

Abstract
Therapeutic resistance is the major challenge in clinic for patients with mantle cell lymphoma (MCL), an aggressive subtype of B-cell lymphoma. In addition to the FDA-approved Bruton’s tyrosine kinase (BTK) inhibitors, multiple clinical trials have demonstrated clinical benefits in targeting BCL-2 by venetoclax and reported to greatly improve clinical outcome for refractory/relapsed patients with MCL alone or in combination with BTK inhibitors. However, resistance to venetoclax is no exception and marks as a new clinic challenge. To decode the underlying mechanisms driving venetoclax resistance, we established two MCL cell lines, Mino-Re and Rec1-Re, with acquired resistance to venetoclax from sensitive Mino and Rec-1. Using reverse phase protein assay (RPPA), an agnostic proteomic approach, we identified targetable signaling pathways that are associated with acquired venetoclax resistance in Mino-Re and Rec1-Re cells. A panel of pro-survival signals was identified to correlate well with venetoclax-resistance, including increased expression of MCL-1, BCL-xL and AKT phosphorylation, and decreased expression of BIM, BAX and PTEN. Based on a high throughput drug screening of over 320 FDA-approved/investigational drugs in the paired venetoclax-sensitive and -resistant cell lines Mino-Re and Rec1-Re, we identified the top candidates that are capable to overcome acquired venetoclax resistance in these cells. The best candidate is PIK-75, a dual inhibitor targeting both PI3K and CDK9. Its action to overcome venetoclax resistance was further confirmed in additional cell lines with primary venetoclax resistance (n=4) and primary patient samples (n=21). Mechanistically, PIK75 treatment potently diminished the elevated MCL-1 expression and AKT activation in cells with acquired or primary venetoclax resistance and resulted in potent anti-MCL activity to overcome these resistances. In addition, PIK75 is also potent in overcoming tumor microenvironment (TME)-associated venetoclax resistance. Furthermore, PIK-75 treatment is efficacious in overcoming primary and acquired venetoclax resistance in xenograft models and inhibited tumor cell dissemination to spleen in mice. Altogether, our data demonstrated that PIK-75 is highly potent in overcoming primary, acquired, or stromal cells-induced venetoclax resistances in MCL cells and revealed a new tumor vulnerability that can be exploited clinically in difficult to treat MCL cases, especially those with venetoclax resistance.
Keywords: PIK75, venetoclax resistance, PI3K, MCL-1, AKT, mantle cell lymphoma
Introduction
Targeting the B-cell leukemia/lymphoma-2 (BCL-2) protein has shown potent therapeutic effect in hematologic malignancies [1-3]. BCL-2 specific inhibitor venetoclax has been approved for treating chronic lymphocytic leukemia (CLL) and has been utilized alone or in combination with BTK inhibitors in numerous clinical trials and demonstrates clinical benefits in other hematologic cancers [4], including mantle cell lymphoma (MCL) [5,6]. However, drug resistance develops frequently and is one of the major challenges for relapsed/refractory (R/R) patients. Although resistance to venetoclax has been studied in several hematologic cancers, its mechanism has not been well defined in MCL yet. Discovering the potential mechanisms of venetoclax resistance lays a foundation for preventing its occurrence or developing new therapeutic strategies to overcome it. Three main mechanisms have been reported to contribute to venetoclax resistance: 1) acquired mutations in BCL-2 family proteins with proliferation-enhancing effects under the selection pressure or aberrant expression of antiapoptotic genes, such as MCL-1 and BCL-xL; 2) dysregulated signaling pathways, such as the PI3K-AKT pathway; and 3) interaction with TME such as stromal cells, which protect cancer cells through cell-cell interaction and cytokine secretion [7-9].
Importantly, many of these dysregulated proteins that play crucial roles in the acquired resistance, including PI3K-AKT/mTOR and B-cell receptor signaling, are also targetable and thus offer potential strategies for overcoming venetoclax resistance. Particularly, novel compounds that inhibit MCL-1 and BCL-xL have shown great efficacy in targeting venetoclax resistance [10-12]. Additionally, TME-associated venetoclax resistance can be counteracted by CD20-directed antibodies or by PI3K-AKT kinase dependent inhibitors.
To uncover the mechanisms of venetoclax resistance and develop a strategy that is tailored to the MCL characteristics, we first generated two venetoclax-resistant MCL cell lines and patient-derived primary cells. RPPA analysis as an agnostic approach was used to investigate the mechanism of acquired venetoclax resistance in these cells, and high throughput drug screening was applied to find candidates to potentially overcome this resistance. The potency of the best candidates was further confirmed in MCL cell lines with primary resistance and primary patient samples with primary resistance or TME-associated resistance. We found that PIK-75, a PI3K/CDK9 dual inhibitor, had great efficacy in inhibiting cell growth in vitro and in vivo not only in MCL cells with acquired resistance, but also in those with primary or TME-associated resistance in vitro and in vivo. Mechanistically, we demonstrated that PIK-75 actions through inhibiting PI3K-AKT signaling and blocking MCL-1 expression to induce cell apoptosis and thus overcomes venetoclax resistance in MCL cells.
Materials and methods
MCL primary tumor samples and cancer cell lines
Twenty-one MCL patient samples were collected after obtaining informed consent and approval from the Institutional Review Board at The University of Texas MD Anderson Cancer Center (MDACC). The patient samples were processed and cryopreserved before use.
MCL cell lines Mino, Rec-1, Maver-1, and Z-138 and human stromal cell line HS-5 were purchased from ATCC (Manassas, VA, USA). Granta-519 cells were kindly provided by Dr. Felipe Samaniego from MDACC (Houston, TX, USA). JeKo-R cells with acquired ibrutinib resistance were generated from ibrutinib-sensitive JeKo-1 cells and provided by Dr. Lan Pham at MDACC. SP-49 cells were kindly provided by Dr. Jianguo Tao at Moffitt Cancer Center (Tampa, FL, USA). JeKo-luc cells were derived from JeKo-1 cells to stably express the luciferase protein for bioluminescence in vivo studies. All MCL cell lines were cultured in RPMI 1640 medium with 10% FBS and 1% penicillin/streptomycin. HS-5 cells were maintained in DMEM medium with 10% FBS and 1% penicillin/streptomycin.
Drugs
PIK-75 (S1205), GSK1059615 (S1360), venetoclax (S8048), and a drug library of 320 drugs (FDA-approved/investigational) were purchased from Selleckchem (Houston, TX, USA).
Induction of venetoclax resistance in cell lines and primary samples in vitro
Two venetoclax-sensitive MCL cell lines Mino and Rec-1 were subjected to long-term exposure to venetoclax using a stepwise dose-increase approach from 1 nM to 1000 nM (Figure 1A). The stable resistant cells Mino-Re and Rec1-Re were cryopreserved for future use.
Figure 1.
