

Abstract

Cyanine dyes represent a family of organic fluorophores with widespread utility in biological-based applications ranging from real-time PCR probes to protein labeling. One burgeoning use currently being explored with indodicarbocyanine (Cy5) in particular is that of accessing exciton delocalization in designer DNA dye aggregate structures for potential development of light-harvesting devices and room-temperature quantum computers. Tuning the hydrophilicity/hydrophobicity of Cy5 dyes in such DNA structures should influence the strength of their excitonic coupling; however, the requisite commercial Cy5 derivatives available for direct incorporation into DNA are nonexistent. Here, we prepare a series of Cy5 derivatives that possess different 5,5′-substituents and detail their incorporation into a set of DNA sequences. In addition to varying dye hydrophobicity/hydrophilicity, the 5,5′-substituents, including hexyloxy, triethyleneglycol monomethyl ether, tert-butyl, and chloro groups were chosen so as to vary the inherent electron-donating/withdrawing character while also tuning their resulting absorption and emission properties. Following the synthesis of parent dyes, one of their pendant alkyl chains was functionalized with a monomethoxytrityl protective group with the remaining hydroxyl-terminated N-propyl linker permitting rapid, same-day phosphoramidite conversion and direct internal DNA incorporation into nascent oligonucleotides with moderate to good yields using a 1 μmole scale automated DNA synthesis. Labeled sequences were cleaved from the controlled pore glass matrix, purified by HPLC, and their photophysical properties were characterized. The DNA-labeled Cy5 derivatives displayed spectroscopic properties that paralleled the parent dyes, with either no change or an increase in fluorescence quantum yield depending upon sequence.
Introduction
The family of cyanine dyes has and continues to see extensive use as molecular probes and biolabels since several reactive versions were first developed more than 30 years ago.1,2 Their application space has covered a broad range of utility including roles as multicolor nucleic acid array probes, generalized protein and antibody labels, real-time PCR probes, within numerous biosensors, and as Förster resonance energy transfer (FRET) donors (D) and/or acceptors (A).3−7 Their latter use within FRET configurations has recently been extended beyond the realm of biosensing to incorporation within designer DNA assemblies in pursuit of two new research foci, namely, for designer light-harvesting and energy transfer (ET) systems along with molecular-scale devices that seek to access and exploit exciton delocalization phenomena. In terms of ET, various D–A cyanine dyes displaying a range of absorption and emission maxima have been used to label oligonucleotides that are then assembled into designer DNA structures allowing them to function as so-called molecular photonic wires.8 Within these structures, a variety of parameters can be iteratively modified and tested either jointly or independently including that of D/A ratios, D/A spacing, number of ET steps, relative dye orientation, interactions with other dyes, structural dimensionality (planar vs 3D), dye density, and so forth.8−10 Cumulatively, this provides a powerful system to develop design strategies to optimally harvest light and then focus it on the nanoscale. To exploit exciton delocalization, DNA structures such as Holliday junctions allow the cyanine dyes to strongly interact and provide access to intriguing optical phenomena such as displaying J- and H-aggregate behavior along with large Davydov splitting.11−14 Harnessing such exciton delocalization properties, especially at room temperature, is of interest for the development of new types of hyperefficient light harvesters and sensors along with excitonic devices that exploit coherence for quantum information processing. Due to its inherently high molecular extinction coefficient (∼2.5 × 105 M–1·cm–1), indodicarbocyanine (Cy5) is a particularly appealing dye for the latter application and Cy5 aggregates on DNA scaffolds have received recent attention in this context.12−20
Beyond the presence of cyanine dyes, the other common enabling element among both the systems described above is that of the underlying DNA architecture, which not only functions to place the dyes in the correct position, and sequential order if that is required, but also provides the dyes sufficient (sub)nanometer proximity relative to each other to engage in efficient excitonic coupling. The inherent modularity of DNA assembly also allows experimental structures and the necessary controls to be easily assembled in a parallel side-by-side format. Indeed, reflecting the power of DNA nanotechnology’s inherently parallel reaction assembly chemistry, some reports have assembled hundreds of different targets and control structures in total to provide necessary data on all potentially contributing ET processes by making almost every possible combination.8−10 Several cyanine dye analogues are available through commercial sources which display reactive functional groups that enable dye incorporation both during and post DNA synthesis, for example, phosphoramidite and N-hydroxysuccinimidyl ester/maleimide derivatives, respectively.21−23 Due to the chemical nature of these functional groups, longer attachment linkers are also often required, which can allow significant conformational freedom of individual dyes within DNA constructs. Excessive freedom of movement can be detrimental to excitonic coupling applications on DNA scaffolds due to positional uncertainty and lack of control over orientation and dye-to-dye interactions. To circumvent this geometrical uncertainty and allow more precise dye placement, the two-point internal insertion approach is preferred, where, in essence, the dye molecule physically replaces a nucleoside within the DNA, see Figure 1. This type of insertion is, however, chemically dependent on having access to the requisite phosphoramidite dye derivative(s) for incorporation during automated DNA synthesis.
Figure 1.

Structures of the four substituted Cy5 dye derivatives for phosphoramidite conversion and insertion in DNA. (A) Structures of the homo-substituted Cy5-hex, Cy5-Peg, Cy5-tBu, and Cy5-Cl derivative dyes synthesized in this study. MMTr is the 4-monomethoxytrityl protecting group. These were prepared in the iodide salt form. (B) General structure of the Cy5 dye derivatives inserted internally into DNA oligonucleotides during phosphoramidite synthesis and attached to the DNA at both the 3′ and 5′ ends. The commercial Cy5 dye used for the same type of DNA labeling has the same structure but is not substituted (R = H).
Recent experimental results in conjunction with theoretical studies have strongly suggested that modifying Cy5 to display various types of substituent groups that alter hydrophobicity/hydrophilicity could further augment its ability to engage in delocalized excitonic coupling in the context of DNA scaffolds.12,24 However, within the available cyanine dyes, beyond a single unsubstituted Cy5 dye version, other types of functionalized Cy5 phosphoramidite derivatives are neither commercially available nor reported. We thus undertook the synthesis of a series of novel Cy5 analogues, with different substituents [n-hexyloxy (hex), triethylene glycol monomethylether (Peg), tert-butyl (tBu), and chloro (Cl)] at the 5,5′-positions (Figure 1). In addition to tuning the hydrophobicity and potential intermolecular interactions of these dyes with each other, variation of these substituents potentially enables tuning of absorption and emission properties, including relative absorption/emission maxima, Stokes shift, and fluorescence quantum yield (QY).12,24 The as-synthesized series of substituted Cy5 dyes displaying short linkers were functionalized with monomethoxytrityl- and phosphoramidite-terminated N-propyl linkers to enable two-point 3′-5′ insertion into nascent oligonucleotides during automated DNA synthesis. Each of the novel dyes was internally incorporated into several different DNA sequences (chosen for downstream excitonic coupling studies) and the basic photophysical properties of each were characterized.
Results and Discussion
Molecular Design
The design of the current Cy5 phosphoramidite derivatives was based on that of the commercially available Cy5 phosphoramidite designated for incorporation internally within oligonucleotides during automated DNA synthesis.25,26 This structure was of relevance as a parent compound because it features two 3-hydroxypropyl linkers originating at the indole’s nitrogen atoms. Upon incorporation into the DNA scaffold, these short linkers limit conformational variability and can ultimately enforce close proximity of dyes placed opposite to each other on complementary strands. To have the ability to later probe and understand the influence of hydrophobicity on dye–dye interactions in DNA scaffolds, we chose to modify the 5,5′-peripheral positions of the parent Cy5 with various substituents.12,24 In contrast to this approach, substituents placed directly on the polymethine chain may result in either undesired steric repulsion between dyes when incorporated into arrays or other undesired changes to relevant photophysical properties.27 Additionally, the electron-donating or electron-withdrawing character of substituents placed at the 5,5′-positions enables the tuning of photophysical properties, such as absorption and emission maxima.24 Thus, we designed and synthesized a series of Cy5 derivatives ready for phosphoramidite conversion with different peripheral substituents including n-hexyloxy (Cy5-hex), 2-[2-(2-methoxyethoxy)ethoxy]ethoxy (Cy5-Peg), tert-butyl (Cy5-tBu), and chloro (Cy5-Cl) groups, see Figure 1. Hexyloxy and Peg substituents possess similar electron-donating capacities but are hydrophobic and hydrophilic, respectively. The tert-butyl group is also hydrophobic and an electron-donating group but to a lesser extent than the hexyloxy, while Cl is a weak acceptor. Finally, the chloro modification was chosen because it has been previously shown that chloro-substituted cyanine derivatives are prone to aggregation,28,29 which could facilitate excitonic interactions within downstream DNA assemblies.
