

Summary
Humans have domesticated diverse species from across the plant kingdom; however, our current understanding of plant domestication is largely founded on major cereal crops. Here, we examine the evolutionary processes and genetic basis underlying the domestication of water caltrop (Trapa spp., Lythraceae), a traditional, yet presently underutilized non‐cereal crop that sustained early Chinese agriculturalists. We generated a chromosome‐level genome assembly of tetraploid T. natans, and then divided the allotetraploid genome into two subgenomes. Based on resequencing data from 57 accessions, representing cultivated diploid T. natans, wild T. natans (2x and 4x) and diploid T. incisa, we showed that water caltrop was likely first domesticated in the Yangtze River Valley as early as 6300 yr BP, and experienced a second improvement c. 800 years ago. We also provided strong support for an allotetraploid origin of T. natans within the past 230 000–310 000 years. By integrating selective sweep and transcriptome profiling analyses, we identified a number of genes potentially selected and/or differentially expressed during domestication, some of which likely contributed not only to larger fruit sizes but also to a more vigorous root system, facilitating nutrient uptake, environmental stress response and underwater photosynthesis. Our results shed light on the evolutionary and domestication history of water caltrop, one of the earliest domesticated crops in China. This study has implications for genomic‐assisted breeding of this presently underutilized aquatic plant, and improves our general understanding of plant domestication.
Keywords: water caltrop, genome sequencing, allopolyploid, domestication, selection, gene expression
Introduction
Plant domestication fundamentally altered the course of human history, causing a shift from hunter–gatherer to agricultural societies and greatly advancing human civilization (Gepts, 2004; Olsen and Wendel, 2013; Purugganan, 2019). During the domestication process, humans modified wild species through breeding to improve a number of agronomic traits (e.g. the size, shape and colour of the plant organs). Elucidating the genetic basis and history of domestication is of great interest as it informs our understanding of early human societies and facilitates the continued improvement of our crops (Meyer and Purugganan, 2013) . However, our current understanding of plant domestication is founded on major cereal crops (e.g. wheat, barley, rice, corn) (Von Wettberg et al., 2018), while little attention has been paid to traditional, yet presently underutilized non‐cereal crop species that sustained early agriculturalists and have been cultivated for millennia (Jarvis et al., 2017). In fact, the cultivation and commercialization of underutilized species is increasingly recognized as a viable development strategy with multiple benefits, such as managing climate risk, enhancing agrobiodiversity and improving rural livelihoods (Meyer et al., 2012).
Trapa L. (Lythraceae), also known as water caltrop, is an annual herbaceous, floating‐leaf aquatic genus. It is widely distributed in temperate and subtropical regions of Europe, Asia and Africa, and is invasive in North America (Chen et al., 2007; Hummel and Kiviat, 2004). The fruits of water caltrop have a high starch content and are an important source of food, whereas the stems and leaves are used as vegetables (Hoque et al., 2009). Based on many archaeological excavations in Eurasia, human consumption of water caltrop dates back to the Neolithic period (3000–9000 years before present, yr BP) (Guo et al., 2017; Hummel and Kiviat, 2004; Karg, 2006) . In China, numerous fruit remains of water caltrop are known from at least 21 Neolithic sites along the Yellow and Yangtze Rivers, often associated with the remains of rice, wild soybean and other plants bearing edible fruits (e.g. Quercus spp., Prunus persica, Euryale ferox) (Guo et al., 2017). Thus, water caltrop and other non‐cereal starch crops were critical calorie sources that sustained early agriculturalists in eastern Asia before the widespread adoption of rice and millet farming. Recent excavations at Tianluoshan site (Lower Yangtze Region; Figure 1a) of the Neolithic period (6300–7000 yr BP) have provided evidence that water caltrop was deliberately altered or selected for fruit size and shape (Guo et al., 2017). The agricultural cultivation history of water caltrop dates back at least to the Tang (618–907 AD) and Song (916–1279 AD) Dynasties, when rivers and lakes in the lower Yangtze River Valley were segmented as farms, and many common cultivars (e.g. ‘Wuling’ and ‘Nanhuling’) were recorded (Zhou, 2012). These historical accounts suggest that water caltrop cultivation was once a major agricultural practice for the ancient Chinese.
Figure 1.
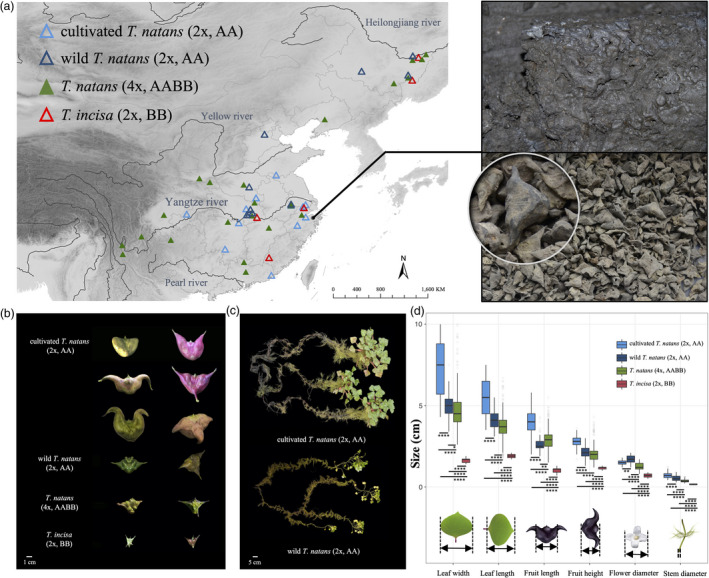
(a) Geographic locations of diploid and tetraploid T. natans as well as T. incisa sampled in this study; the enlarged photograph on the right shows fruit remains of water caltrop that preserved in storage pits at the Tianluoshan site (7000–5800 yr BP), an important settlement of the Neolithic Culture (Guo et al., 2017). (b) Differences in fruit morphology between diploid cultivated and wild Trapa natans (2x, AA), tetraploid T. natans (4x, AABB) and diploid T. incisa (2x, BB). (c) Morphological differences in the root system between wild and cultivated T. natans (2x, AA). (d) Boxplots showing differences in the length and width of leaves, the length and height of fruits as well as the diameters of flowers and stems between diploid (cultivated, wild) and tetraploid T. natans and T. incisa.
During domestication, cultivars were selected for larger fruit size and fewer but stouter spines (Figure 1b) (Kluyver et al., 2017). In addition, compared to wild accessions, cultivars usually have a more vigorous root system (Figure 1c), resulting in improved anchorage of plants in the aquatic sediment and higher rates of dissolved‐nitrogen absorption and underwater photosynthesis (Rich et al., 2012). Water caltrop was harvested wildly by European prehistoric populations to supplement their normal diet between 6000 and 3000 yr BP; however, it has never been domesticated in Europe (Karg, 2006), where it is presently rare and even regionally extinct (Frey et al., 2017). Trapa is now considered to contain two species, i.e., T. natans L., with both diploid (2n = 2x = 48) and tetraploid (2n = 4x = 96) cytotypes, and diploid T. incisa Sieb. & Zucc. (2n = 2x = 48) (Chen et al., 2007; Hoque et al., 2009; Takano and Kadono, 2005) . Previous cytological (Oginuma et al., 1996) and molecular genotyping analyses (allozymes: Takano and Kadono, 2005; AP2 and trnL–F: Kim et al., 2010) suggested that tetraploid T. natans might be of allopolyploid origin, resulting from hybridization between diploid T. natans and T. incisa. Trapa natans mainly differs from T. incisa by having larger fruits (single‐seeded drupes) and a more diverse number of fruit spines (four, two or occasionally no spines vs. four) (Hummel and Kiviat, 2004) (Figure 1b). The two species exhibit a high degree of autogamy (Arima et al., 1999).
In this study, we generated a chromosome‐level genome assembly of tetraploid T. natans, and resequenced 57 accessions, representing cultivated diploid T. natans, wild T. natans (both 2x and 4x) and diploid T. incisa across their distributional ranges from the Pearl River in South China to the Heilongjiang River in Northeast China (Figure 1a). We conducted a comprehensive population genomic survey to reveal genetic relationships among wild and cultivated water caltrop, and to infer their demographic histories. In addition, we identified selection signatures that may be involved in domestication, and compared differences in gene expression profiles between cultivated and wild diploid T. natans. Specifically, our analyses focus on three sets of questions. First, what do the data reveal about the evolutionary history of polyploidy in Trapa? Second, when and how was diploid T. natans brought into cultivation? Finally, what genes bear signatures of selection, and do they provide insights into the trait shifts associated with domestication? Together, our study provides a valuable resource to facilitate comparative genomics, adaptive evolution studies and genomic‐assisted breeding for water caltrop, and improves our general understanding of plant domestication.
Results
Distribution ranges and morphological divergence in Trapa
The distribution range of Trapa in China can be divided into two major regions: (i) South‐to‐Central China, including the freshwater systems of the Pearl, Yangtze and Yellow Rivers (PYY region); and (ii) Northeast China, mainly including the Heilongjiang River (HLR region). Diploid individuals of T. natans and T. incisa are more or less clustered in the eastern portion of China, while tetraploid individuals of T. natans have a much wider distribution range. The diploid cultivars of T. natans were only found in the PYY region (Figure 1a). To evaluate morphological divergence, we compared the sizes of major plant organs, including the length and width of leaves, the length and height of fruits and the diameters of flowers and stems (Figure 1d). In all six morphological measurements, T. incisa was significantly smaller compared to both diploid and tetraploid T. natans (t‐test, P values < 3.4 × 10−12) (Figure 1d). Individuals of cultivated diploid T. natans had the largest leaves, stems and fruits, while those of wild diploid T. natans had the largest flowers on average (P values < 8.8 × 10−3). For almost all of these measurements (except fruit size), individuals of tetraploid T. natans were significantly smaller than those of diploid T. natans (P values = 1.6 × 10−9 – 0.02) (Figure 1d).
Assembly and annotation of the tetraploid T. natans genome
Flow cytometry analysis indicated that tetraploid T. natans had an estimated genome size of ~981 Mb, consistent with k‐mer‐based estimations with five different k‐mer sizes (k = 17: 926.48 Mb; k = 21: 989.93 Mb; k = 31: 1076.76 Mb; k = 51: 1138.70 Mb; k = 71: 1160.24 Mb; Figure S1). For each k, we observed a bimodal distribution of k‐mer frequency in tetraploid T. natans: one major peak represented the unique part of the genome; and the second peak pointed to a twofold higher depth of the major peak, which is expected when sequences are identical between the two subgenomes (Figure S1). Compatible with the high degree of self‐pollination in Trapa (Hummel and Kiviat, 2004; Mahto et al., 2018), we did not observe any peak corresponding to half of the diploid genome depth, which otherwise would have been expected in the presence of substantial levels of heterozygosity. We sequenced and assembled the reference genome using a hybrid approach that combined Pacific Biosciences SMRT sequencing (PacBio), 10X Genomics linked read sequencing (10X Genomics), Illumina sequencing and a Hi‐C chromatin interaction map (see details in Supplementary Methods). The contig‐level assembly was first performed on PacBio long reads (58.66 Gb), after which errors in the assembly were corrected with Illumina short reads (63.11 Gb). The error‐corrected assembly was then scaffolded using 10X Genomics linked reads (72.48 Gb) (Table S1). This yielded a genome assembly of ~1057 Mb with a contig N50 of 3.19 Mb and a scaffold N50 of 20.8 Mb (Table S2). The chromosome‐scale scaffolds were finally assembled based on Hi‐C data (58.12 Gb). A total of 944.34 Mb scaffolds (94% of the estimated genome size) were assembled into 48 chromosome‐scale pseudomolecules (Figure S2, Table S3). The strong Hi‐C signals observed between each pair of homoeologous chromosomes (see Hi‐C linkage plot in Figure S2) were very likely caused by their high levels of genome similarity (95.15%–96.84%, Table S4).
