


Abstract
Super enhancers (SEs) are broad enhancer domains usually containing multiple constituent enhancers that hold elevated activities in gene regulation. Disruption in one or more constituent enhancers causes aberrant SE activities that lead to gene dysregulation in diseases. To quantify SE aberrations, differential analysis is performed to compare SE activities between cell conditions. The state-of-art strategy in estimating differential SEs relies on overall activities and neglect the changes in length and structure of SEs. Here, we propose a novel computational method to identify differential SEs by weighting the combinatorial effects of constituent-enhancer activities and locations (i.e. internal dynamics). In addition to overall activity changes, our method identified four novel classes of differential SEs with distinct enhancer structural alterations. We demonstrate that these structure alterations hold distinct regulatory impact, such as regulating different number of genes and modulating gene expression with different strengths, highlighting the differentiated regulatory roles of these unexplored SE features. When compared to the existing method, our method showed improved identification of differential SEs that were linked to better discernment of cell-type-specific SE activity and functional interpretation.
INTRODUCTION
Super enhancers (SEs) were proposed as broad regulatory domains on genome, usually spanning a minimum of thousands of base pairs and consisting of multiple constituent enhancers (CEs) (1). The CEs work together as a unit, instead of separately, to facilitate high enhancer activity, observed as dense enrichment of cell master regulators, coactivators, mediators and chromatin factors at SEs (2). These characteristics were further demonstrated by the fact that, distinct from regular enhancers, SE is specifically linked to gene regulation associated with cell identity and disease mechanisms (3,4).
Recent studies further showed that, beyond the elevated activity, the internal mechanics of SEs also paly critical roles in defining their prominent roles in gene regulation, known as multi-promoter targeting and long-range interactions (5–8). Some SEs form a clear hierarchical structure where hub CEs are responsible for the functional and structural organization of the whole SEs (6,9). Other SEs, in contrary, receive relative balanced contribution from the CEs. In addition, CEs could establish an open chromatin interaction network within individual SEs (7), indicating the internal crosstalk across CEs in orchestrating SEs’ unique functions.
The activity and relations of individual CEs were well appreciated during computational identification of SEs. Existing algorithms usually contain two processing steps (2,10). First, the activity and locations of genome-wide enhancers are inferred through peak detection using chromatin immunoprecipitation sequencing (ChIP-seq) data (11), particularly that measuring the binding of mediators, master regulators, or active histone mark H3K27Ac. Second, the inferred activity and locations are summarized linearly to prioritize broad enhancer regions (2,3), i.e. SEs, that contain densely enriched enhancers with high activities, i.e. the CEs.
However, the organization of CEs were not considered by current approaches in differential analysis of SEs, a key aspect of research interest when comparing across biological conditions (12–16). The alteration of SEs has been found to be highly associated with disease dysregulation and could be used for drug target identification (14–16). These approaches, in which SEs are treated as individual entities, usually identify differential SEs based on binary strategy, which compares the presence and absence of SEs between biological conditions, neglecting the constituent enhancer statuses. Consequently, differential SEs are generated largely depending on the parameters that algorithms utilized to detect super enhancers (2,3). In addition, the sensitivity to detect changes in the local enhancer organization are downplayed within the broad genomic regions occupied by SEs.
Here, we propose a novel computational method to identify differential SEs by summarizing the combinatorial effects of constituent-enhancer activities and locations. In addition to overall activity changes, our method detects four extra differential categories specifically pointing to the internal structural alterations of SEs. We demonstrate the unique characteristics of these differential SE categories using public datasets by linking their altered activity to transcription factor (TF) binding and gene expression with 3D chromatin interactions (17–19). The results indicate that each SE category regulates distinct sets of gene targets and their expression. Further, we show that our method maximizes the discernment of cell identities when comparing SE profiles of cell lines from the same cancer type. Our method provides sensitive and biologically meaningful identification of differential SEs, which complements existing understanding of SE dynamics. We implemented an R package, DASE (Differential Analysis of Super Enhancers: https://github.com/tenglab/DASE), to facilitate the use of our method.
MATERIALS AND METHODS
Data acquisition
H3K27ac enrichment, gene expression and 3D interaction data were downloaded from ENCODE data portal (20) and GEO repositories (21). Specifically, quality-controlled alignment files of H3K27Ac ChIP-seq and RNA-seq, and chromatin contacts files of POLR2A ChIA-PET were downloaded from ENCODE for six selected cancer cell lines (A549—Lung Cancer, HCT116—Colorectal Cancer, HepG2—Liver Cancer, K562—Leukemia, MCF7—Breast Cancer and SK-N-SH—Neuroblastoma) (accession ID documented in Supplementary Table S1). Raw sequencing files of H3K27ac ChIP-seq, RNA-seq and H3K27ac HiChIP for BC1 and BC3 cell lines were downloaded from GEO with accession IDs GSE136090 (16) & GSE114791 (22) (Supplementary Table S1).
H3K27Ac ChIP-seq data pre-processing
Raw ChIP-seq data from GEO was first aligned to human genome using Bowtie2 (23). Then, all alignment files were processed for peak calling using MACS2 (11), followed by SE detection using ROSE (2). All tools were applied with default parameters. ChIP-seq blacklist regions were excluded for downstream analysis (24).
RNA-seq data analysis
RNA-seq alignment files downloaded from ENCODE were quantified for gene expression using featureCount (25) based on GENCODE annotations. Raw FASTQ files from GEO were processed with semi-alignment and quantification tool Salmon (26) to generate gene expression count table based on GENCODE transcriptome. Then, differential analysis of gene expression was estimated using DESeq2 (27) for all two-condition comparisons. The shrunk fold-changes were extracted to represent gene expression differences (28).
3D chromatin contacts analysis
The chromatin contacts generated by ENCODE project from POLR2A ChIA-PET data were directly adapted to link genes and super enhancers for ENCODE cancer cell lines. Basically, ENCODE project applied strict quality controls, and filtered confident chromatin contacts with at least three normalized interactions (29). H3K27Ac HiChIP data of BC1 and BC3 cell lines are analyzed the same as previously described (16). In brief, reads were aligned to human genome using HiC-Pro (30). Sequencing replicates were merged to call chromatin contacts using hichipper (31) with confident interactions defined as at least three normalized interactions.