Dye Synthesis
In-depth detail of each synthetic step and the corresponding chromatographic separation, NMR results, and mass spectral analysis (where available) are provided in the Supporting Information (Sections 1 and 2). The following represents an abbreviated overview of key steps. The entire synthetic scheme was initiated from the synthesis of 5-substituted indole precursors via Fischer indole synthesis.30−32 For the 5-alkoxyindoles (2-hex and 2-Peg), the synthesis began by preparing 5-methoxyindole (1-OMe) from 4-methoxyphenylhydrazine hydrochloride and 3-methyl-2-butanone in refluxing ethanol (Scheme 1A).31,32 The methoxy group in compound 1-OMe was then hydrolyzed in 48% aqueous hydrobromic acid, yielding the common precursor 1-OH.33 Next, 5-hydroxyindole 1-OH was utilized in Williamson-type ether synthesis, yielding either 5-hexyloxy (2-hex)- or 5-triethyleneglycol monomethyl ether (2-Peg)-substituted indoles from bromohexane and 1-bromo-2-[2-(2-methoxyethoxy)ethoxy]ethane (S1),34 respectively. In parallel, 5-tert-butylindole (2-tBu) and 5-chloroindole (2-Cl) were also prepared from their respective 4-substituted hydrazine hydrochlorides via Fisher indole synthesis (Scheme 1B). For compound 2-Cl, sulfuric acid was added to expedite the slow reaction; otherwise, the HCl inherent to the hydrazine salts was sufficient to catalyze the reaction. The 5-substituted indoles were N-alkylated to the corresponding [(3-acetoxy)propyl]indolinium iodide derivatives by heating to 100 °C in neat,26 freshly prepared 3-iodopropyl acetate S2 (Scheme 1C),26,35 providing good to nearly quantitative yields. Next, the series of indolinium iodide salts were coupled with malonaldehyde dianilide hydrochloride to form N,N-bis(3-acetoxypropyl)indodicarbocyanine derivatives (Scheme 2). Note that the solvent and base varied depending upon the availability at the given time,27,36,37 and yields were comparable nonetheless. Subsequently, the acetoxy groups were deprotected by either HCl or acetyl chloride in the presence of methanol to obtain the N,N-bis(3-hydroxypropyl)indodicarbocyanine series. In the cases of 4-hex and 4-tBu, purification was carried out prior to hydrolysis, while 4-Peg and 4-Cl were hydrolyzed as crude materials. The final, stable dyes (monomethoxytrityl-substituted Cy5 derivatives, Scheme 3A) were then prepared by the substitution reaction of the terminal hydroxyl group with 4-monomethoxytrityl chloride (MMTr-Cl). Due to the statistical nature of this substitution, both mono- and bis-tritylation occur, and the separation of the two products is nontrivial. We found that sacrificing target yield (32% and below) using excess Cy5 significantly limited the formation of the bis-trityl byproduct while also making subsequent purification much more facile. Furthermore, any unreacted bis-hydroxy cyanine was simply recovered and reused, as opposed to the bis-trityl byproduct, which would require additional hydrolysis for its recycling. We note that the 4,4′-dimethoxytrityl (DMTr, shown in Figure S1) group is more common as the protective group of primary alcohols when used for DNA synthesis; however, this protective group is less robust.38
Scheme 1. Synthetic Schemes for Obtaining Cy5 Precursor Indoles and Indolinium Salts,

(A) Synthesis of 5-alkoxy indoles; (B) Synthesis of 5-tert-butyl and 5-chloro indoles; (C) Synthesis of 5-substituted indolinium iodide salts.
Yields are indicated for each step (italicized bold) following compound number; corresponding NMR spectra and mass spectra of all products are shown in the supporting information, in the order of appearance. Details for preparation of compounds S1 and S2 are found in the Supporting Information.
Scheme 2. Synthesis of N,N-Bis(3-hydroxypropyl)indodicarbocyanines.

Individual procedures vary slightly for each cyanine depicted in this scheme; details can be found in the Supporting Information. Yields indicated for each step (italicized bold) following compound number. The corresponding NMR spectra and mass spectra of all products are shown in the Supporting Information in the order of appearance.
Scheme 3. Preparation of Cyanine Dye Derivatives for Internal Incorporation into DNA during Automated Synthesis,

(A) Tritylation of the indodicarbocyanine series; (B) Indodicarbocyanine phosphoramidite conversion/phosphytilation, product yield was assumed to be quantitative based upon TLC. Following a quick purification, the product was immediately dissolved in anhydrous acetonitrile under N2 gas in a reagent bottle and then installed into the DNA synthesizer.
Yields are indicated for each step (italicized bold) following compound number; corresponding NMR spectra and mass spectra of all products are shown in the Supporting Information, in the order of appearance. Details for preparation of compounds S1 and S2 are found in the Supporting Information.
Thus, the more stable 4-monomethoxytrityl (MMTr) group was used to protect the primary alcohol in this work as is also done with commercially available cyanine phosphoramidites. Importantly, exchanging the DMTr with the MMTr group does not impact the automated DNA synthesis protocol and had no apparent effect on overall yields.39 Finally, the MMTr-protected cyanine derivatives were each coupled with 2-cyanoethyl N,N-diisopropylchlorophosphoramidite to yield indodicarbocyanine phosphoramidites (Scheme 3B). Once prepared, the Cy5-phosphoramidites were purified and then immediately dried down. Initial successful Cy5-phosphoramidite syntheses were confirmed by the presence of 31P NMR resonances in the range of 140–150 ppm;40 however, we found that thin-layer chromatography (TLC) provided sufficient confirmation of the product for later trials. Additionally, we modified the scale of the reactions such that all phosphoramidite could be consumed by the DNA synthesizer within two days. Once thoroughly dried, the phosphoramidite samples were reconstituted in dry acetonitrile and immediately used for DNA synthesis.
We attribute our successful DNA synthesis trials to two factors in preparation of the final phosphoramidites. First, we utilize only the iodide salts. The iodide salts, as opposed to the more commonly found commercial Cy5-phosphoramidite chloride salts, have improved solubility and larger retention factor, making their synthesis easier to monitor by TLC. Comparatively, the chloride salts required a much more polar TLC eluent (methanol/dichloromethane as opposed to acetonitrile/dichloromethane), which additionally undergoes substitution on the phosphoramidite moiety complicating TLC interpretation. Second, during initial, poor yielding DNA synthesis trials, we struggled to purify the Cy5-phosphoramidites in a timely manner prior to placing the samples into the DNA synthesizer. We found that concentrating the Cy5-phosphoramidite reaction crude, in an attempt to go directly to column chromatography, results in significant decomposition of the crude product presumably due to the presence of acid when the amine was evaporated. This decomposition was circumvented by washing with saturated sodium bicarbonate. However, the bicarbonate wash converted the excess 2-cyanoethyl N,N-diisopropylchlorophoramidite to cyanoethyl-N,N-diisopropyl-H-phosphonamidate (see Figure S2 for structure, characterization data available in Section 3, and NMR spectra available in Supporting Information Figures S48–S50)41 that unfortunately coeluted with Cy5-phosphoramidites when attempting column chromatography. We opted not to reduce the molar equivalents of 2-cyanoethyl N,N-diisopropylchlorophoramidite, as some amount is inevitably consumed by moisture in the reaction solvent, despite our best efforts to dry the solvent over activated 3 Å molecular sieves.42 Ultimately, we were able to remove the impurity through iterative powderization (see General Procedure for Cy5-Phosphoramidite Synthesis), tracking its removal through TLC upon staining with ninhydrin (impurity shows bright red-orange upon heating the stained TLC). Once removed, we observed significantly improved yields in our DNA synthesis trials.
DNA Synthesis, Oligonucleotide Purification, and Overall Yield
For each new Cy5 dye analogue prepared above, five different DNA oligonucleotide sequences were synthesized with each dye internally incorporated into the target oligo during automated synthesis. Full sequences with the dye insertion locations are listed in Table 1. Our selection of oligonucleotide sequences for synthesis is motivated by the Holliday junction (HJ) DNA structure that is widely applied in the field as a platform for studying dye excitonic coupling and illustrated in Figure S3.11,13,14,18 Sequences HJA-HJD are programmed to self-assemble into this tetrameric HJ structure. The fifth strand—HJAcomp—is designed to be complementary to HJA and assemble to form a linear duplex DNA for any comparative studies with the HJ structure. Each sequence was prepared in multiple copies with at least 3 and up to 8 replicates at the 1 μmol scale undertaken.
Table 1. DNA Sequences Synthesized in This Studya.
| designation | oligonucleotide sequence (5′-3′) |
|---|---|
| HJA | ATATAATCGCTCG-X-CATATTATGACTG |
| HJB | CAGTCATAATATG-X-TGGAATGTGAGTG |
| HJC | CACTCACATTCCA-X-CTCAACACCACAA |
| HJD | TTGTGGTGTTGAG-X-CGAGCGATTATAT |
| HJAcomp | CAGTCATAATATG-X-CGAGCGATTATAT |
Note: ATCG are the naturally occurring nucleotides. X denotes the site of Cy5 analogue incorporation.
DNA synthesis utilized an automated Applied Biosystems Expedite 8909 DNA Oligo Synthesizer (supplied by Biolytic Lab Performance, Inc., Fremont, CA) using solid-phase phosphoramidite coupling chemistry carried out at the 1 μmol scale on controlled-pore glass (CPG) columns that contained the initial 3′ starting base in the protected form. Unsubstituted commercial nucleoside phosphoramidites were coupled at a concentration of 70 mM while the novel Cy5 phosphoramidites were coupled at a concentration of 80 mM (note that commercial Cy5 phosphoramidites recommend coupling at 70 mM).43 DNA oligonucleotides were synthesized following the instrument’s standard coupling protocols, with the exception of the Cy5-phosphoramidite insertion step.44 For coupling of the novel Cy5 analog phosphoramidites (monomer for these purposes), the coupling time was modified such that three pulses of monomer + activator (Act, 0.25 M 5-ethylthio-1H-tetrazole in anhydrous acetonitrile) were pushed into the column by flushing 9–10 pulses (optimized based on the volume of the tubing from the monomer reservoir to the synthesis column) of acetonitrile wash immediately after. The monomer + Act was then allowed to react with the column by flushing seven pulses of wash over the course of 150 s. Next, the unreacted monomer + Act was rapidly flushed out of the column using eight pulses of wash. The entire process was repeated three times to achieve a total of nine pulses of monomer + Act per coupling. The end of the coupling was the same as the standard protocol, which included additional activator and wash steps. See Supporting Information Section 4 for more information on the standard coupling protocols utilized.
All solutions and reagents used with the system were purchased from Glen Research (Sterling, VA) and used in accordance with their instructions and that of the DNA synthesizer. For incorporation of naturally occurring nucleotides, we utilized nucleoside phosphoramidites possessing standard protecting groups (as opposed to ultramild protecting groups) due to supply-line/chemical ordering delays (protecting group information and catalogue numbers for each can be found in Supporting Information Section 4, Table S1). The only exception is acetyl-protected cytidine phosphoramidite (Glen Research, Sterling, VA., Cat. no. 10-1015-1C), which is suitable for ultramild deprotection. Typically, one would prefer to use nucleoside phosphoramidites with ultramild compatible protecting groups, as their deprotection conditions (methanolic potassium carbonate) minimize cyanine hydrolysis.45 Once synthesized, the crude DNA sequences still attached to the CPG columns were stored at 4 °C in dry and dark conditions until a sufficient number of synthetic replicates had been collected (20+) for bulk processing in parallel. The latter began with ammonolysis (see Ammonolysis of DNA Sequences in the Experimental Section) which both frees the DNA sequences from the CPG beads and deprotects the individual nucleobases, but it can also lead to hydrolysis of the cyanines. The manufacturer suggested concentration of 30% ammonia44 resulted in rapid degradation of all novel cyanine-containing sequences. We carefully screened conditions to find that optimal yield and near complete deprotection (>95%) of nucleobases was attained at 7% NH4OH with 1 week reaction time, when using Cy5-hex, Cy5-Peg, and Cy5-tBu. In the case of electron-deficient Cy5-Cl, nearly complete hydrolysis of Cy5 was observed during the 1 week long exposure time; thus, it was tuned back to 48 h. This decreased reaction time results in 5–10% of DNA failing to undergo complete deprotection; however, those species were separable from the target sequence by HPLC.
