

ABSTRACT
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers because of diagnosis at late stage and inherent/acquired chemoresistance. Recent advances in genomic profiling and biology of this disease have not yet been translated to a relevant improvement in terms of disease management and patient’s survival. However, new possibilities for treatment may emerge from studies on key epigenetic factors. Deregulation of microRNA (miRNA) dependent gene expression and mRNA splicing are epigenetic processes that modulate the protein repertoire at the transcriptional level. These processes affect all aspects of PDAC pathogenesis and have great potential to unravel new therapeutic targets and/or biomarkers. Remarkably, several studies showed that they actually interact with each other in influencing PDAC progression. Some splicing factors directly interact with specific miRNAs and either facilitate or inhibit their expression, such as Rbfox2, which cleaves the well-known oncogenic miRNA miR-21. Conversely, miR-15a-5p and miR-25-3p significantly downregulate the splicing factor hnRNPA1 which acts also as a tumour suppressor gene and is involved in processing of miR-18a, which in turn, is a negative regulator of KRAS expression. Therefore, this review describes the interaction between splicing and miRNA, as well as bioinformatic tools to explore the effect of splicing modulation towards miRNA profiles, in order to exploit this interplay for the development of innovative treatments. Targeting aberrant splicing and deregulated miRNA, alone or in combination, may hopefully provide novel therapeutic approaches to fight the complex biology and the common treatment recalcitrance of PDAC.
KEYWORDS: PDAC, splicing deregulation, miRNA, interaction, splicing modulation
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers worldwide [1,2]. Although its incidence and prevalence are lower than several other cancers, such as lung, head and neck, colorectal and breast cancer, the mortality rate almost matches the incidence rate [3]. Furthermore, early detection is difficult in PDAC because of the lack of accurate biomarkers [3,4]. Current biomarkers still have low sensitivity and specificity and, therefore, are not suitable as screening methods [5]. The problem becomes even more complicated as this type of cancer is hard to treat or to manage. PDAC has a rapid progression and only 20% of newly diagnosed patients are eligible for surgical resection, the most effective treatment option for this disease [1]. In addition, PDAC is highly resistant to any therapy upfront and tends also to rebound rapidly after first response/stabilization [3].
Recent studies provided new insights into the underlying mechanism of PDAC evolution, suggesting that, in addition to the specific mutational load, including the concurrent mutations in KRAS, TP53, p16, and DPC4, and to the tumour and stromal heterogeneity, microRNAs (miRNAs) and splicing deregulation could be major players in directing tumorigenesis and tumour evolution [6–9].
MiRNAs have been studied extensively and there is a wide array of functionally relevant miRNAs that play an important role in PDACs [7]. For instance, miR-21 has been implicated in carcinogenesis and tumour progression in many types of solid cancers, including PDAC [10,11]. It may be a strong biomarker for early detection, but lacks specificity as it is upregulated in many types of solid cancers and other diseases [12]. Another well-known miRNA in PDAC is miR-155 that is important for inflammation and metastatic processes, while miR-121 and miR-21 contribute to chemoresistance [13].
Contrary to miRNA, splicing deregulation is a relatively new concept in cancer progression, especially in solid cancers [8]. The discovery of mutations in genes encoding splicing factors increased the interests in this topic, prompting several recent studies. Splicing deregulation due to mutation of splicing factors is especially important in non-solid cancers, such as leukaemia, but their overexpression is widely observed also in solid cancers [14,15]. Despite a low mutational rate of splicing factor 3B subunit 1 (SF3B1) in mesothelioma, its overexpression is diffusely prevalent and significantly associated with increased malignant characteristics and patients survival [16]. In lung cancer, Serine and Arginine Rich Splicing Factor 2 (SRSF2) has been implicated in patient’s survival and tumour progression while Serine/arginine-rich splicing factor 7 (SRSF7) is highly expressed in chemoresistant colorectal cancer [17].
Although miRNA and splicing deregulation draw extensive interest among cancer scientists, the possibility of their interaction emerged only recently. Rodriguez-Aguayo et al. [18] showed that miR-15a-5p and miR-25-3p significantly downregulate tumour suppressor gene splicing factor hnRNPA1 which is a key player in the processing of miR-18a, the negative regulator of KRAS expression. On the other hand, splicing factors themselves could affect miRNA expression as shown by Chen et al., who reported a inhibition of miR-21 by Rbfox-2 [18]. These evidences indicate that there could be a relationship between miRNA and gene splicing, which might influence different oncogenic processes and provide new crucial concepts to be exploited towards more efficient cancer treatments. Therefore, this review discusses this intricate relationship, with a focus on the basic concept of miRNA and splicing deregulation, and on how miRNA and splicing factors affect each other. Additionally, this review concludes with consideration on how to exploit this relationship for future strategies in PDAC management and treatment.
Biology of miRNA and its relevance in PDAC
By definition, a miRNA is a non-coding RNA which typically consists of 19–24 nucleotides with a pivotal role in post-transcriptional regulation [7]. MiRNAs were first discovered in Caenorhabditis elegans and thousands more have been identified in all kinds of organisms [19]. In humans, around 2500 different miRNAs have been identified along with their sequence, transcript annotation, and their location within the genome [20]. MiRNAs biogenesis begins with their transcription by RNA polymerase II which generates long precursors known as primary miRNA transcripts (pri-miRNAs) (Figure S1). This transcript has a wide variability in their length, but it typically ranges between 100 and 1000 base pairs. The pri-miRNAs are then processed by Drosha-DGCR8 ribonuclease complex in the nucleus, producing 70–100 nucleotides long intermediate pre-miRNAs with hairpin shape. Then, this intermediate will be transported into the cytoplasm by Exportin-5 and RanGTP6 where it will further be processed by the endoribonuclease Dicer (also known as endoribonuclease RNase III). Dicer cleaves the terminal loop and produces mature double stranded 19–24 nts RNA. One strand of this mature miRNA will be degraded, while the other is incorporated into Argonaute heteromultimer protein to form highly specialized RNA Induced Silencing Complex (RISC) [21]. The seed sequence in miRNA leads to the complex towards target mRNA by means of RNA-RNA base pairing. The target mRNAs can either be degraded or translationally repressed, which depends on whether the seed sequence matches the target sequence within the mRNA. Complete base pairing between the seed sequences and target mRNAs usually leads to degradation of mRNA while incomplete pairing results in translational suppression [22]. However, this phenomenon also underlies the reason why miRNA is so versatile. It has been shown that a single miRNA can indeed control mRNAs from several genes, while a single mRNA can be targeted by several different miRNAs [23]. In addition, it is estimated that 60% of all genes are controlled by miRNAs which further underscores their importance in the control of gene expression [24]. MiRNAs are also involved in important cellular processes, such as cell proliferation, metabolism, differentiation, apoptosis, and cell signalling[25].
Extensive studies on miRNA gave insight in its important role in many types of cancer, including PDAC. Cancer cells are known to have aberrant miRNA expression: where tumour suppressors’ miRNAs are often downregulated, miRNAs promoting carcinogenesis or tumour progression is usually over-expressed [25]. This deregulation often leads to aberrant cellular processes, such as uncontrolled mitosis, apoptosis, drug resistance, invasion, metastasis and angiogenesis [7,13,14,25].
The first evidence of miRNA dysregulation in PDAC was reported by Poy et al. [26] through a profiling study using mouse pancreas. Follow-up studies using different types of samples confirmed the initial finding that PDAC has a specific miRNA expression profile [17,27,28]. In particular, a study comparing PDAC tissue with adjacent normal pancreatic tissue reported a total of 158 miRNAs differentially expressed [29]. Fifty-one miRNAs were upregulated including miR-196, miR-200a, miR-21, and miR-27a, while 107 miRNAs were downregulated, with miR-96, miR-200, and miR-217 being the most significant [29].
In Table 1 we report an overview of the clinical evidence on miRNA deregulation in PDAC as well as the most interesting preclinical findings on candidate miRNAs emerging from these studies. Schultz and colleagues [28] reported that 43 miRNAs were upregulated while 41 were downregulated when comparing paraffin-embedded PDAC tissue samples with normal ones. The expression of key miRNAs was also different between resectable and non-resectable PDAC patients as reported by Calatayud et al. [30]. Around 22 miRNAs were differentially expressed with miR-64, miR-136, miR-196, miR-492, and miR-622 being the most significant. A separate study by Papaconstantinou et al. showed a different but also some consistent results [31]; miR-21, miR-155, miR-205, miR-221, and miR-222 were consistently overexpressed while miR-31, miR-122, miR-146, and miR-375 were downregulated in PDAC samples. Preclinical and functional analysis showed that miR-21 and miR-155 are the only two miRNAs that were consistently overexpressed and linked to cancer progression [17]. However, several profiling studies showed consistent overexpression of miR-21, miR-155, and miR-221, while miR-34 and miR-145 were downregulated [32]. Remarkably, miR-21 and miR-155 obtained from pancreatic tissue could differentiate malignant from benign lesions with high accuracy [33] and have both been proven to be able to differentiate between pancreatic intra-epithelial neoplasia (PanIN) with normal pancreatic [34,35]. Clinically, miRNAs have been assessed to differentiate benign and malignant lesions, determining the stage of PDAC, as a biomarker for metastasis, and to predict the therapeutic outcome, as illustrated in Figure 1. The potential role of specific miRNA in early diagnostics is particularly important in PDAC since screening modalities are very limited and the disease tends to be diagnosed in advanced stage which has a high risk of metastasis and low therapeutic response [3]. Furthermore, miR-155 is increasingly expressed as early as PanIN-1 while miR-21 is beginning to be abundant in PanIN-2 and −3, suggesting that miR-21 is more suitable for advanced disease marker. Another microRNA that has increased expression in advanced PanIN (PanIN-3) is miR-196b while the expressions of miR-133, miR-185, miR-200 c, and miR-34 c are higher in low-grade neoplasia [36].
Table 1.
MiRNAs aberrantly expressed in PDAC samples
| miRNA | Expression level (N) |
Tissue type | N Samples | Method | Reference | |||
|---|---|---|---|---|---|---|---|---|
| Normal Tissue | PDAC | |||||||
| Low | High | FFPE | Pancreatic cancer (n = 165); Normal Pancreas (n = 35) | RT-PCR | [30] | |||
| Low | High | Fresh Tissue | Pancreatic Cancer (n = 88); Normal Pancreas (n = 98) | qRT-PCR | [31] | |||
| miR-21 | Low | High | Biopsy | Metastatic (n = 31); Non-metastatic (n = 50) | q-PCR | [43] | ||
| Low | High | FFPE | Adjuvant therapy (n = 52); Non-adjuvant therapy (n = 27) | qRT-PCR | [45] | |||
| Low | High | FFPE | Normal pancreas (n = 12); Pancreatitis (n = 45); PDAC (n = 80) | In-situ Hybridization | [171] | |||
| Low | High | Plasma | PDAC (n = 32); Normal healthy (n = 30) | qRT-PCR | [9] | |||
| Low | High | Fresh Tissue | Pancreatic cancer (n = 88); Normal pancreas (n = 98) | qRT-PCR | [31] | |||
| miR-155 | Low | High | FFPE | Pancreatic lesions (n = 55) | qRT-PCR | [33] | ||
| Low | High | Plasma | Pancreatic cancer (n = 40); Normal pancreas (n = 25) | q-PCR | [172] | |||
| Low (N = 80) | High (N = 80) | FFPE | Pancreatic cancer (n = 80); Normal pancreas (n = 80) | In-situ Hybridization | [173] | |||
| Low (N = 98) | High (N = 88) | Fresh Tissue | Pancreatic cancer (n = 88); Normal pancreas (n = 98) | qRT-PCR | [30] | |||
| miR-205 | Low (N = 17) | High (N = 34) | FFPE | Pancreatic cancer (n = 34); Normal pancreas (n = 17) | qRT-PCR | [173] | ||
| Low (N = 17) | High (N = 47) | Serum | Pancreatic cancer (n = 47); Normal pancreas (n = 17) | qRT-PCR | [173] | |||
| miR-205 | Low (N = 5) | High (N = 5) | Fresh Tissue | Pancreatic cancer (n = 5); Normal pancreas (n = 5) | MiRNA Microarray |
[174] | ||
| miR-196b, miR-217, miR-411, miR-198 | High (N = 28) | Low (N = 170) | FFPE | Pancreatic cancer (n = 170); Normal pancreas (n = 28) | RT-PCR | [28] | ||
| miR-210, miR-222 | Low (N = 98) | High (N = 88) | Fresh Tissue | Pancreatic cancer (n = 88); Normal pancreas (n = 98) | qRT-PCR | [31] | ||
| miR-375 | High (N = 35) | Low (N = 165) | FFPE | Pancreatic cancer (n = 165); Normal pancreas (n = 35) | RT-PCR | [30] | ||
| miR-377 | High (N = 30) | Low (N = 30) | Snap-frozen sample | Pancreatic cancer (n = 30); Normal adjacent tissue (n = 30) | qRT-PCR | [175,176] | ||
| miR-127 | High (N = 42) | Low (N = 42) | Snap-frozen sample | Pancreatic cancer (n = 42); Normal adjacent tissue (n = 42) | qRT-PCR | [158] | ||
| miR-181d | Low (N = 37) | High (N = 37) | Snap-frozen sample | Pancreatic cancer (n = 37); Normal adjacent tissue (n = 37) | qRT-PCR | [177] | ||
| miR-107 | Low (N = 80) | High (N = 100) | Plasma | Pancreatic cancer (n = 100); Normal pancreas (n = 80) | qRT-PCR | [178] | ||
| miR-1290 | Low (N = 267) | High (N = 167) | Plasma | Pancreatic cancer (n = 167); Normal pancreas (n = 267) | ddPCR | [179] | ||
Figure 1.
