

Abstract
Colon cancer is initiated under inflammatory conditions associated with upregulation of immune checkpoint proteins. We evaluated immune modulation induced by non-steroidal anti-inflammatory agents used for colon cancer prevention. Both celecoxib and naproxen inhibited polyp growth in APC Min mice. Treatment of mice with either drug significantly decreased PD-L1 expression on polyps in a dose dependent manner (p<0.0001 for both). The decrease in PD-L1 was associated with an influx of CD8+ T-cells into polyps (p<0.0001, celecoxib; p=0.048, naproxen) compared to lesions from untreated animals and correlated with disease control. Naproxen is a nonselective inhibitor of both COX-1 and COX-2, and we questioned the role of the different cyclooxygenases in PD-L1 regulation. Silencing either COX-2 or COX-1 RNA in the murine colon cancer cell line MC38, reduced PD-L1 expression by 86% in COX-2-silenced cells (p<0.0001) while there was little effect with COX-1 siRNA compared to control. Naproxen could inhibit the growth of MC38 in vivo. Naproxen treated mice demonstrated a significant reduction in MC38 growth as compared to control (p<0001). Both Tbet+ CD4 and CD8 tumor infiltrating T-cells (TIL) were significantly increased (p=0.04 and p=0.038 respectively) without a concurrent increase in GATA3+ TIL (p>0.05). CD8+ TIL highly expressed the activation marker, CD69. Not only was PD-L1 expression decreased on tumors, but LAG3+CD8+ T-cells and PD-1 and LAG3 expression on T-regulatory cells was also reduced (p=0.008 and p=0.002 respectively). These data demonstrate COX-2 inhibitors significantly decrease PD-L1 in colonic lesions and favorably impact the phenotype of tumor infiltrating lymphocytes to control tumor growth.
Keywords: COX-2, PD-L1, T-cell, polyps, colon cancer, naproxen, NSAID
INTRODUCTION
Colon cancer offers an ideal model for cancer chemoprevention. A variety of agents that have shown some level of activity in the prevention of progression of high-risk lesions to invasive cancer in clinical and pre-clinical models(1). Combination chemoprevention approaches can be synergistic in the ability to prevent colon cancer growth (2, 3). We are interested in developing vaccines designed to intercept and prevent high-risk colon lesions (4). Non-steroidal anti-inflammatory drugs (NSAIDS), such as celecoxib and naproxen, have already demonstrated some benefit in colorectal cancer prevention with toxicity profiles that would be amenable to combination with antigen specific vaccines (5). COX-2 inhibitors have been shown to decrease the expression of PD-L1 on cells of the innate immune system such as neutrophils, macrophage, and myeloid derived suppressor cells, all of which can inhibit T-cell cytolytic function (6, 7). In preparation for developing combination chemoprevention with NSAIDs and vaccines, we questioned to what extent NSAIDS, as single agents, alter the adaptive immune microenvironment in intestinal polyps and colon cancer in murine models.
MATERIALS AND METHODS
Animal models.
Work was performed in accordance with the University of Washington Animal Care and Use Committee guidelines in a Specific Pathogen Free environment. Animals were purchased from Jackson Laboratory. APC Min [Strain name: C57BL/6J-ApcMin/J] male mice and AKR/J female mice were bred to produce “F1 Min”. Offspring from breeder pairs were genotyped by PCR for the Min mutation using primers: Wild-Type: 5’-GCCATCCCTTCACGTTAG-3’, Common: 5’-TTCCACTTTGGCATAAGGC-3’, Mutant: 5’-TCCTGAGAAAGACAGAAGTTA-3’ (8). Both male and female F1 Min mice were included in the study. Female C57BL/6 mice were used for MC38 (cell line derived from a spontaneous azoxymethane induced colon cancer) implant studies.
Study design.
Animals were randomized into treatment groups sequentially at 6±2 weeks of age. Studies were terminated when the F1 Min mice were 7 months old and when the MC38-implanted tumor volume of the naproxen-treated group was statistically significantly less than the control for a minimum of two measurements. With five F1 Min mice/group, we calculated an 83% power to detect a significant pairwise difference in the number of small intestinal polyps at the two-sided alpha level of 0.05. Three F1 Min mice died before study termination and were excluded (Fig. 1). With 10 C57BL/6/group implanted with MC38 cells, we estimated an 80% power to detect a significant pairwise difference in tumor size at the two-sided alpha level of 0.05. In vitro data was generated from a minimum of three independent experiments. All data points for these experiments are included in the analysis.