Differential analysis of CEs and binary SE differences
For each comparison between two cell lines that both have two ChIP-seq replicates, a uniform peak list was first created by merging overlapped peaks across the compared samples. ChIP-seq reads were then quantified using featureCount (25) to generate a read count table for the peak list. Differential peak analysis was performed by adapting DESeq2 (27) (with parameter type = ‘mean’) to account for the varied dispersion between peaks with low and high read counts. The differential statuses of CEs (H3K27Ac peaks within SEs) were extracted based on their estimated log2 fold-changes and corresponding q-values. We also extracted the normalized coverage for CEs as the weight inputs for SE differential analysis.
Binary SE differences were estimated based on the presence and absence of SEs between compared conditions. Basically, if a SE presents in both compared conditions at the same given location regardless size or activity changes, it will be identified as non-differential. In contrast, if a SE only presents in one condition at a given location, it will be identified as differential.
Modeling differential SEs with SE internal dynamics by DASE
DASE identifies differential SEs by accounting for the combinatorial effects of CEs weighted with their activities and locations. In detail, the methods include the following steps (Supplementary Figure S1).
Input preparation
A uniform list of SEs is generated by merging overlapped SEs between compared conditions. The differential statuses (log2 fold-change) of all CEs located within SEs are extracted as well as their activities (ChIP-seq coverage) and locations (genomic coordinates), as calculated above. In practice, we select the maximum ChIP-seq coverage between compared conditions for each CE to provide better weights in the spline model below.
Weighted spline model
For each SE, the log2 fold-change values of CEs stratified by their genomic locations are fitted using b-spline model, where the importance of CEs is weighted by their relative activities. As a result, CEs show less impacts on the spline fitting if they have low activities and stay close to other CEs. We implement the spline model using R package splines. In addition, to ensure the robustness of b-splines in the case of too many low- or mild-activity CEs, we pre-estimate the polynomial degree for each fitting based on the number of top ranked CEs in each SEs. We choose top ranked CEs as the minimum number of enhancers that build up at least 95% of total SE activity. In detail, we set the polynomial degree of b-spline as 2, 3 and 4 if this number of top ranked CEs is <4, between 4 and 6, and >6, respectively.
Significance estimation
We use permutations to define significant fitted values by b-spline. In brief, we randomly shuffle enhancer activities in each compared sample, re-estimate the differential statuses of all CEs and re-fit splines for all SEs. As a result, we generate a null distribution of fitted b-spline values for all CEs. We repeat the processes 10 times for a stable null distribution. Significant fitted values are defined as those having greater or smaller values than the upper or lower inflection points (significant thresholds) of the null distribution (Supplementary Figure S1).
Status summarization for SE sub-regions
We divide each SE into multiple sub-regions using the intersects of b-spline curves and the significant thresholds (Supplementary Figure S1). For instance, the curve located above the upper threshold indicates an up-altered partial region within the SE, while the curve located below the lower threshold indicates a down-altered partial region. The curves in between indicate non-altered SE sub-regions. To decrease potential noises in SE segmentation, we ignore sub-regions in which CEs account for <1% of the SE activities.
Overall differential status
We further summarize the overall differential statuses for SEs with heuristic approaches based on the altering directions, locations, and activity occupancies (i.e. the percentage of the contained CE activity over the total activity of SEs) of the segmented sub-regions from the previous step. Specifically, if only one region is resulted from segmentation of b-spline curve of a SE, the SE will be identified as either overall-change or non-differential depending on the altering directions of that segment: up/down-altered or non-altered. If two segments are resulted (i.e. one segment is altered and the other is non-altered), we determine differential SEs based on the activity occupancy of the altered segment. In detail, two-segment SEs are identified as non-differential, shortened or overall-change if the altered segment occupies less than 10%, between 10 and 90%, and >90% of total SE activities. For a three-segment SE, we first check if it is hollowed based on whether the middle segment is up/down-altered. If not, we check if it is shifted based on whether the three segments cover three different altering directions consecutively (i.e. up-altered, non-altered and down-altered). Otherwise, the remaining three-segment SEs fall into the following situation: the middle segment is non-altered while the left and right segments are both altered with the same altering direction. We then identify the overall SE statues as non-differential, shortened and overall-change based on the total activity occupancies of the left and right segments as <10%, between 10 and 50%, and >50%. It is noted that the overall-change are filtered with different criteria (break points at 90% versus 50%) between two-segment and three-segment SEs, to account for the total size impacts from the altered segments. For a SE with more than three segments, it is identified as other complex scenario except that a four-segment SE holding all three statuses is defined as hollowed. Finally, we rank the significance of differential SEs using the activity occupancies of the altered segments separately for overall-change, shortened, hollowed and shifted SEs. In summary, overall-change SEs are resulted from one-, two- or three-segment splines; shortened SEs are resulted from two- or three-segment splines; shifted can only be estimated from three-segment splines; hollowed can be estimated from three- or four-segment splines; and all other altering situations are assigned as other complex scenario.
Transcription factor enrichment analysis
ChIP-seq bam files for 78 documented TFs in both K562 and MCF7 cell lines were downloaded from ENCODE with accession ID provided in Supplementary Table S1. TF occupancies are normalized between the two cell lines using RLE algorithm (27) based on TF peaks. After calling differential SEs between MCF7 and K562 with DASE, we extracted TF occupancy from ChIP-seq data for significant differential (fold-change > 4 & q-value < 0.05) CEs that locate within differential SE categories: overall-change, shortened, hollowed, and shifted. The occupancy heatmap for TFs were generated with Deeptools v3.5.1 (32).
SE-gene targeting
We identify SE-gene targeting relationship using 3D chromatin contacts generated from POLR2A ChIA-PET or H3K27Ac HiChIP. Basically, a valid targeting is defined if one end of chromatin contacts is overlapped with SEs, while the other end is overlapped with gene promoters (selected as −3–1 kb from genes’ transcription start sites). Targeting relations are ignored if the SE-promoter distances are less than 20 kb or greater than 500 kb.
