

Abstract
Objectives:
LMNA variants have been previously associated with cardiac abnormalities independent of lipodystrophy. We aimed to assess cardiac impact of familial partial lipodystrophy (FPLD) to understand the role of laminopathy in cardiac manifestations.
Study design:
Retrospective cohort study.
Methods:
Clinical data from 122 patients (age range: 13–77, 101 females) with FPLD were analysed. Mature human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) from a patient with an LMNA variant were studied as proof-of-concept for future studies.
Results:
Subjects with LMNA variants had a higher prevalence of overall cardiac events than others. The likelihood of having an arrhythmia was significantly higher in patients with LMNA variants (OR: 3.77, 95% CI: 1.45–9.83). These patients were at higher risk for atrial fibrillation or flutter (OR: 5.78, 95% CI: 1.04–32.16). The time to the first arrhythmia was significantly shorter in the LMNA group, with a higher HR of 3.52 (95% CI: 1.34–9.27). Non-codon 482 LMNA variants were more likely to be associated with cardiac events (vs. 482 LMNA: OR: 4.74, 95% CI: 1.41–15.98 for arrhythmia; OR: 17.67, 95% CI: 2.45–127.68 for atrial fibrillation or flutter; OR: 5.71, 95% CI: 1.37–23.76 for conduction disease). LMNA mutant hiPSC-CMs showed a higher frequency of spontaneous activity and shorter action potential duration. Functional syncytia of hiPSC-CMs displayed several rhythm alterations such as early afterdepolarizations, spontaneous quiescence and spontaneous tachyarrhythmia, and significantly slower recovery in chronotropic changes induced by isoproterenol exposure.
Conclusions:
Our results highlight the need for vigilant cardiac monitoring in FPLD, especially in patients with LMNA variants who have an increased risk of developing cardiac arrhythmias. In addition, hiPSC-CMs can be studied to understand the basic mechanisms for the arrhythmias in patients with lipodystrophy to understand the impact of specific mutations.
Keywords: arrhythmia, atrial fibrillation, conduction disease, lipodystrophy, LMNA
1 |. INTRODUCTION
Familial partial lipodystrophy (FPLD) is a heterogeneous rare disease characterized by selective fat loss, mainly affecting the limbs.1 The most common type, FPLD2 (Dunnigan variety), is attributed to LMNA pathogenic variants.2 The LMNA gene produces lamin A and C by alternative splicing of exon 10. LMNA pathogenic variants demonstrate remarkable allelic heterogeneity and pleiotropy, and can give rise to more than 16 different diseases (laminopathies).3 Mutations in the N terminal region usually lead to a more severe phenotype, because both lamins A and C are affected, unlike C-terminal mutations where only lamin A is involved. In general, cardiolaminopathies mostly arise from pathogenic variants in the amino-terminal and central rod domain, 5′ to the nuclear localization signal (NLS). Conversely, lipodystrophy mainly originates from C-terminal domain mutations, 3′ to the NLS.4 Variants in the hot-spot (codon 482 in C-terminal domain) make up approximately 80% of all pathogenic variants in known patients with lipodystrophy.1
LMNA is a well-known genetic cause of dilated cardiomyopathy.5 A few notable N terminal variants present overlap between lipodystrophy and cardiac disease: in exon 1 (R28W,6,7 R60G8 and R62G7 reported with FPLD, atrioventricular (AV) block and cardiomyopathy), exon 2 (R131L9 reported with generalized lipodystrophy, cardiomyopathy, and valvular disease), exon 3 (D192V8 presented with FPLD and cardiomyopathy), exon 6 (R349W10,11 associated with cardiomyopathy and arrhythmias), and cardiomyopathy resulting in cardiac transplants in T10I carriers.12 A recent study reported a higher prevalence of arrhythmia and cardiac disease in non-482 LMNA variants (including non-lipodystrophy patients).13 A large study of 444 subjects with LMNA variants (including 72 with lipodystrophy) reported that male sex, non-missense LMNA variants, first degree and higher atrioventricular block, non-sustained ventricular tachycardia and left ventricular ejection fraction were predictors of life-threatening ventricular tachyarrhythmias.14 However, the exact degree of cardiac disease among the different subtypes of FPLD (specifically in those with LMNA variants versus those that do not harbour LMNA variants) still remains unknown. Also, specific evidence documenting the presence of atherosclerotic heart disease in lipodystrophy syndromes is insufficient.15,16
In this retrospective multicentre cohort study, we aimed to (i) characterize the cardiac disease phenotype of FPLD2 and other FPLD types; (ii) define the risk for developing cardiac disease and (iii) explore the association of molecular aetiology and cardiac manifestations. Also, spontaneous rhythm and action potential duration of functional syncytia of mature human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) from a patient with a pathogenic variant at codon 349 of the LMNA gene were assessed in optical mapping experiments using a voltage-sensitive dye to provide the proof-of-concept that these cells can become important tools in understanding the pathophysiology of cardiac disease and arrhythmias in patients with genetic heart diseases.
2 |. METHODS
2.1 |. Description of the cohort and clinical data
One hundred twenty-two patients (median age 49 years, IQR 38–59 years, age range: 13–77; males/females: 21/101) with FPLD who have been followed at the University of Michigan (n = 83) and major academic centres in Turkey (n = 39) with an available cardiac evaluation were included. The diagnosis of lipodystrophy was established based on comprehensive clinical assessment and genetic testing by experienced clinicians at referral centres for this rare disease.
In this study, we specifically inquired the impact of genotype, BMI and glucose control on the occurrence of cardiac events. Data were collected on demographics, past medical history, genotype, body mass index (BMI), laboratory assessments (specifically lipid and glucose control parameters such as HbA1c, triglyceride levels, liver function tests, etc), medications, comorbidities, cardiac manifestations and cardiac disease dates. Cardiac examination was done as part of standard medical care. Typically, patients are seen at least once a year and cardiac examination is part of their visits. Further tests are ordered based on clinical symptoms and findings in the system review, and physical examination. Baseline ECG and echocardiogram are typically obtained at least when patients are evaluated for the first time and studies repeated based on findings and follow-up symptoms/clinical events. ECGs and echocardiograms were reviewed by an experienced electrophysiology subspecialist (H O) and cardiac imaging subspecialist (N B), respectively. Both experts were blinded to any clinical data.
The primary end point was the first record of a cardiac event. Events included arrhythmias, conduction disease, cardiomyopathy, ischaemic cardiac disease and any other cardiac disease. Arrhythmias included sinus arrhythmia, premature ventricular contractions (PVC), premature atrial contractions (PAC), supraventricular tachycardia (SVT), atrial fibrillation or flutter and ventricular tachycardia or fibrillation. Conduction abnormality included atrioventricular block and intraventricular conduction delay.
2.2 |. Proof-of-concept hiPSC studies
Dermal fibroblasts were isolated from a skin biopsy of a patient with LMNA variant R349W. Briefly, the skin sample was minced into ≈1 mm3 fragments under sterile conditions and plated on a six-well plate with 1 mL of fibroblast media (DMEM: F12, 10% FBS, 1%NEAA, 200|μM of L-Glutamine) for 24 hours. Fibroblast outgrowth was monitored, and media was changed every other day until fibroblasts reached confluence and were passed with trypsin for expansion and reprogramming into hiPSC with a commercially available non-integrational vector (Cytotunes 2.0, Thermo, USA). Control fibroblasts (BJ foreskin fibroblasts; ATCC® CRL-2522™) were also reprogrammed as described before.17 In general, we prefer to perform comparisons between genetically-related (parents and/ or normal siblings) and genetically-unrelated control cell lines; nevertheless, the patient that donated the disease-specific fibroblasts did not have close relatives alive that could be recruited to provide genetically-related control samples and the feasible alternative was to perform the comparison with hiPSC generated from a commercially available normal fibroblast line.
