

Abstract
The molecular characterization of tumors now informs clinical cancer care for many patients. This advent of molecular oncology is driven by the expanding number of therapeutic biomarkers that can predict sensitivity to both approved and investigational agents. Beyond its role in driving clinical trial enrollments and guiding therapy in individual patients, large-scale clinical genomics in oncology also represents a rapidly expanding research resource for translational scientific discovery. Here, we review the progress, opportunities, and challenges of scientific and translational discovery from prospective clinical genomic screening programs now routinely conducted in cancer patients.
Introduction
There is widespread enthusiasm for prospective tumor sequencing to guide therapy selection in patients with cancer1–3. Yet, the utility of clinical sequencing in oncology beyond the cancer types in which it is a standard of care has been much debated. Indeed, the number of genomic alterations clinically validated as predictive biomarkers of drug response is relatively small when compared to the number of all mutant genes implicated in cancer4–9. Consequently, simply expanding the adoption of clinical genomics is unlikely to address the unmet needs for the majority of cancer patients given the current milieu of available therapies. Clinical sequencing has therefore begun to inform other aspects of oncology care. An emerging lesson from clinical cancer genomics, defined herein as the prospective clinical sequencing of tumor specimens to guide treatment decisions for active disease, is that these initiatives are generating large-scale data resources that can also be leveraged for scientific discovery across patients, especially when integrated with clinical annotation and treatment data. Here, we review progress in such efforts to uncover fundamental scientific discoveries that ultimately feed the clinical enterprise. We also review the unique challenges posed by this new scientific resource and strategies to overcome them to facilitate rigorous, robust, and reproducible translational science.
Evolution of genomic testing
Next-generation sequencing has become the foundational technology for modern clinical diagnostic testing in oncology, with several laboratory-developed tests recently achieving FDA recognition10,11. NGS testing has become standard of care in many cancer types as a method to identify therapeutically actionable alterations in tumor DNA. Most FDA-approved and/or standard of care biomarker-drug associations are disease-specific and are designed to capture all oncogenic mutations in a gene (such as those in BRCA1/2, IDH1, KIT) or be mutant allele-specific (BRAF V600E, FGFR3 R248C, S249C etc.) or alteration type-specific (ERBB2 amplifications, ALK fusions)2. Newer tumor-agnostic indications are emerging with the approval of immune checkpoint blockade (ICB) therapy in cancers with microsatellite instability (MSI)12,13 and larotrectinib in solid tumors with NTRK fusions14. Testing can also identify patients unlikely to respond to certain therapies, as in the case of KRAS mutations and cetuximab in metastatic colorectal cancers15. Ultimately, given the current class of therapeutic options available, larger next-generation sequencing panels can (with notable exceptions discussed below) detect the majority of relevant genomic biomarkers across a range of abnormality types more efficiently than can conventional single-gene testing while also capturing many potentially emerging associations.
The merits and technical aspects of different clinical sequencing tests in oncology have been reviewed elsewhere1,16–19. The most broadly used strategy by both academic and commercial laboratories has been targeted gene panels for bulk tumor tissue DNA sequencing20–22. These efforts have sought to maximize applicability in the patient population, uptake, and cost effectiveness while leveraging assay designs that balance competing factors in a manner consistent with community standards and best practices23–25 (Box 1). This has necessarily limited the utility of clinical sequencing data for pure discovery (Fig. 1). Multiple cross-cutting forces shape these design choices, including the 1) regulatory approval process, 2) evolving landscape of financial reimbursement, 3) size of the intended patient population, 4) clinical volume at treating centers, 5) breadth of clinical intent, 6) consent structure and complexity, and 7) institution-specific issues involved in operationalizing clinical sequencing as a routine test in a long-established clinical cancer care workflow. While our focus here is DNA and tissue-based clinical genomics in oncology, many of these issues and a host of new challenges must also be overcome for next-generation testing of circulating tumor-derived cell-free DNA (cfDNA), which is discussed elsewhere26.
Box 1: The operational equipoise of modern clinical sequencing in oncology.
Most active cancer patients for whom prospective DNA sequencing is performed to guide treatment decisions receive some form of large-panel targeted sequencing. Assay design23–25 choices regarding panel size and content, depth of sequencing, and specimen type balance competing factors which impact the value of the resulting data for biological and translational discovery science. Key issues and subsequent considerations include:
Panel size and content
- One larger panel, solid tumor versus hematological panels, multiple disease-specific panels?
- Choice depends on breadth of utility
- Can affect the complexity of testing and operations
- Has implications for clinical and scientific discoveries
- Ease of integrative analysis increased with a single test
- Genes and non-genic content
- Standards of clinical significance and actionability can vary by institution
- Adaptability to unanticipated alterations, new discoveries, and advances in treatment easier with a larger genomic footprint
- Identification of complex alterations, mutational signatures, and other molecular phenotypes with clinical relevance
Depth of sequencing
At fixed cost, breadth and depth (sensitivity) of sequencing are inversely correlated
- High depth valuable for sensitivity in suboptimal clinical specimens of low tumor content
- Early detection and identification of mechanisms of resistance152
- Quantitative accuracy
- Clonal hematopoiesis (CH) detection
- Inference of clonality and zygosity
Matched germline sequencing
- Germline pathogenic variant discovery
- Simplifies clinical testing for many cancer patients153 when combined with consent and counseling infrastructure.
Determines origins of oncogenic events between somatic, germline, or CH154.
CH detection and screening for early detection of myeloid disease127
The direct inference of somatic zygosity changes32
Community standards and guidelines in clinical sequencing
DNA requirements
Library qualification and quantification requirements
Proficiency testing: inter-laboratory test performance
Validation: use of reference materials and identification of limits of detection
Quality control metrics: depth and uniformity of coverage, quality scores for base calling and alignment, allelic read percentage, strand and GC bias
Fig. 1: Balancing accessibility, utility, and discovery in clinical genomics.