Pathway enrichment analysis
For pathway enrichment in genes linked by different SE categories, gene sets were first identified for each SE categories based on SE-gene targeting relations in both compared conditions. Then, only uniquely linked genes by each SE category were selected for pathway enrichment using DAVID Bioinformatics Resources v6.8 (33) based on KEGG database (34). For pathway enrichment in genes linked by cell-type-specific SEs, genes were selected as those only identified by DASE compared to the binary strategy. Significant pathways were selected to have P-value <0.05.
RESULTS
Internal dynamics underlie genome-wide SE differences
SEs usually contain multiple constituent enhancers (CEs) located in close genomic proximity along the genome (2,35). It is important to understand the roles of CEs in contributing to the SE lineage-specificity. We explored the SE profiles for six cancer cell types (Figure 1 and Methods) using ChIP-seq data of H3K27Ac histone modification from ENCODE project (36). We found that CEs within the same SEs could alter differently across cell types. For example, the CEs, located at two previous reported SE loci responsible for MYC regulation in multiple cancers (37–40), showed divergent activity patterns across the six cell types (Figure 1A). We term such divergent alterations between CEs as the internal dynamics of SEs, which underline the individual CE effects on determining the cell-type-specific SE activity.
Figure 1.

Internal dynamics of super enhancers. (A) CEs show frequent alterations across cancer cell types at two reported SE loci associated to MYC regulation. ChIP-seq coverage of H3K27Ac are shown. SEs and CEs are labeled at the top as red and grey bars, separately. (B) Frequencies of differential CEs (fold-change > 4 & q-value < 0.05) and SEs (based on binary strategy). (C) Relative width of differential CEs from non-differential and differential SEs (Student's t-test P < 2.2e–16). Relative width is defined as the percentage of CE width over the summed width from all CEs within a SE. (D) Relative coverage of differential CEs from non-differential and differential SEs (Student's t-test P < 2.2e–16). Relative coverage is defined as the percentage of CE coverage over the summed coverage from all CEs within a SE.
We then performed pair-wise comparisons of SE profiles across the six cancer cell types. On average, over 40% of CEs showed significant differential activity (fold-change > 4 and q-value < 0.05) that accounts for over 80% of total SEs in these cell types (Figure 1B). This indicates that SEs undergo frequent internal alterations across cell types. We further estimated how the alteration of CEs contributed to the overall differences of SEs. Here, we identified differential SEs between cancer cell types using the presence/absence (binary) strategy. A small portion of SEs (∼10%) with significantly altered CEs did not show overall differences, while the majority of SEs changed in the same directions as their altered CEs (Figure 1B). This implies the divergent influences of CEs on the overall differential statuses of SEs genome-widely.
Next, we examined the characteristics of CEs that might affect their contribution to the overall SE differences. Not surprisingly, the spanning width and regulatory activity of CEs, indicated by H3K27Ac ChIP-seq coverage, showed significant associations with overall SE differences (Figure 1C and D). It is noted that the values of spanning width and regulatory activity are correlated since wider CEs tend to have higher sequencing coverage. Nevertheless, we demonstrated both metrics here as individual CEs can have quite discrepant values for the two metrics (Supplementary Figure S2). In brief, differential CEs with smaller width or lower activity presented less impacts on the overall statuses of their corresponding SEs. Therefore, we built a model to summarize SE internal dynamics by accounting for these characteristics.
Modeling internal dynamics leads to distinct patterns of differential SEs
We developed a weighted spline model, implemented as an R package DASE, to summarize the internal dynamics into the overall differential statuses of SEs (Figure 2 and Materials and Methods). In brief, differential CEs were first evaluated using existing strategies on detecting differential ChIP-seq peaks (41). Then, a spline was fitted, stratified by enhancer positions, to smooth the differential signals for consecutive CEs. In the smoothing, the activities and width of CEs were taken as fitting weights. Finally, the spline curves were evaluated with permutations to determine reliable differential sub-regions within the SEs, which were further summarized towards the overall differential statuses of SEs.
Figure 2.

Differential SEs modeled with DASE. SE examples are listed with overall-change (A), shortened (B), shifted (C), hollowed (D) and non-differential (E). In each sub-figure, the upper panel lists in order the SE regions, CEs, H3K27Ac ChIP-seq coverage in two cell types with two replicates. The lower panel shows the fitted b-splines in addition to the original log2 fold-change values for CEs (points). Dashed lines indicate the estimated thresholds from permutation to define differential segments within SE regions. In (E), red text indicates the cell line where SE was detected at this locus by the binary strategy.
To illustrate the utility of DASE, we compared SE profiles between two cancer cell lines, K562 and MCF7. These two cell lines have high-quality annotation datasets on ENCODE data portal, including TF binding, 3D chromatin interactions and gene expression, to help evaluate the identified differential SEs. DASE detected an overall-change as well as four novel patterns of differential SEs highlighting the structural alterations within SEs, denoted as shortened, shifted, hollowed and other complex scenarios, separately (Figure 2). Overall-change SEs represent significant overall activity alterations (as captured by the binary strategy) as well as consistent altering behavior among CEs. (Figure 2A). Shortened SEs have significant changes in their sizes by gaining or dismissing CEs on one or both ends of the SEs (Figure 2B). Shifted SEs have migrated genomic locations without significant size changes (i.e. CEs gained on one end of the SEs and dismissed on the other end) (Figure 2C). Hollowed SEs represent those with altered CEs in the middle while the two ends remain intact (Figure 2D). Other complex scenario SEs represent all other complicated or rare cases (Supplementary Figure S3). The quantitative definition of SE categories is detailed in the Methods. Examples of SE structural alterations reveal that structural differences do not necessarily accompany overall activity differences (Figure 2A–D). Together, they provide novel insights to understand SE dynamics between cell conditions. In addition, we note that marginal activity differences that were over-claimed as differential SEs by the binary strategy could be properly corrected by DASE (Figure 2E).