Figure 1.
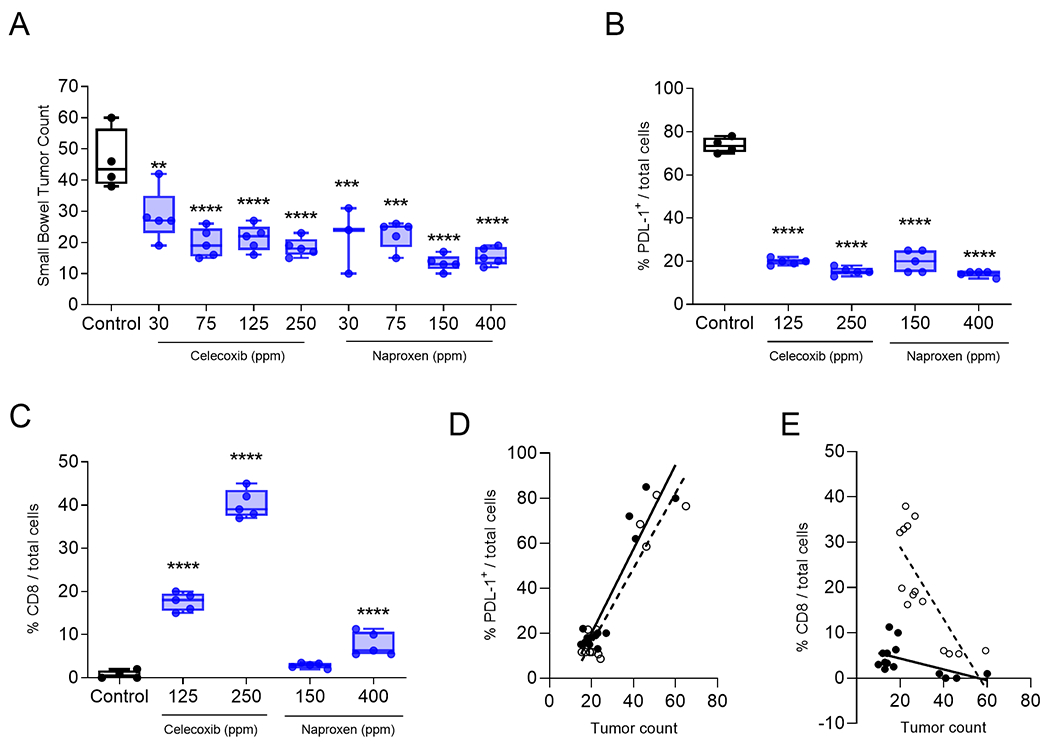
Treatment with either naproxen or celecoxib significantly inhibited the development of intestinal tumors, decreased tumor PD-L1 expression, and increased infiltrating CD8+ T cells. (A) The number of small bowel tumors after treatment with the indicated dose (parts per million, ppm) of celecoxib or naproxen. Percent (B) PD-L1 or (C) CD8+ T-cells per total cells after treatment with normal chow (control) or chow containing the indicated dose of celecoxib or naproxen. All data are presented as box and whisker plots, horizontal line at median and whiskers minimum to maximum, showing all points. Linear regression of percent (D) PD-L1 or (E) CD8 and tumor count for mice untreated or treated with celecoxib (open circles and dotted line) or naproxen (closed circles and solid line). **p<0.01, ***p<0.001, ****p<0.0001. n=3-5 mice/group.
NSAID administration and assessment of tumor growth.
Celecoxib was provided at 30, 75, 125 or 250 ppm and naproxen at 30, 75, 150 or 400 ppm daily, mixed with meal-form, irradiated chow (PicoLab Rodent Diet 20). Samples were confirmed to have ≥94% of the intended concentration of compound by high pressure liquid chromatography. The small intestine was cut longitudinally and tissues fixed in formalin. Tumors were counted under a Nikon SMZ645 microscope by the same operator. Data are expressed as the total number of tumors in the small intestine.