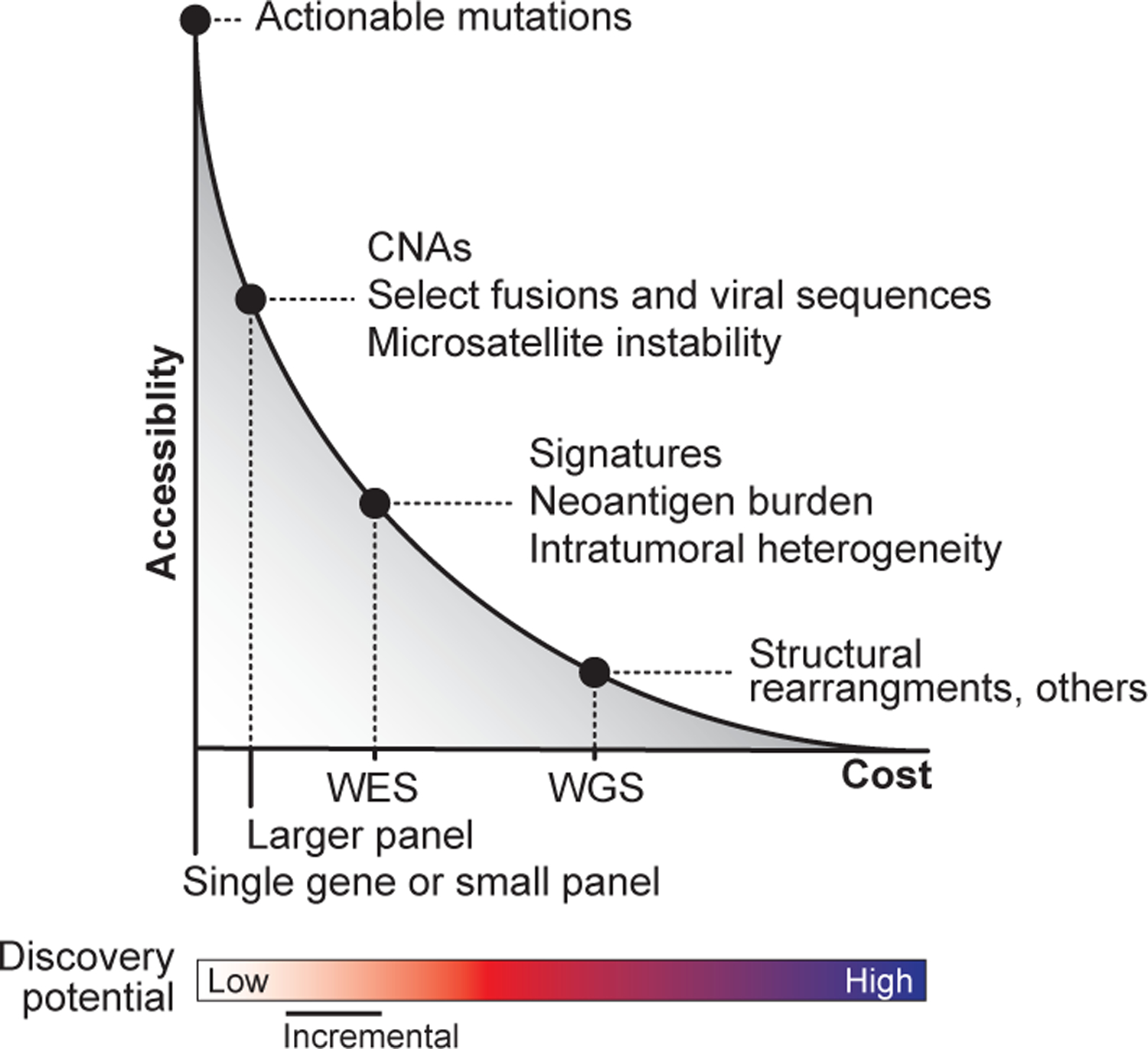
At present, there is a trade-off between cost, accessibility, and the opportunity for novel discovery afforded by different clinical sequencing platforms. The model depicts the current inverse relationship between accessibility and cost. As the cost (x-axis) of different sequencing strategies increases (driven by the size of the genomic footprint and depth of sequencing) accessibility in the community decreases (y-axis). The most common modalities of sequencing are labeled as are examples of the additional types of information enabled possible by adopting the indicated sequencing strategy. The discovery potential of a given strategy typically tracks with cost (bottom). At one end of the spectrum, small gene panels have the lowest cost but have limited discovery potential. In contrast, at the other end of the spectrum, WGS has both the highest cost and greatest discovery potential. WES, whole-exome sequencing; WGS, whole-genome sequencing; CNAs, copy number alterations.
As the clinical scope and intent of prospective sequencing in oncology is fundamentally different from discovery-driven retrospective research sequencing, targeted sequencing remains the methodology of greatest uptake. This is because, given the current treatment landscape in oncology, the majority of therapeutically relevant variants exist in a relatively small subset of all genes. Indeed, the most systematic effort to date to harmonize knowledge about cancer-associated mutations included 3437 unique variants across 357 diseases and 791 drug associations, yet encompassed just 415 genes27. Consequently, if the goal were to match patients to one of the current class of approved or investigational agents, broader scale sequencing such as whole-exome (WES) or genome sequencing (WGS) may provide limited increase in clinical benefit when compared to carefully designed large-panel targeted sequencing assays16. In fact, the detection of cryptic oncogenic events such as promoter mutations or exon skipping28,29, broader allele-specific DNA copy number30, and microsatellite instability31 can be achieved without the reduced sensitivity associated with lower coverage WES or WGS. Nevertheless, areas for which WES and WGS have potentially greater clinical utility due to the more comprehensive sequencing of passenger mutations are the identification of key mutational and genomic signatures such as homologous recombination deficiency32,33 as well as the identification of neoantigens, discrete subclones, and unexpected mechanisms of acquired resistance to therapy. As such, many clinical implementations of WES, WGS, and multimodality DNA and RNA sequencing exist33–40.
Therefore, despite the value of continued larger-panel targeted clinical sequencing, the drift toward clinical whole-genome sequencing (WGS) is likely inevitable. The development of new classes of drugs that target molecular aberrations not accessible by panel sequencing approaches and uniquely detected by WGS could motivate the substantial cost reductions and operational improvements that broad-based implementation of clinical WGS will require in our current health systems. The advent of clinical WGS in oncology care will likely result in a hybrid testing environment. WGS is likely to be used early in clinical care to establish a vital baseline of germline pathogenicity, somatic changes, and their actionability along with broader signatures of genomic changes41. Later in disease management, however, there will be clinical context-specific needs for far more sensitive testing (tissue or plasma-based) for disease monitoring or to identify emerging subclonal resistance for which clinical WGS is suboptimal. No single test will ultimately prove sufficient to address the many clinical needs in oncology care and structural considerations such as the reimbursement landscape must adapt as the utility of such hybrid testing is proven. Notwithstanding the exact configuration of clinical sequencing in oncology, it has recently become clear that the accrual of sequencing information is driving the creation of an entirely new institutional research resource that can catalyze new science and discovery.
New disease biology and richer clinical interpretation
Efforts to pool together prospective sequencing data within and across institutions are generating increasingly larger cohorts of molecularly profiled cancer patients, with opportunities for discovery science. Multiple aspects of clinical sequencing data make it favorable for discovery compared with retrospective research data in cancer. Among these are 1) larger cohort sizes and greater disease diversity than exist in the retrospective research domain; 2) specific quantitative attributes of clinical sequencing such as a greater depth of sequencing; 3) a distinct disease profile often comprised of advanced and post-treatment metastatic disease; and 4) the potentially transformative opportunity for clinical data integration with maturing outcomes and therapeutic phenotypes. Indeed, the analysis of clinical sequencing data has demonstrated that a broad set of genomic features can be identified from existing testing modalities, and that when understood at a basic and translational level, can be fed back into the clinical enterprise to facilitate richer reporting and a deeper understanding of a patient’s disease. This includes, but is not limited to, novel mutant allele and broader driver genetic defect discovery and functional validation; disease-specific genomics; basic mechanisms of tumor evolution; germline genetics; biomarker discovery and validation; mechanisms of therapeutic resistance; and immunogenomics. Each is catalyzed to a different degree by key aspects of clinical sequencing data (Fig. 2). These findings are not limited to individual genes and mutations, but broader genome- and disease-level (including etiological) discoveries42.
Fig. 2: Facets of clinical sequencing that drive translational discovery science.
