

Abstract
Aging is a risk factor for major central nervous system (CNS) disorders. More specifically, aging can be inked to neurodegenerative diseases (NDs) because of its deteriorating impact on neurovascular unit (NVU). Metformin, a first line FDA-approved anti-diabetic drug, has gained increasing interest among researchers for its role in improving aging-related neurodegenerative disorders. Additionally, numerous studies have illustrated metformin’s role in ischemic stroke, a cerebrovascular disorder in which the NVU becomes dysfunctional which can lead to permanent life-threatening disabilities. Considering metformin’s beneficial preclinical actions on various disorders, and the drug’s role in alleviating severity of these conditions through involvement in commonly characterized cellular pathways, we discuss the potential of metformin as a suitable drug candidate for repurposing in CNS disorders.
Keywords: Metformin, Anti- Aging, Neurodegenerative diseases, Ischemic Stroke, Tobacco smoking, Electronic cigarette vaping, Diabetes
Graphical abstract
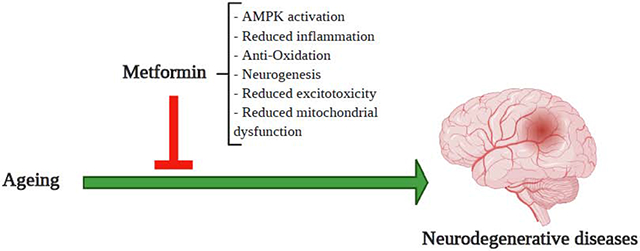
1. Introduction
Aging, a major risk factor for neurodegenerative diseases (NDs), is time dependent anatomical and physical change that reduces the functional and physiological capacity of a living organism [1]. NDs represent a wide number of diseases among which Alzheimer’s disease, Parkinson’s disease and Multiple Sclerosis are the commonly encountered ones that affect more than 6 million people in the United States [2–6]. They are generally associated with microvascular degeneration, neurovascular disintegration and blood brain barrier (BBB) dysfunction [7–9], and represent a substantial danger to human health because of their complex pathophysiology involving loss in memory and cognition which can affect a person’s ability to speak, walk and even breathe [10, 11]. Another central nervous system (CNS) disorder, ischemic stroke is one of the leading causes for mortality and long-term disability in the US with limited therapeutic approaches [12]. The most common cause for ischemic stroke is the partial or complete blockade of blood flow to the brain from a clot occlusion resulting in loss of neurological function [13, 14] Unfortunately, there are few or no efficient treatments available for slowing down the progress of these diseases, which can be attributable to age as one of their major risk factors.
Over the last two decades, there have been limited drugs approved as a first line treatment by the Food and Drug Administration (FDA) for NDs and ischemic stroke with probability of just 3% for CNS drugs getting launched after entry into the phase-I clinical trial [15–18]. Therefore, development of effective therapies is pivotal but only will arise from extensive understanding of mechanism of each disease. Additionally, repurposing of drugs that have shown significant results in pre-clinical studies could accelerate developing effective therapies [19]. Drug repurposing is a strategy that involves searching for new applications of prevailing therapeutics that will allow drugs to reach a greater number of patients for a wider indication. This procedure can bypass several steps of drug development including determination of mechanism of action, formulation and pharmacokinetics which would take on average 10-17 years in contrast to development time of 3-12 years for repurposed drugs [20, 21]. Interestingly, there are 103 compounds listed by geroprotectors.org to facilitate drug repurposing approaches in targeting aging related CNS diseases that have already been approved for use in humans [22]. This database compiles data of existing substances through pharmacological modeling and biostatistical analysis. One of them is the most promising anti-diabetic drug, metformin, because of substantial experimental studies supporting its beneficial effects in CNS diseases.
2. Metformin
Metformin, class biguanide, is a synthetic derivative of French Lilac (Galega Officinalis), a herbal plant traditionally employed in Europe for diabetes treatment [23]. In 1957 French diabetologist Jean Sterne first published the drug’s properties and result of administration in humans for diabetes [24]. The drug was first approved for use in UK in 1958 and became available in the British National Formulary in the same year [25]. Metformin was first approved by the FDA in the year 1994 for treatment of type 2 diabetes mellitus (T2D). In 2009, metformin got approval by American Diabetic Association (ADA) and the European Association for the Study of Diabetes (EASD) as a first line oral anti-diabetic drug because studies showed drastic improvement in morbidity and mortality [26]. Metformin lowers the glucose level in the blood not by sensitizing insulin secretion from beta cells of the pancreas but inhibition of peripheral and hepatic glucose production. Thus, the drug does not cause hypoglycemia [27]. Moreover, there have not been any major safety concerns on the usage of the drug for over sixty years [28]. The only exception is that the drug has been reported to cause lactic acidosis in high doses. As a result, the drug is not recommended in severe liver impairment and chronic kidney disease patients [29]. Outside of its application in the treatment of diabetes, metformin has anti-inflammatory, anti-cancer, cardioprotective, hepatoprotective and antioxidant properties and currently it is being investigated as a drug directly acting on the CNS [30–32]. With its mild side effects and a multi-action drug profile, metformin is a favorable candidate for repurposing.
Considering metformin’s pharmacokinetic profile, there have been numerous studies that are able to demonstrate a clear picture of how the human body pharmacokinetically processes this drug [33–36]. Metformin is slowly and incompletely absorbed from the gastrointestinal tract at doses of 0.5-2 grams per day and the bioavailability after oral administration is 50-60%. Additionally, the drug is not bound to plasma proteins and has an apparent volume of distribution (Vd) of approximately 600L after an oral administration of 2 grams. Moreover, this high value of Vd of metformin suggests that the drug can be absorbed by various tissues across the body. The drug is not affected by any significant biotransformation by liver or biliary secretion and finally it is eliminated through active renal tubular secretion with an elimination half-life of 4-5 hours.
Metformin has capacities to balance survival and death signaling in the primary neurons which improves energy metabolism, oxidative stress and proteostasis [37]. Pre-clinical studies have suggested that the AMP-activated protein kinase (AMPK) dependent mechanisms of the drug are responsible for the antioxidative effects in aging related CNS disorders [31, 38, 39]. Basically, the drug exerts its activities with two proposed mechanisms; AMP-activated protein kinase (AMPK) dependent and AMPK independent manner [40]. Firstly, Metformin inhibits the complex I in mitochondrial respiratory chain and decreases cellular respiration. This increases ADP:ATP and AMP:ATP ratios which activates the AMPK and leads to increased production of ATP and decreased consumption [41]. Also, AMPK activation by metformin inhibits phosphorylation of acetyl-CoA carboxylase (ACC) that increases β oxidation and fatty acid uptake which ultimately results in improved lipid metabolism and insulin sensitivity [42]. Furthermore, AMPK activation promotes glucose consumption and inhibits glucose output [43]. Secondly, an AMPK- independent mechanism of metformin inhibits mitochondrial glycerophosphate dehydrogenase which decreases gluconeogenesis [44]. Moreover, metformin’s role in reducing hyperglycemia as well as hyperinsulinemia, known accelerators of aging, makes it an attractive anti-aging drug [31].
The NF-κB pathway is one of the most prominent inflammatory mediators of aging. A bioinformatic analytical study from various aged tissues showed that NF-κB is the most associated transcription factor altered in gene expression during aging [45]. Similarly, reducing NF-κB activity has been reported to decrease accelerated aging in a mouse model of progeria [46]. In fact, its activation leads to transcription of gene coding for survival signals, inflammatory cytokines, cell cycle modulators and finally angiogenic and growth factors, which eventually creates a replenishing tumor growth environment [47]. Metformin inhibits the phosphorylation of IκB and IKKα/β by preventing the translocation of NF-κB to the nucleus [48]. These effects support AMPK independent activation and further provide evidence for metformin’s potential as an anti-aging drug and possibly as an anti-neoplastic agent.
The mechanistic target of rapamycin (mTOR) is a nutrient response pathway whose inhibition extends lifespan in animal models and ensures protection against age related conditions. Thus, drugs that target the mTOR pathway can be possible therapeutic options in treatment of aging related diseases [49]. Metformin has been demonstrated to depress the mTOR pathway in both an AMPK dependent, where it inhibits protein synthesis via mTOR and suppresses ATP consumption, and an AMPK independent manner through downregulation of insulin like growth factor-1 (IGF-1). This leads to cell growth, proliferation and autophagy [50].
3. The Neurovascular Unit (NVU) and CNS disorders
The BBB is the major site of blood and CNS exchange at the brain microvessel endothelium level [51]. It is a dynamic structure regulates several mechanisms within the brain that involve changes in tight junctional function and activity of enzymes and transporters present at the brain microvascular endothelium. This dynamic barrier accounts for synaptic signaling, neurotransmission, restricted entry of macromolecules into the brain, protection from neurotoxins and selectively provide the necessary supply of solutes and nutrients to the brain [52]. Brain endothelial cells (BECs) are the primary anatomical unit of the BBB, providing a physical separation between the blood and CNS. The “barrier” function of the NVU is conferred by four distinct phenotypes of the endothelial cells that include expression of efflux transporters, metabolic enzymes, tight junctional proteins and reduced levels of pinocytosis [53, 54]. This BBB phenotype of BECs, is greatly influenced by the surrounding cells that function collectively as the NVU. The other important cell types for the BBB induction and maintenance are pericytes, which share a basement membrane with the brain endothelium and astrocytes, and reside in close proximity to the BBB [55]. Neurons, the extracellular matrix, microglia and oligodendrocytes are the other components of the NVU that are responsible for maintenance of normal BBB function under both physiological and inflammatory conditions [56]. Figure 1 illustrates the paracrine interactions between NVU cells and their effects on each other.
Figure 1: Functional interactions among neurovascular unit components.
