

Abstract
The stimulator of interferon genes (STING) cellular signaling pathway is a promising target for cancer immunotherapy. Activation of the intracellular STING protein triggers the production of a multifaceted array of immunostimulatory molecules, which, in the proper context, can drive dendritic cell maturation, antitumor macrophage polarization, T cell priming and activation, natural killer cell activation, vascular reprogramming, and/or cancer cell death, resulting in immune-mediated tumor elimination and generation of antitumor immune memory. Accordingly, there is a significant amount of ongoing preclinical and clinical research towards further understanding the role of the STING pathway in cancer immune surveillance as well as the development of modulators of the pathway as a strategy to stimulate antitumor immunity. Yet, the efficacy of STING pathway agonists is limited by many drug delivery and pharmacological challenges. Depending on the class of STING agonist and the desired administration route, these may include poor drug stability, immunocellular toxicity, immune-related adverse events, limited tumor or lymph node targeting and/or retention, low cellular uptake and intracellular delivery, and a complex dependence on the magnitude and kinetics of STING signaling. This review provides a concise summary of the STING pathway, highlighting recent biological developments, immunological consequences, and implications for drug delivery. This review also offers a critical analysis of an expanding arsenal of chemical strategies that are being employed to enhance the efficacy, safety, and/or clinical utility of STING pathway agonists and lastly draws attention to several opportunities for therapeutic advancements.
Keywords: biomaterials, cancer immunotherapy, cyclic guanosine monophosphate–adenosine monophosphate synthase, drug delivery, immuno-engineering, immuno-oncology, nanomedicine, stimulator of interferon genes
Graphical Abstract

1. Introduction
The stimulator of interferon genes (STING) cellular signaling pathway has profound importance for the health and survival of a large diversity of organisms (e.g. humans, sea anemones, fruit flies, etc.)1, due to its critical role in the immune-mediated elimination of numerous pathogens and diseases2. Accordingly, elements of the STING pathway have been evolutionarily conserved within metazoans for over 600 million years through natural selection3–5. Since the relatively recent scientific discovery of the STING protein in 2008, the pathway has been extensively characterized, and a growing number of infectious pathogens and diseases have been found to stimulate host immune responses by initiating STING signaling6–10.
The STING pathway continuously monitors the cytosol of cells for certain “danger signals” (i.e. anomalies that are indicative of cellular distress) as part of a network of cytosolic pattern recognition receptors of the innate immune system – referred to as cytosolic immune surveillance. Molecular recognition of such irregularities within the cytosol initiates STING signaling (Figure 1), which then propagates a coordinated distress signal that is primarily directed by the cellular production of various proinflammatory cytokines1,11,12. The distress signal ultimately summons an innate immune response that can galvanize the immune system to address a myriad of potential threats. Notably, the immunostimulatory attributes of STING signaling distinguish the pathway as a prime target for applications in cancer immunotherapy (i.e. therapies that either involve or use components of the immune system for the treatment of cancer patients).
Figure 1: The stimulator of interferon genes (STING) cellular signaling pathway.

The cGAS enzyme surveils the cytosol of cells for the accumulation of double-stranded DNA, which serves an indicator of cellular malfunction or infection. Notably, cytosolic double-stranded DNA may arise intrinsically (e.g. self-DNA leakage from nucleus or mitochondria) or extrinsically (e.g. pathogen-derived). Upon recognition (i.e. binding) of double-stranded DNA in the cytosol, cGAS oligomerizes into liquid-like droplets and catalyzes the production of 2′3′-cGAMP, which can bind and activate the STING protein on the endoplasmic reticulum to initiate downstream signaling, primarily through TBK1 and IKK. Notably, STING activation typically leads to the activation of the transcription factors, IRF3 and NF-κB1 as well as NF-κB2, which is known to partially inhibit the activity of NF-κB1. STING signaling results in the production of IFN-I and various other proinflammatory cytokines, the profile of which largely depends on context. Lastly, 2′3′-cGAMP can also vacate its cell of origin through various transport mechanisms and function as an immunotransmitter that can locally propagate STING signaling in neighboring cells. To pharmacologically activate the signaling pathway, STING pathway agonists (i.e. cGAS agonists and STING agonists) must cross the cell membrane, access the cytosol, and evade degradation by various deoxyribonucleases (DNases) and phosphatases. Due to its relatively large size and negative charge, exogenous DNA requires assistance (e.g. pathogen-mediated delivery) to penetrate cellular membranes and gain access the cytosol. Furthermore, DNA is highly susceptible to degradation by DNase I in the extracellular space, DNase II (i.e. Acid DNase) during natural endolysosomal trafficking, and DNase III (i.e. TREX1) in cytosols. Alternatively, CDNs can utilize various membrane channels and transporters to access the cytosol, though the use of such transfer modalities is relatively inefficient and typically requires high local concentrations of CDNs. Moreover, certain naturally occurring CDNs, including 2′3′-cGAMP, are highly susceptible to degradation by ENPP1 in the extracellular space. Figure created with biorender.com.
The specific downstream effects of STING pathway activation can be largely variable, as they depend heavily on cellular context as well as signal intensity and duration13. However, a distinctive feature of mammalian STING signaling is the secretion of interferons (IFNs)14, especially type I IFNs (IFN-I) such as IFN-β15,16, which is known to exhibit pleiotropic effects on cell function17–19. Notably, the type I IFN signature of STING activation has been linked to enhanced antigen-specific T cell responses14,17,18 and natural killer (NK) cell responses20 that collectively drive cell-mediated immunity. In certain settings, STING signaling can also induce various forms of programmed cell death, such as autophagy, apoptosis, necroptosis, and lysosomal cell death21,22. Thus, the versatile nature of downstream STING signaling imparts cells with the ability to elicit a context-dependent immune response that can ultimately result in the clearance of diseased cells15,23,24.
In 2012, it was discovered that the therapeutic efficacy of the small molecule cancer therapeutic, 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) was STING-dependent, establishing that pharmacological activation of STING signaling in solid tumors could promote antitumor responses in mice with established cancer25. Shortly thereafter, in 2014, the STING pathway was found to have a central role in preventing the onset of cancer in mice through tumor immune surveillance26. The STING pathway was thus identified as a promising target for cancer immunotherapy owing to its natural role in initiating and propagating endogenous immune responses to cancer. Moreover, it has now also been shown that many standard-of-care cancer treatments (e.g. DNA-damaging chemotherapies and radiotherapy) may promote additional therapeutic benefits through iatrogenic STING pathway activation27–29. Collectively, these findings have inspired the development of synthetic STING pathway agonists for cancer immunotherapy. Preclinical research using STING agonists to treat cancer has been exceptionally successful for generating antitumor immunity against a wide range of cancer types, which has prompted numerous clinical trials, many of which are ongoing (Table 1).
Table 1:
Clinical Trials of STING agonists for Cancer Therapy.
| Phase 2 Clinical Trials: | Active Compound | Route of Delivery | Sponsor and Collaborators | Trial Identifier | Status |
|---|---|---|---|---|---|
| MIW815 +/− Pembrolizumab in Head and Neck Cancer | MIW815 (ADU-S100): Synthetic CDN STING Agonist | Intratumoral | Aduro Biotech, Inc. | NCT03937141 | Active; Not Recruiting |
| MK-1454 +/− Pembrolizumab in Head and Neck Cancer | MK-1454: Synthetic CDN STING Agonist | Intratumoral | Merck Sharp & Dohme Corp. | NCT04220866 | Active; Not Recruiting |
| Phase 1/2 Clinical Trials: | Active Compound | Route of Delivery | Sponsor and Collaborators | Trial Identifier | Status |
| CDK 002 in Advanced/Metastatic, Recurrent, Injectable Solid Tumors | CDK 002 (exoSTING): PTGFRN-Targeted Exosome containing Synthetic CDN STING Agonist | Intratumoral | Codiak BioSciences | NCT04592484 | Recruiting |
| Phase 1 Clinical Trials: | Active Compound | Route of Delivery | Sponsor and Collaborators | Trial Identifier | Status |
| MIW815 +/− Spartalizumab in Advanced Solid Tumors or Lymphomas | MIW815 (ADU-S100): Synthetic CDN STING Agonist | Intratumoral | Novartis Pharmaceuticals |
NCT03172936 | Completed |
| MIW815 +/− Ipilimumab in Advanced Solid Tumors or Lymphomas | MIW815 (ADU-S100): Synthetic CDN STING Agonist | Intratumoral | Aduro Biotech, Inc. Novartis Pharmaceuticals | NCT02675439 | Active; Not Recruiting |
| E7766 in Non-muscle Invasive Bladder Cancer | E7766: Synthetic CDN STING Agonist | Intravesical | Eisai Inc. H3 Biomedicine Inc. | NCT04109092 | Withdrawn |
| E7766 in Advanced Solid Tumors or Lymphomas | E7766: Synthetic CDN STING Agonist | Intratumoral | Eisai Inc. H3 Biomedicine Inc. | NCT04144140 | Recruiting |
| MK-1454 +/− Pembrolizumab in Advanced Solid Tumors or Lymphomas | MK-1454: Synthetic CDN STING Agonist | Intratumoral | Merck Sharp & Dohme Corp. | NCT03010176 | Active; Not Recruiting |
| MK-2118 +/− Pembrolizumab in Advanced Solid Tumors or Lymphomas | MK-2118: STING Agonist | Intratumoral / Subcutaneous | Merck Sharp & Dohme Corp. | NCT03249792 | Recruiting |
| SB 11285 +/− Nivolumab in Advanced Solid Tumors | SB 11285: Synthetic CDN STING Agonist | Intravenous | Spring Bank Pharmaceuticals, Inc. | NCT04096638 | Recruiting |
| GSK3745417 in Advanced Solid Tumors | GSK3745417: Small Molecule STING Agonist | Intravenous | GlaxoSmithKline | NCT03843359 | Recruiting |
| BMS-986301 +/− Nivolumab or Ipilimumab in Advanced Solid Cancers | BMS-986301: Small Molecule STING Agonist | Intratumoral / Intramuscular | Bristol-Myers Squibb | NCT03956680 | Recruiting |
| SYNB1891 +/− Atezolizumab in Advanced Solid Tumors and Lymphoma | SYNB1891: E. coli STING Agonist | Intratumoral | Synlogic IQVIA Biotech | NCT04167137 | Recruiting |
| Bl 1387446 +/− Ezabenlimab in Advanced Solid Tumors | Bl 1387446 (BI-STING): Synthetic CDN STING Agonist | Intratumoral | Boehringer Ingelheim | NCT04147234 | Recruiting |
| TAK-676 +/− Pembrolizumab in Advanced Solid Tumors | TAK-676: Small Molecule STING Agonist | Intravenous | Takeda | NCT04420884 | Recruiting |
| SNX281 +/− Pembrolizumab in Advanced Solid Tumors | SNX281: Small Molecule STING Agonist | Intravenous | Stingthera, Inc. | NCT04609579 | Recruiting |
| IMSA101 +/− Immune Checkpoint Inhibitor in Advanced Treatment- Refractory Malignancies | IMSA101: Synthetic CDN STING Agonist | Intratumoral | ImmuneSensor Therapeutics Inc. | NCT04020185 | Recruiting |
While STING pathway agonists offer considerable promise for cancer immunotherapy as both a monotherapy and an adjunct to current standard-of-care cancer treatments, none have yet reached the pharmaceutical market. As we will describe, the clinical landscape of STING pathway agonists is rapidly evolving with a number of promising candidates in clinical trials that may soon yield the first approval of a STING agonist for cancer immunotherapy by the US Food and Drug Administration (FDA). Nonetheless, both the efficacy and safety of STING-activating therapeutics are restricted by many drug delivery and pharmacological challenges, including poor drug stability, immunocellular toxicity, immune-related adverse events, limited tumor or lymph node (LN) targeting and/or retention, low cellular uptake and intracellular delivery, and a complex dependence on the magnitude and kinetics of STING signaling30,31. In this review, a detailed summary of the STING pathway as well as a synopsis of chemical strategies to enhance the efficacy, safety, and/or clinical utility of STING pathway agonists are presented.
2. Biochemistry and Biology of the STING Pathway
There are a number of ways through which STING signaling can be initiated. However, activation of the intracellular STING protein, or more specifically, translocation of STING to the Golgi is invariably required for the downstream STING signaling that can trigger innate immune activation32–35. In its resting state, the STING protein is localized on the surface of the endoplasmic reticulum36 and is canonically activated by cyclic dinucleotides (CDNs)37. Alternatively, STING can also be directly bound and activated by several other chemical agents, many of which will be discussed in detail in this review.
Endogenous activation of the STING protein is largely dependent upon the recognition (i.e. binding) of the self-derived CDN, 2′3′-cyclic guanosine monophosphate – adenosine monophosphate (2′3′-cGAMP)38,39. At the forefront of the STING pathway, 2′3′-cGAMP is produced intracellularly by cGAMP synthase (cGAS) after the enzyme detects the aberrant presence of double-stranded DNA (dsDNA) in the cytosol of cells. Thus, both cGAS and STING act as general sensors (i.e. pattern recognition receptors) for pathogens and pathologies that induce the cytosolic accumulation of such danger signals40.
2.1. Recognition of Cytosolic DNA by cGAS
Under normal conditions, the cytosol of cells is largely DNA free, and any nominal amount of DNA that may be present is rapidly degraded by cytosolic nucleases. Accordingly, the accumulation of DNA within the cytosol is indicative of pathogenic threats or compromised cellular states. Mammals express numerous DNA sensors that are capable of detecting and communicating such breaches in cellular homeostasis. Many of these DNA sensors can provoke IFN-I responses to activate innate immunity in response to the abnormal accumulation of either extrinsic or misplaced-self dsDNA within the cytosol41. Extrinsic DNA can infiltrate the cytosol through a variety of mechanisms (e.g. tumor-derived exosomes, viral infection, etc.), while intrinsic, self-DNA derived from mitochondria, chromosomes, or endogenous retroelements can accumulate in the cytosol in response to cellular stress or genetic mutation (Figure 1)10,42–45. Notably, many cancerous cells have an established capacity for releasing endogenous nuclear DNA into the cytosol46–48, which likely contributes to the natural role of the cGAS/STING pathway in both tumor immune surveillance and spontaneous antitumor immunity.
Upstream of STING in the pathway, cGAS is considered to be the predominant contributor to endogenous STING activation following the detection of cytosolic DNA. However, some of the other cytosolic DNA sensors (e.g. DDX41, IFI16, DAI, RNA pol III, LRRFIP1, etc.) can also initiate IFN-I responses through STING signaling49, either in conjunction with cGAS or even in the absence of cGAS50. Notably, cGAS is itself an IFN-stimulated gene (ISG)51, and therefore, in cells with low baseline cGAS expression, cytosolic DNA can initially trigger other DNA sensors. The resultant IFN-I response can then lead to local cGAS production and subsequent cGAS activation if the DNA persists long enough within the cytosol (e.g. prolonged viral challenge), thereby increasing the magnitude of the IFN-I response in a positive feedback manner.
The activation of cGAS by dsDNA has been extensively characterized through many structural and biochemical studies52–57. Briefly, cGAS exhibits an autoinhibited conformation in its unbound, monomeric form. Positively charged sites on the C-terminal domain (CTD) of cGAS bind the sugar-phosphate backbone of dsDNA. Steric interactions between cGAS and the bound DNA induce conformational transitions in cGAS that open the nucleotide binding pocket, which is also located on the CTD. The DNA strands serve as natural crosslinkers to promote cGAS oligomerization54. The dsDNA/cGAS oligomeric complexes undergo liquid–liquid phase separations within the cytosol, forming liquid-like droplets that function as intracellular microreactors for 2′3′-cGAMP production12,58. The activated cGAS enzymes catalyze the production of 2′3′-cGAMP from intracellular adenosine triphosphate (ATP) and guanosine triphosphate (GTP)38,39. The enzymatic synthesis occurs in a stepwise manner through the initial generation of 5′-pppG(2′,5′)pA prior to cyclization to c[G(2′,5′)pA(3′,5′)p]57. Notably, 2′3′-cGAMP has mixed 2′,5′ and 3′,5′ phosphodiester bonds (c[G(2′,5′)pA(3′,5′)p]) in contrast to bacteria-derived CDNs, which exclusively have two uniform 3′,5′ phosphodiester bonds57,59,60 (Figure 2). The biological consequences of CDN linkage orientation are discussed in detail in Section 4.1.
Figure 2: Chemical structures of cyclic dinucleotide (CDN) STING agonists.

(A) Mammalian 2′3′-cGAMP. (B) Various naturally occurring or synthetic CDNs with the noncanonical 2′3′ linkage orientation that is produced by mammals. (C) Various naturally occurring CDNs with the canonical 3′3′ linkage orientation that is produced by bacteria. (D) Synthetic 2′2′-cGAMP with the noncanonical 2′2′ linkage orientation that has not yet been found in nature. (E) Naturally occurring 3′2′-cGAMP with the noncanonical 3′2′ linkage orientation that is produced by Drosophila melanogaster (i.e. fruit flies).
The recognition of dsDNA by cGAS is largely sequence-independent, and the length of dsDNA that is empirically required in vitro for minimal cGAS activation in cell-based assays varies by species (e.g. ~ 45 base pairs (bp) in humans, ~ 20 bp in mice)61,62. With only a few exceptions63, short strands of dsDNA under these length thresholds cannot activate cGAS in any meaningful way, as they are unable to induce the formation of the liquid-like droplets that stabilize the dsDNA/cGAS complex through multivalent interactions58. This is largely due to the relatively low affinity of dsDNA for cGAS, the dissociation constant (KD) of which has been estimated to be ~ 1–2 μM52,54. Notably, the phase-separation of the liquid-like droplets stabilizes the dsDNA/cGAS complexes through more than just enhanced colocalization. The liquid-like droplets sequester the cGAS and dsDNA molecules, thereby providing a barrier that limits the physical access of DNA nucleases that would otherwise degrade the dsDNA ligands64. Prolonged protection from such negative regulators is especially important for cGAS, as it is considered an unusually slow enzyme with one round of cGAMP synthesis taking ~ 20 seconds65.
The cGAS enzyme is allosterically activated by dsDNA in a length-dependent manner, such that binding longer strands of dsDNA increases the presence and stability of the active dsDNA/cGAS biocondensates and thereby increases the local production of 2′3′-cGAMP66,67. Accordingly, the length of cytosolic dsDNA is a critically important determinant of both the magnitude and profile of the resultant immune response. The length-dependent cGAS activation is most pronounced at physiologically relevant low dsDNA concentrations that are comparable to that of self dsDNA sensing and viral infection (e.g. ~ 17 fg/cell for herpes simplex virus 1)13,66. At low dsDNA concentrations (e.g. 15 ng/mL), which are representative of natural exposure, dsDNA that is technically above the length threshold for activation (e.g. 100 bp) fails to induce a measurable response, while much longer dsDNA (e.g. 2000 bp) is still capable of efficiently inducing STING signaling66. Notably, at the high dsDNA concentrations (e.g. 1 μg/mL or greater) that are often assessed in vitro, cGAS activity can be saturated using a relatively low molecular weight dsDNA (e.g. ~ 60 kDa), likely through substrate exhaustion (i.e. depletion of cellular ATP and/or GTP)66.
Cytosolic dsDNA can also activate the protein known as absent in melanoma 2 (AIM2)68, which has noteworthy implications for STING signaling. AIM2 is another prominent pattern recognition receptor for cytosolic dsDNA and is known to modulate STING signaling69–74. Activation of AIM2 characteristically results in pyroptosis-mediated cell death and the release of IL-1β and IL-18 via the AIM2 inflammasome. Concurrent activation of AIM2 and cGAS in antigen presenting cells (APCs) broadens the resultant cytokine response, but it also reduces the magnitude of STING-specific cytokines produced70. The dampened STING signaling caused by simultaneous AIM2 activation is largely due to the pyroptosis induced by AIM2. At the onset of AIM2-induced pyroptosis, gasdermin D pokes small holes in the cellular membrane. The pores in the cellular membrane enable a potassium efflux from the cell, which then inhibits cGAS activation prior to cell death74.
AIM2 evolved as an innate immune sensor much more recently than cGAS (i.e. ~ 110 million years ago75 versus ~ 600 million years ago3) and is entirely orthologous between murine and human species76. Notably, AIM2 is minimally activated by relatively longer dsDNA (i.e. ~ 80 bp)77,78; robust activation of cGAS and AIM2 at in vitro concentrations of ~ 1 μg/mL generally requires dsDNA lengths of at least ~ 100 bp and ~ 200 bp, respectively67,79–83. Though, as previously stated, dsDNA length thresholds for in vitro activation do not necessarily directly correspond with thresholds for in vivo activation, because cells within a living organism do not naturally experience such high cytosolic dsDNA concentrations even under stressed cellular conditions. Future research investigating the interplay between cGAS/STING signaling and the AIM2 inflammasome in a cancer setting will be necessary to define the impact of such dual activation on antitumor immunity.
The primary effector function of AIM2 activation is to induce cell-death, which is a digital (non-tunable) process that does not depend on an allosteric equilibrium84,85. The AIM2 inflammasome does not disassemble after it has formed on sufficiently long cytosolic dsDNA, and the assembly of the AIM2 inflammasome is reinforced by multiple positive feedback loops, which supports a binary signaling response83. Conversely, cGAS activation is tunable and the downstream response can be quite variable and setting specific13,86,87. STING signaling can evoke diverse stress responses that range from the suppression of viral replication to apoptosis depending on signal strength, signaling duration, and cellular context10,13,15,88–90.
2.2. Regulation of cGAS
Mammalian DNA is primarily packaged and compartmentalized inside the nuclei and mitochondria of cells and therefore typically avoids contact with cGAS91. However, nominal amounts of self dsDNA routinely enter the cytosol under normal cellular conditions10,42–45. Mammals have evolved to locally restrict intrinsic activation of pattern recognition receptors to a baseline level by constitutively expressing deoxyribonucleases (DNases)10,92,93. DNase I, DNase II, and TREX1 (i.e. DNase III) actively degrade dsDNA in systemic circulation, lysosomes, and cytosols, respectively44,94–96. The cytosolic exonuclease, TREX1 directly affects the length, concentration, and persistence of dsDNA within the cytosol, and consequently, is critically important for negatively regulating cGAS activity97–100.
TREX1 deficiency has been linked to many type I interferonopathies caused by overactive STING signaling. Most notably, mutations in the TREX1 gene cause Aicardi-Goutières syndrome (AGS) and have also been associated with many other autoimmune diseases, including both familial chilblain lupus and systemic lupus erythematosus100. Interestingly, the genes encoding cGAS and TREX1 are both prominent ISGs and thus they contribute to local regulatory feedback loops that can either amplify or restrict the subsequent immune response in various settings51,101. Recently, the intratumoral inhibition of TREX1 has even been proposed as a novel immunotherapeutic strategy to promote local STING signaling for the treatment of cancer102. Notably, radiotherapy-induced tumor immunogenicity is strongly negatively regulated by TREX1 at high doses of radiation (i.e. 12–18 gray)103,104. It has been shown that reactive oxygen species (ROS), a biproduct of ionizing radiation105 can oxidize intracellular DNA bases106, which can then partially inhibit TREX1-mediated degradation through steric hindrance to perpetuate STING signaling during radiotherapy107,108. However, TREX1 inhibition via oxidized bases is contingent upon low TREX1 concentrations (e.g. ~ 50 nM or less); high concentrations of TREX1 (e.g. ~ 200 nM or greater) can efficiently degrade DNA containing oxidized bases109. Thus, the observed dose-dependent regulation of radiotherapy-induced tumor immunogenicity by TREX1 may be explained by dose-dependent ISG expression, where higher doses of radiation lead to higher concentrations of TREX1, which can then degrade oxidized dsDNA and thereby limit the extent of cGAS activation. In support of this theory, it was determined that consecutive low doses of radiation (i.e. 3× 8 gray) could circumvent TREX1-mediated cGAS inhibition103. Nevertheless, TREX1 represents a formidable obstacle for all DNA-based cGAS-activating cancer therapies and must therefore be given careful consideration when designing such therapeutic approaches.
In addition to TREX1, there are numerous other factors that can significantly influence the intensity of STING signaling in a particular tissue and therefore alter the nature of the resultant immune response. The activity of cGAS is known to be intricately regulated by many different post-translational modifications of cGAS, such as acetylation, glutamylation, phosphorylation, sumoylation, and ubiquitination110–115. Post-translational modifications are heavily dependent on environmental conditions and therefore likely contribute to cell-type specific STING signaling. cGAS activation is also vitally dependent on the ability of cGAS to encounter its dsDNA substrate, which is undoubtedly a function of the protein’s spatiotemporal distribution within cells.
The subcellular localization of cGAS is currently a subject of controversy and seems to be quite dynamic in nature depending on cell cycle phase, cell type, and environmental conditions116. Until recently, cGAS has generally been regarded as a strictly cytosolic protein38; however, recent studies have challenged this theory. In murine bone marrow-derived macrophages (BMDMs) and in human THP1 monocytes, it was determined that cGAS primarily resides on the interior of the plasma membrane due to the electrostatic interactions of the N terminus of cGAS with the membrane-bound PI(4,5)P2 phospholipid117. The intracellular localization of cGAS to the plasma membrane was found to limit the recognition of self dsDNA by spatial segregation from the nucleus and simultaneously maximize the potential response to viral infection by allowing for a more rapid encounter with exogenous DNA.
cGAS has also been identified within the nuclei of mammalian cells38,118–120. Outside of the canonical STING signaling axis, cGAS has an established secondary function, where it operates as a negative regulator of DNA repair, inhibiting homologous recombination in the nucleus121,122. Several research groups currently contend that cGAS is constitutively present in the nuclei of cells at steady state122–124. One study found that the non-catalytic N terminal domain of cGAS was responsible for an association of cGAS with the centromeres of chromosomes within the nuclear compartment123. More recently, another study has asserted that cGAS is predominantly a nuclear protein that is tethered tightly to intact chromatin by a salt-resistant interaction in its resting state124. The researchers found that cGAS was resistant to standard salt-based elution, requiring relatively high salt concentrations for complete solubilization (e.g. 0.75 M NaCl compared to the 420 mM NaCl that is typically used to isolate nuclear proteins). They have suggested that the observed tight interactions of cGAS in the nucleus cannot be explained by its relatively low intrinsic affinity for DNA (e.g. KD ~ 1–2 μM).
These disparate findings indicated that the N terminus of cGAS was dispensable for nuclear localization; instead, the core of human cGAS, composed of a bilobed nucleotidyltransferase structure bridged by an alpha-helical spine, was required for the observed nuclear tethering. It was noted that the amino acid residues, which are important for nuclear tethering, partially overlap with one of the DNA-binding surfaces of cGAS. Consequently, a model of “regulated desequestration” was proposed, which proclaims that cGAS is inactive while chromatin-bound and that there exists an unknown regulated step prior to the assembly of cGAS onto dsDNA that enables its release from chromatin and subsequent activation.
Chromatin tethering is indeed one of several regulatory mechanisms that can inhibit cGAS activation at times where immune activation is unnecessary (e.g. cell division)110,125,126. Specifically, chromatin tethering can prevent the oligomerization of cGAS that is necessary for liquid-like droplet formation and efficient 2′3′-cGAMP synthesis126. Accordingly, the tethering of cGAS to chromatin actually increases during mitosis when the nuclear envelope breaks down, so as to prevent spurious activation of cGAS while DNA is exposed to the cytosol119,127. Further research in this area may lead to the discovery and characterization of the aforementioned unknown regulatory mechanism that is responsible for the release of cGAS from nuclear chromatin, which may thereby enable targeted strategies for controlling the degree of cGAS activation to enhance cancer therapies.
2.3. Regulation of STING
After cGAS catalyzes the synthesis of 2′3′-cGAMP, the CDN acts as a second messenger that binds and activates STING proteins on the endoplasmic reticulum36,39,59. STING comprises four transmembrane helices coupled to a cytoplasmic ligand-binding and signaling domain128. The transmembrane and cytoplasmic regions naturally interact to form a domain-swapped homodimer in its resting form129. Two intertwined STING molecules take the shape of an opened butterfly with the head toward the membrane (Figure 3A)130. Upon binding 2′3′-cGAMP, the STING homodimer undergoes extensive conformational rearrangements. While 2′3′-cGAMP induces closure of the ligand-binding domain, it is important to note that not all agonists of STING provoke a closed lid confirmation (Figure 3B). Indeed, several STING agonists (e.g. the bacteria-derived CDN, cyclic di-guanosine monophosphate (c-di-GMP)) promote STING oligomerization and exhibit immunostimulatory activity without rearrangement of the lid region (Figure 3C)131–133.
Figure 3: Crystal Structures of symmetrical human STING dimers.

(A) The resting ‘Open Lid’ configuration of an apo (i.e. unbound) human STING dimer. Adapted with permissions from reference145. Copyright © 2020 American Association for the Advancement of Science; permission conveyed through Copyright Clearance Center, Inc. (PDB ID: 4F9E)131. Copyright © 2012 Elsevier Science & Technology Journal; permission conveyed through Copyright Clearance Center, Inc. (B) The ‘Closed Lid’ configuration of a holo (i.e. ligand bound) human STING dimer bound to 2′3′-cGAMP. Adapted with permissions from reference145. Copyright © 2020 American Association for the Advancement of Science; permission conveyed through Copyright Clearance Center, Inc. (PDB ID: 4KSY)59. Copyright © 2013 Elsevier Science & Technology Journal; permission conveyed through Copyright Clearance Center, Inc. (C) The ‘Open Lid’ configuration of a holo (i.e. ligand bound) human STING dimer bound to 3′3′-diGMP. Adapted with permissions from reference145. Copyright © 2020 American Association for the Advancement of Science; permission conveyed through Copyright Clearance Center, Inc. (PDB ID: 4F9G)131. Copyright © 2012 Elsevier Science & Technology Journal; permission conveyed through Copyright Clearance Center, Inc.
Activated STING proteins oligomerize, are ubiquitinated, and then traverse the Golgi apparatus, whereupon they are palmitoylated and traffic to submicrometer-sized perinuclear vesicles (i.e. STING translocators)32,134–138. Following translocation though the Golgi body, TANKbinding kinase 1 (TBK1) binds and phosphorylates STING139,140. Notably, TBK1 recruitment to STING has been identified as essential for STING-mediated antitumor immunity141. The STING/TBK1 complex phosphorylates interferon regulatory factor 3 (IRF3), which then homodimerizes and navigates into the nucleus to induce target gene expression136,142–144.
Similar to the liquid phase condensation of cGAS that is triggered by activation of the endogenous STING pathway58, untranslocated ER-resident STING can also undergo a liquid-liquid phase separation146. However, unlike the liquid-like droplets of activated cGAS that enhance STING signaling, the STING condensates contain inactive STING proteins and negatively regulate the pathway by preventing the translocation of STING that is necessary for downstream signaling147. When intracellular 2′3′-cGAMP concentrations reached a certain threshold (e.g. 1 μg/mL in vitro), which is above the threshold for the initial activation of STING by 2′3′-cGAMP (e.g. KD ~ 4.59 nM), STING condensates form as micrometer-sized granules that colocalize with the ER. Additionally, when present in an exceptionally high concentration (e.g. 6 μg/mL in vitro), 2′3′-cGAMP also further induces a fluid-to-gel transition of the STING condensates that significantly decreases their internal molecular mobility. Notably, the STING condensates also formed in response to the bacterial CDN, c-di-GMP. It is currently unclear whether the phase separation of STING occurs in response to all of the known STING agonists or just CDNs. It is also unknown if constitutively active STING mutants trigger the assembly of the STING phase-separator.
While most hydrogels of biocondensates formed by protein liquid-liquid phase separation are largely disordered or assemble into polymeric fibrils148,149, the STING condensates, now termed the STING phase-separator, surprisingly comprise a highly organized membranous structure that resembles jigsaw puzzles. Following DNA virus infection, formation of active STING translocators occurred 3 hours post infection and peaked at 8 hours, whereas inhibitory STING condensates peaked at 20 hours. Thus, formation of the STING phase-separator is a partially delayed response and serves to prevent overactivation of STING and inhibit excessive innate immune signaling.
Additional transcription factors synergize with IRF3 to direct context-dependent antiviral gene expression150. In various settings, STING signaling has been associated with the activation of canonical and non-canonical nuclear factor κB (NF-κB), mitogen-activated protein (MAP) kinases, and signal transducer and activator of transcription (STAT) transcription factors36,151–154. Notably, efficient production of IFN-β, a hallmark of STING signaling, relies on the cooperative assembly of the enhanceosome, a higher order transcription enhancer complex155,156. Individual transcription factors of the enhanceosome, such as IRF3 and canonical NF-κB, cannot initiate IFN-β gene expression by themselves157,158. Instead, they must work in conjunction with each other and several other enhancer components for maximal gene transcription16. Indeed, a 50% decrease in IFN-β production was observed in primary mouse embryonic fibroblast cells when canonical NF-κB expression was partially silenced via RNA interference (RNAi)151.
The intricacy of the enhanceosome elegantly highlights the importance of synergy between multiple inducible transcription factors159. Thus, in addition to post-translational modifications of STING pathway constituents, the combinatorial regulation of gene transcription likely contributes to cell-type specific STING signaling, as it is largely responsible for the selective protein expression that occurs in various environmental conditions. Accordingly, a better understanding of the transcriptional regulation that ensues STING activation in various cell types could lead to more efficacious cancer immunotherapies designed to differentially regulate the expression of certain STING-stimulated proteins to enhance antitumor effects and minimize unnecessary off-target effects.
Single nucleotide polymorphisms in the STING protein are responsible for existence of distinct human STING (hSTING) isoforms that exhibit variable intrinsic activity as well as distinctive reactivity to various STING agonists57,160,161. The five most prominent haplotypes of hSTING are known as WT (R232), HAQ (R71H, G230A, R293Q), REF (R232H), AQ (G230A, R293Q), and Q (R293Q), and their allelic frequencies in the human population are 57.9%, 20.4%, 13.7%, 5.2%, and 1.5%, respectively15,160. Relative to the other major variants, hSTINGHAQ generally exhibits lower intrinsic IFN-I and NF-κB activity, which has been attributed to the R71H substitution that likely affects the protein’s resting localization to the endoplasmic reticulum160,162,163.
There are many agonist-specific differences in the recognition and activation of the various STING isoforms that can be attributed to the unique chemical structures of the STING agonists. While bacteria-derived CDNs can activate murine STING (mSTING) and certain hSTING variants, they do not appreciably activate the hSTINGREF or hSTINGQ isoforms15,160,161. Alternatively, endogenous 2′3′-cGAMP can activate mSTING as well as all 5 of the major hSTING variants15,59. However, whether 2′3′-cGAMP is a weak agonist for certain hSTING isoforms is currently a controversial topic. Some researchers have reported that for hSTINGREF, 2′3′-cGAMP is weaker agonist, exhibiting reduced IFN-I activity, despite generating comparable NF-κB activity160,164. Conversely, others have shown that 2′3′-cGAMP engenders no significant difference in its inducible IFN-I activity with the hSTINGREF isoform15. Furthermore, the small molecule, DMXAA potently activates mSTING, but is unable to activate any of the hSTING variants165. Thus, the isoforms of STING represent a crucial design consideration for the clinical development of any STING agonist, as translatability will favor universal STING agonists.
2.4. Regulation of cGAMP
In addition to binding STING within the cell of origin, endogenous 2′3′-cGAMP can also vacate the native cell and thereby function as an immunotransmitter to neighboring cells166,167. The accumulation of intracellular cGAMP that follows robust cGAS activation creates a strong electrochemical gradient that promotes cGAMP expulsion168,169. The distribution of cGAMP to nearby cells can occur in several different ways, either directly (e.g. cell-to-cell) or indirectly (e.g. secretion followed by proximal cellular uptake). Direct cell-to-cell transfer of cGAMP may occur through connexin-dependent intercellular gap junctions, cellular fusion, and phagocytosis of dead or dying cells47,170–176. Notably, the predominant gap junction protein involved in cGAMP transfer, connexin-43 (Cx43) is also established as a tumor suppressor in many types of cancer177–179. Although cGAMP transfer has not yet been directly linked to the anticancer role of Cx43, facilitating STING signaling in a time of cellular stress could potentially support a tumor suppressor function via the activation of innate immunity. In contrast to the direct transfer of cGAMP, indirect transfer may be mediated by ion channels, transport proteins, virions, and extracellular vesicles released from infected or apoptotic cells167–169,180–187.
Many of these cGAMP transfer modalities have limited functionality in various settings, as several are largely dependent on cellular context, viability, and/or infection status. Indeed, the unidirectional cell membrane transporter, SLC19A1 was shown to be important for cGAMP import in U937 monocyte-derived cells and monocytic THP1 cells, but was also found to be minimally expressed in many other cell types167,181. Additionally, SLC46A2 has more recently been identified as the dominant cGAMP importer in primary human monocytes and monocyte-derived macrophages187. Conversely, gap junctions containing Cx43 and volume-regulated anion channels (VRACs) have important roles in cell survival and are therefore ubiquitously expressed in human cells179,188–190.
Gap junctions form intercellular channels in appositional cellular membranes and thereby promote direct cellular communication and nutrient exchange, both of which are essential to cellular physiology179. VRACs also help maintain cellular homeostasis, though they do so by counteracting dynamic cytoplasmic pressures190–192. Gap junctions193,194 and VRACs168,169 are both capable of two-way molecular transit, unlike some transporters that are simply unidirectional (e.g. the cell-specific cGAMP importers, SLC19A1167,181 and SLC46A2187). Thus, gap junctions and VRACs represent the main cGAMP transfer mechanisms in humans, though the contribution of each is likely tissue specific. Gap junctions were recently found to be essential for cGAMP transfer in lungs upon nanoparticulate STING agonist administration and also in livers following alcohol-induced hepatocyte injury173,174. Alternatively, VRACs were identified as the dominant cGAMP importer in telomerase-immortalized human microvascular endothelial cells, which are characteristic of many tumor microenvironments (TMEs)169.
Notably, gap junctions enable transfer of cGAMP to a limited number of connected cells, while VRACs allow for secretion into the extracellular space and likely enable cGAMP transmission to a larger number of cells via paracrine signaling. Indeed, VRACs were found to be responsible for ~ 50–70% of cGAMP uptake in a wide variety of cell types168. While cGAMP and other CDN STING agonists may enter cells through these portals, the efficiency of cellular import appears to be quite low for these compounds. Notably, when cells are treated in vitro with cGAMP or other CDNs, dose-response studies for STING pathway activation typically yield values for the half-maximal effective concentration (EC50) in the high micromolar range, suggesting inefficient CDN entry into the cytosol via the membrane transporters as well as poor cell membrane permeability due to their polar nature and negative charge195. This cytosolic delivery barrier has inspired the development of nanotechnology to enhance the intracellular delivery of exogenous STING agonists196, which we discuss in detail below (Figure 4).
Figure 4: Intracellular delivery challenges for STING pathway agonists.

Exogenous DNA and CDNs are negatively charged and hydrophilic and consequently cannot readily access the cytosol to activate the STING pathway. While both natural and synthetic CDNs are small enough to infiltrate the cytosol through the use of membrane channels and transporters, these transport modalities are inefficient. Furthermore, extracellular nuclease and phosphatases quickly degrade exogenous DNA and natural CDNs, respectively. Accordingly, relatively high concentrations of CDNs are required to elicit measurable STING activation. Non-nucleotide, small molecule agonists of the STING pathway have potential to passively diffuse across the cell membrane and therefore are an attractive alternative to the natural agonists. Lastly, certain nanocarriers can improve the efficacy and safety of STING pathway agonists by promoting intracellular delivery. Figure created with biorender.com.
Currently, there is no indication that extracellular cGAMP preferentially spreads into any particular cell type, since gap junctions and VRACs are so broadly expressed. Rather, cGAMP likely distributes indiscriminately, but predominantly enters local cells due to the presence of ecto-nucleotide pyrophosphatase phosphodiesterase 1 (ENPP1) in the extracellular space166,197. ENPP1 hydrolyzes extracellular cGAMP and thus prevents extensive spread197,198. Elevated expression of ENPP1 has even been correlated with tumor development in several cancer types166,199,200. Accordingly, inhibitors of ENPP1 are currently being developed for cancer immunotherapy166,201. Synthetic STING agonists without phosphodiester bonds have also been engineered to avoid degradation by ENPP1 and thereby enhance drug stability. Phosphorothioate modifications are commonly employed as they are resistant to ENPP1 degradation and may even enhance cellular uptake and cytosolic delivery15,197,202,203. Though the development of nonhydrolyzable analogs of cGAMP has circumvented the issue of extracellular degradation, evading ENPP1 remains an important design criterion for therapies that exploit natural cGAMP from endogenous STING signaling (e.g. radiotherapy).
The manner in which cGAMP is transferred also uniquely affects the mechanism of action for subsequent STING signaling. Unlike intracellular CDNs that trigger classical cGAS/STING signaling, extracellular CDNs can activate an alternative cGAS/STING signaling pathway204. Liu et al. found that cells primarily endocytose extracellular CDNs in a clathrin-dependent manner. Endocytosed CDNs were released into the cytosol through an unidentified mechanism that required endosome maturation and acidification, whereupon the internalized extracellular CDNs bound cGAS directly. A CDN/cGAS/STING complex was subsequently formed and ultimately activated IRF3. Exceptionally similar downstream effects have been observed between this alternative pathway and the classical pathway, though overall protein expression seems to differ in magnitude with intracellular CDNs and the classical pathway evoking a greater response196,204. In vivo cancer therapies that use CDN STING agonists without a cytosolic delivery agent likely activate this alternative STING signaling pathway and consequently may not maximize their immunostimulatory potential.
The duration of STING pathway stimulation is also a critically important consideration, as it can dramatically influence the balance between immunological outcomes (Figure 5). While, acute and localized activation of the STING pathway generally supports an appropriate level of immune activation for disease eradication, chronic STING signaling can elicit many inflammation-driven diseases. Such diseases include monogenic autoinflammatory syndromes (e.g. STIN-Gassociated vasculopathy with onset in infancy (SAVI), AGS, familial chilblain lupus, etc.), autoimmune diseases (e.g. systemic lupus erythematosus and rheumatoid arthritis), neurological disorders (e.g. ischaemic brain injury, Parkinson disease, Huntington disease, age-dependent macular degeneration, etc.), metabolic diseases (e.g. nonalcoholic steatohepatitis (NASH), alcoholic liver disease, etc.), inflammatory diseases (e.g. sepsis), cardiovascular diseases (e.g. myocardial infarction), cancer (e.g. metastases), as well as senescence and aging205.
Figure 5: The importance of STING signaling kinetics.