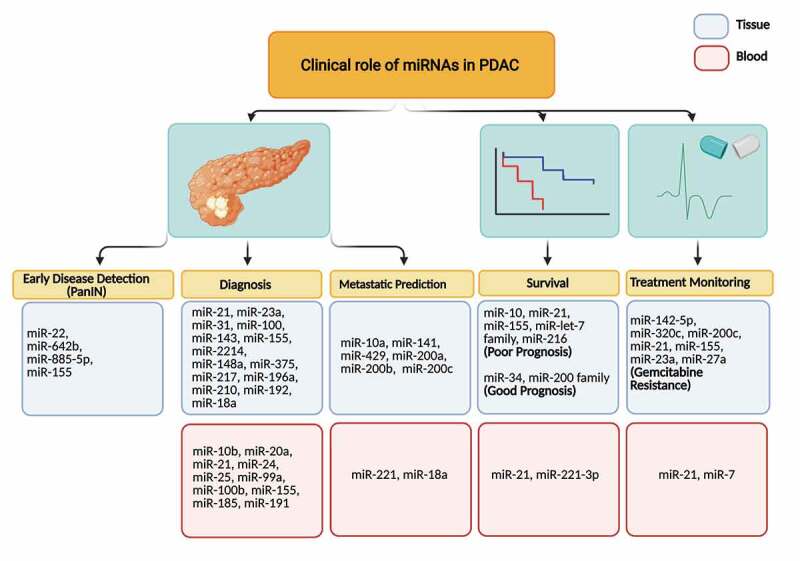
Clinical role of miRNAs in early PDAC detection, diagnosis, metastatic prediction, survival and treatment monitoring. The scheme shows different miRNAs, tissue and blood-derived, which could serve as biomarkers for discriminating the different stages of the disease, as well as for early diagnosis and metastasis prediction. Furthermore, some miRNAs could be associated with prognosis and monitoring of PDAC patients.
The expression levels of miR-21 and miR-155 were also up-regulated also in invasive intraductal papillary mucinous neoplasms (IPMNs) of the pancreas, compared to non-invasive IPMNs, as well as in non-invasive IPMNs compared with normal tissues. Conversely, miR-101 levels were significantly higher in non-invasive IPMNs and normal tissues compared with invasive IPMNs. Furthermore, miR-21 emerged as an independent prognostic biomarker in invasive IPMNs [37].
The information on circulating miRNAs in preneoplastic lesions is limited while circulating miR-10b, miR-20a, miR-21, miR-24, miR-25, miR-99a, miR-100b, miR-155, miR-185, and miR-191 showed a high accuracy in differentiating PDAC with pancreatitis and normal pancreas. Regarding metastasis, both miR-21 and miR-155 have been proven to actively play important role in inducing cell migration and metastasis [38]. However, other microRNA, such as miR-10b, miR-200b, and miR-200 c, miR-218, miR-194, and miR-429 are also emerging biomarkers for metastasis in PDAC [39]. In blood, only miR-221 and miR-18a had been evaluated to be significantly associated with metastasis [40,41]. Unsurprisingly, all of the aforementioned miRNAs are also associated with patient’s prognosis. The tissue expression of miR-10, miR-21, miR-155, miR-let-7 family, and miR-216 are known to predict an unfavourable prognosis, while expression of miR-34 and miR-200 family correlated with a better prognosis. For circulating miRNAs, only miR-21 and miR-221-3p have been consistently proven as indicators of poor prognosis. Despite the urgency in detecting metastasis in pancreatic cancer, there are only limited number of studies regarding the predictive value of microRNA in metastasis which limit their clinical validation and application.
MiR-21 in combination with miR-23a and miR-27a was also associated with more malignant PDAC phenotype and shorter overall survival after tumour resection [42]. In addition, expression of miR-21 determines PDAC response against gemcitabine with a lower expression correlating with better treatment outcome [43–45]. However, tumoural miR-21 overexpression emerged in a pooled meta-analysis assessing miRNAs as prognostic biomarkers in PDAC, independent of other clinicopathologic factors, including adjuvant chemotherapy use [44].
Differential expression of miRNA is not only observed in tissue samples but also in blood. For instance, miR-18a, miR-21, miR-22, miR-24, miR-25, miR-27a, miR-155, miR-185, miR-191, miR-196a, miR-642b and miR-885-5p were significantly upregulated in PDAC patients’ blood plasma [17,41]. Most importantly, blood-based miRNA profiling not only helps to differentiate PDAC patients from healthy individuals but also from other conditions that usually are considered as differential diagnosis such as acute or chronic pancreatitis and benign pancreatic tumours[32]. Another recent study showed that miR-486-5p and miR-938 could differentiate patients with PDAC from those who were healthy or had pancreatitis [46]. Additionally, circulating miRNAs can also be used as therapeutic biomarker. For example, downregulation of miR-181a-5p after FOLFIRINOX therapy correlates with better survival in PDAC but not in those who were treated with nab-paclitaxel and gemcitabine [47].
MiRNA dysregulation drives tumorigenesis through a close link with cellular signalling and metabolism. Several studies demonstrated that miR-21 enhanced PI3K/AKT and MAPK/ERK signalling that promote cell proliferation [48–50]. MiR-21 also suppresses the expression of phosphatase and tensin homolog (PTEN) and programmed cell death protein 4 (PDCD4) which facilitate cellular invasion induced by TGF-β signalling [51,52]. MiR-21 is also known to activate pancreatic stellate cells and cancer associated fibroblasts (CAF) to actively produce extracellular matrix proteins which contribute to its dense stroma [53,54]. On the other hand, miR-155 suppresses suppressor of cytokine signalling 1 (SOCS1) and MLH1 expression within cancer cells and enhance cancer invasion [55,56]. MiR-155 knock down is known to reduce membrane-type 1 matrix metalloproteinase (MT1-MMP), EGFR, and K-Ras expression in PDAC cell lines which led to lower proliferation rates and colony formation [57]. The functionality of other miRNAs has also been studied but seems less clear compared to miR-21 and miR-155.
Splicing factors and alternative splicing in PDAC
During malignant transformation, cancer cells experience aberrant splicing processes which result from mutations at the splice sites, mutations of splicing factors, and/or over/under expression of certain splicing factors [14]. Splicing deregulation could suppress protein expression by directing the inappropriately spliced mRNAs towards non-sense mediated decay or producing more active splice variants of oncogenic proteins [8,9,14]. The clinical application and implication of this process have also been studied both as therapeutic targets and biomarkers [8,15,58] and only recently received attention in PDAC.
Normal splicing is a post-transcriptional process where the introns are removed from primary transcripts, leaving only exons in the final transcript [14] (Figure 2a). In alternative splicing, exons can also be removed and different transcripts can be produced from a single gene [8]. RNA splicing occurs in the nucleus and is facilitated by splicing factors (SFs) which will assemble themselves in a sequential manner during the splicing process. The typical process is initiated by binding of a small nuclear ribonucleoprotein (snRNPs) to the primary transcript. Initially, U1 snRNP binds to the 5’ splice site (5’SS) while U2 snRNP binds to a branch point at the other end of the intron. U1 and U2 then attract more snRNPs (U5, U4/U6) which then form a complete spliceosome and bend the intronic section, forming a lariat-like structure in which the 5’ side of the intron is ligated to the branch point. Then, U4 is removed while the 5’SS site is hydrolysed [15]. In this process, the two extremities of the exons are held together by the spliceosome complex. Consecutively, the 3’SS is cut and the two exons are ligated, forming the final transcript that will be transported to the cytoplasm for translation [8,15,58].
Figure 2.

The mechanism of splicing mediated by splicing factors. The splicing is initiated by binding of U1 at 5’SS and U2 at 3’SS, bending the intron segment. Both SFs (U1 and U2) then recruit another SF which induced loop formation, cleaved the intron segment and ligated the exons [14,15]. Splicing is regulated by splicing factors which bind the primary transcript at several regulatory sites. Several essential regulatory sites and splicing factors are presented in the right part. SRSF2 is particularly important in initiating splicing by facilitating U1 and U2 binding to the primary transcript while SF3B1 mediates U2 binding to BPS. After U1 and U4 detached from spliceosome, only SRSF2 and SF3B1 remain in the complex while the other SFs detached [59]. ESE: Exonic splicing enhancer; ESS: Exonic splicing suppressor; ISE and ISS: Intronic splicing enhancer/suppressor; 5’SS and 3’SS: 5’ or 3’ splice site; BPS: Branch point site; Py-tract: Polypyrimidine-tract.
Splicing is regulated by a wide array of splicing factors which bind to specific sites in primary transcripts [15,59]. The binding sites of those factors can be located in an exon or intron and can induce or repress the splicing process. The most important splicing factors and their binding sites are presented in Figure 2b. Splicing factors act early in the splicing process, facilitating snRNPs binding to primary transcripts [59]. Typically, SRSF2 binds the exonic splicing enhancer in exons flanking the intron and facilitate U1 and U2 binding. It connects to U1 by 70 K linker protein while its interaction with U2 is much more complex. It interacts with the U2 Small Nuclear RNA Auxiliary Factors U2AF1 and U2AF2, as well as with Zinc Finger CCCH-Type, RNA Binding Motif and Serine/Arginine Rich 2 (ZRSR2) and RNA-binding motif 10 (RBM10) in facilitating U2 binding. Additionally, SF3B1 facilitates U2 binding by interacting with a branch point site [15]. After U1 and U4 dissociate from spliceosome, all of the splicing factors are also dissociated except SF3B1 which firmly binds to U2 and SRSF262.
RNA splicing and alternative splicing are crucial steps in protein expression and the isoforms of the proteins that are expressed by a certain gene are determined by these processes [8]. In cancer, these processes can be altered and this alteration can drive carcinogenesis [60]. In fact, in many types of cancer, splicing factors are either mutated or overexpressed, which strongly indicates an aberrant splicing process in cancer [8,15].
Mutations in splicing factors have been identified in several types of cancer as the driving force of carcinogenesis, most notably in haematologic cancers. SF mutations are detected in 78% of refractory anaemia with ringed sideroblasts and 60% of chronic myelomonocytic leukaemia (CMML) while it only happens in less than 5% in pancreatic, lung, breast, and head and neck cancer [15]. However, despite a lower mutational frequency, several SFs are over/under expressed in solid cancer including PDACs [8,14]. For example, SF3B1 and heterogeneous nuclear ribonucleoprotein K (HNRNPK) are consistently overexpressed in PDAC and are linked to an unfavourable prognosis [61–64]. Several important SFs in PDAC and their functions are summarized in Table 2.