For tumor challenge, the syngeneic murine colon cancer tumor cell line, MC38 (0.5 × 106 cells; RRID:CVCL_B288; kindly provided by Dr. David Threadgill and validated by IDEXX testing) was implanted into the flank. On the same day as implant, mice were provided 400 ppm naproxen mixed with normal chow. Tumors were measured every two days as previously described during and after naproxen treatment (9). Data are expressed as mean tumor volume ±SEM.
Immunohistochemistry.
Tumors removed from the intestine were covered with Tissue-Tek O.C.T Compound in a cryomold and stored at −80°C. Frozen blocks were sectioned and fixed with 75% acetone/25% methanol for 5 minutes. The slides were washed thrice with PBS and 10% goat serum was added for 1 hour at room temperature followed by an anti-mouse PD-L1 (abcam; clone: MIH6; 1/100 dilution) or anti-mouse CD8 (AbD Serotec; clone KT15; 1/200 dilution) overnight at 4°C. After washing, the slides were incubated with AlexaFluor 488 anti-rat IgG (abcam; 1:1000 dilution) for 1 hour at room temperature. Positive cells were counted in three 20X microscopic fields and expressed as the mean percent of total cells.
Cell culture and RNA silencing.
MC38, the microsatellite unstable human colon adenocarcinoma cell lines, LOVO (ATCC) and SB10 (Dr. Paraskeva, Bristol, UK), and the CpG island methylator phenotype human colon carcinoma cell, RKO, (ATCC) were used. Cells were seeded at 106 cells/well in six-well plates at 37°C for 24 hours, then treated with naproxen at 200, 400 or 1000 μM or celecoxib at 50 or 100 μM. PD-L1 expression was examined by flow cytometry at 24, 48 and 72 hours. The maximum decrease of expression in each cell line was determined to be at the naproxen 1000 μM and celecoxib 100 μM dose at 72 hours. Data are presented as percent PD-L1 expression in total viable cells. PGE2 levels were measured in the cell culture supernatant by ELISA (R&D Systems) as per the manufacturer’s directions. Results are reported as total PGE2 levels (pg/ml; Suppl. Fig. 1).
For RNA silencing, MC38 cells were seeded at 1.5 X105 cells/well in 6-well plates. After an overnight incubation, cells were transfected with a pool of four siRNA specific for COX1 or COX2 (Qiagen) at 10 nM/siRNA using Dharmfect (GE Heathcare). Maximum gene silencing occurred at 48 hours incubation. Results are reported as percent expression as compared to mock transfected cells (Suppl. Fig. 2).
Immunophenotyping.
For tumor infiltrating lymphocyte (TIL) isolation, tumors were cut into 10-mm pieces, incubated in the manufacturer’s enzyme mix from the Tumor Dissociation kit (Miltenyi Biotec) then applied to the “37°C mouse TDK1” program on the gentleMACs dissociator. After dissociation, cells were washed with DMEM/10% FBS and resuspended in flow cytometry buffer (PBS/1% FBS/2mM EDTA), applied to a 70 μm filter, washed through with flow cytometry buffer and red cells lysed. 0.5×106 viable cells from samples were blocked with anti-mouse CD16/32 (Mouse BD Fc Block, clone 2.4G2). Prior to surface staining, cells were stained with Fixable viability dye 450 (eBiosciences; 1:1000) in PBS and then washed in flow cytometry buffer. Intracellular staining was performed using the FOXP3 buffer set. Antibodies were obtained from BD Bioscience or Thermo Fisher Scientific. Receptor expression was documented in the cells or TIL by incubating with anti-mouse-CD3e (BV510; clone 145-2C11), -TIGIT (BV605; clone 1G9), -Tbet (PerCP-Cy5.5; clone 4B10), -PD-L1 (PE, clone MIH5), -CD45 (PE-Cy5; clone 30-F11), -CD4 (PE-Cy5.5; clone RM4-5), -CD69 (PE-Cy7; clone H1.2F3), -FOXP3 (AlexaFluor488; clone MF14), -LAG3 (APC; clone C9B7W), -CD8a (AlexaFluor700; clone 53-6.7), -PD-1 (APC-Cy7, clone J43), -F480 (PE-CF594; clone T45-2342), -CD11b (BV711; clone M1/70), -Ki67 (AlexaFluor488; clone B56) or the appropriate isotype control for 1 hour at 2-8°C protected from light. Analysis was performed on FACS Canto RUO and data analyzed using FlowJo software (BD Biosciences). Typically, 2×105 events were recorded from the viability gate per sample. Results are reported as a percentage of total cell number or a percentage of a specific cell population.
