

Abstract
Although many individuals achieve weight loss of 10% or more, the ability to maintain a reduced body mass over months and years is much rarer. Unfortunately, our understanding of the adverse consequences of having overweight and obesity argues that long-term maintenance of a reduced weight provides the greatest health benefit. However, to achieve long-term weight reduction requires overcoming neuroendocrine systems that favor restoration of one’s initial weight. Identifying and characterizing the components of these systems will be important if we are to develop therapies and strategies to reduce the rates of obesity and its complications in our modern society. During this session, Eric Ravussin and Steven R. Smith, respectively, discussed the physiology of the weight-reduced state that favors weight regain and a molecular component that contributes to this response.
Introduction
In mammals, the weight-reduced state elicits a complex response of hunger, increased metabolic efficiency, and reduced energy expenditure (EE), which together favor weight regain (1). The evolutionary benefits of such a response, dubbed metabolic adaptation, would appear to be great: the conservation of energy to protect against starvation when food is scarce and the provision of sufficient nutrients for reproduction and lactation. However, similarly apparent are the challenges that such a response creates for individuals who attempt to lose weight and maintain a reduced body mass.
Any homeostatic system requires at least three components: (1) a quantitative variable to maintain or defend, (2) a signal that provides a measure of that variable, and (3) systems that can return the variable to its “equilibrium” when perturbed. Evidence accumulated over the last century argues that body weight, and specifically adipose tissue mass, is defended via a homeostatic system (2). During the last 30 years, studies in humans and rodent models have identified key anatomical structures, aspects of the physiology and molecular components that respond to perturbations in weight and fat mass, that together are helping to define the metabolic adaptations to weight perturbations (3–5).
That body weight is a variable under homeostatic control was postulated as early as the 19th century when Rudolf O. Neumann noted that, despite no conscious effort and the large and varied amount of food consumed annually, his weight did not change significantly over the course of a year (6). Early experimental evidence that such a system exists came from studies functionally mapping brain regions in rats. Lesions to regions of the hypothalamus led to rapid changes in feeding behavior: ablation of nuclei in the medial hypothalamus increases feeding and the development of a form of severe obesity, whereas lesioning of the lateral hypothalamus causes anorexia and, in some instances, starvation and death (7,8). These findings were consistent with distinct areas of the brain being part of a counterbalanced system that regulates feeding behavior. This led several investigators to postulate that these feeding circuits were subservient to a regulatory system, a rheostat, that matched food intake with need and defended body weight against perturbations (9,10).
While the observations of weight stability and identification of feeding circuits were consistent with a homeostatic system of weight control, direct evidence that body weight is “measured” by a signal remained elusive for decades. Indirect measures of body mass, including circulating glucose concentration and temperature, were proposed to be measured variables that led to metabolic adaptation (10–12). The molecular characterization of the obese mutation and identification of leptin, however, identified such a signal that serves as a direct measure of fat mass (4,13,14). With the discovery of leptin, it was hoped that the homeostatic signal that provides a measure of fat mass had been revealed. Although subsequent studies and the failure of leptin as a stand-alone therapy for weight loss suggest the existence of multiple signals, the discovery of leptin as a hormone that circulates in proportion to fat mass did provide direct validation of the “lipostatic” model of weight regulation in which fat mass is “measured” by peripheral signals (15).
While components of metabolic adaptation were being uncovered in the laboratory, in the world at large, an epidemic of obesity was developing (16). The paradox of the discovery of a homeostatic system of weight regulation during a period in which average adiposity in society at large increased revealed that aspects of the physiology and components of the metabolic adaptation system have yet to be discovered. Among unanswered questions raised are these: How is the homeostatic “set point” of adiposity set? Is it modified by external factors and time? What are the genetic and epigenetic components of the weight regulatory system? In what way do environmental and external forces shape or modify the homeostatic levers? While answers to these and other questions remain elusive, they offer the possibility of developing strategies to treat the personal and public health problems that obesity poses. Among the many questions for which the answers would form the basis for mapping out preventive and therapeutic approaches to obesity is this one: What are the components, both physiologic and cellular, that constitute the metabolic adaptation? Providing historical context and key evidence, Ravussin describes the basic physiology of and evidence for metabolic adaptation in humans, while Smith reviews cellular mechanisms by which metabolic adaptation is mediated, focusing on EE.
Metabolic Adaptations in the Weight-Reduced State: An Overview
More than 70 years ago, Dr. Albert Stunkard stated that weight loss treatments often fail owing, in part, to the difficulty in maintaining the weight loss. He famously stated, “Most people will not stay in treatment for obesity. Of those who stay in treatment, most will not lose weight, and of those who do lose weight most will regain it (17).” Since then, weight loss strategies have improved, and many lifestyle interventions are now yielding levels of weight loss that are sufficient to confer significant improvements in cardiometabolic health (18,19). Even more encouraging are the follow-up studies from the Diabetes Prevention Program and Action for Health in Diabetes (Look AHEAD). These studies indicate that intensive lifestyle interventions can produce reasonably long-lasting weight loss in populations known to be resistant to weight loss, such as people with type 2 diabetes or prediabetes (20). Nonetheless, after weight loss, many individuals regain their weight over months and years. In fact, it is now well accepted that decreased EE and increased physiological and psychological drivers of food intake actually undermine the ability to maintain weight loss.
From early studies in the Pima Indians, a population known to be prone to obesity, we determined that both resting metabolic rate (RMR) and fat oxidation are familial traits (likely genetically determined), with siblings being more alike for these traits than unrelated individuals (21,22). Prospective studies in the Pima Indians demonstrated that a low metabolic rate for a given body weight and body composition and/or low fat oxidation were both risk factors for body weight gain (22,23). These data suggested, for the first time, that the physiological variability in energy metabolism (low metabolic rate and low fat oxidation) was an important biological factor underlying the propensity for weight gain and likely the resistance to weight loss in response to caloric restriction. Importantly, we suggested that weight gain and obesity is the price to pay to achieve “normal” metabolic rate and “normal” fat oxidation at a new higher energy flux (24).
