting that MYC utilizes other mediators to control the deposition of O-GlcNAc in target proteins. To address this possibility, we tested the impact of IDH2, D2HGDH and L2HGDH on MYC-mediated O-GlcNAcylation. Genetic deletion of any of these three mitochondrial enzymes blunted the positive influence of MYC on O-GlcNAcylation (Figure 5B, Supplementary Figure S3A). Notably, the modest increase in OGT levels associated with MYC expression was detected equally in all models but it was not sufficient to overcome the negative consequences that deleting IDH2 or D2HGDH and L2HGDH had on O-GlcNAcylation (Figure 5B). These data raised the possibility that MYC-driven elevation of αKG, secondary to the transcription activation of IDH2, D2HGDH and L2HGDH (Figure 2A and5), could be associated with the increase in O-GlcNAcylation. To test this idea, we supplemented αKG to IDH2 or D2HGDH and/or L2HGDH KO cells and were able to rescue their defective O-GlcNAcylation (Figure 5C, Supplementary Figure 3A) - densitometric quantification is shown below the WB panels 5B and 5C. Importantly, neither IDH2, D2HGDH/L2HGDH KO, nor αKG supplementation, modified OGT and OGA expression as to explain their impact on O-GlcNAcylation (Supplementary Figure S3B). UDP(uridine diphosphate)-GlcNAc, the donor substrate for O-GlcNAcylation, is the final product of the nutrient sensing hexosamine biosynthetic pathway (HBP)39. Two enzymes control the first and rate-limiting step in the HBP, glutamine-fructose-6-phosphate transaminase 1 and 2 (GFPT1 and GFPT2)39. Recently, in KRAS/LKB1 dependent lung cancer models, GFPT2 was found to be abnormally expressed and to drive heightened O-GlcNAcylation40. For this reason, we tested whether increased GFPT1 or GFPT2 expression could contribute to our findings. GFPT1 was the only isoform expressed in P493-6 cells, and its expression was not meaningfully modified by MYC expression, IDH2 KO, D2HGDH/L2HGDH KO, or by exposure to αKG. Thus, modulation of GFPT1 and GFPT2 is not responsible for the MYC-IDH2-αKG-mediated increase in O-GlcNAcylation (Supplementary Figure S3C).
Figure 5. O-GlcNAcylation and the modulation of TET enzymes and RNA demethylases FTO and ALKBH5.

A) WB of global O-GlcNAc in P493-6 cells with MYC OFF and ON (various time points post tetracycline withdrawal). Also shown are MYC, OGT and OGA expression. B) WB of global O-GlcNAc in WT P493-6 cells, IDH2 KO or D2HGDH/L2HGDH double KO all with MYC OFF and ON (24h post tetracycline withdrawal). Also show are WBs for MYC and OGT. The WB-based confirmation of the IDH2 and D2HGDH/L2HDH KO is shown in figure 2. C) WB of global O-GlcNAc in WT P493-6 cells, IDH2 KO or D2HGDH/L2HGDH double KO; all with MYC OFF or MYC ON and KO cells with MYC ON supplemented with dimethyl αKG (5mM for 24h post tetracycline withdrawal). D) WBs of TET1-3, FTO and ALKBH5 in subcellular fractions and WCL of P493-6 parental cells (top) or HEK-293T parental (bottom), exposed to DMSO control (ctrl), OSMI-1 (OGTi, 20μM for 16h) or Thiamet-G (5μM for 16h). Purity of the fractions is confirmed by Lamin A and Tubulin expression. WB quantification of global O-GlcNAc, OGT and OGA are shown to the right. E) WBs of TET1, TET3, FTO and ALKBH5 in nuclear fractions of WT, IDH2 KO or D2HGDH/L2HGDH double KO P493-6 cells all with MYC OFF, MYC ON or MYC ON with supplementation of Thiamet-G (OGAi 5μM for 24h post tetracycline withdrawal). WB of global O-GlcNAc, quantification of 5hmC and m6A levels are shown in the bottom panels; p values for are from two-tailed Student’s t test (n=3). Densitometric measurements of protein levels are shown at the bottom of the WB displays. All values are relative to controls and already corrected by WCL abundance, which is shown for reference. All assays were subjected to multiple biological replicates.
