

Abstract
Mutations of RAS oncogenes are responsible for about 30% of all human cancer types, including pancreatic, lung, and colorectal cancers. While KRAS1 is a pseudogene, mutation of KRAS2 (commonly known as KRAS oncogene) is directly or indirectly associated with human cancers. Among the RAS family, KRAS is the most abundant oncogene related to uncontrolled cellular proliferation to generate solid tumors in many types of cancer such as pancreatic carcinoma (over 80%), colon carcinoma (40–50%), lung carcinoma (30–50%), and other types of cancer. Once described as ‘undruggable’, RAS proteins have become ‘druggable’, at least to a certain extent, due to the continuous efforts made during the past four decades. In this account, we discuss the chemistry and biology (wherever available) of the small-molecule inhibitors (synthetic, semi-synthetic, and natural) of KRAS proteins that were published in the past decades. Commercial drugs, as well as investigational molecules from preliminary stages to clinical trials, are categorized and discussed in this study. In summary, this study presents an in-depth discussion of RAS proteins, classifies the RAS superfamily, and describes the molecular mechanism of small-molecule RAS inhibitors
Keywords: pancreatic cancer, lung cancer, colon cancer, small-molecule inhibitors, heterocycles, KRAS, HRAS, NRAS, RAS subfamily, mechanism of action, cell signaling
1. Introduction
The human body is one of the most complicated biological systems, where hundreds to thousands of biochemical, biophysical, and physicochemical transformations occur at every moment. Most of these biotransformations are catalyzed by enzymes or associated with enzymes/proteins. Several studies found that a set of genes responsible for cancer pathogenesis originated from the transforming activity of the Harvey and Kirsten murine sarcoma retroviruses and referred to them as RAS (rat sarcoma virus) in the early 1980s [1]. The initial study of RAS identified them as viral genes. They were then attributed to the rodent genomes and thought to be the cause of the oncogenic properties of tumor viruses [2]. Later, in 1982, the first human RAS genes and RAS proteins were identified, and their mutational activation was observed in human cancer cell lines. Three of the four deadliest cancers in the US, lung, colon, and pancreatic cancer, show RAS mutations’ prevalence, which also extends to other human cancers [3].
The human RAS protein family now includes more than 150 proteins encoded by genes: HRAS, KRAS, and NRAS. It is known that 86% of mutations in the RAS protein, which constitutes mutated RAS proteins in cancer, come from the one most closely related to the KRAS genes. KRAS genes are responsible for several types of cancer, and 90% of pancreatic cancer cases are directly attributable to KRAS mutations [2,4]. Along with the specific types of cancers, RAS proteins are also responsible for other diseases such as RASopathy, Capillary Malformations, and Psychiatric and Neurodevelopmental Disorders [5].
Studies found that aberrant RAS mutation is associated with hyperproliferation developmental disorder and specific mutations in codons 12, 13, and 61. GTP-binding occurs within the particular site and activates RAS signaling. Importantly, all RAS isoforms share sequence identity in all the regions responsible for GDP/GTP binding, GTPase activity, and effector interactions, which suggests functional redundancy. Several synthetic and natural small molecules were evaluated in RAS-dependent cell lines, and mechanistic investigations were carried out in many instances. Despite unceasing progress, many miles remain in the development of drugs with higher potency and lower toxicity.
2. Classification of RAS Protein Superfamily
The RAS protein superfamily comprises small guanosine triphosphatase chemicals (GTPases) that act as molecular on/off switches for numerous cellular activities in response to extracellular signals. The RAS family of proteins functions as a set of signaling nodes triggered by various extracellular stimuli. They also control intracellular signaling. Such signaling eventually regulates gene transcription and influences fundamental processes such as cell growth and differentiation [6,7,8,9,10]. Based on their genetic sequences and biological activities, we found five distinct groups of the RAS superfamily: RAS, Rho, Arf/Sar, Ran, and Rab (Figure 1) [6,8].
Figure 1.

2.1. RAS Subfamily
There are 36 proteins, collectively called RAS oncoproteins, that are the original members of the RAS family. This subfamily has been the topic of much research. The RAS family of human oncogenic members has been intensively investigated, and they modulate cell proliferation, differentiation, morphology, and apoptosis in general. In 1993, the protein kinase Raf was studied and recognized as a RAS effector. RAS is activated when it binds to the Raf serine/threonine kinase. Activated RAS facilitates translocation to the plasma membrane, where additional phosphorylation events enhance Raf kinase activation in its entirety [1,9]. The RAS proteins HRAS, KRAS, and NRAS (H, K, NRAS) are RAS oncoproteins that cause human cancers. The transfusion-based assay can isolate them [10,12,13] from human tumor cells. These three isoforms are most prevalent in human cancers. In addition, the same proteins have recently been linked to central nervous system neuronal plasticity. Among the signaling molecules that are indirectly linked to various cell surface receptors, RAS proteins play a vital role in responding to a wide range of extracellular observations [14].
Most RAS subfamily proteins are primarily found in the plasma membrane area. Therefore, C-terminal prenylation contributes to membrane localization. There is a common Caax (a = aliphatic, x = terminal amino acid) pattern that most prenylation signals follow. This pattern guides cysteine farnesylation (except when x = L or F, which leads to the geranylgeranylation process, commonly seen on RRAS and some other RAP proteins). During the prenylation reaction, proteolysis of the three C-terminal residues (aax) occurs, and methylation of the lipid-modified cysteine is completed [15,16]. RAS subfamily proteins such as the Rap, RRAS, Ral, and Rheb proteins help control the signaling network. RRAS participate in various complex activities such as angiogenesis, regeneration, vascular homeostasis, cell adhesion, and neuronal axon guidance, but their mutation causes invasive cancers [17,18]. Mitogenic stimuli activate Rap proteins, which operate as the controllers of integrin-mediated cell adhesion as well as cell spreading [19]. Ral proteins’ functions include mitogenic responses, protein trafficking, differentiation, and cytoskeleton dynamics. Investigations found that the two Ral proteins play independent and complementary roles in cell transformation [20]. RalA promotes anchorage-independent cell proliferation, but RalB is required to survive against tumor cells [21]. The Rheb protein is largely involved in regulating the cell cycle [22]. Some early studies found that the Rheb protein also performs two additional functions: blocking of MAPK or mitogen-activated protein kinase signaling; inhibiting the RAS-mediated transformation of cultured fibroblasts [23].
2.2. Rho Subfamily
The RAS homolog (Rho) subfamily is closely related to the RAS subgroup. The Rho family mainly operates in signaling networks that subsequently regulate three processes: actin, cell cycle progression, and gene expression [24]. In addition, Rho GTPases are critical cytoskeletal regulators that influence various cellular activities, including cell polarity, migration, vesicle trafficking, and cytokinesis. Rho GTPases have been investigated extensively in several mammalian cell types, with dominant-negative and constitutively active mutants primarily being employed [25]. Rho GTPases have also been identified as crucial regulators of cell polarity. They act as molecular regulators between two states: active GTP–bound and inactive GDP–bound [26]. In addition, many studies have implicated members of the Rho family in hematopoiesis. Multiple human hematologic disorders, including neutrophil dysfunction, leukemia, and Fanconi anemia, have been associated with the deregulation of Rho GTPase family members, increasing the possibility that Rho GTPases and their downstream signaling pathways could be helpful therapeutically [27]. RAC proteins have emerged as essential mediators of Wnt signals, mainly in noncanonical and canonical pathways leading to β-catenin-dependent transcription. Wnt-induced signals appear to promote morphological and transcriptional alterations that affect cell behavior spatially and temporally [28].
To understand the Rho family’s evolutionary history, there have been investigations on over 20 species, including major eukaryotic clades, unicellular organisms, various mammals, platypus, opossum, and so on. Studies include the reconstruction of the ontogeny and chronology of the development of distinct Rho subfamilies. There are eight subfamilies among the 20 mammalian Rho members. The Rac, Rho, and Cdc42 subgroups have been extensively studied among the eight Rho subfamilies [29]. Most likely, 20 canonical Rho members (Rho GTPases) are regulated by four guanine nucleotide dissociation inhibitors (GDIs), 85 guanine nucleotide exchange factors (GEFs), and 66 GTPase-activating proteins (GAPs) [30].
2.3. Rab Subfamily
Rab proteins are small GTP-binding proteins, first characterized as RAS-like genes exhibited in rat brains [31], that comprise the biggest subfamily of the RAS protein superfamily. The yeast Saccharomyces cerevisiae expresses eleven Rab (Yptp/Sec4p) proteins. However, based on the expressed-sequence tags (ESTs) and the sequenced human genome, there could be as many as 63 Rab (Yptp/Sec4p) proteins in humans. Rab proteins act as membrane-associated molecular switches between different organelles to regulate vesicular trafficking pathways. The Rab subfamily promotes vesicular or tubular carriers’ budding in further steps such as the donor-compartment, transport-to-acceptor, vesicle-fusion, and load-release steps [32,33].
Rab GTPases are connected in reverse to membranes by hydrophobic geranylgeranyl groups attached to one or (usually) two carboxy-terminal Cys residues, which is essential for controlling the membrane traffic. Rab proteins are regulated by their ability to act as molecular switches, similar to the other GTPases, that wiggle between GTP- and GDP-bound conformations.
Each transport step starts with the binding of activated Rab proteins to soluble factors that function as ‘effectors’, allowing the transduction of the Rab GTPase signal. Several RAB effectors have been identified. Rab effectors’ structural heterogeneity suggests a collection of highly specialized molecules that function exclusively on specific organelles and transport systems [34]. Rab pathway dysfunction has been linked to diseases such as immunodeficiencies, cancer, and neurological disorders.
2.4. Ran Subfamily
Ran is a member of the RAS superfamily, which is the most abundant small GTP-binding protein. Ran proteins are localized mainly within the nucleus [35]. These proteins regulate the nuclear import/export of materials. In addition, DNA replication, mitotic spindle assembly, and nuclear envelope assembly [36] are also controlled by these proteins. Although the RAN protein is related to the Rab subfamily, it has characteristics that set it apart. Ran function depends on a spatially bounded form of the GTP of Ran, unlike other small GTPases. Nuclear trafficking studies identified the switching behavior of Ran. It was found that Ran functions as a molecular switch and controls nuclear transport receptor–cargo complexes [37,38]. The assembly and disassembly of nuclear transport receptors depend on the guanine-nucleotide-bound state of Ran.
2.5. Arf Subfamily
Similar to the Rab protein family, the Arf family of proteins is another diverse group of proteins that are involved in vesicle trafficking [6]. This subfamily’s first members function as ADP ribosylation factors, and they were named accordingly. Arf proteins play a role in vesicle formation and structure, as well as intracellular vesicular transport, endocytosis, and exocytosis [39,40,41]. The Arf family consists of 30 genes, divided into four groups:
The Arfs
The Arf-like (Arls) proteins
The Secretion-associated RAS-related (SARs) proteins
The single Tripartite protein 23, also known as TRIM23.
ARF GTPases constitute a significant family of eukaryote-specific GTPases. They regulate membrane traffic, actin dynamics, tubulin assembly, and cilia-related functions. Every Arf isoform has several biochemical effects that can be put together to elicit a specific cellular behavior or event. This phenomenon helps researchers assess the pathway’s potential as a target in disorders such as cancer [42,43].
2.6. KRAS, HRAS, and NRAS
Three RAS proteins, HRAS, KRAS, and NRAS, are expressed in all mammal cells and, thus, are closely related. Hence, the aberrant mutation of these three proteins can contribute to the development of oncogenesis, which is often attributed to a unique mutation, and this typically occurs at codons 12, 13, or 61. Investigations have found RAS mutations in 30% of human cancers. Though isoforms have a lot in common, each isoform prefers to couple to specific cancer types. Therefore, there has been much analysis of the mutational spectra of RAS isoforms. One such study shows that factors other than the differences in mutagen exposure exist that contribute to the heterogeneity observed across a single isoform.
2.6.1. KRAS
The KRAS gene is found to be mutated in various types of tumors, such as pancreatic carcinoma (over 80%), colon carcinoma (40–50%), lung carcinoma (30–50%), and others [11,43,44,45,46]. The KRAS isoform causes about 85% of RAS mutations. Human KRAS oncogenes are activated in two ways: when oncogene copies increase abruptly or when the tumor suppressor gene p53 is lost abnormally. When oncogenic KRAS is activated, a mutant KRAS protein remains actively GTP-bound for an extended period. KRAS cycles between two states: an inactive bound GDP state, and an active GTP-bound state. However, only the GTP-bound state of KRAS is necessary for Raf-kinases, PI3K, and RalGDS to bind and consequently activate their effector proteins [47].
2.6.2. HRAS
HRAS mutation comprises 12% of overall RAS mutations. HRAS is one of three human RAS proteins that encode the GTPase HRAS protein. HRAS mutations are found in bladder urothelial carcinoma, breast-invasive ductal carcinoma, lung adenocarcinoma, prostate adenocarcinoma, and colon adenocarcinoma, with these having the most significant prevalence of alterations. However, HRAS mutations are also prevalent in the bladder (4.5%), thyroid (3.2%), and head-and-neck carcinoma (5.1%) [48].
2.6.3. NRAS
NRAS is the minor cancer-causing mutant among the three human RAS proteins, and approximately 2.3% of overall RAS mutations occur because of the NRAS mutant. Oncogenic mutations of NRAS genes in human tumors follow a different distribution pattern, with the highest rates of mutation found at codon Q61 (about 60% of total NRAS mutations) and lower percentages detected at G12 (24.4%) and G13 (12.7%) [49]. Moreover, it was found that NRAS mutations are prominent in hematopoietic and skin (melanocyte) malignancies and myelomas [1].
3. RAS and Cancers
3.1. Pancreatic Cancer
Most cases of pancreatic cancer have mutations in the KRAS gene at the time of diagnosis (>80% of cases). It was found that 90% of pancreatic cancers are developed in the exocrine compartment of the pancreas and cause pancreatic ductal adenocarcinoma (PDAC). Codon 12 is accountable for 98% of PDAC mutations. Research found that codon G12D (51%) is the most aggressive PDAC subtype, followed by G12V (30%), G12A/C/S (2% each), and G12L/F (<1%) (Figure 2) [50]. KRAS plays a vital role in developing PDAC-type pancreatic cancer [51]. The progression of PDAC occurs due to the loss of the tumor-suppressor gene cyclin-dependent kinase inhibitor 2A (CDKN2A) [52]. Although KRAS mutations are undruggable and no anti-KRAS therapies exist for them, some inhibitors show effects against cancers. Inhibition of the RAF-MEK-ERK protein kinase pathway is one of the most potent factors that work against KRAS mutation. This pathway has an important role in the progression of PDAC [53]. It is also reported that iExosomes inhibit PDAC in mice by delivering RNAi. However, exosomes have therapeutic potential to control KRAS-dependent pancreatic cancer [54,55]. Additionally, the KRAS mutation can be inhibited by using a slow-released biodegradable polymer matrix. This process works by delivering siRNA as an extended-release drug to mutated KRAS [56]. Another method for suppressing the KRAS mutation is anti-RAS vaccination. These vaccines contain different mutated genes that work against KRAS mutations [57,58].
Figure 2.

Frequency of KRAS, HRAS, and NRAS in different types of cancer [2,50,59,60].
3.2. Lung Cancer
Lung cancer ranks as the primary cause of cancer-related death among men and women worldwide. Researchers first identified activation of KRAS mutations to cause lung cancers. KRAS mutations are responsible for the majority of lung adenocarcinoma (LADC) and are also seen in non-small-cell lung cancer (NSCLC). KRAS mutations cause 30% of lung carcinoma cases. Significantly, 90% of RAS mutations in LDAC are KRAS mutations, and this figure is around 97% in NSCLC [61,62]. Furthermore, the study suggests that KRAS mutations are more frequent in African American patients than in Asian patients (Figure 2) [63,64]. An investigation of restricted NSCLC tumors, in particular, indicated that KRAS mutations are more common in younger patients, especially young women [65].
Researchers determined that smoking history is essential in the regulation of KRAS mutation in lung cancer. Studies found that a significant amount of lung cancer patients are cigarette smokers, and KRAS mutation is also frequent in adenocarcinomas. Furthermore, it was also identified that 30% of current and former smokers exhibit lung adenocarcinoma. However, KRAS mutation is also found in never-smokers, but the percentage of KRAS mutations is higher in smokers [50,66].
3.3. Colorectal Cancer
Colorectal cancer (CRCs) is one of the principal causes of death in men and women in the United States [67]. Multiple genetic alterations make colon carcinoma more fatal. About 50% of KRAS mutations are responsible for codon 13,14, and 64 in colon adenomas [68,69]. Nearly 40% of KRAS mutation occurs for G12D, G12V, and G13V that activate the MAPK signaling pathway (Figure 2) [70]. Although extensive studies have been conducted to find the therapeutics in CRC treatment, there is still no direct inhibitor of KRAS mutations [71]. More effective treatment and biomarkers are needed because a large percentage of colon cancer is associated with the KRAS oncogenes.
3.4. Other Cancers
RAS mutations comprise a vast portion of cancer-causing diseases. RAS mutations also occur in other cancers such as breast cancer, head-and-neck cancer, bladder tumors, skin cancer, prostate cancer, etc. More attention is needed to identify the RAS inhibitors.
4. Small-Molecule RAS Inhibitors
Small molecules are drugs that impede the cellular pathway or molecules within the pathway. Small-molecule drugs/inhibitors can enter the cell easily because of their low molecular weight and balanced ADME properties. They can target the extracellular cell surface receptor as well as intracellular proteins. After entering the cells, they inhibit other molecules such as proteins or enzymes, which causes various types of disease. For example, gefitinib is a small-molecule anticancer drug used to treat non-small-cell lung cancer [72]. Gefitinib blocks the signaling pathway of epidermal-growth-factor receptors (EGRF), which are responsible for cell proliferation, invasion, etc. RAS proteins contribute considerably to many types of cancer. There is no more effective treatment for RAS mutations. In terms of finding an effective means of treating undruggable RAS mutation, small-molecule drugs or inhibitors can be an effective option for cancer treatment, inspired by robust investigation and research.
