

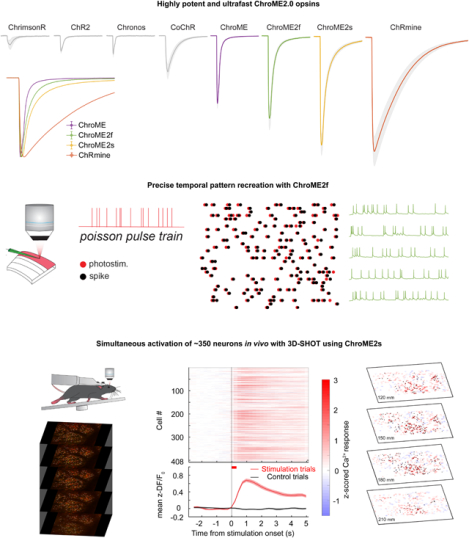
Summary
The biophysical properties of existing optogenetic tools constrain the scale, speed, and fidelity of precise optogenetic control. Here we use structure-guided mutagenesis to engineer opsins that exhibit very high potency while retaining fast kinetics. These new opsins enable large-scale, temporally and spatially precise control of population neural activity. We extensively benchmark these new opsins against existing optogenetic tools and provide a detailed biophysical characterization of a diverse family of opsins under two-photon illumination. This establishes a resource for matching the optimal opsin to the goals and constraints of patterned optogenetics experiments. Finally, by combining these new opsins with optimized procedures for holographic photo-stimulation, we demonstrate the simultaneous co-activation of several hundred spatially defined neurons with a single hologram, and nearly double that number by temporally interleaving holograms at fast rates. These newly engineered opsins substantially extend the capabilities of patterned illumination optogenetic paradigms for addressing neural circuits and behavior.
Graphical Abstract
eTOC blurb
Using structure-guided design, the authors develop second-generation ChroME-based cation channelrhodopsins that exhibit extremely high potency while preserving fast kinetics, thereby expanding the optogenetic toolbox. ChroME2.0 opsins permit spatially and temporally precise two-photon holographic neural control at unprecedented scales, a key technological step forward for understanding brain circuits and behavior.
Introduction
Microbial opsins, which flux ionic current in response to illumination, have empowered neuroscientists to causally perturb brain circuits yielding fundamental insight into brain function(Fenno, Yizhar and Deisseroth, 2011). Optogenetics with spatiotemporally patterned illumination enables investigators to causally relate precise features of neural activity with specific aspects of sensation, cognition and action(Ronzitti, Ventalon, et al., 2017, Marshel et al., 2019, Carrillo-Reid et al., 2019a, Gill et al., 2020, Daie, Svoboda and Druckmann, 2021, Anselmi et al., 2011, Dhawale et al., 2010, Lutz et al., 2008, Fan et al., 2020, Packer et al., 2012, Russell et al., 2019, Zhang et al., 2018, Packer et al., 2014, Rickgauer, Deisseroth and Tank, 2014, Mardinly et al., 2018, Pégard et al., 2017, Yang et al., 2018, Papagiakoumou et al., 2010, dal Maschio et al., 2017, Blumhagen et al., 2011, Farah, Reutsky and Shoham, 2007, Fenno, Yizhar and Deisseroth, 2011, Vaziri and Emiliani, 2012). However, the biophysical properties of the opsins employed, including their conductance, kinetics, and sensitivity, constrain the type and scale of perturbations that can be made(Cardin et al., 2009, Jun and Cardin, 2020, Hass and Glickfeld, 2016, Yu et al., 2020). Although many different opsins have been identified or engineered, writing in precise spatiotemporal patterns of neural activity requires opsins that enable the control of large groups of neurons with high temporal fidelity (Mardinly et al., 2018, Marshel et al., 2019, Prakash et al., 2012, Forli et al., 2018). Thus, opsins that are more potent and have kinetic properties enabling high frequency control of spikes trains can substantially expand the capabilities of optogenetic paradigms, particularly in challenging contexts like the mammalian brain. Additionally, since strong over-expression of microbial opsins can alter neuronal morphology(Miyashita et al., 2013), achieving high performance neural control with lower levels of opsin expression is desirable. Finally, activating large groups of neurons with multiphoton optogenetics requires high potency opsins due to the much higher energies needed to activate opsins with two-photon excitation while avoiding brain heating or tissue damage(Picot et al., 2018, Mardinly et al., 2018). Furthermore, even one-photon optogenetics applications can benefit owing to thermal constraints during visible light illumination (Owen, Liu and Kreitzer, 2019).
Engineering an opsin that provides both high potency and fast closing kinetics is challenging since properties like light sensitivity generally scale inversely with closing kinetics (Mattis et al., 2012). For example, the ultrafast ChR2 mutant CheTa(Gunaydin et al., 2010) or the ultrafast, red-shifted vfChrimson mutant of Chrimson(Mager et al., 2018) exhibit very fast kinetics but smaller photo-currents. Conversely, extremely potent opsins such as the chimeric opsin ReaChR(Lin et al., 2013) or the naturally occurring opsin ChRmine(Marshel et al., 2019) generate large ionic fluxes but have very slow closing kinetics. A point mutant of Chronos(Klapoetke et al., 2014) named ‘ChroME’ somewhat breaks this trend, retaining very fast kinetics while still exhibiting relatively high potency(Mardinly et al., 2018). Thus, if one could engineer more potent variants of ChroME without substantially slowing its kinetics, such improved opsins will better enable the control of large-scale population activity involved in complex perceptions, cognitive functions and motor actions across species.
We thus set out to engineer new opsins based on the ChroME backbone with significantly enhanced potency while retaining fast kinetics. We then benchmarked these new variants against existing tools with a broad set of electrophysiological and optical approaches.
In particular, we present two new mutants (‘ChroME2f’ and ‘ChroME2s’) with enhanced properties that can support large scale, temporally precise multiphoton excitation in the intact brains of awake animals. These enhanced opsins provide up to a 4-6-fold increase in the number of simultaneously controllable neurons compared to ChroME, yet still provide sub-millisecond temporal control at high firing rates.
All-optical perturbation experiments, where the experimenter both controls and measures the activity of neurons with light, are emerging as a powerful tool in neuroscience(Spampinato et al., 2019, Gill et al., 2020, Robinson et al., 2020, Carrillo-Reid et al., 2019b, Daie, Svoboda and Druckmann, 2021, Marshel et al., 2019). However, a main challenge for these experiments is the possibility that the laser used for two-photon imaging of neural activity (e.g., via GCaMP sensors) might incidentally depolarize the neurons of interest through activation of the opsin molecules (Packer et al., 2014, Mardinly et al., 2018, Forli et al., 2018), which we refer to as ‘optical cross-talk’. Since opsins absorb at the typical wavelength for GCaMP imaging (~920–930 nm), this is an important concern for any all-optical experiment using this or other green sensors. We therefore also carefully quantify unwanted ‘optical cross-talk’ at 920 nm across a broad range of conditions to define the limits under which functional imaging can be conducted while generating minimal unwanted depolarization. Furthermore, we provide a comprehensive spectral survey of opsins under two-photon illumination across a wide band (750–1300 nm) that can help experimenters choose among these potent opsins depending on the imaging wavelength to minimize or reduce spectral overlap with the activity sensor. These data provide an essential knowledge base for the further refinement of these opsins and their future use with the ever-expanding toolbox of activity indicators for specific ions, voltage, and neurotransmitters. Finally, by leveraging ChroME2.0 we demonstrate the simultaneous optogenetic perturbation of 300–400 of cortical pyramidal neurons at a time, and >600 neurons per second in the brains of awake mice with multiphoton holographic optogenetics.
Results
Structure-guided design of ultra-potent, ultra-fast opsins based on ChroME
To engineer an ultrapotent opsin that retains fast kinetics we exploited the opsin ChroME, which is a point mutant of the ultra-fast opsin Chronos. Using ChroME as a backbone we targeted several key residues based on structural homology modeling that are likely to either line the channel pore and/or interact with the retinal chromophore, and thus could strongly influence channel biophysics(Kato and Nureki, 2013, Kato et al., 2012) (Fig. 1A). We generated 9 single point mutants, and first characterized each one in cultured CHO cells with visible light illumination to rapidly assay their estimated potency, kinetics, and spectral properties (Fig. 1B–D, 1I, n = 3–30 cells).
Figure 1. Design and characterization of second generation ChroME variants under visible and two-photon excitation.
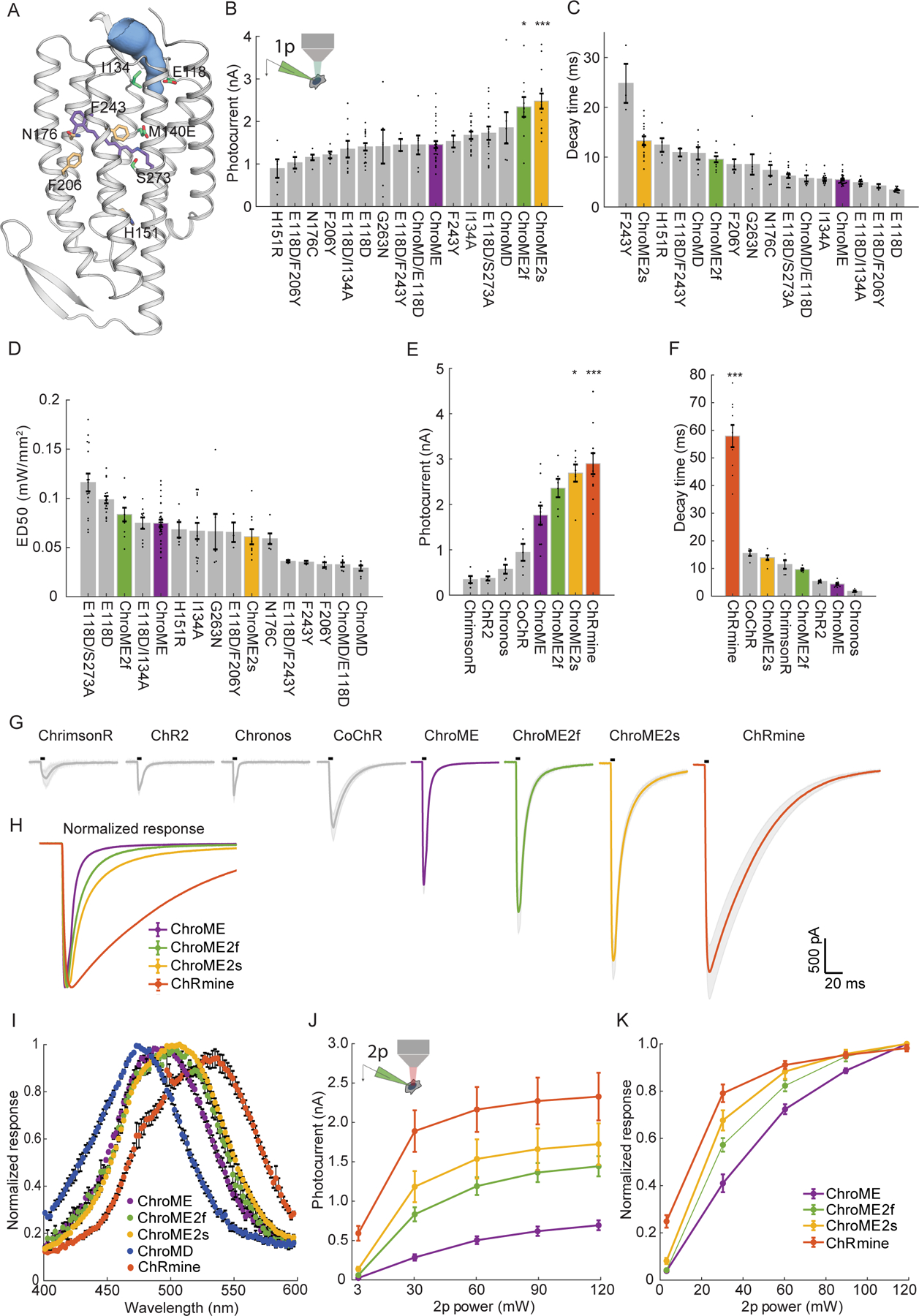
A) Model of ChroME showing some of the amino acids that were targeted for mutagenesis. The retinal chromophore is shown in purple and the putative ion channel pore is indicated in blue.
B) Inset: schematic of a transfected cultured Chinese hamster ovary (CHO) cell under whole cell patch clamp. Peak photocurrents recorded under for each opsin mutant at (power = 0.45 mW at 510 nm unless otherwise indicated). For ChroMD, ChR2 and CoChR, 470 nm was used and for ChrimsonR, 630nm was used).
C) Decay time from monoexponential fits and D) Sensitivity (ED50) of opsin mutants from B)
E) Peak photocurrents of ChroME2f and ChroME2f compared against a panel or previously characterized opsins.
F) Estimated decay times from monoexponential fits for the same panel of opsins as in E).
G) Average photocurrent traces for indicated opsins obtained from data shown in E). The black bar above the trace indicates a 5 ms pulse of light.
H) Peak-normalized traces of the indicated opsins obtained from data shown in E.
I) Visible wavelength spectra of the indicated opsins at the specified wavelengths.
J) Photocurrents at the indicated powers under two-photon illumination at 1040 nm for the indicated opsins.
K) Normalized two-photon photocurrent response for the photocurrents show in J). P < 0.001, Two-way ANOVA, all opsins significantly different.
Data represent the mean ± s.e.m. Statistics: *P < 0.05, and ***P < 0.001 with ChroME as reference; One-way ANOVA with multiple comparisons test.