Upon completion of the ammonolysis, the crude oligo solution underwent salt exchange (see Salt Exchange of DNA Sequences in the Experimental Section for the procedure) using triethylammonium acetate (TEAA) buffer followed by deionized water and was then concentrated to dryness. Dried crude samples can be stored long term at −20 °C if necessary at this stage. We note that this buffer exchange is critical as concentration of the sequences directly from basic solution leads to nearly complete decomposition of the cyanines.
Desalted oligo solutions were then analyzed by liquid chromatography–mass spectrometry (LCMS, see Characterization in the Experimental Section for more details) to quickly confirm the presence of the target sequence in the crude product mixture. Samples containing the confirmed dye-labeled DNA sequences were then pooled and purified by preparatory-scale reverse-phase HPLC (see Oligonucleotide Purification in Experimental Section) with a gradient of increasing methanol in 0.1M TEAA (aq). For Cy5-hex-, Cy5-Peg-, and Cy5-tBu-containing sequences, LC fractions were concentrated to dryness directly from the 0.1 M TEAA buffer/methanol solution. In the case of Cy5-Cl-containing oligonucleotides, individual LC fractions were again subjected to salt exchange to remove the excess buffer, as the direct concentration from TEAA solution resulted in nontrivial (10–20%) decomposition. Dried LC fractions were reconstituted in small volumes of water (Optima Grade), to keep concentration sufficiently high (25–50 μM) to visualize and separate low percentage impurities on LCMS. Purified fractions were combined, concentrated, and then reanalyzed via LCMS for a final purity assessment.
The predicted and observed masses (as m/z, where z is the charge) for each dye-containing sequence can be found in Supporting Information Table S2, and detailed information on LCMS acquisition can be found in Supporting Information Section 5. The primary impurity observed closely follows the target sequence in reverse-phase chromatography and cannot be fully separated; it was identified in each case as the target sequence missing the last nucleobase (denoted as N – 1 sequences). For the Cy5-Cl sequences, there was also an impurity present (1–2%) in all cases which precedes the target band during reverse-phase chromatography, and it was identified as the half target sequence in which the Cy5-Cl had been hydrolyzed (absorption band is observed centered at approximate 430 nm). We accepted all final materials when they achieved greater than 90% purity, with an average sample purity of 95%. Overall, the final yields varied from 4.5 up to 24.3% (Table 3, based on an expected maximum yield of 1 μmole per CPG column at this synthetic scale). The large deviations are due in part to the time a particular sequence was synthesized during a given dye-analogue’s in-use reagent lifetime (e.g., synthesis number 10 from a given Cy5-phosphoramidite sample versus synthesis number 1 will inherently exhibit lower yield due to hydrolysis of the Cy5-phosphoramidite over time). To understand this lowered efficiency, we analyzed a portion of Cy5-phosphoramidite solution 1 day post-installation on the DNA synthesizer and noted the appearance (by TLC, not shown) of two polar spots (low retention factor). 31P NMR analysis of the crude material shows the appearance of multiple resonances at or below 25 ppm.40,41 These low Rf spots have been tentatively assigned as the partially hydrolyzed Cy5-cyanoethylphosphonate and fully hydrolyzed Cy5-H-phosphonate, according to the general phosphoramidite hydrolysis pathway described by Krotz46 and Hargreaves.47 Another factor contributing to overall yield was the varying levels of difficulty with purification encountered, which was found to be both dye- and sequence-dependent.
Table 3. Selected Properties of the Cy5 Analogue-Labeled DNA Sequencesa.
| DNA sequence | av. yieldb (%) | λmax abs (nm) | ε260 (M–1.cm–1) | εCy5 (M–1.cm–1) | λmax em (nm) | Stokes shift (ν, cm–1) | ΦF | ΔΦFc (%) | τavg (ns)d | τ1 (ns)/contribution | τ2 (ns)/contribution | Δτe | krad (ns–1)/knr (ns–1) | τradf (ns) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cy5-hex Series | ||||||||||||||
| HJA | 10.3 | 673 | 260,900 | 216,000 | 696 | 491 | 0.08 | 0.14 | 0.59 | 0.50/0.87 | 1.18/0.13 | 0.40 | 0.14/1.56 | 7.35 |
| HJB | 7.5 | 670 | 272,100 | 219,800 | 696 | 558 | 0.07 | 0 | 0.64 | 0.47/0.65 | 0.96/0.35 | 0.52 | 0.11/1.45 | 9.15 |
| HJC | 12.0 | 674 | 248,700 | 208,100 | 696 | 469 | 0.09 | 0.29 | 0.67 | 0.54/0.77 | 1.10/0.23 | 0.57 | 0.14/1.38 | 7.31 |
| HJD | 8.9 | 673 | 259,100 | 200,900 | 696 | 491 | 0.07 | 0 | 0.57 | 0.47/0.80 | 0.96/0.20 | 0.36 | 0.12/1.63 | 8.16 |
| HJAcomp | 10.5 | 671 | 268,900 | 216,700 | 697 | 556 | 0.07 | 0 | 0.64 | 0.43/0.52 | 0.87/0.48 | 0.50 | 0.11/1.48 | 9.00 |
| Cy5-Peg Series | ||||||||||||||
| HJA | 11.9 | 673 | 265,200 | 214,500 | 694 | 450 | 0.10 | 0.43 | 0.83 | 0.57/0.48 | 1.08/0.52 | 0.89 | 0.12/1.08 | 8.33 |
| HJB | 6.7 | 670 | 276,300 | 213,800 | 693 | 495 | 0.09 | 0.29 | 0.72 | 0.55/0.64 | 1.03/0.36 | 0.64 | 0.13/1.26 | 8.00 |
| HJC | 12.9 | 670 | 253,000 | 209,500 | 692 | 475 | 0.10 | 0.43 | 0.67 | 0.49/0.63 | 0.98/0.37 | 0.52 | 0.15/1.34 | 6.67 |
| HJD | 7.8 | 674 | 264,000 | 221,400 | 693 | 407 | 0.09 | 0.29 | 0.79 | 0.55/0.45 | 0.99/0.55 | 0.79 | 0.11/1.15 | 8.78 |
| HJAcomp | 4.5 | 671 | 273,400 | 219,000 | 693 | 473 | 0.09 | 0.29 | 0.64 | 0.54/0.83 | 1.13/0.17 | 0.45 | 0.14/1.00 | 7.11 |
| Cy5-tBu Series | ||||||||||||||
| HJA | 16 | 659 | 261,800 | 261,100 | 679 | 447 | 0.26 | 0.24 | 1.44 | 0.69/0.15 | 1.57/0.85 | 0.53 | 0.18/0.51 | 5.56 |
| HJB | 13.7 | 656 | 272,700 | 250,000 | 677 | 473 | 0.20 | 0 | 1.07 | 0.77/0.56 | 1.46/0.44 | 0.14 | 0.19/0.74 | 5.26 |
| HJC | 24.3 | 659 | 249,500 | 249,300 | 678 | 425 | 0.26 | 0.24 | 1.41 | 0.64/0.18 | 1.58/0.82 | 0.50 | 0.19/0.52 | 5.26 |
| HJD | 17.2 | 657 | 260,300 | 260,000 | 676 | 428 | 0.21 | 0 | 1.13 | 0.67/0.39 | 1.42/0.61 | 0.20 | 0.19/0.69 | 5.26 |
| HJAcomp | 12.3 | 657 | 270,000 | 269,600 | 678 | 471 | 0.21 | 0 | 1.11 | 0.68/0.40 | 1.39/0.60 | 0.18 | 0.19/0/71 | 5.26 |
| Cy5-Cl Series | ||||||||||||||
| HJA | 5.0 | 656 | 267,568 | 274,200 | 674 | 407 | 0.35 | 0.30 | 1.89 | 1.89 | 0.71 | 0.19/0.34 | 5.26 | |
| HJB | 10.2 | 653 | 279,236 | 288,400 | 672 | 433 | 0.27 | 0 | 1.78 | 1.78 | 0.60 | 0.15/0.41 | 6.67 | |
| HJC | 16.9 | 654 | 255,372 | 269,300 | 673 | 432 | 0.37 | 0.37 | 1.98 | 1.98 | 0.78 | 0.19/0.32 | 5.26 | |
| HJD | 12.7 | 654 | 265,944 | 271,100 | 671 | 387 | 0.28 | 0 | 1.63 | 0.55/0.12 | 1.78/0.88 | 0.47 | 0.17/0.44 | 5.88 |
| HJAcomp | 10.8 | 654 | 275,640 | 276,000 | 673 | 432 | 0.29 | 0 | 1.47 | 0.84/0.28 | 1.71/072 | 0.32 | 0.20/0.36 | 5.00 |
All values determined in neat water.
As compared to an expected 1 μmol maximum.
Percent change of ΦF compared to the parent dye in MeOH.
Average of τ1 and τ2, accounting for weighted contributions.
Percent change of τavg relative to the parent dye in MeOH.