The distinct outcomes of STING activation are balanced by signal persistence. Chronic STING signaling, which is quite often the result of genetic mutations, can lead to numerous IFN-driven inflammatory diseases, autoimmunity, and even cancer metastasis. Conversely, transient STING signaling, which can be induced by the acute STING activation from STING pathway agonists, can galvanize robust antiviral and/or anticancer immunity. Figure created with biorender.com.
Since prolonged stimulation of the STING pathway can lead to lethal inflammatory disease151 as well as cancer development and metastasis in certain settings206–208, the degree and persistence at which cGAMP is able to spread and activate STING in neighboring cells can play a large role in disease pathogenesis. Indeed, STING-induced metastasis in the context of brain cancer has been observed and attributed to the continuous transfer of cGAMP from cancerous cells to neighboring astrocytes via gap junctions47. Therefore, in order to avoid promoting disease progression, careful thought should be given to treatment regimen and the cellular context of the treatment location when designing cancer therapies that exploit cellular transfer of cGAMP.
3. STING and the Cancer Immunity Cycle
3.1. Intrinsic STING Signaling and Innate Antitumor Immunity
The main process through which the immune system recognizes and eliminates cancer has been described as the Cancer Immunity Cycle (CIC)209. The CIC summarizes how antitumor cellular immune responses are initiated and propagated through cooperation between the innate and adaptive immune systems. In principal, the cycle perpetually functions to inhibit cancer formation and growth through the following major steps: 1) Antigen Processing and Presentation, 2) Lymphatic Trafficking, 3) T Cell Priming and Activation, 4) Systemic Trafficking of T Cells, 5) Infiltration of T Cells into Tumors, 6) Immune Recognition of Cancer Cells, and 7) Killing of Cancer Cells / Antigen Release.
Spontaneous CIC operations that prevent the immune escape of pre-cancerous cells can be largely dependent on STING signaling210,211. Mechanistic studies using genetically engineered mouse models of immunodeficiencies have identified STING signaling as an integral mechanism for innate immune sensing of immunogenic cancers. Notably, wildtype mice with functional STING signaling exhibited attenuated tumor growth relative to mice that were deficient in various STING pathway components23,26. In accordance with the CIC, the innate antitumor effects of intrinsic STING signaling have been primarily attributed to enhanced tumor antigen-specific T cell responses212,213. While the STING pathway was found critical to the spontaneous priming of antitumor T cells in certain murine tumor models, several other pattern recognition receptor pathways, including RIG-I and various Toll-like receptors (TLRs) were less essential for generating cell-mediated antitumor immunity despite their conserved ability to induce the production of type I IFNs26. Additionally, in accordance with the dependence of immune checkpoint blockade (ICB) on spontaneous T cell responses, it has been established that functional STING signaling is critical for the maximal efficacy of ICB in murine tumors26,214,215.
The development of cancer is often the result of immunosuppression that impedes the favorable progression of the CIC. Indeed, selective pressure can lead to the deregulation of STING signaling, a prevalent mechanism by which cancer cells evade tumor immune surveillance6,7. In two seminal reports that characterized the functionality of STING signaling in human colon cancer and human melanoma, Barber and colleagues discovered that cGAS and/or STING expression was absent in ~ 54% of colon cancers examined (i.e. 21/39 patient samples)6 and ~ 54% of melanomas examined (i.e. 30/56 patient samples)7 as determined by immunohistochemistry analysis, and greater silencing of cGAS and/or STING expression was observed in the late stages of both cancers relative to their respective earlier stages. Interestingly, the genes encoding cGAS and STING were found to be seldom mutated in pan-cancer (i.e. less than 1% of documented human tumors exhibit missense, nonsense, or frame shift mutations in the cGAS or STING gene)216,217. Instead, epigenetic silencing of cGAS and/or STING is considered the predominant cause of the STING signaling dysfunction that is observed in the immune escape of various cancers6,7,11,216. Accordingly, epigenetic modifications (e.g. hypermethylation of promoter regions, histone modifications, etc.) of the cGAS and/or STING loci, in addition to the possible deregulation of essential signaling partners downstream of STING activation, are likely responsible for poor expression of cGAS and/or STING in as much as 50% of human tumors, though the exact frequency of tumors that have effectively silenced the STING pathway has not yet been reported for pan-cancer and is likely to be tumor-type specific. Furthermore, while many cancers can deregulate STING signaling in the cancer cell compartment, immune cells that are present in those tumors are unlikely to lose their capacity for STING signaling and therefore make ideal targets for STING pathway agonists in such cancers. Thus, cancers with deregulated STING are not necessarily precluded from the therapeutic benefits of STING pathway agonists.
The cellular transfer of cGAMP and/or tumor-derived dsDNA to stromal cells (e.g. myeloid cells, endothelial cells) becomes particularly important for tumor immune surveillance when STING signaling becomes deregulated in cancer cells.104,176. Extrinsic STING signaling may then be employed to promote immune recognition, generation of antitumor immunity, and subsequent immune-mediated elimination of such cancer cells. Notably, it has been suggested that the STING protein may facilitate the intracellular clearance of cGAMP93. Therefore, cGAMP could be prone to accumulate more rapidly in the cytosol when expression of the STING protein is suppressed in cancer cells. Such accumulation of cGAMP in tumor cells could generate high intracellular concentrations that would promote cGAMP transfer to surrounding cell populations. Thus, tumorigenesis could be prevented by activation of antitumor immunity, provided the degree of cGAMP spread was sufficiently high to stimulate innate immune activation. However, this is clearly insufficient to prevent the development of all cancers, since deregulated STING is a common feature of many immune-evasive tumors. Factors such as ENPP1 may critically inhibit the degree of extrinsic STING signaling despite an increased efflux of cGAMP from cancer cells. Restricted cGAMP transfer in such cases might even contribute to the development of tumors with deregulated STING, as sustained low-level STING signaling may actually promote tumor growth and metastasis47,152,206,218, especially for tumors with low antigenicity219.
3.2. Therapeutic Effects of Type I Interferons
As previously mentioned, generation of antitumor innate and adaptive immunity is considered the primary mechanism by which STING activation can combat cancers155. Indeed, in response to STING agonist treatment, antitumor immunity is mainly responsible for the tumor regression observed in murine tumor models as well as the sustained protection against disease recurrence demonstrated by efficacy in tumor rechallenge experiments23,24,220. Such therapeutic responses have been largely attributed to type I IFN signaling in addition to other proinflammatory cytokines (e.g. TNF-α) downstream of STING activation.
Type I IFNs (i.e. IFN-α and IFN-β) are signature cytokines of STING activation and are considered a primary effector induced by STING signaling14. Type I IFNs directly regulate the transcription of over 100 genes that influence protein synthesis, autophagy, apoptosis, angiogenesis, and immunity17. Notably, the direct administration of type I IFNs into solid tumors has demonstrated clinical efficacy, and in 1986, recombinant IFN-α2 became the first immunotherapeutic approved by the FDA for the treatment of cancer. Many mechanisms of action have been proposed for the therapeutic effect of type I IFNs in the treatment of cancer, including both immune-mediated and immune-independent mechanisms. In various settings, type I IFNs have been found to directly inhibit tumor cell proliferation221–223, disrupt tumor vasculature224,225, prompt the maturation of various APCs226–228, induce CTL responses229,230, and activate NK cells223,231,232.
For any IFN-driven cancer therapy (e.g. targeted STING pathway activation), the dosing of type I IFN and/or type I IFN inducers is a critically important therapeutic design consideration, as they can directly influence the mechanism of antitumor activity18,233. Cancer treatments that implement high levels of intratumoral type I IFN can result in significant tumor regression that is largely independent of host adaptive immunity and instead depends heavily upon disruption of the tumor vasculature224. This high-dose ablative effect on tumors has also been observed with STING agonists234 and may be related to the type I IFN component of downstream STING signaling. Similar to the dose-dependence of type I IFN treatment, robust antitumor T cell responses are achieved in murine tumor models with lower, more immunogenic doses of STING agonists, and excessive STING activation fails to sufficiently generate the antitumor immunity that can prevent tumor growth upon rechallenge.
In addition to dosing, timing of intratumoral type I IFN administration and/or induction can affect the development of antitumor immunity, which is essential for durable responses and long-term survival18,233. As stated previously, type I IFNs induce DC maturation226–228. When DCs undergo maturation, they lose their phagocytic ability, thereby preventing the capture of new antigens in favor of an increased ability to cross-prime naïve CD8+ T cells that are specific for antigens previously internalized by the DCs235. Accordingly, Tzeng et al. found that generation of antigenic tumor debris must precede the induction of type I IFNs in order to efficiently prime long-term antitumor immunity236.
Though type I IFNs have demonstrated efficacy in the treatment of cancer, they have also been associated with systemic adverse effects, which have limited their clinical use. The observed side effects for type I IFN therapies included fever and chills upon initial administration and fatigue, depression, and anorexia with continued treatment237,238. While the production of type I IFNs is a critically important component of STING signaling for promoting antitumor immunity, other IFN-independent signaling pathways downstream of STING activation (e.g. NF-κB signaling) are also important for immune regulation239 and can act to balance and resolve the resultant immune response240.
3.3. Immunological Effects of STING Activation
The CIC can be considered as the “central dogma” of cancer immunotherapy; in order to work effectively, cancer immunotherapies must harness the CIC and promote it, either by pushing the cycle forward or by removing the restraints that impede the proper operation of the cycle. Accordingly, the great potential of STING pathway agonists for cancer immunotherapy arises from their exceptional capacity to bolster antitumor immune responses by promoting each phase of the CIC (Figure 6). Indeed, STING has been described as a master regulator of the CIC241.
Figure 6: STING and the Cancer Immunity Cycle.

STING can promote antitumor immunity via the Cancer Immunity Cycle by promoting each of the following steps: 1) Antigen processing and presentation, 2) Lymphatic trafficking, 3) T cell priming and activation, 4) Systemic trafficking of T cells, 5) Infiltration of T cells into tumors, 6) Immune recognition of cancer cells, and 7) Killing of cancer cells / antigen release. Figure created with biorender.com.
3.2.1. Tumor Antigen Processing and Presentation
The production of type I IFNs is essential for the STING-mediated propagation of the CIC. Type I IFNs prompt the maturation of various APCs, promoting the expression of major histocompatibility complex (MHC) molecules, costimulatory molecules, and various other proinflammatory cytokines that are required for T cell priming and activation242. Indeed, STING signaling has been found to stimulate antigen processing and presentation in a manner that is dependent on type I IFN243,244.
A particular subset of DCs known as CD8α+ Batf3 DCs have been described as the main APC responsible for generating antitumor T cells245,246. CD8α+ Batf3 DCs typically reside in secondary lymphoid tissues and are characterized by an exceptional capacity for antigen cross-presentation (i.e. the process of antigen internalization and subsequent antigen presentation in complex with MHC-I to CD8+ T cells). Notably, type I IFN production within solid tumors, like that induced by STING activation, promotes the intratumoral accumulation of CD8α+ Batf3 DCs from surrounding tissues230. Additionally, interferon-alpha/beta receptor (IFNAR) signaling within tumor-infiltrating CD8α+ Batf3 DCs is required for successful cross-priming of tumor antigen–specific CD8+ T cells and subsequent immune control of tumor growth230,247.
Matured APCs, especially matured DCs, upregulate CC-chemokine receptor 7 (CCR7), causing them to enter the lymphatic vasculature, which expresses the CCR7 ligand, CC-chemokine ligand 21 (CCL21)248,249. The APCs then further migrate to the tumor draining lymph nodes (tdLNs), where they can interact with naïve T cells. While T cell activation is thought to primarily occur in tdLNs, it has been suggested that intratumoral expression of type I IFNs may also prompt tumor-infiltrating CD8α+ Batf3 DCs to cross-prime CD8+ T cells within the TME, thus bypassing the need for migration to the tdLNs245. Indeed, the direct activation of naïve T cells in tumors has been observed in mice that were treated with a T cell recirculation blocker250 as well as in mice that were devoid of LNs and spleens251. Furthermore, targeted STING activation within B16-F10 murine melanoma tumors has been reported to induce the intratumoral formation of tertiary lymphoid structures, which may also serve as a local site for T cell priming to occur252.
In addition to the type I IFN effects of STING signaling, there are other downstream effects of STING activation that can also enhance tumor antigen processing and presentation. Notably, STING activation typically results in the production of other proinflammatory cytokines (e.g. IL-6 and TNF-α) and reactive oxygen and nitrogen species that can promote M1-like polarization of macrophages253,254. Moreover, STING signaling can even repolarize the phenotype of existing tumor resident macrophages from M2 to M1254,255. While M2 macrophages tend to be immunosuppressive and protumor, M1 macrophages are more conducive to effective cancer treatments, as they can inhibit the proliferation of surrounding cells via paracrine signaling253 and also induce lysis in various types of cancer cells256,257.
3.2.2. T Cell Priming and Activation
Generally, three signals are required from APCs to activate naïve T cells: peptide antigen displayed on MHC molecules for recognition by the T cell receptor (TCR), co-stimulatory molecules, and certain proinflammatory cytokines, all of which can be enhanced by STING activation as just described. Thus, T cell priming and activation in the tdLNs naturally follow the STING-mediated APC response.
The two major types of effector T cells are MHC-I–restricted CD8+ T cells, which are known as cytotoxic T lymphocytes (CTLs) and MHC-II–restricted CD4+ T cells, which are known as helper T lymphocytes. A main function of CTLs is to directly kill diseased cells that express and present their cognate antigen258, while helper T lymphocytes tend to regulate the function of other immune cells via paracrine signaling259. Notably, functional APC responses from STING signaling can enhance the activation of both CTLs230,244 and helper T lymphocytes243,260. The CTLs are generally considered to be the primary driver of the antitumor immune responses that are stimulated by STING signaling212,213. However, the helper T lymphocytes are known to support CTL function and cytolytic activity. Indeed, in response to STING signaling, the helper T lymphocytes exhibit a balanced Type 1 / Type 2 (Th1/Th2) phenotype, with slightly greater Th1 activity261, which promotes M1 macrophage polarization262,263.
In addition to stimulating T cell responses through the activation of innate immunity, STING signaling can also directly influence antitumor T cell function. STING signaling within T cells has been shown to have varied effects depending on the degree and duration of the stimulus. Hyperactivation of STING can drive antiproliferative and apoptotic signaling within T cells86,264,265. Some lesser degree of STING signaling within T cells does however maintain CD8+ T cell stemness, which can improve T cell-mediated tumor clearance213. In light of this dichotomous role of STING signaling in T cells, careful evaluation of how STING pathway agonists impact antitumor T cell viability and effector function will be critical to maximizing immunotherapeutic responses.
3.2.3. Systemic Trafficking and Tumor Infiltration of T Cells
Before CTLs can recognize and kill cancer cells, they must egress the tdLNs and traffic to tumor sites. Like matured DCs, naïve T cells are largely attracted to and retained within LNs through their expression of CCR7266. Activated T cells migrate out of LNs and into systemic circulation by downregulating CCR7 and simultaneously upregulating the receptor for sphingosine1-phosphate (S1P)267, which is a signaling sphingolipid that is present in the blood at much higher concentrations than in lymphoid organs268,269. Once activated T cells accumulate in the bloodstream, they require additional signals for direction to their effector site. STING signaling generates a chemokine gradient (e.g. CXCL9 and CXCL10) that can guide T cell extravasation into solid tumors230,246,270. Notably, CXCL9 and CXCL10 are also capable of driving NK cell recruitment, activation, and maturation271. Moreover, activated NK cells can augment adaptive antitumor immunity by recruiting additional DCs to the TME272.
Despite the powerful effects of chemokine gradients, dysfunctional tumor vasculature, a common feature of many cancers, can still act as a major barrier to immune cell infiltration and function273. However, vascular normalization, a reversal of tumor vessel abnormalities, has been shown to increase T cell infiltration and restore T cell function273. In addition to promoting chemokine gradients, STING activation can also normalize tumor vasculature and thereby further enhance T cell infiltration into tumors. Specifically, the direct injection of STING agonists into solid murine tumors results in reduced blood vessel density and vascular sprouts as well as an increase in pericyte coverage and an upregulation of endothelial-leukocyte adhesion molecules274. The normalized tumor vasculature that ensues STING activation has been found to facilitate the intratumoral trafficking of effector T cells across the endothelial barrier and condition the TME to enhance antitumor immunity252,274. Notably, while other agents can also normalize tumor vasculature, STING-activating therapeutics offer the potential for coordinating vascular remodeling with reprogramming of the immune microenvironment, which can allow T cells to more efficiently home to tumor sites and perform their effector function.
3.2.4. Recognition and Killing of Cancer Cells / Cancer Antigen Release
STING signaling can trigger tumor elimination either by directly inducing cell death programs in cancer cells275 or indirectly via mechanisms involving the immune system, particularly CTLs26 and NK cells20,276. Notably, the direct induction of cell death programs in cancer cells appears to be most pronounced in hematopoietic malignant cells, such as B cell and T cell lymphomas86. As demonstrated by numerous murine tumor models where immune cells have been knocked out or inhibited, antitumor immune responses are the primary cancer elimination mechanism promoted by STING signaling155.
Antitumor immunity can be enhanced by intratumoral STING signaling in a multitude of ways. STING signaling can promote the expression of MHC-I on the surface of cancer cells to enhance the recognition of cancer cells by CTLs, which promotes CTL-mediated cancer cell death277. Some tumors can however evade this cellular response through loss of MHC-I expression or lack of tumor antigens278–280. NK cells can act to overcome such evasion mechanisms by recognizing stress-induced cells, particularly those that have lost MHC-I, and eliciting a cytotoxic response278,279. NK cells have also been reported to drive tumor cell killing in cancers with poor antigenicity19. Indeed, it has recently been described that NK cells mediate the clearance of CTL-resistant tumors in response to STING agonists281. Furthermore, STING signaling within cancer cells has also been shown to upregulate ligands for the NK cell-specific immunoreceptor, NKG2D, which increases NK cell recognition and elimination of cancer cells282. Cancer cell death can result in the release of additional tumor antigens, which leads to epitope spreading and recommencement of the CIC.
3.4. Iatrogenic STING Activation by Classical Cancer Therapies
As previously mentioned, indirect STING activation is a consequence of many classical cancer treatments (Figure 7), including many DNA-damaging chemotherapies (e.g. cisplatin283, camptothecin284, doxorubicin285, paclitaxel127,286, etoposide287–290, etc.), radiotherapy291, and therapies that compromise the DNA damage response (e.g. poly(ADP)-ribose polymerase 1 (PARP) inhibitors292–296, ataxia telangiectasia and Rad3-related protein (ATR) inhibitors297, etc.). The inadvertent STING activation within tumor cells from such cancer therapies is induced by cellular dsDNA that becomes accessible for cGAS recognition. Many researchers have suggested that the recognition of dsDNA by cGAS in such cases is primarily mediated by micronuclei formation27,28. However, recent work refutes micronuclei as the primary source of dsDNA for the cGAS activation that ensues drug-induced mitotic errors and instead finds that chromatin bridges are mainly responsible for the associated cGAS activation298. Notably, inhibitors of DNA methyltransferases are another class of cancer therapeutics, which have been approved for the treatment of acute myeloid leukemia299 and are known to work well in combination with radiation and various chemotherapies in preclinical cancer models300. Recent findings suggest that the pharmacological inhibition of DNA methylation caused by a DNA methyltransferase inhibitor (i.e. 5-aza-2’-deoxycytidine) can also promote STING signaling by reversing the epigenetic silencing of both cGAS and STING that is commonly observed in a variety of cancer types301.
Figure 7: Cancer therapies that can iatrogenically activate the STING pathway.

STING activation is a known biological consequence of many classical cancer treatments, including DNA-damaging chemotherapies, therapies that compromise the DNA damage response, and radiotherapy. While the effects of classical cancer treatments are multifaceted, therapies that also induce STING signaling have potential to enhance overall therapeutic efficacy by providing a supportive inflammatory context for generating antitumor immunity. Figure created with biorender.com.
While the effects of classical cancer treatments are multifaceted, therapies that also induce STING signaling have potential to enhance overall therapeutic efficacy by providing a supportive inflammatory context for generating antitumor immunity. Indeed, it has been reported that STING signaling actively contributes to immune-mediated tumor growth inhibition in murine tumor models treated with a growing number of cancer treatments, notably including topotecan302, viral oncolytic therapy6, PARP inhibition292,294,303, and radiotherapy291. Additionally, STING agonists were found to synergize well with radiotherapy in murine pancreatic tumors by promoting inflammatory pathways following tumor antigen release by radiotherapy304.
STING signaling has also been implicated in the response to classical cancer treatments even in the absence of immune-mediated mechanisms. STING activation in cancer cells induced by antimitotic chemotherapies (e.g. taxane drugs) has been shown to trigger a proapoptotic secretory phenotype, which promotes BCL-xL-dependent apoptotic priming in untreated cancer cells286. It was confirmed that the STING-dependent apoptotic effects are required for the antitumor response to paclitaxel in vivo. Additionally, autophagy caused by STING-activating chemotherapies can clear diseased cells directly in addition to promoting desirable antitumor immune responses by triggering ATP release and immunogenic cell death (ICD)305,306. In the context of radiotherapy, the cGAS protein can also directly contribute to cancer cell clearance by initiating cell death programs and accelerating γ-irradiation-induced cell ablation122.
The functional significance of iatrogenic STING activation in human cancer patients is currently unclear. As previously discussed, the magnitude and context of STING signaling are critically important determinants of antitumor immune responses, and therefore iatrogenic STING activation may not be optimal for maximizing therapeutic impact. Furthermore, many classical cancer treatments target tumors indiscriminately and thus likely also impact immune cells within the TME. Therefore, the balance of STING activation, degree and type of tumor cell death, and the effect of the treatment on immune cells are all important variables to consider, as they will likely influence therapeutic outcomes307. Nevertheless, research has already begun to explore the employment of nanotechnology for enhancing STING-activating chemotherapies, strategies that not only address drug delivery challenges but also seek to simultaneously reinforce antitumor immunity within the TME308.
4. STING Pathway Agonists
The development of STING pathway agonists as a cancer therapy long preceded the discovery of the STING pathway, beginning with the therapeutic characterization of flavone acetic acid (FAA). FAA was initially described as a vascular-disrupting agent and showed promise as a potential cancer therapeutic, inducing hemorrhagic necrosis in murine tumor models309–311. However, the narrow therapeutic window and poor pharmacokinetic properties of FAA led to the chemically-optimized design of 5,6-Dimethylxanthenone-4-acetic acid (DMXAA)312–315. Over a decade after their initial discovery, both FAA and DMXAA were identified as potent mSTING agonists25,316. The robust antitumor activity of DMXAA in murine tumor models, which is now known to involve STING activation, advanced the compound to clinical testing. However, DMXXA failed in late-stage clinical trials for the treatment of non-small cell lung cancer due to a lack of efficacy317,318.
The negative results from the clinical trials involving DMXAA have since been largely attributed to the species-specific differences in the STING protein that render DMXAA incapable of binding (i.e. activating) any of the major hSTING isoforms165. Chimeric molecules comprised of mSTING with a hSTING CTD did not respond to DMXAA, while a chimeric hSTING molecule with a mSTING CTD resulted in signaling319. Indeed, a specific isoleucine residue of mSTING that is not present in any of the hSTING isoforms is critically involved in the recognition of DMXAA320. Nonetheless, studies involving DMXAA have significantly contributed to a fundamental understanding of the STING pathway in cancer therapy. DMXAA served as the first direct evidence for the existence of non-nucleotide, small molecule STING agonists and also demonstrated their potential for immunotherapy as alternatives to CDNs. A comparable molecule, 10-carboxymethyl-9-acridanone (CMA) was also developed as a STING-targeting antiviral drug, but similarly suffered from an inability to activate hSTING, further guiding the field to develop hSTING agonists321.
4.1. Cyclic Dinucleotide STING Agonists
As the biology of the STING pathway became more defined, the focus of STING-related cancer research and therapeutic development shifted to CDNs (Figure 2), the natural ligands of STING37,322,323. Canonical CDNs, originating in bacteria, comprise a 3′3′ linkage orientation (i.e. two uniform 3′,5′ phosphodiester bonds) and can activate certain hSTING variants60,210. Since they do not activate all hSTING isoforms, canonical CDNs have been largely dismissed as potential drug candidates for lack of translatability15,160,161. Alternatively, noncanonical CDNs possess 2′2′, 3′2′, or 2′3′ linkage orientations. 2′2′-cGAMP, which contains two uniform 2′,5′ phosphodiester bonds, is a synthetic CDN that has not yet been found in nature324. 3′2′-cGAMP, which contains mixed 3′,5′ and 2′,5′ phosphodiester bonds, has recently been discovered in Drosophila melanogaster (i.e. fruit flies) as an intracellular product of cGAS-like receptors that recognize cytosolic double-stranded RNA325,326. 2′3′-cGAMP, which contains mixed 2′,5′ and 3′,5′ phosphodiester bonds, is produced intracellularly by mammalian cGAS and exhibits some level of affinity for all of the major hSTING variants15,59. Notably, the relative hSTING-binding affinities for the various CDN linkage orientations are 2′3′-cGAMP > 2′2′-cGAMP > 3′3′-cGAMP ~ 3′2′-cGAMP59,324. However, as discussed earlier, all natural CDNs are poor drug candidates as they experience inefficient cytosolic delivery and are susceptible to hydrolytic degradation by ENPP1.
Due to the poor drug-like properties of natural CDNs, second-generation STING agonists are now being developed. Both chemically-modified CDNs and non-nucleotide, small molecules are being explored as synthetic hSTING ligands. These compounds have enhanced drug-like qualities and are currently navigating the pharmaceutical pipeline (Table 1). The majority of these new STING agonists are synthetic, non-hydrolyzable CDN analogues with mixed 2′,5′ and 3′,5′ phosphorothioate bonds207. For example, ML RR-S2 CDA (now known as ADU-S100) is a dithio-substituted cyclic di-adenine with mixed phosphorothioate bonds that has been designed to increase stability and lipophilicity and therefore promote enhanced STING signaling. By substituting the non-bridging oxygen atoms at the phosphate bridge with sulfur atoms, the CDN is less susceptible to degradation by phosphodiesterases (e.g. ENPP1) and may even promote cellular uptake and cytosolic delivery15,197,202,203. Additionally, the phosphate bridge configuration, containing both 2′,5′ and 3′,5′ bonds, mimics that of noncanonical CDNs (e.g. endogenous 2′3′-cGAMP), which can activate all of the major hSTING isoforms15. This drug, and similar CDNs, are currently being investigated in clinical trials as an intratumorally administered treatment for head and neck squamous cell carcinoma and other solid tumors as well as lymphomas327,328. Macrocyclized STING agonists, such as E7766, have also been developed and are now being implemented in phase I clinical trials. These aim to improve therapeutic efficacy by utilizing transannular macrocyclic bridges to lock the CDN in its bioactive, “U” conformation329.
4.2. Non-nucleotide, Small Molecule STING Agonists
Although DMXAA lacked a capacity for hSTING activation, it inspired the development of non-nucleotide, small molecule STING agonists (Figure 8). Non-nucleotide, small molecules have potential to exhibit advantageous drug-like properties as well as improved access to the cytosol compared to anionic and highly water-soluble CDNs. Sali et al. developed a small molecule innate immune activator as an antiviral drug for the alphaviruses, Chikungunya (CHIKV) and Venezuelan Encephalitis (VEEV)330. Both viruses are lethal with little to no treatment options, but have been found to be sensitive to the antiviral effects of type I IFN responses331,332. After high throughput screening, they arrived at the molecule, 4-(2-chloro-6-fluorobenzyl)-N-(furan-2-ylmethyl)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxamide, denoted “G10.” G10 demonstrated high potency and low toxicity in human fibroblasts. Using reporter cell assays and quantitative PCR (qPCR), the authors demonstrated G10’s ability to trigger IRF3-dependent IFN signaling. The molecule also reduced replication of both CHIK and VEEV in vitro (IC90 values of 8.01 and 24.57 μM, respectively). Interestingly, STING deletion eliminated the G10-mediated IRF3 S386 phosphorylation and the ability to block viral replication, indicating that G10 acts through a STING-dependent pathway. However, this molecule does not directly bind to STING, but activates STING in an indirect manner, which is still to be explored. G10 demonstrated minimal activity in murine myeloid-derived cells, categorizing it as a hSTING-specific agonist.
Figure 8:

Chemical structures of non-nucleotide, small molecule STING agonists.
Liu et al. also developed a hSTING-specific agonist333. They identified a dispiro diketopiperzine compound, 2,7,2″,2″-dispiro[indene-1″−3″-dione]-tetrahydrodithiazolo[3,2-a:3′,2′-d]pyrazine-5,10(5aH,10aH)-dione (DSDP) through a high throughput screening assay. DSDP induced an IFN-dominant cytokine response in peripheral blood mononuclear cells (PBMCs) and human skin fibroblasts, suppressing the replication of yellow fever virus, dengue virus, and Zika virus. Although this agent shows promise for activating hSTING, it does not efficiently bind mSTING, which limits the use of mouse models for preclinical studies. Similar hSTING specificity was also observed with a well-known antiviral/anti-tumor agent, α-Mangostin, which induced type I IFN responses in 293T cells transfected with hSTING plasmids, but exhibited minimal activity in mSTING334. Although all of these molecules appear to hold great merit, a small-molecule candidate ideally would potently activate both hSTING and mSTING, allowing studies to span various tumor models in immunocompetent mice and enabling preclinical studies that can evaluate toxicity, pharmacodynamics, treatment regimen, and drug combinations.
Bicyclic benzamides and benzothiophene derivatives have also shown hSTING specific activity. Scientists at Curadev Pharma discovered that certain bicyclic benzamides could target the STING pathway as the compounds demonstrated the ability to induce the production of STING-associated cytokines (e.g. type I IFNs, CXCL10, and TNF-α) in human PBMCs335–337. Notably, a set of three intratumoral injections of their lead compounds administered every other day led to significant suppression of tumor growth in BALB/c mice with hSTING-expressing CT26 tumors. Investigators at Merck explored benzothiophene derivatives in the context of STING activation and identified compounds with micromolar potency in both direct STING binding and a cellular reporter assay for type I IFN production338. However, the multi-substituted benzothiophene lacked the ability to generate significantly higher type I IFN secretion compared to 2′3′-cGAMP. In mice with advanced MC38 tumors, the lead compounds were able to stimulate tumor regression when injected intratumorally every 3 to 7 days for up to 30 days, demonstrating their potential for clinical development339.
One of the most promising non-nucleotide, small molecule STING agonists has recently been described by Ramanjulu and collaborators at GlaxoSmithKline. A series of amidobenzimidazole (ABZI) STING agonists was identified using a high throughput screening assay to monitor a library of small molecules competing with the binding of radio-labeled 2′3′-cGAMP to the CTD of hSTING132. The lead compound (i.e. Compound 1 – Figure 9A), which emerged from the in vitro screen, could bind to one subunit of the hSTING homodimer with significant, but relatively low potency (e.g. IC50 ~ 14 μM) compared to the natural STING ligand, 2′3′-cGAMP (e.g. IC50 ~ 200 nM). In order to improve potency, the researchers created a dimer of the lead compound, connecting two molecules together by replacing an N1-hydroxyphenethyl moiety with a four carbon linker. The resulting dimeric amidobenzimidazole ligand (diABZI) (i.e. Compound 2 – Figure 9B) could target homodimeric STING and improved its competitive binding by 1000-fold (e.g. IC50 ~ 20 nM). All key contacts from the monomeric compound and STING were conserved and the linker was shown to have no interactions with the protein. Compound 2 induced dose-dependent secretion of IFN-β in human PBMCs at a level 18-fold higher than cGAMP. The molecule was further optimized through chemical substitution, yielding a diABZI (i.e. Compound 3 – Figure 9C) with greatly improved in vitro activity. Compound 3 was 400-fold more potent than cGAMP in human PBMCs with wildtype STING and was also active in human PBMCs expressing other STING isoforms (e.g. HAQ/HAQ, R232H/R232H) as well as in murine PBMCs. Unlike cGAMP and DMXAA, this compound efficiently activated STING while maintaining an open STING confirmation, indicating that conformational change and lid interactions are not always necessary for STING activation (Figure 9D). Importantly, this diABZI compound was found to bind to both hSTING and mSTING, enabling preclinical analysis of pharmacological properties and antitumor efficacy in mouse tumor models. When administered intravenously using a three-dose regimen to mice with established CT26 colorectal tumors, diABZI resulted in a significant inhibition of tumor growth and an increased overall survival time with 8/10 mice in the study remaining tumor free. This finding was particularly notable as it represented the first published report of a non-nucleotide, small molecule STING agonist with both hSTING and mSTING binding capacity that could activate antitumor immunity and inhibit tumor growth when administered via an intravenous route.
Figure 9: Development of the dimeric amidobenzimidazole (diABZI) STING agonist.

(A) Chemical structure of the monomeric ABZI STING agonist, Compound 1. (B) Chemical structure of the dimeric ABZI (diABZI) STING agonist, Compound 2. (C) Chemical structure of the fully optimized diABZI STING agonist, Compound 3. (D) The ‘Open Lid’ configuration of a holo (i.e. ligand bound) hSTING dimer bound to diABZI Compound 2. Adapted with permissions from reference145. Copyright © 2020 American Association for the Advancement of Science; permission conveyed through Copyright Clearance Center, Inc. (PDB ID: 6DXL)132. Copyright © 2018 Springer Nature BV; permission conveyed through Copyright Clearance Center, Inc.
More recently, two reports have described non-nucleotide, small molecule STING agonists with potential to be delivered via multiple administration routes, including orally. Pan and co-workers at Merck identified MSA-2 (i.e. benzothiophene oxobutanoic acid – Figure 10A) as a STING agonist using a high-throughput, cell-based phenotypic screen that measured IFN-β secretion from THP1 monocytes treated with a diverse library of ~ 2.4 million compounds340. MSA-2 was found to have high cell permeability, STING selectivity, and resulted in phosphorylation of both TBK1 and IRF3 in a dose-dependent manner. The mechanism through which MSA-2 activates STING is unique in that it forms a non-covalent dimer (Figure 10B) in solution and fills the CDN binding pocket with high affinity (e.g. KD = 8 nM) and a slow off-rate (e.g. half-life of 1.3 hours). Interacting with each other through their aromatic cores, two molecules of MSA-2 predimerize in solution before binding to STING in the same site as 2′3′-cGAMP. The dimerized agonist creates a bridge across the STING homodimer, which noncovalently crosslinks the two STING subunits and allows for the stabilization of a closed lid conformation (Figure 10C), similar to that of 2′3′-cGAMP-bound STING. To further support this binding mechanism, a covalently linked monomer was synthesized by replacing both 5-methoxy groups with a propane linker, which also demonstrated potent STING binding activity. Interestingly, MSA-2 is currently the only small molecule reported to undergo such a reversible, noncovalent dimerization in solution to become a pharmacologically active ligand, highlighting the novelty of using MSA-2 rather than the covalent dimer. Computational modeling predicted that the hydrophobicity of MSA-2 (pKa ~ 4.7) increases in acidic conditions due to protonation of the carboxylic acid group with an attendant increase in the fraction of uncharged molecules, which exhibit higher membrane permeability. This suggests that the molecule may have higher cellular membrane permeability in the TME, which tends to be slightly acidic341,342. Indeed, the investigators demonstrated higher MSA-2 concentrations in subcutaneous MC38 tumors than in plasma or in nontumor tissue (e.g. spleen, muscle), resulting in elevated IFN-β and proinflammatory cytokine production at the tumor site. As a result, treatment with MSA-2 resulted in complete tumor regression in 80–100% of mice bearing MC38 colon carcinoma tumors when delivered through IT, subcutaneous, and oral routes. Notably, MSA-2 administered orally in mice exhibited equal or better efficacy than a cGAMP analog dosed by intratumoral or subcutaneous routes. Treatment with MSA-2 also resulted in long-term antitumor immunity and synergized with ICB (i.e. anti-PD-1) in MC38 colorectal, CT26 colorectal, B16-F10 melanoma, and LL-2 lung cancer models. This study highlights the promise of designing STING agonists that exploit biochemical signatures of the tumor microenvironment (e.g. pH, redox, etc.) to preferentially enrich STING activation at tumor sites with potential to reduce systemic inflammation and mitigate associated toxicities.
Figure 10: Development of the MSA-2 (i.e. benzothiophene oxobutanoic acid) STING agonist.

(A) Chemical structure of MSA-2. (B) Self-dimerization of MSA-2 (PDB ID: 6UKM)340. (C) The ‘Closed Lid’ configuration of a holo (i.e. ligand bound) hSTING dimer bound to MSA-2 (PDB ID: 6UKM)340. Adapted with permission from reference340. Copyright © 2020 American Association for the Advancement of Science; permission conveyed through Copyright Clearance Center, Inc.
In a parallel effort, Chin et al. discovered other non-nucleotide, small molecule STING agonists (Figure 11A) with potential for systemic and oral administration through a series of cGAS/STING pathway–targeted cell-based phenotypic screens and subsequent structure-function analysis145. The investigators derived the non-nucleotide, small molecule, SR-717 from SR-001, which was one of the ~ 100,000 compounds in their starting library of commercially available compounds. The commercially acquired SR-001 exhibited a high level of activity in THP1 reporter cells for type I IFN (e.g. EC50 ~ 1.1 μM) and was found to increase the thermal stability of the soluble CDN-binding CTD of recombinant hSTINGREF in a STING thermal shift binding assay. When the researchers chemically resynthesized SR-001 in-house, they were surprised to find that it was no longer active in the STING-binding assay, but was still consistently active in cell-based assays. Analytical characterization of the vendor-bought material revealed the presence of a small but significant amount of the de-esterified derivative, SR-012. This suggested that SR-001 was acting as a prodrug, with the ester being necessary for cell permeability and the active STING-binding species being that with the free carboxylic acid. In accord with this hypothesis, synthetic SR-012 could bind both mSTING and hSTING, but was inactive in cell-based assays due to poor cytosolic delivery owing to its higher water solubility and low membrane permeability.
Figure 11: Development of the SR-717 STING agonist.

(A) Chemical structures of SR-001 (i.e. the prodrug screening hit), SR-012 (i.e. the elucidated STING agonist), SR-717 (i.e. the optimized STING agonist), and SR-301 (i.e. orally bioavailable analog of SR-717)145. (B) The ‘Closed Lid’ configuration of a holo (i.e. ligand bound) hSTING dimer bound to SR-717. Adapted with permission from reference145 (PDB ID: 6XNP)145. Copyright © 2020 American Association for the Advancement of Science; permission conveyed through Copyright Clearance Center, Inc.
To address the low cell permeability of SR-012, Chin et al. synthesized SR-717 – another carboxylic acid–containing analog of SR-001145, which includes a difluoro-substitution of the aniline ring system to increase membrane permeability. Crystallographic analysis demonstrated that two molecules of SR-717 bind at the base of the STING dimer intersubunit cleft, inducing a closed lid conformation (Figure 11B) that closely mimics the binding mode of 2′3′-cGAMP. The paired SR-717 molecules share similar contact residues as observed with 2′3′-cGAMP, which enable it to competitively bind to STING. Importantly, SR-717 was found to bind to all common human alleles of hSTING as well as mSTING, allowing in vivo analysis of pharmacological properties and efficacy in mouse tumor models. Therapeutic doses (e.g. 15 mg/kg or 30 mg/kg) of SR-717 administered intraperitoneally resulted in elevated plasma cytokine concentrations and STING-associated antitumor effects in syngeneic B16-F10 melanoma and MC38 colorectal adenocarcinoma mouse tumor models. intraperitoneal administration of SR-717 also inhibited the formation of pulmonary nodules following intravenous delivery of B16-F10 melanoma cells, suggesting its ability to combat metastasis. Surprisingly, unlike the study of Pan et al. that used MSA-2 with anti-PD-1 in MC38 colorectal, CT26 colorectal, B16-F10 melanoma, and LL-2 lung cancer tumors340, Chin et al. did not observe a benefit of combining SR-717 with ICB (i.e. anti-PD-1 or anti-PD-L1) in the B16-F10 tumor model145. Finally, using an analog of SR-717 with improved bioavailability, SR-301, they demonstrated a modest inhibition of tumor growth using an oral administration route.
4.3. cGAS Agonists
While the STING protein has appropriately garnered much interest as a druggable target for cancer immunotherapy, cGAS has been largely overlooked despite the potential of cGAS activation to more closely mimic the endogenous STING pathway343. It is possible that agonists of cGAS may offer more control over the level and kinetics of local STING signaling, which may be tailored to optimize antitumor immunity79. As mentioned previously, dsDNA in the cytosol can elicit tiered immune responses, the phenotype of which is determined by the physicochemical composition of the dsDNA79. The molecular weight of the cGAS-bound dsDNA (i.e. bp length) influences the prevalence and size of the resultant liquid-like droplets, which function as miniature bioreactors for the efficient production of 2′3′-cGAMP58. Accordingly, the localized production of 2′3′-cGAMP is tightly regulated, and the liquid-like droplets essentially act as in situ drug delivery depots that confer tunability over the degree of 2′3′-cGAMP production, which may be useful for promoting and controlling antitumor immunity.
The lack of development behind cGAS agonists might be attributable to the complexity of cGAS activation combined with the many challenges facing the therapeutic delivery of nucleic acids. Indeed, since freely administered dsDNA is rapidly cleared and degraded with minimal cellular uptake344, dsDNA-based cGAS agonists will require molecular engineering approaches to protect dsDNA from degradation and promote cytosolic delivery of the dsDNA. In theory, small molecule cGAS agonists could circumvent the delivery issues that are associated with the negative charge, hydrophilicity, and relatively large molecular weight of dsDNA. While no small molecule cGAS agonists have been reported to date, Hall et al. have identified a potential small molecule binding site on the cGAS enzyme that may cause catalytic activation of cGAS and is certainly worth investigation345. However, like with most small molecule drugs, an effective small molecule activator of cGAS would need to demonstrate pathway specificity and not significantly affect other cellular processes via off-target activity. Furthermore, phase separation requires multivalent interactions for the assembly of macromolecular complexes346, and therefore a small molecule activator of cGAS may not be able to induce the same liquid-like phase transition that is dependent on DNA-bridging. Indeed, a small molecule agonist would likely have to exhibit self-multimerization to achieve efficient 2′3′-cGAMP production via cGAS oligomerization, which adds another level of complex requirements for the design and development of a small molecule cGAS agonist. Thus, most small molecules would seemingly be unlikely to generate the intracellular microreactors for 2′3′-cGAMP production without assistance from other molecules, and this is perhaps the primary reason that the development of small molecule cGAS agonists has not yet been reported. Notably, DNA-bridging is not required for cGAS inactivation, and there already exist several small molecule cGAS inhibitors that have been developed for applications outside of cancer (e.g. autoimmunity)110,343,347,348.
While not a canonical small molecule per se, the metal ion, manganese(2+) (Mn2+) is worth discussing as it has recently been shown to be capable of independently activating monomeric cGAS in the absence of dsDNA without the need for oligomerization65,349. Notably, Mn2+ can affect STING signaling in several unique ways, and this will be discussed in greater detail in Section 7 along with other potentiators of the STING pathway. Interestingly, Mn2+ enhances the catalytic activity of cGAS and can also allosterically enhance the dsDNA binding activity of cGAS in conjunction with the ATP/GTP substrate pair, which sensitizes cGAS to oligomerization by lowering the threshold for cGAS activation in regard to both cytosolic dsDNA length and local dsDNA concentration in the cytosol65. Thus, in addition to oligomerization-free cGAS activation, Mn2+ may also trigger cGAS activation in cells by promoting the recognition of dsDNA already present in the cytosol at low concentrations and/or low molecular weights (e.g. short dsDNA lengths less than ~ 45 bp) that are ordinarily below the natural threshold for cGAS activation. Accordingly, the recently reported therapeutic efficacy of Mn2+ as a monotherapeutic STING pathway potentiator for cancer therapy350 cannot be solely attributed to oligomerization-free cGAS activation. It remains to be determined whether any cGAS activator that does not somehow induce cGAS oligomerization and droplet formation could achieve therapeutically relevant STING activation and whether they would allow for control over the degree of STING signaling.
In addition to Mn2+-encompassing therapies, DNA-based cGAS agonists that employ delivery technologies to achieve cytosolic accumulation of exogenous dsDNA can also facilitate the pharmacological activation of cGAS. Indeed, targeting cGAS activation via intracellular delivery of dsDNA has now been explored by several research teams including our group351–355. Notably, Garland et al. developed NanoISD, a DNA-based cGAS agonist designed for use as an intratumoral immunotherapy355. NanoISD is a nanoparticle formation that is assembled by complexing an exonuclease-resistant cGAS ligand (i.e. 95 bp phosphorothioate-capped dsDNA) with endosomolytic polymer micelles that can simultaneously enable cytosolic delivery of nucleic acids and inhibit endonuclease degradation of loaded nucleic acids via steric interference. The resultant DNA/polymer nanoparticle complexes are ~ 60–90 nm in diameter and have a positive surface charge of +14.87 mV. It was demonstrated that NanoISD confers deoxyribonuclease resistance, enhances cellular uptake, and promotes endosomal escape of the 95 bp phosphorothioate-capped dsDNA into the cytosol of cells, resulting in potent activation of the STING pathway via cGAS. Furthermore, NanoISD relayed many of the same antitumor effects established for agonists of the STING protein; NanoISD was shown to induce proinflammatory cytokine production, prompt the maturation of antigen presenting cells, promote tumor infiltration of NK cells and CD8+ T cells, reduce tumor burden, and enhance responses to ICB (i.e. anti-PD-1 and anti-CTLA-4). Moreover, when administered at the same dose and with the same treatment regimen in the B16-F10 tumor model, the therapeutic benefit of NanoISD was comparable to that of CpG DNA (i.e. ODN 1826), a well-established innate immune activator, analogues of which are currently being investigated in human clinical trials for the treatment of cancer356. While CpG DNA relies on the cellular expression of TLR9, which is mostly restricted to plasmacytoid dendritic cells and B cells in humans357, cGAS and STING proteins are more ubiquitously expressed in mammalian cells358–360. Additionally, TLR9 signaling can only occur in cells that are directly exposed to CpG DNA, in contrast to STING signaling, which can be locally propagated from cell-to-cell through the transfer of endogenous 2′3′-cGAMP following DNA-induced cGAS activation. Therefore, cGAS agonists may represent a more accessible treatment for promoting antitumor immunity via DNA sensing. Regardless, novel agonists of cGAS increase the arsenal of potential cancer immunotherapies, providing additional opportunities for immune modulation. Accordingly, NanoISD is a promising nucleic acid therapy with clear indications for the treatment of immunologically cold cancers.
5. Drug Delivery Barriers and Pharmacological Challenges
5.1. Intracellular Delivery Barriers for Cyclic Dinucleotides
CDNs have advanced into clinical trials for intratumoral administration and have demonstrated a favorable safety profile and evidence of STING pathway activation. However, initial phase I data for ADU-S100 and MK-1454 demonstrated limited efficacy, though responses in several tumor types were observed when the STING agonists were administered in combination with anti-PD-1 monoclonal antibody treatment (spartalizumab and pembrolizumab, respectively)328,361. Unfortunately, despite very promising pre-clinical data, the lack of impressive clinical responses has recently dampened enthusiasm for intratumoral administration of CDN STING agonists. Indeed, clinical trials of ADU-S100 and Merck’s CDN STING agonist (MK-1454) are no longer recruiting (Table 1). While a limited clinical response may be attributed to a multitude of factors (e.g. study design and outcomes, dose and regimen selection, patient cohort selection, use of combinations, etc.) that may be independent of the activity or immunostimulatory effects of the agent per se, these early clinical studies nonetheless motivate the importance of considering potential drug delivery and pharmacological barriers that may limit the therapeutic efficacy of CDNs (Figure 4).
STING is localized on the endoplasmic reticulum with the ligand binding domain facing cytosolically, and, hence, access of CDNs to the cytosol is thought to be a critical for their activity. However, intracellular delivery of CDNs via passive diffusive transport across the plasma membrane is limited by their negative charge and high aqueous solubility362. CDNs are also relatively large in size (e.g. ~ 700 Da) compared to traditional small molecule drugs typically designed to be less than 500 Da, further limiting their ability to diffuse passively through the membrane197. Current evidence suggests that the activity of free cGAMP can likely be attributed the aforementioned membrane transport processes (e.g. VRACs, connexin gap junctions, SLC19A1, etc.) or via cellular uptake through pinocytosis or endocytosis, but the efficiency of these processes appears limited214 given the relatively high EC50 values (e.g. high micromolar) for free cGAMP typically measured in cell culture assays. Beyond its low cellular permeability, 2′3′-cGAMP activity is also hindered by the hydrolase, ENPP1, which cleaves phosphodiester bonds in the extracellular space197. However, as mentioned previously, nonhydrolyzable analogs can mitigate this effect and have shown improved potency with EC50 values ~ 10-fold higher than 2′3′-cGAMP as determined by measuring dose-dependent IFN-β production in a human THP1 monocyte cell line197. While a significant improvement, the potency of these analogs is still limited by their relatively poor cell membrane permeability. Thus, comparable to nucleic acid-based therapeutics (e.g. siRNA, mRNA, miRNA, etc.), the activity of CDNs is limited by intracellular delivery barriers, prompting the recent development of new chemical strategies for circumventing this challenge. In order to improve membrane permeability and stability against enzymatic degradation, some research groups have modified current CDNs to include fluorine substitutions for 2′-hydrogens or 2′-hydroxyl groups on the pentose rings363–365 (Figure 12). Lioux et al. developed a non-canonical CDN, adenosine-inosine monophosphate (cAIMP) that includes both fluorine and thiophosphate substitutions (Dithio-2′-F-cAIMP – Compound 53) to address both of these delivery challenges364. Similarly, Pimková Polidarová et al. utilized phosphorothioate-linked 3′3′-ci-di(2′F,2′dAMP) to develop phosphoester CDN prodrugs that further enhance permeability by using biolabile protecting groups that mask the negatively charged phosphate groups and release the free, parent drug under intracellular conditions (e.g. exposure to intracellular enzymes)365. Beyond chemically modifying CDNs, nanotechnology has also been developed to circumvent intracellular delivery challenges which we describe in detail in Section 6.
Figure 12: Chemical structures of modified CDN STING agonists.

