

Abstract
Introduction:
A major focus of interstitial lung disease (ILD) has centered on disorders termed idiopathic interstitial pneumonias (IIPs) which include, among others, idiopathic pulmonary fibrosis, idiopathic nonspecific interstitial pneumonia, cryptogenic organizing pneumonia, and respiratory bronchiolitis-interstitial lung disease.
Areas Covered:
We review the radiologic and histologic patterns for the nine disorders classified by multidisciplinary approach as IIP, and describe the remarkable amount of published epidemiologic, translational, and molecular studies demonstrating their associations with numerous yet definitive environmental exposures, occupational exposures, pulmonary diseases, systemic diseases, medication toxicities, and genetic variants.
Expert Opinion:
In the 21st century, these disorders termed IIPs are rarely idiopathic, but rather are well-described radiologic and histologic patterns of lung injury that are associated with a wide array of diverse etiologies. Accordingly, the idiopathic nomenclature is misleading and confusing, and may also promote a lack of inquisitiveness, suggesting the end rather than the beginning of a thorough diagnostic process to identify ILD etiology and initiate patient-centered management. A shift towards more etiology-focused nomenclature will be beneficial to all, including patients hoping for better life quality and disease outcome, general medicine and pulmonary physicians furthering their ILD knowledge, and expert ILD clinicians and researchers who are advancing the ILD field.
Keywords: idiopathic interstitial pneumonia, interstitial lung disease, idiopathic pulmonary fibrosis, usual interstitial pneumonia, nonspecific interstitial pneumonia, organizing pneumonia
1.0. Introduction
Interstitial lung disease (ILD), or interstitial pneumonia, comprises a wide array of diffuse parenchymal lung diseases that encompasses close to one hundred disorders generally characterized by a combination of pulmonary inflammation and pulmonary fibrosis. The classification system, nomenclature, and acronyms used for ILD are complex, and are often confusing to general medicine physicians and pulmonary disease specialists alike. Part of the complexity is related to an incomplete understanding of several of the disease entities, whereas much of the complexity relates likely to the nomenclature used for multidisciplinary diagnoses in ILD. Confusion and complexity surrounding ILD nomenclature applies perhaps most particularly to the group of disorders termed the idiopathic interstitial pneumonias (IIPs).
The term idiopathic has been used for millennia, and has been defined within the medical field as either a disease which arises spontaneously, based on the Greek origin of idios, meaning from one’s own, or in more recent times, a disease that has an unknown cause or mechanism [1, 2]. Use of the idiopathic term gained prominence in pulmonary medicine in the 1970s with the disorder termed idiopathic pulmonary fibrosis (IPF) [3], and the term gained further prominence with the American Thoracic Society/European Respiratory Society classifications of IIPs in 2002 and 2013, in which a group of idiopathic disorders in addition to IPF was described [4, 5]. These classifications certainly acknowledged the secondary forms of ILD as well as their importance, but the idiopathic terminology has since become a cornerstone of the ILD community and has been in the forefront of the ILD medical literature, with thousands of publications over the last several decades related to disorders classified within the IIP spectrum of disease.
The most recent multidisciplinary clinical-radiologic-pathologic classification of the IIPs from 2013 is shown in Table 1 [5]. Almost 50 years have now passed since the first descriptions of IPF, and a remarkable amount of epidemiologic, translational, and molecular studies have accumulated revealing strong associations between these disorders termed IIPs and numerous yet definitive environmental exposures, occupational exposures, pulmonary diseases, systemic diseases, medication toxicities, infectious disorders, and sporadic or inherited genetic variants. What becomes apparent is these disorders termed IIPs are not disorders unto themselves, but rather are well-described radiologic and histologic patterns of lung injury that result from a myriad of diverse etiologies. Thus, in the 21st-century, it appears that these IIP disorders are rarely idiopathic if a comprehensive assessment of the medical, environmental, occupational, and family history is undertaken in conjunction with broad autoimmune serologic testing and selective gene sequencing.
Table 1.
Disorders classified by multidisciplinary approach as idiopathic interstitial pneumonias (IIPs) as outlined by the American Thoracic Society and European Respiratory Society in 2013*
| Idiopathic pulmonary fibrosis (IPF) |
| Idiopathic nonspecific interstitial pneumonia (idiopathic NSIP) |
| Respiratory bronchiolitis-interstitial lung disease (RB-ILD) |
| Desquamative interstitial pneumonia (DIP) |
| Cryptogenic organizing pneumonia (COP) |
| Acute interstitial pneumonia (AIP) |
| Idiopathic lymphoid interstitial pneumonia (idiopathic LIP) |
| Idiopathic pleuroparenchymal fibroelastosis (idiopathic PPFE) |
| Unclassifiable idiopathic interstitial pneumonia |
adapted from the American Thoracic Society/European Respiratory Society Statement from 2013 [5].
In this Review, we will discuss the radiologic and histologic patterns for each of the described IIPs, and will subsequently describe the numerous well-described risk-factors and associations with each of these patterns of lung injury. The term IPF is perhaps the term most in need of change [6, 7], due to its frequency of use, the severity of the disease phenotype, and by the much-often quoted statement that IPF is the most common and severe form of the IIPs. As we describe the numerous risk-factors and associations for each of the IIPs, it will become apparent that the idiopathic terminology is misleading and confusing, and we propose that a shift towards more etiology-focused nomenclature in ILD will be beneficial to all.
2.0. Disorders termed idiopathic interstitial pneumonias (IIPs)
2.1. Idiopathic nonspecific interstitial pneumonia
Nonspecific interstitial pneumonia (NSIP) came into common usage in the early 1990s in which histologic findings in a group of ILD patients that did not fulfill criteria for usual interstitial pneumonia (UIP), desquamative interstitial pneumonia, or acute interstitial pneumonia were described [8]. The histologic pattern of NSIP is characterized by geographically uniform thickening of the alveolar septae (interstitium) throughout the lung parenchyma with overall preservation of the lung architecture [8, 9, 10]. Most patients have both a cellular and fibrotic component, though some reports distinguish histologically cellular NSIP from fibrotic NSIP [9]. Following the histologic descriptions, radiologic findings which correlate to histologic NSIP have been well-described, and consist of reticular and groundglass opacities (GGO) occurring in a lower lobe-predominant distribution, with or without associated signs of fibrosis including traction bronchiectasis and lower lobe volume loss, and often with sparing of the extreme periphery of the lung [9, 10, 11].
Since its original description, numerous disorders and exposures have been associated with an NSIP pattern of injury. The most common association is with systemic autoimmune diseases, including anti-synthetase syndrome, systemic sclerosis (SSc), Sjogren’s syndrome, and rheumatoid arthritis (RA) [12, 13, 14, 15], and it is often stated that an NSIP pattern of injury is the most common pattern of lung disease in patients with autoimmune-related ILD [12, 13]. The strong association of NSIP with systemic autoimmune diseases has almost certainly been strengthened by the wide array of comprehensive autoimmune serologies now available to clinicians, including the broad spectrum of myosotis-specific and myositis-associated antibodies. Numerous other associations with histologic or radiologic NSIP have additionally been well-described, and include cigarette smoking [16, 17, 18], pulmonary drug toxicity [19, 20], pulmonary infections [21], hypersensitivity pneumonitis [22, 23, 24], inflammatory bowel disease [25, 26], common variable immunodeficiency syndrome [27, 28], familial interstitial pneumonia [29, 30, 31], graft-versus-host disease following allogeneic stem cell transplantation [32, 33], and chronic lung allograft dysfunction [34, 35]. It should be noted that although NSIP is commonly observed with numerous conditions, the underlying pathobiologic mechanisms leading to an NSIP pattern of injury are likely diverse given the diversity of the disorders and exposures.
Although many original research reports and consensus statements have supported the concept of idiopathic NSIP as a well-defined clinical entity [4, 5, 11], the numerous diseases and exposures associated with NSIP over the past two decades would now suggest otherwise, and alternatively suggest that NSIP is merely one of many nonspecific patterns of lung injury [36]. Several recent multidisciplinary discussion (MDD) reports further support this concept, indicating that idiopathic NSIP is in fact quite a rare entity, being either completely absent or present in a very small minority of patients presented for MDD [37, 38, 39].
2.2. Cryptogenic organizing pneumonia
Organizing pneumonia (OP) was described histologically at least as early as 1922 and was described as a pattern of injury secondary to pulmonary infection [40]. The entity of OP gained more widespread recognition in pulmonary medicine following several reports in the 1980s which described bronchiolitis obliterans with organizing pneumonia (BOOP) and COP [41, 42]. Histologically, OP is characterized by round- or oval-shaped basophilic staining deposits of extracellular matrix containing spindle-shaped fibroblasts or myofibroblasts [10, 43, 44]. These OP deposits may be located intraluminally within air spaces or within the interstitium following septal incorporation [45, 46]. Following the histologic descriptions, numerous reports have described the radiologic spectrum of findings in OP, which include peribronchiolar consolidation, sparing of the extreme periphery of the lung, GGO, peri-lobular pattern with “arcading”, reticulation, focal nodules, and the reversed halo (atoll) sign [10, 44, 47].
Since its original description, countless disorders and exposures have been associated with an OP pattern of injury. The most common association is with pulmonary infections, being described with numerous bacterial, viral, and fungal infections, and has not surprisingly been described with SARS-coronavirus-2 infection (Covid-19) [40, 43, 44, 48]. The association of OP with respiratory viral infection has almost certainly been strengthened by the advancement of detection methods for respiratory viral pathogens over the past two decades. Another common association of OP is with systemic autoimmune diseases, including anti-synthetase syndrome, SSc, systemic lupus erythematosus (SLE), RA, and Sjogren’s syndrome [12, 43, 44]. Numerous other associations with histologic or radiologic OP have additionally been well-described, and include cigarette smoking [49, 50], pulmonary drug toxicity [51, 52], inflammatory bowel disease [25, 26], organizing diffuse alveolar damage [53, 54], common variable immunodeficiency syndrome [27, 28], external beam radiation therapy [55, 56], graft-versus-host disease following allogeneic stem cell transplantation [57, 58], chronic lung allograft dysfunction [59, 60], hypersensitivity pneumonitis [61, 62], and familial interstitial pneumonia [29, 30]. As with NSIP, it should be noted that although OP is commonly observed with numerous conditions, the underlying pathobiologic mechanisms leading to an OP pattern of injury are likely diverse given the diversity of the disorders and exposures.
