

Abstract
Fragile X Mental Retardation protein (FMRP), widely known for its role in hereditary intellectual disability, is an RNA‐binding protein (RBP) that controls translation of select mRNAs. We discovered that endoplasmic reticulum (ER) stress induces phosphorylation of FMRP on a site that is known to enhance translation inhibition of FMRP‐bound mRNAs. We show ER stress‐induced activation of Inositol requiring enzyme‐1 (IRE1), an ER‐resident stress‐sensing kinase/endoribonuclease, leads to FMRP phosphorylation and to suppression of macrophage cholesterol efflux and apoptotic cell clearance (efferocytosis). Conversely, FMRP deficiency and pharmacological inhibition of IRE1 kinase activity enhances cholesterol efflux and efferocytosis, reducing atherosclerosis in mice. Our results provide mechanistic insights into how ER stress‐induced IRE1 kinase activity contributes to macrophage cholesterol homeostasis and suggests IRE1 inhibition as a promising new way to counteract atherosclerosis.
Keywords: atherosclerosis, cholesterol homeostasis, efferocytosis, ER stress, translational regulation
Subject Categories: Cardiovascular System, Immunology, Metabolism
Targeting IRE1 function and substrate(s) provides a novel therapeutic approach to atherosclerosis. We found a key role for IRE1‐mediated FMRP phosphorylation that suppresses the expression of cholesterol transporters and efferocytosis receptors in macrophages and promotes atherosclerosis progression.
The paper explained.
Problem
A maladaptive immune response to lipid imbalance drives atherosclerosis. Lipid accumulation in plaque‐infiltrating macrophages stresses the ER and promotes atherosclerosis progression. Alleviating ER stress by modulating IRE1 reduces atherosclerosis in a murine model. However, whether IRE1 kinase activity contributes to the atherosclerotic process has remained elusive. Targeting chronic ER stress in atherosclerosis is challenging due to the essential cellular homeostatic functions governed by IRE1. A meticulous exploration of IRE1 kinase function in macrophages and mechanistically authenticated IRE1 kinase substrate(s) can develop into novel therapeutic approaches in atherosclerosis.
Results
Here, we identify FMRP as a kinase substrate of IRE1. We show that IRE1 kinase activation leads to FMRP phosphorylation and suppression of macrophage cholesterol efflux and apoptotic cell clearance in a mouse model of atherosclerosis.
Impact
Altogether, our findings provide mechanistic insight into the translational regulation of cholesterol efflux and efferocytosis in ER‐stressed macrophages and highlight IRE1 kinase domain and its effector, FMRP, as novel therapeutic targets for atherosclerosis.
Introduction
Atherosclerosis is a chronic inflammatory disease triggered by imbalanced lipid metabolism (Rocha & Libby, 2009; Weber & Noels, 2011). In atherosclerotic plaques, macrophages ingest lipoproteins and transform into lipid‐laden foam cells. The foamy macrophages lose their ability to migrate away from the plaques, where they sustain a local state of sterile inflammation, which occurs in the absence of pathogens and is typically associated with the release of immune‐recognizable cellular content from damaged or dying cells (Randolph, 2014). Cholesterol efflux and efferocytosis (the engulfment and clearance of apoptotic cells (AC)) by macrophages, on the other hand, help to resolve inflammation and contribute to plaque stability as counterbalancing mechanisms that oppose plaque rupture (Khera et al, 2011; Westerterp et al, 2013, 2018; Kojima et al, 2017; Yurdagul et al, 2017). Cholesterol efflux by plaque macrophages is the first step in a multistep process, referred to as “reverse cholesterol transport” (RCT), that reduces lipid accumulation in plaques. Macrophages efflux intracellular cholesterol using their plasma membrane cholesterol transporters (such as the ATP‐binding cassette (ABC) transporters subfamily A member‐1 (ABCA1) and subfamily G member‐1 (ABCG1)), which in turn hand the exported cholesterol over to lipid‐poor apolipoproteins, forming high‐density lipoprotein (HDL) particles (Costet et al, 2000; Oram et al, 2000). The cholesterol efflux pathway is transcriptionally activated by the metabolic by‐products of AC‐derived cholesterol in the efferocytic macrophages. As such, efferocytosis and RCT synergize to reduce necrosis and resolve inflammation in plaques (Klucken et al, 2000; Joyce et al, 2002; Tall & Yvan‐Charvet, 2015; Zimmer et al, 2016; Guo et al, 2018).
Transcriptional and post‐transcriptional regulation play critical roles to set sterile inflammation in motion but also in its resolution and, eventually, plaque regression. Post‐transcriptional regulatory steps often involve RNA‐binding proteins (RBPs) that interact with mRNA to alter its processing, transport, translation, and degradation. Several RBPs have been shown to alter the mRNA stability and translation of cytokines to turn‐off the inflammatory response and key molecular regulators of cholesterol homeostasis and some have been linked to atherosclerosis development (atherogenesis) (Chiu et al, 1997; Kang et al, 2011; Zhang et al, 2013; Ramírez et al, 2014; Mobin et al, 2016; Haneklaus et al, 2017). Fragile X Mental Retardation Protein (FMRP) is an RBP that has been widely studied in neurons due to its causal role in the Fragile X Mental Retardation Syndrome (FXS) (Hersh & Saul, 2011; Bagni et al, 2012; Hunter et al, 2014). In FXS, a hypermethylated CGG repeat expansion in the 5′ untranslated region (5′ UTR) of the FMR1 mRNA results in its transcriptional silencing. A subpopulation of the individuals afflicted with FXS and FMRP‐deficient (Fmr1−/−) mice have lower cholesterol levels (Leboucher et al, 2019b). While a prior study has shown FMRP protein expression is induced in macrophage‐enriched areas of the human atherosclerotic plaque (Hansmeier et al, 2018), FMRP’s contribution to the atherosclerotic process has not been investigated directly, and our knowledge of FMRP function has remained largely limited to studies in neurons (Darnell et al, 2011). A key serine phosphorylation (S500 in human; S499 in mouse) on FMRP triggers hierarchical phosphorylation of surrounding serines and threonines, while enhancing FMRP’s translation‐repressing activity on many synaptic function‐linked mRNAs bound by it. The identity of the kinase(s) that phosphorylates FMRP has remained subject to intense discussion (Ceman et al, 2003; Narayanan et al, 2008; Coffee et al, 2012; Niere et al, 2012; Bartley et al, 2014, 2016; Prieto et al, 2020).
In this study, we present evidence that ER stress and hypercholesterolemia in mice induces the phosphorylation of macrophage FMRP on S500 by the inositol‐requiring enzyme‐1 (IRE1), a conserved endoplasmic reticulum (ER) stress‐sensing kinase/endoribonuclease (RNase). Metazoans have two IRE1 paralogues, IRE1α (referred to as IRE1 in this paper) and IRE1β. While IRE1α is ubiquitously expressed, IRE1β expression is restricted to gastrointestinal epithelium (Cloots et al, 2021). To date, IRE1 has been described to trans‐autophosphorylate as a first step in the activation of its RNase modality, which initiates a nonconventional RNA‐splicing reaction and the production of the transcription factor known as spliced X box protein‐1 (XBP1s), which is one of the key drivers of the unfolded protein response (UPR) (Walter & Ron, 2011; Çimen et al, 2016a; Robblee et al, 2016). IRE1 senses both protein folding stress induced by an accumulation of unfolded proteins in the ER lumen and ER membrane lipid bilayer stress induced by an accumulation of cholesterol or saturated fatty acids (SFA) (Seimon et al, 2010; Sukhorukov et al, 2020). While previous studies by us and others have shown that ER stress and subsequent IRE1 activation are causally associated with atherosclerosis progression, the mechanism by which IRE1 contributes to the disease pathogenesis has remained elusive (Erbay et al, 2009; Zhou & Tabas, 2013; Tufanli et al, 2017). Here, we show that IRE1 phosphorylates FMRP on S500, which in turn leads to post‐transcriptional suppression of cholesterol efflux and efferocytosis by macrophages. FMRP deficiency and IRE1 kinase inhibition both enhance RCT and efferocytosis in vivo, reducing foam cell formation and atherosclerosis progression in mice. These findings reveal a novel role for FMRP in macrophages in the regulation of cholesterol homeostasis and efferocytosis and provide mechanistic insight into IRE1‐driven atherosclerotic processes during hypercholesterolemia.
Results
Lipids induce FMRP phosphorylation in IRE1 kinase‐dependent manner
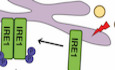
Previously published mass spectrometry‐based IRE1 interactome data revealed multiple FMRP‐interacting proteins potentially associating with IRE1 (Fig 1A) (Acosta‐Alvear et al, 2018). These proteins share significant sequence homology with FMRP and are usually found in homo/heteromeric complexes with FMRP (Zhang et al, 1995). Based on these observations, we reasoned that FMRP might also physically interact with IRE1. Indeed, FMRP co‐immunoprecipitated with IRE1 in non‐stress and ER stress conditions induced by thapsigargin (TG; an inhibitor of the ER Ca2+ pump) and tunicamycin (TM; an inhibitor of N‐linked glycosylation) from human embryonic kidney cell line (HEK293T) cells that were transiently transfected with plasmids encoding both proteins (Fig 1B).
Figure 1. FMRP is a novel IRE1 kinase substrate.
- STING analysis of published IRE1 interactome proteins in relation to FMRP (Acosta‐Alvear et al, 2018).
- HEK293T cells were co‐transfected with IRE1 and FMRP plasmids and stimulated with TG (600 nM) or TM (1 mg/ml) for 2 h. Protein lysates were immunoprecipitated (IP) with anti‐IRE1 or IgG (control) antibodies and analyzed by Western blotting using specific antibodies for FMRP and IRE1 (n = 3 biological replicates).
- RAW 264.7 mouse macrophages were treated with either oxLDL (50 µg/ml) or TG (300 nM) for 6 h. Protein lysates were treated with λ Phosphatase (PPase) for 30 min and analyzed by Western blotting using specific antibodies for pFMRP, FMRP, pIRE1, IRE1, and β‐Actin. pFMRP/FMRP fold induction is depicted above the blots (n = 6 biological replicates).
- Apoe−/− mice were fed with chow diet (CD) or western diet (WD) for 16 weeks followed by peritoneal macrophage (PM) isolation. Protein lysates were analyzed by Western blotting using specific antibodies for pFMRP, FMRP, pIRE1, IRE1, and β‐Actin. pFMRP/FMRP‐fold induction is depicted above the blots (n = 5 mice per group).
- Control‐ or IRE1‐siRNA transfected HEK293T cells were stimulated by either PA (500 µM) or TG (600 nM) for 4 h. Protein lysates were analyzed by Western blotting using specific antibodies for pFMRP, FMRP, pIRE1, IRE1, and β‐Actin. pFMRP/FMRP fold induction is depicted above the blots (n = 4 biological replicates).
- Protein lysates of thioglycolate‐elicited PM from IRE1α+/+ and IRE1α−/− mice (after 16 weeks on WD) were analyzed by Western blotting using specific antibodies for pFMRP, FMRP, pIRE1, IRE1, and β‐Actin. pFMRP/FMRP fold induction is depicted above the blots (n = 4 mice per group).
- MEF cells were transfected with either empty vector, EGFP‐FMRP or 3xFLAG‐IRE1 plasmids then pre‐treated either with vehicle (dimethyl sulfoxide, DMSO) or AMG‐18 (25 µM; 1 h) followed by TG (600 nM) stimulation for 4 h. Protein lysates were analyzed by Western blotting using specific antibodies for pFMRP, FMRP, pIRE1, IRE1, and β‐Actin. pFMRP/FMRP fold induction is depicted above the blots (n = 4 biological replicates).
- C57BL/6 were injected either with DMSO or AMG‐18 (30 mg/kg; 8 h), followed by TM injection (1 mg/kg; 8 h). Protein lysates of thioglycolate‐elicited PM were analyzed by Western blotting using antibodies for pFMRP, FMRP, pIRE1, IRE1, and β‐Actin. pFMRP/FMRP fold induction is depicted above the blots (n = 4 mice per group).
- HEK293T cells were transfected with either empty vector (EV), IRE1‐WT, or IRE1–KD plasmids and stimulated by TG (600 nM; 1 h). Protein lysates from each transfection were separately immunoprecipitated (IP) with anti‐IRE1 antibody and subjected to a kinase reaction with purified hFMRP protein and ATP‐γ‐S (100 µM) in kinase buffer. The IP protein were analyzed by Western blotting using specific antibodies for thiophosphate esters (ThioP), IRE1, and FMRP (n = 3 biological replicates).
- Purified FMRP and IRE1 kinase (activated) proteins were subjected to kinase assay and analyzed by Western blotting using specific antibodies for ThioP, IRE1, and FMRP (n = 3 biological replicates) and with LC‐MS/MS. Identified IRE1 kinase‐mediated FMRP phosphorylation sites (bottom).
- Fmr1−/− mouse embryonic fibroblasts (MEF) were transfected either with EV, WT‐FMRP, SA‐FMRP, or STSA‐FMRP plasmids followed by PA treatment (500 µM; 6 h). Protein lysates were analyzed by Western blotting using specific antibodies for FMRP, pFMRP, pIRE1, and β‐Actin (n = 3 biological replicates).
Data information: A representative blot is shown. In D, E, G, and H data are cumulative results of two independent experiments. Data are mean ± SEM. Unpaired t‐test with Welch’s correction or paired t‐test.
Source data are available online for this figure.
Since FMRP phosphorylation is critical for translation suppression and the association with IRE1 juxtaposes it to a kinase whose substrate is unclear, we wondered whether ER stress alters FMRP phosphorylation state. To assess this possibility, we treated cultured macrophages with known ER stressors, such as TG and oxidized low‐density lipoprotein (oxLDL, another inducer of ER stress). Using specific antibodies that recognize S724 phosphorylation on IRE1 and S500 phosphorylation on FMRP (Reynolds et al, 2015; Wang et al, 2021), we found that TG and oxLDL significantly induced IRE1 autophosphorylation and FMRP phosphorylation but did not affect levels of FMRP protein or Fmr1 mRNA (Fig 1C and Appendix Fig S1A and B). Phosphatase treatment of the samples partially reversed the ER stress‐induced increase in the pFMRP/FMRP ratio (Fig 1C, Appendix Fig S1B), implying FMRP phosphorylation is enhanced by these ER stressors.
