


Abstract
Genome instability has been recognized as a key driver for microbial and cancer adaptation and thus plays a central role in many diseases. Genome instability encompasses different types of genomic alterations, yet most available genome analysis software are limited to just one type of mutation. To overcome this limitation and better understand the role of genetic changes in enhancing pathogenicity we established GIP, a novel, powerful bioinformatic pipeline for comparative genome analysis. Here, we show its application to whole genome sequencing datasets of Leishmania, Plasmodium, Candida and cancer. Applying GIP on available data sets validated our pipeline and demonstrated the power of our tool to drive biological discovery. Applied to Plasmodium vivax genomes, our pipeline uncovered the convergent amplification of erythrocyte binding proteins and identified a nullisomic strain. Re-analyzing genomes of drug adapted Candida albicans strains revealed correlated copy number variations of functionally related genes, strongly supporting a mechanism of epistatic adaptation through interacting gene-dosage changes. Our results illustrate how GIP can be used for the identification of aneuploidy, gene copy number variations, changes in nucleic acid sequences, and chromosomal rearrangements. Altogether, GIP can shed light on the genetic bases of cell adaptation and drive disease biomarker discovery.
INTRODUCTION
In recent years, the field of genomics has rapidly expanded with a fast increase in the number of newly sequenced genomes (1). This surge is a direct consequence of the development of new and ever more efficient, high-throughput capable sequencing technologies (2). On the one hand, the improvement in long reads technology allowed the generation of high-quality genome assemblies (3,4). On the other hand, the decreasing costs for short-reads sequencing and the parallel increase in sequencing throughput propelled the exponential increase of available whole genome sequencing (WGS) data (5). Thanks to these advances one can reasonably expect WGS to rapidly become a key component of personalized medicine and clinical applications.
Indeed, WGS technologies have revolutionized many applications in the fields of medicine and microbiology. WGS can be used for clinical sample screenings in the diagnosis, classification and surveillance of microbial pathogens (6). WGS allows strain identification with better resolution compared to genetic marker-based methods and can inform on the accessory genome of microbes undergoing horizontal gene transfer (6,7). Other important applications of WGS include antimicrobial resistance (AMR) profiling (8), the identification of candidate antigens in vaccine development (9,10), the tracking of outbreaks within hospitals and communities (6,11–15) and microbial evolutionary adaptation (16–18), which can have profound effects on human infections. In this adaptation process, cycles of genetic mutation and environmental selection lead to microbial fitness gain, resulting in drug resistance and shifts in tropism or virulence. As such, genome instability often determines disease outcome (19–22) and is a key driver of phenotypic and genetic variability of microbes and other pathogenetic systems that rely on genome instability for adaptation, such as cancer.
In this context, several consortia-based projects have been established with the goal to produce WGS for the study of different biological systems (5), and a number of publicly available databases have been compiled or updated (23). Parallel to data availability, many bioinformatics tools have been developed to perform specific genome analysis tasks (24,25). For instance, tools such as Freebayes (26), CNVnator (27) and DELLY (28) have been respectively used for the detection or characterization of DNA single nucleotide variants (SNVs), copy number variations (CNVs), and structural variations (SVs), but their scope is limited to the analysis of one genomic feature at the time. A number of integrative WGS pipelines and workflows have been established combining the execution of multiple bioinformatics algorithms serving different analysis steps (29). Even though continuous progress has been made, there is no standardized or unified approach for genomic investigation. For the development of improved WGS data analysis pipelines, several important requirements need be considered, including portability, reproducibility, scalability and compatibility with high-performance computing (HPC) clusters and remote cloud computing. Here we introduce a novel genome instability pipeline (GIP) that fulfills all these requirements. GIP facilitates the genome-wide detection, quantification, comparison and visualization of chromosome aneuploidies, gene CNVs, SNVs and SVs. GIP is implemented in Nextflow (30), a workflow language that allows to execute GIP seamlessly in local workstation, on an HPC or remotely in the cloud. All required environment and software dependencies of GIP are fulfilled and provided with a Singularity container, thus making GIP reproducible, easy-to-install and easy-to-use. GIP allows the use of giptools, a tool-suit of R-based modules for genome data exploration, enabling the comparison of sample sub-sets. GIP and giptools generate a summary report with publication-quality figures and spreadsheet tables. GIP and giptools constitute a single framework for WGS analysis suitable both for large scale batch analysis of individual genomes and comparison of samples from different experimental conditions or origins. Lastly, a key strength of GIP and giptools is the general applicability to different eukaryotic species. We already successfully applied GIP on the analysis of Leishmania genomes (16,31,32). In this study, we validate the use of GIP and giptools using WGS data from published datasets of the three major human pathogens Leishmania infantum, Plasmodium vivax and Candida albicans and as well as three human cancer cell lines. Furthermore, we demonstrate how the extensive and powerful analytical approach operated by GIP and giptools can be used to find new biological signal that escaped previous analyses.
MATERIALS AND METHODS
Genome sequencing and genome assembly data
WGS reads were downloaded from the Sequence Read Archive (SRA) (33), the European Nucleotide Archive (ENA) (34) repositories and the Encyclopedia of DNA Elements (ENCODE) dashboard (35) (Supplementary Table S1). For L. infantum the GCA_900500625 genome reference and gene annotations available from the ENSEMBL protists server (release-48) were used (36). For C. albicans the assembly 21 of the SC5314 strain genome reference and gene annotations available from the Candida Genome Database (CGD) were used (37). For P. vivax the P01 reference genome and gene annotations available from PlasmoDB (release-50) were used (38). For the cancer cell lines, the human genome GRCh38 primary assembly and gene annotations available from ENSEMBL (release-102) were used (36).
GIP and giptools
All results presented in this study were generated using GIP and giptools version 1.0.9. GIP code is maintained and freely distributed at the github page: https://github.com/giovannibussotti/GIP. Figure 1 and Supplementary Figure S1 provide a schematic representation of the GIP workflow. The giptools container is accessible from the Singularity cloud at https://cloud.sylabs.io/library/giovannibussotti/default/giptools. The GIP configuration files (Supplementary Data 1) and the giptools command options used to generate all results (Supplementary Data 2) are provided. The full documentation of GIP and giptools including a description of all options is available from https://gip.readthedocs.io/en/latest/.
Figure 1.

GIP workflow. The schema on the left recapitulates the GIP inputs, processes and outputs (see Materials and Methods). Blue, orange, green and purple boxes indicate genome reference pre-processing, read mapping, quantification and variants computation modules, respectively. The panels on the right demonstrate example plots included in the GIP report computed for individual samples. The ‘Karyotype’ plot shows the coverage distributions for each chromosome (y-axis). The ‘Genomic bins’ plot shows the genomic position (x-axis) and the normalized genomic bin sequencing coverage (y-axis). The ‘Gene CNVs’ plot shows the normalized gene sequencing coverage. The ‘Structural variants’ panel shows a Circos plot representing translocations (black links in the inmost part of the plot), and other possible structural variations in the outer tracks, including insertions, duplications, deletions and inversions. The outmost track shows the normalized sequencing coverage. The ‘Single nucleotide variants’ plot shows on the x and y axes respectively the genomic position and variant allele frequency of detected SNVs.
Read alignment
The repetitive elements of reference genomes were soft-masked by GIP using Red (39). WGS reads were mapped by GIP using BWA-mem (version 0.7.17) (40,41) run with option -M to label shorter split hits as secondary. Then the alignment files were sorted, indexed and reformatted by GIP using Samtools (version 1.8) (42). Finally, read duplicates were removed by GIP using Picard MarkDuplicates (http://broadinstitute.github.io/picard) (version 2.18.9) with the option ‘VALIDATION_STRINGENCY = LENIENT’. In the four considered datasets, WGS reads were aligned against full assemblies, including unsorted contigs if present. However just the canonical assembled chromosomes were considered for all downstream analyses (‘chrs’ option, Supplementary Data 1). A minimum read alignment MAPQ score was adopted to select genes for cluster analysis, and to call for SNVs and SVs (‘MAPQ’ option, Supplementary Data 1). Altogether a total of 6 306 951 266 reads were aligned to the respective reference genomes. The ‘giptools overview’ module was run to gather the alignment statistics as estimated by Picard CollectAlignmentSummaryMetrics (Supplementary Table S2).