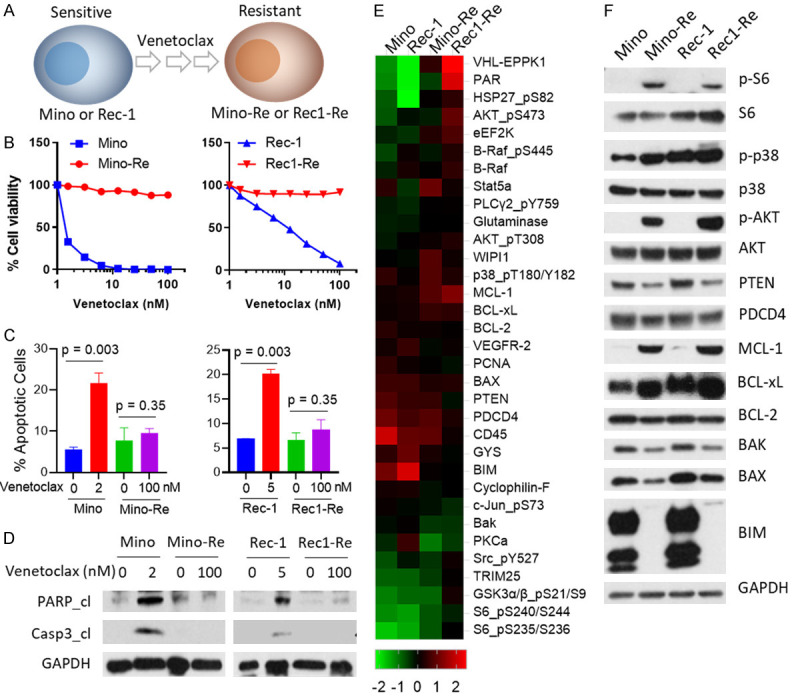
Establishment of venetoclax-resistant cell lines and differential gene expression associated with acquired venetoclax resistance. (A) Two sensitive MCL cell lines (Mino and Rec-1) were exposed to increasing doses of venetoclax stepwise up to 1000 nM. (B) 8-dose cell viability was performed in both parental and resistant cell lines for 72 h. Each treatment for the cell viability assay was set up in triplicate. (C) Mino cells were treated with 2 nM venetoclax, Rec-1 cells were treated with 5 nM venetoclax, the corresponding resistant cells were treated with 100 nM venetoclax for 24 h, and apoptosis was measured by annexin-V/PI assay. Annexin-V+PI- subpopulation and annexin-V+PI+ subpopulations were summed up as total apoptosis. Each treatment for the cell apoptosis assay was set up in triplicate. (D) Cells with the same treatments as described in (C) were collected for western blot. Total PARP, cleaved PARP, total caspase-3, and cleaved caspase-3 were detected in these cell lines. GAPDH was used as internal loading control. Each experiment in (B-D) was repeated three times. (E) Heatmaps of dysregulated proteins. 5×106 cells of each cell line (Mino, Mino-Re, Rec-1 and Rec1-Re) were collected and subjected to RPPA analysis. 425 proteins in total were detected, and proteins with at least two-fold change were selected for generating the heatmap. High-expressing proteins are shown in red, and low-expressing proteins are shown in green. Each sample for RPPA analysis was set up in triplicate. (F) Western blotting was used to confirm dysregulated protein expression comparing parental and resistant cell lines.
To establish venetoclax-resistant primary samples, primary cells from two venetoclax-sensitive patients were co-cultured with the stromal cell line HS-5 and cytokine cocktail (50 ng/ml of IL-10, 50 ng/ml of BAFF, 1 ng/ml of IGF-1, 1 ng/ml of IL-6, and 50 ng/ml CD40L). The fibroblast-like HS-5 cells secrete significant levels of cytokines to promote the survival and growth of macrophages, including granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage-CSF (GM-CSF), macrophage-CSF (M-CSF) and macrophage-inhibitory protein-1 alpha [13]. IL-10, BAFF and CD40L can activate macrophages [14]. Tumor associated macrophages (TAMs) have been shown to correlate with poor prognosis in various types of cancer including solid cancer and hematologic malignancies [15]. Together with the supplemental cytokines, TAMs can be activated to promote the survival and growth of primary MCL cells. HS-5 cells were pre-treated with 10 μg/ml mitomycin-C for 2 h before the co-culture system set-up. The ratio of HS-5: primary MCL cells was 1:10. Primary cells were isolated from the stromal cells once a week and diluted in fresh medium with HS-5 cells and a cytokine cocktail.
High throughput drug screening
A library of 320 FDA-approved/investigational drugs from Selleckchem (Houston, TX) was used for a high throughput drug screening. Four cell lines (Mino, Rec-1, Mino-Re and Rec1-Re) were treated with each drug at 5 µM for 72 h, and cell viability was determined as described previously.
Cell viability and cell apoptosis assay
These assays were performed as described previously [16].
RPPA analysis
5×106 cells of each cell line with/without treatment with vehicle or 50 nM PIK-75 in triplicate for 24 h were subjected to RPPA analysis as described previously [16]. In total, 425 antibodies and four secondary antibodies (as negative controls) were included in the analysis. NormLog2_MedianCentered values were selected for heatmap generation. We set up the parental cell lines or untreated cell lines as “1” (upregulated as compared with resistant or treated cell lines) or “-1” (downregulated as compared with resistant or treated cell lines). The differences in proteins values between parental (or untreated) samples and resistant (or treated) samples < and >0.5 were selected for the heatmap. The heatmap was generated in Cluster 3.0, visualized in Treeview, and presented as a high-resolution Bmp format. Each treatment was set up in triplicate.
Western blotting
Western blotting was performed as described previously [16]. All primary antibodies against PARP, cleaved PARP, Caspase-3, cleaved Caspse-3, BIM, BAX, BAK, BCL-2, BCL-xL, MCL-1, p-S6, S6, p-AKT, AKT, p-p38, p38, PDCD4, PTEN, PDCD4, and GAPDH and secondary antibodies (anti-mouse or anti-rabbit) were purchased from Cell Signaling Technology (Danvers, MA, USA) and used at the manufacturer’s suggested concentration.
Animal studies
All the animal studies were performed after obtaining approval from the Institutional Animal Care and Use Committee, at MDACC.
In vivo long-term drug efficacy evaluation
Approximately 5×106 cells of Rec1-Re or luciferase-engineered JeKo-luc were injected into each 6-8-week-old female NSG mice. Once the tumors became palpable, the mice (n=5 per group) were treated with vehicle, venetoclax (10 mg/kg, oral, daily), or PIK-75 (10 mg/kg, IP, daily). Mouse blood was collected weekly. Human beta-2-microglobulin (B2M) level was detected by ELISA (Thermo Fisher Scientific) according to the manufacturer’s instructions. Tumor burden was measured weekly by caliper or by IVIS Luminescence In Vivo Imaging System (IVIS100, PerkinElmer, USA). Tumor volume was calculated using the following formula: tumor volume (mm3) = length (mm) × width (mm)2/2. Total flux was calculated based on live imaging data. Tumor weights were measured at the end of experiments.
Statistical analysis
The IC50 values were calculated from dose-dependent drug inhibition values using GraphPad Prism-8. Statistically significant differences among treatments were determined by 2-tailed Student t test. Experiments were repeated three times and representative results were presented, as appropriate.
Results
Establishment of MCL cell lines with acquired resistance to venetoclax
To understand the mechanism of venetoclax resistance, we first established two venetoclax-resistant cell lines Mino-Re and Rec1-Re to have acquired resistance to venetoclax at 1000 nM in culture from parental venetoclax-sensitive cell lines Mino (IC50=1.5 nM) and Rec-1 (IC50=10.9 nM), respectively (Figure 1A, 1B and Table 1). Venetoclax treatment at 2 nM (P=0.003) or 5 nM (P=0.003) induced robust cell apoptosis (Figure 1C), PARP cleavage and caspase-3 cleavage in Mino and Rec-1 cells, respectively, but not in Mino-Re and Rec1-Re cells with acquired resistance (Figure 1D). The resistant phenotype was stable even in the absence of venetoclax for >20 passages indicating that the developed venetoclax-resistant phenotype was stable and acquired survival and growth advantage changes without the need for venetoclax for cell re-charging. Therefore, these paired cell lines were ideal cellular models to study the mechanism underlying venetoclax resistance.