Chemical structures of various CDN STING agonists that have been chemically modified for improved stability, activity, and cell permeability. The CDNs modified to include fluorine substitutions for 2′-hydrogens or 2′-hydroxyl groups on the pentose rings (i.e. 2′-F-c-di-GMP363, Dithio-2′-F-cAIMP (Compound 53)364, and the 3′3′-c-Di(2′F,2′dAMP) Prodrug365) exhibit improved membrane permeability as well as stability against enzymatic degradation. The carbocyclic STING agonist, 15a, which comprises carbocyclic nucleotides, cyclopentane instead of ribose, and the imidazole portion of adenine replaced with a pyrimidine ring, exhibits significantly improved STING binding, cellular activity, and membrane permeability366.
Current CDNs, and all STING pathway agonists more generally, also lack cell and tissue specificity. STING is expressed in many cell types including immune, non-immune (e.g. endothelial cells), and cancer cells367, that may respond differently to STING activation with potential to impact therapeutic outcomes. Notably, there is significant evidence that STING overstimulation can be toxic to lymphocytes. For example, T cells have been found to have high STING expression, but in general, exhibit low type I IFN responses86. Instead, the IFN-independent activities of STING can trigger cell death in T cells86,239,368, which may prove problematic for cancer immunotherapy where a central goal is to enhance T-cell infiltration and function in immunogenically “cold” tumors. Interestingly, tumors have been shown induce STING-mediated T cell death as an immune evasion mechanism239. On the other hand, exploiting the sensitivity of T cells to STING-mediated cell death could be advantageous for the treatment of T cell lymphomas. Similarly, STING activation can trigger apoptosis in both normal and malignant murine B cells275,369,370 and may also reduce responsiveness to B cell receptor (BCR) activation, resulting in reduced antibody responses371, though this has not been fully resolved369. Notably, STING is also known to be poorly expressed and partly dysfunctional in resting human B cells372.
Hence, an emerging and important area of research that could address both the challenge of intracellular delivery and cell-specificity is the development of targeting strategies to deliver CDNs to specific cell types based on differential expression of internalizing receptors. In particular, considering the toxicities involved with STING hyperactivation in lymphocytes, it may be particularly advantageous to target tumor-associated myeloid cells, tumor cells, or endothelial cells to improve immunotherapy responses to CDNs. To our knowledge, there are no reports of directly targeted CDNs, though the drug carrier technologies we discuss below are poised to enable more selective targeting to specific cell types with potential to enhance delivery efficiency and efficacy.
5.2. Utility and Challenges with Intratumoral Administration of STING Pathway Agonists
Intratumoral injections offer the advantage of directly targeting a tumor site with relatively well-defined initial concentrations of a therapeutic, while reducing systemic drug exposure and associated risks of toxicity373. Indeed, the intratumoral administration of STING agonists has performed exceptionally well for murine transplantable tumor models, mediating the rejection of many locally-injected tumors and significantly enhancing the therapeutic benefit of ICB and radiotherapy291,374. Accordingly, clinical evaluation of innate immune agonists, including CDNs, has primarily employed an intratumoral administration route. In addition to treating the injected tumor, such intralesional therapy can also act as an in situ cancer vaccine that primes and/or activates a peripheral tumor antigen-specific CTL response capable of eliminating distal disease (i.e. abscopal effect). Although STING activation via intratumoral administration of CDNs is still being explored clinically as a therapeutic strategy for treating solid tumors, there are several challenges that are inherent to this administration route (Figure 13), which may contribute to the underwhelming clinical responses observed in recent clinical trials.
Figure 13: Utility and challenges facing the administration of STING pathway agonists.

The route of therapeutic administration can significantly impact the efficacy of STING pathway agonists. Administration can be local (e.g. intratumoral) or systemic (e.g. intravenous, oral), and each delivery route is associated with unique utility and challenges. Figure created with biorender.com.
The properties of solid tumors can vary widely between patients and cancer types and tumor injection techniques lack standardization, making it difficult to develop an overarching administration and dosing scheme. Parameters such as tumor size, morphology, anatomical location, degree of vascularization, interstitial pressure, mechanical properties, among other variables, can cause significant deviations in intratumoral drug concentration and/or distribution with potential to impact immune responses and therapeutic outcomes375. Moreover, cellular composition and level of STING expression can be variable between tumors, which may significantly affect a STING agonist’s mechanism of action375.
Due to their small size and high water solubility, CDNs rapidly diffuse from the injection site, and therefore, concentrations of CDNs in tumors following intratumoral administration can be transient and highly variable, potentially leading to different CDN dose-dependent effects260,375,376. Indeed, the immune and therapeutic effects elicited by intratumorally administered CDNs have been reported to have a bell-shaped dose-response relationship in mouse tumor models. In recent work by Sivick et al., high intratumoral doses of CDNs resulted in an ablative response, that efficiently inhibited growth of the injected tumor, but also resulted in systemic drug and cytokine exposure that resulted in immune cell death in the tdLNs, thereby impairing priming of anti-tumor adaptive immunity234. By contrast, lower doses of CDN were slightly less effective at inhibiting injected tumor growth, but resulted in a more immunogenic antitumor response characterized by a stronger and more durable peripheral tumor antigen-specific CTL response. As a primary goal of intratumoral administration is to induce a systemic adaptive antitumor immunity, careful selection of local CDN dose appears critical to generating a robust T cell response via in situ vaccination. Therefore, an uneven dispersion of CDNs following injection may result in different responses within the same tumor (i.e. one Section may receive an ablative dose while another receives an immunogenic dose), further complicating dose selection and potentially confounding interpretation of clinical outcomes. Indeed, it has recently been shown that variations in needle design (i.e. the use of a multi-side hole needle) can have a profound effect on intratumoral drug deposition and therapeutic efficacy in mouse tumor models377.
The rapid clearance of CDNs from the injection site also limits the efficiency at which they accumulate and activate STING in the tdLNs, which are prime targets for immunotherapeutic intervention due to their important role in initiating T cell responses to tumor antigens376,378. Since blood capillaries are permeable to molecules smaller than ~ 5–10 nm in diameter, CDNs rapidly clear from interstitial space and primarily partition into the blood stream instead of distributing into the lymphatics. This is supported by studies in mice by Sivick et al., who demonstrated that intratumorally administered CDNs rapidly distribute systemically234 as well as by plasma pharmacokinetic analysis in patients following local CDN administration328. By contrast, nanoparticles (NPs) between 10–100 nm in diameter preferentially drain into lymphatic vessels, resulting in increased accumulation in LNs and uptake by APCs.379–381. This has motivated the design of nanocarriers for CDNs that can exploit lymphatic transport to promote CDN delivery to LNs196,376,382. Indeed, this approach has been leveraged to enhance the vaccine adjuvant properties of CDNs376,382 and could similarly enhance immune responses to tumor antigens by stimulating STING activation in tdLNs, which may also be immunosuppressed. However, lymph vessels might also be dysfunctional in some tumors383, potentially restricting lymphatic drainage of nanoscale STING pathway agonists.
Though there are many advantages to direct intratumoral injection, some tumors are not readily accessible without the aid of advanced image-guided administration techniques and, in certain cases, intratumoral injection may simply not be feasible. Furthermore, STING agonists normally require multiple, repeated intratumoral injections to achieve therapeutic efficacy in murine tumor models, but such dose regimens may not be possible and/or practical for many clinical cases384. For example, in an important recent study by Brody and colleagues, patients with indolent non-Hodgkin’s B cell lymphoma were administered intratumoral injections of Flt-3 ligand (Flt3L) daily for nine days, followed by two days of radiation therapy, followed by eight injections of the immunostimulatory adjuvant poly-ICLC385. While the results of this study offer compelling evidence in support of in situ vaccination, such frequent dosing schedules may also limit patient compliance, which is known to be dependent on the complexity of treatment386. Notably, adherence rates for medication have been reported as low as 52% for patients in the United States with at least one chronic disease387. Additionally, repeated injections can cause problematic local inflammation and associated complications (e.g. vascular catastrophe)375. Indeed, one clinical trial combining ADU-S100 and spartalizumab ICB (i.e. anti-PD-1 monoclonal antibody therapy) required weekly intratumoral injections and resulted in several side effects, including injection site pain, diarrhea, fatigue, and pyrexia328.
The timing, kinetics, and intensity of immunotherapeutic interventions can also significantly affect the generation and maintenance of antitumor immunity, and therefore suboptimal therapeutic dosing regimens can lead to poor therapeutic responses and/or resistance to immunotherapy388,389. Accordingly, there is an opportunity for strategies that modulate the intratumoral delivery of STING pathway agonists to improve therapeutic responses by enabling more precise control over local drug concentrations and pharmacokinetic profiles. Future strategies involving intratumoral delivery of STING pathway agonists could also focus on improving and more tightly regulating tdLN accumulation to maximize antitumor T cell priming and activation. Notably, drug depot technologies, which will be discussed in detail in Section 6.2, can address many of the challenges facing intratumoral administration of STING pathway agonists as they can be delivered locally and facilitate controlled and sustained release to precisely regulate local dose and STING signaling kinetics, while also potentially reducing the need for multiple injections.
5.3. Utility and Challenges with Systemic Administration of STING Pathway Agonists
Systemic (e.g. IV, oral) administration of STING pathway agonists has the potential to mitigate many of the aforementioned challenges of intratumoral administration. However, this route of administration still presents many key barriers that can limit the efficacy of STING-activating therapeutics (Figure 13). Due to their size and hydrophilicity, intravenously administered CDNs have short serum half-lives (e.g. ~ 2 minutes in mice), which, along with their poor drug-like properties, significantly limits their overall exposure, tissue distribution, and activity328,390,391. Accordingly, systemically administered CDNs have proven largely ineffective in limiting tumor growth in mouse tumor models24,196,390. To improve efficacy for intravenous administration, Vyskocil et al. approached some of these limitations by developing a novel carbocyclic STING agonist (i.e. 15a), which comprises carbocyclic nucleotides, cyclopentane instead of ribose, and the imidazole portion of adenine replaced with a pyrimidine ring366 (Figure 12). 15a exhibited a half-life of ~ 23.4 minutes in BALB/c mice when administered intravenously at 1 mg/kg with systemic exposure (i.e. 3.9 μM in plasma collected 5 minutes after in injection) at or above cellular concentrations required for its activity. Furthermore, the unique structure of 15a resulted in significantly improved STING binding, cellular activity, and membrane permeability, leading to a robust antitumor effect in CT-26 tumor models. Although this agent seems promising, it is also a relatively new molecule, and therefore further work will be necessary to determine its efficacy across a larger range of tumor types and treatment regimes.
The barriers listed above, coupled with the preclinical efficacy of DMXAA, a molecule with more canonical drug-like properties, motivated the recent development of the non-nucleotide, small molecule STING agonists that are described in Section 4.1.2. Such agents have enhanced cellular permeability and would ideally also exhibit improved pharmacokinetic properties relative to CDNs, allowing for optimal exposure and STING activation. One significant recent example of a non-nucleotide, small molecule STING agonist is an amidobenzimidazole compound described by investigators at GlaxoSmithKline132. The lead compound (i.e. Compound 3) exhibited a half-life of 1.4 hours and an AUC of 3 μg h−1 ml−1 when administered intravenously at 3 mg/kg in healthy BALB/c mice, and concentrations in the blood exceeded the in vitro EC50 as determined in murine PBMCs (e.g. ~ 200 ng/mL)132. Notably, intravenous administration of Compound 3 resulted in significant tumor growth inhibition in BALB/c mice with subcutaneous CT26 colorectal tumors132.
Two additional non-nucleotide, small molecule STING agonists that have recently been described, MSA-2 and SR-717, have been shown to exert therapeutic effects when administered orally, subcutaneously, or intraperitoneally. In MC38 tumor-bearing C57BL/6 mice, MSA-2 had a plasma half-life of ~ 14 minutes when delivered subcutaneously at 50 mg/kg and ~ 1.7 hours when given orally at 60 mg/kg (note that these half-lives are estimates from published pharmacokinetic data, where calculated half-lives were not described)340, whereas in healthy C57BL/6 mice, SR-717 and its analog for oral delivery (i.e. SR-301) had half-lives of 6.37 hours when administered intraperitoneally at 3 mg/kg and 11.11 hours when dosed orally at 5 mg/kg, respectively145.
However, unlike more conventional cancer therapeutics, relationships between pharmacokinetics, efficacy, and toxicity have not yet been fully defined for STING pathway agonists, and therefore, what the ideal pharmacokinetic properties should be is currently unknown. Given the complex relationships between the kinetics and magnitude of STING activation on immunity and toxicity, it will be important to better understand the underlying pharmacokinetic-pharmacodynamic relationships that govern the efficacy of STING pathway agonists.
In clinical trials of intratumorally administered ADU-S100, a maximum tolerated dose was not reached up to 800 μg in dose-escalation studies328, consistent with the relatively low cellular uptake of CDNs. However, primary concerns underlying the systemic administration of STING pathway agonists are the lack of tumor or cellular specificity and the potential for inducing a toxic systemic inflammatory response. While STING activation in extratumoral cell populations (e.g. myeloid cell populations in secondary lymphoid organs) may exert antitumor effects, STING activation would ideally occur primarily within tumor sites to generate the local inflammatory context and chemokine gradient necessary for the recruitment of T-cells into the TME375 while restricting STING activation in the blood stream and at other organ sites to minimize systemic inflammatory side effects. Currently, the therapeutic window for systemically administered STING agonists appears to be limited by non-specific systemic STING activation that can induce a cytokine storm30, also known as cytokine release syndrome, similar to the response observed in sepsis392. Indeed, there is evidence linking overactivation of STING signaling in macrophages and monocytes to sepsis in mouse models393,394. Symptoms from cytokine storm can range from mild fever, fatigue, headache to more serious physical outcomes such as hypotension, neurotoxicity, and multi-organ system failure392. STING induces a broad spectrum of cytokines, and the predominant mediators of efficacy and toxicity have not been fully resolved. However, defects in type I IFN signaling can result in excessive and unbalanced production of proinflammatory IL-6 and TNF-α, which has been linked to severe COVID-19 disease395 and may also be involved in mediating inflammatory toxicities induced by STING agonists. Moreover, with the recent reports describing MSA-2 and SR-301 as orally available STING agonists, it will also be important to understand the potential implications of STING activation in the gastrointestinal tract, which has a complex role in mediating homeostasis in the gut with oral administration of STING agonists previously being reported to cause or exacerbate colitis396,397.
As results from clinical trials of other STING agonists emerge, particularly for non-nucleotide, small molecule agonists that are likely to activate STING indiscriminately and systemically, it will be critical to more fully understand the determinants of toxicity and efficacy to inform the design of strategies to widen the therapeutic window. One promising strategy may be to achieve more tumor selective STING activation using tumor-targeting agents (e.g. antibodies), prodrug strategies, and/or environmentally-responsive agonists. For example, MSA-2 bears a carboxylate with a pKa of 4.78 and, therefore, upon reaching an acidified TME (e.g. pH ~ 6.0–6.5) that occurs in some cancers341,342, the carboxylic acid protonates, reducing its negative charge and aqueous solubility and thereby increasing its cell membrane permeability340. To our knowledge, the MSA-2 small molecule agonist offers the only published report of an environmentally-responsive or targeted STING agonist, though antibody-targeted agonists of other pattern recognition receptors (e.g. TLR-7 agonists) have been described to preferentially trigger innate immunity at tumor sites398. An important and unresolved question is to which cell population(s) STING pathway agonists should be targeted either within tumors, in the circulation, and/or residing in secondary lymphoid tissue. For example, targeting of tumor-associated macrophages with STING pathway agonists has potential to promote an M1-like phenotype that would create an antitumor immune environment by promoting antigen presentation, secretion of proinflammatory cytokines, and infiltration of CTLs. Nanoparticle platforms for CDN delivery have recently emerged (discussed in Section 6) and a large tool box of strategies exist for integrating targeting ligands onto nanocarriers, which offers promise for enhancing tumor targeting of STING pathway agonists.
It is also important to consider the possibility that STING activation can potentially cause immune tolerance and even the onset of autoimmune disease12. Notably, STING signaling can induce production of indoleamine 2,3 dioxygenase (IDO), which is both counter-regulatory to anti-tumor T cell function and has broad tolerogenic effects399. IDO activation induces downstream effects that drive immunosuppressive regulatory T cell (TREG) differentiation and push macrophages and DCs towards an immunosuppressive phenotype, limiting immune response and promoting tumorigenesis399. Interestingly, intratumoral administration of STING agonists has resulted in more tolerogenic responses in a Lewis lung carcinoma mouse model219. There is also some evidence that suggests that certain factors linked to STING pathway activation, such as chromosomal instability, STING activation in mesenchymal stromal cells, and cGAMP transfer via the astrocyte gap-junctional network may contribute to metastasis, suggesting the need to further explore proper dosing regimens and targeting strategies that may avoid these adverse effects.47,152,400 Beyond tumorigenesis, dysregulation of STING can also contribute the development of inflammatory and autoimmune disease such as vascular and pulmonary syndrome, lupus-like syndromes, and STING-associated vasculopathy401. Researchers have even shown that cGAS/STING activation or increased expression can induce acute pancreatitis, colitis, and liver fibrosis397,402,403. However, it should be noted that most of these arise from sustained or chronic STING activation and, hence, may not be manifested in an immune-oncology setting. Nonetheless, they reflect the dichotomous role of STING signaling and the importance of carefully regulating the dose and kinetics of STING pathway agonists in promoting immunity instead of tolerance or chronic inflammation.
With a number of clinical trials for a diverse array of STING agonists underway or ongoing (Table 1), it will be interesting to learn in the coming months and years to what extent systemically administered agents have a therapeutic window and what acute and chronic side effects, if any, may manifest. Regardless, the potential for inflammatory side effects associated with systemic delivery of STING pathway agonists, and innate immune agonists more generally, motivates the need for tunable drug delivery technologies and/or novel agents that can help mitigate toxicity while still promoting the desired anti-tumor immune response. Such strategies potentially include nanocarriers that can improve the pharmacokinetic properties of STING pathway agonists such as half-life and AUC while exploiting dysfunctional tumor vasculature to enhance tumor accumulation, environmentally-responsive pro-drugs that activate selectively at tumor sites, and molecularly-targeted STING pathway agonist that enrich STING activation at tumor or immune priming sites and/or within specific cell populations. This is a nascent but important and rapidly expanding area of research with vast potential for expanding the therapeutic window of STING pathway agonists, and below we discuss emergent drug delivery strategies for improving the efficacy of STING pathway agonists for both systemic and local administration.
6. Delivery Technologies for STING Pathway Agonists
6.1. Nanotechnology for the Delivery of Cyclic Dinucleotides
Although CDNs exhibit great therapeutic promise for eliciting antitumor immunity, a variety of delivery challenges remain and restrain their potential. Much of this can be attributed to the anionic phosphate groups on CDNs that significantly limits their passive diffusion across the lipophilic plasma membrane and restricts their access to the cytosol for STING binding195,362,404. Thus far, clinical responses to intratumorally administered CDNs have proven modest due, in part, to the delivery barriers described above, including rapid clearance, lack of cytosolic delivery, and nonuniform drug distribution362,376,404,405. Similarly, systemic administration of CDNs is typically ineffective even in preclinical models due to a number of pharmacological shortcomings.
NP delivery platforms have great potential for improving the efficacy of STING pathway agonists as they offer numerous opportunities for enhancing CDN activity (Figure 14). First, NPs provide a strategy for modulating pharmacokinetic and biodistribution properties of drug cargo. A large and well-established tool box of chemical strategies exists for modulating the key physicochemical properties of NPs (e.g. size, shape, surface chemistry, stability, mechanical properties, permeability, etc.) and can be employed to modulate the pharmacokinetic and distribution behavior of STING pathway agonists to optimize immunotherapeutic benefit and minimize off-target inflammatory effects406,407. For example, systemically administered NPs have a well-established capacity for passively targeting solid tumors, which provides a facile and promising approach for increasing drug accumulation in tumors. However, it should be noted that while it is clear that NPs can preferentially accumulate in human metastatic tumors408–412, the primary mechanism of tumor accumulation is controversial413–415. The enhanced permeability and retention (EPR) effect, which was first proposed in 1986, describes how preferential tumor accumulation of NPs can be attributed to hypervascularity, defective vasculature, and poor lymphatic drainage in solid tumors416,417. The EPR effect has been a guiding principle of NP delivery for over 30 years, but recent work has challenged the importance and relative contribution of the EPR effect. Indeed, Sindhwani et al. found that NPs enter tumors using an active process through endothelial cells418, suggesting that physical gaps in tumor vasculature may not be as important as previously thought. Regardless, NP size and surface properties remain important design criteria that can be tuned to promote tumor accumulation.
Figure 14: Opportunities for nanotechnology in the delivery of STING pathway agonists.

Nanotechnology can be employed to overcome many drug delivery challenges and therefore has potential to greatly improve the therapeutic efficacy of STING pathway agonists. Notably, nanotechnology can exploit dysfunctional tumor vasculature, protect drug cargo, promote lymphatic drainage, enable cellular targeting, facilitate cytosolic delivery, and allow for cellular co-delivery of various drugs. Figure created with biorender.com.
Second, NPs can be designed with environmentally-responsive functionalities to enhance cytosolic CDN delivery and/or trigger drug release in response to a specific microenvironmental stimuli (e.g. pH, hypoxia, reactive oxygen species, etc.) for tumor-selective drug release or activation419. Third, NPs allow for co-packaging of multiple agents into a single particle at defined ratios, with potential to enable synergy between STING pathway agonists and other agents (e.g. chemotherapeutics, STING pathway potentiators) to widen the therapeutic window. Finally, NPs provide a versatile platform for introducing targeting ligands (e.g. antibodies, peptides, glycans) with potential to enhance tumor, lymphoid organ, and/or cell-specific delivery of STING pathway agonists. Below we describe recent advancements in NPs for the delivery of STING pathway agonists, which has primarily been directed at improving the delivery of CDNs.
6.1.1. Lipid-based Delivery Systems
Many research groups have sought to address some of the barriers to CDN delivery through the development of liposomal NP formulations (Figure 15)173,254,362,376,420–428. Liposomes are logical candidates for CDN delivery since they have an aqueous core for loading of hydrophilic cargo429–431. Koshy et al. utilized a cationic liposome formulation (Figure 15A) consisting of varying amounts of 2 kDa polyethylene glycol (PEG) along with the cationic lipid, 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), and cholesterol, repurposing a formulation that has been used in gene delivery applications and has been evaluated in cancer patients362. While more cytotoxic than their charge neutral counterparts, cationic liposomes can interact with the anionic cell membrane, allowing for enhanced internalization, and can also promote endosomal escape into the cytosol432. In these studies, the authors chose to load a phosphorothioated cGAMP analog into the liposomes, as that chemical modification confers resistance to degradation by ENPP1 and therefore can elicit a greater degree of STING signaling by avoiding rate-limiting CDN degradation197. A primary goal of the work was to evaluate the effect of surface PEGylation density on particle properties and attendant effects on antitumor efficacy. The liposome formulations used in the study contained 0, 5, and 10 mol% 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(PEG)-2000] (DSPE-PEG(2000)) with a 1:1 DOTAP:cholesterol ratio. Non-PEGylated liposomes resulted in an increased diameter compared to PEGylated particles, likely due to anionic serum protein binding on the cationic liposome surface causing liposome aggregation. While all NP formulations enhanced CDN uptake by BMDCs and increased STING-mediated proinflammatory cytokine expression relative to free cGAMP, PEGylation reduced the positive zeta-potential of the liposomes and thereby reduced CDN uptake by BMDCs and lowered STING activation relative to the non-PEGylated NPs, reflecting the familiar interplay between charge, uptake, toxicity, and activity that has also been described for nucleic acid therapeutics (e.g. siRNA)433. They also evaluated the effect of PEG surface density on inflammatory gene expression and inhibition of tumor growth using an intratumoral administration route in an orthotopic B16-F10 melanoma model. While there were relatively modest differences between the formulations on Ifnb1 and Cxcl9 expression, formulations lacking PEG (e.g. 0% PEG) were significantly less effective than both PEGylated liposomes as well as the free CDN, which led to complete regression of tumors in half of the mice. By evaluating the intratumoral distribution of a fluorescently-labeled CDN, this was attributed to the very poor tumor penetration of the CDN when loaded into the PEG-free liposome owing to aggregation at the injection site. Interestingly, though similar responses were observed in primary tumor regression between free cGAMP and the PEGylated liposomal formulations, the later appeared to confer greater protection from tumor rechallenge. Importantly, the authors also evaluated intravenous administration of the 5% PEGylated liposome in a lung metastatic B16-F10 model. Consistent with the capacity for DOTAP/cholesterol liposomes to passively target the lungs, they demonstrated increased inflammatory gene expression in tumor-bearing lung tissue, but a very modest effect on lung metastatic burden even when combined with ICB (i.e. anti-PD-1 and anti-CTLA-4). Hence, while further optimization of the delivery technology appears to be necessary for treatment of lung metastasis, this work highlights the importance of NP corona chemistry on tumor distribution and antitumor immunity.
Figure 15: Lipid-based CDN delivery systems.

(A) Schematic of a cationic liposomal 2′3′-cGAMP formulation, which comprises DOTAP, cholesterol, and DSPE-PEG(2000), and the proposed mechanism of intracellular 2′3′-cGAMP delivery. Reproduced with permission from reference362. Copyright © 2017 John Wiley & Sons - Books; permission conveyed through Copyright Clearance Center, Inc. (B) Schematic demonstrating that nanoparticles can enhance drug delivery to draining lymph nodes via lymphatic transport. Figure created with biorender.com. (C) By exploiting the lymphatic transport of nanocarriers, CDN (i.e. cdGMP) concentration increases in the draining lymph nodes of the injection site and decreases in the blood stream when delivered with a lipid nanoparticle. Reproduced with permission from reference376. Copyright © 2015 American Society for Clinical Investigation; permission conveyed through Copyright Clearance Center, Inc.
Similarly, Cheng et al. recently developed a non-PEGylated liposomal NP using a mixture of hydrogenated (soy)L-α-phosphatidylcholine (Soy-PC) and DOTAP to deliver 3’3’-cGAMP (referred to as NP-cGAMP)254, resulting in a formulation that could be delivered IV. Liposomes were made using a 100:1 ratio of Soy-PC to DOTAP to encapsulate cGAMP through thin-film hydration followed by membrane extrusion. This method resulted in particles that were ~ 85 nm in diameter with a +15 mV zeta-potential and a cGAMP encapsulation efficiency of ~ 43%. They evaluated their formulation in basal-like triple negative breast cancer C3(1) breast cancer models that are resistant to anti-PD-L1 ICB. Of high translational significance, they also evaluated their approach in a genetically engineered mouse C3(1) Tag model that generates spontaneous primary and secondary tumors with diverse immunosuppressive microenvironments that more accurately reflects human breast cancer. Impressive antitumor effects were observed in both an orthotopic transplant and the genetically engineered mouse model, with NP-cGAMP outperforming free cGAMP in terms of controlling tumor growth and prolonging survival. Notably, the formulation was well tolerated as indicated by minimal mouse weight loss following intravenous administration, and a single dose of cGAMP-loaded liposomes was enough to suppress tumor growth. The antitumor effect of NP-cGAMP was shown to be dependent on both CD4+ and CD8+ T cells as well as macrophages and was correlated with reprogramming of protumorigenic M2-like macrophages towards a more M1-like phenotype that support T cell infiltration and antitumor effector function. Additionally, by inducing a type I IFN response, NP-cGAMP may also have induced tumor cell apoptosis and inhibited proliferation. While less striking results were observed in a B16-F10 model, potentially reflecting differences in immunogenicity, the high degree of efficacy and safety in the C3(1) Tag model afforded by this liposomal formulation offers translational promise for breast cancer immunotherapy.
An alternative cationic lipid to DOTAP was described by Miyabe et al. who developed a synthetic, pH-responsive lipid, YSK05, which has optimal membrane fusogenic activity at pH 6.4 and therefore high endosomal escape activity420. In their initial report, they evaluated several helper lipids and lipid compositions for enhancing the in vitro activity of the bacterial CDN, c-di-GMP, ultimately arriving at a liposomal formulation comprising YSK05:POPE:cholesterol:DMG-PEG at a 40:25:35:1 ratio. Upon demonstrating that the formulation increased expression of CD80, CD86 and MHC class I in murine macrophages in vitro, they immunized mice with a model antigen (i.e. ovalbumin) mixed with c-di-GMP/YSK05 liposomes and demonstrated an enhanced CTL response and protection against challenge with an ovalbumin-expressing E.G7 thymoma cancer cell line. As a follow up to this study, the group demonstrated that intravenous administration of c-di-GMP/YSK05 liposomes reduced lung tumor burden in a metastatic B16-F10 melanoma model434 and found that the antitumor effect was primarily mediated by infiltrating NK cells that destroyed tumor cells due to their low levels of MHCI-I expression. While other reports have implicated T cells as the critical effectors in response to STING agonists, the findings of Miyabe et al. are consistent with those of recently described by Nicolai et al., who found intratumoral CDN administration promoted NK cell activation and antitumor function281. This is significant as strategies to bolster NK-based tumor immunity may be critical for cancers that are adept at evading T cell recognition via MHC-I loss or downregulation.
Liposomal systems have also been developed for dual-delivery of both CDNs and monophosphoryl lipid A (MPLA), an agonist of TLR-4, another important pattern recognition receptor that can lead to IFN-I and proinflammatory responses, as a strategy to more effectively activate local APCs at tumor sites421,422. Here, Karathanasis and co-workers utilized liposome formulations that lacked cationic lipids. Their first formulation consisted only of 1,2 dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), and methoxy-PEG-2000 1,2 distearoyl-sn-glycero-3-phosphoethanolamine (mPEG2000-DSPE) at 48.5, 48.5, and 3 mole percent, respectively, and generated relatively small liposomal particles (i.e. 60 nm in diameter)421. By co-encapsulating c-di-GMP and MPLA they were able to ensure delivery of both agonists to the same cell, resulting in synergistic activation of both STING and TLR-4 signaling. Owing to a lack of cationic lipids, this formulation was particularly well suited for systemic delivery, which resulted in liposome accumulation in APC-rich perivascular regions of tumors, primarily stimulating IFN-I responses within tumor-infiltrating APCs rather than the cancer cells. Impressively, intravenous administration of liposomes loaded with both CDN and MPLA significantly reduced the growth of 4T1 mammary tumors and prevented lung metastasis. In a subsequent study, a similar liposomal formulation consisting of c-di-GMP and MPLA encapsulated in 77:20:3 DPPC, cholesterol, and mPEG2000-DSPE was used to systemically treat Panc20 pancreatic ductal adenocarcoma tumors422. An important aspect of this subsequent work was the finding that synergy between c-di-GMP and MPLA can be increased by systematically adjusting the MPLA/c-di-GMP ratio in the formulation. Specifically, the authors fabricated and tested an MPLAhi (300 μg MPLA per 42 μmol lipids) and MPLAlo (300 μg MPLA per 42 μmol lipids) with a constant amount of CDN loading. They found that the MPLAhi variant generated a 1.6-fold higher IFN-β response than the MPLAlo formulation and, importantly, that dual-encapsulated particles enhanced IFN-β production relative to dose-matched single-agent particles (c-di-GMP-NPs and MPLA-NPs) by 11- and 22-fold. Importantly, this also manifested in vivo, with the MPLAhi formulation tending to increase the number of IFN-β secreting immune cells, and impressively, also inhibited Panc02 tumor growth to a greater extent than free agonists or the MPLAlo formulation. This study nicely illustrates the potential to potentiate STING pathway activation via coordinated delivery of another agent at a defined ratio, a distinctive advantage of leveraging a nanocarrier platform.
While an important advantage of nanocarriers for CDNs has been their ability to enable intravenous administration, they can also be harnessed to improve efficacy via other administration routes. An excellent example of this is the work by Liu et al. who designed a very elegant liposomal cGAMP formulation to specifically target pulmonary APCs to enhance anticancer immunity against lung metastases, which are common in many cancer types423. Because phagocytes such as macrophages and DCs can recognize membrane exposed phosphatidylserine (PS) on apoptotic cells, they integrated PS into a liposomal cGAMP formulation to provide an “eat me” signal for APCs. Their formulation was fabricated through a two-step water-in-oil reverse microemulsion and incorporated calcium phosphate (CaP) to improve cGAMP encapsulation and provide a release and endosomal escape mechanism triggered by a decrease in endo/lysosomal pH. Using a 5:4:1 ratio of phosphatidylserine:1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC):cholesterol resulted in NP-cGAMP particles that were anionic (i.e. −40 mV) and ~ 120 nm in diameter, yet still achieved a 72% cGAMP encapsulation efficiency. Both layers of liposome membrane contained anionic PS, which allowed for both APC targeting and neutralization of excessive cationic Ca2+ of CaP not complexed with cGAMP in the core. Using an aerosolized NP-cGAMP formulation to achieve delivery to the deep lungs via inhalation, particles rapidly distributed throughout both lungs and were efficiently endocytosed by pulmonary APCs. Inhalation of NP-cGAMP alone led to a decrease in the number of metastatic foci in both lungs in a melanoma model of lung metastasis. Importantly, combining NP-cGAMP with radiotherapy further increased therapeutic efficacy in both melanoma and breast cancer metastasis models, even leading to complete regression of lung metastases in some mice. This synergistic effect was attributed to enrichment of STING activation in pulmonary APCs that therefore more efficiently cross-primed antitumor CD8+ T cells and generated a proinflammatory TME that supported T cell infiltration and inhibited immunosuppressive TREG cells.
While the use of targeted nanoparticles for cell- or tissue-specific delivery of STING activation is still in its infancy, this work highlights the potential merits of targeting specific cell populations within a specific tissue, which may also minimize undesired inflammatory side effects. For example, Li et al. have also leveraged the idea of APC targeting by incorporating a DSPE-PEG-mannose conjugate into a liposomal cGAMP formulation as a strategy to target dendritic cells424; however, this was only evaluated using an intratumoral delivery route and the therapeutic impact of mannose-targeting was not established.
In a comprehensive evaluation of STING agonist targeting using nanocarriers, Covarrubia et al. developed a NP platform for systemic c-di-GMP delivery that was designed to target APCs within the perivascular niche of the TME through both passive accumulation and direct ligand binding428. The researchers sought to characterize NP accumulation and cellular uptake in mouse models that mimicked different cancer landscapes, including primary tumors, early metastasis, and late metastasis. The differential uptake by specific immune cell subsets was also examined. The nanoparticles were ~ 60 nm in diameter and consisted of 48.5 mol% DOPC, 48.5 mol% DPPC, and 3 mol% mPEG2000-DSPE. The c-di-GMP was loaded into the particle core through film hydration. For the ligand-functionalized particles, 3 mol% mPEG2000-DSPE was replaced with 3 mol% 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(PEG)-2000] (DSPE-PEG2000 amine) to allow for sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) coupling between amines on the particle surface and thiol groups on the targeting peptides. Three peptides were explored as targeting ligands for their ability to bind fibronectin (CREKA), P-selectin (CDAEWVDVS), and αvβ3 integrin (c(RGDfC)) to preferentially direct the particles to the TME and certain immune cells. Fibronectin is overexpressed on the perivascular extracellular matrix, while P-selectin is overexpressed by endothelial cells in the remodeled tumor vasculature. Additionally, αvβ3 integrin is typically expressed by dendritic cells and macrophages. Although this design offers potential for next-generation, targeted STING technologies, the untargeted NPs demonstrated the highest immune cell uptake and tumor accumulation with the exception of early stage metastasis of 4T1 tumors to the lung and liver, where integrin-targeting NPs had the highest uptake by lung DCs and liver macrophages. These findings are consistent with the increased circulation time of non-targeted nanoparticles and the ability of nanoparticles to exploit dysfunctional vasculature of established tumors for passive targeting. These results also demonstrate the potential impact of molecular targeting in enhancing CDN accumulation at sites of early metastasis, where the vascular endothelium might be less permeable. Notably, in a neoadjuvant therapy model using the 4T1 murine breast cancer and co-administered ICB (i.e. anti-PD-1), the untargeted NPs demonstrated significant therapeutic efficacy and outperformed the integrin-targeted NPs. Though not examined in this report, the authors mention that in future work, they will explore whether targeted immunostimulatory NPs are more effective in a purely metastatic setting. Collectively, this comprehensive study nicely demonstrates the potential of leveraging targeted STING activation for specific clinical scenarios and also highlights the importance of identifying immune and cancer-specific biomarkers towards enabling enhanced delivery of STING agonists and improved therapeutic results.
In another recent example of a targeted NP for CDN delivery, Gou et al. discovered that STING agonists significantly stimulated type I IFN secretion in Clec9a+ dendritic cells and therefore designed a peptide-expressed biomimetic cancer cell membrane (EPBM)-coated nanovaccine platform to preferentially deliver STING agonists and tumor antigens to this particular cell population435. Clec9a is a C-type lectin endocytosis receptor that is responsible for antigen uptake and cross-presentation. Notably, Clec9a has been utilized for targeted tumor vaccines, which motivated this group to develop a 12-mer Clec9a binding peptide (CBP-12) that could be incorporated into various delivery technologies. A retroviral vector encoding CBP-12 was constructed and used to incorporate this peptide into cancer cell membranes, which were then extracted and fused with a PLGA NP core containing 2′3′-cGAMP to form PLGA/STING@EPBM particles, which were ~ 160 nm in diameter. This resulted in enhanced CDN uptake in DCs, expression of ISGs, antigen cross-presentation, and T cell proliferation in the B16-OVA tumor model, which collectively inhibited tumor growth and prolonged survival. This platform also inhibited both tumor growth and lung metastasis in an anti-PD-1-resistant 4T1 tumor model. Additionally, this treatment strategy was further improved by co-administering radiotherapy, which was shown to increase the amount of Clec9a+ DCs within the tumor microenvironment.
While not evaluated in the context of cancer immunotherapy, Wang et al. developed a novel liposomal cGAMP formulation for intranasal delivery and evaluated this as an adjuvant for vaccines to prevent respiratory viral infections, such as influenza173. CD8+ tissue resident memory T-cells (TRM) are essential immune effectors in viral infections, specifically those located within the lung436–438. Therefore, to generate a TRM response, the investigators aimed to design a delivery platform that would not only target pulmonary APCs, but also alveolar epithelial cells without breaching the integrity of the pulmonary surfactant layer. To accomplish this, they designed pulmonary surfactant-biomimetic liposomes (PS-GAMP) to deliver cGAMP along with vaccine antigen (e.g. inactivated influenza virus) into the lung via intranasal immunization. The liposomes were considered PS-mimetic because they were based on PS constituents and ultimately an anionic formulation comprising DPPC:1,2-dipalmitoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DPPG):cholesterol:1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(PEG)-2000] (DPPE-PEG2000) at a mass ratio of 10:1:1:1 was found to most closely mimic the PS and demonstrated the strongest adjuvant properties of the formulations tested. Importantly, the authors also investigated non-PEGylated and cationic variants, which showed less adjuvancy and significantly more toxicity, implicating the negative charge and surface PEGylation as important to the activity and function of their liposomal cGAMP formulation. When used to adjuvant an H1N1 influenza vaccine, PS-GAMP generated a robust CTL response within the respiratory tract, which remained for 6 months post-vaccination, and also elicited cross-protection against other influenza strains. A particularly important result of this work of potential relevance to cancer immunotherapy was their discovery that PS-cGAMP uptake by alveolar macrophages required the surfactant proteins A and D and that cGAMP released into the cytosol of alveolar macrophages was transferred to alveolar epithelial cells through gap junctions, activating STING in both cell types. As cGAMP is known to spread via gap junctions, the design of delivery systems capable of exploiting such transfer mechanisms may prove valuable for improving tumor penetration of CDNs with potential to improve immunotherapeutic efficacy.
LNs act as command centers for orchestrating adaptive immune responses and are therefore another important target tissue for the delivery of STING pathway agonists in addition to tumor sites. Specifically, tdLNs are important sites for priming and expansion of antitumor T cells, yet, like tumor sites, may also be highly immunosuppressed and therefore contribute to T cell dysfunction439,440. Additionally, cancer vaccines are a promising and re-emerging class of cancer immunotherapy, and owing to the critical role of STING activation in generating antitumor immunity, CDNs hold great potential as adjuvants for cancer vaccines382,441. However, due to the rapid clearance of CDNs, their delivery to LNs is inefficient, a challenge that has partly motivated the development of strategies for targeting STING agonists to LNs. NPs of ~ 20–100 nm in diameter can passively target cargo to local draining LNs through drainage from interstitial space into the lymphatic system (Figure 15B)442,443. In a seminal paper, Hanson et al. sought to exploit this transport phenomenon by encapsulating c-di-GMP into phosphatidylcholine liposomes with 5% PEGylation (NP-cdGMP)376, electing to incorporate PEG, since it had been shown to enhance LN trafficking of liposomes in previous studies444,445. Primarily due to its low molecular weight, they reported that subcutaneously administered free c-di-GMP inefficiently trafficked to the LN and instead distributed to the blood, resulting in minimal cellular uptake by APCs in the draining LN. By contrast, NP-cdGMP preferentially accessed the LN and improved CDN uptake by APCs (Figure 15C). Using NP-cdGMP as an adjuvant and ovalbumin (OVA) as a model subunit antigen, they found that liposomal CDN delivery elicited an ~ 3-fold greater CD4+ and CD8+ T cell response relative to a soluble mixture of OVA and c-di-GMP. Accordingly, immunization with NP-cdGMP as an adjuvant reduced tumor growth and prolonged survival in mice inoculated with an OVA-expressing EG.7 tumor. They also evaluated responses to the poorly immunogenic antigen MERP from HIV gp41 and further demonstrated the importance of LN targeting in optimizing CDN adjuvancy. Use of NP-cdGMP as an adjuvant significantly increased the expansion of helper T lymphocytes, induced germinal center formation, and generated a robust and durable humoral response, while mitigating systemic cytokine production.
Finally, a unique approach to enhancing STING activation is to express activated STING itself. Tse et al. formulated lipid NP (LNP) mRNA vaccines to deliver the mRNA transcript of STING with a dominant gain-of-function mutation (V155M) that renders STING constitutively active even in the absence of cGAMP or other STING ligands425. The mRNA was synthesized in vitro by T7 RNA polymerase-mediated transcription with N1-methylpseudouridine in place of uridine. mRNA-loaded LNPs were formed by combining an ionizable lipid:DSPC:cholesterol:PEG-lipid at a ratio of 50:10:38.5:1.5 in ethanol with mRNA in aqueous buffer through synchronized syringe pumps at a 1:2 ratio. After filtration, the particles were 80–100 nm in diameter with greater than 80% mRNA encapsulation. Mice inoculated with TC-1 tumors transformed with HPV16 E6 and E7 oncoproteins were vaccinated with mRNA encoding antigen co-formulated with STING(V155M) mRNA in LNPs. This elicited E7-specific CTLs which suppressed tumor growth and prolonged survival. Similar results were found in a murine lung metastasis model using luciferase-expressing TC-1 cells. This innovative strategy has potential advantages over liposomal delivery of STING ligands, particularly for local administration, including efficient mRNA loading into LNPs, an established safety profile for mRNA-based vaccines, and the possibility of modulating STING activation kinetics through control of intracellular mRNA stability and degradation rate, which may be employed to maximize efficacy and mitigate undesired inflammation.
6.1.2. Polymeric Delivery Systems
While the use of liposomal and lipid-based carriers may provide translational advantages owing to the approval of other lipid-based drug formulations (e.g. Doxil, Patisiran), polymeric carriers afford a greater degree of synthetic control over key physicochemical properties and can confer integration of unique functionalities and, hence, polymers also have great potential for enhancing CDN delivery (Figure 16). In one of the first examples of a polymeric CDN delivery system, Lee et al. developed submicron-sized hydrogel particles loaded with both 3′,3′-cGAMP and 2′,3′-cGAMP (Figure 16A). The particles were assembled using thiol-modified linear polyethyleneimine (LPEI-SH) that was mixed with a solution of hyaluronic acid (HA) and cGAMP to form electrostatic complexes that were then added to an organic solvent and emulsified with surfactants via water-in-oil emulsion446. To introduce thiol groups and, therefore enable crosslinking, the secondary amines on LPEI were reacted with propylene sulfide to achieve ~ 5% thiol backbone modification. Although cGAMP would typically be too small to be stably encapsulated into such porous hydrogels, the intermolecular disulfide linkages and ionic interactions between LPEI and HA and LPEI and cGAMP allowed for a 47% entrapment efficiency. This formulation strategy yielded spherical particles designed to be relatively large in size (i.e. diameter ~ 455 nm) with a positive zeta-potential of +49 mV to more specifically target phagocytotic macrophages and dendritic cells. Upon internalization, intracellular reduction of the disulfide bonds allowed for triggered cGAMP release. Hydrogel particles displayed higher cytocompatibility than LPEI or LPEI/HA complexes and, importantly, enhanced STING activation as measured by induction of IFN-β and IL-6 secretion relative to empty particles, free cGAMP, or LPEI complexed with cGAMP. The authors tested the efficacy of this system as a vaccine platform with OVA injected intramuscularly into C57BL/6 mice and demonstrated that the particle significantly elevated levels of anti-OVA total IgG in serum. While promising as a vaccine platform, further studies to test efficacy as a cancer immunotherapeutic are necessary.
Figure 16: Polymeric CDN delivery systems.