New hiPSC lines were expanded with StemMACS iPS-Brew XF on matrigel for the production of hiPSC-derived cardiomyocytes (hiPSC-CMs). Cardiac directed differentiation was performed with a high-efficiency small molecule protocol.18 Cardiomyocytes were prepared as high-purity functional syncytia of mature hiPSC-CMs as described previously.19,20 After cardiac directed differentiation, the cells were submitted to magnetic assisted cell sorting (MACS) negative selection with PSC-derived cardiomyocyte isolation kit (Miltenyi sBiotec). Briefly, cells were dissociated with trypsin and the enzyme was neutralized with medium containing high serum (20% FBS). Cells were pelleted down by centrifugation and washed with HBSS prior to incubation with non-cardiomyocyte depletion antibody cocktail (1:5 dilution) for 10 minutes. Primary antibody cocktail was removed by dilution followed by centrifugation for cell separation. After removal of supernatant, the cell pellets were resuspended and incubated with magnetic bead-conjugated secondary antibodies (1:5 dilution) included in the kit. Volume of each tube was adjusted to 2 mL with MACS separation buffer (Miltenyi), and cell suspensions were individually applied to magnetic separation columns. Flow-through separation buffer containing cardiomyocytes was centrifuged prior to discard of supernatant. Cardiomyocytes were resuspended in plating medium and cardiomyocyte concentration was adjusted to 1.106 cardiomyocytes/mL. Purified cardiomyocytes were plated as functional syncytia on Matrigel coated PDMS to induce maturation of cardiomyocytes. Optical mapping of voltage changes was used to investigate cardiac electrophysiology. Action potential duration at 80% of repolarization during spontaneous depolarization and paced depolarization frequency was calculated by using the Scroll software. These parameters were also assessed after treatment of the cells with 100 nmol/L isoproterenol.20 Isoproterenol recovery rate was defined as the difference in APD80% immediately after and 5 minutes after isoproterenol treatment divided by APD80% before drug treatment.
2.3 |. Statistical methods
The nature of data distribution was assessed with the Kolmogorov-Smirnov test. Fisher’s exact test or Mann-Whitney U test was used as appropriate to compare groups. Unadjusted and adjusted odds ratios with 95% confidence interval (CI) were calculated for arrhythmia, atrial fibrillation or flutter, and conduction disease using a logistic regression model. Models were adjusted for age, body mass index (BMI, kg/m2) and comorbidities (such as chronic obstructive pulmonary disease (COPD), thyroid disease, chronic kidney disease, amyloidosis, renal tubular acidosis, sleep apnea and lung cancer). For the subgroup analysis (non-482 and 482 LMNA), models were only adjusted for age as a continuous variable due to the limited sample size and number of events. Data were then analysed with a time to event analysis approach where the reference date was the year of lipodystrophy diagnosis. Survival time was calculated from the reference date to end-point date or end of study date (2019) or death. Kaplan-Meier curves were plotted for conduction disease, and arrhythmia for the entire cohort, the log-rank test was used to compare between LMNA and non-LMNA groups. Two groups of patients were excluded from the study: patients who developed an event before lipodystrophy diagnosis and those diagnosed with lipodystrophy before 2004 (due to limited data accessibility). A cox proportional hazards model was used to drive hazard ratios with 95% CI of arrhythmia and conduction disease. When the proportionality assumption was met, the model was tested with time-dependent covariates. Provided P < .05, multivariable models were conducted to adjust for age at the cardiac examination (continuous variable) and comorbidity. Analyses were performed in SAS version 9.4 (SAS Institute Inc, Cary, NC), SPSS v.20, and Prism version 7 (GraphPad Software Inc, San Diego, CA). A P value < .05 was considered statistically significant.
3 |. RESULTS
3.1 |. Genotypic and clinical characteristics
The main characteristics of the study population are reported in Table S1. The majority of patients first underwent hot-spot screening for codon 482 of the LMNA gene followed by targeted sequencing of genes of interest, and whole-exome sequencing (WES) if the previous studies were negative. Seventy-one patients (58%) had pathogenic variants in known lipodystrophy genes (Table S2). LMNA pathogenic variants were found in 60 patients. Eight patients had pathogenic or likely pathogenic variants in the PPARG gene. Also, pathogenic variants were confirmed in the POLD1 (2 patients) and MFN2 (1 patient) genes. WES identified variants of unknown significance (VUS) in novel genes in 15 patients. We were not able to identify any pathogenic variant or VUS in 19 patients. The genetic characterization was incomplete in 17 patients with no causative genetic mutation for lipodystrophy on targeted clinical testing (none of them underwent WES). Of these 17 patients, 9 underwent sequencing of LMNA and PPARG genes, 7 were sequenced only for LMNA codons 10 and 482 and one patient had clinical lipodystrophy panel testing from the University of Chicago Genetics Laboratory that includes 13 genes associated with lipodystrophy
3.2 |. Cardiac phenotype
Table 1 shows the cardiac characteristics of our study population. The median (IQR) age at the cardiac examination was 46 years (34–55). Among 122 patients with FPLD, 95 (78%) were diagnosed with cardiac disease in their lifetime. Of those, 30 (25%) were diagnosed with ischaemic heart disease, 45 (37%) with an arrhythmia, 20 (16%) with conduction disease, 24 (20%) with prolonged QT interval, 9 (7%) with axis deviation and 13 (11%) with cardiomyopathy (CMP).
TABLE 1.
Cardiovascular manifestations
| Total (n = 122) | Michigan (n = 83) | Turkey (n = 39) | P value | |
|---|---|---|---|---|
| Age when cardiac examination, y | 46 (34–55) | 46 (35–57) | 44 (30–53) | 0.284 |
| Sex, Females | 101 (83) | 68 (82) | 33 (85) | 0.802 |
| BMI, kg/m2 | 27.2 (22.6–32.4) | 30 (24.1–34.4) | 24.6 (20.6–26.2) | <0.001 |
| LMNA variant | 60 (49) | 33 (40) | 27 (69) | 0.011* |
| Any cardiac issue | 95 (78) | 69 (83) | 26 (67) | 0.060 |
| Ischaemic heart disease | 30 (25) | 19 (23) | 11 (28) | 0.653 |
| Heart catheterization | 35 (29) | 23 (28) | 12 (31) | 0.830 |
| Balloon angioplasty | 15 (12) | 7 (8) | 8 (21) | 0.077 |
| Cardiac stent | 16 (13) | 8 (10) | 8 (21) | 0.148 |
| Myocardial infarction | 16 (13) | 13 (16) | 3 (8) | 0.383 |
| CABG | 6 (5) | 5 (6) | 1 (3) | 0.663 |
| Stroke | 11 (9) | 9 (11) | 2 (5) | 0.500 |
| PR interval, ms | 156 (142–168) | 158 (146–168) | 153 (136–180) | 0.234 |
| QRS duration, ms | 88 (82–98) | 88 (82–98) | 88 (80–102) | 0.761 |
| QTC, ms | 442 (421–462) | 445 (430–468) | 429 (400–455) | 0.001 |
| Arrhythmia | 45 (37) | 36 (43) | 9 (23) | 0.044 |
| Atrial fib/flutter | 12 (10) | 6 (7) | 6 (15) | 0.196 |
| PVC | 10 (8) | 10 (12) | 0 | 0.030 |
| PAC/SVPC | 7 (6) | 5 (6) | 2 (5) | 1.000 |
| Sinus arrhythmia | 21 (17) | 18 (22) | 3( 8) | 0.072 |
| Conduction abnormality | 20 (16) | 17 (20) | 3 (8) | 0.114 |
| Prolonged QT | 24 (20) | 19 (23) | 5 (13) | 0.229 |
| Axis deviation | 9 (7) | 6 (7) | 3 (8) | 1.000 |
| Cardiomyopathy | 13 (11) | 9 (11) | 4 (10) | 1.000 |
| Congestive heart failure | 18 (15) | 15 (18) | 3 (8) | 0.175 |
| LV hypertrophy† | 29 (32) | 21 (40) | 8 (21) | 0.068 |
| Diastolic dysfunction† | 21 (23) | 14 (27) | 7 (18) | 0.451 |
| Valvular heart disease† | 27 (30) | 13 (25) | 14 (36) | 0.354 |
| LVEF†, % | 60 (60–65) | 63 (57–65) | 60 (60–65) | 0.369 |
Note: Values are median (interquartile range) or n (%).