Genomic bins and genes quantification
GIP was used to evaluate the mean sequencing coverage and the mean read MAPQ of genomic bins and genes. For genomic bins, GIP partitioned the input genomes into adjacent windows of user defined lengths (‘binSize’ option, Supplementary Data 1). The coverage GC-content score bias was corrected (‘CGcorrect’ option, Supplementary Data 1) fitting a LOESS regression with a 5-fold cross validation to optimize the model span parameter. A larger window length was utilized to bin the reference genomes for circos plot representations (‘binSizeCircos’ option, Supplementary Data 1). In Figure 1 (‘Genomic bins’ and ‘Gene CNVs’ plots), Figure 2, Supplementary Figures S2, S3 and Figure 4 bins and genes coverage scores were normalized by median chromosome coverage to highlight amplifications or depletions with respect to the chromosome copy number. In Figure 1 (‘Structural variants’ plot), Figure 3E–G, Figure 5B, Supplementary Figure S6B, C, and Supplementary Figure S7 bins and genes coverage scores were normalized by median genome coverage to account for sequencing library size differences. GIP evaluated statistically significant copy number variant bins and genes (Figure 1 ‘Genomic bins’ and ‘Gene CNVs’ plots) using a P-value threshold of 0.001 (‘covPerBinSigOPT’ and ‘covPerGeSigOPT’ options, Supplementary Data 1). Estimated P-values for bins and genes CNVs were corrected for multiple testing using the Benjamini – Yekutieli (‘–padjust BY’) and the Benjamini – Hochberg (‘–padjust BH’) methods. The somy scores shown in Figure 1 (‘Karyotype’ plot) and Figure 5A were computed multiplying the median genome coverage normalized bin coverage by 2. GIP enabled the CNV analysis of genes sharing high sequence identity by clustering the nucleotide sequences of the genes with low mean MAPQ score into groups with cd-hit-est (version 4.8.1) (43) with options ‘-s 0.9 -c 0.9 -r 0 -d 0 -g 1’. Then for each gene cluster GIP computed the mean gene coverage normalized by median chromosome coverage (Figure 2E, Supplementary Figure S3B). The predictions of CNV regions returned by GIP for the P. vivax dataset were compared to the ones available from previously published work (44). For the analysis we utilized the 178 available samples not affected by national export restrictions. The sample list is available from https://www.malariagen.net/sites/default/files/PvGV_May2016_sample_data_2.xlsx. To enable the comparison, GIP was re-executed considering the same genome refence (Pvax_Sal1) and CNV length cutoff (>3 kb) used in the published work.
Figure 2.

Comparing Leishmania infantum genomes with giptools. (A) Density plot representing the genomic coverage of the seven L. infantum isolates. The x-axis shows the log10 normalized coverage of genomic bins. The y-axis reflects the genomic position. The thirty-six different chromosomes are materialized as separate panels. The blue shading indicates the (2D) kernel density estimates of genomic bins. The two red vertical lines mark the 1.5 and 0.5 coverage values. A selection of 50 000 bins with coverage >1.5 or <0.5 is shown as black dots. (B) Scatterplot of the genomic bin normalized sequencing coverage ratio of samples LIPA83 over ZK43. The x any y axes show the ratio score and the genomic position respectively. Ratio scores >1.25 are labelled in orange and indicate genomic bin amplification. Ratio scores <0.75 are labelled in blue and indicate genomic bin depletion. (C) Ternary comparison showing the relative abundance of the genomic in samples LIPA83, ZK43 and ZK28. The axes report the fraction of the bins normalized sequencing coverage in the three strains. The blue contour indicates the log10 bin density. A subset of 5,000 bins is shown as black dots. Each given point in the plot is adding up to 100. The density area at the center of the plot indicates bins with equal copy number and thus a ∼33 distribution across the three axes. (D) Scatterplot showing the log10 normalized sequencing coverage of annotated genes in ZK43 (x-axis) and LIPA83 (y-axis). The red line indicates the bisector. Dots represent individual genes. (E) Sequencing coverage ratio of gene clusters in samples LIPA83 and ZK43. Dots represent gene clusters. For plots D and E, the ratio scores >1.5 or <0.5 are labelled in orange and blue respectively.
Figure 4.

Gene CNVs interactions. (A) All-vs-all normalized sequencing coverage correlation heatmap. The heatmap is symmetrical along its diagonal and reports both on the rows and the columns the detected gene CNVs. The colour scale indicates with green and pink high levels of positive and negative Pearson correlation, respectively. The side ribbons demonstrate in different colours the chromosome and the correlation cluster of each gene. (B) Gene CNV heatmap. The columns and the rows report respectively the samples and the detected gene CNVs. The colour scale indicates the normalized sequencing coverage of the genes. To ease visualization, coverage values greater than 3 are reported as 3 (red). Black boxes highlight the genomic regions shown in Supplementary Figure S6B (panels 1, 2 and 3) and Supplementary Figure S6C. The ribbons on the left indicate the chromosome and the correlation cluster of each gene. Top ribbons indicate the genotype and the strains resulting from the different evolutionary experiments. (C) Gene interaction network. Nodes indicate gene CNVs. Edges reflect the absolute Pearson correlation value. The closer the nodes are, the higher is the correlation. Only significant interactions (Benjamini–Hochberg adjusted P-value < 0.01) are shown. The colour of the edges indicates in red and blue respectively positive and negative correlations. The colour of the nodes denotes the predicted network cluster for each gene.
Figure 3.

Plasmodium vivax genomic diversity. (A) Predicted maximum likelihood phylogenetic tree reconstruction. (B) PCA analysis of the phylogenetic distances estimated from the tree in (A). Each dot indicates a sample. The colour code reflects the geographic origin of the samples and matches with the colours of the legend in (A). (C) Venn diagram comparing the SNVs of five representative Ethiopian strains. (D) Pairwise scatterplot comparing the variant allele frequency of all detected SNVs in the five Ethiopian strains. (E) Gene panel analysis. The x-axis reports a set of genes of interest. The y-axis indicates the normalized mean gene coverage. The boxplots demonstrate the coverage values distributions for each gene across all samples. Each dot represents the coverage of the indicated gene in a given sample. Dot colours reflect the sample geographic origin as in (A). (F) Reads per kilo base per million mapped reads (RPKM) normalized sequencing coverage density track of sample PD0689_C. The boundaries of the 14 chromosomes are shown on the bottom. (G) Comparison of non-synonymous (N) and synonymous (S) mutations between Ethiopia and Cambodia sample groups. Dots represent genes. The x-axis represents the difference between the mean non-synonymous mutation count in the two sample groups. The y-axis represents the difference between the mean synonymous mutation count in the two sample groups. The dot size demonstrates the ratio of the mean normalized sequencing coverage between the two sample groups for each gene. Red and blue dot colors indicate genes belonging to the 43 genes panel (52) and the custom 6 genes panel respectively.
Figure 5.

Cancer cell lines genome instability. Green, brown and grey colours indicate respectively samples K562, NCI_H460 and T47D. (A) Chromosome coverage analysis. The x-axis reports the chromosomes. The y-axis reports the estimated somy score. The boxes show the somy score distributions. (B) Sub-chromosomal copy number variation. Dots indicate genomic bins. Different panels indicate different chromosomes. The panel columns indicate from left to right four selected chromosomes: 6, 9, 10 and 16. The panel rows show top to bottom the samples K562, NCI_H460 and T47D. The x-axis indicates the genomic position. The y-axis indicates the normalized genomic bin sequencing coverage values. Coverage values greater than 5 are reported as 5. (C) SNV frequency density plots. The four different panels represent different selected chromosomes: 6, 9, 10 and 16. The x-axis reports the variant allele frequency. The y-axis the estimated kernel density between 0 and 3. (D) SNV frequency scatter plots. The four different panels represent different selected chromosomes: 6, 9, 10 and 16. The x-axis indicates the genomic position. The y-axis indicates the variant allele frequency. (E) Chromosome 11 combined SNV and bin coverage plot. To ease visualization, giptools allows the simultaneous displaying of variant allele frequencies (y-axis, left) and sequencing coverage (y-axis, right). Dots represent SNVs. The lines represent the normalized bin sequencing coverage. The x-axis indicates the genomic position. Coverage values >5 are shown as 5.
Gene ontology and metabolic pathway enrichment
The FungiDB online tool (Release 52, 20 May 2021) (45) was used to evaluate the functional enrichment of network gene clusters. For the gene ontology analysis, the biological process (BP), molecular function (MF) and cellular compartment (CC) terms enrichments were tested, considering both computed and curated evidences and a P-value cutoff of 0.05. For the metabolic pathway enrichment, both KEGG (46) and MetaCyc (47) pathway sources were considered with a P-value cutoff of 0.05. Terms and pathways with Benjamini – Hochberg adjusted P-values <0.05 were considered statistically significant.