(A) Formulation of cGAMP and ovalbumin (OVA) into linear polyethyleneimine / hyaluronic acid (LPEI/HA) hydrogels for enhanced STING activation and antigen presentation. Adapted with permission from reference446. Copyright © 2015 Elsevier Science & Technology Journals; permission conveyed through Copyright Clearance Center, Inc. (B) Nanoparticle assembly and cytosolic delivery of CDNs using cationic poly(beta-amino ester) (PBAE) nanoparticles to induce STING activation. Reproduced with permission from reference447. Copyright © 2017 Elsevier Science & Technology Journals; permission conveyed through Copyright Clearance Center, Inc. (C) Chemical structure, formulation strategy, and intracellular delivery mechanism for STING-NPs (i.e. endosomolytic polymersomes for cytosolic delivery of 2′3′-cGAMP). Reproduced with permission from reference196. Copyright © 2019 Springer Nature BV; permission conveyed through Copyright Clearance Center, Inc. (D) Antitumor effect and prolonged survival of mice with B16-F10 melanoma treated with intravenous administration of STING-NPs alone and in combination with ICB (i.e. anti-PD-1 and anti-CTLA-4). Reproduced with permission from reference196. Copyright © 2019 Springer Nature BV; permission conveyed through Copyright Clearance Center, Inc.
Another early example of a polymeric CDN delivery system was described by Junkins et al. who employed an acid-sensitive microparticle platform for intracellular delivery of 3′3′-cGAMP448 as a strategy to improve its activity as a vaccine adjuvant. To formulate particles, the researchers used acetalated dextran (Ace-DEX), which can be solubilized and electrosprayed to fabricate microparticles with high CDN encapsulation efficiency and excellent stability via an industrially scalable process. Notably, organic soluble Ace-DEX can be synthesized through one-step synthesis that converts the pendant hydroxyl groups of FDA-approved, water-soluble, 70 kDa dextran homopolysaccharide into acetal groups, which enhances their organic solubility and enables formulation into polymeric microparticles. Microparticles, having a diameter of ~ 1.54 ± 0.47 μm and zeta-potential of −32.0 ± 0.7, were formed through a coaxial electrohydrodynamic spraying method with Ace-DEX in an ethyl acetate/butanol/ethanol co-solvent mixed with cGAMP in molecular-grade water, achieving 90% cGAMP encapsulation efficiency. Acetylated microparticles provide a mechanism of cargo release that is triggered by a decrease in pH within the endo-lysosome owing to pH-dependent hydrolysis of acetals that results in regeneration of hydroxyl groups, aqueous chain solubility, dissolution of particles, and drug release449. The particles displayed an initial burst release followed by a controlled release profile of CDNs for 28 days. Importantly, these particles were of optimal size to be endocytosed by APCs, and the acid sensitivity allowed for intracellular release of CDNs within the phagolysosome. How the CDNs escaped the endosome into the cytosol to access STING was not described, though it is reasonable to suspect that a high concentration of CDN is achieved in the phagolysosome, which could drive diffusion of CDN across the phagosomal membrane and/or the use of membrane transporters. They found that loading of CDNs into Ace-DEX microparticles resulted in significantly enhanced proinflammatory cytokines and type I IFN responses, providing 1000-fold dose-sparing compared to soluble cGAMP for pro-inflammatory cytokines and type I IFN responses in primary APCs. The microparticles also significantly enhanced local type I IFN and IL-6 responses and preferentially trafficked to the LN while mitigating systemic inflammation when injected intramuscularly. CDN-loaded Ac-DEX particles were then evaluated as an adjuvant for an influenza vaccine and were found to enhance the proinflammatory cytokine response while significantly reducing the required dose of CDN needed to stimulate anti-influenza immunity. This vaccine platform also increased anti-hemagglutinin antibody titers, specifically Th1 IgG response, resulting in complete protection against H1N1 influenza challenge.
In the first application of a polymeric CDN carrier for a cancer application, Wilson et al. employed biodegradable NPs composed of a poly(beta-amino ester) (PBAE) to improve cytosolic delivery of the phosphodiesterase resistant CDNs, ML-RR-CDA and RR-CDG (Figure 16B)447. PBAEs, which have been widely employed for delivery of nucleic acids (e.g. DNA, mRNA), was selected due to its biodegradability, structural diversity, ease of synthesis, ability to electrostatically bind nucleic acids, and endosomolytic activity. The PBAEs were synthesized at a molar ratio of 1.1:1 from monomers 1,4-butanediol diacrylate and 4-amino-1-butanol and then end-capped with a 0.2 M solution of 1-(3-aminopropyl)-4-methylpiperazine. A 500:1 polymer to CDN ratio was used for formulation, resulting in particles that had a diameter of ~ 100 nm with a positive zeta-potential of +10 mV. It is presumed that the CDNs were loaded via electrostatic interactions between phosphate groups on CDNs and cationic amino groups on PBAE, but the overall stability of this loading method is unclear as mono- or divalent electrostatic interactions may be insufficient to confer stability in physiological media. Nonetheless, this strategy increased CDN endocytosis by THP1 human monocytes and RAW 264.7 murine macrophages and was able to enhance activation of IRF3 in vitro relative to free CDN. When combined with ICB (i.e. anti-PD-1), Intratumoral administration of CDNs loaded into PBAEs strongly inhibited tumor growth in a B16-F1 melanoma model relative to dose matched free CDN combined with ICB, and also reduced the dose of CDN required exert comparable therapeutic activity by 10-fold. Of translational significance, it was demonstrated that the formulation could be lyophilized and stored for over 9 months without a significant loss of biological activity. This was an important aspect of this work as translational considerations such as stability and scalability have typically been ignored in the development of nanocarriers for STING pathway agonists.
While an abundance of cationic polymers (e.g. PEI, PBAE, etc.) have been developed for the delivery of macromolecular nucleic acids (e.g. siRNA, mRNA, etc.), CDNs bear only two negative charges and therefore cannot exploit the binding capacity afforded by multivalent interactions with polycationic carriers. Accordingly, our group has postulated that polymeric carriers for CDNs have distinctive design requirements and therefore require new drug delivery approaches196,382,390,450,451. Indeed, Shae et al. recently designed polymeric vesicles (i.e. polymersomes) that have an aqueous core for efficient CDN loading and a vesicle membrane comprising amphiphilic diblock copolymer chains with pH-responsive endosomal membrane-destabilizing activity (Figure 16C)196. Critical to the structure and function of these polymersomes are well-defined mPEG2kDa-block-[(2-diethylaminoethyl methacrylate)-co-(butyl methacrylate)-co-(pyridyl disulfide ethyl methacrylate)]5kDa (PEG-DBP) copolymers synthesized using reversible addition-fragmentation transfer (RAFT) polymerization. The carrier was designed such that at physiologic pH, the membrane-destabilizing DEAMA-co-BMA polymer segments are sequestered in the polymersome bilayer, shielded by a 2 kDa PEG corona to confer colloidal stability and increase circulation half-life. In response to endosomal acidification, the polymersomes disassemble to release CDNs and reveal membrane lytic domains that mediate endosomal escape of CDNs to the cytosol. An important aspect of the design is the copolymerization of thiol-reactive PDSMA groups into the second block for post-assembly crosslinking of the vesicle membrane. This increases chain molecular weight, and consequently endosomolytic activity, while also yielding less toxic, low molecular weight unimers upon reduction of disulfide crosslinks in the cytosol. Polymersomes were formulated via a modified direct hydration method, resulting in ~ 40% cGAMP encapsulation efficiency and yielding surface charge-neutral NPs with a median hydrodynamic diameter of ~ 100 nm, comparable properties to approved liposomal drug formulations, but with potent endosomolytic activity. As a result, these STING-activating NPs (STING-NPs) dramatically increased the immunostimulatory potency of cGAMP in monocyte, macrophage, dendritic cell, and melanoma cell lines, as well as human metastatic melanoma tissue. Although increased CDN cellular uptake contributed to increased activity, this was mostly attributed to potent endosomal escape as analogs with less endosomolytic activity were less efficient at STING activation. Consequently, in an aggressive and poorly immunogenic B16-F10 murine melanoma model, intratumoral administration of STING-NPs converted the TME to an inflamed and tumoricidal microenvironment, with significant upregulation of IFN-I and ISGs, pro-inflammatory cytokines, leukocyte-recruiting chemokines, pro-apoptotic mediators, and markers of DC maturation and T cell activation. In mice bearing two tumors, intratumoral treatment of one tumor (i.e. primary) resulted in a significant decrease in tumor growth rate for both primary and distal tumors, indicative of an abscopal effect. Intratumoral administration of STING-NPs was also applied for the treatment of neuroblastoma and was shown to trigger immunogenic cell death and inflame the TME to inhibit primary and distal tumor growth and improve response to ICB (i.e. anti-PD-L1)450. This study was also the first to evaluate STING activation as a therapeutic target in neuroblastoma, a pediatric cancer that is poorly responsive to immunotherapy and for which new treatment options are urgently needed.
Significantly, STING-NPs opened a therapeutic window for systemic, intravenous delivery of cGAMP, inhibiting tumor growth and increasing mean survival time in mouse models of melanoma (B16-F10 and YUMM1.7) and breast cancer (E0771)196,390. Strikingly, a 40% complete response rate was observed in mice given STING-NPs in combination with ICB (i.e. anti-PD-1 and anti-CTLA-4), whereas neither free cGAMP nor ICB alone demonstrated a therapeutic benefit. It was further found that intravenously administered STING-NPs enhance the half-life of cGAMP by 40-fold from ~ 2 minutes to 90 minutes, resulting in increased tumor accumulation of cGAMP, elevated expression of IFN-I and proinflammatory cytokines in the TME, and a dramatic increase in the number of CD8+ and CD4+ T cells that infiltrated poorly immunogenic B16-F10 melanoma tumors390. Unsurprisingly, and consistent with the behavior of most nanoparticle delivery systems, STING-NPs also accumulated in the liver and spleen, resulting in activation of STING in these tissues and a transient elevation of serum cytokines that peaked around 4 hours and rapidly subsided 8–24 hours later. Importantly, intravenous administration of STING-NPs was safe and well-tolerated, resulting in only mild and transient weight loss, insignificant effects on blood chemistry, including markers of liver (e.g. ALT, AST) and kidney (e.g. creatinine) damage, and no signs of organ damage via histopathology. Nonetheless, splenic and hepatic toxicities are likely to be dose limiting for many nanoparticle-based STING pathway agonists, and therefore, strategies to minimize STING activation in the liver and spleen may allow for improved efficacy and safety.
A distinctive advantage of using nanocarriers for the delivery of STING pathway agonists is the opportunity to co-package additional cargo, which our group has exploited to achieve dual-delivery of CDNs and tumor peptide antigens to enhance response to cancer vaccines. Shae and Baljon et al. demonstrated that endosomolytic polymersomes could be co-loaded with cGAMP and several different peptide antigens, including murine tumor neoantigens382. Through co-loading of both tumor antigen and cGAMP in a common particle, this design mimics the natural immunological cues that underlie spontaneous antitumor immunity. Indeed, the vaccine platform helped ensure synchronous delivery of cGAMP and antigens to APCs in draining LNs, resulting in increased antigen-specific CD8+ T cell responses that inhibited tumor growth and increased survival with combined with ICB in mouse models of B16-F10 melanoma and MC38 colon cancer.
Immunogenic cell death (ICD) is an inflammatory form of cell death that has been implicated in the generation of antitumor adaptive immunity in response to various cancer therapies452. Direct STING activation can trigger ICD in some, but not all, cancer types, and some chemotherapy agents can induce ICD453, whereas others do not. In order to mimic the sequence of events in the process of STING-mediated ICD, Chattopadhyay et al. aimed to devise a platform to deliver CDN STING agonists prior to treatment with cytotoxic chemotherapy agents. This process stimulates STING activation immediately prior to chemotherapy-induced cell death, effectively adjuvating tumor cell debris to stimulate antitumor immunity454. To achieve this, they utilized poly(lactic-co-glycolic) acid (PLGA) nanoshells (NS), which allow for encapsulation of cGAMP (NS-cGAMP) in a large aqueous core and pH-responsive release following internalization455. Although most other CDN carrier systems have been designed to passively target APCs, the authors used this technology to deliver a STING agonist directly to cancer cells followed by subsequent treatment with a cytotoxic agent, a process they refer to as “synthetic ICD” with the “immunogenic” component conferred via exogenous CDN delivery. These particles were formulated through a double emulsion process with low-viscosity, carboxyl-terminated PLGA (50:50 ratio, 0.15–0.25 dL/g), resulting in particles that were ~ 100 nm in diameter with 10 nm thick shells that were able to encapsulate cGAMP at 42% efficiency and enhance STING activation in both immune and cancer cells relative to free CDN. The investigators found that while Intratumoral administration of NS-cGAMP had a modest tumor inhibitory effect in multiple mouse models, therapeutic responses were improved when combined with chemotherapy agents (i.e. irinotecan, doxorubicin, cisplatin). This work offers evidence that coordination of tumor cell death and liberation of tumor antigen (here, via chemotherapy) and activation of innate immunity via the STING pathway can act as an in situ vaccine that enhances priming and activation of tumor antigen-specific T cells. However, how to optimally sequence and/or coordinate chemotherapy with administration of STING agonists to maximize antitumor immunity has not been widely explored and will likely depend on the tumor type and type of chemotherapy agent.
Towards answering this question, Liang and Wang et al. developed a polymeric carrier for dual-delivery of the chemotherapeutic agent SN38 and the murine STING agonist DMXAA456. To achieve this, they synthesized a triblock copolymer with a 5 kDa PEG first block, a second block comprising a redox-responsive prodrug monomer of SN38, and a third block of diethylamino-ethyl methacrylate (DEAMA), which enabled electrostatic interactions with the carboxylic acid group of DMXAA. Through control of block molecular weight, the group established a formulation that assembled into particles that were ~ 30 nm in diameter with greater than 80% loading of DMXAA. Using an intravenous administration route in multiple tumor models, it was demonstrated that NPs containing both SN38 and DMXAA were most effective in inhibiting tumor growth and, notably, also more effective than a formulation comprising a mixture of SN38-loaded NPs and free DMXAA. This improved response was attributed to coordinated tumor cell killing and increased infiltration and activation of APCs in the TME capable of processing and presenting liberated tumor antigen for cross-priming of CD8+ T cells. While alternative sequencing regimens were not considered, these studies suggest that dual-loading of chemotherapy and STING pathway agonists may improve therapeutic responses and they also motivate the design of carrier systems that are optimized to maximize synergy between cytotoxic agents and STING pathway agonists.
To avoid use of chemotherapy while still aiming to enhance tumor antigen uptake by APCs, Lu et al. also leveraged a polymeric carrier for dual-delivery of cGAMP and an siRNA to inhibit expression of the phagocytosis checkpoint, signal regulatory protein α (SIRPα)457. SIRPα is expressed by APCs and binds to the “don’t eat me signal,” CD47 expressed by cancer cells as an immune evasion mechanism to prevent phagocytosis. The group co-loaded cGAMP and siRNA into particles, which had a diameter of ~ 100 nm and were comprised of PEG-block-PLGA copolymers and the cationic lipid DOTAP. The researchers demonstrated the capacity of the system to both enhance cGAMP activity as well as knock down SIRPα in dendritic cells. Using an intravenous administration route in a B16-F10-OVA melanoma model, some enhancement in tumor growth inhibition was demonstrated when both cGAMP and SIRPα siRNA were co-delivered relative to formulations containing just cGAMP or just siRNA. The improved therapeutic response was attributed to enhanced priming and infiltration of antigen-specific T cells. Similar to the work by Liang and Wang et al., this study adds further support for the concept of coordinating STING activation with strategies to increase the amount of tumor antigen available for uptake, processing, and cross-presentation by activated APCs in tumors and tdLNs.
6.1.3. Inorganic Delivery Systems
Inorganic materials have also been utilized for CDN delivery and STING activation, and offer potential advantages such as access to particles of very small size, low polydispersity, and the potential to leverage imaging modalities to evaluate tumor accumulation. An et al. was among the first to leverage an inorganic carrier, using cationic, amine-modified silica NPs (CSiNPs) that allowed for the electrostatic interaction with CDNs while also increasing endocytosis via interactions with anionic cell membranes458. Notably, due to their cationic nature, CSiNPs have intrinsic cytotoxicity, which the investigators postulated could promote the generation of tumor-associated antigens from necrotic tumor cells to improve response to in situ vaccination (i.e. intratumoral immunotherapy). Using silica NPs with a diameter of 30 nm and a 240:1 NP to CDN mass ratio, they achieved 65% CDN encapsulation efficiency, yielding particles that were 35 nm in diameter and had a positive +18 mV zeta-potential. In a B16-F10 melanoma model, intratumoral injection of CSi-c-di-GMP-NPs resulted in an increase in necrotic tumor cells while also prolonging CDN release and achieving a more uniform distribution compared to free CDN. The combination of CSiNPs and CDN, but not individually, activated local APCs and triggered expansion of antigen-specific CTLs, resulting in reduction in tumor growth and prolonged survival. Significantly, an ~ 40% complete response rate was observed and surviving mice were fully protected against tumor rechallenge.
Similarly, Park et al. developed biodegradable mesoporous silica NPs (bMSNs) to enhance the cellular uptake and cytosolic delivery of cyclic-di-adenosine monophosphate (c-di-AMP)459. By reducing the density of the Si-O-Si matrix to increase the pore size and accelerate biodegradation rate compared to conventional MSNs, and also surface-modifying bMSNs with amino groups to allow for electrostatic loading of CDNs, the authors were able to achieve particles that were ~ 80 nm in diameter with over 90% CDN loading efficiency via simple mixing. At a physiological pH of 7.4, CDNs were released from the particle very quickly (i.e. within an hour), but were released slowly over the course of several hours at pH 6.0, mimicking the acidic conditions of some tumors. Despite such rapid release of CDN, bMSNs increased cellular uptake of CDNs by bone marrow-derived dendritic cells (BMDCs), resulting in increased CD40 and CD86 expression and release of cytokines and chemokines including IL-6, IL-12p40, IFN-β, CXCL10, CCL2, CCL3, and CCL5. In a B16-F10-OVA model, a single intratumoral injection of CDN loaded bMSNs completely inhibited tumor growth, whereas only 50% of mice survived following injection of free CDN.
A similar approach was pursued by Bielecki et al. who used mesoporous silica NPs (MSNs) as a nanocarrier for c-di-GMP to boost antitumor immunity in glioblastoma multiforme (GBM)460. The group generated high-surface area, mesoporous structures from tetraethylorthosilicate particle nucleation with cetyltrimethylammonium bromide and functionalized MSNs with N1-(3-trimethoxysilylpropyl) diethylenetriamine to generate amino groups for electrostatic loading of c-di-GMP as well as reactive handles for surface PEGylation. This process yielded PEGylated NPs with a diameter of ~ 60 nm that transitioned from cationic (i.e. +59.1 mV) to nearly charge neutral zeta-potential following loading of c-di-GMP, which was highly efficient (i.e. 99.2% loading efficiency). In contrast to Park et al., CDN release was slow at neutral pH, but accelerated at pH 5.5 due to deprotonation of surface amino groups and reduced electrostatic interactions, allowing for release of CDNs under acidic conditions. The investigators evaluated their platform in an orthotopic GL261 GBM model using an intravenous administration route. They found that MSNs were able to cross the compromised blood–brain barrier in the GBM model to directly access APC rich near-perivascular regions of the brain tumor where they were efficiently taken up by APCs. This triggered a reprogramming of the immunosuppressive GBM TME, resulting in the recruitment of inflammatory macrophages and DCs and to tumor sites, while sparing healthy brain tissue, and, importantly, inhibiting tumor growth.
Chen et al. also employed a similar MSN system to deliver c-di-GMP461, using N-trimethoxysilylpropyl-N,N,N-trimethylammonium chloride to generate cationic particles for electrostatic loading of c-di-GMP; Rhodamine B isothiocyanate (RITC) was integrated to allow for fluorescent imaging. Consistent with other reports, cationic MSNs efficiently loaded c-di-GMP (i.e. greater than 95% encapsulation efficiency), resulting in slightly smaller particles (i.e. ~ 47 nm in diameter) that enhanced CDN uptake and activity. Efficacy was evaluated via intratumoral administration in a 4T1 breast cancer model where MSN-mediated c-di-GMP delivery increased tumor infiltration of macrophages, dendritic cells, and T cells and inhibited tumor growth to a greater degree than free cGAMP. Although not investigated in this series of studies using MSNs, the tunable pore diameter and a large internal surface area of this platform offers the possibility of co-loading different therapeutics, such as chemotherapies or ICB to further enhance efficacy.
6.1.4. Biologically-Derived Carriers
Biologically-derived drug carriers, including cells441, bacteria462, virus-like particles463, extracellular vesicles464–467, and proteins468,469, have also been explored to enhance STING activation in cancer immunotherapy (Figure 17). In one of the first uses of a CDN for a cancer immunotherapy application, Fu et al. developed STINGVAX, a cell-based cancer vaccine system that combined the synthetically-derived, phosphodiesterase-resistant, (RP,RP) dithio CDA diastereomer (RR-S2 CDA) and irradiated GM-CSF-secreting tumor cells as a source of tumor antigens and evaluated vaccine efficacy in SCCFVII, TRAMP, Panc02, CT26, and B16 tumor models441. Vaccines were prepared by pre-incubating the CDN with lethally irradiated GM-CSF-secreting cancer cell lines prior to subcutaneous injection. Whether or not this resulted in CDN uptake by cancer cells was not directly evaluated, but it is conceivable that cells acted as a CDN carrier in these studies and served to co-localize STING agonist with tumor-derived antigens. The investigators found that this cell-based vaccine platform activated DCs in the tdLNs, induced naïve CD8+ T cell priming, significantly reduced tumor growth, and increased survival in multiple mouse tumor models. Notably, when combined with ICB (i.e. anti-PD-1), STINGVAX treatment not only led to cancer regression in both B16 and CT26 tumor models, but also long-term tumor-specific memory as demonstrated in CT26 rechallenge studies. Due to the proven safety profile of both GM-CSF-secreting whole cell vaccines and locally administered CDNs, this cancer vaccine platform appears well positioned for being translated into clinical trials.
Figure 17: Biologically-derived CDN carriers.
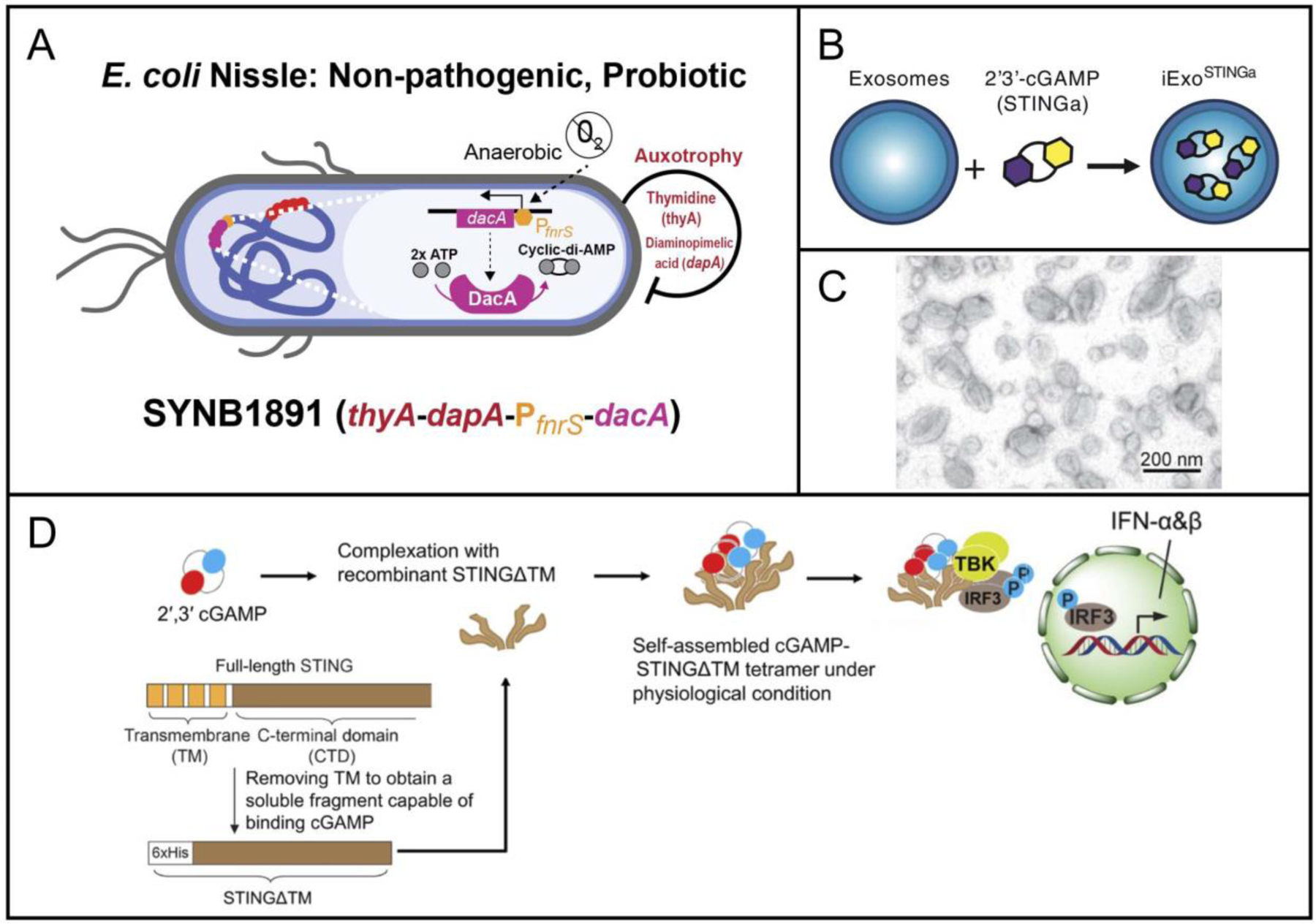
(A) Schematic of the SYNB1891 bacteria strain, which has been engineered to localize in the hypoxic tumor environment, activate STING in tumor APCs through enzymatic production of c-di-AMP, and trigger complementary proinflammatory pathways through additional PRR activation. Reproduced with permission from reference462. Copyright © 2020 Springer Nature. Distributed under a CC BY 4.0 license http://creativecommons.org/licenses/by/4.0/. (B) Schematic of iExoSTINGa exosomes, which have been engineered to deliver 2′3′-cGAMP. Reproduced with permission from reference465. Copyright © 2021 The Authors. Published by Elsevier Inc. on behalf of American Society for Biochemistry and Molecular Biology. Distributed under a CC BY 4.0 license http://creativecommons.org/licenses/by/4.0/. (C) Representative TEM image of extracellular vesicles used for CDN delivery. Reproduced with permission from reference467. Copyright © 2021 Codiak BioSciences, Inc. Distributed under a CC BY-NC-ND 4.0 license http://creativecommons.org/licenses/by-nc-nd/4.0/. (D) Schematic illustrating the delivery of 2′3′-cGAMP using recombinant, transmembrane-deficient STING to induce type I interferon responses. Reproduced with permission from reference468. Copyright © 2020 The Authors, some rights reserved; exclusive licensee AAAS. Distributed under a CC BY-NC 4.0 license http://creativecommons.org/licenses/by-nc/4.0/.
Engineered bacteria have also recently been explored as vectors for CDN delivery, and offer some unique advantages over synthetic systems, since bacteria can endogenously produce CDNs, target APCs via active phagocytosis, trigger complementary pattern recognition receptors, and selectively colonize in tumors462. Leventhal et al. engineered bacteria capable of tumor-selective production of CDNs (Figure 17A), selecting E. coli Nissle 1917 as the bacterial vector since it has increased serum sensitivity, has a well-defined genome, and is susceptible to a broad range of antibiotics, making it a good candidate for engineering of gene circuits to regulate CDN production. Diadenylate cyclase is an enzyme that produces high amounts c-di-AMP (CDA) and was selected as the CDA-producing enzyme. To construct a gene circuit to allow for tumor-selective CDA production, they incorporated a hypoxia-inducible promoter, which bypasses the need for the delivery of exogenous inducers and allows for site-specific activation due to the hypoxic nature of the TME. By introducing 4-hydroxy-tetrahydropicolinate synthase gene and thymidylate synthase gene deletions as a method of biocontainment, they could prevent intratumoral and extratumoral bacterial proliferation, increasing safety to enable possible translation of this living therapeutic. The removal of antibiotic resistance genes resulted in the finalized strain, SYNB1891, which was STING-inducing, tumor-specific, safe, and compliant with manufacturing regulatory guidelines. Type I IFNs were produced in a phagocytosis-dependent manner in both mouse and human APCs. SYNB1891 also activated parallel innate immune signaling pathways, such as TLR-4, resulting in the expression of complementary proinflammatory cytokines to improve immune response. SYNB1891 was delivered to B16-F10 tumor-bearing mice through three intratumoral injections during the span of a week, which delayed tumor growth in a dose-dependent manner and lead to complete tumor rejection in 30–40% of mice. Significant tumor rejection was also seen in A20 B cell lymphoma tumors, further illustrating the therapeutic efficacy and versatility of this system. Overall, this work offers an elegant example of how rationally-designed microorganisms can potentially be leveraged for conditional activation of STING signaling at tumor sites as well as the promise of synthetic biology approaches for regulating cGAS/STING signaling to maximize efficacy and safety.
Extracellular vesicles (EVs) are endogenously generated, lipid-bound nano- and microparticles that are secreted by cells as a natural mechanism for shuttling of diverse cargo, including nucleic acids, proteins, and metabolites, between cells470. Accordingly, there has been considerable recent interest in the production and engineering of EVs as therapeutics and/or drug carriers due to their versatility and high degree of tropism for specific cell and tissue targets471,472. Moreover, EVs have also been identified as a mechanism of transfer of DNA and/or CDNs between cells104,182,302,473, offering a bioinspired strategy for delivery of cGAS/STING agonists that has recently been pursued by several groups in the context of vaccines and cancer immunotherapy (Figures 17B and 17C)464–467. This is exemplified by the work of Jang and colleagues at Codiak BioSciences466 who have recently developed exoSTING, an EV that is exogenously loaded with a synthetic CDN (cAIM(PS)2 Difluor (RP,SP)), which is InvivoGen’s cyclic adenine monophosphate-inosine monophosphate bisphosphorothioate with a fluorine atom at the 2’ position of each nucleoside. EVs isolated from suspension culture-adapted HEK293 cells were loaded with CDNs via simple incubation with sufficiently high concentrations of CDN to allow for passive loading into or onto EVs, followed by washing away of unloaded CDN. It was found that exoSTING was ~ 100–200-fold more potent than free CDN in human PBMC, resulting in significant improvements in antitumor activity when administered intratumorally in a B16-F10 melanoma model. Robust therapeutic effects were observed using very low CDN doses (0.01–0.1 μg), which could be attributed to improved preferential uptake and intracellular delivery in APCs as well as prolonged CDN retention within the TME, with exoSTING increasing CDN half-life by ~ 5-fold and reducing clearance rate by ~ 10-fold. Importantly, it was also reported that exoSTING circumvented the immune ablative effects associated with intratumoral delivery of free CDNs, likely due to the preferential uptake and STING activation in APCs with reduced STING activation in T cells. They also evaluated intravenous administration of exoSTING in a mouse model of hepatocellular carcinoma and demonstrated a significant improvement in antitumor efficacy relative to free CDN, including a ~ 38% complete response rate. Based on these preclinical studies, Codiak has recently initiated a phase 1/2 clinical trial (i.e. NCT04592484) to evaluate intratumorally administered exoSTING in patients with injectable solid tumors.
Finally, the STING protein itself has been used as a CDN carrier468,469. As discussed in Section 3.1, STING is commonly epigenetically silenced in cancer cells and, therefore, CDNs and other STING pathway agonists may not result in STING activation in the tumor cell compartment474. Previous studies have shown that the transmembrane (TM) domain of STING is essential for its translocation, oligomerization, and signaling475, and, therefore, TM-deficient STING would not be expected respond to CDNs or other agonists. However, He et al. discovered that the titration of cGAMP into TM-deficient STING (STINGΔTM) protein under physiological conditions triggered the formulation of self-assembled tetrameric structures that were ~ 30 nm in diameter and comprised of cGAMP and STINGΔTM (Figure 17D)468. When delivered in vitro with commercial transfection reagents, the cGAMP-STINGΔTM complex triggered STING signaling and cytokine production even in STING-deficient cell lines. Therefore, the ribonucleoprotein complex is not only a strong affinity carrier for cGAMP (e.g. KD ~ 73 nM), but it is also able to initiate oligomerization and provide a scaffold for TBK1 recruitment and downstream signaling when cytosolically delivered regardless of STING expression or haplotype. Importantly, the cGAMP-STINGΔTM complexes coupled with commercial transfection reagents were able to activate STING signaling in vivo, both when used as a vaccine adjuvant that enhanced antigen-specific humoral and T-cell responses to a model antigen (i.e. OVA) as well as when administered IT, which inhibited tumor growth in a CT26 colon cancer model. The researchers subsequently furthered the therapeutic development and translatability of STINGΔTM by incorporating a peptide-based cell-penetrating moiety via genetic fusion with a known cell-penetrating domain to bypass the need for any synthetic delivery material469.
6.2. Delivery Platforms for Controlled and Sustained Release of Cyclic Dinucleotides
While direct intratumoral injection of CDN STING agonists has demonstrated an excellent safety profile in patients with evidence of on-target STING activation, results emerging from these clinical trials has been largely underwhelming so far. While there a multitude of factors that likely contribute to these disappointing outcomes, this can be at least partially attributed to the delivery and pharmacological challenges discussed in Section 5, including rapid clearance from the injection site, inconsistent tumor penetration, and lack of control over the magnitude and kinetics of local STING activation. These challenges have inspired the development of injectable or implantable controlled release depot technologies that can enable spatiotemporal control over the delivery of STING pathway agonists with potential to improve the efficacy, safety, and clinical feasibility of intratumoral immunotherapy (Figure 18).
Figure 18: Controlled-release delivery systems for local delivery of STING pathway agonists.

(A) Schematic and fabrication of PLGA microparticles for temporally programable pulsatile cargo release. Reproduced with permission from reference476. Copyright © 2020 American Association for the Advancement of Science; permission conveyed through Copyright Clearance Center, Inc. (B) Cumulative in vivo release of AF647 from microparticles in the B16-F10 tumor model. Reproduced with permission from reference476. Copyright © 2020 American Association for the Advancement of Science; permission conveyed through Copyright Clearance Center, Inc. (C) A nanotube hydrogel for TME regulation and chemoimmunotherapy tumor sensitization. A peptidedrug conjugate was created by linking the hydrophilic tumor-penetrating peptide, iRGD to the hydrophobic anti-cancer drug, camptothecin (CPT). The diCPT-iRGD conjugates self-assembled into cationic supramolecular nanotubes, which electrostatically bound anionic c-di-AMP (i.e. CDA) and enabled localized and sustained drug release within the tumor microenvironment for a combination of cancer immunotherapy and chemotherapy. Reproduced with permission from reference477. Copyright © 2020 Springer Nature BV; permission conveyed through Copyright Clearance Center, Inc.
An excellent and early example of this was the work of Leach et al. who developed STINGel – a peptide hydrogel-based delivery platform for intratumoral administration of CDNs478. They utilized a positively charged, MultiDomain Peptide (MDP) that self-assembles to form a hydrogel that effectively mimics the extracellular matrix, is biodegradable, and can be strategically functionalized479. The group used the MDP hydrogel denoted, K2(SL)6K2, for intratumoral delivery of the CDN, ML-RR-S2 CDA. The cationic lysine groups of K2(SL)6K2 allowed for electrostatic interactions between the hydrogel carrier CDN as well as with surrounding cells and tissue following injection. Importantly, MDPs demonstrate a time and stress-dependent viscosity, allowing them to be delivered through a syringe and localized at the injection site in vivo. Under buffered conditions in vitro, they found that MDP hydrogels sustained the release of CDN for ~ 15 hours, whereas CDN was released from a collagen gel within 5 hours, suggesting that MDPs could allow for higher sustained doses of CDN at the injection site. Utilizing a murine MOC2-E6E7 oral cancer model, they found that a single injection of STINGel significantly reduced tumor growth relative to free CDN as well as CDN administered using a collagen hydrogel, resulting in a 60% complete response rate and induction of immunity that protected from tumor rechallenge. The improved efficacy observed with the MDP hydrogel relative to collagen is notable as this suggests that hydrogel properties and/or CDN release rate are important variables that may be further optimized to maximize antitumor effects. Overall, the use of the MDP hydrogel improves the localized delivery of CDNs and has potential to allow for fewer intratumoral injections.
Although surgical reSection is a common and effective treatment for operable tumors, it can also remove neoantigens and effector immune cells that are necessary for proper immunosurveillance and antitumor mechanisms; additionally, the wound healing process can result in local immunosuppression that may inhibit antitumor immunity480. In order to improve the immunogenicity of the tumor reSection site, Park et al. developed an intraoperative scaffold for sustained and localized release of the CDN, 2′3′-c-di-AM(PS)2 (RP,RP) (STING-RR), R848 (i.e. agonist of TLR-7 and TLR-8), and other immunomodulators481. They selected hydraulic acid (HA) as their hydrogel platform due to its biocompatibility and biodegradability, and leveraged a thiol-modified HA that could be crosslinked via Michael addition using a PEG diacrylate crosslinker. They evaluated over 20 formulations to establish an optimal crosslink density that allowed for fabrication of a sufficiently stiff scaffold for surgical implantation, while still allowing for biodegradation. In mice, scaffold degradation began 5 weeks after implantation, with complete resorption by week 12. After resecting primary 4T1 breast tumors in mice, they locally administered the therapeutic hydrogel, which stimulated a type I IFN response and promoted the infiltration of NK cells, dendritic cells, and T-cells in the TME. This treatment was superior to intratumoral injection of either free STING-RR or R848, and was able to eradicate metastatic tumors that had already developed within the lung prior to surgery. Intraoperative placement of the immunotherapeutic hydrogel was required for therapeutic benefit as systemically administered STING-RR or STING-RR injected locally along with an empty hydrogel had no effect, highlighting the importance of implanting the biomaterial delivery system during this critical intraoperative window.
Adoptive T cell and CAR-T cell therapy is an emerging treatment for many solid tumors; however, their efficacy has been limited due to inefficient lymphocyte trafficking from the circulation into tumors and inhibition of T-cell expansion and function in immunosuppressive “cold” TMEs482,483. To circumvent these barriers, Stephan et al. developed a biodegradable, microporous scaffold for efficient local delivery of tumor-targeting T-cells and immune activators484 and subsequently applied this technology to also deliver CDNs485. The scaffold matrix consisted of polymerized alginate that incorporated GFOGER peptides through carbodiimide chemistry with embedded mesoporous silica microparticles loaded with CDN (i.e. c-di-GMP) and displaying anti-CD3, anti-CD28 and anti-CD137 antibodies. GFOGER peptides mimicking collagen, which lymphocytes naturally utilize for migration, were strategically integrated to allow for binding and migration of lymphocytes in the scaffold while antibodies displayed on microparticles provided immunostimulatory cues for T cells, which could be loaded into the scaffold by hydrating it in the presence of T cells. When implanted into a surgical cavity, T cells robustly expanded and migrated out of the scaffold into the tumor reSection site and to tdLNs, eliminating residual tumor and preventing relapse. By also incorporating c-di-GMP into the microparticles, the biomaterial implant also acted as a “self” vaccine site, with STING activation providing a local immunostimulatory milieu that supported T cell priming and activation, including de novo priming of endogenous T cells capable of eliminating tumor cells not recognized by the CAR T cells. As a result, scaffold co-delivery of c-di-GMP along with tumor-specific CAR T cells stimulated strong antitumor responses in both melanoma and pancreatic tumor models, and elicited long-term immunity against tumor rechallenge.
As discussed previously, most current intralesional STING agonist–based therapies require frequent injections, which can lead to complications and low patient compliance. Recently, a PLGA microparticle platform was developed to allow for long-term, pulsatile release of cGAMP within the tumor site following local injection476. Lu et al. used soft lithography techniques to fabricate arrays of cubic (400 × 400 × 300 μm) PLGA microparticles that can be filled with an aqueous solution and then sealed with a lid to form a closed cavity, resulting in 100% drug encapsulation, high loading capacity, and minimal leakage (Figure 18A). By fabricating particles using PLGA with different properties (e.g. molecular weight, lactide/glycolide ratio), the biodegradation rate of the walls could be tuned, allowing for a time-dependent burst release of capsule contents (Figure 18B). By mixing populations of particles designed with different release rates, a pulsatile release profile of cGAMP could be finely tuned to mimic dosing regimens commonly used for repeated intratumoral injection (e.g. four injections with 3–4 days between). Particles could be administered using a syringe and aggregated at the tumor injection site of B16-F10 melanoma and orthotopic 4T1 breast tumor models, where they were able to release cargo in a programmed, pulsatile manner; particles could be directly injected into a tumor or into a surgical bed following tumor reSection. A single injection of cGAMP-loaded microparticles almost exactly mirrored the therapeutic effect of four injections of free cGAMP, highlighting the capacity of the approach to reduce or minimize the need for multiple intratumoral injections. This was associated with a conversion to a more immunogenic TME, characterized by the infiltration of CTLs, NK cells, DCs, memory T cells, and macrophages with a shift from an M2 to an M1 macrophage phenotype. They further demonstrated that their approach could reduce lung metastasis, enhance responses to ICB, and generate protection against tumor rechallenge. As an example of the potential clinical utility of their approach, they also evaluated the depots in an orthotopic pancreatic tumor model, which is not readily accessible for repeated injection, and demonstrated reduced tumor burden with a single injection of cGAMP-loaded microparticles.
Another recently described depot technology was specifically focused on harnessing synergy between STING agonists and chemotherapy since some chemotherapeutic drugs trigger cell death by inducing DNA damage and activating cGAS/STING signaling.477 Wang et al. developed a drug-loaded supramolecular hydrogel system composed of a self-formulating peptide–drug conjugate, di-camptothecin–iRGD, which comprised a neuropilin-1-binding, tumor-penetrating iRGD peptide and the chemotherapeutic, camptothecin (Figure 18C). By linking the hydrophobic camptothecin to the water soluble peptide through a disulfanyl-ethyl carbonate linkage, which is susceptible to reduction via glutathione, a drug amphiphile was synthesized. Peptide-drug conjugates spontaneously assembled into supramolecular nanotubes in situ with a positively charged surface that enabled electrostatic complexation of the CDN, c-di-AMP. After intratumoral injection, a hydrogel forms immediately and acts as a depot that enables release both camptothecin and c-di-AMP over time. The hydrogel degraded with a near linear profile and sustained local release of CDN over at least two weeks following injection. Local intratumoral administration induced STING-dependent activation of type I IFNs and CXCL10, resulting in infiltration of NK, DCs, and CTLs. In multiple tumor models (CT26 colon cancer, 4T1 breast cancer, GL-261 glioma), a single injection of the hydrogel strongly inhibited tumor growth and increased survival compared to a soluble mixture of CDN and camptothecin, the hydrogel lacking CDN, or a similar hydrogel lacking camptothecin. This offered a compelling demonstration of both the utility and improved efficacy achieved using a sustained CDN release depot as well as the potential to leverage biomaterial design to harness synergy between chemotherapy and STING activation.
6.3. Biomaterials with Intrinsic STING Activity
In addition to CDN delivery vehicles, researchers have also developed and/or used biomaterials that can intrinsically activate the cGAS/STING pathway, either directly or indirectly (Figure 19). Some of the first evidence that biomaterials could stimulate inflammation via cGAS/STING came from Carrol et al., who demonstrated that the cationic polysaccharide, chitosan exerted vaccine adjuvant properties via the cGAS/STING pathway486. Chitosan has been known for its promising adjuvant capabilities (e.g. promotion of DC activation, enhancement of adaptive immunity, etc.), though its mechanisms of action have remained undefined487,488. Carrol et al. utilized a Mycobacterium tuberculosis vaccine construct to demonstrate the ability of chitosan, comprising randomly distributed D-glucosamine and N-acetyl glucosamine, to enhance surface expression of CD40 and CD86 by DCs and elicit highly polarized Th1 and IgG2c antibody responses. However, these responses, as well as the secretion of CXCL10, were mitigated in the absence of IFNAR, showing that type I IFN signaling was essential for this immune activation. They also demonstrated that these responses were specifically dependent on the cGAS/STING pathway due to reduction of IFN-β and CXCL10 production in cGAS/STING deficient mice. Interestingly, they found that chitosan induced the production of mitochondrial-specific ROS, suggesting that cGAS/STING activation occurred in response to the release of mitochondrial DNA into the cytosol. In total, this study defined chitosan as an effective adjuvant able to bolster adaptive immune response through the cGAS/STING pathway.
Figure 19: Biomaterials that can intrinsically activate the STING pathway.