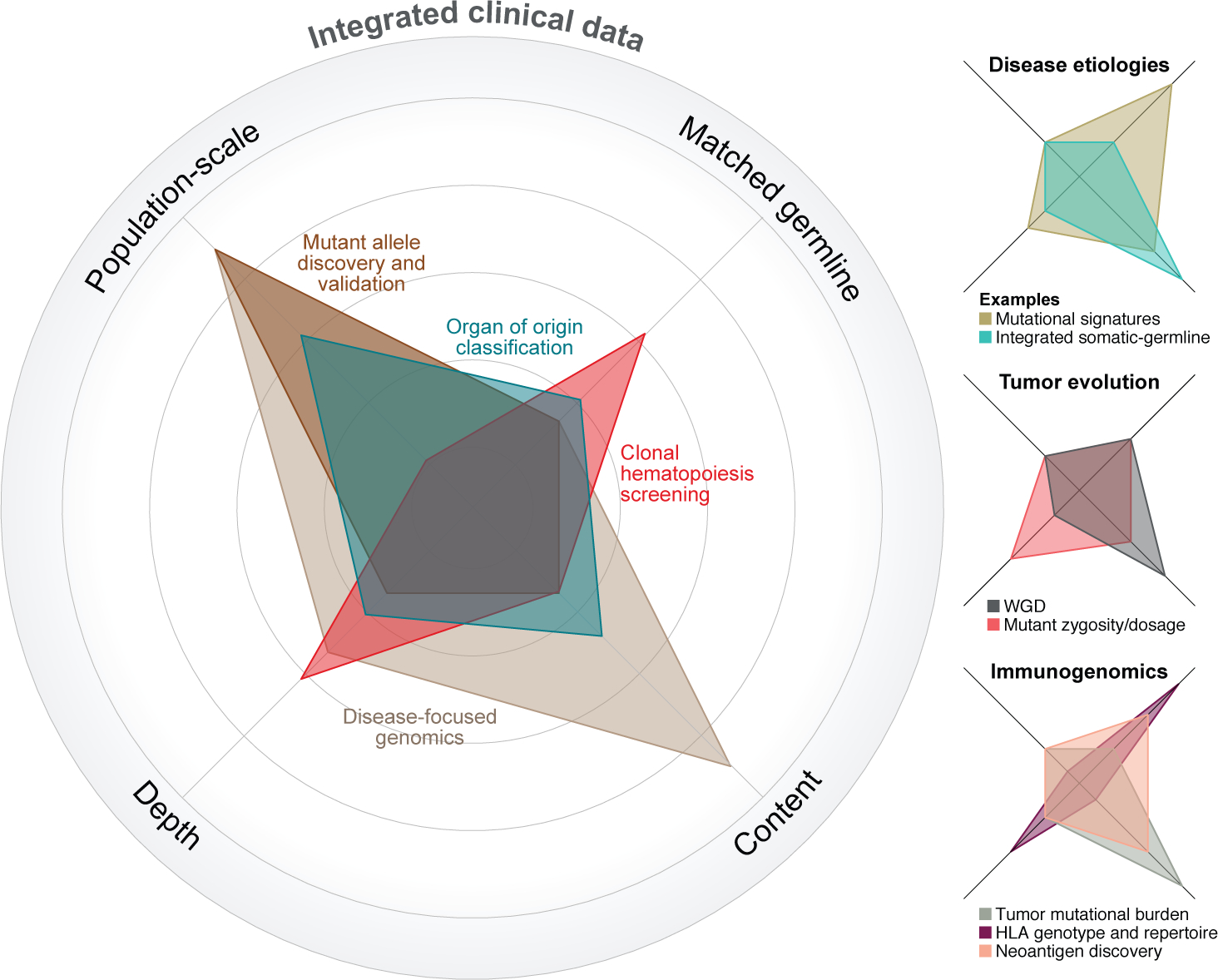
Critical aspects of clinical sequencing that can impact various discovery efforts are its 1) population-scale, 2) use of matched germline sequencing, 3) depth of sequencing, and 4) genomic content (see axes). Each of these facets can also drive different types of discoveries. The relative importance of each axis is displayed for various analysis types, the further away from the center the greater the importance of that particular axis is to the analysis in question.
At the scale of individual alterations
One of the most attractive aspects of clinical sequencing in oncology from the perspective of discovery is the population sizes now being characterized. These sample sizes, albeit from sequencing of already established cancer-associated genes, are driving efforts to mine the long right tail of the frequency distribution of driver mutations across cancer patients. This seeks to overcome a major hurdle in effective precision oncology, of understanding the biological and therapeutic importance of the multitude of variants of uncertain significance identified from such sequencing. To this end, a variety of computational and medium- to high-throughput functional screening approaches, reviewed in part elsewhere2, have been developed to mine this long tail. These efforts are fueled in part by access to population-scale sequencing cohorts and have revealed increasingly rare mutations that provide foundations for accelerated clinical translation2,43, extend biomarkers of therapeutic sensitivity to a greater number of patients44–49, inform drug development50, and uncover new tumor biology 51,52. Indeed, emerging evidence whereby different mutations in the same actionable cancer gene can have distinct biochemical consequences has underscored additional layers of complexity in our mechanistic understanding of pathway signaling53. This can dictate different therapeutic strategies, as has now emerged for mutant BRAF54,55 and MEK156. There now exists multiple decision support systems and emerging community standards to turn this knowledge into practice8,9,27,57,58.
There is a growing appreciation that the mere presence of an oncogenic driver mutation is not sufficient to convey its role in tumorigenesis, cancer progression, prognosis, or response to treatment. Indeed, context matters. For example the importance of serial evolution of oncogenic alterations has been realized in part by the ability to infer changes in mutant zygosity from the combination of allele-specific DNA copy number data and the quantitative accurate mutant allele frequencies from high depth of coverage in panel-based clinical sequencing data. The dosage of mutant alleles can modulate distinct molecular phenotypes59,60, drive tumorigenesis and disease progression61, and shape prognosis and therapeutic sensitivity62. Mutant oncogene dose-dependent sensitivity to targeted therapies has already been demonstrated in patients with AKT1 E17K mutations treated with AKT inhibitors63 and in BRAF V600-mutant metastatic melanomas treated with RAF inhibitor therapy62. Other cryptic mechanisms of serial evolution of mutant oncogenes, such as cis-acting double mutations in PIK3CA, have been linked to greater therapeutic sensitivity64, and additional associations may emerge for new investigational therapies such as inhibitors of KRAS G12C65 and recently approved therapies targeting mutant oncogenes frequently affected by zygosity changes66. Information about zygosity, clonality, co-mutations, disease lineage, and germline status,among other factors, all play a vital part in understanding the role of any individual oncogenic variant in an affected tumor. More comprehensive panel clinical sequencing facilitates the feed-forward loop that propels new discoveries back into the clinical enterprise to enrich clinical reporting and inform cancer care.
Broader genome-wide alterations
Beyond the DNA copy number alterations that span discrete mutant loci and genes frequently targeted by focal amplifications or deletions, the ability to infer genome-wide allele-specific copy number is facilitating discoveries at the genome-scale in clinical sequencing data. Whole-genome doubling (WGD) 67,68, for example, is a tetraploidization of the tumor genome and is among the most common molecular abnormalities in cancer, albeit varying across tumor types and disease states69. WGD is associated with worse clinical outcomes both pan-cancer and in specific disease contexts70. The identification of WGD in clinical sequencing data can facilitate evolutionary analysis of individual patient tumors by revealing the order of acquisition of key alterations70–72 that can aid biological and clinical interpretation and may ultimately help to predict chemo-sensitivity73.