We reasoned that the positive effects of MYC and αKG on O-GlcNAcylation may be mechanistically linked to the modulation of the subcellular localization of TET-1-2-3 and FTO/ALKBH5 (Figure 4, and 5) by the MYC-IDH2-D2/L2HGDH-αKG axis. To test this proposition, we exposed lymphoid and epithelial cell models to OGT and OGA inhibitors (OSMI-1 and Thiamet-G, respectively), isolated nuclear and cytoplasmic fractions, and determined the effects of global O-GlcNAcylation on the subcellular localization of DNA hydroxylases and RNA demethylases. We found that pharmacological modulation of O-GlcNAc levels markedly influenced the subcellular compartment in which these enzymes were located. In brief, TET1, TET3, FTO and ALKBH5 were enriched or depleted in the nucleus when O-GlcNAcylation was elevated or suppressed, respectively, in most instances in association with corresponding change in cytoplasmic levels (Figure 5D). This observation recapitulated the effects of MYC expression and αKG exposure in promoting both O-GlcNAcylation (Figure 5A–B) and nuclear accumulation of these proteins (Figure 4 and5). However, remarkably, TET2 subcellular movement occurred in the opposite direction, with increase and decrease in O-GlcNAcylation resulting in the nuclear depletion and accumulation, respectively, of this enzyme (Figure 5D) - densitometric quantification is shown below the WB panels 5B and 5C. We also verified if modulation of global O-GlcNAcylation levels modified the expression or sub-cellular localization of OGT and OGA. In agreement with earlier evidence41, 42, we detected a marked increase or decrease in OGT levels in cells with low or high O-GlcNAcylation, respectively (Figure 5D, Supplementary Figure 4). OGA levels were also modulated, in the opposite direction, with decrease or increase in cells with low or high O-GlcNAcylation levels, respectively (Figure 5D, Supplementary Figure 4). Notably, the changes in OGA levels were cell specific and consistently detected in HEK-293T but not in the B lymphoid P493-6 model. Lastly, the degree of global O-GlcNAcylation did not alter the subcellular localization of OGT and OGA – the former was found in both nuclear and cytosolic fractions, whereas OGA was primarily detected in the cytosol41, 42 (Supplementary Figure 4).
We also attempted to rescue the phenotype of IDH2 and D2HGDH/L2HGDH KO by increasing O-GlcNAcylation with the OGA inhibitor Thiamet-G. In these assays, we found that the defective MYC-driven nuclear accumulation of TET1, TET3, FTO and ALKBH5 could be restored with exposure to Thiamet-G and associated increase in O-GlcNAcylation (Figure 5E). Importantly, these changes were functionally relevant as exposure to Thiamet-G rescued the defective MYC-driven increase and decrease in 5hmC and m6A marks, respectively, in the KO cells (Figure 5E). We concluded that MYC, at least in part via modulation of IDH2 and D2HGDH/L2HGDH expression and activity, and in an αKG-dependent manner, modulates O-GlcNAcylation of cellular proteins. Moreover, pharmacological disturbance of this post-translation modification directly impacts the sub-cellular localization of DNA and RNA demethylases and it can correct the effects of a defective intermediary metabolism.
Discussion:
In this work, we showed that MYC binds to the IDH2 promoter, transcriptionally activates it, and increases the cellular levels of αKG. We also demonstrated that downstream to the MYC-IDH2-αKG interplay, DNA and RNA are demethylated in association with an increase in activity and nuclear accumulation of the TET DNA hydroxylases and the RNA demethylases FTO and ALKBH5. The increased activity of these enzymes can be ascribed to the elevation of the rate limiting co-substrate αKG secondary to IDH2 transcriptional induction. However, the mediators of the nuclear enrichment of TETs and FTO/ALKBH5 that followed MYC induction and αKG accumulation were less obvious. We discovered that MYC and αKG increased O-GlcNAcylation and that this ubiquitous post-translational modification could drive the subcellular movement of DNA hydroxylases and RNA demethylases.
