

Abstract
DNA replication is a highly precise process which usually functions in a perfect rhythm with cell cycle progression. However, cells are constantly faced with various kinds of obstacles such as blocks in DNA replication, lack of availability of precursors and improper chromosome alignment. When these problems are not addressed, they may lead to chromosome instability and the accumulation of mutations, and even cell death. Therefore, the cell has developed response mechanisms to keep most of these situations under control. Of the many factors that participate in this DNA damage response, members of the family of phosphatidylinositol 3-kinase-related protein kinases (PIKKs), orchestrate the response landscape. Our understanding of two members of the PIKK family, human ATR (yeast Mec1) and ATM (yeast Tel1), and their associated partner proteins, has shown substantial progress through recent biochemical and structural studies. Emerging structural information of these unique kinases show common features that reveal the mechanism of kinase activity.
Keywords: ATR, ATM, Mec1, Tel1, PIKK, DNA damage response, DNA replication, checkpoint
PIKKs (Phosphatidylinositol 3-kinase-related protein kinases) make up a small family of atypical protein kinases that respond to various kinds of stress and are conserved in all eukaryotic cells (Lempiainen and Halazonetis 2009). In yeast, four members of the PIKK family have been identified. Mec1, Tel1, Tor1 and Tor2 (which result from the yeast genome duplication event), and Tra1. Mammals have the Mec1 ortholog ATR, the Tel1 ortholog ATM, the Tor1/2 ortholog mTOR, and the Tra1 ortholog TTRAP. They have two additional members, SMG-1 and DNA-PKcs, which are not found in yeast. While mTOR and Tor1/2 are considered as central regulators of cell growth (Beck and Hall 1999; Yang et al. 2013), and SMG-1 and TTRAP (Tra1) function in nonsense-mediated mRNA decay (Yamashita et al. 2009) and in histone acetylation (Grant et al. 1998), respectively, ATR (Mec1), ATM (Tel1) and DNA-PKcs are required for proper DNA damage response (DDR) and cell cycle checkpoint function. Yeast lacks a DNA-PKcs, which functions in double-stranded DNA break (DSB) repair by nonhomologous end-joining (NHEJ). This review will focus primarily on the checkpoint kinases Tel1ATM and Mec1ATR, their mode of action and their mechanism of activation, as revealed by recent cryoEM studies. Recent reviews delineate the pathways in which these proteins function in cell cycle checkpoints and DNA repair in eukaryotic cells (Paull 2015; Blackford and Jackson 2017; Saldivar et al. 2017; Williams and Zhang 2020). Other recent reviews focus on structural aspects of PIKKs (Baretic et al. 2019; Wu Q et al. 2019; Williams et al. 2020).
Mec1 and Tel1 show the typical architecture of PIKKs in general. The kinase domain constitutes only a minor region in these extremely large proteins, ~250–450 kDa in size (Figure 1). Besides the kinase domain, they share a highly conserved C-terminal regulatory domain, called FATC (FRAP, ATM, TTRAP C-terminus) and an N-proximal regulatory domains called FAT. The remainder of the protein consists mostly of α-helical regions, which have been called HEAT repeats (Huntington, Elongation factor 3, A subunit of protein phosphatase 2A, and TOR1). Although the HEAT repeats are very poorly conserved, they are essential for the unique architecture of PIKKs and they provide binding sites for regulatory proteins (Lovejoy and Cortez 2009; Baretic and Williams 2014).
Figure 1.
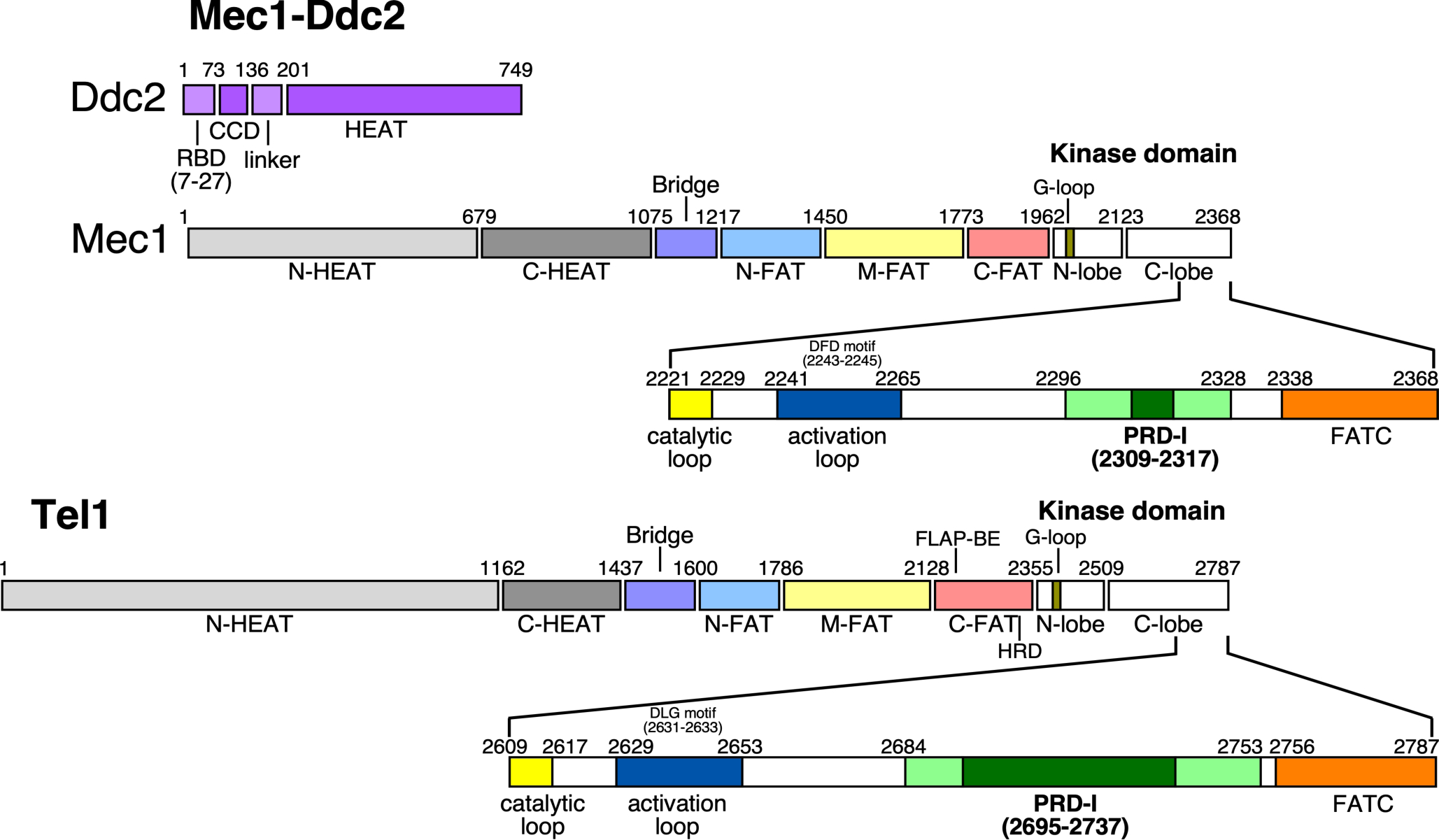
Domain architectures of yeast Mec1-Ddc2 and Tel1. Ddc2 is an integral partner of the Mec1-Ddc2 complex. Similar colors indicate analogous regions between Mec1 and Tel1. Part of the C-lobe region of the kinase domains is expanded to highlight critical regions that play a role in the activation mechanism (discussed in text). The PRD-I (PIKK Regulatory Domain - Insertion) (dark green) varies in size between PIKK members; it plays an important role in auto-inhibiting and/or restricting substrate access to the kinase active site. Residue numbers of S. cerevisiae Mec1-Ddc2 and Tel1 are shown.
Protein kinases are generally activated by two distinct mechanisms. Most kinases are activated by phosphorylation of a critical residue, generally threonine, in the activation loop (Adams 2001). Other kinases, such as cyclin-dependent kinases and most tyrosine kinases are activated by an activator protein, which can be achieved by kinase dimerization as observed with the epidermal growth factor receptor (Jura et al. 2009). PIKKs fall in the second group of protein kinases. They have developed complex activation mechanisms in which several enzymes interplay to produce an amplified response commensurate to the gravity of the activating signal, e.g. DNA damage.
The multifaceted roles of human ATR and ATM in responding to intrinsic as well as extrinsic DNA damaging sources have added a challenging complication to the treatment of cancer patients with radiation or chemotherapy. In order to make such treatments more effective, tweaking the efficiency of the checkpoint machinery of cancer cells by inhibiting ATR or ATM is a desirable goal. This inhibition will make cancer cells more susceptible to cell death at low dosages of ionizing radiation or chemotherapeutic drugs (Karnitz and Zou 2015; Lecona and Fernandez-Capetillo 2018). As an example, the inhibition of ATR has been shown to be particularly effective in tumor cells that are deficient for ATM or the homologous recombination pathway (Mei et al. 2019). In addition, combining ATR inhibitors with other inhibitors that target the homologous recombination pathway show promise in cancer treatment. Several therapeutic inhibitors with varying specificity to ATR have been developed and some are undergoing clinical trials (Foote et al. 2018). Recent advancements in determining the structure of the yeast ortholog of ATR, Mec1, at 2.8 Å resolution (Tannous et al. 2021), may help drive the design of selective inhibitors to another level.
In this review, we will focus primarily on the roles of the yeast cell cycle checkpoint PIKKs, Mec1 and Te11. Specific emphasis will be given to recent biochemical and structural advances in this field. We will describe the kinase activation mechanism of each and discuss their relevance in understanding the mechanism of their mammalian orthologs.