Similar to NSIP, many original research reports and consensus statements have supported the concept of idiopathic OP (COP) as a well-defined clinical entity, but the numerous diseases and exposures associated with OP over the past two decades would now suggest otherwise, and alternatively suggest that OP is merely one of many nonspecific patterns of lung injury [43, 44, 63]. Recent MDD reports similarly indicate that idiopathic OP is in fact quite a rare entity, being either completely absent or present in a very small minority of patients presented for MDD [37, 38, 39].
2.3. Respiratory bronchiolitis-interstitial lung disease and desquamative interstitial pneumonia
Respiratory bronchiolitis (RB) and desquamative interstitial pneumonia (DIP) are often listed as separate disorders [4, 5], but it is now recognized that almost certainly these two disorders represent a spectrum of the same pathobiologic process [64, 65]. Histologically, both are characterized by the accumulation of finely pigmented alveolar macrophages in the lung, often referred to as smokers’ macrophages, in which the pigmented cytoplasm appears golden-brown on H&E staining [64, 65]. In RB, the pigmented macrophages are centered around small airways and the bronchovascular bundle, whereas in DIP, the pigmented macrophages are widespread diffusely throughout the alveolar spaces and lung parenchyma [64, 65, 66]. It should be noted that the term desquamative, originally coined in 1965, was applied since the observed mononuclear cells were considered to represent desquamated epithelial cells [67], and even though the term DIP remains in use today, it is thus considered to be a misnomer.
The substances responsible for the pigmented cytoplasm of macrophages in RB/DIP are particulate matter from the incomplete combustion of inorganic and organic compounds contained in smoke [64, 65, 68, 69]. The composition of these substances consists of numerous elemental metals, carbon black, and hemosiderin [64, 69, 70, 71, 72]. The presence of excessive iron (hemosiderin) within smokers’ macrophages likely results from the high iron content of tobacco smoke [68, 71]. It should be mentioned that pathologists often distinguish finely pigmented macrophages in RB/DIP from more coarsely pigmented macrophages termed hemosiderin-laden macrophages observed with alveolar hemorrhage [64]. However, hemosiderin is present in smokers’ macrophages in RB/DIP as well, indicating this distinction may be related merely to greater accumulation of macrophage hemosiderin in the presence of alveolar hemorrhage.
The radiologic patterns of RB and DIP have been well-characterized. Both generally manifest as GGO, but RB demonstrates small groundglass nodules most often in an upper-lung zone distribution, whereas DIP generally manifests more widespread GGO in a lower-lung zone distribution [66, 67, 73]. The DIP pattern additionally may demonstrate radiologic changes of fibrosis with traction bronchiectasis and lower lobe volume loss [65, 73, 74]. Precisely speaking, the term RB refers to the histologic abnormality whereas RB-ILD refers to concurrent chest CT abnormalities, but often these two terms are used interchangeably.
The histologic pattern of DIP has rarely been attributed to etiologies other than smoke inhalation [75, 76, 77, 78]. However, in some reports patients were concurrent cigarette smokers, and in others, it is not completely clear as to whether the observed clusters of macrophages were pigmented (as DIP is defined) or non-pigmented. Clusters of non-pigmented macrophages within alveolar spaces are a nonspecific finding, being observed with bronchiolar obstruction, pulmonary infection, aspiration, dust exposure, and HP [64].
The ATS/ERS statement in 2013 did describe RB and DIP as smoking-related, but both remained included within the group of idiopathic disorders. Based on decades of observations, the patterns of RB and DIP are specifically associated with smoke inhalation, and continuing to classify them as IIPs is confusing and misleading.
2.4. Acute interstitial pneumonia
The term acute interstitial pneumonia (AIP) is used by the ILD community to imply idiopathic disease, even though the word idiopathic is not included [4, 5]. AIP is at times referred to as Hamman-Rich syndrome, referencing patients described by Hamman and Rich with a form of interstitial fibrosis that was rapidly progressive and fulminant [79]. In AIP, the lung injury is acute, in contrast to the other described IIPs which are manifested in general by subacute or chronic forms of lung injury. Histologically, AIP is characterized by diffuse alveolar damage (DAD), a histopathologic pattern identical to that seen in the acute respiratory distress syndrome (ARDS), manifested by neutrophilic inflammation, noncardiogenic edema, reactive type II pneumocytes, hyaline membranes, and areas of organizing lung injury [80, 81]. Radiologically, AIP is characterized by widespread GGO, patchy areas of consolidation, and often with changes of fibrosis (traction bronchiectasis, volume loss) as the disease progresses [82, 83]. Since DAD is the pathologic hallmark of AIP, AIP is thus considered to represent idiopathic DAD.
It appears that very little published literature has been directed towards AIP as entity over the past 15 years, likely due to the gradual accumulation of numerous well-established risk factors, associations, and causes that have been described for the past several decades with the DAD pattern of injury [84, 85]. These observations have been strengthened almost certainly by the substantial advancement in detection methods for respiratory viral pathogens. Additionally, patients designated as having AIP are rare in published reports utilizing MDD, although admittedly, MDD in ILD may be aimed much more towards patients with subacute or chronic disease than those with acute fulminant disease [37, 38, 39]. Overall, AIP (idiopathic DAD) is likely a rare disorder in the 21st century.
2.5. Idiopathic lymphoid interstitial pneumonia
Lymphoid (or lymphocytic) interstitial pneumonia (LIP) gained prominence in ILD following its description in 1973 by Liebow and Carrington [86]. Histologically, LIP is characterized by an accumulation of lymphocytes, plasma cells, and macrophages throughout the lung interstitium, with well-formed lymphocyte aggregates with germinal centers often observed, and occasionally with associated poorly-formed granulomas [86, 87, 88, 89, 90]. Histologically, LIP is considered to represent a spectrum of disease with follicular bronchiolitis (FB), in which FB demonstrates localized accumulation of lymphoid follicles around small airways, and LIP demonstrates widespread accumulation of lymphocytes and plasma cells throughout the lung parenchyma. There has been past discussion as to whether LIP represented a malignant process, but currently LIP is considered to be a benign reactive disorder, malignant transformation is thought to be rare, and flow cytometry can assist with differentiating LIP from low-grade lymphoid neoplasms [87, 89, 90]. Radiologically, LIP has varied patterns, which have included GGO, small centrilobular nodules, prominent septal lines, and lower lobe-predominant cystic lung disease [87, 89, 91]. Explanations for the cystic abnormalities seen on CT imaging stem from mechanical compression effects of the lymphoid follicles, and include small vessel compression with subsequent parenchymal ischemia, or small airway compression with subsequent airway dilation resulting from a check-valve mechanism [87, 89].
Since its original description, LIP has been associated with numerous pulmonary and systemic processes. The two most common associations with LIP are systemic autoimmune diseases, in particular Sjogren’s syndrome and systemic lupus erythematosus (SLE) [12, 63, 87, 89, 90], and human immunodeficiency virus (HIV) infection, in which the association is observed more often in children [92, 93]. Other described associations include Epstein-Barr virus infection [94], familial interstitial pneumonia [95, 96], common variable immunodeficiency syndrome [97, 98], hypo- or hyper-gammaglobulinemia [86, 87], graft-versus-host disease following allogeneic stem cell transplantation [33, 99], adverse effect of phenytoin [100], and pulmonary amyloidosis [101]. These observations would suggest that LIP is a pattern of lung injury that is associated with numerous diseases and processes, and as stated even as early as 2002, “idiopathic LIP is exceptionally rare” [63].
2.6. Idiopathic pleuroparenchymal fibroelastosis
Descriptions of pleuroparenchymal fibroelastosis (PPFE) first began to be used with ILD terminology in 1992 [102]. As its name suggests, PPFE involves both the pleura and the lung parenchyma, and is a mid- and upper-lung zone predominant process [103, 104]. On chest CT imaging, there is pulmonary fibrosis with reticulation, small areas of consolidation, architectural distortion, and traction bronchiectasis in the most peripheral, subpleural areas of the upper-zone lung parenchyma, which is associated with thickening of the juxtaposed upper-zone pleura and with upper lobe volume loss [103, 104]. Histologically, PPFE is characterized by thickening of the visceral pleura from collagen deposition (fibrosis) and architectural distortion of the lung parenchyma from both collagen and elastin deposition (fibroelastosis) [103, 105]. There are occasionally microscopic areas of organization (fibroblastic foci) and poorly-formed granulomas associated with the lung parenchymal component [105].
Since its original description, numerous disorders and exposures have been associated with a PPFE pattern. These have included systemic autoimmune diseases [103, 106, 107, 108, 109], graft-versus-host disease following allogeneic stem cell transplantation [110, 111], chronic lung allograft dysfunction [112, 113], familial interstitial pneumonia and genetic variants of the telomerase complex [114, 115], external beam radiation therapy [103, 116], occupational exposures to silicates [117, 118], chronic hypersensitivity pneumonitis [119, 120], adverse effect of antineoplastic alkylating agents [121, 122], following liver transplantation [123], and prior pulmonary infections [124, 125]. These observations suggest that PPFE is merely one of many nonspecific patterns of lung injury that is associated with numerous diseases and processes.