Table 2.
Splicing factors in PDAC and their biological and clinical effects in preclinical studies
| Splicing Factors | Biological and Clinical Significance | Reference |
|---|---|---|
| Upregulated in PDAC | [76,80] | |
| Upregulated by Myc | [76,80] | |
| SRSF1 | Promote resistance to gemcitabine | [76] |
| Promote oncogenic splice variant of Bcl-xs, ΔRON and MCL-1s alternative splicing preferring their oncogenic variant | [77–79] | |
| SRSF6 | Increased proliferation and cellular transformation | [72] |
| Prognostic factor for PDAC | [75] | |
| SF3B1 | Overexpression is associated with poor prognosis Mutated in 4% PDAC with mutation associated with better survival |
[15,79 ][65] |
| Important in branch point regulation and alters the splicing process of several oncogenes and tumour suppressor genes | [8,14] | |
| Often downregulated as its control cellular proliferation | [97] | |
| Rbfox2 | Moderate upregulation in cancer tissue increased invasive potential | [98] |
| May specify the mesenchymal tissue- specific splicing profiles both in normal and in cancer tissues | [8] | |
| Superfamily of RNA-binding proteins | [8] | |
| HnRNPs | hnRNPA2B1 and hnRNPA1 altered Bcl-x alternative splicing and facilitate KRAS expression | [8,112] |
| Higher expression associated with poor survival | [8] | |
| HnRNPK | Wide range of effect including alteration in alternative splicing, gene transcription and RNA stability | [8,59,63,106,108,113] |
| Altered PKM expression by favouring PKM2 and induces a Warburg effect | [70,124] | |
| PTBP1 | Upregulation of PTBP1 after chronic exposure to gemcitabine, conferring resistance against the drug | [70] |
Of note, in PDAC, SF3B1 and U2AF1 are the only known SFs with mutations and occur at a very low frequency [15,65]. However, these mutations are interesting because they can be targeted and induce synthetic lethality [66]. Furthermore, it appeared that PDAC relies on the normal form of SFB31, U2AF1, and RBM10 since patients with these mutations tend to have better prognosis compared to the wild types [9,14,15].
A more frequent form of splicing deregulation in PDAC consists in the overexpression of SFs [8]. Several SFs are upregulated in PDAC such as SF3B1, SRSF1, SRSF6, hnRNPK, Heterogeneous Nuclear Ribonucleoprotein A1 (hnRNPA1), and Polypyrimidine Tract Binding Protein 1 (PTBP1), while Rbfox2 tends to be downregulated . These splicing factors are thought to mediate many of PDACs unique characteristics, such as dense stromal, low immunogenicity, immune avoidance, as well as early metastasis, and invasion [9,64,65]. The pivotal role of splicing deregulation in PDAC was described by Wang et al. [8,58,59,61,64–66] who compared alternative splicing in PDAC to normal pancreatic tissue through Affymetrix exon array. Alternative splicing tends to occur in genes encoding extracellular matrix (ECM), ECM-receptor interaction, and focal adhesion protein. In addition, pyruvate kinase and acyl-CoA synthetase long-chain family member 5 (ACSL5) were also present, which suggests that alternative splicing may have an impact on tumour metabolism.
Splicing deregulation drives pancreatic carcinogenesis by shifting the expression of pivotal oncogene and tumour suppressor proteins [5,8,14]. A clear example is the shifting of RON isoform expression of the tyrosine kinase receptor recepteur d’origine nantais (RON) [67,68]. Normally, RON has a low expression in normal pancreatic epithelial cells, but its expression increases gradually from low to high grade pancreatic intra-epithelial neoplasia [68]. In PDAC, it is expressed in 69–96% of cases [68]. However, it is not only its higher expression that makes RON so important in PDAC; RON has indeed also several splicing alterations which lack exon 10, 11, or have 5 + 6 exon skipping [67]. These isoforms are constitutively active and, therefore, have more oncogenic potential compared to native isoform [67].
Another example is Pyruvate Kinase M2 (PKM2) whose expression has been observed in almost all types of tumours [69]. Normally, pancreatic epithelial cells express PKM1 instead of embryonic PKM2. This shift is mediated by PTBP1 which is also overexpressed in PDAC [70]. PTBP1 associates directly to intron 8 of PKM mRNA and induces alternative splicing. Therefore, the higher expression of PKM2 facilitates an oncogenic glycolytic metabolism and increases cancer cell resistance towards genotoxic drugs. This effect was confirmed by a knock-down study where PDAC cell lines with suppressed expression of PTBP1 or PKM2 are much more sensitive to Gemcitabine.
Deregulated splicing factors in PDAC and their interaction with miRNA expression
Several important splicing factor aberrations have been identified in PDACs and, most recently, their association with miRNA expression has been described in different cancer types. Although many known splicing factors are deregulated in cancer, PDAC is relatively unexplored and tends to be limited to SF3B1. Overall, there are four splicing factors which have been studied in more detail and which will be discussed in detail.
SRSF6
SRSF6 is one of the most important splicing factors in PDAC and is often upregulated but not mutated [8,71]. SRSF6 is classified as an oncogenic splicing factor since it enhances cellular proliferation. Jensen et al. [72] reported that SRSF6 overexpression induces excessive keratinocyte hyperplasia in sensitized skin. Cohen-Eliav et al. [73] reported that SRSF6 overexpression increased proliferation rate of immortalized lung epithelial cells, transforming these into malignant cells. In addition, the human protein atlas considers SRSF6 as a potential prognostic marker in renal cancer, liver cancer, and PDAC [74]. Interestingly, and in contrast with the other two above-mentioned cancers, lower SRSF6 expression is correlated with poor prognosis in PDAC.
Keeping with these findings, Li et al. [75] showed that miR-193a-5p downregulates SRSF6, increasing the metastatic potential of PDAC cell lines. Apparently, SRSF6 downregulation was beneficial for cancer cells because it enhanced the invasive properties through the alteration of oxoglutarate dehydrogenase-like and ECM1 protein by alternative splicing. Thus, SRSF6 has a dual, apparently contradictive function since high expression of SRSF6 induces PDAC while a downregulation promotes tumour invasiveness. SRSF6 downregulation probably occurs late in the PDAC evolution, whereas at an early stage of carcinogenesis SRSF6 upregulation is preferred due to its beneficial effect on promoting cellular proliferation and survival. However, further studies in primary cells as well as in patient samples are needed to confirm this hypothesis.
SRSF1
SRSF1 is a well-characterized SR protein in cancer and one of SR proteins that is overexpressed in different cancers, including PDAC [8,59,71]. SRSF1 is a versatile protein, promoting carcinogenesis through several important mechanisms including increased proliferation rate and apoptosis resistance [76]. Furthermore, SRSF1 is a known splicing factor that influences the expression of Bcl-x, RON and MCL-1 isoform expression, changing their anti-apoptotic to pro-mitotic variants in cancer [77–79]. High expression of SRSF1 is induced by the Myc oncogene which is commonly upregulated in many cancers including PDAC [80].
Interestingly, besides its role in processing mRNA splicing, SRSF1 facilitates miRNA processing. SRSF1 is indeed involved in the final cleaving process mediated by Drosha, serving as an auxiliary factor [81]. The miR-7 family depends on SRSF1 for its maturation. However, miR-7 itself would suppress the expression of SRSF1, forming a negative feedback loop. Other miRNAs that depend on SRSF1 include miR-221, miR-222 and miR-17-9286[82]. MiR-221/222 contribute to the progression of PDAC by increasing the expression of MMP-2 and MMP-9, increasing stromal remodelling and the invasive properties of the cancer cells [83]. For miR-17-92 which consists of four members, namely miR-17, miR-18, miR-19, and miR-92, an oncogenic effect was shown [84–87]. In PDAC, miR-19 actually promoted invadopodia and increased the invasiveness [88]. Therefore, it is important to further investigate the interaction between SRSF1 with miR-7, miR-221/222, and miR-17-92 to better understand the feedback loops that exist among them and also to assess why their overexpression favour carcinogenesis instead of tumour suppression.
SF3B1
SF3B1 is a well-known protein with a key role in PDAC [8,14,15]. It has the highest mutation rate among splicing factors and is also often overexpressed in PDAC [15]. Furthermore, this is one of the only three splicing factors that can be inhibited by small-molecule inhibitors so far, making its therapeutic potential higher than other SFs [89].
However, despite the wealth of information regarding the biological role of SF3B1 and its modulation, its relationship with miRNA is poorly understood. Their association has only been studied by Aslan et al. [90] in myelodisplastic syndrome in which they reported that the SF3B1 mutation was associated with global downregulation of tumour suppressor miRNAs from the let-7 family, especially miR-103a and miR-423. However, the mechanism of downregulation was not evaluated and there is still the possibility that this downregulation is not directly related to a SF3B1 mutation but might relate to other signalling aberrations.
Pianigiani et al. [91] demonstrated that there is a relationship between SF3B1 and splice site overlapping miRNAs (SO-miRNA). The precursors of these miRNAs are generated on the intron-exon junctions, from which the name ‘splice-site’ belongs. SF3B1 knockdown did not affect SO-miRNAs in HeLa and HaCAT cells, but increased the level of 52 SO-miRNA including miR-636, miR-6510-5p, miR-3614-3p, miR-3655, miR-3656, miR-4260, miR-5187-3p, miR-7109-5p, and miR-8069. Some of these miRNAs are classified as tumour suppressors [92–95]. However, these in-vitro results cannot be generalized to PDAC and gene silencing using siRNA is different than protein inhibition by a small molecule because there might be a different active site involved in miRNA processing than the inhibited site. Nevertheless, this finding suggests that SF3B1 modulation could have an additional beneficial effect by enhancing tumour suppressor miRNA expression. In addition, when these miRNAs are secreted to the extracellular compartment, they could also serve as biomarker for therapeutic monitoring.
Rbfox2
The RNA-binding Fox (Rbfox) proteins (Rbfox1, Rbfox2 and Rbfox3) constitute an important class of regulators of alternative splicing, and Rbfox2 (RBM9) can influence small and non-coding RNA in PDAC [8,59,96]. This RNA-binding protein is highly conserved in mammals [97], and it is different from Rbfox1 and Rbfox3, whose expression is limited to neuron and muscle cells. Rbfox2 is indeed widely expressed, especially in stem cells, haematopoietic stem cells, and embryos, where it regulates cellular proliferation [8]. In PDAC, Rbfox2 is downregulated, similar to other types of cancer [96]. These findings seem controversial because Rbfox2 is essential for cancer cell invasion, and the level of Rbfox2 increased moderately after the induction of EMT [98]. However, the same study showed that after the initial induction the levels of Rbfox2 were decreased. Notably, Rbfox2 is also subject to regulation by other proteins, and this might also momentary increase its expression [99]. Moreover, this mechanism might enable cancer cells to exploit the EMT promoting ability while evading excessive tumour suppressing effect by Rbfox2.
Rbfox2 is known to upregulate tumour suppressor miRNA, such as miR-20b and miR-107 while cleaving the oncomiR-21 [18–100]. The regulation of miRNA expression by Rbfox2 is mediated by direct binding to cognate sequences in miRNA or indirectly affects miRNA expression by altering Dicer expression. Of note, mutations in the DICER gene as well as in other components of the miRNA biogenesis pathway are not commonly detected in PDAC, and miRNA upregulation is more common than downregulation [100–102]. Additionally, several studies showed that miRNAs are broadly required for the development and maintenance of pancreatic cell lineages and play a role in carcinogenesis [103–105]. These findings suggest that miRNAs play a pivotal role in pancreatic tumorigenesis, and that loss of function mutations in the miRNA processing machinery are selected against during tumour evolution, but the impact of Dicer in later stages of pancreatic tumorigenesis or progression remains limited.