Overview of Mec1/ATR function in cell cycle checkpoints
In the yeast S. cerevisiae, Mec1 is the master checkpoint kinase, which monitors the accurate progression of the cell cycle (Figure 2). Together with its integral partner Ddc2, Mec1 plays an essential role in DNA damage signaling and repair as well as DNA replication. Both Mec1 and Ddc2 are essential genes and the loss of their function in yeast causes cell death. However, this lethality can be rescued by deleting the ribonucleotide reductase inhibitor SML1 (Zhao et al. 1998; Chabes et al. 1999). The upregulation of dNTP levels in sml1Δ rescues the growth defect in the absence of Mec1 or Ddc2, but cells remain highly sensitive to agents or conditions that cause replication fork stalling or DNA damage. The loss of Mec1’s catalytic function yields a similar DNA damage response phenotype as deletion of MEC1 or DDC2 (Cortez et al. 2001; Paciotti et al. 2001). Patients with Seckel syndrome are hypomorphic for ATR. This developmental disorder is typically characterized by dwarfism, mental retardation and defects in DDR (O’Driscoll et al. 2003). Additionally, the complete loss of ATR in mouse causes chromosomal fragmentation and early embryonic lethality (Brown and Baltimore 2000).
Figure 2.
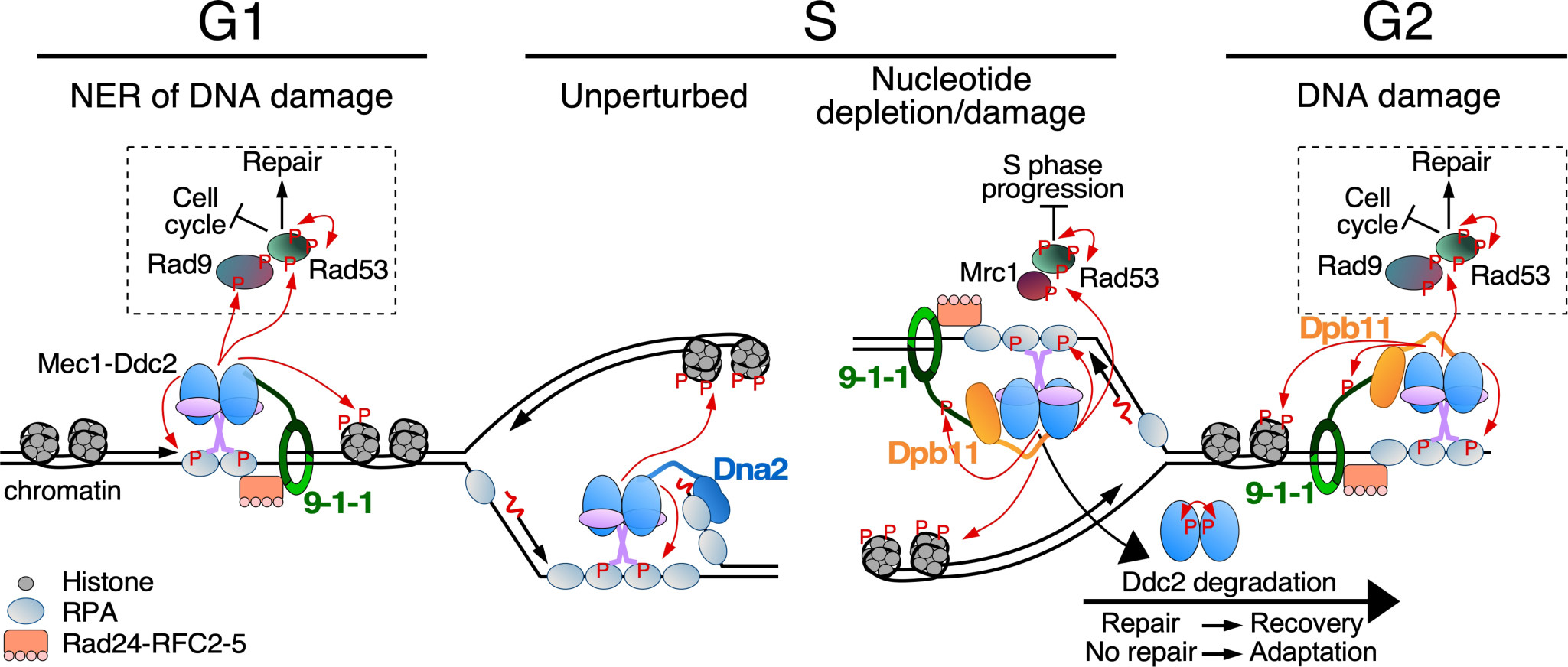
Overview of Mec1-initiated cell cycle checkpoints. Indicated are Mec1 activity (1) during G1 at RPA-coated ssDNA formed as a result of nucleotide excision repair (NER); (2) during S at an unperturbed DNA replication fork or (3) at a stalled fork due to nucleotide depletion or DNA damage; (4) during G2 in response to DNA damage. Mec1-Ddc2 binds RPA-ssDNA and localizes together with Mec1 cell cycle specific activators at the sites of lesions. The 9–1-1 checkpoint clamp is loaded by the Rad24-RFC2–5 clamp loader (human Rad17-RFC2–5) onto gapped DNA, and stimulates Mec1. Phosphorylation of 9–1-1 in S/G2 in response to DNA damage promotes recruitment of Dpb11. Among the many targets of Mec1/ATR is histone H2A (a hallmark of DNA damage), the effector kinase Rad53, and mediator scaffold proteins Rad9 (for DNA damage) or Mrc1 (for replication stress). These scaffold proteins promote auto-hyperphosphorylation of Rad53, promoting downstream pathways including cell cycle arrest and DNA repair. During unperturbed DNA replication, Mec1 may be localized to RPA-coated ssDNA on the lagging strand, and be activated by the nuclease-helicase Dna2, which localizes to DNA flaps. Mec1 auto-phosphorylation and Ddc2 degradation contribute to checkpoint inactivation and adaptation.
Mec1ATR is recruited to sites where stretches of single stranded DNA (ssDNA), coated with replication protein A (RPA) are formed (Zou and Elledge 2003). RPA-coated ssDNA is generated either during DNA damage repair, e.g. nucleotide excision repair or double-strand break repair, or when replication forks stall at natural or damage-induced DNA blocks, or when nucleotide precursors are limited and the coordination between the replication DNA helicase and the DNA polymerases is disrupted (Friedel et al. 2009). In addition, the presence of a compromised machinery caused by genetic defects can also result in the generation of ssDNA, and elicit Mec1ATR recruitment. The interaction between Mec1 and RPA is mediated by the Ddc2ATRIP subunit, through an N-terminal domain referred to as the RPA-binding domain (Zou and Elledge 2003; Ball et al. 2007; Deshpande I et al. 2017).
Recent progress on the structure of Mec1-Ddc2 and of individual domains has advanced our understanding of its architecture, activation mechanism, and the mode of interaction with RPA (Sawicka et al. 2016; Deshpande I et al. 2017; Wang X et al. 2017; Tannous et al. 2021). These include very recent high-resolution structures of Mec1-Ddc2 in both an auto-inhibited and activated state (3.4 and 2.8 Å, respectively). Mec1-Ddc2 exists as a dimer of heterodimers with several dimer interfaces formed between Mec1-Mec1, Mec1-Ddc2, and Ddc2-Ddc2 making up its core architecture, resembling that of a butterfly (Figure 3). A medium resolution cryoEM structure (4.7 Å) of ATR-ATRIP shows a similar envelope as the Mec1-Ddc2 core structure (Rao et al. 2018). All available Mec1-Ddc2 structures lack density for the N-terminal ~185 amino acids of the Ddc2 subunit, indicating that this region assumes conformational flexibility.
Figure 3.