Table 1.
IC50 levels of venetoclax, GSK1059615 and PIK-75 treatments in MCL cell lines
| Cell Line | Venetoclax (nM)* | GSK1059615 (nM)* | PIK-75 (nM)* |
|---|---|---|---|
| Mino | 1.50 | 3.35 | 3.82 |
| Mino-Re | >100 | 5.71 | 1.50 |
| Rec-1 | 10.94 | 5.82 | 6.09 |
| Rec1-Re | >100 | 6.59 | 10.90 |
| Maver-1 | 2.92 | 7.68 | 7.15 |
| Granta519 | 5.76 | 15.21 | 17.19 |
| JeKo-1 | 147.50 | 7.03 | 9.56 |
| Z-138 | 163.90 | 7.05 | 8.09 |
| JeKo-R | >100 | 4.97 | 5.62 |
| SP-49 | >100 | 5.27 | 5.14 |
IC50 values were calculated 72 h post-treatment.
Dysregulated apoptosis pathway and PI3K-AKT signaling confer venetoclax resistance in MCL
The mechanism of venetoclax resistance may vary from one cancer type to another, but factors disturbing the balance between pro- and anti-apoptotic proteins always play a major role in driving venetoclax resistance. RPPA is an unbiased proteomics method that can simultaneously characterize hundreds of proteins by their expression and phosphorylation status, and therefore emerged as our preferred method for interrogating the status of protein profiles and signaling networks. We found that both the pro-apoptotic and anti-apoptotic family members were significantly off balance in resistant cell lines Mino-Re and Rec1-Re compared to parental cell lines Mino and Rec-1 (Figure 1E). For example, anti-apoptotic proteins such as MCL-1 and BCL-xL were significantly up-regulated, while pro-apoptotic proteins such as BIM, BAX, and BAK were down-regulated in the venetoclax-resistant cell lines. The BCL-2 itself was slightly decreased in the resistant cells. Of interest, pro-survival and stress-related proteins such as AKT, S6, and p38 were also activated in venetoclax-resistant cell lines. These findings were further validated by western blotting in these paired venetoclax-sensitive and -resistant cell lines (Figure 1F).
High throughput drug screening identified PIK-75 and GSK1059615 as the top candidates that overcome venetoclax resistance
To find candidate compounds that can overcome venetoclax resistance, we performed a high throughput drug screening over a library consisting of 320 drugs FDA-approved or under clinical investigation in the paired venetoclax-sensitive and -resistant cell line models (Mino vs Mino-Re and Rec-1 vs Rec1-Re) (Figure 2A). Forty-four drugs (at 5 µM) were identified to inhibit at least 80% cell viability compared to the DMSO control (Figure 2A, 2B). These drugs were further grouped based on their targeted signaling pathways, and the five most effective groups targeted PI3K-AKT, DNA topoisomerase, Aurora, VEGFR, and mTOR signaling pathways. The top six drugs (4 targeting PI3K and 2 targeting Aurora kinase) were chosen for validation via a four-dose-dependent cell viability assay (Figure 2C). As a result, PIK-75 and GSK1059615 showed the best efficacy in the paired venetoclax-sensitive and -resistant cell lines. PIK-75 is a dual inhibitor targeting PI3K and CDK9, and has been shown to be selectively potent in targeting AML cells but with significantly less sensitivity to CD34+ normal bone marrow progenitor cells [17]. GSK1059615 is a dual inhibitor targeting PI3K and mTOR pathways [18].
Figure 2.
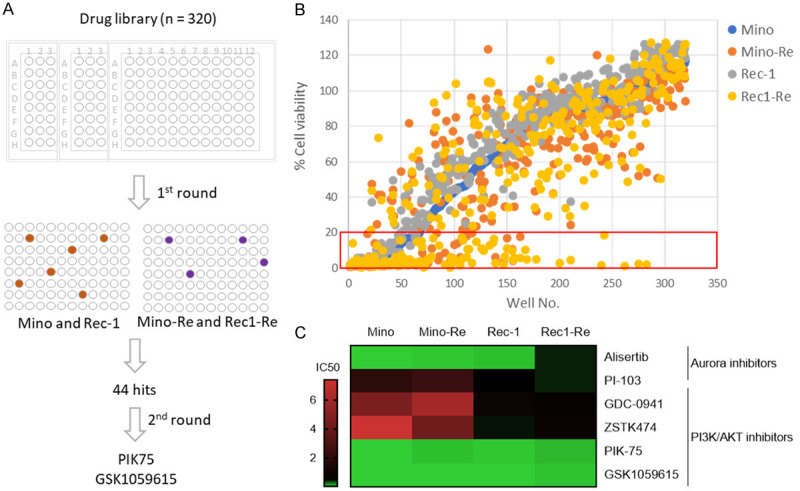
High throughput drug screening to identify candidates for overcoming venetoclax resistance. (A) Schematic illustration of the high-throughput drug screen. The drug screen was performed over a library consisting of 320 drugs in two parental and venetoclax-resistant paired cell lines by a 72 h cell viability test with one dose (5 μM) as first round screen followed by 2nd round drug validation via 4-dose viability assay in the same cell lines to identify the top candidates in overcoming venetoclax resistance. (B) 1st round drug screen at 5 μM by a cell viability inhibition assay in Mino, Mino-Re, Rec-1 and Rec1-Re cells. Each treatment for the cell viability assay was set up in triplicate. Relative cell viability was normalized to DMSO control. Cell viability less than 20% was considered as positive (red box). (C) 2nd round drug validation for the top candidates (six drugs) was performed by a four-dose (three-fold dilution) cell viability assay at 72 h in the same cell lines. The heatmap shown represents the IC50 values (µM). Each treatment for the cell viability assay was set up in triplicate and the experiment was repeated three times.
PIK-75 is highly potent in overcoming a variety of venetoclax resistances
To further evaluate the potency and potential of PIK-75 and GSK1059615 in overcoming venetoclax resistance, we performed an eight-dose cell viability assay. Interestingly, both PIK-75 and GSK1059615 were potent inhibitors with IC50=1.5-10.9 nM and 3.4-6.6 nM at 72 hours upon treatment, respectively, in the paired venetoclax-sensitive and -resistant cell lines (Figure 3A and Table 1). This demonstrated that both compounds are potent in overcoming acquired venetoclax resistance.
Figure 3.