Statistical analysis.
The unpaired, two-tailed Student’s t-test was used to evaluate differences between two groups. To compare more than three groups, a One-way ANOVA with Tukey’s Post-Hoc test was used when there was one variable and a Two-way ANOVA with Bonferoni’s post-test was used when there were two variables. p<0.05 was considered significant. All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software).
DATA AVAILIBILITY
The data generated in this study are available within the article and its supplementary data files.
RESULTS
Treatment with either naproxen or celecoxib significantly inhibited the development of intestinal tumors, decreased tumor PD-L1 expression, and increased infiltrating CD8+ T cells.
Fewer small bowel tumors were observed in mice treated with 30 (mean, 28.6±8.3), 75 (mean, 19.8±4.6), 125 (mean, 21.4 ±1.2) or 250 ppm (mean, 4.7±0.7) celecoxib as compared to the control (mean, 46.2±9.7; p<0.01 for all; Fig. 1A). Similar results were observed when the mice were treated with naproxen. Compared to the control, fewer small bowel tumors were identified in mice treated with 30 (mean, 21.7±10.6), 75 (mean, 22.6±4.5), 150 (mean, 13.4 ±1.1) or 400 ppm (mean, 15.6±1.2) naproxen (p<0.001 for all; Fig. 1A).
We analyzed tumors from the higher doses of NSAID as these concentrations would be more consistent with the human dose of drug. Tumor cell expression of PD-L1 was reduced when mice were treated with 125 or 250 ppm of celecoxib (p<0.0001; Fig. 1B; Suppl Fig. 3). Similarly, tumor cell expression of PD-L1 was reduced when mice were treated with 150 or 400 ppm of naproxen (p<0.0001 for both doses; Fig. 1B; Suppl Fig. 3). Moreover, celecoxib treatment significantly increased tumor infiltrating CD8+ T-cells as compared to the control (p<0.0001 for both doses; Fig. 1C). Naproxen treatment also increased tumor infiltrating CD8+ T-cells as compared to the control (p<0.0001 for the 400 ppm dose; Fig. 1C). A greater number of lesions present in the small bowel was positively correlated with increased PD-L1 expression (r=0.928; p<0.0001 for celecoxib and r=0.928; p<0.0001 for naproxen; Fig. 1D) and negatively correlated with increased infiltrating CD8+ (r=−0.777, p=0.001 for celecoxib and r=−0.547, p=0.042 for naproxen; Fig. 1E).
PD-L1 expression is decreased by COX-2 but not COX-1 inhibition.
Human colon cancer cells express PD-L1 (LOVO: mean, 83±2%, RKO: mean, 93±4% and SB10: mean, 87±2%, Fig. 2A). Naproxen treatment significantly decreased PD-L1 expression on LOVO cells, by 35% (p<0.0001), RKO cells, by 28% (p<0.0001) and SB10 cells, by 16% (p<0.0001; Fig. 2A). MC38 cells also express PD-L1 (mean 44±5%; Fig. 2B). Naproxen treatment significantly reduced PD-L1 expression (mean, 24±5%; p=0.0006) as compared to untreated cells. Celecoxib treatment similarly decreased PD-L1 expression (mean, 32±3%; p=0.007; Fig. 2B) in MC38 cells. We verified that PGE2 levels were reduced by a mean of 85±2% in the naproxen-treated and a mean of 80±7% in the celecoxib-treated cells (p<0.0001 for both; Suppl Fig. 1).
Figure 2.