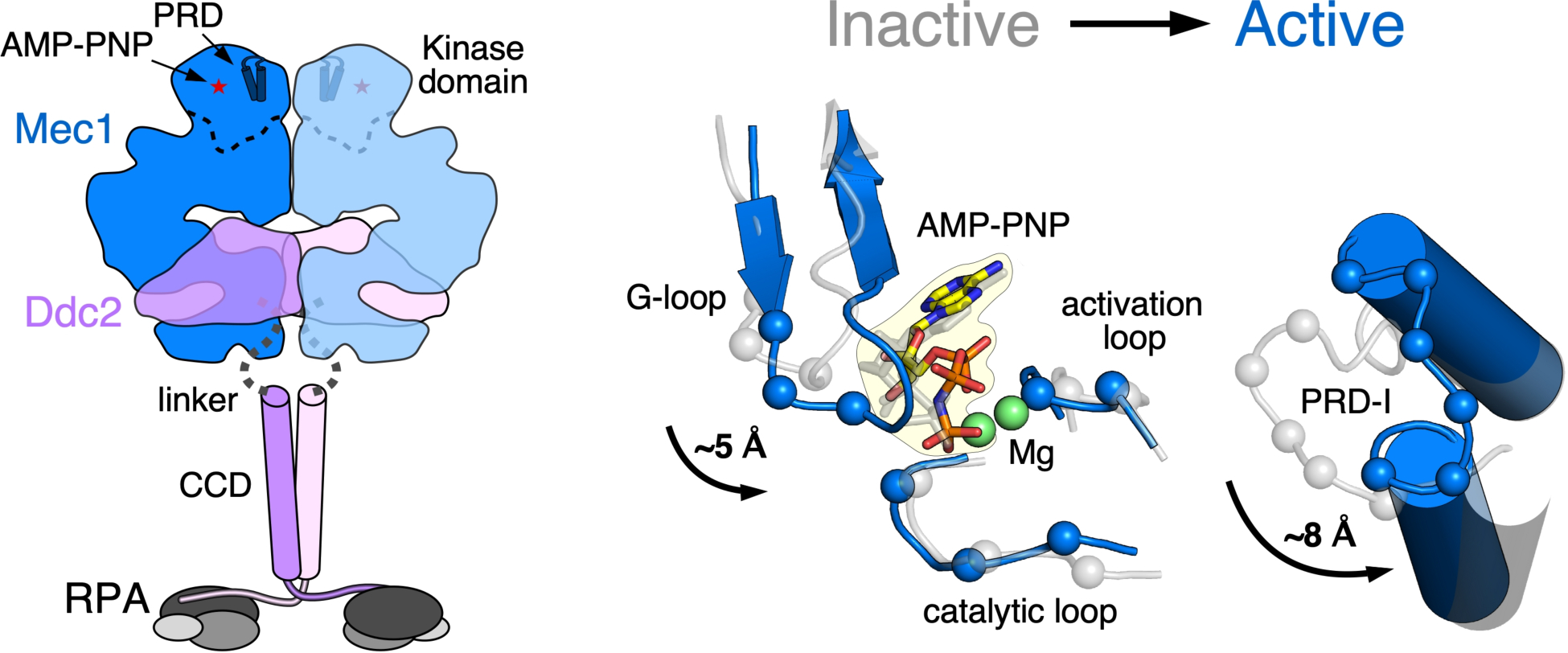
Activation mechanism of Mec1 kinase. Left, model of the Mec1-Ddc2-RPA complex, derived from the cryoEM structure of Mec1-Ddc2 (PDB:6Z2W, 6Z2X, 6Z3A) and the crystal structure of NTD of Ddc2, complexed with the NTD of RPA70 (PDB:5OMB, 5OMD). Ddc2 (1–185) are unresolved in the Mec1-Ddc2 structure. A long, structurally disordered linker of ~ 60 aa between the Mec1-Ddc2 structure and the Ddc2NTD-RPA70NTD structure likely allow vastly different orientations of these two structured complexes. Right, activation of Mec1 is mediated by conformational changes in and around the active site between wild-type Mec1 (auto-inhibited, gray) and Mec1-F2244L (constitutively active, blue). Residues of the G-loop, catalytic loop, activation loop, and the PRD-I rearrange in respect to AMP-PNP to release inhibition and promote catalytic efficiency.
Biochemical and genetic studies of the N-terminal region of Ddc2/ATRIP suggest the presence of a coiled-coil domain and an RPA-interaction domain (Ball and Cortez 2005; Itakura et al. 2005; Kim et al. 2005). The recent crystal structure of the NTD of Kluyveromyces lactis Ddc2 in a complex with the NTD of S. cerevisiae RPA70 (Rfa1 subunit) confirms and extends these biochemical studies (Deshpande I et al. 2017). The structure is extremely elongated, assuming a coiled coil domain, which corresponds to residues 73–136 of S. cerevisiae Ddc2 and ~110-~220 of human ATRIP (Ball and Cortez 2005). Very close to the amino terminus of Ddc2/ATRIP is a characteristic RPA binding domain, typified by the presence of a number of consecutive acidic residues (Ball et al. 2007; Xu et al. 2008). The coiled-coil plus RPA-binding domain region of Ddc2 is separated from the body of the Mec1-Ddc2 structure by a variable unstructured linker of about 50–70 amino acids (in fungi-metazoa). This domain separation endows flexibility to allow the kinase domain of Mec1/ATR to reach DNA-bound targets held in proximity by the DNA-RPA-Ddc2/ATRIP complex. Hundreds of Mec1/ATR targets have been identified by proteomics in yeast and in human cells (Matsuoka et al. 2007; Smolka et al. 2007; Bastos de Oliveira et al. 2015; Lanz et al. 2018).
The current structural model, based on the crystal structure of the Ddc2 NTD, suggests that the two Ddc2 NTDs interact with two RPA70 NTDs in a crisscross manner (Figure 3). Since only a small region of RPA70 was crystallized, it remains to be investigated whether the coiled-coil dimer spatially allows placement of two entire heterotrimeric RPAs. In addition, the symmetry of the crystal structure imposes a head-to-head arrangement of RPAs which may clash with the unidirectionality of the ssDNA to which they bind. Surprisingly, truncation of the RPA-binding and coiled-coil domains of Xenopus laevis ATRIP show no ATR kinase defects in an egg extract assay (Kim et al. 2005). In one study, overexpression in a yeast ddc2Δ mutant of a Ddc2 truncation, in which the entire RPA-binding plus coiled-coil domains were deleted, supported efficient growth in the presence of low levels of damage (Bandhu et al. 2014). However, in another study, a similar deletion of both domains showed sensitivity to DNA damaging agents that activate nucleotide excision repair (Deshpande I et al. 2017). In addition, when also the second checkpoint kinase TEL1 was deleted, the double tel1Δ ddc2-ΔNTD mutant was extremely sensitive to both DNA damage and to hydroxyurea. These genetic studies show the importance of the Ddc2 NTD, particularly when the second, partially compensatory damage sensor kinase Tel1 is missing. In human cells, the coiled-coil domain of ATRIP is important for localization to DNA damage foci (Ball and Cortez 2005). Finally, the unstructured linker region, which may play a role in juxtaposing DNA-bound substrates near the kinase domain, has also been implicated in checkpoint adaptation. Phosphorylation of key serines in the linker region of Ddc2 mediates its degradation and thereby adaptation (Memisoglu et al. 2019). One of the consequences of Ddc2 depletion is the dephosphorylation of the Rad53 effector kinase (the functional homolog of human CHK1 and CHK2), which turns off the checkpoint (Tsabar et al. 2016).
Activators of Mec1/ATR
Yeast Mec1 maintains a low basal kinase activity, which is crucial for cell growth and routine genome maintenance. Mec1 phosphorylates numerous substrates that are important for proper DNA replication in the absence of damage (Lanz et al. 2018). As an example, Mec1 is one of multiple kinases that primes the Mcm2–7 helicase for phosphorylation by Cdc7, thus playing a role in origin firing (Randell et al. 2010). In the event of DNA damage, Mec1’s basal kinase activity is strongly stimulated by the recruitment of the Mec1-Ddc2 complex to sites of lesion and by the interaction with additional factors referred to as Mec1/ATR activators. The formation of a Mec1-Ddc2-ssDNA-RPA complex alone already results in about two-fold kinase stimulation (Biswas et al. 2019). This activity is then further amplified ~20–50 fold by the presence of direct activators. However, while most studies of Mec1/ATR activators have focused on DNA damage-induced activation, it is likely that even in the absence of applied damage, Mec1 functions together with one or more of its activators to mediate proper cell cycle progression. This can be concluded from the observation that yeast cells lacking the backup PIKK Tel1 plus all known Mec1 activators progress very poorly through the cell cycle in otherwise undisturbed cells (Kumar and Burgers 2013). In addition, such cells accumulate gross chromosomal rearrangements at a high frequency (Lanz et al. 2018). Nevertheless, while basal Mec1 shows poor functionality, this low activity does suppress the lethality of a MEC1 deletion.
In yeast, the three direct activators of Mec1 are the Ddc1 subunit of the 9–1-1 checkpoint clamp, the replisome assembly protein Dpb11 (S. pombe Cut5, human TopBP1), and the multi-functional nuclease/helicase Dna2 (Kumar and Burgers 2013). These factors respond to various forms of DNA damage that result in the generation of RPA-coated ssDNA, and activate Mec1 in a cell cycle-dependent manner. In G1, the 9–1-1 (Ddc1-Mec3-Rad17) clamp is the sole activator of Mec1 (Navadgi-Patil and Burgers 2009). It is loaded by the Rad24-RFC clamp loader onto 5’-single stranded-double stranded junctions of RPA-coated ssDNA gaps, which are formed as intermediates during nucleotide excision repair of DNA damage, e.g. UV damage (Giannattasio et al. 2004). The Ddc1Rad9 subunit not only mediates activation of Mec1, which is localized in its unstructured C-terminal tail, but it also interacts in a phosphorylation-dependent manner with the second activator Dpb11Cut5/TopBP1, in budding and fission yeast and in mammals (Wang H and Elledge 2002; Greer et al. 2003; Furuya et al. 2004; Delacroix et al. 2007; Lee J et al. 2007; Puddu et al. 2008). Dpb11 and 9–1-1 act together as a Mec1 checkpoint activation complex in the S and G2 cell cycle phases (Wang H and Elledge 2002; Navadgi-Patil and Burgers 2009). In addition to its checkpoint function, Dpb11 plays an essential role in DNA replication initiation (Araki et al. 1995; Tak et al. 2006). Dna2 is a lagging strand maturation factor with both nuclease and helicase activity (Budd et al. 1995; Bae et al. 1998). It is also involved in double strand break repair and mitochondrial DNA maintenance and, for the present discussion, in the S-phase checkpoint (Wanrooij and Burgers 2015; Zheng et al. 2020). Dna2 is responsible for Mec1 activation in the S-phase, with partial redundancy to the activities of 9–1-1 and Dpb11 (Kumar and Burgers 2013). The assignment of these factors to the different phases of the cell cycle rests in the structured domains of these proteins and not in the unstructured tails. C-terminal truncation of the activation tail of Ddc1Rad9 eliminates the G1 DNA damage checkpoint. But this G1 checkpoint can be restored by fusing the N-terminal activation tail of Dna2, which does not function as an activator in G1, onto the truncated Ddc1 subunit (Kumar and Burgers 2013).