To evaluate the genome assembly quality, we remapped the Illumina short reads to the assembled genome, resulting in a mapping rate of 99.4%. The extent of comprehensive gene coverage was further evaluated using Benchmarking Universal Single‐Copy Orthologs (BUSCO) and the Core Eukaryotic Genes Mapping Approach (CEGMA). BUSCO analyses showed that 1578 (97.7%) out of 1614 universal single‐copy genes could be fully annotated in the assembled genome (Table S5). CEGMA analyses revealed that 236 out of 248 core eukaryotic genes (95.2%) were present in complete length. Taken together, these results indicate a high‐quality assembly and a high level of completeness.
Using both homology‐based searching in known repeat databases and de novo predictions, about 62.31% of the reference genome sequences were identified as repetitive elements. Among these repeats, the vast majority were transposable elements (TEs; 61.49%), while tandem repeats represented only a minor fraction (0.82%). Similar to most genomes, long terminal repeat (LTR) retroelements formed the most abundant category of TEs (56.99%), followed by DNA TEs (2.98%) (Table S6). A total of 68 946 protein‐coding genes were predicted by integrating results of three approaches, including protein‐based homology searching, de novo gene prediction and transcriptome‐based gene prediction (Table S7). Gene annotation was successfully performed on 96% of protein‐coding genes by similarity search against six functional databases (SWISSPROT, NR, KEGG, INTERPRO, PFAM and GO) (Table S8).
Subgenome discrimination and divergence time estimation
Previous cytological (Oginuma et al., 1996) and molecular genotyping analyses (allozymes: Takano and Kadono, 2005; AP2 and trnL–F: Kim et al., 2010) suggested that tetraploid T. natans is of allotetraploid hybrid origin, involving diploid T. natans and T. incisa. This hypothesis was also supported by our morphological analyses, revealing several traits of tetraploid T. natans that were intermediate between diploid T. natans and T. incisa (e.g. length/width of leaves, diameters of flowers and stems; Figure 1d). Although these inferences provided no more detailed information on the evolutionary history of tetraploid T. natans, they gave us clues on how to separate the two subgenomes of allotetraploid T. natans. By mapping whole‐genome resequencing reads of diploids (diploid T. natans and T. incisa) onto the 48 chromosomes of our reference genome, we noted that the allele‐specific mapping ratios of diploid individuals of T. natans and T. incisa to their respective (sub)genome were >60% (22.09–39.75× depth) but <20% (3.88–8.88× depth) to the other (sub)genome (Table S9). For each pair of homoeologous chromosomes, the proportion of covered bases and mean depth of coverage were extraordinarily higher in one chromosome than in the other (Figures 2a,b; see details in Table S10). However, no such significant coverage and depth differences on each homoeologous chromosome pair were observed when the resequencing reads of tetraploid T. natans individuals were mapped to our reference genome (Figure 2a,b; see details in Table S10). We, thus, divided the tetraploid T. natans genome into two distinct subgenomes, hereafter referred to as the ‘A’ subgenome (chromosomes A1–A24) and the ‘B’ subgenome (chromosomes B1–B24) (Figure 2c). Among a total of 1614 universal single‐copy genes, 1541 (95.4%) and 1558 (96.6%) were identified in the A and B subgenomes, respectively, and 1537 (95.2%) were shared between them (Table S5). FASTANI analysis showed a relatively high genome similarity between each chromosome pair, with levels of average nucleotide identity ranging from 95.15% to 96.84% (Table S4). Of the 64 926 protein‐coding genes identified, 57 674 (88.79%) showed high synteny between the A and B subgenomes (Figure 2c). We dated this subgenome divergence to c. 0.9 (0.3–1.4) million years ago (Ma), based on phylogenetic analysis of 337 orthologous genes, retrieved from these two subgenomes together with those of 12 other angiosperm species (Figure 2d). Based on orthologous gene pairs between the A and B subgenomes of tetraploid T. natans, the frequency distribution of synonymous substitutions per site (Ks) exhibited a recent peak, with Ks values having a modal value at 0.009. Given the formula μ = Ks/2T (Tajima, 1989), we, thus, obtained a mutation rate of ~5.0 × 10−9 per site per generation for Trapa.
Figure 2.
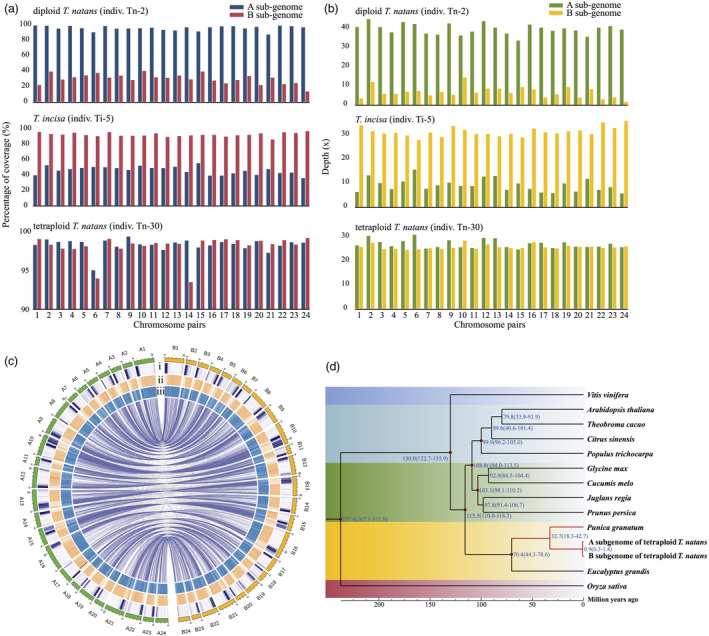
(a, b) The genome mapping coverage (a) and depth (b) of resequenced individuals (one representative each for diploid T. natans, T. incisa and tetraploid T. natans) on each chromosome pair of tetraploid T. natans reference genome (see details in Table S10). (c) Circos plot of the multidimensional topography of the 24 chromosome pairs of the tetraploid Trapa natans (4x, AABB) reference genome. Concentric circles, from outermost to innermost, show (i) gene density, (ii) repeat element density and (iii) GC content. The three metrics are calculated in 0.1 Mb sliding windows. Homoeologous gene blocks between chromosomes are connected with lines. Chromosome IDs have been re‐assigned to represent the two sets of homoeologous chromosomes (A vs. B). (d) Dated phylogeny for the two subgenomes and 12 other angiosperm species based on 337 orthologous genes. Blue numbers at each node represent the inferred divergence times (in million years ago). Red dots represent calibration times of divergence between Oryza sativa and Vitis vinifera, V. vinifera and Populus trichocarpa, Eucalyptus grandis and Prunus persica, Arabidopsis thaliana and P. persica, Theobroma cacao and P. trichocarpa and Glycine max and Cucumis melo. Calibration times were obtained from the Timetree database (http://www.timetree.org/).
Phylogenetic inference and genetic admixture analyses
The proportions of properly paired‐end reads for diploid T. natans (AA), tetraploid T. natans (AABB) and T. incisa (BB) ranged from 69.81% to 96.48%, 77.73% to 97.81% and 82.02% to 95.01% respectively (Table S9). Using the A subgenome of tetraploid T. natans as the reference, we obtained 9 133 926 high‐quality single‐nucleotide polymorphisms (SNPs) across 29 diploid T. natans individuals, 23 tetraploid T. natans individuals and five T. incisa individuals, with a strict filtering standard. It is worth emphasizing that here the A and B subgenomes of tetraploid T. natans individuals were treated separately [termed T. natans (4x, AA) and T. natans (4x, BB) respectively]. After filtering SNPs to a minimum distance of 1000 bp, and with minor allele frequency (MAF) > 0.1, 339 956 high‐quality SNPs were kept for population genomic analyses. The neighbour‐joining (NJ) tree recovered two distinct and well‐supported clusters corresponding strictly to the A and B (sub)genomes, with each clade further divided into three (I–III) and two (IV, V) genetic groups respectively (Figure 3a). Group I included all cultivated and wild T. natans (2x, AA) from the PYY region, while Group II included T. natans (2x, AA) from the HLR region. Both groups formed a monophyletic cluster that was sister to Group III, which consisted of the A subgenomes of tetraploid T. natans (4x, AA). For cultivated T. natans, two accessions (‘XBB’ and ‘NHL’) from the Yangtze River Valley occupied an early diverging (‘basal’) position within cultivars (Figure 3a). Likewise, maximum likelihood (ML) analysis of diploid T. natans from the PYY region also confirmed the basal position of cultivars from the Yangtze River Valley within cultivated T. natans (Figure S3). The B subgenomes of tetraploid T. natans (4x, BB) formed Group IV, with two T. incisa accessions (2x, BB) from the HLR region, while three other T. incisa accessions from the PYY region formed Group V. The principal component analysis (PCA) results were broadly consistent with the NJ tree, but further separated all cultivars from wild T. natans accessions (2x, AA) of the PYY region (Figure 3b).
Figure 3.
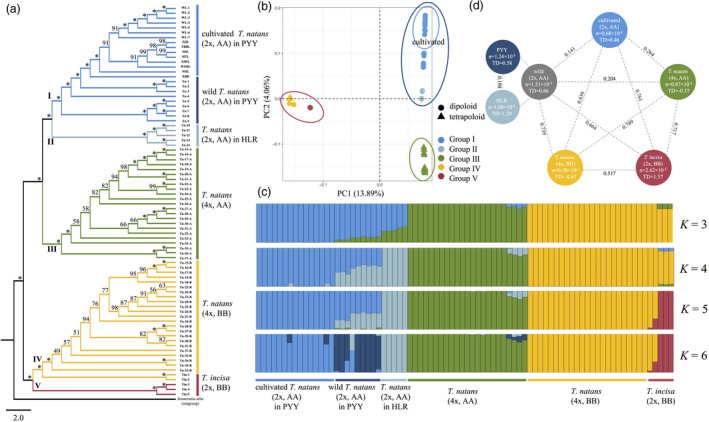
(a) Phylogenetic (NJ) tree, (b) principal component analysis (PCA) and (c) analyses of genetic structure/admixture (for K = 3–6), based on 339 956 single‐nucleotide polymorphisms (SNPs) obtained from the 57 resequenced genomes of diploid Trapa natans (2x, AA; n = 29, including 14 wild and 15 cultivated individuals), tetraploid T. natans (4x, AABB; n = 23) and diploid T. incisa (2x, BB; n = 5). Note that the A and B subgenomes of tetraploid T. natans individuals were treated separately (4x, AA vs. BB). (d) Corresponding estimates of nucleotide diversity (π) and Tajima’s D (TD; see circles) and F ST summary statistics (see values above stippled lines) for each group. PYY, Pearl–Yangtze–Yellow River region; HLR, Heilongjiang River region.
The ADMIXTURE results for K = 3 genetic clusters (Figure 3c) distinguished: (i) diploid T. natans (2x, AA) (blue); (ii) the A subgenomes of tetraploid T. natans (4x, AA) (green) and (iii) the B (sub)genomes [orange: T. incisa (2x, BB) and B subgenomes of tetraploid T. natans (4x, BB)]. Notably, the green cluster (or ‘ancestry component’) of the A subgenomes of tetraploid T. natans (4x, AA) was also present in wild diploid T. natans (2x, AA), but lacking in cultivated T. natans (2x, AA). At the optimal number of clusters, K = 5 (yielding the lowest cross‐validation error, Figure S4), the same clustering patterns of Groups I–V were recovered as in the phylogenetic and PCA analyses. Again, T. incisa (2x, BB) from the HLR region showed higher similarity to the B subgenomes of tetraploid T. natans (4x, BB) than to T. incisa from the PYY region (red). Moreover, at K = 6, an additional ancestry component (dark blue) further differentiated wild diploid T. natans (2x, AA) from the PYY region (Figure 3c).