Sequencing coverage density estimates
GIP was used to convert the read alignment files (.bam format) in binary data files reflecting sequencing coverage (.bigWig format). The coverage file were produced using bamCoverage from the deepTools2 suite (48) (version 3.5.1) with options ‘–normalizeUsing RPKM –ignoreDuplicates –binSize 10 –smoothLength 30’ (‘bigWigOPT’ option, Supplementary Data 1). The coverage track of sample PD0689_C was visualized with IGV using the ‘Bar Chart’, ‘Autoscale’ and windowing function ‘Mean’ options.
Single-nucleotide variant analysis
GIP was used to call SNVs using Freebayes (version 1.3.2) (‘freebayesOPT’ option, Supplementary Data 1) and filter its output (‘filterFreebayesOPT’ option, Supplementary Data 1). Filters included the minimum allele frequency (‘–minFreq’), the minimum number of reads supporting the alternative alleles (‘–minAO’) and minimum mean mapping quality of the reads supporting the reference (‘–minMQMR’) or the alternative allele (‘–minMQM’). A higher number of reads supporting the variants was requested for predictions positioned inside simple repeats of the same nucleotide (homopolymers) (‘–minAOhomopolymer’). The homopolymers were defined as the DNA region spanning ±5 bases from the SNV (‘–contextSpan 5’), with over 40% of identical nucleotides (‘–homopolymerFreq 0.4’). Further, GIP discarded SNVs with sequencing coverage above or below 4 median absolute deviations (MADs) from the median chromosome coverage (‘–MADrange’). SnpEff (version 4.3t) (49) was used to predict the impact of SNVs on coding sequence. The predicted effects that GIP considered synonymous mutations are: ‘synonymous_variant’, ‘stop_retained_variant’ and ‘start_retained’. The predicted effects that GIP considered non-synonymous mutations are: ‘missense_variant’, ‘start_lost’, ‘stop_gained’, ‘stop_lost’ and ‘coding_sequence_variant’. The phylogenetic tree was computed by the giptools module ‘phylogeny’ using IQtree2 (version 2.1.2) (50,51) with options ‘–seqtype DNA –alrt 1000 -B 1000’. The Venn-diagram comparison considered the strains QS0044_C, QS0001_C, QS0037_C, QS0016_C and SGH_358 that were sampled from different locations in Ethiopia, respectively Habala, Badowacho, Arbaminch, Hawassa and Jimma. The strains were selected to have comparable average genome coverage (52). To infer the tree GIP considered the set of filtered SNV and adopted the IUPAC ambiguous notation for the positions with allele frequency <70%. The tree was visualized by giptools using the ggtree R-package (53).
Analysis of structural variants
GIP was used to detect structural variants including insertions, tandem duplications, deletions, inversions and translocations with DELLY (version 0.8.7) (28). SVs predictions were performed on individual samples separately. To reduce incorrect predictions the DELLY output was additionally filtered (‘filterDellyOPT’ option, Supplementary Data 1). GIP discarded poor predictions with DELLY label ‘LowQual’ (‘–rmLowQual’) and low median MAPQ score of mapping reads (‘–minMAPQ’). SVs positioned in proximity of chromosome ends were removed (‘–chrEndFilter’) to limit false predictions caused by potential misassembled regions close to the telomeric ends. To ease visualization and limit the analysis only to best supported SVs GIP restricted the output only to the top predictions (‘–topHqPercentIns’, ‘–topHqPercentDel’, ‘–topHqPercentDup’ and ‘–topHqPercentInv’) based on the SV support score as in Formula (1), where DV, DR, RV and RR are respectively the number of high-quality variant pairs, reference pairs, variant junction reads and reference junction reads.
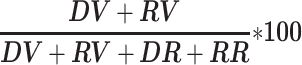 |
Formula 1: SV support score.
The predicted structural variants were represented with Circos (version 0.69–9) (54).
Comparison of CNV callers
GIP was compared to CNVnator (version 0.4.1) (27) and cn.MOPS (version 1.36.0) (55) to predict gene CNVs in the L. infantum dataset (Supplementary Table S1). To run CNVnator the commands ‘-tree’, ‘-his’, ‘-stat’, ‘-partition’ and ‘-call’ were used. Following the authors’ recommendations, the ‘binSize’ parameter was benchmarked using the ‘-eval’ command optimizing the ratio of average read depth signal to its standard deviation. A common optimal ‘binSize’ value of 150 was selected considering the read length and the read depth of the sequencing libraries. Cn.MOPS was run using a windows length (WL) of 150. The WL value was chosen accounting for read depth differences between samples to have approximately an average of 50–100 reads per segment. To account for ploidy differences between samples cn.MOPS was run applying separate normalizations for each chromosome. The estimated chromosome somy score rounded to its closest integer was used as normalization factor with the ‘normalizeChromosomes’ function.
GIP and giptools running time and computational resources
The ‘-with-timeline’ Nextflow option was used to render HTML timelines for all GIP executed processes. The ‘-with-report’ Nextflow option was used to summarize GIP computational resources. A detailed description of each of the two options is available from Nextflow documentation at https://www.nextflow.io/docs/latest/tracing.html#. The giptools computational requirements were estimated using the Linux ‘time’ command (URL https://man7.org/linux/man-pages/man1/time.1.html) together with the ‘-v’ option to obtain a verbose output.
RESULTS
The GIP workflow
GIP is a tool for scientific investigation compatible with Linux systems, requiring minimal configuration and distributed as a self-contained package. Our integrative approach provides a broad range of genomic data analyses and visualizations, and combines new and existing bioinformatic methods (see Materials and Methods). GIP consists of three files: the Nextflow pipeline code, the configuration and the Singularity container files. The pipeline container conveniently provides a working environment with 19 off-the-shelf applications for genomic analyses and 40 R packages listed at https://gip.readthedocs.io/en/latest/software/index.html. The minimum required input is a paired-end WGS data set and a reference genome assembly in the standard fastq and FASTA formats, respectively. GIP analyses include (i) extracting genomic features such as assembly gaps or repetitive elements, (ii) mapping the reads, (iii) evaluating chromosome, gene and genomic bin copy numbers, (iv) identifying and visualizing copy number variation with respect to the reference genome, (v) identifying and quantifying gene clusters, (vi) detecting and annotating SNVs, (vii) measuring non-synonymous (N) and synonymous (S) mutations for all genes, (viii) detecting SVs including tandem duplications, deletions, inversions and break-ends translocations using split-read and read-pair orientation information and (ix) producing a report file providing summary statistics, tables and visualizations (Figure 1, Supplementary Figure S1). For SNVs, calling GIP relies on Freebayes, a popular tool that resolves the issue of having multiple potential ambiguous alignments between the read and the homologous genomic region by examining the whole haplotype of the read independently of the precise alignment positions. It was shown that compared to alternative variant calling software Freebayes demonstrates good performance across different aligners and Illumina platforms (56). For SV detection GIP runs DELLY, a method combining short insert paired-ends, long-range mate-pairs and split-read alignments to precisely define balanced and unbalanced forms of genomic rearrangements at single-nucleotide resolution. DELLY’s prediction accuracy was previously validated by PCR (28) and its performance compared to other structural variant calling algorithms on simulated data under different sequencing parameter settings (28). The benchmark results indicate that DELLY possess very high-positive predictive value and a robust performance across the simulated sequencing parameter space (28). GIP allows to customize filtering and visualization options via the configuration file (see methods). The output of GIP can be used as input for giptools, a tool-suite to compare sample sub-sets and highlight chromosome copy number, gene copy number and SNV differences.
Applying giptools on a Leishmania infantum case study
GIP permits the batch analysis of a set of individual samples, where each sample is considered separately and compared only with respect to the provided reference genome assembly. As a consequence, all variants and copy number alterations detected in a sample merely reflect the differences between the sequenced and the reference genomes. While this application may be sufficient in some circumstances, research projects often involve downstream comparison between samples. Examples include the comparison of gene or chromosome copy variation number between for example drug resistant and drug susceptible samples, or the juxtaposition of SNVs detected in isolates from different geographic areas. For this purpose, we developed giptools, a suit of thirteen modules that allows to compare samples processed by GIP (Table 1). All modules in giptools are fully embedded in the Singularity container and they are provided with their own documentation.