Many mutations of ChroME diminished the effective photo-conductance and/or slowed channel properties. However, several mutants substantially either sped up channel closing or increased whole-cell photocurrents which are both desirable characteristics. Three mutants had notable qualities: a glutamate to aspartate mutation at site 118 in ChroME substantially sped up the kinetics (decay time ChroME: 5.5 ± 0.2 ms, ChroME E118D: 3.4 ± 0.1 ms, Fig. 1C), while an isoleucine to alanine at 134 increased peak photocurrents (ChroME: 1.5 ± 0.08 nA; ChroME I134A: 1.7 ± 0.1 nA, Fig. 1B). A serine to alanine mutation at site 273 likewise significantly potentiated whole-cell photocurrents (ChroME: 1.5 ± 0.1 nA; ChroME S273A: 2.5 ± 0.2 nA, Fig. 1B) but at the expense of a slowing of channel kinetics (decay time ChroME: 5.5 ± 0.2 ms, ChroME S273A 13 ± 1 ms, Fig. 1C). We refer to ChroME S273A as ‘ChroME2s’ since it exhibited the highest potency (i.e., largest maximal photocurrents) of all the mutants we tested as well as improved sensitivity (quantified as the light power to drive the half-maximal response, ‘ED50’, Fig. 1D), but with somewhat slower kinetics.
We therefore hypothesized that combining these mutations could yield an opsin that is substantially more potent than ChroME while retaining very fast kinetics. Indeed, we found that the ChroME triple mutant S273A, E118D, I134A had substantially stronger currents (ChroME: 1.5 ± 0.1 nA; ChroME S273A, E118D, I134A: 2.3 ± 0.2 nA, 1B) and partially rescued the decay time increased by the S273A mutation (ChroME-S273A: 13 ± 1 ms; ChroME S273A, E118D, I134A: 9.6 ± 0.6 ms, 1C). We hereafter refer to this opsin as ‘ChroME2f’ (‘f’, for ‘fast’). Finally, we identified several other mutants with unique and potentially advantageous properties, including a variant termed ‘ChroMD’ (Chronos with a methionine to aspartate mutation at site 140) that exhibited a substantial blue-shift in its absorbance peak (Fig. 1I, n = 3–4 cells) as well as a strong increase in light sensitivity (Fig 1D). We chose to focus our efforts on ChroME2f and ChroME2s owing to their high potency, overall optimal kinetics and similar absorbance spectra with ChroME (Fig 1I) under visible illumination.
We then sought to compare these new ChroME variants with the standard opsins that are currently in use such as Chronos, ChR2, ChrimsonR, CoChR and ChRmine under one-photon excitation in cell culture. The ChroME variants, ChroME2f and ChroME2s, were among the most potent opsins with respect to photocurrents in our comparison (Fig. 1E, n = 5–11 cells). ChRmine, which provides greater photocurrents than ChroME (Marshel et al., 2019) (Fig. 1E), was stronger than even the ChroME2f/s variants (Fig. 1E). However, the closing kinetics of ChRmine were much slower: about 6-fold slower than ChroME2f and 4-fold slower than ChroME2s (Fig. 1F–H). The ChroME2f/s variants also outperformed ChroME under two-photon excitation (Fig. 1J–K).
To test how ChroME2f and ChroME2s would perform in neurons, we next compared photocurrents and light-evoked spiking in L2/3 cortical neurons expressing ChroME, ChroME2s or ChroME2f fused to mRuby2 expressed in mouse L2/3 cortical pyramidal neurons by in utero electroporation. All opsins throughout the study were targeted to the soma (ST) and proximal dendrites by fusing to the proximal-clustering domain of the potassium channel Kv2.1. Quantification of mRuby2 fluorescence in high resolution confocal images from mice expressing each opsin showed that expression across opsins was comparable, albeit slightly higher for ChroME compared to ChroME2f or ChroME2s (Fig. 2A and Fig. S1B). These new soma-targeted variants were trafficked to the plasma membrane of neuronal soma as well as ChroME (without the need for fusion to ER-export and trafficking motifs), as almost no mRuby2 was internal to the cell membrane (Fig. S1C). These results imply that the larger photocurrents in ChroME2f and ChroME2s cells were not a result of greater protein expression or more efficient targeting to the plasma membrane. We then activated them with both one-photon (Fig. 2B, C, n = 6–15 cells) and two-photon excitation (Fig. 2D, E). Whole-cell recordings confirmed ChroME2s and ChroME2f both provided substantially larger photocurrents than ChroME (Fig. 2B, D) and closely recapitulate decay times that were observed in our original screen by cell culture (Fig. 2C, E). ChRmine-expressing neurons exhibited the largest photocurrents, but these currents had dramatically slower decay kinetics (Fig. 2C, E).
Figure 2. Validation of ChroME variants in acute brain slices under visible and two-photon excitation.
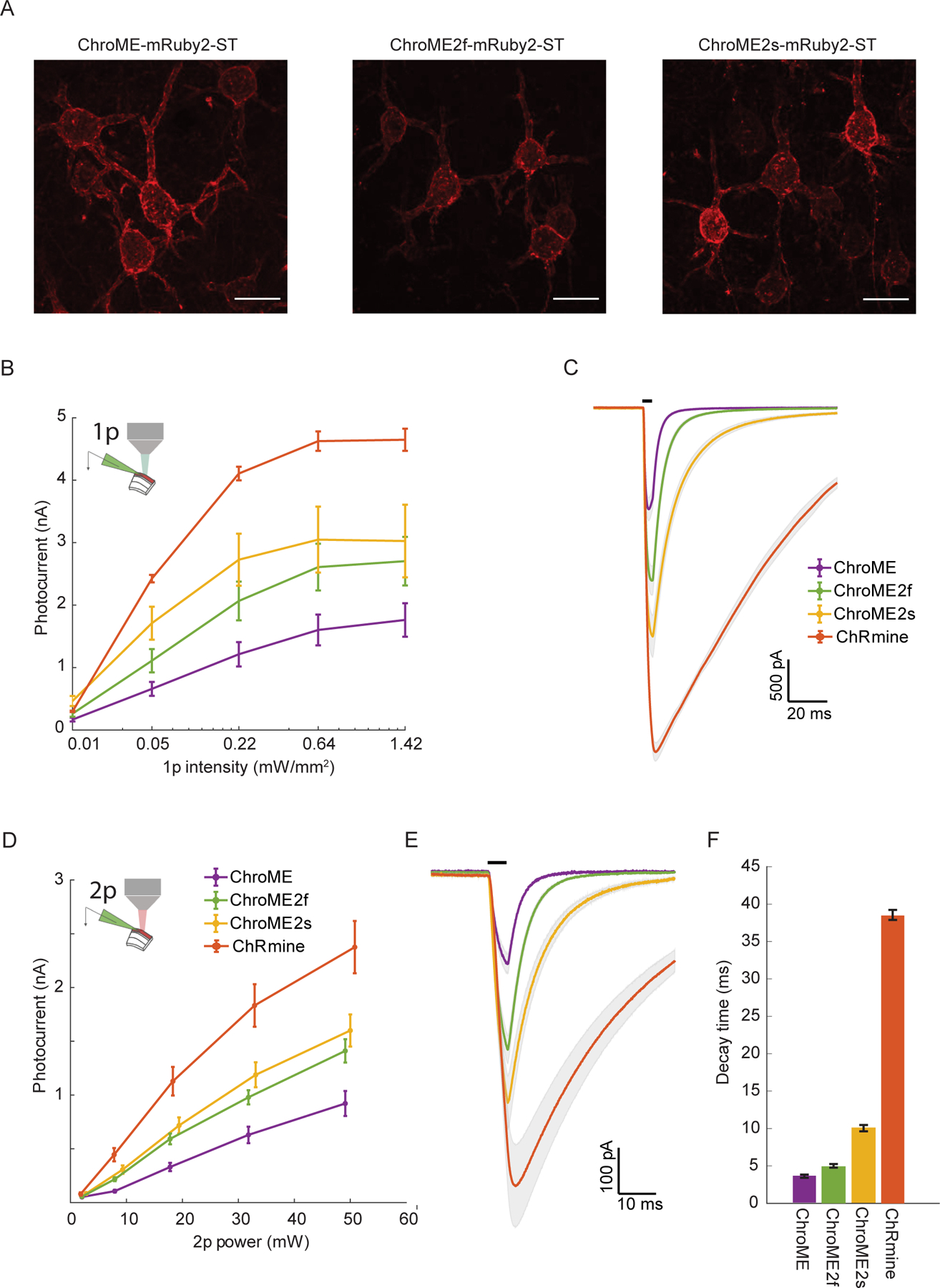
A) Max projections of confocal images of in utero electroporated L2/3 pyramidal neurons expressing ChroME, ChroME2f or ChroME2s, soma-targeted (ST) and fused to mRuby2 (opsin-mRuby2-ST). Scale bar represents 20 μm.
B) Photocurrents under full-field 1p illumination at 510 nm and the indicated powers from L2/3 pyramidal neurons expressing the indicated opsins via in utero electroporation. ChroME: n = 11 cells, 3 mice; ChroME2f: n =17 cells, 2 mice; ChroME2s: n = 12 cells, 2 mice; ChRmine: n = 6 cells, 1 mouse. P<0.005, Two-way ANOVA, all groups significantly different.
C) Average photocurrent traces for indicated opsins obtained from data shown in B). The black bar above the traces indicates a 5 ms pulse of light.
D) As in B) but with two-photon excitation and a ~12.5 μm diameter spot. P<0.005, Two-way ANOVA, all groups significantly different. ChroME: n=8 cells, 1 mouse; ChroME2f: n =14 cells, 2 mice; ChroME2s: n = 10 cells, n = 1 mouse; ChRmine: n = 10 cells, 2 mice.
E) Average photocurrent traces for indicated opsins obtained from data shown in D). The black bar above the traces indicates a 5 ms pulse of light.
F) Decay times of the three opsins measured with monoexponential fits (P<0.05, One-way ANOVA, all groups significantly different except ChroME and ChroME2f with multiple comparisons correction).
Data represent the mean ± s.e.m.
ChroME2.0 variants provide high potency, high temporal fidelity control over neural activity
Based on these results, we next compared the light powers needed to reliably drive neurons to action potential threshold across a range of specific frequencies. We activated L2/3 pyramidal neurons with pulse trains (pulse width = 5 ms) of varying frequency and power, as well as compared the electrophysiological response to single pulses of increasing power and duration (Fig. 3). For these experiments we also included ChRmine since it is among the most potent opsins identified yet(Marshel et al., 2019). Although all four opsins could reliably drive all recorded neurons to fire, we observed substantial differences in the power and frequency response of cortical neurons depending on the opsin used. Both ChroME2f and ChroME2s could drive neurons to spike at substantially lower powers than could ChroME, yet readily maintained high frequency firing up to the highest frequency tested (40 Hz, Fig. 3B, C, E). Importantly, for all ChroME-based opsins, successful light pulses generated close to one spike per pulse (extra spikes per pulse: ChroME: 0.1±0.2%; ChroME2f: 1.1±0.5%; ChroME2s: 1.7±0.7%, Fig. S2A). ChRmine, in contrast, could evoke spiking at even lower power levels than the ChroME2 variants, but the spiking response was not a monotonic function of power, response levels were less reliable across frequencies (Fig. 3D, E), and a significant fraction of spikes (15±3%) came as more than one spike per pulse (Figure S2A). Since a prior study employed shorter pulses to activate ChRmine-expressing neurons(Marshel et al., 2019), we collected an additional data set with pulse trains set using 0.5 ms pulses. Under these conditions the dynamic range of activating ChRmine-expressing neurons appeared larger with respect to laser power, and the response function was monotonic with power (Fig. S2B, n = 5 cells). However, more power per neuron was needed to generate these spikes than when using 5 ms pulses, and at low frequencies (5 Hz) high power 0.5 ms pulses still generated more than one spike (Fig. S2B). Finally, we compared how each opsin could drive cells as a function of illumination time for single pulses ranging between 1–30 ms (Fig. S2C–E). For all opsins, increasing the duration substantially increased spike probability of expressing neurons as a function of power.
Figure 3. Comparison of two-photon light-evoked spiking of L2/3 pyramidal neurons expressing ChroME2.0 variants to ChroME and ChRmine.
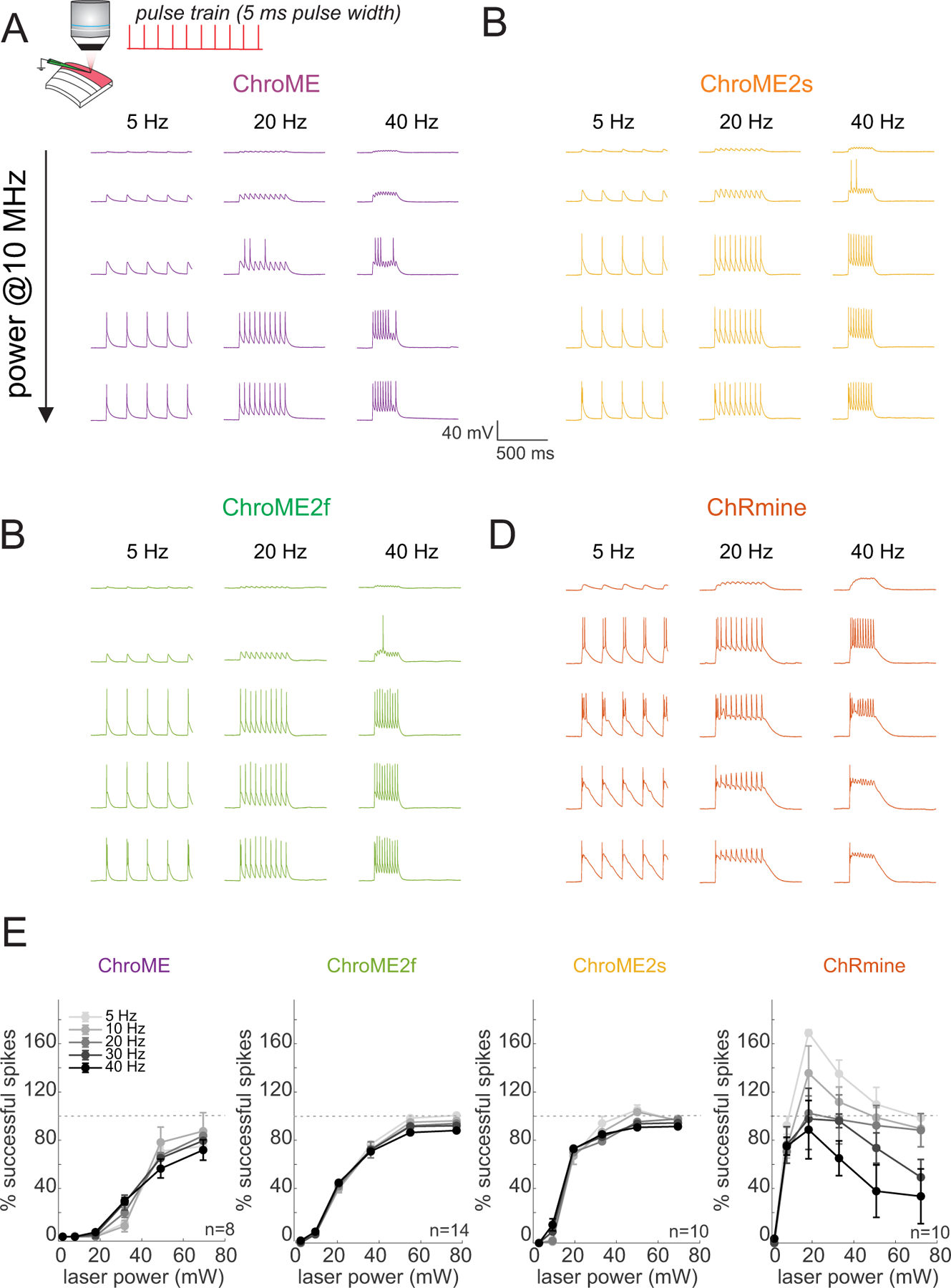
A) Top: Schematic the experiment. A single L2/3 neuron is patched in current clamp mode and illuminated with fixed frequency trains of 5 ms pulses at 1040 nm, 10 MHz repetition rate. Bottom: example traces from a ChroME-expressing neuron,
B-D) Example traces from representative ChroME2f, ChroME2s, and ChRmine-expressing cells.