4.1. Classification of Small-Molecule RAS Inhibitors Based on Their Structure
Nowadays, structure-based drug discovery is becoming an essential strategy for treating various diseases, especially cancer. This strategy is now in vogue because of its cost-effectiveness and faster results compared to traditional drug-discovery methods. However, structural data provide the molecular mechanisms and fundamental functions of a compound. Structural studies have provided hundreds of new targets, and when the target is identified, it is essential to obtain accurate structural information.
This study classified and discussed small-molecule RAS inhibitors according to their structure. We found that most of the RAS inhibitors are heterocyclic compounds. The cyclic part (from the Greek “kyklos”, meaning “circle”) of “heterocycle” indicates that at least one ring structure is present in such a compound, while the prefix hetero- (from the Greek “heteros”, meaning “other” or “different”) refers to any atoms other than carbon (heteroatoms) [73]. The most common heterocycles have four- or five-membered rings containing nitrogen, oxygen, and a sulfur atom. These diverse heterocyclic compounds have found applications as pharmacophores. These pharmacophores are often found in synthetic and natural product drugs. Researchers and pharmaceutical companies are interested in heterocyclic pharmacophores due to their fascinating medicinal properties. We also found that the carbocyclic compound also has RAS-inhibitory properties. Thus, studying the heterocyclic and carbocyclic RAS inhibitors might be an excellent resource for finding potent small-molecule inhibitors for cancer treatment.
Based on the structure aza heterocyclic small-molecule RAS inhibitors with one nitrogen atom are shown in Table 1. After careful review, the mechanistic properties, cancer type and major pharmacophores are also described.
Table 1.
Aza heterocyclic small-molecule RAS inhibitors with one nitrogen atom.
| Compound Name | Structure | Major Pharmacophore(s) | Cancer Type | Targeted Enzyme M/A |
References |
|---|---|---|---|---|---|
| Ganetespib |
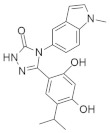
|
Indole, Triazolone, Resorcinol | Lung cancer/colorectal cancer | Inhibits heat-shock protein 90 (Hsp90) | [74] [75] |
| Apatinib |
|
Pyridine | Metastatic lung cancer/breast cancer/gastric cancer | Inhibits vascular endothelial growth factor receptor-2 (VEGFR-2) | [76] |
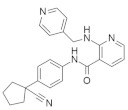
| Oncrasin-1 |
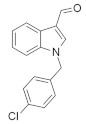
|
Indole, Chlorophenyl | Lung cancer | Inhibits K-RAS/PKCι pathway | [77] |
| † |
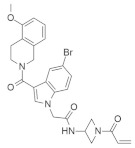
|
Indole, Azetidine, Isoquinoline | Unknown | Inhibits KRASG12C mutation | [78] |
| †† |
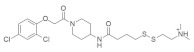
|
Piperidine, Disulfide linkage | Unknown | Inhibits KRASG12C mutation | [79] |
| GDC-0449 (Vismodegib) combined with miRNA |
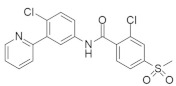
|
Pyridine, Chlorobenzene, Sulfonyl | Pancreatic cancer | Hedgehog (Hh) inhibitor | [80] |
† = N-(1-acryloylazet-id-in-3-yl)-2-(5-bromo-3-(5-methoxy-1,2,3,4-tetrahydroi-soquino-line-2-carbonyl)-1H-indol-1-yl) acetamide; †† = 2-((4-((1-(2-(2,4-dichlorophenoxy) acetyl) piperidin-4-yl) amino)-4-oxobutyl) disulfaneyl)-N,N-dimethylethan-1-aminium.
4.1.1. Ganetespib
The heat-shock protein 90 (Hsp90) is inhibited by ganetespib (Table 1) leading to the degradation of many oncogenic proteins such as v-Raf murine sarcoma viral oncogene homolog B, VEGF receptor, EGFR, Cyclin D1, and mutant p53 [74]. Acquaviva et al. [75] found that ganetespib, a small-molecule inhibitor, suppresses human lung cancer in individualized therapy. Ganetespib showed a potent cytotoxic effect in both in vitro and in vivo studies by inhibiting Hsp90. A set of twenty NSCLC cell lines with known KRAS mutations, including the G12, G13, and Q61, variants, were targeted to identify the cytotoxic activity of ganetespib. Cell lines H727 and H441 showed IC50 values of 28 and 14 nmol/L against the KRASG12V mutant, respectively, which indicates that the small-molecule inhibitor ganetespib is responsive to KRAS mutation. Additionally, ganetespib and the PI3K/mTOR inhibitor BEZ235 showed potential effects in a A549 xenograft model. Finally, a phase II clinical study specified 47% tumor shrinkage in the KRAS-harbored mutation. In 2017, Cercek et al. [74] hypothesized that ganetespib might be an excellent small-molecule inhibitor in colorectal cancer as KRASG12V is one of the main mutants responsible for apoptosis, cell cycle control, and angiogenesis [81,82]. However, in a phase II study with seventeen patients, significant antitumor activity was not shown with Ganetespib (200 mg/m2) treatment when administered intravenously. Interestingly, out of nine patients (3 G12D, 4 G12V, 1 G12S, and 1 G12C), two patients, harboring G12V mutations showed stable conditions.
4.1.2. Apatinib
Apatinib (Table 1) is a small-molecule tyrosine kinase inhibitor that significantly works against metastatic breast cancer and gastric cancer by selectively inhibiting vascular endothelial growth factor receptor-2 (VEGFR-2) [83,84]. In 2017, it was reported that the dosing effect might vary the effectiveness of apatinib in different types of cancer [76]. Their results for four advanced-stage (stage-IV) patients with KRAS mutation, aged 56–81 years, who were diagnosed with metastatic lung adenocarcinoma indicated the potential positive effect of apatinib. Among the four patients, three showed progression-free survival (PFS) (1.5 months, 4.5 months, and 5.5 months) with a small dose. Only one patient was diagnosed with manageable side effects (hoarseness and hemoptysis). Their implemented dosing amount (250 mg/d oral) was significantly lower than the previously studied large-scale trial (500–850 mg/d), in which significant side effects were observed.
4.1.3. Oncrasin-1
A small-molecule compound, oncarsin-1 (Table 1) was identified by synthetic lethality screening and was found to significantly suppress various lung cancers and might inhibit the novel KRAS/PKCι pathway [77]. Although oncarcin-1 has a similar core structure to indole-3-carbinol and lonidamine, an investigation found that it might follow different anticancer mechanisms. This is because indole-3-carbinol [85] and lonidamine [85] have no cytotoxic effect on T29, T29Kt1, T29Ht1, and H460 lung cancer cell lines, whereas oncarcin-1 shows anticancer effects in these cell lines. When T-29 (or T29Kt1) cells were treated with 10 μmol/L oncarsin-1 or H460 cells were treated with 1 μmol/L of oncarsin-1 (equivalent to approximately IC80 doses) the apoptotic cell counts were 33.2%, and 47.2% respectively, which indicated that oncarsin-1 has an effective apoptosis-induction mechanism. Additionally, to identify the KRAS-mutated cell death in H460 cells, orcarsin-1 treatment with control siRNA-treated cells and KRAS siRNA-treated cells showed approximately 17% and 3% more apoptosis, respectively, when compared to treatment with DMSO + siRNA-treated cells. In an in vivo study, oncarsin-1 treatment was compared to solvent-only treatment, and the result showed 75.4% more tumor suppression with oncarsin-1 therapy [77].
4.1.4. N-(1-Acryloylazetidin-3-yl)-2-(5-bromo-3-(5-methoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)-1H-indol-1-yl) Acetamide
Shin et al. found a series of novel KRASG12C inhibitors by exploring the chemotype evolution (custom library synthesis accompanied by subsequent screening) and structure-based design strategies [78]. The newly synthesized compound (†) (Table 1) showed the most potent and selective inhibition against KRASG12C mutants. The path of optimization of this series of small molecules identifies the hidden surface groove bordered by the side chain of Y96/H95/Q99 amino acids. Mechanistically, N-(1-acryloylazetidin-3-yl)-2-(5-bromo-3-(5-methoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)-1H-indol-1-yl) acetamide improves covalent interaction with specifically targeted proteins by means of submolecular inhibition [78]. The first covalent irreversible interaction of KRASG12C was identified by application of tethering, and allosteric binding of the compound in the P2 pocket (near switch II region) resulted in the inhibition of RAS mutation [86]. Further study found more successful results using electrophile screening. An irreversible interaction was produced by means of chemotype evaluation of the anchor molecule (bait), which helped to inhibit the KRASG12C mutation. Shin et al. reported an N-(1-acryloylazetidin-3-yl)-2-(1H-indol-1-yl) acetamide that modifies KRASG12C by most actively hitting with the indole bait and covalently binding with the switch II region. After checking the cytotoxic effect on MIA PaCa-2 cell lines, the compound demonstrated an IC50 value of 0.638 μM. Further investigation proved that adding methyl group in two positions of the indole group increased the activity two-fold (IC50 = 0.299 μM) and two-fold (IC50 = 1.68 μM) in the GTP 2 h exchange and cellular potency assays, respectively, in comparison to the previous compound [78]. A recent report showed that Y96/H95/Q99 are mutated in cancer cells under the influence of KRASG12C inhibitors [87].
4.1.5. 2-((4-((1-(2-(2,4-Dichlorophenoxy) acetyl) piperidin-4-yl) amino)-4-oxobutyl) disulfaneyl)-N,N-dimethylethan-1-aminium
A newly developed small molecule (Table 1) exerts a therapeutic effect by inhibiting KRASG12C mutants and binding to the allosteric pocket. This small-molecule inhibitor is attached to the Cys of KRAS. It binds the allosteric binding site very close to the switching II pocket (SW IIP), resulting in the disruption of GTP-state RAS conformation and the impairment of Raf activity [79]. Although it is difficult to identify the specifically targeted allosteric binding site for a specific protein, cystine-dependent small molecules can irreversibly bind with the allosteric binding site of the KRASG12C oncoprotein.
4.1.6. GDC-0449 (Vismodegib)
The combination therapy of a small-molecule hedgehog (Hh) inhibitor (GDC-0449) (Table 1) (small hydrophobic molecule) and miRNA (miR-let7b) (oligonucleotide) can inhibit pancreatic cancer growth both in vitro and in vivo [80]. Researchers found that increasing the level of Hh signaling promotes cell proliferation by controlling EMT and PI3 in a kinase-dependent manner, which results in different types of cancer (pancreatic cancer, breast cancer, colon cancer, prostate cancer, brain tumor, and basal cell carcinomas). Hh signaling is also responsible for CSCs proliferation and decreases apoptosis by regulating Bcl-2 and Bcl-X. However, GDC-0449 in monotherapy is efficient for tumor cell proliferation in vitro via the inhibiting of Hh signaling [88]. While, due to the lack of efficient delivery, miR-let7b cannot exert an effect on PDAC, it was reported to cause pancreatic cancer cell growth inhibition [89]. Therefore, cationic chains of mPEG-b-PCC-g-DC-g-TEPA copolymeric micelles are used as a suitable delivery system for miR-let7b. Finally, mPEG-b-PCC-g-DC-g-TEPA copolymeric micelles encapsulate GDC-0449 and form a complex with miR-let7b. The synergistic effect of these two inhibitors combined inhibited pancreatic cancer in an in vitro and in vivo study [80].
4.2. Aza Heterocyclic Small-Molecule RAS Inhibitors with More Than One Nitrogen Atom
In Table 2 Aza heterocyclic small-molecule RAS inhibitors with more than one nitrogen atom are classified.
Table 2.
Aza heterocyclic small-molecule RAS inhibitors with more than one nitrogen atom.
| Compound Name | Structure | Major Pharmacophore(s) | Cancer Type | Targeted Enzyme (M/A) | References |
|---|---|---|---|---|---|
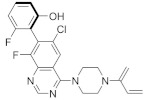
| ARS-1620 |
|
6-chloro-8-fluoroquinazoline, Piperizine | Unknown | KRASG12C inhibitor | [90] |
| ARS-853 |
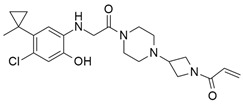
|
Chlorophenol, Piperazine, Azetidine | Unknown | KRASG12C inhibitor | [91] |
| AMG 510 (Lumakras or Sotorasib) |
|
Piperazine, Fluorophenol, Fluoropyrimidinone | Pancreatic cancer/Lung cancer | KRASG12C inhibitor | [92] |
| MRTX849 (Adagrasib) |
|
Piperizine, Chloronaphthalene, Tetrahydropyrido[3,4-d]pyrimidine |
Pancreatic cancer/Lung cancer | KRASG12C inhibitor | [93] |
| ††† |
|
Piperazine, Pyrazolopyrimidine | Pancreatic cancer | Inhibits MAPK/RAF signaling | [94] |
| BGB324 (Bemcentinib) |
|
Cycloheptapyridazine, Pyrrolidine, Triazole | Pancreatic cancer | Axl kinase inhibitor | [95] |
| ABT-737 |
|
Chlorobiphenyl, Piperazine | Colon cancer | Represses Bcl-2/Bcl-XL, resulting in inhibition of the RNAi of PAK4 and PAK1 | [96] |
| AZD6244 (Selumetinib) |
|
Quinoline, Imidazoquinoline | Colorectal cancer | Downregulation of MEK1/2 pathway inhibits KRAS mutation | [97] |
| NVP-BEZ235 (Dactolisib) in Combination with AZD6244 (Selumetinib) |
|
Quinoline, Phenylpropanenitrile, Imidazoquinolinone | Lung cancer | Dual pan PI3K/MEK inhibitor | [98] |
| R115777 (Zarnestra or Tipifarnib) |
|
Chlorophenyl, Imidazole, Quinolinone | Myeloma | Inhibits farnesyl transferase signaling | [99] |
| PPIN-1 PPIN-2 |
|
Biphenyl, Imidazole, N-propyldiazepanone, Tetrahydropyran | Unknown | PPI inhibitor | [100] |
| pan-RAS inhibitor 3144 (RAS-IN-3144) |
|
Indole, Piperazine, Trifluoromethoxyphenyl | Unknown | Downregulates PI3K/AKT, RAF/MEK/ERK signaling | [101] |
| Deltarasin |
|
Benzimidazole, Piperidine | Pancreatic cancer/Lung cancer | Downregulates RAS/RAF signaling pathway | [102] |
| †††† |

|
Fluoroquinoline, Fluorophenol, Piperazine, Piperazinone | Unknown |
KRASG12C inhibitor |
[103] |
| SML-8-73-1 SML-10-70-1 |
 SML-8-73-1 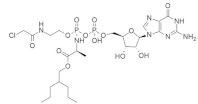 SML-10-70-1 |
Purine, Tetrahydrofuran | Unknown | Inhibits KRASG12C binding with guanine-binding site | [104] |
††† = 1-(2-hydroxyethyl)-4-(2-methyl-3,5-diphenylpyrazolo[1,5-a] pyrimidin-7-yl) piperazin-1-ium. †††† = (2R,4aR)-3-acryloyl-11-chloro-9-fluoro-10-(6-fluoro-2-hydroxycyclohexa-2,4-dien-1-yl)-2,6-dimethyl-2,3,4,4a-tetrahydro-1H-pyrazino [1’,2’:4,5] pyrazino[2,3-c] quinolin-5(6H)-one.
4.2.1. ARS-1620
The current study was successful in considering KRAS as a therapeutic target. The new investigation might be a promising therapeutic approach for KRAS-dependent cancer treatment. Article [90], which explored the KRASG12C S-IIP-binding site in a structure-based design study, showed KRAS inhibition through the invention of a new covalent compound (ARS-1620). ARS-1620 (Table 2) is the first orally bioavailable, potent, and selective small-molecule KRASG12C inhibitor. Moreover, it also provides evidence of therapeutic inhibition for both in vitro and in vivo studies. ARS-1620 treatment showed drastically different results with 2D monolayer adherent and 3D ultra-low adherent suspension in in vitro and in vivo studies [90]. Three-dimensional ULA spheroid culture showed a much more effective result than 2D monolayer culture. This result suggested an inherent difference in KRAS dependence in different types of culture cells.
4.2.2. ARS-853
Further studies were conducted to increase the potency of ARS 1620. An investigation found that this small-molecule compound did not show any cellular response when bound with non KRASG12C, even with 10-fold higher concentrations, but targeted the GDP-bound protein ARS-853 (Table 2) and inactivated the signaling of KRASG12C with an IC50 value of 1 μmol/L, indicating the selectivity of ARS-853 [91]. As a result, a more potent and selective covalently bounded KRASG12C inhibitor was identified.
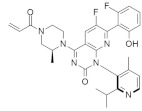
4.2.3. AMG 510 (Lumakras or Sotorasib)
To make the ARS-1620 more efficient and draggable, Canon et al. [92] developed a new small-molecule inhibitor named AMG 510. This compound is superior to ARS-1620, and they have minimal structural differences. The use of an irreversible strategy of the His95 groove, which is close to the cystine pocket AMG 510 (Table 2), produces robust inhibition of the KRASG12C oncogene. It also downregulates the MAPK signaling pathway in both pancreatic and lung cancer. However, it does not affect wild-type KRASG12C mutations, which proves the specificity of AMG 510. After the consequent success, a further study was conducted to obtain a more efficacious result. Combinatorial therapy of AMG 510 with a MAPK inhibitor (carboplatin) created a more potent outcome than monotherapy. These effective outcomes induced researchers to move forward. As a result, synergistic effects were tested in a mice model, and 90% of mice showed complete tumor suppression. Finally, a preliminary clinical study on the human model provided 50% tumor regression. However, most of the success of these comprehensive studies relates only to the KRASG12C oncogene, mostly found in lung cancer [105]. Only 2% of PDAC are responsible for the KRASG12C oncogene [50]. Nevertheless, after reviewing this success, it was suggested that the KRASG12D and KRASG12V mutations, responsible for approximately 80% of pancreatic cancer, might be the primary targets for future studies [50].