CABG indicates coronary artery bypass graft surgery; PVC, premature ventricular complex; PAC, premature atrial complex; SVPC, supraventricular premature complex; LVEF, left ventricular ejection fraction.
Echocardiogram is available in 91 patients (52 Michigan and 39 Turkey).
Seven patients with no complete gene sequencing for LMNA are excluded.
Of those with ischaemic heart disease, 15 (12%) patients had balloon angioplasty, 16 (13%) had a cardiac stent, 16 (13%) had myocardial infarction, and 6 (5%) had a history of CABG. Of those with arrhythmia, 12 (10%) had atrial fibrillation or flutter, 10 (8%) PVCs, 7 (6%) PAC/SVPC and 21 (17%) had sinus arrhythmia. For those with CMP diagnosis, 7 (6%) patients had dilated CMP, 3 (3%) hypertrophic, 2 (2%) ischaemic, and 1 (1%) postpartum CMP. Clinically, 18 (15%) patients developed congestive heart failure in their lifetime. Among the 90 patients who had an echocardiogram (74%), 29 (32%) had left ventricular hypertrophy, 21 (23%) had diastolic dysfunction and 27 (30%) had valvular heart disease.
There were only 4 observed deaths caused by cardiac events during the study observation period. Sudden cardiac death occurred in a 41-year-old woman with LMNA pathogenic variant R349W. The cause of death was myocardial infarction in a 72-year-old man with LMNA R482W variant and heart failure in 2 patients (a 58-year-old woman with the LMNA pathogenic variant R62G and a 77-year-old man with LMNA pathogenic variant R482Q). Dual-chamber cardiac defibrillators were implanted in 5 patients (4 with non-482 LMNA pathogenic variants, and in another patient from the non-LMNA group).
3.3 |. Impact of glycemic control and BMI on cardiac disease presentation
Overall, ischaemic heart disease was more prevalent among patients with HbA1c > 7.5% versus those with lower HbA1c [23 subjects (34.8%) vs 6 subjects (10.9%); P = .003; one subject excluded as no HbA1c level was available at the time of cardiac evaluation]. Also, percutaneous coronary interventions [13 subjects (19.7%) vs 3 subjects (5.5%), P = .030] were more commonly performed in the group with higher HbA1c. No significant association was found between BMI (adjusted for age) and cardiac events.
3.4 |. Presence of a pathogenic variant in LMNA gene versus other causes
The study cohort was stratified by genotype into two groups, those harbouring pathogenic variants in LMNA (LMNA group, n = 60) vs those that do not (non-LMNA group, n = 55) groups (Table 2). Seven patients were excluded from this analysis because they did not have complete gene sequencing for LMNA (they were only tested for 482 hotspots). Patients in the non-LMNA group had a significantly higher median BMI, glucose, HbA1c, triglycerides, total cholesterol and non-HDL cholesterol levels than patients in the LMNA group. Their unfavourable metabolic profile was also reflected in medication use as patients in the non-LMNA group required significantly higher doses of insulin, more frequent use of concentrated insulin and high dose statins (Table S3). Leptin levels were lower in the LMNA group.
TABLE 2.
Clinical characteristics and the LMNA variant
| LMNA (n = 60) | Non-LMNA (n = 55) | P value | |
|---|---|---|---|
| Cardiac examination age, y | 46 (33–54) | 51 (36–57) | 0.259 |
| Sex, Females | 48 (80) | 46 (84) | 0.638 |
| BMI, kg/m2 | 24.7 (21.7–27.6) | 31.5 (25.9–35.6) | <0.001 |
| Diabetes Mellitus | 50 (83) | 52 (95) | 0.078 |
| Hypertension | 42 (70) | 36 (66) | 0.691 |
| Pancreatitis | 12 (20) | 20 (36) | 0.062 |
| Glucose, mg/dL | 123 (93–181) | 165 (128–239) | 0.006 |
| HbA1c, % | 7.0 (6.0–8.6) | 8.3 (7.1–9.2) | 0.009 |
| Triglycerides, mg/dL | 279 (174–48 5) | 342 (246–896) | 0.038 |
| Total cholesterol, mg/dL | 193 (160–228) | 226 (174–293) | 0.007 |
| LDL cholesterol, mg/dL | 93 (64–129) | 106 (65–145) | 0.367 |
| HDL cholesterol, mg/dL | 38 (31–44) | 33 (29–42) | 0.138 |
| Non-HDL cholesterol, mg/dL | 149 (116–192) | 187 (138–254) | 0.003 |
| Leptin†, ng/mL | 3.2 (1.5–7.78) | 12 (5.29–18.50) | <0.001 |
| Ischaemic heart disease | 14 (23) | 16 (29) | 0.528 |
| Stroke | 4 (7) | 7 (13) | 0.348 |
| Arrhythmia | 27 (45) | 12 (22) | 0.011 |
| Atrial fib/flutter | 10 (17) | 2 (4) | 0.031 |
| PVC | 7 (12) | 1 (2) | 0.063 |
| PAC/SVPC | 5 (8) | 2 (4) | 0.442 |
| Sinus arrhythmia | 10 (17) | 7 (13) | 0.607 |
| Conduction abnormality | 13 (22) | 7 (13) | 0.229 |
| Axis deviation | 6 (10) | 3 (6) | 0.494 |
| Prolonged QT | 13 (22) | 10 (18) | 0.816 |
| Cardiomyopathy | 10 (17) | 3 (6) | 0.078 |
| Congestive heart failure | 11 (18) | 7 (13) | 0.451 |
| LV hypertrophy‡ | 13 (27) | 16 (40) | 0.256 |
| Diastolic dysfunction‡ | 9 (18) | 12 (30) | 0.219 |
| Valvular heart disease‡ | 18 (37) | 9 (23) | 0.170 |
Note: Values are median (interquartile range) or n (%).