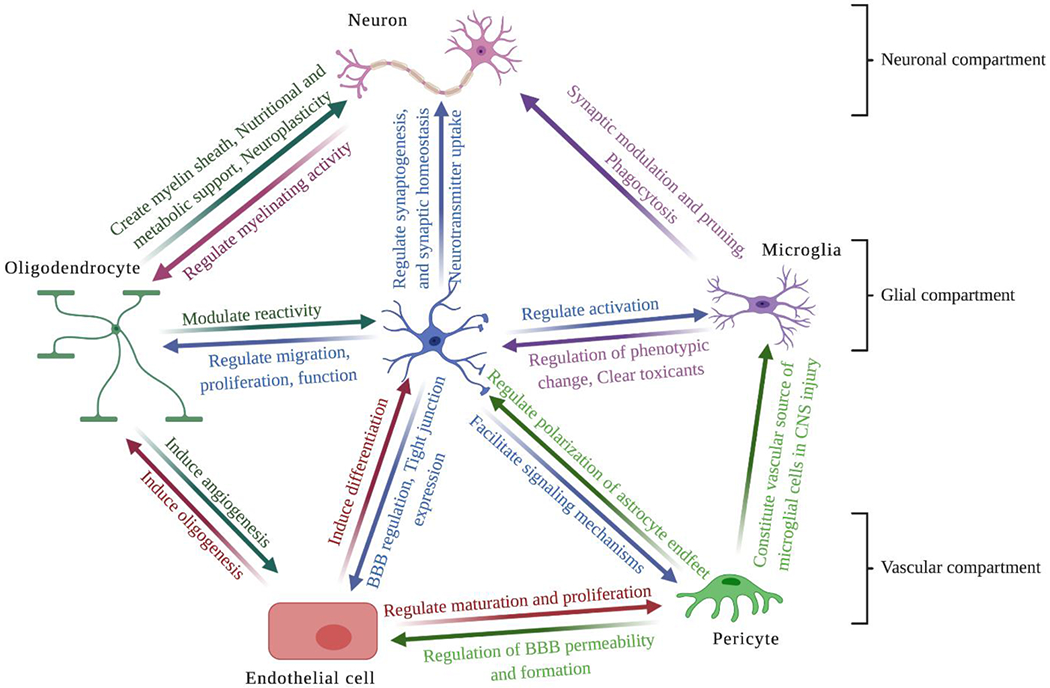
Interactions between the neuronal, glial and vascular compartments within the NVU are crucial for maintaining normal function of the brain. Also, different cells within each compartment are functioning and interacting cooperatively to regulate brain homeostasis.
3.1. Pathophysiological changes in the components of NVU with aging in CNS diseases
The global decrease in cerebral blood flow as well as neurovascular uncoupling are considered as hallmarks of normal aging and neurodegenerative diseases [57]. There is reduced expression of specialized tight junction proteins such as occludin, claudin-5 and occludens-1 in the NVU that causes BBB dysfunction [58–60]. Additionally, pericyte deficiency results from their detachment from endothelial cells [9]. This causes neurotoxic macromolecules to leak or accumulate into the brain. Moreover, there is accumulation of iron in astrocytes which reduces expression of ceruloplasmin in the CNS [61]. Excessive iron can generate free radicals and cause BBB alteration. Furthermore, detachment of astrocytic end foot from the BECs results in weak communication between neurons and the endothelium, eventually causing neurovascular uncoupling and hence neurodegeneration [62].
Data from a human study showed significant increase in albumin leakage through the BBB in aged healthy individuals compared to young healthy population [63]. Similarly, morphological changes in the BBB and leakage of albumin and IgG to the brain parenchyma were confirmed for brain regions that encompass cognitive functions, such as the hippocampus [64]. Furthermore, some brain regions of old senescence-accelerated prone mouse strain 8 (SAMP8), showed increased transport of TNF-α [65]. Also, in both physiologically aged and SAMP8 mice, the BBB has decreased expression of GLUT-1 transporter, which results in decreased transport of necessary glucose to the brain [66]. The activity of efflux transporter, P- glycoprotein, is also decreased in experimental models of ageing which results in reduced elimination of toxins from brain to the blood capillaries [67].
Hypoxia increases the permeability of various macromolecules such as albumin and dextran across the BBB [68, 69]. This occurs as a result of increased exposure to free radicals and/or inflammatory cytokines at the levels of tight junction proteins. Angiogenic factors like vascular endothelial growth factor (VEGF) and hypoxia-induced factor 1-α decrease with age. Brain-derived neurotrophic factor, a growth factor secreted by both neurons and BECs, which contributes to synaptic plasticity, decreases with age. Furthermore, age-related decreases in nitric oxide bioavailability alters BECs sensitivity to VEGF. Figure 2 illustrates the effects of ageing on the NYU components and the subsequent outcomes.
Figure 2:
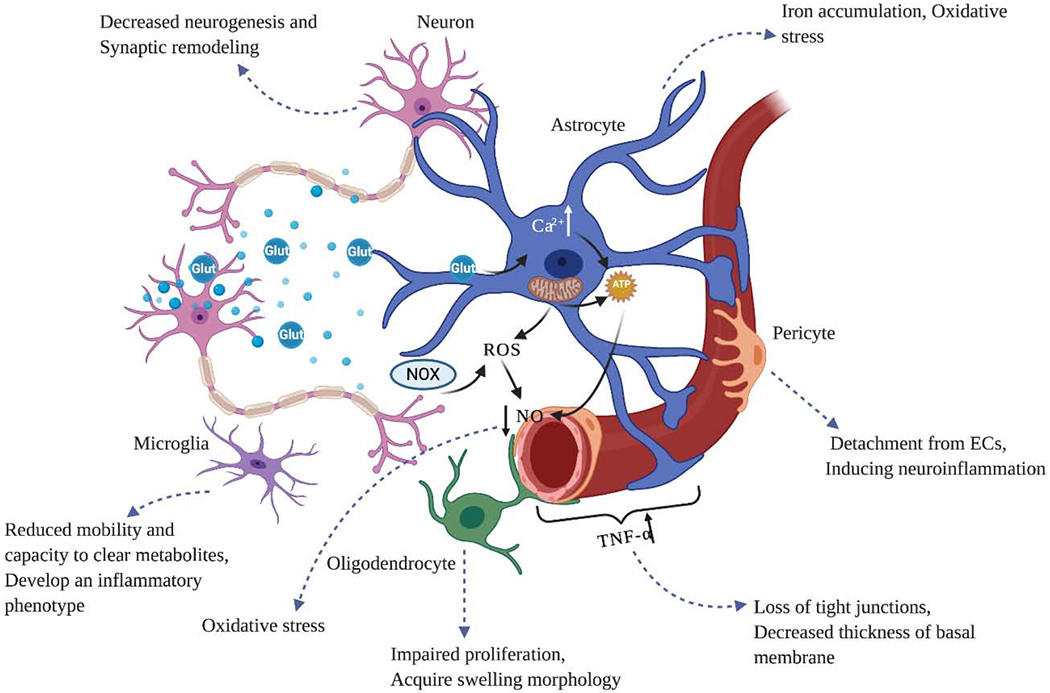
Effects of ageing on the NVU components and their interactions. Ageing can influence all the cells within the NVU and results in neurovascular uncoupling and neurodegeneration. In endothelial cells, ageing induces the secretion of pro-inflammatory factors such as TNF-a, IL-1B and VCAM-1, tight junctional protein disruption as a result of chronic neuroinflammation, and BBB impairment. It also increases astrocytic reactivity and impairs the role of astrocytic endfoot in connecting neurons and endothelial cells. BBB disruption and loss of communication with other cells result in neuronal dysfunction and neurodegeneration. Therefore, the regenerative capacities of neurons including synaptic plasticity, neurogenesis and synaptogenesis decrease significantly.
3.2. Regional brain distribution of metformin
Pre-clinical studies have shown that with a single oral dose of 50-150 mg/kg, metformin can cross the BBB [70, 71] and it was able to restore brain AMPK activity [72, 73]. However, because of disturbances of AMPK activity in inflammatory CNS conditions such as NDs and ischemic stroke, it is important to determine the drug’s specific pharmacological target within the brain. The analysis of brain specific distribution of metformin under normal and inflammatory conditions in one study showed a higher level of drug accumulation in pituitary gland, olfactory bulb, striatum and hypothalamus [74]. In contrast, the hippocampus, cerebellum and frontal cortex showed low accumulation of the drug in inflamed brain samples. Furthermore, it was suggested that various concentrations of the drug in different parts of brain may result from either up or down regulation of determinants of intracellular accumulation of the drug, such as membrane-bound organic cationic transporters or mitochondria. It is apparent that future studies are required to determine the regional brain distribution of metformin and decipher the drug’s action in molecular level.
4. CNS disorders and role of Metformin
4.1. Alzheimer’s disease
Alzheimer’s disease (AD) is the most common form of dementia and progressive neurodegenerative disorder that causes memory loss and cognitive dysfunction. In fact, with growing evidences of AD’s pathogenesis it could be suggested that impaired insulin signaling is responsible for neurodegeneration [75]. Interestingly, studies have shown that metformin has potent anti-inflammatory actions and it ameliorates AD-associated neuropathological changes and decreases cognitive impairment in diabetic mice model and also in patients with T2D [76, 77]. A study showed that oral and intranasal metformin administration can improve memory and cognition in AD patients [78]. Furthermore, another study compared data of the pro-neurogenic potential of metformin to that of donepezil, a first-line acetylcholinesterase inhibitor, in an aluminum chloride induced mouse model of neurodegeneration [79]. The study concluded that the mice treated with metformin exhibited an enhanced number of post-mitotic NeuN positive cells compared to that treated with donepezil. Therefore, metformin mediated neurogenesis could eventually be a potential treatment option in neurodegenerative diseases that cause cognitive impairment.
Pathologically, AD is characterized by deposition of intracellular neurofibrillary tangles and extracellular amyloid-β(Aβ) plaques. The extracellular deposition of Aβ plaque surrounded by microglia and astrocytes is reported to be due to overactivation of glia which causes excessive release of pro-inflammatory factors such as IL-1β and TNF-α [80, 81]. However, activation of glia is important for the clearance of Aβ deposition through phagocytosis, linking dual role for neuroinflammation on Aβ pathology [82, 83]. Epidemiological studies have shown that long term use of anti-inflammatory agents has helped in reducing risk of developing AD [84]. However, with these agents, contradictory results in AD treatment have been observed, highlighting the need for more specific anti-inflammatory therapies. Interestingly, metformin administration was shown to attenuate spatial memory deficits, decrease Aβ plaque load and chronic inflammation via activation of microglia and astrocytes as well as pro-inflammatory mediators in the hippocampus and cortex of APP/PS1 mice [82, 83, 85, 86]. These studies suggested that metformin can enhance functional recovery via regulating AMPK, mTOR and NF-kB signaling pathways in hippocampus, thus improving neurologic defects.
4.2. Parkinson’s Disease
Parkinson’s disease (PD) is one of the most common NDs that is accompanied by progressive loss of dopaminergic neurons in the substantia nigra compacta and aggregation of Lewy bodies and Lewy neurites in various parts of the affected brains [87, 88]. Mitochondrial dysfunction, oxidative stress, neuroinflammation, ubiquitin proteasome system and disturbed proteostasis due to impaired autophagy-lysosomal pathway are the hallmarks of PD [89, 90]. A previous study demonstrated that metformin reduces the number of nonfunctional mitochondria and ROS generation by promoting mitophagy in an AMPK-dependent manner [91]. Additionally, metformin was shown to activate the ATF2/CREB pathway in an AMPK-independent manner and stimulate peroxisome proliferator-activated receptor gamma coactivator-1a (PGC-1a) and its target genes [92]. PGC-1a is a transcriptional cofactor that is actively involved in regulation of mitochondrial anti-oxidative defense mechanism [92]. Moreover, in the mid brain of MPTP mouse model of PD, metformin restored anti-oxidative mediators such as superoxide dismutase, catalase and glutathione [93]. PD is characterized by various motor and non-motor symptoms such as rigidity, bradykinesia, freezing of gait, cognitive abnormalities and depression [94].