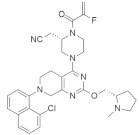
4.2.4. MRTX849 (Adagrasib)
Another structurally modified small-molecule inhibitor, MRTX849 (Table 2), developed by Jill Hallin et al. [93], was found to be a very potent, selective, and covalent KRASG12C inhibitor in pancreatic and lung cancer, and is structurally and functionally close to the AMG 510. This compound exerts effectiveness in combination therapy and has remarkable potency when administered alone. Thus, a phase I clinical study was conducted with the single agent in two patients with final-stage lung and colon carcinoma. The results showed partial responses to these two critical types of cancer. However, the return of ERK signaling and the lack of inhibition of mTOR-S6 signaling made the MRTX849 response short-lasting and ineffective. The results from this study influence future expectations for pancreatic cancer treatment.
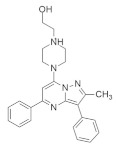
4.2.5. 1-(2-Hydroxyethyl)-4-(2-Methyl-3,5-Diphenylpyrazolo[1,5-a] Pyrimidin-7-yl) Piperazin-1-ium
Recently, investigations found that AMG 510 is not the only small-molecule inhibitor that is potent in terms of the KRAS mutation. In 2019, McCarthy et al. developed a new small-molecule inhibitor [94], which has the affinity to bind to the allosteric binding site. This allosteric pyrazolopyrimidine-based inhibitor (†††) (Table 2) binds with the allosteric p1 pocket of both wild-type and KRAS mutants. This new small molecule blocks the MAPK pathway by downregulating the Raf signaling towards RAS mutation. Another advantage of this allosteric inhibition is that it is not limited to only this specific KRAS-type allele. It shows benefits for wild-type and GTP-bound subtypes of the KRAS oncogene. This remarkable positive result makes this allosteric inhibitor a first-line therapy for tumor treatment in a different range. This small-molecule allosteric inhibitor showed notably good results in lung and oral cancer cell lines that make the allosteric pyrazolopyrimidine-based inhibitor applicable for PDAC treatment.
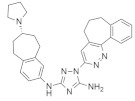
4.2.6. BGB324 (Bemcentinib)
PDAC progression is directly interlinked with Axl, where the TBK1–NFkB pathway and innate immune suppression are caused by Axl kinase signaling [106,107]. BGB324 (Table 2) is an Axl kinase inhibitor that inhibits colony formation in the PDAC cancer cell line, and gemcitabine produces a moderate therapeutic effect in vitro. BGB324 works by decreasing the phosphorylation of Akt, and TBK1 causes inhibition of Axl signaling [95]. An investigation showed that treatment with BGB324 in six human and three mice PDAC cell lines achieved IC50 values ranging from 1 to 4 μmol/L. When BGB324 was administered in combination with gemcitabine, the therapeutic efficacy was identified for a prolonged time. A study on the murine model showed that control drugs or only gemcitabine therapy provide therapeutic effects for not more than one day. Still, applying combination therapy (BGB324+ gemcitabine) exerted a prolonged effect and extends the possibility of novel treatments against PDAC [95].
4.2.7. ABT-737
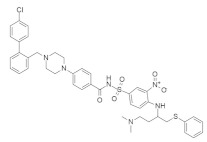
p21-activated kinase (PAK) is an effector of small GTPase Rac and cdc42 proteins. These proteins are responsible for cell-cycle regulation, cell division, and transformation [108,109]. Studies found that inhibition of PAK4 and PAK1 suppresses the KRAS and BRAF mutation in colon cancer in vitro. ABT-737 (Table 2) is a small-molecule inhibitor that represses Bcl-2/Bcl-XL, resulting in the inhibition of RNAi of PAK4 and PAK1 in HCT116 colon cancer cells [96]. Although PAK1 and PAK4 phosphorylate are common substrates in phosphorylation pathways (RAF/MEK/ERK) [110,111,112,113], they did not show any consistent inhibition of the RAF1, MEK, or ERK signaling pathways in HCT116 colon cancer cells. The results showed that ABT-737 treatment in PAK4 led to 43–89% reductions in 6 of 7 cells, and the suppression of PAK1 caused 45–50% decreases in 2 of 7 cells according to the cell proliferation assay. Interestingly, when PAK4 and PAK1 were treated simultaneously, only PAK4 responded for seven colon cancer cell lines in the cell proliferation assay. As a result, further investigation identified that PAK4 and PAK1 suppression were responsible for 95% and 80% cell proliferation, respectively, in HCT116 colon cancer cell lines alone [96].
4.2.8. AZD6244 (Selumetinib)
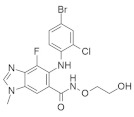
The identification of specific inhibitors for specific biomarkers is an important strategy in cancer treatment. Tentler et al. [97] classified the biomarkers that showed therapeutic responses to the MEK1/2 pathway using the small-molecule inhibitor AZD6244 (Table 2) in KRAS-mutated colorectal cancer. They tested AZD6244 in 37 CRC cell lines with KRAS/BRAF mutation status. They found IC50 values of ≤0.1 mmol/L in AZD6244-sensitive (7 of 27 cells) and of >1 mmol/L in AZD6244-resistant (11 of 27 cells) CRC cell lines. A further study was conducted to classify the genomic target for AZD6244. Tests conducted using in vivo mouse model, and human model found that the KRAS mutants had limited therapeutic options and expressed different gene array and pathway analysis [97]. The Wnt signaling pathway was overexpressed in AZD6244-resistant CRC cell lines, indicating that this pathway might be responsible for resistance in the MEK inhibitor and, furthermore, the Wnt pathway might be a potential target for MEK inhibitors via modification of their structure or therapeutic activity [97].
4.2.9. NVP-BEZ235 (Dactolisib)
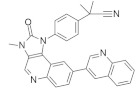
A small-molecule compound, NVP-BEZ235 (Table 2), was identified as a potent dual pan PI3K/MEK inhibitor in lung cancer. Doxycycline induces p110α H1047R mutation in human lung cancer cells [98]. P110α is a catalytic subunit that is encoded by the PIK3CA gene and activates PI3K [114]. NVP-BEZ235 inhibits p110α H1047R mutation by blocking the kinase activity of PI3K. A further in vivo study was conducted in the mice model to identify the efficacy of NVP-BEZ235 by inhibiting TORC1. However, combination therapy of NVP-BEZ235 and rapamycin successfully decreased the S6 phosphorylation of TORC1, resulting in significant tumor suppression. However, the single treatment of NVP-BEZ235 did not respond in an in vivo study [98]. An investigation of previous research revealed that PI3K caused KRAS mutation in the mouse model [115,116]. As a result, different genetic alteration approaches were used in further investigations, and it was confirmed that the downregulation of PI3K suppresses KRAS-mutated lung carcinoma, but the effect was not so significant. Recent studies found that the inhibition of both PI3K and ERK signaling can be highly effective in cancer treatment. As a result, the combination treatment of NVP-BEZ235 (PI3K inhibitor) and ARRY-142886 (MEK Inhibitor) [117] showed significant tumor suppression in KRASG12D lung cancer cells, whereas ARRY-142886 alone exerted mild effect [98].
4.2.10. R115777 (Zarnestra or Tipifarnib)
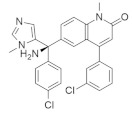
A potent small-molecule inhibitor, R115777 (Table 2), can inhibit farnesyl transferase signaling by inducing apoptosis in myeloma cells. RAS mutation is commonly seen in myeloma and is activated by interleukin receptor 6 [118]. Previous studies found that another FTI inhibited RAS prenylation by activating growth arrest in different myeloma cells, whereas R115777 also showed a similar mechanism in inhibiting myeloma [118]. On the contrary, further investigation showed that R115777 inhibited cell growth following another mechanism that did not involve inhibiting RAS prenylation. These results indicate that R115777 follows an independent RAS mechanism [99]. Apoptosis activity was investigated in Mcl-1, Bcl-XL, and Bcl-2 antiapoptotic proteins [119], which are overexpressed in myeloma [120,121]. It was found that R115777 suppresses Bax overexpression by disputing mitochondrial membrane and activating endoplasmic reticulum stress. To identify the inhibition of caspase-9-induced apoptosis, the activity of R115777 was compared to known caspase-9 inhibitors, and the results showed a dose-dependent inhibition of caspase-9, which indicates that R115777 induces apoptosis by inhibiting caspase-9 [99].
4.2.11. PPIN-1, PPIN-2
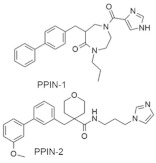
Targeting protein–protein interaction (PPI) is an attractive way to treat RAS mutation. Cruz-Migoni et al. [100] identified two PPI inhibitors, PPIN-1 and PPIN-2 (Table 2), which bind to pocket 1 near the effective binding site. However, after binding to KRAS166G12D, these inhibitors cannot impair any RAS mutation due to the presence of intracellular anti-RAS antibody fragments. Nevertheless, using a synthetic method, a new compound can be developed by combining the two classes of RAS-binding compounds. As a result, the inhibitor PPIN was combined with one of the RAS-binding intracellular antibody derivatives (Abd-7), and the RAS inducer PPI was inhibited. To bind these RAS inhibitors together, PPIN biphenyl group acted as the bait, and three crossover compounds, Ch-1, Ch-2, and Ch-3, were synthesized. The combination of PPIN-2 and these newly synthesized compounds bound smoothly in pocket 1, and Ch-1 and Ch-3 showed better IC50 values (5.3 and 4.5 µM, respectively) than Abd-7 [100].
4.2.12. pan-RAS Inhibitor 3144 (RAS-IN-3144)
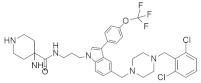
Investigations found that it is tough to identify specific inhibitors that bind to the RAS protein in the appropriate binding site [122,123]. Sometimes interacting with allosteric or adjacent binding sites, small-molecule inhibitors can produce therapeutic effects on RAS mutation. Welsch et al. [101] synthesized the pan-RAS inhibitor 3144 (Table 2), which showed a binding effect with the adjacent site of the KRAS oncoprotein. After passing different experimental conditions, 3144 was found to bind with KRASG12D and KRASG12V. Further investigation found that 3144 induced caspase activity, increased apoptosis, and mechanistically downregulated the overexpression of the RAS effectors PI3K/AKT in addition to RAF/MEK/ERK signaling [101]. Additionally, 3144 inhibited progression and tumor growth in the case of RAS mutation both in vitro and in vivo. A further experiment showed its effectiveness on KRAS-mutated pancreatic cancer in a mouse model, but significant side effects were also observed in this case [101].
4.2.13. Deltarasin
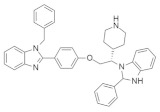
Recent studies found that Deltarasin (Table 2) downregulates the RAS/RAF signaling pathway by inhibiting Phosphodiesterase-δ (PDEδ) binding with the hydrophobic pocket of PDEδ, resulting in the inhibition of KRAS-harbored pancreatic ductal adenocarcinoma (PDAC) [124,125]. As KRAS mutation is responsible for different types of cancers—lung cancer, colorectal cancer, etc.—more investigation is desirable for inhibiting KRAS oncogenes. The majority of lung cancer incidents happen for KRAS mutants [126]. As a result, Leung et al. [127] first identified that deltarasin induces apoptosis significantly, both in vitro and in vivo, in lung cancer cells. It also induces autophagy in lung cancer cells by inhibiting the MAPK/mTOR signaling pathway. It was also shown that when deltarasin is treated with 3-MA (autophagy inhibitor), it increases autophagic properties and produces more intracellular ROS levels, thus protecting against further autophagy.
4.2.14. (2. R,4aR)-3-Acryloyl-11-chloro-9-fluoro-10-(6-fluoro-2-hydroxycyclohexa-2,4-dien-1-yl)-2,6-dimethyl-2,3,4,4a-tetrahydro-1H-pyrazino [1’,2’:4,5] Pyrazino[2,3-c] Quinolin-5(6H)-one
Kettle et al. [103] identified a potent and selective KRASG12C inhibitor (Table 2) by introducing a key methyl group to the piperazine. The KRASG12C allele constitutes an “Achilles heel” where the small-molecule inhibitors can bind covalently, bind the mutant cysteine, and create an allosteric pocket on GDP-bound RAS. The strategy modifies the weak KRASG12C inhibitor that binds to this specific site, increasing the potency of the newly synthesized compound. The in vivo study showed significant tumor regression in KRASG12C mutant Miapaca2 xenografts.
4.2.15. SML-8-73-1 and SML-10-70-1
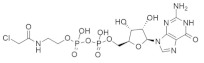
Min Lim et al. [104] found that guanine-based small-molecules covalently bind in the guanine-binding site and that binding with covalent inhibitors can irreversibly inhibit KRAS signaling. KRAS mutations occur near the usual position of the gamma phosphate of the GTP-binding site. After conducting the X-ray crystallography and molecular docking study, a promising candidate (SML-8-73-1) was synthesized, which binds the specific guanine-binding site. To improve the potency, a new analog of SML-8-73-1 (Table 2) was developed by modifying the beta phosphate as an alanine ester phosphoramidite, resulting in SML-10-70-1(Table 2). Moreover, the new derivative, SML-8-73-1, produced antiproliferative effects on the KRASG12C-mutated cell line.
4.3. Oxoheterocyclic Small-Molecule RAS Inhibitors
Identification of major pharmacophore(s) and biomolecular target based on the structure of oxoheterocyclic RAS inhibitors are shown in Table 3.
Table 3.
Oxo heterocyclic small-molecule RAS inhibitors.
| Cancer Type | Compound Name | Structure | Major Pharmacophore(s) | Biomolecular Target (M/A) | Reference |
|---|---|---|---|---|---|
| Lung cancer | NHTD |
|
Tetrahydrodibenzofuran, 2,4-Dihydroxybenzohydrazide | Inhibits tumor progression, decreasing CRAF, ERK, and AKT phosphorylation | [102] |
| Unknown | PD98059 |
|
Chromenone | MEK inhibitor | [128] |
| Colorectal cancer | Wortmannin |
|
Furoindenoisochromene | Suppresses upregulation of PI3K | [129] |
4.3.1. NHTD
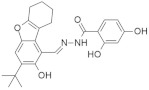
The binding of KRAS with the prenyl-binding protein (PDEδ) produces its oncogenic property. Therefore, inhibition of PDEδ can be an effective path for KRAS-induced cancer treatment [125,130]. The small-molecule prenyl inhibitor NHTD (Table 3) blocks the prenyl-binding pocket as well as localization to the plasma membrane, resulting in the disruption of oncogenic KRAS-PDEδ binding [102]. NHTD also can induce apoptosis in different types of lung cancer cells. In the in vivo study, NHTD inhibited tumor progression in xenograft and mouse models by decreasing CRAF, ERK, and AKT phosphorylation. Computational molecular docking studies identified a specific binding site for a particular small molecule.
4.3.2. PD98059
It was reported that colon cancer is associated with two genetic events: the Wnt signaling pathway and KRAS mutation. Additionally, vascular endothelial growth factor (VEGF) is operated by both genetic factors [128]. KRAS exerts an oncogenic effect by activating multiple Raf/MEK/ERK and PI3K pathways. An experiment was conducted with LY294002 and PD98059 (Table 3) on the ERK and PI3K pathways to determine if the Wnt signaling pathway follows similar pathways for the association of colon cancer. The results indicated that the PI3K effector pathway was critical for the KRASval12-mediated stimulation of Wnt signaling.
Additionally, further investigation was conducted to determine if both KRAS and Wnt signaling regulate the phosphorylation of TCF4. The results showed that β catenin formed a complex in both the phosphorylated and unphosphorylated form, but KRAS did not control the phosphorylation of TCF4. β catenin is the key regulator for Wnt signaling; KRAS regulates the stability of β catenin. Moreover, KRAS controls GSK-3 β Activity independently of Serine-9 Phosphorylation. Finally, these two genetic factors follow a unique interaction between two oncogenic pathways by which KRAS increases signaling via the Wnt pathway in colon cancer [128].
4.3.3. Wortmannin
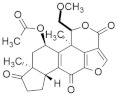
Wortmannin (Table 3) is a PI3K inhibitor that covalently binds to the p110 subunit of PI3K; it irreversibly inhibits PI3K and decreases AKT phosphorylation [131,132]. Bialkowska and colleagues [129] identified that wortmannin decreased pAKT levels in DLD-1 cells via a reduction in KLF5 levels. Recent studies found that PI3K/AKT activation is often associated with colorectal cancer and enhanced colorectal cancer development [133]. Wortmannin inhibits colorectal cancer by suppressing the upregulation of PI3K in a dose-dependent manner.
4.4. Mixed Heterocyclic Small-Molecule RAS Inhibitors
According to the structure, mixed heterocyclic small-molecule RAS inhibitors are classified in Table 4. The bio-molecular targets and mechanistic studies are also described.
Table 4.