E) Plots of the fraction of 5 ms light pulses that drove spikes across laser powers and stimulation frequencies for the four opsins. Sample size (cells) is indicated in the panels. The two-photon excitation spot size was 12.5 um in diameter. Data represent the mean ± s.e.m. ChroME: n=8 cells, 1 mouse; ChroME2f: n =14 cells, 2 mice; ChroME2s: n = 10 cells, n = 1 mouse; ChRmine: n = 10 cells, 2 mice. >100% successful spikes indicates more than one spike generated per light pulse (‘extra spikes’).
To obtain a better understanding of how these different opsins would influence the temporal fidelity of light induced spike trains in more physiological conditions, we stimulated opsin-expressing neurons with broadband (‘Poisson’) trains of light pulses (pulse width = 5 ms) while injecting noisy sub-threshold currents to mimic the synaptic bombardment neurons experience under in vivo conditions (Fig. 4). This approach allowed us to quantify how spike latency, jitter, and the probability of spike success varied with instantaneous spike frequency. The laser power for each neuron was set above the level needed to reliably evoke an action potential with a single isolated 5 ms pulse to control for variation in opsin expression and intrinsic excitability across neurons. We found that ChroME2f- or ChroME2s-expressing neurons could reliably follow pulse trains with sub-millisecond jitter across a wide range of frequencies, similar to previously collected data on ChroME-expressing neurons (Mardinly et al., 2018). (Fig. 4A–F), while ChRmine-expressing neurons dropped to about 50% spike success even at low frequencies (Fig. 4F). We retested ChRmine-expressing neurons with a pulse width of 0.5 ms or at higher laser powers, but under these conditions the neurons still did not reliably follow the random, broadband pulse trains (Fig. S2G–L). These data demonstrate that ChroME2f and ChroME2s can enable the precise reproduction of physiological-like spike trains across a broad range of frequencies.
Figure 4. Temporal fidelity of light evoked spiking with broadband ‘poisson’ stimulus trains.
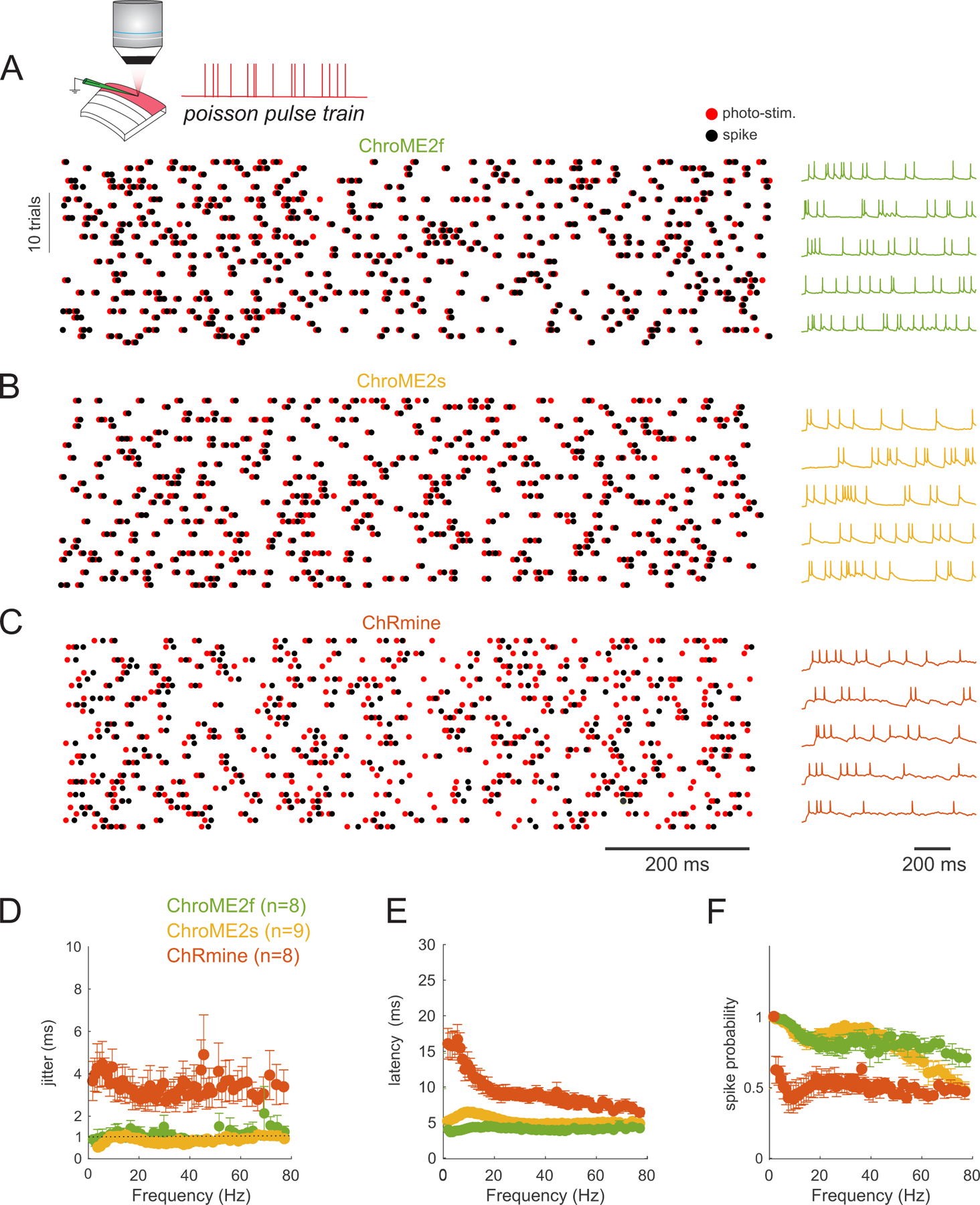
A) Top: Schematic of the experiment. A single L2/3 neuron is patched in current clamp mode and illuminated with poisson-like trains of 5 ms pulses at 1040 nm, 10 MHz repetition rate. Bottom: example raster plot of light pulses (red) and spikes (black) from a ChroME2f-expressing neurons. Right: five example traces from the recording.
B,C) Example spike rasters (left) and traces from neurons expressing ChroME2s or ChRmine.
D-F) Plot of the jitter, latency and spike probability of light-evoked spikes during the broadband stimulation across the three indicated opsins as a function of instantaneous pulse frequency. Sample size is indicated in the panel. Data represent the mean ± s.e.m. ChroME2f: n =8 cells, 2 mice; ChroME2s: n = 9 cells, n = 2 mice; ChRmine: n = 8 cells, 3 mice.
Optical cross-talk and spectral characteristics of the new and existing opsins
The spectral absorbance of opsins delineates the optimal optical wavelength to use for their photo-excitation, but also critically determines how strongly they are incidentally activated by a second two-photon imaging laser(Forli et al., 2018, Chen, Papagiakoumou and Emiliani, 2018, Mardinly et al., 2018, Packer et al., 2014, Gill et al., 2020, Soor et al., 2019). Few opsins have been fully spectrally characterized in the two-photon regime since most prior studies have employed Ti:Sapphire lasers that have a limited tuning range (Shemesh et al., 2017). Thus, we acquired absorbance spectra (820–1300 nm, Insight X3) for a large panel of opsins with diverse biophysical features to provide a critical knowledge base for the future selection of opsins for two-photon optogenetics experiments. We expressed 13 different opsins in cell culture and illuminated the cells with a soma-sized spot of light calibrated for the intrinsic power spectrum of the laser source and transmission of the microscope (Fig. 5 and S3). ChroME, ChroME2f, and ChroME2s all exhibited similar two-photon excitation spectra with a peak of ~1000 nm, substantial absorbance at 920 nm, and minimal absorbance beyond 1200 nm. We characterized other well-known opsins including ChRmine, ChrimsonR, Chronos, CoChR, ReaChR and ChR2 that have also been used for two-photon excitation (Fig. S3). In general, their two-photon excitation matched well with partial spectra taken in prior work where available, (Shemesh et al., 2017), see Fig. 1I above). The blue-shifted ChroME mutant, ChroMD, showed a strong blue shift consistent with the 1p absorbance data described above. Additionally, we characterized the more recently identified PsChR, which is perhaps the most blue-shifted cation opsin yet described (Govorunova et al, 2013). Finally, we obtained complete spectra for the potent anion opsins GtACR1 and GtACR2. These data establish a palette of opsins for diverse experiments and help outline possible multi-spectral combinations for bidirectional optogenetics.
Figure 5. Two-photon excitation spectra of the ChroME2.0 variants and ChRmine.
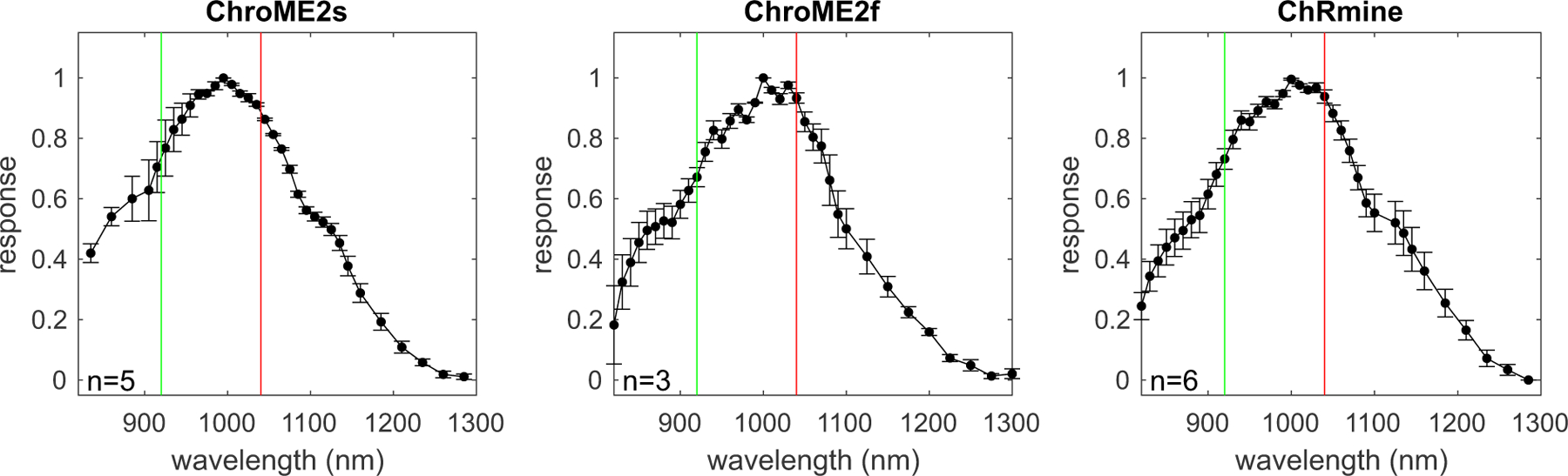
For each opsin the plot shows the peak-normalized photocurrent recorded in CHO cells transfected with the indicated opsin using an Insight X3 tunable laser (80 MHz). The green vertical line marks 920 nm, while the red line indicates 1040 nm. Data represent the mean ± s.e.m. ‘n’ = cells.
Next, to quantify optical cross-talk that could occur during all-optical experiments, we made whole-cell current-clamp recordings of cortical neurons expressing each opsin, while imaging the slice with a resonant-scanning galvo/galvo system (Fig. 6A). We systematically varied three key determinants of optical cross-talk: imaging power, field of view, and frame rate. We found that under imaging conditions that are commonly used to sample cortical neurons volumetrically (wavelength = 920 nm, FOV = 980 × 980 microns, frame rate = 6 Hz) neurons expressing ChroME or ChroME2f showed minimal scan-induced depolarization (peak = 1.1 ± 0.26 mV, mean: 0.27 ± 0.08 mV @25 mW for ChroME, peak = 0.73 ± 0.08 mV, mean: 0.29 ± 0.05 mV @25 mW for ChroME2f) (Fig. 6B, D, F, Figure S4, S5). Neurons expressing ChroME2s and ChRmine showed somewhat larger depolarization (ChroME2s: peak = 2 ± 0.33 mV, mean: 1.05 ± 0.2 mV @25 mW, ChRmine: peak = 2.4 ± 0.4 mV, mean: 1.1 ± 0.2 mV @25 mW, one-way analysis of variance (ANOVA) p=0.0002, Fisher’s Least Significant Difference tests used for all multiple comparisons: p<0.05 for all pairs besides ChRmine and Chrome2s, ChroME and ChroME2f) (Fig. 6B, D, F, Fig. S4, S5).