In total, about 47% of SEs showed overall-change between K562 and MCF7, reflecting the distinct chromatin structure underlying each cancer type (Supplementary Figure S4). Shortened SEs dominated among all types of structural differences (65%), indicating the wide spreading of SE size changes. The other types of structural differences, although not prevailing, represent the diverse dynamics of SE profiles responsible for cell-type-specific gene regulation. We show that those structural differences consistently present in comparisons across more cancer cell types in later sections.
Diverse differential SEs synergistically build up gene regulation
We further characterized the functional roles of the differential SE patterns in gene regulation. SEs are usually enriched with various transcriptional regulators and cofactors (8), which play critical roles in supporting SE interactions with gene targets (Figure 3A). We examined the protein binding profiles across the differential SE patterns. In total, we analyzed 78 TFs that have ChIP-seq data available for K562 and MCF7 cell lines by ENCODE project. TFs showed a high correlation with CE activities (Figure 3B and Supplementary Figure S5), regardless of the differential patterns of the corresponding SEs (Supplementary Figure S6), suggesting different patterns of SE alterations share similar mechanisms in recruiting TFs. Among these TFs, two clear modes of enrichment were identified (Figure 3B and Supplementary Figure S5): (i) those enriched at active CEs in both K562 and MCF7 cell lines (e.g. MBD2), suggesting that these TFs are involved in maintaining key cell functions; (ii) those enriched at active CEs in only one cell type but not the other (e.g. YBX1 and ZKSCAN1), indicating their roles in cell-type-specific gene regulation. This suggests different types of SE alterations are involved in both cell-type-specific and housekeeping-related regulation.
Figure 3.

Differential SE categories linked to distinct regulatory features. (A) An example shows TF in aiding SE-gene regulation. (B) Differential CEs enriched with cell-type-specific (YBX1 & ZKSCAN1) and key-function (MBD2) related TFs. Top panels are TF signals at CEs active in K562. Bottom panels are TF signals at CEs enriched in MCF7. Middle panels are the aggregated binding signals from the top and bottom panels with blue and orange lines indicating signals in K562 and MCF7, separately. The signs of y-axises in the middle panels represent enrichment directions. (C) Differences on the linked gene numbers between MCF7 and K562 by differential CEs, which are grouped by their altering statuses. Gene targets are identified based on chromatin contacts from POLR2A ChIA-PET Data. (D) Differences on the linked gene numbers between MCF7 and K562 by differential SEs, which are grouped by their differential patterns. For each differential SE category, SEs are separated into two sub-groups based on their enrichment directions. (E) log2 fold-change of the linked genes between MCF7 and K562 by differential CEs. (F) log2 fold-change of the linked genes between MCF7 and K562 by differential SEs. For each differential SE category, genes are separated into two groups by the enrichment directions of their linked SEs. P-values were calculated with Wilcoxon rank-sum test (C, D) and Student's t-test (E, F) with notations as *P < 0.05, ***P < 0.001 and n.s. = not significant. (G) Overlaps of the linked genes by four differential SE categories between MCF7 and K562. (H) KEGG signaling pathways (P < 0.05) uniquely associated to each differential SE category.
Beyond TF binding, we examined the downstream effects of SE alterations on gene expression. We identified SE target genes in each cell type using 3D chromatin interactions based on POLR2A targeted ChIA-PET data. As expected, the gained CEs usually establish new gene targets, while the dismissed CEs remove existing targets (Figure 3C). Consequently, SEs with increased activity (e.g. strengthened or lengthened with gained CEs) in one cell type usually target more genes compared to their altered forms (e.g. weakened or shortened with dismissed CEs) in the other cell type (Figure 3D). Interestingly, we observed that such effects differed across the differential SE patterns, with heavier effects presented by overall-change, shortened and hollowed SEs, and nearly no effects by shifted SEs (Figure 3D). The marginal effects by shifted SEs are expected as they provide no signs of the altering directions of SE activities. Here, to minimize the sequencing coverage effects on gene target counting with ChIA-PET data, we normalized the count differences by subtracting the median count difference (i.e. 1) of the control SE group (i.e. the non-differential SEs).
Differential analysis of gene expression between K562 and MCF7 cell lines indicated that the gained CEs between the two cell types were significantly associated with upregulated gene expression (Figure 3E). A similar association was also observed at the SE level, with increased SE activity presenting higher amplification on gene expression (Figure 3F). Again, overall-change and shortened SEs showed higher regulatory effects, while shifted SEs presented nearly no effects. Here, hollowed SEs showed no impact on gene expression, indicating their functions might be limited to maintaining the proper number of gene targets (Figure 3D). As a control, we observed no significant effects on gene expression by the non-differential SEs (Figure 3F).
We then performed pathway enrichment analysis on genes linked by different types of SE alterations. The majority of genes (∼75%) are linked by only one type of differential SEs (Figure 3G), with the overall-change and shortened SEs linked with the most and comparable number of genes. We focused on these genes linked by only one type of SEs and identified distinct sets of signaling pathways uniquely associated with each type of differential SEs (Figure 3H). For instance, FoxO Signaling Pathway (42–44), cAMP Signaling Pathway (45), and AMPK signaling pathway (46) are enriched with genes linked to overall-change, shortened and hollowed SEs, respectively. To further understand SE functions in the context of the compared cell lines, we separated each differential SE category into two sub-groups: up-altered in K562 and up-altered in MCF7. We repeated pathway enrichment analysis on these two sub-groups. Interestingly, overall-increased SEs drive the signaling pathway enrichment in K562, while lengthened SEs drive the signaling pathway enrichment in MCF7 (Supplementary Figure S7). This highlights the divergent regulatory mechanisms by different SE categories in different cell lines. Pathway enrichment was also performed between other cell lines, with different SE categories showing varied importance in different comparisons (Supplementary Figure S8). In summary, different patterns of SE alterations synergistically build up gene regulation by playing distinct roles in modulating gene expression and cellular functions.