Mixed heterocyclic small-molecule RAS inhibitors.
| Compound Name | Structure | Major Pharmacophore(s) | Cancer Type | Targeted Enzyme (M/A) |
Reference |
|---|---|---|---|---|---|
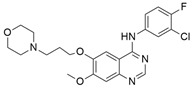
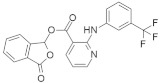
| Talniflumate + Gefitinib |
|
Chlorofluorophenyl, Morpholine, Quinazoline, Pyridine, Benzofuranone | Pancreatic cancer | Inhibition of 2 β-1,6 N-acetylglucosaminyltransferase (GCNT3) | [134] |
| CPD-0857 and KY1022 |
 & 
|
Pyrazole, Pyran, Thienopyrimidine | Colorectal cancer | Inhibition of Wnt/β-catenin, RAS/ERK, and PI3K/AKT | [135] |
| KYA1797K (ab229170) |
|
Nitrophenyl, Furan, Thioxothiazolidin-4-one | Colorectal cancer | Unknown | [136] |
| 0375-0604 (KRAS inhibitor-9 or DUN09716) |
|
Benzothiazolylthio-3-chloroaniline | Unknown | Inhibition of KRAS mutation by downregulating RAF/MEK/ERK and RAF/PI3K/AKT signaling pathways | [137] |
| 7773 |
|
Tetrazole, Piperidine, Benzodioxole | Lung cancer | Binding with hydrophobic surface of Igf2bp1 in KH3 and KH4 domain inhibits KRAS mutation. | [138] |
| NSC-658497 |
|
Thioxothiazolidine, Chromane-2,4-dione, Nitrophenyl | Unknown | Binding with hydrophobic pocket downregulates pERK1/2 and pAKT signaling | [139] |
| JNJ-74699157 |

|
Pyrazolopyrimidine, Benzocyclononaphanone | Unknown | KRASG12C inhibitor | Phase I Clinical trial completed |
4.4.1. Talniflumate + Gefitinib
Mucin is one of the major culprits that hinder drug delivery, and several clinical studies identified that mucin is overexpressed in KRAS-driven pancreatic cancer in mouse and human models [140,141,142,143]. Enzyme 2 β-1,6 N-acetylglucosaminyltransferase (GCNT3) is recognized as a novel core mucin-synthesized enzyme and targeting this enzyme could decrease the overexpression of mucin [134]. KRAS mutation with p48Cre/+-LSL-KRASG12D/+ GEM upregulates the mucin concentration in pancreatic intraepithelial neoplasia (PanIN) and PDAC. Further study found that GCNT3 enzyme is abnormally expressed from GEM in pancreatic cancer as compared to the pancreases under normal conditions, resulting in high mucin formation. Therefore, GCNT3 is used as a novel target to inhibit the overexpression of mucin in pancreatic cancer. Talniflumate (Table 4) is a mucin inhibitor with a good binding capacity with GCNT3 following in-silico validation. The docking score of talniflumate is very impressive compared to the known ligand GALB1, 3GALNAC. Further study confirmed that talniflumate, after binding with GCNT3, inhibits the protein expression of GNCT3 and significantly decreases the overexpression of mucin. Further investigation of the EGFR inhibitor (gefitinib) (Table 4) confirmed the occurrence of remarkable tumor regression in PDAC and PanIN, with a significant decrease in mucin expression.
4.4.2. CPD0857 and KY1022
Wnt/β-catenin and RAS-MAPK signaling are the central pathways for KRAS, and APC mutation causes drug-resistant colorectal cancer (CRC) [144]. Recently, Jung Kyu Choi and colleagues [135] identified a novel compound with inhibitory capacity against both the Wnt/β-catenin and RAS-MAPK signaling pathways in drug-resistant CRC. Mechanistically, CPD0857 (Table 4) does not downregulate β-catenin and RAS protein signaling, indicating that it might follow a different mechanism. After observing the apoptotic activity of CPD0857, the results suggested that the inhibition of cell growth and HCT116 happened due to PI3K/AKT suppression, and in vivo tumor suppression occurred following the inhibition of Wnt/β-catenin and RAS/ERK signaling [135]. A previous study found that another small-molecule inhibitor, KY1022 (Table 4), destabilizes both Wnt/β-catenin and RAS/MAPK signaling. This study explicitly targeted KRASG12D in a mouse model, and the results showed a significant decrease in tumor growth [145].
4.4.3. KYA1797K
Another small-molecule KYA1797K (Table 4), was identified as a potent inhibitor of cetuximab-resistant KRAS-driven colorectal cancer [137]. This small-molecule inhibitor functions by disrupting β-catenin and RAS via GSK3β activation. KYA1797K overcame the cetuximab-resistant KRAS mutation when applied to the cetuximab-resistant CRC cell, but it showed dose-dependency in inhibiting the mutation. In vivo analysis was also performed to observe the cetuximab-resistant KRAS-harboring CRC tumor in the Xenograft mouse model [137]. There was no tumor suppression when cetuximab was solely applied to the mouse model. On the other hand, KYA1797K alone or combination with cetuximab showed significant regression of tumor growth. A further experiment conducted on an ApcMin/+/KRASG12DLA2 mouse model showed tumor growth inhibition, but also demonstrated cetuximab resistance [137].
4.4.4. 0375-0604
Xie et al. [146] synthesized a new small-molecule inhibitor, 0375-0604 (Table 4), which exerts a binding effect with the switch region (switch-I, II) of the KRAS-harbored cell line. The significant characteristic of 0375-0604 is that it comprises two hydrogen bonds that bind with the backbone of Met67 and the side chain of Glu37 in the switch I and switch II pocket, respectively. Moreover, after binding to the pocket, 0375-0604 inhibits KRAS mutation by downregulating RAF/MEK/ERK and RAF/PI3K/AKT signaling pathway. It induces apoptosis, leading to the arrest of the G2/M cell cycle in the KRAS-harbored NSCLC cell line.
4.4.5. 7773
Oncofetal RNA-binding protein (Igf2bp1) is responsible for different types of cancer [138] and can synergize mutation to KRAS. Researchers synthesized the small-molecule inhibitor 7773 (Table 4), which can bind with the hydrophobic surface of Igf2bp1 in the KH3 and KH4 domain and inhibit the binding of KRAS 6 RNA in vitro. Targeting Igf2bp1, this small-molecule inhibitor can reduce KRAS mRNA, resulting in a decrease in KRAS protein and downregulation of signaling, thereby decreasing cell growth. Thus, it might be a potential therapeutic target in cancer treatment.
4.4.6. NSC-658497
RAS signaling is activated by SOS1 (guanine nucleotide exchange factors) [139], which is responsible for receptor tyrosine kinase signaling to RAS. A small-molecule inhibitor, NSC-658497 (Table 4), was identified as interacting with SOS1, competitively inhibiting SOS1–RAS interaction, and suppressing SOS1 GEF activity following dose-dependency. Mechanistically, the structure of NSC-658497 comprises aromatic benzopyran and the polar nitrophenyl moiety. A benzopyran derivative interacts with the hydrophobic pocket of SOS1, which acts as bait within the active site of SOS1. In contrast, the polar nitrophenyl moiety may interact with the outside of the hydrophobic pocket of SOS1. Following this mechanism, NSC-658497 showed an inhibitory effect against RAS oncogenic protein. NSC-658497 was also found to inhibit cell proliferation in a murine model by downregulating the pERK1/2 and pAKT signaling pathways.
4.4.7. JNJ-74699157
JNJ-74699157 (Table 4) was identified as a KRASG12C inhibitor and was found to covalently bind to KRASG12C at the critical mutant residue cysteine 12, locking the GTPase in an inactive state and inhibiting KRAS signaling [147]. This small-molecule inhibitor is now in a phase-I clinical trial. The clinical trial of JNJ-74699157 was reported to be completed within one year, with only 10 participants and no results published [148].
4.5. Carbocyclic Small-Molecule RAS Inhibitors
Carbocycles are also very important as heterocycles in drug discovery research. After meticulous investigation, small-molecule RAS inhibitors with the carbocyclic structure are shown in Table 5.
Table 5.
Carbocyclic small-molecule RAS inhibitors.
| ID | Structure | Major Pharmacophore(s) | Cancer Type | Biomolecular Target (M/A) | Reference |
|---|---|---|---|---|---|
| PKF115-584 (Calphostin C) |
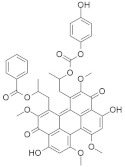
|
Tetramethoxy-3,10-dioxo-3,10-dihydroperylen-1-yl) propan-2-yl benzoate | Colorectal cancer | Downregulates MAPK and RalA signaling | [149] |
| Kobe0065 + Kobe2602 |
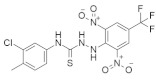 Kobe0065 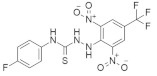 Kobe2602 |
2,6-dinitro-4-(trifluoromethyl)phenyl, Carbothioamide, Halophenyl | Unknown | Inhibits HRASG12V and KRASG12V mutation | [150] |
| ††††† |
|
Phenylacetamide | Myeloid leukemia | Inhibits isoprenylcysteine carboxylmethyltransferase (ICMT) | [151] |
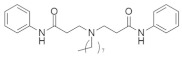
| Salirasib + FTS, Salirasib |
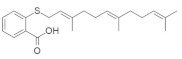 Salirasib 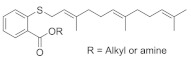 FTS, Salirasib |
Trimethyldodecatrienylthiobenzoic acid | Bladder cancer (salirasib) | Inhibits glycolysis and oxidative phosphorylation pathways | [152] [153] |
††††† = 3,3’-(ethylazanediyl)bis(N-phenylpropanamide).
4.5.1. PKF115-584 (Calphostin C)
Combination therapy of PKF115-584 (Table 5) (β-catenin inhibitor) and transfarnesylthiosalicylic acid (FTS, salirasib) (RAS inhibitor) can inhibit both Wnt-associated and KRAS-associated colorectal cancer [154,155]. Although both inhibitors can solely suppress colorectal cancer, combination therapy synergizes their inhibitory effect. The investigation found that when this combination drug therapy was applied to two colorectal cancer cells carrying both β-catenin and the KRAS mutation, the Ls174T cells showed inhibition of MAPK signaling while the DLD-1 cells inhibited FOS expression, which downregulates RalA [149]. This combination therapy also showed significant cell growth-inducing apoptosis, which occurred because of the downregulation of survivin activity. In addition, a combination of these two inhibitors suppressed tumor growth, and their synergistic effect was specific to the KRAS and Wnt mutant as they did not show any effect on wild-type KRAS. It was also identified that treatment with these combined inhibitors requires a small dose and exerts minimal side effects [149].
4.5.2. Kobe0065 + Kobe2602
Identification of a suitable or specific binding pocket on the surface of the RAS proteins where the therapeutically active small-molecule drug can bind is a promising strategy for treating RAS-related cancer. Thus, Shima et al. [150] identified a therapeutic family of small-molecule inhibitors (Kobe0065 and its analog Kobe2602) (Table 5) that can inhibit multiple types of RAS mutation. A previous study found that small-molecule inhibitors can inhibit SOS expression by binding with KRAS-GDP [156]. After in silico validation, it was identified that Kobe0065 and Kobe2602 could inhibit RAS activity both in vitro and in vivo, binding with HRAS·GTP-c-Raf-1. Both inhibitors efficiently downregulated the phosphorylation of MEK and ERK. These downregulations inhibited the HRASG12V mutation in NIH 3T3 cells. In addition, they showed antitumor activity on KRASG12V mutated SW480 colon cancer cells when orally administered.
4.5.3. 3,3’-(Ethylazanediyl)bis(N-phenylpropanamide)
Methylation of the carboxy-terminal amino acid is one of the post-transcriptional modifications of oncogenic RAS protein that is activated by isoprenylcysteine carboxylmethyltransferase (ICMT) [157]. The inhibition of isoprenylcysteine carboxylmethyltransferase (ICMT) can be an effective pathway for RAS-associated cancer treatment. Marín-Ramos et al. [151] identified a potent small-molecule inhibitor (Table 5) that can impair four RAS isoforms (HRAS, KRAS4A, and -4B, and NRAS). The activity of compound 3 was tested on KRAS (G12C, G12D, G12V, and G13D) isoforms and two NRAS-mutated cell lines (significantly found in myeloid leukemia and myeloma [158]); compound 3 demonstrated significant cytotoxic activity for all the isoforms, with the most potent IC50 value being observed in the NRAS (Q61K) isoform [151].
4.5.4. Salirasib + FTS, Salirasib
HRAS upregulation is frequently identified in bladder cancer (BC) [159]. Salirasib (Table 5) has been identified as a potent RAS inhibitor in different types of cancer but requires a high concentration for inhibition. Sugita et al. [152] investigated the therapeutic activity of salirasib in HRAS-mutated BC. The authors investigated the binding of salirasib with siRNA targets HRAS, and it was found to result in the inhibition of cell proliferation invasion. Proteomic analysis showed that salirasib inhibits the glycolysis and oxidative phosphorylation pathways. Another series comprising the RAS inhibitor S-trans,trans-farnesylthiosalicylic acid (FTS salirasib) (Table 5), which interacted with the RAS membrane, showed a transformation. Goldberg and co-workers developed [153] a new derivative of FTS salirasib where the carboxyl group was modified by esterification and amidation. They identified all the modified FTS salirasib amides, and it was found that two esters significantly inhibited the growth of Panc-1 and of U87 cells, where at least one RAS isoform showed a chronic effect. They also identified that the modified FTS salirasib did not produce any toxicity in Panc-1 and U87 cells.
4.6. Miscellaneous Small-Molecule RAS Inhibitors
There are some small-molecule RAS inhibitors that are in clinical trials. Details about these RAS inhibitors have not been published yet. We also identified one thioheterocyclic small molecule RAS inhibitor. In Table 6, we classified these RAS inhibitors as miscellaneous small-molecule RAS inhibitors.
Table 6.
Miscellaneous small-molecule RAS inhibitors.
| ID | Structure | Major Pharmacophore(s) | Cancer Type | Targeted Enzyme/ (M/A) |
References |
|---|---|---|---|---|---|
| ML264 |
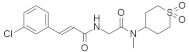
|
Dioxidotetrahydro-2H-thiopyran, Chlorophenyl | Colorectal cancer | KLF5 inhibitor | [160] |
| GDC-6036 | Unpublished | Unpublished | KRASG12C inhibitor | Phase I Clinical trial (Genentech Inc) |
|
| LY3499446 | Unpublished | Unpublished | KRASG12C inhibitor | Phase I and II Clinical trial (Eli Lilly) |
|
| D-1553 | Unpublished | Unpublished | KRASG12C inhibitor | Phase I Clinical trial (InventisBio Co., Ltd., Shanghai, China) |
4.6.1. ML264
Krüppel-like factor 5 (KLF5) is a transcription factor that is overexpressed in proliferating intestinal crypt epithelial cells. KLF5 acts as a mediator of the RAS/MAPK and WNT signaling pathways under homeostatic conditions, and it increases their oncogenic function in intestinal adenomas. Sabando et al. [160] identified a novel KLF5 inhibitor with a therapeutic effect in colorectal cancer. Treatment with ML264 (Table 6) in DLD-1 and HCT116 colorectal cell lines showed a regression in the protein levels of KLF5 that paralleled a reduction in the levels of the transcription factor early growth response 1 (EGR1), which is a direct effector of KLF5 expression. The application of ML264 in a nude mice model inhibited proliferation in colorectal cancer.
4.6.2. GDC-6036
GDC-6036 is a c inhibitor. The combination of GDC-6036 with atezolizumab causes intracellular interactions (ICI), its combination with cetuximab or erlotinib blocks EGFR, and the combination of GDC-6036 with bevacizumab inhibits VEGF. GDC-6036 is also in phase I clinical trial. However, the structure of this compound is not disclosed [148].
4.6.3. LY3499446
LY3499446 is another KRASG12C inhibitor that is combined with abemaciclib, and it inhibits the regulation of CKD4. This small-molecule inhibitor showed promising results in phase I clinical trials. Unfortunately, in 2020, LY3499446 was terminated due to unexpected toxicity [148,161].
4.6.4. D-1553
D-1553 also inhibits KRASG12C mutants. This inhibitor is in a phase I clinical trial. Details about these inhibitors have not been published, and data from ongoing clinical trials have not been reported [148,161].
5. Small-Molecule Natural Products as RAS Inhibitors
Various anticancer compounds have been identified from natural sources. Researchers identified that from 1981 to 2019, the US federal drug administration (FDA) approved 185 small-molecule anticancer drugs to treat various types of cancer. Among these 185 small-molecule anticancer drugs, 43% are from natural product derivatives, and 18% are unaltered natural product anticancer drugs [162,163,164]. These data highlight that natural products comprise a vast research area in cancer treatment. Herein, we describe natural product RAS inhibitors and their molecular mechanisms of action.
5.1. Natural Product RAS Inhibitors with Heterocyclic Skeleton
5.1.1. Quercetin
Quercetin (Figure 3) is a dietary flavonoid found in tea, onions, grapes, wines, and apples [165]. Investigations found that quercetin can inhibit human colorectal cancer by inhibiting the expression of the p21 RAS mutant. An immunocytochemical study found that the administration of 10 μM quercetin reduces p21 KRAS in colon cancer cells and in initial colorectal cancer. This quercetin treatment is time and concentration-dependent. At twenty hours, 10 μM quercetin treatment induces 50% p21 KRAS inhibition. Interestingly, quercetin produces a similar therapeutic effect for KRAS, HRAS, and NRAS oncoproteins, and their effect does not depend on the cell cycle position of colon cancer [165]. Zhang and co-workers [166] investigated the possibility that quercetin also inhibits the cell growth and metastasis of osteosarcoma. Although quercetin administration solely decreases osteosarcoma, its combination with cisplatin, a well-known chemotherapeutic agent, synergizes with the quercetin activity in osteosarcoma. Additionally, treatment with 5 μM quercetin in 143B cells did not show cell viability, whereas significant cell viability was observed with 10 μM quercetin. Furthermore, when 5 μM cisplatin was administrated to 143B cells, the IC50 value was 6.12 M. In the meantime, cotreatment with 5 μM of quercetin and cisplatin showed an IC50 of 4.21, which indicates that quercetin enhanced the cisplatin sensitivity of 143B. Cisplatin showed resistance towards osteosarcoma, and MiR-217 decreased the resistance of osteosarcoma [167]. Treatment with quercetin and/or cisplatin upregulates MiR-217, but treatment with KRAS downregulates mRNA and protein levels.
Figure 3.

Heterocyclic natural product RAS inhibitors.
5.1.2. Artemidolide C
Artemidolide is a dimeric sesquiterpene lactone and FPTase inhibitor. It is an (A-D) series that is isolated from Artemisia spp. [168]. In vitro, artemidolide C (Figure 3) treatment showed dose-dependent inhibition, and approximate growth inhibition followed the administration of 1.3–8.1 μM. The growth inhibition in three human cancer cell lines (colon, breast, and CNS) showed sensitivity when treated with artemidolide C, but the renal tumor cell line A498 showed resistance [168]. An earlier study identified that, due to the presence of the α-methylene-γ- lactone group, sesquiterpene lactones exhibit antitumor activity [169]. However, the α-methylene-γ- lactone unit in artemidolide did not affect the inhibition of FPTase activity, but the problem was solved when hydrogenated artemidolides exerted low antiproliferative activity. Furthermore, in vivo antiproliferative activity in nude mice showed that artemidolide C inhibits tumor growth [168]. Another study showed that treatment with artemidolide C in a HRAS-mutated cell line resulted in degradation of the HRAS protein [170].