BMI indicates body mass index; LDL, low density lipoprotein; HDL, high density lipoprotein; HbA1c, haemoglobin A1c; PVC, premature ventricular complex; PAC, premature atrial complex; SVPC, supraventricular premature complex.
Leptin levels are before metreleptin treatment closest to cardiac evaluation.
Echocardiogram is available in 88 patients (48 LMNA and 40 non-LMNA). Seven patients with no complete gene sequencing for LMNA are excluded.
Although patients with LMNA variants had lower BMI, and better metabolic control than patients in the non002DLMNA group, both groups had similar ratios of ischaemic heart disease, myocardial infarction or stroke events. On the other hand, patients with pathological LMNA variants had a higher prevalence of arrhythmia, specifically atrial fibrillation or flutter (Table 2). The odds ratio for arrhythmia and atrial fibrillation or flutter were higher in patients with LMNA pathogenic variants (Table 3). After adjusting for age at the cardiac examination, BMI and comorbidities, the odds of arrhythmia and atrial fibrillation or flutter were still significantly higher among patients with LMNA variants. The median time to first recorded arrhythmic event from the diagnosis of lipodystrophy was significantly shorter for patients with LMNA variants compared with the non-LMNA group (Figure 1). The hazard rate of arrhythmia was 3.52 times (95% CI: 1.34–9.27) higher among patients with LMNA variants compared to those with no LMNA variant.
TABLE 3.
Cardiac events by LMNA pathogenic variant
| Odds Ratio (95% CI); P value | Odds Ratio (95% CI); P value * | |
| LMNA variant compared to non-LMNA | ||
| Arrhythmia | 2.93 (1.29–6.64); 0.010 | 3.77 (1.45–9.83); 0.007 |
| Atrial fibrillation/ Atrial flutter | 5.30 (1.11–25.39); 0.037 | 5.78 (1.04–32.16); 0.045 |
| Conduction abnormality | 1.90 (0.70–5.17); 0.211 | 2.20 (0.71–6.85); 0.173 |
| Odds Ratio (95% CI); p value | Odds Ratio (95% CI); P value † | |
| Non-482 LMNA variant compared to 482 | ||
| Arrhythmia | 3.37 (1.12–10.08); 0.030 | 4.74 (1.41–15.98); 0.012 |
| Atrial fibrillation/ Atrial flutter | 5.44 (1.24–23.95); 0.025 | 17.67 (2.45–127.68); 0.004 |
| Conduction abnormality | 3.77 (1.05–13.57); 0.042 | 5.71 (1.37–23.76); 0.017 |
Adjusted for age at cardiac examination, BMI, comorbidities. Sex distribution and diabetes were not included in the model since >85% are diabetics and >80% are females. In addition, sex distribution was not different between LMNA and non-LMNA group, and the sample size did not allow for extra parameters in the model. Seven patients with no complete gene sequencing for LMNA are excluded.
Adjusted only for age at cardiac examination due to limited sample size. Seven patients with no complete gene sequencing for LMNA are excluded.
FIGURE 1.
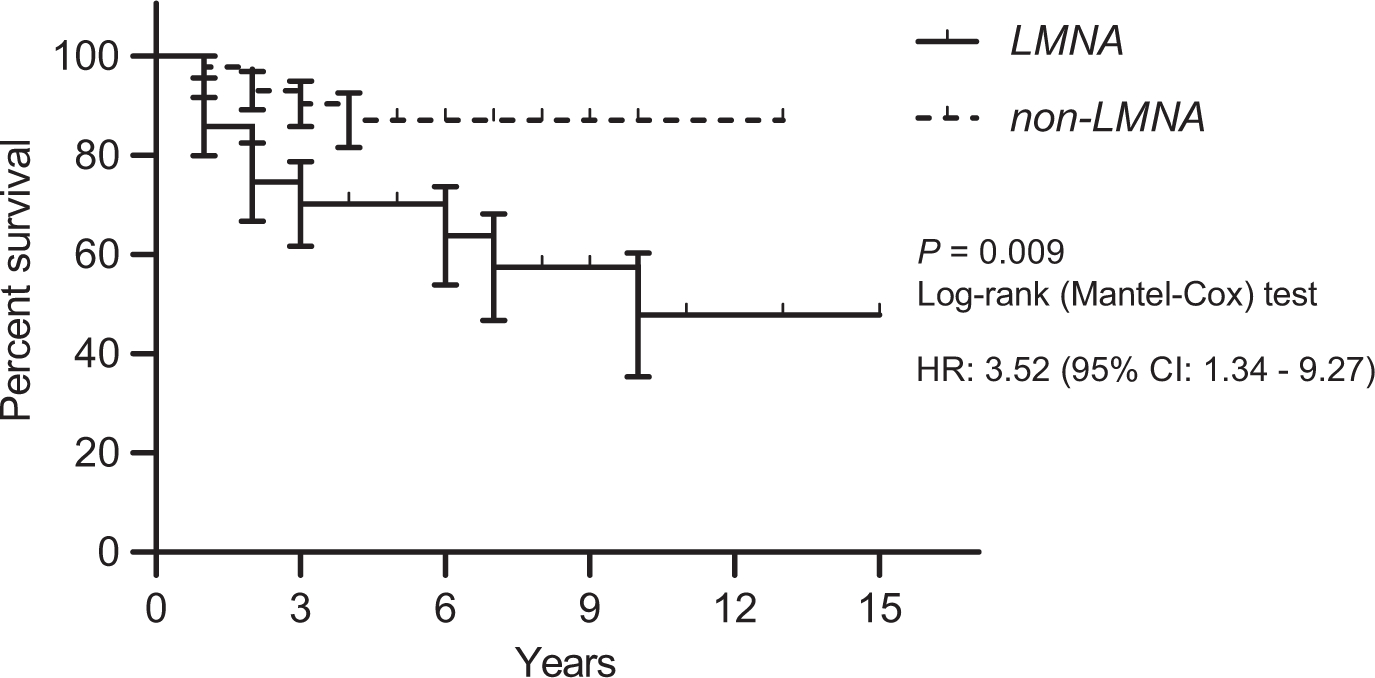
Kaplan-Meier survival curve showing arrhythmia for LMNA and non-LMNA patients
3.5 |. ‘Hot spot’ versus other pathogenic LMNA variants
LMNA group was then subdivided according to the most frequent hotspot mutation site, exon 8 codon 482. Table 4 shows the characteristics of both groups. There was no difference in age, sex, BMI, presence of diabetes, hypertension, hepatic steatosis, and pancreatitis, lipid profiles and HbAlc levels; however, patients with non-482 LMNA variants were more likely to have myocardial infarction, arrhythmia, atrial fibrillation/flutter, axis deviation, cardiomyopathy and congestive heart failure. Among patients with LMNA pathogenic variants, those with non-482 codon variants had higher odds of arrhythmia, atrial fibrillation or flutter and conduction abnormality. The higher risk persisted after adjusting for age at the cardiac examination (Table 3). Patients with non-482 LMNA pathogenic variants also had higher odds of arrhythmia and atrial fibrillation or flutter compared to the rest of the cohort (data not shown).
TABLE 4.