Table 1.
giptools modules
| Module name | Purpose |
|---|---|
| karyotype | Compare chromosome sequencing coverage distributions |
| binCNV | Compare bin sequencing coverage in two samples |
| geCNV | Compare gene sequencing coverage in two samples |
| ternary | Compare gene sequencing coverage in three samples |
| ternaryBin | Compare bin sequencing coverage in three samples |
| SNV | Compare SNVs in multiple samples |
| binDensity | Density plot of bin sequencing coverage of multiple samples |
| geInteraction | Detect CNV genes in multiple samples and produce correlation-based networks |
| genomeDistance | Compare sample genomic distances |
| phylogeny | Extract SNV union and infer phylogenetic tree |
| convergentCNV | Detect convergent CNV gene amplifications |
| overview | Overview of sequencing coverage of chromosomes, genomic bins and genes |
| panel | Extract genomic information of a gene panel |
To illustrate the type of exploratory data analyses and the biological questions that can be addressed, we tested giptools on a previously analyzed dataset of seven clinical L. infantum isolates from Tunisia (31). Leishmania is the etiological agent of leishmaniasis, a life-threatening human and veterinary disease affecting 12 million people worldwide (57). Parasites were derived from seven patients affected with visceral leishmaniasis, expanded in cell culture and their genomes were sequenced. This dataset includes four Glucantime drug susceptible isolates and three isolates from relapsed patients, and their comparison may inform on genetic factors resulting in treatment failure. Giptools allowed the detection and visualization of pervasive intra-chromosomal CNVs across the thirty-six Leishmania chromosomes (Figure 2A). Additionally, giptools enables targeted comparison of normalized genomic bin sequencing coverage of sample pairs. We used giptools’ ‘binCNV’ module to compute the ratio between corresponding genomic bins of the strains LIPA83 over ZK43, which correspond to a first-episode and a relapse leishmaniasis isolate, respectively. Giptools represents different chromosomes as separate panels (Figure 2B), as part of single genome-wide overview (Supplementary Figure S2A) or as distinct plots (Supplementary Figure S2B). This analysis allowed the identification of 2,905 and 2,208 bins that were respectively amplified or depleted in LIPA83 with respect to ZK43. The results are returned by giptools as a Microsoft Excel table (.xlsx format) providing ratio scores at each genomic position (Supplementary Table S3). Likewise, giptools permits three-way comparisons of normalized genomic bin sequencing coverage with ternary plots (Figure 2C). We used this representation to display the genomic bin relative abundance in samples ZK43, LIPA83 and ZK28. The analysis shows important strain-specific differences in bin copy number that are visualized by shifts of the signals out of the center. Similar to genomic bin analysis, giptools makes it possible to compare the sequencing depth of annotated genes and thus the copy number in two or three samples (Figure 2D and Supplementary Figure S3). However, the determination of gene copy number might be impeded by (i) short read length or fragment insert size, (ii) the complexity of the target genome, or (iii) the presence of repetitive elements. Together with the coverage, GIP also computes a mean read map quality (MAPQ) score for each gene and allows a different strategy to determine the copy number of low MAPQ genes (see methods). This gene MAPQ score is a measure that reflects how much each gene is supported by unambiguously mapped reads (high MAPQ) in contrast to multimapping reads (low MAPQ). The evaluation of LIPA83 and ZK43 gene coverage ratio scores revealed 13 gene CNVs with a stringent MAPQ cutoff of 50 (Supplementary Table S4). The maximum normalized coverage value of 6.5 was observed for a putative amastin surface glycoprotein (LINF_310009800). Other examples of gene CNVs in this set include the putative surface antigen protein 2 (LINF_120013500) and the heat shock protein HSP33 (LINF_300021600) (Supplementary Table S4). Genes falling below a user defined MAPQ score and sharing high level of sequence similarity are assigned to the same gene cluster, and their measured coverage scores are averaged across all members of the group. Low MAPQ scores can also be associated with single genes, e.g. in the case of internal repetitive elements that cause multiple ambiguous alignments inside the gene itself, or if mapping occurs in possibly misannotated intergenic regions. The LIPA83/ZK43 comparison showed 27 CNV gene clusters, including cluster cl303 (three genes annotated as ‘amastin-like’) and cluster cl16 (2 tb-292 membrane-associated protein-like proteins) (Figure 2E, Supplementary Table S4). These results demonstrate the power of GIP and giptools to detect and compare intra-chromosomal CNVs in Leishmania at genomic bin level. Conveniently, analogous two- or three-ways comparisons can be applied to reveal copy number variations at individual gene or gene cluster levels.
Benchmarking of prediction tools
GIP uses Freebayes and DELLY to perform SNVs and SVs calls while providing additional options to filter and reformat their output for downstream analyses. Freebayes and DELLY predictions were already extensively benchmarked against other tools and validated in previous reports (28,56,58). GIP implements its own approach to quantify, normalize, compare and visualize genomic bins and genes. One gene CNV predicted using GIP was experimentally tested in a separate study where we confirmed by PCR the deletion of a NIMA-like kinase gene (59). In the following we used the L. infantum dataset to compare the gene CNV predictions returned by GIP with the ones of two popular CNV detection tools, CNVnator (27) and cn.MOPS (55). The three methods use sequencing coverage depth information extracted from DNA read alignments. The comparison indicates that most of GIP predictions overlap with the ones of both CNVnator and cn.MOPS (169 or 61.4%) or with CNVnator only (96 or 34.9%) (Supplementary Figure S4A). Overall CNVnator and cn.MOPS predict 13.4 and 3.2 times more CNV genes than GIP, respectively. While this result could reflect a superior sensitivity, it suggests a high rate of false positive predictions caused by confounding chromosome aneuploidy. The chromosomes with the greatest number of CNVnator and cn.MOPS predictions are the ones which are amplified in at least one of the considered isolates (Supplementary Figure S4B). CNVnator can be run on individual samples only, therefore it is not suited to account for potential between-samples chromosome copy number differences. cn.MOPS can be run on multiple samples together and allows the possibility to apply adapted normalizations on each chromosome separately. This approach alleviates the problem of aneuploidy-induced false gene CNV predictions, demonstrating to be effective for some chromosomes (e.g. LinJ12 or LinJ13, Supplementary Figure S4B) but still failing for chromosomes LinJ06, LinJ16, and partially for LinJ23 and LinJ24. In cn.MOPS the normalization factor is an integer value representing the chromosome ploidy. Conceivably, the normalization in cn.MOPS suffers from the within-sample population mosaicism, in which individual cells present chromosome copy number differences. On the contrary, to call bin or gene CNVs GIP normalizes by the actual measured median chromosome sequencing coverage, thus accounting for ‘partial’ chromosome copy number shifts. Overall, GIP uses a maximum of 1.057 Gigabytes of random access memory (RAM) in the ‘covPerGe’ step to calculate normalized gene sequencing coverage values, plus 0.39236 Gigabytes in the ‘geInteraction’ giptools step to evaluate the copy number variant genes. CNVnator and cn.MOPS require 2.727132 and 0.791924 Gb respectively. The cumulative running time to execute the GIP ‘covPerGe’ step on each individual sample and to execute the ‘geInteraction’ giptools step is of 18 min and 59.06 s. For comparison CNVnator employs 12 min and 46.53 s, while and cn.MOPS takes 6 min and 21.04 s. Altogether, these results show that to compute gene CNVs on the L. infantum dataset our pipeline has similar memory requirements but longer execution times compared to CNVnator and cn.MOPS. The analysis confirms that the CNV gene predictions produced by our pipeline largely recapitulate the ones produced by CNVnator or cn.MOPS. Furthermore, GIP provides the added benefit of a higher robustness when predicting intra-chromosomal CNVs in the event of chromosomal aneuploidy.
Comparative genomics of a Plasmodium vivax WGS dataset
We next applied GIP and giptools on other biological systems to demonstrate its broad applicability outside the Leishmania field, including the human apicomplexan parasite P. vivax. Plasmodium vivax is a protist parasite and a human pathogen causing malaria. Plasmodium vivax gives rise every year to 130 million clinical cases (60), and it is estimated that 2.5 billion people are at risk of infection worldwide (61–63). We applied GIP and giptools to investigate genomic variations across a sizeable dataset of 222 P. vivax genomes isolated from clinical samples of 14 countries worldwide (44,52) (Supplementary Table S1). The GIP quantification analysis was able to recover 5 out of the 11 previously reported CNV regions and predicted 73 new large (>3 kb) copy number variant areas (Supplementary Figure S5, Supplementary Table S5). Unmatched CNVs could be explained by sample sets differences. The published study considered a set of 228 samples from which two unspecified samples were removed showing excessive variation in read coverage. Conversely, in our analysis we considered the set of 178 available samples not affected by national export restrictions. The phylogenetic tree reconstruction and PCA analyses (Figure 3A and B) showed a high correlation between genotypes and the geographic origin of the samples. However, we detected substantial genomic variability between isolates collected at smaller geographical scale, with 14 555 SNVs (∼42% of the total) uniquely characterizing representative samples from five Ethiopian study sites (52) (Figure 3C and D). This result may reflect diverging evolutionary trajectories radiating from few founder strains. At gene level we profiled the copy number variations of two gene panels. The first panel accounts for 43 previously described genes encoding for potential erythrocyte binding proteins suggested to operate at the interface of the parasite-host invasion process (52). The second panel includes two drug resistance markers comprising the chloroquine resistance transporter PVP01_0109300 and the multidrug resistance protein 1 PVP01_1010900, and four proteins implicated in red blood cell invasion, such as the merozoite surface protein gene MSP7 PVP01_1219700, the reticulocyte binding protein gene 2c PVP01_0534300, the serine-repeat antigen 3 PVP01_0417000, and the reticulocyte binding protein 2b PVP01_0800700) (64–73). Read depth analysis indicated that four genes in the first panel (PVP01_0623800, PVP01_1031400, PVP01_1031200, PVP01_1031300) show a high degree of variability, with amplifications observed in samples from distinct geographic (Figure 3E). This convergence is sign of strong natural selection, which further sustains the functional importance of these genes in the infection process. Furthermore, six genes positioned on chromosome 14 are absent in the Thai strain PD0689_C as a result of the loss of this chromosome (nullisomy) (Figure 3E and F). Finally, the comparison of synonymous and non-synonymous SNVs in the panel of genes revealed important differences between sample groups. Our analysis indicates an overall higher number of non-synonymous mutations in Ethiopian compared to Cambodian isolates, therefore suggesting a stronger evolutionary pressure acting on the African strains (Figure 3G). Taken together these analyses well illustrate how GIP and giptools can be readily applied for bulk analysis of P. vivax genomes to assess genome diversity, extract evolutionary information and identify potential disease biomarkers.