HnRNPs
Heterogeneous nuclear ribonucleoproteins (HnRNPs) are essential members of the RNA-binding proteins (RBPs) that act as regulators of alternative splicing, particularly, in linking the primary transcript with splicing machinery [106]. Several of its family members have been studied in relation to their role in carcinogenesis [106–108]. In PDAC, HnRNPA2B1 and HnRNPA1 are known for their role in tumour progression by shifting the Bcl-x isoform expression and facilitating KRAS expression, respectively [18,109]. In addition, HnRNPs are prognostic factors in PDAC with a higher expression associated with significantly shorter survival [108].
While there is no direct evidence of HnRNP and miRNA interaction in PDAC, their interaction has been identified in ovarian cancer. Aguayo et al. [18] reported that HnRNPA1 suppresses miR-18a expression in docetaxel-resistant ovarian cancer cell lines. MiR-18a normally suppresses KRAS expression, but its downregulation enhanced KRAS expression and facilitated resistance to docetaxel [110,111]. MiR-18a has also been investigated in PDAC and elicited the same effect towards KRAS [112]. HnRNPA1 expression was also suppressed by miR-15a-5p and miR-25-3p [18]. These two miRNAs are also known as tumour suppressor miRNAs in PDAC [7]. Therefore, the same molecular mechanism might exist in PDAC, and inhibiting or blocking HnRNPs could be explored as a new way to fight chemoresistance in this disease.
Another member of HnRNPs that was recently investigated regarding its role in PDAC is HnRNPK [6,8,14,106]. Much of the biology of HnRNPKs is still under investigation because these proteins are not only involved in RNA splicing but also in DNA transcription and RNA stability [113]. They are also responsible for the downregulation of some tumour suppressor genes in PDAC [63,64]. Remarkably, HnRNPKs interact with miR-223, an oncomiR that enhances cell proliferation and migration [63]. These effects have been attributed to downregulation to miR-223 targets FBXW7 and PDS5B, two tumour suppressor proteins which inhibit cellular migration and induce apoptosis [63,114]. A similar finding on the importance of miR-223 was also found in pancreatic cancer cells when using the naturally occurring isoflavonic phytoestrogen genistein that inhibited miR-223 expression which in turn enhanced FBXW7 expression [115]. These effects resulted in inhibition of cell growth and induction of apoptosis.
PTBP1
PTBP1 has been investigated for its role in PDAC metabolism [70,108]. The expression of PTBP1 was increased in two Gemcitabine resistant PDAC cell lines (PANC-1 and Pt45P1) where it modulated alternative splicing alteration of PKM, resulting in overexpression of the cancer-related PKM2 isoform, whose high expression also correlated with worse prognosis in PDAC patients [70]. PTBP1 is also considered a prognostic factor and a potential therapeutic target due to its role in enhancing PDAC metabolism [108].
The only proven miRNA that directly interacts with PTBP1 in PDAC is miR-124, which directly downregulates PTBP1 mRNA and shifts PKM isoform expression from PKM2 to PKM1. The importance of miR-124 and PTBP1 interaction was shown by ectopic expression of miR-124 or administration of PTBP1 siRNA which increased sensitivity to gemcitabine and relieved autophagy in gemcitabine resistant PDAC cell lines [116]. However, in PDAC, miR-124 is mostly downregulated which facilitates increased PTBP1 expression, favouring Warburg effect [117,118]. In neural differentiation, Yeom et al. [119] observed that PTBP1 could repress miR-124 maturation by directly binding to pri-miR-124 and blocked transcript cleavage by DROSHA. Therefore, we can assume that a low expression of miR-124 could also result from increased expression of PTBP1 and this potential feedback loop adds to the complexity of splicing factors-miRNA interaction in cancer.
Another miRNA known to interact with PTBP1 is miR-133b [119]. Although there are no data regarding their interaction in PDAC, miR-133b is downregulated in PDAC and has been considered as a tumour suppressor miRNA based on findings in other cancer types [120–123]. In colorectal cancer, miR-133b silenced PTBP1 expression and inhibited the Warburg effect by promoting the expression of the PKM1 isoform [124]. Due to its low expression in PDAC, miR-133b could also exert a similar effect in PDAC. Despite the limited direct evidence, there is a strong indication of interaction between splicing deregulation and miRNA in PDAC. A summary of relevant splicing factors and the miRNAs that interact with each other as well as their main biological effects is presented in Table 3. Remarkably, further studies exploring this field of research are now extremely timely since splicing inhibitors are becoming available as novel anticancer drugs and could offer a new therapeutic strategy for PDAC.
Table 3.
Relevant splicing factors and miRNAs affecting key aggressive biological features of PDAC in preclinical studies
| Main Effect | Splicing Factor | Associated miRNA | Interaction | Biological Impact | Reference |
|---|---|---|---|---|---|
| PROLIFERATION | |||||
| SF3B1 | miR-636, miR-6510-5p, miR-3614-3p, miR-3655, miR-3656, miR-4260, miR-5187-3p, miR-7109-5p, miR-8069, miR-155-3p, miR-148a-3p, miR-98-5p, and miR-21-3p | Upregulation miR-636, miR-6510-5p, miR-3614-3p, miR-3655, miR-3656, miR-4260, miR-5187-3p, miR-7109-5p, and miR-8069 Downregulation miR-155-3p, miR-148a-3p, miR-98-5p, and miR-21-3p |
Decreased cellular proliferation Enhanced keratinocyte differentiation |
[43,91] | |
| HnRNPs | miR-18a, miR-15a-5p, miR-25-3p | Downregulation of miR-18a by HnRNPA1 Suppression of HnRNPA1 by miR-15a-5p and miR-25-3p |
Increased KRAS activation resulted from miR-18a downregulation | [18] | |
| miR-223 | Increased expression of miR-223 by HnRNPK | Increased cancer cell proliferation by FBXW7 and PDS5B suppression | [63] | ||
| EPITHELIAL-TO-MESENCHYMAL TRANSITION AND METASTASIS | |||||
| SRSF1 | miR-7 family, miR-17, miR-18 | Facilitate miR-7 family biosynthesis Increased miR-221/222 expression |
Increased invasion and metastasis through MMP-2 and −9 upregulation | [81,83] | |
| SRSF1 | miR-19, miR-92 | Facilitate miR-17-92 family biosynthesis | Increased invasion and metastasis through MMP-2 and −9 upregulation | [82,83] | |
| SRSF6 | miR-193a-5p | SRSF6 downregulation by miR-193a-5p | Facilitate metastasis by alteration in oxoglutarate dehydrogenase-like (OGDHL) and extracellular matrix protein 1 (ECM1) alternative splicing | [75] | |
| Rbfox2 | miR-20b, miR-21 | Upregulation of miR-20b Suppressing miR-21 expression |
Considered as anti-cancer splicing factors; suppressing cellular proliferation at normal tissue | [180] | |
| miR-107 | Upregulation of miR-107 | Considered as anti-cancer splicing factors; suppressing cellular proliferation at normal tissue Enhancing EMT in PDAC when moderately increased in PDAC |
[181] | ||
| TUMOUR METABOLISM | |||||
| PTBP1 | miR-124 | In neuron: PTBP1 suppress miR-124 cleavage (not confirmed in cancer) | Enhanced resistance against gemcitabine | [116,119] | |
| miR-124, miR-133b | PTBP1 downregulation by miR-124 and miR-133b | Altered cancer metabolism favouring Warburg effect by promoting PKM2 expression | [124] | ||
5 miRNA profiling methods
Accurate detection and quantification of miRNAs represent a major challenge due to the small size of miRNAs (approximately 22 nucleotides), the high sequence homology among members of the same family and the low abundance in biofluids. Currently, miRNAs profiling is a growing field of study, although conventional methods for detecting miRNAs still remain the gold-standards used to confirm the results of new detection techniques [125].
Northern blot is a widely used historical method to measure the expression of miRNAs ranging from the primitive miRNA to the mature form. It is based on molecular hybridization and gel electrophoresis and is able to simultaneously determine the size of miRNAs. However, Northern blot has several disadvantages: it is a time-consuming technique, requires large amounts of samples and reagents, with low sensitivity (pM-nM range) and low throughput [126].
Current miRNA detection strategies include reverse transcription-quantitative polymerase chain reaction (RT-qPCR), which is so far the undeniably gold-standard method for routine testing, especially for diagnostic purposes. It is commonly used to detect miRNAs at any stage of maturation, but does not allow the identification of new miRNAs. RT-qPCR is less time-consuming technique than Northern blotting and displays higher sensitivity, specificity and reproducibility than Northern blot. In addition, it converts small miRNA sequences into longer sequences by adding a poly(A) tail (poly(A)-tailed RT-qPCR) or a stem-loop structure (stem-loop RT-qPCR) overcoming the primer design limitation [127]. An innovation is represented by the ddPCR (droplet digital PCR) which offers greater performance, improved sensitivity, and accuracy as it allows for absolute quantification of miRNAs without the need for a reference gene [128].
PCR techniques cannot detect nucleotide sequences in cells and tissue sections, while in situ hybridization (ISH) can visualize miRNAs within cells and can determine the spatiotemporal expression of miRNAs, elucidating their biological role as well as their pathologic involvement in numerous diseases [129]. This technique is labour intensive and is limited by its low-throughput nature but the recent development of directly labelled fluorescence probes and multiplexed miRNA ISH methods allowed to detect multiple miRNAs per reaction.
Microarray is a hybridization-based method suitable for relative quantification. Locked nucleic acids (LNAs) can be incorporated into capture probes to normalize the melting temperature (Tm) whose variance is related to miRNA GC content [128,130]. The strength of this method is the multiplexed detection of multiple miRNAs in a single reaction, although it cannot discriminate between miRNA variants and has poor sensitivity compared to RNA-seq because it lacks the amplification step. On the other hand, microarray assays are fast, expensive and high-throughput [131].
Finally, Next-Generation Sequencing (NGS) is a highly accurate miRNA profiling technique that can simultaneously measure expression level and sequence changes, as well as detect unknown miRNAs. It should be noted that NGS has the highest multiplexing capability as specific primers are not required for each targeted miRNA detection [125,132]. Drawbacks to NGS include time-consuming for converting a sample into a library for sequencing, expensive analyses due to sophisticated software and qualified personnel for data analysis and it is not a fully automated technique as well [126].
Since multiplexing capability plays a crucial role in miRNAs detection, in addition to the above-mentioned multiplexing approaches, it is worth citing the suspension arrays (i.e., on-particle), which represent promising emerging methods for highly multiplex analysis of complex samples due to the versatility of the encoded microspheres used in conjunction with flow cytometry [133]. Furthermore, Rondelez et al. recently reported an isothermal amplification mechanism for multiplex and digital detection of miRNAs using the rational building of a molecular circuit that suppresses non-specific amplification due to cross-talk reactions [134]. In conclusion, extensive efforts have been made so far to develop efficient and sensitive methods for miRNA detection, but there still remains a need for a standardized method that should be highly sensitive, specific and multiplexable.
Predicting the effect of splicing modulation and its effect towards miRNA profile of PDAC
Splicing modulation is a new emerging therapeutic approach that had been tested in several types of cancer either pre-clinically or clinically [15,66,89,135–137]. Splicing modulation is promising because of its high potency to induce apoptosis and suppress cellular migration. Cancer cells harbouring mutations in genes encoding splicing factors are the most promising targets [15,89,136,137]. However, splicing modulation could also be applied in cancer cells with splicing factors overexpression [9,89,135].
The important role of splicing deregulation in PDAC carcinogenesis and the potency of several splicing modulators might increase the potential application of splicing inhibitors in PDAC. However, there have not yet been studies evaluating the efficacy of splicing inhibitors/modulators in PDAC. Preclinical studies have demonstrated that the SF3B1 inhibitors pladienolide B and E7107 were effective in gastric cancer, cervical cancer, and peritoneal mesothelioma [135,138–140]. In particular, pladienolide B has high efficacy in gastric cancer with complete tumour elimination in SCID mice in just 2 weeks and has an IC50 in the nanomolar range. Similar findings were observed in peritoneal mesothelioma where pladienolide B and E7107 inhibited cell proliferation and migration [135]. Remarkably, in vivo treatment with E7107 resulted in complete regression of peritoneal tumours in the second week. SF3B1 inhibition also showed similar efficacy in cervical cancer and cutaneous squamous cell carcinoma but, in these tumours they apparently showed more efficacy towards cells with mutated p53[140].