Figure 6. Activation of opsin variants by resonant galvo scanning.
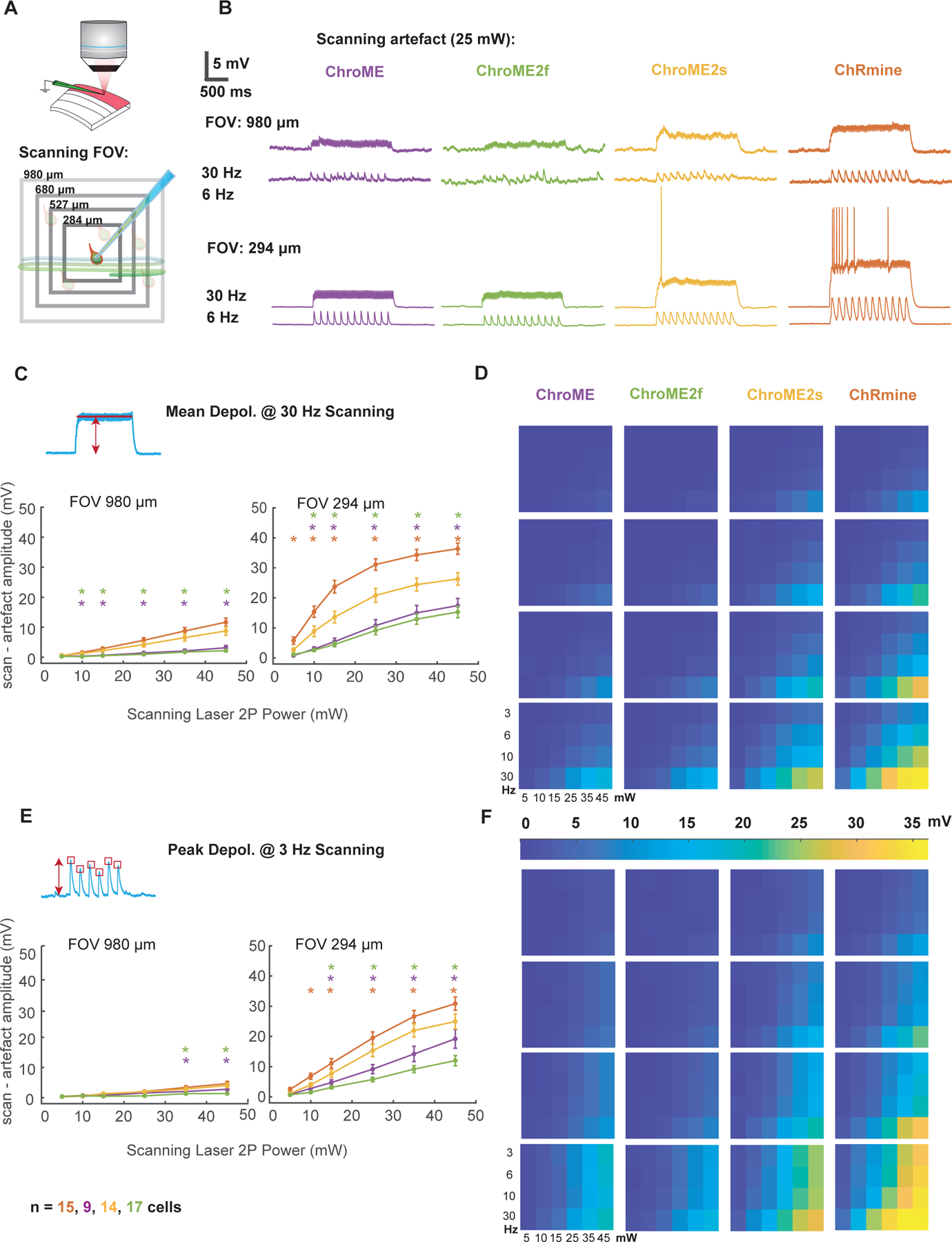
A) Schematic of the experiment indicating a patched neuron expressing opsin and raster scanning the imaging laser at different optical zooms.
B) Example membrane potential traces from four neurons expressing ChroME, ChroME2f, ChroME2s, and ChRmine at two different zoom levels and two different imaging rates. Laser power = 25 mW, 920 nm.
C) Plot of the mean scanning artifact amplitude calculated as mean membrane potential during the scanning period across a sample of neurons. For each of the four opsins two different zoom levels and six different laser powers at 30 Hz frame rate are displayed to visualise differences in artifact on both sides of the explored parameters spectrum. One-way ANOVA p<0.05 for the majority of conditions tested. Stars mark conditions for which post-hoc tests p values were <0.05 between ChroME2s and ChRmine (red stars), ChroME2s and ChroME (purple stars), ChroME2s and ChroME2f (green stars).
D) Heat map plot for scanning artefact across all 96 conditions.
E, F) Same as for C), D) but for the peak depolarization artifact displayed at 3 Hz. Colored stars indicate post hoc test p-value as described in C). Sample size: cell number per opsin indicated at the bottom (the same for all the figures). Recorded cells come from 6,4,5and 7 animals expressing ChroME, ChroMe2f, ChorME2s, and ChRmine. Error bars are s.e.m.
In smaller FOVs at higher magnifications (294 × 294 microns) which are often used for imaging sub-cellular structures such as dendrites, spines and axonal boutons, depolarizations caused by the scanning beam were substantially larger (Fig. S4A). In many ChRmine-expressing and some ChroME2s-expressing neurons the scanning directly evoked spiking from resting potential, but this did not occur in neurons expressing ChroME or ChroME2f (Fig. S5A, B). We also measured the intrinsic physiological properties of the neurons under study but did not find substantial differences (Fig. S5C). Finally, we measured 2p-scanning evoked currents in voltage clamp directly (Fig. S4B, C), which better reveals the kinetic differences in the scanning-induced photo-conductances between the different opsins. These data provide critical details that can guide which of these opsins to choose for distinct classes of all-optical multiphoton optogenetics experiments and reflect the tradeoffs between opsin potency and kinetics and optical cross-talk during two photon imaging.
In vivo performance and large-scale control of neural activity with ChroME2.0 variants
Next, we sought to compare the performance of the most potent opsins identified above for in vivo holographic two-photon optogenetic control. We expressed ChroME, ChroME2s, ChroME2f, ChRmine and ChrimsonR in L2/3 pyramidal neurons via AAV viral transfection (using the template vector AAV-CAG-DIO-[Opsin]-ST-P2A-H2B-mRuby3) in mice expressing GCaMP6s transgenically in cortical excitatory neurons (Camk2a-tTa;tetO-GCaMP6s;emx-IRES-Cre mice). Calcium imaging was performed at 920 nm and photo-stimulation at 1030 nm. We found that their potency measured in brain slices was largely consistent with their potency for photo-stimulation in vivo (Fig. 7). ChroME2s and ChroME2f-expressing neurons were both substantially more sensitive than ChroME, with a strong decrease in their ED50s: ChroME 88 ± 10 mW, ChroME2f 22 ± 1 mW, ChroME2s 12 ± 3 mW (assessed by linear interpolation of the power curves; *p<0.05 across all comparisons, multiple t-tests, n ≥ 3 mice with ≥ 70 targeted cells per opsin). The highest difference was observed at 25 mW/cell, where the fraction of neurons that could be photoactivated increased ~4-fold for ChroME2f and ~5-fold for ChroME2s; at 50 mW/cell, the increase was 2–2.5-fold compared to ChroME (Fig. 7 C,D). ChRmine exhibited the highest potency among this panel, slightly outperforming ChroME2s, while ChrimsonR exhibited the weakest potency (fraction photoactivable at 50 mW, ChrimsonR: 0.06 ± 0.04, ChroME: 0.33 ± 0.05, ChroME2f: 0.76 ± 0.07, ChroME2s: 0.84 ± 0.01, Chrmine: 0.90 ± 0.04, ***p<0.001, one-way ANOVA, n= 4 mice for ChroME and 3 mice for all other opsins with ≥ 40 targeted cells each, Fig. 7C, Fig. S6. These results indicate that new generation opsins present robust advantages for in vivo large-scale activation of cells.
Figure 7. In vivo all-optical comparison of opsin potency for photo-stimulation.
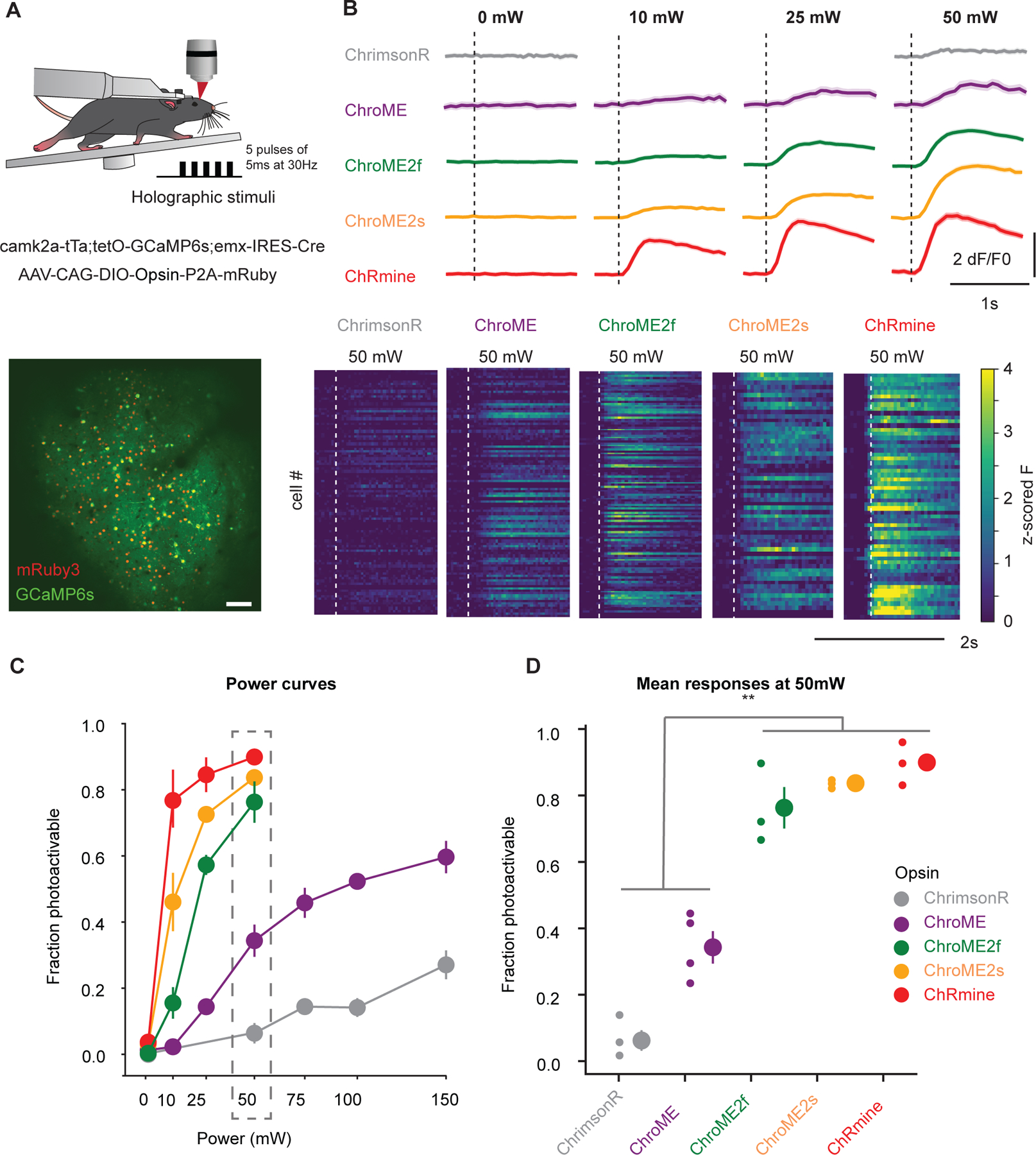
A) Top: Experimental schematic. Adult transgenic mice virally expressing each of the five opsins in excitatory cells were holographically stimulated using 3D-SHOT (Mardinly et al., 2018). Individual cells were excited with a train of 5 pulses of 5ms duration at 30Hz at 1030 nm. Calcium imaging is performed at 920 nm. Bottom: In vivo two-photon image taken at 1020 nm of a representative FOV with all excitatory neurons expressing GCaMP6s (green). Opsin-expressing neurons are labeled with nuclear mRuby3 (red), which is used to automatically detect target cells. Scale bar: 100mm.
B) Top: Example traces (recorded at 920 nm imaging wavelength) of mean population responses of cells expressing each of the opsins to different power intensities. Bottom: Representative mean z-scored fluorescence peristimulus time histograms (PSTHs) for opsin-expressing cells stimulated at 50mW. Dashed lines indicate the onset of photostimulation.
C) Fraction of photoactivable cells as a function of power for each opsin (***p<0.001 for opsin effect, Two-way ANOVA). Post-hoc Tukey’s multiple comparison test was applied to ChroME2f, ChroME2s and ChRmine across all powers: ChroME2f < ChroME2s < ChRmine, *p<0.05 for all comparisons, n>=3 mice with >= 40 cells each per opsin. Error bars indicate s.e.m. Total number of mice were: 3, 4, 3, 3, 3, for ChrimsonR, ChroME, ChroME2f, ChroME2s and ChRmine, respectively.
D) Mean (± s.e.m.) fraction of successfully photoactivated cells (relative to all automatically segmented cells targeted in a FOV) for each opsin at 50mW. New generation opsins (ChroME2f, ChroMe2s and ChRmine) were significantly more potent than the previously generated ones (***p<0.001, One-way ANOVA, post-hoc multiple t-test with holm correction: Chrimson < ChroME < ChroME2f ≃ ChroME2s ≃ ChRmine, n ≥ 3 mice per opsin, with ≥40 cells per mice). Total number of mice were: 3, 4, 3, 3, 3, for ChrimsonR, ChroME, ChroME2f, ChroME2s and ChRmine, respectively.