5.1.3. Statins
Mevalonate intermediates, including farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), which are responsible for activating RAS proteins, were potentially inhibited by statins in pancreatic cancer [171]. Simvastatin (Figure 3) treatment in MiaPaCa-2 human pancreatic cancer cells showed that 200 genes were affected by simvastatin treatment. This was due to the interaction between FPP and simvastatin. However, it was observed that the normalization of the expression of KRAS-related genes and the GFP-KRAS protein trafficking was partially prevented by the addition of any of the mevalonate pathways intermediates. Finally, the addition of FPP or GGPP normalized simvastatin-treated altered genes. Therefore, KRAS protein trafficking can be successfully inhibited by statin treatment in pancreatic cancer [171].
5.1.4. Manumycin A
Exosomes are necessarily involved in the trafficking of oncogenic factors and the neoplastic alteration of stem cells, resulting in cancer progression [172,173]. Manumycin A (Figure 3) (MA) is a natural macrolide antibiotic isolated from Streptomyces parvulus and has been identified as an exosome that can be targeted therapeutically. To determine the nontoxic dose of manumycin A and evaluate the effect of exosome biogenesis, MA was applied to RWPE-1 and PC-3 cells, and no effect was observed, while approximately 8% and 10% cell death was observed on C4-2B and 22Rv1 cells, respectively [174]. However, using the EV analysis based on TRPS, the Nanosight300 and NanoFACS analyses reported that MA selectively inhibits the biogenesis and secretion of exosomes in some castration-resistant prostate cancer (CRPC) cells. In addition, MA inhibited exosome biogenesis and secretion by downregulating the RAS/Raf/ERK1/2 pathway in CRPC cells. Finally, MA inhibits exosome biogenesis and secretion, suppressing RAS signaling pathways (RAS/Raf/ERK).
5.1.5. Gliotoxin
Liver injury happens due to an overdose of analgesic and antipyretic acetaminophen (APAP). Article [175] described that hepatotoxicity due to an excessive dose of APAP activates RAS mutation in mice models. Gliotoxin (Figure 3) is a sulfur-containing mycotoxin produced by various pathogenic fungi, including Aspergillus fumigatus [176]. As a potent farnesyl transferase inhibitor (FTI), Gliotoxin decreased RAS overexpression upon APAP overdosing. It was also identified that RAS activation concomitantly increased with hepatotoxicity [175].
Furthermore, APAP dosing reduces the hepatic glutathione amount, but manumycin A (inhibiting the RAS GTP and ALT interaction) treatment does not affect this. Additionally, the results suggested that RAS activation is regulated by JNK phosphorylation because APAP dosing induces JNK phosphorylation, and the application of manumycin A significantly decreases JNK phosphorylation. After APAP dosing, gliotoxin treatment reduces serum amounts of ALT and IFN-γ and inhibits RAS activation [175].
5.1.6. Preussomerin G
In 1991, Holly A. and colleagues isolated a group of preussomerins (A–F) (preussomerin G, Figure 3) from the coprophilous fungus Preussia isómera [177]. Farnesyl protein transferase is responsible for RAS farnesylation (p21), which is associated with the RAS plasma membrane and helps to produce RAS signaling. Singh et al. [178] isolated preussomerins and deoxipreussomerins from the extract of a dung-inhabiting coelomycetous fungus from Chaco Province, Argentina, which has inhibitory function towards RAS farnesylation.
5.1.7. Pepticinnamin E
Pepticinnamin E (Figure 3) is another FPTase inhibitor. A series of Pepticinnamins were isolated from the culture broth of Streptomyces sp. OH-4652(18). Among them, pepticinnamin E is the primary product that contains a rare N-terminal cinnamoyl moiety and several nonproteinogenic amino acids. All isolated pepticinnamin showed a potent inhibitory property towards FTase with IC50 values 0.1–1.0 μM, but pepticinnamin C produced the strongest inhibition. Furthermore, pepticinnamin E exerts an effect of competitively binding with the p21 RAS protein and non-competitively binding to the substrate farnesyl pyrophosphate [179]. Another study identified a strong reduction in FPTase E, which is expected to treat cancer and malaria [180]. Thutewohl et al. [181] found pepticinnamin E induces apoptosis in RAS-mutated cell lines, which is connected to the inhibition of FTase activity. They identified 51 analogs of pepticinnamin. Overall, twenty compounds showed an inhibitory effect with an IC50 value of 1 μM.
5.1.8. Bryostatin-1
Protein kinase C (PKC) phosphorylates S181 into the polybasic region that promotes the rapid dissociation of KRAS in the plasma membrane and has an association with the outer membrane of mitochondria where phospho-KRAS interact with Bcl-XL. The PKC agonist accelerates the apoptosis of cells altered with the KRAS oncogene. KRAS, with a phosphomimetic residue at position 181, induces apoptosis via a pathway that requires Bcl-XL [182]. Bryostatin-1 (Figure 3) is a cyclic macrolide that is isolated from the marine bryozoan Bugula neritina [183]. Moreover, Bryostatin-1 is a PKC agonist [184] that inhibits KRAS-dependent cell transformation and growth in an S181-dependent manner. An in vivo study showed that bryostatin-1 was efficient against tumors in nude mice containing oncogenic KASG12V, but in KRASG12V181A-driven tumors, it had reduced activity [182].
5.1.9. Piperlongumine
Piperlongumine (PL) (Figure 3), a natural alkaloid present in Piper longum Linn, has been reported to exhibit notable anticancer effects in various in vitro studies. Kumar and colleagues [185] first identified a chemo-preventive effect of PL in colon cancer-infected animal models. PL showed significant antineoplastic activity towards colon cancer cell growth by targeting RAS proteins and the PI3K/Akt signaling pathway. It was also identified that PL blocked the cell cycle progression at the G2/M phase and increased the mitochondrial apoptotic pathway by downregulating Bcl-2 levels. Moreover, PL showed liver and kidney toxicity.
5.1.10. Confluentin
Yaqoob et al. [186] isolated three compounds, confluentin (Figure 3), grifolin, and neogrifolin, from a methanolic extract of the terricolous polypore of Albatrellus flettii (British Columbian mushroom), which showed cell viability. However, the cell viability mechanism of this novel molecule is not very well established. In a cytotoxic study, the confluent showed IC50 values of 25.9 ± 2.9 μM (n = 3), 33.5 ± 4.0 μM (n = 3), and 25.8 ± 4.1 μM (n = 3) against HeLa, SW480, and HT29 cells, respectively [186]. Moreover, they identified confluentin-induced apoptosis for the first time, with a cell-cycle arrest at the G2/M phase in SW480 human colon cancer cells. Additionally, KRASG12D, containing the SW480 human colon cancer cell line, was used to investigate the KRAS inhibition of these three molecules. Confluentin showed a more significant effect than grifolin and neogrifolin, but in the HT29 cell line, which contains wild-type KRAS, a similar inhibitory effect against the KRAS mutation was observed for all three compounds. Finally, using an in vitro fluorescence polarization method, it was found that confluentin inhibits the physical interaction between IMP1 and KRAS RNA [186].
5.1.11. Swinhopeptolides
Two new cyclic depsipeptides named swinhopeptolides A and B were isolated from the marine sponge Theonella swinhoei cf. verrucosa, collected from Papua New Guinea [187]. Both compounds have 11 diverse amino acid residues and 13-carbon polyketide moieties attached at the N-terminus. To identify the effect on RAS-Raf protein–protein interactions of Swinhopeptolides A (Figure 3) and B, a cell-based Bioluminescence Resonance Energy Transfer (BRET) was performed. The results showed that swinhopeptolides A and B effectively inhibited RAS-Raf protein–protein interaction with IC50 values of 5.8 and 8.5 μM, respectively [187]. Moreover, Swinhopeptolides A and B exhibited potential RAS/Raf signaling pathway suppression.
5.1.12. Avicin G
Avicin G (Figure 3) is a plant-derived triterpenoid saponin from Acacia victoriae; it mislocalizes KRASG12V from the Plasma membrane (PM) and disrupts the PM spatial organization of oncogenic KRAS and HRAS by depleting their phosphatidylserine (PtdSer) and cholesterol contents, respectively, at the inner PM leaflet [188]. Further investigation suggested that avicin G inhibits KRASG12V and HRASG12V mutation by downregulating the pERK signaling pathway and pAkt, but more inhibition was observed for KRASG12V cells. Next, a cell proliferation assay was performed on human pancreatic ductal adenocarcinoma (PDAC) cells and non-small-cell lung carcinoma (NSCLC) cells. The results showed significant growth inhibition for both types of the cancer cell line. Previous research identified that lysosome activity is vital for KRAS-driven cancer growth, but avicin G [189,190] blocks the lysosome activity by elevating lysosomal PH and inhibiting the phosphorylation of ERK signaling. It was also identified that Avicin G is a potent SMase inhibitor. It exerts a potential inhibitory effect on acid and neutral SMase [188].
5.2. Natural Product RAS Inhibitor with Carbocyclic Skeleton
5.2.1. Prostratin
Prostratin (Figure 4) is a phorbol ester, which was first isolated from the bark of the mamala tree of Samoa, Homalanthus nutans (Euphorbiaceae) [191]. Prostratin inhibits KRAS and HRAS tumor growth by suppressing non-canonical Wnt/Ca2+ signaling. Comparing KRAS and HRAS, KRAS produces more tumorigenesis, although they have a comparable level of canonical RAS signaling. The reason for this is the ability to induce tumor initiation that is directly related to the ability of KRAS to suppress Fzd8-mediated non-canonical Wnt/Ca2+ signaling. In the KRASG12V mutation, Fzd8 was downregulated, and the CaMKii level was decreased more significantly than that of HRASG12V, resulting in the phosphorylation of threonine 286. Therefore, prostratin (PKC activator) interrupts KRAS calmodulin-binding that increases the level of Fzd8, thereby inhibiting KRAS mutation in pancreatic cancer cells [191].
Figure 4.

Carbocyclic natural product RAS inhibitors.
5.2.2. D-Limonene
Farnesyl protein transferase (FPTase) is an intermediate of the mevalonate pathway, which contains the isopropyl group. The interaction of FPTase with a potent inhibitor can repress malevolent pathways, resulting in the inhibition of transforming characteristics of the RAS mutant. D-Limonene (Figure 4) is a common monoterpene that can inhibit FPTase, and has been identified in essential oils of orange, lime, grapefruit, lemon, mandarin, and many other plants. To determine the FTPase inhibition activity, D-Limonene and some other compounds were applied to partially purified FPTase from LLC-PK 1 cells [192]. The results suggested that among all the tested compounds, D-Limonene, TD-I, TD-I1, and gallotannin produce an inhibitory effect towards FPTase but in a dose-dependent manner. To identify the cell proliferation activity, D-Limonene, TD-I, TD-I1, and gallotannin were tested on W1-38, CACO, A549, and PaCa cells; significant cell proliferation was identified for all the cell lines and the effects on PaCa cells were due to the inhibition of P21RAS membrane association. Additionally, the administration of D-Limonene on A549 cells showed moderate HRAS inhibition. Moreover, western blot analysis showed that D-Limonene also has an increased cytotoxic effect [192].
Another study found that D-Limonene significantly affected skin tumor growth in mouse models by inhibiting 12-O-tetradecanoylphorbol-13-acetate (TPA) [193]. The development of mouse skin tumor was initiated with 7,12-dimethylbenz[a]anthracene (DMBA) and went to the progression state because of 12-O-tetradecanoylphorbol-13-acetate (TPA). In comparison to TPA treatment [194] (previously studied), topical treatment with D-Limonene (12.30 h) in a mouse model showed only 27% and 9% increases in the weight of the skin paunches, whereas TPA (12 h) showed an increase of 48% [193]. Similarly, D-Limonene treatment significantly reduced COX-2-positive cells in the epidermis. Additionally, the application of D-Limonene suppresses the RAS, Raf signaling, and phosphorylation of extracellular protein kinase in the case of DMBA-/TPA-induced tumors [193].
5.2.3. Methyl Linderone
Methyl linderone (ML) (Figure 4) is a cyclo-pentenedione isolated from the fruits of Lindera erythrocarpa (family Lauraceae). Yoon et al. [195] identified that treatment with ML inhibits breast cancer. After performing a cell viability assay, they found that 10 μM is the nontoxic dose of ML. To study the effect of ML in invasion and migration, 12-O-tetradecanoyl phorbol-13-acetate (TPA)-stimulated MCF-7 cells were used. The finding was that TPA-stimulated invasion and migration were inhibited by MA, but in a dose-dependent manner. Furthermore, it was identified that ML inhibited two mRNA—MMP-9 and IL-8—and the protein expression in TPA-treated MCF-7 cells. Moreover, the mechanism of inhibition of MA on MCF-7 cells was followed by the targeting of the TPA-stimulated phosphorylation of ERK and the downstream factors, AP-1 and STAT3.
5.2.4. Antroquinonol
Antroquinonol (Figure 4) extracted from the mycelium of Antrodia camphorate, was identified as the smallest anticancer molecule [196]. Although the mechanism of action of Antroquinonol remains unclear, some evidence suggested that antroquinonol inhibits RAS- and RAS-mediated protein function by blocking the expression of isopropyl protein transferase. Moreover, it was identified that antroquinonol selectively binds with FPTase and geranyl transferase 1, the most RAS-activating enzyme. Antroquinonol effectively inhibits the RAS activity in vitro. It was also determined that antroquinonol can induce apoptosis by blocking the PI3K/mTOR pathway [197]. Moreover, antroquinonol treatment in lung cancer cells induces autophagic property of LC3B-1 to LC3B-II.
5.2.5. 5-CQA (5-O-Caffeoylquinic Acid)
Chlorogenic acid (5-O-caffeoylquinic acid, 5-CQA) was identified in coffee beans used to make green coffee and, after roasting, black coffee, a widespread drink worldwide. 5CQA (Figure 4) was the first natural compound targeting RAS mutation [198]. A current study found that intake of healthy food, abundant in polyphenols, decreases neurovegetative diseases, with 5-CQA being one of them [199]. Moreover, 5CQA affects inflammation, cancer, and neurovegetative diseases. Although the molecular mechanism of biological effectiveness of 5CQA is not investigated, an investigation found that it inhibits RAS-dependent breast cancer. After evaluating the standard NMR result, it was identified that 5-CQA reduces cancer growth by modulating p42/p44 MAPKs activation/phosphorylation. A molecular docking study found four binding sites (S-I-P and S-II-P for both complexes, and S-I-II-P for the HRASGDP complex only) in oncogenic HRAS protein where 5-CQA can bind and determined the FullFitness scores. It was also identified that 5-CQA noncompetitively interacted with the binding site because the computationally found 5-CQA binding site was distinct from the active site.
5.2.6. Lupeol
Lupeol (Figure 4) is a triterpenoid, a FPTase inhibitor, and specifically inhibits KRAS mutants, but not wt KRAS [200]. After analyzing its crystal structure, Ganaie et al. identified 20 terpenoids for their KRAS-binding affinity. In a comprehensive study on lup-20 (29)- en-3b-ol (lupeol) as a KRAS inhibitor, differential scanning fluorimetry, immunoprecipitation assays, isothermal titration calorimetry were performed, and lupeol was identified as a potent KRAS inhibitor. Lupeol has a potential inhibitory effect on mutant KRASG12V. In vivo lupeol administration in a mouse model showed inhibition of the development of pancreatic intraepithelial neoplasia.
5.2.7. Grifolin and Neogrifolin
Grifolin (7a) and neogrifolin (7b) (Figure 4) were isolated from the ethanolic extract of a bioactive mushroom that is indigenous to British Columbia [186]. One more compound, confluentin, was isolated. We discussed the biological activity of confluentin in an earlier section. The aforementioned investigation showed that grifolin and neogrifolin could inhibit oncogenic KRAS in human colon cancer. They also exerted their inhibitory effect in the presence of wild KRAS [186].
5.3. Natural Product RAS Inhibitor with Acyclic Skeleton
Chaethomellic Acids A
Chaethomellic acids are a class of alkyl dicarboxylic acids isolated from Chaetomella acutiseta [201]. An investigation identified two novel compounds, chaethomellic acid A (Figure 5) and B, identified three structural types of FPTase inhibitors—phosphonic acids, chaethomellic acids, and zaragozic acids—that produced weak inhibition to geranyl protein transferase, but each of them showed competitive effects towards farnesyl diphosphate when inhibiting FPTase. Among these three classes, phosphonic acid and chaethomellic acid showed outstanding selectivity towards FPTase and geranyl transferase activity, while zaragozic acids showed less selectivity. Mechanistically, chaethomellic acid A competitively inhibits FPTase and noncompetitively inhibits RAS protein. To identify the FPTase activity, all the compounds were assayed in Ha-RAS containing NIH3T3 fibroblast cells; chaethomellic acids and zaragozic acids had no effect in this assay.
Figure 5.

Acyclic natural product RAS inhibitor Chaetomellic acid A.
Nogueira et al. [202] identified that long-term treatment with chaethomellic acid A reduces renal mass reduction-induced renal fibrosis in a rat model by selectively inhibiting Ha-Ras farnesylation. After analyzing the previous studies [203,204], it was hypothesized that chaethomellic acid A treated CKD by altering the Raf/MEK/ERK and PI3K-Akt pathways. Chaethomellic acid A was administered in three different rat models (23 µg/Kg), three times a week for 6 months, resulting in significantly decreased renal fibrosis, mainly glomerulosclerosis and arteriolosclerosis.