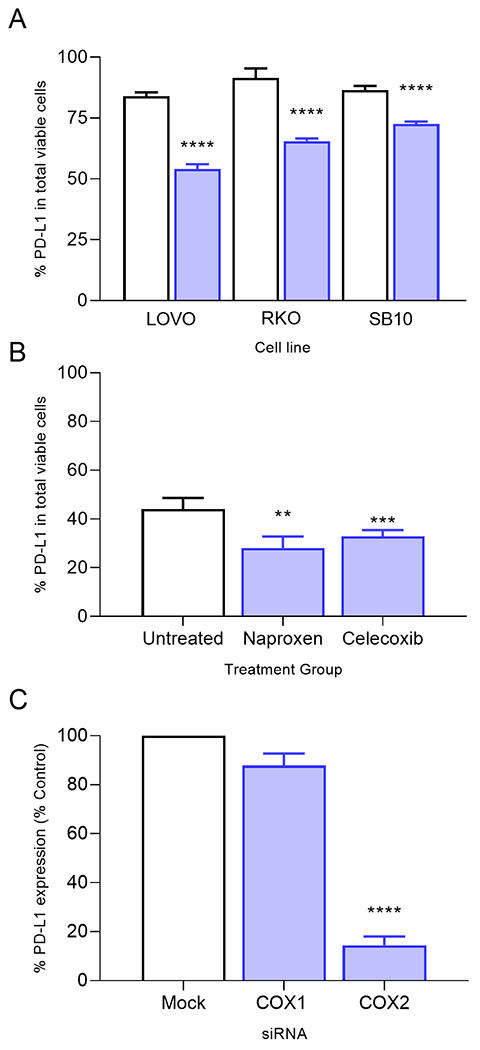
PD-L1 expression is decreased by COX-2 but not COX-1 inhibition. (A) Percent PD-L1 on viable cells on LOVO, RKO or SB10 cells untreated (white bars) or treated with naproxen, 1000 μM (blue bars). (B) Percent PD-L1 on viable cells in MC38 cells untreated (white bar) or treated with naproxen (1000 μM) or celecoxib (100 μM) (blue bars). (C) Percent PD-L1 expression relative to the mock control transfected cells after silencing of the indicated gene in MC38 cells. **p=0.01, ****p<0.0001; n=3-4 independent experiments.
We silenced expression of COX-1 by 72% (Suppl. Fig. 2A) and COX-2 by 82% with specific siRNA (Suppl. Fig. 2B) as compared to a mock siRNA transfection (p<0.0001 for both) in MC38 colon cancer cells. Silencing COX-1 did not affect PD-L1 expression (Fig. 2C). The percentage of cells expressing PD-L1 in the COX-1-silenced cells (mean, 87±5%), was similar to that observed on the mock transfected cells (p=0.07). However, PD-L1 expression was reduced by 86% in COX-2-silenced cells (p<0.0001; Fig. 2C).
Naproxen treatment inhibited the growth of MC38 in vivo and increased the influx of activated Tbet+ tumor infiltrating lymphocytes.
MC38 implanted tumors provided a uniform in vivo model to evaluate the anti-tumor effect of naproxen and the adaptive immune infiltrate associated with tumor control. Naproxen-treated mice demonstrated a significant reduction in tumor growth (mean, 180±114 mm3) as compared to mice fed normal chow (mean, 1174±125 mm3; p<0.0001; Fig. 3A). The inhibitory effect was transient, however, with disease control continuing in only 30% of mice (3/10) twenty days after cessation of NSAID treatment (Suppl Fig. 4).
Figure 3.