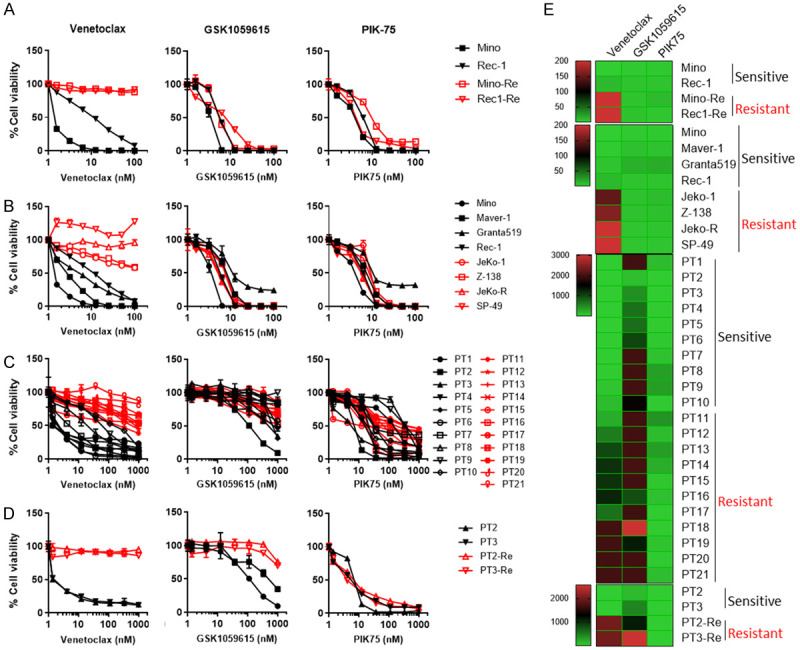
PIK-75 is potent in overcoming a variety of venetoclax resistances. (A, B) In vitro efficacy of BCL2i venetoclax, PI3Ki PIK-75 and GSK1059615 in paired venetoclax-sensitive (Mino and Rec-1) and -resistant (Mino-Re and Rec1-Re) cell lines (A), and additional 8 MCL cell line panel (B) by eight-dose cell viability assay at 72 hours upon treatment. (C) In vitro efficacy of venetoclax, PIK-75 and GSK1059615 in 21 primary MCL patient samples at 24 hours upon treatment. (D) Two venetoclax-sensitive (ex vivo) patient samples (PT2 and PT3) were co-cultured with HS-5 stromal cells and supplemental cytokines for two weeks. An eight-dose 24-hour cell viability assay was conducted. Each treatment for the cell viability assay was set up in triplicate. (E) Heatmaps of IC50 of venetoclax, GSK1059615 and PIK-75 in MCL cell lines and primary patient samples with/without coculture with HS-5 cells based on the data (A-D). Sensitivity to venetoclax was indicated in the right for each cell line or patient sample.
To test whether PIK-75 is also capable to overcome primary venetoclax resistance, in addition to acquired venetoclax resistance, we screened an additional panel of eight MCL cell lines for cell viability to venetoclax and PIK-75. Two cell lines, Maver-1 and Granta-519, were identified as venetoclax-sensitive (IC50=2.9 and 5.8 nM, respectively), in addition to Mino and Rec-1. Four cell lines JeKo-1, JeKo R, Z138 and SP-49 were venetoclax-resistant (IC50>100 nM). Consistently, both PIK-75 (IC50=1.5-17.2 nM) and GSK1059615 (IC50=3.4-15.2 nM) were potent in targeting all these MCL cell lines (Figure 3B, 3E and Table 1).
To ascertain the clinical potential of PIK-75 and GSK1059615, we next asked if this also applies to primary patient samples. Among a panel of 21 primary MCL patient samples, PT 1-10 were determined to be venetoclax sensitive (IC50=5.2-125.1 nM) and PT 11-21 were resistant (IC50>244.8 nM), based on IC50 values at 24 hours upon treatment (Figure 3C, 3E and Table 2). Intriguingly, PIK-75 showed higher potency in targeting primary patient samples ex vivo (IC50=6.3-425.2 nM) while only a partial efficacy was observed for GSK1059615 (IC50>110.7 nM) (Figure 3C, 3E and Table 2). These data suggested that PIK-75 may have more clinical potential than GSK1059615 in targeting MCL cells and in overcoming venetoclax resistance in primary MCL patient samples.
Table 2.
IC50 levels of venetoclax, GSK1059615 and PIK-75 treatments in patient samples
| Patient ID | Venetoclax (nM) | GSK1059615 (nM) | PIK-75 (nM)* |
|---|---|---|---|
| PT1 | 5.2 | >2000 | 121.2 |
| PT2 | 2.6 | 110.7 | 6.8 |
| PT3 | 2.8 | 458.4 | 5.9 |
| PT4 | 4.6 | 687.1 | 16.4 |
| PT5 | 2.2 | 707.0 | 20.9 |
| PT6 | 2.4 | 1003.0 | 35.2 |
| PT7 | 5.7 | >2000 | 84.9 |
| PT8 | 22.0 | >2000 | 335.8 |
| PT9 | 58.4 | >2000 | 347.1 |
| PT10 | 125.1 | 1574.0 | 28.8 |
| PT11 | 244.8 | >2000 | 428.1 |
| PT12 | 596.2 | >2000 | 123.0 |
| PT13 | 986.1 | >2000 | 344.9 |
| PT14 | 1159.0 | 2024.0 | 231.7 |
| PT15 | 1141.0 | >2000 | 52.0 |
| PT16 | 1243.0 | 943.0 | 92.2 |
| PT17 | 685.9 | 1971.0 | 20.9 |
| PT18 | >2000 | 3030.0 | 16.4 |
| PT19 | >2000 | 1311.0 | 74.5 |
| PT20 | >2000 | >2000 | 29.9 |
| PT21 | >2000 | >2000 | 119.4 |
IC50 values were calculated 24 h post-treatment.
It is well known that TME plays a critical role in drug resistance in many cancer types [19]. To mimic TME-associated venetoclax resistance, we co-cultured two primary patient samples PT2 (from a clinically venetoclax resistant patient) and PT3 (from a venetoclax-naïve patient), with human stromal cell line (HS-5) and a panel of supplemental cytokine cocktail (see Materials and Methods). Both PT2 and PT3 were sensitive to venetoclax ex vivo (IC50=3.1 and 3.4 nM, respectively) in the absence of co-culture and became highly resistant to venetoclax (IC50>2000 nM) after two weeks of co-culture (Figure 3D, 3E, left panel). Interestingly, PIK-75 showed high potency at similar levels in targeting both parental venetoclax-sensitive patient samples and their derived samples with TME-associated venetoclax resistance (IC50=5.8-9.0 nM) (Figure 3D, 3E, right panel). In contrast, GSK1059615 was only slightly effective in the parental samples (IC50=110.7 and 458.4 nM, respectively) and much less potent in those with TME-associated venetoclax resistance (IC50=1112 and 2566 nM, respectively) (Figure 3D, 3E, middle panel). Together, these data support the notion that PIK-75 is more potent than GSK1059615 in overcoming primary, acquired, and TME-associated venetoclax resistance in primary patient samples and therefore, PIK-75 was chosen for further investigation.