τrad = krad–1. Extinction coefficients were determined utilizing nearest-neighbor approximation for DNA absorbance at 260 nm55−57 while also accounting for Cy5 contribution. Cy5 percent contribution (0.02–0.04) was based on the ratio of absorbance at 260 nm and at Cy5 λmax for each of the parent dyes obtained in methanol. It was assumed that Cy5 percent contribution at 260 nm is constant between MeOH and H2O. Fluorescence QY (ΦF) determined against 5,10,15,20-tetraphenylporphyrin standard (ΦF = 0.07 in toluene).54
Dye Properties
Due to the overall poor solubility of the parent cyanine dye derivatives in water, we were unable to determine the octanol–water partition coefficient (log P) by the traditional “shake flask” method.48−50 In lieu of this approach, we utilized modeling software developed by Advanced Chemistry Development, Inc. to predict the partition coefficient and solubility in water (log S0), as shown in Table 2. As an additional means of comparison with direct relevance to the final intended utility of these oligonucleotides, we determined the average concentration of methanol required to elute the DNA sequences containing the given dye from the reverse-phase HPLC column (the greater the methanol content required to elute, the more hydrophobic the dye). The DNA–dye sequences were utilized for this instead of the free dyes because they are capable of eluting at lower methanol content, but the hydrophobic character of the dye still dominates the elution profile. The calculated log P and log S0 values suggest that the order of increasing hydrophobicity, according to the functional group, is as follows: Peg < Cl ≪ tBu ≪ hex. This is in close agreement with the relative “stickiness” of the DNA–dye sequences toward the C18 stationary phase. The slightly greater methanol content required to elute Cy5-Peg-containing sequences relative to Cy5-Cl can be attributed to the amphiphilic nature of the Peg chains, where the increased number of hydrogen bond-accepting O atoms favor interaction with the aqueous phase. This tendency is, however, offset by the increased number of C–H bonds, which promote interaction with the hydrophobic stationary phase.51−53
Table 2. Select Properties of the Novel Indodicarbocyanine Dyes.
| dye | log P | log S0 | av. % MeOH for DNA-dye elution | λmax abs (nm) | λmax em (nm) | Stokes’ shift (cm–1) | ΦF | τ (ns) |
|---|---|---|---|---|---|---|---|---|
| Cy5-hex | 5.66 | –9.94 | 51 | 670 | 699 | 619 | 0.07 | 0.42 |
| Cy5-Peg | 1.58 | –7.88 | 31.5 | 668 | 693 | 540 | 0.07 | 0.44 |
| Cy5-tBu | 3.43 | –8.25 | 41 | 654 | 678 | 541 | 0.21 | 0.94 |
| Cy5-Cl | 2.35 | –7.94 | 30.5 | 648 | 670 | 507 | 0.27 | 1.11 |
| Cy5a | 1.73 | –6.25 | 30.0 |
Cy5 refers to the unsubstituted Cy5. For some comparative values of this dye as incorporated into DNA, see Table S3. Absorption and emission properties were determined in methanol. QY (ΦF) was determined against the standard TPP (ΦF = 0.07 in toluene).54 Acetate-functionalized cyanines (4) were utilized for the log S0 and log P as this more closely matches the cyanine structure once imbedded within DNA. log P and log S0 were determined using the Molecular Property Calculations on the Percepta Platform from Advanced Chemistry Development, Inc. (Toronto, Canada). Average % methanol for DNA–dye elution based on elution time for each sequence (HJA-D and HJAcomp) containing the indicated dye.
Relative absorption and emission properties of the novel cyanine dyes are also summarized in Table 2. We present the data acquired for those possessing pendant hydroxyl substituents (5), as the diacetoxy cyanines (4) were not cleanly isolated in all cases. Figure 2 shows the normalized absorption and emission spectra of the cyanines, respectively, as acquired in methanol. Here, the wavelength of the maximum absorbance shifts with respect to the electron-withdrawing/donating character of the 5,5’-substituents, spanning the window of 648–670 nm. The greater the electron-donating character, the greater the bathochromic shift, and vice versa. Thus, the order of increasing shift of the absorbance maxima is Cy5-Cl < Cy5-tBu < Cy5-Peg ≈ Cy5-hex. The emission maxima of this series, spanning 670–699 nm, follows the same order with each Cy5 possessing a moderate Stokes shift in the range of 507–619 cm–1. The mirror images between absorption and fluorescence spectra along with moderate Stokes shifts suggest that the peripheral substituents used in the present study have only minimal effect on the excited-state structural relaxation relative to the ground-state conformation for each Cy5 derivative. We also note that the Cy5-Peg and Cy5-hex have broader absorption spectra than the other two derivatives (Figure 2A). The fluorescence QY (ΦF) and the excited-state fluorescence lifetimes (τ) of these dyes follow an inverse trend as a function of the absorption peak maxima, where the weaker the electron-donating character of the 5,5′-substituents, the larger the ΦF exhibited. Thus, the largest determined ΦF was 0.27 for Cy5-Cl, decreasing to 0.21 for Cy5-tBu, and decreasing further to 0.07 for Cy5-hex and Cy5-Peg. The corresponding τ values ranged from 0.42 up to 1.11 ns and appeared to predominantly consist of a single exponential decay. In addition, we determined ΦF for 4-hex and 4-tBu (not shown), and these were found to be identical to those of Cy5-hex and Cy5-tBu, respectively, indicating that the free hydroxyl substituents stemming from the indolenine nitrogens did not influence our measurements.
Figure 2.

Absorption and emission spectra for the as-synthesized dye series. (A) Normalized absorption spectra (in terms of extinction coefficient) of Cy5-hex (red), Cy5-Peg (blue), Cy5-tBu (pink), and Cy5-Cl (green) acquired in methanol with concentration approximated to 4–5 μM (Abs ∼ 1.0 AU). (B) Corresponding normalized emission spectra for the same sample series acquired in methanol with concentration approximated to 0.5 μM (Abs < 0.1 AU). For emission, samples were excited at 600 nm and spectra were collected from 620 to 850 nm.
Photophysical Properties of the Dye-Labeled Oligonucleotides
Table 3 summarizes the absorption and emission properties of the 20 novel Cy5-containing DNA sequences in H2O (HPLC). The corresponding absorption and emission spectra collected from each of the single-stranded (ss) oligonucleotides are presented in Figure 3. For comparative purposes, the properties of unsubstituted Cy5 oligonucleotides (R = H) are summarized in Supporting Information Section 6, Table S3. From this point on, use of Cy5-hex, Cy5-Peg, Cy5-tBu or Cy5-Cl refers to the given dye-labeled oligonucleotide. Upon incorporation into DNA, the absorption maxima measured in water for these Cy5 derivatives are identical or slightly red-shifted (0–3 nm for Cy5-hex, 2–6 nm for Cy5-Peg, 2–5 nm for Cy5-tBu, and 5–8 nm for the Cy5-Cl oligonucleotides), compared to the parent dyes measured in methanol. The absorption maxima vary within 1–4 nm on a sequence-to-sequence basis within each HJ-Cy5 series and there is no discernible trend as to which HJ sequence produces the greatest bathochromic shift. For example, HJC-Cy5-tBu is the most red-shifted of its series, while HJC-Cy5-Peg is the most blue-shifted of its series. The incorporation of the various Cy5 analogues does not significantly influence the shape of the DNA absorption band in any case (see Figures S72–S76). Since the DNA and Cy5 analogue absorbance behaved independently, we determined the molar absorptivity (ε, M–1·cm–1) of the Cy5 analogue in each sequence based on the calculated absorbance of the DNA at 260 nm. For this, we used the nearest-neighbor approximation55−57 and assumed that the percent contribution of the Cy5 analogue at 260 nm was constant between the parent Cy5 in methanol and the DNA-incorporated Cy5 analogue in water. Thus, the ε at 260 nm values found in Table 3 is the sum of the calculated value and the Cy5 analogue contribution. For Cy5 analogues incorporated into sequences, the molar absorptivity at the wavelength of maximum absorbance (εCy5) falls within a narrow window for each DNA–dye series: 200,900–219,800 (M–1·cm–1) for Cy5-hex sequences, 209,500–221,400 (M–1·cm–1) for Cy5-Peg sequences, 249,300–269,600 (M–1·cm–1) for Cy5-tBu sequences, and 269,300–288,400 (M–1·cm–1) for the Cy5-Cl sequences. The deviation of εCy5 between sequences, and also within the same series of a given dye, is likely due to small differences in microenvironments and their relative purities. The significantly lower εCy5 (∼20% average decrease) observed for Cy5-hex and Cy5-Peg is attributed to the increased width of the collective vibronic absorption bands (A0–0 and A0–1) relative to those of Cy5-tBu and Cy5-Cl. Note that we are unable to determine full-width at half-maximum (FWHM) as the A0–0 and A0–1 vibronic bands begin to overlap before the A0–0 falls below 50% maximum intensity, but the relative broadness can nonetheless be seen in Figure 3A–D.
Figure 3.

Absorption and emission spectra for the Cy5 dye series incorporated into the 5 DNA oligonucleotides. Absorption spectra (expressed according to extinction coefficient, ε) of the Cy5-hex- (A), Cy5-Peg- (B), Cy5-tBu- (C), and Cy5-Cl (D)-containing DNA sequences acquired in water at 4–5 μM. The inset highlights the range of observed absorption maxima as also indicated in purple. The corresponding normalized emission spectra (arbitrary units, AU) for Cy5-hex (E), Cy5-Peg (F), Cy5-tBu (G), and Cy5-Cl (H) acquired in water at a concentration of 0.4–0.5 μM. For emission, samples were excited at 600 nm and spectra collected from 620 to 850 nm. Averaged emission maxima are shown in purple.
The emission maxima observed in the DNA-Cy5 analogue sequences parallel the trend observed for the unincorporated dyes—the order of shortest to longest wavelength-emitting dye does not change and the Stokes’ shifts remain in a similarly moderate range. As with the absorption maxima, the emission maxima vary slightly on a per-sequence basis, and the specific sequence with the greatest bathochromic shift within each series is dye- and sequence-dependent. As shown in Table 3, the fluorescence QYs in the HJ Cy5-hex series are the weakest of all sequences studied, ranging from 0.07–0.09, followed closely by the HJ Cy5-Peg series (0.09–0.10). The QY values of the HJ Cy5-tBu series are next in the range of 0.20–0.26 and the largest fluorescence QYs belong to the HJ Cy5-Cl series ranging from 0.27–0.37. On a per dye basis, these results, as anticipated, parallel those of the non-DNA-conjugated parent dyes. In all cases, the fluorescence QYs of DNA-incorporated Cy5s are either unchanged or slightly enhanced relative to those of the parent dyes. For the HJ Cy5-Peg series, QY as a whole increases between 29 and 43% (see ΔΦF values in Table 3); for the remaining series, only HJA and HJC exhibit enhanced emission. For the HJA/C Cy5-hex series, the increase is 14–29%, for the HJA/C Cy5-tBu, the increase is 24%, and for the HJA/C Cy5-Cl, an increase of 30–37% is seen.