There is a growing list of biomaterials known to activate the STING pathway, either directly or indirectly. Direct activation of the STING pathway involves molecules that can bind to and functionally activate either cGAS or STING proteins. Indirect activation of the STING pathway most commonly involves endogenous cGAS activation and is typically achieved by inducing the cytosolic relocation of DNA from mitochondria and/or nuclei. Figure created with biorender.com.
More recently, Turley et al. have identified optimal characteristics of chitin-derived polymers for the activation of DCs and the induction of antigen-specific cellular immune responses489. The researchers found that degree of chitin deacetylation, acetylation pattern, and its regulation of mitochondrial ROS are the key determinants of its immune enhancing effects. Notably, only chitin-derived polymers with a high degree of deacetylation enhanced the generation of mitochondrial ROS and thereby the STING-mediated induction of type I IFN. It was determined that chitin-derived polymers with a degree of deacetylation less than 80% are poor adjuvants, while a fully deacetylated polyglucosamine polymer is most effective as a vaccine adjuvant. Furthermore, for the chitin-derived polymers that are not fully deacetylated, a heterogenous acetylation pattern (i.e. clustering of the remaining acetyl groups) was favorable to a homogenous acetylation pattern (i.e. even distribution of the remaining acetyl groups), which was likely due to charge distribution and its effect on mitochondrial stress. Indeed, when packed closely together, positively charged species, such as the free amines generated by deacetylation, might more efficiently promote mitochondrial association and mitochondrial membrane disruption490–492.
Since the initial characterization of chitosan as a cGAS/STING pathway activator, Qiutong et al. has applied chitosan in a nano-complex with anti-PD-L1 antibodies to achieve pulmonary delivery of ICB through inhalation in lung metastasis493. Direct pulmonary delivery of therapeutics allows for a reduction in systemic distribution and high, localized drug concentration494; however, antibody delivery is normally limited by alveolar mucosal barriers that inhibit the permeation of large, hydrophilic, and anionic biomacromolecules495. The direct complexation of positively-charged chitosan to antibodies allows for interactions with anionic sialic acid of mucins to enhance permeability and delivery of macromolecular therapeutics directly into the lung. The innate adjuvant activity of chitosan also allows for complimentary STING activation to synergize and improve ICB therapy. The nanocomplex was prepared in one step by mixing positively-charged chitosan and negatively-charged antibodies, allowing for polyelectrolyte complexation. At a 1:1 ratio, this nanocomplex formed particles with a diameter of ~ 60 nm and surface charge of +24 mV that could stick to the negatively-charged mucus lung epithelium to prolong retention and reversibly open tight junctions of lung mucosa to increase the penetration of ICB. Repeated inhalation of this complex promoted the infiltration of immune cells, especially CTLs, at and around tumor lesions of B16-F10 melanoma lung metastases. The enhanced retention and penetration of ICB and additional STING-activating adjuvant effects allowed for prolonged survival over 60 days.
There have been several other positively charged molecules that have been identified as indirect activators of the cGAS/STING pathway via mitochondrial DNA release. Positively charged molecules are attracted to the negative membrane potential of mitochondria (i.e. ~ −170 mV)491,492 and are therefore more likely to associate with mitochondrial membranes and cause membrane disruption. Notably, cationic nanocarriers (e.g. transfection agents) can activate the cGAS/STING pathway via mitochondrial damage and the subsequent release of mitochondrial DNA into the cytosol496,497. Furthermore, the most prevalent adjuvant in licensed human vaccines, aluminum hydroxide (i.e. alum)498 also exhibits a positive surface charge at physiological pH499 and can similarly activate the cGAS/STING pathway via the release of self dsDNA500–502. However, STING-driven gene expression appears to be heavily restricted and often undetectable in many cell types in response to alum-induced activation503, which suggests that alum is a relatively poor STING pathway agonist and also highlights how indirect activation of the cGAS/STING pathway can be quite multifaceted.
There is some evidence that certain non-positively charged biomaterials are also capable of indirectly activating cGAS/STING via the release of self dsDNA. Indeed, Benmerzoug et al. observed that silicosis patients with a history of intense silica (i.e. silicon dioxide) exposure to the lungs exhibit increased circulating dsDNA in their plasma and increased expression of CXCL10 in their sputum50. The researchers then identified the mechanisms behind silica-induced lung inflammation, determining that silica can intrinsically induce self-dsDNA release and a subsequent STING-mediated IFN-I response. Notably, the mechanism of silica-induced self-dsDNA release was not determined. As silica tends to be charge-neutral to slightly negatively charged504, it is unlikely that silica triggers mitochondrial dsDNA release in the same manner as the positively charged species that indirectly activate cGAS. Alternatively, the researchers proposed that mitochondrial DNA may accumulate in the cytosol in response to silica exposure through the initiation of apoptosis in a BAX and BCL-2-dependent manner505,506, activation of mitochondrial permeability transition pore507,508, or deficient control by transcription factor A mitochondria (TFAM)506. Interestingly, in vitro studies suggested that the mechanism of STING activation varied by cell type. After silica exposure, mitochondrial dsDNA in dendritic cells was released into the cytosol and activated the cGAS/STING signaling axis in a conventional manner, while macrophages exhibited an alternative and less common form of STING activation. The activation of STING in macrophages following silica exposure was cGAS-independent and instead required the accumulation of extracellular dsDNA, which was then internalized and processed through DDX41 and IFI204 (i.e. the murine ortholog of IFI16) DNA sensors. Nonetheless, STING pathway activation was identified as essential for the onset of silica-induced lung inflammation, and DNase I was proposed as a potential therapeutic treatment to clear extracellular dsDNA and thereby attenuate STING signaling.
While biomaterials that indirectly activate cGAS/STING signaling can be quite useful, they primarily depend on the induction of cell-stress mechanisms, which may not be suitable for certain applications. Indeed, many of the biomaterials that indirectly activate the cGAS/STING pathway also stimulate other immunomodulatory pathways. For example, chitosan also triggers the cellular production of IL-1β and IL-18 via activation of the NLRP3 inflammasome509. Furthermore, the biomaterials that trigger the intracellular release of mitochondrial DNA can indirectly activate several different DNA sensors (e.g. cGAS, AIM2, TLR9, etc.)510,511 due to the relatively large size (i.e. ~ 16.5 kb in length) and nucleotide composition of mitochondrial DNA512. Accordingly, the downstream signaling cascades induced by such biomaterials are heavily context-dependent.
Immunomodulatory biomaterials with pathway specificity do not involve pathway cross-talk and can therefore offer enhanced control over therapeutically programming immune responses. Investigators have recently sought to develop synthetic biomaterials that can directly activate STING via direct binding interactions with the STING protein (Figure 20). This approach was first elegantly demonstrated by the group of Jinming Gao, who synthesized a library of ultra-pH sensitive NPs comprised of a PEG first block and a second, pH-responsive block containing tertiary amines with linear or cyclic side chains513. Having demonstrated that all polymers could efficiently load the model antigen OVA, they screened the ability of carriers to elicit an OVA-specific CTL response and identified a lead polymer, PC7A, that contained a cyclic amine side chain and was selected due to its ability to induce strong CTL responses without the use of any exogenous adjuvants. Using OVA, they demonstrated that OVA-PC7A NP was capable of eliciting a 20-fold higher CTL response compared to commonly used adjuvants including alum, LPS, and CpG. Th1 and Th2 responses assessed via measurement of IgG1 and IgG2c titers were also shown to be higher or comparable to these common adjuvants. Following subcutaneous injection, OVA-PC7A NPs accumulated in the peripheral LNs and significantly increased OVA-positive CD8α+ DC cells. Interestingly, these effects were reduced in cGAS−/− mice, demonstrating a dependence on cGAS/STING signaling to exert adjuvant effects, and additional pull-down methods using the CTD of STING elucidated that PC7A could bind to STING directly. Collectively, these findings suggested that PC7A was acting as a direct STING agonist. The therapeutic efficacy of the vaccine was observed in B16-F10, MC38, and HPV E6/7 TC-1 tumor models. The vaccine also demonstrated synergy with anti-PD-1 antibody therapy in B16-OVA and TC-1 models. PC7A-NPs were also used to deliver cGAMP in models of HIV-1 infection, which can inhibit activation of the STING pathway514. Through IFN-I signaling, this NP system for cGAMP delivery was able to elicit strong, long-acting antiretroviral responses to HIV-1.
Figure 20: Biomaterials that can directly bind and activate STING.

(A) Relative IFNB1 and CXCL10 mRNA levels over time in THP1 cells treated with 2′3′-cGAMP or the synthetic diblock copolymer, PC7A, along with the chemical structure of PC7A. Reproduced with permission from reference164. Copyright © 2021 Springer Nature BV; permission conveyed through Copyright Clearance Center, Inc. (B) PC7A led to sustained TBK1/IRF3 phosphorylation and slower STING degradation compared to 2′3′-cGAMP in THP1 cells. Reproduced with permission from reference164. Copyright © 2021 Springer Nature BV; permission conveyed through Copyright Clearance Center, Inc. (C) Schematic of STING oligomerization and the uncharacteristic immunostimulatory condensation (i.e. unlike that of the natural STING phase-separator, which negatively regulates STING-driven gene expression146) induced by PC7A. Reproduced with permission from reference164. Copyright © 2021 Springer Nature BV; permission conveyed through Copyright Clearance Center, Inc. (D) Schematic of mRNA-encapsulating LNPs incorporating STING-activating ionizable lipidoids. A18 was selected as the lead cyclic lipid candidate. Reproduced with permission from reference515. Copyright © 2019 Springer Nature BV; permission conveyed through Copyright Clearance Center, Inc.
In an important follow-up study, the researchers identified an alternative STING binding site for PC7A, indicating that PC7A does not compete with 2′3′-cGAMP in STING binding164. Hence, PC7A could be combined with 2′3′-cGAMP for synergistic STING activation, which is particularly useful for STING variants that exhibit reduced activity to 2′3′-cGAMP stimulation (e.g. the REF (R232H) STING isoform, present in ~ 14% of humans)160. Indeed, synergy between co-delivered 2′3′-cGAMP and PC7A NPs was demonstrated in MC38 and TC-1 tumors. Notably, they found that PC7A deterred lysosomal degradation of STING by buffering endosomal pH and induced a unique liquid-liquid phase condensation of STING, both of which led to enhanced and sustained downstream signaling as compared to 2′3′-cGAMP treatment (Figures 20A–C). The formation of the PC7A-induced STING condensates was found to correlate with activity, unlike the inhibitory STING phase-separator that negatively regulates the pathway upon substantial pathway activation146. Though not addressed in their work, PC7A may inhibit the STING phase-separator in addition to promoting its own immunostimulatory biocondensates. Thus, the prolonged activity of PC7A-induced STING signaling could potentially be attributed to PC7A-mediated inhibition of the STING phase-separator in addition to the established ability of the polymer to prevent lysosomal degradation of STING.
Taking a similar approach, Miao et al. synthesized a combinatorial library of over 1,000 ionizable lipid-like materials and screened the ability of lipid nanoparticle formulations to stimulate potent immune responses to mRNA-based vaccines515. Although mRNA vaccines are advantageous due to their ability to intracellularly express whole protein antigens, their efficacy is challenged by mRNA hydrolysis and inadequate antigen loading and APC maturation516. After synthesizing and optimizing functional lipid libraries through a three-dimensional multi-component reaction system, they found that lipids containing cyclic-amino headgroups, unsaturated lipid tail, and a dihydroimidazole linker (Figure 20D) were not only highly efficient at delivering mRNA, but also stimulated type I IFNs and ISGs in a STING-dependent manner as evidenced by reduced innate and antigen-specific adaptive immune responses in STING KO cells and mice. Using a lipid pulldown assay, they also demonstrated that lipids with cyclic-amino headgroups, in contrast to those with linear structures, could directly associate with STING, which was further supported by dynamic molecular docking simulations that estimated a KD of ~ 50 μM for A18, the lead cyclic lipid candidate from the library. By formulating antigen-encoding mRNA into LNPs comprising A18, the fusogenic helper lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), C14-PEG, and cholesterol at an undisclosed molar ratio, the investigators generated a potent mRNA cancer vaccine capable of stimulating strong CTL and Th1 CD4+ T cell responses. Using mRNA encoding the tumor antigen tyrosine-related protein 2 (mTRP2), the A18 mRNA LNP vaccine inhibited tumor growth and increased survival in mice with B16-F10 tumors to a greater degree than an analogous LNP formulated using a lipid with a linear headgroup (i.e. A25), or using the established MC3 lipid, demonstrating the importance of head group design and resulting STING activation in therapeutic vaccine efficacy. To further define the efficacy of the A18 mRNA vaccine, it was formulated with mRNA encoding the human papillomavirus E7 protein and co-administered with anti-PD-1 in a TC-1 tumor model, resulting in robust cures in these mice, further demonstrating its efficacy as an mRNA delivery vehicle and intrinsic STING stimulator.
7. Therapeutic Potentiators of the STING Pathway
There is a growing list of known STING pathway potentiators (Figure 21), which currently includes certain metal ions (e.g. Mn2+ and Mg2+), cGAS-binding proteins, inhibitors of DNA methyltransferases, various inhibitors of NF-κB signaling, as well as ENPP1 inhibitors for therapies that utilize endogenous 2′3′-cGAMP (e.g. radiotherapy). These therapeutic agents have potential to improve the efficacy and/or safety of STING pathway activation for cancer immunotherapy and could be utilized in combination with STING pathway agonists either through local co-administration or by rational co-incorporation into drug delivery platforms.
Figure 21: Strategies for potentiating STING signaling.

The magnitude of STING-driven gene expression and/or profile of the resultant immune response can be modulated by many different biochemical agents (i.e. potentiators). Depicted in this figure are some notable potentiators of the cGAS/STING pathway. These potentiators include: certain metal ions (e.g. Mn2+ and Mg2+), which can amplify STING signaling through a variety of mechanisms; cGAS-binding proteins, which can augment 2′3′-cGAMP production by sensitizing cGAS to dsDNA; inhibitors of DNA methyltransferases, which can restore the activity of epigenetically silenced cGAS and STING proteins; various inhibitors of NF-κB signaling, which can influence downstream gene expression; inhibitors of the STING phase-separator, which have potential to prevent the inhibition of STING signaling induced by the liquid-liquid phase condensation of STING; VRAC agonists, which may enable enhanced transmission of CDN STING agonists; and inhibitors of ENPP1, which can be used to increase local 2′3′-cGAMP concentrations by deterring the degradation of 2′3′-cGAMP. Figure created with biorender.com.
It is well established that transition metals can regulate the function of enzymes, as nearly half of all enzymes utilize metal cofactors517,518. Manganese, which is one of the most abundant metals within mammals, was recently identified as a natural, triggerable potentiator of the STING pathway in 2018519. Indeed, the majority of manganese(2+) (Mn2+) within cells is confined to membrane-enclosed organelles (e.g. Golgi and mitochondria) at cellular steady-state and therefore avoids innate immune sensors in the cytosol. However, upon viral infection, Mn2+ is released into the cytosol, where it can promote STING pathway activation519.
Over the past several years, cytosolic Mn2+ has been reported to potentiate STING signaling in several unique ways: 1) Mn2+ can independently activate monomeric cGAS in the absence of dsDNA without the need for cGAS oligomerization65,349; 2) In conjunction with the ATP/GTP substrate pair, Mn2+ allosterically enhances the dsDNA binding capacity of cGAS. Conversely, dsDNA enhances the Mn2+ binding capacity of cGAS, which is also amplified by larger molecular weight dsDNA. Accordingly, coupling Mn2+ with cytosolic dsDNA can lower the threshold for STING pathway activation by several orders of magnitude519 as they both act in a concerted manner for maximal cGAS-substrate recognition65; 3) Mn2+ accelerates the overall catalytic activity of dsDNA-bound cGAS resulting in much greater production of cGAMP65,519; 4) Mn2+ may increase the binding affinity of cGAMP to STING. Some reports suggest that Mn2+ can augment cGAMP-STING binding affinity146,519, though a more recent publication found that Mn2+ does not affect the binding affinity between STING and STING agonists520. Thus, this particular point remains to be clarified; 5) Mn2+ induces the phosphorylation of both TBK1 and p65 in a STING-independent manner and when in the presence of STING agonists, Mn2+ enhances the assembly of the enhanceosome, resulting in greatly increased production of IFN-β520.
Collectively, all these attributes make cytosolic Mn2+ an exceptionally potent and noteworthy STING pathway potentiator. Additionally, it has also recently been determined that Mn2+ is essential in the innate immune sensing of tumors and that combining Mn2+ with ICB can synergistically boost antitumor immunity350. Furthermore, a phase 1 clinical trial investigating the combination of Mn2+ and anti-PD-1 antibody yielded promising efficacy in patients with advanced metastatic solid tumors350 (NCT03991559).
Several research groups have already begun to develop nanotechnology and/or depots for Mn2+ delivery to promote enhanced pharmacological STING pathway activation for cancer immunotherapy354,521–525. Wang et al. reported a biomaterial-based delivery approach that coupled the divalent cation chelator, alginate with Mn2+ in the context of radiotherapy521. The researchers found that intratumoral injections of Mn2+ by itself could indeed enhance the antitumor immune response following RT, but that the timing of administration was critical for efficacy. Free Mn2+ was metabolized out from tumors within minutes and DNA did not accumulate in the cytosol of cells until ~ 24 hours post RT treatment. Accordingly, Mn2+ injected intratumorally immediately after RT was unable to enhance the therapy, while intratumoral injection 24 hours after RT did demonstrate efficacy. They subsequently employed alginate to act as a depot to control the release of Mn2+ for up to 72 hours. Administration of the alginate-manganese complexes 24 hour after RT lead to 90% tumor inhibition rate and a significantly extended average survival time.
Hou et al. created a multifaceted NP for STING pathway activation in tumors522. Doxorubicin (DOX) was encapsulated within amorphous porous manganese phosphate (APMP) NPs, which were then coated them with phospholipids (PL) for improved stability in systemic circulation and triggerable phospholipase-mediated degradation within tumor cells. When administered IV, the resultant PL/APMP-DOX NPs navigated to tumors, released DOX to induce DNA damage and subsequent cGAS activation, and released Mn2+ to augment cGAS/STING activity. The PL/APMP-DOX NP treatment boosted DC maturation and increased tumor infiltration of both cytotoxic T cells and NK cells in the 4T1 murine breast cancer model.
Zhou et al. also developed a multifunctional NP platform, which likely operates in a similar manner to the PL/APMP-DOX NPs (i.e. delivering DOX and potentiating the STING pathway with manganese)354. Their NP platform was prepared by co-assembling dsDNA-gold conjugates and DOX onto Mn3O4 nanoflowers. 59 bp poly(dA):poly(dT) was chosen as the dsDNA to activate the STING pathway. The poly(dT) single-stranded DNA was pre-conjugated onto AuNP through an Au-S bond and then annealed with complementary strand. This was then loaded onto Mn3O4 nanoflowers via a noncovalent attachment method. Finally, DOX was loaded onto the complex, resulting in a final particle diameter of ~ 354 nm and a surface charge of −7.7 mV. Following intravenous administration, manganese and gold from the nanoflower NPs were detected in B16-F10 tumors, suggesting some level of passive targeting. It was reported that the dsDNA stimulated the immune response by activating the STING pathway via cGAS, while the DOX exerted its chemotherapeutic antitumor activity. Though not addressed by the authors, it is likely that the DOX also contributed indirectly to the STING pathway activation via its DNA-damaging capacity and that the Mn2+ degradation product of nanoflower enhanced the STING signaling within the tumors. The combination particles significantly inhibited tumor growth and prolonged survival in the 4T1 tumor model and successfully demonstrated potential for synergy between a STING-pathway agonist and a chemotherapy.
Chen et al. reported a thiolated and Mn2+ coordinated CDN nanovaccine (termed Mn-cGAMP NVs) that facilitates the cytosolic co-delivery of 2′3′-cGAMP and Mn2+ to potentiate an antitumor immune response against B16-F10 murine melanoma following intratumoral administration523. They utilized polymerized guanidine-containing disulfides to assemble with 2′3′-cGAMP and then coordinate with Mn2+ ions, forming particles that were ~ 176 nm in diameter. The Mn-cGAMP NVs attenuated primary tumor growth, inhibited distal tumor growth, and improved responses when administered in combination with anti-PD-L1 monoclonal antibody treatment.
Yang et al. engineered a biomimetic nanoplatform using cancer cell membranes extracted from B16-F10 cells to co-encapsulate manganese dioxide (MnO2) NPs and the established photothermal therapy sensitizer, 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR)524. Interestingly, manganese was the sole adjuvant in their system and it was used to induce STING signaling via cGAS activation. The resultant vesicles had a diameter of ~ 125 nm and displayed a negative surface charge of −19 mV. Notably, the researchers found that slightly acidic conditions (e.g. pH ~ 6.8) with high concentrations of hydrogen peroxide (e.g. 2.5 mM H2O2) triggered the release of Mn2+ from the vesicles and that the vesicles promoted the tumor accumulation of both Mn2+ and DiR following intravenous injection. The systemic administration of their construct coupled with targeted photothermal therapy enabled partial tumor regression in primary tumors, multinodular tumors, metastatic tumors, and recurrent tumors. Additionally, transcriptomic analysis of the tumors following treatment demonstrated the upregulation of STING-driven genes, supporting on-target STING activation in vivo.
Gao et al. described the development and characterization of PEGylated manganese phosphate (MnP-PEG) nanoclusters for cancer immunotherapy525. The particles were fabricated by mixing Mn2+ and PO43− ions in solution followed by the addition of a phosphate-functionalized 5 kDa PEG polymer. The MnP-PEG nanoclusters were ~ 150 nm in diameter with a negative surface charge of −11 mV. It was determined that the nanoparticles could mediate endocytosis, acid-triggered Mn2+ release, and STING signaling. Furthermore, intratumoral administration of the MnP-PEG nanoclusters in the B16-F10 tumor model enhanced the tumor infiltration of DCs and macrophages as well as activated (i.e. CD69+) tumor-infiltrating CD8+ and CD4+ T cells and NK cells. The treatment also resulted in antitumor efficacy as a monotherapy and improved responses to ICB (i.e. anti-PD-1 therapy).
Sun et al. developed a new cancer immunotherapeutic that co-delivers Mn2+ and CDA in coordination nanoparticles520. After screening various metal ions for potential synergy with STING agonists, they identified Mn2+ as a noteworthy potentiator of the STING pathway. Indeed, Mn2+-mediated potentiation of the STING pathway was found to be independent of STING variants and STING agonist structures, and the intratumoral treatment of mice bearing CT26 tumors with a soluble mixture of CDA and Mn2+ resulted in a significant increase in antigen-specific T cells and an attendant inhibition of tumor growth compared to either monotherapy. Interestingly, it was subsequently determined that Mn2+ can self-assemble with CDA in methanol to form coordination polymers with diameters ranging from nanometers to micrometers; however, these complexes were shown to be unstable under physiological conditions. In light of these physicochemical properties, the researchers developed a nanoparticle system that could stabilize the CDA–Mn2+ coordination polymers. Dioleoyl-sn-glycero-3-phosphoethanolamine-N-(histidine)11 was added as an additional coordination ligand to promote the formation of a stabilizing hydrophobic core, which was then coated with an outer PEG-lipid layer by resuspending in a solution of DOPC:cholesterol:DSPE-PEG5000 (4:1:1 molar ratio), thereby allowing for aqueous suspension of the resultant particles. The final platform, denoted CMPCDA, comprised particles that were ~ 118 nm in diameter with a neutral charge, and the loading efficiencies of CDA and Mn2+ were 39.6% and 25.3%, respectively. Notably, this new immunotherapeutic could be delivered intravenously, where a significant increase in serum IFNβ, TNFα, CXCL-9 and CXCL-10 was observed in mice with CT26 tumors compared to those treated with a soluble mixture of CDA and Mn2+. Systemic treatment with CMPCDA significantly decreased CT26 tumor growth, eliminated established tumors in 50% of mice, and conferred resistance to tumor rechallenge, whereas treatment with soluble agents had no response. This platform was also validated in tobacco carcinogen-associated syngeneic squamous cell carcinoma and B16-F10 tumor models, where it was found to outperform the highly potent diABZI small molecule agonist, demonstrating the significant potential of metalloimmunotherapy in nano-based cancer therapeutics.
There exist several known cGAS-binding proteins that also bind DNA and thereby promote cGAS activity526–529. By providing additional binding sites for cytosolic DNA, these cGAS-binding proteins enhance the recognition of DNA by cGAS, which augments 2′3′-cGAMP production and STING signaling. Polyglutamine binding protein 1 (PQBP1) has been described as a proximal innate sensor of a human immunodeficiency virus type 1 (HIV-1) infection, as it was found to enhance the IRF3-dependent innate response in primary human monocyte-derived DCs (MDDCs) by directly binding reverse-transcribed HIV-1 DNA and cGAS526. The CCHC-type zinc-finger (ZF) protein, ZCCHC3 was similarly reported as a co-sensor of cGAS, capable of improving the innate immune response to cytosolic dsDNA and the DNA viruses, herpes simplex virus 1 (HSV-1) and vaccinia virus527. GTPase-activating protein SH3 domain-binding protein 1 (G3BP1) was identified as another positive regulator of cGAS activity with the inhibition of G3BP1 partially rescuing cGAS-mediated autoinflammation in a Trex1−/− mouse model528. Lastly, the secreted bacterial protein, streptavidin was recently reported to bind both DNA and cGAS to promote cGAS-dependent immune responses against the DNA virus, HSV-1529. Notably, streptavidin exhibits exceptionally strong noncovalent interactions with biotin and has accordingly been extensively used for many biotechnological applications, such as molecular purification, molecular detection, and drug delivery. Therefore, the unique interaction of streptavidin with cGAS and DNA, which can lead to immunostimulation, complicates the clinical and biotechnological usage of streptavidin. Indeed, careful consideration should be given when choosing to use streptavidin in certain applications. However, since enhanced STING signaling is beneficial for many cancer types, these cGAS-binding proteins have potential for therapeutic use in combination with cGAS-activating cancer therapies, though molecular engineering or nanotechnology would likely need to be employed for in vivo delivery of these molecules.
As briefly mentioned in Section 3.4, inhibitors of DNA methyltransferases are approved for the treatment of certain cancers and are also capable of improving intratumoral STING signaling and tumor immunogenicity299,301. Indeed, Falahat et al. recently determined that promoter hypermethylation of cGAS and STING genes mediates transcriptional silencing and impairs STING signaling function in melanoma, which disrupts tumor antigen presentation and the accumulation of tumor infiltrating lymphocytes301. By inhibiting DNA methylation with a clinically available DNA methyltransferase inhibitor (i.e. 5-aza-2’-deoxycytidine), the researchers were able to restore the activity of cGAS and STING and thereby improve antigenicity through the augmentation of MHC class I surface expression and antigen presentation. This ultimately resulted in enhanced T cell recognition of melanoma. Therefore, inhibitors of DNA methyltransferases could possibly be used along with STING pathway agonists to improve antitumor immune responses in cancers where STING is epigenetically silenced.
Recent studies conducted by Hou et al. have demonstrated that irradiation-induced STING signaling activates both canonical NF-κB (i.e. NF-κB1) and noncanonical NF-κB (i.e. NF-κB2) in tumor-localized DCs240. Interestingly, the researchers also found that the NF-κB2 pathway negatively regulates NF-κB1–mediated gene transcription and that they could enhance the antitumor effect of irradiation in murine models by inhibiting downstream signaling of the noncanonical pathway with intratumoral injections of a specific NF-κB2 inhibitor (i.e. SN52). Thus, targeted inhibition of NF-κB2 represents another possible strategy for potentiating the therapeutic effects of STING signaling in cancer. Tuning the downstream signaling that follows STING activation holds tremendous promise, because it may yield outcomes where beneficial effects of STING signaling (e.g. antitumor immunity) are maximized and negative effects (e.g. toxicity, immune regulation, etc.) are minimized. Interestingly, an inhibitor of downstream NF-κB1 signaling (i.e. SN50) has recently been characterized in combination with vaccine adjuvants and was described as an immune potentiator capable of decreasing markers associated with poor tolerability and improving the protective response of vaccination530, which suggests that therapeutic context is certainly important as well.
Carozza et al. found that many cancer cells continuously export endogenous 2′3′-cGAMP and that 2′3′-cGAMP is rapidly degraded by ENPP1 in the extracellular space166. They also determined that depletion of extracellular 2′3′-cGAMP by intratumoral injection of wildtype STING decreased the tumor infiltration of immune cells and eliminated the curative effects of tumor irradiation. Moreover, intratumoral administration of ENPP1 inhibitors elevated extracellular 2′3′-cGAMP concentrations and promoted improved responses to radiation therapy as demonstrated by delayed tumor growth. Notably, ENPP1 inhibitors would also limit levels of immunosuppressive adenosine in addition to elevate the levels of 2′3′-cGAMP200. Accordingly, ENPP1 inhibitors are currently being explored in preclinically with cGAS-activating therapies531, as they are likely to synergize with therapies that involve endogenous 2′3′-cGAMP.
In addition to the established potentiators of the STING pathway, there are still many other possible agents that might also propagate STING signaling, such as inhibitors of the STING phase-separator and VRAC agonists, both of which could synergize with cGAS-activating therapies by enhancing the production and spread of 2′3′-cGAMP. Though such agents have not yet been directly explored in the context of STING signaling and cancer immunotherapy, future investigation is certainly warranted.
8. Summary, Perspectives, and Future Directions
Since elucidating its critical role as a central link between innate and adaptive immunity in cancer immune surveillance, the cGAS/STING pathway has emerged as one of the most exciting and promising targets in immuno-oncology. Indeed, as indicated by the rate of publications, academic interest is increasing exponentially (Figure 22), and many pharmaceutical companies are developing STING pathway agonists and racing to translate them into the clinic. This fervent research-and-development activity is motivated by the clear and growing need for new immunotherapeutic strategies to increase immune recognition and eradication of tumors, particularly those that do not, or only poorly, respond to currently FDA-approved ICB antibodies. While an expanding number of therapeutic candidates are being developed to address this challenge (e.g. alternative checkpoint inhibitors, cytokine therapeutics, cell-based therapies, etc.), the multimodal activity of the STING pathway to “jump-start” and propagate the cancer immunity cycle offers compelling rationale for its enormous potential as an immunotherapy target. Indeed, preclinical studies of an increasing number of STING agonists have demonstrated remarkable results in many tumor models, sometimes resulting in complete and durable therapeutic responses in a majority of treated mice, even in models of highly immunosuppressive tumors.
Figure 22: Rate of Publications.