PIK-75 overcomes venetoclax resistance by blocking inhibiting PI3K-AKT signaling and MCL-1 expression
We showed by RPPA and subsequent western blot that up-regulation of pro-survival molecules MCL-1 and BCL-xL and activation of PI3K-AKT signaling were the major events in driving venetoclax-acquired resistance (Figure 1E, 1F), indicating that targeting these pathways has the potential to overcome the resistance. To understand the mechanism of PIK-75 action, we treated both Rec-1 and Rec1-Re cells with 50 nM PIK-75 for 24 h and performed RPPA analysis again as an unbiased proteomics approach. Multiple proteins were dysregulated in both cell lines upon PIK-75 treatment. In particular, PIK-75 induced loss of MCL-1 protein expression in both MCL-1 high-expressing Rec1-Re cells (venetoclax-resistant) and MCL-1 low-expressing Rec-1 cells (venetoclax-sensitive) (Figure 4A). AKT phosphorylation was also dramatically reduced in PIK-75-treated cells compared to the DMSO control, in both Rec-1 and Rec1-Re cells (Figure 4A). To verify this, we treated both Rec-1 and Rec1-Re cells with designated concentrations of venetoclax and PIK-75 for 24 h and then tested the expression of MCL-1 and AKT phosphorylation by western blotting. As expected, PIK-75 potently blocked MCL-1 expression and reduced AKT phosphorylation in both cell lines (Figure 4B). Of note, expression of another pro-apoptosis protein BAK was not dramatically affected by PIK-75 treatment. Based on RPPA analysis, caspase-3 and -7 cleavage were highly induced upon PIK-75 treatment. Consistently, PIK-75 at 10 and 50 nM induced dose-dependent cell apoptosis in both Rec-1 (P=0.0030 and 0.0002, respectively) and Rec1-Re cells (P=0.0014 and 0.0002, respectively). PIK-75 at either dose was more potent in targeting Rec1-Re cells than venetoclax at 100 nM (P=0.031 and 0.0003, respectively) (Figure 4C). Consistently, higher levels of cleaved PARP and caspase-3 were observed in Rec1-Re cells treated with PIK-75 compared to venetoclax (Figure 4B). Together, these data demonstrated that PIK-75 induces cell apoptosis and overcomes venetoclax resistance via inhibiting PI3K-AKT signaling and blocking MCL-1 expression.
Figure 4.
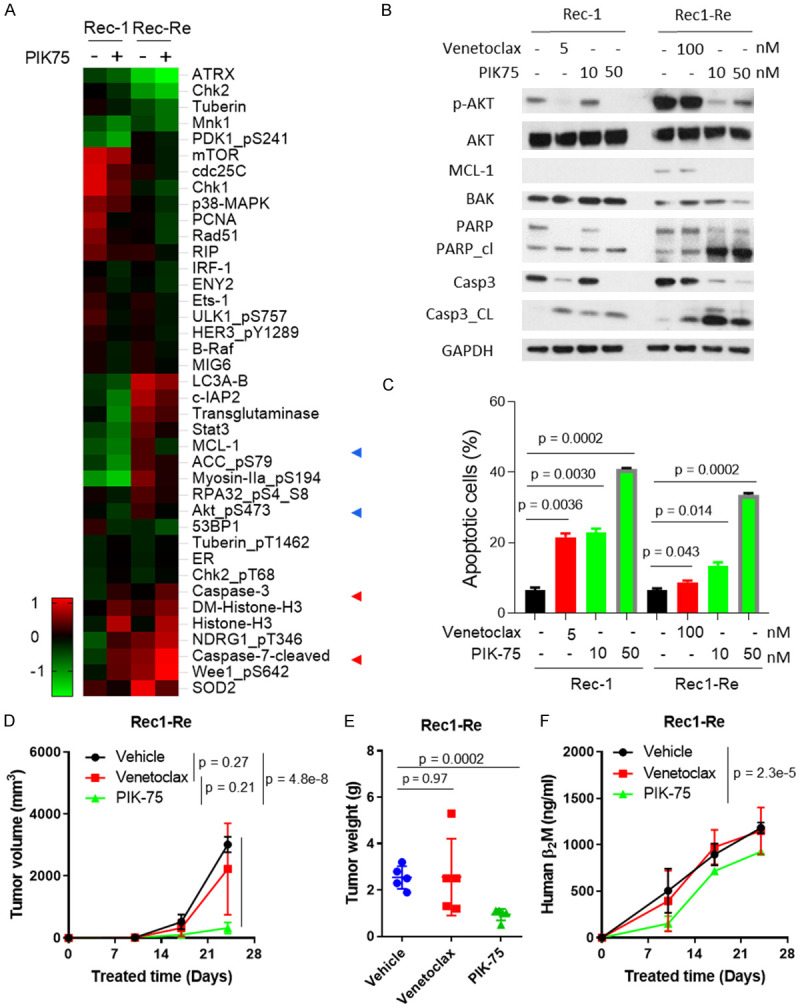
PIK-75 inhibits PI3K-AKT activity and overcomes acquired venetoclax resistance in vitro and in vivo. (A) Rec-1 and Rec1-Re cells were treated with 50 nM PIK-75 for 24 h and subjected to RPPA analysis. A heatmap was made with NormLog2_MedianCentered values that were < and >0.5 times between untreated and treated samples. (B, C) Rec-1 and Rec1-Re cells were treated with designated venetoclax or PIK-75 concentrations for 24 h. Cells were harvested and analyzed by western blotting (B) and cell apoptosis assay (C) to confirm the RPPA data and biological effects. (D-F) 5×106 Rec1-Re cells were injected subcutaneously into each NSG mouse and treated with vehicle, 50 mg/kg venetoclax, or 10 mg/kg PIK-75, daily. Tumor volumes were measured weekly (D), and mouse blood was collected weekly; human B2M in mouse serum was measured by ELISA assay (F). Tumor weight was measured at the end of experiment (E). Student t test was used for statistical analysis, n=5.
PIK-75 blocks in vivo homing of MCL cells to the mouse spleen
PI3K-AKT signaling plays a crucial role in regulating the tumor-TME interplay and targeting PI3K-AKT signaling has been shown to block malignant B-cell homing [20,21]. Therefore, we hypothesized that PIK-75 may also interfere with the malignant B-cell homing to different sites via targeting PI3K. To address this, we performed a short-term in vivo B-cell homing assay using a clinically-assessed venetoclax-resistant primary patient sample (PT2) (Figure 3D). The MCL cells homed predominantly to the spleen instead of the bone marrow site (Figure 5A, 5B). A 30 min pretreatment with PIK-75 at 100 nM inhibited MCL homing to mouse spleen at 16 h post-treatment (P=0.011) (Figure 5A, 5B). We also observed a concomitant of increased retention of circulating tumor cells in the blood of mice injected with PIK-75-pretreated MCL cells, compared to vehicle-pretreated cells (P=0.025). As expected, PIK-75 pretreatment did not induce cell apoptosis in ex vivo culture (data not shown). Therefore, these data may suggest that ex vivo short-term PIK-75 pretreatment blocks MCL homing to mouse spleen, likely via targeting PI3K-AKT mediated tumor-TME interplay.
Figure 5.
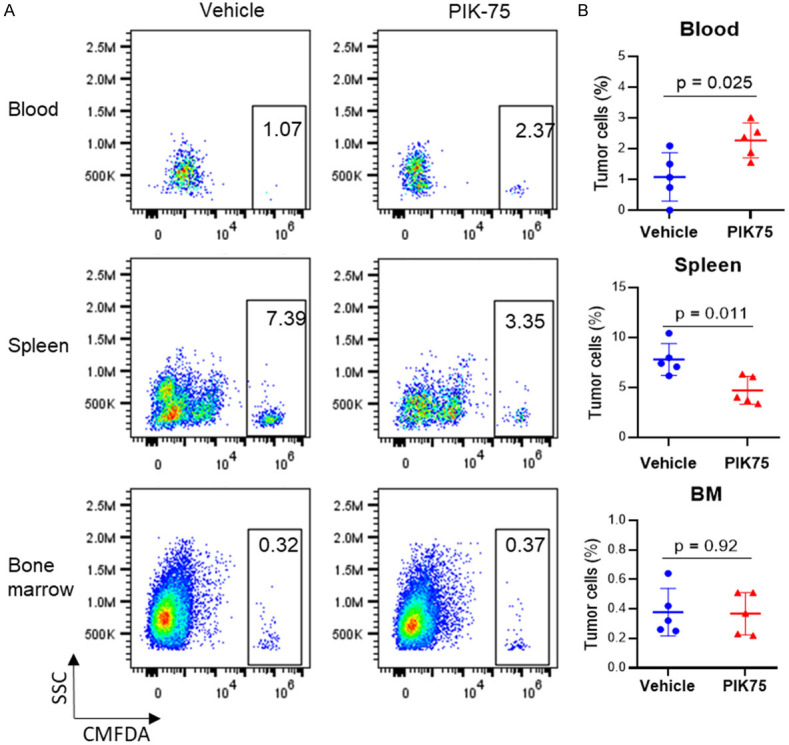
PI3K inhibitor PIK-75 blocks in vivo homing of MCL cells to mouse spleen. (A) CMFDA-labelled patient cells (PT2) were pre-treated with vehicle (DMSO) or PIK-75 at 100 nM for 30 min before tail vein inoculation into NSG mice. Blood, spleen, and bone marrow were collected at 16 hours post inoculation for tumor cell detection by flow cytometry based on CMFDA labeling (x-axis). (B) Percentage of tumor cells was quantified based on flow cytometry analysis. Student t test was used for statistical analysis, n=5.