Previous studies showed that hyperlipidemia induces ER stress in plaque macrophages in vivo (Moore et al, 2013; Kim et al, 2016). We next investigated whether hyperlipidemia also has an effect on FMRP phosphorylation on S499 (mouse S499 corresponds to human S500). To this end, we used mice deficient in apolipoprotein E (Apoe−/−) as this is a protein found in plasma lipoprotein particles and facilitates cholesterol clearance from the circulation (Davignon et al, 1999). In agreement with previous reports, we observed that ER stress (as monitored by IRE1 autophosphorylation) was induced in the peritoneal macrophages (PM) obtained from hyperlipidemic, Apoe−/− mice that were fed with a Western diet (WD) for 16 weeks when compared to Apoe−/− mice fed with chow diet (CD) (Fig 1D). We found that FMRP S499 phosphorylation, which leads to FMRP‐mediated translational suppression, was 1.4‐fold elevated by a chronic exposure to hypercholesterolemia, whereas FMRP protein and Fmr1 mRNA expression levels remained unchanged (Fig 1D; Appendix Fig S1C and D).
We next asked what role IRE1 plays in ER stress‐induced FMRP phosphorylation. To address this question, we transfected IRE1‐specific siRNA to suppress IRE1 expression in a human cell line, followed by ER stress induction by a saturated fatty acid, palmitate (PA), that is known to induce ER stress, or TG. While both ER stressors induced the phosphorylation of IRE1 and FMRP, this was prevented in IRE1 knock‐down cells (Fig 1E and Appendix Fig S1E). In addition, we analyzed hypercholesterolemia‐induced FMRP phosphorylation in PMs obtained from Apoe−/− mice with a genetic deletion of IRE1α in the myeloid lineage (IRE1−/−) (see Methods) after feeding with WD for 16 weeks. FMRP phosphorylation, but not FMRP protein or Fmr1 mRNA, was reduced in PM isolated from IRE1−/− mice when compared to those isolated from IRE1+/+ mice (Fig 1F, Appendix Fig S1F and G). While these results show a clear reduction in FMRP phosphorylation upon siRNA treatment or gene knock‐out, we observed residual signal in both cases (Fig 1E and F). As we confirm below (Fig 1K), the antibody used in these experiments is phosphorylation specific. We, therefore, surmise that partial phosphorylation on S500/S499 may also be mediated by the other kinases that are known to phosphorylate this residue on FMRP (Narayanan et al, 2008; Bartley et al, 2016), in addition to IRE1 as we show here. Collectively, our in vitro and in vivo results strongly support that ER stress‐induced activation of IRE1 kinase leads to enhanced phosphorylation of FMRP on S500/S499.
IRE1 phosphorylates FMRP
To begin investigating the role of IRE1’s kinase activity in ER stress‐induced FMRP phosphorylation, we expressed a 3xFLAG‐tagged IRE1 (FLAG‐IRE1) and/or EGFP‐FMRP in wild‐type mouse embryonic fibroblasts (MEFs). Endogenous FMRP and IRE1 migrated faster in SDS–PAGE gel than the epitope tagged EGFP‐FMRP and FLAG‐IRE1, respectively. In all conditions, the MEFs were TG‐treated (to stimulate IRE1 kinase activity) in the absence or presence of an IRE1 kinase‐specific inhibitor (AMG‐18) (Papandreou et al, 2011; Ghosh et al, 2014; Tufanli et al, 2017; Harnoss et al, 2019). In the absence of AMG‐18, both IRE1 and FMRP were phosphorylated. AMG‐18 treatment prevented IRE1 phosphorylation while clearly reducing FMRP phosphorylation (Fig 1G and Appendix Fig S1H). While these data further support the notion that IRE1 phosphorylates FMRP during ER stress, alternative kinase(s) appears to mediate the same reaction. This is consistent the published data that several kinases can phosphorylate the same residue on FMRP (Narayanan et al, 2008; Bartley et al, 2016). Furthermore, in an in vivo setting where mice were injected with TM to induce IRE1 kinase activity, treatment with AMG‐18 inhibited IRE1 kinase activity and reduced FMRP phosphorylation in PM (Fig 1H and Appendix Fig S1I). Taken together, our findings support the notion that IRE1 kinase activity makes an important contribution to ER stress‐induced FMRP S499 phosphorylation in mouse macrophages and MEFs as well as S500 in HEK293T cells. Our data also support that other known or unknown FMRP kinase(s) are responsible for basal FMRP phosphorylation that is observed in non‐stress conditions.
To ask whether IRE1 can phosphorylate FMRP directly, we employed in vitro assays. To this end, we immunoprecipitated wild type (WT) or kinase dead (KD) mutant IRE1 from HEK293T cells (after treating with ER stressor) and incubated the immunoprecipitates with purified, recombinant human FMRP protein in a kinase reaction. The reaction included ATP‐γ‐S instead of ATP, which allows kinases to thio‐phosphorylate their substrates. The resultant kinase reaction was analyzed by Western blotting using an anti‐thiophosphate ester antibody. In the reaction containing IRE1‐WT, both proteins were thio‐phosphorylated, but not in the reaction containing the IRE1‐KD mutant (Fig 1I).
To determine the specific amino acids phosphorylated by IRE1, we performed the kinase reaction using purified, recombinant human IRE1‐kinase/RNase domains (amino acids 468–977) and human FMRP proteins. Both IRE1 and FMRP were phosphorylated in this reaction in an AMG‐18‐sensitive manner (Fig 1J). Liquid chromatography‐mass spectrometry (LC‐MS/MS)‐based analysis of the phosphorylated FMRP residues in this kinase reaction revealed eight IRE1‐induced phosphorylation sites on FMRP at serine (S362, S494, S497, S500, S504) and threonine (T502, T592, T594) (Fig 1J, Appendix Fig S1J and K). Importantly, S500 in human FMRP is the previously identified FMRP phosphorylation site that was shown to enhance FMRP‐mediated translational suppression (Ceman et al, 2003; Narayanan et al, 2008; Coffee et al, 2012; Niere et al, 2012; Bartley et al, 2014, 2016; Prieto et al, 2020).
Using site‐directed mutagenesis, we engineered two mutant versions of FMRP, in which we either changed S500 to alanine (SA mutant) or S500, T502, and S504 to alanine (STSA triple mutant) to block phosphorylation at S500 and amino acids in its proximity. We then induced ER stress with PA in Fmr1−/− MEFs transiently transfected with WT FMRP and FMRP mutants (SA and STSA). PA induced FMRP phosphorylation in WT FMRP‐reconstituted cells but failed to do so in cells expressing the SA and STSA mutants of FMRP (Fig 1J). These data confirm the specificity of the FMRP antibody for phosphorylated S500 and is consistent with the notion that IRE1 kinase directly phosphorylates human FMRP protein on S500.
IRE1‐FMRP signaling induces foam cell formation while suppressing RCT
We next wondered what role FMRP plays in macrophage biology that is relevant to atherosclerotic plaque development. We performed an in vivo macrophage foam cell formation assay in the peritoneum of Fmr1−/− and Fmr1+/+ mice, using a well‐established method in which we induced hyperlipidemia using a combination of adenoviral‐delivery of proprotein convertase subtilisin kexin 9 (AAV_PCSK9), a protein that directs hepatic low‐density lipoprotein (LDL) receptors for degradation, and feeding with WD for 16 weeks (Li et al, 2007; Tsimikas et al, 2011; Peled et al, 2017). FMRP deficiency significantly reduced foam cell formation in vivo (Fig 2A). Next, we fed Apoe−/− mice with a WD (12 weeks) and injected them daily with the IRE1 kinase inhibitor, AMG‐18, or vehicle for the last 4 weeks. AMG‐18 reduced foam cell formation in the peritoneum in vivo (Fig 2B). This result indicates that IRE1‐FMRP signaling axis enhances foam cell formation.
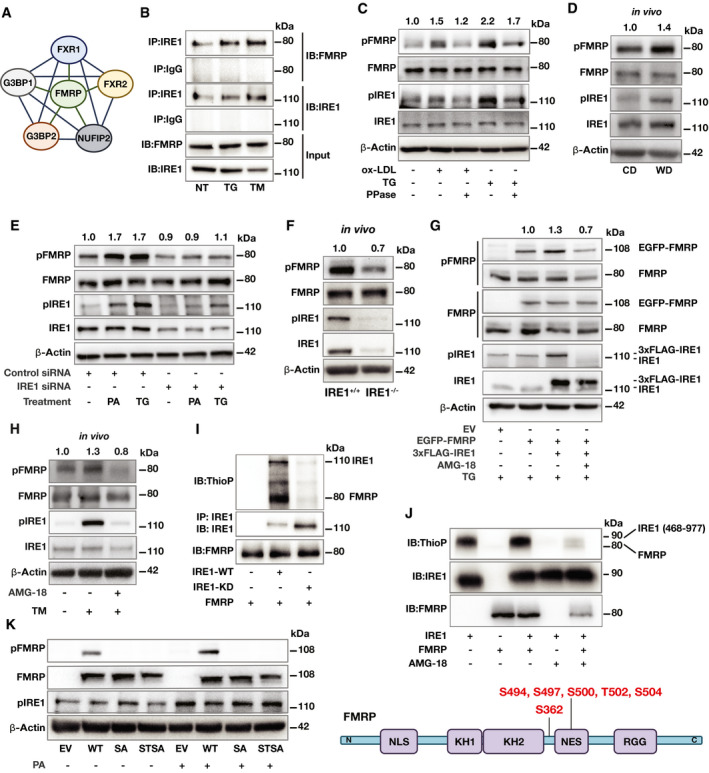
Figure 2. FMRP deficiency enhances RCT while reducing foam cell formation in vivo .
-
AFmr1+/+ and Fmr1−/− mice were injected with AAV_PCSK9 and fed with 16 weeks of WD. Residential PM were stained with Oil Red O (ORO) and imaged (n = 7 mice per group; Scale bar = 50 µm).
-
BApoe−/− mice were fed with WD (12 weeks) and injected with vehicle (DMSO) or AMG‐18 (30 mg/kg/day) in the last 4 weeks of WD. Residential PM were stained with ORO and imaged (n = 5 mice per group; Scale bar = 50 µm).
-
C–EFlow cytometry analysis of BMDMs after dil‐ac‐LDL (25 µg/ml) loading for 24 h; (C) control‐ or Fmr1‐siRNA transfected BMDM (n = 4 biological replicates), (D) Fmr1+/+ and Fmr1−/− BMDM (n = 4 biological replicates), (E) BMDM pre‐treated with either vehicle (DMSO) or AMG‐18 (5 µM; 1 h) (n = 6 biological replicates).
-
F, GMacrophages were pre‐loaded with fluorescently labeled cholesterol (16 h) followed by incubation in efflux medium including APOA1 (25 µg/ml) or HDL (50 µg/ml) as acceptors for 6 h. % Efflux was calculated as cholesterol signal in medium/cholesterol signal in medium and cell: Cholesterol efflux in (F) Fmr1+/+ and Fmr1−/− BMDM (n = 4 biological replicates) and in (G) BMDM that were pre‐treated either with DMSO or AMG‐18 (5 µM; 1 h) (n = 4 biological replicates).
-
H–KRCT experiment: (H) Schematic representation of C57BL6 mice were injected with [3H]‐cholesterol‐loaded foamy Fmr1+/+ and Fmr1−/− BMDM, (I) plasma cholesterol levels after 24 and 48 h, (J) liver cholesterol levels after 48 h, and (K) feces cholesterol levels after 48 h (n = 12 mice per group).
Data information: Data are mean ± SEM. Unpaired t‐test with Welch’s correction.
Source data are available online for this figure.
We also transfected wild type BMDMs with either Fmr1‐specific or control siRNA and followed by loading the cells with 3,3′‐dioctadecylindocarbocyanine (dil)‐labeled acetylated LDL (Ac‐LDL) (Carotti et al, 2021). Flow cytometry analysis revealed that Fmr1 silencing significantly reduced % dil‐acLDL internalized in macrophages (Fig 2C, Appendix Fig S2A). Likewise, Fmr1−/− BMDM displayed reduced % dil‐acLDL internalization when compared to Fmr1+/+ BMDM (Fig 2D). A similar reduction in foam cell formation was observed with the IRE1 kinase inhibitor (Fig 2E). These results indicate that both the inhibition of IRE1 kinase activity and the genetic deletion of its proposed substrate, FMRP, reduce foam cell formation in vitro and in vivo. Reduced foam cell formation could be explained with less cholesterol uptake, however, neither FMRP knock down nor IRE1 kinase inhibition altered cholesterol uptake in macrophages (Appendix Fig S2B). We reasoned that this observation is most likely related to an increase in cholesterol export (RCT; due to increased translation of cholesterol exporters) from Fmr1−/− macrophages. Indeed, FMRP deficiency led to an increase in cholesterol efflux coupled with its loading onto the cholesterol carriers, apolipoprotein‐A1 (APOA1) and HDL (Fig 2F), and, likewise, the IRE1 kinase inhibitor enhanced cholesterol efflux (Fig 2G). Thus, ER stress‐associated reduction in cholesterol efflux is dependent on both IRE1 kinase activity and FMRP.
Next, we determined whether the absence of FMRP in BMDMs also enhances RCT in vivo. To this end, we pre‐loaded Fmr1+/+ and Fmr1−/− BMDMs with [3H]‐cholesterol and injected the cells subcutaneously into WT mice (Fig 2H). FMRP deficiency in macrophages significantly increased radioactivity counts ([3H]‐cholesterol) in the plasma, liver, and feces of the recipient mice, when compared to recipient mice that received Fmr1+/+ macrophages (Fig 2I–K), demonstrating that FMRP deficiency in macrophages enhances RCT in mice.