The contribution of MYC to metabolism in normal and neoplastic tissues is extensive43, 44. The data presented here add a new facet to “MYC’s metabolic toolkit”, the transcriptional targeting of IDH2 and attendant contribution to the cellular pool of αKG. Notably, MYC did not target the cytoplasmic IDH1, which suggests a more dominant role for MYC in coordinating mitochondrial energy metabolism; the relationship between MYC and the evolutionary distinct NAD-dependent IDH3 remains to be investigated. We recently reported that MYC transcriptionally activates D2HGDH and L2HGDH, mitochondrial dehydrogenases which perform the essential oxidation of the toxic metabolite 2-HG into αKG, and showed that that interplay also influenced the epigenome and epitranscriptome5. Herein, in multiple assays, we utilized a double D2HGDH and L2HGDH KO model, which thoroughly validated the earlier data in single D2HGDH or L2HGDH KO cells. In particular, these data reinforced the concept that 2-HG oxidation can significantly influence the αKG pool, perhaps in part because a variety of environmental factors (e.g., hypoxia and acidosis) can promote the promiscuous generation and abnormally elevate the levels of the precursor metabolite 2-HG6, 7. Thus, MYC controls at least three cellular systems that produce αKG: reactions that promote 2-HG oxidation5, the anaplerotic events derived from glutaminolysis44, and, as reported here, transcriptional activation of IDH2. We suggest that MYC is a master node for the generation of mitochondrial αKG.
There is ample recognition that mitochondria function as a signaling organelles2, 45 and that part of these activities include epigenetic remodeling46. We added to this knowledge by showing that MYC, in a IDH2-αKG dependent manner activate TET DNA hydroxylases and RNA demethylases and promote remodeling of the epigenome and epitranscriptome. Remarkably, we discovered that these effects were not exclusively associated with the actual or relative availability of αKG as a co-substrate to these enzymes, but that it also involved modulation of their subcellular localization, namely a movement from the cytosol to the nucleus, as we recently reported in the context of D2HGDH and L2HGDH expression and activity5. In this previous report, we demonstrated that the effects occur rapidly following exposure to ectopic αKG and that they were not accompanied of changes in the total levels of these enzymes, suggesting that a covalent post-translational modification may participate in this process5.
Here, we found that MYC and αKG increased O-GlcNAcylation and showed that this post-translational modification could independently drive the subcellular movement of DNA hydroxylases and RNA demethylases. We focused on this ubiquitous, non-canonical, mono-glycosylation as a putative mechanistic link between MYC/αKG and sub-cellular movement of TETs and FTO/ALKBH5 for two main reasons: 1) UDP-GlcNAc, the donor substrate for O-GlcNAcylation, is the final product of the nutrient sensing HBP, which integrates glucose, glutamine, amino acid, fatty acid and nucleotide metabolism, much of which is also positive modulated by MYC activity21, 39, 44; 2) O-GlcNAcylation has been shown earlier to influence subcellular compartmentalization of proteins21, including that of TET347. Further, TET1, 2 and 3 have been identified as binding partners of OGT48–50. In the latter context, the binding appears to primarily bring OGT to chromatin, promote O-GlcNAcylation of histone 2B’s serine 112 and subsequently activate transcription, although the role of OGT in the O-GlcNAcylation of TET proteins and on modulation of their stability has not been excluded21, 51. In addition, O-GlcNAcylation has been linked to the control of subcellular movement and activation of the NF-κB pathway52, 53. Gratifyingly, the focus on this post-translational modification was rewarding and we added several novel concepts at the interface of intermediate metabolism, O-GlcNAcylation and subcellular localization of αKG-dependent dioxygenases: First, in a B lymphocyte model, we showed that MYC expression is a major positive regulator of global O-GlcNAcylation. Although it is tempting to link this enhanced global O-GlcNAcylation to a putative MYC-driven higher flux through the HBP, recent evidence suggest that nutrient availability and O-GlcNAcylation are not always directly correlated, but instead O-GlcNAcylation levels vary according to the overall metabolic needs of the cells54. Perhaps in agreement with this line of thought, we found that MYC positive effects on O-GlcNAcylation are at least in part dependent on the expression of the mitochondrial dehydrogenases IDH2, D2HGDH, and L2HGDH, and appear independent of GFPT1. Second, in agreement with the role of IDH2, D2HGDH and L2HGDH in generating αKG, synthetic cell permeable αKG rescued their deletion and, in the context of MYC re-expression, significantly increased O-GlcNAc deposition. The