Clinical Characteristics and Codon 482 Variants of the LMNA Gene
| Non-482 variant (n = 22) | 482 variant (n = 38) | P value | |
|---|---|---|---|
| Cardiac examination age, y | 42 (31–49) | 47 (33–57) | 0.094 |
| Sex, Females | 17 (77) | 31 (82) | 0.744 |
| BMI, kg/m2 | 22.9 (19.6–27.3) | 24.8 (21.9–28.3) | 0.123 |
| Diabetes Mellitus | 19 (86) | 31 (82) | 0.732 |
| Hypertension | 15 (68) | 27 (71) | 1.000 |
| Pancreatitis | 6 (27) | 6 (16) | 0.327 |
| Glucose, mg/dL | 114 (93–171) | 142 (101–184) | 0.193 |
| HbA1c, % | 6.8 (5.8–8.1) | 7.7 (6.0–8.7) | 0.337 |
| Triglycerides, mg/dL | 190 (126–410) | 323 (196–485) | 0.079 |
| Total cholesterol, mg/dL | 202 (159–268) | 193 (160–226) | 0.544 |
| LDL cholesterol, mg/dL | 96 (75–130) | 93 (63–125) | 0.520 |
| HDL cholesterol, mg/dL | 39 (35–45) | 36 (30–44) | 0.167 |
| Non-HDL cholesterol, mg/dL | 149 (116–207) | 149 (116–187) | 0.713 |
| Leptin†, ng/mL | 4.2 (1.5–8.5) | 3.1 (1.4–7.4) | 0.773 |
| Ischaemic heart disease | 6 (27) | 8 (21) | 0.753 |
| Myocardial infarction | 5 (23) | 1 (3) | 0.021 |
| Stroke | 2 (9) | 2 (5) | 0.619 |
| Arrhythmia | 14 (64) | 13 (34) | 0.034 |
| Atrial fib/flutter | 7 (32) | 3 (8) | 0.029 |
| PVC | 2 (9) | 5 (13) | 1.000 |
| PAC/SVPC | 3 (14) | 2 (5) | 0.346 |
| Sinus arrhythmia | 4 (18) | 6 (16) | 1.000 |
| Conduction abnormality | 8 (36) | 5 (13) | 0.052 |
| Axis deviation | 5 (23) | 1 (3) | 0.021 |
| Prolonged QT | 6 (27) | 7 (18) | 0.520 |
| Cardiomyopathy | 8 (36) | 2 (5) | 0.003 |
| Congestive heart failure | 8 (36) | 3 (8) | 0.012 |
| LV hypertrophy‡ | 3 (17) | 10 (32) | 0.282 |
| Diastolic dysfunction‡ | 5 (28) | 4 (13) | 0.400 |
| Valvular heart disease‡ | 10 (56) | 10 (33) | 0.441 |
Note: Values are median (interquartile range) or n (%).
BMI indicates body mass index; LDL, low density lipoprotein; HDL, high density lipoprotein; HbA1c, haemoglobin A1c; PVC, premature ventricular complex; PAC, premature atrial complex; SVPC, supraventricular premature complex.
Leptin levels are before metreleptin treatment closest to cardiac evaluation.
Echocardiogram is available in 48 patients (18 with non-codon 482 variant and 30 patients with codon 482 variant).
We also evaluated how patients with the hot-spot variants compared to those with the non-LMNA genotypes. Premature ventricular contractions were more commonly observed in patients with codon 482 LMNA variants than in the group with no LMNA pathogenic variants (Table S4). Numerically, patients with codon 482 LMNA variants had arrhythmia and atrial fibrillation/flutter at a higher rate, but these differences were not statistically significant. Additionally, BMI and leptin levels were lower in patients with codon 482 LMNA variants than those with no LMNA variants.
3.6 |. Clinical characteristics of the LMNA R349W variant
We had the opportunity to study the hiPSCs of a patient harbouring a pathogenic variant (R349W) in the LMNA gene. The cell samples were obtained from this case when she was 39. This patient had multiple comorbidities including diabetes, hyperlipidemia, hypertension, hyperandrogenism and NASH. She presented with a unique form of lipodystrophy at age 36, previously completely undiagnosed, with profound fat loss from her extremities, neck and face and anterior trunk, but with increased fat deposition in the back and specifically with an exaggerated buffalo hump. She had acrolysis and underdeveloped breasts, but high circulating testosterone levels without aberrant signs of hirsutism. She developed progressive proteinuria while she was followed. She had history of infertility and had one pregnancy that resulted in foetal loss at 35 weeks. The patient’s cardiac history included stent implantation for coronary artery disease, myocardial infarction (non-ST-elevation MI), cardiomyopathy and conduction abnormality all of which were uncovered after we initiated cardiac workup for the observed variant. The patient had sudden cardiac death at age 41, 2 years after we obtained her skin biopsy.
We also had the opportunity to observe additional patients harbouring the same mutation in an unrelated pedigree from Turkey and a completely unrelated male case originally from England. This latter patient with LMNA R349W from England had diabetes, hyperlipidemia and hepatic steatosis. He was diagnosed with supraventricular tachycardia when he was 28 years old. He also had aortic stenosis. Other pertinent clinical features included anxiety, partial alopecia and proteinuria.
The Turkish LMNA R349W pedigree had previously been reported.10 Data from three living members of this pedigree are included in this study. In contrast to codon 482 LMNA variants, these patients had fat loss affecting the face and neck, and limb lipoatrophy was prominent distally. Paroxysmal atrial fibrillation/flutter was detected in the proband (42-year-old woman) who later developed an episode of stroke. She had additional clinical features such as hearing loss, micrognathia, scoliosis, partial alopecia, skin atrophy and proteinuria. Her son and daughter were also studied. Among these younger generation members, the older brother developed episodes of atrial tachycardia. There was further cardiac history in the family. Although not included in this analysis, her father’s medical records indicate that he died at age 44 after being admitted with coronary artery disease and cardiac arrhythmias to the emergency room. Her brother died at age 33 due to heart failure. His medical records indicate that he had atrial fibrillation, severe mitral and tricuspid valve regurgitation, and pulmonary hypertension.
3.7 |. Cellular Electrophysiologic Characteristics in the cells harbouring LMNA R349W variant
Functional syncytia of mature hiPSC-CMs from a patient with LMNA R349W variant were prepared for the study of action potential and rate of spontaneous activation.
Affected patient’s cardiomyocytes had a higher frequency of spontaneous depolarizations (Figure 2A), and shorter uncorrected APD80% (action potential duration at 80% repolarization) at 1 Hz compared with control cardiomyocytes (Figure 2B). Action potential duration was adjusted with Fridericia correction formula because of differences in spontaneous depolarization rhythm and showed that corrected APD80% is shorter in the patient’s cardiomyocytes than normal cardiomyocytes (Figure 2C). Furthermore, cardiomyocytes from the patient with LMNA R349W variant also demonstrated several rhythm alterations such as early afterdepolarizations, spontaneous quiescence and spontaneous tachyarrhythmia (Supplemental data; Movie 1); none of those were observed in the control cell lines (Figure 2D). Cardiomyocytes differentiated from hiPSC-CMs carrying the R349W variant presented heterogeneous rhythm of spontaneous repolarization varying from normal rhythm to Torsade de Pointes-like activation and quiescence. Therefore, we have obtained several recordings per functional mature syncytia to be able to document rhythm abnormalities in these cells (6 syncytia and 65 optical mapping recordings). We also have obtained multiple optical mapping recordings from control cardiomyocytes (6 syncytia and 17 optical mapping recordings). LMNA R349W cardiomyocytes had preserved chronotropic response to isoproterenol, although chronotropic changes were greater in control than affected cardiomyocytes. Additionally, the recovery rate was significantly lower in affected than in control cardiomyocytes (Figure 2E), which indicates disrupted beta-adrenergic response.