2.7. Unclassifiable idiopathic interstitial pneumonia
Unclassifiable interstitial pneumonia, or unclassifiable ILD, has been used over the past few decades to denote interstitial pneumonia in which one of the more precise clinical forms of ILD was not able to be identified [4, 5]. It has additionally become more commonly used as MDD for ILD has become more widely implemented, with recognition that a subset of ILD patients remain with unclassifiable disease even with MDD and a comprehensive clinical evaluation [38, 39, 126, 127]. Three scenarios regarding the unclassifiable terminology seem plausible. First, a comprehensive assessment suggests more than one possible etiology for ILD, thus making a single diagnosis challenging [39]. Second, chest CT and SLB may demonstrate widespread abnormalities indicative of ILD and pulmonary fibrosis, but the more precise patterns of UIP, NSIP, or OP are absent, thus making the ILD “difficult to classify” [128]. Third, unclassifiable ILD could be used to indicate the process is “not yet classifiable” [38], as would often be applied in an MDD setting in which further clinical testing is recommended or is pending, and which perhaps could lead to a more definitive ILD diagnosis at a later time. In any of these instances, the term unclassifiable ILD seems appropriate [38, 39, 126, 127].
In the past, the term unclassifiable has at times been suggested to be not particularly helpful, but in our judgment, it seems appropriate and honest to use this term in selected instances, as it demonstrates some of the uncertainty that can exist despite a comprehensive evaluation within MDD by ILD experts [128]. However, labeling the process as idiopathic seems unnecessary and confusing, and we propose utilization of the terms unclassifiable IP or unclassifiable ILD when appropriate following MDD.
2.8. Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is perhaps the term most in need of change [6, 7] due to its frequency of use and the severity of this disease phenotype. IPF has often been described as the most common consensus diagnosis following MDD [38], and is likely the form of pulmonary fibrosis most frequently identified for research funding opportunities within the ILD research community. The prognosis for individual patients with IPF may vary significantly, but despite the imprecise terminology, many patients with the IPF phenotype of disease will have a devastating disorder with a poor prognosis and a modest response to therapeutic interventions.
Fibrosis or scarring of the lungs has been described for millennia, gained further recognition in the early part of the 20th century [129, 130], and subsequently the term IPF came into usage in the 1970s [3]. When examining each of the words individually - idiopathic, pulmonary, fibrosis - this would seem to imply an umbrella-type term encompassing patients with numerous radiologic and histologic patterns of fibrosis without an identified etiology. However, the designation IPF has evolved over the past 15 years to represent a narrow phenotype of disease, with rather precise clinical, radiologic, and histologic findings [5, 131, 132]. Histologically, the lungs show a UIP pattern of injury, characterized by heterogenous areas of densely fibrotic lung interspersed with areas of relatively normal appearing lung, honeycomb cysts lined by mature respiratory epithelium and filled with mucin, and juxtaposed fibroblast foci [5, 80, 81, 131, 132]. On imaging, the histologic term UIP has been gradually extrapolated to chest CT, and is characterized by reticular opacities in a peripheral-, posterior- and basilar-predominant distribution, and often accompanied by traction bronchiectasis and honeycombing [131, 132]. The clinical designation of IPF thus requires a radiologic pattern of typical or probable UIP for diagnostic confidence, and although SLB is generally not performed in patients meeting established clinical and radiologic criteria, the designation IPF implies that lung histology will demonstrate a UIP pattern if obtained by SLB or at the time of lung transplantation.
Throughout this manuscript, we have demonstrated that the various described forms of IIP are rarely idiopathic, and the same applies for patients designated as having IPF. First, numerous epidemiologic studies have demonstrated cigarette smoking as a major risk factor for the development of the IPF phenotype, and in the recent pirfenidone and nintedanib clinical trials, the majority of enrolled patients were current or prior cigarette smokers [133, 134, 135, 136, 137, 138, 139]. Second, exposure to various forms of dust, particularly in the occupational setting and including metal dust, wood dust, and paper dust, have been strongly linked epidemiologically to the development of pulmonary fibrosis with an IPF phenotype of disease [108, 134, 135, 136, 137, 140, 141, 142]. Third, a variety of environmental and occupational exposures in addition to dust have been described to result in the IPF phenotype of disease, including agriculture/farming [135, 140, 142], livestock [135], stone/sand/silica [135, 140, 141, 142], and fumes/gases/vapors [108, 136, 140]. Fourth, epidemiologic studies have demonstrated that male gender is associated with development of the IPF phenotype, and in the recent pirfenidone and nintedanib trials, the majority of enrolled patients were male [136, 138, 139, 143, 144]. This association may be related merely to occupational and environmental risk factors, but may be perhaps related to hormonal differences affecting pulmonary fibrosis pathobiology [144, 145]. Fifth, many patients with the IPF phenotype have gastroesophageal reflux disease (GERD), and chronic aspiration related to GERD has been postulated to have a pathobiologic role in IPF [146, 147]. Lastly, the past decade has seen an extensive number of molecular studies indicating the role that genetic variants have in the development of the IPF phenotype, including both common and rare genetic variants in sporadic and familial disease, respectively. Variants have been described in genes related to telomere biology, surfactant protein biology, mucin production, inflammation and immunity, collagen synthesis, and cell cycle progression [30, 148, 149, 150, 151]. Rare genetic variants in patients with the IPF phenotype may be the etiology of disease pathobiologically particularly in patients with no evidence of concerning environmental, occupational, or avocational exposures.
In addition to these numerous risk factors and associations, an alternative interpretation of the IPF term would be idiopathic UIP, since IPF is defined by radiologic and histologic UIP. A UIP pattern has been associated with many diverse etiologies, including systemic autoimmune diseases (particularly RA and SSc) [13, 15, 39, 152], pulmonary drug toxicity [39, 51, 153], chronic hypersensitivity pneumonitis [39, 62, 153], inflammatory bowel disease [25, 154], occupational exposures [137, 141, 153], and combined pulmonary fibrosis and emphysema (CPFE) [155].
Overall, it is not the narrow phenotype described as IPF that is misleading or confusing, as general pulmonary clinicians and ILD experts can recognize the radiologic and histologic patterns of UIP, but rather it is perplexing that this group of patients continues to be designated as having idiopathic disease. As early as 2006, it was stated that IPF is likely a heterogeneous disorder caused by a number of environmental and occupational exposures [135], and the substantial observed contributions over the past decade of common and rare genetic variants to this disease process further strengthens these thoughts. In the early 21st century, it is apparent that the term IPF no longer accurately reflects the current epidemiologic and genetic understanding of this disease process [6].
3.0. Conclusion
In this Review, we have discussed the radiologic and histologic patterns for each of the disorders currently classified by a multidisciplinary approach as IIPs, a group of disorders within the ILD spectrum of disease addressed by two detailed consensus statements and thousands of published manuscripts over the past 30 years. Additionally, we have discussed the extensive well-described risk-factors and associations with each of these patterns of lung injury, and it has become apparent that in the 21st century, these disorders are rarely idiopathic. The patterns of RB and DIP are specifically associated with smoke inhalation, most commonly with cigarette smoke. If we remove the prefix “idiopathic” from the remaining IIPs, we have well-described radiologic and histologic patterns of lung injury (NSIP, OP, DAD, LIP, PPFE, UIP) that rather than being idiopathic, are lung injury patterns that are associated with countless yet definitive environmental exposures, occupational exposures, pulmonary diseases, systemic diseases, medication toxicities, infectious disorders, and common or rare genetic variants. We do fully recognize that the majority of the epidemiologic, translational, and molecular studies that have demonstrated these risk factors and associations do not prove disease causality, yet the sheer volume and solid consistency of the data indicate that these associations are very strong. The stronger the association, the likelier the possibility of a causal link, and moving from association to causation is a critical step for taking preventive actions and devising therapeutic interventions [156]. In our judgement, the greater depth in which a patient’s medical, environmental, occupational, and family history are explored along with autoimmune serologic testing and selective gene sequencing, it appears substantially more likely that a multidisciplinary diagnosis other than idiopathic disease will be rendered [39, 141, 142].
As a result, continuing to label these disorders as idiopathic is misleading and confusing. Furthermore, perpetuating the idiopathic nomenclature potentially promotes a lack of inquisitiveness to clinicians. Once these disorders are labeled as idiopathic, this is often perceived as the end of the diagnostic and evaluation process rather than the beginning, the beginning of a comprehensive diagnostic evaluation utilizing all tools available in the 21st century to identify the likely etiology of ILD. These tools include experienced history-taking skills by ILD clinicians, ILD questionnaires to supplement environmental and occupational exposure assessment, a comprehensive array of autoimmune serologies, a diverse array of microbiologic techniques for bacterial, mycobacterial, fungal and viral pathogens, and an increasing ability to pursue genetic counseling and concurrent gene sequencing in patients with a family history of ILD. Identifying the likely etiology of ILD provides clinicians an opportunity to initiate patient-centered medical management, including possible environmental remediation, avocational behavior modification, change in medication management strategy, occupation-related strategies, genetic counseling, or lung transplantation evaluation, all which may improve a patient’s quality of life and hopefully disease outcome.
Even with accurate identification of ILD etiology and the best efforts of patients and clinicians to maximize environmental modifications and treatment strategies, some patients will continue to have progressive and relentless fibrotic lung disease. These observations suggest that irrespective of etiology, there may be a threshold of lung injury beyond which common mechanisms of fibrosis progression occur. In these patients, treatment with anti-fibrotic therapy, either pirfenidone or nintedanib, should be considered, as these therapies have been shown to slow the decline in pulmonary function in patients with the IPF phenotype as well as other subgroups of patients with pulmonary fibrosis [138, 139, 157, 158].
Based upon the overall evidence summarized above, we have proposed an alternative etiology-focused nomenclature classification for multidisciplinary diagnoses in ILD, which is shown in Table 2, and which is not too dissimilar from other alternative classification systems that have been recently proposed by other investigators [6]. We fully recognize that any classification system is likely to be imperfect, but we believe that an etiology-focused classification will provide much needed clarity and understanding as to the diverse nature of ILD. For patients with the IPF phenotype of disease, we have used in Table 2 an alternative term “progressive age-dependent fibrosis (PADF)”, which was proposed in 2018 given the inadequacy of the term IPF to characterize this group of patients [6]. Given the substantial accumulation of scientific evidence, it would likely be beneficial to all if the term idiopathic is completely removed from ILD nomenclature over the next several years.