6. Conclusions
Up- and down-regulations of proteins are crucial in terms of causing diseases, including cancer. RAS family proteins, particularly KRAS, play central roles in different stages of cancer development, invasion, propagation, metathesis, and angiogenesis, leading to the formation of solid tumors [205,206,207]. Previously, there was a myth that KRAS is undruggable, but continuous efforts from different parts of the world have established that it is druggable. Despite the progress, the current protocols encounter several limitations, including the loss of selectivity, the causing of toxicity, and the production of incongruent patient outcomes. Various genomic and histologic mechanisms promote resistance to covalent KRASG12C inhibitors. For example, the development of heterogeneous responses to some KRAS inhibitors, such as MRTX849 or AMG510, bypass their anti-growth effects by producing more KRAS variants, to which these inhibitors do not bind. The developed KRAS alterations include G12D/R/V/W, G13D, Q61H, R68S, H95D/Q/R, Y96C, and high-level amplification of the KRASG12C allele. The attained bypass mechanisms of resistance include MET amplification, activating mutations in NRAS, BRAF, MAP2K1, and RET, oncogenic fusions involving ALK, RET, BRAF, RAF1, and FGFR3, and loss-of-function mutations in NF1 and PTEN. Accordingly, more in-depth research is required to overcome the resistance towards KRAS inhibitors. In conclusion, in spite of there being unceasing progress, many miles remain in the development of RAS inhibitors with higher potency and selectivity, lower toxicity, and apprehended drug-resistance.
Acknowledgments
This paper is respectfully dedicated to Subrata Laskar in honoring his lifelong commitment and devotion to Chemical Sciences. The authors are grateful to the Department of Chemistry and the School of Earth Environment & Marine Sciences (SEEMS) of the University of Texas Rio Grande Valley for start-up funding (to DB) and extending facilities for this study. The Department of Chemistry at the University of Texas Rio Grande Valley is grateful for the generous support provided by a Departmental Grant from the Robert A. Welch Foundation (Grant No. BX-0048).
Author Contributions
Conceptualization and supervision, D.B.; writing—review and editing S.A.S. and D.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Karnoub A.E., Weinberg R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox A.D., Der C.J. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics. CA Cancer J. Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 4.Liu P., Wang Y., Li X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B. 2019;9:871–879. doi: 10.1016/j.apsb.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simanshu D.K., Nissley D.V., McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wennerberg K., Rossman K.L., Der C.J. The Ras superfamily at a glance. J. Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 7.Rojas A.M., Fuentes G., Rausell A., Valencia A. The Ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012;196:189–201. doi: 10.1083/jcb.201103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colicelli J. Human RAS superfamily proteins and related GTPases. Sci. STKE. 2004;250:13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Repasky G.A., Chenette E.J., Der C.J. Renewing the conspiracy theory debate: Does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639–647. doi: 10.1016/j.tcb.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Perucho M., Goldfarb M., Shimizu K., Lama C., Fogh J., Wigler M. Human-tumor-derived cell lines contain common and different transforming genes. Cell. 1981;27:467–476. doi: 10.1016/0092-8674(81)90388-3. [DOI] [PubMed] [Google Scholar]
- 11.Wittinghofer A., Vetter I.R. Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 2011;80:943–971. doi: 10.1146/annurev-biochem-062708-134043. [DOI] [PubMed] [Google Scholar]
- 12.Krontiris T.G., Cooper G.M. Transforming activity of human tumor DNAs. Proc. Natl. Acad. Sci. USA. 1981;78:1181–1184. doi: 10.1073/pnas.78.2.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih C., Padhy L.C., Murray M., Weinberg R.A. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature. 1981;290:261–264. doi: 10.1038/290261a0. [DOI] [PubMed] [Google Scholar]
- 14.Nakhaei-Rad S., Haghighi F., Nouri P., Rezaei Adariani S., Lissy J., Kazemein Jasemi N.S., Dvorsky R., Ahmadian M.R. Structural fingerprints, interactions, and signaling networks of RAS family proteins beyond RAS isoforms. Crit. Rev. Biochem. Mol. Biol. 2018;53:130–156. doi: 10.1080/10409238.2018.1431605. [DOI] [PubMed] [Google Scholar]
- 15.Clarke S., Tamanoi F. Fighting cancer by disrupting C-terminal methylation of signaling proteins. J. Clin. Investig. 2004;113:513–515. doi: 10.1172/JCI200421059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silvius J.R. Mechanisms of Ras protein targeting in mammalian cells. J. Membr. Biol. 2002;190:83–92. doi: 10.1007/s00232-002-1026-4. [DOI] [PubMed] [Google Scholar]
- 17.Kwong L., Wozniak M.A., Collins A.S., Wilson S.D., Keely P.J. R-Ras promotes focal adhesion formation through focal adhesion kinase and p130(Cas) by a novel mechanism that differs from integrins. Mol. Cell. Biol. 2003;23:933–949. doi: 10.1128/MCB.23.3.933-949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuhjelm J., Peränen J. The C-terminal end of R-Ras contains a focal adhesion targeting signal. J. Cell Sci. 2003;116:3729–3738. doi: 10.1242/jcs.00689. [DOI] [PubMed] [Google Scholar]
- 19.Reedquist K.A., Ross E., Koop E.A., Wolthuis R.M., Zwartkruis F.J., van Kooyk Y., Salmon M., Buckley C.D., Bos J.L. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J. Cell Biol. 2000;148:1151–1158. doi: 10.1083/jcb.148.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chien Y., White M.A. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 2003;4:800–806. doi: 10.1038/sj.embor.embor899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shipitsin M., Feig L.A. RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol. Cell. Biol. 2004;24:5746–5756. doi: 10.1128/MCB.24.13.5746-5756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark G.J., Kinch M.S., Rogers-Graham K., Sebti S.M., Hamilton A.D., Der C.J. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J. Biol. Chem. 1997;272:10608–10615. doi: 10.1074/jbc.272.16.10608. [DOI] [PubMed] [Google Scholar]
- 23.Im E., von Lintig F.C., Chen J., Zhuang S., Qui W., Chowdhury S., Worley P.F., Boss G.R., Pilz R.B. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene. 2002;21:6356–6365. doi: 10.1038/sj.onc.1205792. [DOI] [PubMed] [Google Scholar]
- 24.Etienne-Manneville S., Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 25.Heasman S.J., Ridley A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 26.Vogt N., Seiler S. The RHO1-specific GTPase-activating Protein LRG1 Regulates Polar Tip Growth in Parallel to Ndr Kinase Signaling in Neurospora. Mol. Biol. Cell. 2008;19:4554–4569. doi: 10.1091/mbc.e07-12-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulloy J.C., Cancelas J.A., Filippi M.D., Kalfa T.A., Guo F., Zheng Y. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115:936–947. doi: 10.1182/blood-2009-09-198127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlessinger K., Hall A., Tolwinski N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009;23:265–277. doi: 10.1101/gad.1760809. [DOI] [PubMed] [Google Scholar]
- 29.Boureux A., Vignal E., Faure S., Fort P. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol. Biol. Evol. 2007;24:203–216. doi: 10.1093/molbev/msl145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mosaddeghzadeh N., Ahmadian M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells. 2021;10:1831. doi: 10.3390/cells10071831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Touchot N., Chardin P., Tavitian A. Four additional members of the ras gene superfamily isolated by an oligonucleotide strategy: Molecular cloning of YPT-related cDNAs from a rat brain library. Proc. Natl. Acad. Sci. USA. 1987;84:8210–8214. doi: 10.1073/pnas.84.23.8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 33.Pereira-Leal J.B., Seabra M.C. Evolution of the Rab family of small GTP-binding proteins. J. Mol. Biol. 2001;313:889–901. doi: 10.1006/jmbi.2001.5072. [DOI] [PubMed] [Google Scholar]
- 34.Zerial M., McBride H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 35.Moore M.S., Blobel G. A G protein involved in nucleocytoplasmic transport: The role of Ran. Trends Biochem. Sci. 1994;19:211–216. doi: 10.1016/0968-0004(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 36.Weis K. Regulating access to the genome: Nucleocytoplasmic transport throughout the cell cycle. Cell. 2003;112:441–451. doi: 10.1016/S0092-8674(03)00082-5. [DOI] [PubMed] [Google Scholar]
- 37.Li H.Y., Cao K., Zheng Y. Ran in the spindle checkpoint: A new function for a versatile GTPase. Trends Cell Biol. 2003;13:553–557. doi: 10.1016/j.tcb.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Joseph J. Ran at a glance. J. Cell Sci. 2006;119:3481–3484. doi: 10.1242/jcs.03071. [DOI] [PubMed] [Google Scholar]
- 39.Kahn R.A., Gilman A.G. Purification of a protein cofactor required for ADP-ribosylation of the stimulatory regulatory component of adenylate cyclase by cholera toxin. J. Biol. Chem. 1984;259:6228–6234. doi: 10.1016/S0021-9258(20)82130-9. [DOI] [PubMed] [Google Scholar]
- 40.Sewell J.L., Kahn R.A. Sequences of the bovine and yeast ADP-ribosylation factor and comparison to other GTP-binding proteins. Proc. Natl. Acad. Sci. USA. 1988;85:4620–4624. doi: 10.1073/pnas.85.13.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price S.R., Nightingale M., Tsai S.C., Williamson K.C., Adamik R., Chen H.C., Moss J., Vaughan M. Guanine nucleotide-binding proteins that enhance choleragen ADP-ribosyltransferase activity: Nucleotide and deduced amino acid sequence of an ADP-ribosylation factor cDNA. Proc. Natl. Acad. Sci. USA. 1988;85:5488–5491. doi: 10.1073/pnas.85.15.5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vargová R., Wideman J.G., Derelle R., Klimeš V., Kahn R.A., Dacks J.B., Eliáš M. A Eukaryote-Wide Perspective on the Diversity and Evolution of the ARF GTPase Protein Family. Genome Biol. Evol. 2021;13:evab157. doi: 10.1093/gbe/evab157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen P.W., Gasilina A., Yadav M.P., Randazzo P.A. Control of cell signaling by Arf GTPases and their regulators: Focus on links to cancer and other GTPase families. Biochim. Biophys. Acta Mol. Cell Res. 2022;1869:119171. doi: 10.1016/j.bbamcr.2021.119171. [DOI] [PubMed] [Google Scholar]
- 44.Muzny D.M., Bainbridge M.N., Chang K., Dinh H.H., Drummond J.A., Fowler G., Kovar C.L., Lewis L.R., Morgan M.B., Newsham I.F., et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey P., Chang D.K., Nones K., Johns A.L., Patch A.M., Gingras M.C., Miller D.K., Christ A.N., Bruxner T.J., Quinn M.C., et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 46.Campbell J.D., Alexandrov A., Kim J., Wala J., Berger A.H., Pedamallu C.S., Shukla S.A., Guo G., Brooks A.N., Murray B.A., et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016;48:607–616. doi: 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keeton A.B., Salter E.A., Piazza G.A. The RAS-Effector Interaction as a Drug Target. Cancer Res. 2017;77:221–226. doi: 10.1158/0008-5472.CAN-16-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adderley H., Blackhall F.H., Lindsay C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine. 2019;41:711–716. doi: 10.1016/j.ebiom.2019.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernández-Medarde A., Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2:344–358. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bryant K.L., Mancias J.D., Kimmelman A.C., Der C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lanfredini S., Thapa A., O’Neill E. RAS in pancreatic cancer. Biochem. Soc. Trans. 2019;47:961–972. doi: 10.1042/BST20170521. [DOI] [PubMed] [Google Scholar]
- 52.Schutte M., Hruban R.H., Geradts J., Maynard R., Hilgers W., Rabindran S.K., Moskaluk C.A., Hahn S.A., Schwarte-Waldhoff I., Schmiegel W., et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126–3130. [PubMed] [Google Scholar]
- 53.Waters A.M., Der C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018;8:a031435. doi: 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kordelas L., Rebmann V., Ludwig A.K., Radtke S., Ruesing J., Doeppner T.R., Epple M., Horn P.A., Beelen D.W., Giebel B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014;28:970–973. doi: 10.1038/leu.2014.41. [DOI] [PubMed] [Google Scholar]
- 55.Kamerkar S., LeBleu V.S., Sugimoto H., Yang S., Ruivo C.F., Melo S.A., Lee J.J., Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zorde Khvalevsky E., Gabai R., Rachmut I.H., Horwitz E., Brunschwig Z., Orbach A., Shemi A., Golan T., Domb A.J., Yavin E., et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA. 2013;110:20723–20728. doi: 10.1073/pnas.1314307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mosolits S., Ullenhag G., Mellstedt H. Therapeutic vaccination in patients with gastrointestinal malignancies. A review of immunological and clinical results. Ann. Oncol. 2005;16:847–862. doi: 10.1093/annonc/mdi192. [DOI] [PubMed] [Google Scholar]
- 58.Quandt J., Schlude C., Bartoschek M., Will R., Cid-Arregui A., Schölch S., Reissfelder C., Weitz J., Schneider M., Wiemann S., et al. Long-peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation-specific effector as well as regulatory T cell responses. Oncoimmunology. 2018;7:e1500671. doi: 10.1080/2162402X.2018.1500671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu G., Pei L., Xia H., Tang Q., Bi F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer. 2021;20:143. doi: 10.1186/s12943-021-01441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karachaliou N., Mayo C., Costa C., Magrí I., Gimenez-Capitan A., Molina-Vila M.A., Rosell R. KRAS Mutations in Lung Cancer. Clin. Lung Cancer. 2013;14:205–214. doi: 10.1016/j.cllc.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 61.Santos E., Martin-Zanca D., Reddy E.P., Pierotti M.A., Della Porta G., Barbacid M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science. 1984;223:661–664. doi: 10.1126/science.6695174. [DOI] [PubMed] [Google Scholar]
- 62.Rodenhuis S., van de Wetering M.L., Mooi W.J., Evers S.G., van Zandwijk N., Bos J.L. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N. Engl. J. Med. 1987;317:929–935. doi: 10.1056/NEJM198710083171504. [DOI] [PubMed] [Google Scholar]
- 63.Dearden S., Stevens J., Wu Y.L., Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: Meta-analyses by ethnicity and histology (mutMap) Ann. Oncol. 2013;24:2371–2376. doi: 10.1093/annonc/mdt205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang H., Liang S.Q., Schmid R.A., Peng R.W. New Horizons in KRAS-Mutant Lung Cancer: Dawn After Darkness. Front. Oncol. 2019;9:953. doi: 10.3389/fonc.2019.00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matikas A., Mistriotis D., Georgoulias V., Kotsakis A. Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity. Crit. Rev. Oncol. Hematol. 2017;110:1–12. doi: 10.1016/j.critrevonc.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 66.Ahrendt S.A., Decker P.A., Alawi E.A., Zhu Yr Y.R., Sanchez-Cespedes M., Yang S.C., Haasler G.B., Kajdacsy-Balla A., Demeure M.J., Sidransky D. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525–1530. doi: 10.1002/1097-0142(20010915)92:6<1525::AID-CNCR1478>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 67.Siegel R.L., Miller K.D., Goding Sauer A., Fedewa S.A., Butterly L.F., Anderson J.C., Cercek A., Smith R.A., Jemal A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020;70:145–164. doi: 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 68.Bos J.L., Fearon E.R., Hamilton S.R., Verlaan-de Vries M., van Boom J.H., van der Eb A.J., Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 69.Vogelstein B., Fearon E.R., Hamilton S.R., Kern S.E., Preisinger A.C., Leppert M., Nakamura Y., White R., Smits A.M., Bos J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 70.Neumann J., Zeindl-Eberhart E., Kirchner T., Jung A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol. Res. Pract. 2009;205:858–862. doi: 10.1016/j.prp.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 71.Sveen A., Kopetz S., Lothe R.A. Biomarker-guided therapy for colorectal cancer: Strength in complexity. Nat. Rev. Clin. Oncol. 2020;17:11–32. doi: 10.1038/s41571-019-0241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.. Lavanya V., Mohamed A.A.A., Ahmed N., Rishi A.K., Jamal S. Small molecule inhibitors as emerging cancer therapeutics. Integr. Cancer Sci. Ther. 2014;1:39–46. doi: 10.15761/ICST.1000109. [DOI] [Google Scholar]
- 73.Bandyopadhyay D., Banik B.K. Microwave-assisted synthesis of medicinally privileged heterocycles. In: Brahmachari G., editor. Green Synthetic Approaches for Biologically Relevant Heterocycles. 2nd ed. Volume 1. Elsevier; Oxford, UK: 2021. pp. 49–110. Advanced Synthetic Techniques. Chapter 3. [Google Scholar]