In human cells, two direct activators of ATR have been identified, TopBP1 and ETAA1 (Makiniemi et al. 2001; Kumagai et al. 2006; Bass et al. 2016; Lee YC et al. 2016). The latter activator does not have a yeast homolog. TopBP1’s checkpoint activity is essential as a point mutation in the activation domain (W1147R), which falls outside its essential replication initiation domain, causes mouse embryonic lethality during the blastocyst stage (Zhou et al. 2013). TopBP1Dpb11/Cut5 is recruited to sites of lesions in a manner similar to yeast. The checkpoint clamp 9–1-1 (Rad9-Rad1-Hus1), which is loaded by its loader Rad17-RFC onto RPA-coated ssDNA-dsDNA 5’-junctions (Ellison and Stillman 2003), is responsible for recruiting TopBP1 in a manner that is regulated by phosphorylation of Rad9 (Delacroix et al. 2007). No direct activation of ATR by human 9–1-1 has been demonstrated (Delacroix et al. 2007). Apparently, the main checkpoint function of human 9–1-1 is in localizing TopBP1 to sites of DNA damage. Consistent with this model, the artificial localization of the activating domain of TopBP1 to chromatin, e.g. through fusion with histone H2B, obviates the need for 9–1-1 in ATR activation, although DNA damage is still required for full activation of ATR (Delacroix et al. 2007). Taking this approach one step further in yeast, the artificial colocalization of LacI-Ddc1 and LacI-Ddc2-Mec1 to Lac repressor arrays causes Mec1 hyperactivation and gratuitous checkpoint activity independent of DNA damage (Bonilla et al. 2008).
Whereas TopBP1Dpb11,Cut5 is the common, evolutionary conserved Mec1/ATR activator, the second human activator for ATR, ETAA1 has no known ortholog in yeast. ETAA1−/− homozygous mice largely die (~75%) during late embryonic development. Those homozygous mice that do survive are smaller in size and show defects in the T-cell response, but they are otherwise healthy and fertile (Miosge et al. 2017). Consistent with these results, ETAA1-deficient human cells are viable. They show hypersensitivity to hydroxyurea and mitomycin C, but not to ionizing radiation or cisplatin (Bass et al. 2016; Haahr et al. 2016; Lee YC et al. 2016). Moreover, the combination of defects in TopBP1 and ETAA1 shows synthetic lethality, suggesting that they have, at least in part, distinct functions.
ETAA1 is recruited to replicating DNA via its binding to RPA-coated ssDNA, which is a common intermediate at replication forks. Deletion of the ATR activation domain of ETAA1 greatly reduces ATR activity in the S-phase in unperturbed cells, providing additional evidence that ETAA1 functions during normal DNA replication (Saldivar et al. 2018). This property of ETAA1 resembles that of yeast Dna2, which, among its other varied functions, is required for normal DNA replication during the maturation of Okazaki fragments (Zheng et al. 2020).
Mec1/ATR activator domains contain two aromatic residues, which are essential for activating the kinase (Wanrooij et al. 2016; Thada and Cortez 2019). These aromatic residues are frequently flanked by several hydrophobic residues. In yeast, the aromatics reside in structurally disordered regions that can vary in length up to several hundreds of amino acids, in which the key aromatics are separated from just a few to over one hundred residues (Kumar and Burgers 2013). While one of the aromatics in the metazoan ATR activators is also localized in an unstructured region (Kumagai et al. 2006), the second aromatic residue is localized in a coiled-coil domain (Thada and Cortez 2019). Mutation of these aromatics eliminate activator activity without disrupting the coiled-coil structure. Predicted coiled-coil domains are lacking for the yeast activators. Small peptides, 9–30 amino acids in length, derived from the yeast Ddc1 or Dna2 unstructured regions are sufficient to stimulate the in vitro kinase activity of Mec1, although the apparent binding affinities are much lower than those of the intact domains (Wanrooij et al. 2016). Peptides in which the key aromatic residues or flanking hydrophobics were replaced by Ala showed no or severely reduced activity. Remarkably, a peptide derived from fission yeast Rad9 also stimulated budding yeast Mec1, suggesting the possibility that, unlike human 9–1-1, fission yeast 9–1-1 may also activate its Rad3Mec1,ATR checkpoint kinase (Wanrooij et al. 2016). The observation that the Mec1 activation function resides in long unstructured regions of these activator proteins does not exclude the possibility of additional activities in these unstructured regions. For instance, the unstructured C-terminal tail of Ddc1/Rad9 interacts with Dpb11/Cut5/TopBP1, and the N-terminal tail of Dna2 binds hairpin DNA structures (Park et al. 2020).
Recent mutational studies of Mec1 identified a mutation in the activation loop of Mec1, which caused constitutive high activity of the enzyme, obviating the need for activators (Tannous et al. 2021). Genetic studies revealed that this constitutive, high kinase activity inhibits cell growth and cell cycle progression and causes damage-sensitivity. In contrast, when Mec1 activators are missing in a mutant strain lacking also Tel1, which is highly deleterious for growth, the constitutive mec1 mutant suppresses the growth defect and damage sensitivity, indicating the importance of an optimal Mec1 activity for both normal cell growth and DDR. In human, ATR activity is important in insuring the proper completion of the S-phase of the cell cycle. It is marked by increased H2AX phosphorylation which peaks in the mid S-phase, as it is in yeast (Saldivar et al. 2018; Tannous et al. 2021). However, the down regulation of ATR activity is crucial to mark the end of the S-phase and for cells to progress into the G2 phase (Saldivar et al. 2018). Notably, a yeast mutant with constitutive Mec1 activity shows a continued elevated level of phospho-H2A and a delay in the progression of the cell cycle, reaffirming the importance of a well-regulated Mec1/ATR kinase activity.
Overview of ATM/Tel1 checkpoint kinase
ATM (Ataxia telangiectasia mutated) regulates the initiation of checkpoint activation in response to DNA double strand breaks or to oxidative stress, coordinating it to DNA repair, cell cycle progression and overall cellular metabolism (Paull 2015). ATM is an important cancer marker and target for cancer therapy (Jette et al. 2020; Lavin and Yeo 2020). AT cells not only show cell cycle checkpoint defects in response to agents that induce double-stranded breaks, but they are also sensitive to oxidative stress and show mitochondrial dysfunction (Guleria and Chandna 2016; Choi and Chung 2020). Its yeast ortholog Tel1 is a cell cycle checkpoint kinase with overlapping functions to the major checkpoint kinase Mec1. Tel1 is non-essential for cell growth, responds mainly to DNA double strand breaks and is involved in telomere length regulation (Greenwell et al. 1995; Mallory and Petes 2000). Surprisingly, given our knowledge of ATM biology, the yeast cell cycle checkpoint in response to DSBs is mainly mediated by Mec1 (Grenon et al. 2006), whereas Tel1’s function is only invoked in response to a large number of DSBs or “dirty” DSBs, e.g. those with a covalently attached protein (Mantiero et al. 2007; Fukunaga et al. 2011). An additional function of yeast Tel1 is to promote efficient resection at DSBs and thereby promote subsequent checkpoint activation on the resulting ssDNA by Mec1 (Mantiero et al. 2007) (Figure 4). Furthermore, Tel1 participates in stabilizing replication forks near DSBs (Doksani et al. 2009), and Tel1 in part suppresses the checkpoint defects of MEC1 mutants when replication forks stall (Clerici et al. 2001). Interestingly, dominant, hyperactive TEL1 mutants that show increased suppression of Mec1 defects, also show an over-elongation of telomeres, indicating that Tel1 activity positively regulates telomere length (Baldo et al. 2008).
Figure 4. Role of Tel1 at double-stranded DNA breaks and telomeres.