Unfortunately, clinical trials with splicing modulators have been limited by toxicity. Indeed, in a phase I pharmacokinetic and pharmacodynamic study of E7107 in advanced solid tumours, when using doses above 4.3 mg/m2, several patients suffered from gastrointestinal side effects, such as diarrhoea, vomiting, dehydration, and in two cases there was vision loss [141].
H3B-8800 is another SF3B1 inhibitor and entered phase I clinical trial in 2016, with a focus on patients with MDS, AML and CMML (NCT02841540). Initial results revealed dose-dependent target engagement, a predictable pharmacokinetic profile and a favourable safety profile, even with prolonged dosing. Although objective therapeutic responses have not been achieved to date, 14% of patients had reduced requirements for red blood cell or platelet transfusions [142].
A number of other drugs targeting splicing factors have shown encouraging preclinical effects in mouse models of cancer, such as inhibitors of SRPK and CLK protein kinases that phosphorylate SR proteins and thereby inhibit angiogenesis by inducing changes in the alternative splicing of VEGF [143,144]. Other splicing inhibitors targeting a variety of spliceosomal components also reduce cancer cell proliferation in vitro [145–148], but their effects in animal models of cancer are not yet known.
However, toxicity may be prevented by the use of a lower dose of splicing modulators, and the risk of reduced efficacy can be avoided by rationale combinations with different antitumor strategies, including modulation of selected miRNAs.
There are no data yet on the potential effect of splicing inhibitors on miRNA in PDAC. The most plausible candidates as therapeutic targets are PTBP1 and HnRNPK because their role has been already established in PDAC [63,70]. Moreover, SF3B1 has the advantage as a therapeutic target due to the availability of small-molecule inhibitors [89,137,139,140,149]. Of note, SF3B1 inhibition resulted in upregulation of tumour suppressor SO-miRNAs in a cervical cancer cell line [91]. Therefore, similar studies should be performed to demonstrate this effect in PDAC cell lines.
The effect of splicing inhibitors targeting those three splicing factors might however be predicted using available data. Calabreta and colleagues [70] provided initial evidence that targeting the splicing factor PTBP1 in gemcitabine resistant PDAC cell line by siRNA shifted PKM isoform expression towards PKM1 which was accompanied by increased sensitivity towards gemcitabine and an enhanced level of cleaved caspase-3. Li et al. [116] studied the long non-coding ROR in PDAC and found that PTBP1 was the target of tumour suppressor miR-124 which could effectively block its expression. However, in PDAC, long non-coding ROR acts as sponge that binds miR-124, preventing it to regulate PTBP1 expression and increasing PKM2 expression. This study suggested that miR-124 could be used as a marker for Warburg effect in PDAC as well as a therapeutic agent candidate to target PTBP1 in gemcitabine resistant PDAC. However, the other targets of miR-124 should be elucidated to minimize unfavourable off-target effects.
Another potential SF target candidate is HnRNPK which is known for its role in enhancing cancer cellular proliferation, invasion and metastasis in PDAC [63]. HnRNPK is associated with miR-223 which suppressed FBXW7 as previously described. However, the sister chromatid cohesion protein PDS5 homolog B (PDS5B) is another important target of miR-223. The downregulation of miR-223 led to increased expression of PDS5B which resulted in inhibition of cellular proliferation and migration [114].
Inhibition of SF3B1 could possibly be effective and may produce the most pronounced miRNA profile changes in PDAC. In cervical cancer, SF3B1 inhibition resulted in an increase of several tumour suppressor miRNA, most notably miR-636, miR-6510-5p, miR-3614-3p, miR-3655, miR-3656, miR-4260, miR-5187-3p, miR-7109-5p, and miR-8069 [96]. In addition, four miRNAs were downregulated, namely miR-155-3p, miR-148a-3p, miR-98-5p, and miR-21-3p. Apparently, SF3B1 inhibition can suppress the expression of miR-155 and miR-21 which play important roles in PDAC. However, this should be further investigated in PDAC preclinical models.
The effect of upregulation of tumour suppressor miRNAs or downregulation of oncogenic miRNAs is expected to have a wide impact [25]. A summary of potential effects of splicing modulation on the miRNA profile in PDAC as well as their biological effects is depicted in Figure 3. For example, miR-21 and miR-155 have many targets that are involved in carcinogenesis and metastasis [11,12,50,53,55,56]. Suppression of these oncogenic miRNAs can thus potentially lead to tumour suppression and inhibition of metastasis. However, these miRNAs can also serve as potential biomarkers of tumour progression or response to treatment, and could improve the clinical management of PDAC patients by monitoring the modulation of these miRNAs in samples that can be collected during treatment/follow-up, such as in liquid biopsy studies.
Figure 3.

The interaction of relevant splicing factors in PDAC and their associated miRNA. PTBP1 and HnRNPK are considered as the relevant targets in pancreatic cancer and have demonstrated their interaction with miRNAs in PDAC (miR-124 and miR-223, respectively). Despite lack of evidence in PDAC, cervical cancer experiment demonstrated that SF3B1 inhibition resulted in extensive change in miRNA expression and potentially brings more profound effects than PTBP1 and HnRNPK [63,70,89,91].
Bioinformatic tools to predict the effect of splicing modulation towards miRNA profiles
Bioinformatics uses advanced computing, mathematics and biological knowledge to store, manage, analyse and get insights into biological data. In recent years, there has been a boom of publicly available computational tools, online data analysis modules, biological data repositories, and bioinformatics workflow management systems [150]. In order to assess how splicing modulation can affect miRNA profiles, alternative splicing detection tools such as rMATS [151], SUPPA2 [152] or MISO [153] can first detect differential splicing between conditions, after which splicing motif analysis tools like MEME [154] or RNAContext [155] can use these splicing motifs to identify regulators of alternatively spliced junctions. Lastly, potential miRNA targets of splicing modulation can be detected using miRNA-target databases such as mirTarBase [156] where experimentally validated miRNA-target interactions are curated, or mirDB [157] where the predictive algorithm MirTarget is used to analyse thousands of miRNA-target interactions from high-throughput experiments. An example of analysis pipeline to identify splicing factors modulated by miRNAs is reported in Figure 4.
Figure 4.

Bioinformatic pipeline for splicing factor and miRNA modulation discovery with RNA-seq data. Raw data or mapped BAM files are used as input for alternative splicing (AS) tools such as MISO, rMATs and SUPPA2 to identify differential alternative splicing events. Next, a motif analysis is performed to identify the splicing factor (SF) specific for that RNA isoform. Lastly, miRNA-target databases are used to retrieve possible miRNA targeting SF. Additional miRNA profile can be useful to detect miRNA modulation through SFs.
The first step is the identification of alternative spliced (AS) events. While there is a plethora of available tools, no current tool can be regarded as the golden standard and the matter of choice strictly depends on the research question and familiarity. Research into which tools are superior is currently incomplete. One example of a tool that detects differential alternative spliced RNA transcripts is MISO, published in 2010 [157]. This statistical model estimates expression of alternatively spliced exons and isoforms using mapped reads as input format. MISO then uses Bayesian inference to compute the probability that an RNA-seq read originated from a particular isoform. Despite being the most cited and used tool for alternative spliced differential analysis, it has no longer been maintained since its publication, and it has high computational time. rMATS [151] is a more recent and often-cited tool used in differential splicing analysis, it analyses replicates and includes a function to handle paired and unpaired replicates. Another common tool recently published is SUPPA2156. This type of algorithm requires two biological replicates because it accounts for biological variability, which is important for the reliability of the estimations that are drawn from the data. However, it can work with multiple conditions and includes the possibility to perform hierarchical clustering on differentially spliced events to identify common regulatory mechanisms. Other tools capable of detecting AS and compare AS patterns between sample groups are DEXSEq, SplicingCompass, Altanalyze, BitSeq, EBSeq, and Cuffdiff2 whose performances are extensively reviewed in Lahat and Grellsheid [158].
Once differentially expressed AS events are identified, prediction of splicing factors is the second step of the analysis. Motif analysis tools may be used to identify the direct regulators of alternative spliced junctions. For example, MEME suite [154] provides a unified portal of online discovery tools for DNA binding sites and protein interaction domains. MEME is an online web-based module that includes three sequence scanning algorithms that allow to scan different DNA and protein databases. Next, transcription factor motif can be further analysed for putative functions using GOMo tool [159,160]. Previous tools such as RNAcontext [155] and GraphProt [143] work with classification and regression model settings and are not capable of de novo sequence-structure motifs. Other tools for motif analysis recently published are SSMART [161] and TrawlerWeb [162] which is a web version of the previous published standalone tool. Trawlerweb is currently the fastest online de novo motif discovery tool and it displays resulting scores allowing the user to prioritize the choice for validation experiments. Validation analysis, such as in vitro/in vivo binding assays, cross-linking immunoprecipitation sequencing (CLIP-seq), minigene splicing reporter assays (invitro) or anti-sense oligonucleotides which block splicing factor-binding sites are needed to validate results from the (splicing) motif analysis.
The last step of the analysis is the identification of putative miRNAs that are targeting the splicing factors based on previously performed motif analysis. Nowadays there is a plethora of miRNAs-target databases available on the web, such as miRTarBase [156], mirDB [157], miRBase [163] and TarBase [164]. In addition, a new R package multimiR [165] includes a compilation of around 50 million records in human and mouse from 14 different databases and it expands on miRNAs involved in drug response and disease annotation.
However, an integrative analysis with miRNA profile can be useful to detect miRNA modulation (e.g., inhibition) through the predicted SFs. Nowadays, there are several ways to analyse miRNA-seq profiles, and here we describe the general downstream analysis pipeline with the most commonly used tools.
After trimming and quality check, the resulting reads are aligned to a reference database containing miRNA sequences. miRbase [163] is the primary database of published miRNA sequences which is often used for miRNA mapping. A broader database of small-RNA and miRNA sequences is mirGeneDB 2.0 [166] resulting in more precise annotation while avoiding misleading miRNA annotation from other types of small-RNAs. The read sequences are mapped to reference databases through mapping tools. There is an increasing number of mapping tools for small-RNA sequences and the most used are: miRanalyzer [167], miRDeep2 [168] and sRNAbench [169]. All these tools rely on Bowtie algorithm [170] (allowing mismatches and improving speed of alignment).
sRNAbench and its downstream analysis tool sRNAtoolbox includes an automatic processing of the five most used library preparation protocols (including new reference genomes from Ensembl, NCBI and MirGeneDB), a consensus differential expression analysis, target prediction, analysis of unmapped reads, batch mode to profile several samples at once with the same set of parameters and improved visualization and mapping statistics. This also enables users with a ‘non-bioinformatics’ background to analyse small-RNA high-throughput data from raw fastq files (standard output files from sequencing machines) to post-processed data for differential analysis and miRNA-target prediction.
The ever-expanding field of bioinformatic studies and the enormous availability of wet-lab data has given rise to several predictive models that are extremely useful for target prediction and to prioritize experimental validation targets.
Conclusions and Future Perspectives
The interaction of miRNAs and splicing deregulation is an understudied field, but evidence of their close interconnection is increasing. Currently, the application of miRNAs is focused on their role as biomarker while splicing inhibitors are under investigation as a novel therapeutic strategy.
Increasing evidence shows that splicing deregulation resulting from mutation or overexpression can produce a pronounced aberration in miRNA expression in different cancer types, including PDAC. For instance, the upregulation of tumour suppressing miRNAs may mediate an anti-cancer effect of splicing modulation as was shown by the inhibition of PTBP1 and SF3B1 in cervical cancer. Potentially similar inhibitory effects and the impact of other SFs on cancer progression and miRNA profile still need to be investigated in PDAC. Moreover, specific miRNAs could be used as a target to downregulate specific SFs and also for combined therapeutic approaches.
Remarkably, novel bioinformatics tools are providing extensive data that can be used to deepen our knowledge in the biological effects of the interplay between splicing and miRNAs, as well as several predictive models for target prediction in order to prioritize future experimental and clinical validation. In-depth analysis of PDAC aberrant splicing patterns associated with miRNA profiling may indeed further provide mechanistic insight to successfully target key PDAC drivers. Targeting aberrant splicing and the reciprocal interaction with deregulated miRNA could therefore provide more effective therapeutic approach to combat the complex biology of PDAC and its chemoresistant features.