The accurate inference of mutational signatures74 in clinical genomic data can reveal aspects of inherited susceptibility, disease etiology and pathogenesis, environmental exposures, and therapeutic sensitivity. Notably among these signatures is microsatellite instability (MSI), which is characterized by hypermutation at repetitive sequence motifs due to DNA mismatch repair (MMR) dysfunction. MSI is a tumor-agnostic biomarker of sensitivity to ICB therapy13. The ability to infer MSI from larger-panel clinical sequencing without assay iteration has facilitated its rapid clinical validation31 and uptake. MSI represents a critical link between germline pathogenicity (MMR defects and predisposition to multiple cancer types75) and somatic phenotypes that can drive further consolidation of clinical testing by motivating simultaneous germline and somatic testing (described below). However, challenges remain for identifying and interpreting other mutational signatures76. Despite the development of algorithms for their inference from targeted sequencing data77, not all mutational signatures are sufficiently specific in their nucleotide pattern to be accurately inferred from such data or may require the detection of multiple orthogonal signatures to reduce false positives, even when using broader sequencing strategies78,79. Moreover, incorrect attribution of such mutational signatures can complicate the clinical interpretation of key lesions and therapeutic vulnerabilities, perhaps best typified by homologous recombination deficiency and PARP inhibitor use80,81. The specificity and accuracy of mutational signature inference, whether driven by substitutions, DNA copy number changes82, structural rearrangements, or the combination thereof33,78, will improve with the clinical use of the aforementioned WGS sequencing modalities78.
Integrated germline and somatic reporting
With adoption of routine mutational signature and zygosity inference from matched germline profiling in the context of clinical sequencing, there exists an opportunity for fully integrated germline and somatic reporting to more completely understand disease pathogenesis and inform treatment. Such an approach may ultimately go beyond addressing ambiguities regarding the origin of somatic phenotypes, but define a more comprehensive view of disease as arising from the interaction of heritable and somatic factors. This shift toward integrated germline-somatic reporting is motivated in part by the rate at which pathogenic variants areidentified from broad-based clinical genomic profiling in both unselected patient cohorts and those with cancer37,83–87. Placing germline pathogenic variants into their somatic context, be it their tumor-specific zygosity or association with broader somatic mutational signatures, is increasingly important to guide the interpretation of their biological significance. Indeed, there is emerging evidence that zygosity changes that accompany germline pathogenic variants in even classical cancer predisposition genes are variable depending on cancer type, and may reflect differences in subsequent somatic mutational signatures of that lesion and even therapy response32.
Such discovery-based science at the intersection of germline and somatic cancer genetics is revealing new complexities in the interpretation of individual variants (Box 2). These complexities are therapeutically relevant today, such as in the case of a cancer patient found to harbor a pathogenic variant in a MMR gene but whose tumor lacks MSI and therefore would not be predicted sensitive to ICB therapy12,13. Richer clinical reporting could streamline testing for patients and inform multiple aspects of clinical care; 88, however, to our knowledge routine fully integrated clinical reporting is not presently performed at scale. This approach remains complicated by privacy considerations, cost concerns, and the complexity of incidental findings89, which have potential consequences for genetic counseling90 and triggering cascades of subsequent care of uncertain value91. These are complex and both labor- and expertise-intensive endeavours92 that require substantial infrastructure and investment - likely explaining, at least in part, the prevalence of tumor-only sequencing. Addressing the complexities for integrated germline and somatic reporting is urgent as the community is already seeking to incorporate somatic features into the classification of germline variants of uncertain significance93.
Box 2: Unified clinical germline and somatic reporting.
Concurrent clinical evaluation of germline pathogenicity and somatic tumor genomics from prospective clinical sequencing will reveal new complexities that affect our ability to interpret the significance of germline findings.
The hitchhiker effect
Knowing the target. Is somatic LOH targeting the chromosomal locus spanning a germline pathogenic variant selecting for its biallelic inactivation, or does this reflect the selective pressure for the loss of a proximal somatically mutant tumor suppressor gene elsewhere on the same chromosome?
One or both alleles?
Is a somatic truncating mutation in a tumor suppressor gene not accompanied by a second hit truly heterozygous? Or does this represent the second hit in a carrier of a germline pathogenic allele in the same gene not simultaneously reported?
The significance of apparent dispensability
Widespread genomic losses typical of advanced cancer genomes can delete the germline pathogenic allele somatically. Was the germline event incidental to tumor development? Or initially essential, but now dispensable to advanced disease?
Prospective immunogenomics
A perhaps surprising area of innovation for larger-panel clinical sequencing has been immunogenomics. Principle among these advances is the routine calculation of tumor-intrinsic biomarkers of response to ICB therapy such as tumor mutational burden (TMB)94–97. However, TMB can be affected by tumor purity98, intratumoral heterogeneity99, disease subtype100, mutational signatures101–103, and from the perspective of clinical genomics, panel size and content104–106. Overall, considerable work remains to refine the clinical utility of TMB107 and to better understand both the factors by which it co-varies and how it affects and confounds the identification of single-gene or pathway-based biomarkers of (ICB) therapy.
As an adjunct to TMB, other factors essential for modern immunogenomics can be readily inferred from the current generation of clinical sequencing assays such as the genotypes of human leukocyte antigen genes (HLA-A, B, and C). These genes encode MHC class I proteins that present intracellular peptides on the cell surface to the immune system. Overall, the diversity and evolutionary divergence of the HLA class I repertoire has been associated with differences in the efficacy of ICB therapy108,109. In tumors, somatic mutations in these genes and related antigen processing machinery have been correlated with immune cell infiltration110, and somatic loss-of-heterozygosity (LOH) of the HLA locus is associated with high subclonal neoantigen burden and is a mechanism of immune escape111,112. Notably, accurate HLA class I genotyping 113,114 and somatic HLA zygosity inference requires patient-matched germline DNA sequencing, reinforcing its importance as part of the routine clinical workflow. This can further consolidate clinical testing modalities and inform novel enrichment and accrual strategies for the next generation of clinical trials testing HLA-specific neoantigen-directed cancer vaccines among others115,116.
The inclusion of HLA class I genes in paired tumor and matched germline clinical sequencing also facilitates neoantigen discovery117. Tumor-specific neoantigens result from somatic mutations and are foreign peptides absent from normal tissue. Clonal neoantigen burden is a biomarker of response to ICB therapy99. Looking beyond just the burden of neoantigens, fitness models have been proposed to measure neoantigen quality, which is also associated with longer term survival in pancreatic cancer patients118,119. Personalized vaccine strategies are being developed to target neoantigens that autologously bind to HLA genes within cancer patients120–123. However, targeted clinical sequencing can only identify a small fraction of the potential neoantigens present in individual tumors. As such, WES has been primarily used to identify and prioritize putative neoantigens and vaccine targets via in silico approaches, often in conjunction with RNA sequencing to assess peptide expression121,123. By contrast, other neoantigen-based strategies can directly benefit from the current generation of largely targeted sequencing in oncology. One such strategy is leveraging shared or ‘public’ neoantigens that bind common HLA subtypes and can potentially benefit a larger population of patients and avoid much of the complexity and cost of developing personalized therapies from private neoantigens124–126.
Early detection and disease classification
Clinical genomics has proven enormously valuable for identifying patients with disease states that inform screening and early detection for other malignancies. For instance, prospective clinical sequencing can identify patients with solid tumors as well as clonal hematopoiesis (CH)127. CH is characterized by somatic mutations in hematopoietic stem and progenitor cells in patients without hematologic disease128,129. CH mutations typically arise in genes associated with myeloid disease and other malignancies and therefore are readily detected in prospective sequencing of cancer patients, especially those for whom matched germline sequencing is performed at high depth of coverage. As cancer patients with CH are at greater risk of developing hematologic malignancies such as AML and MDS127, it may serve as an increasingly important facet of early detection and screening130,131, with future clinical validation particularly dependent on large-scale clinical sequencing initiatives. Finally, broader efforts exist to integrate multiple types of alterations that can be readily inferred from targeted clinical sequencing data of individual patient tumors, coupled to novel computational methods development including those in machine learning. One such example is the development of algorithms to predict tissue of origin for cancers from clinical genomics data alone132 that were themselves trained on large-scale clinical sequencing data. Such point-of-care decision support can serve as a useful adjunct to conventional histologic diagnosis and is another example of the value of discovery in clinical sequencing feeding back into the clinical enterprise, in this case potentially providing integrated pathological diagnoses.