Accounting for internal dynamics improves identification and interpretation of differential SEs
Besides the characterization of SE structural alterations, DASE presents an overall improvement on differential SE identification over the existing binary strategy (12–16). We summarized the improvements based on pair-wise comparisons across the six cancer cell types. These cells have H3K27Ac ChIP-seq, 3D chromatin interaction and gene expression data available which enable the functional assessment on targeting gene expression. On average, the discrepant identification between DASE and the binary strategy accounts for ∼18% of the total SEs and covers all patterns of SE alterations (Figure 4A). For example, overall-change SEs by DASE might be identified as non-differentials by the binary strategy if SEs were detected at the same region in both MCF7 and K562 cell lines (Figure 4B). Most newly identified differential SEs by DASE have structural alterations (∼91%). The H3K27Ac and TF binding show stronger signal differences at the differential SEs newly identified by DASE compared to those only identified by binary strategy (Figure 4C and Supplementary Figure S9). Among all discrepant differential SEs, the overall-change SEs newly identified by DASE showed the strongest impact on altering the numbers and expression of the gene targets (Figure 4D), suggesting they were falsely identified as non-differential by the binary strategy. The other newly identified SEs by DASE presented relatively higher gene effects compared to the differential SEs only identified by the binary strategy (Figure 4D), suggesting the overall improved sensitivity and specificity by the DASE identification. Here, we didn’t assess the shifted and other complex scenario SEs in this analysis as they provided no signs of the altering direction for gene expression. To avoid confounding effects from genes targeted by multiple SEs, we left out genes that were also linked by the common differential or non-differential SEs in the analysis.
Figure 4.
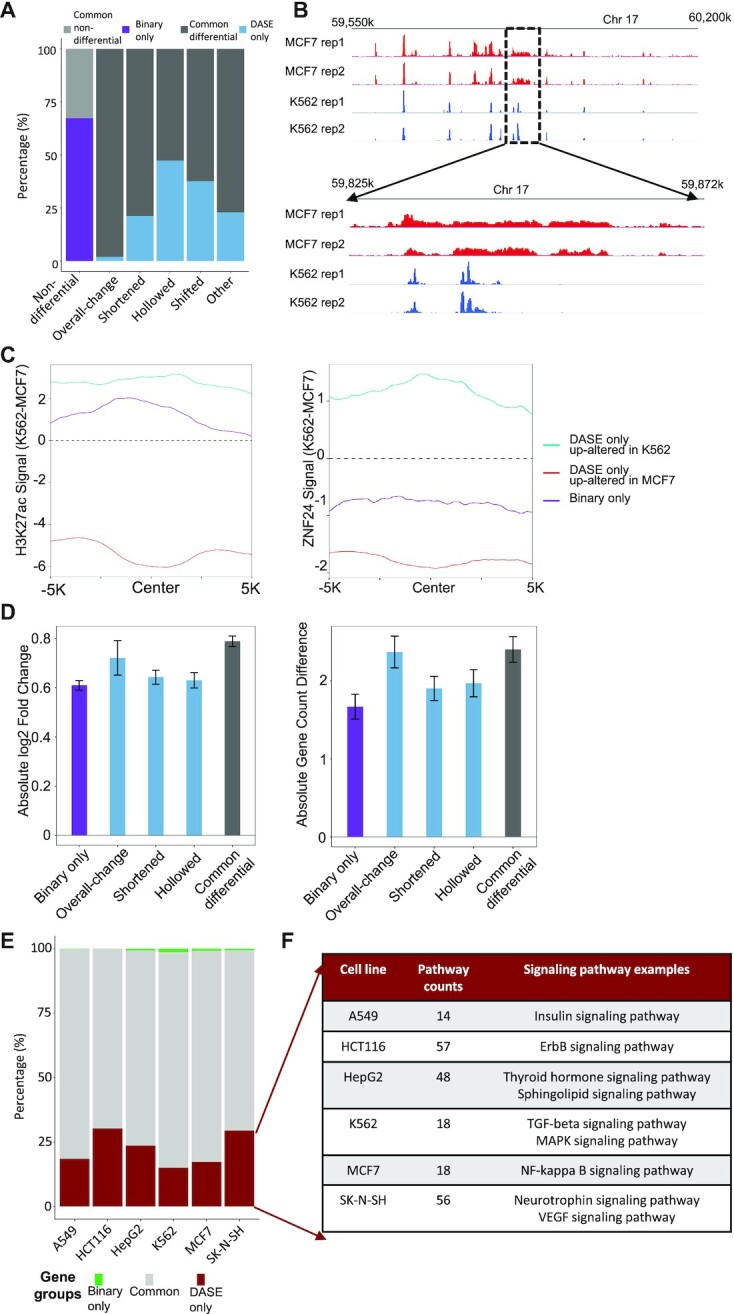
Comparisons between DASE and the binary strategy. (A) Average discrepancies between DASE and the binary strategy across pair-wise comparisons of the six cancer cell types. Light grey: common non-differentials by DASE and the binary strategy; Dark grey: common differentials; purple: differential only by the binary strategy; blue: differential only by DASE. X-axis is labels with SE categories based on DASE. (B) An example region showing overall-change SE by DASE but no change by the binary strategy. (C) H3K27Ac and TF binding signals at the CEs of the discrepant differential SEs. Lines indicate the differences of binding signals between cell lines. (D) Impact on gene expression and linked gene numbers by the discrepant differential SEs between DASE and the binary strategy. Bars indicate the mean of log2 fold-change of gene expression or changes of linked gene numbers in each pair-wise comparison and error bars represent the standard error of the means. Results from the two altering directions of the gene effects are merged based on absolute values. (E) Genes uniquely linked to SEs with increased activity (overall increased, lengthened or hollowed with increased CEs) in one cancer cell type compared to the other five cells. Green: genes uniquely found by binary strategy; red: genes uniquely found by DASE; grey: genes found by both methods. (F) KEGG pathways (P < 0.05) enriched in the uniquely linked genes by DASE.