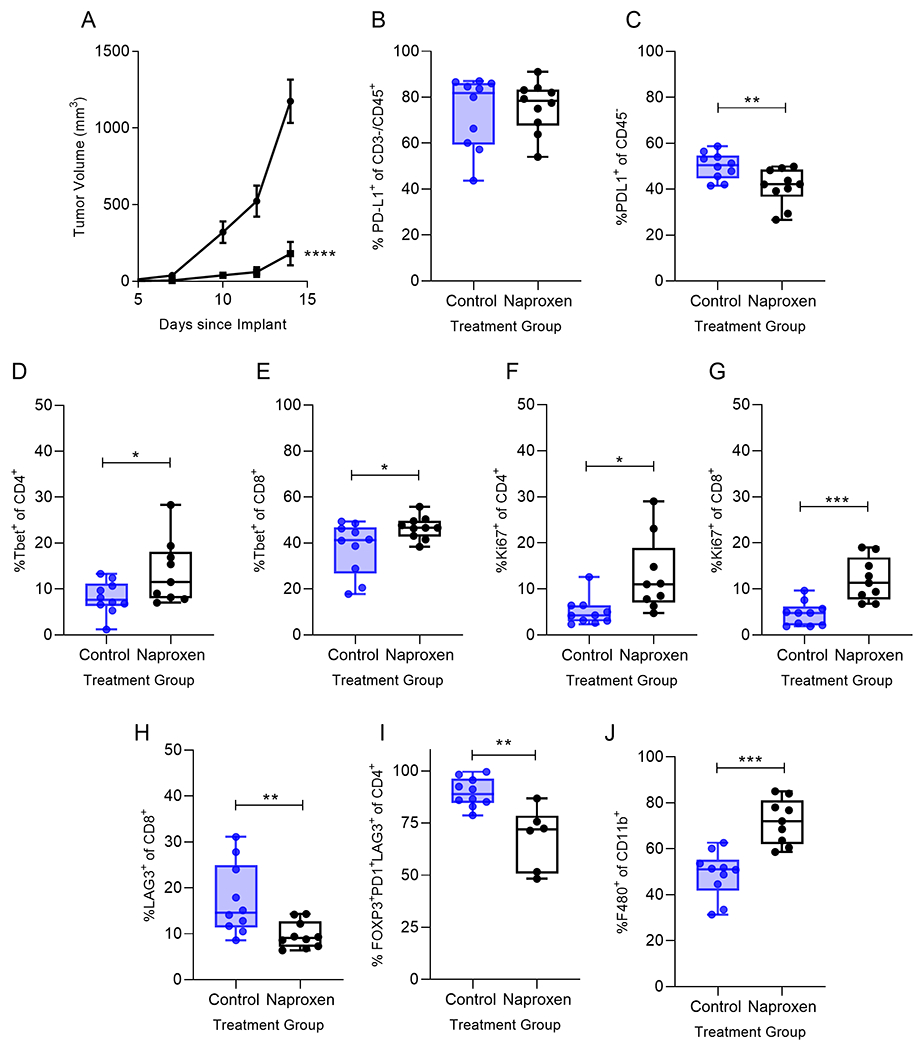
Naproxen treatment inhibited the growth of MC38 in vivo and increased the influx of activated Tbet+ tumor infiltrating lymphocytes. (A) Tumor volume of mice treated with control diet (●) or diet containing 400 ppm Naproxen (■); n=10 mice/group; ****p<0.0001. Percent of infiltrating (B) CD45+CD3-PD-L1+ (C) CD45−PD-L1+, (D) Tbet+CD4+ (E) Tbet+CD8+, (F) Ki67+CD4+, (G) Ki67+CD8+, (H) LAG3+CD8+ (I) CD4+FOXP3+PD-1+LAG3+ and (J) F480+CD11b+ for the indicated treatment group presented as box and whisker plots, horizontal line at median and whiskers minimum to maximum, showing all points. *p<0.05; **p<0.01; ***p<0.001 n=6-10 mice/group.
We harvested tumors from naproxen treated and control mice at the timepoint when tumor growth was significantly inhibited in the treatment group to evaluate tumor immune infiltrates elicited by naproxen treatment. While PD-L1 levels were not different between the two groups for CD45+CD3− cells (p=0.70; Fig. 3B), naproxen treatment significantly reduced PD-L1 expression on CD45− tumor cells compared to controls (p=0.009; Fig. 3C). TIL derived from naproxen treated mice demonstrated a significant increase in Tbet+CD4+ (p=0.04, Fig. 3D) and Tbet+CD8+ T-cells (p=0.038, Fig. 3E). The Tbet+CD8+ T-cells expressed the activation marker CD69 compared to controls (p=0.044). There was no difference between naproxen treated and control groups in CD4+GATA3+ (p=0.570) and CD8+GATA3+ (p=0.816) T-cells. Naproxen treatment induced proliferation of both CD4 (p=0.011, Fig. 3F) and CD8 T-cells (p=0.0004, Fig. 3G) in the tumor, an effect that was not observed in controls.