Funding Statement
This work was supported by the CCA Foundation 2012, 2015 and 2018 grants (G.J.P., E.G.,); KWF Dutch Cancer Society grants [KWF project#19571] (E.G.); AIRC/IG-grant (E.G.); Indonesian Endowment Fund Scholarship (I.G.P.S.); KWF Kankerbestrijding [19571]; Indonesian endowment fund scholarship;AIRC/IG-grant;
Authors’ contribution
D.I.S, G.M., I.B.M., O.R., G.J.P, E.G. wrote the manuscript; D.I.S., O.R. and G.M. designed figures and tables; G.J.P, E.G., P.D. and S.C. extensively revised the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- [2].The Netherland Cancer Registry [Internet]. Dutch Cancer Figure: Pancreatic Cancer2018 [cited 2019. Feb 22]; Available from: https://www.cijfersoverkanker.nl/selecties/Dataset_1/img5c8269c859485
- [3].McGuigan A, Kelly P, Turkington RC, et al. Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. 2018;24(43):4846–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fahrmann JF, Bantis LE, Capello M, et al. A Plasma-Derived Protein-Metabolite Multiplexed Panel for Early-Stage Pancreatic Cancer. J Natl Cancer Inst. 2019;111(4):372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Orth M, Metzger P, Gerum S, et al. Pancreatic ductal adenocarcinoma: biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat Oncol. 2019;14:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yang C, Wu Q, Huang K, et al. Genome-Wide Profiling Reveals the Landscape of Prognostic Alternative Splicing Signatures in Pancreatic Ductal Adenocarcinoma. Front Oncol. 2019;9:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Słotwiński R, Lech G, Słotwińska SM.. MicroRNAs in pancreatic cancer diagnosis and therapy. Cent Eur J Immunol. 2018;43(3):314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Anczuków O, Krainer AR. Splicing-factor alterations in cancers. RNA. 2016;22(9):1285–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang J, Dumartin L, Mafficini A, et al. Splice variants as novel targets in pancreatic ductal adenocarcinoma. Sci Rep. 2017;7(1):2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hu G, Tao F, Wang W, et al. Prognostic value of microRNA-21 in pancreatic ductal adenocarcinoma: a meta-analysis. World J Surg Oncol. 2016;14(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sicard F, Gayral M, Lulka H, et al. Targeting miR-21 for the therapy of pancreatic cancer. Mol Ther. 2013;21(5):986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Feng Y-H, Tsao C-J. Emerging role of microRNA-21 in cancer. Biomed Rep. 2016;5(4):395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Daoud AZ, Mulholland EJ, Cole G, et al. MicroRNAs in Pancreatic Cancer: biomarkers, prognostic, and therapeutic modulators. BMC Cancer. 2019;19(1):1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Escobar-Hoyos L, Knorr K, Abdel-Wahab A-WO. Aberrant RNA Splicing in Cancer. Annu. Rev. Cancer Biol. 2019;3(1):167–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dvinge H, Kim E, Abdel-Wahab O, et al. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer. 2016;16(7):413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kahn T, Bosch J, Giovannetti E, et al. Effect of sodium nitrate loading on electrolyte transport by the renal tubule. The American Journal of physiology. 1975;229(3):54714. [DOI] [PubMed] [Google Scholar]
- [17].Ali S, Almhanna K, Chen W, et al. Differentially expressed miRNAs in the plasma may provide a molecular signature for aggressive pancreatic cancer. Am J Transl Res. 2010;3(1):28–47. [PMC free article] [PubMed] [Google Scholar]
- [18].Rodriguez-Aguayo C, Monroig P, Del C, et al. Regulation of hnRNPA1 by microRNAs controls the miR-18a–K-RAS axis in chemotherapy-resistant ovarian cancer. Cell Discov. 2017;3:17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. [DOI] [PubMed] [Google Scholar]
- [20].Juzenas S, Venkatesh G, Hübenthal M, et al. A comprehensive, cell specific microRNA catalogue of human peripheral blood. Nucleic Acids Res. 2017;45(16):9290–9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. [DOI] [PubMed] [Google Scholar]
- [22].Winter J, Jung S, Keller S, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11(3):228–234. [DOI] [PubMed] [Google Scholar]
- [23].Zhang F, Wang D. The Pattern of microRNA Binding Site Distribution. Genes (Basel). 2017;8(11):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hogg DR, Harries LW. Human genetic variation and its effect on miRNA biogenesis, activity and function. Biochem Soc Trans. 2014;42(4):1184–1189. [DOI] [PubMed] [Google Scholar]
- [25].Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther. 2016;1(1):15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Poy MN, Eliasson L, Krutzfeldt J, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432(7014):226–230. [DOI] [PubMed] [Google Scholar]
- [27].Olson P, Lu J, Zhang H, et al. MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev. 2009;23(18):2152–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schultz NA, Werner J, Willenbrock H, et al. MicroRNA expression profiles associated with pancreatic adenocarcinoma and ampullary adenocarcinoma. Mod Pathol. 2012;25(12):1609–1622. [DOI] [PubMed] [Google Scholar]
- [29].Hong TH, Park IY. MicroRNA expression profiling of diagnostic needle aspirates from surgical pancreatic cancer specimens. Ann Surg Treat Res. 2014;87(6):290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Calatayud D, Dehlendorff C, Boisen MK, et al. Tissue MicroRNA profiles as diagnostic and prognostic biomarkers in patients with resectable pancreatic ductal adenocarcinoma and periampullary cancers. Biomark Res. 2017;5(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Papaconstantinou IG, Manta A, Gazouli M, et al. Expression of microRNAs in patients with pancreatic cancer and its prognostic significance. Pancreas. 2013;42(1):67–71. [DOI] [PubMed] [Google Scholar]
- [32].Rawat M, Kadian K, Gupta Y, et al. MicroRNA in Pancreatic Cancer: from Biology to Therapeutic Potential. Genes (Basel). 2019;10(10):752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Frampton AE, Krell J, Prado MM, et al. Prospective validation of microRNA signatures for detecting pancreatic malignant transformation in endoscopic-ultrasound guided fine-needle aspiration biopsies. Oncotarget. 2016;7(19):28556–28569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Du Rieu MC, Torrisani J, Selves J, et al. MicroRNA-21 Is Induced Early in Pancreatic Ductal Adenocarcinoma Precursor Lesions. Clin Chem. 2010;56(4):603–612. [DOI] [PubMed] [Google Scholar]
- [35].Ryu JK, Hong S-M, Karikari CA, et al. Aberrant MicroRNA-155 Expression Is an Early Event in the Multistep Progression of Pancreatic Adenocarcinoma. Pancreatology. 2010;10(1):66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yu J, Li A, Hong S-M, et al. MicroRNA Alterations of Pancreatic Intraepithelial Neoplasias. Clin Cancer Res. 2012;18(4):981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Caponi S, Funel N, Frampton AE, et al. The good, the bad and the ugly: a tale of miR-101, miR-21 and miR-155 in pancreatic intraductal papillary mucinous neoplasms. Ann Oncol. 2013;24(3):734–741. [DOI] [PubMed] [Google Scholar]
- [38].Wei X, Wang W, Wang L, et al. Micro RNA -21 induces 5-fluorouracil resistance in human pancreatic cancer cells by regulating PTEN and PDCD4. Cancer Med. 2016;5(4):693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mees ST, Mardin WA, Wendel C, et al. EP300-A miRNA-regulated metastasis suppressor gene in ductal adenocarcinomas of the pancreas: role of EP300 in Metastasis of Pancreatic Cancer. Int J Cancer. 2010;126:114–124. [DOI] [PubMed] [Google Scholar]
- [40].Kawaguchi T, Komatsu S, Ichikawa D, et al. Clinical impact of circulating miR-221 in plasma of patients with pancreatic cancer. Br J Cancer. 2013;108(2):361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Morimura R, Komatsu S, Ichikawa D, et al. Novel diagnostic value of circulating miR-18a in plasma of patients with pancreatic cancer. Br J Cancer. 2011;105(11):1733–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Frampton AE, Castellano L, Colombo T, et al. Integrated molecular analysis to investigate the role of microRNAs in pancreatic tumour growth and progression. Lancet. 2015;385:S37. [DOI] [PubMed] [Google Scholar]
- [43].Giovannetti E, Funel N, Peters GJ, et al. MicroRNA-21 in pancreatic cancer: correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res. 2010;70(11):4528–4538. . [DOI] [PubMed] [Google Scholar]
- [44].Frampton AE, Krell J, Jamieson NB, et al. microRNAs with prognostic significance in pancreatic ductal adenocarcinoma: a meta-analysis. Eur J Cancer. 2015;51(11):1389–1404. [DOI] [PubMed] [Google Scholar]
- [45].Hwang J-H, Voortman J, Giovannetti E, et al. Identification of microRNA-21 as a biomarker for chemoresistance and clinical outcome following adjuvant therapy in resectable pancreatic cancer. PloS One. 2010;5(5):e10630–e10630. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tys LL, Meijer LL, Prado MM, et al. Circulating microRNAs as diagnostic biomarkers for pancreatic cancer. Expert Rev Mol Diagn. 2015;15(12):1525–1529. [DOI] [PubMed] [Google Scholar]
- [47].Meijer LL, Garajova I, Caparello C, et al. Plasma miR-181a-5p Downregulation Predicts Response and Improved Survival After FOLFIRINOX in Pancreatic Ductal Adenocarcinoma. Annals of surgery; 2018. [DOI] [PubMed]
- [48].Park J-K, Lee EJ, Esau C, et al. Antisense inhibition of microRNA-21 or −221 arrests cell cycle, induces apoptosis, and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma. Pancreas. 2009;38(7):e190–9. [DOI] [PubMed] [Google Scholar]
- [49].Toste PA, Li L, Kadera BE, et al. p85α is a microRNA target and affects chemosensitivity in pancreatic cancer. J Surg Res. 2015;196(2):285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhao Q, Chen S, Zhu Z, et al. miR-21 promotes EGF-induced pancreatic cancer cell proliferation by targeting Spry2. Cell Death Dis. 2018;9(12):1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhao M, Wang L, Liu J, et al. MiR-21 Suppresses Anoikis through Targeting PDCD4 and PTEN in Human Esophageal Adenocarcinoma. Curr Med Sci. 2018;38(2):245–251. [DOI] [PubMed] [Google Scholar]
- [52].Garajová I, Le Large TY, Frampton AE, et al. Molecular mechanisms underlying the role of microRNAs in the chemoresistance of pancreatic cancer. Biomed Res Int. 2014;2014:678401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen S, Chen X, Shan T, et al. MiR-21-mediated Metabolic Alteration of Cancer-associated Fibroblasts and Its Effect on Pancreatic Cancer Cell Behavior. Int J Biol Sci. 2018;14(1):100–110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [54].Kunita A, Morita S, Irisa TU, et al. MicroRNA-21 in cancer-associated fibroblasts supports lung adenocarcinoma progression. Sci Rep. 2018;8(1):8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Czochor JR, Sulkowski P, Glazer PM. miR-155 Overexpression Promotes Genomic Instability by Reducing High-fidelity Polymerase Delta Expression and Activating Error-Prone DSB Repair. Mol Cancer Res. 2016;14(4):363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ye J, Guo R, Shi Y, et al. miR-155 Regulated Inflammation Response by the SOCS1-STAT3-PDCD4 Axis in Atherogenesis. Mediators Inflamm. 2016;2016:8060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ali S, Banerjee S, Logna F, et al. Inactivation of Ink4a/Arf leads to deregulated expression of miRNAs in K-Ras transgenic mouse model of pancreatic cancer. J Cell Physiol. 2012;227(10):3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [58].Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013;3(11):1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Urbanski LM, Leclair N, Anczuków O. Alternative-splicing defects in cancer: splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip Rev RNA. 2018;9:e1476–e1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sciarrillo R, Wojtuszkiewicz A, Assaraf YG, et al. The role of alternative splicing in cancer: from oncogenesis to drug resistance. Drug Resist Updat. 2020;53:100728. [DOI] [PubMed] [Google Scholar]