Gene CNV analysis of Candida albicans evolutionary adapted strains
We next applied GIP and giptools to the human fungal pathogen C. albicans, an opportunistic yeast exhibiting major genome plasticity (74–83) and causing hundreds of thousands of severe infections each year (84). Candidemia, a bloodstream infection with Candida, are often associated with high rates of morbidity and mortality (15–50%) notwithstanding existing antifungal treatments (85,86). We applied GIP and giptools to a C. albicans WGS dataset described in a recent study that covers five different progenitor strains (P75063, P75016, P78042, SC5314, AMS3050) and investigates CNVs driving tolerance and resistance to anti-fungal azole drugs (87) (Supplementary Table S6). We analyzed nineteen samples, including (i) four clinical isolates (P75063, P75016, P78042, SC5314), (ii) seven strains selected in vitro against the anti-fungal drug fluconazole (FLC) (AMS4104, AMS4105, AMS4106, AMS4107, AMS4397, AMS4444, AMS4702), (iii) four isogenic colonies adapted to the drug miconazole (AMS3051, AMS3052, AMS3053 and AMS3054) together with their progenitor (AMS3050) and (iv) three colonies derived from a miconazole-adapted population and isolated on a rich medium (AMS3092, AMS3093 and AMS3094) (87–89). GIP and giptools were able to reproduce previous observations of the amplification of the genes for the drug efflux pumps TAC1 (orf19.3188) and ERG11 (orf19.922), for the stress response proteins HSP70 (orf19.4980), CGR1 (orf19.2722), ERO1 (orf19.4871), TPK1 (orf19.4892), ASR1 (orf19.2344), PBS2 (orf19.7388) and CRZ1 (orf19.7359), and for proteins involved in membrane and cell wall integrity, including CDR3 (orf19.1313), NCP1 (orf19.2672), ECM21 (orf19.4887), MNN23 (orf19.4874), RHB1 (orf19.5994) and KRE6 (orf19.7363) (Supplementary Table S6). Furthermore, the powerful comparative approach of our pipeline permitted the discovery of 1505 genes showing correlating or anti-correlating copy number variations (Figure 4A and B, Supplementary Table S6), which could be assigned to nine distinct correlation clusters (CC) (Supplementary Figure S6A, Supplementary Table S7) that escaped previous analyses. We verified the sequencing coverage of genomic regions encompassing gene CNVs, including three regions amplified in fluconazole resistant strains (Figure 4B, Supplementary Figure S6B) (87) and a region whose amplification correlates with the level of miconazole resistance (87) (Supplementary Figure S6C), as well as the loss of heterozygosity associated to the depletion of chromosome 3 left arm in sample AMS3051 (Supplementary Figure S6D) (87). Eventually, by representing genes and absolute correlation respectively as nodes and edges of a network, we identified nine highly interconnected network clusters (NC) (Figure 4C, Supplementary Table S8). NC7, NC8 and NC9 embody genes from individual chromosomes, respectively chromosomes 1, 3 and 4. The most parsimonious explanation for the high levels of correlation observed in these NCs (Figure 4C) is the occurrence of sub-chromosomal amplifications affecting several adjacent genes. A different scenario is pictured for each of the remaining NCs (NC1–6) where the genes are located on different chromosomes thus suggesting genetic interactions that causes coordinated changes in gene copy number. The gene ontology (GO) and metabolic pathway analyses revealed a significant functional enrichment of genes expressed on the cell surface and involved in the interaction with the host (NC2), gibberellin biosynthesis (NC3), transmembrane nucleobase transporters (NC4) and gluconeogenesis (NC5) (Supplementary Table S9). Altogether, GIP and giptools are validated by reproducing previously published results, and beyond that can drive new biological findings as documented by the discovery of a network of epistatic CNV interactions supporting genomic adaptation in C. albicans populations under drug selection.
Exploring instability of larger genomes using cancer cell lines as a benchmark
The larger genome size, and the higher number of genes and WGS reads can represent a challenge when working with higher eukaryotes. For the purpose of comparison, the human genome is ∼216 times larger than the one of C. albicans we analyse in this study. Therefore, we sought to evaluate the applicability of the GIP and giptools framework to human data and utilized a panel of genomes from cancer cell lines as a test set. In our analyses we considered publicly available WGS data of the cell lines T47D, NCI_H460 and K562 (90), which respectively derive from human breast, lung and blood cancers. The karyotype analysis revealed aneuploidy for all chromosomes except chromosome 4 (Figure 5A). The observed heterogeneity in read depth across chromosomes, illustrated by large interquartile range in the boxplot, suggests sub-chromosomal or episomal copy number variations, or the co-existence of karyotypically different sub-populations. Indeed, the coverage analysis confirmed the pervasive presence of CNVs both at chromosomal and sub-chromosomal levels (Supplementary Figure S7) with remarkable instability observed for specific chromosomes, e.g. chromosomes 6, 9, 10 and 16 (Figure 5B). Overall, we detected 1 647 016 SNVs (Supplementary Data 3) and allele frequency shifts with respect to the reference genome, suggesting haplotype selection and the preferential expression of distinct alleles in different cell lines (Figure 5C and Supplementary Figure S8). Furthermore, we identified repeated loss of heterozygosity events and uneven distribution of SNVs that form ‘patches’ of high frequency correlating with chromosomal and sub-chromosomal CNVs (Figure 5D, E and Supplementary Figure S9). These results identify GIP and giptools as a powerful new platform to reveal loci, genes or alleles that are under natural selection in cancer cells, thus allowing important new insight into the genetic basis of tumor development, cancer cell evolution and drug resistance.
Running times and computational resources
We used the Nextflow built-in options to render the process execution time and the computational resources of each task executed by GIP for each sample considered in this study. As exemplified for the cancer cell line dataset, the GIP read mapping process accounts for the longest execution time for all three samples (Supplementary Figure S10A) and the highest I/O (Input/Output) data access, with peaks of read and written bytes of 773.4 and 598.8 Gb, respectively (Supplementary data 4). The process requiring the highest physical memory is ‘bigWigGenomeCov’ (mean usage 37.18 Gb) (Supplementary Figure S10B). This step runs deepTools2 and bedGraphToBigWig (91) to create wiggle (wig) type files representing continuous-valued sequencing coverage data in indexed binary format (bigWig). The time line and the computational resources usage including CPU, memory, job duration and I/O of all datasets are provided (Supplementary data 4). The computational resources and process execution statistics to run all giptools commands described in this study are provided (Supplementary Table S10). The longest running time (474 578.57 and 4 417.61 s in user and system time, respectively) and the maximum required RAM size (9 465 100 Kb) were measured for the estimation of P. vivax phylogeny (Supplementary Table S10), and largely explained by the execution of IQtree2.