The effect of metformin on the substantia nigra, a critical brain structure affected in PD, has not been studied in detail. Nevertheless, data from a study suggests effect of metformin on microglial activation in the substantia nigra in a pro-inflammagen lipopolysaccharide (LPS) at both the cellular and molecular levels [95]. This study supports that metformin inhibits microglia activation as measured by immunoreactivity markers and it minimizes expression of pro and anti-inflammatory cytokines by phosphorylation of mitogen activated protein kinase (MAPK) as well as by ROS generation through inhibition of NADPH enzyme. However, metformin failed to protect the dopaminergic neurons of the substantia nigra in response to intranigral LPS.
4.3. Multiple sclerosis
Multiple sclerosis (MS) is a chronic demyelinating disease characterized by delayed remyelination of axons making it susceptible to irreversible degeneration. This substantiates progressive neurologic decline as associated with later stages of MS and can extend over decades [96]. The delayed remyelination occurs with aging due to delayed differentiation of oligodendrocytes progenitor cells (OPCs) to oligodendrocytes, the myelin forming cells of the CNS [97, 98]. In fact, studies demonstrate that regulatory mechanism that control OPC differentiation are nonfunctional in aging brain [99, 100]. Additional studies found that chronically demyelinated MS lesions contain OPCs that were undifferentiated and increase in number of OPCs in white matter lesions of aged animals [101, 102]. These OPCs did not contribute in remyelination, indicating that differentiation of OPCs into oligodendrocytes is crucial for remyelination. It has also been shown that, remyelination could be enhanced by adding pro differentiating factors lacking in aged brain [103, 104]. However, aged OPCs differentiate very slowly and become unresponsive to pro-differentiation signals. The diminished functional capacity is associated with hallmarks of cellular aging such as mitochondrial dysfunction, unfolded protein response, autophagy, NF-kB and p-38 MAPK signaling. When metformin was used as a pharmacological approach targeting endogenous OPCs, it stimulated remyelination via AMPK-dependent mechanism and led to increased mitochondrial function as required for differentiation of OPCs. Also, metformin reduced oxidative stress with activation of antioxidative defense in oligodendrocytes exposed to cytokines via AMPK activation. Hence, metformin has potential to limit neurologic deficits in MS and related neurodegenerative diseases. In Table 1 we have summarized some of the effects of metformin treatment in NDs from experimental studies.
Table 1:
Effect of Metformin treatment in Neurodegenerative diseases in experimental animal models and cultured cells
| Neurodegenerative disease | Description of study | Outcome(s) | References |
|---|---|---|---|
| Alzheimer’s disease | -Male db/db mice -200 mg/kg/day intraperitoneal metformin for 18 weeks | -Metformin mitigated the increase of total and phosphorylated tau and decreased the JNK activation. -Attenuated the reduction of synaptophysin |
[77] |
| -Primary cortical neurons and Neuro2a neuroblastoma cells treated with metformin | -Metformin significantly increased the generation of intracellular and extracellular AB species -Upregulated transcription of B-secretase (BACE1) -In combination with insulin, metformin enhances insulin’s effect in reducing AB level |
[71] | |
| -In vitro model of “type 3 diabetes” -Differentiation of neuronal cell line Neuro-2a under prolonged presence of insulin and treatment with metformin |
Metformin ameliorated neuronal insulin resistance Insulin sensitization by metformin prevented AD- associated neuropathological changes |
[105] | |
| -db/db mice treated with 200 mg/kg metformin by oral gavage | -Metformin decreased AB influx across the BBB and decreased level of AB in hippocampus -Significant reduction of nuclear NF-kB p65 of brain microvessel endothelial cells -Suppression of caspase-3 activity and inhibited neuronal apoptosis |
[106] | |
| -Primary cortical neurons treated with up to 2.5 mM metformin for 1-24 hours | -Metformin induces PP2A activity by inhibiting mTOR activity and reduces tau phosphorylation | [107] | |
| Human neural stem cells exposed to AB and treated with metformin | -Decreased Aβ-mediated mitochondrial deficiency -Significant restoration of mitochondrial morphology -Rescued cell viability through AMPK pathway activation |
[108] | |
| Parkinson disease |
-Adult male swiss albino mice - Induction of parkinsonism in mice -Post metformin 500mg/kg treatment for 21 days |
-Long-term metformin treatment resulted in significant improvement of the locomotor and muscular activities - Brain-derived neurotrophic factor significantly increased in metformin treatment group |
[93] |
| -Ten-week-old male C57BL/6 mice Induction of parkinsonism in mice Treatment with metformin 5mg/ml in drinking water for 5 weeks |
Attenuated degeneration of substantia nigra compacta dopaminergic neurons by inhibiting microglial overactivation induced neuroinflammation Elevated striatal dopaminergic levels and improved motor impairment |
[91] | |
| -10-week-old adult male C57BL/6 mice -MPTP injection (30 mg/kg/day) for the first 7 days to induce PD -Post metformin (200 mg/kg/day) for the next 7 days |
-Metformin rescued tyrosine hydroxylase-positive neurons and attenuated astroglial activation in the nigrostriatal pathway. -Metformin restored dopamine depletion and behavioral impairments exerted by MPTP -Metformin ameliorated MPTP-induced synuclein phosphorylation which was accompanied by increased methylation of protein phosphatase 2A (PP2A), a phosphatase related to α-synuclein dephosphorylation |
[109] | |
| -In-vitro pre-metformin treatment in SH-SY5Y cells | -Metformin’s AMP activation induced microtubule mediated autophagy and eliminated mitochondrial reactive oxygen species - Reduced MPP+ induced cytotoxicity and neuronal apoptosis |
[91] | |
| Multiple Sclerosis | - C57BL/6 J mice were administered with 0.2% cuprizone for 5 weeks for demyelination induction. -Post metformin treatment of 50 mg/kg/day for 1 weeks |
-Increased localization of precursor oligodendrocytes and their renewal in the corpus callosum via AMPK/mTOR pathway -Reduced brain apoptosis markers and attenuated motor dysfunction |
[110] |
| Male C57BL/6 mice were fed with 0.2% cuprizone plus metformin by oral gavage of 100 mg/kg body weight in saline every day from day 0 until to the end of 6 weeks one time in a day |
-The myelinated axons within corpus callosum of cuprizone-induced demyelination animals increased after administration of metformin -Metformin ameliorated the oxidative stress induced by cuprizone -Metformin upregulated expression of mitochondrial biogenesis genes -Metformin ameliorated the oxidative stress induced by cuprizone -Astrogliosis and microgliosis were decreased after metformin administration while it enhanced the number of oligodendrocytes |
[111] | |
| -Female C57BL/6 wild-type mice, 8– 10 weeks of age -Induction of experimental autoimmune encephalomyelitis induced by subcutaneous injection of 250 μg of MOG35-55 peptide -100 mg/kg Metformin dissolved in saline solution was intraperitonially administered for 20 days |
-Metformin reduced Th17 and increased Treg cell percentages along with the levels of associated cytokines -Metformin inhibited activation of mTOR and its downstream target, HIF-1α |
[112] |
4.4. Ischemic stroke
Ischemic stroke can result in cerebral ischemia/reperfusion (I/R) injury, which is characterized by transient loss followed by rapid return of the blood flow to the brain and can lead to a higher amount of neuronal death, enhanced brain infarction and substantial cognitive dysfunction compared to ischemia alone [113, 114]. The ischemic cascade follows in seconds to minutes after the blockage and is comprised of multiple, continuous biochemical events leading to severe focal hypoperfusion, excitotoxicity and oxidative damage eventually leading to BBB dysfunction and inflammation [115]. White matter injury, as well as damage and death of astrocytes, also contribute to cerebral impairment [116, 117]. Inflammation initially causes release of harmful radicals and cytokines; however, it also enables synaptic remodeling that helps to remove damaged tissues [118]. Similarly, glial cells promote angiogenesis and neurogenesis to regulate the BBB, but conversely forms glial scar that may further prevent neuronal plasticity [119]. Below we have discussed the pathways through which metformin exerts its protective effect in ischemic stroke.
4.4.1. AMPK activation
As with NDs, AMPK activation is believed to be metformin’s key pathway involved in ischemic stroke. There are several studies suggesting that metformin acts through the AMPK signaling pathway in reduction of the cerebral infarct area and neuronal apoptosis in ischemic rat models [39, 120, 121]. Metformin’s AMPK activation in suppression of post ischemic neuroinflammation has been reported to be either because of NrF2 anti-oxidant pathway activation or inhibition of NF-κB cascade [38]. An in-vitro study showed that metformin treatment following oxygen-glucose deprivation and reperfusion (OGD/R) reduces NF-κB and intercellular adhesion molecule-1(ICAM-1) in BECs [38]. Similarly, metformin reduced TNF-a induced inflammation in BECs by activation of AMPK [122]. Furthermore, metformin treatment improved apoptosis of primary fetal rat derived hippocampal neurons that were subjected to OGD [123]. An in-vitro study showed metformin’s effect in primary rat cortical astrocytes subjected to OGD and found out that AMPK is critical for the regulation of apoptosis in a caspase independent manner [124].
AMPK consists of an α catalytic subunit and a β and γ regular subunits and all of them play important roles in regulation of energy metabolism [125]. All these subunits are highly expressed in neurons, glia and astrocytes of the NVU. Studies have shown that diverse cellular combination of AMPK subunits may account for various effects of AMPK activation following stroke. For instance, in an experimental stroke model, systemic gene depletion of α-2 subunit confers neuroprotection while cell-specific deletion of α-2 subunit in astrocytes after cerebral ischemia has been found to be detrimental [126]. Additionally, administration of metformin caused increased neuronal damage in post-ischemic conditions [127]. The differential effects of metformin are contributable to several causes that include type and length of ischemia, dosage, duration and route and timing of administration of metformin and co-existing conditions like diabetes [128]. chronic kidney diseases [129] and tobacco smoking [130]. Figure 3 illustrates the neuroprotective effects of metformin in ischemic stroke through AMPK signaling pathway.