The average excited-state fluorescence lifetimes (τavg) along with the corresponding radiative and nonradiative rate constants for the substituted Cy5-labeled DNA oligonucleotides are collected and presented in Table 3. In contrast to the free substituted Cy5 dyes in methanol, the fluorescence decays of the dyes incorporated into DNA were generally found to be nonexponential and well fit by a biexponential decay function. This observation suggests that the substituted Cy5 dyes experience different local environments and may assume multiple configurations in the excited state when attached to DNA strands in the aqueous environment. Similar to the free dyes in methanol, the τavg increases in the order Cy5-hex < Cy5-Peg < Cy5-tBu < Cy5-Cl. For each substituted Cy5, the τavg depends on the particular DNA sequence. The variation in τavg with the DNA sequence is the greatest for the Cy5-tBu and Cy5-Cl series, where the longest and shortest fluorescence lifetimes differ by as much as 26%. The radiative rate for each substituted Cy5 DNA oligo is estimated from58
| 1 |
where ΦF is the fluorescence QY and results in the trend krad,Cy5-hex ∼ krad,Cy5-Peg < krad,Cy5-Cl ∼ krad,Cy5-tBu. Thus, the red-shifted Cy5-hex and Cy5-Peg oligonucleotides, which have relatively broad absorption bands, generally show smaller radiative rates than the Cy5-Cl- and Cy5-tBu-labeled oligonucleotides. The radiative rate was also found to be dependent on the DNA sequence for each substituted Cy5. We note that the radiative rate for cyanine dyes,59 and also other dyes,60 has been observed to depend on the environment surrounding the dye. The nonradiative rate is determined from
| 2 |
where 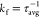 . The results in Table 3 show that knr generally increases in
the order knr,Cy5-Cl < knr,Cy5-tBu < knr,Cy5-Peg < knr,Cy5-hex, and there is variation in knr with the DNA sequence for each substituted
Cy5. As there are a number of possible decay pathways that may contribute
to the substantially larger knr for Cy5-hex
and Cy5-Peg, determining the primary contributor is beyond the scope
of this work.
. The results in Table 3 show that knr generally increases in
the order knr,Cy5-Cl < knr,Cy5-tBu < knr,Cy5-Peg < knr,Cy5-hex, and there is variation in knr with the DNA sequence for each substituted
Cy5. As there are a number of possible decay pathways that may contribute
to the substantially larger knr for Cy5-hex
and Cy5-Peg, determining the primary contributor is beyond the scope
of this work.
Figure 4 compares the absorption and emission profiles of all the HJA and HJAcomp sequences for each dye derivative along with plotting the corresponding spectra for the unsubstituted Cy5 in the same sequences. As is clearly seen, all the substituted analogues possess longer wavelength maxima than the unsubstituted Cy5. This is not surprising behavior as the substituents do not equally influence the HOMO and LUMO energies in dyes. For electron-withdrawing groups, the decrease to HOMO energy is less than the decrease to LUMO energy, and for electron-donating groups, the increase to HOMO is greater than the increase to LUMO. Cumulatively, in both scenarios, the HOMO–LUMO gap is shrinking which causes the observed red-shift to emission.
Figure 4.

Absorption and emission spectra for select Cy5 dye series DNA oligonucleotides. Normalized absorption spectra of HJA (A) and HJAcomp (B) sequences incorporating Cy5-hex Cy5-Peg, Cy5-tBu, Cy5-Cl, and Cy5 dye acquired in water at a concentration of 4–5 μM. The corresponding normalized emission spectra for HJA (C) and HJAcomp (D) sequences acquired in water at a concentration of 0.4–0.5 μM. For emission, samples were excited at 600 nm and spectra were collected from 620 to 850 nm.
Here, we have described the preparation of four different indodicarbocyanine (Cy5) derivatives that possess different 5,5′-substituents including hexyloxy, triethylene glycol monomethyl ether, tert-butyl, and chloro groups. These substituents were chosen primarily to vary the inherent electron-donating/withdrawing—hydrophobicity/hydrophilicity characteristics of the final dye construct. Studies to confirm these particular characteristics can become quite complex and still remain to be performed. However, analysis of the substituted dye solubility and hydrophobicity, as estimated from modeling and HPLC analysis, suggest that they are indeed demonstrating the physicochemical properties that were expected for each derivative. Phosphoramidite derivatives of each dye were subsequently prepared and then internally incorporated into a series of DNA oligonucleotides using two-point insertion via automated DNA synthesis. The average yield observed per 1 μmol CPG cartridge during synthesis ranged from 4.5 up to nearly 25%. Given that a number of replicate syntheses were carried out for each oligo, the overall yield of each sequence was in the hundreds of nanomoles despite the relative lower yield for some on a per CPG cartridge basis.
Overall, we were pleased to find that even after undergoing conversion to the final phosphoramidite, exposure to all the highly reactive automated DNA chemistry reactions/environments, extended purification protocols with multiple harsh chemical treatments, and multiple HPLC procedures with subsequent concentration, the dye derivatives mostly maintained the photophysical properties of the parent dye in terms of excited-state fluorescence lifetime and QY. Coming back to the applications posited in the Introduction that are based on excitonic delocalization,12−16,18−20 studies examining how each of these Cy5 derivatives is capable of engaging in excitonic coupling with itself and perhaps the other dyes in this series are currently underway. These will require assembling the oligonucleotides into appropriate DNA scaffolds such as direct dimers and Holliday junction-like structures which the current synthetic series is meant to specifically facilitate. It is hoped that these efforts will provide strong insights with respect to designing better dye derivatives for such purposes along with optimizing the mechanisms that underpin this promising room-temperature optical phenomena.
Experimental Section
Materials and Methods
Chemical Synthesis
Triphenylphosphine and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (97%) were purchased from Acros Organics. 4-Methoxyphenylhydrazine hydrochloride (95%), malonaldehyde dianilide hydrochloride, and triethyleneglycol monomethyl ether were purchased from TCI America. 3-Methyl-2-butanone (98%) and 3-chloropropyl acetate (98%) were purchased from Alfa Aesar. Cesium carbonate (99.9%) was purchased from Chem-Impex International. 4-Methoxytriphenylmethyl chloride (4-monomethoxytrityl chloride, 97%) and 4-chlorophenylhydrazine hydrochloride (93%) were purchased from Oakwood Chemical. Hydrobromic acid (HBr, 48%), N-bromosuccinimide (99%), sodium iodide (NaI, 98%), triethylamine (99.5%), N,N-diisopropylethylamine (99.5%), and sulfuric acid (H2SO4, reagent grade, 95–98%) were purchased from Sigma-Aldrich. Bulk chromatography solvents were purchased from Pharmco/Greenfield Global. All the other chemicals, including reaction solvents were purchased from Millipore-Sigma or Fisher Scientific and used as received, except for CH2Cl2, CH3CN, and N,N-diisopropylethylamine, which were dried over freshly activated 3 Å molecular sieves (Sigma-Aldrich) before use for phosphoramidite coupling reaction. For the unsubstituted Cy5 dye, we used commercially available Cy5-H (R = H) as the standard for comparison of the DNA-Cy5 sequences. Standard sequences were prepared by automated DNA synthesis in the same manner using the commercially available Cy5 phosphoramidite 1-[3-(4-monomethoxytrityloxy)propyl]-1′-[3-[(2-cyanoethyl)-(N,N-diisopropylphosphoramidityl)]propyl]-3,3,3′,3′-tetramethylindodicarbocyanine chloride (Glen Research, Sterling VA).