PIK-75 overcomes acquired and primary venetoclax resistance in mouse xenograft models
To test the in vivo potency of PIK-75 in overcoming venetoclax resistance, we generated cell line-derived xenograft (CDX) models by subcutaneous injection of Rec1-Re cells. The mice bearing xenografts were treated with vehicle, venetoclax (10 mg/kg, oral, daily), or PIK-75 (10 mg/kg, IP, daily). As expected, venetoclax failed to inhibit tumor growth in Rec1-Re CDXs (P=0.21) confirming the in vivo venetoclax-resistant phenotype (Figure 4D-F). In contrast, PIK-75 reduced tumor burden (P=0.0002) and production of B2M significantly (P=0.000002) as compared to the vehicle-treated group (Figure 4D-F). In addition, no gross abnormality was detected in mice treated with PIK-75. This demonstrated that PIK-75 is safe and highly potent in overcoming acquired venetoclax resistance in vivo. Next, we tested if this also applies to primary resistance. JeKo-1, Z138, JeKo R and SP-49 were intrinsically resistant to venetoclax while highly sensitive to PIK-75 treatment (IC50=5.1-9.6 nM) (Figure 3B, 3E and Table 1). Similar to the Rec1-Re venetoclax resistance model system, PIK-75 at 10 nM induced robust cell apoptosis in JeKo-1 as evident by PARP cleavage and caspase-3 cleavage, and diminished expression of MCL-1 and AKT phosphorylation (Figures 4B and 6A). Furthermore, PIK-75 (P<0.05), but not venetoclax (P=0.64), effectively inhibited tumor growth of luciferase-expressing JeKo-luc derived xenografts in vivo, compared to vehicle-treated group (Figure 6B-D). Again, no gross abnormality was detected in mice treated with PIK-75. Altogether, these data demonstrated that PIK-75 is a safe and potent candidate for overcoming primary and acquired venetoclax resistance in MCL.
Figure 6.
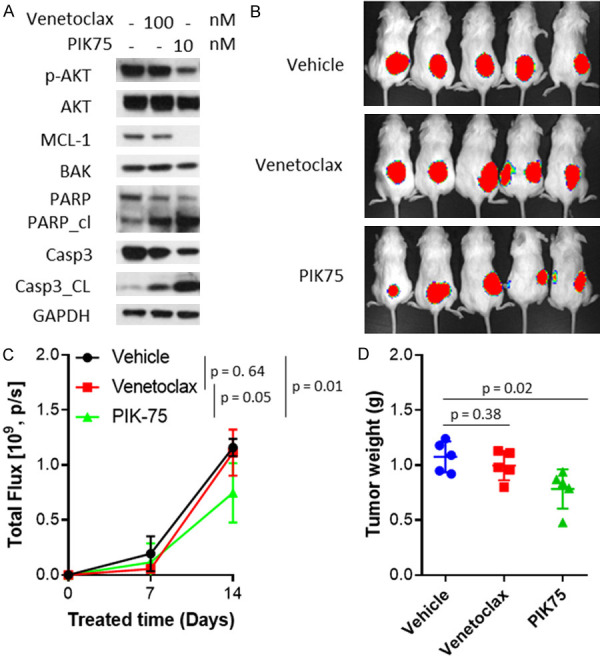
PIK-75 overcomes primary venetoclax resistance in mouse xenograft models. (A) JeKo-1 cells were treated with venetoclax at 100 nM or PIK-75 at 10 nM for 24 hours. Cells were harvested and analyzed by western blotting. (B-D) 5×106 luciferase-expressing JeKo-luc cells were injected subcutaneously into each NSG mouse and treated with vehicle, 50 mg/kg venetoclax, or 10 mg/kg PIK-75 daily. Tumor load was measured weekly by bioluminescence imaging (B) and total flux was calculated based on live imaging data (C). Tumor weight was measured at the end of the experiment (D). Student t test was used for statistical analysis, n=5.
Discussion
In this study, we demonstrated that PIK75 targeting both PI3K and CDK9, is potent for overcoming primary, acquired and TME-associated venetoclax resistance in MCL by suppressing PI3K-AKT signaling and blocking MCL-1 expression (Supplementary Figure 1). PIK-75 therapeutic efficacy has been reported in acute myeloid leukemia (AML); the mechanism involves primarily dual inhibition of the PI3K pathway and CDK9-mediated MCL-1 transcription [22]. PIK-75 targets PI3Kα isoform and dissociates the interaction between BCL-xL and BAK [23]. In this study, PIK-75 was one of the top hits from the high throughput drug screening, for the first time, it was shown to overcome venetoclax resistance. AKT phosphorylation was significantly diminished in both venetoclax-sensitive and -resistant cells, and MCL-1 expression elevated in venetoclax-resistant cells was effectively blocked upon PIK75 treatment. This demonstrates its potency in overcoming venetoclax resistance. The tumor microenvironment plays an important role in drug resistance and subsequently, drugs that are capable of targeting tissue homing, such as the BTK inhibitor ibrutinib, have an increased therapeutic efficacy [24,25]. Interestingly, we showed that PIK-75 is also able to inhibit B-cell homing to the spleen in a short-term in vivo homing assay. This indicates that PIK-75 may overcome tissue niche-specific drug resistance by also targeting the tumor-microenvironment crosstalk. Taken together, PIK-75 has potential to overcome venetoclax resistance in MCL by dually targeting PI3K and MCL-1 and by interfering with the tumor microenvironment.
Cell apoptosis is tightly regulated by a balance between pro-apoptotic and anti-apoptotic proteins. Sensitivity to venetoclax relies not only on BCL-2 expression, but also on the expression of other BCL2 family proteins, as they form complexes to coordinately mediate cell apoptosis [3,26]. In CLL, phosphorylation of BCL-2 indicates the status of venetoclax resistance, as BCL-2 phosphorylation inhibits interaction between venetoclax and the BCL-2/BIM/BAX complex [27]. In multiple myeloma (MM), sensitivity to venetoclax is correlated with high BCL-2 and low BCL-xL or MCL-1 expression [28]. We have previously reported that sensitivity of MCL or DLBCL to venetoclax correlated strongly with the BCL-2 protein expression level, although other BCL-2 family members also modulate the therapeutic response [29]. For example, BCL-xL and MCL-1 expression correlated with venetoclax resistance, while BIM and BAK levels were negatively correlated with the resistance. Taken together, aberrant expression of BCL-2 family proteins may contribute to primary resistance and acquired resistance to venetoclax.