Table 2.
Proposed etiology-focused nomenclature and classification for multidisciplinary diagnoses in interstitial lung disease (ILD)
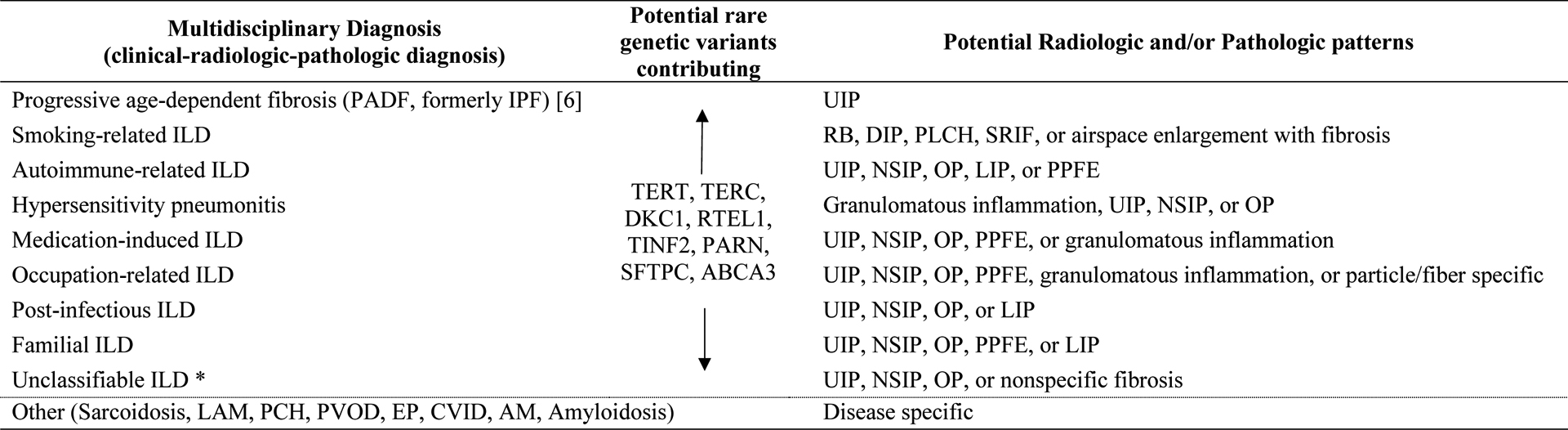
|
Definition of abbreviations: IPF, idiopathic pulmonary fibrosis; UIP, usual interstitial pneumonia; RB, respiratory bronchiolitis; DIP, desquamative interstitial pneumonia; PLCH, pulmonary Langerhans cell histiocytosis; SRIF, smoking-related interstitial fibrosis; NSIP, nonspecific interstitial pneumonia; OP, organizing pneumonia; LIP, lymphoid interstitial pneumonia; PPFE, pleuroparenchymal fibroelastosis; TERT, telomerase reverse transcriptase; TERC, telomerase RNA component; DKC1, dyskerin pseudouridine synthase 1; RTEL1, regulator of telomere elongation helicase 1; TINF2, TERF1 interacting nuclear factor 2; PARN, poly(A)-specific ribonuclease; SFTPC, surfactant protein C; ABCA3, ATP binding cassette subfamily A member 3; LAM, lymphangioleiomyomatosis; PCH, pulmonary capillary hemangiomatosis; PVOD, pulmonary veno-occlusive disease; EP, eosinophilic pneumonia; CVID, combined variable immunodeficiency; AM, alveolar microlithiasis.
see text for unclassifiable ILD inclusion criteria
One could perhaps ask why put forth so much time and effort on nomenclature. However, throughout clinical medicine and biomedical research, utilization of precise terminology is expected and is insightful. Not infrequently, a precise association or etiology for one of the IIPs has been identified clinically or in a published study, but yet the disorder continues to be labeled by multidisciplinary diagnosis as idiopathic or cryptogenic disease. One can thus understand how many general medicine and pulmonary physicians may find the terminology and language used in ILD complex and even impenetrable. The pulmonary and ILD community will benefit from a shift away from labeling these disorders as idiopathic or cryptogenic and towards nomenclature which is more etiology-focused. This would be similar to other specialties and subspecialities of medicine in which disorders previously classified as idiopathic have been renamed recognizing their diverse environmental, immune, or genetic etiologies [159, 160, 161, 162]. A shift in ILD nomenclature will not be easy, as the idiopathic terminology is long-standing and has been embedded in the ILD community and literature for some time. But beginning to utilize more etiology-focused ILD nomenclature will be beneficial to everyone, including patients hoping for a better quality of life and disease outcome, trainees, general medicine and pulmonary physicians furthering their knowledge and experience in ILD, and expert ILD clinicians and researchers who are defining and advancing the ILD field.
4.0. Expert Opinion
Interstitial lung disease (ILD) comprises a wide array of diseases that are generally characterized by a combination of pulmonary inflammation and pulmonary fibrosis. A major emphasis in ILD over the past two decades has focused on a group of disorders classified by multidisciplinary approach as idiopathic interstitial pneumonias (IIPs), which have become a cornerstone of the ILD nomenclature. Substantial scientific efforts to understand the underlying epidemiology, pathobiology, and causality of these IIP disorders have transpired, and what has emerged is that these disorders, rather than being idiopathic, are well-described radiologic and histologic patterns of lung injury that are driven by a remarkable amount of diverse environmental and occupational exposures, autoimmune mechanisms, pulmonary and systemic diseases, medication toxicities, and genetic variants. The idiopathic nomenclature is thus misleading and confusing, and may additionally promote a lack of inquisitiveness, suggesting the end rather than the beginning of a thorough diagnostic process to identify ILD etiology and initiate patient-centered management.
Based upon this scientific evidence, a new etiology-focused nomenclature classification for multidisciplinary diagnoses in ILD is needed. It is likely difficult for any single ILD classification system to be perfect, but an etiology-focused classification will provide much needed clarity and understanding as to the diverse etiologies of ILD. Additionally, new nomenclature is needed for patients meeting current diagnostic criteria for idiopathic pulmonary fibrosis (IPF), as the IPF term no longer accurately describes the broad epidemiologic and genetic features of this group of patients. The specialty of pulmonary medicine and the field of ILD will likely benefit if the term idiopathic is completely removed from ILD nomenclature over the next five years.
Despite advances in ILD over the past two decades, timely and accurate ILD diagnoses remain as significant challenges. For the general medicine physician, diagnosing ILD is challenging since the nonspecific symptoms of ILD (cough, dyspnea, inspiratory crackles) are often observed in a variety of alternative cardiopulmonary disorders. Perhaps the increasing utilization of computed tomography (CT) of the chest across the medical community can assist in this regard. For the pulmonary specialist, arriving at accurate multidisciplinary ILD diagnoses is likewise challenging, as individual patients often have complex past medical histories and exposures, and determining the most important causal factor can be complex. Likely of benefit in this regard are an increasing array of tools in the 21st century to assist, including experienced history-taking skills by ILD clinicians, ILD questionnaires to supplement environmental and occupational exposure assessment, a comprehensive array of autoimmune serologies, a diverse array of microbiologic techniques for bacterial, mycobacterial, fungal and viral pathogens, and an increasing ability to pursue genetic counseling and gene sequencing in patients with a family history of ILD.
What will the ILD landscape look like over the next decade? As long as smoke inhalation in various forms remains prevalent, there will likely continue to be varying manifestations of smoking-related ILD. The pace of new pharmaceutical drug development will likely give rise to new classes of medications that despite their clinical efficacy may be associated with medication-induced ILD in some patients. Traditional occupation-related ILD (e.g., pneumoconioses) will likely reduce in incidence as fewer of these occupations remain and improved industrial safety measures are implemented, but perhaps evolving occupational exposures may result in newer forms of occupation-related ILD. The COVID-19 pandemic has certainly informed all physicians as to the role that infectious pathogens can play in the development of progressive fibrotic ILD. As genetic sequencing becomes more logistically achievable, this will likely increase our understanding of the role of genetic variants across all groups of patients with ILD. For all forms of ILD, we will hopefully see an improvement in treatment options over the next decade as well as biomedical research continues in this field.
At present, what should be the best approach by ILD clinicians? Multidisciplinary ILD diagnoses should best be ascertained by a collegial multidisciplinary team involving specialists from pulmonary medicine, rheumatology, thoracic radiology, and thoracic pathology, and when appropriate involving additional expertise from occupational and environmental physicians and medical geneticists. A nomenclature and classification system for multidisciplinary clinical-radiologic-pathologic diagnoses in ILD that is etiology-focused will provide much needed clarity and understanding. Identifying the likely etiology of ILD provides clinicians an opportunity to initiate patient-centered medical management, including possible environmental remediation, avocational behavior modification, change in medication management strategy, occupation-related strategies, genetic counseling, or lung transplantation evaluation. As an ILD community, our overall goals should continue be to promote an environment that allows expert ILD clinicians and researchers to advance the ILD field, to assist general medicine and pulmonary physicians in furthering their ILD knowledge, and most importantly, to improve the quality of life and disease outcome of patients with ILD.
Article highlights.