Overall, the breadth of these discovery efforts emphasize that, when properly controlling for the effect of prior therapy, even foundational aspects of tumorigenesis and cancer biology can be revealed from the sequencing of largely advanced and metastatic specimens.
Implementation and downstream uses
As clinical genomics has emerged as standard-of-care testing for many indications, this has led to a proliferation of different assays at major academic medical centers, which has both benefits and drawbacks. Complexities include the substantial and ongoing investments in assay development, clinical validation, and infrastructure as well as the variability in test content and capability that can complicate care, reimbursement, and trial accrual and design. By contrast, advantages include control over gene panel content, access to deep clinical annotation, sample and assay quality assessment data to drive iterative assay improvement (of diminishing returns as the feasibility of broad-based clinical WGS grows), customizable integration of reports into the electronic medical record, a unified platform for clinical use and discovery, and long-term cost effectiveness16. Test proliferation at academic medical centers also indirectly fosters innovation for the broader field that may not otherwise happen, through responses to institution-specific needs. These advantages are often seen only with economies of scale, so the field must democratize the expertise and benefits from such test development to the broader clinical oncology community. This could dovetail with other models that seek to build partnership with the pharmaceutical industry and government with a network of centers to facilitate information, data, and resource exchange133.
Translational science in clinical trials
Target-driven clinical trials were an early crucible through which key lessons were learned for the implementation and use of prospective sequencing in oncology2. Challenges abound for the translational science incorporated into such trials. Significant variability in clinical sequencing test capabilities exist as discussed above between sites enrolling patients in multi-center trials. This effectively limits the sample size for correlative genomics to only those sites with sufficient capabilities unless trial agreements break down the barriers typical of competitive academic sites to facilitate the transfer of materials for study. In addition, the desire in many target-driven trials for central tissue testing to support companion diagnostic test development that may be redundant with the activities of the individual sites can further limit high-quality genomic studies by depleting key tissue resources. Many of these issues can be overcome with cross-laboratory concordance testing134,135 and compliance with guidelines and standards for next-generation sequencing23,25,136. Also playing an important role is community-wide harmonization of key biological features across various clinical sequencing assays, such as recent efforts for tumor mutational burden105–107. While challenging, these efforts are likely more straightforward than harmonizing the real-time interpretation of candidate enrolling and sensitizing lesions27 across sites without some degree of central review or formal FDA recognition137
As biomarker hypotheses and enrichment strategies to guide trial enrollment become more complex, some enrolling sites and institutional tests will have the necessary capabilities, while others may not. These differences in testing capability may in turn influence accrual, trial design and availability, and ultimately drug approval. Even if such challenges were overcome, many key scientific and clinical questions remain that are difficult to address in the setting of a target-driven clinical trial, where the populations are necessarily small and uniform molecular profiling is absent. This has led to tremendous enthusiasm for real-world data and evidence to supplement the lessons learned from clinical trials138,139. Prospective studies using real-world data, defined here as population-level clinical and molecular data acquired outside the context of a clinical trial, provide an avenue to address such questions, but these will require careful clinical data homogenization, the selection of adequate control populations, and more to ensure that clinical practice realities do not limit robust results.
Retrospective biomarker analyses
The greatest value of clinical sequencing for discovery is arguably the real-time integration with clinical data including maturing outcomes and treatment phenotypes. This permits both an initial discovery and its clinical cross-validation to take place at the same time and in the same cohort, which is a strategy leveraged for many of the aforementioned discoveries. Clinical and treatment annotation will be of increasing importance for key retrospective biomarker questions that are impossible to answer in the setting of a clinical trial. However, a multitude of challenges complicate rigorous retrospective analyses, which can risk erroneous clinical findings (Fig. 3a).
Fig. 3: The known unknowns and potential pitfalls of retrospective biomarker analyses.
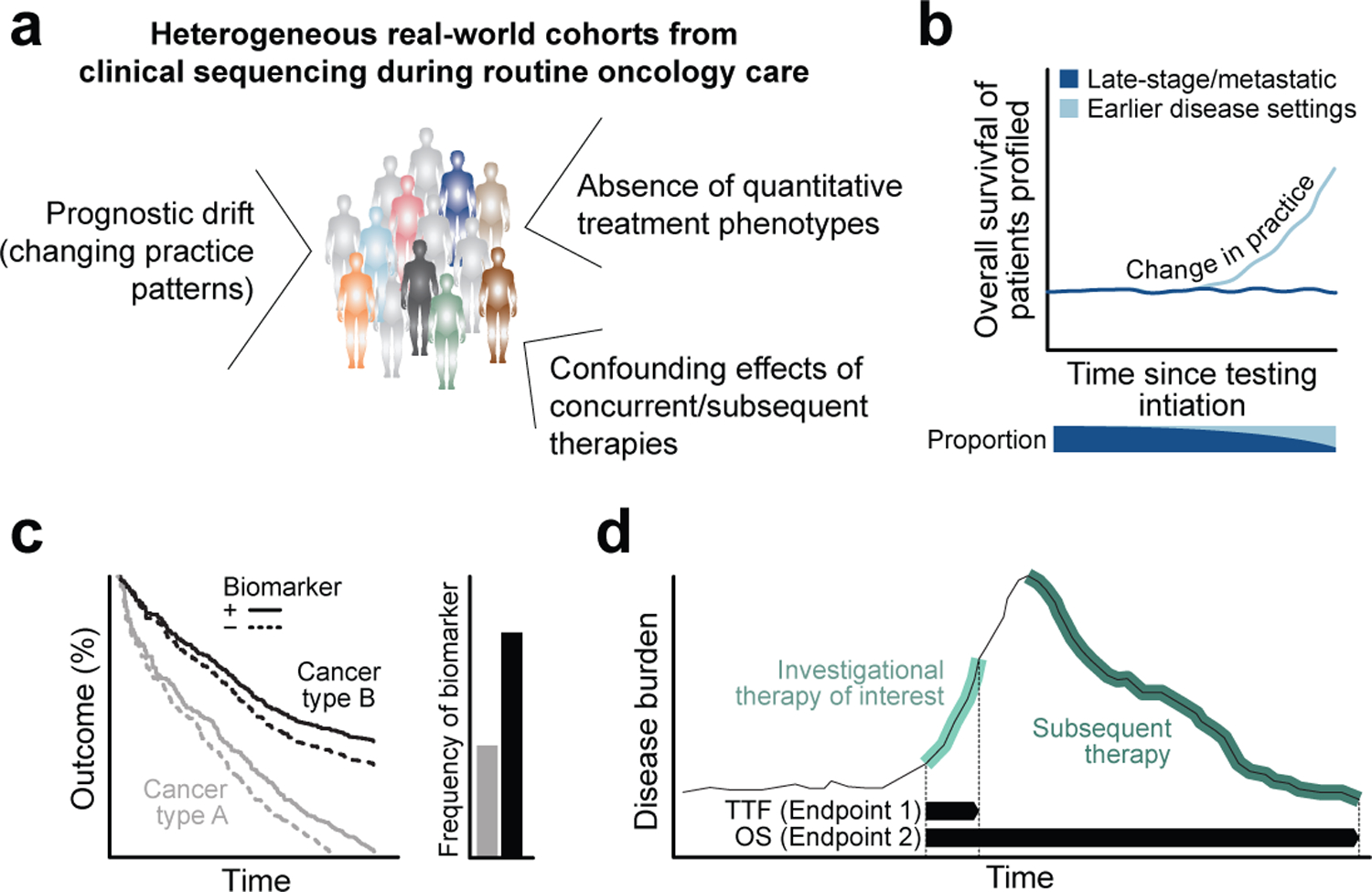
a) Various potential confounding factors that complicate the current generation of rigorous retrospective biomarker analyses using real-world clinical sequencing data in oncology. b) Over time, as clinical practice patterns change and the patient population for which clinical sequencing is routinely performed expands beyond late-stage and treatment refractory disease, the prognostic composition of the cohort will shift, leading to potentially spurious associations with outcome. c) Biomarker analysis pan-cancer can fail to discriminate between survival differences driven by underlying biology versus therapeutic intervention when affected cancer types have very different natural histories or the biomarker itself bestows favorable or worse prognosis. d) The effect of subsequent lines of therapy can confound key clinical endpoints in clinical sequencing cohorts composed of patients with heterogeneously administered therapies. TTF, time to treatment failure; OS, overall survival.