- 74.Cercek A., Shia J., Gollub M., Chou J.F., Capanu M., Raasch P., Reidy-Lagunes D., Proia D.A., Vakiani E., Solit D.B., et al. Ganetespib, a novel Hsp90 inhibitor in patients with KRAS mutated and wild type, refractory metastatic colorectal cancer. Clin. Colorectal Cancer. 2014;13:207–212. doi: 10.1016/j.clcc.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Acquaviva J., Smith D.L., Sang J., Friedland J.C., He S., Sequeira M., Zhang C., Wada Y., Proia D.A. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol. Cancer Ther. 2012;11:2633–2643. doi: 10.1158/1535-7163.MCT-12-0615. [DOI] [PubMed] [Google Scholar]
- 76.Zeng D.-X., Wang C.-G., Huang J.-A., Jiang J.-H. Apatinib in the treatment of advanced lung adenocarcinoma with KRAS mutation. Onco. Targets Ther. 2017;10:4269–4272. doi: 10.2147/OTT.S139520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo W., Wu S., Liu J., Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–7408. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shin Y., Jeong J.W., Wurz R.P., Achanta P., Arvedson T., Bartberger M.D., Campuzano I.D.G., Fucini R., Hansen S.K., Ingersoll J., et al. Discovery of N-(1-Acryloylazetidin-3-yl)-2-(1H-indol-1-yl)acetamides as Covalent Inhibitors of KRASG12C. ACS Med. Chem. Lett. 2019;10:1302–1308. doi: 10.1021/acsmedchemlett.9b00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ostrem J.M., Peters U., Sos M.L., Wells J.A., Shokat K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar V., Mondal G., Slavik P., Rachagani S., Batra S.K., Mahato R.I. Codelivery of small molecule hedgehog inhibitor and miRNA for treating pancreatic cancer. Mol. Pharm. 2015;12:1289–1298. doi: 10.1021/mp500847s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Al-Mulla F., Milner-White E.J., Going J.J., Birnie G.D. Structural differences between valine-12 and aspartate-12 Ras proteins may modify carcinoma aggression. J. Pathol. 1999;187:433–438. doi: 10.1002/(SICI)1096-9896(199903)187:4<433::AID-PATH273>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 82.Monticone M., Biollo E., Maffei M., Donadini A., Romeo F., Storlazzi C.T., Giaretti W., Castagnola P. Gene expression deregulation by KRAS G12D and G12V in a BRAF V600E context. Mol. Cancer. 2008;7:92. doi: 10.1186/1476-4598-7-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li J., Qin S., Xu J., Guo W., Xiong J., Bai Y., Sun G., Yang Y., Wang L., Xu N., et al. Apatinib for chemotherapy-refractory advanced metastatic gastric cancer: Results from a randomized, placebo-controlled, parallel-arm, phase II trial. J. Clin. Oncol. 2013;31:3219–3225. doi: 10.1200/JCO.2013.48.8585. [DOI] [PubMed] [Google Scholar]
- 84.Hu X., Zhang J., Xu B., Jiang Z., Ragaz J., Tong Z., Zhang Q., Wang X., Feng J., Pang D., et al. Multicenter phase II study of apatinib, a novel VEGFR inhibitor in heavily pretreated patients with metastatic triple-negative breast cancer. Int. J. Cancer. 2014;135:1961–1969. doi: 10.1002/ijc.28829. [DOI] [PubMed] [Google Scholar]
- 85.Chinni S.R., Li Y., Upadhyay S., Koppolu P.K., Sarkar F.H. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20:2927–2936. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 86.Erlanson D.A., Braisted A.C., Raphael D.R., Randal M., Stroud R.M., Gordon E.M., Wells J.A. Site-directed ligand discovery. Proc. Natl. Acad. Sci. USA. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Awad M.M., Liu S., Rybkin I., Arbour K.C., Dilly J., Zhu V.W., Johnson M.L., Heist R.S., Patil T., Riely G.J., et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021;384:2382–2393. doi: 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Giroux-Leprieur E., Costantini A., Ding V.W., He B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018;19:2835. doi: 10.3390/ijms19092835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Torrisani J., Bournet B., du Rieu M.C., Bouisson M., Souque A., Escourrou J., Buscail L., Cordelier P. let-7 MicroRNA transfer in pancreatic cancer-derived cells inhibits in vitro cell proliferation but fails to alter tumor progression. Hum. Gene Ther. 2009;20:831–844. doi: 10.1089/hum.2008.134. [DOI] [PubMed] [Google Scholar]
- 90.Janes M.R., Zhang J., Li L.S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S.J., Darjania L., et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172:578–589. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 91.Patricelli M.P., Janes M.R., Li L.S., Hansen R., Peters U., Kessler L.V., Chen Y., Kucharski J.M., Feng J., Ely T., et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 92.Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C.G., Koppada N., et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 93.Hallin J., Engstrom L.D., Hargis L., Calinisan A., Aranda R., Briere D.M., Sudhakar N., Bowcut V., Baer B.R., Ballard J.A., et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McCarthy M.J., Pagba C.V., Prakash P., Naji A.K., van der Hoeven D., Liang H., Gupta A.K., Zhou Y., Cho K.J., Hancock J.F., et al. Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega. 2019;4:2921–2930. doi: 10.1021/acsomega.8b03308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ludwig K.F., Du W., Sorrelle N.B., Wnuk-Lipinska K., Topalovski M., Toombs J.E., Cruz V.H., Yabuuchi S., Rajeshkumar N.V., Maitra A., et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res. 2018;78:246–255. doi: 10.1158/0008-5472.CAN-17-1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tabusa H., Brooks T., Massey A.J. Knockdown of PAK4 or PAK1 inhibits the proliferation of mutant KRAS colon cancer cells independently of RAF/MEK/ERK and PI3K/AKT signaling. Mol. Cancer Res. 2013;11:109–121. doi: 10.1158/1541-7786.MCR-12-0466. [DOI] [PubMed] [Google Scholar]
- 97.Tentler J.J., Nallapareddy S., Tan A.C., Spreafico A., Pitts T.M., Morelli M.P., Selby H.M., Kachaeva M.I., Flanigan S.A., Kulikowski G.N., et al. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol. Cancer Ther. 2010;9:3351–3362. doi: 10.1158/1535-7163.MCT-10-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Engelman J.A., Chen L., Tan X., Crosby K., Guimaraes A.R., Upadhyay R., Maira M., McNamara K., Perera S.A., Song Y., et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beaupre D.M., Cepero E., Obeng E.A., Boise L.H., Lichtenheld M.G. R115777 induces Ras-independent apoptosis of myeloma cells via multiple intrinsic pathways. Mol. Cancer Ther. 2004;3:179–186. [PubMed] [Google Scholar]
- 100.Cruz-Migoni A., Canning P., Quevedo C.E., Bataille C.J.R., Bery N., Miller A., Russell A.J., Phillips S.E.V., Carr S.B., Rabbitts T.H. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc. Natl. Acad. Sci. USA. 2019;116:2545–2550. doi: 10.1073/pnas.1811360116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Welsch M.E., Kaplan A., Chambers J.M., Stokes M.E., Bos P.H., Zask A., Zhang Y., Sanchez-Martin M., Badgley M.A., Huang C.S., et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell. 2017;168:878–889.e829. doi: 10.1016/j.cell.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leung E.L., Luo L.X., Li Y., Liu Z.Q., Li L.L., Shi D.F., Xie Y., Huang M., Lu L.L., Duan F.G., et al. Identification of a new inhibitor of KRAS-PDEδ interaction targeting KRAS mutant nonsmall cell lung cancer. Int. J. Cancer. 2019;145:1334–1345. doi: 10.1002/ijc.32222. [DOI] [PubMed] [Google Scholar]
- 103.Kettle J.G., Bagal S.K., Bickerton S., Bodnarchuk M.S., Breed J., Carbajo R.J., Cassar D.J., Chakraborty A., Cosulich S., Cumming I., et al. Structure-Based Design and Pharmacokinetic Optimization of Covalent Allosteric Inhibitors of the Mutant GTPase KRASG12C. J. Med. Chem. 2020;63:4468–4483. doi: 10.1021/acs.jmedchem.9b01720. [DOI] [PubMed] [Google Scholar]
- 104.Lim S.M., Westover K.D., Ficarro S.B., Harrison R.A., Choi H.G., Pacold M.E., Carrasco M., Hunter J., Kim N.D., Xie T., et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. Engl. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Román M., Baraibar I., López I., Nadal E., Rolfo C., Vicent S., Gil-Bazo I. KRAS oncogene in non-small cell lung cancer: Clinical perspectives on the treatment of an old target. Mol. Cancer. 2018;17:33. doi: 10.1186/s12943-018-0789-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Song X., Wang H., Logsdon C.D., Rashid A., Fleming J.B., Abbruzzese J.L., Gomez H.F., Evans D.B., Wang H. Overexpression of receptor tyrosine kinase Axl promotes tumor cell invasion and survival in pancreatic ductal adenocarcinoma. Cancer. 2011;117:734–743. doi: 10.1002/cncr.25483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leconet W., Larbouret C., Chardès T., Thomas G., Neiveyans M., Busson M., Jarlier M., Radosevic-Robin N., Pugnière M., Bernex F., et al. Preclinical validation of AXL receptor as a target for antibody-based pancreatic cancer immunotherapy. Oncogene. 2014;33:5405–5414. doi: 10.1038/onc.2013.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wells C.M., Jones G.E. The emerging importance of group II PAKs. Biochem. J. 2010;425:465–473. doi: 10.1042/BJ20091173. [DOI] [PubMed] [Google Scholar]
- 109.Li L.H., Zheng M.H., Luo Q., Ye Q., Feng B., Lu A.G., Wang M.L., Chen X.H., Su L.P., Liu B.Y. P21-activated protein kinase 1 induces colorectal cancer metastasis involving ERK activation and phosphorylation of FAK at Ser-910. Int. J. Oncol. 2010;37:951–962. [PubMed] [Google Scholar]
- 110.Park E.R., Eblen S.T., Catling A.D. MEK1 activation by PAK: A novel mechanism. Cell Signal. 2007;19:1488–1496. doi: 10.1016/j.cellsig.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang J., Wang J., Guo Q., Wang Y., Zhou Y., Peng H., Cheng M., Zhao D., Li F. LCH-7749944, a novel and potent p21-activated kinase 4 inhibitor, suppresses proliferation and invasion in human gastric cancer cells. Cancer Lett. 2012;317:24–32. doi: 10.1016/j.canlet.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 112.Zang M., Hayne C., Luo Z. Interaction between active Pak1 and Raf-1 is necessary for phosphorylation and activation of Raf-1. J. Biol. Chem. 2002;277:4395–4405. doi: 10.1074/jbc.M110000200. [DOI] [PubMed] [Google Scholar]
- 113.Koh W., Sachidanandam K., Stratman A.N., Sacharidou A., Mayo A.M., Murphy E.A., Cheresh D.A., Davis G.E. Formation of endothelial lumens requires a coordinated PKCepsilon-, Src-, Pak- and Raf-kinase-dependent signaling cascade downstream of Cdc42 activation. J. Cell Sci. 2009;122:1812–1822. doi: 10.1242/jcs.045799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Samuels Y., Wang Z., Bardelli A., Silliman N., Ptak J., Szabo S., Yan H., Gazdar A., Powell S.M., Riggins G.J., et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 115.She Q.B., Solit D.B., Ye Q., O’Reilly K.E., Lobo J., Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8:287–297. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Engelman J.A. The role of phosphoinositide 3-kinase pathway inhibitors in the treatment of lung cancer. Clin. Cancer Res. 2007;13:4637s–4640s. doi: 10.1158/1078-0432.CCR-07-0653. [DOI] [PubMed] [Google Scholar]
- 117.Yeh T.C., Marsh V., Bernat B.A., Ballard J., Colwell H., Evans R.J., Parry J., Smith D., Brandhuber B.J., Gross S., et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 118.Beaupre D.M., McCafferty-Grad J., Bahlis N.J., Boise L.H., Lichtenheld M.G. Farnesyl transferase inhibitors enhance death receptor signals and induce apoptosis in multiple myeloma cells. Leuk. Lymphoma. 2003;44:2123–2134. doi: 10.1080/1042819031000116652. [DOI] [PubMed] [Google Scholar]
- 119.Hengartner M.O. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 120.Harada N., Hata H., Yoshida M., Soniki T., Nagasaki A., Kuribayashi N., Kimura T., Matsuzaki H., Mitsuya H. Expression of Bcl-2 family of proteins in fresh myeloma cells. Leukemia. 1998;12:1817–1820. doi: 10.1038/sj.leu.2401168. [DOI] [PubMed] [Google Scholar]
- 121.Miguel-García A., Orero T., Matutes E., Carbonell F., Miguel-Sosa A., Linares M., Tarín F., Herrera M., García-Talavera J., Carbonell-Ramón F. bcl-2 expression in plasma cells from neoplastic gammopathies and reactive plasmacytosis: A comparative study. Haematologica. 1998;83:298–304. [PubMed] [Google Scholar]
- 122.Cox A.D., Fesik S.W., Kimmelman A.C., Luo J., Der C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gysin S., Salt M., Young A., McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zimmermann G., Schultz-Fademrecht C., Küchler P., Murarka S., Ismail S., Triola G., Nussbaumer P., Wittinghofer A., Waldmann H. Structure guided design and kinetic analysis of highly potent benzimidazole inhibitors targeting the PDEδ prenyl binding site. J. Med. Chem. 2014;57:5435–5448. doi: 10.1021/jm500632s. [DOI] [PubMed] [Google Scholar]
- 125.Zimmermann G., Papke B., Ismail S., Vartak N., Chandra A., Hoffmann M., Hahn S.A., Triola G., Wittinghofer A., Bastiaens P.I., et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 126.Ghimessy A., Radeczky P., Laszlo V., Hegedus B., Renyi-Vamos F., Fillinger J., Klepetko W., Lang C., Dome B., Megyesfalvi Z. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev. 2020;39:1159–1177. doi: 10.1007/s10555-020-09903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Leung E.L.H., Luo L.X., Liu Z.Q., Wong V.K.W., Lu L.L., Xie Y., Zhang N., Qu Y.Q., Fan X.X., Li Y., et al. Inhibition of KRAS-dependent lung cancer cell growth by deltarasin: Blockage of autophagy increases its cytotoxicity. Cell Death Dis. 2018;9:216. doi: 10.1038/s41419-017-0065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li J., Mizukami Y., Zhang X., Jo W.S., Chung D.C. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3beta. Gastroenterology. 2005;128:1907–1918. doi: 10.1053/j.gastro.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 129.Bialkowska A.B., Du Y., Fu H., Yang V.W. Identification of novel small-molecule compounds that inhibit the proproliferative Kruppel-like factor 5 in colorectal cancer cells by high-throughput screening. Mol. Cancer Ther. 2009;8:563–570. doi: 10.1158/1535-7163.MCT-08-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Papke B., Murarka S., Vogel H.A., Martín-Gago P., Kovacevic M., Truxius D.C., Fansa E.K., Ismail S., Zimmermann G., Heinelt K., et al. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat. Commun. 2016;7:11360. doi: 10.1038/ncomms11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Geltz N.R., Augustine J.A. The p85 and p110 subunits of phosphatidylinositol 3-kinase-alpha are substrates, in vitro, for a constitutively associated protein tyrosine kinase in platelets. Blood. 1998;91:930–939. doi: 10.1182/blood.V91.3.930. [DOI] [PubMed] [Google Scholar]
- 132.Chan T.O., Rittenhouse S.E., Tsichlis P.N. AKT/PKB and other D3 phosphoinositide-regulated kinases: Kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 133.Michl P., Downward J. Mechanisms of disease: PI3K/AKT signaling in gastrointestinal cancers. Z. Gastroenterol. 2005;43:1133–1139. doi: 10.1055/s-2005-858638. [DOI] [PubMed] [Google Scholar]
- 134.Rao C.V., Janakiram N.B., Madka V., Kumar G., Scott E.J., Pathuri G., Bryant T., Kutche H., Zhang Y., Biddick L., et al. Small-Molecule Inhibition of GCNT3 Disrupts Mucin Biosynthesis and Malignant Cellular Behaviors in Pancreatic Cancer. Cancer Res. 2016;76:1965–1974. doi: 10.1158/0008-5472.CAN-15-2820. [DOI] [PubMed] [Google Scholar]
- 135.Choi J.K., Cho H., Moon B.S. Small Molecule Destabilizer of β-Catenin and Ras Proteins Antagonizes Growth of K-Ras Mutation-Driven Colorectal Cancers Resistant to EGFR Inhibitors. Target. Oncol. 2020;15:645–657. doi: 10.1007/s11523-020-00755-5. [DOI] [PubMed] [Google Scholar]
- 136.Lee S.K., Cho Y.H., Cha P.H., Yoon J.S., Ro E.J., Jeong W.J., Park J., Kim H., Il Kim T., Min D.S., et al. A small molecule approach to degrade RAS with EGFR repression is a potential therapy for KRAS mutation-driven colorectal cancer resistance to cetuximab. Exp. Mol. Med. 2018;50:1–12. doi: 10.1038/s12276-018-0182-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xie C., Li Y., Li L.L., Fan X.X., Wang Y.W., Wei C.L., Liu L., Leung E.L.H., Yao X.J. Identification of a New Potent Inhibitor Targeting KRAS in Non-small Cell Lung Cancer Cells. Front. Pharmacol. 2017;8:823. doi: 10.3389/fphar.2017.00823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wallis N., Oberman F., Shurrush K., Germain N., Greenwald G., Gershon T., Pearl T., Abis G., Singh V., Singh A., et al. Small molecule inhibitor of Igf2bp1 represses Kras and a pro-oncogenic phenotype in cancer cells. RNA Biol. 2022;19:26–43. doi: 10.1080/15476286.2021.2010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Evelyn C.R., Duan X., Biesiada J., Seibel W.L., Meller J., Zheng Y. Rational design of small molecule inhibitors targeting the Ras GEF, SOS1. Chem. Biol. 2014;21:1618–1628. doi: 10.1016/j.chembiol.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kaur S., Kumar S., Momi N., Sasson A.R., Batra S.K. Mucins in pancreatic cancer and its microenvironment. Nat. Rev. Gastroenterol. Hepatol. 2013;10:607–620. doi: 10.1038/nrgastro.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nagata K., Horinouchi M., Saitou M., Higashi M., Nomoto M., Goto M., Yonezawa S. Mucin expression profile in pancreatic cancer and the precursor lesions. J. Hepatobiliary Pancreat. Surg. 2007;14:243–254. doi: 10.1007/s00534-006-1169-2. [DOI] [PubMed] [Google Scholar]