Genetic diversity and linkage disequilibrium
Among the different genetic groups examined, T. incisa (2x, BB) had the highest genetic diversity (π = 2.62 × 10−3). In diploid T. natans (2x, AA), genetic diversity in wild (PYY + HLR) accessions was about two times higher than in cultivars (π = 1.51 × 10−3 vs. 0.68 × 10−3), and slightly higher in PYY than HLR accessions (π = 1.24 × 10−3 vs. 1.00 × 10−3). The A subgenomes of tetraploid T. natans (4x, AA) harboured only half of the genetic diversity (π = 0.87 × 10−3) of diploid T. natans, while the B subgenomes had the lowest genetic diversity (π = 0.50 × 10−3) (Figure 3d). Significantly reduced diversity as well as negative Tajima’s D values (−0.37 and −0.67) suggested that tetraploid T. natans had suffered more severe population contraction compared to diploid T. natans and T. incisa.
Genetic differentiation (F ST) between T. incisa (2x, BB) and the B subgenomes of tetraploid T. natans (4x, BB) was 0.52, while F ST varied from 0.20 to 0.26 between diploid T. natans (2x, AA; cultivated and wild accessions) and the A subgenomes of tetraploid T. natans (4x, AA) (Figure 3d). Differentiation between cultivated and wild accessions of T. natans (both 2x, AA) was moderate (0.14), and lower than that between wild PYY and HLR accessions (0.20) (Figure 3d).
Linkage disequilibrium (LD), estimated by r 2 values, decayed at a much lower rate in T. incisa (2x, BB) and the B subgenomes of tetraploid T. natans (4x, BB) than in diploid T. natans (2x, AA) from the PYY region and the A subgenomes of tetraploid T. natans (4x, AA) (Figure S5). However, diploid T. natans from the HLR region had the largest r 2 values and LD hardly decayed within 500 Kb. The cultivars of T. natans (2x, AA) had larger r 2 values, on average, than the wild accessions, and LD decayed much slower in the former group, possibly due to domestication (Figure S5). A close look at the chromosome level revealed that five chromosomes (i.e. Chr4A, Chr8A, Chr17A, Chr19A and Chr21A) were most responsible for this slow LD decay in the cultivars (Figure S6).
Inference of demographic history and domestication events
We performed pairwise sequentially Markovian coalescent (PSMC) analyses (Li and Durbin, 2011) to estimate population size changes in diploid T. natans (including wild and cultivated accessions) and T. incisa, using combined haplotypes of two individuals from the same group. Two bottlenecks were identified and dated in diploid T. natans: the first at c. 0.4 Ma (mid‐Pleistocene), which reduced N e from 10−14 × 104 to c. 4 × 104, and the second, c. 20 thousand years ago (Kya), coinciding with the Last Glacial Maximum, and which further reduced N e to c. 2 × 104. In T. incisa, the first bottleneck occurred much further back in time, c. 0.9 Ma (Figure 4a). In addition, species divergence can be inferred from the pseudodiploid PSMC profiles as a signal of population size increase in the distant past, because the distinct genomic haplotypes from separated populations accumulate mutations independently (Li and Durbin, 2011). We, thus, also used PSMC profiles to infer the timing of divergence between diploid T. natans (2x, AA) and T. incisa (2x, BB), which revealed their split during the late Early Pleistocene, c. 1.0 Ma (Figure 4a; but see below). Although the PSMC profiles ostensibly showed rapid population growth within the last c. 6000 generations, this likely represents an artifact, because PSMC has difficulties in estimating N e on very recent timescales (Li and Durbin, 2011).
Figure 4.
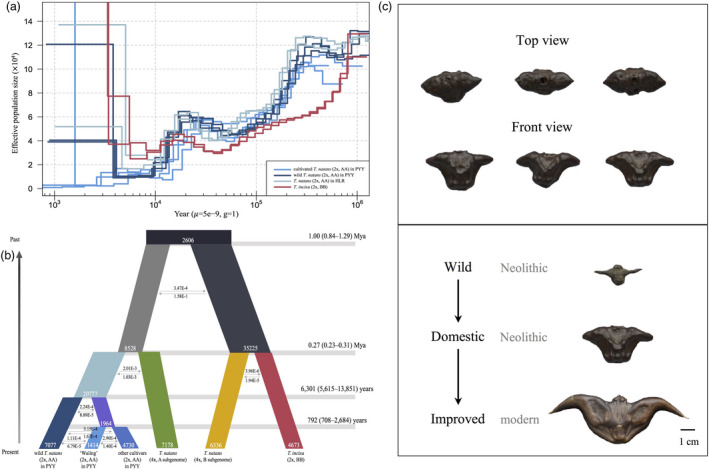
Inference of demographic and domestication history of Trapa, using PSMC and FASTSIMCOAL2. (a) Population size histories of diploid T. natans and T. incisa. (b) Schematic of the best demographic scenario modelled using FASTSIMCOAL2. The numbers on the right axis indicate the split time. Column width represents the relative effective population size. Estimates of gene flow among species, cultivars and subgenomes are given as the migration fraction per generation. (c) Fruits of domestic water caltrop excavated from the Tianluoshan site (upper figure), and a simple schematic illustrating the process of water caltrop domestication (lower figure).
According to the best‐supported demographic (FASTSIMCOAL2) model (AIC = 0, ω = 1; Table S11), diploid T. natans and T. incisa diverged c. 1 Ma (95% confidence interval, CI: 0.84–1.29 Ma) (Figure 4b), hence well in accord with the PSMC estimate (c. 1 Ma). The simultaneous divergence of the A and B subgenomes of tetraploid T. natans from their ancestral genome (i.e. its allopolyploid origin from diploid T. natans and T. incisa) was dated to the mid‐to‐late Pleistocene, c. 0.27 (0.23–0.31) Ma (Figure 4b; Table S12). The domestication of diploid T. natans was dated to the Neolithic period, c. 6.30 (5.62–13.85) Kya, while further improvement of the cultivar ‘Wuling’ took place in historical times, c. 792 (708–2684) years ago; both events were associated with population contractions. The current N e of wild T. natans was estimated to be larger (~7000) than those of cultivars (~1400 for ‘Wuling’ and ~4700 for other cultivars; Figure 4b; Table S12). For this best‐supported model, a goodness‐of‐fit test showed that the estimation of this model fit the observed data well (Figure S7).
Identification of genes under selection during domestication and their functions
To detect genome‐wide selective sweeps in cultivated relative to wild accessions of T. natans (2x, AA) from the PYY region, we sought to identify regions with (i) allele frequency differentiation between cultivated and wild T. natans, using the cross‐population composite likelihood ratio (XP‐CLR) method (Chen et al., 2010); (ii) low levels of polymorphism in cultivated relative to wild T. natans and (iii) large negative Tajima’s D values in cultivated T. natans. Using a cut‐off of normalized XP‐CLR scores ≥3.29 (i.e. P ≤ 0.001) combined with π wild/π cult ≥ 2 and Tajima’s D ≤ −1, a total of 126 genomic regions were identified to be under selective sweeps during domestication (Table S13). Sixty‐six regions (52%) were located in five chromosomes (i.e. Chr4A, Chr8A, Chr17A, Chr19A and Chr21A), where an above‐average LD was found (Figure S6), and another 15 and 10 regions (20%) were located in Chr10A and Chr24A respectively. Within those 126 regions, we identified 205 protein‐coding genes, 44 of which were located in Chr4, 23 in Chr10A, 22 in Chr19A, 17 in Chr21A and 14 in Chr17A (Table S13).
Of all potentially selected genes during domestication, 111 could be assigned to three biological processes/physiological pathways (Figure S8; Table S14): (i) growth and development of seed (endosperm, embryo, cotyledon), root (root hair, lateral root and adventitious root), shoot apex, flower and leaf; (ii) biosynthetic, photosynthetic and metabolic processes, including starch, pectin, sucrose biosynthesis and turnover and (iii) response to abiotic stress tolerance (drought, salinity, temperature). Many of those genes were related to phytohormones, such as auxin, abscisic acid (ABA) and brassinosteroids (Table S14).
Multiple candidate genes were identified that could be essential for the development of morphological traits in cultivated T. natans (2x, AA). For example, at least 19 genes (e.g. SAC7, RPD1, OCT1) were identified potentially related to root development (apex; lateral or adventitious root) (Table S15). Four genes (i.e. PID, PIN1, SUS3 and CRA1) could be related to asymmetric cotyledon development and the transformation of sucrose to starch storage in the cotyledons (Table S14). Five genes involved in the metabolism of pectin (a major cell wall component) were also found to be under selection (i.e. RHM1, PME7, G9, At1g02816 and At4g02250), and four of them (except RHM1) were located in a particular region (5.79–5.86 Mb) of Chr19A (Figures 5a,b; Table S14).
Figure 5.
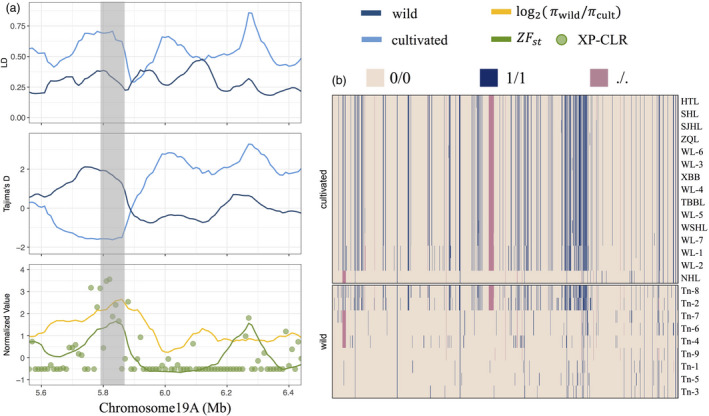
Genes under selective sweeps during domestication of diploid Trapa natans (2x, AA) in Chr19A. (a) Differences in linkage disequilibrium (LD), Tajima’s D, XP‐CLR values, Z‐score transformed F ST estimates and π ratios between cultivated and wild T. natans (from top to bottom panels). (b) Composite haplotypes in all cultivated and wild T. natans from the PYY region.
Gene expression profile differences between cultivated and wild T. natans in the PYY region
To comprehensively understand the genetic basis underlying domestication‐associated phenotype changes in Trapa (especially the differences in fruit size), we generated RNA sequencing (RNA‐seq) data to compare gene expression differentiation between cultivated and wild T. natans (2x, AA) from the PYY region. One wild accession of T. natans and two most common cultivars, ‘Wuling’ and ‘Nanhuling’, were selected as typical materials. As pre‐anthesis factors and early fruit growth are important in determining final fruit size and shape (Cruz‐Castillo et al., 2002; Yang et al., 2021), we collected five samples of flower/fruit tissues that represented five developmental stages (F0: flower bud; F1: fertilized flower; F2: fruit before sepal abscission; F3: fruit after sepal abscission and F4: juvenile fruit), as well as one sample of leaf tissue (L) from each accession (Figure 6a; see details in Table S16). Based on cultivar–wild and inter‐cultivar comparisons, we identified 25 078 expressed genes, on average, for each comparison, using a threshold of ≥10 read counts per gene. When averaged across the six tissues examined, the differentially expressed genes (DEGs) in each of the two cultivar–wild comparisons (mean = 3255 genes) were four times higher than in the inter‐cultivar comparison (mean = 740 genes, Figure 6b). In the cultivar–wild comparisons, and across all six tissues, the largest number of DEGs (5000–7900) were identified at flower/fruit developmental stages F1 and F3 (Figure 6b). In all subsequent analyses, we focused only on those DEGs that were shared by the two cultivar–wild comparisons (i.e. 8621 and 10 257 DEGs at F1 and F3, respectively, and 962–4433 DEGs at other stages; Figure S9). Based on this dataset, fuzzy c‐means clustering revealed four major expression reaction norms (‘increasing’, ‘decreasing’, ‘Z‐shape’ and ‘S‐shape’) from stage F0 to F4 in both cultivars and wild accessions (Figure 6c). In wild accessions, reaction norm curves tended to have a turning point of expression at stage F1, while in the cultivars the turning point usually appeared at later (F2 or F3) stages (Figure 6c). Based on Gene Ontology (GO) enrichment analysis, and taking all six tissue types into account, these shared DEGs were mainly involved in (i) development (e.g. leaf, root, shoot, flower, fruit) and cell wall organization (459–287 DEGs); (ii) biosynthesis and metabolism (719–5287 DEGs) and (iii) stress/immune responses (327–2062 DEGs; Figure S10; Table S17).