Characterization
1H and 13C NMR spectra were recorded on a Bruker SpectroSpin or Bruker Ascend 400 MHz spectrometer (Bruker Corporation, Billerica, MA). Chemical shifts for 1H NMR spectra are reported relative to the tetramethylsilane (TMS) signal in deuterated solvent (TMS, δ = 0.00 ppm). All J values are reported in hertz. Chemical shifts for 13C NMR spectra are reported relative to the chloroform-d (TMS, δ = 0.00 ppm) or the residual solvent peak in DMSO-d6 (δ = 2.52 ppm)61 in the case of 5-tBu. All coupling constants (J) are reported in hertz. Chemical shifts for 13C NMR spectra are reported relative to the residual solvent signal in chloroform-d (δ = 77.16 ppm) or DMSO-d6 (δ = 39.52 ppm)61 in the case of 5-tBu. Mass spectral analysis for both small molecules and DNA oligonucleotides was performed using an ACQUITY UPLC system equipped with a single quadrupole (SQD2) mass detector (Waters, Inc., Milford, MA) as described.62,63 Additionally, purity of the DNA oligonucleotides was assessed on the ACQUITY UPLC using the photodiode array detector (PDA eλ Detector) and fluorescence detector (FLR Detector) modules. All LCMS samples were injected from Fisher brand optima grade solvents: MeOH/H2O (1/1) for small molecules and neat H2O for DNA oligonucleotides and then eluted using an increasing gradient of methanol in aqueous 0.05 M TEAA buffer (pH 7.0). For small molecules, a Waters BEH C18 column (part no. 186002350) was utilized, and for DNA sequences, a Waters Oligonucleotide BEH C18 column (part no. 186003949) was utilized. Absorption spectra were recorded using a Cary 60 UV–vis (Agilent Technologies, Inc., Santa Clara, CA), and fluorescence spectra were recorded using a FluoroMax-4 spectrofluorometer (Horiba Scientific, Piscataway, NJ). Fluorescence QYs were determined against the 5,10,15,20-tetraphenylporphyrin (TPP) standard (ΦF = 0.07 in toluene),52 with common excitation at 600 nm. The TPP standard was purchased from Frontier Specialty Chemicals (Logan, UT) and then oxidized with 2,3-dichloro-5,6-cyano-p-benzoquinone (DDQ) to remove the stated 1–3% chlorin impurity. TPP was cross-referenced against oxazine 720 perchlorate (Luxottica-Exciton, Lockbourne, OH, ΦF = 0.63 in MeOH),64,65 upon excitation at 580 nm with agreement of ±5%. The obtained fluorescence spectra were corrected for wavelength-dependent instrumental sensitivity. Excited-state fluorescence lifetimes were collected using a system described previously.66−68
Synthetic Procedures
Detailed descriptions for the synthesis of 5-methoxy-2,3,3-trimethylindolenine (1-OMe), 5-hydroxy-2,3,3-trimethylindolenine (1-OH), 5-n-hexyloxy-2,3,3-trimethylindolenine (2-hex), 5-{2-[2-(2-methoxyethoxy)ethoxy]ethoxy}-2,3,3-trimethylindolenine (2-Peg), 5-tert-butyl-2,3,3-trimethylindolenine (2-tBu), 5-chloro-2,3,3-trimethylindolenine (2-Cl), 1-[3-(acetoxy)propyl]-5-n-hexyloxy-2,3,3-trimethylindolinium iodide (3-hex), 1-[3-(acetoxy)propyl]-5-{2-[2-(2-methoxyethoxy)ethoxy]ethoxy}-2,3,3-trimethylindolinium iodide (3-Peg), 1-[3-(acetoxy)propyl]-5-tert-butyl-2,3,3-trimethylindolinium iodide (3-tBu), 5-chloro-1-[3-(acetoxy)propyl]-2,3,3-trimethylindolinium iodide (3-Cl), 1,1′-bis(3-acetoxypropyl)-5,5′-bis(n-hexyloxy)-3,3,3′,3′-tetramethylindodicarbocyanine iodide (4-hex), 1,1′-bis(3-acetoxypropyl)-5,5′-bis(tert-butyl)-3,3,3′,3′-tetramethylindodicarbocyanine iodide (4-tBu), 1,1′-bis(3-hydroxypropyl)-5,5′-bis(n-hexyloxy)-3,3,3′,3′-tetramethylindodicarbocyanine iodide (5-hex), 1,1′-bis(3-hydroxypropyl)-5,5′-bis{2-[2-(2-methoxyethoxy)ethoxy]ethoxy}-3,3,3′,3′-tetramethylindodicarbocyanine iodide (5-Peg), 1,1′-bis(3-hydroxypropyl)-5,5′-bis(tert-butyl)-3,3,3′,3′-tetramethyl-indodicarbocyanine iodide (5-tBu), 5,5′-dichloro-1,1′-bis(3-hydroxypropyl)-3,3,3′,3′-tetramethyl-indodicarbocyanine iodide (5-Cl), 1-(3-hydroxypropyl)-1’-[3-(4-monomethoxytrityloxy)propyl]-5,5′-bis(n-hexyloxy)-3,3,3′,3′-tetramethyl-indodicarbocyanine iodide (Cy5-hex), 1-(3-hydroxypropyl)-1’-[3-(4-monomethoxytrityloxy)propyl]-5,5′-bis{2-[2-(2-methoxyethoxy)ethoxy]ethoxy}-3,3,3′,3′-tetramethyl-indodicarbocyanine iodide (Cy5-Peg), 1-(3-hydroxypropyl)-1’-[3-(4-monomethoxytrityloxy)propyl]-5,5′-bis(tert-butyl)-3,3,3′,3′-tetramethyl-indodicarbocyanine iodide (Cy5-tBu), and 5,5′-dichloro-1-(3-hydroxypropyl)-1’-[3-(4-monomethoxytrityloxy)propyl]-tetramethyl-indodicarbocyanine iodide (Cy5-Cl) are provided in Supporting Information Section 2.
General Procedure for Cy5-Phosphoramidite Synthesis
In preparation for the reaction, an oven-dried round-bottom flask was charged with one MMTr-Cy5 iodide derivative (5, 0.16 mmol) and then coevaporated with dry CH3CN (3 times), dried under vacuum for 2–8 h, and then either stored under nitrogen overnight or used immediately. At the time of reaction, the precharged vessel was flushed/filled with dry nitrogen, then dissolved in dried CH2Cl2 (3.2 mL), and treated with dried N,N-diisopropylethylamine (170 μL, 0.96 mmol). Then, 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (110 μL, 0.48 mmol) was transferred from the glovebox directly to the reaction solution. The reaction mixture was vigorously stirred at room temperature in the dark for 30 min under N2. The reaction completion was assessed by TLC [CH2Cl2/CH3CN, (2:1) or (3:1) depending on Cy5 analogue]. Note that in all cases, the retention factor (Rf) of Cy5-phosphoramidite was slightly larger than that of the hydroxyl-functionalized Cy5, thus cospotting the reaction crude against the starting material is invaluable. Once determination is complete, the solution was washed with saturated aqueous NaHCO3 solution (2 times). The organic layer was dried over Na2SO4, filtered, and concentrated. The crude product was then dried under vacuum for 30 min to ensure that all volatiles are removed before proceeding. The crude product was then dissolved in dry CH2Cl2 (∼1 mL), and dry hexane (2–3 mL) was added to the solution. The solvent was then slowly stripped on a rotary evaporator until the majority of the product precipitated; then, the yellow-green-colored supernatant was discarded. This procedure was repeated until the undesired H-phosphonate byproduct is no longer visible on TLC (bright red-orange following staining with ninhydrin and heating), and the supernatant was the blue color of Cy5. The product was then coevaporated with dry CH3CN (3 times) and dried under vacuum for 30 min. Following this, the product was immediately used for DNA synthesis by resolubilizing to a concentration of 80 mM in anhydrous CH3CN (2 mL), transferring directly to a reagent bottle, attaching that bottle to the DNA synthesizer, priming the bottle lines, and starting the synthesis. See Supporting Information Section 4 for more information on the automated DNA synthesis coupling protocols.
Ammonolysis of DNA Sequences
Following DNA synthesis, the contents of a CPG-bound DNA column are emptied into a clean 20 mL scintillation vial. The residual CPG beads in the column were then flushed with 3 mL of water, into the scintillation vial. Next, 1 mL of 28% NH4OH (aq) was added to the vial, resulting in a final concentration of approximately 7% NH4OH (aq). The contents of the scintillation vial were then shaken, in darkness for 48 h (for Cy5-Cl-containing sequences) or 1 week (for all other Cy5-containing sequences).
Salt Exchange of DNA Sequences
Salt exchange was performed on each DNA sequence following the ammonolysis step. To begin, a Glen-Pak DNA Purification Cartridge (Glen Research, Sterling, VA, catalogue no. 60–5200) was wetted with 5 mL of neat MeCN (HPLC grade) and then flushed with 5 mL of 0.2 M TEAA (in 18 Ω water). The DNA–dye sequence (constituted in ammonia solution) was then passed through the Glen-Pak column via a 10 mL syringe 3–5 times. If blue color remains in the ammonia solution after several passes, the solution was retained and set aside. The Glen-Pak was then flushed/washed with 5 mL of 0.2 M TEAA followed by 5 mL of 18Ω water. The Glen-Pak was next purged of residual water by plunging air until no droplets were observed. DNA–dye sequences were then collected into an Eppendorf tube by eluting from Glen-Pak with ∼1 mL of MeCN/water (80/20). If the original ammonia solution retained color and was set aside, the Glen-Pak was reconstituted by flushing with 5 mL of 0.2 M TEAA, and the loading/washing/eluting process was repeated. The second eluent of DNA–dye solution was combined with the first and then concentrated to dryness on a Speed-Vac SPD 1030 (Thermo Fisher Scientific, Waltham, MA).
Oligonucleotide Purification
Purification was performed on a preparatory LC system composed of the following Waters, Inc. (Milford, MA) components: 2707 Autosampler, 2545 Quaternary Gradient Module, 2998 Photodiode Array Detector, and Fraction Collector III. Individual oligonucleotides were injected from HPLC grade water (500 μL automated injection) onto an XBridge OST C18 OBD 19 × 50 mm column (part no. 186008962, Waters, Inc., Milford, MA) and eluted with a gradient of increasing methanol in 0.1M TEAA (aq.). Individual oligonucleotides required two, half sample injections to reach adequate purity (i.e. the bulk sample was dissolved in 1 mL of water, and purified by two trials, each using a 500 μL injection). Elution gradients reflect those developed to characterize the individual sequences, gradients can be found in Supporting Information, Section 5. Oligonucleotides containing Cy5-hex, Cy5-Peg or Cy5-tBu were concentrated to dryness on a Speed-Vac SPD 1030 (Thermo Fisher Scientific, Waltham, MA) immediately following elution. Oligonucleotides containing Cy5-Cl were desalted following the above procedure, as direct concentration from 0.1 M TEAA resulted in partial (10−20%) decomposition. Following concentration, all fractions were assessed for purity by LCMS (see Characterization, above), combined where appropriate, then assessed again by LCMS to determine final purity. Oligonucleotides were deemed acceptable when purity exceeded 90%. Final samples were again concentrated down then stored at −20 °C in the dark.
Acknowledgments
The authors acknowledge the U.S. Naval Research Laboratory (NRL), the Office of Naval Research, and the NRL’s Nanoscience Institute for programmatic funding. We thank Scott MacDonald from Advanced Chemistry Development, Inc. (Toronto, Canada) for assistance with log P and log S0 determination and Dr. Greg Ellis for his expertise in the interpretation of mass spectra. Research at Boise State University was supported by the Department of the Navy, Office of Naval Research (ONR), under ONR award no. N00014-19-1–2615. D.M. was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under award number K99EB030013. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c06921.
Synthetic schemes and procedures, 1H and 13C NMR spectra, coupling protocols for automated DNA synthesis, confirmatory LCMS traces for DNA-Cy5 analogy sequences, and additional absorption spectra of select labeled DNA oligonucleotides (PDF)
Author Contributions
A.M. and K.S. contributed equally to this work. All authors have given approval for the final version.
The authors declare no competing financial interest.