Finally, we estimated an upper bound on how many neurons we could simultaneously co-activate in a large volume in vivo under these expression conditions (Fig. 8) and with a maximal total instantaneous laser energy at the sample of ~4W (1040 nm). Since ChroME2s is nearly as potent as ChRmine but still provides excellent control over high frequency firing, we focused on this opsin and designed an optogenetic paradigm to maximize the number of neurons that could be simultaneously photo-stimulated at a single moment (with a single phase mask on the spatial light modulator, SLM) or to maximize the number of neurons that could be co-activated within a defined time window (e.g, one second) by rapidly interleaving multiple SLM phase masks (Fig. 8B). We systematically varied pulse duration, frequency, and power to identify the conditions which would optimize the size of the activated ensemble under both conditions (Fig. S7). To further optimize this, we selected opsin-expressing neurons that showed higher light sensitivity. To this end, we systematically probed the optogenetic gain of >1,000 neurons in the brain volume, selected the top 30% of excitable neurons, and then adjusted the power directed to each neuron in a multi-spot hologram to drive a significant increase in calcium signal (Fig. 8D, Fig. S7C). Figure 8C summarizes the hologram sizes used across experiments and the resulting photoactivation rates (n = 6 mice/16 sessions overall). In one example animal under these conditions we could simultaneously increase the activity of 346 neurons for a 408-target hologram (Fig. 8D,E; Wilcoxon rank-sum test, one-tailed p<0.025). The remaining 62 neurons either did not show a detectable change in calcium signal or were suppressed. The light-induced responses in the targeted cells were reliable across repetitions of the same hologram (Fig. S8A–E). We also noted that many non-targeted cells showed detectable modulations of the firing rates, some showing increased activity while others exhibiting suppression. Those with increased activity might either be indirectly activated by the illuminating hologram (‘off-target’) or synaptically activated by the targeted neurons, or a combination of the two. We compared the magnitude of the responses in non-targeted neurons between cells that were <25μm from a target (‘near’) or further away (‘far’) and found that near cells showed a significantly larger modulation than far cells (Fig. S8F–H). This potentially supports the notion that some the response of non-targeted cells was off-target activation, although indirect activation through the network is difficult to rule out.
Figure 8. Large ensemble photo-stimulation in vivo with ChroME2s.
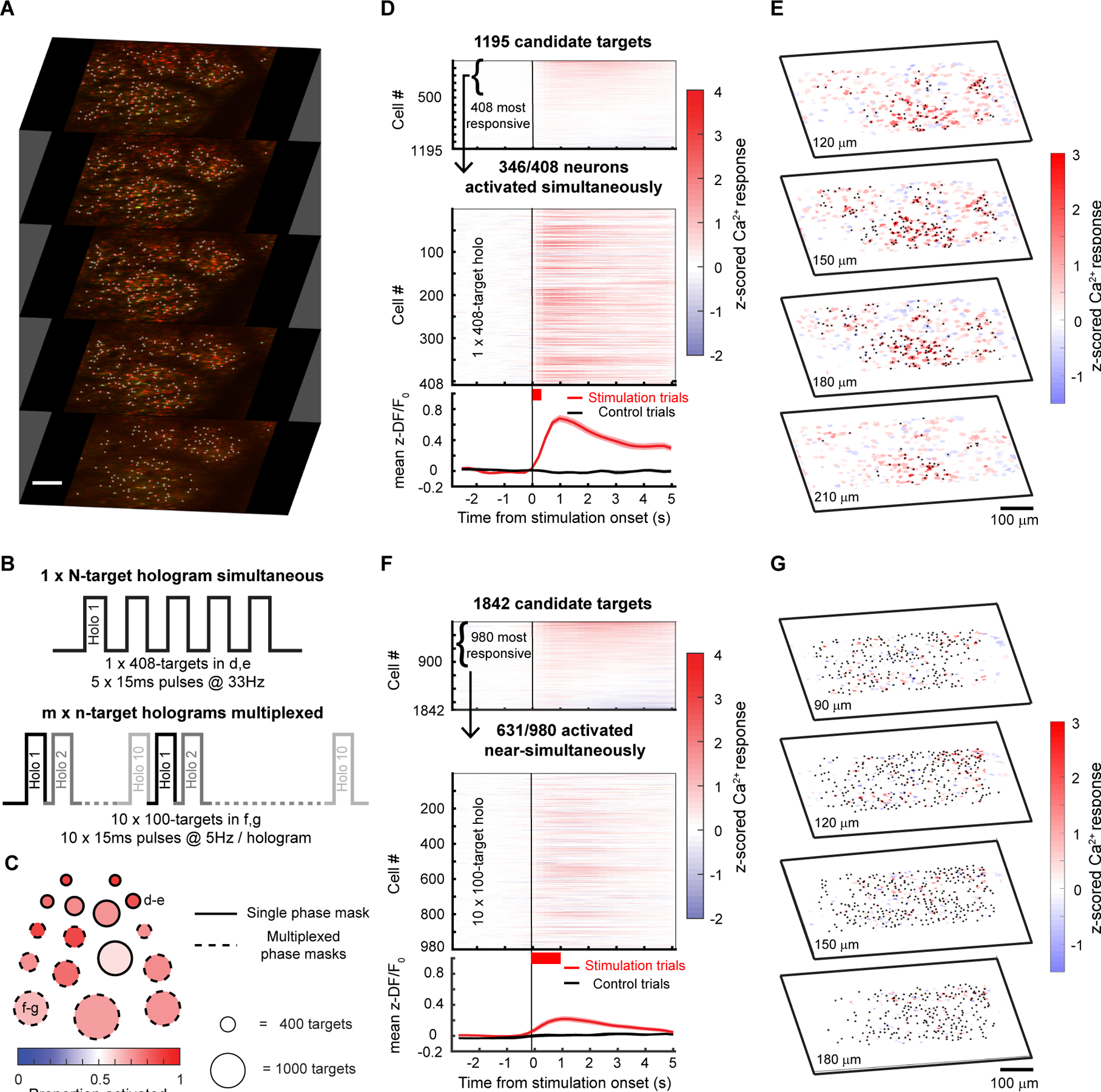
A) Example 5-plane volume stack used for large ensemble photo-stimulation experiments in mice viral expressing soma-targeted-ChroME2s-p2A-H2B-mRuby3. Imaging wavelength: 920 nm, photo-stimulation wavelength: 1040 nm. Initial targets for stimulation were identified based on nuclear mRuby3 expression (gray dots overlaid on red nuclei) and screened in small groups of 20–30 neurons for activation thresholds using 2-photon calcium imaging.
B) Schematic of large ensemble stimulation protocols. Top: A single holographic ensemble comprised of the most activatable neurons in the volume was stimulated at a predetermined frequency (5 × 15 ms pulses at 33 Hz for the 408-target ensemble in D-E). Bottom: The most activatable neurons in the volume were randomly divided into m × n-target ensembles and stimulated in a multiplexed manner by fast switching of SLM frames (5 × 15 ms pulses at 22 Hz for the 10 × 100-target ensembles in F-G).
C) Summary of the size and photoactivation of the targeted ensemble in each experiment using single phase masks (solid edged circles) and multiplexed phase masks (dashed edged circles). The diameter of the circle indicates the size of the ensemble, ranging from 308 targets for the single phase mask experiments to 1300 targets for the multiplexed experiments. The fill color indicates the proportion of targeted neurons successfully activated in each experiment. The example experiments shown in the remaining panels are denoted with the corresponding panel letters. Large ensemble experiments were performed on 6 mice/16 sessions (single phase mask: n=5 mice/7 sessions, multiplexed phase masks: n=5 mice/9 sessions).
D) Top: Raster plot of the response of 1195 candidate neurons across 4 planes to stimulation in the power screening step. In this example, the top 408 highly activated neurons at 20mW (Wilcoxon rank-sum test between z-scored ΔF/F before and after stimulation, p < 0.05) were picked for subsequent large ensemble stimulation. Middle: Raster plot of calcium responses to the 408-target ensemble stimulation at 20mW. The thick red bar below the x-axis indicates the total duration of photostimulation. 346/408 neurons in the ensemble were significantly activated and 51/408 neurons were suppressed by the stimulation (Wilcoxon rank-sum test, p < 0.05). Bottom: mean ± s.e.m calcium response of all neurons in the targeted ensemble on stimulation trials (red trace) and control trials (black trace).
E) 4-plane volumetric maps of the mean calcium responses of all recorded neurons for the 408-target ensemble experiment in D.
F) As in E) but for multiplexed stimulation of 10, 100-target ensembles. 631/980 neurons in the ensemble were significantly activated and 323/980 neurons were suppressed by the stimulation (Wilcoxon rank-sum test, p < 0.05). Bottom: mean ± s.e.m calcium response of all neurons in the targeted ensemble on stimulation trials (red trace) and control trials (black trace).
G) Similar to E but for the multiplexed ensemble stimulation shown in F.
In many cases, simply elevating firing rate across a large population of neurons in a single trial even without strict simultaneity (i.e., coactivation within a few ms) can be highly informative. We generated a series of holograms where each hologram targeted ~100 neurons and updated the SLM at a high rate that our simulations suggested could yield the maximum number of co-activatable neurons on a single one second epoch. Under these constraints, we were able to drive increases in firing rates in up to 631 neurons for 10 × 100-target holograms multiplexed into a single 1-second trial (Fig. 8F,G; Wilcoxon rank-sum test, one-tailed p < 0.025). Again, many of the targeted neurons showed net suppression during this large-scale stimulation, presumably due to local, recurrent inhibition.
Taken together, the engineering and identification of microbial opsins optimized for patterned illumination optogenetics, combined with new paradigms for maximizing neural ensemble control, substantially expands the capabilities of spatially precise population neural control. Our data demonstrate that ChroME2f and ChroME2s are ultra-potent opsins that substantially increase the size of a controllable neural population with patterned illumination optogenetics, yet still provide outstanding millisecond temporal control across a broad array of conditions.
Discussion
The biophysical properties of microbial opsins critically determine the scale, speed, and fidelity of optogenetic experiments, particularly for two-photon optogenetic excitation of neuronal population in vivo. Although numerous opsins have been characterized to date, there is still a need for opsins with enhanced features, specifically for multiphoton approaches where an ever-increasing scale and speed of photo-activation is desirable. We used rational design of the previously characterized opsin ChroME to design and validate a panel of mutants with enhanced properties, notably improved potency but with fast kinetics, and variants that span the spectral range, enabling all-optical experiments with the ever-expanding array of fluorescent optical indicators of neuronal activity, such as for calcium, voltage and neurotransmitter release, that span the two-photon spectrum. Importantly, we identified two ChroME variants, ‘ChroME2f’ and ‘ChroME2s’ that provide markedly enhanced photocurrents when expressed in cell culture or in cortical neurons ex vivo, yielding much greater optical sensitivity while preserving the temporal fidelity afforded by their parent opsin, ChroME. We present detailed two-photon excitation spectral analysis of these opsins, as well as provide the most comprehensive two-photon excitation spectral data on a set of other widely used opsins available to date. This work establishes a critical knowledge base for a wide toolkit of opsins that can be used under a variety of experimental constraints.
In brain slices, ChroME2f and ChroME2s enabled very high-fidelity temporal control across a broad range of frequencies while requiring substantially less power than ChroME. Although ChRmine offers the ability to co-activate the most neurons with a limited power budget, it only provided reliable temporal control under a very narrow range of the conditions we tested, and primarily only with shorter illumination times that negate some of the benefit of its high sensitivity. Previous work has found that opsins with varying kinetic characteristics can enable sub-millisecond control under specific conditions(Chen et al., 2019, Ronzitti, Conti, et al., 2017, Marshel et al., 2019), although other work has demonstrated that under demanding applications opsins with faster closing kinetics more reliably provide high frequency, high fidelity temporal control(Gunaydin et al., 2010, Mager et al., 2018, Mardinly et al., 2018). These studies, consistent with the work here, showed that opsins with fast closing kinetics limit extra spikes and missed spikes. Thus, when an experiment demands faithful reproduction of specific spike trains or frequencies of action potential generation, but when careful tuning of laser power or duration for each neuron is not possible, ChroME2f or ChroME2s are likely to be preferable to slower closing-kinetic opsins, such as ChRmine. Opsin expression levels can vary widely across neurons in a single preparation, and so tuning the light levels delivered to each neuron may be challenging. Conversely, when the specific aim is to activate as many neurons as possible at a time, ChRmine presents a potentially better choice. It is important to note that in this study we only used scanless (soma-sized spot) illumination of opsin-expressing neurons, rather than ‘spiral-scan’ activation. Whether these different modes of two photon optogenetic activation would yield different results may be tested in the future.
A key concern for any opsin in an all-optical experiment, however, is unwanted activation of the opsin by the imaging laser. Under identical conditions and among the four opsins we tested, our data show that ChRmine-expressing neurons exhibited the highest levels of such optical-crosstalk, with direct drive of action potentials depending on the imaging parameters. Although not as severe in all conditions, ChroME2s-expressing were also susceptible to substantial unwanted depolarization. In contrast, ChroME2f and ChroME exhibited similar levels of optical-cross talk, and substantially less than ChRmine or ChroME2s under most conditions. We suspect that a variety of opsin characteristics influence this crosstalk, including the 2p absorbance spectrum, the on and off-kinetics, their sensitivity, and their photo-conductance. Since ChRmine has slower off kinetics and higher light sensitivity but not a substantially difference 2p absorbance spectrum than the ChroME opsins, we tentatively ascribe this difference to these characteristics. Engineering a fast ChRmine mutant or one with lower light sensitivity could address this issue, although this would also compromise its net potency for optical stimulation. Based on our measurements of crosstalk for the various opsins we tested, we propose that when crosstalk must be avoided, ChroME2f should be the opsin of choice. When crosstalk is less of concern, either ChroME2s or ChRmine should suffice, although as noted above, ChroME2s should be chosen for experiment that require the photo-induction of precise numbers of frequencies of action potentials.