Foremost among challenges is the lack of rigorously validated quantitative treatment outcomes. Overall survival (OS) measured from the start of treatment is justifiably the established and preferred endpoint for demonstrating clinical benefit to a therapy of interest in clinical trials. However, to date most patients for whom clinical sequencing is obtained are not part of a therapeutic trial enrolling homogenous patient populations who then receive highly standardized therapy, the response to which could then be measured in a standardized and quantitative manner. Moreover, an increasing number of retrospective biomarker analyses are being performed pan-cancer, driven in part by the recent excitement around tissue-agnostic biomarkers of therapeutic sensitivity13,14. Yet, multiple potential confounding factors can plague different clinical endpoints in retrospectively collected treatment data in a pan-cancer cohort, preventing accurate assessment of clinical benefit from a given line of therapy. This is especially true of the preferred OS endpoint that, when analyzed pan-cancer, does not account for the often-profound prognostic differences between individual cancer types.
First, initial efforts to incorporate clinical genomics into active cancer care focused on the most advanced and treatment-refractory patients in need of novel therapeutic strategies who had the poorest prognosis. Over time, as practice patterns change with the approval of biomarker-driven therapies in earlier settings, clinical sequencing has expanded to patients with earlier-stage disease. This can lead to ‘prognostic drift’ in a cohort that accrues in real-time that, if not correctly adjusted for, can produce spurious associations with outcome (Fig. 3b). Second, prognosis between any two cancer types can vary widely and independently of the candidate biomarker of interest (Fig. 3c). If a cancer type of longer natural history also has an alteration rate of a candidate biomarker that is significantly greater than a poorer prognosis cancer type, simple outcome analyses using a clinical endpoint like OS from the start of therapy may arrive at the wrong conclusion about the effect of the sensitizing biomarker. Third, several key genomic alterations being investigated as therapeutic biomarkers can alone distinguish prognostically distinct subsets within individual cancer types, as is the case with BRCA1/2 mutations in ovarian cancers140. Consequently, any analysis of such a biomarker using OS will inevitably be confounded by the favorable prognosis its presence bestows on the carrier and fail to discriminate between survival differences driven by underlying biology versus therapeutic intervention. Fourth, OS from the start of therapy can be confounded by both therapies received concurrently with, and following the cessation of a given line of treatment (Fig. 3d). The confounding effect of concurrent and subsequent therapy is of particular concern in highly heterogeneous real-world cohorts such as those generated by clinical sequencing in oncology. For these reasons, both routine clinical trial practice and international health authority guidance suggests that OS should only be utilized as the primary endpoint for comparative analyses in large, rigorously designed, adequately powered, and highly controlled randomized phase III trials that enroll homogenous patient populations who then receive highly standardized therapy141–143.
Retrospective therapeutic biomarker analyses must therefore leverage one of multiple potentially suboptimal alternative endpoints that can be subjective or difficult to standardize. Some are more conservative or straightforward to curate than others, such as time to treatment failure or progression-free survival, which may be less prone to confounding in the real-world setting of clinical sequencing in cancer. However, they too can fail to reflect desired therapeutic effects. Time to treatment failure cannot quantify benefit for agents given for fixed durations like platinum-based chemotherapies or anti-CTLA-4 blockade, while progression free survival can be difficult to determine without the regular interval scans and radiographic measurements that standardize determination of disease progression on the majority of clinical trials.
Ultimately, rigorous and independently validated biomarkers of sensitivity to individual therapeutic agents are urgently needed to guide effective cancer care. However, false signals can lead the biomedical community astray at high cost in time and resources for institutions and patients. So, despite the exploratory power of these cohorts and data, key biomarker questions can likely only be tested robustly in a rigorously controlled large-scale prospective clinical trial where all of the aforementioned confounding clinical variables can be annotated, controlled, and duly adjusted for. For others, recognizing that no real-world data approach is perfect can motivate policies that require independent cohorts of clinical cross-validation for individual findings and a focus on more conservative endpoints, which mirrors best practices in the field for signal-seeking clinical trials.
Tragedy of the commons
Atypical of research data, clinical sequencing datasets obtained during the course of oncology care are often not siloed in any single research laboratory and therefore can represent catalyzing institutional resources for discovery if shared broadly. Nevertheless, challenges abound. It is unclear how proper attribution and credit can be ensured for data producers, who may be part of the clinical operational enterprise distinct from the research teams leveraging this data. Nor is it clear how to ensure privacy protections for cohorts composed, in part, of still-active cancer patients, while also meeting the obligations to the broader research community for the public deposition of sensitive data types such as raw sequencing data, germline variant calls, and others. It remains difficult to mediate the use of potentially encumbered or embargoed data, for instance from clinical trials with existing agreements governing the use of data prior to the public reporting of clinical efficacy data. These issues are particularly fraught when it comes to clinical annotation and treatment phenotypes. Navigating the issues around clinical data for integration with molecular information requires buy-in from key stakeholders from the individual treating physicians to the larger research enterprise and potential outside entities such as clinical trial sponsors, among others. Cross-institutional initiatives such as AACR GENIE144 are seeking new ways to address these issues while aggregating clinical sequencing data from many sources for broader utilization145.
At our academic cancer center, we have generated and shared clinical sequencing data from a prospective tumor profiling initiative for tens of thousands of tumors over the last several years to facilitate its use. All analyzed molecular data to date is shared via the cBioPortal for Cancer Genomics146,147. Raw data is available on institutional computing resources and is accessible to any research team at the institution via an IRB-approved process codified in the protocol to which all patients consent for clinical sequencing. These resources are updated nightly to ensure broadest availability and use. Such resources, however, require nimble governance that ensures open, broad, and rigorous utilization while assisting researchers to adjudicate overlapping uses. Questions have arisen such as how best to navigate two different research groups at the institution asking the same question of the same largely unpublished data, or how to handle situations when two research groups, leveraging the same institutional data resource but using slightly different analytical approaches, arrive at different conclusions. Ultimately an open dialogue among the key operational, clinical, and scientific stakeholders is necessary to ensure best use of these newer generations of institutional research resources. Our community must also ensure rigorous meta-data accompanies the publication of clinical sequencing and clinical annotation data to ensure proper use by others in the biomedical community encountering these data types for the first time.