Figure 6.
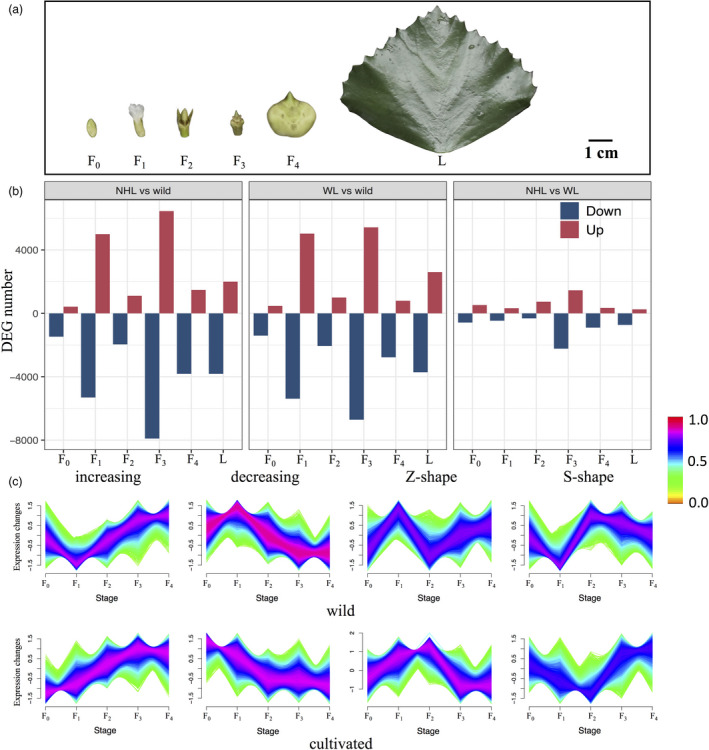
(a) Five samples of flower/fruit tissues representing five developmental stages (F0: flower bud; F1: fertilized flower; F2: fruit before sepal abscission; F3: fruit after sepal abscission and F4: juvenile fruit) and one sample of leaf tissue (L) used for RNA sequencing (exemplified by the ‘Nanhuling’ (NHL) cultivar; see details in Table S16). (b) Numbers of differentially expressed (up‐ or down‐regulated) genes (DEGs) in three pairwise comparisons between cultivars (NHL, ‘Nanhuling’; WL, ‘Wuling’) and wild accessions of diploid Trapa natans (left: NHL vs. wild; middle: WL vs. wild; right: NHL vs. WL) in six tissue samples viz. developmental stages (flower/fruit stages: F0–F4; leaf: L; see text for details). (c) Changes in gene expression in terms of four reaction norms (‘increasing’, ‘decreasing’, ‘Z‐shape’ and ‘S‐shape’) for wild and cultivated (NHL + WL) accessions, respectively, at the five flower/fruit developmental stages (F0–F4). The colour bar represents the scale of membership scores of a given gene in the corresponding cluster (see Materials and Methods for details regarding the calculations). Genes whose expression patterns are very similar to a given centroid will be assigned a high membership in that cluster (maximum 1.0, red), whereas genes that bear little similarity to the centroid will have a low membership.
Expression patterns of genes under selection
Of 205 genes found to be under selection, 137 exhibited significant expression differentiations in at least one tissue between cultivars and wild T. natans from the PYY region (2x, AA) (Table S18). Fuzzy c‐means clustering identified a total of 51 selected genes as cluster representatives in either cultivars or wild accessions of T. natans (Figure 6c). In the cultivars, nine and 11 genes had S‐ and Z‐shaped reaction norm curves, respectively, all with expression turning points at stage F2. By contrast, in wild T. natans, ten and nine genes were found to have S‐shaped and decreasing expression patterns, respectively, all with expression turning points at stage F1. Of the 51 genes, six were located in Chr10A (at 22.16–22.21 Mb), eight in Chr19A (5.24–5.86 Mb) and 11 in Chr4A (11.6–11.82 Mb; Table S19). Overall, these developmental‐dependent expression differentiations indicate that those genes, found to be under selective sweeps during domestication, could be involved in specific physiological processes related to nutrient supply or fruit development, potentially contributing to increased yield performance. For example, the gene encoding sucrose synthase 3 (SUS3), whose activity is associated with starch accumulation in seeds (Table S14), had an expression level maximum (Z‐shape) at fruit developmental stage F2 in cultivars, while in wild accessions the expression maximum appeared earlier at flower stage F1 (Figure 6c; Table S19). Another candidate gene, XTH8, involved in fruit ripening (Table S14) had a higher expression level (S‐shape) at stages F3 and F4 in cultivars than in wild accessions (Figure 6c; Table S19).
Discussion
The early presence of Trapa in China is attested by the numerous fruit fossils reported in Miocene deposits collected in eastern China (Xiao et al., 2020). Consequently, Trapa clearly experienced Quaternary climatic fluctuations in East Asian territories and probably underwent dramatic range contractions and/or expansions across East Asia during the last 2 million years. It is worth noting that our estimated divergence times using FASTSIMCOAL2 should be interpreted with caution, because we did not account for population size (N e) changes and taxon‐specific mutation rates to simplify the FASTSIMCOAL2 models and save computational time, which could have led to biases in our divergence time estimates (DeChaine and Martin, 2006; Momigliano et al., 2021). However, these divergence time estimates correspond relatively well to major biogeographic events in East Asia. FASTSIMCOAL2 simulation dated the divergence between the parental lineages of diploid T. natans and T. incisa to c. 1 Ma (95% CI: 0.84–1.29 Ma) (Figure 4b). Our estimated divergence time, thus, coincides precisely with the Pre‐Pastonian (glacial) stage of the Early Pleistocene (0.8–1.3 Ma) (Hepburn and Radloff, 2011). Accordingly, this time estimate is also consistent with expectations of allopatric species formation of temperate plants in East Asia, resulting from Late Quaternary refugial isolation (see also Qiu et al., 2009, 2011). By contrast, tetraploid T. natans formed very recently, between 0.23 and 0.31 Ma (Figure 4b). Both our PSMC modelling (Figure 4a) and FASTSIMCOAL2 analysis (Figure 4b) indicated an increase in effective population size of its parental lineages during the early‐to‐mid Pleistocene. Changes in the connectivity of the presently discontinuous aquatic habitat systems induced by the Quaternary climatic fluctuations (Volkova et al., 2010; Yasuda et al., 2005) likely promoted population expansions and secondary contacts between diploid T. natans and T. incisa, thus leading to the formation of tetraploid T. natans via hybridization and polyploidization. The capacity of tetraploid T. natans to self‐fertilize and propagate clonally (Mikulyuk and Nault, 2009) might have increased its ability to become established, and thus despite potential reproductive disadvantages in the early stages of polyploid hybrid formation (Herben et al., 2017). This, together with a relatively uniform aquatic environment, favouring dominant, single‐purpose genotypes (Santamaría, 2002), could have allowed the establishment and subsequent range expansion of tetraploid T. natans over the last 230 000–310 000 years. Allopolyploid origins of similar recent, i.e., (Late) Quaternary time scales are likely to hold for several other aquatic taxa (e.g. Isoetes spp.: Liu et al., 2004; Dai et al., 2020; Nymphaea candida: Volkova et al., 2010; Myriophyllum spp.: Lü et al., 2017).
The cultivars of T. natans in China, regardless of their fruit morphological differences (Figure 1b), are all derived from diploid T. natans (2x, AA) inhabiting the freshwater systems of the Pearl–Yangtze–Yellow River (PYY) region. In particular, the Yangtze River Valley is likely the earliest centre of water caltrop domestication (Figures 3a, Figure S3). In fact, our genomic results are in line with recent archaeological findings (Guo et al., 2017), suggesting that the initiation of water caltrop domestication in China began c. 6300 (5600–13900) yr BP (Figure 4b; Table S12). The fruit remains of water caltrop discovered at the Tianluoshan site (Lower Yangtze Region; 6300–7000 yr BP, Guo et al., 2017) are smaller in size than the fruits of modern cultivars, but remarkably larger than those of extant wild accessions (Figure 4c). This indicates that water caltrop was already under intensive domestication and cultivation by approximately 7000 yr BP, and underwent a second round of artificial selection (viz. ‘improvement’) for increased fruit size (Guo et al., 2017). Our results further indicate that the ‘Wuling’ cultivar, one of the most commonly cultivated modern inbred lines of diploid T. natans in the Yangtze River Valley, appeared c. 800 (700–2900) yr BP (Figure 4b; Table S12), suggesting that ‘a second improvement practice’ most likely occurred during the Tang (618–907 AD) and Song (916–1279 AD) Dynasties. This finding also accords with an abundance of new cultivar names and fruit morphological descriptions found in local historical documents from these periods (Zhou, 2012). There is archaeological and genetic evidence suggesting that rice was first domesticated along the Yangtze River Valley (Gross and Zhao, 2014). Together with our present results, this provides strong evidence that the Yangtze River Valley is a major cradle of Chinese agriculture.
The cultivars of T. natans have a more vigorous root system with significantly denser and longer lateral (adventitious) roots and root hairs (Figure 1c). The submerged adventitious roots of aquatic plants play a crucial role in nutrient uptake and photosynthesis (Rich et al., 2012). The roots of water caltrop can absorb dissolved inorganic nitrogen from water and sediment, which has been found to be positively correlated with leaf and fruit biomass (Tsuchiya and Iwakuma, 1993; Yan and Xu, 1992). By searching for signatures of selective sweeps, we identified genes and genomic regions enriched for functional processes of root development and photosynthesis (Tables S14 and S15). Gene Ontology analysis of DEGs confirmed signals of enrichment not only for these two processes, but also for those responding to phytohormones, such as auxin and abscisic acid, which have been shown to regulate adventitious root formation in many studies (Guan et al., 2019; Harris, 2015; Pacurar et al., 2014).
Vigorous adventitious roots can also help to sense and respond to shifting environments in water systems where currents can induce abiotic stress, such as salinity and temperature fluctuations (Bellini et al., 2014; Steffens and Rasmussen, 2016). Indeed, seed germination, growth and development of water caltrop are sensitive to fluctuations in pH, NaCl concentration and temperature fluctuations (Kurihara and Ikusima, 1991; Vuorela and Aalto, 1982). Hence, we also identified abiotic stress‐related genes responding to, e.g., chemical and cold stimulus (Table S14), which were enriched in targets of domestication and expression differentiation between cultivars and wild accessions. In conclusion, as found in many cereal crops, such as maize and rice (Bellini et al., 2014), the adventitious roots of water caltrop were a key target of domestication, given their crucial roles in, e.g., nutrient uptake, environmental stress response and underwater photosynthesis. Our study also revealed many candidate genes involved in seed germination, flowering, as well as starch storage, which thus provide a valuable resource to facilitate genomic‐assisted breeding for water caltrop.