Gene mutations are also considered an important factor for venetoclax resistance. Acquisition of mutations within BCL-2 or BAX results in venetoclax resistance in MCL-like murine lymphoma cells and in a human MCL cell line (HBL-2), respectively [30]. Mutations (F101C or F101L) within the BCL-2 BH3 domain impair venetoclax binding, and BAK mutation interferes with its anchoring to the mitochondrial membrane [31]. However, these mutations were not identified in Rec1-Re and Mino-Re cells with acquired venetoclax resistance (data not shown). Instead, we revealed non-genetic aberration of PI3K signaling and elevated expression of MCL-1 and BCL-xL, which likely drive the venetoclax resistance in our system.
Acquired drug resistance models can be generated by chronic drug exposure or interaction with the microenvironment [7,9,30,32,33]. In MCL, chronic exposure of HBL-2 cells to venetoclax led to an increase in MCL-1 expression and moderate decrease in BIM expression [30]. Similar results were observed in a murine MCL model developed to be resistant to venetoclax. Upon stimulation by CD40 antibody or CD40L-expressed stromal cells, MCL cell lines and patient samples can acquire venetoclax-resistant properties [32]. This phenotype included dramatic up-regulation of BCL-xL and a slight up-regulation of MCL-1. For BCL-2 itself, changes in protein expression were not unanimously shared by all malignant phenotypes. Consistently, our results showed that a dysregulated balance between anti-apoptotic proteins such as MCL-1 and BCL-xL and pro-apoptotic proteins such as BIM, BAX, and BAK is the hallmark of venetoclax-resistant cells in MCL. Additionally, pro-survival and stress-related signaling pathways were elevated in the venetoclax-resistant cells. These included up-regulation of p-S6, p-AKT, and p-p38, also reported previously to contribute to venetoclax resistance [29,34-36]. Of interest, two tumor suppressors, PTEN and PDCD4, showed decreased expression in venetoclax-resistant MCL cells. We speculated that suppression of PTEN could lead to activation of AKT and S6. PDCD4 can alter PI3K-AKT by regulating miR-184 expression [37]. Whether down-regulation of these survival pathway-related tumor suppressors plays a role in venetoclax resistance requires further investigation.
Acknowledgements
We thank the patients and their families who contributed to this research study. We thank Dr. Liana Adam for review and editing, and Dr. Lei Nie for discussion and manuscript revision. This study was supported by philanthropy from Steve and Nancy Fox Cancer Research Fund. The RPPA Core is supported by NCI Grant # CA16672 and Dr. Yiling Lu’s NIH R50 Grant # R50CA221675: Functional Proteomics by Reverse Phase Protein Array in Cancer.
Disclosure of conflict of interest
M.W. is a consultant for: InnoCare, Loxo Oncology, Juno, Oncternal, CStone, AstraZeneca, Janssen, VelosBio, Pharmacyclics, Genentech, Bayer Healthcare. His research is funded by: Pharmacyclics, Janssen, AstraZeneca, Celgene, Loxo Oncology, Kite Pharma, Juno, BioInvent, VelosBio, Acerta Pharma, Oncternal, Verastem, Molecular Templates, Lilly, Innocare. All other authors declare no competing financial interests.
Supporting Information
References
- 1.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, Desai MB, Cerri E, Heitner Enschede S, Humerickhouse RA, Wierda WG, Seymour JF. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan N, Kahl B. Targeting BCL-2 in hematologic malignancies. Targeted Oncol. 2018:257–267. doi: 10.1007/s11523-018-0560-7. [DOI] [PubMed] [Google Scholar]
- 3.Davids MS. Targeting BCL-2 in B-cell lymphomas. Blood. 2017;130:1081–1088. doi: 10.1182/blood-2017-04-737338. [DOI] [PubMed] [Google Scholar]
- 4.Davids MS, Roberts AW, Seymour JF, Pagel JM, Kahl BS, Wierda WG, Puvvada S, Kipps TJ, Anderson MA, Salem AH, Dunbar M, Zhu M, Peale F, Ross JA, Gressick L, Desai M, Kim SY, Verdugo M, Humerickhouse RA, Gordon GB, Gerecitano JF. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J. Clin. Oncol. 2017;35:826–833. doi: 10.1200/JCO.2016.70.4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tam C, Roberts AW, Anderson M, Dawson S, Hicks R, Burbury K, Turner G, Di Iulio J, Bressel M, Westerman D. Combination ibrutinib (Ibr) and venetoclax (Ven) for the treatment of mantle cell lymphoma (MCL): primary endpoint assessment of the phase 2 AIM study. Hematol Oncol. 2017;35:144–145. [Google Scholar]
- 6.Tam C, Rule S, Le Gouill S, Vitolo U, Tsao L, Cavazos N, Beaupre D, Wang M. Phase 3 study of ibrutinib in combination with venetoclax in patients with relapsed/refractory mantle cell lymphoma (MCL) Hematol Oncol. 2017;35:421–422. [Google Scholar]
- 7.Jayappa KD, Portell CA, Gordon VL, Capaldo BJ, Bekiranov S, Axelrod MJ, Brett LK, Wulfkuhle JD, Gallagher RI, Petricoin EF, Bender TP, Williams ME, Weber MJ. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017;1:933–946. doi: 10.1182/bloodadvances.2016004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thijssen R, Slinger E, Weller K, Geest CR, Beaumont T, van Oers MH, Kater AP, Eldering E. Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica. 2015;100:e302–e306. doi: 10.3324/haematol.2015.124560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bojarczuk K, Sasi BK, Gobessi S, Innocenti I, Pozzato G, Laurenti L, Efremov DG. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood. 2016;127:3192–3201. doi: 10.1182/blood-2015-10-675009. [DOI] [PubMed] [Google Scholar]
- 10.Pan R, Ruvolo VR, Wei J, Konopleva M, Reed JC, Pellecchia M, Andreeff M, Ruvolo PP. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (-) BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood. 2015;126:363–372. doi: 10.1182/blood-2014-10-604975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O, Lefebure M, Martin BP, Dawson MA, Johnstone RW, Shortt J. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia. 2015;29:1437–41. doi: 10.1038/leu.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teh TC, Nguyen NY, Moujalled DM, Segal D, Pomilio G, Rijal S, Jabbour A, Cummins K, Lackovic K, Blombery P, Thompson E, Ekert PG, Lessene G, Glaser SP, Huang DCS, Roberts AW, Guthridge MA, Wei AH. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia. 2018;32:303–312. doi: 10.1038/leu.2017.243. [DOI] [PubMed] [Google Scholar]
- 13.Adamo A, Delfino P, Gatti A, Bonato A, Takam Kamga P, Bazzoni R, Ugel S, Mercuri A, Caligola S, Krampera M. HS-5 and HS-27A stromal cell lines to study bone marrow mesenchymal stromal cell-mediated support to cancer development. Front Cell Dev Biol. 2020;8:584232. doi: 10.3389/fcell.2020.584232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogden CA, Pound JD, Batth BK, Owens S, Johannessen I, Wood K, Gregory CD. Enhanced apoptotic cell clearance capacity and B cell survival factor production by IL-10-activated macrophages: implications for Burkitt’s lymphoma. J Immunol. 2005;174:3015–3023. doi: 10.4049/jimmunol.174.5.3015. [DOI] [PubMed] [Google Scholar]