In response to weight loss, RMR is often reduced to a greater extent than would be expected by the loss of fat-free mass and fat mass, a physiological mechanism usually called “metabolic adaptation.” It is now well accepted that such an adaptation is one of the reasons why the body resists further weight loss and individuals regain weight so easily. In lean adults and adults with obesity, Rosenbaum et al. reported significant metabolic adaptation after weight loss (25) that persisted for more than 1 year following weight loss (26). An even larger metabolic adaptation was reported in 16 individuals participating in “Biggest Loser” (NBC) competition. These individuals lost, on average, 40% of their body weight throughout the competition. Compared with individuals of the same sex, weight, body composition, and age who never lost weight, the weight-reduced participants from “The Biggest Loser” were shown to have an RMR that was almost 300 kcal/d lower following their dramatic weight loss (27,28). Fothergill et al. confirmed that such an extreme metabolic adaptation was sustained 6 years later despite an almost 70% regain of the initial weight loss (29).
Interestingly, the magnitude of the observed metabolic adaptation was strongly related to the decrease in plasma leptin concentration (28). This observation suggests that the decline in leptin may be an important biological signal for the resistance to further weight loss (a survival mechanism) that triggers decreased EE and possibly fat oxidation after caloric restriction and weight loss (28,30). Indeed, leptin is a physiological driver of both the sympathetic nervous system and the thyroid axis, both of which are important modulators of EE (31). Other studies conducted in normal-weight volunteers or volunteers with slight overweight have identified similar metabolic adaptation after 1 and 2 years of caloric restriction (32,33). Together, these studies suggest that the degree of metabolic adaptation is related not only to the magnitude of weight loss but also to the speed of the weight loss as indicated by the data in “The Biggest Loser.”
In an impressive series of studies in which metabolic adaptation was measured in response to acute episodes of overfeeding or fasting, Piaggi et al. identified “thrifty” and “spendthrift” phenotypes associated with resistance or ease of losing weight during caloric restriction (34). Recently, the same authors extended these observations to show that changes in fat oxidation (a smaller decrease in respiratory quotient, reflecting a smaller increase in lipid oxidation rate) in response to overfeeding or fasting was an important determinant of spontaneous body weight change (35). Together, these prospective observations emphasize the role of energy metabolism (EE and fat oxidation) in the control of body weight and in the maintenance of weight loss.
Not only do caloric restriction and weight loss induce a “thrifty” metabolism, but they also cause changes in the circulating mediators of appetite with an elevation of orexigenic signals and a reduction of anorexic signals. In a study of 50 participants with overweight or obesity, a 10-week weight loss program with a low-energy diet was accompanied by decreases in circulating levels of anorexigenic signals including leptin, peptide YY, cholecystokinin, insulin, and amylin and increases in circulating levels of orexigenic signals including ghrelin, glucose-dependent insulinotropic polypeptide, and pancreatic polypeptide in parallel to increased appetite (36). Importantly, these signals and feelings of hunger were still present 1 year later after these individuals had maintained ~70% of the initial weight loss. Collectively, the persistence of metabolic adaptation and the increases in physiological appetite signals likely play an important role in the lack of success in maintaining the weight loss.
Besides metabolic adaptation, weight loss is also associated with obligatory decreases in the thermic effect of food (related to decreased food intake) and in the energy cost of weight-bearing physical activity. The decrease in the three components of EE (RMR, thermic effect of food, EE for physical activity) and the increase in orexigenic signals together attenuate weight loss and favor weight regain. Whether these long-lasting adaptations (lower EE and fat oxidation and greater appetite signals) to caloric restriction and weight loss can cause a biological resetting of the defended body weight remains to be proven.
Based on our present knowledge, we can draw distinct lessons that may help those people struggling to maintain a healthier weight in our modern obesogenic environment (37):
Weight loss, and even dramatic weight loss, is indeed possible. However, the kind of weight loss achieved in “The Biggest Loser” (except maybe for those obtained via bariatric surgery) is almost impossible to maintain when individuals return to their natural environment in the absence of constant therapeutic support.
The major drivers of weight regain are the well-established physiological responses to weight loss, i.e., the persistence of a lower RMR, lower energy cost of weight-bearing activities, decreased fat oxidation, and persistence of increased appetite related to long-lasting increased orexigenic and decreased anorexic signals.
Only those who engage in high levels of physical activity after weight loss have been successful in maintaining a reduced weight (38,39).
Importantly, a moderate and realistic weight loss (5%-10%) recommendation can yield significant health benefits (19). Because of such findings, the recommendations should shift from the need for large weight loss (e.g., > 20% of body mass) toward moderate weight loss with a strong emphasis on weight loss maintenance. Planning for weight loss maintenance ideally should begin during the initial period of weight loss. To be successful at maintaining a new lower body weight, a supportive environment that enlists healthy behaviors is imperative. Special attention to lifestyle changes implemented during the weight loss phase such as diet and physical activity will help to counteract the biologic and metabolic adaptation triggered by weight loss and help the maintenance of weight loss (37,39). If weight loss is unsuccessful or if a rebound in weight is unavoidable, medication should be considered in conjunction with continued lifestyle modifications. Recommending that patients weigh themselves frequently is also a useful tool for weight loss and weight maintenance success: if a small weight regain is detected, reinitiating some of the behaviors that produced the initial weight loss is recommended.
We must also stop blaming the individuals who struggle to achieve or maintain weight loss. Laziness is clearly not a factor in the commonly observed weight regain. For instance, it may be challenging to find a more dedicated group of individuals than those who participated in “The Biggest Loser” competition. As a result, we need to better understand the biology driving weight regain if we are to improve treatment. Until then, we need to be more accepting of variations in body size and focus our effort on improving the health of our patients, not weight per se. We must also recognize that human-engineered changes in environmental conditions (the built environment) have resulted in improved health (reduction of infectious diseases and extension of life-span) but have also triggered an epidemic of noncommunicable chronic diseases with obesity at the forefront. Only some reversal of the “obesogenic” built environment will help to reverse this frightening epidemic. In the meantime, it will be necessary for individuals prone to weight gain and resistant to weight loss (those with a “thrifty” genotype/phenotype) to be vigilant and adopt a lifestyle counteracting their biological predispositions.
Potential Mechanisms for Metabolic Adaptation in the Weight-Reduced State
Metabolic adaptation to caloric restriction and the weight-reduced state is defined as a reduction in whole-body EE even after accounting for changes in body composition. This section will cover proposed cellular mechanisms by which metabolic adaptation occurs in the weight-reduced state.