Material and methods
Sampling information and morphological trait analyses
A total of 52 individuals of T. natans (both 2n = 2x = 48 and 2n = 4x = 96) and five individuals of diploid T. incisa (2n = 2x = 48) were collected from 48 sites located in the Pearl–Yangtze–Yellow (PYY) region of South‐to‐Central China and the Heilongjiang River (HLR) region of Northeast China (Figure 1a; Table S9). The 52 samples of T. natans represent 15 accessions of the nine most common cultivars (only 2x, AA) and 37 wild accessions, including 14 diploid (2x, AA) and 23 tetraploid (4x, AABB) individuals. All samples were used for genome resequencing. Here, we identified the ploidy level and genome type for each individual by flow cytometry, with Oryza sativa ssp. japonica as an internal standard, and mapping whole‐genome resequencing reads onto the 48 chromosomes of our reference genome of tetraploid T. natans. To evaluate morphological divergence between wild (2x, 4x) and cultivated (2x) accessions of T. natans and T. incisa (2x), we measured a total of 100 individuals for six morphological traits (Table S20), including the length and width of leaves, the length and height of fruits and the diameters of flowers and stems. Each of three stems, adult leaves, fruits and blooming flowers were measured per individual. Data for all the traits were statistically analysed by using t‐test, implemented in the R software package GGPUBR (https://CRAN.R‐project.org/package=ggpubr).
Genome sequencing and de novo assembly
An individual plant of tetraploid T. natans growing in Hangzhou Botanical Garden, Zhejiang, China (120.12°E, 30.25°N) was used for reference genome sequencing. High‐quality genomic DNA was extracted from fresh leaves using DNAsecure Plant Kit (Tiangen Biotech, Beijing, China). The voucher specimen was identified by the authors and is deposited at the herbarium of Zhejiang University (LCZ180910). We produced genome data using a hybrid approach that combined PacBio, 10X Genomics, Illumina sequencing and a Hi‐C chromatin interaction map (see details in Supplementary Methods). The genome size of this individual was estimated using both flow cytometry and k‐mer frequency distribution analysis. To ensure that the k length had no effect on the estimations, k‐mer count histograms for five k‐mers (i.e. 17, 21, 31, 51 and 71) were generated using Jellyfish v.2.2.4 (Marcais and Kingsford, 2011). Self‐correction and contig‐level assembly were performed on full PacBio long reads using FALCON v.0.3.0 (https://github.com/PacificBiosciences/FALCON/). The draft assembly was further polished using PacBio reads with QUIVER algorithm (Chin et al., 2013) and Illumina paired‐end reads with PILON v.1.24 (Walker et al., 2014). The error‐corrected assembly was then scaffolded by the FRAGSCAFF software (Adey et al., 2014) using 10X Genomics linked reads. To further construct a chromosome‐level assembly of the genome, clean reads from Hi‐C were mapped to scaffolds using BWA v.0.7.12 (Li and Durbin, 2009) and uniquely mapped reads were retained. After filtering invalid read pairs, sorting and quality assessment using HIC‐PRO v.2.8.1 (Servant et al., 2015), uniquely mapped valid reads were used to cluster, order and orient scaffolds onto chromosomes by LACHESIS software (Burton et al., 2013). To evaluate the completeness of the genome assembly, three methods were applied, including mapping the Illumina reads back to the reference genome, BUSCO gene mapping (Simão et al., 2015) and CEGMA (Parra et al., 2007) analyses.
Identification of repetitive genomic elements
Transposable elements in the tetraploid T. natans genome were detected with both homology‐based searching in known repeat database and de novo predictions. For homology‐based detection, REPEATMASKER v.3.3.0 (Tarailo‐Graovac and Chen, 2009) was used to identify TEs against the REPBASE database (Bao et al., 2015) at the DNA level, and REPEATPROTEINMASK v.3.2.2, implemented in REPEATMASKER, was used to detect TEs at the protein level. De novo TEs were detected by REPEATMASKER based on a de novo repeat library constructed by REPEATMODELER v.1.0.4 (http://www.repeatmasker.org/RepeatModeler.html), LTR_FINDER v.1.0.5 (Xu and Wang, 2007), and REPEATSCOUT v.1.0.5 (Price et al., 2005). TANDEM REPEATS FINDER v.4.0.9 (Benson, 1999) was used to identify tandem repeat sequences in the genome.
Annotation of protein‐coding genes
We combined protein homology‐based, de novo and transcriptome‐based prediction methods to annotate the genome assembly of tetraploid T. natans. For the protein homology‐based prediction, protein sequences of Arabidopsis lyrata, A. thaliana, Boechera stricta, Corchorus capsularis and Punica granatum were retrieved from Ensembl Genome Browser (http://www.ensembl.org/index.html) and NCBI (http://www.ncbi.nlm.nih.gov), and then aligned to our genome assembly using TBLASTN (E‐value ≤ 1e‐5). The program SOLAR v.0.0.19 (Yu et al., 2006) was used to concatenate BLAST hits that correspond to reference proteins. Afterwards, GENEWISE v.2.2.0 (Birney et al., 2004) was used to predict gene structures and define gene models contained in each protein region. For the de novo prediction, five prediction tools, including AUGUSTUS v.2.5.5 (Stanke et al., 2006), GENSCAN v.1.0 (Burge and Karlin, 1997), GLIMMERHMM v.3.0.1 (Majoros et al., 2004), GENEID v.1.4.4 (Blanco et al., 2007) and SNAP (Korf, 2004), were used to predict coding regions in the repeat‐masked genome. For the transcriptome‐based prediction, RNA‐seq short reads were firstly mapped to the genome assembly using TOPHAT v.2.0.8 (Trapnell et al., 2009). The alignment results were then used as input for CUFFLINKS v.2.2.1 (Trapnell et al., 2010) with default parameters for genome‐based transcript assembly. Finally, the assembled transcripts were aligned to the genome assembly and filtered with the Program to Assemble Spliced Alignment (PASA) v.2.0.4 (Haas, 2003) to detect likely protein‐coding regions. All gene models predicted from the above approaches were integrated into a comprehensive non‐redundant set of gene structures, using EVIDENCEMODELER v.1.1.1 (Haas et al., 2008). The obtained non‐redundant set was updated by PASA to generate the information about untranslated regions and alternative splicing, and to obtain final gene models.
To achieve the functional annotation, BLASTP was used to compare the protein sequences against SwissProt, Non‐redundant (NR) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases with an E‐value threshold of 1e−5. In addition, INTERPROSCAN v.4.8 (Quevillon et al., 2005) was used to annotate the functional domains of genes by searching against the InterPro and Pfam databases. Gene Ontology IDs for each gene were also determined by the BLAST2GO pipeline (Conesa et al., 2005).
Synteny analysis and divergence time estimation
We successfully divided the chromosomes of allotetraploid T. natans (4x, AABB) into the A and B subgenomes based on the distinct mapping ratio, coverage and depth of short reads from its diploid progenitors T. natans (2x, AA) and T. incisa (2x, BB) (see details below). Average nucleotide identity between each homoeologous chromosome pair was calculated using FASTANI v.1.1 (Jain et al., 2018). The synteny analysis of the A and B subgenomes was assessed and plotted using MCSCANX (Wang et al., 2012) and CIRCOS v.0.69 (Krzywinski et al., 2009). We also used the script “add_ka_and_ks_to_collinearity.pl” in MCSCANX to calculate the Ks values of the collinear orthologous gene pairs between the A and B subgenomes. For phylogenetic analysis, orthologous genes were retrieved from the two subgenomes and 12 other angiosperm species. The single‐copy orthologs were aligned using MUSCLE v.3.8.3 (Edgar, 2004) and then concatenated into a super‐gene alignment matrix. The alignment was trimmed with GBLOCKS v.0.91b (Castresana, 2000). Maximum likelihood analyses were conducted using RAXML‐HPC v.8.2.8 (Stamatakis, 2014) with 1000 bootstrap replicates. Divergence times were estimated by the program MCMCTREE in PAML v.4.7a (Yang, 2007), using six calibration points (see details in Supplementary Methods).
Resequencing, short read mapping and SNP calling
To distinguish the A and B subgenomes in our reference genome of allotetraploid T. natans (4x, AABB) and to identify SNPs from all accessions, we applied a three‐step allele‐specific mapping protocol, using resequencing short reads of both diploid progenitors, T. natans (2x, AA) and T. incisa (2x, BB). First, we prepared paired‐end genome resequencing libraries for each individual with an insert size of 350 bp and sequenced those on the Illumina HiSeq X Ten platform (Illumina, Inc., San Diego, CA, United States) to generate raw sequences with a read length of 150 bp. The clean reads of each diploid individual were then mapped onto all 48 chromosomes of the tetraploid T. natans reference genome, using default settings of BWA‐MEM v.0.7.17 (Li, 2013). Mapping statistics were computed from the BAM files using the FLAGSTAT function of SAMTOOLS v.1.9 (Li et al., 2009) to determine the ratio of mapped reads for each individual. The proportion of covered bases and mean depth of coverage for each chromosome were also calculated using BAMCOV (https://github.com/fbreitwieser/bamcov).
Second, paired‐end short reads of tetraploid individuals of T. natans (4x, AABB) were prepared and sequenced, and the reads were aligned to the tetraploid reference genome using the approach described above. For each individual, reads that were aligned specifically to the A or B subgenomes were separated into two groups. For diploid T. natans and T. incisa, only reads aligned to, respectively, the A and B subgenomes were retained.
Reads retained after allele‐specific mapping filtering were again aligned to the A subgenome using BWA‐MEM v.0.7.17 (Li, 2013). Resulting Sequence Alignment/Map (SAM) format files were sorted, converted to Binary Alignment/Map (BAM) format and indexed using SAMTOOLS v.1.9 (Li et al., 2009). Potential PCR duplicates were marked using PICARD v.2.1.9 (http://picard.sourceforge.net) and were ignored during genotyping. Genotypes for each individual were called using HAPLOTYPECALLER from the Genome Analysis Toolkit (GATK) v.4.1.2 (Depristo et al., 2011), and then combined by GENOTYPEGVCFS (GATK‐GVCF workflow). This approach produced a single variant calling file (VCF) containing all called (polymorphic and monomorphic) sites.
We also generated another VCF file that contained only SNPs and carried out stringent filtering based on the following parameters: ‘QD < 5.0 || MQ < 50.0 || FS > 10.0 || SOR > 2 || MQRankSum < −2 || MQRankSum > 2 || ReadPosRankSum < −3 || ReadPosRankSum > 3 || InbreedingCoeff > −0.5 || ExcessHet > 3’. These filtering thresholds were defined following the GATK best practice pipeline (Van der Auwera et al., 2013) with some adjustment according to the obtained distributions of the GATK annotation scores.
Phylogenetic inference and population structure analyses
For phylogenetic inference and population structure analyses, SNPs were further thinned using a distance filter of > 1000 bp between SNPs, and a rare SNP filter of MAF > 0.1. A NJ tree was constructed to investigate genetic relationships among the A vs. B subgenomes of tetraploid T. natans individuals as well as diploid individuals of T. natans and T. incisa, using TREEBEST v.1.9.2 (Vilella et al., 2009), with 1000 bootstrap replicates. Based on previous phylogenetic analyses (Berger et al., 2016), the tree was rooted with Sonneratia alba (Lythraceae) (the sequence data were downloaded from NCBI Short Read Archive under accession number: SRS1179875). In addition, a PCA was performed to assess the relatedness and clustering of subgenomes and diploid individuals using VCFTOOLS v.0.1.16 (Danecek et al., 2011) and PLINK v.1.90 (Purcell et al., 2007). We also used ADMIXTURE v.1.3.0 (Alexander et al., 2009) to infer the number of ancestral genetic clusters (K) and their levels of admixture; the optimal K (set to vary from 1 to 10) was determined with the lowest value of cross‐validation (CV) error. To further infer the most likely geographical origin of cultivars, a phylogenetic analysis was conducted independently on diploid T. natans from the PYY region using two accessions of diploid wild T. natans from the HLR region (Tn‐10, Tn‐13; Table S9) as outgroups. The ML tree was generated in RAXML‐HPC v.8.2.8 (Stamatakis, 2014) under the GTR+G+I model, with 1000 bootstrap replicates.