Google Scholar search results for: “stimulator of interferon genes” “cancer immunotherapy”.
Unfortunately, early clinical studies evaluating STING agonists in cancer patients have been less successful and arguably disappointing relative to initial, though perhaps unrealistic, expectations. Notably, data from the Aduro/Novartis (ADU-S100)328,532 and Merck (MK-1454)361 clinical trials of intratumorally administered CDNs (Table 1) were underwhelming with low response rates observed in treated patients (e.g. 2.1% overall response for MIW815 (ADU-S100) monotherapy, 0% overall response for MK-1454 monotherapy, and 24% overall response for MK-1454 in combination with pembrolizumab (i.e. anti-PD-1 monoclonal antibody therapy)). Additionally, neither study demonstrated consistent abscopal effects (i.e. shrinkage of non-injected distal tumors), which is a primary goal of an intralesional therapy (i.e. in situ vaccination). While not uncommon in drug development, such a gap between the remarkable preclinical efficacy, which was observed across many investigators, types of STING agonists, and tumor models, and these initial clinical outcomes motivate the need to better understand both the biological and pharmacological mechanisms that are restraining efficacy and to develop new agents, delivery systems, and/or drug combinations to more fully realize the immunotherapeutic potential of the STING pathway in patients. Below we offer additional perspective into emerging chemical and materials-based strategies for addressing known and putative barriers to the efficacy of STING pathway agonists, many of which appear poised for future clinical evaluation.
In considering the clinical trial data from the recent Aduro/Novartis and Merck studies, it is important to recognize that these studies enrolled patients with a range of solid tumor types, and many patients also had advanced disease that had progressed following other treatments. Additionally, the trials did not involve focused biomarker screening (i.e. testing for specific gene expression and/or protein signatures that are known to be more conducive to a given therapy), which can help to identify patients that are more likely to respond to treatment. Moreover, clinical observation of an abscopal response following intratumoral administration currently remains the exception rather than the norm373, and therefore it is perhaps unrealistic to expect robust therapeutic responses from a single intratumorally injected agent. This is noteworthy, as 11/15 of the ongoing trials (Table 1) appear to be focused on intratumoral administration of STING agonists. Thus, care should be taken when interpreting the results of these trials, as multiple factors have likely contributed to the unfavorable clinical outcomes observed thus far, a number of which may not be attributed to STING as a drug target or even the agents themselves.
As described in Section 5.2, while intratumoral administration clearly has potential, especially for some cancer types and clinical scenarios, free CDNs (e.g. ADU-S100) rapidly clear from intratumoral injection sites and display poor pharmacokinetic properties with a short half-life (e.g. 10–20 minutes). Considering the complex temporal relationships among innate immunity, antigen processing, and T cell priming384,389, such transient drug exposure, and the resultant burst of local STING activation at the tumor site, is likely suboptimal for in situ vaccination to generate systemic T cell responses that are capable of mediating abscopal responses. While this challenge could perhaps be addressed with repeated administration, the short half-life of CDNs (and most STING agonists described to date), coupled with the intrinsically transient nature of the resulting inflammatory response, may require a frequency of local administration that would not be feasible, particularly in clinical scenarios that require image-guided injection of tumors. This motivates the need for drug delivery technologies that allow for improved and tunable control over the retention and/or distribution of STING agonists following intratumoral administration as well as a deeper understanding of the interplay among STING agonist pharmacokinetics, pharmacodynamics, and antitumor immunity that can inform the design of local delivery strategies to improve clinical responses. Notably, delivery vehicles have been employed to improve the therapeutic efficacy of two other leading innate immune agonists, CpG ODN356 (i.e. agonist of TLR-9) and poly-IC533 (i.e. agonist of TLR-3 and MDA-5), which has led to ongoing clinical trials for intratumoral administration; CMP-001 is a CpG ODN encapsulated into a virus-like particle (NCT04695977), and poly-ICLC is an electrostatic formulation comprising poly-IC stabilized with poly-L-lysine and carboxymethylcellulose (NCT03789097).
Considering the significant recent advancements in nanoparticles for CDN delivery (Section 6.1) that can improve cellular uptake, promote more efficient cytosolic delivery, harness lymphatic drainage to reprogram tdLNs, and/or increase local retention of CDNs, clinical investigation of these technologies for intratumoral administration should be a priority, as they offer a relatively simple approach for addressing the pharmacological shortcomings of locally administered CDNs. Indeed, the results of the clinical trials by Codiak Biosciences using an exosome-based delivery system for CDN delivery and by Synlogic with a bacterial-based delivery system (Table 1) are much anticipated as they may help in assessing the extent to which the delivery barriers facing freely administered CDNs have impeded clinical efficacy.
Comparable to local oncolytic virus therapy (e.g. T-VEC534), nanoparticle-based STING agonists appear to be ideal for direct injection into solid tumor sites. However, in post-surgical settings, implantable and/or injectable biomaterial scaffolds may be better suited for administration into reSection cavities to control the release of STING agonists to boost antitumor immunity. In addition to allowing for sustained and tunable drug release that may reduce the necessary number of local injections for efficacy, biomaterial scaffolds and depots also afford important opportunities for protecting drug cargo from clearance and/or degradation, promoting cell-specific interactions via targeted (e.g. chemokine-induced) cellular infiltration, and programming the coordinated (e.g. combinatorial, pulsatile) release of multiple chemically-distinct agents. As the cancer immunity cycle requires the cooperation and coordination of multiple cell types, the use of scaffolds or gels for cell-specific orchestration has potential to improve therapeutic responses for STING pathway agonists. Notably, DCs, and conceivably other cell types, can be directed into gels for targeted, in situ manipulation as demonstrated by Mooney and co-workers, who have reported the development of injectable cryogels loaded with a chemoattractant (i.e. GM-CSF) to enhance the local accumulation of specialized antigen-presenting cells (e.g. DCs)535. Additionally, maximizing antitumor responses via intratumoral delivery (and immunotherapy more generally) is likely to require use of multiple agents that are properly sequenced. This is nicely illustrated in the work by Brody et al. described above (Section 5.2) where image-guided injection of Flt-3 ligand for nine days, two days of radiotherapy, and eight injections of poly-ICLC were employed to generate an abscopal response in lymphoma patients385. Likewise, the seminal studies by Wittrup and co-workers demonstrated the importance of sequencing a tumor-targeted IgG and IL-2 prior to IFNα in maximizing antitumor immunity236.
While much work remains to be done to understand how to best combine STING pathway agonists with other therapeutics, both for local and systemic administration, major advancements in designer biomaterials for local drug delivery will enable the continued development of technologies for intratumoral immunotherapy that allow for optimal control over the release and/or activation of multiple agents in a spatiotemporally programmable manner. Such opportunities are nicely exemplified by the recent work described above in Section 6.2 from the groups of Honggang Cui477, Matthias Stephan485, and the team of Robert Langer, Daniel Anderson, and Ana Jaklenc at MIT476. In particular, the work by Lu et al. from the MIT team is very promising as it uniquely allows for pulsatile, programmed release of not only STING agonists but also a diverse range of other molecules476. This presents an important opportunity to coordinate STING agonist delivery with other therapeutics agents (e.g. chemotherapy, ICB, etc.) to optimize antitumor immunity using fewer injections, and perhaps only a single administration, which would dramatically improve clinical utility and expand the number of patients that would be eligible for intratumoral administration.
Nonetheless, the translational challenges and the initial low clinically efficacy of intratumoral administration has motivated recent advancements in drug delivery systems for CDNs as well as other STING agonists that exert therapeutic effects when administered systemically, including both modified CDNs and non-nucleotide, small molecules. These medicinal chemistry developments address some, but not all, of the intracellular delivery and pharmacological limitations of natural CDNs (e.g. 2′3′-cGAMP) and first-generation synthetic CDNs (e.g. ADU-S100). While efficacy data for many of these agents has not been published in the academic literature, compounds such as MSA-2, SR-717, diABZIs, and 15a appear promising in preclinical studies, and a number of these have recently entered the clinical pipeline (Table 1), with data expected in the next several years. The outcome of these trials will be important for informing the continued development of STING pathway agonists and for identifying immunopharmacological barriers that limit their safety and efficacy.
The critical question for systemically administered STING agonists, regardless of type of STING agonist or formulation method, will be the width of the therapeutic window. Systemic administration of STING agonists can result in a transient systemic inflammatory response132,145,340,390 that can resemble a cytokine storm similar to that of other innate immune activators (e.g. PEG-Intron536, CMP-001356, etc.), which have been known to cause patients to temporarily experience flu-like symptoms. Indeed, systemic inflammation could very well prove to be dose limiting in patients receiving an intravenous or oral administration of STING agonists. Accordingly, as systemically administered STING agonists move forward, an important consideration will be how to expand their therapeutic window by minimizing inflammatory side effects. In addition to a growing clinical arsenal of approaches to combat cytokine storm and other deleterious systemic inflammation (e.g. anti-IL-6 monoclonal antibody therapy537–539), a multitude of exciting chemical, biomolecular, and pharmaceutical engineering strategies can be envisioned to address this critical challenge, including some already in development. For example, the non-nucleotide STING agonist MSA-2 described in Section 5.3 leverages a protonizable carboxylic acid group that increases cell membrane permeability in the acidic microenvironment associated with some tumors340. Nonetheless, systemic administration of MSA-2 still induces STING activation in other tissues, and therefore, the degree to which exploiting the acidic TME drug widens the therapeutic window relative to other STING agonists remains to be seen in human clinical studies. Furthermore, it will also be important to consider that the pH of tumors (as well as other microenvironmental factors) can vary significantly between cancer types, patients, and tumor sites341. Thus, it will be important to continue to develop STING agonists and/or drug carriers capable of selectively targeting tumor sites via other environmentally-responsive mechanisms (e.g. redox, protease expression levels, etc.). Fortunately, an expansive tool box of environmentally-responsive drug carriers and conditionally-cleavable chemical linkers have already been developed, primarily for chemotherapeutics, to enhance drug accumulation at tumor sites540. Leveraging such chemical strategies to exploit microenvironmental signatures to enrich STING activation at tumor sites has not been widely explored, but holds much potential for improving the efficacy and safety of systemically administered STING agonists.
Likewise, harnessing molecular targeting strategies such as antibodies, peptides, and glycans for STING agonists also holds significant promise for achieving more tumor selective activation of innate immunity and minimizing inflammatory side effects, provided an appropriate selective target can be identified. While no published reports are available, antibody drug conjugates (ADCs) for targeted STING agonist delivery are being developed by several companies, including Mersana (i.e. XMT-2056)541,542, Takeda (i.e. TAK-500)210, and Curadev (i.e. CRD5500)543. Based on recent reports leveraging ADC technology for the delivery of other innate immune agonists398, such targeting strategies appear well poised to be a major advancement in the field. However, this also raises the important and unknown question as to which cell type(s) in the tumor should STING agonists be targeted. Mechanistic preclinical studies, which have almost exclusively utilized intratumoral administration of CDNs, have implicated a number of different cell populations (e.g. cancer cells, endothelial cells, macrophages, dendritic cells) as being important contributors to STING agonist activity and therapeutic efficacy. This is perhaps not surprising given the relatively ubiquitous expression profile STING across cell populations as well as the multifaceted paracrine effects exerted by downstream innate effectors (e.g. type I IFNs and other proinflammatory cytokines) on many cell types within the TME. Which cell type(s) to target may also depend on the cancer type and/or the stromal composition of TME, as STING activation can trigger distinctive effects in different cell populations. A still unresolved question is the extent to which cancer cell–specific STING activation is important for therapeutic efficacy; that is, are there advantages (or potentially disadvantages) to activating STING signaling in cancer cells or is targeting stromal populations (e.g. macrophages, dendritic cells) sufficient or superior, with cancer cells primarily acting as bystanders during the initial phase of the innate immune response. This will be an important question to resolve, as STING signaling is often suppressed and dysfunctional in cancer cells474. Accordingly, in cases where cancer cell–intrinsic STING activation is critical to efficacy, careful patient selection and/or adjunctive therapies to enhance STING expression in cancer cells (e.g. treatment with DNA methyl transferase inhibitors301) will be required. Clearly, more detailed knowledge of how specific cell types contribute to the therapeutic efficacy of STING agonists, and how this may also vary between cancer types, will be critical to the future design of molecularly-targeted STING agonists. Notably, in addition to their potential therapeutic utility, ADCs may also be of use as a research tool for addressing these outstanding questions. Indeed, Cetinbas et al. are pursuing such a strategy to dissect the importance of cell-specific STING pathway activation in tumors and have identified distinct differences in resultant immune profiles generated by non-targeted STING agonists and cancer cell–targeted STING agonists, which suggested that cancer cells can positively contribute to antitumor immunity in certain cases544.
Another concern facing systemic administration of STING agonists, particularly non-nucleotide, small molecules, is the potential for inducing toxicity in T cells, which are important for therapeutic efficacy212. Indeed, T cells express high levels of the STING protein and appear to be highly susceptible to STING-induced apoptosis86,239,368. Many non-nucleotide, small molecule STING agonists directly access the cytosol via passive diffusion across cellular plasma membranes, resulting in indiscriminate STING activation in T cells with potentially deleterious effects on antitumor adaptive immunity. The use of nanoparticle-based drug carriers, including many described above, offer the possibility of minimizing such effects, since T cells have a relatively low capacity for endocytosis of nanoparticles545. Indeed, in our studies using STING-NPs, we have observed negligible uptake of nanoparticles (i.e. CDNs) by T cells in the TME, whereas we and others have demonstrated that nanocarriers can enhance uptake by cancer cells and myeloid cells in the tumor196,546.
While this represents an important advantage of using NP-based STING agonists, this is counterbalanced by the likelihood that a large fraction of administered nanoparticles, typically the vast majority regardless of nanoparticle properties, will accumulate in the liver with potential for hepatic STING activation. In our recent analysis of CDN biodistribution following intravenous delivery with a therapeutic dose of STING-NPs (i.e. polymersomes), we unsurprisingly observed a high degree of CDN accumulation in the liver, but a disproportionally low degree of hepatic STING activation as measured by inflammatory gene expression. We postulate that Kupffer cells, which have a well-established role in clearing nanoparticles from the circulation, were the primary contributors to STING activation in the liver as hepatocytes have been reported to have low levels of STING expression547. Hence, clearance of nanoparticles by the liver may not impose as significant of a barrier to intravenously administered nanoparticulate STING agonists as might otherwise be anticipated. Nonetheless, evidence is also emerging that chronic STING activation in the liver can promote nonalcoholic fatty liver disease (NAFLD)548. Therefore, STING-induced liver damage is a valid safety consideration for any nanoparticle-based STING agonist and motivates the design of carriers that minimize liver accumulation and/or hepatic STING activation. For example, the biodistribution profile of NPs can be tuned to some degree to bias delivery to other organ sites (e.g. spleen, lungs, bone marrow) by altering the physicochemical properties of the particles (e.g. charge, size, etc.)549,550 or through the addition of targeting ligands551, and liver preconditioning strategies have also been employed to reduce Kupffer cell uptake and liver accumulation of nanomedicines552. It will be interesting and important to determine if such approaches can widen the therapeutic window of nanoparticle STING agonists. Nonetheless, developing nanoparticle-based platforms that allow for more tumor-selective STING activation, either through molecular targeting or environmentally-responsive release, will be an important future direction to pursue.
Another important consideration in using nanoparticles for intravenous delivery of STING agonists, is their inefficient capacity to deliver drug cargo to tumor sites. Though nanoparticles can preferentially accumulate in human metastatic tumors408–412, only a small percentage of an intravenously injected drug dose reaches the tumor553,554. Indeed, in their now notorious article, Chan and co-workers have estimated that less than 1% of nanoparticles reach tumor sites in preclinical tumor models555. However, the shortcomings of inefficient tumor delivery have largely been manifested in applications where efficacy is dependent on delivery of high drug doses to the vast majority of tumor cells at a majority of tumor sites (e.g. chemotherapy, siRNAs against cancer cell targets). We maintain that this may not be as critical of a barrier, and may even be an important opportunity, for nanoscale STING agonists and other nanoparticle-based immunostimulants, where robust therapeutic responses may be achieved via delivery to a relatively small subset of cancer or stromal cells capable of initiating endogenous programs of systemic antitumor immunity556–559. This important distinction has been nicely highlighted by the Karathanasis group, who have demonstrated that intravenous administration of both liposomal421,422 and silica-based nanocarriers460 results in CDN accumulation primarily in perivascular regions where STING activation in local cell populations results in the recruitment of antitumor effectors into the TME. Nonetheless, nanoparticle delivery strategies that can further enhance the tumor accumulation and penetration of STING agonists require continued development, particularly for cases where the passive targeting may not be appreciable.
While targeting STING agonists to the TME is a rational and likely effective strategy for expanding the therapeutic window, this may not be a necessity provided systemic inflammatory effects can be adequately controlled. While their pharmacological mechanisms have not been fully described, it is likely that systemically administered small molecule STING agonists work at least in part via the induction of a peripheral inflammatory response initiated by diverse cell populations. A STING-driven cocktail of circulating proinflammatory cytokines can act on the TME via multiple mechanisms in manners comparable to systemic cytokine therapies. Moreover, such a systemic response can mobilize antitumor effector cells in immune reservoirs (e.g. bone marrow, spleen, etc.), causing them to migrate into tumors. For example, using a cationic liposomal formulation, Nakamura et al. demonstrated that cytokines secreted into the circulation by liver macrophages triggered the activation of NK cells in the spleen, which then led to the elimination of melanoma metastases in the lung following the migration of the activated NK cells560. Therefore, a complementary approach to tumor targeting is to develop molecules and/or delivery platforms for optimizing the magnitude and duration of systemic STING activation. However, as discussed in Section 5.3, it is also not yet known what the ideal pharmacokinetics for STING agonists (and innate immune agonists more generally) should be, nor is it known whether a slow and sustained or a fast “on/off” profile is optimal for therapeutic efficacy. Notably, while chronic STING activation is often associated with certain autoimmune and chronic inflammatory diseases205, acute STING activation might also be problematic if the magnitude and distribution of STING signaling is too intense. The half-life of STING agonists is relatively short, ranging from several minutes for CDNs to several hours for SR-717, which results in a spike in blood levels of proinflammatory cytokines typically 2–8 hours post administration340,390. The pharmacokinetics of nanoparticle-based STING agonists has not been widely described beyond our report on STING-NPs390, but the half-life of PEGylated liposomes, for example, can be on the order of several days561. Therefore, it will be important to better understand the pharmacokinetic-pharmacodynamic relationships that underlie antitumor immunity and therapeutic efficacy of STING agonists and to develop strategies to more precisely tune their circulation half-life and other key pharmacological properties.
In addition to strategies for tumor targeting and controlling systemic inflammation, a third approach to improving the safety and efficacy of STING agonists is to deploy adjunctive therapies that do not directly activate the pathway, but instead increase sensitivity to STING agonists and/or modulate the inflammatory response. While the discovery of STING pathway potentiators, such as those described in Section 7, is still in its infancy, therapeutic strategies that co-deliver STING pathway agonists with Mn2+ have already emerged and have demonstrated preclinical efficacy354,520,523. Likewise, the studies by Hou et al., which demonstrate that selective inhibition of specific NF-κB pathway components (e.g. non-canonical NF-κB) can increase the STING-driven IFN-I response240, offer a pharmacologically tractable target for potentiating STING signaling. Intriguingly, though not yet explored for STING agonists, the Esser-Kahn group has demonstrated that inhibiting non-canonical NF-κB signaling can minimize overproduction of TNF-α and IL-6 (i.e. “wasted inflammation”) induced by CpG ODN (i.e. agonist of TLR-9)530,562. This not only reduced the toxicity associated with excessive systemic release of these particular cytokines, but also boosted antibody responses elicited via vaccination. Therefore, it will be important to elucidate whether such “wasted inflammation” exists for STING agonists in the context of cancer immunotherapy, and if so, how to target the signaling pathway to maximize production of factors that are critical for efficacy while minimizing those that contribute to inflammatory toxicities. As these and other pathway modulators are identified and further defined, it will be increasingly important to develop therapeutics as well as drug delivery systems that maximizes their ability to synergize with STING agonists. Indeed, an important advantage of drug carriers is their capacity to deliver multiple agents in precisely balanced ratios563, a strategy that has achieved clinical success in liposomal delivery of chemotherapeutics (e.g. Vyxeos)564. Applying similar strategies for co-delivery of STING agonists and pathway modulators holds promise for further improving their efficacy and safety when administered systemically.
Finally, as is often the case in immunology, the STING pathway is a double-edged sword with potential to exert dichotomous effects that are dictated by numerous factors (e.g. biological context, magnitude of STING-driven gene expression, kinetics of signaling, etc.) and must be considered when optimizing the design and/or delivery of STING agonists. In response to STING activation, a number of immunosuppressive factors may also increase as a regulatory mechanism to dampen the inflammatory response. STING activation can result in the production of IDO-1399, the infiltration of immunosuppressive MDSCs565, and the upregulation of PD-L1450 and other immune checkpoints (e.g. B7-H3566), amongst other counter regulatory mechanisms11 that may inhibit antitumor immunity or even drive tumor progression. Accordingly, the development of rationally designed immunotherapy combinations that target these acquired resistance mechanisms is likely to be necessary to fully realize the potential of STING agonists. Which mechanisms to target, however, may also depend on specific cancer types or patient subpopulations, and additional research is needed to more completely understand these mechanisms and to develop biomarkers to predict which patients may be more likely to respond to STING agonists. Such efforts are ongoing in the setting of other immunotherapy agents and combinations, and the clinical data emerging from the ongoing and future clinical trials will be critical for beginning to fill this knowledge gap. It will be important to consider this emerging information in the design of next-generation STING agonists and delivery technologies. Several groups have already developed systems that allow for co-delivery of STING agonists and therapeutics that can target other therapeutic pathways354,421,457,477,522,567. While such dual-delivery approaches may prove critical for optimally combining STING agonists with other agents, caution should be taken in considering whether or not two agents that target different pathways and/or cell types need to be chemically or physically coupled, especially in cases where one of the agents is already approved for clinical use. This not only adds additional complexity to manufacturing and regulatory approval, but may also not yield the optimal sequencing or dose of each agent, which may be more readily achieved by simply adjusting the administration regimen of each component independently.
Research over the past decade has led to an enormous leap in our knowledge of the cGAS/STING pathway and its complex but critical role in cancer immune surveillance. Despite early clinical setbacks, the importance of the STING pathway in antitumor immunity is increasingly clear, and STING agonists continue to hold great promise as pharmacological agents for the treatment of cancer, with many now entering clinical trials. The outcomes of these trials, coupled with continued mechanistic investigations in preclinical models, is expected to accelerate our understanding of the immunopharmacological mechanisms and shortcomings of a diversity of STING agonists as well as drug delivery technologies. As this information emerges, the recent and continued advancement in chemical and biomolecular strategies for STING pathway activation, including many described herein, will be critical to realizing the full clinical potential of this promising immunotherapeutic target.
Supplementary Material
Acknowledgements:
We thank Dr. Karan Arora (Vanderbilt University) for his assistance in reviewing and generating the chemical structures presented in the manuscript. This work was supported by grants from the National Science Foundation (CBET-1554623 and MCB-2036809 to JTW), a Vanderbilt Ingram Cancer Center (VICC) Ambassador Discovery Grant (JTW), the Melanoma Research Alliance (503565 to JTW), Susan G. Komen (CCR19609205 to JTW), the Congressionally-Directed Medical Research Program (KC170091 to JTW), the National Institutes of Health (R01-CA245134 to JTW), and a Stand Up To Cancer Innovative Research Grant (SU2C-AACR-IRG 20-17 JTW). Stand Up To Cancer (SU2C) is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. KG acknowledges a Pediatric Oncology Student Training (POST) grant from Alex’s Lemonade Stand Foundation. TS acknowledges a Pediatric Oncology Student Training (POST) grant from Alex’s Lemonade Stand Foundation and a National Science Foundation Graduate Research Fellowship under Grant Number 1937963.
Author Information:
Kyle M. Garland received his B.S. degree in Bioengineering with a focus in Biopharmaceutical Engineering at Lehigh University in 2016. He then joined the Department of Chemical & Biomolecular Engineering at Vanderbilt University, where he studied Immunoengineering under the direction of Dr. John T. Wilson. In 2017, he was awarded the Pediatric Oncology Student Training (POST) Grant from Alex’s Lemonade Stand Foundation, which allowed him to pursue STING activation strategies in the context of pediatric neuroblastoma. In 2020, he received the Russell G. Hamilton Graduate Leadership Institute Dissertation Enhancement Grant, which enabled him to conduct research that focused on developing a novel cGAS agonist. He earned his Ph.D. degree in Chemical Engineering at Vanderbilt University in 2021. His dissertation describes the development of innate immune activators for local cancer therapy applications.
Taylor L. Sheehy received a B.S. degree in Biochemistry in 2019 at Duquesne University, where she pursued research studying the effects of macromolecular crowding on enzyme kinetics. Upon graduation, she began pursuing her Ph.D. in Biomedical Engineering under the direction of Dr. John T. Wilson. Taylor received a National Science Foundation Graduate Research Fellowship Program (GRFP) award in 2020 to support her work in molecularly engineered STING agonist delivery technologies to improve cancer immunotherapy. These strategies focus on tumor and immune cell-specific targeting regimes. Although this work can be applied to numerous cancer models, Taylor also received a Pediatric Oncology Student Training (POST) award from Alex’s Lemonade Stand Foundation in 2020 to pursue projects that concentrate on treatments specifically for pediatric neuroblastoma.
John T. Wilson is an Associate Professor of Chemical & Biomolecular Engineering at Vanderbilt University. Dr. Wilson earned his Ph.D. in Bioengineering from the Georgia Institute of Technology and completed his postdoctoral training at the University of Washington prior to launching his independent Immunoengineering Laboratory in 2014. His research lies at the interface of molecular engineering and immunology, bringing together polymer and organic chemistry, nanotechnology, pharmaceutical science, and immunobiology to develop new technologies for immune modulation, with applications in next-generation vaccines for infectious diseases, cancer immunotherapies, and therapies for autoimmune and chronic inflammatory diseases.
Footnotes
Conflicts of Interest:
J.T.W. is an inventor on U.S. Patent 10,696,985 “Reversibly Crosslinked Endosomolytic Polymer Vesicles for Cytosolic Drug Delivery” and on U.S. Patent Application PCT/US2019/058945 “Graft Copolymers, Methods of Forming Graft Copolymers, and Methods of Use Thereof” which both describe drug delivery technologies that have been used for STING agonist delivery.
References:
- (1).Barber GN STING: infection, inflammation and cancer. Nat. Rev. Immunol 2015, 15, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ahn J; Barber GN STING signaling and host defense against microbial infection. Exp. Mol. Med 2019, 51, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wu X; Wu FH; Wang X; Wang L; Siedow JN; Zhang W; Pei ZM Molecular evolutionary and structural analysis of the cytosolic DNA sensor cGAS and STING. Nucleic Acids Res 2014, 42, 8243–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Margolis SR; Wilson SC; Vance RE Evolutionary origins of cGAS-STING signaling. Trends Immunol 2017, 38, 733–743. [DOI] [PubMed] [Google Scholar]
- (5).Kranzusch PJ; Wilson SC; Lee AS; Berger JM; Doudna JA; Vance RE Ancient origin of cGAS-STING reveals mechanism of universal 2’,3’ cGAMP signaling. Mol. Cell 2015, 59, 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Xia T; Konno H; Ahn J; Barber GN Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep 2016, 14, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Xia T; Konno H; Barber GN Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res 2016, 76, 6747–6759. [DOI] [PubMed] [Google Scholar]
- (8).Xiao N; Wei J; Xu S; Du H; Huang M; Zhang S; Ye W; Sun L; Chen Q cGAS activation causes lupus-like autoimmune disorders in a TREX1 mutant mouse model. J. Autoimmun 2019, 100, 84–94. [DOI] [PubMed] [Google Scholar]
- (9).Yu Y; Liu Y; An W; Song J; Zhang Y; Zhao X STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Invest 2019, 129, 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ablasser A; Chen ZJ cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [DOI] [PubMed] [Google Scholar]
- (11).Chen Q; Sun L; Chen ZJ Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol 2016, 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- (12).Motwani M; Pesiridis S; Fitzgerald KA DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet 2019, 20, 657–674. [DOI] [PubMed] [Google Scholar]
- (13).Hooy RM; Sohn J The allosteric activation of cGAS underpins its dynamic signaling landscape. Elife 2018, 7, e39984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wu J; Chen ZJ Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol 2014, 32, 461–488. [DOI] [PubMed] [Google Scholar]
- (15).Corrales L; Glickman LH; Mcwhirter SM; Kanne DB; Sivick KE; Katibah GE; Woo SR; Lemmens E; Banda T; Leong JJ et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep 2015, 11, 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Honda K; Takaoka A; Taniguchi T Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [DOI] [PubMed] [Google Scholar]
- (17).Borden EC Interferons alpha and beta in cancer: therapeutic opportunities from new insights. Nat. Rev. Drug Discov 2019, 18, 219–234. [DOI] [PubMed] [Google Scholar]
- (18).Gajewski TF; Corrales L New perspectives on type I IFNs in cancer. Cytokine Growth Factor Rev 2015, 26, 175–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Benci JL; Johnson LR; Choa R; Xu Y; Qiu J; Zhou Z; Xu B; Ye D; Nathanson KL; June CH et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 2019, 178, 933–948 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Marcus A; Mao AJ; Lensink-Vasan M; Wang L; Vance RE; Raulet DH Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity 2018, 49, 754–763 e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Paludan SR; Reinert LS; Hornung V DNA-stimulated cell death: implications for host defence, inflammatory diseases and cancer. Nat. Rev. Immunol 2019, 19, 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Murthy AMV; Robinson N; Kumar S Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ 2020, 27, 2989–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Demaria O; De Gassart A; Coso S; Gestermann N; Di Domizio J; Flatz L; Gaide O; Michielin O; Hwu P; Petrova TV et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 15408–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li T; Cheng H; Yuan H; Xu Q; Shu C; Zhang Y; Xu P; Tan J; Rui Y; Li P et al. Antitumor activity of cGAMP via stimulation of cGAS-cGAMP-STING-IRF3 mediated innate immune response. Sci. Rep 2016, 6, 19049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Prantner D; Perkins DJ; Lai W; Williams MS; Sharma S; Fitzgerald KA; Vogel SN 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J. Biol. Chem 2012, 287, 39776–39788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Woo SR; Fuertes MB; Corrales L; Spranger S; Furdyna MJ; Leung MY; Duggan R; Wang Y; Barber GN; Fitzgerald KA et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yum S; Li M; Chen ZJ Old dogs, new trick: classic cancer therapies activate cGAS. Cell Res 2020, 30, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Storozynsky Q; Hitt MM The Impact of radiation-induced DNA damage on cGAS-STING-mediated immune responses to cancer. Int. J. Mol. Sci 2020, 21, 8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Le Naour J; Zitvogel L; Galluzzi L; Vacchelli E; Kroemer G Trial watch: STING agonists in cancer therapy. Oncoimmunology 2020, 9, 1777624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Su T; Zhang Y; Valerie K; Wang X-Y; Lin S; Zhu G STING activation in cancer immunotherapy. Theranostics 2019, 9, 7759–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wu JJ; Zhao L; Hu HG; Li WH; Li YM Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med. Res. Rev 2019, 40, 1117–1141. [DOI] [PubMed] [Google Scholar]
- (32).Dobbs N; Burnaevskiy N; Chen D; Gonugunta VK; Alto NM; Yan N STING activation by translocation from the ER Is associated with infection and autoinflammatory disease. Cell Host Microbe 2015, 18, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lepelley A; Martin-Niclos MJ; Le Bihan M; Marsh JA; Uggenti C; Rice GI; Bondet V; Duffy D; Hertzog J; Rehwinkel J et al. Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J. Exp. Med 2020, 217, e20200600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Deng Z; Chong Z; Law CS; Mukai K; Ho FO; Martinu T; Backes BJ; Eckalbar WL; Taguchi T; Shum AK A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J. Exp. Med 2020, 217, e20201045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mukai K; Ogawa E; Uematsu R; Kuchitsu Y; Kiku F; Uemura T; Waguri S; Suzuki T; Dohmae N; Arai H et al. Homeostatic regulation of STING by retrograde membrane traffic to the ER. Nat. Commun 2021, 12, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ishikawa H; Barber GN STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Burdette DL; Monroe KM; Sotelo-Troha K; Iwig JS; Eckert B; Hyodo M; Hayakawa Y; Vance RE STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sun L; Wu J; Du F; Chen X; Chen ZJ Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wu J; Sun L; Chen X; Du F; Shi H; Chen C; Chen ZJ Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cai X; Chiu YH; Chen ZJ The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [DOI] [PubMed] [Google Scholar]
- (41).Cavlar T; Ablasser A; Hornung V Induction of type I IFNs by intracellular DNA-sensing pathways. Immunol. Cell Biol 2012, 90, 474–482. [DOI] [PubMed] [Google Scholar]
- (42).Ahn J; Ruiz P; Barber GN Intrinsic self-DNA triggers inflammatory disease dependent on STING. J. Immunol 2014, 193, 4634–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ahn J; Barber GN Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr. Opin. Immunol 2014, 31, 121–126. [DOI] [PubMed] [Google Scholar]
- (44).Pandey S; Kawai T In Biological DNA Sensor; Ishii KJ;Tang CK, Eds.; Academic Press: Amsterdam, 2014. [Google Scholar]
- (45).Zierhut C; Funabiki H Regulation and consequences of cGAS activation by self-DNA. Trends Cell Biol 2020, 30, 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ho SS; Zhang WY; Tan NY; Khatoo M; Suter MA; Tripathi S; Cheung FS; Lim WK; Tan PH; Ngeow J et al. The DNA structure-specific endonuclease MUS81 mediates DNA sensor STING-dependent host rejection of prostate cancer cells. Immunity 2016, 44, 1177–1189. [DOI] [PubMed] [Google Scholar]
- (47).Chen Q; Boire A; Jin X; Valiente M; Er EE; Lopez-Soto A; Jacob L; Patwa R; Shah H; Xu K et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Vanpouille-Box C; Demaria S; Formenti SC; Galluzzi L Cytosolic DNA sensing in organismal tumor control. Cancer Cell 2018, 34, 361–378. [DOI] [PubMed] [Google Scholar]
- (49).Bhat N; Fitzgerald KA Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur. J. Immunol 2014, 44, 634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Benmerzoug S; Rose S; Bounab B; Gosset D; Duneau L; Chenuet P; Mollet L; Le Bert M; Lambers C; Geleff S et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat. Commun 2018, 9, 5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ma F; Li B; Liu SY; Iyer SS; Yu Y; Wu A; Cheng G Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J. Immunol 2015, 194, 1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Civril F; Deimling T; De Oliveira Mann CC; Ablasser A; Moldt M; Witte G; Hornung V; Hopfner KP Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kranzusch PJ; Lee AS; Berger JM; Doudna JA Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep 2013, 3, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Li X; Shu C; Yi G; Chaton CT; Shelton CL; Diao J; Zuo X; Kao CC; Herr AB; Li P Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 2013, 39, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhang X; Wu J; Du F; Xu H; Sun L; Chen Z; Brautigam CA; Zhang X; Chen ZJ The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep 2014, 6, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Xie W; Lama L; Adura C; Tomita D; Glickman JF; Tuschl T; Patel DJ Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 11946–11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Gao P; Ascano M; Wu Y; Barchet W; Gaffney BL; Zillinger T; Serganov AA; Liu Y; Jones RA; Hartmann G et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013, 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Du M; Chen ZJ DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zhang X; Shi H; Wu J; Zhang X; Sun L; Chen C; Chen ZJ Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Danilchanka O; Mekalanos JJ Cyclic dinucleotides and the innate immune response. Cell 2013, 154, 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zhou W; Whiteley AT; De Oliveira Mann CC; Morehouse BR; Nowak RP; Fischer ES; Gray NS; Mekalanos JJ; Kranzusch PJ Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell 2018, 174, 300–311 e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Karayel E; Burckstummer T; Bilban M; Durnberger G; Weitzer S; Martinez J; Superti-Furga G The TLR-independent DNA recognition pathway in murine macrophages: Ligand features and molecular signature. Eur. J. Immunol 2009, 39, 1929–1936. [DOI] [PubMed] [Google Scholar]
- (63).Herzner AM; Hagmann CA; Goldeck M; Wolter S; Kubler K; Wittmann S; Gramberg T; Andreeva L; Hopfner KP; Mertens C et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol 2015, 16, 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Zhou W; Mohr L; Maciejowski J; Kranzusch PJ cGAS phase separation inhibits TREX1-mediated DNA degradation and enhances cytosolic DNA sensing. Mol. Cell 2021, 81, 739–755 e737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hooy RM; Massaccesi G; Rousseau KE; Chattergoon MA; Sohn J Allosteric coupling between Mn2+ and dsDNA controls the catalytic efficiency and fidelity of cGAS. Nucleic Acids Res 2020, 48, 4435–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Luecke S; Holleufer A; Christensen MH; Jonsson KL; Boni GA; Sorensen LK; Johannsen M; Jakobsen MR; Hartmann R; Paludan SR cGAS is activated by DNA in a length-dependent manner. EMBO Rep 2017, 18, 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Andreeva L; Hiller B; Kostrewa D; Lassig C; De Oliveira Mann CC; Jan Drexler D; Maiser A; Gaidt M; Leonhardt H; Hornung V et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017, 549, 394–398. [DOI] [PubMed] [Google Scholar]
- (68).Lugrin J; Martinon F The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev 2018, 281, 99–114. [DOI] [PubMed] [Google Scholar]
- (69).Hornung V; Ablasser A; Charrel-Dennis M; Bauernfeind F; Horvath G; Caffrey DR; Latz E; Fitzgerald KA AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Corrales L; Woo SR; Williams JB; Mcwhirter SM; Dubensky TW Jr.; Gajewski TF Antagonism of the STING pathway via activation of the AIM2 inflammasome by intracellular DNA. J. Immunol 2016, 196, 3191–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Liu C; Yue R; Yang Y; Cui Y; Yang L; Zhao D; Zhou X AIM2 inhibits autophagy and IFN-beta production during M. bovis infection. Oncotarget 2016, 7, 46972–46987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Yan S; Shen H; Lian Q; Jin W; Zhang R; Lin X; Gu W; Sun X; Meng G; Tian Z et al. Deficiency of the AIM2-ASC signal uncovers the STING-driven overreactive response of type I IFN and reciprocal depression of protective IFN-gamma immunity in mycobacterial infection. J. Immunol 2018, 200, 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Nakaya Y; Lilue J; Stavrou S; Moran EA; Ross SR AIM2-like receptors positively and negatively regulate the interferon response induced by cytosolic DNA. mBio 2017, 8, e00944–00917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Banerjee I; Behl B; Mendonca M; Shrivastava G; Russo AJ; Menoret A; Ghosh A; Vella AT; Vanaja SK; Sarkar SN et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity 2018, 49, 413–426 e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Cridland JA; Curley EZ; Wykes MN; Schroder K; Sweet MJ; Roberts TL; Ragan MA; Kassahn KS; Stacey KJ The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol. Biol 2012, 12, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Gaidt MM; Ebert TS; Chauhan D; Ramshorn K; Pinci F; Zuber S; O’duill F; Schmid-Burgk JL; Hoss F; Buhmann R et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell 2017, 171, 1110–1124 e1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Jin T; Perry A; Jiang J; Smith P; Curry JA; Unterholzner L; Jiang Z; Horvath G; Rathinam VA; Johnstone RW et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 2012, 36, 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Li H; Wang J; Wang J; Cao LS; Wang ZX; Wu JW Structural mechanism of DNA recognition by the p202 HINa domain: insights into the inhibition of Aim2-mediated inflammatory signalling. Acta Crystallogr. F Struct. Biol. Commun 2014, 70, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Emming S; Schroder K Tiered DNA sensors for escalating responses. Science 2019, 365, 1375–1376. [DOI] [PubMed] [Google Scholar]
- (80).Muruve DA; Petrilli V; Zaiss AK; White LR; Clark SA; Ross PJ; Parks RJ; Tschopp J The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [DOI] [PubMed] [Google Scholar]
- (81).Morrone SR; Matyszewski M; Yu X; Delannoy M; Egelman EH; Sohn J Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun 2015, 6, 7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Morrone SR; Wang T; Constantoulakis LM; Hooy RM; Delannoy MJ; Sohn J Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Matyszewski M; Morrone SR; Sohn J Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E1963–E1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Liu T; Yamaguchi Y; Shirasaki Y; Shikada K; Yamagishi M; Hoshino K; Kaisho T; Takemoto K; Suzuki T; Kuranaga E et al. Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Rep 2014, 8, 974–982. [DOI] [PubMed] [Google Scholar]
- (85).Roberts TL; Idris A; Dunn JA; Kelly GM; Burnton CM; Hodgson S; Hardy LL; Garceau V; Sweet MJ; Ross IL et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [DOI] [PubMed] [Google Scholar]
- (86).Gulen MF; Koch U; Haag SM; Schuler F; Apetoh L; Villunger A; Radtke F; Ablasser A Signalling strength determines proapoptotic functions of STING. Nat. Commun 2017, 8, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Li T; Chen ZJ The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med 2018, 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Liu S; Guan W STING signaling promotes apoptosis, necrosis, and cell death: an overview and update. Mediators Inflamm 2018, 2018, 1202797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Sokolowska O; Nowis D STING signaling in cancer cells: Important or not? Arch. Immunol. Ther. Exp. (Warsz) 2018, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Hu Q; Wu J; Ren Y; Wu X; Gao L; Wang G; Gu G; Ren H; Hong Z; Slade DA et al. Degree of STING activation is associated with disease outcomes. Gut 2020, 69, 792–794. [DOI] [PubMed] [Google Scholar]
- (91).Roers A; Hiller B; Hornung V Recognition of endogenous nucleic acids by the innate immune system. Immunity 2016, 44, 739–754. [DOI] [PubMed] [Google Scholar]
- (92).Miyake K; Shibata T; Ohto U; Shimizu T Emerging roles of the processing of nucleic acids and toll-like receptors in innate immune responses to nucleic acids. J. Leukoc. Biol 2017, 101, 135–142. [DOI] [PubMed] [Google Scholar]
- (93).Gao D; Li T; Li XD; Chen X; Li QZ; Wight-Carter M; Chen ZJ Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. U. S. A 2015, 112, E5699–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Baranovskii AG; Buneva VN; Nevinsky GA Human deoxyribonucleases. Biochemistry (Mosc) 2004, 69, 587–601. [DOI] [PubMed] [Google Scholar]
- (95).Benmerzoug S; Ryffel B; Togbe D; Quesniaux VFJ Self-DNA sensing in lung inflammatory diseases. Trends Immunol 2019, 40, 719–734. [DOI] [PubMed] [Google Scholar]
- (96).Wang Q; Liu X; Zhou Q; Wang C Cytosolic sensing of aberrant DNA: arming STING on the endoplasmic reticulum. Expert Opin. Ther. Targets 2015, 19, 1397–1409. [DOI] [PubMed] [Google Scholar]