We will first cover a preferred approach to calculate metabolic adaptation and the concept of muscular efficiency. Next, we will review potential mechanisms of efficiency in energy use as it relates to energetics of muscular contraction, hormonal control, mitochondrial efficiency, and two potential futile cycles: calcium cycling and the Masaro substrate cycle. Each of these areas may impact metabolic adaptation. It should be noted that nutrient digestion and/or absorption may also alter energy balance and have been hypothesized to influence EE. Alterations in the gut microbiome occur with weight loss (39), but the contribution of gut digestive processes and the gut microbiome to the propensity to conserve energy and increase body weight have not been well explored. This topic will therefore not be covered in this section. Lastly, we will discuss the design of research protocols to study metabolic adaptation including the integration of multiple data streams.
Calculation of metabolic adaptation
In the context of weight loss, Galgani and Santos (40) have reviewed the implications of these differences and made recommendations for the calculation of metabolic adaptation following weight loss. Specifically, they highlight the importance of measuring the residual difference of predicted EE against actual EE before and after a weight loss intervention (Figure 1).
Figure 1.
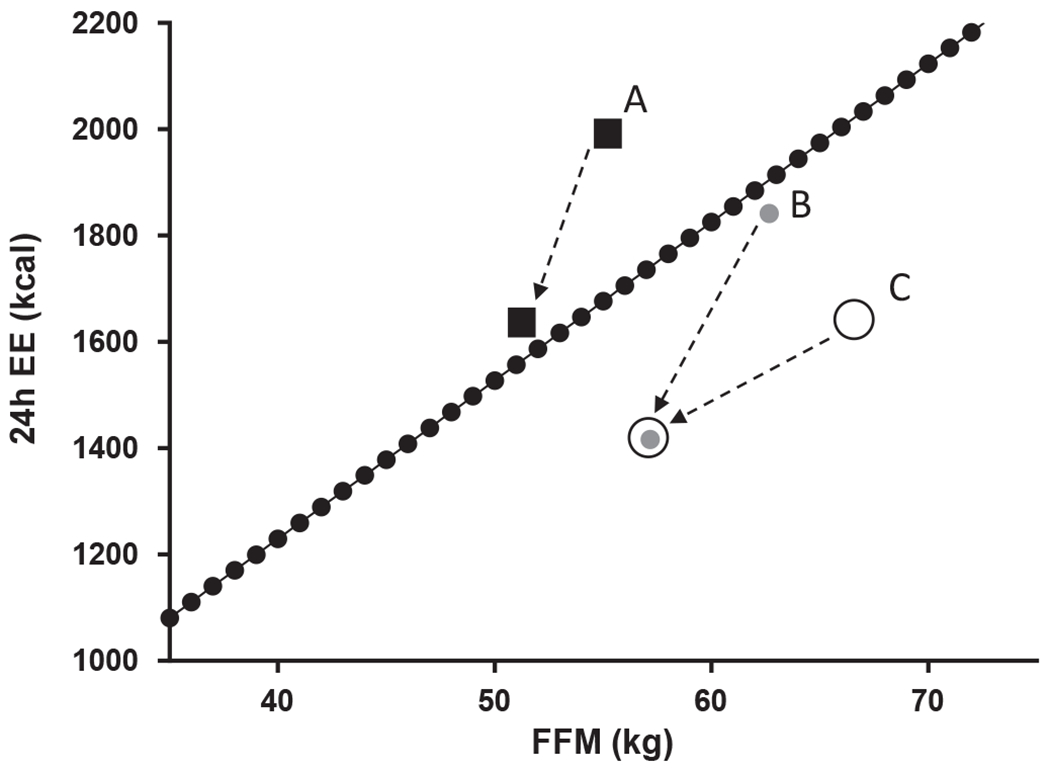
A novel method to calculate weight-loss-induced metabolic adaptation defined by Galgani and Santos (40). Metabolic adaptation should be calculated as the difference in residuals (i.e., actual minus predicted energy expenditure [EE]) between post- and pre-weight-loss intervention. If only the postintervention residual was calculated, participant A would be considered a non-metabolically adapted participant when actually he/she had a substantial drop in thermogenesis. If only the postintervention residual of participants B and C was calculated, it would be concluded that he/she had a similar metabolic adaptation. By considering the preintervention residual, it is clear that participant B has become more metabolically adapted than participant C. Fat-free mass (FFM) is the major determinant of resting EE.
Variation in organ size and the calculation of metabolic adaptation
Interindividual variation in body composition, specifically organ size, can influence EE relative to basic measures of body composition. Bosy-Westphal and colleagues (41) have reported that obesity per se influences the relative contributions of organs to overall EE. How this affects the calculation of metabolic adaptation is rarely discussed and should be considered. Race was once believed to influence gross body-composition-adjusted EE. Using magnetic resonance imaging to measure organ size, Gallagher and colleagues found that interindividual variation in the mass of select high-metabolic-rate organs mediated variability in resting EE but not race per se (42,43). More work should be done on how the genetic or environmental regulation of organ development (44) influences EE.
Gross muscle efficiency
Gross efficiency is the ratio of work completed to the amount of energy expended. In 2015, Broskey et al. (45) demonstrated that muscular efficiency was positively correlated to maximal ATP flux measured by dynamic magnetic resonance spectroscopy or biopsy-measured mitochondrial content (Figure 2). Important to this discussion, Amati and colleagues (46) found that gross efficiency improved with weight loss and that exercise training has an additive effect on improving gross efficiency. How these changes and the interindividual differences in efficiency affect the propensity to regain weight is unknown. Together, these studies suggest that individuals who lose weight become more efficient, expending less energy for a given amount of work. This increase in efficiency might predispose them to regain weight.
Figure 2.