Population genetic analyses and linkage disequilibrium
For each group inferred from the population structure analyses [i.e. wild diploid T. natans (including wild‐PYY and wild‐HLR populations) (2x, AA); cultivated T. natans (2x, AA); the A (4x, AA) and B subgenomes (4x, BB) of tetraploid T. natans; and T. incisa (2x, BB)], we calculated Tajima’s D and population fixation statistics (F ST) with 100‐kb sliding windows and a step size of 10 kb, using VCFTOOLS v.0.1.16 (Danecek et al., 2011). To avoid systematic bias generated by missing data, especially for the B subgenomes of tetraploid T. natans and T. incisa (the proportion of missing data for these two groups was about 28.05% and 34.19%, respectively, compared to 12.64%, 13.06%, 5.87% for wild diploid T. natans, cultivated T. natans, and the A subgenomes of tetraploid T. natans respectively), genome‐wide nucleotide diversity (π) was calculated with PIXY v.1.2.4 (Korunes and Samuk, 2021) using the VCF file that included invariant sites. Genome‐wide decay of LD was determined by the software POPLDDECAY (Zhang et al., 2019).
Inference of demographic history
Demographic histories were reconstructed for diploid T. natans (including wild and cultivated accessions) and T. incisa, using PSMC analysis (Li and Durbin, 2011). This method estimates changes in effective population size (N e) over time by measuring the rate of decrease in heterozygosity across regions of the genome. Due to the predominance of self‐pollination in Trapa (Hummel and Kiviat, 2004; Mahto et al., 2018), the genome sequence for each diploid individual is effectively homozygous, providing a single haploid genome. We, thus, randomly paired haplotypes from two individuals within the same genetic group by a customized Perl script, to construct pseudodiploid genomes for PSMC runs, as defined in Kryvokhyzha et al. (2019). The analysis was performed with the following parameters: ‐N30 ‐t15 ‐r5 ‐p "4+25*2+4+6". The variance of the inferred N e trajectories was assessed using three different pseudodiploids for each genetic group. The mutation rate of 5.0 × 10−9 per site per generation (See Results) and a generation time of 1 year were used for Trapa species.
To infer the evolutionary and domestication history of water caltrop, we estimated various demographic parameters [i.e. divergence time (T), N e and gene flow (m)] from the SNP data of species, cultivars and subgenomes [i.e. wild‐PYY T. natans (2x, AA); cultivar ‘Wuling’ (2x, AA); other cultivars (2x, AA); the A (4x, AA) and B (4x, BB) subgenomes of tetraploid T. natans; and T. incisa (2x, BB)], using two‐dimensional (2D) joint site frequency spectra (SFS; python script available at https://github.com/isaacovercast/easySFS), as implemented in FASTSIMCOAL2 (Excoffier et al., 2013). This procedure uses coalescent simulations to calculate the likelihoods of observed allele frequency spectra (see Nielsen, 2000) under user‐specified demographic models. Based on our phylogenetic and population genetic structure results (Figure 4a–c), our demographic models aimed at investigating the following events and divergence times within the genus: (i) the split of diploid T. natans and T. incisa from their ancestral population at T AB; (ii) the origin of allotetraploid T. natans at T tetraploid (i.e. onset of simultaneous divergence of the A and B subgenomes from their respective parental gnomes); (iii) the origin of cultivated T. natans from wild T. natans in the PYY region at T domestication and (iv) the ‘improvement’ of the ‘Wuling’ cultivar at T improve. To further refine this demographic scenario with more complex scenarios of asymmetric gene flow, four alternative models with different sets of migration directions were investigated (Figure S11). One hundred replicates were performed for each model with 100 000 coalescent simulations and 40 cycles of the likelihood maximization algorithm for each replicate. The best model was determined based on Akaike’s information criterion (AIC) and Akaike’s weight of evidence (ω). Point estimates of demographic parameters for the best‐supported model were selected based on the highest maximum composite likelihood of the 100 replicate runs. Finally, we generated 100 parametric bootstrap replicates with starting values initialized from the point estimates of the best‐supported model to obtain confidence intervals for all parameters.
Genomic scan of selective sweeps associated with domestication
To identify genome‐wide selective sweeps in cultivated relative to wild accessions of T. natans (2x, AA) from the PYY region, we used three approaches, i.e., XP‐CLR test, π ratios (π wild/π cult) and Tajima’s D values. The XP‐CLR test was performed in the program XP‐CLR v.1.0 (Chen et al., 2010) with the following parameters: window size of 100 kb, step size of 10 kb, maximum number of SNPs within a window 200 and correlation level for two SNPs weighted with a cut‐off of 0.95. The π ratios and Tajima’s D values were calculated by sliding window analysis with a window size of 100 kb and a step size of 10 kb. In the cultivated accessions, genomic sweep regions were identified based on normalized XP‐CLR scores ≥ 3.29 (i.e. P ≤ 0.001) combined with π wild/π cult ≥ 2 and Tajima’s D ≤ −1. Genes within these regions were subjected to GO enrichment analysis using Arabidopsis thaliana orthologues at AGRIGO v.2.0 (Tian et al., 2017) with default parameters.
RNA‐seq data generation and gene expression analysis
We generated RNA sequencing (RNA‐seq) data to compare gene expression differentiation between cultivated and wild T. natans from the PYY region. To this aim we collected five samples of flower/fruit tissues and one sample of leaf tissue (L) from wild accessions of diploid T. natans and two most common cultivars, ‘Wuling’ and ‘Nanhuling’. The flower/fruit tissues represented five developmental stages (F0: flower bud; F1: fertilized flower; F2: fruit before sepal abscission; F3: fruit after sepal abscission and F4: juvenile fruit; see Figure 6a; Table S16). Three biological replicates were performed for each tissue sample and developmental stage. RNA‐seq libraries were prepared using the TruSeq RNA Library Kit (Illumina, NEB) and sequenced on a NovaSeq 6000 platform with paired‐end reads 150 bp in length (see details in Table S16). Clean reads from each sample were mapped to the A subgenome of the tetraploid T. natans reference genome using HISAT2 v.2.0.4 (Kim et al., 2015). Only correctly paired reads with a unique mapping position were retained for read counting of the annotated genes using FEATURECOUNTS v.1.4.6 (Liao et al., 2014).
For library size normalization, we applied the trimmed mean of M values (TMM) method (Robinson and Oshlack, 2010), as implemented in the R package ‘edgeR’ (Anders et al., 2013). Read counts were used as expression level for each gene, with mean and variance based on the negative binomial distribution. A generalized linear model was used to evaluate the effect of domestication (‘Wuling’ vs. wild and ‘Nanhuling’ vs. wild) or the difference between cultivars (‘Wuling’ vs. ‘Nanhuling’). Genes with false discovery rate adjusted P values < 0.05 were considered as significant DEGs. Up‐ or down‐regulated DEGs were defined depending on whether the log2 fold change (log2FC) was ≥1 or ≤−1 respectively. DEGs shared between each cultivar–wild comparison (i.e. ‘Wuling’ vs. wild and ‘Nanhuling’ vs. wild) were selected for functional GO enrichment analysis. The latter was performed for both up‐ and down‐regulated DEGs using AGRIGO v.2.0 (Tian et al., 2017) with default parameters.
To better understand changes in gene expression across the five developmental stages (F0 to F4), expression reaction norms of all DEGs were summarized by fuzzy c‐means clustering, using the R package ‘MFUZZ’ (Kumar and Futschik, 2007). The sum of squared errors (SSEs), defined as the sum of squared distances between each member of a cluster and its cluster centroid, was calculated for each number of clusters (from 1 to 20). The optimal number of clusters was determined when the decrease of SSEs was lower than 10% of the total SSEs and the SSEs would not significantly decrease with each new addition of a cluster (Figure S12). Four clusters of expression reaction norm were identified in terms of ‘increasing’, ‘decreasing’, ‘S‐shape’ and ‘Z‐shape’ respectively. Membership scores were calculated for all genes within each cluster and genes with top 30% membership scores were selected as cluster representatives.
Conflict of interest
All authors confirm that they have no conflict of interest.
Author contributions
YQ conceived the project. YQ and JC designed the experiments and coordinated research activities. RL, YC, XZ, ZZ, YY, LW and ZH collected samples and phenotypic data. RL, YC and XZ performed the research. RL, YC, XZ, YF, ZL and JC analysed data. YG and GS contributed to the interpretation of results. RL, JC and YQ wrote the manuscript. HPC and KMO revised the manuscript. All authors read and approved the manuscript. RL, YC and XZ contributed equally to this work.
Supporting information
Figure S1 K‐mer‐based genome size estimates.
Figure S2 Genome‐wide analysis of chromatin interactions in the tetraploid T rapa natans genome based on Hi‐C data.
Figure S3 Phylogenetic relationships of diploid Trapa natans (2x, AA) from the PYY region inferred from maximum likelihood (ML) analysis based on SNP data.
Figure S4 Plot of ADMIXTURE cross‐validation error across values of K.
Figure S5 LD decay determined by correlation of allele frequencies (r 2) against distance (kb) in six Trapa populations.
Figure S6 LD decay distances in cultivars of Trapa natans for each of the 24 chromosomes.
Figure S7 The goodness‐of‐fit of the best scenario summarized by the observed 2D‐SFS, modelled 2D‐SFS, residuals between the model and the data and marginal SFS.
Figure S8 Significantly enriched biological process GO categories of 205 candidate genes under positive selection.
Figure S9 Heatmaps and Venn diagrams of common differentially expressed (up‐ or down‐regulated) genes (DEGs) shared by the two cultivar–wild comparisons (i.e. ‘Wuling’ vs. wild and ‘Nanhuling’ vs. wild).
Figure S10 Significantly enriched GO categories under biological process of differentially expressed (up‐ or down‐regulated) genes (DEGs) shared by the two cultivar–wild comparisons (i.e. ‘Wuling’ vs. wild and ‘Nanhuling’ vs. wild).
Figure S11 Test of alternative demographic models, with different sets of migration directions using FASTSIMCOAL2.
Figure S12 The relationship between the sum of squared error (SSE) and the number of clusters.
Table S1 Statistics of sequencing data used for the tetraploid Trapa natans genome assembly.
Table S2 Statistics of the reference genome assembly of tetraploid Trapa natans.
Table S3 The length of each chromosome.
Table S4 Average nucleotide identity for each chromosome pair of the tetraploid Trapa natans reference genome.
Table S5 Validation of the completeness of the reference genome of tetraploid Trapa natans using the BUSCO approach.
Table S6 Statistics of the transposable elements (TEs) in the tetraploid Trapa natans genome.
Table S7 Prediction of protein‐coding gene models in the reference genome of tetraploid Trapa natans.
Table S8 Function annotation of genes in the tetraploid Trapa natans genome.
Table S9 Sample information used in this study, and mapping ratios and depth of resequenced individuals.
Table S10 The genome mapping coverage and depth of resequenced individuals on each chromosome pair of tetraploid Trapa natans reference genome.
Table S11 Comparison of demographic models analysed with FASTSIMCOAL2.
Table S12 Parameter estimates of effective population sizes, divergence time and migration events for species, cultivars and subgenomes of Trapa.