Ultimately, unfettered data access and sharing is critical for the biomedical enterprise, catalyzing a greater body of science than can be achieved by any one group. However, such sharing raises the risk of scientific overlap and even misinterpretation that can lead to incorrect findings and stifle progress in clinical genomics. Responsibility ultimately lies both with data producers and users to ensure shared data are analyzed in a manner that is rigorous, well-documented, and trustworthy to ensure progress in improving human health and oncology.
Outlook
Significant progress remains to be made in extending the clinical benefit of prospective molecular characterization to more cancer patients. In parallel, molecular profiling initiatives will continue to grow as an increasing component of oncology care - especially as entirely new modalities of characterization, such as cell-free DNA and single-cell sequencing, mature toward clinical utilization148–151. While each of these new technologies will come with their own regulatory, ethical, and practical considerations and complexities, together they represent an unprecedented opportunity for scientific discovery. Our health systems must therefore mature to support this degree of routine molecular profiling, enable seamless and structured data sharing, and ensure real-time integration with deep clinical phenotyping. This will accelerate the discovery of clinical phenotypes associated with alterations in the cancer genome and drive expanded use of real-world evidence to aid in clinical and regulatory decision making.
Acknowledgements
We thank M.F. Berger and D.B. Solit for discussions. This work was supported by National Institutes of Health awards P30 CA008748, U54 OD020355 (B.S.T.), R01 CA207244 (D.M.H., B.S.T.), R01 CA204749 (B.S.T.), and R01 CA245069 (B.S.T.)
Footnotes
Competing Financial Interests
D.M.H. reports receiving research funding from AstraZeneca, Puma Biotechnology, Loxo Oncology and personal fees from Atara Biotherapeutics, Chugai Pharma, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer, Debiopharm Group, and Genentech. B.S.T. reports receiving honoria and research funding from Genentech and Illumina and advisory board activities for Boehringer Ingelheim and Loxo Oncology, a wholly owned subsidiary of Eli Lilly. All stated activities were outside of the work described herein. No other disclosures were noted.
Bibliography
- 1.Horak P, Fröhling S & Glimm H Integrating next-generation sequencing into clinical oncology: strategies, promises and pitfalls. ESMO Open 1, e000094 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyman DM, Taylor BS & Baselga J Implementing Genome-Driven Oncology. Cell 168, 584–599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger MF & Mardis ER The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol 15, 353–365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamborero D et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 10, 25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patterson SE et al. The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Hum Genomics 10, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang L et al. The cancer precision medicine knowledge base for structured clinical-grade mutations and interpretations. J. Am. Med. Inform. Assoc 24, 513–519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vogelstein B et al. Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffith M et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet 49, 170–174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakravarty D et al. Oncokb: A precision oncology knowledge base. JCO Precis. Oncol 2017, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ratner M First multi-gene NGS diagnostic kit approved. Nat. Biotechnol 35, 699 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Allegretti M et al. Tearing down the walls: FDA approves next generation sequencing (NGS) assays for actionable cancer genomic aberrations. J. Exp. Clin. Cancer Res 37, 47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le DT et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le DT et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drilon A et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med 378, 731–739 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Cutsem E et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med 360, 1408–1417 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Damodaran S, Berger MF & Roychowdhury S Clinical tumor sequencing: opportunities and challenges for precision cancer medicine. Am. Soc. Clin. Oncol. Educ. Book e175–82 (2015). doi: 10.14694/EdBook_AM.2015.35.e175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shevchenko Y & Bale S Clinical versus research sequencing. Cold Spring Harb. Perspect. Med 6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw KRM & Maitra A The status and impact of clinical tumor genome sequencing. Annu. Rev. Genomics Hum. Genet 20, 413–432 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Gagan J & Van Allen EM Next-generation sequencing to guide cancer therapy. Genome Med. 7, 80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sholl LM et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frampton GM et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol 31, 1023–1031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zehir A et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med 23, 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gargis AS et al. Good laboratory practice for clinical next-generation sequencing informatics pipelines. Nat. Biotechnol 33, 689–693 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gargis AS et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol 30, 1033–1036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jennings LJ et al. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn 19, 341–365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cescon DW, Bratman SV, Chan SM & Siu LL Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 1, 276–290 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Wagner AH et al. A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat. Genet 52, 448–457 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang FW et al. Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paik PK et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 5, 842–849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng DT et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn 17, 251–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Middha S et al. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precis. Oncol 2017, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonsson P et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 571, 576–579 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Staaf J et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat. Med 25, 1526–1533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beltran H et al. Whole-Exome Sequencing of Metastatic Cancer and Biomarkers of Treatment Response. JAMA Oncol. 1, 466–474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roychowdhury S et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci. Transl. Med 3, 111ra121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rusch M et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat. Commun 9, 3962 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parsons DW et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol. 2, 616–624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghazani AA et al. Assigning clinical meaning to somatic and germ-line whole-exome sequencing data in a prospective cancer precision medicine study. Genet. Med 19, 787–795 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Berner AM, Morrissey GJ & Murugaesu N Clinical analysis of whole genome sequencing in cancer patients. Curr. Genet. Med. Rep 7, 136–143 (2019). [Google Scholar]
- 40.Pleasance E et al. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat. Cancer (2020). doi: 10.1038/s43018-020-0050-6 [DOI] [PubMed] [Google Scholar]
- 41.Nangalia J & Campbell PJ Genome Sequencing during a Patient’s Journey through Cancer. N. Engl. J. Med 381, 2145–2156 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Touat M et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang MT et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 8, 174–183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Razanamahery J et al. Erdheim-Chester disease with concomitant Rosai-Dorfman like lesions: a distinct entity mainly driven by MAP2K1. Haematologica 105, e5–e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durham BH et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat. Med 25, 1839–1842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hyman DM et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 554, 189–194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diamond EL et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 567, 521–524 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smyth LM et al. Efficacy and Determinants of Response to HER Kinase Inhibition in HER2-Mutant Metastatic Breast Cancer. Cancer Discov. 10, 198–213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Slotkin EK et al. Patient-Driven Discovery, Therapeutic Targeting, and Post-Clinical Validation of a Novel AKT1 Fusion-Driven Cancer. Cancer Discov. 9, 605–616 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drilon A et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov. 7, 963–972 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saito Y et al. Landscape and function of multiple mutations within individual oncogenes. Nature (2020). doi: 10.1038/s41586-020-2175-2 [DOI] [PubMed] [Google Scholar]
- 52.Gorelick AN et al. Phase and context shape the function of composite oncogenic mutations. Nature 582, 100–103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brenan L et al. Phenotypic characterization of a comprehensive set of MAPK1/ERK2 missense mutants. Cell Rep. 17, 1171–1183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao Z et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 28, 370–383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao Z et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao Y et al. Allele-Specific Mechanisms of Activation of MEK1 Mutants Determine Their Properties. Cancer Discov. 8, 648–661 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mateo J et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol 29, 1895–1902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li MM et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the association for molecular pathology, american society of clinical oncology, and college of american pathologists. J. Mol. Diagn 19, 4–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Madsen RR et al. Oncogenic PIK3CA promotes cellular stemness in an allele dose-dependent manner. Proc Natl Acad Sci USA 116, 8380–8389 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burgess MR et al. KRAS allelic imbalance enhances fitness and modulates MAP kinase dependence in cancer. Cell 168, 817–829.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mueller S et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 554, 62–68 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bielski CM et al. Widespread selection for oncogenic mutant allele imbalance in cancer. Cancer Cell 34, 852–862.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hyman DM et al. AKT inhibition in solid tumors with AKT1 mutations. J. Clin. Oncol 35, 2251–2259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasan N et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science 366, 714–723 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Canon J et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019). [DOI] [PubMed] [Google Scholar]