We further evaluated DASE by gene functions linked to the differential SEs. We identified cell-type-specific regulated genes that were linked to the SEs with increased activity (i.e. overall increased, lengthened, or hollowed with increased CEs) in one cancer cell type compared to the other five cell types. We then compared the obtained gene list between DASE and the binary strategy. Surprisingly, DASE recovered nearly all the cell-type-specific regulated genes by the binary strategy and found additional genes mainly linked to the SE structural alterations (Figure 4E). We examined the pathways enriched in these additionally identified genes and found a number of cancer-associated pathways, indicating the critical roles of the novel structural alterations (Figure 4F). For example, Insulin Signaling Pathway (47), ErbB Signaling Pathway (48), Thyroid Hormone Signaling Pathway (49), TGF-beta Signaling Pathway (50), NF-kappa B signaling pathway (51) and Neurotrophin Signaling Pathway (52), linked to SEs that DASE uniquely identified in A549, HCT116, HepG2, K562, MCF7 and SK-N-SH, respectively. In addition, eight DASE-detected differential SEs, that showed cell-specific enriched activity in one of these cancer cell lines, were previously reported by ENdb database (53) as disease enhancers in the corresponding cancers. This highlights the capability of DASE to prioritize functional SEs. In summary, DASE showed improved sensitivity in linking differential SEs to cell-type-specific regulation, particularly through the consideration of internal structural alterations.
Accounting for internal dynamics maximize the discerning of cell identity
Given the improved sensitivity in the cross-cancer analysis above, we further evaluate DASE by within-cancer comparison. We applied DASE to compare SE profiles between two similar cancer cell lines, BC1 and BC3, that are B lymphocyte cells derived from Lymphoma under different viral infections. We previously demonstrated that different viral infections led to a distinct enhancer connectome on these cell lines (16).
Overall, the two similar cell lines presented much higher similarity of SE profiles (Figure 5A). We linked the differential SEs to their target genes using chromatin interactions identified by H3K27Ac HiChIP datasets. Similar gene effect patterns were observed across differential SE patterns, as we found previously (Figure 3C–F). The linked genes were enriched in both frequency and expression in the same direction as CEs/SEs altering between BC1 and BC3 cell lines (Figure 5B–E). Specifically, such effects are stronger by overall-change SEs, followed by shortened SEs, consistent with the findings in cross-cancer analysis (Figure 3C–F). Finally, we extracted the uniquely linked genes by the differential SE patterns (Figure 5F) and performed pathway enrichment analysis. We found unique pathways such as Viral Carcinogenesis particularly linked to the shortened SEs (Figure 5G). This suggests shortened SEs play key roles in gene dysregulation in response to the different viral carcinogenesis between BC1 and BC3 cell lines (54,55). We then split shortened SEs into those lengthened in BC1 and those lengthened in BC3 and performed pathway enrichment on the two groups. We found that Viral Carcinogenesis pathway only enriched in genes linked to lengthened SEs in BC1 (Figure 5G), indicating the complexity of viral infection by two virus settings in BC1 as compared to single viral infection in BC3. Therefore, by accounting for the SE internal dynamics, we found that cell-line-specific gene regulation linked to differential SEs, particularly those with structural alterations, highlighting the differentiated roles of SE categories and the importance of featuring internal dynamics in SE differential analysis.
Figure 5.

Identifying SE differences between similar cancer cell lines. (A) Proportions of differential SE categories identified from within-cancer (BC1 versus BC3) and cross-cancer comparisons (pairwise between six cancer cell types from ENCODE). Count differences (B, C) and log2 fold-changes (D, E) of linked genes by differential CEs (B, D) and SEs (C, E) between BC1 and BC3 cell lines. Gene targets are identified based on chromatin contacts from H3K27Ac HiChIP data. P-values were calculated with Wilcoxon rank-sum test (B, C) and Student's t-test (D, E) with notations as *P < 0.05, **P < 0.01, *** P < 0.001 and n.s. = not significant. (F) Overlaps of SE-linked genes across four differential SE categories between BC1 and BC3. (G) KEGG pathways (P < 0.05) that uniquely associated to each differential SE category. Pathways enriched in shortened SEs are highlighted separately for two sub-groups: those lengthened in BC1 and those lengthened in BC3.
DISCUSSION
In this manuscript, we proposed a novel computational method DASE to identify differential SEs by summarizing the internal dynamics. We categorized differential SEs into five major groups based on their overall activity and structural alterations: overall-change, shortened, hollowed, shifted and others. By assessing differential SEs with the enriched TFs and linked target genes, we found distinct characteristics associated with different groups of SEs, such as linking with different numbers of genes and affecting gene expression at divergent impact. When compared with the widely adapted binary approach, DASE found an improved list of differential SEs which are linked to cell-type-specific gene function. This highlighted the elevated performance by DASE identification. It further demonstrated the increased power in identifying cell-line-specific SE regulation when applied to similar cell lines.
Specifically, our improved performance is powered by the consideration of SE internal dynamics. For instance, SEs might show frequent internal alterations yet with no overall activity changes, as shown in our study. These differences, however, if not accounted for, could under-estimate the genome-wide variation of SE profiles and consequentially bias the evaluation of SE effects on gene regulation. On the other side, significant activity changes of SEs are usually combined with structural alterations, either globally or partially, indicating modeling structural differences won’t lose specificity in detecting true SE differences. However, we did notice that some SEs hold marginal activity changes which were weighted differently as discrepant calls between the binary strategy and our methods. Nevertheless, these SEs usually showed lower effects on gene expression compared to other differential SEs. Especially, those discrepant differential SEs could regulate genes in alternative way by altering the number of linked gene targets if they present significant structural alterations.
One limitation of our methods is that we cannot identify structural differences when a SE contains only one CE. We proposed a weighted spline model to account for the contribution of CEs by their width and activities. Thus, the model requires at least two CEs within a SE to generate a confident estimation. In practice, we identified SEs with only one CE as either non-differentials or overall-change if their activities are significantly altered. In addition, we identified differential SEs as other complex scenarios if their internal patterns cannot be attributed to all other categories. We detailed this in the Materials and Methods section. In practice, we found this category only accounts for a small portion of SEs (Figure 5A). We leave a closer interpretation of such complexity to future work.