- 142.Moniaux N., Andrianifahanana M., Brand R.E., Batra S.K. Multiple roles of mucins in pancreatic cancer, a lethal and challenging malignancy. Br. J. Cancer. 2004;91:1633–1638. doi: 10.1038/sj.bjc.6602163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Torres M.P., Chakraborty S., Souchek J., Batra S.K. Mucin-based targeted pancreatic cancer therapy. Curr. Pharm. Des. 2012;18:2472–2481. doi: 10.2174/13816128112092472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Moon B.S., Jeong W.J., Park J., Kim T.I., Min D.S., Choi K.Y. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/β-catenin signaling. J. Natl. Cancer Inst. 2014;106:djt373. doi: 10.1093/jnci/djt373. [DOI] [PubMed] [Google Scholar]
- 145.Cho Y.H., Cha P.H., Kaduwal S., Park J.C., Lee S.K., Yoon J.S., Shin W., Kim H., Ro E.J., Koo K.H., et al. KY1022, a small molecule destabilizing Ras via targeting the Wnt/β-catenin pathway, inhibits development of metastatic colorectal cancer. Oncotarget. 2016;7:81727–81740. doi: 10.18632/oncotarget.13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Degrauwe N., Suvà M.L., Janiszewska M., Riggi N., Stamenkovic I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016;30:2459–2474. doi: 10.1101/gad.287540.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen H., Smaill J.B., Liu T., Ding K., Lu X. Small-Molecule Inhibitors Directly Targeting KRAS as Anticancer Therapeutics. J. Med. Chem. 2020;63:14404–14424. doi: 10.1021/acs.jmedchem.0c01312. [DOI] [PubMed] [Google Scholar]
- 148.Ning W., Yang Z., Kocher G.J., Dorn P., Peng R.W. A Breakthrough Brought about by Targeting KRAS(G12C): Nonconformity Is Punished. Cancers. 2022;14:390. doi: 10.3390/cancers14020390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mologni L., Brussolo S., Ceccon M., Gambacorti-Passerini C. Synergistic effects of combined Wnt/KRAS inhibition in colorectal cancer cells. PLoS ONE. 2012;7:e51449. doi: 10.1371/journal.pone.0051449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shima F., Yoshikawa Y., Ye M., Araki M., Matsumoto S., Liao J., Hu L., Sugimoto T., Ijiri Y., Takeda A., et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA. 2013;110:8182–8187. doi: 10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Marín-Ramos N.I., Balabasquer M., Ortega-Nogales F.J., Torrecillas I.R., Gil-Ordóñez A., Marcos-Ramiro B., Aguilar-Garrido P., Cushman I., Romero A., Medrano F.J., et al. A Potent Isoprenylcysteine Carboxylmethyltransferase (ICMT) Inhibitor Improves Survival in Ras-Driven Acute Myeloid Leukemia. J. Med. Chem. 2019;62:6035–6046. doi: 10.1021/acs.jmedchem.9b00145. [DOI] [PubMed] [Google Scholar]
- 152.Sugita S., Enokida H., Yoshino H., Miyamoto K., Yonemori M., Sakaguchi T., Osako Y., Nakagawa M. HRAS as a potential therapeutic target of salirasib RAS inhibitor in bladder cancer. Int. J. Oncol. 2018;53:725–736. doi: 10.3892/ijo.2018.4435. [DOI] [PubMed] [Google Scholar]
- 153.Goldberg L., Haklai R., Bauer V., Heiss A., Kloog Y. New derivatives of farnesylthiosalicylic acid (salirasib) for cancer treatment: Farnesylthiosalicylamide inhibits tumor growth in nude mice models. J. Med. Chem. 2009;52:197–205. doi: 10.1021/jm801165r. [DOI] [PubMed] [Google Scholar]
- 154.Wood L.D., Parsons D.W., Jones S., Lin J., Sjöblom T., Leary R.J., Shen D., Boca S.M., Barber T., Ptak J., et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 155.Sjöblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., Mandelker D., Leary R.J., Ptak J., Silliman N., et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 156.Maurer T., Garrenton L.S., Oh A., Pitts K., Anderson D.J., Skelton N.J., Fauber B.P., Pan B., Malek S., Stokoe D., et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang M., Casey P.J. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016;17:110–122. doi: 10.1038/nrm.2015.11. [DOI] [PubMed] [Google Scholar]
- 158.Stephen A.G., Esposito D., Bagni R.K., McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 159.Pandith A.A., Shah Z.A., Khan N.P., Baba K.M., Wani M.S., Siddiqi M.A. HRAS T81C polymorphism modulates risk of urinary bladder cancer and predicts advanced tumors in ethnic Kashmiri population. Urol. Oncol. 2013;31:487–492. doi: 10.1016/j.urolonc.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 160.Ruiz de Sabando A., Wang C., He Y., García-Barros M., Kim J., Shroyer K.R., Bannister T.D., Yang V.W., Bialkowska A.B. ML264, A Novel Small-Molecule Compound That Potently Inhibits Growth of Colorectal Cancer. Mol. Cancer Ther. 2016;15:72–83. doi: 10.1158/1535-7163.MCT-15-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bannoura S.F., Uddin M.H., Nagasaka M., Fazili F., Al-Hallak M.N., Philip P.A., El-Rayes B., Azmi A.S. Targeting KRAS in pancreatic cancer: New drugs on the horizon. Cancer Metastasis Rev. 2021;40:819–835. doi: 10.1007/s10555-021-09990-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Debasish Bandyopadhyay V.G., Gonzales F. Anti-Cancer Agents from Natural Sources. In: Dutt R., Sharma A.K., Keservani K.R., Garg V., editors. Promising Drug Molecules of Natural Origin. 1st ed. Apple Academic Press; New York, NY, USA: 2020. p. 80. Bioscience, Medicine, Dentistry, Nursing & Allied Health. [Google Scholar]
- 163.Debasish Bandyopadhyay V.G., Gonzales F. Heterocyclic Drugs from Plants. In: Dutt R., Sharma A.K., Keservani K.R., Garg V., editors. Promising Drug Molecules of Natural Origin. 1st ed. Apple Academic Press; New York, NY, USA: 2020. p. 56. Bioscience, Medicine, Dentistry, Nursing & Allied Health. [Google Scholar]
- 164.Newman D.J., Cragg G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- 165.Ranelletti F.O., Maggiano N., Serra F.G., Ricci R., Larocca L.M., Lanza P., Scambia G., Fattorossi A., Capelli A., Piantelli M. Quercetin inhibits p21-RAS expression in human colon cancer cell lines and in primary colorectal tumors. Int. J. Cancer. 2000;85:438–445. doi: 10.1002/(SICI)1097-0215(20000201)85:3<438::AID-IJC22>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 166.Zhang X., Guo Q., Chen J., Chen Z. Quercetin Enhances Cisplatin Sensitivity of Human Osteosarcoma Cells by Modulating microRNA-217-KRAS Axis. Mol. Cells. 2015;38:638–642. doi: 10.14348/molcells.2015.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Guo J., Feng Z., Huang Z.a., Wang H., Lu W. MicroRNA-217 functions as a tumour suppressor gene and correlates with cell resistance to cisplatin in lung cancer. Mol. Cells. 2014;37:664–671. doi: 10.14348/molcells.2014.0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee S.H., Lee M.-Y., Kang H.-M., Han D.C., Son K.-H., Yang D.C., Sung N.-D., Lee C.W., Kim H.M., Kwon B.-M. Anti-tumor activity of the farnesyl-protein transferase inhibitors arteminolides, isolated from Artemisa. Bioorg. Med. Chem. 2003;11:4545–4549. doi: 10.1016/j.bmc.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 169.Gibbs J.B., Oliff A. The potential of farnesyltransferase inhibitors as cancer chemotherapeutics. Annu. Rev. Pharmacol. Toxicol. 1997;37:143–166. doi: 10.1146/annurev.pharmtox.37.1.143. [DOI] [PubMed] [Google Scholar]
- 170.Lee S.H., Kim H.K., Seo J.M., Kang H.M., Kim J.H., Son K.H., Lee H., Kwon B.M., Shin J., Seo Y. Arteminolides B, C, and D, new inhibitors of farnesyl protein transferase from Artemisia argyi. J. Org. Chem. 2002;67:7670–7675. doi: 10.1021/jo020299z. [DOI] [PubMed] [Google Scholar]
- 171.Gbelcová H., Rimpelová S., Knejzlík Z., Šáchová J., Kolář M., Strnad H., Repiská V., D’Acunto W.C., Ruml T., Vítek L. Isoprenoids responsible for protein prenylation modulate the biological effects of statins on pancreatic cancer cells. Lipids Health Dis. 2017;16:1–10. doi: 10.1186/s12944-017-0641-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Minciacchi V.R., Freeman M.R., Di Vizio D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015;40:41–51. doi: 10.1016/j.semcdb.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Stoorvogel W. Resolving sorting mechanisms into exosomes. Cell Res. 2015;25:531–532. doi: 10.1038/cr.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Datta A., Kim H., Lal M., McGee L., Johnson A., Moustafa A.A., Jones J.C., Mondal D., Ferrer M., Abdel-Mageed A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017;408:73–81. doi: 10.1016/j.canlet.2017.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Saha B., Nandi D. Farnesyltransferase inhibitors reduce Ras activation and ameliorate acetaminophen-induced liver injury in mice. Hepatology. 2009;50:1547–1557. doi: 10.1002/hep.23180. [DOI] [PubMed] [Google Scholar]
- 176.Angamuthu V., Shanmugavadivu M., Nagarajan G., Velmurugan B.K. Pharmacological activities of antroquinonol- Mini review. Chem. Biol. Interact. 2019;297:8–15. doi: 10.1016/j.cbi.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 177.Weber H.A., Gloer J.B. The preussomerins: Novel antifungal metabolites from the coprophilous fungus Preussia isomera Cain. J. Org. Chem. 1991;56:4355–4360. doi: 10.1021/jo00014a007. [DOI] [Google Scholar]
- 178.Singh S.B., Zink D.L., Liesch J.M., Ball R.G., Goetz M.A., Bolessa E.A., Giacobbe R.A., Silverman K.C., Bills G.F. Preussomerins and Deoxypreussomerins: Novel Inhibitors of Ras Farnesyl-Protein Transferase. J. Org. Chem. 1994;59:6296–6302. doi: 10.1021/jo00100a035. [DOI] [Google Scholar]
- 179.Õmura S., Tomoda H. Microbial metabolites affecting lipid biosynthesis. Pure Appl. Chem. 1994;66:2267–2270. doi: 10.1351/pac199466102267. [DOI] [Google Scholar]
- 180.Santa Maria K.C., Chan A.N., O’Neill E.M., Li B. Targeted Rediscovery and Biosynthesis of the Farnesyl-Transferase Inhibitor Pepticinnamin E. Chembiochem. 2019;20:1387–1393. doi: 10.1002/cbic.201900025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Thutewohl M., Kissau >L., Popkirova B., Karaguni I.M., Nowak T., Bate M., Kuhlmann J., Müller O., Waldmann H. Identification of mono- and bisubstrate inhibitors of protein farnesyltransferase and inducers of apoptosis from a pepticinnamin E library. Bioorg. Med. Chem. 2003;11:2617–2626. doi: 10.1016/S0968-0896(03)00160-3. [DOI] [PubMed] [Google Scholar]
- 182.Bivona T.G., Quatela S.E., Bodemann B.O., Ahearn I.M., Soskis M.J., Mor A., Miura J., Wiener H.H., Wright L., Saba S.G., et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell. 2006;21:481–493. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 183.Pettit G.R., Herald C.L., Doubek D.L., Herald D.L., Arnold E., Clardy J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982;104:6846–6848. doi: 10.1021/ja00388a092. [DOI] [Google Scholar]
- 184.Kortmansky J., Schwartz G.K. Bryostatin-1: A novel PKC inhibitor in clinical development. Cancer Investig. 2003;21:924–936. doi: 10.1081/CNV-120025095. [DOI] [PubMed] [Google Scholar]
- 185.Kumar S., Agnihotri N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed. Pharmacother. 2019;109:1462–1477. doi: 10.1016/j.biopha.2018.10.182. [DOI] [PubMed] [Google Scholar]
- 186.Yaqoob A., Li W.M., Liu V., Wang C., Mackedenski S., Tackaberry L.E., Massicotte H.B., Egger K.N., Reimer K., Lee C.H. Grifolin, neogrifolin and confluentin from the terricolous polypore Albatrellus flettii suppress KRAS expression in human colon cancer cells. PLoS ONE. 2020;15:e0231948. doi: 10.1371/journal.pone.0231948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kim C.-K., Wang D., Bokesch H.R., Fuller R.W., Smith E., Henrich C.J., Durrant D.E., Morrison D.K., Bewley C.A., Gustafson K.R. Swinhopeptolides A and B: Cyclic Depsipeptides from the Sponge Theonella swinhoei That Inhibit Ras/Raf Interaction. J. Nat. Prod. 2020;83:1288–1294. doi: 10.1021/acs.jnatprod.0c00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Garrido C.M., Henkels K.M., Rehl K.M., Liang H., Zhou Y., Gutterman J.U., Cho K.J. Avicin G is a potent sphingomyelinase inhibitor and blocks oncogenic K- and H-Ras signaling. Sci. Rep. 2020;10:9120. doi: 10.1038/s41598-020-65882-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Perera R.M., Stoykova S., Nicolay B.N., Ross K.N., Fitamant J., Boukhali M., Lengrand J., Deshpande V., Selig M.K., Ferrone C.R., et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–365. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Petersen N.H., Olsen O.D., Groth-Pedersen L., Ellegaard A.M., Bilgin M., Redmer S., Ostenfeld M.S., Ulanet D., Dovmark T.H., Lønborg A., et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–393. doi: 10.1016/j.ccr.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 191.Wang M.T., Holderfield M., Galeas J., Delrosario R., To M.D., Balmain A., McCormick F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell. 2015;163:1237–1251. doi: 10.1016/j.cell.2015.10.041. [DOI] [PubMed] [Google Scholar]
- 192.Chen X., Yano Y., Hasuma T., Yoshimata T., Yinna W., Otani S. Inhibition of farnesyl protein transferase and P21ras memebrane association by d-limonene in human pancreas tumor cells in vitro. Chin. Med. Sci. J. 1999;14:138–144. [PubMed] [Google Scholar]
- 193.Chaudhary S.C., Siddiqui M.S., Athar M., Alam M.S. D-Limonene modulates inflammation, oxidative stress and Ras-ERK pathway to inhibit murine skin tumorigenesis. Hum. Exp. Toxicol. 2012;31:798–811. doi: 10.1177/0960327111434948. [DOI] [PubMed] [Google Scholar]
- 194.Afaq F., Saleem M., Krueger C.G., Reed J.D., Mukhtar H. Anthocyanin- and hydrolyzable tannin-rich pomegranate fruit extract modulates MAPK and NF-kappaB pathways and inhibits skin tumorigenesis in CD-1 mice. Int. J. Cancer. 2005;113:423–433. doi: 10.1002/ijc.20587. [DOI] [PubMed] [Google Scholar]
- 195.Yoon J.H., Pham T.H., Lee J., Lee J., Ryu H.W., Oh S.R., Oh J.W., Yoon D.Y. Methyl Linderone Suppresses TPA-Stimulated IL-8 and MMP-9 Expression Via the ERK/STAT3 Pathway in MCF-7 Breast Cancer Cells. J. Microbiol. Biotechnol. 2020;30:325–332. doi: 10.4014/jmb.1911.11068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ho C.L., Wang J.L., Lee C.C., Cheng H.Y., Wen W.C., Cheng H.H., Chen M.C. Antroquinonol blocks Ras and Rho signaling via the inhibition of protein isoprenyltransferase activity in cancer cells. Biomed. Pharmacother. 2014;68:1007–1014. doi: 10.1016/j.biopha.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 197.Kumar V.B., Yuan T.C., Liou J.W., Yang C.J., Sung P.J., Weng C.F. Antroquinonol inhibits NSCLC proliferation by altering PI3K/mTOR proteins and miRNA expression profiles. Mutat. Res. 2011;707:42–52. doi: 10.1016/j.mrfmmm.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 198.Palmioli A., Sacco E., Abraham S., Thomas C.J., Di Domizio A., De Gioia L., Gaponenko V., Vanoni M., Peri F. First experimental identification of Ras-inhibitor binding interface using a water-soluble Ras ligand. Bioorg. Med. Chem. Lett. 2009;19:4217–4222. doi: 10.1016/j.bmcl.2009.05.107. [DOI] [PubMed] [Google Scholar]
- 199.Palmioli A., Ciaramelli C., Tisi R., Spinelli M., De Sanctis G., Sacco E., Airoldi C. Natural Compounds in Cancer Prevention: Effects of Coffee Extracts and Their Main Polyphenolic Component, 5-O-Caffeoylquinic Acid, on Oncogenic Ras Proteins. Chem. Asian J. 2017;12:2457–2466. doi: 10.1002/asia.201700844. [DOI] [PubMed] [Google Scholar]
- 200.Ganaie A.A., Siddique H.R., Sheikh I.A., Parray A., Wang L., Panyam J., Villalta P.W., Deng Y., Konety B.R., Saleem M. A novel terpenoid class for prevention and treatment of KRAS-driven cancers: Comprehensive analysis using in situ, in vitro, and in vivo model systems. Mol. Carcinog. 2020;59:886–896. doi: 10.1002/mc.23200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gibbs J.B., Pompliano D.L., Mosser S.D., Rands E., Lingham R.B., Singh S.B., Scolnick E.M., Kohl N.E., Oliff A. Selective inhibition of farnesyl-protein transferase blocks ras processing in vivo. J. Biol. Chem. 1993;268:7617–7620. doi: 10.1016/S0021-9258(18)52998-7. [DOI] [PubMed] [Google Scholar]
- 202.Nogueira A., Vala H., Vasconcelos-Nóbrega C., Faustino-Rocha A.I., Pires C.A., Colaço A., Oliveira P.A., Pires M.J. Long-term treatment with chaethomellic acid A reduces glomerulosclerosis and arteriolosclerosis in a rat model of chronic kidney disease. Biomed. Pharmacother. 2017;96:489–496. doi: 10.1016/j.biopha.2017.09.137. [DOI] [PubMed] [Google Scholar]
- 203.Krepinsky J.C., Li Y., Chang Y., Liu L., Peng F., Wu D., Tang D., Scholey J., Ingram A.J. Akt mediates mechanical strain-induced collagen production by mesangial cells. J. Am. Soc. Nephrol. 2005;16:1661–1672. doi: 10.1681/ASN.2004100897. [DOI] [PubMed] [Google Scholar]
- 204.Rodríguez-Peña A.B., Grande M.T., Eleno N., Arévalo M., Guerrero C., Santos E., López-Novoa J.M. Activation of Erk1/2 and Akt following unilateral ureteral obstruction. Kidney Int. 2008;74:196–209. doi: 10.1038/ki.2008.160. [DOI] [PubMed] [Google Scholar]
- 205.Bandyopadhyay D., Lopez G., Cantu S., Balboa S., Garcia A., Silva C., Valdes V. Chemistry Research and Applications: Organic and Medicinal Chemistry. Volume 1. Nova Science Publishers; New York, NY, USA: 2018. Chapter 9. [Google Scholar]
- 206.Bandyopadhyay D. Preparation of Substituted Pyridinyl Azetidinone Derivatives for Use in Treating Cancer and Other Diseases. WO2019227040A1. 2018 May 25;
- 207.Bandyopadhyay D. Farmer to pharmacist: Curcumin as an anti-invasive and antimetastatic agent for the treatment of cancer1. Front. Chem. 2014;2:113. doi: 10.3389/fchem.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not Applicable.