- [61].The Human Protein Atlas . SF3B1: pathology Atlas Pancreatic Cancer [Internet]. 2019. [cited 2019 Feb 22]; Available from: https://www.proteinatlas.org/ENSG00000115524-SF3B1/pathology/tissue/pancreatic+cancer#Quantity
- [62].The Human Protein Atlas. HNRNPK expression in Pancreatic Cancer [Internet] . 2019; Available from: https://www.proteinatlas.org/ENSG00000165119-HNRNPK/pathology/pancreatic+cancer
- [63].He D, Huang C, Zhou Q, et al. HnRNPK/miR-223/FBXW7 feedback cascade promotes pancreatic cancer cell growth and invasion. Oncotarget. 2017;8(12):20165–20178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhou R, Shanas R, Nelson MA, et al. Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int J Cancer. 2010;126(2):395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lee SC-W, North K, Kim E, et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell. 2018;34(225–241.e8):225–241.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chakedis J, French R, Babicky M, et al. A novel protein isoform of the RON tyrosine kinase receptor transforms human pancreatic duct epithelial cells. Oncogene. 2016;35(25):3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Thomas RM, Toney K, Fenoglio-Preiser C, et al. The RON Receptor Tyrosine Kinase Mediates Oncogenic Phenotypes in Pancreatic Cancer Cells and Is Increasingly Expressed during Pancreatic Cancer Progression. Cancer Res. 2007;67(13):6075 LP – 6082. [DOI] [PubMed] [Google Scholar]
- [69].Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. [DOI] [PubMed] [Google Scholar]
- [70].Calabretta S, Bielli P, Passacantilli I, et al. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene. 2016;35(16):2031–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Karni R, De Stanchina E, Lowe SW, et al. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14(3):185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol. 2014;21(2):189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cohen-Eliav M, Golan-Gerstl R, Siegfried Z, et al. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol. 2013;229(4):630–639. [DOI] [PubMed] [Google Scholar]
- [74].The Human Protein Atlas. SRSF6 : Pancreatic cancer pathology [Internet]. 2019; Available from: https://www.proteinatlas.org/ENSG00000124193-SRSF6/pathology/pancreatic+cancer
- [75].Li M, Wu P, Yang Z, et al. miR-193a-5p Promotes Pancreatic Cancer Cell Migration and Invasion Through SRSF6-Mediated Alternative Splicing of OGDHL and ECM1. Lancet. 2019;10(1):38–59. [PMC free article] [PubMed] [Google Scholar]
- [76].Gonçalves V, Jordan P. Posttranscriptional Regulation of Splicing Factor SRSF1 and Its Role in Cancer Cell Biology. Biomed Res Int. 2015;2015:287048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Anczuków O, Rosenberg AZ, Akerman M, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19(2):220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gautrey HL, Tyson-Capper AJ. Regulation of Mcl-1 by SRSF1 and SRSF5 in cancer cells. PloS One. 2012;7(12):e51497–e51497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Leu S, Lin Y-M, Wu C-H OP. Loss of Pnn expression results in mouse early embryonic lethality and cellular apoptosis through SRSF1-mediated alternative expression of Bcl-xS and ICAD. J Cell Sci. 2012;125(13):3164 LP – 3172. [DOI] [PubMed] [Google Scholar]
- [80].Das S, Anczuków O, Akerman M, et al. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012;1(2):110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wu H, Sun S, Tu K, et al. A splicing-independent function of SF2/ASF in microRNA processing. Mol Cell. 2010;38(1):67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Woods K, Thomson JM, Hammond SM. Direct Regulation of an Oncogenic Micro-RNA Cluster by E2F Transcription Factors. J Biol Chem. 2007;282(4):2130–2134. [DOI] [PubMed] [Google Scholar]
- [83].Xu Q, Li P, Chen X, et al. miR-221/222 induces pancreatic cancer progression through the regulation of matrix metalloproteinases. Oncotarget. 2015;6(16):14153–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol. 2010;42(8):1348–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Murphy BL, Obad S, Bihannic L, et al. Silencing of the miR-17∼92 Cluster Family Inhibits Medulloblastoma Progression. Cancer Res. 2013;73(23):7068–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Olive V, Bennett MJ, Walker JC, et al. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009;23(24):2839–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kort EJ, Farber L, Tretiakova M, et al. The E2F3-Oncomir-1 axis is activated in Wilms’ tumor. Cancer Res. 2008;68(11):4034–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Quattrochi B, Gulvady A, Driscoll DR, et al. MicroRNAs of the mir-17~92 cluster regulate multiple aspects of pancreatic tumor development and progression. Oncotarget. 2017;8(22):35902–35918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lin J-C. Therapeutic Applications of Targeted Alternative Splicing to Cancer Treatment. Int J Mol Sci. 2017;19(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Aslan D, Garde C, Nygaard MK, et al. Tumor suppressor microRNAs are downregulated in myelodysplastic syndrome with spliceosome mutations. Oncotarget. 2016;7(9):9951–9963. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Pianigiani G, Licastro D, Fortugno P, et al. Microprocessor-dependent processing of splice site overlapping microRNA exons does not result in changes in alternative splicing. RNA. 2018;24(9):1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jang J-Y, Lee Y-S, Jeon Y-K, et al. ANT2 suppression by shRNA restores miR-636 expression, thereby downregulating Ras and inhibiting tumorigenesis of hepatocellular carcinoma. Exp Mol Med. 2013;45(1):e3–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wang Z, Tong D, Han C, et al. Blockade of miR-3614 maturation by IGF2BP3 increases TRIM25 expression and promotes breast cancer cell proliferation. EBioMedicine. 2019;41:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yang R-M, Zhan M, Xu S-W, et al. miR-3656 expression enhances the chemosensitivity of pancreatic cancer to gemcitabine through modulation of the RHOF/EMT axis. Cell Death Dis. 2017;8(10):e3129–e3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Itami-Matsumoto S, Hayakawa M, Uchida-Kobayashi S, et al. Circulating Exosomal miRNA Profiles Predict 1570 the Occurrence and Recurrence of Hepatocellular Carcinoma in Patients with Direct-Acting Antiviral-Induced Sustained Viral Response. Biomedicines. 2019;7(4):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, et al. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015;43(10):5130–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kuroyanagi H. Fox-1 family of RNA-binding proteins. Cell Mol Life Sci. 2009;66(24):3895–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Braeutigam C, Rago L, Rolke A, et al. RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene. 2014;33(9):1082–1092. [DOI] [PubMed] [Google Scholar]
- [99].Janakiraman H, House RP, Gangaraju VK, et al. (lncRNA) and Short (miRNA) of It: tGFβ-Mediated Control of RNA-Binding Proteins and Noncoding RNAs. Mol Cancer Res. 2018;16(4):567 LP – 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zhang Y, Li M, Wang H, et al. Profiling of 95 MicroRNAs in Pancreatic Cancer Cell Lines and Surgical Specimens by Real-Time PCR Analysis. World J Surg. 2009;33(4):698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Frampton AE, Castellano L, Colombo T, et al. MicroRNAs Cooperatively Inhibit a Network of Tumor Suppressor Genes to Promote Pancreatic Tumor Growth and Progression. Gastroenterology. 2014;146(1):268–277.e18. [DOI] [PubMed] [Google Scholar]
- [102].Bloomston M, Frankel WL, Petrocca F, et al. MicroRNA Expression Patterns to Differentiate Pancreatic Adenocarcinoma From Normal Pancreas and Chronic Pancreatitis. JAMA. 2007;297(17):1901. [DOI] [PubMed] [Google Scholar]
- [103].Wang YJ, McAllister F, Bailey JM, et al. Is Required for Maintenance of Adult Pancreatic Acinar Cell Identity and Plays a Role in Kras-Driven Pancreatic Neoplasia. PLoS ONE. 2014;9(11):e113127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Morris JP, Greer R, Russ HA, et al. Dicer Regulates Differentiation and Viability during Mouse Pancreatic Cancer Initiation. PLoS ONE. 2014;9(5):e95486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Rachagani S, Macha MA, Menning MS, et al. Changes in microRNA (miRNA) expression during pancreatic cancer development and progression in a genetically engineered KrasG12D;Pdx1-Cre mouse (KC) model. Oncotarget. 2015;6(37):40295–40309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kędzierska H, Piekiełko-Witkowska A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017;396:53–65. [DOI] [PubMed] [Google Scholar]
- [107].Gonçalves V, Pereira JFS, Jordan P. Signaling Pathways Driving Aberrant Splicing in Cancer Cells. Genes (Basel). 2017;9(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Qiao L, Xie N, Bai Y, et al. Identification of Upregulated HNRNPs Associated with Poor Prognosis in Pancreatic Cancer. Biomed Res Int. 2019;2019:5134050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Chen Z-Y, Cai L, Zhu J, et al. Fyn requires HnRNPA2B1 and Sam68 to synergistically regulate apoptosis in pancreatic cancer. Carcinogenesis. 2011;32(10):1419–1426. [DOI] [PubMed] [Google Scholar]
- [110].Shen K, Cao Z, Zhu R, et al. The dual functional role of MicroRNA-18a (miR-18a) in cancer development. Clin Transl Med. 2019;8(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tsang WP, Kwok TT. The miR-18a* microRNA functions as a potential tumor suppressor by targeting on K-Ras. Carcinogenesis. 2009;30(6):953–959. [DOI] [PubMed] [Google Scholar]
- [112].Cogoi S, Rapozzi V, Cauci S, et al. Critical role of hnRNP A1 in activating KRAS transcription in pancreatic cancer cells: a molecular mechanism involving G4 DNA. Biochim Biophys Acta Gen Subj. 2017;1861(5):1389–1398. [DOI] [PubMed] [Google Scholar]
- [113].Mikula M, Dzwonek A, Karczmarski J, et al. Landscape of the hnRNP K protein–protein interactome. PROTEOMICS. 2006;6(8):2395–2406. [DOI] [PubMed] [Google Scholar]
- [114].Ma J, Cao T, Cui Y, et al. miR-223 Regulates Cell Proliferation and Invasion via Targeting PDS5B in Pancreatic Cancer Cells. Mol Ther Nucleic Acids. 2019;14:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Ma J, Cheng L, Liu H, et al. Genistein Down-Regulates miR-223 Expression in Pancreatic Cancer Cells. Curr Drug Targets. 2013;14(10):1150–1156. [DOI] [PubMed] [Google Scholar]
- [116].Li C, Zhao Z, Zhou Z, et al. Linc-ROR confers gemcitabine resistance to pancreatic cancer cells via inducing autophagy and modulating the miR-124/PTBP1/PKM2 axis. Cancer Chemother Pharmacol. 2016;78(6):1199–1207. [DOI] [PubMed] [Google Scholar]
- [117].Wu D-H, Liang H, Lu S-N, et al. miR-124 Suppresses Pancreatic Ductal Adenocarcinoma Growth by Regulating Monocarboxylate Transporter 1-Mediated Cancer Lactate Metabolism. Cell Physiol Biochem. 2018;50(3):924–935. [DOI] [PubMed] [Google Scholar]
- [118].Wang P, Chen L, Zhang J, et al. Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene. 2014;33(4):514–524. [DOI] [PubMed] [Google Scholar]
- [119].Yeom K-H, Mitchell S, Linares AJ, et al. Polypyrimidine tract-binding protein blocks miRNA-124 biogenesis to enforce its neuronal-specific expression in the mouse. PNAS. 2018; 115:E11061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wu D, Pan H, Zhou Y, et al. microRNA-133b downregulation and inhibition of cell proliferation, migration and invasion by targeting matrix metallopeptidase-9 in renal cell carcinoma. Mol Med Rep. 2014;9(6):2491–2498. [DOI] [PubMed] [Google Scholar]