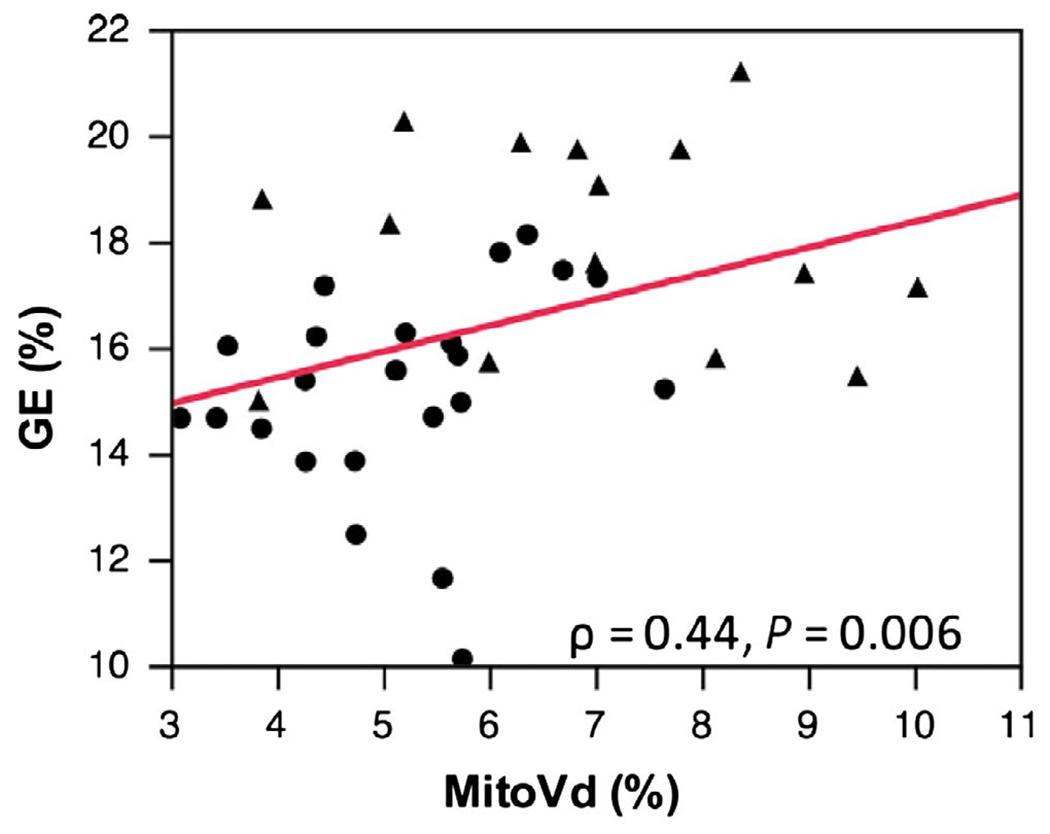
Association between mitochondria volume density and gross efficiency. Mitochondrial volume density (MitoVd%) was measured by electron microscopy of skeletal muscle representing the mass of mitochondria in the muscle. Gross efficiency (GE%) is total energy consumed (oxygen consumption measured by indirect calorimetry) divided by the work produced as measured by cycle ergometry (47).
Molecular targets and mechanisms of energy wasting
Rolfe and Brown (47) have thoroughly reviewed the cellular components of EE, providing an excellent framework to help understand metabolic adaptation of EE to weight loss. We argue that understanding the underlying cellular and molecular mechanisms will provide novel opportunities for intervention to augment an individual’s capacity for weight loss and reduce one’s propensity for weight regain. In addition to addressing EE and oxidative efficiency, we will review general considerations on the measurement of changes in EE and adjustments for changes in body mass.
Endocrine adaptations to caloric restriction
Caloric restriction is known to increase the circulating levels of cortisol and decrease the concentration of growth hormone and bioactive triiodothyronine (T3) (48). Studies from the Dulloo lab provide evidence that local thyroid hormone production declines in the weight-reduced state in rats (49). Muscle deiodinase type 3 (DIO3) increases, which inactivates T3 and the expression of T3-activating enzyme and reduces deiodinase type 2 (DIO2), leading to a slower net formation of T3 from its thyroxine (T4) precursor. The latter studies need to be proven in humans but highlight the concept of tissue level control of the hormonal effects on muscular efficiency.
Mitochondrial efficiency
Studies have demonstrated extensive variation in mitochondrial coupling measured in vivo in human muscle (50,51) (Figure 3). These studies and others suggest that this variation might contribute to the reduced EE and propensity to regain weight in the weight-reduced state. Sparks et al. (52) found that mitochondrial efficiency, measured as ATP generated per molecule of O2 utilized (P/O), increases with caloric restriction and is dependent upon the baseline P/O. Individuals in whom mitochondria ATP production is tightly coupled to electron transport at baseline had more significant improvement in muscle in ATP maximal synthetic rates and P/O following calorie restriction (52).
Figure 3.
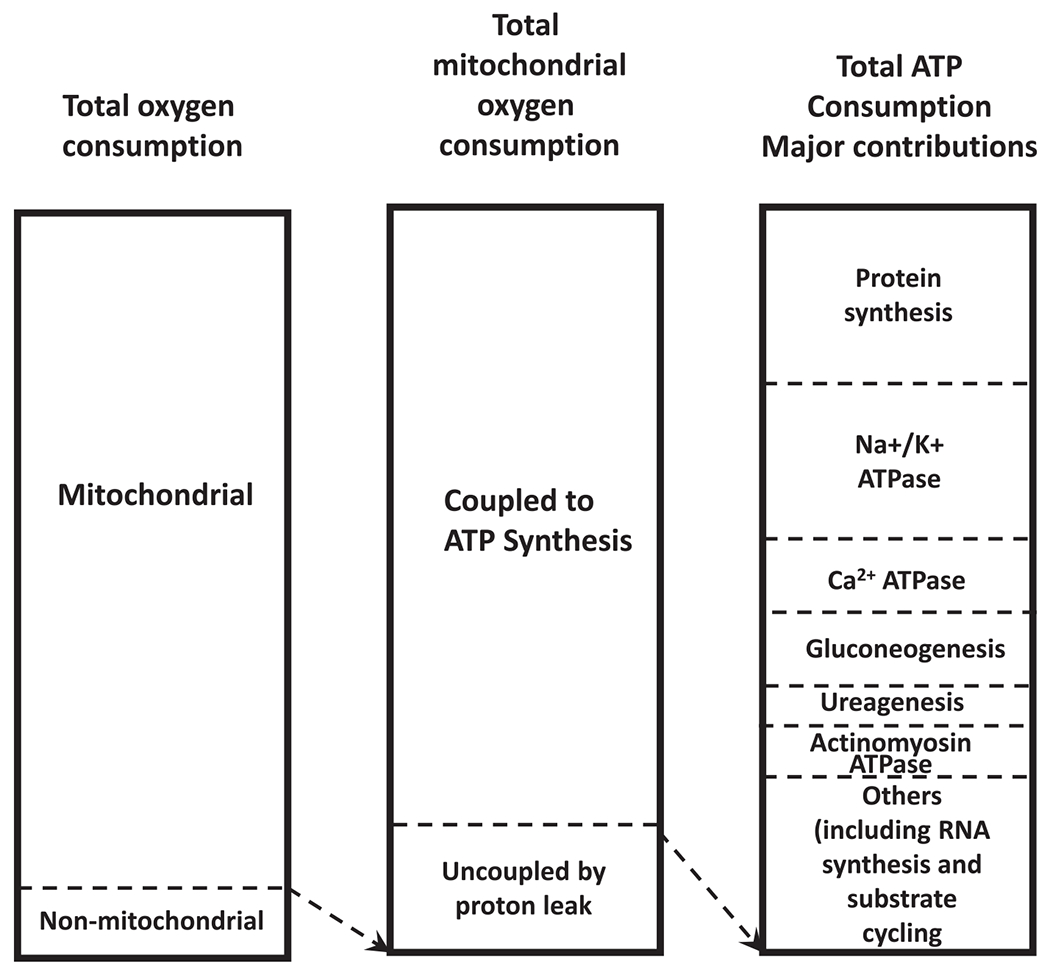
Estimated contribution of processes to energy utilization. The first column shows the contribution of mitochondrial to nonmitochondrial oxygen consumption to total respiration. Second column shows the proportion of used to drive ATP synthesis versus proton leak. The third column represents the contribution of ATP consuming processes to total ATP production.