A major focus of interstitial lung disease (ILD) centers on nine disorders termed idiopathic interstitial pneumonias (IIPs), which include idiopathic pulmonary fibrosis
Since their original descriptions, substantial epidemiologic, translational, and molecular studies have demonstrated that IIPs are driven by combinations of environmental and occupational exposures, autoimmune mechanisms, pulmonary and systemic diseases, medication toxicities, and genetic variants
The IIPs are thus rarely idiopathic in the 21st century, and the idiopathic nomenclature is often misleading and confusing
Rather than being idiopathic, the IIPs are well-described radiologic and histologic patterns of lung injury that are associated with a wide array of diverse etiologies
A shift towards more etiology-focused nomenclature will provide better clarity and understanding in ILD, and will be beneficial to all, including patients, general medicine and pulmonary physicians, and expert ILD clinicians and researchers
Funding
This work was supported by the United States National Institutes of Health NIH-NIAMS R01AR077562 and NIH-NHLBI R01HL126897, and United States Veterans Affairs Merit Award I01BX002499.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Liddell K Choosing a dermatological hero for the millennium. Hippocrates of Cos (460–377 BC). Clin Exp Dermatol. 2000. Jan;25(1):86–8. doi: 10.1046/j.1365-2230.2000.0580d.x. [DOI] [PubMed] [Google Scholar]
- 2.Cottin V Pulmonary fibrosis: “idiopathic” is not “cryptogenic”. Eur Respir J. 2019. Mar;53(3). doi: 10.1183/13993003.02314-2018. [DOI] [PubMed] [Google Scholar]
- 3.Crystal RG, Fulmer JD, Roberts WC, et al. Idiopathic pulmonary fibrosis. Clinical, histologic, radiographic, physiologic, scintigraphic, cytologic, and biochemical aspects. Ann Intern Med. 1976. Dec;85(6):769–88. doi: 10.7326/0003-4819-85-6-769. [DOI] [PubMed] [Google Scholar]
- 4.American Thoracic S, European Respiratory S. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002. Jan 15;165(2):277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 5.Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013. Sep 15;188(6):733–48. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolters PJ, Blackwell TS, Eickelberg O, et al. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir Med. 2018. Feb;6(2):154–160. doi: 10.1016/S2213-2600(18)30007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** discusses rationale for renaming (rebranding) IPF
- 7.Thannickal VJ, Wells A, Kolb M. Idiopathic pulmonary fibrosis: idiopathic no more? Lancet Respir Med. 2018. Feb;6(2):84–85. doi: 10.1016/S2213-2600(18)30022-5. [DOI] [PubMed] [Google Scholar]; ** discusses rationale for nomenclature change for IPF
- 8.Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol. 1994. Feb;18(2):136–47. [PubMed] [Google Scholar]
- 9.Sumikawa H, Johkoh T, Ichikado K, et al. Nonspecific interstitial pneumonia: histologic correlation with high-resolution CT in 29 patients. Eur J Radiol. 2009. Apr;70(1):35–40. doi: 10.1016/j.ejrad.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Todd NW, Marciniak ET, Sachdeva A, et al. Organizing pneumonia/non-specific interstitial pneumonia overlap is associated with unfavorable lung disease progression . Respir Med 2015. Nov;109(11):1460–8. doi: 10.1016/j.rmed.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Travis WD, Hunninghake G, King TE Jr., et al. Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am J Respir Crit Care Med. 2008. Jun 15;177(12):1338–47. doi: 10.1164/rccm.200611-1685OC. [DOI] [PubMed] [Google Scholar]
- 12.Arrossi AV. Pulmonary Pathology in Rheumatic Disease. Clin Chest Med. 2019. Sep;40(3):667–677. doi: 10.1016/j.ccm.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 13.Mira-Avendano I, Abril A, Burger CD, et al. Interstitial Lung Disease and Other Pulmonary Manifestations in Connective Tissue Diseases. Mayo Clin Proc. 2019. Feb;94(2):309–325. doi: 10.1016/j.mayocp.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Romagnoli M, Nannini C, Piciucchi S, et al. Idiopathic nonspecific interstitial pneumonia: an interstitial lung disease associated with autoimmune disorders? Eur Respir J. 2011. Aug;38(2):384–91. doi: 10.1183/09031936.00094910. [DOI] [PubMed] [Google Scholar]
- 15.Solomon JJ, Olson AL, Fischer A, et al. Scleroderma lung disease. Eur Respir Rev. 2013. Mar 1;22(127):6–19. doi: 10.1183/09059180.00005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franks TJ, Galvin JR. Smoking-Related “Interstitial” Lung Disease. Arch Pathol Lab Med. 2015. Aug;139(8):974–7. doi: 10.5858/arpa.2013-0384-RA. [DOI] [PubMed] [Google Scholar]
- 17.Marten K, Milne D, Antoniou KM, et al. Non-specific interstitial pneumonia in cigarette smokers: a CT study. Eur Radiol. 2009. Jul;19(7):1679–85. doi: 10.1007/s00330-009-1308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaherty KR, Fell C, Aubry MC, et al. Smoking-related idiopathic interstitial pneumonia. Eur Respir J. 2014. Sep;44(3):594–602. doi: 10.1183/09031936.00166813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skeoch S, Weatherley N, Swift AJ, et al. Drug-Induced Interstitial Lung Disease: A Systematic Review. J Clin Med. 2018. Oct 15;7(10). doi: 10.3390/jcm7100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalisz KR, Ramaiya NH, Laukamp KR, et al. Immune Checkpoint Inhibitor Therapy-related Pneumonitis: Patterns and Management. Radiographics. 2019. Nov-Dec;39(7):1923–1937. doi: 10.1148/rg.2019190036. [DOI] [PubMed] [Google Scholar]
- 21.Azadeh N, Limper AH, Carmona EM, et al. The Role of Infection in Interstitial Lung Diseases: A Review. Chest. 2017. Oct;152(4):842–852. doi: 10.1016/j.chest.2017.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasakova M, Morell F, Walsh S, et al. Hypersensitivity Pneumonitis: Perspectives in Diagnosis and Management. Am J Respir Crit Care Med. 2017. Sep 15;196(6):680–689. doi: 10.1164/rccm.201611-2201PP. [DOI] [PubMed] [Google Scholar]
- 23.Churg A, Sin DD, Everett D, et al. Pathologic patterns and survival in chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2009. Dec;33(12):1765–70. doi: 10.1097/PAS.0b013e3181bb2538. [DOI] [PubMed] [Google Scholar]
- 24.Vourlekis JS, Schwarz MI, Cool CD, et al. Nonspecific interstitial pneumonitis as the sole histologic expression of hypersensitivity pneumonitis. Am J Med. 2002. Apr 15;112(6):490–3. doi: 10.1016/s0002-9343(02)01046-x. [DOI] [PubMed] [Google Scholar]
- 25.Majewski S, Piotrowski W. Pulmonary manifestations of inflammatory bowel disease. Arch Med Sci. 2015. Dec 10;11(6):1179–88. doi: 10.5114/aoms.2015.56343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cozzi D, Moroni C, Addeo G, et al. Radiological Patterns of Lung Involvement in Inflammatory Bowel Disease. Gastroenterol Res Pract. 2018;2018:5697846. doi: 10.1155/2018/5697846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cinetto F, Scarpa R, Rattazzi M, et al. The broad spectrum of lung diseases in primary antibody deficiencies. Eur Respir Rev. 2018. Sep 30;27(149). doi: 10.1183/16000617.0019-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bierry G, Boileau J, Barnig C, et al. Thoracic manifestations of primary humoral immunodeficiency: a comprehensive review. Radiographics. 2009. Nov;29(7):1909–20. doi: 10.1148/rg.297095717. [DOI] [PubMed] [Google Scholar]
- 29.Steele MP, Speer MC, Loyd JE, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005. Nov 1;172(9):1146–52. doi: 10.1164/rccm.200408-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borie R, Le Guen P, Ghanem M, et al. The genetics of interstitial lung diseases. Eur Respir Rev. 2019. Sep 30;28(153). doi: 10.1183/16000617.0053-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]; * broad discussion of genetic variants in ILD
- 31.Bennett D, Mazzei MA, Squitieri NC, et al. Familial pulmonary fibrosis: Clinical and radiological characteristics and progression analysis in different high resolution-CT patterns. Respir Med. 2017. May;126:75–83. doi: 10.1016/j.rmed.2017.03.020. [DOI] [PubMed] [Google Scholar]
- 32.Miyagawa-Hayashino A, Sonobe M, Kubo T, et al. Non-specific interstitial pneumonia as a manifestation of graft-versus-host disease following pediatric allogeneic hematopoietic stem cell transplantation. Pathol Int. 2010. Feb;60(2):137–42. doi: 10.1111/j.1440-1827.2009.02492.x. [DOI] [PubMed] [Google Scholar]
- 33.Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med. 2014. Oct;108(10):1525–33. doi: 10.1016/j.rmed.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 34.Scallan C, Venado A, Han L, et al. Recurrent Pulmonary Fibrosis in a Lung Allograft Secondary to De Novo Antisynthetase Syndrome. Ann Am Thorac Soc. 2020. Jul;17(7):901–904. doi: 10.1513/AnnalsATS.202002-126RL. [DOI] [PubMed] [Google Scholar]
- 35.Hinze AM, Lin CT, Hussien AF, et al. Longitudinal assessment of interstitial lung disease in single lung transplant recipients with scleroderma. Rheumatology (Oxford). 2020. Apr 1;59(4):790–798. doi: 10.1093/rheumatology/kez341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wells AU, Cottin V. Nonspecific interstitial pneumonia: time to be more specific? Curr Opin Pulm Med. 2016. Sep;22(5):450–5. doi: 10.1097/MCP.0000000000000302. [DOI] [PubMed] [Google Scholar]