Under imaging parameters than can be used for large scale imaging of neuronal activity with calcium indicators - namely very large fields of view (~1 mm × 1 mm), low frame rates (3–6 Hz), and modest imaging powers, unwanted depolarization for ChroME and ChroME2f was negligible, while for ChroME2s and ChRmine might still be deemed acceptable (~1–5 mV). In contrast, when imaging smaller fields of view (~0.3 mm 0.3 mm) at high frame rates (30 Hz), conditions that are also commonly used, the calcium imaging laser will likely directly drive action potentials in neurons expressing ChRmine and ChroME2s which may alter network physiology and thus compromise the interpretations of the experiment. These experiments argue that opsins and imaging conditions must be chosen with care to avoid unwanted sub-threshold, and occasionally supra-threshold, activation of opsin-expressing neurons by imaging lasers (Packer et al., 2014). Notably, we only tested such ‘optical cross-talk’ in L2/3 pyramidal cells. Other classes of neurons which may exhibit significantly higher intrinsic excitability (such as neurons with higher input resistance, lower action potential thresholds, or resulting from higher opsin-expression levels) might be activated under a broader array of conditions. Thus, it may be important for any given experimental paradigm to directly test this in the cell types studied and under the imaging conditions used.
In vivo we found that ChroME2f and ChroME2s substantially outperformed ChroME with respect to the power it took to photoactivate neurons. Correspondingly, we found that with ChroME2s we could simultaneously activate very large populations of neurons in the brains of awake mice. Brain heating places an upper limit on the scale of such experiments (Picot et al., 2018, Owen, Liu and Kreitzer, 2019, Mardinly et al., 2018, Podgorski and Ranganathan, 2016), and thus further engineering or identification of even more potent opsins could still be advantageous depending on the goals of an experiment. Another constraint in computer generated holography is that as the number of target spots increases in a hologram, the contrast ratio of the hologram decreases(Pégard et al., 2017). This can be addressed by using sparser holograms and rapidly multiplexing between phase masks with ultrafast SLMs, using multiple conventional SLMs(Marshel et al., 2019), or by scanning the excitation laser across a single SLM that is split along its length into separate phase masks(Parot et al., 2020)(Faini et al., 2021). A related problem is separating ‘off-target’ direct activation of non-holographically targeted neurons, from indirect effects driven through the network. This is not trivial when all neurons express opsin. Indeed, in our experiments, 67% of all non-targeted neurons were near (within 25um of) a targeted neuron, substantially increasing the prevalence of off-target activation. One potential solution would be to express opsin in most neurons except for a sparse random subset. Then, by comparing the induced response in non-targeted cells expressing the opsin to those that don’t, one could rigorously estimate the fraction of the response in non-targets that was direct versus indirect. To achieve this one might co-inject an AAV that drives the opsin in a ‘Cre-off’ manner, mixed with a dilute AAV driving Cre and an AAV driving a Cre-on fluorophore (e.g, mRuby3). In this context, mRuby3-expressing cells would not express opsin and any effects on the activity must be synaptically driven. While future work must overcome these challenges, the high-performance opsins we present here provide the ability to co-activate much larger ensembles but still with very high temporal fidelity than previously possible. This opens the door to a much broader array of perturbations that should help elucidate the neural basis of perception, cognition and behavior.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to the Lead Contact, Hillel Adesnik (hadesnik@berkeley.edu).
Materials availability
All animal strains used in this study are available from Jackson Laboratories. All plasmids and viral vectors generated for the study have been made available from Addgene or can be obtained from the lead contact upon request. Accession numbers are listed in the key resources table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| AAV9-CAG-DIO-ChroME-ST-P2A-H2B-mRuby3 | Mardinly et al., 2018 | N/A |
| AAV9-CAG-DIO-ChroME2s-ST-P2A-H2B-mRuby3 | This article | N/A |
| AAV9-CAG-DIO-ChroME2f-ST-P2A-H2B-mRuby3 | This article | N/A |
| AAV9-CAG-DIO-ChRmine-ST-P2A-H2B-mRuby3 | This article | N/A |
| AAV9-CAG-DIO-ChrimsonR-ST-P2A-H2B-mRuby3 | This article | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: CD1 (ICR) | Charles River | RRID: MGI:5659424 |
| Mouse: B6;CBA-Tg(Camk2a-tTA)1Mmay/J | Jackson Laboratory | RRID: IMSR_JAX: 003010 |
| Mouse: B6;DBA-Tg(tetO-GCaMP6s)2Niell/J | Jackson Laboratory | RRID: IMSR_JAX: 024742 |
| Mouse: B6.129S2-Emx1tm1(cre)Krj/J | Jackson Laboratory | RRID: IMSR_JAX: 005628 |
| Recombinant DNA | ||
| pCAG-ChroME-mRuby2-ST | Mardinly et al., 2018 | Addgene_108902 |
| pAAV-CAG-DIO-ChroME-ST-P2A-H2B-mRuby3 | Mardinly et al., 2018 | Addgene_108912 |
| pAAV-hSyn-DIO-ChrimsonR-mRuby2-ST | Pégard et al., 2017 | Addgene_105448 |
| pCAG-Chronos-mRuby2-ST | Pégard et al., 2017 | Addgene_105446 |
| pCAG-ChrimsonR-mRuby2-ST | Pégard et al., 2017 | Addgene_105447 |
| pCAG-ChR2-mRuby2-ST | Mardinly et al., 2018 | Addgene_109125 |
| pCAG-ChroME(F243Y)-mRuby2-ST | Mardinly et al., 2018 | Addgene_109127 |
| pCAG-ChroME2s-mRuby2-ST | Mardinly et al., 2018 | Addgene_109128 |
| pCAG-CoChR-mRuby2-ST | Mardinly et al., 2018 | Addgene_109136 |
| pCAG-ChroME2f-mRuby2-ST | This article | Addgene_170160 |
| pAAV-CAG-DIO-ChroME2s-ST-P2A-H2B-mRuby3 | This article | Addgene_170163 |
| pAAV-CAG-DIO-ChroME2f-ST-P2A-H2B-mRuby3 | This article | Addgene_171150 |
| pCAG-ChroMD-mRuby2-ST | This article | Addgene_171158 |
| pCAG-ChRmine-mRuby2-ST | This article | N/A |
| pAAV-CAG-DIO-ChRmine-ST-P2A-H2B-mRuby3 | This article | N/A |
| pCAG-PsChR-mRuby2-ST | This article | N/A |
| pCAG-GtACR1-mRuby2-ST | Mardinly et al., 2018 | Addgene_109136 |
| pFUGW-hGtACR2-EYFP | Govorunova et al., 2013 | Addgene_67877 |
| pAAV-ReaChR-mCherry | This article | N/A |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Cell Profiler | Carpenter et al., 2006 | https://cellprofiler.org/ |
| MATLAB | MathWorks | RRID: SCR_001622 |
| Python | Python Software Foundation | RRID: SCR_008394 |
| Suite2p | Pachitariu et al., 2016 | RRID: SCR_016434 |
| ScanImage | Vidrio Inc. | RRID: SCR_014307 |
| Adobe Illustrator | Adobe | RRID: SCR_010279 |
| Other | ||
| 1p excitation – Spectra X light engine | Lumencor | https://lumencor.com/products/spectra-x-light-engine |
| Tunable laser – Insight X3 | Spectra-Physics | https://www.spectra-physics.com/f/insight-x3-tunable-laser |
| 2p stimulation laser – Femtotrain | Spectra-Physics | https://www.spectra-physics.com/f/femtotrain-femtosecond-oscillator |
| 2p stimulation laser -Monaco | Coherent | https://www.coherent.com/lasers/ultrashort-pulse/monaco |
| 2p stimulation laser- Femtosecond | Satsuma HP2, Amplitude Systems | https://amplitude-laser.com/products/femtosecond-lasers/satsuma/ |
| 2p imaging laser – Mai Tai | Spectraphysics | https://www.spectra-physics.com/f/mai-tai-ultrafast-laser |
Data and code and availability
All data and analysis software are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experiments on animals were conducted with approval of the Animal Care and Use Committee of the University of California, Berkeley. In all experiments we attempted to use male and female mice equally. Mice used for experiments in this study were either wild-type (CD-1 (ICR) white strain, obtained from Charles River), or triple transgenic mice EMX1-Cre;CaMK2-tTA;tetO-GCaMP6s obtained by crossing the corresponding lines in-house (JAX stock# 005628, Jax stock# 003010, Jax stock # 024742, respectively). Mice were housed in cohorts of five or fewer in a reverse light:dark cycle of 12:12 hours, with experiments occurring during the dark phase.
METHOD DETAILS
Plasmid construction and Mutagenesis
Mutations in Chronos and ChroME were introduced by either site-directed mutagenesis or overlap extension PCR and verified by DNA sequencing. ChrimsonR, ChR2, CoChR, and ChRmine were obtained from Addgene. All opsins were fused to mRuby2 at their C-terminus and sub-cloned into the pCAGGS (pCAG) expression vector by In-Fusion cloning (Clontech, Mountain View, CA). In order to target the opsins to the soma and proximal dendrites of neurons, the sequence encoding the proximal restriction and clustering domain of the Kv2.1 voltage-gated potassium channel consisting of amino acids 536–600 (soma-targeting; ST) was codon optimized, synthesized (Integrated DNA Technologies, Coralville, IA) and inserted at the C-terminus of mRuby2 by In-Fusion cloning. For Adeno Associated virus (AAV) preparations, soma-targeted opsin cDNA was fused to a FLAG tag for immunofluorescence and a nuclear mRuby3 via a P2A self-cleaving peptide into pAAV and virus was prepared either at the Penn Vector Core or at the UC Berkeley Vision Core’s Gene Delivery Module.
In Utero Electroporations and brain slice recording
Electroporations were performed on pregnant CD1 (ICR) mice (E15, Charles River). For each surgery, the mouse was initially anesthetized with 5% isoflurane and maintained with 2.5% isoflurane. The surgery was conducted on a heating pad to maintain body temperature, and warm sterile phosphate-buffered saline (PBS) was intermittently perfused over the pups throughout the procedure. A micropipette was used to inject ~2 μl of recombinant DNA at a concentration of 2 μg/μl into the left ventricle of each embryo’s brain (typically DNA encoding opsins with GCaMP6s at a concentration of 2:1). Fast-green (Sigma-Aldrich) was used to visualize a successful injection. Using platinum-plated 5mm Tweezertrodes (BTX Harvard Apparatus) electrodes connected by a Y-connector to the negative pole, both sides of the embryo’s head were gently grabbed and a third electrode connected to the positive pole was placed slightly shifted below lambda to target the visual cortex and electroporated with 6 pulses at 30 V with a 1s delay using an Electro Square Porator (BTX Harvard Apparatus). After the procedure, the mouse was allowed to recover and come to term, and the delivered pups were screened for GCaMP6s expression and allowed to develop normally. Acute coronal slices were prepared and recorded from mice (ages p10–29) as described. Mice were screened with a handheld 300 mW 594 nm laser and filter goggles for expression after decapitation and before slicing. After slicing, recordings were made from the slices with strongest expression from the densest area as judged by fluorescence.
Histology and confocal microscopy
Mice were deeply anesthetized with ketamine/xylazine and transcardially perfused with phosphate-buffered saline (PBS) and 4% paraformaldehyde. Brains were post fixed for at least 2 h. Brains were embedded in 30% sucrose solution overnight, then frozen and 40 μm sections made on a microtome (American Optical Society). All sections were mounted on slides and sealed with Vectashield with DAPI (Vector Laboratories). Confocal images were acquired using an Olympus Fluoview system (Fv1000 Olympus Microscope) running the Fluoview software (Olympus), with 488 and 543nm lasers. For quantification of opsin expression, analysis was performed using the open-source CellProfiler software (Carpenter et al., 2006). An image analysis pipeline (detailed in Fig. S1a) was constructed to detect and measure mRuby2 fluorescence from projections of 0.43 μM stacks of confocal images from multiple sections for each of the three opsins quantified.
In vitro electrophysiology
In vitro slice recordings were performed on 300um -thick coronal slices coming from P12 to P44 (opsin characterization experiments) or 4-6-week-old animals (cross-talk experiments) in utero electroporated at E15.5 with plasmids containing one of the opsin: ChRmine, ChroME, ChroME2s or ChroME2f and GCaMP6s (2:1 proportion). Opsin positive cells in L2/3 were identified under 1P conditions. Whole-cell patch-clamp protocols were performed in ACSF perfusion solution (in mM: NaCL 119, NaHCO3 26, Glucose 20, KCl 2.5, CaCl 2.5, MgSO4 1.3, NaH2PO4 1.3) in temperature-controlled (33°C) conditions.
Patch pipette (4–7 MΩ) were pulled from borosilicate glass filaments (Sutter Instruments) and filled with K-gluconate solution (in mM: 110 K-gluconate, 10, HEPES, 1 EGTA, 20 KCl, 2MgCl2, 2 Na2ATP, 0.25 Na3GTP, 10 Phosphocreatine, 295 mOsm, pH=7.45) mixed with 5uL of 50 μM Alexa hydrazide 488 dye (cross-talk experiments only, Thermo Fisher Scientific). Data was recorded at 20 kHz using 700b Multiclamp Axon Amplifier (Molecular Devices). The headstage with the electrode holder (G23 Instruments) were controlled by Motorized Micromanipulator (MP285A, Sutter Instruments). All data was acquired and analyzed with custom code written in Matlab using the National Instruments Data Acquisition Toolbox.
Viral vectors (for in vivo experiments)
Triple transgenic mice expressing GCaMP6s and Cre recombinase in excitatory neurons were obtained by crossing CaMKII-tTA to teto-GCaMP6s (Mardinly et al., 2018); Emx1-Cre mice. These mice were injected with adenoviral vectors expressing soma-targeted opsin and the red fluorophore mRuby3 in a Cre dependent fashion. All viruses had identical scaffold: AAV9-CAG.DIO.[Opsin]-FLAG-ST.P2A.H2B.mRuby3.WPRE.SV40 where “Opsin” is one of the five opsins shown in Fig. 7: ChrimsonR, ChroME, ChroME2f, ChroME2s or ChRmine. Custom made viral preparations for Chrimson, ChroME and ChroME2s were generated by Addgene, whereas viral preparations for ChroME2f and ChRmine were generated by Berkeley Vision Science Core, Gene Delivery Module Facility and presented as the AAV9-2YF variant serotype (Dalkara et al., 2012).