We observed no difference between naproxen treated and control tumor infiltrating T-cells for PD-1 expression (CD4+PD-1+, p=0.434; CD8+PD-1+, p=0.08) or expression of TIGIT (CD4+TIGIT+, p=0.415; CD8+TIGIT+, p=0.109). There were, however, significantly fewer LAG3+CD8+ T-cells infiltrating the tumors of mice treated with naproxen as compared to control (p=0.008; Fig. 3H). Treatment also significantly reduced the T-regulatory subset expressing both PD-1 and LAG3 (p=0.002; Fig. 3I). While tumor associated macrophages were significantly increased in number after naproxen treatment (p=0.001; Fig. 3J), PD-L1 expression on the macrophage was no different between NSAID treated and control groups (p=0.839).
DISCUSSION
COX-2 inhibitors, such as celecoxib and naproxen, show partial prophylactic efficacy in murine intestinal tumor models (10, 11). The mechanism by which these drugs inhibit tumor development has been attributed to blocking the synthesis of prostaglandins and COX-2 expression both of which have tumor promoting effects (12, 13). Recently, inhibition of prostaglandin E2 or COX-2 has been shown to reduce expression of PD-L1 in innate immune cells, tumor associated macrophage and myeloid derived suppressor cells, indicating an immunologic mechanism of action for NSAIDs (7). The role of COX-2 in the regulation of PD-L1 on tumor cells is less well defined. Investigations of a panel of lung cancer cell lines demonstrated that both COX-2 and PD-L1 were upregulated in malignant cells, but incubation of the cells with celecoxib did not impact the level of PD-L1 expression (14). A study of COX-2 expression in a murine model of malignant glioma showed celecoxib could moderately reduce the level of PD-L1 in tumors, but the addition of an anti-PD-1 monoclonal antibody was required to achieve significant inhibition of COX-2 and an anti-tumor effect (15). Data presented here shows that a primary effect of NSAID treatment in these intestinal tumor models may be reduction of PD-L1 expression on tumor cells rather than innate immune cells, such as macrophage. Further, NSAID could modulate PD-L1 expression in both polyps as well as colon carcinomas to significant therapeutic benefit.
NSAID treatment resulted in an increased influx of Type I T-cells into lesions as defined by CD8 T-cells and the expression of Tbet, a transcription factor required for optimal Type I T-cell function. The presence of increased CD8 T-cells in polyps was associated with disease control. Type II T-cells, defined by GATA3+ or a T-regulatory cell phenotype are associated with colon cancer initiation and progression (16). Cytokines produced by Type I T-cells, such as interferon-gamma, induce the expression of COX-2 in innate immune cells resulting in immune suppression and T-cell anergy (17). Our data suggests that NSAID treatment prevents the development of anergy as evidenced by significant levels of proliferating CD4 and CD8 T-cells in the tumors and evidence of T-cell activation. Further, despite the marked influx of Type I T-cells, there was no evidence of upregulation of PD-1 or other immune checkpoint proteins such as TIGIT. Indeed, LAG-3 an inhibitory receptor which becomes upregulated on activated T-cells was also downregulated with NSAID treatment (18).
The mechanism by which COX-2 modulates PD-L1 is not fully understood, but is most likely related to NF-kB expression. NF-kB regulates PD-L1 gene transcription through p65 binding to the PD-L1 promoter increasing protein expression during an immune response (19). COX-2 inhibitors suppress expression of NF-kB in a dose dependent fashion (19). NSAID therapy, in particular with COX-2 inhibitors, can reverse inflammation-induced immune suppression and support an effective anti-tumor response. Clinical efficacy can be seen in both intestinal polyp and colon cancer models and represents an additional mechanism of action for this class of agents.
Supplementary Material
PREVENTION RELEVANCE.
Non-steroidal anti-inflammatories (NSAIDS) are an essential component of any combination chemoprevention of colon cancer. We show NSAID treatment reduces PD-L1 expression on intestinal tumor cells. NSAID regulation of PD-L1 is dependent on COX-2 expression. These data underscore an important immunologic mechanism of action for NSAID in colon cancer prevention.
Grant support:
NCI C3085101 (ML Disis), American Cancer Society Clinical Research Professorship, CRP-15-106-01-LIB (ML Disis), Helen B. Slonaker Endowed Professor for Cancer Research (ML Disis).