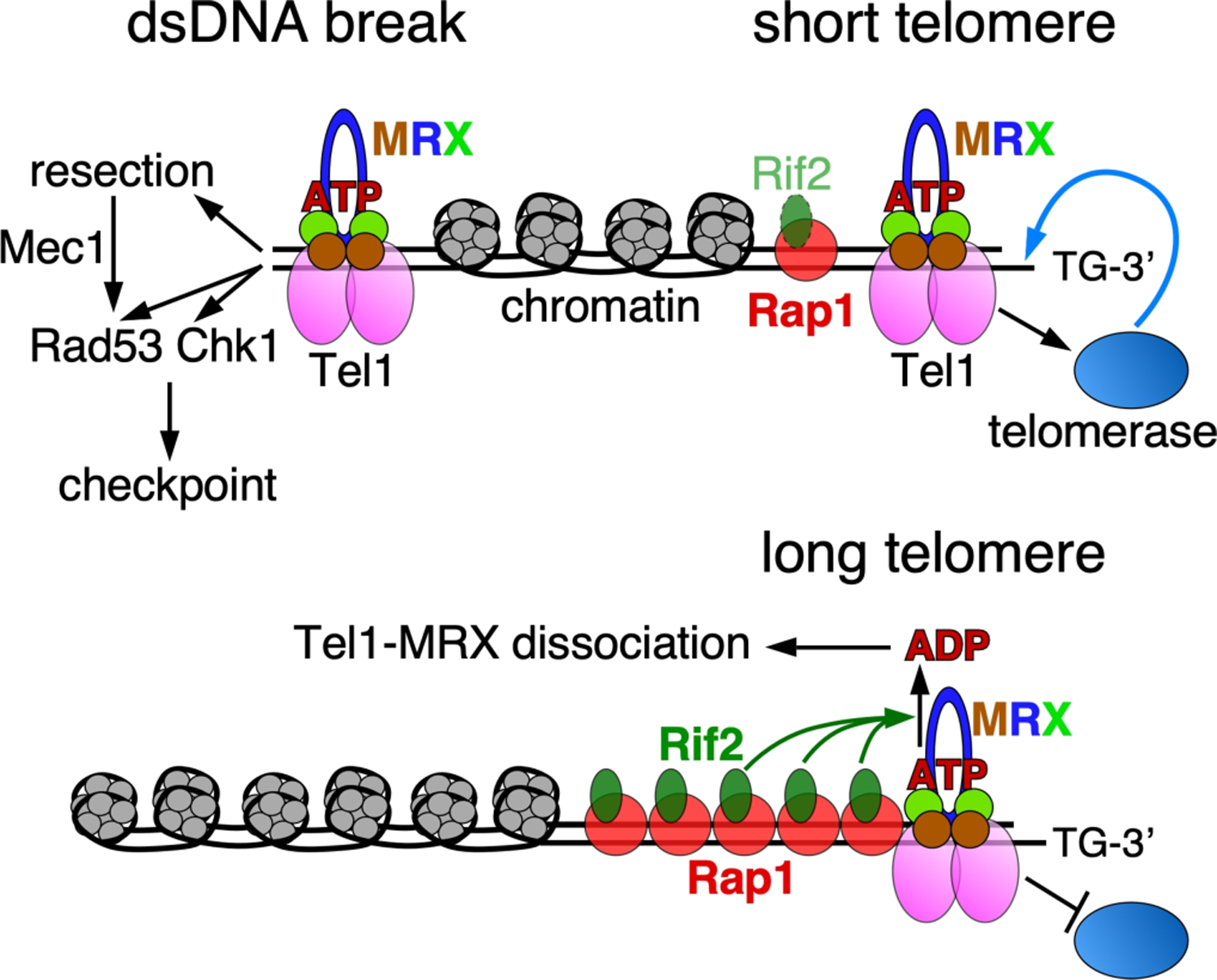
Damage-induced dsDNA breaks signal the recruitment of Tel1 by MRX. Only the ATP-form of Rad50 is competent for activation of Tel1. Activated Tel1 initiates a checkpoint by phosphorylating targets including Rad53 and Chk1. Tel1 additionally targets other factors that initiate DNA resection, which leads to a Mec1-dependent checkpoint response. Rif2 regulates telomere lengths by binding Rap1 and acting on Rad50. At a long telomere (bottom), the increased local concentration of Rif2 discharges the ATP-bound form of Rad50, thereby suppressing Tel1 activity, and promoting dissociation of Tel1 and MRX. At a short telomere (top), the relative lack of Rif2 allows Tel1-MRX activation of telomerase, resulting in telomere elongation.
Recent progress on the structure of Tel1/ATM from different organisms shows that Tel1/ATM mainly exists in dimeric form (Sawicka et al. 2016; Wang X et al. 2016; Baretic et al. 2017; Xin et al. 2019; Jansma et al. 2020; Yates et al. 2020), with the exception of a low resolution structure of a monomeric form of human ATM in a mixed monomeric-dimeric population (Xiao et al. 2019).
The Tel1/ATM dimer interface consists of multiple layers, similar to the Mec1 dimerization interface, with the bottom layer being hydrophobic in nature (Jansma et al. 2020; Yates et al. 2020). This interface is weakened in the presence of magnesium and a non-hydrolysable ATP analog AMP-PNP, an adjustment expected for rearrangement of the catalytic site and for allowing substrate access. However, the change affecting the dimer interface does not lead to a full transition of the dimer into a monomer, which is an energetically expensive process. ATM has been proposed to undergo a dimer to monomer transition upon activation (Bakkenist and Kastan 2003). This transition is achieved by autophosphorylation of ATM on Ser-1981, which is also dependent on the acetylation of Lys-3016 by the Tip60 histone acetyltransferase (Sun et al. 2005). However, this mechanism does not appear to be strictly conserved in mouse ATM (Pellegrini et al. 2006). Furthermore, in Xenopus laevis extracts, the dimer to monomer transition of ATM is facilitated by MRN (see below) binding or by DNA binding, and is independent of phosphorylation (Dupre et al. 2006). Interestingly, the monomeric form of human ATM, overexpressed in a human cell line, was only observed when chromosomal DNA was not removed during ATM purification (Xiao et al. 2019). Possible dimer to monomer transitions of fungal Tel1 and the role of post-translational modifications, remain to be investigated.
ATM/Tel1 activation by MRN/X
The MRN/X complex consists of Mre11 nuclease, Rad50 ATPase and the Nbs1 (Xrs2 in yeast) auxiliary subunit, and this complex is involved in the processing of DSBs for repair by non-homologous end joining or homologous recombination (Oh and Symington 2018; Paull 2018). The MRN/X complex also activates ATM/Tel1 in response to DSBs. Like ATR/Mec1, ATM/Tel1 has a low basal activity and the presence of double-stranded DNA and MRN/X stimulates this activity by two orders of magnitude (Lee JH and Paull 2004; Hailemariam, Kumar, et al. 2019). An interesting property of the system is that only MRN/X with Rad50 in the ATP form, and not in the ADP form, is capable of stimulating Tel1/ATM kinase activity. In an elegant biochemical study, Lee et al. altered the ATP-binding site of ATM such that it could accept N6-(furfuryl)-ATP instead of ATP as a phospho-donor (Lee JH et al. 2013). Then, they showed that binding to Rad50 of the non-hydrolysable ATP analog AMP-PNP, which locks this protein into the ATP form, stimulated ATM protein kinase activity with N6-(furfuryl)-ATP. In support of the model, molecular dynamics simulations of the yeast Mre11-Rad50 complex and additional mutational studies also indicate that only the ATP form of Rad50 is active for Tel1 stimulation (Cassani et al. 2019). These studies are also consistent with biochemical and structural studies of Mre11-Rad50 showing that ATP-Rad50 mediates DNA binding whereas its hydrolysis to ADP causes dissociation (Hopfner et al. 2000; Mockel et al. 2012). The activation of ATM/Tel1 is dependent on its interaction with the MRN/X auxiliary subunit Nbs1/Xrs2 (Nakada et al. 2003; You et al. 2005).
While the activation mechanisms of mammalian and fungal ATM/Tel1 show many of the expected similarities, there are also some significant differences. Optimal stimulation of human ATM requires very long dsDNA of about a kilobase in length (Lee JH and Paull 2005), whereas optimal Tel1 stimulation by MRX is already achieved with ~150 nt of dsDNA (Hailemariam, Kumar, et al. 2019). Furthermore, DNA ends are essential for ATM stimulation by MRN, but they are dispensable for Tel1 stimulation by MRX (Hailemariam, Kumar, et al. 2019). Biophysical studies of yeast MRX show that the complex can load onto internal DNA stretches and subsequently migrate towards DNA ends (Myler et al. 2017). However, while internal DNA can mediate Tel1-MRX activity, nucleosomal DNA is completely inactive (Hailemariam, Kumar, et al. 2019). These studies suggest that only dsDNA breaks where nucleosomes are cleared to produce naked DNA (Tsukuda et al. 2005), provide a suitable platform for active MRX-Tel1, because the existence of ~150 nt long regions on the yeast chromosomes, which are devoid of nucleosomes or other DNA binding proteins, is unlikely.
In addition to this canonical activation to DSBs, ATM is also activated when cells are exposed to oxidative stress (Shackelford et al. 2001). ATM’s in vitro kinase activity is stimulated by oxidizing agents such as hydrogen peroxide, in a manner that is independent of DNA or the MRN complex, suggesting that MRN is not required for the oxidative stress response by ATM (Guo et al. 2010). Interestingly, the oxidized ATM has formed a covalently linked dimer through disulfide linkages.
Role of Tel1 in telomere length elongation in yeast
One of the characteristic features of Tel1/ATM activation by MRX/N is that only the Rad50-ATP form of this heterotrimeric complex is capable of stimulating kinase activity. This property has been exploited by the yeast Rif2 protein to regulate telomere length in yeast (Figure 4). Rap1 protein is a conserved essential transcription factor, which binds double-stranded telomeric repeat sequences in complex, alternative DNA binding modes (Longtine et al. 1989; Wotton and Shore 1997; Li and de Lange 2003; Feldmann et al. 2015). The Rif1 and Rif2 (Rap1 interacting factor) proteins bind Rap1 and are components of the yeast shelterin-like complex (Hardy et al. 1992; Wotton and Shore 1997; Bourns et al. 1998). They are involved in telomere protection from nucleolytic degradation (Bonetti et al. 2010; Ribeyre and Shore 2012), and in telomere length regulation by controlling access of telomerase to telomeres of varying lengths. Specifically, Rif1 and Rif2 inhibit telomerase on long telomeres but not, or less so, on short telomeres (Teixeira et al. 2004; Shi et al. 2013). Yeast rif1Δ and/or rif2Δ mutants have telomeres with longer, but also more variable lengths and show associated telomere dysfunction phenotypes (Bonetti et al. 2010; Kaizer et al. 2015).