Further studies are needed to determine whether improved mitochondrial efficiency in the weight-reduced state contributes directly to regain weight; this is currently unknown. Elegant retrospective studies from the Harper lab have identified differences in skeletal muscle characteristics after weight loss consistent with this proposition (53). Later studies revealed that persons who lost more weight had increased mitochondrial efficiency and reduced proton leak (54). Prospective studies are needed to confirm whether this finding is present before weight loss or a response to caloric restriction and the weight-reduced state.
Calcium cycling
Another key cellular system for the control of cellular efficiency in muscle is the sarco/endoplasmic reticulum CA2+ ATPase pump. In a series of elegant rodent studies, Perisamy and colleagues showed that this system, via the modulator sarcolipin, is a key controller of muscle thermogenesis (55,56). The dwarf open reading frame peptide (DWORF) modulates the sarco/endoplasmic reticulum CA2+ ATPase (57) (Figure 4), but whether this pathway is or is not regulated in the weight-reduced state is currently unknown. Future studies should explore this question.
Figure 4.
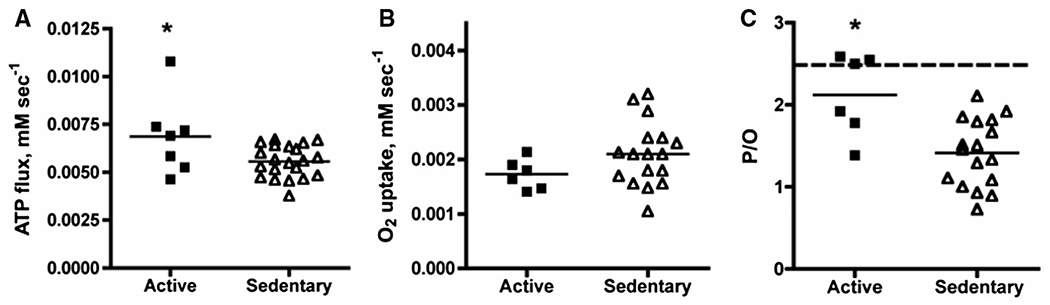
Wide variation in mitochondrial coupling measured in vivo in human muscle. (A) ATP flux, (B) O2 flux, and (C) mitochondrial coupling are calculated from P/O in individuals from the active and sedentary groups. The horizontal dashed line in panel C represents theoretical value for mitochondrial coupling with glucose as a substrate. Resting ATP turnover was calculated from the rate of decline in phosphocreatine in skeletal muscle during lower limb ischemia. Oxygen consumption was measured using optical spectroscopy of the dynamic fall in muscle deoxymyoglobin during a second bout of ischemia. P/O was calculated as ATP flux/oxygen consumption. Figure is taken from (50).
The Masaro substrate cycle
Several additional futile cycles have been identified and could serve to increase or decrease efficiency of energy conservation during caloric restriction and in the weight-reduced state. One cycle of particular interest is the simultaneous induction of fatty acid synthesis and beta-oxidation, first identified by Masoro as part of the heat production machinery in activated brown adipose tissue. This cycle was rediscovered in the modern era by Adams, also in brown adipose tissue, and independently by Dulloo and colleagues in skeletal muscle (Figure 5) (58–60). Future studies should explore whether this pathway differs in the weight-reduced state.
Figure 5.