- 37.Chai GT, Tan TC, Lee YS, et al. Impact of an interstitial lung disease service in the diagnosis and management of interstitial lung disease in Singapore. Singapore Med J. 2020. Jun;61(6):302–307. doi: 10.11622/smedj.2019069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Sadeleer LJ, Meert C, Yserbyt J, et al. Diagnostic Ability of a Dynamic Multidisciplinary Discussion in Interstitial Lung Diseases: A Retrospective Observational Study of 938 Cases. Chest. 2018. Jun;153(6):1416–1423. doi: 10.1016/j.chest.2018.03.026. [DOI] [PubMed] [Google Scholar]
- 39.Dodia N, Amariei D, Kenaa B, et al. A comprehensive assessment of environmental exposures and the medical history guides multidisciplinary discussion in interstitial lung disease. Respir Med. 2021. Feb 11;179:106333. doi: 10.1016/j.rmed.2021.106333. [DOI] [PMC free article] [PubMed] [Google Scholar]; * discusses impact of medical history and exposures assessment in multidisciplinary ILD diagnoses
- 40.Floyd R Organization of pneumonic exudates. Am J Med Sci. 1922;163(4):527–548. [Google Scholar]
- 41.Davison AG, Heard BE, McAllister WA, et al. Cryptogenic organizing pneumonitis. Q J Med. 1983. Summer;52(207):382–94. [PubMed] [Google Scholar]
- 42.Epler GR, Colby TV, McLoud TC, et al. Bronchiolitis obliterans organizing pneumonia. N Engl J Med. 1985. Jan 17;312(3):152–8. doi: 10.1056/NEJM198501173120304. [DOI] [PubMed] [Google Scholar]
- 43.Cordier JF. Organising pneumonia. Thorax. 2000. Apr;55(4):318–28. doi: 10.1136/thorax.55.4.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberton BJ, Hansell DM. Organizing pneumonia: a kaleidoscope of concepts and morphologies. Eur Radiol. 2011. Nov;21(11):2244–54. doi: 10.1007/s00330-011-2191-6. [DOI] [PubMed] [Google Scholar]
- 45.Myers JL, Katzenstein AL. Ultrastructural evidence of alveolar epithelial injury in idiopathic bronchiolitis obliterans-organizing pneumonia. Am J Pathol. 1988. Jul;132(1):102–9. [PMC free article] [PubMed] [Google Scholar]
- 46.Todd NW, Atamas SP, Luzina IG, et al. Permanent alveolar collapse is the predominant mechanism in idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2015. Aug;9(4):411–8. doi: 10.1586/17476348.2015.1067609. [DOI] [PubMed] [Google Scholar]
- 47.Lee JW, Lee KS, Lee HY, et al. Cryptogenic organizing pneumonia: serial high-resolution CT findings in 22 patients. AJR Am J Roentgenol. 2010. Oct;195(4):916–22. doi: 10.2214/AJR.09.3940. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Jin C, Wu CC, et al. Organizing pneumonia of COVID-19: Time-dependent evolution and outcome in CT findings. PLoS One. 2020;15(11):e0240347. doi: 10.1371/journal.pone.0240347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romero S, Barroso E, Rodriguez-Paniagua M, et al. Organizing pneumonia adjacent to lung cancer: frequency and clinico-pathologic features. Lung Cancer. 2002. Feb;35(2):195–201. doi: 10.1016/s0169-5002(01)00405-6. [DOI] [PubMed] [Google Scholar]
- 50.Pardo J, Panizo A, Sola I, et al. Prognostic value of clinical, morphologic, and immunohistochemical factors in patients with bronchiolitis obliterans-organizing pneumonia. Hum Pathol. 2013. May;44(5):718–24. doi: 10.1016/j.humpath.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 51.Schwaiblmair M, Behr W, Haeckel T, et al. Drug induced interstitial lung disease. Open Respir Med J. 2012;6:63–74. doi: 10.2174/1874306401206010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishino M, Hatabu H, Hodi FS, et al. Drug-Related Pneumonitis in the Era of Precision Cancer Therapy. JCO Precis Oncol. 2017;1. doi: 10.1200/PO.17.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castro CY. ARDS and diffuse alveolar damage: a pathologist’s perspective. Semin Thorac Cardiovasc Surg. 2006. Spring;18(1):13–9. doi: 10.1053/j.semtcvs.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 54.Cardinal-Fernandez P, Lorente JA, Ballen-Barragan A, et al. Acute Respiratory Distress Syndrome and Diffuse Alveolar Damage. New Insights on a Complex Relationship. Ann Am Thorac Soc. 2017. Jun;14(6):844–850. doi: 10.1513/AnnalsATS.201609-728PS. [DOI] [PubMed] [Google Scholar]
- 55.Otani K, Seo Y, Ogawa K. Radiation-Induced Organizing Pneumonia: A Characteristic Disease that Requires Symptom-Oriented Management. Int J Mol Sci. 2017. Jan 27;18(2). doi: 10.3390/ijms18020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murai T, Shibamoto Y, Nishiyama T, et al. Organizing pneumonia after stereotactic ablative radiotherapy of the lung. Radiat Oncol. 2012. Aug 1;7:123. doi: 10.1186/1748-717X-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adachi Y, Ozeki K, Ukai S, et al. Patterns of onset and outcome of cryptogenic organizing pneumonia after allogeneic hematopoietic stem cell transplantation. Int J Hematol. 2019. Jun;109(6):700–710. doi: 10.1007/s12185-019-02643-9. [DOI] [PubMed] [Google Scholar]
- 58.Haider S, Durairajan N, Soubani AO. Noninfectious pulmonary complications of haematopoietic stem cell transplantation. Eur Respir Rev. 2020. Jun 30;29(156). doi: 10.1183/16000617.0119-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Costa AN, Carraro RM, Nascimento EC, et al. Acute Fibrinoid Organizing Pneumonia in Lung Transplant: The Most Feared Allograft Dysfunction. Transplantation. 2016. Mar;100(3):e11–2. doi: 10.1097/TP.0000000000001088. [DOI] [PubMed] [Google Scholar]
- 60.Vanstapel A, Verleden SE, Weynand B, et al. Late-onset “acute fibrinous and organising pneumonia” impairs long-term lung allograft function and survival. Eur Respir J. 2020. Sep;56(3). doi: 10.1183/13993003.02292-2019. [DOI] [PubMed] [Google Scholar]
- 61.Myers JL. Hypersensitivity pneumonia: the role of lung biopsy in diagnosis and management. Mod Pathol. 2012. Jan;25 Suppl 1:S58–67. doi: 10.1038/modpathol.2011.152. [DOI] [PubMed] [Google Scholar]
- 62.Raghu G, Remy-Jardin M, Ryerson CJ, et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2020. Aug 1;202(3):e36–e69. doi: 10.1164/rccm.202005-2032ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nicholson AG. Classification of idiopathic interstitial pneumonias: making sense of the alphabet soup. Histopathology. 2002. Nov;41(5):381–91. doi: 10.1046/j.1365-2559.2002.01421.x. [DOI] [PubMed] [Google Scholar]
- 64.Rossi G, Cavazza A, Spagnolo P, et al. The role of macrophages in interstitial lung diseases: Number 3 in the Series “Pathology for the clinician” Edited by Peter Dorfmuller and Alberto Cavazza. Eur Respir Rev. 2017. Sep 30;26(145). doi: 10.1183/16000617.0009-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tazelaar HD, Wright JL, Churg A. Desquamative interstitial pneumonia. Histopathology. 2011. Mar;58(4):509–16. doi: 10.1111/j.1365-2559.2010.03649.x. [DOI] [PubMed] [Google Scholar]
- 66.Ryu JH, Myers JL, Capizzi SA, et al. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest. 2005. Jan;127(1):178–84. doi: 10.1378/chest.127.1.178. [DOI] [PubMed] [Google Scholar]
- 67.Liebow AA, Steer A, Billingsley JG. Desquamative Interstitial Pneumonia. Am J Med. 1965. Sep;39:369–404. doi: 10.1016/0002-9343(65)90206-8. [DOI] [PubMed] [Google Scholar]
- 68.Ghio AJ, Hilborn ED, Stonehuerner JG, et al. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am J Respir Crit Care Med. 2008. Dec 1;178(11):1130–8. doi: 10.1164/rccm.200802-334OC. [DOI] [PubMed] [Google Scholar]
- 69.You R, Lu W, Shan M, et al. Nanoparticulate carbon black in cigarette smoke induces DNA cleavage and Th17-mediated emphysema. Elife. 2015. Oct 5;4:e09623. doi: 10.7554/eLife.09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brody AR, Craighead JE. Cytoplasmic inclusions in pulmonary macrophages of cigarette smokers. Lab Invest. 1975. Feb;32(2):125–32. [PubMed] [Google Scholar]
- 71.Mohan S, Ho T, Kjarsgaard M, et al. Hemosiderin in sputum macrophages may predict infective exacerbations of chronic obstructive pulmonary disease: a retrospective observational study. BMC Pulm Med. 2017. Apr 12;17(1):60. doi: 10.1186/s12890-017-0408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pappas RS. Toxic elements in tobacco and in cigarette smoke: inflammation and sensitization. Metallomics. 2011. Nov;3(11):1181–98. doi: 10.1039/c1mt00066g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar A, Cherian SV, Vassallo R, et al. Current Concepts in Pathogenesis, Diagnosis, and Management of Smoking-Related Interstitial Lung Diseases. Chest. 2018. Aug;154(2):394–408. doi: 10.1016/j.chest.2017.11.023. [DOI] [PubMed] [Google Scholar]; * broad discussion of smoking-related ILD
- 74.Godbert B, Wissler MP, Vignaud JM. Desquamative interstitial pneumonia: an analytic review with an emphasis on aetiology. Eur Respir Rev. 2013. Jun 1;22(128):117–23. doi: 10.1183/09059180.00005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ishii H, Iwata A, Sakamoto N, et al. Desquamative interstitial pneumonia (DIP) in a patient with rheumatoid arthritis: is DIP associated with autoimmune disorders? Intern Med. 2009;48(10):827–30. doi: 10.2169/internalmedicine.48.1876. [DOI] [PubMed] [Google Scholar]
- 76.Swartz JS, Chatterjee S, Parambil JG. Desquamative interstitial pneumonia as the initial manifestation of systemic sclerosis. J Clin Rheumatol. 2010. Sep;16(6):284–6. doi: 10.1097/RHU.0b013e3181eed86d. [DOI] [PubMed] [Google Scholar]