Yeast Rif2’s inhibition of MRX is intimately connected to the mechanism of Tel1 kinase activation. MRX(N)-stimulated Tel1/ATM activity is essential for recruitment and activity of telomerase at telomeres (Tsukamoto et al. 2001; Wu Y et al. 2007; Martina et al. 2012). Biochemical studies have shown that Rif2 protein stimulates the ATPase activity of Rad50 (Cassani et al. 2016; Hailemariam, De Bona, et al. 2019). Interestingly, a single point mutation in the unstructured N-terminal tail of Rif2 (F8A), fails to stimulate the Rad50 ATPase and rif2-F8A is a phenotypic null mutant for telomere length regulation (Kaizer et al. 2015; Hailemariam, De Bona, et al. 2019). These studies have suggested a model in which Rif2 discharges the ATP state of Rad50, making MRX inactive for Tel1 kinase stimulation and in addition causing dissociation of MRX together with its associated Tel1 (Hopfner et al. 2000; Hirano et al. 2009; Deshpande RA et al. 2014; Cassani et al. 2019). Long telomeres have a higher local concentration of Rif2 and show more severe inhibition of MRX-Tel1 than short telomeres. The important result of such a sliding scale inhibition of telomerase is that telomeres have a more uniform length distribution.
A common mechanism for PIKK activation through movement of the PRD-I - a signature motif for kinase activation
While the key interactions between Mec1/ATR and its activators have been mapped to the critical aromatic residues in the unstructured region of the activators, the interaction sites on Mec1/ATR remain to be determined. From a structural aspect, the interaction between Mec1/ATR and its activators may be transient, making it challenging to isolate a stable complex for structural studies. In one structural study of a PIKK, the activated complex between mTORC1 and its activator RHEB could only be stabilized for cryoEM analysis by crosslinking (Yang et al. 2017). Fortunately, insights in the structure of the activated state of Mec1-Ddc2 could be obtained by the study of a Mec1 mutant with constitutive activity (Tannous et al. 2021). A single mutation (F2244L) in the activation loop of Mec1 yields a kinase in the “ON” state, without substantial further stimulation by any of the three activators. The 2.8 Å resolution cryoEM structure of this mutant kinase, compared with the 3.8 Å structure of the wild-type enzyme, both in a complex with a non-hydrolysable ATP analog, has yielded valuable information regarding the conformational changes in the kinase domain that turn on the enzyme (Figure 3) (Tannous et al. 2021).
Activation of mTORC1 by RHEB introduces a series of conformational changes that bring the N-terminals of N-HEAT, M-HEAT, and FAT closer together in addition to minor movements in the kinase domain (Yang et al. 2017). In contrast, activating Mec1 by the F2244L mutation only results in conformational changes in the kinase and FAT domains. Additional conformational changes upon binding of an activator protein (Ddc1, Dpb11, or Dna2) to Mec1-Ddc2 cannot be discounted, and remain to be investigated. The most dramatic conformational changes in Mec1 were those of the PRD-I (PIKK Regulatory Domain - Insertion) loop, which links two conserved α-helices in the PRD (Figure 3). In Tel1, the PRD-I plays a role in maintaining low basal activity by blocking substrate binding (Jansma et al. 2020; Yates et al. 2020). Unfortunately, the PRD-I was not resolved in cryoEM structures of mTOR (Yang et al. 2017). Access to the active site of Mec1 and Tel1, and likely also in other PIKKs, appears to be blocked by the PRD-I loop. Upon activation, the movement of the PRD-I allows the reconfiguration of the active site to coordinate ATP in an active state and allow access of protein substrates. Interestingly, the substantial rearrangement of the kinase domain and the C-FAT rotation makes the existence of a mixed dimer (active:inhibited) unlikely, because it would result in serious steric clashes. This suggests that both Mec1-Ddc2 half sites undergo these conformational transitions, or that the dimer dissociates upon activation of one half site.
Previous to the structural studies, the PRD was described for human ATR after a single mutation in the C-terminal end of the kinase domain, K2589. The mutant yielded a kinase that failed to respond to TopBP1 stimulation but maintained its basal kinase activity (Mordes et al. 2008). The analogous lysine K3016 in the ATM PRD region plays an important role in ATM activation upon acetylation (Sun et al. 2005). A recent report shows that the common human cancer-associated mutation R3008H in the PRD region, when modeled in mouse ATM (R3016) fails to show ATM activation in response to oxidative stress or DNA damage (Milanovic et al. 2020).
The PRD-I, which links the two conserved α-helices, collectively referred to as PRD, varies in size between different members of PIKKs and can range from 9–27 residues in Mec1/ATR to 43–50 residues in Tel1/ATM, and to ~70–75 residues in DNA-PKcs and mTOR. Possibly, its divergence in size and sequence may reflect their unique responses to their activating partners in response to various cellular events. Additional high resolution structural data will further elucidate the role of these signature motifs in regulating the kinase activity of PIKK family.
Table 1.
Core proteins involved in the DNA damage and replication checkpoint pathways in budding yeast and human.
| PIKK |
Activator |
Mediator |
Effector kinase |
|||||
|---|---|---|---|---|---|---|---|---|
| human | yeast | Sensor | human | yeast | human | yeast | human | yeast |
|
| ||||||||
| ATR-ATRIP | Mec1-Ddc2 | RPA-ssDNA | TopBP1 ETAA1 |
Dpb11 9-1-1 Dna2 |
Claspin 53BP1 Mdc1 |
Mrc1 Rad9 |
Chk1 | Rad53 |
| ATM | Tel1 | dsDNA break | MRN | MRX | CtIP. | Sae2 | Chk2 | Rad53 Chk1 |
Acknowledgments
We thank the members of our laboratory and Roberto Galletto for input and critical reading during the progress of this work.
Funding
This work was supported in part by a grant from the National Institutes of Health (GM-118129) to P.B.
Footnotes
Disclosure statement
The authors report no conflict of interest.
References
- Adams JA. 2001. Kinetic and catalytic mechanisms of protein kinases. Chem Rev. 101(8):2271–2290. [DOI] [PubMed] [Google Scholar]
- Araki H, Leem SH, Phongdara A, Sugino A. 1995. Dpb11, which interacts with DNA polymerase II(epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc Natl Acad Sci USA. 92(25):11791–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SH, Choi E, Lee KH, Park JS, Lee SH, Seo YS. 1998. Dna2 of Saccharomyces cerevisiae possesses a single-stranded DNA-specific endonuclease activity that is able to act on double-stranded DNA in the presence of ATP. J Biol Chem. 273(41):26880–26890. [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 421(6922):499–506. [DOI] [PubMed] [Google Scholar]
- Baldo V, Testoni V, Lucchini G, Longhese MP. 2008. Dominant TEL1-hy mutations compensate for Mec1 lack of functions in the DNA damage response. Mol Cell Biol. 28(1):358–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball HL, Cortez D. 2005. ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J Biol Chem. 280(36):31390–31396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. 2007. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol Cell Biol. 27(9):3367–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandhu A, Kang J, Fukunaga K, Goto G, Sugimoto K. 2014. Ddc2 mediates Mec1 activation through a Ddc1- or Dpb11-independent mechanism. PLoS Genet. 10(2):e1004136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baretic D, Maia de Oliveira T, Niess M, Wan P, Pollard H, Johnson CM, Truman C, McCall E, Fisher D, Williams R et al. 2019. Structural insights into the critical DNA damage sensors DNA-PKcs, ATM and ATR. Prog Biophys Mol Biol. 147:4–16. [DOI] [PubMed] [Google Scholar]
- Baretic D, Pollard HK, Fisher DI, Johnson CM, Santhanam B, Truman CM, Kouba T, Fersht AR, Phillips C, Williams RL. 2017. Structures of closed and open conformations of dimeric human ATM. Sci Adv. 3(5):e1700933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baretic D, Williams RL. 2014. PIKKs--the solenoid nest where partners and kinases meet. Curr Opin Struct Biol. 29:134–142. [DOI] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, Cortez D. 2016. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol. 18(11):1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Cussiol JR, Das J, Jeong MC, Doerfler L, Schmidt KH, Yu H, Smolka MB. 2015. Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol Cell. 57(6):1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T, Hall MN. 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 402(6762):689–692. [DOI] [PubMed] [Google Scholar]
- Biswas H, Goto G, Wang W, Sung P, Sugimoto K. 2019. Ddc2ATRIP promotes Mec1ATR activation at RPA-ssDNA tracts. PLoS Genet. 15(8):e1008294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP. 2017. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 66(6):801–817. [DOI] [PubMed] [Google Scholar]
- Bonetti D, Clerici M, Anbalagan S, Martina M, Lucchini G, Longhese MP. 2010. Shelterin-like proteins and Yku inhibit nucleolytic processing of Saccharomyces cerevisiae telomeres. PLoS Genet. 6(5):e1000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla CY, Melo JA, Toczyski DP. 2008. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol Cell. 30(3):267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourns BD, Alexander MK, Smith AM, Zakian VA. 1998. Sir proteins, Rif proteins, and Cdc13p bind Saccharomyces telomeres in vivo. Mol Cell Biol. 18(9):5600–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. 2000. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 14(4):397–402. [PMC free article] [PubMed] [Google Scholar]
- Budd ME, Choe W-C, Campbell J. 1995. DNA2 Encodes a DNA Helicase Essential for Replication of Eukaryotic Chromosomes. J Biol Chem. 270(45):26766–26769. [DOI] [PubMed] [Google Scholar]
- Cassani C, Gobbini E, Wang W, Niu H, Clerici M, Sung P, Longhese MP. 2016. Tel1 and Rif2 Regulate MRX Functions in End-Tethering and Repair of DNA Double-Strand Breaks. PLoS Biol. 14(2):e1002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassani C, Vertemara J, Bassani M, Marsella A, Tisi R, Zampella G, Longhese MP. 2019. The ATP-bound conformation of the Mre11-Rad50 complex is essential for Tel1/ATM activation. Nucleic Acids Res. 47(7):3550–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A, Domkin V, Thelander L. 1999. Yeast Sml1, a protein inhibitor of ribonucleotide reductase. J Biol Chem. 274(51):36679–36683. [DOI] [PubMed] [Google Scholar]