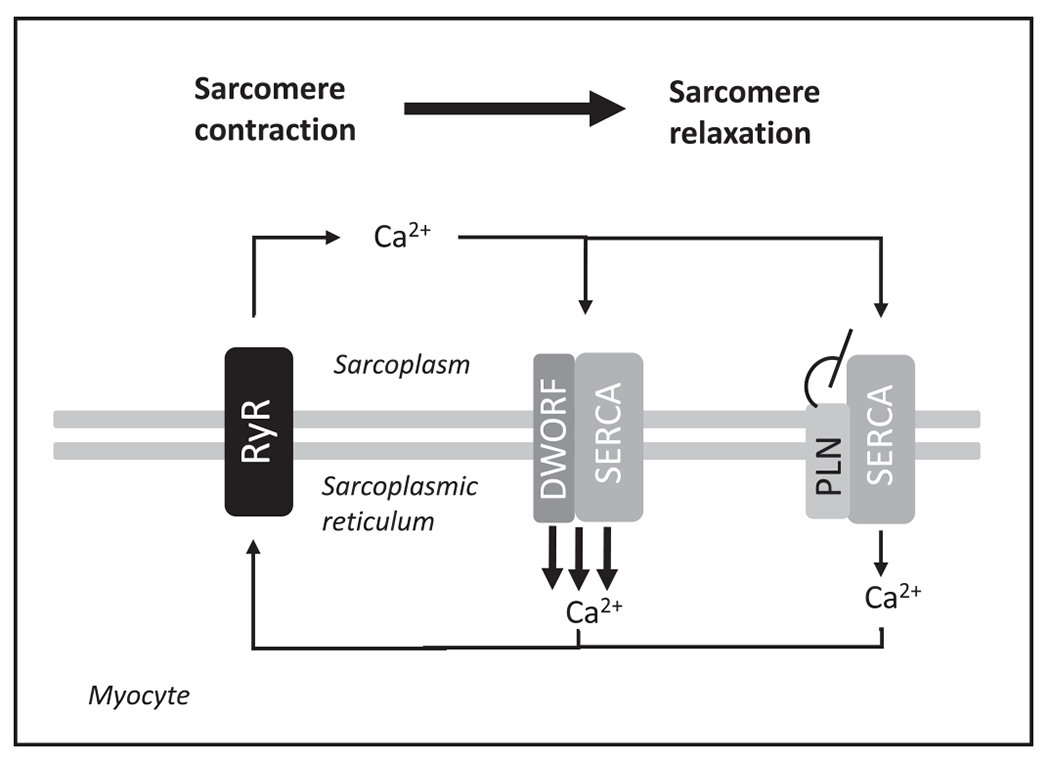
Working model of the effects of dwarf open reading frame peptide (DWORF) see Science 2016 Jan 15;351(6270):271-5 foarity function in myocytes. The relative efficiency of calcium recycling is hypothesized to regulate body temperature. The discovery of DWORF suggests that efficiency can be actively regulated and is a potential mechanism by which caloric restriction might change resting metabolic rate or the efficiency of muscular contraction. Specifically, DWORF is a micropeptide that enhances SERCA activity by displacing PLN, a potent SERCA inhibitor. Figure redrawn and adapted from (57). PLN, phospholamban; RyR, ryanodine receptors; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase.
These are just some of the areas that should be explored that might explain the overall effects and interindividual variation in changes in EE in the weight-reduced state (Figure 6).
Figure 6.
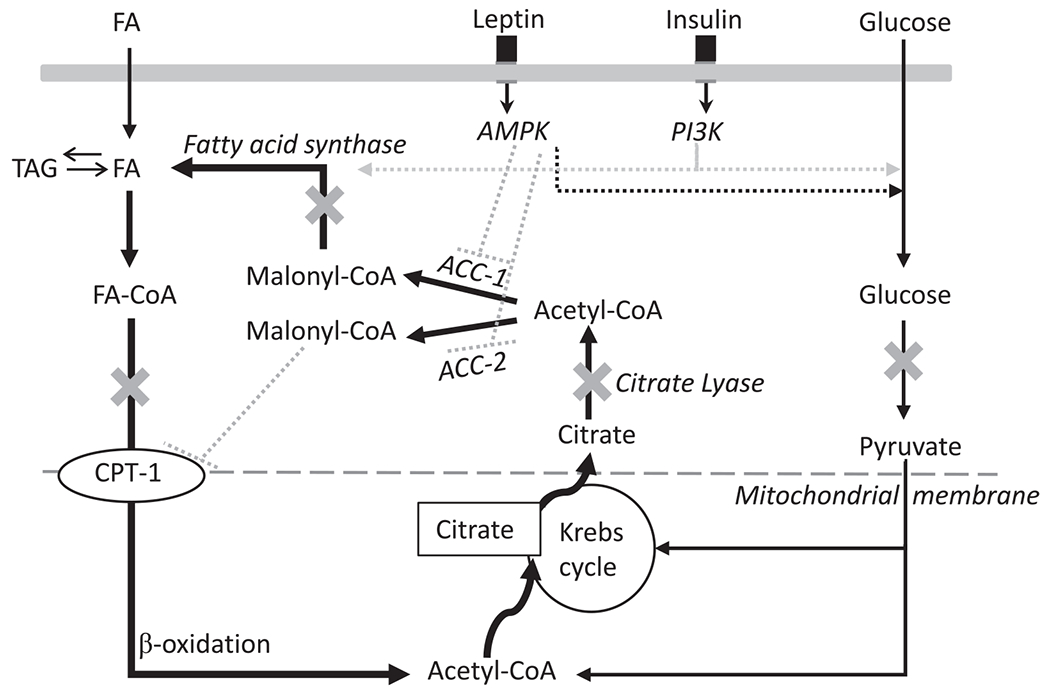
Model of thermogenesis in skeletal muscle caused by substrate cycling between de novo lipogenesis and lipid oxidation orchestrated by PI3K and AMPK. Evidence for this substrate cycling stems from calorimetric studies showing leptin induces thermic effects in skeletal muscle, which is enhanced with insulin but inhibited with pharmacological compounds targeting crucial points in the substrate flux as denote by “X” on the diagram. Specifically, 2-deoxyglucose is a glucose analog that binds to hexokinase and inhibits glycolysis; hydroxy-citrate inhibits citrate lyase and therefore the conversion of citrate to acetyl-CoA; cerulenin inhibits FA synthase and, consequently, the conversion of malonyl-CoA to FA; and etomoxir inhibits CPT-1 and the entry of FA into the mitochondria for beta-oxidation. Repeated substrate recycling of acetyl-CoA through lipogenesis and β-oxidation would be ATP dependent and therefore drive thermogenesis. Figure amended from (58). AMP-activated protein kinase (AMPK), Coenzyme A (CoA), Carnitine palmitoyltransferase I (CPT-1), fatty acid (FA), Phosphoinositide 3-kinase (PI3K).
The design of research protocols to study metabolic adaptation
Studies of EE in humans is subject to genetic variations, the environment in which measurements are made, and the metabolic history of individuals. Some consideration should be given to the baseline “state” in the study of metabolic adaptation (40). Performing measurements in weight-stable individuals (i.e., without antecedent weight loss or gain) is important; prior weight loss will reduce EE relative to body composition, and conversely, prior overfeeding will increase EE relative to body composition. The literature does not speak to how much variability in body weight, and for how long, is acceptable. Interindividual variation in EE unrelated to feeding status also produces variation at baseline in a research study. Environmental factors such as stress (catecholamines), medications such as beta blockers (61), and ambient temperature (62) alter EE adjusted for body composition as well. Controlling for these factors is critical for meaningful analyses.