- 15.Le K, Sun J, Khawaja H, Shibata M, Maggirwar SB, Smith MR, Gupta M. Mantle cell lymphoma polarizes tumor-associated macrophages into M2-like macrophages, which in turn promote tumorigenesis. Blood Adv. 2021;5:2863–2878. doi: 10.1182/bloodadvances.2020003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li CJ, Jiang C, Liu Y, Bell T, Ma W, Ye Y, Huang S, Guo H, Zhang H, Wang L, Wang J, Nomie K, Zhang L, Wang M. Pleiotropic action of novel Bruton’s Tyrosine kinase inhibitor BGB-3111 in mantle cell lymphoma. Mol Cancer Ther. 2019;18:267–277. doi: 10.1158/1535-7163.MCT-18-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas D, Powell JA, Vergez F, Segal DH, Nguyen NY, Baker A, Teh TC, Barry EF, Sarry JE, Lee EM, Nero TL, Jabbour AM, Pomilio G, Green BD, Manenti S, Glaser SP, Parker MW, Lopez AF, Ekert PG, Lock RB, Huang DC, Nilsson SK, Recher C, Wei AH, Guthridge MA. Targeting acute myeloid leukemia by dual inhibition of PI3K signaling and Cdk9-mediated Mcl-1 transcription. Blood. 2013;122:738–748. doi: 10.1182/blood-2012-08-447441. [DOI] [PubMed] [Google Scholar]
- 18.Carnero A. Novel inhibitors of the PI3K family. Expert Opin Investig Drugs. 2009;18:1265–1277. doi: 10.1517/13543780903066798. [DOI] [PubMed] [Google Scholar]
- 19.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 20.Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, Mathew S, Vijay P, Eng K, Ali S, Johnson A, Chang B, Ely S, Elemento O, Mason CE, Leonard JP, Chen-Kiang S. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4:1022–1035. doi: 10.1158/2159-8290.CD-14-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piddock RE, Loughran N, Marlein CR, Robinson SD, Edwards DR, Yu S, Pillinger GE, Zhou Z, Zaitseva L, Auger MJ, Rushworth SA, Bowles KM. PI3Kδ and PI3Kγ isoforms have distinct functions in regulating pro-tumoural signalling in the multiple myeloma microenvironment. Blood Cancer J. 2017;7:e539. doi: 10.1038/bcj.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas D, Powell JA, Vergez F, Segal DH, Nguyen NY, Baker A, Teh TC, Barry EF, Sarry JE, Lee EM, Nero TL, Jabbour AM, Pomilio G, Green BD, Manenti S, Glaser SP, Parker MW, Lopez AF, Ekert PG, Lock RB, Huang DC, Nilsson SK, Récher C, Wei AH, Guthridge MA. Targeting acute myeloid leukemia by dual inhibition of PI3K signaling and Cdk9-mediated Mcl-1 transcription. Blood. 2013;122:738–748. doi: 10.1182/blood-2012-08-447441. [DOI] [PubMed] [Google Scholar]
- 23.Berghausen EM, Behringer A, Vantler M, Baldus S, Rosenkranz S. The p110 alpha[[unable to display character: &] ] selective PI3K inhibitor PIK75 reverses SU5416/hypoxia-induced PH. Circulation. 2014:130. [Google Scholar]
- 24.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O’Brien S, Chiorazzi N, Burger JA. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–1189. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, Blum KA. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, Caldwell JT, Xiang S, Zhang X, Chu R, Wang ZJ, Lin H, Taub JW, Ge Y. Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. 2016;22:4440–51. doi: 10.1158/1078-0432.CCR-15-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song T, Chai G, Liu Y, Yu X, Wang Z, Zhang Z. Bcl-2 phosphorylation confers resistance on chronic lymphocytic leukaemia cells to the BH3 mimetics ABT-737, ABT-263 and ABT-199 by impeding direct binding. Br J Pharmacol. 2016;173:471–483. doi: 10.1111/bph.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Punnoose EA, Leverson JD, Peale F, Boghaert ER, Belmont LD, Tan N, Young A, Mitten M, Ingalla E, Darbonne WC, Oleksijew A, Tapang P, Yue P, Oeh J, Lee L, Maiga S, Fairbrother WJ, Amiot M, Souers AJ, Sampath D. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther. 2016;15:1132–1144. doi: 10.1158/1535-7163.MCT-15-0730. [DOI] [PubMed] [Google Scholar]
- 29.Pham LV, Huang S, Zhang H, Zhang J, Bell T, Zhou S, Pogue E, Ding Z, Lam L, Westin J, Davis RE, Young KH, Medeiros LJ, Ford RJ, Nomie K, Zhang L, Wang M. Strategic therapeutic targeting to overcome venetoclax resistance in aggressive B-cell lymphomas. Clin Cancer Res. 2018;24:3967–3980. doi: 10.1158/1078-0432.CCR-17-3004. [DOI] [PubMed] [Google Scholar]
- 30.Fresquet V, Rieger M, Carolis C, García-Barchino MJ, Martinez-Climent JA. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014;123:4111–4119. doi: 10.1182/blood-2014-03-560284. [DOI] [PubMed] [Google Scholar]
- 31.Stephen KT, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, Leverson JD, Lam LT. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer. 2017;17:399. doi: 10.1186/s12885-017-3383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiron D, Dousset C, Brosseau C, Touzeau C, Maïga S, Moreau P, Pellat-Deceunynck C, Le Gouill S, Amiot M. Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget. 2015;6:8750. doi: 10.18632/oncotarget.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiron D, Bellanger C, Papin A, Tessoulin B, Dousset C, Maiga S, Moreau A, Esbelin J, Trichet V, Chen-Kiang S, Moreau P, Touzeau C, Le Gouill S, Amiot M, Pellat-Deceunynck C. Microenvironment-dependent proliferation and mitochondrial priming loss in mantle cell lymphoma is overcome by anti-CD20. Blood. 2016;128:2808–2818. doi: 10.1182/blood-2016-06-720490. [DOI] [PubMed] [Google Scholar]
- 34.Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, Hsi ED, Almasan A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6:e1593. doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bodo J, Zhao X, Durkin L, Souers AJ, Phillips DC, Smith MR, Hsi ED. Acquired resistance to venetoclax (ABT-199) in t (14; 18) positive lymphoma cells. Oncotarget. 2016;7:70000–70010. doi: 10.18632/oncotarget.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huelsemann MF, Patz M, Beckmann L, Brinkmann K, Otto T, Fandrey J, Becker HJ, Theurich S, von Bergwelt-Baildon M, Pallasch CP, Zahedi RP, Kashkar H, Reinhardt HC, Hallek M, Wendtner CM, Frenzel LP. Hypoxia-induced p38 MAPK activation reduces Mcl-1 expression and facilitates sensitivity towards BH3 mimetics in chronic lymphocytic leukemia. Leukemia. 2015;29:981. doi: 10.1038/leu.2014.320. [DOI] [PubMed] [Google Scholar]
- 37.Zhen Y, Liu Z, Yang H, Yu X, Wu Q, Hua S, Long X, Jiang Q, Song Y, Cheng C, Wang H, Zhao M, Fu Q, Lyu X, Chen Y, Fan Y, Liu Y, Li X, Fang W. Tumor suppressor PDCD4 modulates miR-184-mediated direct suppression of C-MYC and BCL2 blocking cell growth and survival in nasopharyngeal carcinoma. Cell Death Dis. 2013;4:e872. doi: 10.1038/cddis.2013.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.