- [121].Wang Q-Y, Zhou C-X, Zhan M-N, et al. MiR-133b targets Sox9 to control pathogenesis and metastasis of breast cancer. Cell Death Dis. 2018;9(7):752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Li D, Xia L, Chen M, et al. miR-133b, a particular member of myomiRs, coming into playing its unique pathological role in human cancer. Oncotarget. 2017;8(30):50193–50208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Khan MA, Zubair H, Srivastava SK, et al. Insights into the Role of microRNAs in Pancreatic Cancer Pathogenesis: potential for Diagnosis, Prognosis, and Therapy. Adv Exp Med Biol. 2015;889:71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Taniguchi K, Sakai M, Sugito N, et al. PTBP1-associated microRNA-1 and −133b suppress the Warburg effect in colorectal tumors. Oncotarget. 2016;7(14):18940–18952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Siddika T, Heinemann IU. Bringing MicroRNAs to Light: methods for MicroRNA Quantification and Visualization in Live Cells. Front Bioeng Biotechnol. 2021;8:619583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Dave VP, Ngo TA, Pernestig A-K, et al. MicroRNA amplification and detection technologies: opportunities and challenges for point of care diagnostics. Lab Invest. 2019;99(4):452–469. [DOI] [PubMed] [Google Scholar]
- [127].Ye J, Xu M, Tian X, et al. Research advances in the detection of miRNA. J Pharm Anal. 2019;9(4):217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Saliminejad K, Khorram Khorshid HR, Soleymani Fard S, et al. An overview of microRNAs: biology, functions, therapeutics, and analysis methods: SALIMINEJAD et al. J Cell Physiol. 2019;234(5):5451–5465. [DOI] [PubMed] [Google Scholar]
- [129].Chu Y-H, Hardin H, Zhang R, et al. In situ hybridization: introduction to techniques, applications and pitfalls in the performance and interpretation of assays. Semin Diagn Pathol. 2019; 36:336–341. [DOI] [PubMed] [Google Scholar]
- [130].Kilic T, Erdem A, Ozsoz M, et al. microRNA biosensors: opportunities and challenges among conventional and commercially available techniques. Biosens Bioelectron. 2018;99:525–546. [DOI] [PubMed] [Google Scholar]
- [131].Ouyang T, Liu Z, Han Z, et al. MicroRNA Detection Specificity: recent Advances and Future Perspective. Anal Chem. 2019;91(5):3179–3186. [DOI] [PubMed] [Google Scholar]
- [132].Gines G, Menezes R, Xiao W, et al. Emerging isothermal amplification technologies for microRNA biosensing: applications to liquid biopsies. Mol Aspects Med. 2020;72:100832. [DOI] [PubMed] [Google Scholar]
- [133].Jet T, Gines G, Rondelez Y, et al. Advances in multiplexed techniques for the detection and quantification of microRNAs. Chem Soc Rev. 2021;10:1039. [DOI] [PubMed] [Google Scholar]
- [134].Rondelez Y, Gines G. Multiplex Digital MicroRNA Detection Using Cross-Inhibitory DNA Circuits. ACS Sensors. 2020;5(8):2430–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Sciarrillo R, Wojtuszkiewicz A, El Hassouni B, et al. Splicing modulation as novel therapeutic strategy against diffuse malignant peritoneal mesothelioma. EBioMedicine; 2018. [DOI] [PMC free article] [PubMed]
- [136].Dehm SM. Test-Firing Ammunition for Spliceosome Inhibition in Cancer. Clin Cancer Res. 2013;19(22):6064 LP – 6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Hong DS, Kurzrock R, Naing A, et al. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs. 2014;32(3):436–444. [DOI] [PubMed] [Google Scholar]
- [138].Zhang Q, Di C, Yan J, et al. Inhibition of SF3b1 by pladienolide B evokes cycle arrest, apoptosis induction and p73 splicing in human cervical carcinoma cells. Artif Cells Nanomed Biotechnol. 2019;47(1):1273–1280. [DOI] [PubMed] [Google Scholar]
- [139].Zhang Y, Yuan Z, Jiang Y, et al. Inhibition of Splicing Factor 3b Subunit 1 (SF3B1) Reduced Cell Proliferation, Induced Apoptosis and Resulted in Cell Cycle Arrest by Regulating Homeobox A10 (HOXA10) Splicing in AGS and MKN28 Human Gastric Cancer Cells. Med Sci Monit. 2020;26:e919460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Hepburn LA, McHugh A, Fernandes K, et al. Targeting the spliceosome for cutaneous squamous cell carcinoma therapy: a role for c-MYC and wild-type p53 in determining the degree of tumour selectivity. Oncotarget. 2018;9(33):23029–23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Eskens FALM, Ramos FJ, Burger H, et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the First-in-Class Spliceosome Inhibitor E7107 in Patients with Advanced Solid Tumors. Clin Cancer Res. 2013;19(22):6296–6304. [DOI] [PubMed] [Google Scholar]
- [142].Steensma DP, Wermke M, Klimek VM, et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood. 2019;134(Supplement_1):673. [Google Scholar]
- [143].Amin E, Oltean S, Hua J, et al. WT1 Mutants Reveal SRPK1 to Be a Downstream Angiogenesis Target by Altering VEGF Splicing. Cancer Cell. 2011;20(6):768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Hatcher JM, Wu G, Zeng C, et al. SRPKIN-1: a Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem Biol. 2018;25(4):460–470.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Iwatani-Yoshihara M, Ito M, Klein MG, et al. Discovery of Allosteric Inhibitors Targeting the Spliceosomal RNA Helicase Brr2. J Med Chem. 2017;60(13):5759–5771. [DOI] [PubMed] [Google Scholar]
- [146].Jagtap PKA, Garg D, Kapp TG, et al. Rational Design of Cyclic Peptide Inhibitors of U2AF Homology Motif (UHM) Domains To Modulate Pre-mRNA Splicing. J Med Chem. 2016;59(22):10190–10197. [DOI] [PubMed] [Google Scholar]
- [147].Effenberger KA, James RC, Urabe VK, et al. The Natural Product N-Palmitoyl-l-leucine Selectively Inhibits Late Assembly of Human Spliceosomes. J Biol Chem. 2015;290(46):27524–27531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Sidarovich A, Will CL, Anokhina MM, et al. Identification of a small molecule inhibitor that stalls splicing at an early step of spliceosome activation. Elife. 2017;6:e23533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Kotake Y, Sagane K, Owa T, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3(9):570. [DOI] [PubMed] [Google Scholar]
- [150].Teufel A, Krupp M, Weinmann A, et al. Current bioinformatics tools in genomic biomedical research (Review). International Journal of Molecular Medicine [Internet] 2006. [cited 2020 Oct 20]; Available from: http://www.spandidos-publications.com/10.3892/ijmm.17.6.967 [PubMed]
- [151].Shen S, Park JW, Lu Z, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. PNAS. 2014;111:E5593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Trincado JL, Entizne JC, Hysenaj G, et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biology [Internet] 2018. [cited 2020 Oct 20]; 19. Available from: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1417-1 [DOI] [PMC free article] [PubMed]
- [153].Katz Y, Wang ET, Airoldi EM, et al. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7(12):1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server):W202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Kazan H, Ray D, Chan ET, et al. RNAcontext: a New Method for Learning the Sequence and Structure Binding Preferences of RNA-Binding Proteins. PLoS Comput Biol. 2010;6(7):e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Huang H-Y, Lin Y-C-D, Li J, et al. miRTarBase 2020: updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res 2019. gkz896 10.1093/nar/gkz896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Lahat A, Grellscheid SN. Differential mRNA Alternative Splicing [Internet]. In: Aransay AM, Lavín Trueba JL, editors. Field Guidelines for Genetic Experimental Designs in High-Throughput Sequencing. Cham: Springer International Publishing; 2016. [cited 2020 Oct 20]. page 105–119.Available from: http://link.springer.com/10.1007/978-3-319-31350-4_5 [Google Scholar]
- [159].Bodén M, Bailey TL. Associating transcription factor-binding site motifs with target GO terms and target genes. Nucleic Acids Res. 2008;36(12):4108–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Maticzka D, Lange SJ, Costa F, et al. GraphProt: modeling binding preferences of RNA-binding proteins. Genome Biol. 2014;15(1):R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Munteanu A, Mukherjee N, Ohler U. SSMART: sequence-structure motif identification for RNA-binding proteins. Bioinformatics [Internet] 2018 [cited 2020. Oct 20]; Available from: https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/bty404/5035778 [DOI] [PMC free article] [PubMed]
- [162].Dang LT, Tondl M, Chiu MHH, et al. TrawlerWeb: an online de novo motif discovery tool for next-generation sequencing datasets. BMC Genomics [Internet] 2018. [cited 2020 Oct 20]; 19. Available from: https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4630-0 [DOI] [PMC free article] [PubMed]
- [163].Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2018;46(D1):D239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Ru Y, Kechris KJ, Tabakoff B, et al. The multiMiR R package and database: integration of microRNA–target interactions along with their disease and drug associations. Nucleic Acids Res. 2014;42(17):e133–e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Fromm B, Domanska D, Høye E, et al. MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res. 2020;48(D1):D132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Hackenberg M, Rodriguez-Ezpeleta N, Aransay AM. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 2011;39(suppl):W132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Friedländer MR, Mackowiak SD, Li N, et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Aparicio-Puerta E, Lebrón R, Rueda A, et al. sRNAbench and sRNAtoolbox 2019: intuitive fast small RNA profiling and differential expression. Nucleic Acids Res. 2019;47(W1):W530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Dillhoff M, Liu J, Frankel W, et al. MicroRNA-21 is Overexpressed in Pancreatic Cancer and a Potential Predictor of Survival. J Gastrointestinal Surg. 2008;12(12):2171–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Cote GA, Gore JA, McElyea SD, et al. Study to Develop a Diagnostic Test for Pancreatic Ductal Adenocarcinoma Based on Differential Expression of Select miRNA in Plasma and Bile. Am J Gastroenterol. 2014;109(12):1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Michael Traeger M, Rehkaemper J, Ullerich H, et al. The ambiguous role of microRNA-205 and its clinical potential in pancreatic ductal adenocarcinoma. J Cancer Res Clin Oncol. 2018;144(12):2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Qin R-F, Zhang J, Huo H-R, et al. MiR-205 mediated APC regulation contributes to pancreatic cancer cell proliferation. World J Gastroenterol. 2019;25(28):3775–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Chang W, Liu M, Xu J, et al. MiR-377 inhibits the proliferation of pancreatic cancer by targeting Pim-3. Tumor Biol. 2016;37(11):14813–14824. [DOI] [PubMed] [Google Scholar]
- [176].Yu Y, Liu L, Ma R, et al. MicroRNA-127 is aberrantly downregulated and acted as a functional tumor suppressor in human pancreatic cancer. Tumor Biol. 2016;37(10):14249–14257. [DOI] [PubMed] [Google Scholar]
- [177].Zhang G, Liu D, Long G, et al. Downregulation of microRNA-181d had suppressive effect on pancreatic cancer development through inverse regulation of KNAIN2. Tumor Biol. 2017;39:101042831769836. [DOI] [PubMed] [Google Scholar]
- [178].Imamura T, Komatsu S, Ichikawa D, et al. Depleted tumor suppressor miR-107 in plasma relates to tumor progression and is a novel therapeutic target in pancreatic cancer. Scientific Reports [Internet] 2017. [cited 2020 Oct 29]; 7. Available from: http://www.nature.com/articles/s41598-017-06137-8 [DOI] [PMC free article] [PubMed]
- [179].Tavano F, Gioffreda D, Valvano MR, et al. Droplet digital PCR quantification of miR-1290 as a circulating biomarker for pancreatic cancer. Scientific Reports [Internet] 2018. [cited 2020 Oct 29]; 8. Available from: http://www.nature.com/articles/s41598-018-34597-z [DOI] [PMC free article] [PubMed]
- [180].Chen Y, Yang F, Zubovic L, et al. Targeted inhibition of oncogenic miR-21 maturation with designed RNA-binding proteins. Nat Chem Biol. 2016;12(9):717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Chen Y, Zubovic L, Yang F, et al. Rbfox proteins regulate microRNA biogenesis by sequence-specific binding to their precursors and target downstream Dicer. Nucleic Acids Res. 2016;44(9):4381–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]