FMRP regulates macrophage efferocytosis
We next investigated the impact of FMRP deficiency on efferocytosis, a primary process that promotes atherosclerotic plaque regression by removing apoptotic macrophages in the lesion area. To this end, we transfected BMDM (red fluorescent stained) with Fmr1‐specific or control siRNA and incubated them with green fluorescent‐labeled apoptotic cells (ACs), in which apoptosis was induced by ultraviolet (UV) irradiation. FMRP knock‐down increased efferocytosis of ACs (as measured by colocalization of the fluorescent markers), when compared to control (Fig 3A). FMRP‐deficient BMDMs also increased efferocytosis when compared to wild type BMDMs under both no‐stress and PA‐induced ER stress conditions (Fig 3B). Next, we induced hyperlipidemia in Fmr1−/− and Fmr1+/+ mice as described above using a combination of AAV_PCSK9 injection and feeding with a WD. We then injected the mice intraperitoneally with green fluorescent‐labeled ACs and harvested PM macrophages. FMRP‐deficient PMs displayed enhanced efferocytosis compared to WT PMs (Fig 3C).
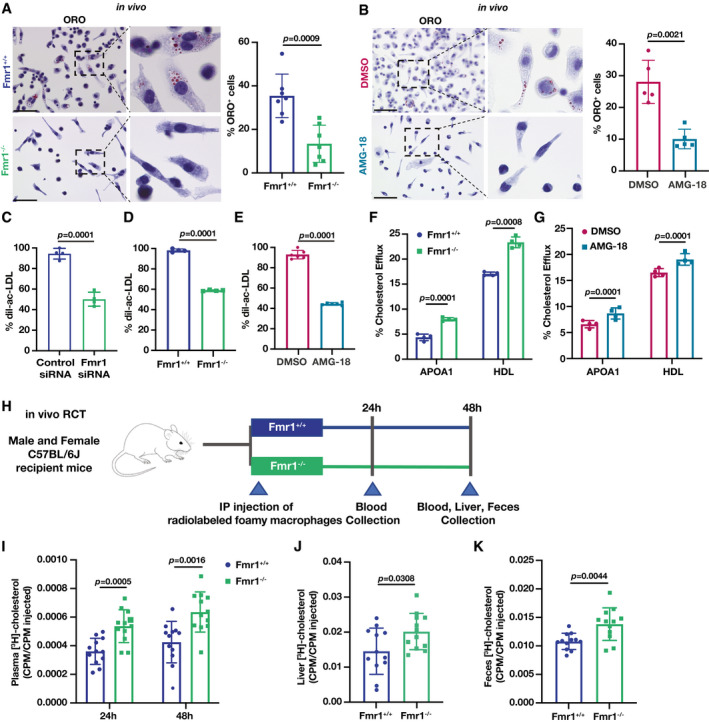
Figure 3. FMRP deficiency increases efferocytosis in vivo .
-
A–EIn vitro and in vivo efferocytosis experiments, where percentage of macrophages F4/80+ (red) that ingested apoptotic cells (AC) labeled with carboxyfluorescein succinimidyl ester (CFSE)+ (green) were reported as % efferocytosis. (A) BMDMs were transfected with Fmr1‐ or control‐siRNA and incubated CFSE‐labeled AC for the indicated hours (n = 4 biological replicates). (B) Fmr1+/+ and Fmr1−/− BMDMs were treated with PA (500 µM) for 6 h and then incubated with CFSE‐labeled ACs for 4 h (n = 4 biological replicates). (C) Fmr1+/+ and Fmr1−/− mice were fed WD (16 weeks) and injected intraperitoneally with CFSE‐labeled AC (1.5 h), followed by PM elicitation (n = 4–5 mice per group). (D) BMDM were pre‐treated either with vehicle (DMSO) or AMG‐18 (5 µM) for 1 h then incubated with CFSE‐labeled Acs for 4 h (n = 3 biological replicates). (E) C57BL/6 mice were injected with AMG‐18 (30 mg/kg) or vehicle (DMSO) for 8 h, followed by intraperitoneal injection with CFSE‐labeled ACs for 1.5 h and PM elicitation (n = 4 mice per group).
-
F, GIn vitro continuous efferocytosis experiments, where macrophages were stained for F4/80+ (red), AC were labeled with CFSE (AC‐1; green) or Violet (AC2; violet). % continuous efferocytosis was determined by the ratio of F4/80+, CFSE+, and Violet+ (triple positive) cells to total F4/80+ and CFSE+ (double positive) cells. (F) Fmr1+/+ and Fmr1−/− BMDM were incubated with AC‐1 for 2 h, and after 2 h interval, incubated with AC‐2 for 2 more hours (n = 4–3 biological replicates). (G) BMDM were pre‐treated either with vehicle (DMSO) or AMG‐18 (5 µM) for 1 h, incubated with CFSE‐labeled AC‐1 for 2 h, followed by incubation with Violet‐labeled AC‐2 for 2 h and PM collection (n = 4 biological replicates).
Data information: For all images scale bar = 50 µm; Red: Macrophages, Green: AC/AC‐1, Violet: AC‐2. Data are mean ± SEM. Unpaired t‐test with Welch’s correction.
Source data are available online for this figure.
We next asked how IRE1 kinase activity impacts efferocytosis by macrophages. We observed that AMG‐18 increased efferocytosis of ACs in BMDMs (Fig 3D). Next, we injected wild type mice with AMG‐18 (for 8 h) followed by injection of green fluorescent‐labeled ACs. AMG‐18 also induced efferocytosis of ACs by PM in vivo (Fig 3E).
Continued clearance of ACs by macrophages prevents the accumulation of necrotic cells and is an important process that promotes atherosclerosis regression (Yurdagul et al, 2017). To determine whether Fmr1−/− macrophages can efficiently internalize multiple ACs over consecutive rounds of engulfment, we incubated macrophages with green fluorescent‐labeled AC for 2 h, followed by second incubation with violet fluorescent‐labeled AC for 2 more hours. FMRP‐deficient BMDMs displayed an increase in continued efferocytosis when compared to wild type BMDMs (Fig 3F). Likewise, treatment of wild type BMDM with AMG‐18 enhanced continued efferocytosis of ACs (Fig 3G). Collectively, these results demonstrate that the ablation of IRE1 kinase activity and its proposed substrate, FMRP, enhances efferocytosis in vitro and in vivo.
Translational suppression of cholesterol transporters and efferocytosis regulators by FMRP during ER stress
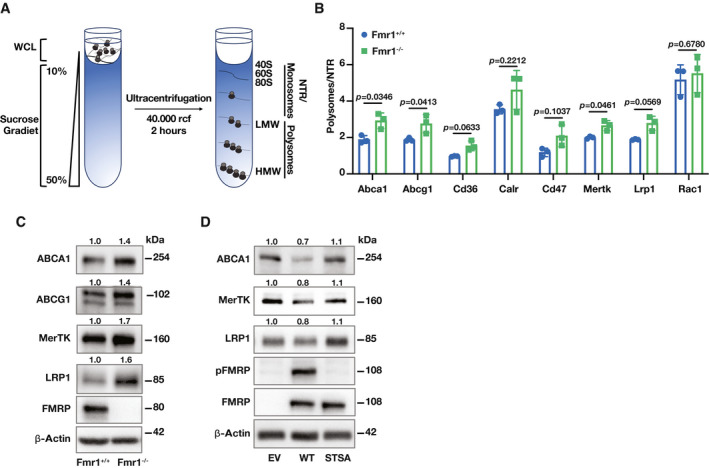
Consistent with our observations that macrophage FMRP plays a role in suppressing cholesterol efflux and efferocytosis, published data describing the FMRP‐RNA interactome also suggest that FMRP interacts with mRNA‐encoding proteins involved in cholesterol trafficking (such as Abca1 and Abcg1) and efferocytosis receptors (such as c‐Mer tyrosine kinase (Mertk) and LDL receptor‐related protein 1 (Lrp1)), suggesting FMRP may impair their translation (Darnell et al, 2011; Ascano et al, 2012). To assess this notion in macrophages, we next performed polyribosome profiling in Fmr1+/+ and Fmr1−/− BMDMs under PA‐induced ER stress conditions (Fig 4A and B, Appendix Fig S3A). Indeed, the mRNA abundance for the cholesterol transporters, Abca1 and Abcg1, and the mRNA abundance for efferocytosis regulators, Mertk, Lrp1, and Cd36, were increased in translating polysome fractions and decreased in non‐translating (NTR) fractions in the Fmr1−/− BMDMs when compared to Fmr1+/+ BMDMs (Fig 4B), while the abundance of these mRNAs in the total cell lysate was unchanged (Appendix Fig S3B).
Figure 4. FMRP targets in macrophages.
-
A, BRNA lysates from Fmr1+/+ and Fmr1−/− BMDM that were treated with PA (500 µM; 6 h) were fractionated using a 10–50% sucrose gradient and separated to polysome, monosome/NTR fractions. The absorbance (260 nm) of RNA was measured and plotted as a function of time (n = 3 biological replicates). (A) Representative profile for RNA distribution from genotypes based on UV absorbance readings after sucrose gradient fractionation. (B) The ratio of the Abca1, Abcg1, Mertk, Lrp1, Cd36, Cd47, and Rac1 mRNA in polysome to NTR fraction (n = 3 biological replicates).
-
CBMDM were isolated from Fmr1+/+ and Fmr1−/−, and protein lysates were analyzed by Western blotting using specific antibodies for ABCA1, ABCG1, MerTK, LRP1, FMRP, and β‐Actin antibodies and fold inductions relative to β‐Actin are depicted above the blots (n = 6 biological replicates).
-
DFmr1−/− MEF cells were transfected with EV, WT‐FMRP, or STSA‐FMRP plasmids followed by PA treatment (500 µM; 6 h). Protein lysates were analyzed by Western blotting using specific antibodies for ABCA1, MerTK, LRP1, pFMRP, FMRP, and β‐Actin and fold inductions relative to β‐Actin are depicted above the blots (n = 5 biological replicates).
Data information: A representative blot is shown. In C and D, data are cumulative results of 2 and 3 independent experiments, respectively. In Western blots, the protein expression fold change was calculated relative to β‐Actin and depicted above the blots and a representative blot was shown. Data are mean ± SEM. Unpaired t‐test with Welch’s correction.
Source data are available online for this figure.
We next analyzed the corresponding protein expression changes for the FMRP‐regulated mRNA targets from the same experiment in Fig 4A and B. As expected, ABCA1, ABCG1, MerTK, and LRP1 protein expression levels were induced in Fmr1−/− BMDMs (Fig 4C and Appendix Fig S3C). To further assess the impact of IRE1‐mediated FMRP phosphorylation on the translation of FMRP’s targets, we overexpressed the FMRP phosphorylation‐deficient mutant (STSA) or WT‐FMRP in Fmr1−/− MEFs and treated with PA to induce ER stress. ABCA1, LRP1, and MerTK expression levels were reduced in the Fmr1−/− MEF expressing WT‐FMRP, but not in Fmr1−/− MEFs expressing STSA‐FMRP (Fig 4D and Appendix Fig S3D). Taken together, our data demonstrate that IRE1‐mediated FMRP phosphorylation tunes cholesterol transporters and efferocytosis regulators expression in macrophages.
FMRP knock‐down and IRE1 kinase inhibition alleviates atherosclerosis
Our findings demonstrate that both IRE1 kinase inhibition and FMRP deficiency result in increased RCT, reduced foam cell formation, and enhanced efferocytosis in vivo, suggesting that FMRP deficiency in mice leads to protection from atherosclerosis. We tested this notion using Fmr1−/− and Fmr1+/+ mice in which hyperlipidemia was induced by combining AAV_PCSK9 injection with a WD as described above (Fig 5A). Although there was a very slight decrease in body weight ratio; the plasma glucose, total plasma cholesterol (TPC), lipoprotein levels, and the number of circulating, major type of immune cells were indistinguishable between the Fmr1−/− and Fmr1+/+ genotypes (Appendix Fig S4A–F). Yet, FMRP deficiency resulted in a significant reduction in atherosclerotic lesions in en face aorta preparations (Fig 5B). FMRP deficiency did not alter aortic root lesion area despite a significantly decreased foam cell area (as assessed by Oil Red O staining) (Fig 5C and D). We next asked what may be contributing to this phenotype. The necrotic core area in the lesions from Fmr1−/− mice was significantly less than in Fmr1+/+ lesions (Fig 5E), indicating improved AC clearance by Fmr1−/− macrophages in plaques. We also observed a significant reduction in lesion macrophages (as assessed by the anti‐monocyte macrophage 2 (MOMA‐2)‐stained area) and the number of apoptotic cells (TUNEL‐stained) per macrophage area (Appendix Fig S4G and H). We further investigated whether apoptosis is altered in Fmr1−/− and AMG‐18‐treated macrophages. There was no significant change between the groups (Appendix Fig S4I), supporting the notion that the primary consequence of inhibiting IRE1‐FMRP signaling is efficient clearance of apoptotic cells through increasing efferocytosis capacity. Additionally, we detected no change in smooth muscle area (stained with smooth muscle actin (SMA)), but there was a significant increase in the collagen content of Fmr1−/− lesions when compared to Fmr1+/+ lesions, suggesting this could be the reason why aortic root lesion area is not significantly altered (Appendix Fig S4J and K).
Figure 5. FMRP‐deficiency alleviates atherosclerosis.