Supplementary Material
References
- Ernst L. A.; Gupta R. K.; Mujumdar R. B.; Waggoner A. S. Cyanine Dye Labeling Reagents for Sulfhydryl-Groups. Cytometry 1989, 10, 3–10. 10.1002/cyto.990100103. [DOI] [PubMed] [Google Scholar]
- Mujumdar R. B.; Ernst L. A.; Mujumdar S. R.; Lewis C. J.; Waggoner A. S. Cyanine Dye Labeling Reagents – Sulfoindocyanine Succinimidyl Esters. Bioconjugate Chem. 1993, 4, 105–111. 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- Gerowska M.; Hall L.; Richardson J.; Shelbourne M.; Brown T. Efficient Reverse Click Labeling of Azide Oligonucleotides with Multiple Alkynyl Cy-Dyes Applied to the Synthesis of HyBeacon Probes for Genetic Analysis. Tetrahedron 2012, 68, 857–864. 10.1016/j.tet.2011.11.041. [DOI] [Google Scholar]
- Holzhauser C.; Berndl S.; Menacher F.; Breunig M.; Göpferich A.; Wagenknecht H.-A. Synthesis and Optical Properties of Cyanine Dyes as Fluorescent DNA Base Substitutions for Live Cell Imaging. Eur. J. Org. Chem. 2010, 2010, 1239–1248. 10.1002/ejoc.200901423. [DOI] [Google Scholar]
- Massey M.; Algar W. R.; Krull U. J. Fluorescence Resonance Energy Transfer (FRET) for DNA Biosensors: FRET Pairs and Forster Distances for Various dye-DNA Conjugates. Anal. Chim. Acta 2006, 568, 181–189. 10.1016/j.aca.2005.12.050. [DOI] [PubMed] [Google Scholar]
- Medintz I. L.; Matthew Mauro J. Use of a Cyanine Dye as a Reporter Probe in Reagentless Maltose Sensors Based on E-coli Maltose Binding Protein. Anal. Lett. 2004, 37, 191–202. 10.1081/al-120027785. [DOI] [Google Scholar]
- Schobel U.; Egelhaaf H.-J.; Brecht A.; Oelkrug D.; Gauglitz G. New-Donor-Acceptor Pair for Fluorescent Immunoassays by Energy Transfer. Bioconjugate Chem. 1999, 10, 1107–1114. 10.1021/bc990073b. [DOI] [PubMed] [Google Scholar]
- Buckhout-White S.; Brown III C. W.; Hastman D. A.; Ancona M. G.; Melinger J. S.; Goldman E. R.; Medintz I. L. Expanding Molecular Logic Capabilities in DNA-Scaffolded MultiFRET Triads. RSC Adv. 2016, 6, 97587–97598. 10.1039/c6ra23079b. [DOI] [Google Scholar]
- Cunningham P. D.; Spillmann C. M.; Melinger J. S.; Ancona M. G.; Kim Y. C.; Mathur D.; Buckhout-White S.; Goldman E. R.; Medintz I. L. Forster Resonance Energy Transfer in Linear DNA Multifluorophore Photonic Wires: Comparing Dual versus Split Rail Building Block Designs. Adv. Opt. Mater. 2021, 9, 2100884. 10.1002/adom.202100884. [DOI] [Google Scholar]
- Mathur D.; Samanta A.; Ancona M. G.; Díaz S. A.; Kim Y.; Melinger J. S.; Goldman E. R.; Sadowski J. P.; Ong L. L.; Yin P.; Medintz I. L. Understanding Förster Resonance Energy Transfer in the Sheet Regime with DNA Brick-Based Dye Networks. ACS Nano 2021, 15, 16452–16468. 10.1021/acsnano.1c05871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Patten L. K.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121, 6905–6916. 10.1021/acs.jpca.7b04344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fothergill J. W.; Hernandez A. C.; Knowlton W. B.; Yurke B.; Li L. Ab Initio Studies of Exciton Interactions of Cy5 Dyes. J. Phys. Chem. A 2018, 122, 8989–8997. 10.1021/acs.jpca.8b05237. [DOI] [PubMed] [Google Scholar]
- Huff J. S.; Davis P. H.; Christy A.; Kellis D. L.; Kandadai N.; Toa Z. S. D.; Scholes G. D.; Yurke B.; Knowlton W. B.; Pensack R. D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett. 2019, 10, 2386–2392. 10.1021/acs.jpclett.9b00404. [DOI] [PubMed] [Google Scholar]
- Huff J. S.; Turner D. B.; Mass O. A.; Patten L. K.; Wilson C. K.; Roy S. K.; Barclay M. S.; Yurke B.; Knowlton W. B.; Davis P. H.; Pensack R. D. Excited-State Lifetimes of DNA-Templated Cyanine Dimer, Trimer, and Tetramer Aggregates: The Role of Exciton Delocalization, Dye Separation, and DNA Heterogeneity. J. Phys. Chem. B 2021, 125, 10240–10259. 10.1021/acs.jpcb.1c04517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham P. D.; Khachatrian A.; Buckhout-White S.; Deschamps J. R.; Goldman E. R.; Medintz I. L.; Melinger J. S. Resonance Energy Transfer in DNA Duplexes Labeled with Localized Dyes. J. Phys. Chem. B 2014, 118, 14555–14565. 10.1021/jp5065006. [DOI] [PubMed] [Google Scholar]
- Cunningham P. D.; Kim Y. C.; Díaz S. A.; Buckhout-White S.; Mathur D.; Medintz I. L.; Melinger J. S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. 10.1021/acs.jpcb.8b02134. [DOI] [PubMed] [Google Scholar]
- Rolczynski B. S.; Díaz S. A.; Kim Y. C.; Medintz I. L.; Cunningham P. D.; Melinger J. S. Understanding Disorder, Vibronic Structure, and Delocalization in Electronically Coupled Dimers on DNA Duplexes. J. Phys. Chem. A 2021, 125, 9632–9644. 10.1021/acs.jpca.1c07205. [DOI] [PubMed] [Google Scholar]
- Cannon B. L.; Patten L. K.; Kellis D. L.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122, 2086–2095. 10.1021/acs.jpca.7b12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova L. I.; Malinovskii V. L.; Patsenker L. D.; Häner R. J- vs. H-type Assembly: Pentamethine Cyanine (Cy5) as a Near-IR Chiroptical Reporter. Chem. Commun. 2013, 49, 5298–5300. 10.1039/c3cc42103a. [DOI] [PubMed] [Google Scholar]
- Mass O. A.; Wilson C. K.; Roy S. K.; Barclay M. S.; Patten L. K.; Terpetschnig E. A.; Lee J.; Pensack R. D.; Yurke B.; Knowlton W. B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124, 9636–9647. 10.1021/acs.jpcb.0c06480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostov O.; Liboska R.; Páv O.; Novák P.; Rosenberg I. Solid-Phase Synthesis of Phosphorothioate/Phosphonothioate and Phosphoramidate/Phosphonamidate Oligonucleotides. Molecules 2019, 24, 1872. 10.3390/molecules24101872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuri S.; Church G. M. Large-scale de novo DNA Synthesis: Technologies and Applications. Nat. Methods 2014, 11, 499–507. 10.1038/nmeth.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen M.; Gothelf K. V. Chemistries for DNA Nanotechnology. Chem. Rev. 2019, 119, 6384–6458. 10.1021/acs.chemrev.8b00570. [DOI] [PubMed] [Google Scholar]
- Biaggne A.; Knowlton W. B.; Yurke B.; Lee J.; Li L. Substituent Effects on the Solubility and Electronic Properties of the Cyanine Dye Cy5: Density Functional and Time-Dependent Density Functional Theory Calculations. Molecules 2021, 26, 524. 10.3390/molecules26030524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brush C. K.; Anderson E. D.. Indocarbocyanine-linked phosphoramidites. U.S. Patent 5,556,959 A, 1996.
- Brush C. K.; Anderson E. D.. Indocarbocyanine and benzindocarbocyanine phosphoramidites. U.S. Patent 5,808,044 A, 1998.