DISCUSSION
Genome instability is a key driver of evolution for microbial pathogens and cancer cells (92) and a major source of human morbidity. Here we introduce GIP and giptools, an integrated framework for the genotype profiling of biological systems exploiting genome instability for adaptation. While our pipeline relies on existing genome analysis tools (Supplementary Figure S1), it also implements its own algorithm that quantifies, corrects by GC content, normalizes, compares, estimates significance and visualizes coverage of genomic bins and genes. Furthermore, it defines and quantifies gene clusters, detects chromosome aneuploidies and discovers gene interactions. We document the power and versatility of GIP and giptools by performing genomic screenings of three major pathogenic eukaryotes and human cancer cell lines. While originally deployed for Leishmania genome analysis, in this study we validate the use of our pipeline on other organisms reproducing expected results. For example, in C. albicans we confirmed the CNVs correlating to drug resistance as well as a loss of heterozygosity event (Supplementary Figure S6B–D). Parallel to this we also show how GIP and giptools can be used for data mining and discovery of new signals that escaped existing tools. New findings include (i) the discovery of the convergent amplification of erythrocyte binding proteins in P. vivax strains sampled from distinct geographic areas (Figure 3E), (ii) the detection of a nullisomic strain (Figure 3F), (iii) the identification of correlated copy number variations between genes positioned on separate chromosomes of C. albicans adapting strains, and (iv) the functional association of such genes, strongly supporting a mechanism of epistatic interactions exerted through gene-dosage changes, and corroborating previous reports on adapting Leishmania populations (59).
Importantly, GIP and giptools overcome key limitations of current analysis tools, such as the breadth of analysis that is often limited to individual types of mutations, and the lack of genome-wide, comprehensive reports. To ease genome instability investigations our pipeline offers a single solution to karyotype, gene CNV, SNV and SV batch analyses, providing summary reports and high-quality, genome-wide visualizations. Furthermore, some tools (27,93–95) identify variations with respects to a reference assembly only, which leaves the between samples comparisons to external tools that need installing, may be incompatible in terms of file format, and may rely on different analytical assumptions. To address this limitation giptools enables custom sample comparisons, to explore differences and common features between genomes, and provides a vast choice of analytical tools with compatible features. Likewise, current tools are often restricted to the analysis of data from one or few species only (96–100), but are not generally applicable to different biological systems, which interferes with the investigation of genome variations across multiple species and the exploration potentially conserved genomic adaptation mechanisms. By contrast, our pipeline limits as much as possible the use of hardcoded parametrization, which could limit its use to a specific organism. Therefore, GIP’s flexible design makes it adapted for the genome analysis of both model and non-model organisms, including Leishmania or human.
Many current tools are further limited in software portability and reproducibility across different computer environments, which can produce faulty results calling their clinical application into question. Conversely, thanks to the Singularity implementation all required software are embedded and provided within the software container. As a consequence, users can easily recreate the same work environment just by downloading the pipeline container, and reproduce exactly the same publication-quality plots and tables presented in this study. Lastly, one more common limitation is posed by software scalability. In the WGS domain, with the rapid increase new samples made available and the enormous amount of data generated in each sequencing run, the CPU and memory resources of local workstation risk to quickly become inadequate for data analysis. Therefore, it is paramount that WGS tools are implemented to run on high-performance computing (HPC) clusters and feature remote cloud computing solutions. Because of its Nextflow implementation GIP can be executed on a local machine, on cluster resource manager or the cloud. GIP can be applied on individual samples and without additional effort on large WGS data sets for batch computation as shown for the 222 P. vivax genomes.
These results well illustrate how GIP and giptools can be applied to perform extended genomic analyses in different biological systems and drive biomedical discovery. To conclude, we believe that GIP and giptools represent a step forward toward reproducible research in genomics, and provide a robust computational framework to study how microbes and tumor cells harness genome instability for environmental adaptation and fitness gain.
DATA AVAILABILITY
All data is available in the main text or the supplementary materials.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge Dr Antonio Borderia for kindly discussing with us the aspects relative to pipeline distribution. We also would like to acknowledge Dr Aida Bouratbine for generously providing the Leishmania infantum WGS dataset.
Contributor Information
Gerald F Späth, Institut Pasteur, Université de Paris, INSERM U1201, Unité de Parasitologie moléculaire et Signalisation, Paris, France.
Giovanni Bussotti, Institut Pasteur, Université de Paris, INSERM U1201, Unité de Parasitologie moléculaire et Signalisation, Paris, France; Institut Pasteur, Université de Paris, Bioinformatics and Biostatistics Hub, F-75015 Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institut Pasteur International Department to the LeiSHield Consortium and the EU H2020 project LeiSHield-MATI [REP-778298-1]. Funding for open access charge: Institut Pasteur International Department to the LeiSHield Consortium and the EU H2020 project LeiSHield-MATI [REP-778298-1].
Conflict of interest statement. None declared.
REFERENCES
- 1. Muir P., Li S., Lou S., Wang D., Spakowicz D.J., Salichos L., Zhang J., Weinstock G.M., Isaacs F., Rozowsky J.et al.. The real cost of sequencing: scaling computation to keep pace with data generation. Genome Biol. 2016; 17:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pareek C.S., Smoczynski R., Tretyn A.. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011; 52:413–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chin C.S., Alexander D.H., Marks P., Klammer A.A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E.E.et al.. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 2013; 10:563–569. [DOI] [PubMed] [Google Scholar]
- 4. Chin C.S., Peluso P., Sedlazeck F.J., Nattestad M., Concepcion G.T., Clum A., Dunn C., O’Malley R., Figueroa-Balderas R., Morales-Cruz A.et al.. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods. 2016; 13:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reuter J.A., Spacek D.V., Snyder M.P.. High-throughput sequencing technologies. Mol. Cell. 2015; 58:586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balloux F., Bronstad Brynildsrud O., van Dorp L., Shaw L.P., Chen H., Harris K.A., Wang H., Eldholm V.. From theory to practice: translating whole-genome sequencing (WGS) into the clinic. Trends Microbiol. 2018; 26:1035–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang D.F., Zhi X.Y., Zhang J., Paoli G.C., Cui Y., Shi C., Shi X.. Preliminary comparative genomics revealed pathogenic potential and international spread of Staphylococcus argenteus. BMC Genomics. 2017; 18:808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oniciuc E.A., Likotrafiti E., Alvarez-Molina A., Prieto M., Santos J.A., Alvarez-Ordonez A.. The present and future of whole genome sequencing (WGS) and whole metagenome sequencing (WMS) for surveillance of antimicrobial resistant microorganisms and antimicrobial resistance genes across the food chain. Genes (Basel.). 2018; 9:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fraser C.M., Eisen J.A., Salzberg S.L.. Microbial genome sequencing. Nature. 2000; 406:799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pizza M., Scarlato V., Masignani V., Giuliani M.M., Arico B., Comanducci M., Jennings G.T., Baldi L., Bartolini E., Capecchi B.et al.. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science. 2000; 287:1816–1820. [DOI] [PubMed] [Google Scholar]
- 11. Nanduri S.A., Metcalf B.J., Arwady M.A., Edens C., Lavin M.A., Morgan J., Clegg W., Beron A., Albertson J.P., Link-Gelles R.et al.. Prolonged and large outbreak of invasive group A Streptococcus disease within a nursing home: repeated intrafacility transmission of a single strain. Clin. Microbiol. Infect. 2019; 25:248.e1–248.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kong Z., Zhao P., Liu H., Yu X., Qin Y., Su Z., Wang S., Xu H., Chen J.. Whole-genome sequencing for the investigation of a hospital outbreak of MRSA in China. PLoS One. 2016; 11:e0149844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang Y., Wei Z., Wang Y., Hua X., Feng Y., Yu Y.. Tracking a hospital outbreak of KPC-producing ST11 Klebsiella pneumoniae with whole genome sequencing. Clin. Microbiol. Infect. 2015; 21:1001–1007. [DOI] [PubMed] [Google Scholar]
- 14. Fitzpatrick M.A., Ozer E.A., Hauser A.R.. Utility of whole-genome sequencing in characterizing acinetobacter epidemiology and analyzing hospital outbreaks. J. Clin. Microbiol. 2016; 54:593–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Didelot X., Dordel J., Whittles L.K., Collins C., Bilek N., Bishop C.J., White P.J., Aanensen D.M., Parkhill J., Bentley S.D.et al.. Genomic analysis and comparison of two gonorrhea outbreaks. mBio. 2016; 7:e00525-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bussotti G., Gouzelou E., Cortes Boite M., Kherachi I., Harrat Z., Eddaikra N., Mottram J.C., Antoniou M., Christodoulou V., Bali A.et al.. Leishmania genome dynamics during environmental adaptation reveal strain-specific differences in gene copy number variation, karyotype instability, and telomeric amplification. MBio. 2018; 9:e01399-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dumetz F., Imamura H., Sanders M., Seblova V., Myskova J., Pescher P., Vanaerschot M., Meehan C.J., Cuypers B., De Muylder G.et al.. Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. MBio. 2017; 8: 10.1128/mBio.00599-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwabl P., Boite M.C., Bussotti G., Jacobs A., Andersson B., Moreira O., Freitas-Mesquita A.L., Meyer-Fernandes J.R., Telleria E.L., Traub-Cseko Y.et al.. Colonization and genetic diversification processes of Leishmania infantum in the Americas. Commun. Biol. 2021; 4:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Darmon E., Leach D.R.. Bacterial genome instability. Microbiol. Mol. Biol. Rev. 2014; 78:1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanahan D., Weinberg R.A.. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 21. Hughes D., Andersson D.I.. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat. Rev. Genet. 2015; 16:459–471. [DOI] [PubMed] [Google Scholar]
- 22. McGranahan N., Swanton C.. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017; 168:613–628. [DOI] [PubMed] [Google Scholar]
- 23. Aurrecoechea C., Barreto A., Basenko E.Y., Brestelli J., Brunk B.P., Cade S., Crouch K., Doherty R., Falke D., Fischer S.et al.. EuPathDB: the eukaryotic pathogen genomics database resource. Nucleic Acids Res. 2017; 45:D581–D591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dolled-Filhart M.P., Lee M. Jr, Ou-Yang C.W., Haraksingh R.R., Lin J.C. Computational and bioinformatics frameworks for next-generation whole exome and genome sequencing. ScientificWorld J. 2013; 2013:730210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pabinger S., Dander A., Fischer M., Snajder R., Sperk M., Efremova M., Krabichler B., Speicher M.R., Zschocke J., Trajanoski Z.. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014; 15:256–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garrison E., Marth G.. Haplotype-based variant detection from short-read sequencing. 2012; arXiv doi:20 July 2012, preprint: not peer reviewedhttps://arxiv.org/abs/1207.3907.