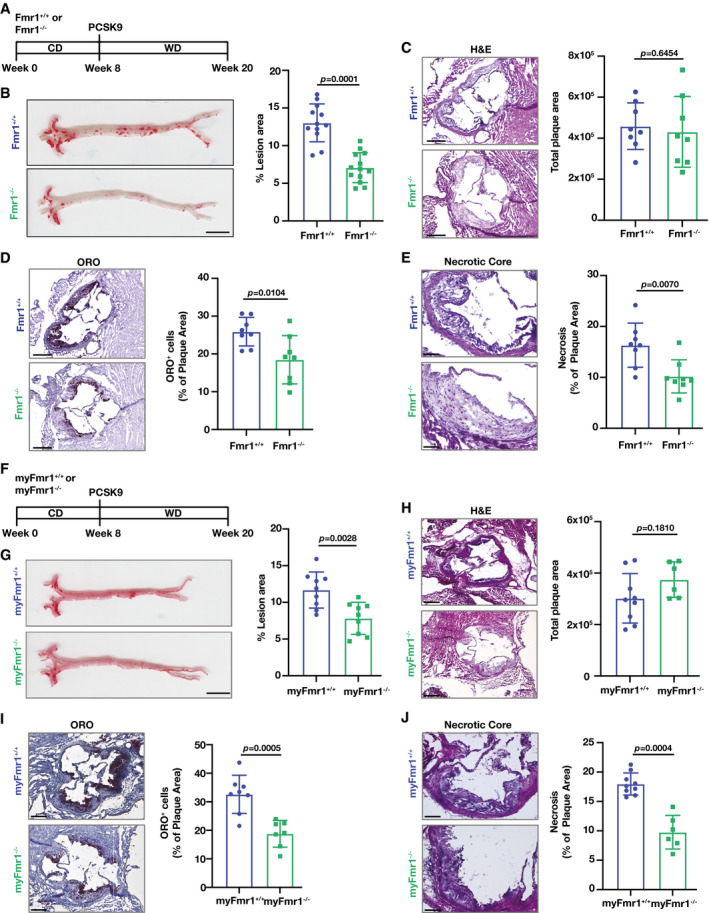
- Atherosclerosis experiment design in Fmr1+/+ and Fmr1−/− mice that were injected with AAV_PCSK9 and fed WD (16 weeks).
- Lesion area calculated from en face aorta, stained with ORO (n = 12–13 mice per group; Scale bar = 5 mm).
- Total plaque area was calculated from hematoxylin & eosin (H&E)‐stained aortic root sections (n = 8 mice per group; Scale bar = 300 µm).
- Foam cell area was calculated from ORO‐stained aortic root sections (n = 8 mice per group; Scale bar = 300 µm).
- Necrotic area was calculated from H&E‐stained aortic root sections (n = 8 mice per group; Scale bar = 100 µm).
- Atherosclerosis experiment design in myFmr1+/+ and myFmr1−/− mice that were injected with AAV_PCSK9 and fed WD (16 weeks).
- Lesion area calculated from en face aorta, stained with ORO (n = 9 mice per group; Scale bar = 5 mm).
- Total plaque area was calculated from H&E‐stained aortic root sections (n = 9–6 mice per group; Scale bar = 300 µm).
- Foam cell area was calculated from ORO‐stained aortic root sections (n = 9–6 mice per group; Scale bar = 300 µm).
- Necrotic area was calculated from H&E‐stained aortic root sections (n = 9–6 mice per group; Scale bar = 100 µm).
Data information: Data are mean ± SEM; Mann Whitney U test.
Source data are available online for this figure.
To approach the role of macrophage FMRP in atherosclerosis, we generated a myeloid Fmr1‐deficient mouse model (myFmr1−/−) and induced hyperlipidemia (Fig 5F). As expected from the systemic deletion data shown in Fig 5A, the body weight, plasma glucose and TPC were indistinguishable between the myFmr1−/− and myFmr1+/+ genotypes (Appendix Fig S4L–N). in general, myeloid‐specific FMRP deficiency paralleled the results obtained for Fmr1−/− mice: it resulted in a significant reduction in atherosclerotic lesions in en face aorta preparations (Fig 5G). Myeloid‐specific FMRP deficiency did not alter aortic root lesion area but significantly reduced foam cell area (Fig 5H and I). The necrotic core area in the lesions from myFmr1−/− mice was also significantly less than in myFmr1+/+ lesions (Fig 5J), indicating improved AC clearance by Fmr1−/− macrophages in plaques and supporting our observations that FMRP deficiency increases macrophage efferocytosis in vitro and in vivo (Fig 3). Thus, comparing the data obtained with the Fmr1−/− mice with those obtained from the myFmr1−/− mice indicates that the atheroprotective effects observed in Fmr1−/− mice are mostly, if not exclusively, due to FMRP’s role in myeloid cells such as macrophages.
To test the notion that IRE1 functions upstream of FMRP, we next investigated the impact of IRE1 kinase inhibition on atherosclerosis. To this end, we fed Apoe−/− mice with WD for 12 weeks and injected them with AMG‐18 or vehicle once daily for the last 4 weeks of WD (Fig 6A). We observed no significant differences in body weight, plasma glucose or TPC, and the number of circulating immune cells between the groups (Appendix Fig S5A–D). Consistent with an earlier publication that determined the effective and non‐toxic dose for AMG‐18 in mice, the inhibitor engaged its molecular target effectively in the treatment group (as assessed by reduced IRE1 autophosphorylation) (Appendix Fig S5E). IRE1 kinase inhibition led to a decrease in atherosclerotic lesions in en face aorta preparations (Fig 6B). As shown above for the Fmr1−/− mice, the aortic root lesion area was not different between the groups (Fig 6C), but foam cell area as well as necrotic core area were significantly decreased (Fig 6D and E).
Figure 6. IRE1 Kinase inhibition alleviates atherosclerosis.
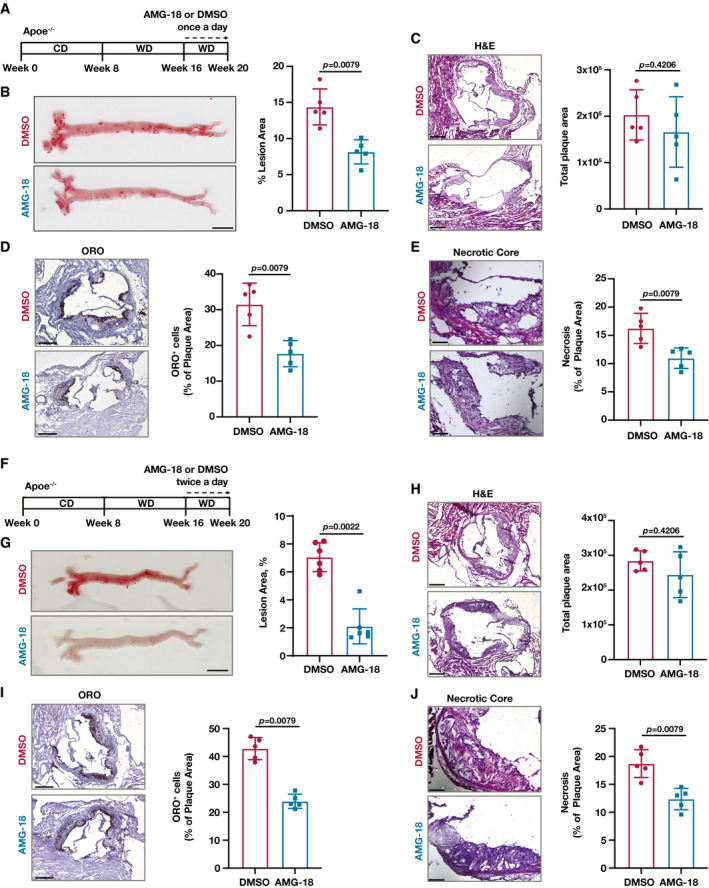
- Atherosclerosis experiment design in Apoe−/− mice were fed with WD (12 weeks) and injected with vehicle (DMSO) or AMG‐18 (30 mg/kg) once a day in the last 4 weeks of WD.
- Lesion area calculated from en face aorta, stained with ORO (n = 5 mice per group; Scale bar = 5 mm).
- Total plaque area was calculated from H&E‐stained aortic root sections (n = 5 mice per group; Scale bar = 300 µm).
- Foam cell area was calculated from ORO‐stained aortic root sections (n = 5 mice per group; Scale bar = 300 µm).
- Necrotic area was calculated from H&E‐stained aortic root sections (n = 5 mice per group; Scale bar = 100 µm).
- Atherosclerosis experiment design in Apoe−/− mice were fed with WD (12 weeks) and injected with vehicle (DMSO) or AMG‐18 (30 mg/kg) twice a day in the last 4 weeks of WD.
- Lesion area calculated from en face aorta, stained with ORO (n = 6 mice per group; Scale bar = 5 mm).
- Total plaque area was calculated from H&E‐stained aortic root sections (n = 5 mice per group; Scale bar = 300 µm).
- Foam cell area was calculated from ORO‐stained aortic root sections (n = 5 mice per group; Scale bar = 300 µm).
- Necrotic area was calculated from H&E‐stained aortic root sections (n = 5 mice per group; Scale bar = 100 µm).
Data information: Data are mean ± SEM; Mann Whitney U test.
Source data are available online for this figure.
We also assessed the impact of twice daily injections of the same dose of AMG‐18 on Apoe−/− mice that were fed with WD for 12 weeks (Fig 6F). While we noted a decrease in body weight ratio and TPC, there were no significant differences in plasma glucose levels similar to once daily injection (Appendix Fig S5F–H), and the inhibitor engaged its molecular target effectively (as assessed by reduced IRE1 autophosphorylation) (Appendix Fig S5I and J). IRE1 kinase inhibition led to a significant decrease in atherosclerotic lesions in en face aorta preparations (Fig 6G). AMG‐18 injection did not alter aortic root lesion area but significantly decreased foam cell area and necrotic core area (Fig 6H–J). Collectively, our findings thus demonstrate that the inhibition of IRE1 kinase activity by a small‐molecule inhibitor or genetic ablation of FMRP, its kinase substrate, in macrophages can reduce the progression of hypercholesterolemia‐induced atherosclerosis.
Discussion
Chronic lipid accumulation in the ER membranes (e.g., during obesity and in hyperlipidemia) has been shown to impair ER functions and activate UPR signaling (Li et al, 2004; Borradaile et al, 2006; Fu et al, 2011; Volmer et al, 2013; Çimen et al, 2016b). Numerous studies have shown that ER stress is an important primer for sterile inflammation that drives insulin resistance and atherogenesis (Hotamisligil, 2017). Moreover, alleviating ER stress by inhibiting either PERK or IRE1 signaling prevents atherosclerosis progression (Erbay et al, 2009; Tabas, 2010; Tufanli et al, 2017; Onat et al, 2019). But how does chronic IRE1 kinase activation contribute to the atherosclerotic process? Our work revealed that in ER‐stressed macrophages phosphorylation of FMRP, an RBP functioning as a translational suppressor, is enhanced in an IRE1 kinase‐dependent manner. This phosphorylation event results in a gain‐of‐function for FMRP, which leads to enhanced translational suppression of cholesterol transporters and efferocytosis receptors. Although ubiquitously expressed, prior work focused almost exclusively on FMRP’s role in neurons in the context of its devastating role in FXS pathology (Sethna et al, 2014; Davis & Broadie, 2017; Leboucher et al, 2019a). Our work reveals a novel function for FMRP in macrophages. Intriguingly, an earlier publication showed that FMRP protein expression is elevated in the macrophage‐enriched area of human atherosclerotic plaques (Tuomisto et al, 2003). Moreover, lower cholesterol levels have been measured in a subpopulation of the individuals afflicted with FXS, as well as in Fmr1−/− mice (Berry‐Kravis et al, 2015; Lisik et al, 2016; Leboucher et al, 2019b). Although these findings foreshadowed a role of FMRP in the regulation of cholesterol homeostasis (Tuomisto et al, 2003; Darnell et al, 2011; Ascano et al, 2012), FMRP’s contribution to atherosclerosis was not investigated prior to this study, perhaps because Fmr1 lies on the X chromosome and most genome‐wide association studies in humans focus on variants on autosomal chromosomes excluding sex chromosomes. While characterizing the impact of IRE1 kinase activity and it’s here proposed kinase substrate, FMRP, on macrophage functions, our current study revealed a role for an “IRE1‐FMRP signaling axis” in the regulation of macrophage cholesterol trafficking and efferocytosis, which are among the primary cellular mechanisms that can regress atherosclerosis (Fig 7).
Figure 7. IRE1‐FMRP signaling controls cholesterol efflux and efferocytosis pathways in macrophages.
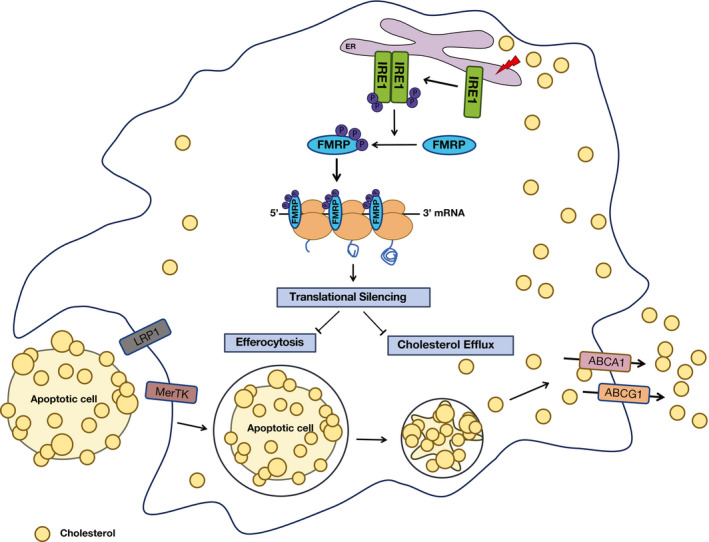
IRE1‐mediated FMRP phosphorylation suppresses translation of mRNA for key cholesterol transporters and efferocytosis receptors in macrophages and promotes atherosclerosis.
To gain insight into the physiological role of macrophage FMRP, we investigated the consequences of FMRP deficiency on macrophage biology that are relevant to atherosclerosis development and plaque regression, such as foam cell formation, cholesterol efflux, and efferocytosis. Our findings show that FMRP has a prominent role in all three processes. Additionally, our observations are consistent with the finding that several mRNAs encoding cholesterol transporters and efferocytosis receptors interact with FMRP in prior RNA crosslinking immunoprecipitation (CLIP) experiments (Darnell et al, 2011; Ascano et al, 2012). Our data show that the absence of FMRP in macrophages increases the occupancy of these mRNAs in the polysome fraction, paralleling the increased expression of their corresponding protein products. In agreement with our observations in cells, FMRP deficiency in macrophages increased RCT and efferocytosis in mice. Moreover, systemwide and myeloid‐specific Fmr1 knock‐out mice were protected from hypercholesterolemia‐induced atherosclerosis progression. In agreement with this notion, the IRE1 kinase‐specific inhibitor, AMG‐18, phenocopies the beneficial impact of FMRP deficiency (such as enhanced cholesterol efflux and efferocytosis) in macrophages. While our studies provide strong support for the role of macrophage FMRP in atherosclerosis progression, we did not examine the contribution of FMRP activity in other lesion cell types, such as endothelial or vascular smooth muscle cells, on atherosclerosis. The impairment of RCT and efferocytosis by IRE1‐FMRP signaling implies that interventions to ablate FMRP in a macrophage‐specific manner and in the adult organism could promote atherosclerotic plaque stabilization and hinder plaque progression, while escaping an adverse impact of FMRP deficiency on neuronal development.