Figure 3:
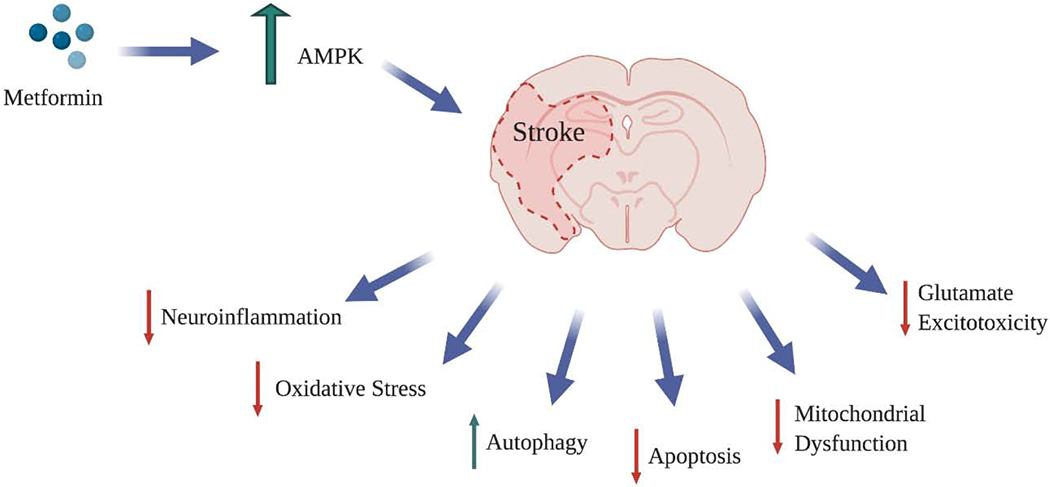
Neuroprotective effects of metformin in ischemic stroke through AMPK signaling pathway. During an ischemic event, a series of neuroprotective mechanisms are stimulated by activated AMPK in order to maintain the energy homeostasis of the brain. Activated AMPK promotes autophagy, reduces neuroinflammation and oxidative stress by decreasing the levels of inflammatory factors (NF-KB, TNF-B, IL-1B, IL-6) and ROS production respectively. Also, it restrains apoptosis, glutamate excitotoxicity through inhibiting glutamate release and mitochondrial dysfunction therefore, promoting energy metabolism and glucose uptake.
4.4.2. Alleviation of oxidative stress mediated inflammation
ROS induced tissue damage is one of the most important components of cerebral I/R injury [131]. An I/R event leads to detrimental effects to the BBB, causing increased leucocyte infiltration into the brain tissue due to the upregulation of ICAM-1 and activation of microglia that leads to increased production of ROS and RNS [132]. Therefore, alleviating the damage caused by I/R injury induced by oxidative stress can be promising in the treatment of ischemic stroke. A study showed that metformin treatment significantly increased endogenous antioxidants enzymes such as superoxide dismutase (SOD) and glutathione (GSH) levels in the brain tissue [133]. This reversed the oxidative injury caused by increased levels of ROS. Moreover, metformin treatment following I/R injury leads to reductions in both the expression of ICAM-1 and subsequent leucocyte infiltration. This was confirmed with decreased myeloperoxidase+ and GrL+ cells after I/R event in brain tissue [38].
An I/R event leads to upregulation of microglia M1 signature genes including IL-1B and CD32, leading to inflammation. It was shown that post metformin treatment reversed the polarization of microglia M1 to M2 signature genes, including CD206 and arginase-1, as well as anti-inflammatory cytokines IL-4 and IL-10, resulting in reduced brain inflammation [120, 134]. Moreover, a study conducted in LPS stimulated microglial cells, used to mimic an I/R event and treated with metformin, showed that the cells produced higher levels of anti-inflammatory cytokine, interleukin-10, compared to control group [120]. This suggests that metformin could have anti-inflammatory activity following I/R injury in microglial cells. Also, elevated numbers of angiogenic structures were seen with bEnd.3 cells when they were exposed to medium that contained metformin-treated microglia.
4.4.3. Neurogenesis
It has been shown that, following an ischemic injury, there is an increased production of neurons and subsequent neuroblast migration to damaged area of the brain as a part of endogenous repair mechanism [135]. Hence, therapies that promote post-ischemic neurogenesis can be a potential target for ischemic stroke. A study tested long-term neuroprotection effects of metformin following hypoxic ischemia injury and found reduced neuronal degeneration in the CA1 region of hippocampal area [136]. The CA1 region of hippocampus is highly sensitive to HI injury. Similarly, metformin treatment reversed increasing levels of cleaved caspase 3, promoted anti-apoptosis protein BCL-2 expression, inhibited pro-apoptosis protein BAX in the cortex and hippocampus.
Another study confirmed that metformin promoted neuroblasts proliferation and differentiation in the hippocampal area [137]. It was supported that metformin enhances regulation of neurogenesis and has a likely contribution in treatment of cerebral ischemic injury. An I/R event can also induce activation of astrocytes leading to increase in production of glial fibrillary acidic protein (GFAP). This causes formation of glial scar in the brain that eventually reduces regeneration of neurons, impeding recovery after an ischemic event [138, 139]. Also, metformin treatment post I/R injury reduces GFAP+ cells, suggesting the regeneration of neurons and thereby functional recovery [140].
4.4.4. Other important pathways
PI3K/Akt1/JNK3/C-Jun pathway is one of the crucial signaling pathways in cell survival. Cellular growth factors such as platelet derived growth factor can stimulate release of phosphatidylinositol 3 Kinase (PI3K) after activation of Akt1, which simultaneously inactivates c-Jun N- terminal kinase-3 (JNK-3) molecule that is found in heart and brain. JNK-3 is associated with expression of apoptotic proteins and is negatively correlated with neuronal survival in stroke model [141]. Interestingly, a study has demonstrated the involvement of this pathway when metformin improved impairment of hippocampal controlled behaviors and reduced cell apoptosis in an I/R rat model.. Thus, PI3K/Akt1/JNK3/C-Jun pathway incorporates metformin’s neuroprotective role in hippocampal deficits caused by I/R injury [142]. Additionally, pre-treatment of BECs isolated from a diabetic rat model with metformin and exposure to OGD/R, resulted in reduced formation of nitrotyrosine and p85 (PI3K regulatory subunit) nitration compared to the control group. The mechanism behind this is reduced reactive nitrogen species in the BECs that would eventually result in restored regulation of the apoptotic pathway and alleviation of I/R injury [143].
Arterial baroreflex pathway is one of the few important pathways for stroke prevention [144] and its dysfunction has been reported to be an important risk factor for the development of stroke [39]. In fact, the α7nAChR pathway, a ligand gated ion channel that is highly expressed in the macrophages of different tissues in the brain, is demonstrated to be the downstream of arterial baroreflex pathway and involved in neuroprotection against ischemic cerebral injury [145, 146].
The activation of this receptor inhibits the production of inflammatory cytokines and thus reduces local inflammatory response. Studies have shown that metformin reduces chronic inflammation by inhibiting proinflammatory cytokines levels, in both animal and human, labelling it as a promising anti-inflammatory agent [39, 147]. Additionally, metformin regulates the action of vagal nervous, one arm of arterial baroreflex in the CNS, which in turn is mediated mainly by α7nACh receptor [148, 149]. Moreover, metformin increases the life span of stroke-prone spontaneously hypersensitive rats, reducing the middle cerebral artery occlusion (MCAO) induced infarct size in the brain and upregulating the expression of α7nACh receptors. This resulted in downregulation of pro-inflammatory cytokines in serum and peri-infarct area of ischemic brain. However, this effect of metformin was markedly attenuated by deactivating the arterial baroreflex by sinoaortic denervation in rats. This further confirms the involvement of the pathway in metformin mediated protection against stroke.
4.4.5. Dose, duration and timing of metformin treatment in ischemic stroke
Investigators have conducted a study based on dose, duration and timing of metformin administration in male C57BL/6 mice that were subjected to both transient and permanent MCAO surgery [150]. A 7-day treatment of 10mg/kg prior to permanent MCAO, showed significant improvement as compared to shorter duration treatments. Therefore, pretreatment time window is an essential factor in metformin administration. In contrast, in transient MCAOs, mice did not show any significant improvement suggesting that metformin may not be beneficial in cases of blood reperfusion.
A different study treated Wistar rats with metformin at 200 mg/kg orally for 7 days and subjected to I/R for the next 7 days [151]. In the same way, the study also conducted both post and continuous (pre-post) administration and compared results with pre-administration. The results showed significant reduction in malonaldehyde level, an oxidative stress marker, in metformin pre-treatment group as compared to continuous group. However, studies have also shown that treatment with 200 mg/kg metformin for a longer duration of 14 days after transient MCAO (tMCAO) reduced the brain atrophy volume [30, 122]. Below we have summarized the results of various studies based on the timing and duration of metformin treatment in experimental stroke models.
5. Metformin’s role in ischemic stroke comorbidities
T2D and smoking are common risk factors, as well as coexisting conditions, that can aggravate ischemic stroke prognosis and recovery [163–165]. People with diabetes have higher risk of developing ischemic stroke and myocardial infarction because of increased atherosclerosis and endothelial cell dysfunction, concomitantly causing slower recovery after cerebral I/R injury [166]. A diabetic situation can promote higher bleeding following an ischemic event slowing down the brain recovery process [167]. In addition, T2D has been shown to impair post stroke reparative neovascularization and impede the recovery due to vascular regression in the brain [161]. One study suggests that metformin reduces post stroke nitrotyrosine levels in the brain parenchyma and cerebrovasculature in a diabetic rat model [143]. Furthermore, these diabetic rats were observed to have increased caspase-3 activation with I/R injury induction and treatment with metformin reduced caspase-3 cleavage causing reduced apoptotic cell death. Interestingly, a recent study has suggested exacerbation of ischemia in chronic kidney disease-induced female mice and reports that chronic pre-treatment with metformin is beneficial in stroke recovery. The underlying mechanism is AMPK phosphorylation and activation of canonical NF-kB pathway, as shown by decreased expression of microglia/macrophages M1 signature genes within the ischemic lesions of CKD-induced mice treated with metformin [129].
Tobacco smoking (TS) is considered as a contributing etiology in some NDs and stroke. Moreover, TS promotes glucose intolerance and increases risk of developing T2D [168]. Several studies have shown that TS is associated with decreased vascular endothelial function in a causative and dose dependent manner [169, 170] which is primarily related to ROS content of tobacco smoke, nicotine and oxidative stress driven inflammation [171, 172]. This suggests that TS shares common pathogenic traits similar to that of T2D in stroke [173]. Furthermore, studies show that the release pattern of angiogenic, oxidative and inflammatory factors of BECs in response to hyperglycemia and TS-induced stroke conditions are similar, which supports common pathogenic involvements in BBB impairment [39]. A study investigated effects of nicotine exposure on neuronal glucose utilization in an in-vitro stroke model and found out that it caused decreased neuronal GLUT-1 uptake, up-regulated α7 nicotinic acetylcholine receptor and eventually led to decreased glycolysis. This caused a state of glucose deprivation at the NVU in the brain and could possibly lead to enhanced ischemic brain injury and/or stroke risk [164] [174]. Furthermore, studies from our group show that metformin also activates the Nrf2 pathway, independent of AMPK phosphorylation, in both in-vitro and in-vivo cigarette smoke-induced cerebrovascular models [175]. Metformin induced renormalization of tight junction proteins, BBB integrity, inflammation and oxidative stress markers, and expression of Glut-1 and thrombomodulin.