- 27. Abyzov A., Urban A.E., Snyder M., Gerstein M.. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011; 21:974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rausch T., Zichner T., Schlattl A., Stütz A.M., Benes V., Korbel J.O.. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012; 28:i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hwang K.B., Lee I.H., Li H., Won D.G., Hernandez-Ferrer C., Negron J.A., Kong S.W.. Comparative analysis of whole-genome sequencing pipelines to minimize false negative findings. Sci. Rep. 2019; 9:3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C.. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017; 35:316–319. [DOI] [PubMed] [Google Scholar]
- 31. Bussotti G., Benkahla A., Jeddi F., Souiai O., Aoun K., Spath G.F., Bouratbine A.. Nuclear and mitochondrial genome sequencing of North-African Leishmania infantum isolates from cured and relapsed visceral leishmaniasis patients reveals variations correlating with geography and phenotype. Microb. Genom. 2020; 6:mgen000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prieto Barja P., Pescher P., bussotti g., Dumetz F., Imamura H., Kedra D., Domagalska M., Chaumeau V., Himmelbauer H., Pages M.et al.. Haplotype selection as an adaptive mechanism in the protozoan pathogen Leishmania donovani. Nat. Ecol. Evol. 2017; 1:1961–1969. [DOI] [PubMed] [Google Scholar]
- 33. Leinonen R., Sugawara H., Shumway M.Collaboration, I.N.S.D. . The sequence read archive. Nucleic Acids Res. 2011; 39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leinonen R., Akhtar R., Birney E., Bower L., Cerdeno-Tarraga A., Cheng Y., Cleland I., Faruque N., Goodgame N., Gibson R.et al.. The European Nucleotide Archive. Nucleic Acids Res. 2011; 39:D28–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sloan C.A., Chan E.T., Davidson J.M., Malladi V.S., Strattan J.S., Hitz B.C., Gabdank I., Narayanan A.K., Ho M., Lee B.T.et al.. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016; 44:D726–D732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Howe K.L., Contreras-Moreira B., De Silva N., Maslen G., Akanni W., Allen J., Alvarez-Jarreta J., Barba M., Bolser D.M., Cambell L.et al.. Ensembl Genomes 2020-enabling non-vertebrate genomic research. Nucleic Acids Res. 2020; 48:D689–D695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arnaud M.B., Costanzo M.C., Skrzypek M.S., Binkley G., Lane C., Miyasato S.R., Sherlock G.. The Candida Genome Database (CGD), a community resource for Candida albicans gene and protein information. Nucleic Acids Res. 2005; 33:D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bahl A., Brunk B., Crabtree J., Fraunholz M.J., Gajria B., Grant G.R., Ginsburg H., Gupta D., Kissinger J.C., Labo P.et al.. PlasmoDB: the Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 2003; 31:212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Girgis H.Z. Red: an intelligent, rapid, accurate tool for detecting repeats de-novo on the genomic scale. BMC Bioinformatics. 2015; 16:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 41. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Subgroup G.P.D.P.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li W., Godzik A.. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- 44. Pearson R.D., Amato R., Auburn S., Miotto O., Almagro-Garcia J., Amaratunga C., Suon S., Mao S., Noviyanti R., Trimarsanto H.et al.. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat. Genet. 2016; 48:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Basenko E.Y., Pulman J.A., Shanmugasundram A., Harb O.S., Crouch K., Starns D., Warrenfeltz S., Aurrecoechea C., Stoeckert C.J. Jr, Kissinger J.C.et al.. FungiDB: an integrated bioinformatic resource for fungi and oomycetes. J. Fungi. (Basel.). 2018; 4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Caspi R., Billington R., Keseler I.M., Kothari A., Krummenacker M., Midford P.E., Ong W.K., Paley S., Subhraveti P., Karp P.D.. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 2020; 48:D445–D453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M.. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012; 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., von Haeseler A., Lanfear R.. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020; 37:1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hoang D.T., Chernomor O., von Haeseler A., Minh B.Q., Vinh L.S.. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018; 35:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ford A., Kepple D., Abagero B.R., Connors J., Pearson R., Auburn S., Getachew S., Ford C., Gunalan K., Miller L.H.et al.. Whole genome sequencing of Plasmodium vivax isolates reveals frequent sequence and structural polymorphisms in erythrocyte binding genes. PLoS Negl. Trop. Dis. 2020; 14:e0008234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu G., Lam T.T., Zhu H., Guan Y.. Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol. Biol. Evol. 2018; 35:3041–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A.. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Klambauer G., Schwarzbauer K., Mayr A., Clevert D.A., Mitterecker A., Bodenhofer U., Hochreiter S.. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012; 40:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hwang S., Kim E., Lee I., Marcotte E.M.. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015; 5:17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alvar J., Velez I.D., Bern C., Herrero M., Desjeux P., Cano J., Jannin J., den Boer M.Team, W.H.O.L.C. . Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012; 7:e35671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gabrielaite M., Torp M.H., Rasmussen M.S., Andreu-Sánchez S., Vieira F.G., Pedersen C.B., Kinalis S., Madsen M.B., Yde C.W., Olsen L.R.et al.. A comparison of tools for copy-number variation detection in germline whole exome and whole genome sequencing data. 2021; bioRxiv doi:30 April 2021, preprint: not peer reviewed 10.1101/2021.04.30.442110. [DOI] [PMC free article] [PubMed]
- 59. Giovanni Bussotti L.P., Pescher P., Domagalska M.A., Shanmugha Rajan K., Doniger T., Hiregange D.G., Myler P.J., Unger R., Michaeli S.J., Spaeth G.F.. Genome instability drives epistatic adaptation in the human pathogen Leishmania. 2021; bioRxiv doi:23 June 2021, preprint: not peer reviewed 10.1101/2021.06.15.448517. [DOI] [PMC free article] [PubMed]
- 60. WHO World Malaria Report. 2018; Geneva: World Health Organization. [Google Scholar]
- 61. Price R.N., Tjitra E., Guerra C.A., Yeung S., White N.J., Anstey N.M.. Vivax malaria: neglected and not benign. Am. J. Trop. Med. Hyg. 2007; 77:79–87. [PMC free article] [PubMed] [Google Scholar]
- 62. Gething P.W., Elyazar I.R., Moyes C.L., Smith D.L., Battle K.E., Guerra C.A., Patil A.P., Tatem A.J., Howes R.E., Myers M.F.et al.. A long neglected world malaria map: Plasmodium vivax endemicity in 2010. PLoS Negl. Trop. Dis. 2012; 6:e1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Battle K.E., Gething P.W., Elyazar I.R., Moyes C.L., Sinka M.E., Howes R.E., Guerra C.A., Price R.N., Baird K.J., Hay S.I.. The global public health significance of Plasmodium vivax. Adv. Parasitol. 2012; 80:1–111. [DOI] [PubMed] [Google Scholar]
- 64. Singh V., Gupta P., Pande V.. Revisiting the multigene families: Plasmodium var and vir genes. J. Vector Borne. Dis. 2014; 51:75–81. [PubMed] [Google Scholar]
- 65. Rayner J.C., Tran T.M., Corredor V., Huber C.S., Barnwell J.W., Galinski M.R.. Dramatic difference in diversity between Plasmodium falciparum and Plasmodium vivax reticulocyte binding-like genes. Am. J. Trop. Med. Hyg. 2005; 72:666–674. [PubMed] [Google Scholar]
- 66. Rahul C.N., Shiva Krishna K., Pawar A.P., Bai M., Kumar V., Phadke S., Rajesh V.. Genetic and structural characterization of PvSERA4: potential implication as therapeutic target for Plasmodium vivax malaria. J. Biomol. Struct. Dyn. 2014; 32:580–590. [DOI] [PubMed] [Google Scholar]
- 67. Rahul C.N., Shiva Krishna K., Meera M., Phadke S., Rajesh V.. Plasmodium vivax: N-terminal diversity in the blood stage SERA genes from Indian isolates. Blood Cells Mol. Dis. 2015; 55:30–35. [DOI] [PubMed] [Google Scholar]