Integration of multiple potential mechanisms in the study of metabolic adaptation
Many studies will only explore one of these mechanisms. Unfortunately, it is difficult to ascertain the independent effects of the myriad of potential factors because human studies often do not integrate whole body, neural, endocrine, and cellular mechanisms. Parsing the relative contribution of each will be necessary, and measuring many components simultaneously is a challenging but necessary pathway forward.
A recent investigation by Vinales et al. (63) exemplifies an integrative approach. They were able to determine an effect of epinephrine on both EE and core body temperature. They found evidence for a ceiling effect for human thermogenic response to diet when epinephrine was increased. This kind of multiparametric investigation will be needed to identify specific mechanisms that contribute to total 24-hour EE and also for studies that aim to identify the contributions to components of total EE, including rest (resting EE), sleep (sleeping EE), diet induced thermogenesis, and physical activity.
Conclusion
We have attempted to briefly review key concepts in the assessment of EE in the weight-reduced state that point to known and potential mechanisms and regulators of efficiency (hormones such as leptin and T3, as well as mitochondrial and nonmitochondrial control systems) as a source of the observed decreased energy waste and decreased heat production. Many other potential mechanisms are still likely to be discovered. The challenge will be to parse out the interconnectedness and the individual contribution of these pathways to the ability to lose weight and to maintain weight loss. Future studies should aspire to collect data on the activity of as many of these systems as possible in order to develop rational strategies to achieve and maintain weight loss when desirable to improve health.
Acknowledgments
Dr. Smith would like to acknowledge Dr. Katie Whytock and Seema Sernovitz for assisting in the preparation of this manuscript.
Footnotes
Disclosure: The authors declared no conflict of interest.
References
- 1.Greenway FL. Physiological adaptations to weight loss and factors favouring weight regain. Int J Obes (Lond) 2015;39:1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Passmore R The regulation of body-weight in man. Proc Nutr Soc 1971;30:122–127. [DOI] [PubMed] [Google Scholar]
- 3.Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med 1995;332:621–628. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994;372:425–432. [DOI] [PubMed] [Google Scholar]
- 5.Flier JS, Maratos-Flier E. Obesity and the hypothalamus: novel peptides for new pathways. Cell 1998;92:437–440. [DOI] [PubMed] [Google Scholar]
- 6.Neumann R Experimentelle Beitrage zur Lehre von dem taglichen Nahrungsbedarf des Menschen unter besonder Berucksichtigung der notwendigen Eiweifsmenge Arc Hyg. 1902;45:1–87. [Google Scholar]
- 7.Hetherington AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Anat Rec 1940;78:149–172. [Google Scholar]
- 8.Brobeck JR. Neural control of hunger, appetite, and satiety. Yale J Biol Med 1957;29:565–574. [PMC free article] [PubMed] [Google Scholar]
- 9.Mayer J Glucostatic mechanism of regulation of food intake. N Engl J Med 1953;249:13–16. [DOI] [PubMed] [Google Scholar]
- 10.Brobeck JR. Food and temperature. Recent Prog Horm Res 1960;16:439–466. [PubMed] [Google Scholar]
- 11.Kennedy GC. The hypothalamus and obesity. Proc R Soc Med 1966;59:1276–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porte D Jr., Woods SC. Regulation of food intake and body weight in insulin. Diabetologia 1981;20:274–280. [DOI] [PubMed] [Google Scholar]
- 13.Friedman JM, Leibel RL, Siegel DS, Walsh J, Bahary N. Molecular mapping of the mouse ob mutation. Genomics 1991;11:1054–1062. [DOI] [PubMed] [Google Scholar]
- 14.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996;334:292–295. [DOI] [PubMed] [Google Scholar]
- 15.Ravussin Y, Leibel RL, Ferrante AW Jr. A missing link in body weight homeostasis: the catabolic signal of the overfed state. Cell Metab 2014;20:565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arroyo-Johnson C, Mincey KD. Obesity epidemiology worldwide. Gastroenterol Clin North Am 2016;45:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stunkard AJ. The management of obesity. NY State J Med 1958;79–87. [PubMed] [Google Scholar]
- 18.Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Magkos F, Fraterrigo G, Yoshino J, et al. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab 2016;23:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Look AHEAD Research Group. Eight-year weight losses with an intensive lifestyle intervention: the Look AHEAD study. Obesity (Silver Spring) 2014;22:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogardus C, Lillioja S, Ravussin E, et al. Familial dependence of the resting metabolic rate. N Engl J Med 1986;315:96–100. [DOI] [PubMed] [Google Scholar]
- 22.Zurlo F, Lillioja S, Esposito-Del Puente A, et al. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol 1990;259:E650–E657. [DOI] [PubMed] [Google Scholar]
- 23.Ravussin E, Lillioja S, Knowler WC, et al. Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med 1988;318:467–472. [DOI] [PubMed] [Google Scholar]
- 24.Ravussin E, Swinburn BA. Metabolic predictors of obesity – cross-sectional versus longitudinal data. Int J Obesity 1993;17:S28–S31. [PubMed] [Google Scholar]
- 25.Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med 1995;332:621–628. [DOI] [PubMed] [Google Scholar]
- 26.Rosenbaum M, Hirsch J, Gallagher DA, Leibel RL. Long-term persistence of adaptive thermogenesis in subjects who have maintained a reduced body weight. Am J Clin Nutr 2008;88:906–912. [DOI] [PubMed] [Google Scholar]
- 27.Johannsen DL, Knuth ND, Huizenga R, Rood JC, Ravussin E, Hall KD. Metabolic slowing with massive weight loss despite preservation of fat-free mass. J Clin Endocr Metab 2016;101:2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knuth ND, Johannsen DL, Tamboli RA, et al. Metabolic adaptation following massive weight loss is related to the degree of energy imbalance and changes in circulating leptin. Obesity (Silver Spring) 2014;22:2563–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fothergill E, Guo J, Howard L, et al. Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring) 2016;24:1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lecoultre V, Ravussin E, Redman LM. The fall in leptin concentration is a major determinant of the metabolic adaptation induced by caloric restriction independently of the changes in leptin circadian rhythms. J Clin Endocrinol Metab 2011;96:E1512–E1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leibel RL. The role of leptin in the control of body weight. Nutr Rev 2002;60:S15–S19. [DOI] [PubMed] [Google Scholar]