- Choi JE, Chung WH. 2020. Functional interplay between the oxidative stress response and DNA damage checkpoint signaling for genome maintenance in aerobic organisms. J Microbiol. 58(2):81–91. [DOI] [PubMed] [Google Scholar]
- Clerici M, Paciotti V, Baldo V, Romano M, Lucchini G, Longhese MP. 2001. Hyperactivation of the yeast DNA damage checkpoint by TEL1 and DDC2 overexpression. EMBO J. 20(22):6485–6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. 2001. ATR and ATRIP: partners in checkpoint signaling. Science. 294(5547):1713–1716. [DOI] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. 2007. The Rad9-Hus1-Rad1 (9–1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 21(12):1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande I, Seeber A, Shimada K, Keusch JJ, Gut H, Gasser SM. 2017. Structural Basis of Mec1-Ddc2-RPA Assembly and Activation on Single-Stranded DNA at Sites of Damage. Mol Cell. 68(2):431–445 e435. [DOI] [PubMed] [Google Scholar]
- Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, Lee JH, Classen S, Guenther G, Russell P, Tainer JA et al. 2014. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 33(5):482–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, Bermejo R, Fiorani S, Haber JE, Foiani M. 2009. Replicon dynamics, dormant origin firing, and terminal fork integrity after double-strand break formation. Cell. 137(2):247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Gautier J. 2006. Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat Struct Mol Biol. 13(5):451–457. [DOI] [PubMed] [Google Scholar]
- Ellison V, Stillman B. 2003. Biochemical Characterization of DNA Damage Checkpoint Complexes: Clamp Loader and Clamp Complexes with Specificity for 5’ Recessed DNA. PLoS Biol. 1:231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann EA, De Bona P, Galletto R. 2015. The wrapping loop and Rap1 C-terminal (RCT) domain of yeast Rap1 modulate access to different DNA binding modes. J Biol Chem. 290(18):11455–11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote KM, Nissink JWM, McGuire T, Turner P, Guichard S, Yates JWT, Lau A, Blades K, Heathcote D, Odedra R et al. 2018. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J Med Chem. 61(22):9889–9907. [DOI] [PubMed] [Google Scholar]
- Friedel AM, Pike BL, Gasser SM. 2009. ATR/Mec1: coordinating fork stability and repair. Curr Opin Cell Biol. 21(2):237–244. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Kwon Y, Sung P, Sugimoto K. 2011. Activation of protein kinase Tel1 through recognition of protein-bound DNA ends. Mol Cell Biol. 31(10):1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya K, Poitelea M, Guo L, Caspari T, Carr AM. 2004. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 18(10):1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Longhese MP, Plevani P, Muzi-Falconi M. 2004. Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. EMBO J. 23:429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant PA, Schieltz D, Pray-Grant MG, Yates JR 3rd, Workman JL. 1998. The ATM-related cofactor Tra1 is a component of the purified SAGA complex. Mol Cell. 2(6):863–867. [DOI] [PubMed] [Google Scholar]
- Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. 1995. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell. 82(5):823–829. [DOI] [PubMed] [Google Scholar]
- Greer DA, Besley BD, Kennedy KB, Davey S. 2003. hRad9 rapidly binds DNA containing double-strand breaks and is required for damage-dependent topoisomerase II beta binding protein 1 focus formation. Cancer Res. 63(16):4829–4835. [PubMed] [Google Scholar]
- Grenon M, Magill CP, Lowndes NF, Jackson SP. 2006. Double-strand breaks trigger MRX- and Mec1-dependent, but Tel1-independent, checkpoint activation. FEMS Yeast Res. 6(5):836–847. [DOI] [PubMed] [Google Scholar]
- Guleria A, Chandna S. 2016. ATM kinase: Much more than a DNA damage responsive protein. DNA Repair (Amst). 39:1–20. [DOI] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. 2010. ATM activation by oxidative stress. Science. 330(6003):517–521. [DOI] [PubMed] [Google Scholar]
- Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Raschle M, Mailand N. 2016. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol. 18(11):1196–1207. [DOI] [PubMed] [Google Scholar]
- Hailemariam S, De Bona P, Galletto R, Hohl M, Petrini JH, Burgers PM. 2019. The telomere-binding protein Rif2 and ATP-bound Rad50 have opposing roles in the activation of yeast Tel1(ATM) kinase. J Biol Chem. 294(49):18846–18852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailemariam S, Kumar S, Burgers PM. 2019. Activation of Tel1(ATM) kinase requires Rad50 ATPase and long nucleosome-free DNA but no DNA ends. J Biol Chem. 294(26):10120–10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CF, Sussel L, Shore D. 1992. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 6(5):801–814. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Fukunaga K, Sugimoto K. 2009. Rif1 and rif2 inhibit localization of tel1 to DNA ends. Mol Cell. 33(3):312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA. 2000. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 101(7):789–800. [DOI] [PubMed] [Google Scholar]
- Itakura E, Sawada I, Matsuura A. 2005. Dimerization of the ATRIP protein through the coiled-coil motif and its implication to the maintenance of stalled replication forks. Mol Biol Cell. 16(12):5551–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansma M, Linke-Winnebeck C, Eustermann S, Lammens K, Kostrewa D, Stakyte K, Litz C, Kessler B, Hopfner KP. 2020. Near-Complete Structure and Model of Tel1ATM from Chaetomium thermophilum Reveals a Robust Autoinhibited ATP State. Structure. 28(1):83–95 e85. [DOI] [PubMed] [Google Scholar]
- Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, Kolinsky M, Williams GJ, Bose P, Lees-Miller SP. 2020. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers (Basel). 12(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. 2009. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 137(7):1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizer H, Connelly CJ, Bettridge K, Viggiani C, Greider CW. 2015. Regulation of Telomere Length Requires a Conserved N-Terminal Domain of Rif2 in Saccharomyces cerevisiae. Genetics. 201(2):573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnitz LM, Zou L. 2015. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 21(21):4780–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Kumagai A, Lee J, Dunphy WG. 2005. Phosphorylation of Chk1 by ATM- and Rad3-related (ATR) in Xenopus egg extracts requires binding of ATRIP to ATR but not the stable DNA-binding or coiled-coil domains of ATRIP. J Biol Chem. 280(46):38355–38364. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. 2006. TopBP1 activates the ATR-ATRIP complex. Cell. 124(5):943–955. [DOI] [PubMed] [Google Scholar]
- Kumar S, Burgers PM. 2013. Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev. 27(3):313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Oberly S, Sanford EJ, Sharma S, Chabes A, Smolka MB. 2018. Separable roles for Mec1/ATR in genome maintenance, DNA replication, and checkpoint signaling. Genes Dev. 32(11–12):822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin MF, Yeo AJ. 2020. Clinical potential of ATM inhibitors. Mutat Res. 821:111695. [DOI] [PubMed] [Google Scholar]
- Lecona E, Fernandez-Capetillo O. 2018. Targeting ATR in cancer. Nat Rev Cancer. 18(9):586–595. [DOI] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG. 2007. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem. 282(38):28036–28044. [DOI] [PubMed] [Google Scholar]
- Lee JH, Mand MR, Deshpande RA, Kinoshita E, Yang SH, Wyman C, Paull TT. 2013. Ataxia telangiectasia-mutated (ATM) kinase activity is regulated by ATP-driven conformational changes in the Mre11/Rad50/Nbs1 (MRN) complex. J Biol Chem 288(18):12840–12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 304(5667):93–96. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 308(5721):551–554. [DOI] [PubMed] [Google Scholar]
- Lee YC, Zhou Q, Chen J, Yuan J. 2016. RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr Biol. 26(24):3257–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lempiainen H, Halazonetis TD. 2009. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 28(20):3067–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, de Lange T. 2003. Rap1 affects the length and heterogeneity of human telomeres. Mol Biol Cell. 14(12):5060–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, Wilson NM, Petracek ME, Berman J. 1989. A yeast telomere binding activity binds to two related telomere sequence motifs and is indistinguishable from RAP1. Curr Genet. 16(4):225–239. [DOI] [PubMed] [Google Scholar]
- Lovejoy CA, Cortez D. 2009. Common mechanisms of PIKK regulation. DNA Repair (Amst). 8(9):1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makiniemi M, Hillukkala T, Tuusa J, Reini K, Vaara M, Huang D, Pospiech H, Majuri I, Westerling T, Makela TP et al. 2001. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J Biol Chem. 276(32):30399–30406. [DOI] [PubMed] [Google Scholar]
- Mallory JC, Petes TD. 2000. Protein kinase activity of Tel1p and Mec1p, two Saccharomyces cerevisiae proteins related to the human ATM protein kinase. Proc Natl Acad Sci U S A. 97(25):13749–13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Clerici M, Lucchini G, Longhese MP. 2007. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 8(4):380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Clerici M, Baldo V, Bonetti D, Lucchini G, Longhese MP. 2012. A balance between Tel1 and Rif2 activities regulates nucleolytic processing and elongation at telomeres. Mol Cell Biol. 32(9):1604–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 316(5828):1160–1166. [DOI] [PubMed] [Google Scholar]