Table S13 Putative regions under positive selection in cultivars of Trapa natans (2x).
Table S14 205 candidate genes under selective sweep and functional classification.
Table S15 Genes identified that are potentially related to the development of root apex, lateral root or adventitious root.
Table S16 List of samples, tissues, read lengths and mapped reads for RNA‐seq and gene expression analyses.
Table S17 The GO analysis of differentially expressed genes between cultivated and wild Trapa natans (2x) across all six tissues.
Table S18 Expression differentiation of 205 genes under selection.
Table S19 Candidate genes under positive selection with a membership score above top 30% in cultivated and wild Trapa natans (2x).
Table S20 Six morphological characters of Trapa natans (2x, 4x) and T. incisa (2x) measured in this study.
Appendix S1 Supplementary methods.
Acknowledgements
We thank Bing‐Yang Ding (Zhejiang Forestry Academy), Chuan Chen, Meng‐Xiao Hu and Hong‐Yi Wang (Hangzhou Botanical Garden), Yu‐Lai Yin and Fang‐Fang Sun (Suzhou Academy of Agricultural Science) and Wei‐Dong Ke and Jing Peng (Wuhan Institute of Vegetable Sciences) for their assistance with the sample collections. Special thanks to Jing‐Xia Wu (University of Chinese Academy of Sciences) for her assistance with the schematic drawing of plant organs. We also gratefully acknowledge helpful comments from Douglas E. Soltis (University of Florida) and two anonymous reviewers on earlier versions of this manuscript. This work was supported by the National Natural Science Foundation of China (31872652, 32161143003), and the collaborative program of Chinese Academy of Agricultural Sciences (CAAS)‐Jinhua Academy of Agricultural Sciences, funded by Jinhua City of Zhejiang Province.
Lu, R.‐S. , Chen, Y. , Zhang, X.‐Y. , Feng, Y. , Comes, H. P. , Li, Z. , Zheng, Z.‐S. , Yuan, Y. , Wang, L.‐Y. , Huang, Z.‐J. , Guo, Y. , Sun, G.‐P. , Olsen, K. M. , Chen, J. and Qiu, Y.‐X. (2022) Genome sequencing and transcriptome analyses provide insights into the origin and domestication of water caltrop (Trapa spp., Lythraceae). Plant Biotechnol. J., 10.1111/pbi.13758
Contributor Information
Jun Chen, Email: cjevol@zju.edu.cn.
Ying‐Xiong Qiu, Email: qyxhero@zju.edu.cn.
Data Availability Statement
The assembled genome of tetraploid Trapa natans and all raw sequencing data have been deposited under NCBI BioProject PRJNA725399 with accession nos. SRR14331438–SRR14331442. The transcriptomic data for genome annotation have been deposited under NCBI BioProject PRJNA732259 with accession nos. SRR14630938–SRR14630942. The whole‐genome resequencing data have been deposited under NCBI BioProject PRJNA727489 with accession nos. SRR14428604–SRR14428660. The transcriptomic data for wild T. natans and two cultivars (‘Wuling’, ‘Nanhuling’) have been deposited under NCBI BioProject PRJNA731291 with accession nos. SRR14597377–SRR14597430.
References
- Adey, A. , Kitzman, J.O. , Burton, J.N. , Daza, R. , Kumar, A. , Christiansen, L. , Ronaghi, M. et al. (2014) In vitro, long‐range sequence information for de novo genome assembly via transposase contiguity. Genome Res. 24, 2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, D.H. , Novembre, J. and Lange, K. (2009) Fast model‐based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, S. , McCarthy, D.J. , Chen, Y. , Okoniewski, M. , Smyth, G.K. , Huber, W. and Robinson, M.D. (2013) Count‐based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- Arima, S. , Daigoho, M. and Hoque, A. (1999) Flower development and anthesis behavior in the water chestnut (Trapa sp.). Bull. Faculty Agric. Saga Univ. 84, 83–92. [Google Scholar]
- Bao, W. , Kojima, K.K. and Kohany, O. (2015) Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA, 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini, C. , Pacurar, D.I. and Perrone, I. (2014) Adventitious roots and lateral roots: similarities and differences. Annu. Rev. Plant Biol. 65, 639–666. [DOI] [PubMed] [Google Scholar]
- Benson, G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, B.A. , Kriebel, R. , Spalink, D. and Sytsma, K.J. (2016) Divergence times, historical biogeography, and shifts in speciation rates of Myrtales. Mol. Phylogenet. Evol. 95, 116–136. [DOI] [PubMed] [Google Scholar]
- Birney, E. , Clamp, M. and Durbin, R. (2004) GeneWise and genomewise. Genome Res. 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco, E. , Parra, G. and Guigo, R. (2007) Using geneid to identify genes. Curr. Protoc. Bioinformatics, 18, 1–4. [DOI] [PubMed] [Google Scholar]
- Burge, C. and Karlin, S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. [DOI] [PubMed] [Google Scholar]
- Burton, J.N. , Adey, A. , Patwardhan, R.P. , Qiu, R. , Kitzman, J.O. and Shendure, J. (2013) Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana, J. (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. [DOI] [PubMed] [Google Scholar]
- Chen, H. , Patterson, N. and Reich, D. (2010) Population differentiation as a test for selective sweeps. Genome Res. 20, 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.R. , Ding, B.Y. and Funston, M. (2007) Trapaceae. In Flora of China, Vol. 13 ( Wu, Z.Y. , Raven, P.H. and Hong, D.Y. , eds), pp. 290–291. Beijing: Science Press; & St. Louis, MO: Missouri Botanical Garden Press. [Google Scholar]
- Chin, C.S. , Alexander, D.H. , Marks, P. , Klammer, A.A. , Drake, J. , Heiner, C. , Clum, A. et al. (2013) Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nat. Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Conesa, A. , Gotz, S. , Garcia‐Gomez, J.M. , Terol, J. , Talon, M. and Robles, M. (2005) Blast2go: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Cruz‐Castillo, J.G. , Woolley, D.J. and Lawes, G.S. (2002) Kiwifruit size and CPPU response are influenced by the time of anthesis. Sci. Hortic. 95, 23–30. [Google Scholar]
- Dai, X. , Li, X. , Huang, Y. and Liu, X. (2020) The speciation and adaptation of the polyploids: a case study of the Chinese Isoetes L. diploid‐polyploid complex. BMC Evol. Biol. 20, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChaine, E.G. and Martin, A.P. (2006) Using coalescent simulations to test the impact of Quaternary climate cycles on divergence in an alpine plant‐insect association. Evolution, 60, 1004–1013. [PubMed] [Google Scholar]
- DePristo, M.A. , Banks, E. , Poplin, R. , Garimella, K.V. , Maguire, J.R. , Hartl, C. , Philippakis, A.A. et al. (2011) A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat. Genet. 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L. , Dupanloup, I. , Huerta‐Sanchez, E. , Sousa, V.C. and Foll, M. (2013) Robust demographic inference from genomic and SNP data. PLoS Genet. 9, e1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey, D. , Reisch, C. , Narduzzi‐Wicht, B. , Baur, E. , Cornejo, C. , Alessi, M. and Schoenenberger, N. (2017) Historical museum specimens reveal the loss of genetic and morphological diversity due to local extinctions in the endangered water chestnut Trapa natans L. (Lythraceae) from the southern Alpine lake area. Bot. J. Linn. Soc. 185, 343–358. [Google Scholar]
- Gepts, P. (2004) Crop domestication as a long‐term selection experiment. Plant Breed. Rev. 24, 1–44. [Google Scholar]
- Gross, B.L. and Zhao, Z. (2014) Archaeological and genetic insights into the origins of domesticated rice. Proc. Natl Acad. Sci. USA, 111(17), 6190–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, L. , Tayengwa, R. , Cheng, Z.M. , Peer, W.A. , Murphy, A.S. and Zhao, M. (2019) Auxin regulates adventitious root formation in tomato cuttings. BMC Plant Biol. 19, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. , Wu, R. , Sun, G. , Zheng, Y. and Fuller, B.T. (2017) Neolithic cultivation of water chestnuts (Trapa L.) at Tianluoshan (7000–6300 cal BP), Zhejiang Province, China. Sci Rep. 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, J. (2015) Abscisic acid: hidden architect of root system structure. Plants, 4, 548–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepburn, H.R. and Radloff, S.E. (2011) Biogeography. In Honeybees of Asia ( Hepburn, H. and Radloff, S. , eds), pp. 51–67. Heidelberg, Germany: Springer. [Google Scholar]
- Herben, T. , Suda, J. and Klimešová, J. (2017) Polyploid species rely on vegetative reproduction more than diploids: a re‐examination of the old hypothesis. Ann. Bot. 120, 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque, A. , Davey, M.R. and Arima, S. (2009) Water chestnut: potential of biotechnology for crop improvement. J. New Seeds, 10, 180–195. [Google Scholar]
- Hummel, M. and Kiviat, E. (2004) Review of world literature on water chestnut with implications for management in North America. J. Aquat. Plant Manag. 42, 17–28. [Google Scholar]
- Jain, C. , Rodriguez‐R, L.M. , Phillippy, A.M. , Konstantinidis, K.T. and Aluru, S. (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis, D.E. , Ho, Y.S. , Lightfoot, D.J. , Schmöckel, S.M. , Li, B.O. , Borm, T.J.A. , Ohyanagi, H. et al. (2017) The genome of Chenopodium quinoa . Nature, 542, 307–312. [DOI] [PubMed] [Google Scholar]
- Karg, S. (2006) The water chestnut (Trapa natans L.) as a food resource during the 4th to 1st millennia BC at Lake Federsee, Bad Buchau (southern Germany). Environ. Archaeol. 11, 125–130. [Google Scholar]
- Kim, C. , Ryun Na, H. and Choi, H. (2010) Molecular genotyping of Trapa bispinosa and T. japonica (Trapaceae) based on nuclear AP2 and chloroplast DNA trnL‐F region. Am. J. Bot. 97, 149–152. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluyver, T.A. , Jones, G. , Pujol, B. , Bennett, C. , Mockford, E.J. , Charles, M. , Rees, M. et al. (2017) Unconscious selection drove seed enlargement in vegetable crops. Evol. Lett. 1, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korunes, K.L. and Samuk, K. (2021) pixy: Unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Mol. Ecol. Resour. 21, 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryvokhyzha, D. , Salcedo, A. , Eriksson, M.C. , Duan, T. , Tawari, N. , Chen, J. , Guerrina, M. et al. (2019) Parental legacy, demography, and admixture influenced the evolution of the two subgenomes of the tetraploid Capsella bursapastoris (Brassicaceae). PLoS Genet. 15, e1007949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , Jones, S.J. et al. (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, L. and Futschik, M.E. (2007) Mfuzz: a software package for soft clustering of microarray data. Bioinformation, 2, 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara, M. and Ikusima, I. (1991) The ecology of the seed in Trapa natans var. japonica in a eutrophic lake. Vegetatio, 97, 117–124. [Google Scholar]
- Li, H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. https://arxiv.org/abs/1303.3997.