- 32.Ravussin E, Redman LM, Rochon J, et al. A 2-year randomized controlled trial of human caloric restriction: feasibility and effects on predictors of health span and longevity. J Gerontol A Biol Sci Med Sci 2015;70:1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Redman LM, Smith SR, Burton JH, Martin CK, Il’yasova D, Ravussin E. Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab 2018;27:805–815.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piaggi P Metabolic determinants of weight gain in humans. Obesity (Silver Spring) 2019;27:691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Begaye B, Vinales KL, Hollstein T, et al. Impaired metabolic flexibility to high-fat overfeeding predicts future weight gain in healthy adults. Diabetes 2020;69:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sumithran P, Prendergast LA, Delbridge E, et al. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med 2011;365:1597–1604. [DOI] [PubMed] [Google Scholar]
- 37.Ravussin E, Ryan DH. Energy expenditure and weight control: is the biggest loser the best loser? Obesity (Silver Spring) 2016;24:1607–1608. [DOI] [PubMed] [Google Scholar]
- 38.Wing RR, Hill JO. Successful weight loss maintenance. Annu Rev Nutr 2001;21:323–341. [DOI] [PubMed] [Google Scholar]
- 39.Krajmalnik-Brown R, Ilhan ZE, Kang DW, DiBaise JK. Effects of gut microbes on nutrient absorption and energy regulation. Nutr Clin Pract 2012;27:201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galgani JE, Santos JL. Insights about weight loss-induced metabolic adaptation. Obesity (Silver Spring) 2016;24:277–278. [DOI] [PubMed] [Google Scholar]
- 41.Bosy-Westphal A, Braun W, Schautz B, Muller MJ. Issues in characterizing resting energy expenditure in obesity and after weight loss. Front Physiol 2013;4:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallagher D, Albu J, He Q, et al. Small organs with a high metabolic rate explain lower resting energy expenditure in African American than in white adults. Am J Clin Nutr 2006;83:1062–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Javed F, He Q, Davidson LE, et al. Brain and high metabolic rate organ mass: contributions to resting energy expenditure beyond fat-free mass. Am J Clin Nutr 2010;91:907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Penzo-Mendez AI, Stanger BZ. Organ-size regulation in mammals. Cold Spring Harb Perspect Biol 2015;7:a019240. doi: 10.1101/cshperspect.a019240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broskey NT, Boss A, Fares EJ, et al. Exercise efficiency relates with mitochondrial content and function in older adults. Physiol Rep 2015;3:e12418. doi: 10.14814/phy2.12418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amati F, Dubé JJ, Shay C, Goodpaster BH. Separate and combined effects of exercise training and weight loss on exercise efficiency and substrate oxidation. J Appl Physiol (1985) 2008;105:825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 1997;77:731–758. [DOI] [PubMed] [Google Scholar]
- 48.Redman LM, Ravussin E. Endocrine alterations in response to calorie restriction in humans. Mol Cell Endocrinol 2009;299:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Andrade PB, Neff LA, Strosova MK, et al. Caloric restriction induces energy-sparing alterations in skeletal muscle contraction, fiber composition and local thyroid hormone metabolism that persist during catch-up fat upon refeeding. Front Physiol 2015;6:254. doi: 10.3389/fphys.2015.00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conley KE, Amara CE, Bajpeyi S, et al. Higher mitochondrial respiration and uncoupling with reduced electron transport chain content in vivo in muscle of sedentary versus active subjects. J Clin Endocrinol Metab 2013;98:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sparks LM, Redman LM, Conley KE, et al. Differences in mitochondrial coupling reveal a novel signature of mitohormesis in muscle of healthy individuals. J Clin Endocrinol Metab 2016;101:4994–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sparks LM, Redman LM, Conley KE, et al. Effects of 12 months of caloric restriction on muscle mitochondrial function in healthy individuals. J Clin Endocrinol Metab 2017;102:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerrits MF, Ghosh S, Kavaslar N, et al. Distinct skeletal muscle fiber characteristics and gene expression in diet-sensitive versus diet-resistant obesity. J Lipid Res 2010;51:2394–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thrush AB, Dent R, McPherson R, Harper ME. Implications of mitochondrial uncoupling in skeletal muscle in the development and treatment of obesity. FEBS J 2013;280:5015–5029. [DOI] [PubMed] [Google Scholar]
- 55.Bal NC, Maurya SK, Sopariwala DH, et al. Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nat Med 2012;18:1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maurya SK, Herrera JL, Sahoo SK, et al. Sarcolipin signaling promotes mitochondrial biogenesis and oxidative metabolism in skeletal muscle. Cell Rep 2018;24:2919–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nelson BR, Makarewich CA, Anderson DM, et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 2016;351:271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dulloo AG, Gubler M, Montani JP, Seydoux J, Solinas G. Substrate cycling between de novo lipogenesis and lipid oxidation: a thermogenic mechanism against skeletal muscle lipotoxicity and glucolipotoxicity. Int J Obes Relat Metab Disord 2004;28:S29–S37. [DOI] [PubMed] [Google Scholar]
- 59.Masoro EJ. Role of lipogenesis in nonshivering thermogenesis. Fed Proc 1963;22:868–873. [PubMed] [Google Scholar]
- 60.Yu XX, Lewin DA, Forrest W, Adams SH. Cold elicits the simultaneous induction of fatty acid synthesis and beta-oxidation in murine brown adipose tissue: prediction from differential gene expression and confirmation in vivo. FASEB J 2002;16:155–168. [DOI] [PubMed] [Google Scholar]
- 61.Monroe MB, Seals DR, Shapiro LF, et al. Direct evidence for tonic sympathetic support of resting metabolic rate in healthy adult humans. Am J Physiol Endocrinol Metab 2001;280:E740–E744. [DOI] [PubMed] [Google Scholar]
- 62.Chen KY, Brychta RJ, Linderman JD, et al. Brown fat activation mediates cold-induced thermogenesis in adult humans in response to a mild decrease in ambient temperature. J Clin Endocrinol Metab 2013;98:E1218–E1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vinales KL, Begaye B, Thearle MS, Krakoff J, Piaggi P. Core body temperature, energy expenditure, and epinephrine during fasting, eucaloric feeding, and overfeeding in healthy adult men: evidence for a ceiling effect for human thermogenic response to diet. Metabolism 2019;94:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]