- 77.Doan ML, Guillerman RP, Dishop MK, et al. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax. 2008. Apr;63(4):366–73. doi: 10.1136/thx.2007.083766. [DOI] [PubMed] [Google Scholar]
- 78.Lougheed MD, Roos JO, Waddell WR, et al. Desquamative interstitial pneumonitis and diffuse alveolar damage in textile workers. Potential role of mycotoxins. Chest. 1995. Nov;108(5):1196–200. doi: 10.1378/chest.108.5.1196. [DOI] [PubMed] [Google Scholar]
- 79.Hamman L, Rich AR. Fulminating Diffuse Interstitial Fibrosis of the Lungs. Trans Am Clin Climatol Assoc. 1935;51:154–63. [PMC free article] [PubMed] [Google Scholar]
- 80.Beasley MB. The pathologist’s approach to acute lung injury. Arch Pathol Lab Med. 2010. May;134(5):719–27. doi: 10.1043/1543-2165-134.5.719. [DOI] [PubMed] [Google Scholar]
- 81.Leslie KO. My approach to interstitial lung disease using clinical, radiological and histopathological patterns. J Clin Pathol. 2009. May;62(5):387–401. doi: 10.1136/jcp.2008.059782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ichikado K, Suga M, Muller NL, et al. Acute interstitial pneumonia: comparison of high-resolution computed tomography findings between survivors and nonsurvivors. Am J Respir Crit Care Med. 2002. Jun 1;165(11):1551–6. doi: 10.1164/rccm.2106157. [DOI] [PubMed] [Google Scholar]
- 83.Mueller-Mang C, Grosse C, Schmid K, et al. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics. 2007. May-Jun;27(3):595–615. doi: 10.1148/rg.273065130. [DOI] [PubMed] [Google Scholar]
- 84.Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019. Mar 14;5(1):18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thompson BT, Chambers RC, Liu KD. Acute Respiratory Distress Syndrome. N Engl J Med. 2017. Aug 10;377(6):562–572. doi: 10.1056/NEJMra1608077. [DOI] [PubMed] [Google Scholar]
- 86.Liebow AA, Carrington CB. Diffuse pulmonary lymphoreticular infiltrations associated with dysproteinemia. Med Clin North Am. 1973. May;57(3):809–43. doi: 10.1016/s0025-7125(16)32278-7. [DOI] [PubMed] [Google Scholar]
- 87.Swigris JJ, Berry GJ, Raffin TA, et al. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002. Dec;122(6):2150–64. doi: 10.1378/chest.122.6.2150. [DOI] [PubMed] [Google Scholar]
- 88.Koss MN, Hochholzer L, Langloss JM, et al. Lymphoid interstitial pneumonia: clinicopathological and immunopathological findings in 18 cases. Pathology. 1987. Apr;19(2):178–85. doi: 10.3109/00313028709077131. [DOI] [PubMed] [Google Scholar]
- 89.Gupta N, Vassallo R, Wikenheiser-Brokamp KA, et al. Diffuse Cystic Lung Disease. Part II. Am J Respir Crit Care Med. 2015. Jul 1;192(1):17–29. doi: 10.1164/rccm.201411-2096CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bradley B, Branley HM, Egan JJ, et al. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008. Sep;63 Suppl 5:v1–58. doi: 10.1136/thx.2008.101691. [DOI] [PubMed] [Google Scholar]
- 91.Johkoh T, Muller NL, Pickford HA, et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology. 1999. Aug;212(2):567–72. doi: 10.1148/radiology.212.2.r99au05567. [DOI] [PubMed] [Google Scholar]
- 92.Travis WD, Fox CH, Devaney KO, et al. Lymphoid pneumonitis in 50 adult patients infected with the human immunodeficiency virus: lymphocytic interstitial pneumonitis versus nonspecific interstitial pneumonitis. Hum Pathol. 1992. May;23(5):529–41. doi: 10.1016/0046-8177(92)90130-u. [DOI] [PubMed] [Google Scholar]
- 93.van Zyl-Smit RN, Naidoo J, Wainwright H, et al. HIV associated Lymphocytic Interstitial Pneumonia: a clinical, histological and radiographic study from an HIV endemic resource-poor setting. BMC Pulm Med. 2015. Apr 22;15:38. doi: 10.1186/s12890-015-0030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prasoppokakorn T, Assanasen T, Chantranuwatana P, et al. EBV-associated lymphoid interstitial pneumonia in IBD patient: Case report and literature review. Respir Med Case Rep. 2020;30:101059. doi: 10.1016/j.rmcr.2020.101059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wright JA, Pennington JE. Familial lymphoid interstitial pneumonitis. J Pediatr. 1987. Oct;111(4):638. doi: 10.1016/s0022-3476(87)80147-6. [DOI] [PubMed] [Google Scholar]
- 96.O’Brodovich HM, Moser MM, Lu L. Familial lymphoid interstitial pneumonia: a long-term follow-up. Pediatrics. 1980. Mar;65(3):523–8. [PubMed] [Google Scholar]
- 97.Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency--histologic and immunohistochemical analyses of 16 cases. Hum Pathol. 2015. Sep;46(9):1306–14. doi: 10.1016/j.humpath.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Davies CW, Juniper MC, Gray W, et al. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin A. Thorax. 2000. Jan;55(1):88–90. doi: 10.1136/thorax.55.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Perreault C, Cousineau S, D’Angelo G, et al. Lymphoid interstitial pneumonia after allogeneic bone marrow transplantation. A possible manifestation of chronic graft-versus-host disease. Cancer. 1985. Jan 1;55(1):1–9. doi: . [DOI] [PubMed] [Google Scholar]
- 100.Chamberlain DW, Hyland RH, Ross DJ. Diphenylhydantoin-induced lymphocytic interstitial pneumonia. Chest. 1986. Sep;90(3):458–60. doi: 10.1378/chest.90.3.458. [DOI] [PubMed] [Google Scholar]
- 101.Avelino EBP, Verza L, Neves T, et al. Lymphocytic interstitial pneumonia and pulmonary amyloidosis in Sjogren’s syndrome. Radiol Bras. 2019. Nov-Dec;52(6):410–411. doi: 10.1590/0100-3984.2017.0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Amitani R, Niimi A, Kuse F Idiopathic Pulmonary Upper Lobe Fibrosis (IPUF). Kokyu. 1992;11:693–699. [Google Scholar]
- 103.Chua F, Desai SR, Nicholson AG, et al. Pleuroparenchymal Fibroelastosis. A Review of Clinical, Radiological, and Pathological Characteristics. Ann Am Thorac Soc. 2019. Nov;16(11):1351–1359. doi: 10.1513/AnnalsATS.201902-181CME. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Frankel SK, Cool CD, Lynch DA, et al. Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest. 2004. Dec;126(6):2007–13. doi: 10.1378/chest.126.6.2007. [DOI] [PubMed] [Google Scholar]
- 105.Khiroya R, Macaluso C, Montero MA, et al. Pleuroparenchymal Fibroelastosis: A Review of Histopathologic Features and the Relationship Between Histologic Parameters and Survival. Am J Surg Pathol. 2017. Dec;41(12):1683–1689. doi: 10.1097/PAS.0000000000000928. [DOI] [PubMed] [Google Scholar]
- 106.Bonifazi M, Sverzellati N, Negri E, et al. Pleuroparenchymal fibroelastosis in systemic sclerosis: prevalence and prognostic impact. Eur Respir J. 2020. Jul;56(1). doi: 10.1183/13993003.02135-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Enomoto Y, Nakamura Y, Colby TV, et al. Radiologic pleuroparenchymal fibroelastosis-like lesion in connective tissue disease-related interstitial lung disease. PLoS One. 2017;12(6):e0180283. doi: 10.1371/journal.pone.0180283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iwai K, Mori T, Yamada N, et al. Idiopathic pulmonary fibrosis. Epidemiologic approaches to occupational exposure. Am J Respir Crit Care Med. 1994. Sep;150(3):670–5. doi: 10.1164/ajrccm.150.3.8087336. [DOI] [PubMed] [Google Scholar]
- 109.Orlandi M, Landini N, Bruni C, et al. Pleuroparenchymal fibroelastosis in rheumatic autoimmune diseases: a systematic literature review. Rheumatology (Oxford). 2020. Dec 1;59(12):3645–3656. doi: 10.1093/rheumatology/keaa451. [DOI] [PubMed] [Google Scholar]
- 110.Bondeelle L, Gras J, Michonneau D, et al. Pleuroparenchymal fibroelastosis after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2020. May;55(5):982–986. doi: 10.1038/s41409-019-0636-8. [DOI] [PubMed] [Google Scholar]
- 111.von der Thusen JH, Hansell DM, Tominaga M, et al. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol. 2011. Dec;24(12):1633–9. doi: 10.1038/modpathol.2011.114. [DOI] [PubMed] [Google Scholar]
- 112.Pakhale SS, Hadjiliadis D, Howell DN, et al. Upper lobe fibrosis: a novel manifestation of chronic allograft dysfunction in lung transplantation. J Heart Lung Transplant. 2005. Sep;24(9):1260–8. doi: 10.1016/j.healun.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 113.Ofek E, Sato M, Saito T, et al. Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Mod Pathol. 2013. Mar;26(3):350–6. doi: 10.1038/modpathol.2012.171. [DOI] [PubMed] [Google Scholar]
- 114.Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016. Dec;48(6):1710–1720. doi: 10.1183/13993003.00308-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Borie R, Bouvry D, Cottin V, et al. Regulator of telomere length 1 (RTEL1) mutations are associated with heterogeneous pulmonary and extra-pulmonary phenotypes. Eur Respir J. 2019. Feb;53(2). doi: 10.1183/13993003.00508-2018. [DOI] [PubMed] [Google Scholar]
- 116.Watanabe K Pleuroparenchymal Fibroelastosis: Its Clinical Characteristics. Curr Respir Med Rev. 2013. Jun;9:299–237. doi: 10.2174/1573398X0904140129125307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu L, Rassaei N, Caruso C. Pleuroparenchymal Fibroelastosis With Long History of Asbestos and Silicon Exposure. Int J Surg Pathol. 2018. Apr;26(2):190–193. doi: 10.1177/1066896917739399. [DOI] [PubMed] [Google Scholar]
- 118.Huang Z, Li S, Zhu Y, et al. Pleuroparenchymal fibroelastosis associated with aluminosilicate dust: a case report. Int J Clin Exp Pathol. 2015;8(7):8676–9. [PMC free article] [PubMed] [Google Scholar]