Two recent studies show that FMRP plays a metabolic role in the liver. For example, systemic FMRP deficiency in mice led to enhanced glucose tolerance and insulin sensitivity, while lowering plasma triglyceride and cholesterol content (Leboucher et al, 2019b). Another study showed combined deficiency of FMRP and its paralog FXR2 (Fragile X mental retardation autosomal homolog 2) resulted in lower body fat and increased sensitivity to insulin (Lumaban & Nelson, 2015). Intriguingly, the FMRP loss‐of‐function in liver resulted in increased translation of hepatic mRNA involved in lipid metabolism (Leboucher et al, 2019b). In contrast to this previous work, we did not find Fmr1 deficiency‐associated changes in plasma cholesterol or lipoprotein levels in our atherosclerosis mouse models, perhaps due to the severe hypercholesterolemia induced by both genetic and dietary interventions.
In an earlier study, we had shown that small‐molecule inhibitors that are specific for IRE1’s RNase activity prevented lipid‐induced inflammasome activation and secretion of mature interleukin‐1β (m‐IL‐1β) and m‐IL‐18 in both mouse and human macrophages while reducing hyperlipidemia‐induced m‐IL‐1β and m‐IL‐18 production and atherosclerotic plaque size in mice (Tufanli et al, 2017). FMRP suppression also reduces m‐IL‐1β secreted from macrophages but without altering inflammasome activation (Appendix Fig S6A–F). Knocking out FMRP from macrophages has no effect on IL‐1β mRNA levels in the whole cell lysate or in the polysomes, suggesting against transcriptional or translational control over these cytokines’ production (Appendix Fig S6G and H). As expected, inhibition of IRE1 kinase also reduces m‐IL‐1β (Appendix Fig S6I–K). Intriguingly, IL‐1β can be secreted through ABCA1 and compete with cholesterol efflux through the same transporter (Tumurkhuu et al, 2018). Since the inhibition of IRE1 kinase‐FMRP axis leads to a marked upregulation of cholesterol efflux in macrophages (as shown in our study), it is plausible that increased demand for ABCA1 for cholesterol efflux could prevent IL‐1β secretion through this route.
Our finding that IRE1 phosphorylates FMRP leads to an important question: Why does the UPR signaling impinge on FMRP‐mediated translational suppression? While intensely debated, multiple mechanisms have been proposed for translational suppression by FMRP including polyribosome stalling, miRNA‐mediated translational silencing, and impairing translational initiation (Napoli et al, 2008; Edbauer et al, 2010; Darnell et al, 2011; Chen et al, 2014). Recently, phosphorylation‐dependent FMRP‐RNA phase separation has been suggested as a concept underlying these diverse regulatory mechanisms (Tsang et al, 2019). It is plausible that during ER stress, IRE1‐induced FMRP phosphorylation, licensed by S500 phosphorylation and then spreading to adjacent serines and threonines, enhances phase separation of FMRP and leads to the redistribution of FMRP‐bound RNA into cytoplasmic granules, where they will not be translated. This would, in effect, reduce the protein folding stress and improve ER homeostasis. Indeed, ER stress induces stress granule formation, which contain mRNAs, RBPs (including FMRP), 40S ribosomal subunits, and translational pre‐initiation factors (Buchan & Parker, 2009).
Finally, our findings demonstrate that IRE1 kinase‐specific inhibitor, AMG‐18, can phenocopy the beneficial impact of FMRP deficiency (such as enhanced cholesterol efflux and efferocytosis) in macrophages. Although we cannot definitively show that FMRP is the only mediator downstream of IRE1 kinase activity in regulating the proposed mechanisms, our data strongly support that this novel IRE1 kinase target’s expression in macrophages plays an important role in cholesterol efflux and apoptotic cell clearance and is an important contributor to atheroprotection offered by IRE1 kinase inhibition.
Our study provides mechanistic insight into the translational regulation of cholesterol efflux and efferocytosis by macrophages during ER stress, highlighting IRE1 and its effector FMRP as a promising molecular target for regulating atherosclerosis. The small‐molecule inhibitors of IRE1 kinase activity represent a translational opportunity in combating atherosclerosis.
Materials and Methods
General study design
Three or more independent replicates were performed for cell‐based experiments. Mice were randomly assigned to independent cohorts, and data analysis was performed blind. The only elimination criteria used for mouse studies was based on health and as advised by a veterinarian.
Reagents and plasmids
WT‐Ire1‐pcDNA3, and Kinase‐dead IRE1 mutant (K599A)‐pcDNA3 plasmids were generated in Dr. Peter Walter’s laboratory (University of California, San Francisco). Fatty acid‐free bovine serum albumin was from Gold Biotechnology (A‐421–250). Lipofectamine 3000 (L3000001) and EDTA (15575020) were purchased from Invitrogen. Polyethylemine (PEI; molecular weight 25,000) was from Polysciences. Neon transfection system kit (MPK10096) was from Thermo Scientific and used with the Neon electroporation system, also from Thermo Scientific, according to previously published protocols for BMDM (Tufanli et al, 2017; Onat et al, 2019). l‐Glutamine, Dulbecco’s modified Eagle’s Medium (DMEM), penicillin/streptomycin (P/S), fetal bovine serum (FBS), Roswell Park Memorial Institute (RPMI)‐1640 medium, phosphate buffer saline (PBS), CellTrace™ CFSE cell proliferation kit (C34554), CellTrace™ Violet Cell Proliferation Kit (C34557), Fmr1 siRNA (AM16708), Control siRNA (negative control, AM4611), Revert Aid First strand cDNA synthesis kit (K1691), and Formaldehyde (31901) were from Thermo Scientific. Protease inhibitor cocktail (P8340), phosphatase inhibitor cocktail‐3 (P0044), Dimethyl sulfoxide (DMSO, D8418), Trypsin, Ampicillin, palmitic acid (PA; P0500), Oil Red O solution (O1391), Hematoxylin (HHS32), Lipoprotein‐deficient serum (S5519), CC mount (C9368), ECL™ Prime Western Blotting Detection Reagent (GERPN2232), fatty acid‐free BSA (A6003), Sucrose (S0389), and thioglycolate solution (70157) were purchased from Sigma. Dil‐labeled ac‐LDL (770201‐7) was from Kalen Biomedical. The IRE1 Kinase inhibitor, AMG‐18 were from Tocris. Primary antibodies used for immunoblotting were from the following companies: anti‐pIRE1 (phsopho‐S724; 124945), anti‐FMRP (ab17722), anti‐LRP1 (ab92544), anti‐α‐SMA (ab5694), Anti‐IL‐1 beta antibody (ab9722), Anti‐pro Caspase1 + p10 + p12 antibody [EPR16883] (ab179515), and anti‐Thiophosphate ester antibody (ab133473) were purchased from Abcam; anti‐pFMRP (phosphor‐S499; p1125‐499) from PhosphoSolutions; anti‐IRE1 (3294), anti‐LRP1 (64099) and anti‐FMRP (4317) from Cell‐Signaling; anti‐FMRP (NBP02‐01770), anti‐ABCA1 (NB400‐105), and anti‐ABCG1 (NB400‐132) from Novus Biologicals; anti‐β‐Actin‐horse radish peroxidase (47778) and Secondary IgG‐Goat (sc‐2354) from Santa Cruz Biotechnology; anti‐MOMA‐2 from Bio‐Rad; anti‐PE‐F4/80 (123110), anti‐FMRP (834601), Propidium Iodide Solution, PI (421301), Cell Staining Buffer (420201), and TruStain FcX (anti‐CD16/32, 101319) from Biolegend. Active ERN1 human recombinant protein (E31‐11G) was from Biotech. FMRP human recombinant protein (TP322699) was from Origene. Cholesterol Uptake Assay Kit (ab236212), cholesterol efflux assay kit (ab196985), Masson Trichrome (04‐010802), Fluoroshield mounting reagent with DAPI (ab104139), ATP‐γ‐S (ab138911), and p‐Nitrobenzyl mesylate (PNBM, ab138910) were purchased from Abcam. Aqua‐Mount (41799‐008) was from VWR. Cremophor EL (238470) and Cycloheximide (239763) were purchased from EMD Millipore. TUNEL (In situ Cell Death detection Kit, Fluorescenin; 11684795910) was purchased from Roshe. Hematoxylin and Eosin Stain Kit (H‐3502) was purchased from Vector Laboratories. Wako Diagnostics total cholesterol E kit (NC9138103) was purchased from Fujifilm Medical Systems USA 99902601. TRIsure (BIO‐38033) was purchased from Bioline. Power‐Up‐SYBR green (A25742) was purchased from Applied Biosystems. 3H‐cholesterol (NET139001MC) was purchased from Perkin Elmer. HBSS (14175095) was purchased from Gibco. Anti‐MERTK (AF‐591‐SP) antibody was purchased from R&D. Secondary IgG‐Rabbit (5220‐0337) and Secondary‐IgG‐Mouse (5220‐0341) were purchased from SeraCare. Lambda phosphatase (λ Phosphatase, PPase) enzyme (sc‐200312) was purchased from Santu Cruz.
Mice studies and treatments
C57BL/6 (WT, Fmr1+/+), Fmr1−/−, and Apolipoprotein E‐deficient (Apoe−/−) mice were purchased from Jackson Laboratory. IRE1 conditional knock out (Ire1αflox/flox) mice were a kind gift from Dr. Takao Iwawaki (Kanazawa Medical University, Japan) and characterized before (Iwawaki et al, 2009). Ire1αflox/flox mice were intercrossed from with LysMcre mice purchased from Jackson Lab (004781) to obtain Ire1αflox/flox, LysMcre mice (IRE1−/−), which had a myeloid‐specific Ire1α gene deletion. Fmr1 conditional knock out (Fmr1flox/flox) mice were a kind gift from Dr. David Nelson. Fmr1flox/flox mice were inter‐crossed with LysMcre mice purchased from Jackson Lab (004781) to obtain myeloid Fmr1‐deficient (myFmr1−/−) mice.
Starting at 8 weeks of age, Fmr1+/+, Fmr1−/−, myFmr1+/+, or myFmr1−/− mice were injected with 1 × 1010 AAV_PCSK9 (AAV8‐D377Y‐mPCSK9, Vector BiolabsAAV‐268246) via tail vein, then fed with normal chow or high cholesterol/high fat atherosclerotic mouse diet from Envigo (TD.88137) for 6–16 weeks. Apoe−/− mice were fed with WD (12 weeks) and intraperitoneally injected with vehicle (DMSO) or AMG‐18 (30 mg/kg) once or twice a day in the last 4 weeks of WD. C57BL/6 were injected with Tunicamycin (TM, 1 mg/kg) and with AMG‐18 (30 mg/kg) or vehicle (DMSO) in 20% vol/vol Cremophor EL saline solution, as described in (Tufanli et al, 2017). Eight hours later, peritoneal macrophages were isolated by thioglycolate elicitation for further analysis.
Husbandry conditions and study approval
Mice were kept under specific pathogen‐free conditions with food and water ad libititum. Both female and male mice were used for experiments. All animal experiments were performed according to protocols approved by the Experimental Animal Ethical Care Committees at Bilkent University, Ankara, Turkey or Cedars Sinai Medical Center, Los Angeles, USA or the University of Ottawa Animal Care Committee, Ottawa, ON K1N 6N5, Canada.
Macrophage isolation
Peritoneal macrophages
3% thioglycolate solution was injected to mice intraperitoneally and peritoneal macrophages were collected 4 days after by washing the peritoneal cavity with ice‐cold PBS (10 ml) as described before (Zhang et al, 2008; Onat et al, 2019). Cells were centrifuged at 500 g for 5 min at 4°C and resuspended in RPMI medium in cell culture plates. Macrophages were incubated at 5% carbon dioxide incubator at 37°C for 30 min to attach and non‐adherent cells were removed along with media. Cells were rinsed with PBS and used for protein isolation or RNA isolation.
Bone Marrow‐Derived Macrophages (BMDM)
Bone marrows were collected form the tibia and femurs of mice into RPMI containing 1% Penicillin/streptomycin (P/S) cocktail as previously described (Tufanli et al, 2017). After filtering through a cell strainer (BD, 352350), cells were centrifuged at 500 g for 5 min and resuspended in RPMI enriched with 20% L929 cells conditioned medium, 10% heat‐inactivated fetal bovine serum (FBS), and 1% P/S cocktail, followed by growth on Petri dishes and differentiation to macrophages for 5–10 days.
Cell lines
Fmr1−/− mouse embryonic fibroblasts (MEF) were generated in Dr. David Nelson’s laboratory (Baylor College of Medicine, Houston, Texas). HEK293T, Jurkat (human T lymphocytes), and L‐929 (mouse fibroblasts) cells were obtained from ATCC. Cells were cultured in RPMI or DMEM supplemented with 10% heat‐inactivated FBS and %1 P/S cocktail. Cells were cultured in a humidified CO2 incubator at 37°C. All cells were regularly tested for mycoplasma contamination.
Transfection
60–80% confluent HEK293T, WT or Fmr1−/− MEF cells were transfected using Lipofectamine 3000 or Polyethylemine (PEI). BMDM and HEK293T cells were electroporated either with IRE1‐, Fmr1‐ (100 nM), or control‐siRNA using Neon electroporator (Thermo Scientific) as per specific conditions provided by the manufacturer and as described earlier (Tufanli et al, 2017). 24–36 h after transfection, cells were treated with PA or TG to induce ER stress.
Palmitate (PA)/bovine serum albumin (BSA) complex preparation
PA was dissolved in absolute ethanol to yield a stock concentration of 500 mM and stored at −80°C. Stock PA was diluted to working concentration and suspended with %1 fatty acid‐free BSA in serum‐free RPMI growth medium by mixing at 55°C for 15 min as described before (Erbay et al, 2009).