FIGURE 2.
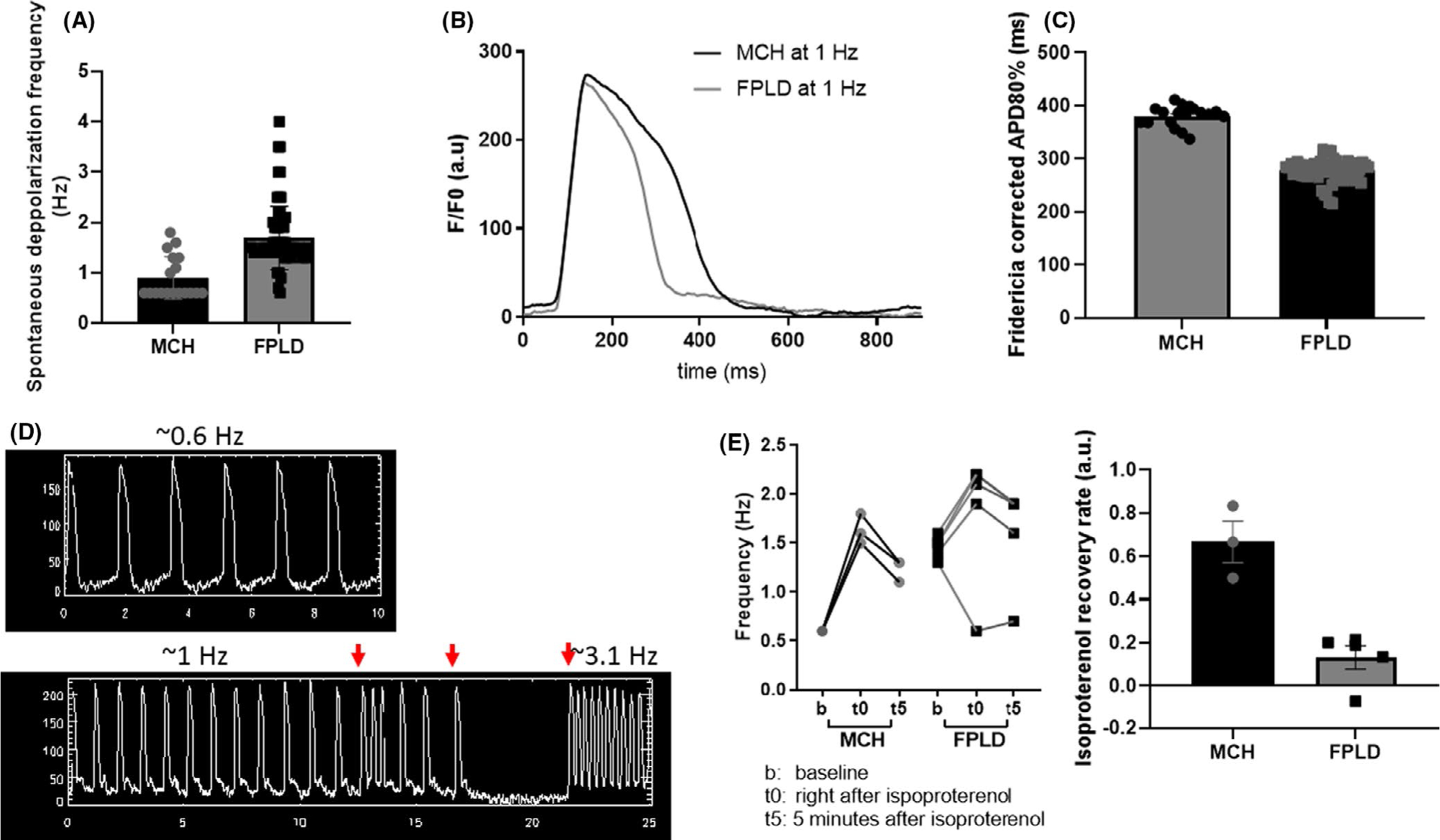
Functional syncytia of mature human induced pluripotent stem cell-derived cardiomyocytes from a patient carrying a variant (LMNA R349W) causative of familial partial lipodystrophy (FPLD) were submitted to optical mapping for assessment of membrane voltage changes. (A) LMNA mutant cardiomyocytes had a higher frequency of spontaneous depolarization in relation to control cell line (MCH) (P < .001). (B) APD80% of repolarization was shorter in cells carrying the LMNA variant. (C) Duration of spontaneous action potential duration at 80% of repolarization was adjusted to the frequency of spontaneous depolarization with Fridericia correction and showed that LMNA mutant cardiomyocytes presented shorter corrected APD80% compared to control cardiomyocytes (P < .001). (D) Additionally, LMNA mutant cardiomyocytes presented several rhythm alterations (red arrows) such as early afterdepolarizations, spontaneous quiescence and spontaneous tachyarrhythmia; none of those were observed in the control cell lines. (E) Finally, both control and LMNA mutant cardiomyocytes showed a positive chronotropic response to isoproterenol. Nevertheless, isoproterenol recovery rate was significantly lower in the LMNA mutant cardiomyocytes
4 |. DISCUSSION
This multicentre study reveals a high prevalence of cardiac events in patients with FPLD that highlights the need for vigilant cardiac monitoring in FPLD, especially, in patients with FPLD2 who exhibited a disproportionately higher risk of developing cardiac arrhythmias such as atrial fibrillation/atrial flutter. A study of hiPSC-CMs from a patient with LMNA pathological variant R349W showed further evidence regarding arrhythmogenic potential of the underlying genetic defect as evidenced by a higher frequency of autonomous activity, shorter action potential duration, slower recovery from chronotropic changes and several rhythm alterations such as early afterdepolarizations, spontaneous quiescence and spontaneous tachyarrhythmia. Although patients with LMNA variants had lower BMI, and better metabolic control than non-LMNA patients, both groups had similar ratios of ischaemic heart disease, myocardial infarction or stroke events. Several factors may have contributed to high prevalence of metabolic disturbances in the patients with no LMNA variants, which include, but are not limited to, diet, lifestyle, and higher BMI.21 We want to note that a similar degree of ischaemic disease in the group with LMNA variants highlights the predisposition of this group despite being in better metabolic state. Overall cardiac events, were more common in the LMNA group with a higher prevalence of arrhythmia, specifically atrial fibrillation or flutter, emphasizing the difference in cardiac phenotypes between the groups.
In addition to higher odds of arrhythmia and atrial fibrillation or flutter, the median time from lipodystrophy diagnosis to arrhythmia was significantly shorter for patients with LMNA variants. The association of the LMNA gene with cardiomyopathy and arrhythmia has been previously demonstrated in several studies.22,23 It is known that these patients may require ICD as a result of high risk of high degree atrioventricular block and ventricular arrhythmias. On the other hand, cardiac manifestations of LMNA variants causing the lipodystrophy phenotype had been only occasionally reported,8,15,24–26 and not systematically studied except in a few clinical case series from France.13,27 The rarity of disorders caused by the LMNA variants makes it difficult to phenotypically classify all variants scattered over the entire LMNA gene. Phenotypic heterogeneity is high among carriers of LMNA variants ranging from lipodystrophy to neuromuscular and cardiac disorders, and overlaps are sometimes observed.1 The severity of cardiac phenotype can be different even in patients presenting with primary cardiac disease. For instance, Hoorntje et al28 reported that the LMNA p.R331G variant was associated with milder clinical events than other LMNA variants causing LMNA-related cardiac disease. One explanation for the rare reporting of cardiac events in patients with LMNA variants causing lipodystrophy might be the presence of relatively milder cardiac disease. Nevertheless, as shown in our study, an increased risk for specific cardiac events exists in patients with LMNA variants compared with other aetiologies of FPLD; thus, rigorous evaluation and follow-up are required. Among these cardiac events, atrial fibrillation is a well- known cardiac disease to be associated with increased morbidity and mortality, in part due to the risk of thromboembolic disease, but further long-term studies are needed to confirm whether LMNA variants increase the risk of clinical outcomes due to atrial fibrillation in patients with FPLD.