Surgery for in vivo experiments
AAV vectors were injected intracortically in V1 and cranial window surgeries were performed immediately after. Briefly, mice were anesthetized with isoflurane (2%) and administered 2 mg/kg of dexamethasone as an anti-inflammatory and 0.05 mg/kg buprenorphine as an analgesic. The scalp was removed, the fascia retracted, and the skull lightly etched. Following application of Vetbond (3M) to the skull surface, a custom stainless steel headplate was fixed to the skull with two dental cements: Metabond (C&B) followed by Ortho Jet (Lang). After the dental cement dried, a 3-mm diameter craniotomy over the left primary somatosensory cortex was drilled, and residual bleeding stopped with repeated wet–dry cycles using sterile artificial cerebrospinal fluid, gauze, and Gelfoam (Pfizer). A window plug consisting of two 3-mm diameter coverslips glued to the bottom of a single 5-mm diameter coverslip (using Norland Optical Adhesive #71) was placed over the craniotomy and sealed permanently using Ortho Jet (Lang). Animals were allowed to recover in a heated recovery cage before being returned to their home cage. Seven days after surgery, animals were habituated to head-fixation under a freely moving circular treadmill, and in vivo all-optical opsin potency estimations were done after 21 days post-surgery to allow for saturated opsin expression.
One-photon and two-photon opsin characterization experimental rig
For slice and cell culture experiments characterizing various opsins features (Figures 1–4) we employed a Scientifica slice scope equipped with Spectra X light engine for one-photon experiments (Lumencor), providing various light wavelengths and a Femtotrain 1040-5 (Spectra Physics, 10 MHz) for two-photon excitation. The laser beam was custom fitted to the scope resulting in a single focused two photon laser spot targeted at the cell. The spot size was ~12.5 μm and generated through conventional achromatic lenses and a 40X Olympus water immersion objective. Power was controlled by a Pockels Cell under computer control and gated by a laser shutter (Thorlabs). Photo-stimulus width was 5 ms unless otherwise indicated.
CHO cell recording
Chinese hamster ovary CHO cells were transfected as above with a pCAG-opsin-mRuby2-Kv2.1 plasmid. 1–2 days after transfection coverslips with transfected cells were transferred to the brain slice rig described above. One-photon photostimulation of cells was performed at 510nm for Chronos, ChroME, ChoME variants, and ChRmine at a power of 0.45 mW 470 nm for ChR2 and CoChR and 630nm for ChrimsonR using a Spectra X light engine (Lumencor). Currents were measured at a holding potential of −60 mV. The time to peak current was measured from average currents, and decay time constants were measured by fitting the traces from stimuli offset to a single exponential.
Two-photon excitation spectra of opsins
Spectra collection experiments were performed on CHO cells prepared as mentioned above with an Insight X3 tunable laser. Power was controlled and carefully calibrated across the entire spectrum with the Pockels cell and set to be below saturation for all opsins. Power calibration was performed with a Thorlabs thermal power meter through the microscope objective to account for all optics coatings in the path and the power spectrum of the laser. Wavelength was controlled by the serial interface via Matlab.
Brain slice characterization of opsin response characteristics
For the broadband ‘poisson’ photo-stimulation tests, photo-stimulus width was 5 ms unless otherwise indicated. On each trial a random pulse train (pulse times drawn from a Poisson distribution with mean rates of 10, 20 and 30 Hz) were delivered to each neuron. At the beginning of the experiment the power of a single 5 ms (or 0.5 ms in Figure S1a, Figure S2) laser exposure was set to reliably drive an action potential across successive trials (inter-trial interval = 2.3 seconds). In each recording, we additionally injected a small, subthreshold noisy current injection that was a fixed amplitude across all experiments. This subthreshold was meant to simulate in vivo conditions where cells experience a barrage of noisy synaptic input. There was no effect of opsin on the impact of this current injection. The mean depolarization due to this current injection for ChroME2f was 7.0±1 mV, n = 5, for ChroME2s was 5.7±0.2 n = 9, and for ChRmine was 7.2±0.4 n = 5, p = 0.08, 1-way-ANOVA. A post-hoc multiple comparison test showed no significant differences between groups. Spike jitter was computed as the standard deviation of spikes times across all light-evoked spiked in the experiment. Spike probability was computed as the fraction of light pulses that drove one or more spikes. Spike latency was the time between the onsite of the laser exposure and the peak of the subsequent action potential prior to any subsequent laser exposure.
Two-photon holographic setups
In vivo opsin characterization, opsin cross-talk experiments and large-scale photostimulation experiments were performed on two 3D-SHOT multiphoton holographic setups (refer to (Mardinly et al., 2018) for detailed description). The setups were both custom built around a commercial Sutter MOM microscope platform (Sutter Instruments) and combined a 3D photostimulation path, a fast resonant-galvo two-photon raster scanning imaging path and a wide-field one-photon epifluorescence/IR transmitted light imaging path. The stimulation and imaging beams were merged together using a polarizing beamsplitter placed before the microscope tube lens.
Femtosecond fiber lasers were used for photostimulation: Satsuma HP2 (1030nm, 2MHz, 350fs, Amplitude Systemes) for in vivo opsin characterization (setup 1) and Monaco 1035-80-60 (1040nm, 1MHz, 300fs, Coherent) for large scale photostimulation (setup 2). On both setups, the stimulation laser was directed onto a blazed diffraction grating (600l/mm, 1000nm blaze, Edmund Optics 49–570 or 33010FL01-520R Newport Corporation) for temporal focusing. In order to be able to utilize the total available laser power on setup 2 (60W laser output), the beam was enlarged by a 2.5 factor to prevent heat damage of the grating surface. The spot on the grating was relayed onto a rotating diffuser where it formed a temporally focused spot. The rotating diffuser was used to both randomize the phase pattern imprinted on the temporally focused spot and to expand the beam in the direction orthogonal to the temporal focusing direction and fully fill the spatial light modulator (HSP1920 192×1152 pixels Meadowlark Optics). The SLM plane was relayed through 4f systems to the back aperture of an Olympus 20x water immersion objective, resulting in custom 2D or 3D distribution of temporally-focused spots at the focus of the objective. Holographic phase masks were calculated using the iterative Gerchber-Saxton algorithm (Gerchberg and Saxton, 1972) and intensity distribution was corrected to accommodate for diffraction efficiencies.
The two-photon imaging paths relied on Ti:sapphire lasers, Chameleon (Coherent, setup 1) or Mai Tai (Spectra Physics, setup 2), with external power control via Pockels cells (Conoptics, Inc). For fast raster scanning, both systems were equipped with conjugated 8 kHz resonant galvo-galvo systems (relayed with either a pair of Plössl lenses – setup1, or a pair of 90° off-axis parabolic mirrors - setup2) (Negrean and Mansvelder, 2014). The imaging path hardware was controlled by ScanImage software and custom Matlab code was used to control the spatial light modulator for targeted photostimulation and synchronize with imaging.
Epifluorescence excitation was via an X-Cite LED (Excelitas Technologies, patch clamp rig for Fig. 6) or Spectra X (Lumencor, patch clamp fig for Fig. 1–5) light source filtered by appropriate excitation filter set. For slice transillumination we used a 750nm and IR diffuser. The image was collected using an Olympus 20 × magnification water-immersion objective and a CCD camera and displayed on a screen enabling targeted patch clamping (Fig. 6) or Olympus 40x water immersion objective (Figs. 1–5).
Cross-talk characterization data collection
Cross-talk characterization was performed in current clamp across 96 different imaging conditions of variable FOV (980, 680, 527,294 μm), imaging frequency (3,6,10,30 Hz) and power (5,10,15,25,35,45 mW). The FOV size was kept constant at 512×512 pixels and a pixel dwell time of 87 ns. Each experimental trace contained a baseline period and 2s scanning period at a given condition. At the beginning of the experiment, we used current injection to characterize each cell’s intrinsic properties. For voltage clamp experiments cells were clamped at −70 mV. Access resistance was monitored throughout the protocol’s duration and only cells in which the Rs/Rm ratio was below 0.3 were accepted (mean and standard error for each opsin group: ChRmine 0.1±0.01, ChroME 0.15±0.02, ChroME2s 0.15±0.02, ChroME2f 0.10±0.01). For current clamp protocols, cells were kept at resting potential and only cells with stable resting potential (<4 mV change) during the entire protocol were accepted for data analysis. Each opsin group was collected using tissue obtained from 4–7 animals and contained: 15, 9, 14, 17 cells for ChRmine, ChroME, ChroME2s, ChroME2f respectively. For all the measurements, data were baseline-subtracted and spikes were removed by applying a median filter. Outliers’ analysis was applied to the 2P evoked photocurrent results to exclude cells whose mean was 2 standard deviations away from the population mean at each power. The same group of cells was taken into account in all the measurements. All measurements are expressed as the grand mean over all the cells and s.e.m. One-way ANOVA across all conditions was calculated to specify statistical significance and post hoc analysis (Fisher’s Least Significant Difference test). For scanning-evoked spike count analysis the scanning period was binned into 5 ms periods. Spikes were detected by crossings above 0 mV. A smaller subset of cells was also recorded in Voltage Clamp configuration using the same imaging conditions.
In vivo all optical experiments:
Triple transgenic mice (camk2a-tTa;tetO-GCaMP6s;emx-IRES-Cre mice) were head-fixed on a freely spinning running wheel. Imaging and photostimulation wavelengths were respectively 920nm and 1030–1040nm. Neurons that co-expressed opsin and nuclear-targeted H2B-mRuby3 and GCaMP6s were automatically segmented based on frame-averaged images taken at 1020nm using custom peak-intensity detection Matlab software. The centroids of the segmented masks were then used to compute holographic phase masks that were loaded sequentially on the spatial light modulator. For experiments in Fig. 7, acquisitions were performed at 15 or 30 Hz frame rate (15 Hz for ChroME2f, ChroME2s and ChRmine, 30 Hz for ChroME and Chrimson) and with ~900 × 900 μm fields of view. The imaging/photostimulation plane was between 100 and 200 μm below the pial surface and the imaging power was below 50mW for all opsins.
Large ensemble stimulation experiments (Fig. 8) were performed on setup 2 (see description above) and involved n = 6 mice. For these experiments, fast multi-plane imaging was achieved with an electrically tunable lens (Optotune AG) in the imaging path just before the scanning unit. Typically, 3–5 planes spaced 30μm apart were acquired at 4–6 Hz frame rate. Each plane covered a ~680 × 680 μm field of view at zoom 1.5x or a ~950 × 950 μm field of view at zoom 1x, and the imaged volume had an axial span of 60–120 μm and was located between 100–300 μm below the pial surface. Stimulation targets were identified across all planes based on frame-averaged images taken at 920 nm, and using the segmentation procedure described above. At the beginning of each experimental session, all identified (typically 1200–2000) holographic targets were included in a screening procedure and putative neurons were screened for photoactivatability by targeting them in groups of 20–30 at several powers between 0 – 80 mW. For each cell, trialwise photoactivation was determined using a Wilcoxon test between the raw calcium fluorescence values before and after stimulation. The proportion of photoactivated trials as a function of power was fit to a logistic function, and the power at which a given cell was photoactivatable on a third of the trials based on the logistic fit was taken as the threshold power for that cell for the remainder of the session. The main experiment involved either holographically targeting neurons in a single large ensemble simultaneously using one phase mask (n = 5 mice, 7 sessions) or temporally multiplexing phase masks for near-simultaneous targeting of smaller groups of neurons part of an overall larger ensemble (n = 5 mice, 9 sessions). Temporal multiplexing was performed in one of two ways - a few sessions had all phase masks exhaustively interleaved (i.e., all phase masks were presented once before any phase mask was repeated; n = 4 sessions), and the remaining sessions had non-exhaustive interleaving of groups of phase masks in order to maintain a high stimulation frequency (i.e., 2–3 phase masks were repeated till completion before the next group was presented, and so on; n = 5 sessions). The size of the ensembles was determined from the distribution of threshold powers computed in the screening step and the total usable power in the setup. In each case, power was distributed to each cell taking into account the threshold power and the diffraction efficiency of the path at the corresponding targeted location.
2P imaging and photo-stimulation data analysis
Opsin comparison experiments
For power curve comparison experiments in Figure 7, motion correction and calcium source extraction were performed using Suite2p (Pachitariu et al., 2016). Briefly, raw calcium videos were motion-corrected using Suite2P with subpixel alignment = 10, and calcium sources were extracted. Calcium sources were then manually examined and accepted or rejected based on their overlap with morphologically identifiable neurons. Neuropil subtracted fluorescence vectors (F) (using a neuropil coefficient of 0.7) were used for downstream analysis. Calcium signals were acquired continuously, and each cell’s fluorescence was z-scored. Holographic targets were aligned to calcium sources by calculating the Euclidean distance between the centroids of all holographic targets and all calcium sources and finding the minimum. Rarely, the automated software assigned targets to calcium sources with distance >10 μm; these were excluded from subsequent analysis. For power curve comparison experiments in Fig. 7, neurons were stimulated in groups of 1 (for ChroME and ChrimsonR) or 10 (for ChroME2s, ChroME2f and ChroME), targets per phase mask. The stimulation consisted in 5 pulses of 5 ms duration at 30 Hz for each hologram, and all cells were stimulated at different powers. The mean z-scored F of each cell was aligned with the time of the stimulation, a baseline of 5–10 frames was calculated and compared to the mean z-scored F in the response window. A trial-wise two-sided Wilcoxon test was used to assess whether each cell was photoactivated. Because the sequence of targeted cells was conserved across trials, occasionally, a neuron would systematically respond to off target light coming from a nearby target during its baseline period (Figure S8). These cells were excluded from the analysis by performing a trial-wise Wilcoxon test during the baseline period at 0 mW vs at each of the powers. The fraction of photoactivated cells was calculated by dividing successfully activated neurons by the total number of automatically targeted neurons.