Western blot analysis
Cells were lysed in lysis buffer (50 mM HEPES pH:7,9, 100 mM NaCl, 10 mM EDTA,10 mM NaF, 4 mM NaPP, 1% Triton, 1 mM phenylmethanesulfonylfluoride (PMSF), 1× phosphatase inhibitor cocktail 3 and 1× (10 µM) protease inhibitor cocktail as described in (Çimen et al, 2016b). After centrifugation, clear lysates were mixed with sodium dodecyl sulfate (SDS) loading dye and heated at 95°C for 5 min before loading on SDS–polyacrylamide gel (SDS–PAGE) gels. After separation according to protein molecular weights on these gels, samples were transferred to polyvinylidene difluoride (PVDF) membrane. Blocking and antibody incubation of the membranes were carried out in tris‐buffered saline (TBS) buffer prepared with 0.1% Tween‐20 (v/v) and 5% (w/v) dry milk or BSA. ECL prime reagent was used to develop the membranes, and images were captured with ChemiDoc (BioRad).
Antibody dilutions
Anti‐pIRE1 1:2,000, anti‐FMRP 1:2,000, anti‐LRP1 1:5,000, anti‐IL‐1 beta 1:500, Anti‐pro Caspase1 + p10 + p12 1:2,000, anti‐Thiophosphate ester 1:5,000, anti‐pFMRP (phosphor‐S499) 1:2,000, anti‐IRE1 1:2,000, anti‐ABCA1 1:1,000, anti‐ABCG1 1:1,000, Anti‐MERTK 1:1,000, anti‐β‐Actin‐horse radish peroxidase 1:5,000, Secondary IgG‐Goat 1:10,000, Secondary IgG‐Rabbit 1:10,000, Secondary‐IgG‐Mouse 1:10,000.
RNA isolation and quantitative reverse transcription polymerase chain reaction (qRT‐PCR)
Total RNA was isolated using TRIsure. RNA extractions were than reverse transcribed by using Revert Aid First strand cDNA synthesis kit to complementary deoxyribonucleic acid (cDNA) according to manufacturer’s protocol. Using specific primers, cDNAs were amplified on Rotor Gene (Qiagen). Power‐Up‐SYBR green (Applied Biosystems, A25742) was used for qRT‐PCR reaction. The following PCR primers were used for mRNA expression analysis:
mmu‐Fmr1‐F 5′ CCGAACAGATAATCGTCCACG 3′
mmu‐Fmr1‐R 5′ ACGCTGTCTGGCTTTTCCTTC 3′
mmu‐Abca1‐F 5′ AAAACCGCAGACATCCTTCAG 3′
mmu‐Abca1‐R 5′ CATACCGAAACTCGTTCACCC 3′
mmu‐Abcg1‐F 5′ GGTCCTGACACATCTGCGAA 3′
mmu‐Abcg1‐R 5′ CAGGACCTTCTTGGCTTCGT 3′
mmu‐Mertk‐F 5′ CAGGGCCTTTACCAGGGAGA 3′
mmu‐Mertk‐R 5′ TGTGTGCTGGATGTGATCTTC 3′
mmu‐Lrp1‐F 5′ GCCTACACCTGGAGAGATAGC 3′
mmu‐Lrp1‐R 5′ GGCAACTTACGAGCAGGCT 3′
mmu‐Cd36‐F 5′ GTGCTCTCCCTTGATTCTGC 3′
mmu‐Cd36‐R 5′ CTGCACCAATAACAGCTCCA 3′
mmu‐Cd47‐F 5′ TGGTGGGAAACTACACTTGCG 3′
mmu‐Cd47‐R 5′ CGTGCGGTTTTTCAGCTCTAT 3′
mmu‐Calr‐F 5′ GCAGACCCTGCCATCTATTTC 3′
mmu‐Calr‐R 5′ TCGGACTTATGTTTGGATTCGAC 3′
mmu‐Rac1‐F 5′ ATGCAGGCCATCAAGTGTG 3′
mmu‐Rac1‐R 5′ TAGGAGAGGGGACGCAATCT 3′
mmu‐IL‐1β ‐F 5′ CAACCAACAAGTGATATTCTCCATG 3′
mmu‐IL‐1β ‐R 5′ GATCCACACTCTCCAGCTGCA 3′
mmu‐Gapdh‐F 5′ ATTCAACGGCACAGTCAAGG 3′
mmu‐Gapdh‐R 5′ TGGATGCAGGGATGATGTTC 3′
The following primers were used to introduce site‐directed mutagenesis on FMRP plasmids:
S500A‐F 5′ GCATCAAATGCTGCTGAAGCAGAAGCTGACCACAGAGAC 3′
S500A‐R‐ 5′ GTCTCTGTGGTCAGCTTCTGCTTCAGCAGCATTTGATGC 3′
S500‐T502‐S504A‐F 5′ GCATCAAATGCTGCTGAAGCAGAAGCTGACCACAGAGAC 3′
S500‐T502‐S504A‐R 5′ GTCTCTGTGGTCAGCTTCTGCTTCAGCAGCATTTGATGC 3′
Identifying phosphorylation sites on hFMRP using Mass spectrometry
Two in vitro kinase reactions of hFMRP and ERN1, worth 4.5 µg protein each were methanol‐chloroform precipitated (Wessel & Flügge, 1984). Dried pellets were dissolved in either [1] 8 M urea/100 mM triethylammonium bicarbonate (TEAB, Thermo Scientific 90114), pH 8.5, or [2] 100 mM ammonium acetate (Sigma‐Aldrich A1542), with or without 8 M urea. Proteins were reduced with 5 mM tris(2‐carboxyethyl) phosphine hydrochloride (TCEP‐HCl, Thermo Scientific C4709) and alkylated with 10 mM 2‐chloroacetamide (Sigma‐Aldrich 22790). Proteins dissolved in urea/TEAB were digested at 37°C in 0.8 M urea/100 mM TEAB, pH 8.5, sequentially with 500 ng Trypsin (Promega V5117) for 17 h, followed by 500 ng Endoproteinase GluC (NEB P8100S) for 4.5 h and quenched with formic acid, 5% final concentration, while proteins dissolved in urea/TEAB or urea/ammonium acetate were digested with 200 ng Proteinase K (Sigma‐Aldrich P2308) at 37°C for 30 min and heat quenched at 90°C for 15 min (similar reactions in ammonium acetate without urea were performed for 30 min or 15 min followed by 16 h digestion with trypsin) (Baboo et al, 2021). The digest was injected directly onto a 20 cm, 100 µm ID column packed with BEH 1.7 µm C18 resin (Waters 186005225). Samples were separated at a flow rate of 400 nl/min on an nLC 1000 (Thermo LC120). Buffer A and B were 0.1% formic acid in 5% acetonitrile and 0.1% formic acid in 80% acetonitrile, respectively. A gradient of 1–25% B over 110 min, an increase to 40% B over next 20 min, an increase to 90% B over another 10 min, and a hold at 90% B for the final 10 min were used for a total run time of 140 min. The column was re‐equilibrated with 20 µl of buffer A prior to the injection of sample. Peptides were eluted directly from the tip of the column and nano‐sprayed into the mass spectrometer by application of 2.8 kV voltage at back of the column. The Orbitrap Fusion Lumos (Thermo) was operated in data‐dependent mode. Full MS1 scans were collected in the Orbitrap at 120K resolution with a mass range of 400–1,500 m/z and an AGC target of 4e5. The cycle time was set to 3 s, and within these 3 s, the most abundant ions per scan were selected for CID MS/MS in the ion trap with an AGC target of 2e4 and minimum intensity of 5,000. Maximum fill times were set to 50 and 35 ms for MS and MS/MS scans, respectively. Quadrupole isolation at 1.6 m/z was used, monoisotopic precursor selection was enabled, charge states of 2–7 were selected, and dynamic exclusion was used with an exclusion duration of 5 s. Samples were also analyzed with HCD fragmentation (35 NCE) and detection at 7,500 resolution.
Protein and peptide identification were done with Integrated Proteomics Pipeline—IP2 (Integrated Proteomics Applications). Tandem mass spectra were extracted from raw files using RawConverter (He et al, 2015) and searched with ProLuCID (Xu et al, 2015) against a concatenated database comprising of amino acid sequences from vendors for FMRP, hERN1, and Endoproteinase GluC, UniProt reference proteome of Escherichia coli K12 (UP000000625) Homo sapiens (UP000005640). The search space included all fully tryptic and half‐tryptic peptide candidates (no enzyme specificity for sample treated with Proteinase K). Carbamidomethylation (+57.02146) was considered a static modification on cysteine, and phosphorylation (+79.966331) was considered a differential modification on serine/threonine/tyrosine. Data were searched with 50 ppm precursor ion tolerance and 500 ppm fragment ion tolerance. Identified proteins were filtered to using DTASelect (Tab b et al, 2002) and utilizing a target‐decoy database search strategy to control the false discovery rate at 1%, at the spectrum level (Peng et al, 2003). A minimum of 1 peptide per protein and 1 tryptic end per peptide (no tryptic ends in case of Proteinase K treatment) were required and precursor delta mass cut‐off was fixed at 10 ppm. Localization scores were assigned to identified sites of phosphorylation using A‐Score (Beausoleil et al, 2006).
Co‐immunoprecipitation and kinase assay
Co‐immunoprecipitation
HEK293T cells were co‐transfected with IRE1 and FMRP plasmids for 24 h followed by TG (600 nM) or TM (1 µg/ml) for 2 h. Equal amounts of protein lysates were precipitated with specific antibodies (anti‐IRE1 1:250 and anti‐FMRP 1:250) at 4°C overnight on a rocker. Protein magnetic beads were added to each sample and incubated at 4°C for overnight. Immunoprecipitates were then analyzed by Western blot.
Kinase assay
HEK293T cells were transfected with either with WT‐ or KD‐IRE1 plasmids for 24 h, followed by TG (600 nM) for 2 h for IRE1 activation. Equal amounts of protein lysates were then precipitated with specific IRE1 antibody‐coated magnetic beads at 4°C overnight on a rocker. Immunoprecipitates were incubated at 30°C for 45 min in kinase assay buffer (SignalChem K01‐09) with specific ATP analogue (ATP‐γ‐S, 100 µM) and purified hFMRP protein. p‐Nitrobenzyl mesylate (PNBM) and 0.5 M ETDA solution were added to reaction after 45 min and incubated for additional 2 h at RT to alkylate the kinase substrate. Samples were boiled with SDS–PAGE loading dye at 95°C for 5 min to release the proteins from magnetic beads. Beads were separated using magnetic rack and supernatants were analyzed by Western blot.
Kinase assay for phospho‐proteomics
Recombinant active IRE1 (500 ng) and FMRP (500 ng) proteins were incubated in kinase buffer at 30°C for 45 min with ATP‐γ‐S (100 µM). Samples were then incubated at 24°C for 1 h with PNBM (2.5 mM). Samples were boiled in SDS loading buffer at 95°C and loaded to SDS–PAGE. Anti‐thiophosphate ester antibody was used to detect alkylated kinase substrate.
Polysome fractionation
Polysome fractionation protocol was adapted from Stastna et al (Stastna et al, 2018). Briefly, Fmr1+/+ and Fmr1−/− BMDM were treated with PA (500 µM) for 6 h followed by cycloheximide (100 µg/ml) for 10 min prior to lysis with buffer (100 mM KCl, 20 mM Tris pH 7.5, 5 mM MgCl2, 0.4% NP‐40, 100 µg/ml cycloheximide, 0.1 U RNase inhibitor and protease inhibitor cocktail). Clear lysates were loaded to 10–50% sucrose gradient (in Beckman Coulter Thinwall, Ultra‐Clear tubes, 344059) and centrifuged (in Beckman LE‐80K) for 120 min at 28,4061 g at 4°C in a swinging bucket rotor (Beckman SW41) with no‐brake. Each gradient was collected as 17 fractions in microcentrifuge tubes with continuous monitoring of absorbance at 254 nm (Biologic LP (pump), Biorad 731‐8300; BioFrac, Biorad 741‐0002) and frozen immediately at −80°C for further analysis.
Cholesterol efflux and foam cell formation assays
Cholesterol efflux assay
Efflux assay were performed according to manufacturer’s instructions (ab196985). Briefly, macrophages were pre‐loaded with fluorescently labeled cholesterol for 16 h in RPMI media including ACAT inhibitor (5 mg/ml), followed by incubation in efflux medium including cholesterol acceptors apolipoprotein A1 (APOA1; 25 µg/ml) or high‐density lipoprotein (HDL; 50 µg/ml) for 6 h. % Efflux was calculated as cholesterol signal in medium/cholesterol signal in medium and cell.
In vitro foam cell formation assay
BMDM were incubated with RPMI containing dil‐labeled ac‐LDL (25 µg/ml), 10% lipoprotein‐deficient serum, and 20% l‐Glutamine for 24 h. After cholesterol loading, cells were rinsed with PBS and collected in 2% BSA in PBS. Flow cytometry was performed on a BD Fortessa using FACSDiva software with single stain compensation controls acquired on the same day.
In vivo foam cell formation assay
Fmr1+/+ and Fmr1−/− mice were injected with a gain‐of‐function mutant (D377Y) of proprotein convertase subtilisin kexin 9 (PCSK9)‐encoding adeno‐associated virus (AAV_mPCSK9) and fed with 16 weeks of WD to induced hypercholesterolemia. Apoe−/− mice were fed with 12 weeks of WD with 4 weeks of AMG‐18 (30 mg/kg, once a day) injection during the last 4 weeks of WD. The peritoneal macrophages were collected and assessed for lipid accumulation by Oil‐Red O and Hematoxylin staining.
Reverse cholesterol transport (RCT) assay
Preparation of radiolabel cholesterol
Ag‐LDL (50 µg/ml, made in house with endotoxin‐free LDL isolated from human plasma) and [3H]‐cholesterol (5 µCi/ml) were incubated for 1 h at 37°C in a sterile endotoxin free bottle.