It is still not clear how aberrant LMNA transcript leads to alterations in cardiac phenotype. Myocardial fibrosis has been previously proposed to be a responsible mechanism for both arrhythmogenesis and cardiomyopathy.29 On the other hand, crucial cellular processes such as structural integrity of the nucleus, the structural integrity of the cell (via interactions between nuclear lamina, cytoskeleton and extracellular matrix) and the stiffness of the cell, regulation of gene expression, and cellular signalling pathways are known to be affected by pathogenic LMNA variants.30
The protocol for differentiation of hiPSCs into cardiomyocytes generates a heterogeneous population of cardiac cells consisting of atrial-, nodal- and ventricular-like cardiomyocytes, and each individual syncytium has a natural pacemaker.31 We found that this heterogeneous population of cardiomyocytes carrying a non-482 LMNA pathogenic variant causative of a lipodystrophy phenotype presented a wide array of arrhythmias, including EADs, quiescence, and tachyarrhythmias. Tachyarrhythmias and quiescence may indicate defective generation of pacemaker activity by nodal-like cells which are governed by the potassium inward funny current (If) and transient and long-acting calcium currents (ICa, T and ICa, L).32 Additionally, with respect to the disturbances in pacemaker activity that may be calcium-mediated, we have observed that studied cardiomyocytes have blunted recovery after an isoproterenol challenge indicating calcium overload or at least delay in the reestablishment of intracellular calcium concentration. Abnormal intracellular calcium concentration and shortened APD have been shown experimentally to induce cardiomyocyte early afterdepolarizations,33 and the presence of EADs in these cardiomyocytes may result from defective intracellular calcium handling as well. The nature of the disturbance in the calcium handling system in affected cardiomyocytes is yet to be determined and future studies should focus on the activity of SERCA2a, phospholamban and other proteins regulating calcium relocation into different intracellular compartments.
Similar to our observation, Kwapich et al13 recently reported more frequent sudden death and more frequent use of cardiac implantable electronic devices in non-482 than codon 482 LMNA pathogenic variant carriers. Also, non-482 LMNA pathogenic variant carriers had more abnormalities on electrocardiography, had greater frequencies of left atrial enlargement, and lower left ventricular ejection fractions than codon 482 pathogenic variant carriers. Although it remains unknown why patients with non-482 LMNA pathogenic variants were more likely to present with cardiac manifestations, overlapping progeroid characteristics (such as stiffness of the extracellular matrix or intracellular architecture abnormalities) might contribute to increased cardiac risk in these patients.1,10,34
Our study has several limitations. First, the retrospective nature of the study makes it liable to unmeasured potential confounding factors. Although extensive efforts have been put into data collection, data were obtained from records, and availability was limited to what already exists. Second, only patients who had an available ECG in the records were included. This might distort the result if more ECGs were clinically indicated in one group compared with another though ECG can be considered a routine test in all patients with lipodystrophy. Sample heterogeneity was another limiting factor. Patients were included from two countries. It is quite possible that patient characteristics might differ between the two countries. For example, Michigan patients had higher BMI, which could be due to the difference in diet in the US compared with the diet in Turkey. There were country-specific differences in treatment algorithms of patients with lipodystrophy and hyperlipidemia. Smoking is an important contributor to cardiac risk. The Turkish data set did not include any smoking history, so we could not account for smoking in this retrospective study. More than 90 percent of the Michigan patients are not current smokers and only a small percentage of the patients had substantial smoking history (more than 15 pack-years that would impact cardiac events). There were also differences in genetic testing algorithms. Besides, no formal family screening algorithm was performed in any of the centres. Nevertheless, lipodystrophy is a rare disease, and achieving the case numbers of this report was only possible with international collaboration. Finally, the hiPSC-CM study lacks an isogenic control to rule out factors related to genetic background which may have contributed to the arrhythmic phenotype. Another issue is that the study remains descriptive rather than mechanistic. We did not conduct a study of potential structural alterations in the hiPSC-CMs, including altered trafficking of ion channels which might have contributed to the differences between affected and control cells in the electrophysiological phenotype. Our studies in the induced cardiomyocytes are intended as preliminary and for proof- of-concept. Our data imply that these cells can be utilized to fully characterize the pathophysiology of arrhythmias and also for screening the best cardiac therapeutics in the future.
In conclusion, our results highlight a high prevalence of cardiac events in patients with FPLD. Patients with FPLD2 caused by LMNA variants are at a higher risk for developing cardiac arrhythmias, especially atrial fibrillation or flutter. Patients with non-482 LMNA pathogenic variants are at further risk. For this reason, early diagnosis and providing adequate screening as well as appropriate interventions for cardiac and metabolic abnormalities is crucial to reduce the occurrence of cardiovascular events in this population. The association of the LMNA gene and arrhythmogenic potential is further supported by data from hiPSC-CMs that may help us better understand the mechanisms of the cardiac phenotype associated with the underlying genetic abnormalities and create precision therapy opportunities in the future.
Supplementary Material
ACKNOWLEDGEMENT
We thank our patients for their willingness to share their clinical data for publication.
CONFLICT OF INTEREST
The authors report following conflicts: BA has attended Scientific Advisory Board Meetings organized by Aegerion Pharmaceuticals (now Amryt Pharma) and Regeneron Pharmaceuticals and has received honoraria as a speaker from AstraZeneca, Lilly, MSD, Novartis, Novo Nordisk, Boehringer-Ingelheim, Servier and Sanofi-Aventis. BA has taken consulting fees from Amryt Pharma and has received writing support from Aegerion Pharmaceuticals (past) and Amryt Pharma (current) in unrelated manuscripts. EAO reports the following conflicts: Grant support: Aegerion Pharmaceuticals (now Amryt Pharma), Ionis Pharmaceuticals, Akcea Therapeutics, Gemphire Therapeutics, GI Dynamics (current), AstraZeneca (past two years). Consultant or Advisor: AstraZeneca, Akcea Therapeutics, Ionis Pharmaceuticals, Thera Therapeutics and BMS (Past), Regeneron, Aegerion (now Amryt Pharma). Drug support: Aegerion Pharmaceuticals (now Amryt Pharma), Rhythm Pharmaceuticals, Regenereon (all current) and Akcea Therapeutics (past), other support: (specifically writing support in unrelated manuscripts) Aegerion Pharmaceuticals (now Amryt Pharma). AMR reports the following conflict: consultant for CARTOX (current).