- (97).Stetson DB; Ko JS; Heidmann T; Medzhitov R Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Gray EE; Treuting PM; Woodward JJ; Stetson DB Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutieres syndrome. J. Immunol 2015, 195, 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Ablasser A; Hemmerling I; Schmid-Burgk JL; Behrendt R; Roers A; Hornung V TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol 2014, 192, 5993–5997. [DOI] [PubMed] [Google Scholar]
- (100).Orebaugh CD; Fye JM; Harvey S; Hollis T; Perrino FW The TREX1 exonuclease R114H mutation in Aicardi-Goutieres syndrome and lupus reveals dimeric structure requirements for DNA degradation activity. J. Biol. Chem 2011, 286, 40246–40254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Xu J; Zoltick PW; Gamero AM; Gallucci S TLR ligands up-regulate Trex1 expression in murine conventional dendritic cells through type I Interferon and NF-kappaB-dependent signaling pathways. J. Leukoc. Biol 2014, 96, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Hemphill WO; Simpson SR; Liu M; Salsbury FR Jr.; Hollis T; Grayson JM; Perrino FW TREX1 as a novel immunotherapeutic target. Front. Immunol 2021, 12, 660184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Vanpouille-Box C; Alard A; Aryankalayil MJ; Sarfraz Y; Diamond JM; Schneider RJ; Inghirami G; Coleman CN; Formenti SC; Demaria S DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun 2017, 8, 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Diamond JM; Vanpouille-Box C; Spada S; Rudqvist NP; Chapman JR; Ueberheide BM; Pilones KA; Sarfraz Y; Formenti SC; Demaria S Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol. Res 2018, 6, 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Azzam EI; Jay-Gerin JP; Pain D Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett 2012, 327, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Yoshida T; Goto S; Kawakatsu M; Urata Y; Li TS Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free Radic. Res 2012, 46, 147–153. [DOI] [PubMed] [Google Scholar]
- (107).Gehrke N; Mertens C; Zillinger T; Wenzel J; Bald T; Zahn S; Tuting T; Hartmann G; Barchet W Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 2013, 39, 482–495. [DOI] [PubMed] [Google Scholar]
- (108).Fang C; Mo F; Liu L; Du J; Luo M; Men K; Na F; Wang W; Yang H; Wei X Oxidized mitochondrial DNA sensing by STING signaling promotes the antitumor effect of an irradiated immunogenic cancer cell vaccine. Cell. Mol. Immunol 2020, 18, 2211–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Huang KW; Liu TC; Liang RY; Chu LY; Cheng HL; Chu JW; Hsiao YY Structural basis for overhang excision and terminal unwinding of DNA duplexes by TREX1. PLoS Biol 2018, 16, e2005653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Dai J; Huang YJ; He X; Zhao M; Wang X; Liu ZS; Xue W; Cai H; Zhan XY; Huang SY et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell 2019, 176, 1447–1460 e1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Xia P; Ye B; Wang S; Zhu X; Du Y; Xiong Z; Tian Y; Fan Z Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol 2016, 17, 369–378. [DOI] [PubMed] [Google Scholar]
- (112).Seo GJ; Yang A; Tan B; Kim S; Liang Q; Choi Y; Yuan W; Feng P; Park HS; Jung JU Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep 2015, 13, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Hu MM; Yang Q; Xie XQ; Liao CY; Lin H; Liu TT; Yin L; Shu HB Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 2016, 45, 555–569. [DOI] [PubMed] [Google Scholar]
- (114).Wang Q; Huang L; Hong Z; Lv Z; Mao Z; Tang Y; Kong X; Li S; Cui Y; Liu H et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog 2017, 13, e1006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Chen X; Chen Y Ubiquitination of cGAS by TRAF6 regulates anti-DNA viral innate immune responses. Biochem. Biophys. Res. Commun 2019, 514, 659–664. [DOI] [PubMed] [Google Scholar]
- (116).Gekara NO; Jiang H The innate immune DNA sensor cGAS: A membrane, cytosolic, or nuclear protein? Sci. Signal 2019, 12, eaax3521. [DOI] [PubMed] [Google Scholar]
- (117).Barnett KC; Coronas-Serna JM; Zhou W; Ernandes MJ; Cao A; Kranzusch PJ; Kagan JC Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell 2019, 176, 1432–1446 e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Xia P; Wang S; Ye B; Du Y; Li C; Xiong Z; Qu Y; Fan Z A circular RNA protects dormant hematopoietic stem cells from DNA sensor cGAS-mediated exhaustion. Immunity 2018, 48, 688–701 e687. [DOI] [PubMed] [Google Scholar]
- (119).Yang H; Wang H; Ren J; Chen Q; Chen ZJ cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Orzalli MH; Broekema NM; Diner BA; Hancks DC; Elde NC; Cristea IM; Knipe DM cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. U. S. A 2015, 112, E1773–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Liu H; Zhang H; Wu X; Ma D; Wu J; Wang L; Jiang Y; Fei Y; Zhu C; Tan R et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [DOI] [PubMed] [Google Scholar]
- (122).Jiang H; Xue X; Panda S; Kawale A; Hooy RM; Liang F; Sohn J; Sung P; Gekara NO Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J 2019, 38, e102718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Gentili M; Lahaye X; Nadalin F; Nader GPF; Puig Lombardi E; Herve S; De Silva NS; Rookhuizen DC; Zueva E; Goudot C et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep 2019, 26, 2377–2393 e2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Volkman HE; Cambier S; Gray EE; Stetson DB Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 2019, 8, e47491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Guey B; Wischnewski M; Decout A; Makasheva K; Kaynak M; Sakar MS; Fierz B; Ablasser A BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science 2020, 369, 823–828. [DOI] [PubMed] [Google Scholar]
- (126).Li T; Huang T; Du M; Chen X; Du F; Ren J; Chen ZJ Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 2021, 371, eabc5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Zierhut C; Yamaguchi N; Paredes M; Luo JD; Carroll T; Funabiki H The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 2019, 178, 302–315 e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Shang G; Zhu D; Li N; Zhang J; Zhu C; Lu D; Liu C; Yu Q; Zhao Y; Xu S et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol 2012, 19, 725–727. [DOI] [PubMed] [Google Scholar]
- (129).Shang G; Zhang C; Chen ZJ; Bai XC; Zhang X Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Huang YH; Liu XY; Du XX; Jiang ZF; Su XD The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat. Struct. Mol. Biol 2012, 19, 728–730. [DOI] [PubMed] [Google Scholar]
- (131).Yin Q; Tian Y; Kabaleeswaran V; Jiang X; Tu D; Eck MJ; Chen ZJ; Wu H Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol. Cell 2012, 46, 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Ramanjulu JM; Pesiridis GS; Yang J; Concha N; Singhaus R; Zhang SY; Tran JL; Moore P; Lehmann S; Eberl HC et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [DOI] [PubMed] [Google Scholar]
- (133).Ergun SL; Fernandez D; Weiss TM; Li L STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell 2019, 178, 290–301 e210. [DOI] [PubMed] [Google Scholar]
- (134).Ni G; Konno H; Barber GN Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci. Immunol 2017, 2, eaah7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Mukai K; Konno H; Akiba T; Uemura T; Waguri S; Kobayashi T; Barber GN; Arai H; Taguchi T Activation of STING requires palmitoylation at the Golgi. Nat. Commun 2016, 7, 11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Ishikawa H; Ma Z; Barber GN STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Saitoh T; Fujita N; Hayashi T; Takahara K; Satoh T; Lee H; Matsunaga K; Kageyama S; Omori H; Noda T et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Luo WW; Li S; Li C; Lian H; Yang Q; Zhong B; Shu HB iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol 2016, 17, 1057–1066. [DOI] [PubMed] [Google Scholar]
- (139).Liu S; Cai X; Wu J; Cong Q; Chen X; Li T; Du F; Ren J; Wu YT; Grishin NV et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- (140).Ogawa E; Mukai K; Saito K; Arai H; Taguchi T The binding of TBK1 to STING requires exocytic membrane traffic from the ER. Biochem. Biophys. Res. Commun 2018, 503, 138–145. [DOI] [PubMed] [Google Scholar]
- (141).Yum S; Li M; Fang Y; Chen ZJ TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2100225118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Tanaka Y; Chen ZJ STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 2012, 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Abe T; Harashima A; Xia T; Konno H; Konno K; Morales A; Ahn J; Gutman D; Barber GN STING recognition of cytoplasmic DNA instigates cellular defense. Mol. Cell 2013, 50, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Stetson DB; Medzhitov R Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2006, 24, 93–103. [DOI] [PubMed] [Google Scholar]
- (145).Chin EN; Yu C; Vartabedian VF; Jia Y; Kumar M; Gamo AM; Vernier W; Ali SH; Kissai M; Lazar DC et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 2020, 369, 993–999. [DOI] [PubMed] [Google Scholar]
- (146).Yu X; Zhang L; Shen J; Zhai Y; Jiang Q; Yi M; Deng X; Ruan Z; Fang R; Chen Z et al. The STING phase-separator suppresses innate immune signalling. Nat. Cell Biol 2021, 23, 330–340. [DOI] [PubMed] [Google Scholar]
- (147).Xiao Q; Mcatee CK; Su X Phase separation in immune signalling. Nat. Rev. Immunol 2021, Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Peran I; Mittag T Molecular structure in biomolecular condensates. Curr. Opin. Struct. Biol 2020, 60, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Kato M; Han TW; Xie S; Shi K; Du X; Wu LC; Mirzaei H; Goldsmith EJ; Longgood J; Pei J et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Balka KR; Louis C; Saunders TL; Smith AM; Calleja DJ; D’silva DB; Moghaddas F; Tailler M; Lawlor KE; Zhan Y et al. TBK1 and IKKepsilon act redundantly to mediate STING-induced NF-kappaB responses in myeloid cells. Cell Rep 2020, 31, 107492. [DOI] [PubMed] [Google Scholar]
- (151).Abe T; Barber GN Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol 2014, 88, 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Bakhoum SF; Ngo B; Laughney AM; Cavallo JA; Murphy CJ; Ly P; Shah P; Sriram RK; Watkins TBK; Taunk NK et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Mcwhirter SM; Barbalat R; Monroe KM; Fontana MF; Hyodo M; Joncker NT; Ishii KJ; Akira S; Colonna M; Chen ZJ et al. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J. Exp. Med 2009, 206, 1899–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Chen H; Sun H; You F; Sun W; Zhou X; Chen L; Yang J; Wang Y; Tang H; Guan Y et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 2011, 147, 436–446. [DOI] [PubMed] [Google Scholar]
- (155).Flood BA; Higgs EF; Li S; Luke JJ; Gajewski TF STING pathway agonism as a cancer therapeutic. Immunol. Rev 2019, 290, 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Thanos D; Maniatis T Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 1995, 83, 1091–1100. [DOI] [PubMed] [Google Scholar]
- (157).Maniatis T; Falvo JV; Kim TH; Kim TK; Lin CH; Parekh BS; Wathelet MG Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol 1998, 63, 609–620. [DOI] [PubMed] [Google Scholar]
- (158).Panne D; Maniatis T; Harrison SC An atomic model of the interferon-beta enhanceosome. Cell 2007, 129, 1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Smale ST Selective transcription in response to an inflammatory stimulus. Cell 2010, 140, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Yi G; Brendel VP; Shu C; Li P; Palanathan S; Cheng Kao C Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One 2013, 8, e77846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Diner EJ; Burdette DL; Wilson SC; Monroe KM; Kellenberger CA; Hyodo M; Hayakawa Y; Hammond MC; Vance RE The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 2013, 3, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Jin L; Xu LG; Yang IV; Davidson EJ; Schwartz DA; Wurfel MM; Cambier JC Identification and characterization of a loss-of-function human MPYS variant. Genes Immun 2011, 12, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Patel S; Jin L TMEM173 variants and potential importance to human biology and disease. Genes Immun 2019, 20, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Li S; Luo M; Wang Z; Feng Q; Wilhelm J; Wang X; Li W; Wang J; Cholka A; Fu YX et al. Prolonged activation of innate immune pathways by a polyvalent STING agonist. Nature biomedical engineering 2021, 5, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Shih AY; Damm-Ganamet KL; Mirzadegan T Dynamic structural differences between human and mouse STING lead to differing sensitivity to DMXAA. Biophys. J 2018, 114, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Carozza JA; Böhnert V; Nguyen KC; Skariah G; Shaw KE; Brown JA; Rafat M; Von Eyben R; Graves EE; Glenn JS et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nat. Cancer 2020, 1, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Ritchie C; Cordova AF; Hess GT; Bassik MC; Li L SLC19A1 is an importer of the immunotransmitter cGAMP. Mol. Cell 2019, 75, 372–381 e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Zhou C; Chen X; Planells-Cases R; Chu J; Wang L; Cao L; Li Z; Lopez-Cayuqueo KI; Xie Y; Ye S et al. Transfer of cGAMP into bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral immunity. Immunity 2020, 52, 767–781.e766. [DOI] [PubMed] [Google Scholar]
- (169).Lahey LJ; Mardjuki RE; Wen X; Hess GT; Ritchie C; Carozza JA; Bohnert V; Maduke M; Bassik MC; Li L LRRC8A:C/E heteromeric channels are ubiquitous transporters of cGAMP. Mol. Cell 2020, 80, 578–591 e575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Ablasser A; Schmid-Burgk JL; Hemmerling I; Horvath GL; Schmidt T; Latz E; Hornung V Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Schadt L; Sparano C; Schweiger NA; Silina K; Cecconi V; Lucchiari G; Yagita H; Guggisberg E; Saba S; Nascakova Z et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep 2019, 29, 1236–1248 e1237. [DOI] [PubMed] [Google Scholar]
- (172).Pepin G; De Nardo D; Rootes CL; Ullah TR; Al-Asmari SS; Balka KR; Li HM; Quinn KM; Moghaddas F; Chappaz S et al. Connexin-dependent transfer of cGAMP to phagocytes modulates antiviral responses. mBio 2020, 11, e03187–03119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Wang J; Li P; Yu Y; Fu Y; Jiang H; Lu M; Sun Z; Jiang S; Lu L; Wu MX Pulmonary surfactant-biomimetic nanoparticles potentiate heterosubtypic influenza immunity. Science 2020, 367, eaau0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Luther J; Khan S; Gala MK; Kedrin D; Sridharan G; Goodman RP; Garber JJ; Masia R; Diagacomo E; Adams D et al. Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 11667–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Xu S; Ducroux A; Ponnurangam A; Vieyres G; Franz S; Musken M; Zillinger T; Malassa A; Ewald E; Hornung V et al. cGAS-mediated innate immunity spreads intercellularly through HIV-1 Env-induced membrane fusion sites. Cell Host Microbe 2016, 20, 443–457. [DOI] [PubMed] [Google Scholar]
- (176).Ahn J; Xia T; Rabasa Capote A; Betancourt D; Barber GN Extrinsic phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell 2018, 33, 862–873 e865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Naser Al Deen N; Abouhaidar M; Talhouk R Connexin43 as a tumor suppressor: Proposed Connexin43 mRNA-circularRNAs-microRNAs axis towards prevention and early detection in breast cancer. Front. Med. (Lausanne) 2019, 6, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Sirnes S; Bruun J; Kolberg M; Kjenseth A; Lind GE; Svindland A; Brech A; Nesbakken A; Lothe RA; Leithe E et al. Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int. J. Cancer 2012, 131, 570–581. [DOI] [PubMed] [Google Scholar]
- (179).Bonacquisti EE; Nguyen J Connexin 43 (Cx43) in cancer: Implications for therapeutic approaches via gap junctions. Cancer Lett 2019, 442, 439–444. [DOI] [PubMed] [Google Scholar]
- (180).Zhou Y; Fei M; Zhang G; Liang WC; Lin W; Wu Y; Piskol R; Ridgway J; Mcnamara E; Huang H et al. Blockade of the phagocytic receptor MerTK on tumor-associated macrophages enhances P2×7R-dependent STING activation by tumor-derived cGAMP. Immunity 2020, 52, 357–373 e359. [DOI] [PubMed] [Google Scholar]
- (181).Luteijn RD; Zaver SA; Gowen BG; Wyman SK; Garelis NE; Onia L; Mcwhirter SM; Katibah GE; Corn JE; Woodward JJ et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 2019, 573, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Gentili M; Kowal J; Tkach M; Satoh T; Lahaye X; Conrad C; Boyron M; Lombard B; Durand S; Kroemer G et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 2015, 349, 1232–1236. [DOI] [PubMed] [Google Scholar]
- (183).Bridgeman A; Maelfait J; Davenne T; Partridge T; Peng Y; Mayer A; Dong T; Kaever V; Borrow P; Rehwinkel J Viruses transfer the antiviral second messenger cGAMP between cells. Science 2015, 349, 1228–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Kalamvoki M; Du T; Roizman B Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E4991–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Torralba D; Baixauli F; Villarroya-Beltri C; Fernandez-Delgado I; Latorre-Pellicer A; Acin-Perez R; Martin-Cofreces NB; Jaso-Tamame AL; Iborra S; Jorge I et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun 2018, 9, 2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Kato Y; Park J; Takamatsu H; Konaka H; Aoki W; Aburaya S; Ueda M; Nishide M; Koyama S; Hayama Y et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis 2018, 77, 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Cordova AF; Ritchie C; Bohnert V; Li L Human SLC46A2 Is the dominant cGAMP importer in extracellular cGAMP-sensing macrophages and monocytes. ACS Cent. Sci 2021, 7, 1073–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Oyamada M; Oyamada Y; Takamatsu T Regulation of connexin expression. Biochimica et biophysica acta 2005, 1719, 6–23. [DOI] [PubMed] [Google Scholar]
- (189).Strange K; Yamada T; Denton JSA 30-year journey from volume-regulated anion currents to molecular structure of the LRRC8 channel. J. Gen. Physiol 2019, 151, 100–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Chen L; Konig B; Liu T; Pervaiz S; Razzaque YS; Stauber T More than just a pressure relief valve: physiological roles of volume-regulated LRRC8 anion channels. Biol. Chem 2019, 400, 1481–1496. [DOI] [PubMed] [Google Scholar]
- (191).Jentsch TJ VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol 2016, 17, 293–307. [DOI] [PubMed] [Google Scholar]
- (192).Osei-Owusu J; Yang J; Vitery MDC; Qiu Z Molecular biology and physiology of volume-regulated anion channel (VRAC). Curr. Top. Membr 2018, 81, 177–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Laird DW Life cycle of connexins in health and disease. Biochem J 2006, 394, 527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Kar R; Batra N; Riquelme MA; Jiang JX Biological role of connexin intercellular channels and hemichannels. Arch Biochem Biophys 2012, 524, 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Hubbell JA; Swartz MA Trojan horses for immunotherapy. Nat. Nanotechnol 2019, 14, 196–197. [DOI] [PubMed] [Google Scholar]
- (196).Shae D; Becker KW; Christov P; Yun DS; Lytton-Jean AKR; Sevimli S; Ascano M; Kelley M; Johnson DB; Balko JM et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol 2019, 14, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Li L; Yin Q; Kuss P; Maliga Z; Millan JL; Wu H; Mitchison TJ Hydrolysis of 2’3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol 2014, 10, 1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Namasivayam V; Lee SY; Muller CE The promiscuous ectonucleotidase NPP1: molecular insights into substrate binding and hydrolysis. Biochim. Biophys. Acta Gen. Subj 2017, 1861, 603–614. [DOI] [PubMed] [Google Scholar]
- (199).Lau WM; Doucet M; Stadel R; Huang D; Weber KL; Kominsky SL Enpp1: a potential facilitator of breast cancer bone metastasis. PLoS One 2013, 8, e66752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Li J; Duran MA; Dhanota N; Chatila WK; Bettigole SE; Kwon J; Sriram RK; Humphries MP; Salto-Tellez M; James JA et al. Metastasis and immune evasion from extracellular cGAMP hydrolysis. Cancer Discov 2020, 11, 1212–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Kawaguchi M; Han X; Hisada T; Nishikawa S; Kano K; Ieda N; Aoki J; Toyama T; Nakagawa H Development of an ENPP1 fluorescence probe for inhibitor screening, cellular imaging, and prognostic assessment of malignant breast cancer. J. Med. Chem 2019, 62, 9254–9269. [DOI] [PubMed] [Google Scholar]
- (202).Yan H; Wang X; Kuolee R; Chen W Synthesis and immunostimulatory properties of the phosphorothioate analogues of cdiGMP. Bioorg. Med. Chem. Lett 2008, 18, 5631–5634. [DOI] [PubMed] [Google Scholar]
- (203).Eckstein F Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther 2014, 24, 374–387. [DOI] [PubMed] [Google Scholar]
- (204).Liu H; Moura-Alves P; Pei G; Mollenkopf HJ; Hurwitz R; Wu X; Wang F; Liu S; Ma M; Fei Y et al. cGAS facilitates sensing of extracellular cyclic dinucleotides to activate innate immunity. EMBO Rep 2019, 20, e46293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Decout A; Katz JD; Venkatraman S; Ablasser A The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol 2021, 21, 548–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Ahn J; Xia T; Konno H; Konno K; Ruiz P; Barber GN Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun 2014, 5, 5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Kwon J; Bakhoum SF The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov 2020, 10, 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Zheng J; Mo J; Zhu T; Zhuo W; Yi Y; Hu S; Yin J; Zhang W; Zhou H; Liu Z Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer 2020, 19, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Chen DS; Mellman I Oncology meets immunology: the cancer-immunity cycle. Immunity 2013, 39, 1–10. [DOI] [PubMed] [Google Scholar]
- (210).Lian Y; Duffy KJ; Yang J STING activation and its application in immuno-oncology. Curr. Top. Med. Chem 2019, 19, 2205–2227. [DOI] [PubMed] [Google Scholar]
- (211).Khoo LT; Chen LY Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep 2018, 19, e46935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Woo SR; Corrales L; Gajewski TF The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol 2015, 36, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Li W; Lu L; Lu J; Wang X; Yang C; Jin J; Wu L; Hong X; Li F; Cao D et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci. Transl. Med 2020, 12, eaay9013. [DOI] [PubMed] [Google Scholar]
- (214).Wang H; Hu S; Chen X; Shi H; Chen C; Sun L; Chen ZJ cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Liu X; Pu Y; Cron K; Deng L; Kline J; Frazier WA; Xu H; Peng H; Fu YX; Xu MM CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med 2015, 21, 1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Konno H; Yamauchi S; Berglund A; Putney RM; Mule JJ; Barber GN Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018, 37, 2037–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Bakhoum SF; Cantley LC The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018, 174, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Lemos H; Huang L; Mcgaha TL; Mellor AL Cytosolic DNA sensing via the stimulator of interferon genes adaptor: Yin and Yang of immune responses to DNA. Eur. J. Immunol 2014, 44, 2847–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Lemos H; Mohamed E; Huang L; Ou R; Pacholczyk G; Arbab AS; Munn D; Mellor AL STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res 2016, 76, 2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Corrales L; Woo S-R; Gajewski TF Extremely potent immunotherapeutic activity of a STING agonist in the B16 melanoma model in vivo. J. Immunother. Cancer 2013, 1, O15. [Google Scholar]
- (221).Qin XQ; Tao N; Dergay A; Moy P; Fawell S; Davis A; Wilson JM; Barsoum J Interferon-beta gene therapy inhibits tumor formation and causes regression of established tumors in immune-deficient mice. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 14411–14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Kim HS; Lee MS STAT1 as a key modulator of cell death. Cell. Signal 2007, 19, 454–465. [DOI] [PubMed] [Google Scholar]
- (223).Ryuke Y; Mizuno M; Natsume A; Suzuki O; Nobayashi M; Kageshita T; Matsumoto K; Saida T; Yoshida J Growth inhibition of subcutaneous mouse melanoma and induction of natural killer cells by liposome-mediated interferon-beta gene therapy. Melanoma Res 2003, 13, 349–356. [DOI] [PubMed] [Google Scholar]
- (224).Spaapen RM; Leung MY; Fuertes MB; Kline JP; Zhang L; Zheng Y; Fu YX; Luo X; Cohen KS; Gajewski TF Therapeutic activity of high-dose intratumoral IFN-beta requires direct effect on the tumor vasculature. J. Immunol 2014, 193, 4254–4260. [DOI] [PubMed] [Google Scholar]
- (225).Indraccolo S Interferon-alpha as angiogenesis inhibitor: learning from tumor models. Autoimmunity 2010, 43, 244–247. [DOI] [PubMed] [Google Scholar]
- (226).Luft T; Pang KC; Thomas E; Hertzog P; Hart DN; Trapani J; Cebon J Type I IFNs enhance the terminal differentiation of dendritic cells. J. Immunol 1998, 161, 1947–1953. [PubMed] [Google Scholar]
- (227).Paquette RL; Hsu NC; Kiertscher SM; Park AN; Tran L; Roth MD; Glaspy J Interferon-alpha and granulocyte-macrophage colony-stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells. J. Leukoc. Biol 1998, 64, 358–367. [DOI] [PubMed] [Google Scholar]
- (228).Radvanyi LG; Banerjee A; Weir M; Messner H Low levels of interferon-alpha induce CD86 (B7.2) expression and accelerates dendritic cell maturation from human peripheral blood mononuclear cells. Scand. J. Immunol 1999, 50, 499–509. [DOI] [PubMed] [Google Scholar]
- (229).Yang X; Zhang X; Fu ML; Weichselbaum RR; Gajewski TF; Guo Y; Fu YX Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 2014, 25, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Fuertes MB; Kacha AK; Kline J; Woo SR; Kranz DM; Murphy KM; Gajewski TF Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med 2011, 208, 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Qin XQ; Beckham C; Brown JL; Lukashev M; Barsoum J Human and mouse IFN-beta gene therapy exhibits different anti-tumor mechanisms in mouse models. Mol. Ther 2001, 4, 356–364. [DOI] [PubMed] [Google Scholar]
- (232).Gidlund M; Orn A; Wigzell H; Senik A; Gresser I Enhanced NK cell activity in mice injected with interferon and interferon inducers. Nature 1978, 273, 759–761. [DOI] [PubMed] [Google Scholar]
- (233).Snell LM; Mcgaha TL; Brooks DG Type I interferon in chronic virus infection and cancer. Trends Immunol 2017, 38, 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Sivick KE; Desbien AL; Glickman LH; Reiner GL; Corrales L; Surh NH; Hudson TE; Vu UT; Francica BJ; Banda T et al. Magnitude of therapeutic STING activation determines CD8(+) T cell-mediated anti-tumor immunity. Cell Rep 2018, 25, 3074–3085 e3075. [DOI] [PubMed] [Google Scholar]
- (235).Wilson NS; Behrens GM; Lundie RJ; Smith CM; Waithman J; Young L; Forehan SP; Mount A; Steptoe RJ; Shortman KD et al. Systemic activation of dendritic cells by toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol 2006, 7, 165–172. [DOI] [PubMed] [Google Scholar]
- (236).Tzeng A; Kauke MJ; Zhu EF; Moynihan KD; Opel CF; Yang NJ; Mehta N; Kelly RL; Szeto GL; Overwijk WW et al. Temporally programmed CD8alpha(+) DC activation enhances combination cancer immunotherapy. Cell Rep 2016, 17, 2503–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Borden EC; Parkinson D A perspective on the clinical effectiveness and tolerance of interferon-alpha. Semin. Oncol 1998, 25, 3–8. [PubMed] [Google Scholar]
- (238).Weber JS; Yang JC; Atkins MB; Disis ML Toxicities of immunotherapy for the practitioner. J. Clin. Oncol 2015, 33, 2092–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Wu J; Dobbs N; Yang K; Yan N Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity 2020, 53, 115–126 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Hou Y; Liang H; Rao E; Zheng W; Huang X; Deng L; Zhang Y; Yu X; Xu M; Mauceri H et al. Non-canonical NF-kappaB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity 2018, 49, 490–503 e494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Zhu Y; An X; Zhang X; Qiao Y; Zheng T; Li X STING: a master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Coffelt SB; De Visser KE Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol 2015, 36, 198–216. [DOI] [PubMed] [Google Scholar]
- (243).Skrnjug I; Guzman CA; Rueckert C Cyclic GMP-AMP displays mucosal adjuvant activity in mice. PLoS One 2014, 9, e110150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Gutjahr A; Papagno L; Nicoli F; Kanuma T; Kuse N; Cabral-Piccin MP; Rochereau N; Gostick E; Lioux T; Perouzel E et al. The STING ligand cGAMP potentiates the efficacy of vaccine-induced CD8+ T cells. JCI Insight 2019, 4, e125107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Corrales L; Matson V; Flood B; Spranger S; Gajewski TF Innate immune signaling and regulation in cancer immunotherapy. Cell Res 2017, 27, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Spranger S; Dai D; Horton B; Gajewski TF Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017, 31, 711–723 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Diamond MS; Kinder M; Matsushita H; Mashayekhi M; Dunn GP; Archambault JM; Lee H; Arthur CD; White JM; Kalinke U et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med 2011, 208, 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Worbs T; Hammerschmidt SI; Forster R Dendritic cell migration in health and disease. Nat. Rev. Immunol 2017, 17, 30–48. [DOI] [PubMed] [Google Scholar]
- (249).Hampton HR; Chtanova T Lymphatic migration of immune cells. Front. Immunol 2019, 10, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Thompson ED; Enriquez HL; Fu YX; Engelhard VH Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J. Exp. Med 2010, 207, 1791–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Ma Y; Adjemian S; Mattarollo SR; Yamazaki T; Aymeric L; Yang H; Portela Catani JP; Hannani D; Duret H; Steegh K et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [DOI] [PubMed] [Google Scholar]
- (252).Chelvanambi M; Fecek RJ; Taylor JL; Storkus WJ STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J. Immunother. Cancer 2021, 9, e001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Wang N; Liang H; Zen K Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol 2014, 5, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Cheng N; Watkins-Schulz R; Junkins RD; David CN; Johnson BM; Montgomery SA; Peine KJ; Darr DB; Yuan H; Mckinnon KP et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1–insensitive models of triple-negative breast cancer. JCI Insight 2018, 3, e120638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Downey CM; Aghaei M; Schwendener RA; Jirik FR DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2’3’-cGAMP, induces M2 macrophage repolarization. PLoS One 2014, 9, e99988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Pan XQ The mechanism of the anticancer function of M1 macrophages and their use in the clinic. Chin. J. Cancer 2012, 31, 557–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Chen Y; Song Y; Du W; Gong L; Chang H; Zou Z Tumor-associated macrophages: an accomplice in solid tumor progression. J. Biomed. Sci 2019, 26, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Wong P; Pamer EG CD8 T cell responses to infectious pathogens. Annu. Rev. Immunol 2003, 21, 29–70. [DOI] [PubMed] [Google Scholar]
- (259).Zhu J; Yamane H; Paul WE Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol 2010, 28, 445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Wang J; Li P; Wu MX Natural STING agonist as an “ideal” adjuvant for cutaneous vaccination. J. Invest. Dermatol 2016, 136, 2183–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Shakya AK; Lee CH; Uddin MJ; Gill HS Assessment of Th1/Th2 bias of STING agonists coated on microneedles for possible use in skin allergen immunotherapy. Mol. Pharm 2018, 15, 5437–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Heusinkveld M; De Vos Van Steenwijk PJ; Goedemans R; Ramwadhdoebe TH; Gorter A; Welters MJ; Van Hall T; Van Der Burg SH M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J. Immunol 2011, 187, 1157–1165. [DOI] [PubMed] [Google Scholar]
- (263).Eisel D; Das K; Dickes E; Konig R; Osen W; Eichmuller SB Cognate interaction with CD4(+) T cells instructs tumor-associated macrophages to acquire M1-like phenotype. Front. Immunol 2019, 10, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Cerboni S; Jeremiah N; Gentili M; Gehrmann U; Conrad C; Stolzenberg MC; Picard C; Neven B; Fischer A; Amigorena S et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med 2017, 214, 1769–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Larkin B; Ilyukha V; Sorokin M; Buzdin A; Vannier E; Poltorak A Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J. Immunol 2017, 199, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Hunter MC; Teijeira A; Halin C T cell trafficking through lymphatic vessels. Front. Immunol 2016, 7, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Pham TH; Okada T; Matloubian M; Lo CG; Cyster JG S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity 2008, 28, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Schwab SR; Pereira JP; Matloubian M; Xu Y; Huang Y; Cyster JG Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [DOI] [PubMed] [Google Scholar]
- (269).Lo CG; Xu Y; Proia RL; Cyster JG Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J. Exp. Med 2005, 201, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Harlin H; Meng Y; Peterson AC; Zha Y; Tretiakova M; Slingluff C; Mckee M; Gajewski TF Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009, 69, 3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Pak-Wittel MA; Yang L; Sojka DK; Rivenbark JG; Yokoyama WM Interferon-gamma mediates chemokine-dependent recruitment of natural killer cells during viral infection. Proc. Natl. Acad. Sci. U. S. A 2013, 110, E50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Barry KC; Hsu J; Broz ML; Cueto FJ; Binnewies M; Combes AJ; Nelson AE; Loo K; Kumar R; Rosenblum MD et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med 2018, 24, 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Munn LL; Jain RK Vascular regulation of antitumor immunity. Science 2019, 365, 544–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Yang H; Lee WS; Kong SJ; Kim CG; Kim JH; Chang SK; Kim S; Kim G; Chon HJ; Kim C STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Invest 2019, 129, 4350–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Tang CH; Zundell JA; Ranatunga S; Lin C; Nefedova Y; Del Valle JR; Hu CC Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res 2016, 76, 2137–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Takashima K; Takeda Y; Oshiumi H; Shime H; Okabe M; Ikawa M; Matsumoto M; Seya T STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem. Biophys. Res. Commun 2016, 478, 1764–1771. [DOI] [PubMed] [Google Scholar]
- (277).Grabosch S; Bulatovic M; Zeng F; Ma T; Zhang L; Ross M; Brozick J; Fang Y; Tseng G; Kim E et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Roemer MG; Advani RH; Redd RA; Pinkus GS; Natkunam Y; Ligon AH; Connelly CF; Pak CJ; Carey CD; Daadi SE et al. Classical hodgkin lymphoma with reduced beta2M/MHC class I expression is associated with inferior outcome independent of 9p24.1 status. Cancer Immunol. Res 2016, 4, 910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Garrido F; Aptsiauri N; Doorduijn EM; Garcia Lora AM; Van Hall T The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol 2016, 39, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Alexandrov LB; Stratton MR Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev 2014, 24, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Nicolai CJ; Wolf N; Chang IC; Kirn G; Marcus A; Ndubaku CO; Mcwhirter SM; Raulet DH NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci. Immunol 2020, 5, eaaz2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Lam AR; Bert NL; Ho SS; Shen YJ; Tang LF; Xiong GM; Croxford JL; Koo CX; Ishii KJ; Akira S et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res 2014, 74, 2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Parkes EE; Walker SM; Taggart LE; Mccabe N; Knight LA; Wilkinson R; Mccloskey KD; Buckley NE; Savage KI; Salto-Tellez M et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J. Natl. Cancer Inst 2017, 109, djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Pepin G; Nejad C; Ferrand J; Thomas BJ; Stunden HJ; Sanij E; Foo CH; Stewart CR; Cain JE; Bardin PG et al. Topoisomerase 1 inhibition promotes cyclic GMP-AMP synthase-dependent antiviral responses. mBio 2017, 8, e01611–01617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Luthra P; Aguirre S; Yen BC; Pietzsch CA; Sanchez-Aparicio MT; Tigabu B; Morlock LK; Garcia-Sastre A; Leung DW; Williams NS et al. Topoisomerase II inhibitors induce DNA damage-dependent interferon responses circumventing ebola virus immune evasion. mBio 2017, 8, e00368–00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Lohard S; Bourgeois N; Maillet L; Gautier F; Fetiveau A; Lasla H; Nguyen F; Vuillier C; Dumont A; Moreau-Aubry A et al. STING-dependent paracriny shapes apoptotic priming of breast tumors in response to anti-mitotic treatment. Nat. Commun 2020, 11, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Wilkinson RD; Johnston DI; Parkes EE; Mccabe N; Kennedy RD Abstract 3787: Exploring the effect of chemotherapies on STING-dependent cytokine release. Cancer Res 2018, 78, 3787. [Google Scholar]
- (288).Maekawa H; Inoue T; Ouchi H; Jao TM; Inoue R; Nishi H; Fujii R; Ishidate F; Tanaka T; Tanaka Y et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep 2019, 29, 1261–1273 e1266. [DOI] [PubMed] [Google Scholar]
- (289).Unterholzner L; Dunphy G cGAS-independent STING activation in response to DNA damage. Mol. Cell. Oncol 2019, 6, 1558682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Wang Z; Chen J; Hu J; Zhang H; Xu F; He W; Wang X; Li M; Lu W; Zeng G et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J. Clin. Invest 2019, 129, 4850–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Deng L; Liang H; Xu M; Yang X; Burnette B; Arina A; Li XD; Mauceri H; Beckett M; Darga T et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Chabanon RM; Muirhead G; Krastev DB; Adam J; Morel D; Garrido M; Lamb A; Henon C; Dorvault N; Rouanne M et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Invest 2019, 129, 1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Ding L; Kim HJ; Wang Q; Kearns M; Jiang T; Ohlson CE; Li BB; Xie S; Liu JF; Stover EH et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep 2018, 25, 2972–2980 e2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Pantelidou C; Sonzogni O; De Oliveria Taveira M; Mehta AK; Kothari A; Wang D; Visal T; Li MK; Pinto J; Castrillon JA et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov 2019, 9, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Reislander T; Lombardi EP; Groelly FJ; Miar A; Porru M; Di Vito S; Wright B; Lockstone H; Biroccio A; Harris A et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun 2019, 10, 3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Shen J; Zhao W; Ju Z; Wang L; Peng Y; Labrie M; Yap TA; Mills GB; Peng G PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res 2019, 79, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Dillon MT; Bergerhoff KF; Pedersen M; Whittock H; Crespo-Rodriguez E; Patin EC; Pearson A; Smith HG; Paget JTE; Patel RR et al. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin. Cancer Res 2019, 25, 3392–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Flynn PJ; Koch PD; Mitchison TJ Chromatin bridges, not micronuclei, activate cGAS after drug-induced mitotic errors in human cells. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2103585118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Dan H; Zhang S; Zhou Y; Guan Q DNA methyltransferase inhibitors: catalysts for antitumour immune responses. Onco Targets Ther 2019, 12, 10903–10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Gravina GL; Festuccia C; Marampon F; Popov VM; Pestell RG; Zani BM; Tombolini V Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol. Cancer 2010, 9, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Falahat R; Berglund A; Putney RM; Perez-Villarroel P; Aoyama S; Pilon-Thomas S; Barber GN; Mule JJ Epigenetic reprogramming of tumor cell-intrinsic STING function sculpts antigenicity and T cell recognition of melanoma. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2013598118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Kitai Y; Kawasaki T; Sueyoshi T; Kobiyama K; Ishii KJ; Zou J; Akira S; Matsuda T; Kawai T DNA-containing exosomes derived from cancer cells treated with topotecan activate a STING-dependent pathway and reinforce antitumor immunity. J. Immunol 2017, 198, 1649–1659. [DOI] [PubMed] [Google Scholar]
- (303).Sen T; Rodriguez BL; Chen L; Corte CMD; Morikawa N; Fujimoto J; Cristea S; Nguyen T; Diao L; Li L et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 2019, 9, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Baird JR; Friedman D; Cottam B; Dubensky TW Jr.; Kanne DB; Bambina S; Bahjat K; Crittenden MR; Gough MJ Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res 2016, 76, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Gui X; Yang H; Li T; Tan X; Shi P; Li M; Du F; Chen ZJ Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Michaud M; Martins I; Sukkurwala AQ; Adjemian S; Ma Y; Pellegatti P; Shen S; Kepp O; Scoazec M; Mignot G et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [DOI] [PubMed] [Google Scholar]
- (307).Landman SL; Ressing ME; Van Der Veen AG Balancing STING in antimicrobial defense and autoinflammation. Cytokine Growth Factor Rev 2020, 55, 1–14. [DOI] [PubMed] [Google Scholar]
- (308).Zhao J; Ma S; Xu Y; Si X; Yao H; Huang Z; Zhang Y; Yu H; Tang Z; Song W et al. In situ activation of STING pathway with polymeric SN38 for cancer chemoimmunotherapy. Biomaterials 2021, 268, 120542. [DOI] [PubMed] [Google Scholar]
- (309).Smith GP; Calveley SB; Smith MJ; Baguley BC Flavone acetic acid (NSC 347512) induces haemorrhagic necrosis of mouse colon 26 and 38 tumours. Eur. J. Cancer Clin. Oncol 1987, 23, 1209–1211. [DOI] [PubMed] [Google Scholar]
- (310).Bibby MC; Phillips RM; Double JA; Pratesi G Anti-tumour activity of flavone acetic acid (NSC 347512) in mice--influence of immune status. Br. J. Cancer 1991, 63, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Kerr DJ; Kaye SB Flavone acetic acid--preclinical and clinical activity. Eur. J. Cancer Clin. Oncol 1989, 25, 1271–1272. [DOI] [PubMed] [Google Scholar]
- (312).Rewcastle GW; Atwell GJ; Li ZA; Baguley BC; Denny WA Potential antitumor agents. 61. Structure-activity relationships for in vivo colon 38 activity among disubstituted 9-oxo-9H-xanthene-4-acetic acids. J. Med. Chem 1991, 34, 217–222. [DOI] [PubMed] [Google Scholar]
- (313).Baguley BC; Ching LM DMXAA: an antivascular agent with multiple host responses. Int. J. Radiat. Oncol. Biol. Phys 2002, 54, 1503–1511. [DOI] [PubMed] [Google Scholar]
- (314).Woon ST; Reddy CB; Drummond CJ; Schooltink MA; Baguley BC; Kieda C; Ching LM A comparison of the ability of DMXAA and xanthenone analogues to activate NF-kappaB in murine and human cell lines. Oncol. Res 2005, 15, 351–364. [DOI] [PubMed] [Google Scholar]
- (315).Philpott M; Baguley BC; Ching LM Induction of tumour necrosis factor-alpha by single and repeated doses of the antitumour agent 5,6-dimethylxanthenone-4-acetic acid. Cancer Chemother. Pharmacol 1995, 36, 143–148. [DOI] [PubMed] [Google Scholar]
- (316).Kim S; Li L; Maliga Z; Yin Q; Wu H; Mitchison TJ Anticancer flavonoids are mouse-selective STING agonists. ACS Chem. Biol 2013, 8, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Mckeage MJ; Reck M; Jameson MB; Rosenthal MA; Gibbs D; Mainwaring PN; Freitag L; Sullivan R; Von Pawel J Phase II study of ASA404 (vadimezan, 5,6-dimethylxanthenone-4-acetic acid/DMXAA) 1800mg/m(2) combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Lung cancer (Amsterdam, Netherlands) 2009, 65, 192–197. [DOI] [PubMed] [Google Scholar]
- (318).Lara PN Jr.; Douillard JY; Nakagawa K; Von Pawel J; Mckeage MJ; Albert I; Losonczy G; Reck M; Heo DS; Fan X et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J. Clin. Oncol 2011, 29, 2965–2971. [DOI] [PubMed] [Google Scholar]
- (319).Conlon J; Burdette DL; Sharma S; Bhat N; Thompson M; Jiang Z; Rathinam VA; Monks B; Jin T; Xiao TS et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J. Immunol 2013, 190, 5216–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Gao P; Zillinger T; Wang W; Ascano M; Dai P; Hartmann G; Tuschl T; Deng L; Barchet W; Patel DJ Binding-pocket and lid-region substitutions render human STING sensitive to the species-specific drug DMXAA. Cell Rep 2014, 8, 1668–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Cavlar T; Deimling T; Ablasser A; Hopfner KP; Hornung V Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J 2013, 32, 1440–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Xiao TS; Fitzgerald KA The cGAS-STING pathway for DNA sensing. Mol. Cell 2013, 51, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Ablasser A; Goldeck M; Cavlar T; Deimling T; Witte G; Rohl I; Hopfner KP; Ludwig J; Hornung V cGAS produces a 2’−5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Shi H; Wu J; Chen ZJ; Chen C Molecular basis for the specific recognition of the metazoan cyclic GMP-AMP by the innate immune adaptor protein STING. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 8947–8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Slavik KM; Morehouse BR; Ragucci AE; Zhou W; Ai X; Chen Y; Li L; Wei Z; Bahre H; Konig M et al. cGAS-like receptors sense RNA and control 3’2’-cGAMP signalling in drosophila. Nature 2021, 597, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Holleufer A; Winther KG; Gad HH; Ai X; Chen Y; Li L; Wei Z; Deng H; Liu J; Frederiksen NA et al. Two cGAS-like receptors induce antiviral immunity in drosophila. Nature 2021, 597, 114–118. [DOI] [PubMed] [Google Scholar]
- (327).Zandberg DP; Ferris R; Laux D; Mehra R; Nabell L; Kaczmar J; Gibson MK; Kim YJ; Neupane P; Bauman J et al. 71P A phase II study of ADU-S100 in combination with pembrolizumab in adult patients with PD-L1+ recurrent or metastatic HNSCC: Preliminary safety, efficacy and PK/PD results. Ann. Oncol 2020, 31, S1446–S1447. [Google Scholar]
- (328).Meric-Bernstam F; Sandhu SK; Hamid O; Spreafico A; Kasper S; Dummer R; Shimizu T; Steeghs N; Lewis N; Talluto CC et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol 2019, 37, 2507–2507. [Google Scholar]
- (329).Kim DS; Endo A; Fang FG; Huang KC; Bao X; Choi HW; Majumder U; Shen YY; Mathieu S; Zhu X et al. E7766, a macrocycle-bridged stimulator of interferon genes (STING) agonist with potent pan-genotypic activity. ChemMedChem 2021, 16, 1740–1743. [DOI] [PubMed] [Google Scholar]
- (330).Sali TM; Pryke KM; Abraham J; Liu A; Archer I; Broeckel R; Staverosky JA; Smith JL; Al-Shammari A; Amsler L et al. Characterization of a novel human-specific STING agonist that elicits antiviral activity against emerging alphaviruses. PLoS Pathog 2015, 11, e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Couderc T; Chrétien F; Schilte C; Disson O; Brigitte M; Guivel-Benhassine F; Touret Y; Barau G; Cayet N; Schuffenecker I et al. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog 2008, 4, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Lukaszewski RA; Brooks TJ Pegylated alpha interferon is an effective treatment for virulent venezuelan equine encephalitis virus and has profound effects on the host immune response to infection. J. Virol 2000, 74, 5006–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Liu B; Tang L; Zhang X; Ma J; Sehgal M; Cheng J; Zhang X; Zhou Y; Du Y; Kulp J et al. A cell-based high throughput screening assay for the discovery of cGAS-STING pathway agonists. Antiviral Res 2017, 147, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (334).Zhang Y; Sun Z; Pei J; Luo Q; Zeng X; Li Q; Yang Z; Quan J Identification of α-Mangostin as an agonist of human STING. ChemMedChem 2018, 13, 2057–2064. [DOI] [PubMed] [Google Scholar]
- (335).Banerjee M; Middya S; Basu S; Ghosh R; Pryde D, Small molecule modulators of human STING. U.S Patent US-2020138827-A1, 2017.
- (336).Banerjee M; Middya S; Basu S; Ghosh R; Pryde D; Yadav D; Shrivastava R; Surya A. G10 is a direct activator of human STING3. PlosOne 2020, 15 (9), e023774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Banerjee M; Middya S; Basu S; Ghosh R; Pryde D; Yadav D; Shrivastava R; Surya A. Small molecule modulators of human STING, US Patent 20200147083, 2020.
- (338).Altman MD; Cash BD; Chang W; Cumming JN; Haidle AM; Henderson TJ; Jewell JP; Larsen MA; Liang R; Lim J, 2019.
- (339).Cemerski S; Cumming J; Kopinja J; Perera S; Trotter BW; Tse AN-C, 2019.
- (340).Pan BS; Perera SA; Piesvaux JA; Presland JP; Schroeder GK; Cumming JN; Trotter BW; Altman MD; Buevich AV; Cash B et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 2020, 369, eaba6098. [DOI] [PubMed] [Google Scholar]
- (341).Vaupel P; Kallinowski F; Okunieff P Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 1989, 49, 6449–6465. [PubMed] [Google Scholar]
- (342).Webb BA; Chimenti M; Jacobson MP; Barber DL Dysregulated pH: a perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [DOI] [PubMed] [Google Scholar]
- (343).Ding C; Song Z; Shen A; Chen T; Zhang A Small molecules targeting the innate immune cGAS‒STING‒TBK1 signaling pathway. Acta Pharm. Sin. B 2020, 10, 2272–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Vanpouille-Box C; Hoffmann JA; Galluzzi L Pharmacological modulation of nucleic acid sensors - therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov 2019, 18, 845–867. [DOI] [PubMed] [Google Scholar]
- (345).Hall J; Ralph EC; Shanker S; Wang H; Byrnes LJ; Horst R; Wong J; Brault A; Dumlao D; Smith JF et al. The catalytic mechanism of cyclic GMP-AMP synthase (cGAS) and implications for innate immunity and inhibition. Protein Sci 2017, 26, 2367–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Hyman AA; Simons K Cell biology. Beyond oil and water--phase transitions in cells. Science 2012, 337, 1047–1049. [DOI] [PubMed] [Google Scholar]