Electronic Cigarettes (E-Cigs), also known as Vapes, Blues or Juuls, have gained popularity among young adults, as they are marketed as a safer alternative to TS. It is also believed to be a useful tool for smoking cessation, however information on nicotine dependence and various cerebrovascular toxicities from e-cigs remain less known [176, 177]. A recent Surgeon General’s Report showed that it is challenging to make conclusions about the efficacy of e-Cigs for cessation based on clinical trials, mainly due to the heterogenous group of products[178]. In addition, there are limited studies on effects of e-cigs on stroke outcome in comparison to that of TS. Investigations from our lab have attempted to decipher the brain effects of nicotine in both adult and adolescent rodent models. We have shown that exposure to nicotine and e-Cig vapor in adult mice causes downregulation of brain GLUT1 and GLUT3 expression and leads to decreased brain glucose uptake under both normoxic and ischemic conditions [164]. Another study compared effects of e-cigs versus that of TS in brain and found out that oxidative stress promoted by e-cigs extract was not dissimilar from that induced by extracts from traditional ones on BECs in a mouse model. In fact, they found out that both of their exposures worsen stroke outcomes in mice that underwent tMCAO by downregulating NrF2 and thrombomodulin. Moreover, animals that received daily doses of metformin with either TS or e-cigs exposure, exhibited better stroke outcome as compared to the untreated counterparts. This was demonstrated by reduced infarct size and better neurological scores in animals receiving metformin. Also, the NrF2 levels in brain of these animals were significantly higher compared to untreated group [130]. Additional studies in our lab have shown that prenatal e-Cig exposure increases the sensitivity of offspring to neonatal hypoxia. This caused motor and cognitive deficits and enhanced edema in adolescent offspring after neonatal hypoxia. Interestingly, prenatal e-Cig exposure decreased brain GLUTs expression in offspring after neonatal hypoxia [179]. Future studies should investigate the protective effects of metformin in the developing as well as aged brain, with respect to NDs and ischemic stroke.
6. Conclusions and future directions
Despite significant advances in the development of therapeutics for symptomatic treatment of neurodegenerative diseases, there is still a critical demand for novel therapeutic agents that can modify diseases and target the contributing pathways. Numerous studies mentioned in this review suggest metformin as a potential new treatment for neurodegenerative diseases and ischemic stroke because it has been demonstrated to improve functional recovery through affecting the injury mechanisms in experimental animal models. It has also been shown that metformin has promising effects on cognition and could be effective in counteracting the deleterious effects of aging which is due to its ability to affect several biological pathways including energy production. Metformin easily crosses the BBB and distributes in several brain regions. Therefore, safety, pharmacokinetic profile and multi-functional properties of metformin, make it a promising neurotherapeutic agent. However, it is still unclear what brain levels are necessary at what time to exert the neuroprotective effects and what type of regional brain distribution is necessary for protective efficacy. Hence, brain pharmacokinetic, biodistribution, time window selection and focused molecular studies are warranted in diseased animal models to further develop metformin as a neurotherapeutic to treat NDs.
Table 2:
Therapeutic effects and underlying mechanism(s) of action of metformin in experimental stroke models based on timing/duration of metformin treatment.
| Mechanism of metformin | outcome | Timing/duration of metformin treatment | References |
|---|---|---|---|
| AMPK activation and AMPK-dependent M2 polarization of microglial cells | -Improved angiogenesis and neurogenesis -Functional recovery | Post- stroke chronic (30 days) treatment | [152] |
| AMPK activation, promoted eNOS phosphorylation | -Reduced ischemia-induced brain atrophy volume -Promoted focal angiogenesis and neurogenesis |
Post- stroke (2 weeks) treatment | [121] |
| AMPK activation, modulating inflammatory and antioxidant pathways | -Attenuated cellular levels of NF-κB, TNF alpha and cyclooxygenase-2, -Increased levels of Nrf2 and heme oxygenase-1 -Enhanced levels of glutathione and catalase activities | Pre-treatment | [39] |
| AMPK activation | Chronic: -Improved stroke-induced lactate generation, -Imeliorated stroke-induced activation of AMPK Acute: -Increased infarct volume -Increased pAMPK levels |
-Pre/Post-stroke chronic (3 weeks) treatment -Pre-stroke acute (3 days) treatment |
[153] |
| AMPK activation | -Improved stroke-induced behavioral deficits -Enhanced angiogenesis |
-Post-stroke chronic (3 weeks) treatment | [140] |
| AMPK phosphorylation, NF-κB inhibition, down-regulation of cytokines and ICAM-1 expression |
-Reduced infarct volume -Improved neurobehavioral outcomes -Decreased BBB permeability |
Post-stroke (2 weeks) treatment | [154] |
| Brain NF-κB suppression, reduction of pro-inflammatory cytokines and iNOS, | - Reduced infarct volume and improved neurological deficits -Ameliorated microgliosis and astrocytosis |
Pre-treatment (3 weeks) | [147] |
| AMPK-dependent autophagy |
Improved sensory motor signs Improved anxiolytic behavior and locomotion -Decreased autophagy factors |
Pre-treatment (2 weeks) | [155] |
| improved the arterial baroreflex function, enhanced cholinergic anti-inflammatory pathway | -Up-regulation of vesicular acetylcholine transporter (VAChT) and α7nAChR - Down-regulated levels of pro-inflammatory cytokines |
Pre-treatment (3 weeks) | [156] |
| AMPK activation | -Enhanced learning and memory -Improved neurological outcomes |
Pre-treatment (2 weeks) | [157] |
| AMPK activation | -Attenuated apoptotic cell death - Induced mitochondrial biogenesis proteins |
Pre-treatment (2 weeks) | [158] |
| Suppression of nitrotyrosine formation and nitration, improving Akt phosphorylation in endothelial cells, direct antioxidative effect |
- Attenuated stroke-induced nitrative signaling -Improved angiogenesis -Prevented vasoregression-Improved functional recovery |
Post-stroke (2 weeks) treatment | [143] |
| Activation of Akt1, reducing phosphorylation of JNK3 and c-Jun, elevation of cleaved caspase-3 |
Reduced cell apoptosis Attenuated the deficits of hippocampal related behaviors |
Post-stroke (1 week) treatment | [142] |
| Pre- activation of AMPK - dependent autophagy | -Reduced infarct volume -Reduced neurological deficits -Reduced cell apoptosis |
Pre-treatment (single dose) | [159] |
| Activation of peripheral AMPK | -Reduced ischemic neuronal damage -Decreased development of post-ischemic glucose intolerance |
Post-stroke (1-3 days) treatment | [160] |
| Glycemic intervention |
Improved neurovascular repair Improved functional outcome |
Post-stroke (2 weeks) treatment | [161] |
| Glycemic intervention, AMPK activation and anti oxidative effects |
-Reduced vascular remodeling -Reduced severity of hemorrhagic transformation -Decreased edema -Improved functional recovery |
Post-stroke (4 weeks) treatment | [162] |
| Decreased expressions of total and phosphorylated AMPK | - Ameliorated brain infarct -Improved neurological scores -Reduced cell apoptosis |
Pre-treatment (1 week) | [150] |
Highlights:
Aging-related CNS disorders have complex pathophysiology with few or no treatments
Studies show that, metformin, an anti-diabetic drug, has neuroprotective action
Consideration of dose, duration and timing of treatment is important for metformin use
Metformin has the cerebroprotective potential for ischemic stroke
Metformin is a suitable candidate for repurposing in aging-related CNS disorders
Acknowledgments
Funding:
This work was supported by NINDS R01 NS#117906 to TJA
Abbreviations:
- NDS
neurodegenerative diseases
- NVU
neurovascular unit
- BBB
blood brain barrier
- CNS
central nervous system
- FDA
Food and Drug Administration
- T2D
type 2 diabetes mellitus
- Vd
volume of distribution
- AMPK
AMP-activated protein kinase
- mTOR
mechanistic target of rapamycin
- IGF-1
insulin like growth factor-1
- BECs
brain endothelial cells
- MMPs
matrix metallopeptidases
- VCAM-1
vascular cell adhesion molecule-1
- SAMP8
senescence-accelerated prone mouse strain 8
- ROS
reactive oxygen species
- VEGF
vascular endothelial growth factor
- AD
Alzheimer’s disease
- Aβ
amyloid-β
- PD
Parkinson’s disease
- PGC-1a
peroxisome proliferator-activated receptor gamma coactivator-1a
- LPS
lipopolysaccharide
- MAPK
mitogen activated protein kinase
- OPCs
oligodendrocytes progenitor cells
- I/R
ischemia/reperfusion
- OGD/R
oxygen-glucose deprivation and reperfusion
- ICAM-1
intercellular adhesion molecule-1
- MCAO
middle cerebral artery occlusion
- eNOS
endothelial nitric oxide synthetase
- GFAP
glial fibrillary acidic protein
- PI3K
phosphatidylinositol 3 Kinase
- JNK-3
c-Jun N- terminal kinase-3
- CKD
Chronic Kidney Disease
- TS
Tobacco smoking
- E-Cigs
Electronic Cigarettes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest:
The authors declare that they have no known competing for financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Uncategorized References
- 1.Hung C-W, et al. , Ageing and neurodegenerative diseases. Ageing research reviews, 2010. 9: p. S36–S46. [DOI] [PubMed] [Google Scholar]
- 2.Association A.s., 2019 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 2019. 15(3): p. 321–387. [Google Scholar]
- 3.Leray E, et al. , Epidemiology of multiple sclerosis. Revue neurologique, 2016. 172(1): p. 3–13. [DOI] [PubMed] [Google Scholar]
- 4.Lill C and Klein C, Epidemiology and causes of Parkinson’s disease. Der Nervenarzt, 2017. 88(4): p. 345–355. [DOI] [PubMed] [Google Scholar]
- 5.Maher P, The potential of flavonoids for the treatment of neurodegenerative diseases. International journal of molecular sciences, 2019. 20(12): p. 3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young JJ, et al. , Frontotemporal dementia: latest evidence and clinical implications. Therapeutic advances in psychopharmacology, 2018. 8(1): p. 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown WR and Thore CR, Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathology and applied neurobiology, 2011. 37(1): p. 56–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong Z, et al. , ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nature neuroscience, 2008. 11(4): p. 420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zlokovic BV, Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature Reviews Neuroscience, 2011. 12(12): p. 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wyss-Coray T, Ageing, neurodegeneration and brain rejuvenation. Nature, 2016. 539(7628): p. 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gitler AD, Dhillon P, and Shorter J, Neurodegenerative disease: models, mechanisms, and a new hope. 2017, The Company of Biologists Ltd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benjamin EJ, et al. , Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation, 2017. 135(10): p. e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brzica H, et al. , Role of transporters in central nervous system drug delivery and blood-brain barrier protection: relevance to treatment of stroke. Journal of central nervous system disease, 2017. 9: p. 1179573517693802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nozohouri S, et al. , Novel approaches for the delivery of therapeutics in ischemic stroke. Drug Discovery Today, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dowden H and Munro J, Trends in clinical success rates and therapeutic focus. Nature Reviews Drug Discovery, 2019. 18(7): p. 495–497. [DOI] [PubMed] [Google Scholar]