role played by αKG (and IDH2 and D2-L2HGDH) in this process is intriguing, and surely in future work it will be important to test the possibility that OGT activity is stimulated by αKG. Third, we demonstrated that TET enzymes are differentially regulated by O-GlcNAcylation, with TET1 and TET3 accumulating in the nucleus and TET2 in the cytosol upon OGA inhibition (increase in O-GlcNAcylation). This observation is notable because in an earlier report of ectopic transient expression of TET1, TET2, and TET3 in HEK-293T cells, only TET3 was influenced by O-GlcNAcylation, which in that instance, contrary to our data, was shown to promote its nuclear export (29). The reasons for this difference are unclear and the cell model is less likely to account for it since we showed that both in lymphoid (P493-6) and epithelial (HEK-293T) models, increase in O-GlcNAcylation retains endogenous TET3 in the nucleus. Thus, we are tempted to ascribe this distinction to the ectopic overexpression used earlier (29). A discrepancy in data related to the interaction of OGT/O-GlcNAcylation with TET has been noted before, i.e., the relative roles of TET1, TET2, and TET3 in modulating OGT48–50, and experimental conditions, in particular ectopic overexpression may have been the reason for some of these differences55. An interesting aspect of our data relates to the distinct effect of O-GlcNAcylation on TET2 vs. TET1/TET3. While the reasons for this difference are not immediately clear, it implies that the TET1 and TET3 may be the main mediators of 5hmC accumulation downstream of the putative MYC/O-GlcNAc interplay. In addition, these data suggest that the accumulation of TETs in the nucleus following MYC induction/αKG supplementation is not simply because these enzymes associate to OGT and chromatin, otherwise TET2 would also be enriched in the nucleus in condition of increased O-GlcNAcylation. Fourth, we showed for that O-GlcNAcylation promoted and FTO and ALKBH5 nuclear localization, suggesting that akin to the association between this mono-glycosylation and epigenetic programs mediated by histone modification and DNA methylation21, deposition of O-GlcNAc may also influence the epitranscriptome. Examination of a putative binding of OGT and/or OGA with FTO and/or ALKBH5 is an important step for future investigation. Likewise, it will important to define if, beyond TETs and FTO/ALKBH5 examined here, O-GlcNAcylation also influences the subcellular localization of other αKG-dependent enzymes. Lastly, we confirmed earlier suggestions that changes if global O-GlcNAc levels can influence the expression of OGT and OGA in a compensatory fashion, which possibly reflects an attempt to restore O-GlcNAcylation homeostasis41, 42.
We are cognizant that this study also has limitations. It is possible that exogenous cell permeable αKG increases O-GlcNAcylation in manner that is independent of the pathway modified by MYC, even though it “rescued” the phenotype of cells lacking dehydrogenases that generate αKG. In addition, the link between MYC/IDH2/αKG, O-GlcNAcylation and nuclear localization of DNA and RNA demethylases remains correlative. The identification and mutation of the serine and threonine residues which are targeted for OGT-mediated mono-glycosylation is necessary to establish a causal link. Also, we primarily modulated global O-GlcNAcylation and, thus, the accompanying changes in the subcellular localization of TETs and FTO/ALKBH5 may be related to the O-GlcNAc deposition or removal in proteins that control nuclear/cytoplasmic shuttling. We find this possibility less likely due to the known direct O-GlcNAcylation of TET enzymes, and the movement in the opposite direction of TET2 vs. TET1 and TET3. Finally, as noted above, two burning questions raised by these data are still to be answered: Is OGT an αKG-dependent enzyme? Does OGT bind to FTO and ALKBH5 and influence their stability/function and the epitranscriptome at large.
In summary, we showed that MYC induces IDH2 transcription, and that this novel interplay, in an αKG-dependent manner, modulates the activity and promotes the nuclear accumulation of DNA and RNA demethylases. We demonstrated that the MYC-αKG axis positively influences global O-GlcNAcylation and correlated it to the subcellular movement of TETs and FTO/ALKBH5. Finally, we showed that pharmacological perturbation of OGT and OGA can be used to control the sub-compartment in which the αKG-dependent TET DNA hydroxylase and RNA demethylases primarily reside, and global 5hmC and m6A levels.
Supplementary Material
Acknowledgments:
This work was funded by grants from the National Institute of Health (R01-ES031522 and R01-GM140456), the Cancer Prevention and Research Institute of Texas (CPRIT - RP190043) and the Veterans Administration (I01BX001882), all to RCTA. We thank Jamie Myers for technical help.
Footnotes
Competing interests: The authors declare that they have no competing interests.