RCT assay
Fmr1+/+ and Fmr1−/− BMDM were incubated with radiolabeled ag‐LDL for 30 h followed by warm HBSS wash and equilibration in 2 mg/ml fatty acid‐free BSA overnight. Cells were washed twice in ice cold HBSS and incubated with EDTA (5 mM) for 20 min at 4°C and spun down at 200 g for 5 min. Cells were resuspended in ice cold DMEM and injected into C57BL6N mice subcutaneously in the scruff of the neck. Blood was collected at 24 h via the saphenous vein and at 48 h via cardiac puncture of anesthetized mice. Plasma was used for liquid scintillation counting. At 48 h, livers were removed for scintillation counting. Feces were collected over a 48‐h period, and total feces radioactivity was measured. All [3H]‐tracer measurements are expressed relative to the injected amount.
In vitro and in vivo efferocytosis
Induction of apoptosis and labeling of Jurkat cells
Jurkat cells were fluorescently labeled with CellTrace CSFE or Violet (2 µM) in PBS for 20 min. Cells were then washed ones with PBS and seeded in conditioned DMEM medium followed by irradiation under a 254 nm UV lamp for 5 min. Cells were incubated under normal cell culture conditions for 3–4 h. Apoptosis was confirmed by Anexin V+ staining (minimum 85% Annexin V+ cells). The apoptotic cells (ACs) were centrifuged at 500 g for 5 min and resuspended in conditioned DMEM for experiments.
In vitro efferocytosis
Bone marrow‐derived macrophages were plated in six‐well dishes at a density of 0.5 × 106 cells per well. CSFE‐labeled ACs were incubated with the macrophages for 2–4 min at a 5:1 AC:macrophage ratio followed by washing three times with PBS. Some groups of macrophages were then incubated for another 2 h in normal cell culture media, followed by the addition of Violet‐labeled ACs. After 2 h, macrophages were washed three times with PBS to remove unbound ACs, and then the macrophages were fixed with 4% formaldehyde for 20 min, rinsed three times with PBS, blocked by TruStain FcX™ (anti‐mouse CD16/32) for 10 min and then stained with PE‐F4/80 o/n. The percentage of PE‐F4/80+ and CFSE+ double positive cells to total PE‐F4/80+ cells was reported as % efferocytosis and PE‐F4/80+, CFSE+ and Violet+ triple positive cells to PE‐F4/80+ and CFSE+ double positive cells was reported as % continuous efferocytosis.
In vivo efferocytosis
Fmr1+/+ or Fmr1−/− mice were fed with WD for 16 weeks and injected with 1 × 106 CFSE‐labeled ACs and 1.5 h later subsequently peritoneal lavages were collected and stained for PE‐F4/80+ resident macrophages. The percentage of PE‐F4/80+ and CFSE+ double positive cells to total PE‐F4/80+ cells was reported as % efferocytosis. Another group of C57BL/6 was injected with AMG‐18 (30 mg/kg) or vehicle (DMSO). After 8 h both groups were interperitoneally injected with CFSE‐labeled ACs and 1.5 h later peritoneal lavages were collected and cultured for 30 min to allow cells to attach. Macrophages were washed three times with PBS to remove unbound ACs, and then the macrophages were fixed with 4% formaldehyde for 20 min, rinsed three times with PBS, blocked by TruStain FcX™ (anti‐mouse CD16/32) for 10 min, and then stained with PE‐F4/80 o/n. The percentage of F4/80+ and CFSE+ double positive cells to total F4/80+ cells was reported as % efferocytosis.
En face Oil‐Red O staining
Aortas opened longitudinally were rinsed with 60% isopropanol for 1 min, stained with Oil‐Red O solution for 20 min, and then distained in 60% isopropanol for 1 min and rinsed in PBS. The lesion area was quantitated as percent of Oil‐Red O staining area in total aorta area.
Immunohistochemistry
7‐μm‐thick aortic root cryosections (from OCT embedded heart tissue) were stained with antibodies for: anti‐MOMA‐2 (1:500) and anti‐α‐SMA (1:500) and images were captured with fluorescent microscope. Cryosections were stained with Masson’s Trichrome, TUNEL, Hematoxylin and Eosin (H&E) according to manufacturer’s instructions. Cryosections were stained with H&E for morphometric lesion analysis. The total lesion area and necrotic area were quantified as previously described from 4 sequential sections (60 µM apart, beginning at the base of the aortic root) as previously described (Çimen et al, 2016b). Foam cell area was calculated from Oil‐Red O stained 4 sequential sections (60 µM apart, beginning at the base of the aortic root) and collagen content from Masson’s Trichrome stained sections using ImageJ as previously described (Çimen et al, 2016b).
The fluorescent immunostainings were carried out on cryosections that were fixed in cold acetone for 10 min, blocked in goat serum/BSA/PBS as previously described. All stained sections were mounted with fluoroshield mounting reagent with DAPI. Fluorescent signal calculations: (i) TUNEL staining: the sections were double stained with MOMA‐2 to mark the macrophage‐enriched area. The Mean Fluorescent Intensity (MFI) corresponding to primary antibody signal was calculated from the MOMA‐2‐positive area. The background fluorescence of the non‐stained area inside the lesion was subtracted from the total MFI corresponding to each signal (ii) α‐SMA staining: α‐SMA positive area was calculated from the plaque area. The background fluorescence of the non‐stained area inside the lesion was subtracted from the total MFI corresponding to each signal. Data were quantified as total MFI signal compared with baseline (Çimen et al, 2016b; Tufanli et al, 2017).
Apoptosis detection by flow cytometry
Apoptosis was induced after treatments by PA (500 µM) treatment for 12 h. Fc receptors were blocked by pre‐incubating cells with 0.25 µg of TruStain FcX™ PLUS (anti‐mouse CD16/32) Antibody per 106 cells for 5–10 min on ice. Cells were then incubated with PI on ice for 20 min in the dark followed by 2× with 2 ml of cell staining buffer. Cells were resuspended in 500 µl of cell staining buffer and analyzed on a BD Fortessa using FACSDiva software with single stain compensation controls acquired on the same day. Data were analyzed using FlowJo analysis software (FlowJo, LLC).
Flow cytometric analysis of peripheral blood
100 µl of blood was collected in EDTA‐coated tubes and red blood cells were removed by incubation (3×) in Ammonium‐Chloride‐Potassium (ACK, Thermo Fisher A1049201) solution for 5 min at room temperature. Peripheral blood mononuclear cells were then resuspended in FACS buffers (2% BSA in PBS) and incubated for 20 min on ice with the following antibodies: anti‐CD45‐Pac. Blue (clone 30‐F11), CD3e‐PE (Clone 145‐2C11), CD11b‐APC (Clone M1/70), CD19‐BV650 (Clone 6D5), Ly6C‐PE/Dazzle (Clone HK1.4), and Ly6G‐PerCP Cy5.5 (1A8) in 1:100 dilution ratio. Stained samples were washed once and resuspended in FACS buffer containing DAPI (4 µg/ml). Flow cytometry was performed on a BD Fortessa using FACSDiva software with single stain compensation controls acquired on the same day. Data were analyzed using FlowJo analysis software (FlowJo, LLC). All antibodies were purchased from Biolegend (San Diego, CA) and used at the manufacturer’s recommended concentrations.
Plasma lipids and lipoprotein analysis
Plasma was analyzed by FPLC in the Department of Internal Medicine/Lipid Science, Wake Forest University School of Medicine Winston‐Salem, NC 27019 as described (Çimen et al, 2016b). The total cholesterol and triglyceride measurement were performed using WAKO Cholesterol E kit according to the manufacturer’s instructions.
Statistics
Results are reported as mean ± SEM and statistical significance was determined with Unpaired t‐test with Welch’s or Mann–Whitney correction test by GraphPad Software, LLC.
Author contributions
Zehra Yildirim: Data curation; Formal analysis; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review & editing. Sabyasachi Baboo: Formal analysis; Investigation; Methodology; Writing—review & editing. Syed Muhammad Hamid: Formal analysis; Investigation; Methodology. Asli Ekin Dogan: Formal analysis; Investigation; Visualization; Methodology. Ozlem Tufanli: Formal analysis; Investigation; Methodology. Sabrina Robichaud: Formal analysis; Investigation; Methodology. Christina Emerton: Formal analysis; Investigation; Methodology. Jolene K Diedrich: Formal analysis; Investigation; Methodology. Hasan Vatandaslar: Formal analysis; Investigation; Methodology. Fotis Nikolos: Formal analysis; Investigation; Methodology. Yanghong Gu: Investigation; Methodology. Takao Iwawaki: Methodology. Elizabeth J Tarling: Formal analysis; Supervision; Funding acquisition; Investigation; Methodology; Writing—review & editing. Mireille Ouimet: Formal analysis; Supervision; Investigation; Methodology; Writing—review & editing. David Nelson: Formal analysis; Supervision; Investigation; Methodology; Writing—review & editing. John R Yates III: Data curation; Formal analysis; Supervision; Investigation; Writing—review & editing. Peter Walter: Formal analysis; Investigation; Writing—review & editing. Ebru Erbay: Conceptualization; Formal analysis; Supervision; Funding acquisition; Investigation; Writing—original draft; Project administration; Writing—review & editing.
Disclosure and competing interests statement
PW is an inventor on U.S. Patent 9708247 held by the Regents of the University of California that describes ISRIB and its analogs. Rights to the invention have been licensed by UCSF to Calico. EE, SMH, OT, and PW will become employees of Altos Labs. All other authors declare that they have no competing interests.
Supporting information
Appendix
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We express our appreciation to Jennifer C. Darnell (The Rockefeller University, New York) for providing and guiding with Fmr1 plasmid and to Roberta Gottlieb and Juliana Germano (Cedars‐Sinai Medical Center) for their guidance with polysome profiling. This study was funded by the following: National Institutes of Health grant R01HL152156 (EE), Cedars‐Sinai Medical Center Internal Support (EE), National Institutes of Health grant R01GM32384 (PW), Investigator of the Howar Hughes Medical Institute (PW), National Institutes of Health grant P41GM103533 (JRY), Vanier Canada Graduate Scholarship (SR), Canadian Institutes for Health Research PJT‐391187 (MO).
EMBO Mol Med (2022) 14: e15344.
Data availability
All data are available in the main text or the supplementary materials. Research materials used in the article can be requested from authors. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (Deutsch et al, 2020) via the PRIDE (Perez‐Riverol et al, 2016, 2019) partner repository with the dataset identifier PXD030594 (http://www.ebi.ac.uk/pride/archive/projects/PXD030594).