- 16.Nozohouri S, Vaidya B, and Abbruscato TJ, Exosomes in Ischemic Stroke. Current Pharmaceutical Design, 2020. [DOI] [PubMed] [Google Scholar]
- 17.Sifat AE, Vaidya B, and Abbruscato TJ, Blood-brain barrier protection as a therapeutic strategy for acute ischemic stroke. The AAPS Journal, 2017. 19(4): p. 957–972. [DOI] [PubMed] [Google Scholar]
- 18.Swalley SE, Expanding therapeutic opportunities for neurodegenerative diseases: A perspective on the important role of phenotypic screening. Bioorganic & Medicinal Chemistry, 2020. 28(3): p. 115239. [DOI] [PubMed] [Google Scholar]
- 19.Alamri FF, et al. , Delayed atomoxetine or fluoxetine treatment coupled with limited voluntary running promotes motor recovery in mice after ischemic stroke. Neural regeneration research, 2021. 16(7): p. 1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dudley JT, Deshpande T, and Butte AJ, Exploiting drug-disease relationships for computational drug repositioning. Briefings in bioinformatics, 2011. 12(4): p. 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallikarjun V and Swift J, Therapeutic manipulation of ageing: repurposing old dogs and discovering new tricks. EBioMedicine, 2016. 14: p. 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moskalev A, et al. , Geroprotectors. org: a new, structured and curated database of current therapeutic interventions in aging and age-related disease. Aging (Albany NY), 2015. 7(9): p. 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodarzi MO and Bryer-Ash M, Metformin revisited: re-evaluation of its properties and role in the pharmacopoeia of modern antidiabetic agents. Diabetes, Obesity and Metabolism, 2005. 7(6): p. 654–665. [DOI] [PubMed] [Google Scholar]
- 24.Bailey CJ and Day C, Metformin: its botanical background. Practical Diabetes International, 2004. 21(3): p. 115–117. [Google Scholar]
- 25.Fladden D, Goat’s rue-French lilac-ltalian fitch-Spanish sainfoin: gallega officinalis and metformin: the Edinburgh connection. JOURNAL-ROYAL COLLEGE OF PHYSICIANS OF EDINBURGH, 2005. 35(3): p. 258. [PubMed] [Google Scholar]
- 26.Nathan DM, et al. , Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes care, 2009. 32(1): p. 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wróbel MP, et al. , Metformin—a new old drug. Endokrynologia Polska, 2017. 68(4): p. 482– 496. [DOI] [PubMed] [Google Scholar]
- 28.Crowley MJ, et al. , Clinical outcomes of metformin use in populations with chronic kidney disease, congestive heart failure, or chronic liver disease: a systematic review. Annals of internal medicine, 2017. 166(3): p. 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flory J and Lipska K, Metformin in 2019. Jama, 2019. 321(19): p. 1926–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calvert JW, et al. , Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS–mediated signaling. Diabetes, 2008. 57(3): p. 696–705. [DOI] [PubMed] [Google Scholar]
- 31.Martin-Montalvo A, et al. , Metformin improves healthspan and lifespan in mice. Nature communications, 2013. 4: p. 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheen A, Esser N, and Paquot N, Antidiabetic agents: potential anti-inflammatory activity beyond glucose control. Diabetes & metabolism, 2015. 41(3): p. 183–194. [DOI] [PubMed] [Google Scholar]
- 33.Foretz M, et al. , Metformin: from mechanisms of action to therapies. Cell metabolism, 2014. 20(6): p. 953–966. [DOI] [PubMed] [Google Scholar]
- 34.Graham GG, et al. , Clinical pharmacokinetics of metformin. Clinical pharmacokinetics, 2011. 50(2): p. 81–98. [DOI] [PubMed] [Google Scholar]
- 35.Markowicz-Piasecka M, et al. , Is metformin a perfect drug? Updates in pharmacokinetics and pharmacodynamics. Current pharmaceutical design, 2017. 23(17): p. 2532–2550. [DOI] [PubMed] [Google Scholar]
- 36.Minamii T, Nogami M, and Ogawa W, Mechanisms of metformin action: in and out of the gut. Journal of diabetes investigation, 2018. 9(4): p. 701–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Isoda K, et al. , Metformin inhibits proinflammatory responses and nuclear factor-κB in human vascular wall cells. Arteriosclerosis, thrombosis, and vascular biology, 2006. 26(3): p. 611–617. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, et al. , Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. Journal of neuroinflammation, 2014. 11(1): p. 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashabi G, et al. , Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metabolic brain disease, 2015. 30(3): p. 747–754. [DOI] [PubMed] [Google Scholar]
- 40.Rena G, Hardie DG, and Pearson ER, The mechanisms of action of metformin. Diabetologia, 2017. 60(9): p. 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piskovatska V, et al. , Metformin as a geroprotector: experimental and clinical evidence. Biogerontology, 2019. 20(1): p. 33–48. [DOI] [PubMed] [Google Scholar]
- 42.Fullerton MD, et al. , Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nature medicine, 2013. 19(12): p. 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johanns M, et al. , AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nature communications, 2016. 7(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madiraju AK, et al. , Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature, 2014. 510(7506): p. 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adler AS, et al. , Motif module map reveals enforcement of aging by continual NF-κB activity. Genes & development, 2007. 21(24): p. 3244–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tilstra JS, et al. , NF-κB inhibition delays DNA damage-induced senescence and aging in mice. The Journal of clinical investigation, 2012. 122(7): p. 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanigur Sultuybek G, Soydas T, and Yenmis G, NF-κB as the mediator of metformin’s effect on ageing and ageing-related diseases. Clinical and Experimental Pharmacology and Physiology, 2019. 46(5): p.413–422. [DOI] [PubMed] [Google Scholar]
- 48.Moiseeva O, et al. , Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κ B activation. Aging cell, 2013. 12(3): p. 489–498. [DOI] [PubMed] [Google Scholar]
- 49.Johnson SC, Rabinovitch PS, and Kaeberlein M, mTOR is a key modulator of ageing and age-related disease. Nature, 2013. 493(7432): p. 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cǎtoi AF, et al. , Metformin Modulates the Mechanisms of Ageing, in Metformin. 2019, IntechOpen. [Google Scholar]
- 51.Nozohouri S, et al. , Estimating Brain Permeability Using In Vitro Blood-Brain Barrier Models. 2020. [DOI] [PubMed] [Google Scholar]
- 52.Abbott NJ, et al. , Structure and function of the blood–brain barrier. Neurobiology of disease, 2010. 37(1): p. 13–25. [DOI] [PubMed] [Google Scholar]
- 53.Bonkowski D, et al. , The CNS microvascular pericyte: pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids and Barriers of the CNS, 2011. 8(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jo DH, et al. , Interaction between pericytes and endothelial cells leads to formation of tight junction in hyaloid vessels. Molecules and cells, 2013. 36(5): p. 465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armulik A, et al. , Pericytes regulate the blood–brain barrier. Nature, 2010. 468(7323): p. 557–561. [DOI] [PubMed] [Google Scholar]
- 56.Muoio V, Persson P, and Sendeski M, The neurovascular unit–concept review. Acta physiologica, 2014. 210(4): p. 790–798. [DOI] [PubMed] [Google Scholar]
- 57.Jia G, et al. , Endothelial cell senescence in aging-related vascular dysfunction. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 2019. 1865(7): p. 1802–1809. [DOI] [PubMed] [Google Scholar]
- 58.Elahy M, et al. , Blood–brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immunity & Ageing, 2015. 12(1): p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Erickson MA and Banks WA, Age-Associated Changes in the Immune System and Blood–Brain Barrier Functions. International journal of molecular sciences, 2019. 20(7): p. 1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamazaki Y, et al. , Vascular cell senescence contributes to blood–brain barrier breakdown. Stroke, 2016. 47(4): p. 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeong SY and David S, Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. Journal of Neuroscience, 2006. 26(38): p. 9810–9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Graves S.l. and Baker DJ, Implicating endothelial cell senescence to dysfunction in the ageing and diseased brain. Basic & Clinical Pharmacology & Toxicology, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Popescu BO, et al. , Blood-brain barrier alterations in ageing and dementia. Journal of the neurological sciences, 2009. 283(1–2): p. 99–106. [DOI] [PubMed] [Google Scholar]
- 64.Pelegrí C, et al. , Increased permeability of blood–brain barrier on the hippocampus of a murine model of senescence. Mechanisms of ageing and development, 2007. 128(9): p. 522–528. [DOI] [PubMed] [Google Scholar]
- 65.Banks WA, Moinuddin A, and Morley JE, Regional transport of TNF-α across the blood-brain barrier in young ICR and young and aged SAMP8 mice. Neurobiology of aging, 2001. 22(4): p. 671–676. [DOI] [PubMed] [Google Scholar]
- 66.Vorbrodt A, et al. , Immunogold study of regional differences in the distribution of glucose transporter (GLUT-1) in mouse brain associated with physiological and accelerated aging and scrapie infection. Journal of neurocytology, 1999. 28(9): p. 711–719. [DOI] [PubMed] [Google Scholar]
- 67.Toornvliet R, et al. , Effect of age on functional P-glycoprotein in the blood-brain barrier measured by use of (R)-[11C] verapamil and positron emission tomography. Clinical Pharmacology & Therapeutics, 2006. 79(6): p. 540–548. [DOI] [PubMed] [Google Scholar]
- 68.Lochhead JJ, et al. , Erratum: Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation (Journal of Cerebral Blood Flow and Metabolism (2010) 30 (1625-1636. Journal of Cerebral Blood Flow and Metabolism, 2011. 31(2): p. 790–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Popescu BO, Triggers and effectors of oxidative stress at blood-brain barrier level: relevance for brain ageing and neurodegeneration. Oxidative medicine and cellular longevity, 2013. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beckmann R, Biguanide (Experimenteller Teil). Oral wirksame Antidiabetika, 1971. 29: p. 439–596. [Google Scholar]