Funding information
Infrastructure and data management support has been provided by the NIH Clinical and Translational Science Awards grant UL1TR000433, the Nutrition Obesity Research Centers grant P30 DK089503, and NIH institutional grant DK034933. Finally, the work was supported by generous gifts to the Lipodystrophy Fund at the University of Michigan made by the Sopha family, and the White Point Foundation of Turkey. JWI acknowledges support from the Morton S. and Henrietta K. Sellner Professorship in Human Genetics.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Akinci B, Meral R, Oral EA. Phenotypic and genetic characteristics of lipodystrophy: pathophysiology, metabolic abnormalities, and comorbidities. Curr Diab Rep. 2018;18(12):143. [DOI] [PubMed] [Google Scholar]
- 2.Shackleton S, Lloyd DJ, Jackson SN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24(2):153–156. [DOI] [PubMed] [Google Scholar]
- 3.Lattanzi G, Maggi L, Araujo-Vilar D. Laminopathies. Nucleus. 2018;9(1):543–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hegele R LMNA mutation position predicts organ system involvement in laminopathies. Clin Genet. 2005;68(1):31–34. [DOI] [PubMed] [Google Scholar]
- 5.Sylvius N, Tesson F. Lamin A/C and cardiac diseases. Curr Opin Cardiol. 2006;21(3):159–165. [DOI] [PubMed] [Google Scholar]
- 6.Turk M, Wehnert M, Schroder R, Chevessier F. Multisystem disorder and limb girdle muscular dystrophy caused by LMNA p. R28W mutation. Neuromuscul Disord. 2013;23(7):587–590. [DOI] [PubMed] [Google Scholar]
- 7.Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med. 2002;112(7):549–555. [DOI] [PubMed] [Google Scholar]
- 8.Subramanyam L, Simha V, Garg A. Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations. Clin Genet. 2010;78(1):66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caux F, Dubosclard E, Lascols O, et al. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J Clin Endocrinol Metab. 2003;88(3):1006–1013. [DOI] [PubMed] [Google Scholar]
- 10.Akinci B, Onay H, Demir T, et al. Clinical presentations, metabolic abnormalities and end-organ complications in patients with familial partial lipodystrophy. Metabolism. 2017;72:109–119. [DOI] [PubMed] [Google Scholar]
- 11.Ajluni N, Meral R, Neidert AH, et al. Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf). 2017;86(5):698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain I, Patni N, Ueda M, et al. A novel generalized lipodystrophy-associated progeroid syndrome due to recurrent heterozygous LMNA p. T10I mutation. J Clin Endocrinol Metab. 2018;103(3):1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwapich M, Lacroix D, Espiard S, et al. Cardiometabolic assessment of lamin A/C gene mutation carriers: a phenotype-genotype correlation. Diabetes Metab. 2019;45(4):382–389. [DOI] [PubMed] [Google Scholar]
- 14.Wahbi K, Ben Yaou R, Gandjbakhch E, et al. Development and validation of a new risk prediction score for life-threatening ventricular tachyarrhythmias in laminopathies. Circulation. 2019;140(4):293–302. [DOI] [PubMed] [Google Scholar]
- 15.Hegele RA. Premature atherosclerosis associated with monogenic insulin resistance. Circulation. 2001;103(18):2225–2229. [DOI] [PubMed] [Google Scholar]
- 16.Hussain I, Patni N, Garg A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology. 2019;51(2):202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.da Rocha AM, Creech J, Thonn E, Mironov S, Herron TJ. Detection of drug-induced torsades de pointes arrhythmia mechanisms using hiPSC-CM syncytial monolayers in a high-throughput screening voltage sensitive dye assay. Toxicol Sci. 2020;173(2):402–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lian X, Hsiao C, Wilson G, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA. 2012;109(27):E18 48–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herron TJ. Calcium and voltage mapping in hiPSC-CM monolayers. Cell Calcium. 2016;59(2–3):84–90. [DOI] [PubMed] [Google Scholar]
- 20.Herron TJ, Rocha AM, Campbell KF, et al. Extracellular matrix-mediated maturation of human pluripotent stem cell-derived cardiac monolayer structure and electrophysiological function. Circ Arrhythm Electrophysiol. 2016;9(4):e003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeru I, Vatier C, Vantyghem MC, Lascols O, Vigouroux C. LMNA-associated partial lipodystrophy: anticipation of metabolic complications. J Med Genet. 2017;54(6):413–416. [DOI] [PubMed] [Google Scholar]
- 22.van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. 2012;59(5):493–500. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Baldinger SH, Gandjbakhch E, et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J Am Coll Cardiol. 2016;68(21):2299–2307. [DOI] [PubMed] [Google Scholar]
- 24.Panikkath R, Panikkath D, Sanchez-Iglesias S, Araujo-Vilar D, Lado-Abeal J. An uncommon association of familial partial lipodystrophy, dilated cardiomyopathy, and conduction system disease. J Investig Med High Impact Case Rep. 2016;4(3):2324709616658495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Kooi AJ, Bonne G, Eymard B, et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59(4):620–623. [DOI] [PubMed] [Google Scholar]
- 26.Haque WA, Vuitch F, Garg A. Post-mortem findings in familial partial lipodystrophy, Dunnigan variety. Diabet Med. 2002;19(12):1022–1025. [DOI] [PubMed] [Google Scholar]
- 27.Vantyghem MC, Pigny P, Maurage CA, et al. Patients with familial partial lipodystrophy of the Dunnigan type due to a LMNA R482W mutation show muscular and cardiac abnormalities. J Clin Endocrinol Metab. 2004;89(11):5337–5346. [DOI] [PubMed] [Google Scholar]
- 28.Hoorntje ET, Bollen IA, Barge-Schaapveld DQ, et al. Lamin A/C-related cardiac disease: late onset with a variable and mild phenotype in a large cohort of patients with the lamin A/C p. (Arg331Gln) Founder Mutation. Circ Cardiovasc Genet. 2017;10(4):e001631. [DOI] [PubMed] [Google Scholar]
- 29.van Tintelen JP, Tio RA, Kerstjens-Frederikse WS, et al. Severe myocardial fibrosis caused by a deletion of the 5’ end of the lamin A/C gene. J Am Coll Cardiol. 2007;49(25):2430–2439. [DOI] [PubMed] [Google Scholar]
- 30.Ho R, Hegele RA. Complex effects of laminopathy mutations on nuclear structure and function. Clin Genet. 2019;95(2):199–209. [DOI] [PubMed] [Google Scholar]
- 31.Bizy A, Guerrero-Serna G, Hu B, et al. Myosin light chain 2-based selection of human iPSC-derived early ventricular cardiac myocytes. Stem Cell Res. 2013;11(3):1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morad M, Zhang XH. Mechanisms of spontaneous pacing: sinoatrial nodal cells, neonatal cardiomyocytes, and human stem cell derived cardiomyocytes. Can J Physiol Pharmacol. 2017;95(10):1100–1107. [DOI] [PubMed] [Google Scholar]
- 33.Tang L, Joung B, Ogawa M, Chen PS, Lin SF. Intracellular calcium dynamics, shortened action potential duration, and late-phase 3 early afterdepolarization in Langendorff-perfused rabbit ventricles. J Cardiovasc Electrophysiol. 2012;23(12):1364–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rivera-Torres J, Calvo CJ, Llach A, et al. Cardiac electrical defects in progeroid mice and Hutchinson-Gilford progeria syndrome patients with nuclear lamina alterations. Proc Natl Acad Sci USA. 2016;113(46):E7250–E7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.