- 68. Luo Z., Sullivan S.A., Carlton J.M.. The biology of Plasmodium vivax explored through genomics. Ann. N. Y. Acad. Sci. 2015; 1342:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lin J.T., Patel J.C., Kharabora O., Sattabongkot J., Muth S., Ubalee R., Schuster A.L., Rogers W.O., Wongsrichanalai C., Juliano J.J.. Plasmodium vivax isolates from Cambodia and Thailand show high genetic complexity and distinct patterns of P. vivax multidrug resistance gene 1 (pvmdr1) polymorphisms. Am. J. Trop. Med. Hyg. 2013; 88:1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gunalan K., Niangaly A., Thera M.A., Doumbo O.K., Miller L.H.. Plasmodium vivax infections of Duffy-negative erythrocytes: historically undetected or a recent adaptation?. Trends Parasitol. 2018; 34:420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Costa G.L., Amaral L.C., Fontes C.J.F., Carvalho L.H., de Brito C.F.A., de Sousa T.N. Assessment of copy number variation in genes related to drug resistance in Plasmodium vivax and Plasmodium falciparum isolates from the Brazilian Amazon and a systematic review of the literature. Malar J. 2017; 16:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cornejo O.E., Fisher D., Escalante A.A.. Genome-wide patterns of genetic polymorphism and signatures of selection in Plasmodium vivax. Genome Biol. Evol. 2014; 7:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen E., Salinas N.D., Huang Y., Ntumngia F., Plasencia M.D., Gross M.L., Adams J.H., Tolia N.H.. Broadly neutralizing epitopes in the Plasmodium vivax vaccine candidate Duffy Binding Protein. Proc. Natl. Acad. Sci U.S.A. 2016; 113:6277–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zolan M.E. Chromosome-length polymorphism in fungi. Microbiol. Rev. 1995; 59:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Suzuki T., Nishibayashi S., Kuroiwa T., Kanbe T., Tanaka K.. Variance of ploidy in Candida albicans. J. Bacteriol. 1982; 152:893–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sionov E., Chang Y.C., Kwon-Chung K.J.. Azole heteroresistance in Cryptococcus neoformans: emergence of resistant clones with chromosomal disomy in the mouse brain during fluconazole treatment. Antimicrob. Agents Chemother. 2013; 57:5127–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shin J.H., Chae M.J., Song J.W., Jung S.I., Cho D., Kee S.J., Kim S.H., Shin M.G., Suh S.P., Ryang D.W.. Changes in karyotype and azole susceptibility of sequential bloodstream isolates from patients with Candida glabrata candidemia. J. Clin. Microbiol. 2007; 45:2385–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Selmecki A., Forche A., Berman J.. Genomic plasticity of the human fungal pathogen Candida albicans. Eukaryot. Cell. 2010; 9:991–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Magee B.B., Magee P.T.. Induction of mating in Candida albicans by construction of MTLa and MTLalpha strains. Science. 2000; 289:310–313. [DOI] [PubMed] [Google Scholar]
- 80. Gerstein A.C., Fu M.S., Mukaremera L., Li Z., Ormerod K.L., Fraser J.A., Berman J., Nielsen K.. Polyploid titan cells produce haploid and aneuploid progeny to promote stress adaptation. mBio. 2015; 6:e01340-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Croll D., McDonald B.A.. The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog. 2012; 8:e1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chibana H., Beckerman J.L., Magee P.T.. Fine-resolution physical mapping of genomic diversity in Candida albicans. Genome Res. 2000; 10:1865–1877. [DOI] [PubMed] [Google Scholar]
- 83. Bravo Ruiz G., Ross Z.K., Holmes E., Schelenz S., Gow N.A.R., Lorenz A.. Rapid and extensive karyotype diversification in haploid clinical Candida auris isolates. Curr. Genet. 2019; 65:1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Brown G.D., Netea M.G.. Exciting developments in the immunology of fungal infections. Cell Host Microbe. 2012; 11:422–424. [DOI] [PubMed] [Google Scholar]
- 85. Pfaller M.A., Diekema D.J., Turnidge J.D., Castanheira M., Jones R.N.. Twenty years of the SENTRY antifungal surveillance program: results for Candida species from 1997-2016. Open Forum Infect. Dis. 2019; 6:S79–S94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pfaller M.A., Castanheira M., Messer S.A., Moet G.J., Jones R.N.. Variation in Candida spp. distribution and antifungal resistance rates among bloodstream infection isolates by patient age: report from the SENTRY Antimicrobial Surveillance Program (2008-2009). Diagn. Microbiol. Infect. Dis. 2010; 68:278–283. [DOI] [PubMed] [Google Scholar]
- 87. Todd R.T., Selmecki A.. Expandable and reversible copy number amplification drives rapid adaptation to antifungal drugs. Elife. 2020; 9:e58349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mount H.O., Revie N.M., Todd R.T., Anstett K., Collins C., Costanzo M., Boone C., Robbins N., Selmecki A., Cowen L.E.. Global analysis of genetic circuitry and adaptive mechanisms enabling resistance to the azole antifungal drugs. PLoS Genet. 2018; 14:e1007319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hirakawa M.P., Martinez D.A., Sakthikumar S., Anderson M.Z., Berlin A., Gujja S., Zeng Q., Zisson E., Wang J.M., Greenberg J.M.et al.. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res. 2015; 25:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dixon J.R., Xu J., Dileep V., Zhan Y., Song F., Le V.T., Yardimci G.G., Chakraborty A., Bann D.V., Wang Y.et al.. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018; 50:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kent W.J., Zweig A.S., Barber G., Hinrichs A.S., Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010; 26:2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yao Y., Dai W.. Genomic instability and cancer. J. Carcinog Mutagen. 2014; 5:1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Boeva V., Popova T., Bleakley K., Chiche P., Cappo J., Schleiermacher G., Janoueix-Lerosey I., Delattre O., Barillot E.. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012; 28:423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wang Z., Hormozdiari F., Yang W.Y., Halperin E., Eskin E.. CNVeM: copy number variation detection using uncertainty of read mapping. J. Comput. Biol. 2013; 20:224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yoon S., Xuan Z., Makarov V., Ye K., Sebat J.. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009; 19:1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Abbey D.A., Funt J., Lurie-Weinberger M.N., Thompson D.A., Regev A., Myers C.L., Berman J.. YMAP: a pipeline for visualization of copy number variation and loss of heterozygosity in eukaryotic pathogens. Genome Med. 2014; 6:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bogaerts B., Delcourt T., Soetaert K., Boarbi S., Ceyssens P.J., Winand R., Van Braekel J., De Keersmaecker S.C.J., Roosens N.H.C., Marchal K.et al.. A bioinformatics WGS workflow for clinical Mycobacterium tuberculosis complex isolate analysis, validated using a reference collection extensively characterized with conventional methods and in silico approaches. J. Clin. Microbiol. 2021; 59:e00202-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bogaerts B., Winand R., Fu Q., Van Braekel J., Ceyssens P.J., Mattheus W., Bertrand S., De Keersmaecker S.C.J., Roosens N.H.C., Vanneste K.. Validation of a bioinformatics workflow for routine analysis of whole-genome sequencing data and related challenges for pathogen typing in a European National Reference Center: Neisseria meningitidis as a proof-of-concept. Front. Microbiol. 2019; 10:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ellison M.A., Walker J.L., Ropp P.J., Durrant J.D., Arndt K.M.. MutantHuntWGS: a pipeline for identifying Saccharomyces cerevisiae mutations. G3 (Bethesda). 2020; 10:3009–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Quijada N.M., Rodriguez-Lazaro D., Eiros J.M., Hernandez M.. TORMES: an automated pipeline for whole bacterial genome analysis. Bioinformatics. 2019; 35:4207–4212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text or the supplementary materials.