References
- Acosta‐Alvear D, Karagöz GE, Fröhlich F, Li H, Walther TC, Walter P (2018) The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife 7: e43036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M et al (2012) FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492: 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baboo S, Diedrich JK, Martínez‐Bartolomé S, Wang X, Schiffner T, Groschel B, Schief WR, Paulson JC, Yates JR (2021) DeGlyPHER: an ultrasensitive method for the analysis of viral spike N‐glycoforms. Anal Chem 93: 13651–13657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Tassone F, Neri G, Hagerman R (2012) Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J Clin Invest 122: 4314–4322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartley CM, O'Keefe RA, Bordey A (2014) FMRP S499 is phosphorylated independent of mTORC1‐S6K1 activity. PLoS One 9: e96956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartley CM, O'Keefe RA, Blice‐Baum A, Mihailescu MR, Gong X, Miyares L, Karaca E, Bordey A (2016) Mammalian FMRP S499 Is phosphorylated by CK2 and promotes secondary phosphorylation of FMRP. eNeuro 3: ENEURO.0092‐16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP (2006) A probability‐based approach for high‐throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24: 1285–1292 [DOI] [PubMed] [Google Scholar]
- Berry‐Kravis E, Levin R, Shah H, Mathur S, Darnell JC, Ouyang B (2015) Cholesterol levels in fragile X syndrome. Am J Med Genet A 167a: 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 47: 2726–2737 [DOI] [PubMed] [Google Scholar]
- Buchan JR, Parker R (2009) Eukaryotic stress granules: the ins and outs of translation. Mol Cell 36: 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carotti S, Zingariello M, Francesconi M, D’Andrea L, Latasa MU, Colyn L, Fernandez‐Barrena MG, Flammia RS, Falchi M, Righi D et al (2021) Fragile X mental retardation protein in intrahepatic cholangiocarcinoma: regulating the cancer cell behavior plasticity at the leading edge. Oncogene 40: 4033–4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceman S, O'Donnell WT, Reed M, Patton S, Pohl J, Warren ST (2003) Phosphorylation influences the translation state of FMRP‐associated polyribosomes. Hum Mol Genet 12: 3295–3305 [DOI] [PubMed] [Google Scholar]
- Chen E, Sharma MR, Shi X, Agrawal RK, Joseph S (2014) Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol Cell 54: 407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu DS, Oram JF, LeBoeuf RC, Alpers CE, O'Brien KD (1997) High‐density lipoprotein‐binding protein (HBP)/vigilin is expressed in human atherosclerotic lesions and colocalizes with apolipoprotein E. Arterioscler Thromb Vasc Biol 17: 2350–2358 [DOI] [PubMed] [Google Scholar]
- Çimen I, Kocatürk B, Koyuncu S, Tufanlı Ö, Onat UI, Yıldırım AD, Apaydın O, Demirsoy Ş, Aykut ZG, Nguyen UT et al (2016a) Prevention of atherosclerosis by bioactive palmitoleate through suppression of organelle stress and inflammasome activation. Sci Transl Med 8: 358ra126 [DOI] [PubMed] [Google Scholar]
- Çimen I, Kocatürk B, Koyuncu S, Tufanlı Ö, Onat UI, Yıldırım AD, Apaydın O, Demirsoy Ş, Aykut ZG, Nguyen UT et al (2016b) Prevention of atherosclerosis by bioactive palmitoleate through suppression of organelle stress and inflammasome activation. Sci Transl Med 8: 358ra126 [DOI] [PubMed] [Google Scholar]
- Cloots E, Simpson MS, De Nolf C, Lencer WI, Janssens S, Grey MJ (2021) Evolution and function of the epithelial cell‐specific ER stress sensor IRE1β. Mucosal Immunol 14: 1235–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee RL Jr, Williamson AJ, Adkins CM, Gray MC, Page TL, Broadie K (2012) In vivo neuronal function of the fragile X mental retardation protein is regulated by phosphorylation. Hum Mol Genet 21: 900–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costet P, Luo Y, Wang N, Tall AR (2000) Sterol‐dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem 275: 28240–28245 [DOI] [PubMed] [Google Scholar]
- Darnell J, Van Driesche S, Zhang C, Hung K, Mele A, Fraser C, Stone E, Chen C, Fak J, Chi S et al (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146: 247–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davignon J, Cohn JS, Mabile L, Bernier L (1999) Apolipoprotein E and atherosclerosis: insight from animal and human studies. Clin Chim Acta 286: 115–143 [DOI] [PubMed] [Google Scholar]
- Davis JK, Broadie K (2017) Multifarious functions of the fragile X mental retardation protein. Trends Genet 33: 703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Bandeira N, Sharma V, Perez‐Riverol Y, Carver JJ, Kundu DJ, García‐Seisdedos D, Jarnuczak AF, Hewapathirana S, Pullman BS et al (2020) The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res 48: D1145–d1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, Tada T, Dolan BM, Sharp PA, Sheng M (2010) Regulation of synaptic structure and function by FMRP‐associated microRNAs miR‐125b and miR‐132. Neuron 65: 373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, Fazio S, Wiest MM, Watkins SM, Linton MF et al (2009) Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med 15: 1383–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473: 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R, Wang L, Wang E, Perera B, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H et al (2014) Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158: 534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Yuan W, Yu B, Kuai R, Hu W, Morin EE, Garcia‐Barrio MT, Zhang J, Moon JJ, Schwendeman A et al (2018) Synthetic high‐density lipoprotein‐mediated targeted delivery of liver X receptors agonist promotes atherosclerosis regression. EBioMedicine 28: 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haneklaus M, O'Neil JD, Clark AR, Masters SL, O'Neill LAJ (2017) The RNA‐binding protein Tristetraprolin (TTP) is a critical negative regulator of the NLRP3 inflammasome. J Biol Chem 292: 6869–6881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansmeier N, Buttigieg J, Kumar P, Pelle S, Choi KY, Kopriva D, Chao TC (2018) Identification of mature atherosclerotic plaque proteome signatures using data‐independent acquisition mass spectrometry. J Proteome Res 17: 164–176 [DOI] [PubMed] [Google Scholar]
- Harnoss JM, Le Thomas A, Shemorry A, Marsters SA, Lawrence DA, Lu M, Chen Y‐C, Qing J, Totpal K, Kan D et al (2019) Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc Natl Acad Sci USA 116: 16420–16429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Diedrich J, Chu YY, Yates JR 3rd (2015) Extracting accurate precursor information for tandem mass spectra by RawConverter. Anal Chem 87: 11361–11367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersh JH, Saul RA (2011) Health supervision for children with fragile X syndrome. Pediatrics 127: 994–1006 [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS (2017) Inflammation, metaflammation and immunometabolic disorders. Nat Rev 542: 177–185 [DOI] [PubMed] [Google Scholar]
- Hunter J, Rivero‐Arias O, Angelov A, Kim E, Fotheringham I, Leal J (2014) Epidemiology of fragile X syndrome: a systematic review and meta‐analysis. Am J Med Genet A 164a: 1648–1658 [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Yamanaka S, Kohno K (2009) Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA 106: 16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce CW, Amar MJA, Lambert G, Vaisman BL, Paigen B, Najib‐Fruchart J, Hoyt RF, Neufeld ED, Remaley AT, Fredrickson DS et al (2002) The ATP binding cassette transporter A1 (ABCA1) modulates the development of aortic atherosclerosis in C57BL/6 and apoE‐knockout mice. Proc Natl Acad Sci USA 99: 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JG, Amar MJ, Remaley AT, Kwon J, Blackshear PJ, Wang PY, Hwang PM (2011) Zinc finger protein tristetraprolin interacts with CCL3 mRNA and regulates tissue inflammation. J Immunol 187: 2696–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Cuchel M, de la Llera‐Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL et al (2011) Cholesterol efflux capacity, high‐density lipoprotein function, and atherosclerosis. N Engl J Med 364: 127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yun EY, Park SW, Goo TW, Seo M (2016) Allomyrina dichotoma larvae regulate food intake and body weight in high fat diet‐induced obese mice through mTOR and Mapk signaling pathways. Nutrients 8: 100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucken J, Buchler C, Orso E, Kaminski WE, Porsch‐Ozcurumez M, Liebisch G, Kapinsky M, Diederich W, Drobnik W, Dean M et al (2000) ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci USA 97: 817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Weissman IL, Leeper NJ (2017) The role of efferocytosis in atherosclerosis. Circulation 135: 476–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboucher A, Bermudez‐Martin P, Mouska X, Amri EZ, Pisani DF, Davidovic L (2019a) Fmr1‐deficiency impacts body composition, skeleton, and bone microstructure in a mouse model of fragile X syndrome. Front Endocrinol 10: 678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboucher A, Pisani DF, Martinez‐Gili L, Chilloux J, Bermudez‐Martin P, Van Dijck A, Ganief T, Macek B, Becker JAJ, Le Merrer J et al (2019b) The translational regulator FMRP controls lipid and glucose metabolism in mice and humans. Mol Metab 21: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tumanut C, Gavigan J‐A, Huang W‐J, Hampton E, Tumanut R, Suen K, Trauger J, Spraggon G, Lesley S et al (2007) Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J 406: 203–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ge M, Ciani L, Kuriakose G, Westover EJ, Dura M, Covey DF, Freed JH, Maxfield FR, Lytton J et al (2004) Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic‐endoplasmic reticulum calcium ATPase‐2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol‐loaded macrophages. J Biol Chem 279: 37030–37039 [DOI] [PubMed] [Google Scholar]
- Lisik MZ, Gutmajster E, Sieroń AL (2016) Low levels of HDL in fragile X syndrome patients. Lipids 51: 189–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumaban JG, Nelson DL (2015) The Fragile X proteins Fmrp and Fxr2p cooperate to regulate glucose metabolism in mice. Hum Mol Genet 24: 2175–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobin MB, Gerstberger S, Teupser D, Campana B, Charisse K, Heim MH, Manoharan M, Tuschl T, Stoffel M (2016) The RNA‐binding protein vigilin regulates VLDL secretion through modulation of Apob mRNA translation. Nat Commun 7: 12848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Sheedy FJ, Fisher EA (2013) Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13: 709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M et al (2008) The fragile X syndrome protein represses activity‐dependent translation through CYFIP1, a new 4E‐BP. Cell 134: 1042–1054 [DOI] [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Thomas G, Ceman S, Bassell GJ, Warren ST (2008) S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis‐dependent mammalian target of rapamycin (mTOR) signaling cascade. J Biol Chem 283: 18478–18482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niere F, Wilkerson JR, Huber KM (2012) Evidence for a fragile X mental retardation protein‐mediated translational switch in metabotropic glutamate receptor‐triggered Arc translation and long‐term depression. J Neurosci 32: 5924–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onat UI, Yildirim AD, Tufanli Ö, Çimen I, Kocatürk B, Veli Z, Hamid SM, Shimada K, Chen S, Sin J et al (2019) Intercepting the lipid‐induced integrated stress response reduces atherosclerosis. J Am Coll Cardiol 73: 1149–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram JF, Lawn RM, Garvin MR, Wade DP (2000) ABCA1 is the cAMP‐inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem 275: 34508–34511 [DOI] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow‐Cordero DE, Bouley DM, Offner F, Niwa M et al (2011) Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117: 1311–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled M, Nishi H, Weinstock A, Barrett TJ, Zhou F, Quezada A, Fisher EA (2017) A wild‐type mouse‐based model for the regression of inflammation in atherosclerosis. PLoS One 12: e0173975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC‐MS/MS) for large‐scale protein analysis: the yeast proteome. J Proteome Res 2: 43–50 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–d450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Xu Q‐W, Wang R, Uszkoreit J, Griss J, Sanchez A, Reisinger F, Csordas A, Ternent T, del‐Toro N et al (2016) PRIDE inspector toolsuite: moving toward a universal visualization tool for proteomics data standard formats and quality assessment of ProteomeXchange datasets. Mol Cell Proteomics 15: 305–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto M, Folci A, Martin S (2020) Post‐translational modifications of the Fragile X mental retardation protein in neuronal function and dysfunction. Mol Psychiatry 25: 1688–1703 [DOI] [PubMed] [Google Scholar]
- Ramírez CM, Lin CS, Abdelmohsen K, Goedeke L, Yoon J‐H, Madrigal‐Matute J, Martin‐Ventura JL, Vo DT, Uren PJ, Penalva LO et al (2014) RNA binding protein HuR regulates the expression of ABCA1. J Lipid Res 55: 1066–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ (2014) Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res 114: 1757–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CD, Smith GD, Jefferson TS, Lugo JN (2015) Comparison of equivalence between two commercially available S499‐phosphorylated FMRP antibodies in mice. PLoS One 10: e0143134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robblee MM, Kim CC, Porter Abate J, Valdearcos M, Sandlund KL, Shenoy MK, Volmer R, Iwawaki T, Koliwad SK (2016) Saturated fatty acids engage an IRE1α‐dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep 14: 2611–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha VZ, Libby P (2009) Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol 6: 399–409 [DOI] [PubMed] [Google Scholar]
- Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S et al (2010) Atherogenic lipids and lipoproteins trigger CD36‐TLR2‐dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab 12: 467–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna F, Moon C, Wang H (2014) From FMRP function to potential therapies for fragile X syndrome. Neurochem Res 39: 1016–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stastna M, Thomas A, Germano J, Pourpirali S, Van Eyk JE, Gottlieb RA (2018) Dynamic proteomic and miRNA analysis of polysomes from isolated mouse heart after langendorff perfusion. J vis Exp 138: 58079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhorukov VN, Khotina VA, Bagheri Ekta M, Ivanova EA, Sobenin IA, Orekhov AN (2020) Endoplasmic reticulum stress in macrophages: the vicious circle of lipid accumulation and pro‐inflammatory response. Biomedicines 8: 210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I (2010) The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res 107: 839–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, Yates JR 3rd (2002) DTASelect and contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1: 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR, Yvan‐Charvet L (2015) Cholesterol, inflammation and innate immunity. Nat Rev Immunol 15: 104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang B, Arsenault J, Vernon RM, Lin H, Sonenberg N, Wang LY, Bah A, Forman‐Kay JD (2019) Phosphoregulated FMRP phase separation models activity‐dependent translation through bidirectional control of mRNA granule formation. Proc Natl Acad Sci USA 116: 4218–4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsimikas S, Miyanohara A, Hartvigsen K, Merki E, Shaw PX, Chou M‐Y, Pattison J, Torzewski M, Sollors J, Friedmann T et al (2011) Human oxidation‐specific antibodies reduce foam cell formation and atherosclerosis progression. J Am Coll Cardiol 58: 1715–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufanli O, Telkoparan Akillilar P, Acosta‐Alvear D, Kocaturk B, Onat UI, Hamid SM, Çimen I, Walter P, Weber C, Erbay E (2017) Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc Natl Acad Sci USA 114: E1395–e1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumurkhuu G, Dagvadorj J, Porritt RA, Crother TR, Shimada K, Tarling EJ, Erbay E, Arditi M, Chen S (2018) Chlamydia pneumoniae hijacks a host autoregulatory IL‐1β loop to drive foam cell formation and accelerate atherosclerosis. Cell Metab 28: 432–448.e434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuomisto TT, Korkeela A, Rutanen J, Viita H, Bräsen JH, Riekkinen MS, Rissanen TT, Karkola K, Kiraly Z, Kölble K et al (2003) Gene expression in macrophage‐rich inflammatory cell infiltrates in human atherosclerotic lesions as studied by laser microdissection and DNA array: overexpression of HMG‐CoA reductase, colony stimulating factor receptors, CD11A/CD18 integrins, and interleukin receptors. Arterioscler Thromb Vasc Biol 23: 2235–2240 [DOI] [PubMed] [Google Scholar]
- Volmer R, van der Ploeg K, Ron D (2013) Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA 110: 4628–4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086 [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang B, Wang J, Wu H, Xu S, Zhang J, Wang L (2021) Discovery of LAMP‐2A as potential biomarkers for glioblastoma development by modulating apoptosis through N‐CoR degradation. Cell Commun Signal 19: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, Noels H (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17: 1410–1422 [DOI] [PubMed] [Google Scholar]
- Wessel D, Flügge UI (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138: 141–143 [DOI] [PubMed] [Google Scholar]
- Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide‐van Gemert S, Wang N et al (2018) Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation 138: 898–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, Murphy AJ, Wang MI, Pagler TA, Vengrenyuk Y, Kappus MS, Gorman DJ, Nagareddy PR, Zhu X, Abramowicz S et al (2013) Deficiency of ATP‐binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res 112: 1456–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X et al (2015) ProLuCID: An improved SEQUEST‐like algorithm with enhanced sensitivity and specificity. J Proteomics 129: 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurdagul A Jr, Doran AC, Cai B, Fredman G, Tabas IA (2017) Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med 4: 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Taylor WR, Joseph G, Caracciolo V, Gonzales DM, Sidell N, Seli E, Blackshear PJ, Kallen CB (2013) mRNA‐binding protein ZFP36 is expressed in atherosclerotic lesions and reduces inflammation in aortic endothelial cells. Arterioscler Thromb Vasc Biol 33: 1212–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Goncalves R, Mosser DM (2008) The isolation and characterization of murine macrophages. Curr Protoc Immunol Chapter 14: Unit 14.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, O'Connor JP, Siomi MC, Srinivasan S, Dutra A, Nussbaum RL, Dreyfuss G (1995) The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2. Embo J 14: 5358–5366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou AX, Tabas I (2013) The UPR in atherosclerosis. Semin Immunopathol 35: 321–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer S, Grebe A, Bakke SS, Bode N, Halvorsen B, Ulas T, Skjelland M, De Nardo D, Labzin LI, Kerksiek A et al (2016) Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med 8: 333ra350 [DOI] [PMC free article] [PubMed] [Google Scholar]