- 66.André F et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med 380, 1929–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Carter SL et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 30, 413–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zack TI et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet 45, 1134–1140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Priestley P et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bielski CM et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet 50, 1189–1195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dewhurst SM et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 4, 175–185 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.López S et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet 52, 283–293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuznetsova AY et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 14, 2810–2820 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Latham A et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol 37, 286–295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Hoeck A, Tjoonk NH, van Boxtel R & Cuppen E Portrait of a cancer: mutational signature analyses for cancer diagnostics. BMC Cancer 19, 457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gulhan DC, Lee JJ-K, Melloni GEM, Cortés-Ciriano I & Park PJ Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet 51, 912–919 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Davies H et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med 23, 517–525 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Degasperi A et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat. Cancer 1, 249–263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma J, Setton J, Lee NY, Riaz N & Powell SN The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun 9, 3292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Swisher EM et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 18, 75–87 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Timms KM et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 16, 475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meric-Bernstam F et al. Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann. Oncol 27, 795–800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schrader KA et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2, 104–111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang K-L et al. Pathogenic germline variants in 10,389 adult cancers. Cell 173, 355–370.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu C et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun 6, 10086 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mandelker D et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA 318, 825–835 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mandelker D & Ceyhan-Birsoy O Evolving Significance of Tumor-Normal Sequencing in Cancer Care. Trends Cancer (2019). doi: 10.1016/j.trecan.2019.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Green RC et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med 15, 565–574 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gray SW et al. Oncologists’ and cancer patients’ views on whole-exome sequencing and incidental findings: results from the CanSeq study. Genet. Med 18, 1011–1019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ganguli I et al. Cascades of care after incidental findings in a US national survey of physicians. JAMA Netw. Open 2, e1913325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johns AL et al. Lost in translation: returning germline genetic results in genome-scale cancer research. Genome Med. 9, 41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Walsh MF et al. Integrating somatic variant data and biomarkers for germline variant classification in cancer predisposition genes. Hum. Mutat 39, 1542–1552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Snyder A et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Van Allen EM et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schrock AB et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol 30, 1096–1103 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Samstein RM et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet 51, 202–206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Anagnostou V et al. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat. Cancer 1, 99–111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McGranahan N et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu D et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med 25, 1916–1927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rizvi NA et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miao D et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet 50, 1271–1281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang S, Jia M, He Z & Liu X-S APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 37, 3924–3936 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chalmers ZR et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fancello L, Gandini S, Pelicci PG & Mazzarella L Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J. Immunother. Cancer 7, 183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vokes NI et al. Harmonization of Tumor Mutational Burden Quantification and Association With Response to Immune Checkpoint Blockade in Non-Small-Cell Lung Cancer. JCO Precis. Oncol 3, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chan TA et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol 30, 44–56 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chowell D et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 359, 582–587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chowell D et al. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat. Med 25, 1715–1720 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Giannakis M et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 15, 857–865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McGranahan N et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 171, 1259–1271.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosenthal R et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shukla SA et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol 33, 1152–1158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Szolek A et al. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chandran SS & Klebanoff CA T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev 290, 127–147 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hollingsworth RE & Jansen K Turning the corner on therapeutic cancer vaccines. npj Vaccines 4, 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jurtz V et al. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol 199, 3360–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Łuksza M et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551, 517–520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Balachandran VP et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sahin U et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 121.Ott PA et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hilf N et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565, 240–245 (2019). [DOI] [PubMed] [Google Scholar]
- 123.Keskin DB et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Klebanoff CA & Wolchok JD Shared cancer neoantigens: Making private matters public. J. Exp. Med 215, 5–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tran E et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med 375, 2255–2262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang QJ et al. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res 4, 204–214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Coombs CC et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 21, 374–382.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Abelson S et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400–404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Desai P et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med 24, 1015–1023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Penson A et al. Development of Genome-Derived Tumor Type Prediction to Inform Clinical Cancer Care. JAMA Oncol. (2019). doi: 10.1001/jamaoncol.2019.3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dalton WS & Friend SH Cancer biomarkers--an invitation to the table. Science 312, 1165–1168 (2006). [DOI] [PubMed] [Google Scholar]
- 134.Merker JD et al. Proficiency Testing of Standardized Samples Shows Very High Interlaboratory Agreement for Clinical Next-Generation Sequencing-Based Oncology Assays. Arch. Pathol. Lab. Med 143, 463–471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tricoli JV et al. Design and development of the molecular analysis for Therapy Choice (NCI-MATCH) Designated Laboratory Network. J. Clin. Oncol 37, 3016–3016 (2019). [Google Scholar]
- 136.Rehm HL et al. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med 15, 733–747 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.FDA Recognition of Public Human Genetic Variant Databases. at <https://www.fda.gov/medical-devices/precision-medicine/fda-recognition-public-human-genetic-variant-databases>
- 138.Sherman RE et al. Real-World Evidence - What Is It and What Can It Tell Us? N. Engl. J. Med 375, 2293–2297 (2016). [DOI] [PubMed] [Google Scholar]
- 139.Booth CM, Karim S & Mackillop WJ Real-world data: towards achieving the achievable in cancer care. Nat. Rev. Clin. Oncol 16, 312–325 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Bolton KL et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 307, 382–390 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics Guidance for Industry. at <https://www.fda.gov/media/71195/download>
- 142.Korn EL, Freidlin B & Abrams JS Overall survival as the outcome for randomized clinical trials with effective subsequent therapies. J. Clin. Oncol 29, 2439–2442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Saad ED & Buyse M Overall survival: patient outcome, therapeutic objective, clinical trial end point, or public health measure? J. Clin. Oncol 30, 1750–1754 (2012). [DOI] [PubMed] [Google Scholar]
- 144.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 7, 818–831 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Smyth LM et al. Characteristics and Outcome of AKT1E17K-Mutant Breast Cancer Defined through AACR Project GENIE, a Clinicogenomic Registry. Cancer Discov. (2020). doi: 10.1158/2159-8290.CD-19-1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rossi G & Ignatiadis M Promises and pitfalls of using liquid biopsy for precision medicine. Cancer Res. 79, 2798–804 (2019). [DOI] [PubMed] [Google Scholar]
- 149.Pantel K & Alix-Panabières C Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat. Rev. Clin. Oncol 16, 409–424 (2019). [DOI] [PubMed] [Google Scholar]
- 150.Jerby-Arnon L et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim C et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879–893.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Holohan C, Van Schaeybroeck S, Longley DB & Johnston PG Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726 (2013). [DOI] [PubMed] [Google Scholar]
- 153.Mandelker D et al. Germline-Focused Analysis of Tumour-Only Sequencing: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol (2019). doi: 10.1093/annonc/mdz136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ptashkin RN et al. Prevalence of Clonal Hematopoiesis Mutations in Tumor-Only Clinical Genomic Profiling of Solid Tumors. JAMA Oncol. 4, 1589–1593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]