Disclosures of Potential Conflicts of Interest:
ML Disis has a commercial research grant from EMD Serono, Precigen, Pfizer, and stock interest in Epithany. She is a patent holder at the University of Washington.
REFERENCES
- 1.Zilli M, Iacobelli S. Chemoprophylaxis in gastrointestinal tumors. Eur Rev Med Pharmacol Sci. 2010;14:285–91. [PubMed] [Google Scholar]
- 2.Zhou M, Zheng J, Bi J, Wu X, Lyu J, Gao K. Synergistic inhibition of colon cancer cell growth by a combination of atorvastatin and phloretin. Oncol Lett. 2018;15:1985–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida T, Horinaka M, Takara M, Tsuchihashi M, Mukai N, Wakada M, et al. Combination of isoliquiritigenin and tumor necrosis factor-related apoptosis-inducing ligand induces apoptosis in colon cancer HT29 cells. Environ Health Prev Med. 2008;13:281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corulli LR, Cecil DL, Gad E, Koehnlein M, Coveler AL, Childs JS, et al. Multi-epitope-based vaccines for colon cancer treatment and prevention. Front Immunol. 2021;12:729809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohammed A, Yarla NS, Madka V, Rao CV. Clinically relevant anti-inflammatory agents for chemoprevention of colorectal cancer: New perspectives. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng H, Kan A, Lyu N, He M, Huang X, Qiao S, et al. Tumor-derived lactate inhibit the efficacy of lenvatinib through regulating PD-L1 expression on neutrophil in hepatocellular carcinoma. J Immunother Cancer. 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U S A. 2017;114:1117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steffensen IL, Alexander J. Impact of genetic background on spontaneous or 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (phip)-induced intestinal tumorigenesis in min/+ mice. Cancer Lett. 2006;240:289–96. [DOI] [PubMed] [Google Scholar]
- 9.Gad E, Rastetter L, Slota M, Koehnlein M, Treuting PM, Dang Y, et al. Natural history of tumor growth and immune modulation in common spontaneous murine mammary tumor models. Breast Cancer Res Treat. 2014;148:501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandhi SR, Tiwari AK, Kunte DP, De la Cruz MA, Stypula Y, Gibson T, et al. Association of stem-like cells in gender-specific chemoprevention against intestinal neoplasia in min mouse. Oncol Rep. 2011;26:1127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan M, Myung SJ, Fink SP, Lawrence E, Lutterbaugh J, Yang P, et al. 15-hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proc Natl Acad Sci U S A. 2009;106:9409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gurpinar E, Grizzle WE, Piazza GA. NSAIDS inhibit tumorigenesis, but how? Clin Cancer Res. 2014;20:1104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou P, Cheng SW, Yang R, Wang B, Liu J. Combination chemoprevention: Future direction of colorectal cancer prevention. Eur J Cancer Prev. 2012;21:231–40. [DOI] [PubMed] [Google Scholar]
- 14.Shimizu K, Okita R, Saisho S, Maeda AI, Nojima Y, Nakata M. Impact of COX2 inhibitor for regulation of PD-L1 expression in non-small cell lung cancer. Anticancer Res. 2018;38:4637–44. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi I, Nakajima K, Shono K, Mizobuchi Y, Fujihara T, Shikata E, et al. Downregulation of PD-L1 via FKBP5 by celecoxib augments antitumor effects of PD-1 blockade in a malignant glioma model. Neurooncol Adv. 2020;2:vdz058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gamez-Belmonte R, Erkert L, Wirtz S, Becker C. The regulation of intestinal inflammation and cancer development by Type 2 immune responses. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong JL, Obermajer N, Odunsi K, Edwards RP, Kalinski P. Synergistic COX2 induction by IFN-gamma and TNF-alpha self-limits Type-1 immunity in the human tumor microenvironment. Cancer Immunol Res. 2016;4:303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chocarro L, Blanco E, Zuazo M, Arasanz H, Bocanegra A, Fernandez-Rubio L, et al. Understanding LAG-3 signaling. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Betzler AC, Theodoraki MN, Schuler PJ, Doscher J, Laban S, Hoffmann TK, et al. NF-kappaB and its role in checkpoint control. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.