- (347).Vincent J; Adura C; Gao P; Luz A; Lama L; Asano Y; Okamoto R; Imaeda T; Aida J; Rothamel K et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun 2017, 8, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Lama L; Adura C; Xie W; Tomita D; Kamei T; Kuryavyi V; Gogakos T; Steinberg JI; Miller M; Ramos-Espiritu L et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat. Commun 2019, 10, 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Zhao Z; Ma Z; Wang B; Guan Y; Su XD; Jiang Z Mn(2+) directly activates cGAS and structural analysis suggests Mn(2+) induces a noncanonical catalytic synthesis of 2’3’-cGAMP. Cell Rep 2020, 32, 108053. [DOI] [PubMed] [Google Scholar]
- (350).Lv M; Chen M; Zhang R; Zhang W; Wang C; Zhang Y; Wei X; Guan Y; Liu J; Feng K et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res 2020, 30, 966–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Huang L; Li L; Lemos H; Chandler PR; Pacholczyk G; Baban B; Barber GN; Hayakawa Y; Mcgaha TL; Ravishankar B et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J. Immunol 2013, 191, 3509–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Zheng M; Xie L; Liang Y; Wu S; Xu H; Zhang Y; Liu H; Lin D; Han J; Lu K Recognition of cytosolic DNA attenuates glucose metabolism and induces AMPK mediated energy stress response. Int. J. Biol. Sci 2015, 11, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Laursen MF; Christensen E; Degn LLT; Jonsson K; Jakobsen MR; Agger R; Kofod-Olsen E CD11c-targeted delivery of DNA to dendritic cells leads to cGAS- and STING-dependent maturation. J. Immunother 2018, 41, 9–18. [DOI] [PubMed] [Google Scholar]
- (354).Zhou M; Wang X; Lin S; Cheng Y; Zhao S; Lin J; Fang Z; Lou Z; Qin L; Wei H Multifunctional STING-activating Mn3 O4 @Au-dsDNA/DOX nanoparticle for antitumor immunotherapy. Adv. Healthc. Mater 2020, 9, e2000064. [DOI] [PubMed] [Google Scholar]
- (355).Garland KM; Rosch JC; Carson CS; Wang-Bishop L; Hanna A; Sevimli S; Van Kaer C; Balko JM; Ascano M; Wilson JT Pharmacological activation of cGAS for cancer immunotherapy. Front. Immunol 2021, 12, 753472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Ribas A; Medina T; Kirkwood JM; Zakharia Y; Gonzalez R; Davar D; Chmielowski B; Campbell KM; Bao R; Kelley H et al. Overcoming PD-1 blockade resistance with CpG-A toll-like receptor 9 agonist vidutolimod in patients with metastatic melanoma. Cancer Discov 2021, Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Hornung V; Rothenfusser S; Britsch S; Krug A; Jahrsdorfer B; Giese T; Endres S; Hartmann G Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol 2002, 168, 4531–4537. [DOI] [PubMed] [Google Scholar]
- (358).Uhlen M; Fagerberg L; Hallstrom BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson A; Kampf C; Sjostedt E; Asplund A et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- (359).Uhlen M; Zhang C; Lee S; Sjostedt E; Fagerberg L; Bidkhori G; Benfeitas R; Arif M; Liu Z; Edfors F et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [DOI] [PubMed] [Google Scholar]
- (360).Verrier ER; Langevin C Cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS), a multifaceted platform of intracellular DNA sensing. Front. Immunol 2021, 12, 637399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Harrington KJ; Brody J; Ingham M; Strauss J; Cemerski S; Wang M; Tse A; Khilnani A; Marabelle A; Golan T Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol 2018, 29, viii712. [Google Scholar]
- (362).Koshy ST; Cheung AS; Gu L; Graveline AR; Mooney DJ Liposomal delivery enhances immune activation by STING agonists for cancer immunotherapy. Adv. Biosyst 2017, 1, 1600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Zhou J; Watt S; Wang J; Nakayama S; Sayre DA; Lam YF; Lee VT; Sintim HO Potent suppression of c-di-GMP synthesis via I-site allosteric inhibition of diguanylate cyclases with 2’-F-c-di-GMP. Bioorg. Med. Chem 2013, 21, 4396–4404. [DOI] [PubMed] [Google Scholar]
- (364).Lioux T; Mauny MA; Lamoureux A; Bascoul N; Hays M; Vernejoul F; Baudru AS; Boularan C; Lopes-Vicente J; Qushair G et al. Design, synthesis, and biological evaluation of novel cyclic adenosine-inosine monophosphate (cAIMP) analogs that activate stimulator of interferon genes (STING). J. Med. Chem 2016, 59, 10253–10267. [DOI] [PubMed] [Google Scholar]
- (365).Pimkova Polidarova M; Brehova P; Kaiser MM; Smola M; Dracinsky M; Smith J; Marek A; Dejmek M; Sala M; Gutten O et al. Synthesis and biological evaluation of phosphoester and phosphorothioate prodrugs of STING agonist 3’,3’-c-Di(2’F,2’dAMP). J. Med. Chem 2021, 64, 7596–7616. [DOI] [PubMed] [Google Scholar]
- (366).Vyskocil S; Cardin D; Ciavarri J; Conlon J; Cullis C; England D; Gershman R; Gigstad K; Gipson K; Gould A et al. Identification of novel carbocyclic pyrimidine cyclic dinucleotide STING agonists for antitumor immunotherapy using systemic intravenous route. J. Med. Chem 2021, 64, 6902–6923. [DOI] [PubMed] [Google Scholar]
- (367).Mabbott NA; Baillie JK; Brown H; Freeman TC; Hume DA An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genet 2013, 14, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (368).Wu J; Chen YJ; Dobbs N; Sakai T; Liou J; Miner JJ; Yan N STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med 2019, 216, 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (369).Walker MM; Crute BW; Cambier JC; Getahun A B cell-intrinsic STING signaling triggers cell activation, synergizes with B cell receptor signals, and promotes antibody responses. J. Immunol 2018, 201, 2641–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Tansakul M; Thim-Uam A; Saethang T; Makjaroen J; Wongprom B; Pisitkun T; Pisitkun P Deficiency of STING promotes collagen-specific antibody production and B cell survival in collagen-induced arthritis. Front. Immunol 2020, 11, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).Tang C-HA; Lee AC; Chang S; Xu Q; Shao A; Lo Y; Spalek WT; Pinilla-Ibarz JA; Del Valle JR; Hu C-CA STING regulates BCR signaling in normal and malignant B cells. Cell. Mol. Immunol 2021, 18, 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Gram AM; Sun C; Landman SL; Oosenbrug T; Koppejan HJ; Kwakkenbos MJ; Hoeben RC; Paludan SR; Ressing ME Human B cells fail to secrete type I interferons upon cytoplasmic DNA exposure. Mol. Immunol 2017, 91, 225–237. [DOI] [PubMed] [Google Scholar]
- (373).Melero I; Castanon E; Alvarez M; Champiat S; Marabelle A Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat. Rev. Clin. Oncol 2021, 18, 558–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Ager CR; Reilley MJ; Nicholas C; Bartkowiak T; Jaiswal AR; Curran MA Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol. Res 2017, 5, 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Marabelle A; Andtbacka R; Harrington K; Melero I; Leidner R; De Baere T; Robert C; Ascierto PA; Baurain JF; Imperiale M et al. Starting the fight in the tumor: expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann. Oncol 2018, 29, 2163–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Hanson MC; Crespo MP; Abraham W; Moynihan KD; Szeto GL; Chen SH; Melo MB; Mueller S; Irvine DJ Nanoparticulate STING agonists are potent lymph node–targeted vaccine adjuvants. J. Clin. Investig 2015, 125, 2532–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Munoz NM; Williams M; Dixon K; Dupuis C; Mcwatters A; Avritscher R; Manrique SZ; Mchugh K; Murthy R; Tam A et al. Influence of injection technique, drug formulation and tumor microenvironment on intratumoral immunotherapy delivery and efficacy. J. Immunother. Cancer 2021, 9, e001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Nakamura T; Harashima H Dawn of lipid nanoparticles in lymph node targeting: Potential in cancer immunotherapy. Adv. Drug Deliv. Rev 2020, 167, 78–88. [DOI] [PubMed] [Google Scholar]
- (379).Reddy ST; Rehor A; Schmoekel HG; Hubbell JA; Swartz MA In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [DOI] [PubMed] [Google Scholar]
- (380).Reddy ST; Van Der Vlies AJ; Simeoni E; Angeli V; Randolph GJ; O’neil CP; Lee LK; Swartz MA; Hubbell JA Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol 2007, 25, 1159–1164. [DOI] [PubMed] [Google Scholar]
- (381).Cabral H; Makino J; Matsumoto Y; Mi P; Wu H; Nomoto T; Toh K; Yamada N; Higuchi Y; Konishi S et al. Systemic targeting of lymph node metastasis through the blood vascular system by using size-controlled nanocarriers. ACS Nano 2015, 9, 4957–4967. [DOI] [PubMed] [Google Scholar]
- (382).Shae D; Baljon JJ; Wehbe M; Christov PP; Becker KW; Kumar A; Suryadevara N; Carson CS; Palmer CR; Knight FC et al. Co-delivery of peptide neoantigens and stimulator of interferon genes agonists enhances response to cancer vaccines. ACS Nano 2020, 14, 9904–9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Stacker SA; Williams SP; Karnezis T; Shayan R; Fox SB; Achen MG Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [DOI] [PubMed] [Google Scholar]
- (384).Rothschilds AM; Wittrup KD What, why, where, and when: Bringing timing to immuno-oncology. Trends Immunol 2019, 40, 12–21. [DOI] [PubMed] [Google Scholar]
- (385).Hammerich L; Marron TU; Upadhyay R; Svensson-Arvelund J; Dhainaut M; Hussein S; Zhan Y; Ostrowski D; Yellin M; Marsh H et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med 2019, 25, 814–824. [DOI] [PubMed] [Google Scholar]
- (386).Osterberg L; Blaschke T Adherence to medication. N. Engl. J. Med 2005, 353, 487–497. [DOI] [PubMed] [Google Scholar]
- (387).Ibrahim KM; Schommer JC; Morisky DE; Rodriguez R; Gaither C; Snyder M The association between medication experiences and beliefs and low medication adherence in patients with chronic disease from two different societies: the USA and the Sultanate of Oman. Pharmacy (Basel) 2021, 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Benci JL; Xu B; Qiu Y; Wu TJ; Dada H; Twyman-Saint Victor C; Cucolo L; Lee DSM; Pauken KE; Huang AC et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 2016, 167, 1540–1554 e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Irvine DJ; Aung A; Silva M Controlling timing and location in vaccines. Adv. Drug Deliv. Rev 2020, 158, 91–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Wehbe M; Wang-Bishop L; Becker KW; Shae D; Baljon JJ; He X; Christov P; Boyd KL; Balko JM; Wilson JT Nanoparticle delivery improves the pharmacokinetic properties of cyclic dinucleotide STING agonists to open a therapeutic window for intravenous administration. J. Control. Release 2020, 330, 1118–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Xi Q; Wang M; Jia W; Yang M; Hu J; Jin J; Chen X; Yin D; Wang X Design, synthesis, and biological evaluation of amidobenzimidazole derivatives as stimulator of interferon genes (STING) receptor agonists. J. Med. Chem 2020, 63, 260–282. [DOI] [PubMed] [Google Scholar]
- (392).Shimabukuro-Vornhagen A; Godel P; Subklewe M; Stemmler HJ; Schlosser HA; Schlaak M; Kochanek M; Boll B; Von Bergwelt-Baildon MS Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Zhang H; Zeng L; Xie M; Liu J; Zhou B; Wu R; Cao L; Kroemer G; Wang H; Billiar TR et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe 2020, 27, 556–570 e556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Zeng L; Kang R; Zhu S; Wang X; Cao L; Wang H; Billiar TR; Jiang J; Tang D ALK is a therapeutic target for lethal sepsis. Sci. Transl. Med 2017, 9, eaan5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Costela-Ruiz VJ; Illescas-Montes R; Puerta-Puerta JM; Ruiz C; Melguizo-Rodríguez L SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev 2020, 54, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Ma C; Yang D; Wang B; Wu C; Wu Y; Li S; Liu X; Lassen K; Dai L; Yang S Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Sci. Adv 2020, 6, eaaz6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Martin GR; Blomquist CM; Henare KL; Jirik FR Stimulator of interferon genes (STING) activation exacerbates experimental colitis in mice. Sci. Rep 2019, 9, 14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (398).Ackerman SE; Pearson CI; Gregorio JD; Gonzalez JC; Kenkel JA; Hartmann FJ; Luo A; Ho PY; Leblanc H; Blum LK et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat. Cancer 2021, 2, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (399).Munn DH; Mellor AL IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol 2016, 37, 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Zheng Z; Jia S; Shao C; Shi Y Irradiation induces cancer lung metastasis through activation of the cGAS–STING–CCL5 pathway in mesenchymal stromal cells. Cell Death Dis 2020, 11, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Kumar V A STING to inflammation and autoimmunity. J. Leukoc. Biol 2019, 106, 171–185. [DOI] [PubMed] [Google Scholar]
- (402).Zhao Q; Wei Y; Pandol SJ; Li L; Habtezion A STING signaling promotes inflammation in experimental acute pancreatitis. Gastroenterology 2018, 154, 1822–1835.e1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (403).Luo X; Li H; Ma L; Zhou J; Guo X; Woo SL; Pei Y; Knight LR; Deveau M; Chen Y et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology 2018, 155, 1971–1984.e1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (404).Mullard A Can innate immune system targets turn up the heat on ‘cold’ tumours? Nat. Rev. Drug Discov 2018, 17, 3–5. [DOI] [PubMed] [Google Scholar]
- (405).Dubensky TW Jr.; Kanne DB; Leong ML Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther. Adv. Vaccines 2013, 1, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Goldberg MS Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [DOI] [PubMed] [Google Scholar]
- (407).Riley RS; June CH; Langer R; Mitchell MJ Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov 2019, 18, 175–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (408).Man F; Lammers T; R TMDR Imaging nanomedicine-based drug delivery: a review of clinical studies. Mol. Imaging Biol 2018, 20, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).Rastinehad AR; Anastos H; Wajswol E; Winoker JS; Sfakianos JP; Doppalapudi SK; Carrick MR; Knauer CJ; Taouli B; Lewis SC et al. Gold nanoshell-localized photothermal ablation of prostate tumors in a clinical pilot device study. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 18590–18596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (410).Lee H; Shields AF; Siegel BA; Miller KD; Krop I; Ma CX; Lorusso PM; Munster PN; Campbell K; Gaddy DF et al. (64)Cu-MM-302 Positron emission tomography quantifies variability of enhanced permeability and retention of nanoparticles in relation to treatment response in patients with metastatic breast cancer. Clin. Cancer Res 2017, 23, 4190–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Ramanathan RK; Korn RL; Raghunand N; Sachdev JC; Newbold RG; Jameson G; Fetterly GJ; Prey J; Klinz SG; Kim J et al. Correlation between ferumoxytol uptake in tumor lesions by MRI and response to nanoliposomal irinotecan in patients with advanced solid tumors: a pilot study. Clin. Cancer Res 2017, 23, 3638–3648. [DOI] [PubMed] [Google Scholar]
- (412).Harrington KJ; Mohammadtaghi S; Uster PS; Glass D; Peters AM; Vile RG; Stewart JS Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin. Cancer Res 2001, 7, 243–254. [PubMed] [Google Scholar]
- (413).Mitchell MJ; Billingsley MM; Haley RM; Wechsler ME; Peppas NA; Langer R Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov 2021, 20, 101–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Wilhelm S; Tavares AJ; Dai Q; Ohta S; Audet J; Dvorak HF; Chan WCW Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater 2016, 1, 16014. [Google Scholar]
- (415).Lammers T Macro-nanomedicine: Targeting the big picture. J. Control. Release 2019, 294, 372–375. [DOI] [PubMed] [Google Scholar]
- (416).Matsumura Y; Maeda H A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- (417).Gerlowski LE; Jain RK Microvascular permeability of normal and neoplastic tissues. Microvasc. Res 1986, 31, 288–305. [DOI] [PubMed] [Google Scholar]
- (418).Sindhwani S; Syed AM; Ngai J; Kingston BR; Maiorino L; Rothschild J; Macmillan P; Zhang Y; Rajesh NU; Hoang T et al. The entry of nanoparticles into solid tumours. Nat. Mater 2020, 19, 566–575. [DOI] [PubMed] [Google Scholar]
- (419).Fernandes C; Suares D; Yergeri MC Tumor microenvironment targeted nanotherapy. Front. Pharmacol 2018, 9, 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Miyabe H; Hyodo M; Nakamura T; Sato Y; Hayakawa Y; Harashima H A new adjuvant delivery system ‘cyclic di-GMP/YSK05 liposome’ for cancer immunotherapy. J. Control. Release 2014, 184, 20–27. [DOI] [PubMed] [Google Scholar]
- (421).Atukorale PU; Raghunathan SP; Raguveer V; Moon TJ; Zheng C; Bielecki PA; Wiese ML; Goldberg AL; Covarrubias G; Hoimes CJ et al. Nanoparticle encapsulation of synergistic immune agonists enables systemic codelivery to tumor sites and IFNbeta-driven antitumor immunity. Cancer Res 2019, 79, 5394–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Lorkowski ME; Atukorale PU; Bielecki PA; Tong KH; Covarrubias G; Zhang Y; Loutrianakis G; Moon TJ; Santulli AR; Becicka WM et al. Immunostimulatory nanoparticle incorporating two immune agonists for the treatment of pancreatic tumors. J. Control. Release 2021, 330, 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Liu Y; Crowe WN; Wang L; Lu Y; Petty WJ; Habib AA; Zhao D An inhalable nanoparticulate STING agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nat. Commun 2019, 10, 5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (424).Li K; Ye Y; Liu L; Sha Q; Wang X; Jiao T; Zhang L; Wang J The lipid platform increases the activity of STING agonists to synergize checkpoint blockade therapy against melanoma. Biomaterials science 2021, 9, 765–773. [DOI] [PubMed] [Google Scholar]
- (425).Tse SW; Mckinney K; Walker W; Nguyen M; Iacovelli J; Small C; Hopson K; Zaks T; Huang E mRNA-encoded constitutively active STING(V155M) is a potent genetic adjuvant of antigen-specific CD8(+) T-cell response. Mol. Ther 2021, 29, 2227–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Chen J; Qiu M; Ye Z; Nyalile T; Li Y; Glass Z; Zhao X; Yang L; Chen J; Xu Q In situ cancer vaccination using lipidoid nanoparticles. Sci. Adv 2021, 7, eabf1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Liu Y; Wang L; Song Q; Ali M; Crowe WN; Kucera GL; Hawkins GA; Soker S; Thomas KW; Miller LD et al. Intrapleural nano-immunotherapy promotes innate and adaptive immune responses to enhance anti-PD-L1 therapy for malignant pleural effusion. Nat. Nanotechnol 2021, Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Covarrubias G; Moon TJ; Loutrianakis G; Sims HM; Umapathy MP; Lorkowski ME; Bielecki PA; Wiese ML; Atukorale PU; Karathanasis E Comparison of the uptake of untargeted and targeted immunostimulatory nanoparticles by immune cells in the microenvironment of metastatic breast cancer. J. Mater. Chem. B 2021, Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Kanasty R; Dorkin JR; Vegas A; Anderson D Delivery materials for siRNA therapeutics. Nat. Mater 2013, 12, 967–977. [DOI] [PubMed] [Google Scholar]
- (430).Tseng Y-C; Mozumdar S; Huang L Lipid-based systemic delivery of siRNA. Adv. Drug Deliv. Rev 2009, 61, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Akbarzadeh A; Rezaei-Sadabady R; Davaran S; Joo SW; Zarghami N; Hanifehpour Y; Samiei M; Kouhi M; Nejati-Koshki K Liposome: classification, preparation, and applications. Nanoscale Res. Lett 2013, 8, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).Liu C; Zhang L; Zhu W; Guo R; Sun H; Chen X; Deng N Barriers and strategies of cationic liposomes for cancer gene therapy. Mol. Ther. Methods Clin. Dev 2020, 18, 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Ikeda Y; Nagasaki Y Impacts of PEGylation on the gene and oligonucleotide delivery system. J. Appl. Polym. Sci 2014, 131, 40293. [Google Scholar]
- (434).Nakamura T; Miyabe H; Hyodo M; Sato Y; Hayakawa Y; Harashima H Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J. Control. Release 2015, 216, 149–157. [DOI] [PubMed] [Google Scholar]
- (435).Gou S; Liu W; Wang S; Chen G; Chen Z; Qiu L; Zhou X; Wu Y; Qi Y; Gao Y Engineered nanovaccine targeting Clec9a(+) dendritic cells remarkably enhances the cancer immunotherapy effects of STING agonist. Nano Lett 2021, 21, 9939–9950. [DOI] [PubMed] [Google Scholar]
- (436).Pizzolla A; Nguyen TH; Sant S; Jaffar J; Loudovaris T; Mannering SI; Thomas PG; Westall GP; Kedzierska K; Wakim LM Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J. Clin. Invest 2018, 128, 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (437).Koutsakos M; Illing PT; Nguyen THO; Mifsud NA; Crawford JC; Rizzetto S; Eltahla AA; Clemens EB; Sant S; Chua BY et al. Human CD8(+) T cell cross-reactivity across influenza A, B and C viruses. Nat. Immunol 2019, 20, 613–625. [DOI] [PubMed] [Google Scholar]
- (438).Knight FC; Wilson JT Engineering vaccines for tissue-resident memory T cells. Adv. Ther. (Weinh) 2021, 4, 2000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Hiam-Galvez KJ; Allen BM; Spitzer MH Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (440).Pereira ER; Jones D; Jung K; Padera TP The lymph node microenvironment and its role in the progression of metastatic cancer. Semin. Cell Dev. Biol 2015, 38, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).Fu J; Kanne DB; Leong M; Glickman LH; Mcwhirter SM; Lemmens E; Mechette K; Leong JJ; Lauer P; Liu W et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med 2015, 7, 283ra252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Irvine DJ; Swartz MA; Szeto GL Engineering synthetic vaccines using cues from natural immunity. Nat. Mater 2013, 12, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Storni T; Kundig TM; Senti G; Johansen P Immunity in response to particulate antigen-delivery systems. Adv. Drug Deliv. Rev 2005, 57, 333–355. [DOI] [PubMed] [Google Scholar]
- (444).Blume G; Cevc G Liposomes for the sustained drug release in vivo. Biochimica et biophysica acta 1990, 1029, 91–97. [DOI] [PubMed] [Google Scholar]
- (445).Cai S; Yang Q; Bagby TR; Forrest ML Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv. Drug Deliv. Rev 2011, 63, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Lee E; Jang H-E; Kang YY; Kim J; Ahn J-H; Mok H Submicron-sized hydrogels incorporating cyclic dinucleotides for selective delivery and elevated cytokine release in macrophages. Acta Biomater 2016, 29, 271–281. [DOI] [PubMed] [Google Scholar]
- (447).Wilson DR; Sen R; Sunshine JC; Pardoll DM; Green JJ; Kim YJ Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomedicine : nanotechnology, biology, and medicine 2018, 14, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Junkins RD; Gallovic MD; Johnson BM; Collier MA; Watkins-Schulz R; Cheng N; David CN; Mcgee CE; Sempowski GD; Shterev I et al. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J. Control. Release 2018, 270, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Murthy N; Xu M; Schuck S; Kunisawa J; Shastri N; Fréchet JM A macromolecular delivery vehicle for protein-based vaccines: acid-degradable protein-loaded microgels. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 4995–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Wang-Bishop L; Wehbe M; Shae D; James J; Hacker BC; Garland K; Chistov PP; Rafat M; Balko JM; Wilson JT Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. J. Immunother. Cancer 2020, 8, e000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Nguyen DC; Shae D; Pagendarm HM; Becker KW; Wehbe M; Kilchrist KV; Pastora LE; Palmer CR; Seber P; Christov PP et al. Amphiphilic polyelectrolyte graft copolymers enhance the activity of cyclic dinucleotide STING agonists. Adv. Healthc. Mater 2021, 10, e2001056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Fucikova J; Kepp O; Kasikova L; Petroni G; Yamazaki T; Liu P; Zhao L; Spisek R; Kroemer G; Galluzzi L Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis 2020, 11, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Gulla A; Morelli E; Samur MK; Botta C; Hideshima T; Bianchi G; Fulciniti M; Malvestiti S; Prabhala RH; Talluri S et al. Bortezomib induces anti–multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov 2021, 2, 468–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Chattopadhyay S; Liu Y-H; Fang Z-S; Lin C-L; Lin J-C; Yao B-Y; Hu C-MJ Synthetic immunogenic cell death mediated by intracellular delivery of STING agonist nanoshells enhances anticancer chemo-immunotherapy. Nano Lett 2020, 20, 2246–2256. [DOI] [PubMed] [Google Scholar]
- (455).Lin LC; Huang CY; Yao BY; Lin JC; Agrawal A; Algaissi A; Peng BH; Liu YH; Huang PH; Juang RH et al. Viromimetic STING agonist-loaded hollow polymeric nanoparticles for safe and effective vaccination against middle east respiratory syndrome coronavirus. Adv. Funct. Mater 2019, 29, 1807616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Liang J; Wang H; Ding W; Huang J; Zhou X; Wang H; Dong X; Li G; Chen E; Zhou F et al. Nanoparticle-enhanced chemo-immunotherapy to trigger robust antitumor immunity. Sci. Adv 2020, 6, eabc3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Lu ZD; Chen YF; Shen S; Xu CF; Wang J Co-delivery of phagocytosis checkpoint silencer and stimulator of interferon genes agonist for synergetic cancer immunotherapy. ACS Appl. Mater. Interfaces 2021, 13, 29424–29438. [DOI] [PubMed] [Google Scholar]
- (458).An M; Yu C; Xi J; Reyes J; Mao G; Wei WZ; Liu H Induction of necrotic cell death and activation of STING in the tumor microenvironment via cationic silica nanoparticles leading to enhanced antitumor immunity. Nanoscale 2018, 10, 9311–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Park KS; Xu C; Sun X; Louttit C; Moon JJ Improving STING agonist delivery for cancer immunotherapy using biodegradable mesoporous silica nanoparticles. Adv. Ther 2020, 3, 2000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Bielecki PA; Lorkowski ME; Becicka WM; Atukorale PU; Moon TJ; Zhang Y; Wiese M; Covarrubias G; Ravichandran S; Karathanasis E Immunostimulatory silica nanoparticle boosts innate immunity in brain tumors. Nanoscale Horiz 2021, 6, 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (461).Chen Y-P; Xu L; Tang T-W; Chen C-H; Zheng Q-H; Liu T-P; Mou C-Y; Wu C-H; Wu S-H STING activator c‑di-GMP-loaded mesoporous silica nanoparticles enhance immunotherapy against breast cancer. ACS Appl. Mater. Interfaces 2020, 12, 56741–56752. [DOI] [PubMed] [Google Scholar]
- (462).Leventhal DS; Sokolovska A; Li N; Plescia C; Kolodziej SA; Gallant CW; Christmas R; Gao J-R; James MJ; Abin-Fuentes A et al. Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat. Commun 2020, 11, 2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Chauveau L; Bridgeman A; Tan TK; Beveridge R; Frost JN; Rijal P; Pedroza-Pacheco I; Partridge T; Gilbert-Jaramillo J; Knight ML et al. Inclusion of cGAMP within virus-like particle vaccines enhances their immunogenicity. EMBO Rep 2021, 22, e52447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Rao L; Wu L; Liu Z; Tian R; Yu G; Zhou Z; Yang K; Xiong HG; Zhang A; Yu GT et al. Hybrid cellular membrane nanovesicles amplify macrophage immune responses against cancer recurrence and metastasis. Nat. Commun 2020, 11, 4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Mcandrews KM; Che SPY; Lebleu VS; Kalluri R Effective delivery of STING agonist using exosomes suppresses tumor growth and enhances anti-tumor immunity. J. Biol. Chem 2021, Epub ahead of print, 100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Jang SC; Economides KD; Moniz RJ; Sia CL; Lewis N; Mccoy C; Zi T; Zhang K; Harrison RA; Lim J et al. ExoSTING, an extracellular vesicle loaded with STING agonists, promotes tumor immune surveillance. Commun. Biol 2021, 4, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Dooley K; Mcconnell RE; Xu K; Lewis ND; Haupt S; Youniss MR; Martin S; Sia CL; Mccoy C; Moniz RJ et al. A versatile platform for generating engineered extracellular vesicles with defined therapeutic properties. Mol. Ther 2021, 29, 1729–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (468).He Y; Hong C; Yan EZ; Fletcher SJ; Zhu G; Yang M; Li Y; Sun X; Irvine DJ; Li J et al. Self-assembled cGAMP-STINGΔTM signaling complex as a bioinspired platform for cGAMP delivery. Sci. Adv 2020, 6, eaba7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (469).Sun X; Ni Y; He Y; Yang M; Tani T; Kitajima S; Barbie DA; Li J Engineering the immune adaptor protein STING as a functional carrier. Adv. Ther 2021, 4, 2100066. [Google Scholar]
- (470).Van Niel G; D’angelo G; Raposo G Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol 2018, 19, 213–228. [DOI] [PubMed] [Google Scholar]
- (471).Valadi H; Ekström K; Bossios A; Sjöstrand M; Lee JJ; Lötvall JO Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 2007, 9, 654–659. [DOI] [PubMed] [Google Scholar]
- (472).Yim N; Ryu SW; Choi K; Lee KR; Lee S; Choi H; Kim J; Shaker MR; Sun W; Park JH et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun 2016, 7, 12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).Nandakumar R; Tschismarov R; Meissner F; Prabakaran T; Krissanaprasit A; Farahani E; Zhang BC; Assil S; Martin A; Bertrams W et al. Intracellular bacteria engage a STING-TBK1-MVB12b pathway to enable paracrine cGAS-STING signalling. Nat. Microbiol 2019, 4, 701–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (474).Ahn J; Konno H; Barber GN Diverse roles of STING-dependent signaling on the development of cancer. Oncogene 2015, 34, 5302–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Zhao B; Du F; Xu P; Shu C; Sankaran B; Bell SL; Liu M; Lei Y; Gao X; Fu X et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019, 569, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Lu X; Miao L; Gao W; Chen Z; Mchugh KJ; Sun Y; Tochka Z; Tomasic S; Sadtler K; Hyacinthe A et al. Engineered PLGA microparticles for long-term, pulsatile release of STING agonist for cancer immunotherapy. Sci. Transl. Med 2020, 12, eaaz6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (477).Wang F; Su H; Xu D; Dai W; Zhang W; Wang Z; Anderson CF; Zheng M; Oh R; Wan F et al. Tumour sensitization via the extended intratumoural release of a STING agonist and camptothecin from a self-assembled hydrogel. Nature biomedical engineering 2020, 4, 1090–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).Leach DG; Dharmaraj N; Piotrowski SL; Lopez-Silva TL; Lei YL; Sikora AG; Young S; Hartgerink JD STINGel: controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials 2018, 163, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (479).Aulisa L; Dong H; Hartgerink JD Self-assembly of multidomain peptides: sequence variation allows control over cross-linking and viscoelasticity. Biomacromolecules 2009, 10, 2694–2698. [DOI] [PubMed] [Google Scholar]
- (480).Macfarlane AWT; Jillab M; Plimack ER; Hudes GR; Uzzo RG; Litwin S; Dulaimi E; Al-Saleem T; Campbell KS PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor reSection. Cancer Immunol. Res 2014, 2, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Park CG; Hartl CA; Schmid D; Carmona EM; Kim H-J; Goldberg MS Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci. Transl. Med 2018, 10, eaar1916. [DOI] [PubMed] [Google Scholar]
- (482).Kershaw MH; Westwood JA; Parker LL; Wang G; Eshhar Z; Mavroukakis SA; White DE; Wunderlich JR; Canevari S; Rogers-Freezer L et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res 2006, 12, 6106–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (483).Lamers CH; Sleijfer S; Van Steenbergen S; Van Elzakker P; Van Krimpen B; Groot C; Vulto A; Den Bakker M; Oosterwijk E; Debets R et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol.Ther 2013, 21, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (484).Stephan SB; Taber AM; Jileaeva I; Pegues EP; Sentman CL; Stephan MT Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol 2015, 33, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (485).Smith TT; Moffett HF; Stephan SB; Opel CF; Dumigan AG; Jiang X; Pillarisetty VG; Pillai SPS; Wittrup KD; Stephan MT Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Investig 2017, 127, 2176–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (486).Carroll EC; Jin L; Mori A; Muñoz-Wolf N; Oleszycka E; Moran HBT; Mansouri S; Mcentee CP; Lambe E; Agger EM et al. The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS-STING-dependent induction of type I interferons. Immunity 2016, 44, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (487).Mori A; Oleszycka E; Sharp FA; Coleman M; Ozasa Y; Singh M; O’hagan DT; Tajber L; Corrigan OI; Mcneela EA et al. The vaccine adjuvant alum inhibits IL-12 by promoting PI3 kinase signaling while chitosan does not inhibit IL-12 and enhances Th1 and Th17 responses. Eur. J. Immunol 2012, 42, 2709–2719. [DOI] [PubMed] [Google Scholar]
- (488).Bueter CL; Lee CK; Wang JP; Ostroff GR; Specht CA; Levitz SM Spectrum and mechanisms of inflammasome activation by chitosan. J. Immunol 2014, 192, 5943–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (489).Turley JL; Moran HBT; Mcentee CP; O’grady K; Munoz-Wolf N; Jin L; Follmann F; Andersen P; Andersson M; Lavelle EC Chitin-derived polymer deacetylation regulates mitochondrial reactive oxygen species dependent cGAS-STING and NLRP3 inflammasome activation. Biomaterials 2021, 275, 120961. [DOI] [PubMed] [Google Scholar]
- (490).Frohlich E The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomedicine 2012, 7, 5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Ma X; Gong N; Zhong L; Sun J; Liang XJ Future of nanotherapeutics: Targeting the cellular sub-organelles. Biomaterials 2016, 97, 10–21. [DOI] [PubMed] [Google Scholar]
- (492).Qin J; Gong N; Liao Z; Zhang S; Timashev P; Huo S; Liang XJ Recent progress in mitochondria-targeting-based nanotechnology for cancer treatment. Nanoscale 2021, 13, 7108–7118. [DOI] [PubMed] [Google Scholar]
- (493).Jin Q; Zhu W; Zhu J; Zhu J; Shen J; Liu Z; Yang Y; Chen Q Nanoparticle-mediated delivery of inhaled immunotherapeutics for treating lung metastasis. Adv. Mater 2021, 33, e2007557. [DOI] [PubMed] [Google Scholar]
- (494).Mehta P; Bothiraja C; Kadam S; Pawar A Probing the influence of lactose fines, a USP modified induction port and modified DDU apparatus on the aerodynamic behavior of a fluticasone propionate dry powder inhaler. New J. Chem 2019, 43, 17327–17339. [Google Scholar]
- (495).Altorki NK; Markowitz GJ; Gao D; Port JL; Saxena A; Stiles B; Mcgraw T; Mittal V The lung microenvironment: an important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (496).Wei X; Shao B; He Z; Ye T; Luo M; Sang Y; Liang X; Wang W; Luo S; Yang S et al. Cationic nanocarriers induce cell necrosis through impairment of Na(+)/K(+)-ATPase and cause subsequent inflammatory response. Cell Res 2015, 25, 237–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (497).Liu L; Liu Y; Xu B; Liu C; Jia Y; Liu T; Fang C; Wang W; Ren J; He Z et al. Negative regulation of cationic nanoparticle-induced inflammatory toxicity through the increased production of prostaglandin E2 via mitochondrial DNA-activated Ly6C(+) monocytes. Theranostics 2018, 8, 3138–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Moyer TJ; Kato Y; Abraham W; Chang JYH; Kulp DW; Watson N; Turner HL; Menis S; Abbott RK; Bhiman JN et al. Engineered immunogen binding to alum adjuvant enhances humoral immunity. Nat. Med 2020, 26, 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (499).Garçon N; Friede M In Plotkin’s Vaccines (Seventh Edition); Plotkin SA;Orenstein WA;Offit PA;Edwards KM, Eds.; Elsevier, 2018. [Google Scholar]
- (500).Mckee AS; Burchill MA; Munks MW; Jin L; Kappler JW; Friedman RS; Jacobelli J; Marrack P Host DNA released in response to aluminum adjuvant enhances MHC class II-mediated antigen presentation and prolongs CD4 T-cell interactions with dendritic cells. Proc. Natl. Acad. Sci. U. S. A 2013, 110, E1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Muñoz-Wolf N; Mccluskey S; Lavelle EC In Immunopotentiators in Modern Vaccines (Second Edition); Schijns VEJC;O’Hagan DT, Eds.; Academic Press, 2017. [Google Scholar]
- (502).Shimada K; Crother TR; Karlin J; Dagvadorj J; Chiba N; Chen S; Ramanujan VK; Wolf AJ; Vergnes L; Ojcius DM et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (503).Riteau N; Sher A Chitosan: an adjuvant with an unanticipated STING. Immunity 2016, 44, 522–524. [DOI] [PubMed] [Google Scholar]
- (504).Barisik M; Atalay S; Beskok A; Qian S Size dependent surface charge properties of silica nanoparticles. J. Phys. Chem. C 2014, 118, 1836–1842. [Google Scholar]
- (505).Rongvaux A; Jackson R; Harman CC; Li T; West AP; De Zoete MR; Wu Y; Yordy B; Lakhani SA; Kuan CY et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (506).West AP; Khoury-Hanold W; Staron M; Tal MC; Pineda CM; Lang SM; Bestwick M; Duguay BA; Raimundo N; Macduff DA et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (507).Patrushev M; Kasymov V; Patrusheva V; Ushakova T; Gogvadze V; Gaziev A Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell. Mol. Life Sci 2004, 61, 3100–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Garcia N; Chavez E Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci 2007, 81, 1160–1166. [DOI] [PubMed] [Google Scholar]
- (509).Bueter CL; Lee CK; Rathinam V. a. K.; Healy GJ; Taron CH; Specht CA; Levitz SM Chitosan but not chitin activates the inflammasome by a mechanism dependent upon phagocytosis. J. Biol. Chem 2011, 286, 35447–35455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Fang C; Wei X; Wei Y Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (511).Riley JS; Tait SW Mitochondrial DNA in inflammation and immunity. EMBO Rep 2020, 21, e49799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Anderson S; Bankier AT; Barrell BG; De Bruijn MH; Coulson AR; Drouin J; Eperon IC; Nierlich DP; Roe BA; Sanger F et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [DOI] [PubMed] [Google Scholar]
- (513).Luo M; Wang H; Wang Z; Cai H; Lu Z; Li Y; Du M; Huang G; Wang C; Chen X et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol 2017, 12, 648–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).Luo M; Liu Z; Zhang X; Han C; Samandi LZ; Dong C; Sumer BD; Lea J; Fu YX; Gao J Synergistic STING activation by PC7A nanovaccine and ionizing radiation improves cancer immunotherapy. J. Control. Release 2019, 300, 154–160. [DOI] [PubMed] [Google Scholar]
- (515).Miao L; Li L; Huang Y; Delcassian D; Chahal J; Han J; Shi Y; Sadtler K; Gao W; Lin J et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol 2019, 37, 1174–1185. [DOI] [PubMed] [Google Scholar]
- (516).Pardi N; Hogan MJ; Porter FW; Weissman D mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov 2018, 17, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (517).Waldron KJ; Rutherford JC; Ford D; Robinson NJ Metalloproteins and metal sensing. Nature 2009, 460, 823–830. [DOI] [PubMed] [Google Scholar]
- (518).Wang C; Zhang R; Wei X; Lv M; Jiang Z Metalloimmunology: the metal ion-controlled immunity. Adv. Immunol 2020, 145, 187–241. [DOI] [PubMed] [Google Scholar]
- (519).Wang C; Guan Y; Lv M; Zhang R; Guo Z; Wei X; Du X; Yang J; Li T; Wan Y et al. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity 2018, 48, 675–687 e677. [DOI] [PubMed] [Google Scholar]
- (520).Sun X; Zhang Y; Li J; Park KS; Han K; Zhou X; Xu Y; Nam J; Xu J; Shi X et al. Amplifying STING activation by cyclic dinucleotide-manganese particles for local and systemic cancer metalloimmunotherapy. Nat. Nanotechnol 2021, 16, 1260–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (521).Wang C; Sun Z; Zhao C; Zhang Z; Wang H; Liu Y; Guo Y; Zhang B; Gu L; Yu Y et al. Maintaining manganese in tumor to activate cGAS-STING pathway evokes a robust abscopal anti-tumor effect. J. Control. Release 2021, 331, 480–490. [DOI] [PubMed] [Google Scholar]
- (522).Hou L; Tian C; Yan Y; Zhang L; Zhang H; Zhang Z Manganese-based nanoactivator optimizes cancer immunotherapy via enhancing innate immunity. ACS Nano 2020, 14, 3927–3940. [DOI] [PubMed] [Google Scholar]
- (523).Chen C; Tong Y; Zheng Y; Shi Y; Chen Z; Li J; Liu X; Zhang D; Yang H Cytosolic delivery of thiolated Mn-cGAMP nanovaccine to enhance the antitumor immune responses. Small 2021, 17, e2006970. [DOI] [PubMed] [Google Scholar]
- (524).Yang X; Yang Y; Bian J; Wei J; Wang Z; Zhou Z; Li Z; Sun M Converting primary tumor towards an in situ STING-activating vaccine via a biomimetic nanoplatform against recurrent and metastatic tumors. Nano Today 2021, 38, 101109. [Google Scholar]
- (525).Gao M; Xie Y-Q; Lei K; Zhao Y; Kurum A; Van Herck S; Guo Y; Hu X; Tang L A manganese phosphate nanocluster activates the cGAS-STING pathway for enhanced cancer immunotherapy. Adv. Ther 2021, 4, 2100065. [Google Scholar]
- (526).Yoh SM; Schneider M; Seifried J; Soonthornvacharin S; Akleh RE; Olivieri KC; De Jesus PD; Ruan C; De Castro E; Ruiz PA et al. PQBP1 Is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 2015, 161, 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Lian H; Wei J; Zang R; Ye W; Yang Q; Zhang XN; Chen YD; Fu YZ; Hu MM; Lei CQ et al. ZCCHC3 is a co-sensor of cGAS for dsDNA recognition in innate immune response. Nat. Commun 2018, 9, 3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Liu ZS; Cai H; Xue W; Wang M; Xia T; Li WJ; Xing JQ; Zhao M; Huang YJ; Chen S et al. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol 2019, 20, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (529).Zhang Y; Ma Z; Wang Y; Boyer J; Ni G; Cheng L; Su S; Zhang Z; Zhu Z; Qian J et al. Streptavidin promotes DNA binding and activation of cGAS to enhance innate immunity. iScience 2020, 23, 101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (530).Moser BA; Escalante-Buendia Y; Steinhardt RC; Rosenberger MG; Cassaidy BJ; Naorem N; Chon AC; Nguyen MH; Tran NT; Esser-Kahn AP Small molecule NF-kappaB inhibitors as immune potentiators for enhancement of vaccine adjuvants. Front. Immunol 2020, 11, 511513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (531).Onyedibe KI; Wang M; Sintim HO ENPP1, an old enzyme with new functions, and small molecule inhibitors-a STING in the tale of ENPP1. Molecules 2019, 24, 4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (532).Meric-Bernstam F; Sweis RF; Hodi FS; Messersmith WA; Andtbacka RHI; Ingham M; Lewis N; Chen X; Pelletier M; Chen X et al. Phase I dose-escalation trial of MIW815 (ADU-S100), an intratumoral STING agonist, in patients with advanced/metastatic solid tumors or lymphomas. Clin. Cancer Res 2021, Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- (533).Levy HB; Baer G; Baron S; Buckler CE; Gibbs CJ; Iadarola MJ; London WT; Rice J A modified polyriboinosinic-polyribocytidylic acid complex that induces interferon in primates. J. Infect. Dis 1975, 132, 434–439. [DOI] [PubMed] [Google Scholar]
- (534).Sun L; Funchain P; Song JM; Rayman P; Tannenbaum C; Ko J; Mcnamara M; Marcela Diaz-Montero C; Gastman B Talimogene laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: a case series. J. Immunother. Cancer 2018, 6, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Bencherif SA; Warren Sands R; Ali OA; Li WA; Lewin SA; Braschler TM; Shih TY; Verbeke CS; Bhatta D; Dranoff G et al. Injectable cryogel-based whole-cell cancer vaccines. Nat. Commun 2015, 6, 7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (536).Bukowski RM; Tendler C; Cutler D; Rose E; Laughlin MM; Statkevich P Treating cancer with PEG Intron: pharmacokinetic profile and dosing guidelines for an improved interferon-alpha-2b formulation. Cancer 2002, 95, 389–396. [DOI] [PubMed] [Google Scholar]
- (537).Tanaka T; Narazaki M; Kishimoto T Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy 2016, 8, 959–970. [DOI] [PubMed] [Google Scholar]
- (538).Le RQ; Li L; Yuan W; Shord SS; Nie L; Habtemariam BA; Przepiorka D; Farrell AT; Pazdur R FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist 2018, 23, 943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (539).Investigators R-C; Gordon AC; Mouncey PR; Al-Beidh F; Rowan KM; Nichol AD; Arabi YM; Annane D; Beane A; Van Bentum-Puijk W et al. Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N. Engl. J. Med 2021, 384, 1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (540).Xue Y; Bai H; Peng B; Fang B; Baell J; Li L; Huang W; Voelcker NH Stimulus-cleavable chemistry in the field of controlled drug delivery. Chem. Soc. Rev 2021, 50, 4872–4931. [DOI] [PubMed] [Google Scholar]
- (541).Bukhalid RA; Duvall JR; Cetinbas NM; Catcott KC; Avocetien K; Bentley KW; Bradley S; Carter T; Chin C-N; Clardy S et al. Abstract 6706: Systemic administration of STING agonist antibody-drug conjugates elicit potent anti-tumor immune responses with minimal induction of circulating cytokines. Cancer Res 2020, 80, 6706. [Google Scholar]
- (542).Duvall JR; Bukhalid RA; Cetinbas NM; Catcott KC; Slocum K; Avocetien K; Bentley KW; Bradley S; Clardy S; Collins SD et al. Abstract 1738: XMT-2056, a well-tolerated, immunosynthen-based STING-agonist antibody-drug conjugate which induces anti-tumor immune activity. Cancer Res 2021, 81, 1738. [Google Scholar]
- (543).Banerjee M; Basu S; Middya S; Shrivastava R; Ghosh R; Pryde DC; Yadav D; Bhattacharya G; Soram T; Puniya K et al. Abstract LB-061: CRD5500: A versatile small molecule STING agonist amenable to bioconjugation as an ADC. Cancer Res 2019, 79, LB–061. [Google Scholar]
- (544).Cetinbas NM; Monnell T; Catcott K; Lee W; Shaw P; Slocum K; Avocetien K; Bentley K; Clardy S; Jones B et al. Abstract 1773: Tumor cell-intrinsic STING pathway activation leads to robust induction of type III interferons and contributes to the anti-tumor activity elicited by STING agonism. Cancer Res 2021, 81, 1773. [Google Scholar]
- (545).Zhao Y; Zheng Z; Cohen CJ; Gattinoni L; Palmer DC; Restifo NP; Rosenberg SA; Morgan RA High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther 2006, 13, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (546).Irvine DJ; Dane EL Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol 2020, 20, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (547).Lei Z; Deng M; Yi Z; Sun Q; Shapiro RA; Xu H; Li T; Loughran PA; Griepentrog JE; Huang H et al. cGAS-mediated autophagy protects the liver from ischemia-reperfusion injury independently of STING. Am. J. Physiol. Gastrointest. Liver Physiol 2018, 314, G655–G667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).Qiao JT; Cui C; Qing L; Wang LS; He TY; Yan F; Liu FQ; Shen YH; Hou XG; Chen L Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 2018, 81, 13–24. [DOI] [PubMed] [Google Scholar]
- (549).Cheng Q; Wei T; Farbiak L; Johnson LT; Dilliard SA; Siegwart DJ Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol 2020, 15, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (550).Kranz LM; Diken M; Haas H; Kreiter S; Loquai C; Reuter KC; Meng M; Fritz D; Vascotto F; Hefesha H et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [DOI] [PubMed] [Google Scholar]
- (551).Yoo J; Park C; Yi G; Lee D; Koo H Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers (Basel) 2019, 11, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (552).Saunders NRM; Paolini MS; Fenton OS; Poul L; Devalliere J; Mpambani F; Darmon A; Bergere M; Jibault O; Germain M et al. A nanoprimer to improve the systemic delivery of siRNA and mRNA. Nano Lett 2020, 20, 4264–4269. [DOI] [PubMed] [Google Scholar]
- (553).Shao K; Singha S; Clemente-Casares X; Tsai S; Yang Y; Santamaria P Nanoparticle-based immunotherapy for cancer. ACS Nano 2015, 9, 16–30. [DOI] [PubMed] [Google Scholar]
- (554).Das M; Shen L; Liu Q; Goodwin TJ; Huang L Nanoparticle delivery of RIG-I agonist enables effective and safe adjuvant therapy in pancreatic cancer. Mol. Ther 2019, 27, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (555).Dai Q; Wilhelm S; Ding D; Syed AM; Sindhwani S; Zhang Y; Chen YY; Macmillan P; Chan WCW Quantifying the ligand-coated nanoparticle delivery to cancer cells in solid tumors. ACS Nano 2018, 12, 8423–8435. [DOI] [PubMed] [Google Scholar]
- (556).Miller MA; Chandra R; Cuccarese MF; Pfirschke C; Engblom C; Stapleton S; Adhikary U; Kohler RH; Mohan JF; Pittet MJ et al. Radiation therapy primes tumors for nanotherapeutic delivery via macrophage-mediated vascular bursts. Sci. Transl. Med 2017, 9, eaal0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (557).Miller MA; Zheng YR; Gadde S; Pfirschke C; Zope H; Engblom C; Kohler RH; Iwamoto Y; Yang KS; Askevold B et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat. Commun 2015, 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Rodell CB; Arlauckas SP; Cuccarese MF; Garris CS; Li R; Ahmed MS; Kohler RH; Pittet MJ; Weissleder R TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nature biomedical engineering 2018, 2, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (559).Zhang F; Stephan SB; Ene CI; Smith TT; Holland EC; Stephan MT Nanoparticles that reshape the tumor milieu create a therapeutic window for effective T-cell therapy in solid malignancies. Cancer Res 2018, 78, 3718–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).Nakamura T; Sato T; Endo R; Sasaki S; Takahashi N; Sato Y; Hyodo M; Hayakawa Y; Harashima H STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J. Immunother. Cancer 2021, 9, e002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (561).Song G; Wu H; Yoshino K; Zamboni WC Factors affecting the pharmacokinetics and pharmacodynamics of liposomal drugs. J. Liposome Res 2012, 22, 177–192. [DOI] [PubMed] [Google Scholar]
- (562).Moser BA; Steinhardt RC; Escalante-Buendia Y; Boltz DA; Barker KM; Cassaidy BJ; Rosenberger MG; Yoo S; Mcgonnigal BG; Esser-Kahn AP Increased vaccine tolerability and protection via NF-kappaB modulation. Sci. Adv 2020, 6, eaaz8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Mu Q; Yu J; Mcconnachie LA; Kraft JC; Gao Y; Gulati GK; Ho RJY Translation of combination nanodrugs into nanomedicines: lessons learned and future outlook. J. Drug Target 2018, 26, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (564).Krauss AC; Gao X; Li L; Manning ML; Patel P; Fu W; Janoria KG; Gieser G; Bateman DA; Przepiorka D et al. FDA approval summary: (daunorubicin and cytarabine) liposome for injection for the treatment of adults with high-risk acute myeloid leukemia. Clin. Cancer Res 2019, 25, 2685–2690. [DOI] [PubMed] [Google Scholar]
- (565).Liang H; Deng L; Hou Y; Meng X; Huang X; Rao E; Zheng W; Mauceri H; Mack M; Xu M et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun 2017, 8, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (566).Wilson JT; Shae D; Gonzalez-Ericsson PI; Sanchez V; Gong J; Liang Y; Hinerfeld D; Beechem JM; Balko JM Abstract 4978: Digital spatial profiling of molecular responses to nanoparticle STING agonists identify S100A9 and B7-H3 as possible escape mechanisms. Cancer Res 2019, 79, 4978.31431460 [Google Scholar]
- (567).Fentahun Darge H; Yibru Hanurry E; Simegniew Birhan Y; Worku Mekonnen T; Tizazu Andrgie A; Chou H-Y; Lai J-Y; Tsai H-C Multifunctional drug-loaded micelles encapsulated in thermo-sensitive hydrogel for in vivo local cancer treatment: Synergistic effects of anti-vascular and immuno-chemotherapy. Chem. Eng. J 2021, 406, 126879. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.