References:
- 1.Legendre F, MacLean A, Appanna VP, Appanna VD. Biochemical pathways to alpha-ketoglutarate, a multi-faceted metabolite. World J Microbiol Biotechnol 2020. Jul 20; 36(8): 123. [DOI] [PubMed] [Google Scholar]
- 2.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Science advances 2016. May; 2(5): e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin AP, Abbas S, Kim SW, Ortega M, Bouamar H, Escobedo Y, et al. D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nature communications 2015; 6: 7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ye D, Guan KL, Xiong Y. Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends in cancer 2018. Feb; 4(2): 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiu Z, Lin AP, Jiang S, Elkashef SM, Myers J, Srikantan S, et al. MYC Regulation of D2HGDH and L2HGDH Influences the Epigenome and Epitranscriptome. Cell Chem Biol 2020. May 21; 27(5): 538–550 e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell metabolism 2015. Aug 4; 22(2): 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell metabolism 2015. Aug 4; 22(2): 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S, He L, Yao K. The Antioxidative Function of Alpha-Ketoglutarate and Its Applications. Biomed Res Int 2018; 2018: 3408467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Losman JA, Koivunen P, Kaelin WG Jr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat Rev Cancer 2020. Dec; 20(12): 710–726. [DOI] [PubMed] [Google Scholar]
- 10.Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014. Jun 19; 510(7505): 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asadi Shahmirzadi A, Edgar D, Liao CY, Hsu YM, Lucanic M, Asadi Shahmirzadi A, et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell metabolism 2020. Sep 1; 32(3): 447–456 e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clinical cancer research : an official journal of the American Association for Cancer Research 2012. Oct 15; 18(20): 5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elkashef SM, Lin AP, Myers J, Sill H, Jiang D, Dahia PLM, et al. IDH Mutation, Competitive Inhibition of FTO, and RNA Methylation. Cancer cell 2017. May 08; 31(5): 619–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abla H, Sollazzo M, Gasparre G, Iommarini L, Porcelli AM. The multifaceted contribution of alpha-ketoglutarate to tumor progression: An opportunity to exploit? Seminars in cell & developmental biology 2020. Feb; 98: 26–33. [DOI] [PubMed] [Google Scholar]
- 15.Savoia M, Cencioni C, Mori M, Atlante S, Zaccagnini G, Devanna P, et al. P300/CBP-associated factor regulates transcription and function of isocitrate dehydrogenase 2 during muscle differentiation. Faseb J 2019. Mar; 33(3): 4107–4123. [DOI] [PubMed] [Google Scholar]
- 16.Yang X, Du T, Wang X, Zhang Y, Hu W, Du X, et al. IDH1, a CHOP and C/EBPbeta-responsive gene under ER stress, sensitizes human melanoma cells to hypoxia-induced apoptosis. Cancer Lett 2015. Sep 1; 365(2): 201–210. [DOI] [PubMed] [Google Scholar]
- 17.Shechter I, Dai P, Huo L, Guan G. IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells: evidence that IDH1 may regulate lipogenesis in hepatic cells. J Lipid Res 2003. Nov; 44(11): 2169–2180. [DOI] [PubMed] [Google Scholar]
- 18.Butler MJ, Aguiar RCT. Biology Informs Treatment Choices in Diffuse Large B Cell Lymphoma. Trends in cancer 2017. Dec; 3(12): 871–882. [DOI] [PubMed] [Google Scholar]
- 19.Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB, Shaw R, et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell systems 2018. Mar 28; 6(3): 282–300 e282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer discovery 2015. Oct; 5(10): 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 2017. Jul; 18(7): 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, et al. Control of cell growth by c-Myc in the absence of cell division. Current biology : CB 1999. Nov 4; 9(21): 1255–1258. [DOI] [PubMed] [Google Scholar]
- 23.Sasi B, Ethiraj P, Myers J, Lin AP, Jiang S, Qiu Z, et al. Regulation of PD-L1 expression is a novel facet of cyclic-AMP-mediated immunosuppression. Leukemia 2021. Jul; 35(7): 1990–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SW, Rai D, McKeller MR, Aguiar RC. Rational combined targeting of phosphodiesterase 4B and SYK in DLBCL. Blood 2009. Jun 11; 113(24): 6153–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suhasini AN, Wang L, Holder KN, Lin AP, Bhatnagar H, Kim SW, et al. A phosphodiesterase 4B-dependent interplay between tumor cells and the microenvironment regulates angiogenesis in B-cell lymphoma. Leukemia 2016. Mar; 30(3): 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouamar H, Jiang D, Wang L, Lin AP, Ortega M, Aguiar RC. MicroRNA 155 Control of p53 Activity Is Context Dependent and Mediated by Aicda and Socs1. Mol Cell Biol 2015. Apr 15; 35(8): 1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu Z, Holder KN, Lin AP, Myers J, Jiang S, Gorena KM, et al. Generation and characterization of the Emicro-Irf8 mouse model. Cancer Genet 2020. Jul; 245: 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang D, Aguiar RC. MicroRNA-155 