- 71.Chen Y, et al. , Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proceedings of the National Academy of Sciences, 2009. 106(10): p. 3907–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang D, et al. , Cytotoxic edema: mechanisms of pathological cell swelling. Neurosurg Focus, 2007. 22(5): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nath N, et al. , Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. The Journal of Immunology, 2009. 182(12): p. 8005–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Łabuzek K, et al. , Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacological Reports, 2010. 62(5): p. 956–965. [DOI] [PubMed] [Google Scholar]
- 75.de la Monte SM, Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Current Alzheimer Research, 2012. 9(1): p. 35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hsu C-C, et al. , Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. Journal of Alzheimer’s Disease, 2011. 24(3): p. 485–493. [DOI] [PubMed] [Google Scholar]
- 77.Li J, et al. , Metformin attenuates Alzheimer’s disease-like neuropathology in obese, leptin-resistant mice. Pharmacology biochemistry and behavior, 2012. 101(4): p. 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alagiakrishnan K, et al. , Montreal Cognitive Assessment is superior to Standardized Mini-Mental Status Exam in detecting mild cognitive impairment in the middle-aged and elderly patients with type 2 diabetes mellitus. BioMed research international, 2013. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahmed S, et al. , Effect of metformin on adult hippocampal neurogenesis: comparison with donepezil and links to cognition. Journal of Molecular Neuroscience, 2017. 62(1): p. 88–98. [DOI] [PubMed] [Google Scholar]
- 80.Lian H, et al. , Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. Journal of Neuroscience, 2016. 36(2): p. 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Strooper B and Karran E, The cellular phase of Alzheimer’s disease. Cell, 2016. 164(4): p. 603–615. [DOI] [PubMed] [Google Scholar]
- 82.Ou Z, et al. , Metformin treatment prevents amyloid plaque deposition and memory Impairment in APP/PS1 mice. Brain, behavior, and immunity, 2018. 69: p. 351–363. [DOI] [PubMed] [Google Scholar]
- 83.Ries M and Sastre M, Mechanisms of Aβ clearance and degradation by glial cells. Frontiers in aging neuroscience, 2016. 8: p. 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patrono C and Baigent C, Coxibs, traditional NSAIDs, and cardiovascular safety post-precision: what we thought we knew then and what we think we know now. Clinical Pharmacology & Therapeutics, 2017. 102(2): p. 238–245. [DOI] [PubMed] [Google Scholar]
- 85.Lu X-Y, et al. , Metformin Ameliorates A6 Pathology by Insulin-Degrading Enzyme in a Transgenic Mouse Model of Alzheimer’s Disease. Oxidative Medicine and Cellular Longevity, 2020. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Picone P, et al. , Metformin increases APP expression and processing via oxidative stress, mitochondrial dysfunction and NF-κB activation: Use of insulin to attenuate metformin’s effect. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 2015. 1853(5): p. 1046–1059. [DOI] [PubMed] [Google Scholar]
- 87.Ishii R, et al. , Decrease in plasma levels of α-synuclein is evident in patients with Parkinson’s disease after elimination of heterophilic antibody interference. PloSone, 2015. 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Savitt JM, Dawson VL, and Dawson TM, Diagnosis and treatment of Parkinson disease: molecules to medicine. The Journal of clinical investigation, 2006. 116(7): p. 1744–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ghavami S, et al. , Autophagy and apoptosis dysfunction in neurodegenerative disorders. Progress in neurobiology, 2014. 112: p. 24–49. [DOI] [PubMed] [Google Scholar]
- 90.Osellame LD and Duchen MR, Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy, 2013. 9(10): p. 1633–1635. [DOI] [PubMed] [Google Scholar]
- 91.Lu M, et al. , Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. International Journal of Neuropsychopharmacology, 2016. 19(9): p. pyw047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kang H, et al. , Activation of the ATF2/CREB-PGC-1α pathway by metformin leads to dopaminergic neuroprotection. Oncotarget, 2017. 8(30): p. 48603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patil S, et al. , Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience, 2014. 277: p. 747–754. [DOI] [PubMed] [Google Scholar]
- 94.Jankovic J, Parkinson’s disease: clinical features and diagnosis. Journal of neurology, neurosurgery & psychiatry, 2008. 79(4): p. 368–376. [DOI] [PubMed] [Google Scholar]
- 95.Tayara K, et al. , Divergent effects of metformin on an inflammatory model of Parkinson’s disease. Frontiers in cellular neuroscience, 2018. 12: p. 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Franklin RJ, Edgar JM, and Smith KJ, Neuroprotection and repair in multiple sclerosis. Nature Reviews Neurology, 2012. 8(11): p. 624–634. [DOI] [PubMed] [Google Scholar]
- 97.Oh J, Lee YD, and Wagers AJ, Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nature medicine, 2014. 20(8): p. 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sim FJ, et al. , The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. Journal of Neuroscience, 2002. 22(7): p. 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cantuti-Castelvetri L, et al. , Defective cholesterol clearance limits remyelination in the aged central nervous system. Science, 2018. 359(6376): p. 684–688. [DOI] [PubMed] [Google Scholar]
- 100.Shen S, et al. , Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nature neuroscience, 2008. 11(9): p. 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Woodruff RH, et al. , Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Molecular and Cellular Neuroscience, 2004. 25(2): p. 252–262. [DOI] [PubMed] [Google Scholar]
- 102.Boyd A, Zhang H, and Williams A, Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta neuropathologica, 2013. 125(6): p. 841–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Neumann B, et al. , Metformin restores CNS remyelination capacity by rejuvenating aged stem cells. Cell stem cell, 2019. 25(4): p. 473–485.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paintlia AS, et al. , AMP-activated protein kinase signaling protects oligodendrocytes that restore central nervous system functions in an experimental autoimmune encephalomyelitis model. The American journal of pathology, 2013. 183(2): p. 526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gupta A, Bisht B, and Dey CS, Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology, 2011. 60(6): p. 910–920. [DOI] [PubMed] [Google Scholar]
- 106.Chen F, et al. , Antidiabetic drugs restore abnormal transport of amyloid-B across the blood–brain barrier and memory impairment in db/db mice. Neuropharmacology, 2016. 101: p. 123– 136. [DOI] [PubMed] [Google Scholar]
- 107.Kickstein E, et al. , Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proceedings of the National Academy of Sciences, 2010. 107(50): p. 21830–21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chiang M-C, et al. , Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Experimental cell research, 2016. 347(2): p. 322–331. [DOI] [PubMed] [Google Scholar]
- 109.Katila N, et al. , Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology, 2017. 125: p. 396–407. [DOI] [PubMed] [Google Scholar]
- 110.Sanadgol N, et al. , Metformin accelerates myelin recovery and ameliorates behavioral deficits in the animal model of multiple sclerosis via adjustment of AMPK/Nrf2/mTOR signaling and maintenance of endogenous oligodendrogenesis during brain self-repairing period. Pharmacological Reports, 2020. 72(3): p. 641–658. [DOI] [PubMed] [Google Scholar]
- 111.Largani SHH, et al. , Oligoprotective effect of metformin through the AMPK-dependent on restoration of mitochondrial hemostasis in the cuprizone-induced multiple sclerosis model. Journal of Molecular Histology, 2019. 50(3): p. 263–271. [DOI] [PubMed] [Google Scholar]
- 112.Sun Y, et al. , Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. Journal of Neuroimmunology, 2016. 292: p. 58–67. [DOI] [PubMed] [Google Scholar]
- 113.Leech T, Chattipakorn N, and Chattipakorn SC, The beneficial roles of metformin on the brain with cerebral ischaemia/reperfusion injury. Pharmacological research, 2019. 146: p. 104261. [DOI] [PubMed] [Google Scholar]
- 114.Lin L, Wang X, and Yu Z, Ischemia-reperfusion injury in the brain: mechanisms and potential therapeutic strategies. Biochemistry & pharmacology: open access, 2016. 5(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lakhan SE, Kirchgessner A, and Hofer M, Inflammatory mechanisms in ischemic stroke: therapeutic approaches. Journal of translational medicine, 2009. 7(1): p. 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ni Y, et al. , RIP1K contributes to neuronal and astrocytic cell death in ischemic stroke via activating autophagic-lysosomal pathway. Neuroscience, 2018. 371: p. 60–74. [DOI] [PubMed] [Google Scholar]
- 117.Wang Y, et al. , White matter injury in ischemic stroke. Progress in neurobiology, 2016. 141: p. 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Angels Font M, Arboix A, and Krupinski J, Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Current cardiology reviews, 2010. 6(3): p. 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.George PM and Steinberg GK, Novel stroke therapeutics: unraveling stroke pathophysiology and its impact on clinical treatments. Neuron, 2015. 87(2): p. 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jin Q, et al. , Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain, behavior, and immunity, 2014. 40: p. 131–142. [DOI] [PubMed] [Google Scholar]
- 121.Liu Y, et al. , Metformin promotes focal angiogenesis and neurogenesis in mice following middle cerebral artery occlusion. Neuroscience letters, 2014. 579: p. 46–51. [DOI] [PubMed] [Google Scholar]
- 122.Hattori Y, et al. , Metformin inhibits cytokine-induced nuclear factor κB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension, 2006. 47(6): p. 1183–1188. [DOI] [PubMed] [Google Scholar]
- 123.Zhao M, et al. , Neuro-protective role of metformin in patients with acute stroke and type 2 diabetes mellitus via AMPK/mammalian target of rapamycin (mTOR) signaling pathway and oxidative stress. Medical science monitor: international medical journal of experimental and clinical research, 2019. 25: p. 2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gabryel B and Liber S, Metformin limits apoptosis in primary rat cortical astrocytes subjected to oxygen and glucose deprivation. Folia neuropathologica, 2018. 56(4): p. 328–336. [DOI] [PubMed] [Google Scholar]
- 125.Hardie DG, Ross FA, and Hawley SA, AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews Molecular cell biology, 2012. 13(4): p. 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li J, et al. , Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke, 2007. 38(11): p. 2992–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hardie DG and Frenguelli BG, A neural protection racket: AMPK and the GABAB receptor. Neuron, 2007. 53(2): p. 159–162. [DOI] [PubMed] [Google Scholar]