Large ensemble stimulation experiments
For the large ensemble stimulation experiments, the simultaneous activation of many neurons introduced large and systematic correlations that led to a miscount of calcium sources based on Suite2P alone. Therefore, we employed a hybrid calcium source extraction procedure that combined Suite2P-based extraction as described above for the opsin comparison experiments (diameter = 14 μm, neuropil/cell diameter ratio = 5) with custom extraction of calcium signals around all previously identified holographic targets using the same parameters for cell and neuropil size that were used for the Suite2P step. To do this, holographic targets were first aligned to Suite2P-identified calcium sources using 2-d cross-correlation of two sets of images – the centroids of holographic targets, and the centroids of Suite2P sources that exceeded a redness probability of 0.33. Following offset correction and alignment, Suite2P calcium sources were matched to the closest target centroid within a 10 μm radius – sources that were greater than this distance away from any target were deemed to be non-targeted neurons. Targets that were not matched to any Suite2P source were subjected to the custom calcium signal extraction procedure described above.
Neuropil subtracted fluorescence vectors (F) were converted to relative fluorescence values (ΔF/F0) on each trial by subtracting out and dividing by the mean F over a 10-frame baseline on that trial. The relative fluorescence traces for each cell were z-scored and all subsequent analyses were performed on the z-scored ΔF/F0 traces (referred to as “activity” below). The trial-averaged activity trace for each cell was aligned to the start of stimulation for that cell to depict stimulation-evoked activity changes. To determine photoactivation or suppression, we performed a one-tailed Wilcoxon rank-sum test between the activity values 2 seconds preceding simulation onset and 2 seconds around activity extrema post- stimulation onset for each cell.
To determine the optimal stimulation parameters, we generated parameter surface fits to the evoked response by sweeping through stimulation power, pulse width, frequency and number of pulses in a separate set of experiments and picked the combination of parameters that produced the highest population responses. For the multiplexed hologram experiments, we also factored in the trade-off between pulse width/frequency and number of interleavable phase masks between pulses targeting the same hologram. Prior to each large ensemble stimulation experiment, we pre-screened targets in the volume of interest to test their optogenetic gain by targeting them in small groups across a range of powers for 20 repetitions each. Then, for each target, we fit a logistic function to the proportion of significantly photoactivated trials at each power (with added anchors of proportion 1 at a power well above the tested range and proportion 0 at a negative power below the tested range). The power at which the neuron was photoactivatable on a third of the trials based on the fit was taken as its threshold power. We then either included the target in or discarded it from the main large ensemble based on the total available power and the distribution of threshold powers across screening targets.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using MATLAB or Python. The analyses performed were ANOVAs, with multiple comparisons. Unless otherwise noted, all plots with error bars were reported as mean ± SEM. Sample size was not predetermined using power analysis.
Supplementary Material
Highlights.
ChroME2.0 opsins enable large-scale, temporally preicse optogenetic control
ChroME2s allows large ensemble 2p stimulation of >600 neurons per second in vivo.
Detailed biophysical analysis of the current opsin toolbox with 2p excitation
Acknowledgements
This work was funded by NIH grant UF1NS107574 (HA), NIH Grant F32-EY031977 (WDH), the Simons Foundation Collaboration for the Global Brain award 415569 and NEI grant K99 EY029758-01 to I.A.O, and the New York Stem Cell Foundation. H.A. and S.B are New York Stem Cell Foundation Robertson Investigators. We thank Spectra Physics and Patrick Kolsch for use of the Insight X3, Karl Deisseroth for the ChRmine sequence and plasmid for cloning, and Janine Beyer for technical support. We thank Mei Li, UC Berkeley Vision Science Core Gene Delivery Module Facility for viral preparations and Silvio Temprana for assistance with confocal microscopy. We thank all the members of the Adesnik lab for comments on the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
H. Adesnik has a patent related to this work: 3D Sparse Holographic Temporal focusing, 2016, L. Waller, N. Pegard, and H. Adesnik, Provisional Patent Application #62-429,017.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
References
- Anselmi F et al. (2011) ‘Three-dimensional imaging and photostimulation by remote-focusing and holographic light patterning’, Proceedings of the National Academy of Sciences of the United States of America. doi: 10.1073/pnas.1109111108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumhagen F et al. (2011) ‘Neuronal filtering of multiplexed odour representations’, Nature. doi: 10.1038/nature10633. [DOI] [PubMed] [Google Scholar]
- Cardin JA et al. (2009) ‘Driving fast-spiking cells induces gamma rhythm and controls sensory responses’, Nature. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter et al. (2006) ‘CellProfiler: image analysis software for identifying and quantifying cell phenotypes’, Genome Biology doi: 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Reid L et al. (2019b) ‘Controlling Visually Guided Behavior by Holographic Recalling of Cortical Ensembles’, Cell. doi: 10.1016/j.cell.2019.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaigneau E et al. Two-photon holographic stimulation of ReaChR Frontiers in Cellular Neuroscience (2016) 10(OCT2016) doi: 10.3389/fncel.2016.00234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IW et al. (2019) ‘In Vivo submillisecond two-photon optogenetics with temporally focused patterned light’, Journal of Neuroscience. doi: 10.1523/JNEUROSCI.1785-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IW, Papagiakoumou E and Emiliani V (2018) ‘Towards circuit optogenetics’, Current Opinion in Neurobiology. doi: 10.1016/j.conb.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daie K, Svoboda K and Druckmann S (2021) ‘Targeted photostimulation uncovers circuit motifs supporting short-term memory’, Nature Neuroscience. doi: 10.1038/s41593-020-00776-3 [DOI] [PubMed] [Google Scholar]
- Daklara D, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Science Translational Medicine.doi: 10.1126/scitranslmed.3005708 [DOI] [PubMed] [Google Scholar]
- dal Maschio M et al. (2017) ‘Linking Neurons to Network Function and Behavior by Two-Photon Holographic Optogenetics and Volumetric Imaging’, Neuron. doi: 10.1016/j.neuron.2017.04.034. [DOI] [PubMed] [Google Scholar]
- Dhawale AK et al. (2010) ‘Non-redundant odor coding by sister mitral cells revealed by light addressable glomeruli in the mouse’, Nature Neuroscience, 13(11), pp. 1404–1412. doi: 10.1038/nn.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faini G, Molinier C, Telliez C, Tourain C, Forget BC, Ronzitti E, Emiliani V ‘Ultrafast light targeting for high-throughput precise control of neuronal networks’. Biorxiv. doi: 10.1101/2021.06.14.448315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan LZ et al. (2020) ‘All-Optical Electrophysiology Reveals the Role of Lateral Inhibition in Sensory Processing in Cortical Layer 1’, Cell. doi: 10.1016/j.cell.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah N, Reutsky I and Shoham S (2007) ‘Patterned optical activation of retinal ganglion cells’, in Annual International Conference of the IEEE Engineering in Medicine and Biology - Proceedings. doi: 10.1109/IEMBS.2007.4353812. [DOI] [PubMed] [Google Scholar]
- Fenno L, Yizhar O and Deisseroth K (2011) ‘The development and application of optogenetics’, Annual Review of Neuroscience. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli A et al. (2018) ‘Two-Photon Bidirectional Control and Imaging of Neuronal Excitability with High Spatial Resolution In Vivo’, Cell Reports. doi: 10.1016/j.celrep.2018.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerchberg RW and Saxton WO (1972) ‘PRACTICAL ALGORITHM FOR THE DETERMINATION OF PHASE FROM IMAGE AND DIFFRACTION PLANE PICTURES.’, Optik (Stuttgart). [Google Scholar]
- Gill JV et al. (2020) ‘Precise Holographic Manipulation of Olfactory Circuits Reveals Coding Features Determining Perceptual Detection’, Neuron. Elsevier. doi: 10.1016/j.neuron.2020.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorunova EG et al. (2013) Characterization of a highly efficient blue-shifted channelrhodopsin from the marine alga Platymonas subcordiformis. Journal of Biological Chemistry. DOI: 10.1074/jbc.M113.505495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin LA et al. (2010) ‘Ultrafast optogenetic control’, Nature Neuroscience. doi: 10.1038/nn.2495. [DOI] [PubMed] [Google Scholar]
- Hass CA and Glickfeld LL (2016) ‘High-fidelity optical excitation of cortico-cortical projections at physiological frequencies’, Journal of Neurophysiology, 116(5). doi: 10.1152/jn.00456.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun NY and Cardin JA (2020) ‘Activation of distinct channelrhodopsin variants engages different patterns of network activity’, eNeuro, 7(1). doi: 10.1523/ENEURO.0222-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HE et al. (2012) ‘Crystal structure of the channelrhodopsin light-gated cation channel’, Nature. doi: 10.1038/nature10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HE and Nureki O (2013) ‘Crystal structure of channelrhodopsin, a light-gated cation channel - All cations lead through the monomer’, Biophysics (Japan). doi: 10.2142/biophysics.9.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke NC et al. (2014) ‘Independent optical excitation of distinct neural populations’, Nature Methods. doi: 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY et al. (2013) ‘ReaChR: A red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation’, Nature Neuroscience. doi: 10.1038/nn.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz C et al. (2008) ‘Holographic photolysis of caged neurotransmitters’, Nature Methods. doi: 10.1038/nmeth.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager T et al. (2018) ‘High frequency neural spiking and auditory signaling by ultrafast red-shifted optogenetics’, Nature Communications, 9(1). doi: 10.1038/s41467-018-04146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinly AR et al. (2018) ‘Precise multimodal optical control of neural ensemble activity’, Nature Neuroscience. Springer; US, 21(6), pp. 881–893. doi: 10.1038/s41593-018-0139-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshel JH et al. (2019) ‘Cortical layer-specific critical dynamics triggering perception’, Science, 365(6453). doi: 10.1126/science.aaw5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis J et al. (2012) ‘Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins’, Nature Methods, 9(2). doi: 10.1038/nmeth.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita T et al. (2013) ‘Long-term channelrhodopsin-2 (ChR2) expression can induce abnormal axonal morphology and targeting in cerebral cortex’, Frontiers in Neural Circuits. doi: 10.3389/fncir.2013.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrean A and Mansvelder HD (2014) ‘Optimal lens design and use in laser-scanning microscopy’, Biomedical Optics Express, 5(5). doi: 10.1364/boe.5.001588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen SF, Liu MH and Kreitzer AC (2019) ‘Thermal constraints on in vivo optogenetic manipulations’, Nature Neuroscience. doi: 10.1038/s41593-019-0422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachitariu M et al. (2016) ‘Suite2p: beyond 10,000 neurons with standard two-photon microscopy’, bioRxiv. doi: 10.1101/061507. [DOI] [Google Scholar]
- Packer AM et al. (2012) ‘Two-photon optogenetics of dendritic spines and neural circuits.’, Nature methods, 9(12), pp. 1202–5. doi: 10.1038/nmeth.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AM et al. (2014) ‘Simultaneous all-optical manipulation and recording of neural circuit activity with cellular resolution in vivo’, 12(2). doi: 10.1038/nmeth.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagiakoumou E et al. (2010) ‘Scanless two-photon excitation of channelrhodopsin-2’, Nature Methods. doi: 10.1038/nmeth.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parot VJ et al. (2020) ‘Microsecond timescale selective access two-photon targeting for functional measurements in tissue’, in Optics InfoBase Conference Papers. doi: 10.1364/BRAIN.2020.BTu1C.6. [DOI] [Google Scholar]
- Pégard NC et al. (2017) ‘Three-dimensional scanless holographic optogenetics with temporal focusing (3D-SHOT)’, Nature Communications, 8(1), p. 1228. doi: 10.1038/s41467-017-01031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picot A et al. (2018) ‘Temperature Rise under Two-Photon Optogenetic Brain Stimulation’, Cell Reports. doi: 10.1016/j.celrep.2018.06.119. [DOI] [PubMed] [Google Scholar]
- Podgorski K and Ranganathan G (2016) ‘Brain heating induced by near-infrared lasers during multiphoton microscopy’, Journal of Neurophysiology, 116(3). doi: 10.1152/jn.00275.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R et al. (2012) ‘Two-photon optogenetic toolbox for fast inhibition, excitation and bistable modulation.’, Nature methods. Nature Publishing Group, 9(12), pp. 1171–9. doi: 10.1038/nmeth.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickgauer JP, Deisseroth K and Tank DW (2014) ‘Simultaneous cellular-resolution optical perturbation and imaging of place cell firing fields’, Nature Neuroscience. doi: 10.1038/nn.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NTM et al. (2020) ‘Targeted Activation of Hippocampal Place Cells Drives Memory-Guided Spatial Behavior’, Cell. doi: 10.1016/j.cell.2020.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronzitti E, Ventalon C, et al. (2017) ‘Recent advances in patterned photostimulation for optogenetics’, Journal of Optics (United Kingdom). doi: 10.1088/2040-8986/aa8299. [DOI] [Google Scholar]
- Ronzitti E, Conti R, et al. (2017) ‘Submillisecond optogenetic control of neuronal firing with two-photon holographic photoactivation of chronos’, Journal of Neuroscience. doi: 10.1523/JNEUROSCI.1246-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell LE et al. (2021) ‘The influence of visual cortex on perception is modulated by behavioural state’, bioRxiv, p. 706010. doi: 10.1101/706010. [DOI] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh OA et al. (2017) Temporally precise single-cell-resolution optogenetics, Nature Neuroscience. Doi: 10.1038/s41593-017-0018-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soor NS et al. (2019) ‘All-optical crosstalk-free manipulation and readout of Chronos-expressing neurons’, Journal of Physics D: Applied Physics, 52(10). doi: 10.1088/1361-6463/aaf944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampinato G et al. (2019) ‘All-optical interrogation of a direction selective retinal circuit by holographic wave front shaping: Supplementary figures’, bioRxiv. doi: 10.1101/513192. [DOI] [Google Scholar]
- Vaziri A and Emiliani V (2012) ‘Reshaping the optical dimension in optogenetics’, Current Opinion in Neurobiology. doi: 10.1016/j.conb.2011.11.011. [DOI] [PubMed] [Google Scholar]
- Yang W et al. (2018) ‘Simultaneous two-photon imaging and two-photon optogenetics of cortical circuits in three dimensions’, eLife. doi: 10.7554/eLife.32671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C et al. (2020) ‘Frequency-specific optogenetic deep brain stimulation of subthalamic nucleus improves parkinsonian motor behaviors’, Journal of Neuroscience, 40(22). doi: 10.1523/JNEUROSCI.3071-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z et al. (2018) ‘Closed-loop all-optical interrogation of neural circuits in vivo’, Nature Methods. doi: 10.1038/s41592-018-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.