controls RB phosphorylation in normal and malignant B lymphocytes via the noncanonical TGF-beta1/SMAD5 signaling module. Blood 2014. Jan 2; 123(1): 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ortega M, Bhatnagar H, Lin AP, Wang L, Aster JC, Sill H, et al. A microRNA-mediated regulatory loop modulates NOTCH and MYC oncogenic signals in B- and T-cell malignancies. Leukemia 2015. Apr; 29(4): 968–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung I, Aguiar RC. MicroRNA-155 expression and outcome in diffuse large B-cell lymphoma. Br J Haematol 2009. Jan; 144(1): 138–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proceedings of the National Academy of Sciences of the United States of America 2009. Mar 3; 106(9): 3384–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 2014. Aug; 11(8): 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nature biotechnology 2014. Sep; 32(9): 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SW, Rai D, Aguiar RC. Gene set enrichment analysis unveils the mechanism for the phosphodiesterase 4B control of glucocorticoid response in B-cell lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2011. Nov 1; 17(21): 6723–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cooney JD, Lin AP, Jiang D, Wang L, Suhasini AN, Myers J, et al. Synergistic Targeting of the Regulatory and Catalytic Subunits of PI3Kdelta in Mature B-cell Malignancies. Clinical cancer research : an official journal of the American Association for Cancer Research 2018. Mar 1; 24(5): 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelly K, Mejia A, Suhasini AN, Lin AP, Kuhn J, Karnad AB, et al. Safety and Pharmacodynamics of the PDE4 Inhibitor Roflumilast in Advanced B-cell Malignancies. Clinical cancer research : an official journal of the American Association for Cancer Research 2017. Mar 01; 23(5): 1186–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouamar H, Abbas S, Lin AP, Wang L, Jiang D, Holder KN, et al. A capture-sequencing strategy identifies IRF8, EBF1, and APRIL as novel IGH fusion partners in B-cell lymphoma. Blood 2013. Aug 1; 122(5): 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sodi VL, Khaku S, Krutilina R, Schwab LP, Vocadlo DJ, Seagroves TN, et al. mTOR/MYC Axis Regulates O-GlcNAc Transferase Expression and O-GlcNAcylation in Breast Cancer. Mol Cancer Res 2015. May; 13(5): 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiaradonna F, Ricciardiello F, Palorini R. The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 2018. Jun 2; 7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Lee HM, Cai F, Ko B, Yang C, Lieu EL, et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat Metab 2020. Dec; 2(12): 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagel AK, Ball LE. O-GlcNAc transferase and O-GlcNAcase: achieving target substrate specificity. Amino Acids 2014. Oct; 46(10): 2305–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ong Q, Han W, Yang X. O-GlcNAc as an Integrator of Signaling Pathways. Front Endocrinol (Lausanne) 2018; 9: 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goetzman ES, Prochownik EV. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front Endocrinol (Lausanne) 2018; 9: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsieh AL, Walton ZE, Altman BJ, Stine ZE, Dang CV. MYC and metabolism on the path to cancer. Seminars in cell & developmental biology 2015. Jul; 43: 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chakrabarty RP, Chandel NS. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell stem cell 2021. Mar 4; 28(3): 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santos JH. Mitochondria signaling to the epigenome: A novel role for an old organelle. Free radical biology & medicine 2021. Jul; 170: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Q, Liu X, Gao W, Li P, Hou J, Li J, et al. Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked beta-N-acetylglucosamine transferase (OGT). The Journal of biological chemistry 2014. Feb 28; 289(9): 5986–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Molecular cell 2013. Feb 21; 49(4): 645–656. [DOI] [PubMed] [Google Scholar]
- 49.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2013. Jan 24; 493(7433): 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. The EMBO journal 2013. Mar 6; 32(5): 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends in genetics : TIG 2014. Oct; 30(10): 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. Activation of the transcriptional function of the NF-kappaB protein c-Rel by O-GlcNAc glycosylation. Sci Signal 2013. Aug 27; 6(290): ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Golks A, Tran TT, Goetschy JF, Guerini D. Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. The EMBO journal 2007. Oct 17; 26(20): 4368–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab 2012. Aug 8; 16(2): 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balasubramani A, Rao A. O-GlcNAcylation and 5-methylcytosine oxidation: an unexpected association between OGT and TETs. Molecular cell 2013. Feb 21; 49(4): 618–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.