- 128.Leech T, Chattipakorn N, and Chattipakorn SC, The beneficial roles of metformin on the brain with cerebral ischaemia/reperfusion injury. Pharmacological research, 2019: p. 104261. [DOI] [PubMed] [Google Scholar]
- 129.Grissi M, et al. , F0081 METFORMIN ALLEVIATES STROKE SEVERITY IN FEMALE MICE WITH CHRONIC KIDNEY DISEASE THROUGH AMPK ACTIVATION AND SUBSEQUENT DECREASE OF MICROGLIA/MACROPHAGES M1 POLARIZATION. Nephrology Dialysis Transplantation, 2019. 34(Supplement_l): p. gfz096. FO081. [Google Scholar]
- 130.Kaisar MA, et al. , Offsetting the impact of smoking and e-cigarette vaping on the cerebrovascular system and stroke injury: Is Metformin a viable countermeasure? Redox biology, 2017. 13: p. 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.DeGracia DJ, Towards a dynamical network view of brain ischemia and reperfusion. Part I: background and preliminaries. Journal of experimental stroke & translational medicine, 2010. 3(1): p. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Surinkaew P, et al. , Role of microglia under cardiac and cerebral ischemia/reperfusion (l/R) injury. Metabolic brain disease, 2018. 33(4): p. 1019–1030. [DOI] [PubMed] [Google Scholar]
- 133.Zeng J, et al. , Metformin Protects against Oxidative Stress Injury Induced by Ischemia/Reperfusion via Regulation of the lncRNA-H19/miR-148a-3p/Rock2 Axis. Oxidative Medicine and Cellular Longevity, 2019. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hu X, et al. Microglial and macrophage polarization—new prospects for brain repair. Nature Reviews Neurology, 2015. 11(1): p. 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Moskowitz MA, Lo EH, and ladecola C, The science of stroke: mechanisms in search of treatments. Neuron, 2010. 67(2): p. 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fang M, et al. , Metformin treatment after the hypoxia-ischemia attenuates brain injury in newborn rats. Oncotarget, 2017. 8(43): p. 75308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yuan R, et al. , Metformin reduces neuronal damage and promotes neuroblast proliferation and differentiation in a cerebral ischemia/reperfusion rat model. NeuroReport, 2019. 30(3): p. 232– 240. [DOI] [PubMed] [Google Scholar]
- 138.Huang L, et al. , Glial scar formation occurs in the human brain after ischemic stroke. International journal of medical sciences, 2014. 11(4): p. 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pekny M, Wilhelmsson U, and Pekna M, The dual role of astrocyte activation and reactive gliosis. Neuroscience letters, 2014. 565: p. 30–38. [DOI] [PubMed] [Google Scholar]
- 140.Venna VR, et al. , Chronic metformin treatment improves post-stroke angiogenesis and recovery after experimental stroke. European Journal of Neuroscience, 2014. 39(12): p. 2129–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Klocke R, et al. , Granulocyte colony-stimulating factor (G-CSF) for cardio-and cerebrovascular regenerative applications. Current medicinal chemistry, 2008. 15(10): p. 968–977. [DOI] [PubMed] [Google Scholar]
- 142.Ge X-H, et al. , Metformin protects the brain against ischemia/reperfusion injury through PI3K/Akt1/JNK3 signaling pathways in rats. Physiology & behavior, 2017. 170: p. 115–123. [DOI] [PubMed] [Google Scholar]
- 143.Abdelsaid M, et al. , Metformin treatment in the period after stroke prevents nitrative stress and restores angiogenic signaling in the brain in diabetes. Diabetes, 2015. 64(5): p. 1804–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Batczewska D, Ptaszyhski P, and Cygankiewicz I, Baroreflex sensitivity: measurement and clinical aspects. Przeglad lekarski, 2015. 72(11): p. 682–689. [PubMed] [Google Scholar]
- 145.Lu B, et al. , α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Molecular medicine, 2014. 20(1): p. 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Meroni E, et al. , Intestinal macrophages and their interaction with the enteric nervous system in health and inflammatory bowel disease. Acta Physiologica, 2019. 225(3): p. e13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhu X-C, et al. , Chronic metformin preconditioning provides neuroprotection via suppression of NF-κB-mediated inflammatory pathway in rats with permanent cerebral ischemia. Molecular neurobiology, 2015. 52(1): p. 375–385. [DOI] [PubMed] [Google Scholar]
- 148.Li D-J, et al. , Dysfunction of the cholinergic anti-inflammatory pathway mediates organ damage in hypertension, hypertension, 2011. 57(2): p. 298–307. [DOI] [PubMed] [Google Scholar]
- 149.Petersen JS and DiBona GF, Effects of central metformin administration on responses to air-jet stress and on arterial baroreflex function in spontaneously hypertensive rats. Journal of hypertension, 1997. 15(3): p. 285–291. [DOI] [PubMed] [Google Scholar]
- 150.Deng T, et al. , Pre-stroke metformin treatment is neuroprotective involving AMPK reduction. Neurochemical research, 2016. 41(10): p. 2719–2727. [DOI] [PubMed] [Google Scholar]
- 151.Karimipour M, et al. , Pre-treatment with metformin in comparison with post-treatment reduces cerebral ischemia reperfusion induced injuries in rats. Bulletin of Emergency & Trauma, 2018. 6(2): p. 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang P-F, et al. , Polyinosinic-polycytidylic acid has therapeutic effects against cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via TLR3. 2014. 192(10): p. 4783–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li J, et al. , Effects of metformin in experimental stroke. Stroke, 2010. 41(11): p. 2645–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Farbood Y, et al. , Targeting adenosine monophosphate-activated protein kinase by metformin adjusts post-ischemic hyperemia and extracellular neuronal discharge in transient global cerebral ischemia. Microcirculation, 2015. 22(7): p. 534–541. [DOI] [PubMed] [Google Scholar]
- 155.Sarkaki A, et al. , Metformin improves anxiety-like behaviors through AMPK-dependent regulation of autophagy following transient forebrain ischemia. Metabolic brain disease, 2015. 30(5): p. 1139–1150. [DOI] [PubMed] [Google Scholar]
- 156.Guo J-M, et al. , Involvement of arterial baroreflex and nicotinic acetylcholine receptor α7 subunit pathway in the protection of metformin against stroke in stroke-prone spontaneously hypertensive rats. European Journal of Pharmacology, 2017. 798: p. 1–8. [DOI] [PubMed] [Google Scholar]
- 157.Ghadernezhad N, et al. , Metformin pretreatment enhanced learning and memory in cerebral forebrain ischaemia: the role of the AMPK/BDNF/P70SK signalling pathway. Pharmaceutical biology, 2016. 54(10): p. 2211–2219. [DOI] [PubMed] [Google Scholar]
- 158.Ashabi G, et al. , Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: interference of AMPK/PGC-1α pathway. Metabolic brain disease, 2014. 29(1): p. 47–58. [DOI] [PubMed] [Google Scholar]
- 159.Jiang T, et al. , Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. British journal of pharmacology, 2014. 171(13): p. 3146–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Harada S, Fujita-Hamabe W, and Tokuyama S, The importance of regulation of blood glucose levels through activation of peripheral 5′-AMP-activated protein kinase on ischemic neuronal damage. Brain research, 2010. 1351: p. 254–263. [DOI] [PubMed] [Google Scholar]
- 161.Prakash R, et al. , Vascularization pattern after ischemic stroke is different in control versus diabetic rats: relevance to stroke recovery. Stroke, 2013. 44(10): p. 2875–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Elgebaly MM, et al. , Vascular protection in diabetic stroke: role of matrix metalloprotease-dependent vascular remodeling. Journal of Cerebral Blood Flow & Metabolism, 2010. 30(12): p. 1928–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kyu HH, et al. , Physical activity and risk of breast cancer, colon cancer, diabetes, ischemic heart disease, and ischemic stroke events: systematic review and dose-response meta-analysis for the Global Burden of Disease Study 2013. bmj, 2016. 354: p. i3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sifat AE, et al. , Nicotine and electronic cigarette (E-Cig) exposure decreases brain glucose utilization in ischemic stroke. Journal of neurochemistry, 2018. 147(2): p. 204–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sifat AE, et al. , Neurovascular unit transport responses to ischemia and common coexisting conditions: Smoking and diabetes. American Journal of Physiology-Cell Physiology, 2019. 316(1): p. C2–C15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fluynh W, et al. , The effect of diabetes on cortical function in stroke: implications for poststroke plasticity. Diabetes, 2017. 66(6): p. 1661–1670. [DOI] [PubMed] [Google Scholar]
- 167.Ergul A, et al. , Increased hemorrhagic transformation and altered infarct size and localization after experimental stroke in a rat model type 2 diabetes. BMC neurology, 2007. 7(1): p. 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Willi C, et al. , Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. Jama, 2007. 298(22): p. 2654–2664. [DOI] [PubMed] [Google Scholar]
- 169.Gill JS, et al. , Cigarette smoking: a risk factor for hemorrhagic and nonhemorrhagic stroke. Archives of Internal Medicine, 1989. 149(9): p. 2053–2057. [DOI] [PubMed] [Google Scholar]
- 170.Naik P, et al. , Oxidative and pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: is smoking reduced or nicotine-free products really safe? BMC neuroscience, 2014. 15(1): p. 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Das S, et al. , Oxidative stress in the brain of nicotine-induced toxicity: protective role of Andrographis paniculata Nees and vitamin E. Applied Physiology, Nutrition, and Metabolism, 2009. 34(2): p. 124–135. [DOI] [PubMed] [Google Scholar]
- 172.Paulson JR, et al. , Nicotine exacerbates brain edema during in vitro and in vivo focal ischemic conditions. Journal of pharmacology and experimental therapeutics, 2010. 332(2): p. 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Giacco F and Brownlee M, Oxidative stress and diabetic complications. Circulation research, 2009. 107(9): p. 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Shah KK, Boreddy PR, and Abbruscato TJ, Nicotine pre-exposure reduces stroke-induced glucose transporter-1 activity at the blood–brain barrier in mice. Fluids and Barriers of the CNS, 2014. 12(1): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Prasad S, et al. , Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox biology, 2017. 12: p. 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bold KW, et al. , Reasons for trying e-cigarettes and risk of continued use. Pediatrics, 2016. 138(3): p. e20160895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Health U.D.o. and Services H, E-cigarette use among youth and young adults. A report of the Surgeon General. Atlanta, GA, 2016. [PubMed] [Google Scholar]
- 178.Adams JM, Smoking Cessation—Progress, Barriers, and New Opportunities: The Surgeon General’s Report on Smoking Cessation. Jama, 2020. 323(24): p. 2470–2471. [DOI] [PubMed] [Google Scholar]
- 179.Sifat AE, et al. , Prenatal electronic cigarette exposure decreases brain glucose utilization and worsens outcome in offspring hypoxic-ischemic brain injury. Journal of Neurochemistry, 2020. 153(1): p. 63–79. [DOI] [PubMed] [Google Scholar]