- 119.Jacob J, Odink A, Brun AL, et al. Functional associations of pleuroparenchymal fibroelastosis and emphysema with hypersensitivity pneumonitis. Respir Med. 2018. May;138:95–101. doi: 10.1016/j.rmed.2018.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sugino K, Ono H, Watanabe N, et al. Acute exacerbation in chronic bird fancier’s lung with pleuroparenchymal fibroelastosis. Respirol Case Rep. 2021. Jan;9(1):e00693. doi: 10.1002/rcr2.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beynat-Mouterde C, Beltramo G, Lezmi G, et al. Pleuroparenchymal fibroelastosis as a late complication of chemotherapy agents. Eur Respir J. 2014. Aug;44(2):523–7. doi: 10.1183/09031936.00214713. [DOI] [PubMed] [Google Scholar]
- 122.Chen F, Matsubara K, Miyagawa-Hayashino A, et al. Lung transplantation for pleuroparenchymal fibroelastosis after chemotherapy. Ann Thorac Surg. 2014. Nov;98(5):e115–7. doi: 10.1016/j.athoracsur.2014.07.045. [DOI] [PubMed] [Google Scholar]
- 123.Goondi D, Franko A, Johannson KA. Pleuroparenchymal Fibroelastosis Post Liver Transplantation. Am J Respir Crit Care Med. 2021. Mar 4. doi: 10.1164/rccm.202009-3432IM. [DOI] [PubMed] [Google Scholar]
- 124.Kurosaki F, Bando M, Nakayama M, et al. Clinical features of pulmonary aspergillosis associated with interstitial pneumonia. Intern Med. 2014;53(12):1299–306. doi: 10.2169/internalmedicine.53.1578. [DOI] [PubMed] [Google Scholar]
- 125.Reddy TL, Tominaga M, Hansell DM, et al. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J. 2012. Aug;40(2):377–85. doi: 10.1183/09031936.00165111. [DOI] [PubMed] [Google Scholar]
- 126.Chaudhuri N, Spencer L, Greaves M, et al. A Review of the Multidisciplinary Diagnosis of Interstitial Lung Diseases: A Retrospective Analysis in a Single UK Specialist Centre. J Clin Med. 2016. Jul 27;5(8). doi: 10.3390/jcm5080066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grewal JS, Morisset J, Fisher JH, et al. Role of a Regional Multidisciplinary Conference in the Diagnosis of Interstitial Lung Disease. Ann Am Thorac Soc. 2019. Apr;16(4):455–462. doi: 10.1513/AnnalsATS.201811-794OC. [DOI] [PubMed] [Google Scholar]
- 128.Jones KD. Unclassifiable interstitial lung disease: a pathologist’s perspective. Eur Respir Rev. 2018. Mar 31;27(147). doi: 10.1183/16000617.0132-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** discussion of concepts related to unclassifiable ILD
- 129.Orr C, Jacobs W. Pulmonary Fibrosis. Radiology. 1926;7:318–325. [Google Scholar]
- 130.Takishima T, Shimura S Definition and Classification of Pulmonary Fibrosis. In: Takishma T, editor. Basic and Clinical Aspects of Pulmonary Fibrosis: CRC Press, Boca Raton, FL.; 1994. p. 293–303. [Google Scholar]
- 131.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011. Mar 15;183(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018. Sep 1;198(5):e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 133.Baumgartner KB, Samet JM, Stidley CA, et al. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997. Jan;155(1):242–8. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 134.Miyake Y, Sasaki S, Yokoyama T, et al. Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Ann Occup Hyg. 2005. Apr;49(3):259–65. doi: 10.1093/annhyg/meh090. [DOI] [PubMed] [Google Scholar]
- 135.Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc. 2006. Jun;3(4):293–8. doi: 10.1513/pats.200512-131TK. [DOI] [PubMed] [Google Scholar]
- 136.Ekstrom M, Gustafson T, Boman K, et al. Effects of smoking, gender and occupational exposure on the risk of severe pulmonary fibrosis: a population-based case-control study. BMJ Open. 2014. Jan 9;4(1):e004018. doi: 10.1136/bmjopen-2013-004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gulati M, Redlich CA. Asbestosis and environmental causes of usual interstitial pneumonia. Curr Opin Pulm Med. 2015. Mar;21(2):193–200. doi: 10.1097/MCP.0000000000000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.King TE Jr., Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014. May 29;370(22):2083–92. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 139.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014. May 29;370(22):2071–82. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 140.Blanc PD, Annesi-Maesano I, Balmes JR, et al. The Occupational Burden of Nonmalignant Respiratory Diseases. An Official American Thoracic Society and European Respiratory Society Statement. Am J Respir Crit Care Med. 2019. Jun 1;199(11):1312–1334. doi: 10.1164/rccm.201904-0717ST. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** discusses the role of occupational exposures in ILD
- 141.Lee CT, Adegunsoye A, Chung JH, et al. Characteristics and Prevalence of Domestic and Occupational Inhalational Exposures Across Interstitial Lung Diseases. Chest. 2021. Feb 20. doi: 10.1016/j.chest.2021.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Copeland CR, Collins BF, Salisbury ML. Identification and Remediation of Environmental Exposures in Patients With Interstitial Lung Disease: Evidence Review and Practical Considerations. Chest. 2021. Feb 18. doi: 10.1016/j.chest.2021.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** discusses importance of environmental remediation in patients with ILD
- 143.Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol. 2013. Nov 25;5:483–92. doi: 10.2147/CLEP.S54815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kalafatis D, Gao J, Pesonen I, et al. Gender differences at presentation of idiopathic pulmonary fibrosis in Sweden. BMC Pulm Med. 2019. Nov 27;19(1):222. doi: 10.1186/s12890-019-0994-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fang C, Huang H, Zhang Q, et al. Relation between sex hormones and leucocyte telomere length in men with idiopathic pulmonary fibrosis. Respirology. 2020. Dec;25(12):1265–1273. doi: 10.1111/resp.13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kreuter M, Raghu G. Gastro-oesophageal reflux and idiopathic pulmonary fibrosis: the heart burn in patients with IPF can no longer be silent. Eur Respir J. 2018. Jun;51(6). doi: 10.1183/13993003.00921-2018. [DOI] [PubMed] [Google Scholar]
- 147.Ghisa M, Marinelli C, Savarino V, et al. Idiopathic pulmonary fibrosis and GERD: links and risks. Ther Clin Risk Manag. 2019;15:1081–1093. doi: 10.2147/TCRM.S184291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kropski JA, Blackwell TS, Loyd JE. The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J. 2015. Jun;45(6):1717–27. doi: 10.1183/09031936.00163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Allen RJ, Guillen-Guio B, Oldham JM, et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2020. Mar 1;201(5):564–574. doi: 10.1164/rccm.201905-1017OC. [DOI] [PMC free article] [PubMed] [Google Scholar]; * role of genetics in IPF
- 150.Schwartz DA. Idiopathic Pulmonary Fibrosis Is a Genetic Disease Involving Mucus and the Peripheral Airways. Ann Am Thorac Soc. 2018. Nov;15(Suppl 3):S192–S197. doi: 10.1513/AnnalsATS.201802-144AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Spagnolo P, Cottin V. Genetics of idiopathic pulmonary fibrosis: from mechanistic pathways to personalised medicine. J Med Genet. 2017. Feb;54(2):93–99. doi: 10.1136/jmedgenet-2016-103973. [DOI] [PubMed] [Google Scholar]
- 152.Kim EJ, Elicker BM, Maldonado F, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. 2010. Jun;35(6):1322–8. doi: 10.1183/09031936.00092309. [DOI] [PubMed] [Google Scholar]
- 153.Wuyts WA, Cavazza A, Rossi G, et al. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014. Sep;23(133):308–19. doi: 10.1183/09059180.00004914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Basseri B, Enayati P, Marchevsky A, et al. Pulmonary manifestations of inflammatory bowel disease: case presentations and review. J Crohns Colitis. 2010. Oct;4(4):390–7. doi: 10.1016/j.crohns.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 155.Jankowich MD, Rounds SIS. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012. Jan;141(1):222–231. doi: 10.1378/chest.11-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lucas RM, McMichael AJ. Association or causation: evaluating links between “environment and disease”. Bull World Health Organ. 2005. Oct;83(10):792–5. doi: /S0042–96862005001000017. [PMC free article] [PubMed] [Google Scholar]
- 157.Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med. 2019. Oct 31;381(18):1718–1727. doi: 10.1056/NEJMoa1908681. [DOI] [PubMed] [Google Scholar]
- 158.Behr J, Prasse A, Kreuter M, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med. 2021. May;9(5):476–486. doi: 10.1016/S2213-2600(20)30554-3. [DOI] [PubMed] [Google Scholar]
- 159.McCrae K Immune thrombocytopenia: no longer ‘idiopathic’. Cleve Clin J Med. 2011. Jun;78(6):358–73. doi: 10.3949/ccjm.78gr.10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mullen SA, Berkovic SF, Commission IG. Genetic generalized epilepsies. Epilepsia. 2018. Jun;59(6):1148–1153. doi: 10.1111/epi.14042. [DOI] [PubMed] [Google Scholar]
- 161.Raj MA, Ranka S, Goyal A. Hypertrophic Obstructive Cardiomyopathy. StatPearls. Treasure Island (FL) 2021. [Google Scholar]
- 162.Ferrario F, Tadros M, Napodano P, et al. Rapidly progressive glomerulonephritis (RPGN): is there still an “idiopathic” subgroup? Adv Exp Med Biol. 1993;336:431–4. doi: 10.1007/978-1-4757-9182-2_76. [DOI] [PubMed] [Google Scholar]