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2011) Inference of human population history from individual whole‐genome sequences. Nature, 475, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Smyth, G.K. and Shi, W. (2014) FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Gituru, W.R. and Wang, Q.F. (2004) Distribution of basic diploid and polyploid species of Isoetes in East Asia. J. Biogeogr. 31, 1239–1250. [Google Scholar]
- Lü, T.F. , Li, X. , Wang, S.N. , Han, L. , Hao, F. , Fu, C.L. , Zhang, P. et al. (2017) Allohexaploid speciation of the two closely related species Myriophyllum spicatum and M. sibiricum (Haloragaceae). Aquat. Bot. 142, 105–111. [Google Scholar]
- Mahto, K.U. , Shaheen, A. , Kumari, S. , Singh, I.S. and Kumar, N. (2018) DNA polymorphism analysis of Indian germplasms of Trapa natans using RAPD molecular Marker. Biocatal. Agric. Biotechnol. 15, 146–149. [Google Scholar]
- Majoros, W.H. , Pertea, M. and Salzberg, S.L. (2004) TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Marcais, G. and Kingsford, C. (2011) A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics, 27, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, R.S. , DuVal, A.E. and Jensen, H.R. (2012) Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol. 196, 29–48. [DOI] [PubMed] [Google Scholar]
- Meyer, R.S. and Purugganan, M.D. (2013) Evolution of crop species: genetics of domestication and diversification. Nat. Rev. Genet. 14, 840–852. [DOI] [PubMed] [Google Scholar]
- Mikulyuk, A. and Nault, M.E. (2009) Water chestnut (Trapa natans): a technical review of distribution, ecology, impacts, and management. Wisconsin, USA: Wisconsin Department of Natural Resources Bureau of Science Servic Madison. [Google Scholar]
- Momigliano, P. , Florin, A.B. and Merilä, J. (2021) Biases in demographic modeling affect our understanding of recent divergence. Mol. Biol. Evol. 38, 2967–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R. (2000) Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics, 154, 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oginuma, K. , Takano, A. and Kadono, Y. (1996) Karyomorphology of some Trapaceae in Japan. APG, 47, 47–52. [Google Scholar]
- Olsen, K.M. and Wendel, J.F. (2013) A bountiful harvest: genomic insights into crop domestication phenotypes. Annu. Rev. Plant Biol. 64, 47–70. [DOI] [PubMed] [Google Scholar]
- Pacurar, D.I. , Perrone, I. and Bellini, C. (2014) Auxin is a central player in the hormone cross‐talks that control adventitious rooting. Acta Physiol. Plant. 151, 83–96. [DOI] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. and Korf, I. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Price, A.L. , Jones, N.C. and Pevzner, P.A. (2005) De novo identification of repeat families in large genomes. Bioinformatics, 21, 351–358. [DOI] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M.A.R. , Bender, D. , Maller, J. et al. (2007) PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am. J. Hum. Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purugganan, M.D. (2019) Evolutionary insights into the nature of plant domestication. Curr. Biol. 29, R705–R714. [DOI] [PubMed] [Google Scholar]
- Qiu, Y.X. , Fu, C.X. and Comes, H.P. (2011) Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 59, 225–244. [DOI] [PubMed] [Google Scholar]
- Qiu, Y.X. , Sun, Y. , Zhang, X.P. , Lee, J. , Fu, C.X. and Comes, H.P. (2009) Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to Quaternary climate change and landbridge configurations. New Phytol. 183, 480–495. [DOI] [PubMed] [Google Scholar]
- Quevillon, E. , Silventoinen, V. , Pillai, S. , Harte, N. , Mulder, N. , Apweiler, R. and Lopez, R. (2005) InterProScan: protein domains identifier. Nucleic Acids Res. 33, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich, S.M. , Ludwig, M. and Colmer, T.D. (2012) Aquatic adventitious root development in partially and completely submerged wetland plants Cotula coronopifolia and Meionectes brownii . Ann. Bot. 110, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M.D. and Oshlack, A. (2010) A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol. 11, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaría, L. (2002) Why are most aquatic plants widely distributed? dispersal, clonal growth and small‐scale heterogeneity in a stressful environment. Acta Oecol. 23, 137–154. [Google Scholar]
- Servant, N. , Varoquaux, N. , Lajoie, B.R. , Viara, E. , Chen, C. , Vert, J. , Heard, E. et al. (2015) HiC‐Pro: an optimized and flexible pipeline for Hi‐C data processing. Genome Biol. 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Schoffmann, O. , Morgenstern, B. and Waack, S. (2006) Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics, 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens, B. and Rasmussen, A. (2016) The physiology of adventitious roots. Plant Physiol. 170, 603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F. (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, A. and Kadono, Y. (2005) Allozyme variations and classification of Trapa (Trapaceae) in Japan. Aquat. Bot. 83, 108–118. [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics, 25, 4–10. [DOI] [PubMed] [Google Scholar]
- Tian, T. , Liu, Y. , Yan, H. , You, Q. , Yi, X. , Du, Z. , Xu, W. et al. (2017) agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , Van Baren, M.J. , Salzberg, S.L. et al. (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya, T. and Iwakuma, T. (1993) Growth and leaf life‐span of a floating‐leaved plant, Trapa natans L., as influenced by nitrogen flux. Aquat. Bot. 46, 317–324. [Google Scholar]
- Van der Auwera, G.A. , Carneiro, M.O. , Hartl, C. , Poplin, R. , Del, A.G. , Levy‐Moonshine, A. , Jordan, T. et al. (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics, 43, 10–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella, A.J. , Severin, J. , Ureta‐Vidal, A. , Heng, L. , Durbin, R. and Birney, E. (2009) EnsemblCompara GeneTrees: Complete, duplication‐aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkova, P.A. , Trávníček, P. and Brochmann, C. (2010) Evolutionary dynamics across discontinuous freshwater systems: Rapid expansions and repeated allopolyploid origins in the Palearctic white water–lilies (Nymphaea). Taxon, 59, 483–494. [Google Scholar]
- von Wettberg, E.J.B. , Chang, P.L. , Başdemir, F. , Carrasquila‐Garcia, N. , Korbu, L.B. , Moenga, S.M. , Bedada, G. et al. (2018) Ecology and genomics of an important crop wild relative as a prelude to agricultural innovation. Nat. Commun. 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorela, I. and Aalto, M. (1982) Palaeobotanical investigations at a Neolithic dwelling site in southern Finland, with special reference to Trapa natans. Ann. Bot. Fenn. 19, 81–92. [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C.A. et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , DeBarry, J.D. , Tan, X. , Li, J. , Wang, X. , Lee, T.‐H. et al. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, L. , Wang, X. , Li, X.M. , Li, X.C. , Jia, H. , Sun, N. , Liang, J.Q. et al. (2020) Numerical taxonomy of Miocene Trapa L. fossil fruits from eastern Zhejiang, China. Earth Sci. Front. 27, 110–121. [Google Scholar]
- Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Res. 35, 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, S.Z. and Xu, S.X. (1992) A study on the root system of Trapa acornis Nakai. Acta Bot Boreali‐occidentalia Sin. 12, 218–223. [Google Scholar]
- Yang, L. , Liu, L. , Wang, Z. , Zong, Y. , Yu, L. , Li, Y. , Liao, F. et al. (2021) Comparative anatomical and transcriptomic insights into Vaccinium corymbosum flower bud and fruit throughout development. BMC Plant Biol. 21, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yasuda, S.P. , Vogel, P. , Tsuchiya, K. , Han, S.H. , Lin, L.K. and Suzuki, H. (2005) Phylogeographic patterning of mtDNA in the widely distributed harvest mouse (Micromys minutus) suggests dramatic cycles of range contraction and expansion during the mid‐ to late Pleistocene. Can. J. Zool. 83, 1411–1420. [Google Scholar]
- Yu, X.J. , Zheng, H.K. , Wang, J. , Wang, W. and Su, B. (2006) Detecting lineage‐specific adaptive evolution of brain‐expressed genes in human using rhesus macaque as outgroup. Genomics, 88, 745–751. [DOI] [PubMed] [Google Scholar]
- Zhang, C. , Dong, S. , Xu, J. , He, W. and Yang, T. (2019) PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics, 35, 1786–1788. [DOI] [PubMed] [Google Scholar]
- Zhou, Q. (2012) The planting of water chestnut and wetland agriculture development in the Huzhou Plain in the Tang‐Song Dynasties. Agric. Hist. China, 3, 11–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 K‐mer‐based genome size estimates.
Figure S2 Genome‐wide analysis of chromatin interactions in the tetraploid T rapa natans genome based on Hi‐C data.
Figure S3 Phylogenetic relationships of diploid Trapa natans (2x, AA) from the PYY region inferred from maximum likelihood (ML) analysis based on SNP data.
Figure S4 Plot of ADMIXTURE cross‐validation error across values of K.
Figure S5 LD decay determined by correlation of allele frequencies (r 2) against distance (kb) in six Trapa populations.
Figure S6 LD decay distances in cultivars of Trapa natans for each of the 24 chromosomes.
Figure S7 The goodness‐of‐fit of the best scenario summarized by the observed 2D‐SFS, modelled 2D‐SFS, residuals between the model and the data and marginal SFS.
Figure S8 Significantly enriched biological process GO categories of 205 candidate genes under positive selection.
Figure S9 Heatmaps and Venn diagrams of common differentially expressed (up‐ or down‐regulated) genes (DEGs) shared by the two cultivar–wild comparisons (i.e. ‘Wuling’ vs. wild and ‘Nanhuling’ vs. wild).
Figure S10 Significantly enriched GO categories under biological process of differentially expressed (up‐ or down‐regulated) genes (DEGs) shared by the two cultivar–wild comparisons (i.e. ‘Wuling’ vs. wild and ‘Nanhuling’ vs. wild).
Figure S11 Test of alternative demographic models, with different sets of migration directions using FASTSIMCOAL2.
Figure S12 The relationship between the sum of squared error (SSE) and the number of clusters.
Table S1 Statistics of sequencing data used for the tetraploid Trapa natans genome assembly.
Table S2 Statistics of the reference genome assembly of tetraploid Trapa natans.
Table S3 The length of each chromosome.
Table S4 Average nucleotide identity for each chromosome pair of the tetraploid Trapa natans reference genome.
Table S5 Validation of the completeness of the reference genome of tetraploid Trapa natans using the BUSCO approach.
Table S6 Statistics of the transposable elements (TEs) in the tetraploid Trapa natans genome.
Table S7 Prediction of protein‐coding gene models in the reference genome of tetraploid Trapa natans.
Table S8 Function annotation of genes in the tetraploid Trapa natans genome.
Table S9 Sample information used in this study, and mapping ratios and depth of resequenced individuals.
Table S10 The genome mapping coverage and depth of resequenced individuals on each chromosome pair of tetraploid Trapa natans reference genome.
Table S11 Comparison of demographic models analysed with FASTSIMCOAL2.
Table S12 Parameter estimates of effective population sizes, divergence time and migration events for species, cultivars and subgenomes of Trapa.
Table S13 Putative regions under positive selection in cultivars of Trapa natans (2x).
Table S14 205 candidate genes under selective sweep and functional classification.
Table S15 Genes identified that are potentially related to the development of root apex, lateral root or adventitious root.
Table S16 List of samples, tissues, read lengths and mapped reads for RNA‐seq and gene expression analyses.
Table S17 The GO analysis of differentially expressed genes between cultivated and wild Trapa natans (2x) across all six tissues.
Table S18 Expression differentiation of 205 genes under selection.
Table S19 Candidate genes under positive selection with a membership score above top 30% in cultivated and wild Trapa natans (2x).
Table S20 Six morphological characters of Trapa natans (2x, 4x) and T. incisa (2x) measured in this study.
Appendix S1 Supplementary methods.
Data Availability Statement
The assembled genome of tetraploid Trapa natans and all raw sequencing data have been deposited under NCBI BioProject PRJNA725399 with accession nos. SRR14331438–SRR14331442. The transcriptomic data for genome annotation have been deposited under NCBI BioProject PRJNA732259 with accession nos. SRR14630938–SRR14630942. The whole‐genome resequencing data have been deposited under NCBI BioProject PRJNA727489 with accession nos. SRR14428604–SRR14428660. The transcriptomic data for wild T. natans and two cultivars (‘Wuling’, ‘Nanhuling’) have been deposited under NCBI BioProject PRJNA731291 with accession nos. SRR14597377–SRR14597430.