- Owens E. A.; Hyun H.; Dost T. L.; Lee J. H.; Park G.; Pham D. H.; Park M. H.; Choi H. S.; Henary M. Near-Infrared Illumination of Native Tissues for Image-Guided Surgery. J. Med. Chem. 2016, 59, 5311–5323. 10.1021/acs.jmedchem.6b00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caram J. R.; Doria S.; Eisele D. M.; Freyria F. S.; Sinclair T. S.; Rebentrost P.; Lloyd S.; Bawendi M. G. Room-Temperature Micron-Scale Exciton Migration in a Stabilized Emissive Molecular Aggregate. Nano Lett. 2016, 16, 6808–6815. 10.1021/acs.nanolett.6b02529. [DOI] [PubMed] [Google Scholar]
- Cao W.; Sletten E. M. Fluorescent Cyanine Dye J-Aggregates in the Fluorous Phase. J. Am. Chem. Soc. 2018, 140, 2727–2730. 10.1021/jacs.7b11925. [DOI] [PubMed] [Google Scholar]
- Mishra A.; Behera R. K.; Behera P. K.; Mishra B. K.; Behera G. B. Cyanines During the 1990s: A Review. Chem. Rev. 2000, 100, 1973–2012. 10.1021/cr990402t. [DOI] [PubMed] [Google Scholar]
- Izuta S.; Yamaguchi S.; Kosaka T.; Okamoto A. Reversible and Photoresponsive Immobilization of Nonadherent Cells by Spiropyran-Conjugated PEG–Lipids. ACS Appl. Bio Mater. 2019, 2, 33–38. 10.1021/acsabm.8b00656. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Hwang S. H. Synthesis and Photostability of Functional Squarylium Dyes. Dyes Pigm. 1997, 35, 111–121. 10.1016/s0143-7208(96)00101-5. [DOI] [Google Scholar]
- Hemmer J. R.; Smith P. D.; van Horn M.; Alnemrat S.; Mason B. P.; de Alaniz J. R.; Osswald S.; Hooper J. P. High Strain-Rate Response of Spiropyran Mechanophores in PMMA. J. Polym. Sci., Part B: Polym. Phys. 2014, 52, 1347–1356. 10.1002/polb.23569. [DOI] [Google Scholar]
- Liu Q.; Kanahashi K.; Matsuki K.; Manzhos S.; Feron K.; Bottle S. E.; Tanaka K.; Nanseki T.; Takenobu T.; Tanaka H.; Sonar P. Triethylene Glycol Substituted Diketopyrrolopyrrole- and Isoindigo-Dye Based Donor-Acceptor Copolymers for Organic Light-Emitting Electrochemical Cells and Transistors. Adv. Electron. Mater. 2020, 6, 1901414. 10.1002/aelm.201901414. [DOI] [Google Scholar]
- Baba A.; Shibata I.; Fujiwara M.; Matsuda H. Novel Use of Organotin Halide-Base Complex in Organic Synthesis. Cycloaddition Reaction of Oxetane with Isocyanates. Tetrahedron Lett. 1985, 26, 5167–5170. 10.1016/s0040-4039(00)98893-2. [DOI] [Google Scholar]
- Owens E. A.; Hyun H.; Tawney J. G.; Choi H. S.; Henary M. Correlating Molecular Character of NIR Imaging Agents with Tissue-Specific Uptake. J. Med. Chem. 2015, 58, 4348–4356. 10.1021/acs.jmedchem.5b00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie M. S.; Götz R.; Franke C.; Bowler M.; Kumari N.; Magidson V.; Levitus M.; Loncarek J.; Sauer M.; Schnermann M. J. Cyanine Conformational Restraint in the Far-Red Range. J. Am. Chem. Soc. 2017, 139, 12406–12409. 10.1021/jacs.7b07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.; Rammler D. H.; Goldberg I. H.; Khorana H. G. Studies on Polynucleotides. XIV. Specific Synthesis of the C3″-C5″ Interribonucleotide Linkage. Syntheses of Uridylyl-(3″→5″)-Uridine and Uridylyl-(3″→5″)-Adenosine. J. Am. Chem. Soc. 1962, 84, 430–440. 10.1021/ja00862a023. [DOI] [Google Scholar]
- Fukui K.; Morimoto M.; Segawa H.; Tanaka K.; Shimidzu T. Synthesis and Properties of an Oligonucleotide Modified with an Acridine Derivative at the Artificial Abasic Site. Bioconjugate Chem. 1996, 7, 349–355. 10.1021/bc960019k. [DOI] [PubMed] [Google Scholar]
- Nielsen J.; Taagaard M.; Marugg J. E.; van Boom J. H.; Dahl O. Application of 2-cyanoethyl N, N,N’,N’-tetraisopropylphosphorodiamidite for in situ Preparation of Deoxyribonucleoside Phosphoramidites and Their Use in Polymer-Supported Synthesis of Oligodeoxyribonucleotides. Nucleic Acids Res. 1986, 14, 7391–7403. 10.1093/nar/14.18.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M.; Ugi I. Chloro-N,N-dialkylamino-2,2,2-trichloro-tert-butoxy-phosphines, New Reagents in the Syntheses of Oligonucleotides. Z. Naturforsch., B: J. Chem. Sci. 1995, 50, 948–952. 10.1515/znb-1995-0617. [DOI] [Google Scholar]
- Williams D. B. G.; Lawton M. Drying of Organic Solvents: Quantitative Evaluation of the Efficiency of Several Desiccants. J. Org. Chem. 2010, 75, 8351–8354. 10.1021/jo101589h. [DOI] [PubMed] [Google Scholar]
- Glen Research (Sterling, VA) . Cyanine 5 Phosphoramidite. https://www.glenresearch.com/10-5915.html (accessed March 1, 2022).
- Biosystems A.Expedite 8900 Nucleic Acid Synthesis System User’s Guide; Applied Biosystems: Foster City, CA USA, 2001; Vol. Part Number PB601306 Rev. 2. [Google Scholar]
- Glen Research . Deprotection Guide; Glen Research: Sterling, VA, 2020. https://www.glenresearch.com/media/folio3/productattachments/application_guides/Deprotection_Guide_20200110.pdf (accessed March 1, 2022).
- Krotz A. H.; Rentel C.; Gorman D.; Olsen P.; Gaus H. J.; McArdle J. V.; Scozzari A. N. Solution Stability and Degradation Pathway of Deoxyribonucleoside Phosphoramidites in Acetonitrile. Nucleosides, Nucleotides Nucleic Acids 2004, 23, 767–775. 10.1081/ncn-120039215. [DOI] [PubMed] [Google Scholar]
- Hargreaves J. S.; Kaiser R.; Wolber P. K. The Degradation of dG Phosphoramidites in Solution. Nucleosides, Nucleotides Nucleic Acids 2015, 34, 691–707. 10.1080/15257770.2015.1071846. [DOI] [PubMed] [Google Scholar]
- Leo A.; Hansch C.; Elkins D. Partition Coefficients and Their Uses. Chem. Rev. 1971, 71, 525–616. 10.1021/cr60274a001. [DOI] [Google Scholar]
- Dearden J. C.; Bresnen G. M. The Measurement of Partition Coefficients. Quant. Struct.-Act. Relat. 1988, 7, 133–144. 10.1002/qsar.19880070304. [DOI] [Google Scholar]
- Sliwoski G.; Kothiwale S.; Meiler J.; Lowe E. W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentell K. B.; Dorsey J. G. Retention Mechanisms in Reversed-Phase Liquid Chromatography. Stationary-Phase Bonding Density and Partitioning. Anal. Chem. 1989, 61, 930–934. 10.1021/ac00184a003. [DOI] [PubMed] [Google Scholar]
- Rafferty J. L.; Zhang L.; Siepmann J. I.; Schure M. R. Retention Mechanism in Reversed-Phase Liquid Chromatography: A Molecular Perspective. Anal. Chem. 2007, 79, 6551–6558. 10.1021/ac0705115. [DOI] [PubMed] [Google Scholar]
- Neue U. D.HPLC Columns, Theory, Technology, and Practice; Wiley, 1998; Vol. 26, pp 439–440. [Google Scholar]
- Mandal A. K.; Taniguchi M.; Diers J. R.; Niedzwiedzki D. M.; Kirmaier C.; Lindsey J. S.; Bocian D. F.; Holten D. Photophysical Properties and Electronic Structure of Porphyrins Bearing Zero to Four meso-Phenyl Substituents: New Insights into Seemingly Well Understood Tetrapyrroles. J. Phys. Chem. A 2016, 120, 9719–9731. 10.1021/acs.jpca.6b09483. [DOI] [PubMed] [Google Scholar]
- Breslauer K. J.; Frank R.; Blocker H.; Marky L. A. Predicting DNA Duplex Stability From the Base Sequence. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 3746–3750. 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto N.; Nakano S.-i.; Yoneyama M.; Honda K.-i. Improved Thermodynamic Parameters and Helix Initiation Factor to Predict Stability of DNA Duplexes. Nucleic Acids Res. 1996, 24, 4501–4505. 10.1093/nar/24.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaLucia J.; Allawi H. T.; Seneviratne P. A. Improved Nearest-Neighbor Parameters for Predicting DNA Duplex Stability. Biochemistry 1996, 35, 3555–3562. 10.1021/bi951907q. [DOI] [PubMed] [Google Scholar]
- Turro N. J.; Ramamurthy V.; Scaiano J. C.. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Melville, NY USA, 2010. [Google Scholar]
- Patra D.; Malaeb N. N.; Haddadin M. J.; Kurth M. J. Influence of Substituent and Solvent on the Radiative Process of Singlet Excited States of Novel Cyclic Azacyanine Derivatives. J. Fluoresc. 2012, 22, 707–717. 10.1007/s10895-011-1006-0. [DOI] [PubMed] [Google Scholar]
- Lewis J. E.; Maroncelli M. On the (Uninteresting) Dependence of the Absorption and Emission Transition Moments of Coumarin 153 on Solvent. Chem. Phys. Lett. 1998, 282, 197–203. 10.1016/s0009-2614(97)01270-0. [DOI] [Google Scholar]
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
- Breger J. C.; Susumu K.; Lasarte-Aragonés G.; Díaz S. A.; Brask J.; Medintz I. L. Quantum Dot Lipase Biosensor Utilizing a Custom-Synthesized Peptidyl-Ester Substrate. ACS Sens. 2020, 5, 1295–1304. 10.1021/acssensors.9b02291. [DOI] [PubMed] [Google Scholar]
- Gemmill K. B.; Díaz S. A.; Blanco-Canosa J. B.; Deschamps J. R.; Pons T.; Liu H.-W.; Deniz A.; Melinger J.; Oh E.; Susumu K.; Stewart M. H.; Hastman D. A.; North S. H.; Delehanty J. B.; Dawson P. E.; Medintz I. L. Examining the Polyproline Nanoscopic Ruler in the Context of Quantum Dots. Chem. Mater. 2015, 27, 6222–6237. 10.1021/acs.chemmater.5b03181. [DOI] [Google Scholar]
- Drexhage K. H. Fluorescence Efficiency of Laser Dyes. J. Res. Natl. Bur. Stand., Sect. A 1976, 80A, 421–428. 10.6028/jres.080a.044. [DOI] [Google Scholar]
- Sens R.; Drexhage K. H. Fluorescence Quantum Yield of Oxazine and Carbazine Laser Dyes. J. Lumin. 1981, 24–25, 709–712. 10.1016/0022-2313(81)90075-2. [DOI] [Google Scholar]
- Boeneman K.; Prasuhn D. E.; Blanco-Canosa J. B.; Dawson P. E.; Melinger J. S.; Ancona M.; Stewart M. H.; Susumu K.; Huston A.; Medintz I. L. Self-Assembled Quantum Dot-Sensitized Multivalent DNA Photonic Wires. J. Am. Chem. Soc. 2010, 132, 18177–18190. 10.1021/ja106465x. [DOI] [PubMed] [Google Scholar]
- Algar W. R.; Khachatrian A.; Melinger J. S.; Huston A. L.; Stewart M. H.; Susumu K.; Blanco-Canosa J. B.; Oh E.; Dawson P. E.; Medintz I. L. Concurrent Modulation of Quantum Dot Photoluminescence Using a Combination of Charge Transfer and Forster Resonance Energy Transfer: Competitive Quenching and Multiplexed Biosensing Modality. J. Am. Chem. Soc. 2017, 139, 363–372. 10.1021/jacs.6b11042. [DOI] [PubMed] [Google Scholar]
- Brown C. W.; Buckhout-White S.; Díaz S. A.; Melinger J. S.; Ancona M. G.; Goldman E. R.; Medintz I. L. Evaluating Dye-Labeled DNA Dendrimers for Potential Applications in Molecular Biosensing. ACS Sens. 2017, 2, 401–410. 10.1021/acssensors.6b00778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.