- Mei L, Zhang J, He K, Zhang J. 2019. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: where we stand. J Hematol Oncol. 12(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memisoglu G, Lanz MC, Eapen VV, Jordan JM, Lee K, Smolka MB, Haber JE. 2019. Mec1(ATR) Autophosphorylation and Ddc2(ATRIP) Phosphorylation Regulates DNA Damage Checkpoint Signaling. Cell Rep. 28(4):1090–1102 e1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanovic M, Houghton LM, Menolfi D, Lee JH, Yamamoto K, Li Y, Lee BJ, Xu J, Estes VM, Wang D et al. 2020. The cancer-associated ATM R3008H mutation reveals the link between ATM activation and its exchange. Cancer Res. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miosge LA, Sontani Y, Chuah A, Horikawa K, Russell TA, Mei Y, Wagle MV, Howard DR, Enders A, Tscharke DC et al. 2017. Systems-guided forward genetic screen reveals a critical role of the replication stress response protein ETAA1 in T cell clonal expansion. Proc Natl Acad Sci U S A. 114(26):E5216–E5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockel C, Lammens K, Schele A, Hopfner KP. 2012. ATP driven structural changes of the bacterial Mre11:Rad50 catalytic head complex. Nucleic Acids Res. 40(2):914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordes DA, Glick GG, Zhao R, Cortez D. 2008. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 22(11):1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myler LR, Gallardo IF, Soniat MM, Deshpande RA, Gonzalez XB, Kim Y, Paull TT, Finkelstein IJ. 2017. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol Cell. 67(5):891–898 e894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Matsumoto K, Sugimoto K. 2003. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 17(16):1957–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. 2009. The unstructured C-terminal tail of the 9–1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell. 36(5):743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. 2003. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 33(4):497–501. [DOI] [PubMed] [Google Scholar]
- Oh J, Symington LS. 2018. Role of the Mre11 Complex in Preserving Genome Integrity. Genes (Basel). 9(12):589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciotti V, Clerici M, Scotti M, Lucchini G, Longhese MP. 2001. Characterization of mec1 kinase-deficient mutants and of new hypomorphic mec1 alleles impairing subsets of the DNA damage response pathway. Mol Cell Biol. 21(12):3913–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Karatayeva N, Demin AA, Munashingha PR, Seo YS. 2020. The secondary-structured DNA-binding activity of Dna2 endonuclease/helicase is critical to cell growth under replication stress. FEBS J. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT. 2015. Mechanisms of ATM Activation. Annu Rev Biochem. 84:711–738. [DOI] [PubMed] [Google Scholar]
- Paull TT. 2018. 20 Years of Mre11 Biology: No End in Sight. Mol Cell. 71(3):419–427. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, Nussenzweig A. 2006. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 443(7108):222–225. [DOI] [PubMed] [Google Scholar]
- Puddu F, Granata M, Di Nola L, Balestrini A, Piergiovanni G, Lazzaro F, Giannattasio M, Plevani P, Muzi-Falconi M. 2008. Phosphorylation of the budding yeast 9–1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol Cell Biol. 28(15):4782–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randell JC, Fan A, Chan C, Francis LI, Heller RC, Galani K, Bell SP. 2010. Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol Cell. 40(3):353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Q, Liu M, Tian Y, Wu Z, Hao Y, Song L, Qin Z, Ding C, Wang HW, Wang J et al. 2018. Cryo-EM structure of human ATR-ATRIP complex. Cell Res. 28(2):143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeyre C, Shore D. 2012. Anticheckpoint pathways at telomeres in yeast. Nat Struct Mol Biol. 19(3):307–313. [DOI] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA. 2017. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 18(10):622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Hamperl S, Bocek MJ, Chung M, Bass TE, Cisneros-Soberanis F, Samejima K, Xie L, Paulson JR, Earnshaw WC et al. 2018. An intrinsic S/G2 checkpoint enforced by ATR. Science. 361(6404):806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka M, Wanrooij PH, Darbari VC, Tannous E, Hailemariam S, Bose D, Makarova AV, Burgers PM, Zhang X. 2016. The Dimeric Architecture of Checkpoint Kinases Mec1ATR and Tel1ATM Reveal a Common Structural Organization. J Biol Chem. 291(26):13436–13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS. 2001. The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. J Biol Chem. 276(24):21951–21959. [DOI] [PubMed] [Google Scholar]
- Shi T, Bunker RD, Mattarocci S, Ribeyre C, Faty M, Gut H, Scrima A, Rass U, Rubin SM, Shore D et al. 2013. Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell. 153(6):1340–1353. [DOI] [PubMed] [Google Scholar]
- Smolka MB, Albuquerque CP, Chen SH, Zhou H. 2007. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci U S A. 104(25):10364–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. 2005. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 102(37):13182–13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak YS, Tanaka Y, Endo S, Kamimura Y, Araki H. 2006. A CDK-catalysed regulatory phosphorylation for formation of the DNA replication complex Sld2-Dpb11. EMBO J. 25(9):1987–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannous EA, Yates LA, Zhang X, Burgers PM. 2021. Mechanism of auto-inhibition and activation of Mec1(ATR) checkpoint kinase. Nat Struct Mol Biol. 28(1):50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MT, Arneric M, Sperisen P, Lingner J. 2004. Telomere length homeostasis is achieved via a switch between telomerase- extendible and -nonextendible states. Cell. 117(3):323–335. [DOI] [PubMed] [Google Scholar]
- Thada V, Cortez D. 2019. Common motifs in ETAA1 and TOPBP1 required for ATR kinase activation. J Biol Chem. 294(21):8395–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsabar M, Waterman DP, Aguilar F, Katsnelson L, Eapen VV, Memisoglu G, Haber JE. 2016. Asf1 facilitates dephosphorylation of Rad53 after DNA double-strand break repair. Genes Dev. 30(10):1211–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto Y, Taggart AK, Zakian VA. 2001. The role of the Mre11-Rad50-Xrs2 complex in telomerase- mediated lengthening of Saccharomyces cerevisiae telomeres. Curr Biol. 11(17):1328–1335. [DOI] [PubMed] [Google Scholar]
- Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. 2005. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 438(7066):379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Elledge SJ. 2002. Genetic and physical interactions between DPB11 and DDC1 in the yeast DNA damage response pathway. Genetics. 160(4):1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chu H, Lv M, Zhang Z, Qiu S, Liu H, Shen X, Wang W, Cai G. 2016. Structure of the intact ATM/Tel1 kinase. Nat Commun. 7:11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ran T, Zhang X, Xin J, Zhang Z, Wu T, Wang W, Cai G. 2017. 3.9 Å structure of the yeast Mec1-Ddc2 complex, a homolog of human ATR-ATRIP. Science. 358(6367):1206–1209. [DOI] [PubMed] [Google Scholar]
- Wanrooij PH, Burgers PM. 2015. Yet another job for Dna2: Checkpoint activation. DNA Repair (Amst). 32:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij PH, Tannous E, Kumar S, Navadgi-Patil VM, Burgers PM. 2016. Probing the Mec1ATR Checkpoint Activation Mechanism with Small Peptides. J Biol Chem. 291(1):393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RM, Yates LA, Zhang X. 2020. Structures and regulations of ATM and ATR, master kinases in genome integrity. Curr Opin Struct Biol. 61:98–105. [DOI] [PubMed] [Google Scholar]
- Williams RM, Zhang X. 2020. Roles of ATM and ATR in DNA double strand breaks and replication stress. Prog Biophys Mol Biol. In press. [DOI] [PubMed] [Google Scholar]
- Wotton D, Shore D. 1997. A novel Rap1p-interacting factor, Rif2p, cooperates with Rif1p to regulate telomere length in Saccharomyces cerevisiae. Genes Dev. 11(6):748–760. [DOI] [PubMed] [Google Scholar]
- Wu Q, Liang S, Ochi T, Chirgadze DY, Huiskonen JT, Blundell TL. 2019. Understanding the structure and role of DNA-PK in NHEJ: How X-ray diffraction and cryo-EM contribute in complementary ways. Prog Biophys Mol Biol. 147:26–32. [DOI] [PubMed] [Google Scholar]
- Wu Y, Xiao S, Zhu XD. 2007. MRE11-RAD50-NBS1 and ATM function as co-mediators of TRF1 in telomere length control. Nat Struct Mol Biol. 14(9):832–840. [DOI] [PubMed] [Google Scholar]
- Xiao J, Liu M, Qi Y, Chaban Y, Gao C, Pan B, Tian Y, Yu Z, Li J, Zhang P et al. 2019. Structural insights into the activation of ATM kinase. Cell Res. 29(8):683–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin J, Xu Z, Wang X, Tian Y, Zhang Z, Cai G. 2019. Structural basis of allosteric regulation of Tel1/ATM kinase. Cell Res. 29(8):655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. 2008. The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol Cell Biol. 28(24):7345–7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Izumi N, Kashima I, Ohnishi T, Saari B, Katsuhata Y, Muramatsu R, Morita T, Iwamatsu A, Hachiya T et al. 2009. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 23(9):1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, Dhar A, Pavletich NP. 2017. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature. 552(7685):368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. 2013. mTOR kinase structure, mechanism and regulation. Nature. 497(7448):217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LA, Williams RM, Hailemariam S, Ayala R, Burgers P, Zhang X. 2020. Cryo-EM Structure of Nucleotide-Bound Tel1(ATM) Unravels the Molecular Basis of Inhibition and Structural Rationale for Disease-Associated Mutations. Structure. 28(1):96–104 e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Chahwan C, Bailis J, Hunter T, Russell P. 2005. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 25(13):5363–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R. 1998. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 2(3):329–340. [DOI] [PubMed] [Google Scholar]
- Zheng L, Meng Y, Campbell JL, Shen B. 2020. Multiple roles of DNA2 nuclease/helicase in DNA metabolism, genome stability and human diseases. Nucleic Acids Res. 48(1):16–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZW, Liu C, Li TL, Bruhn C, Krueger A, Min W, Wang ZQ, Carr AM. 2013. An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genet. 9(8):e1003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 300(5625):1542–1548. [DOI] [PubMed] [Google Scholar]