It is noted that the differential analysis by DASE is performed based on the merged CEs and SEs between conditions, a widely adapted strategy in differential analysis of ChIP-seq (41). However, if the width and coverage of the merged CEs/SEs increase dramatically from the original regions, it might indicate unreasonable merging that causes unfair comparisons between mismatched CEs/SEs. We thus compared the width and coverage between merged CEs/SEs and CEs/SEs detected in individual samples (Supplementary Figure S10) and found that the values of the two metrics were maintained reasonably. This might be partially explained by (i) enhancers usually show sharp peaks based on H3K27Ac signals; (ii) the locations of enhancer and SE regions (regardless inactive and active) are relatively stable across different cells (partially encoded by the underneath DNA sequence motifs) (56,57).
SEs were conceptionally defined based on the intensity and enrichment of consecutive enhancers (2,3). As a result, significant changes of SEs may correspond to two scenarios: activity changing between two SEs or status transitions between SEs and regular enhancers. These scenarios may associate with different functional interpretations since regular enhancers tend to regulate less and closer genes compared to SEs. Although we did not provide approaches to discriminate the two scenarios as that goes beyond the scope of our proposed study, feasible strategies could be implemented in future work to improve the downstream interpretation. For instance, scanning the distances between SEs and gene promoters could help filter regular enhancers as they are usually close to their gene targets (58,59). Also, SE alterations can be linked to the status changes of local chromatin, such as phase separation (60), to help determine if transitions occur between SE and regular enhancers. These require the integration of additional datasets to define chromatin statuses.
DATA AVAILABILITY
All used datasets were obtained from ENCODE and GEO data portal. The dataset accession IDs are reported in Supplementary Table S1. DASE is available as an R package here: https://github.com/tenglab/DASE.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Ann Chen for the insightful discussion on our work.
Contributor Information
Xiang Liu, Department of Biostatistics and Bioinformatics, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA.
Bo Zhao, Division of Infectious Disease, Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA 02115, USA.
Timothy I Shaw, Department of Biostatistics and Bioinformatics, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA.
Brooke L Fridley, Department of Biostatistics and Bioinformatics, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA.
Derek R Duckett, Department of Drug Discovery, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA.
Aik Choon Tan, Department of Biostatistics and Bioinformatics, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA.
Mingxiang Teng, Department of Biostatistics and Bioinformatics, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Biostatistics and Bioinformatics Shared Resource with Moffitt Cancer Center Grant from NCI [P30 CA076292, in part]; NIAID [R01 AI123420]; NCI [R01 CA262530]; DoD [W81XWH-22-1-0025]. Funding for open access charge: Moffitt Cancer Center.
Conflict of interest statement. None declared.
REFERENCES
- 1. Pott S., Lieb J.D.. What are super-enhancers. Nat. Genet. 2015; 47:8–12. [DOI] [PubMed] [Google Scholar]
- 2. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A., Young R.A.. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parker S.C., Stitzel M.L., Taylor D.L., Orozco J.M., Erdos M.R., Akiyama J.A., van Bueren K.L., Chines P.S., Narisu N., Program N.C.S.et al.. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beagrie R.A., Scialdone A., Schueler M., Kraemer D.C., Chotalia M., Xie S.Q., Barbieri M., de Santiago I., Lavitas L.M., Branco M.R.et al.. Complex multi-enhancer contacts captured by genome architecture mapping. Nature. 2017; 543:519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang J., Li K., Cai W., Liu X., Zhang Y., Orkin S.H., Xu J., Yuan G.C.. Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 2018; 9:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li T., Jia L., Cao Y., Chen Q., Li C.. OCEAN-C: mapping hubs of open chromatin interactions across the genome reveals gene regulatory networks. Genome Biol. 2018; 19:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Novo C.L., Javierre B.M., Cairns J., Segonds-Pichon A., Wingett S.W., Freire-Pritchett P., Furlan-Magaril M., Schoenfelder S., Fraser P., Rugg-Gunn P.J.. Long-Range enhancer interactions are prevalent in mouse embryonic stem cells and are reorganized upon pluripotent state transition. Cell Rep. 2018; 22:2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kai Y., Li B.E., Zhu M., Li G.Y., Chen F., Han Y., Cha H.J., Orkin S.H., Cai W., Huang J.et al.. Mapping the evolving landscape of super-enhancers during cell differentiation. Genome Biol. 2021; 22:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loven J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., Bradner J.E., Lee T.I., Young R.A.. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of chip-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amaral P.P., Bannister A.J.. Re-place your BETs: the dynamics of super enhancers. Mol. Cell. 2014; 56:187–189. [DOI] [PubMed] [Google Scholar]
- 13. Allahyar A., Vermeulen C., Bouwman B.A.M., Krijger P.H.L., Verstegen M., Geeven G., van Kranenburg M., Pieterse M., Straver R., Haarhuis J.H.I.et al.. Enhancer hubs and loop collisions identified from single-allele topologies. Nat. Genet. 2018; 50:1151–1160. [DOI] [PubMed] [Google Scholar]
- 14. Jia Y., Chng W.J., Zhou J.. Super-enhancers: critical roles and therapeutic targets in hematologic malignancies. J. Hematol. Oncol. 2019; 12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan J., Jiang Y.Y., Mayakonda A., Huang M., Ding L.W., Lin H., Yu F., Lu Y., Loh T.K.S., Chow M.et al.. Super-Enhancers promote transcriptional dysregulation in nasopharyngeal carcinoma. Cancer Res. 2017; 77:6614–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang C., Zhang L., Ke L., Ding W., Jiang S., Li D., Narita Y., Hou I., Liang J., Li S.et al.. Primary effusion lymphoma enhancer connectome links super-enhancers to dependency factors. Nat. Commun. 2020; 11:6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fullwood M.J., Liu M.H., Pan Y.F., Liu J., Xu H., Mohamed Y.B., Orlov Y.L., Velkov S., Ho A., Mei P.H.et al.. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009; 462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mumbach M.R., Rubin A.J., Flynn R.A., Dai C., Khavari P.A., Greenleaf W.J., Chang H.Y.. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods. 2016; 13:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fullwood M.J., Wei C.L., Liu E.T., Ruan Y.. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res. 2009; 19:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K.et al.. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edgar R., Domrachev M., Lash A.E.. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wong J.P., Stuhlmiller T.J., Giffin L.C., Lin C., Bigi R., Zhao J., Zhang W., Bravo Cruz A.G., Park S.I., Earp H.S.et al.. Kinome profiling of non-Hodgkin lymphoma identifies tyro3 as a therapeutic target in primary effusion lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:16541–16550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Amemiya H.M., Kundaje A., Boyle A.P.. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 2019; 9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 26. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu A., Ibrahim J.G., Love M.I.. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2019; 35:2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Consortium E.P., Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A.et al.. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020; 583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.J., Vert J.P., Heard E., Dekker J., Barillot E.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015; 16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lareau C.A., Aryee M.J.. hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat. Methods. 2018; 15:155–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang da W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 34. Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shin H.Y., Willi M., HyunYoo K., Zeng X., Wang C., Metser G., Hennighausen L.. Hierarchy within the mammary STAT5-driven wap super-enhancer. Nat. Genet. 2016; 48:904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Encode Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schuijers J., Manteiga J.C., Weintraub A.S., Day D.S., Zamudio A.V., Hnisz D., Lee T.I., Young R.A.. Transcriptional dysregulation of MYC reveals common enhancer-docking mechanism. Cell Rep. 2018; 23:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen D., Zhao Z., Huang Z., Chen D.C., Zhu X.X., Wang Y.Z., Yan Y.W., Tang S., Madhavan S., Ni W.et al.. Super enhancer inhibitors suppress MYC driven transcriptional amplification and tumor progression in osteosarcoma. Bone Res. 2018; 6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lancho O., Herranz D.. The MYC Enhancer-ome: long-range transcriptional regulation of MYC in cancer. Trends Cancer. 2018; 4:810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang C., Jiang S., Zhang L., Li D., Liang J., Narita Y., Hou I., Zhong Q., Gewurz B.E., Teng M.et al.. TAF family proteins and MEF2C are essential for epstein-barr virus super-enhancer activity. J. Virol. 2019; 93:e00513-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R.et al.. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pellicano F., Scott M.T., Helgason G.V., Hopcroft L.E., Allan E.K., Aspinall-O’Dea M., Copland M., Pierce A., Huntly B.J., Whetton A.D.et al.. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells. 2014; 32:2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lin C.H., Chang C.Y., Lee K.R., Lin H.J., Chen T.H., Wan L.. Flavones inhibit breast cancer proliferation through the akt/foxo3a signaling pathway. BMC Cancer. 2015; 15:958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hornsveld M., Smits L.M.M., Meerlo M., van Amersfoort M., Groot Koerkamp M.J.A., van Leenen D., Kloet D.E.A., Holstege F.C.P., Derksen P.W.B., Burgering B.M.T.et al.. FOXO transcription factors both suppress and support breast cancer progression. Cancer Res. 2018; 78:2356–2369. [DOI] [PubMed] [Google Scholar]
- 45. Dong H., Claffey K.P., Brocke S., Epstein P.M.. Inhibition of breast cancer cell migration by activation of cAMP signaling. Breast Cancer Res. Treat. 2015; 152:17–28. [DOI] [PubMed] [Google Scholar]
- 46. Mihaylova M.M., Shaw R.J.. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011; 13:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kodama Y., Baxter R.C., Martin J.L.. Insulin-like growth factor-I inhibits cell growth in the a549 non-small lung cancer cell line. Am. J. Respir. Cell Mol. Biol. 2002; 27:336–344. [DOI] [PubMed] [Google Scholar]
- 48. Lee H., Lee H., Chin H., Kim K., Lee D.. ERBB3 knockdown induces cell cycle arrest and activation of bak and Bax-dependent apoptosis in colon cancer cells. Oncotarget. 2014; 5:5138–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liao C.H., Yeh C.T., Huang Y.H., Wu S.M., Chi H.C., Tsai M.M., Tsai C.Y., Liao C.J., Tseng Y.H., Lin Y.H.et al.. Dickkopf 4 positively regulated by the thyroid hormone receptor suppresses cell invasion in human hepatoma cells. Hepatology. 2012; 55:910–920. [DOI] [PubMed] [Google Scholar]
- 50. Dong M., Blobe G.C.. Role of transforming growth factor-beta in hematologic malignancies. Blood. 2006; 107:4589–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu T., Zhang L., Joo D., Sun S.C.. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017; 2:17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Capasso M., Diskin S.J.. Genetics and genomics of neuroblastoma. Cancer Treat. Res. 2010; 155:65–84. [DOI] [PubMed] [Google Scholar]
- 53. Bai X., Shi S., Ai B., Jiang Y., Liu Y., Han X., Xu M., Pan Q., Wang F., Wang Q.et al.. ENdb: a manually curated database of experimentally supported enhancers for human and mouse. Nucleic Acids Res. 2020; 48:D51–D57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cesarman E., Moore P.S., Rao P.H., Inghirami G., Knowles D.M., Chang Y.. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood. 1995; 86:2708–2714. [PubMed] [Google Scholar]
- 55. Arvanitakis L., Mesri E.A., Nador R.G., Said J.W., Asch A.S., Knowles D.M., Cesarman E.. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of epstein-barr virus. Blood. 1996; 88:2648–2654. [PubMed] [Google Scholar]
- 56. Ernst J., Kellis M.. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010; 28:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marco E., Meuleman W., Huang J., Glass K., Pinello L., Wang J., Kellis M., Yuan G.C.. Multi-scale chromatin state annotation using a hierarchical hidden markov model. Nat. Commun. 2017; 8:15011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fulco C.P., Nasser J., Jones T.R., Munson G., Bergman D.T., Subramanian V., Grossman S.R., Anyoha R., Doughty B.R., Patwardhan T.A.et al.. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019; 51:1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moore J.E., Pratt H.E., Purcaro M.J., Weng Z.. A curated benchmark of enhancer-gene interactions for evaluating enhancer-target gene prediction methods. Genome Biol. 2020; 21:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C.et al.. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018; 361:eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All used datasets were obtained from ENCODE and GEO data portal. The dataset accession IDs are reported in Supplementary Table S1. DASE is available as an R package here: https://github.com/tenglab/DASE.