

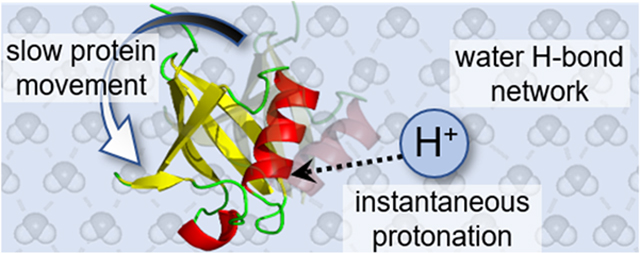
Abstract
We present simple considerations of how differences in time scales of motions of protons, the lightest and fastest chemical moiety, and the much longer time scales associated with the dynamics of proteins, among the heaviest and slowest analytes, may allow many protein conformations from solution to be kinetically trapped during the process of electrospraying protein solutions into the gas phase. In solution, the quantum nature of protons leads them to change locations by tunneling, an instantaneous process; moreover, the Grotthuss mechanism suggests that these small particles can respond nearly instantaneously to the dynamic motions of proteins that occur on much longer time scales. A conformational change is accompanied by favorable or unfavorable variations in the free energy of the system, providing the impetus for solvent ↔ protein proton exchange. Thus, as thermal distributions of protein conformations interconvert, protonation states rapidly respond, as specific acidic and basic sites are exposed or protected. In the vacuum of the mass spectrometer, protons become immobilized in locations that are specific to the protein conformations from which they were incorporated. In this way, conformational states from solution are preserved upon electrospraying them into the gas phase. These ideas are consistent with the exquisite sensitivity of electrospray mass spectra to small changes of the local environment that alter protein structure in solution. We might remember this approximation for the protonation of proteins in solution with the colloquial expression—protons are fast and smart; proteins are slow and dumb.
Graphical Abstract
INTRODUCTION
Fast Migration Rates of Protons in Solution.
The Grotthuss mechanism (Scheme 1) predicts that protons can be transferred through long distances in water with extraordinary speed.1 Unlike all other ions, a proton lacks electronic structure; as the lightest chemical moiety, these particles behave quantum mechanically and can change position by tunneling.2 In solution, protons are conducted nearly instantaneously in a manner that can be thought of in analogy with the conduction of electrons through a wire. The mechanism in Scheme 1 has a proton migration rate that is limited by the vibrational frequency of the conducting water molecules. Perhaps the most remarkable aspect of this mechanism lies not in the profound consequences of such high velocity migration (estimated to be >800 m·s−1; nearly 1 μm in a ns)3 but rather that Grotthuss proposed it in 1806 using OH as the molecular formula of water instead of H2O, as the correct stoichiometry was not determined by Avogadro until 1811.4
Scheme 1.

Description of H+ Transfer through H2O Wire Predicted by Grotthuss in 1806 (albeit for OH instead of H2O)a
aSee ref 1 for details.
Structural Changes in Large Molecules Such As Proteins Are Relatively Slow.
One consequence of high-speed proton motion is that rates of proton-transfer reactions in large flexible molecules such as proteins will be limited by the availability of acidic and basic sites rather than the rate of transferring a proton from solution to these sites. Consider the dynamics of an ensemble of protein conformational states in solution. Interconversion of different structures involves rearrangements of the positions of a plethora of atoms and molecules involved in intramolecular, solvent–protein, and solvent–solvent interactions that frame each structure-coupled processes that require substantial time. For example, secondary structures such as α-helices are often formed on nanosecond to microsecond time scales, whereas tertiary structures often require microsecond to second and quaternary structures even longer.5–9 As these structures are being established, new basic or acidic sites may become solvent exposed many times as conformations fluctuate. Because of the differences in mechanisms of motions (and time scales) of conformational changes and protons, any changes in the solvent accessibility of such sites due to conformer fluctuation would rapidly accommodate changes in protonation state. That is, during a large conformational change, protons may flicker on and off specific sites many times as a new solution structure is established. Crudely speaking, we might say that protons are fast and smart; proteins are slow and dumb.10
Proposed Electrospray Mechanisms.
The differences in proton migration and protein conformation time scales raise interesting possibilities for capturing information about solution structure by nanoelectrospray ionization (n-ESI) paired with mass spectrometry (MS).11 The mechanisms of ESI have been debated extensively, and efforts toward characterizing these processes are still an active area of research. There are three predominant mechanisms that account for experimental ESI observations: the ion evaporation, charge residue, and chain ejection models, all of which have been described in detail elsewhere.12–16 In general,17–20 electrified analyte solutions are sprayed from a small tip (typically nanometer to micrometer) to form a Taylor cone that emits a fine mist of charged droplets containing analyte molecules. In the case of positive-mode n-ESI, each droplet carries positive charges due to the presence of excess ions including H+, Na+, K+, etc. Once emitted from the Taylor cone, the charged droplets undergo rapid solvent evaporation and shrinkage that ultimately increases their charge density. As the droplets reach the Raleigh limit,21 which depends on the solution properties, the droplets can no longer sustain increased Coulomb repulsion. Jet fissioning occurs to produce smaller droplets, and repeated evaporation and fission events yield highly charged, nm-radii droplets that enter the MS instrument.17–19
As the droplet shrinks, its surface-to-volume ratio increases, as does its internal strain energy;22 because of this, we anticipate that small droplets will be less stable and evaporate faster than large droplets. Therefore, the quenching process should speed up as the protein approaches the anhydrous state. There are several factors that influence the droplet size and time scale of solvent evaporation, including emitter size, flow rate, distance from the MS inlet, and solvent. We note that in n-ESI experiments, where emitter sizes are typically >5 μm, droplet effects such as pH changes, recondensation events due to poor desolvation, and salt adduction are diminished.23,24 This is further supported by ion mobility spectrometry (IMS) cross-section measurements made with n-ESI that agree with theoretical measurements of compact, dehydrated structures.25–27 In n-ESI, it is estimated that protein ions are evaporated and quenched (i.e., kinetically trapped as gaseous ions) on the order of 1–100 μs.28–30 This suggests that under some conditions the key step of establishing the charge state by n-ESI is much faster than protein conformational changes.
Are Unique Solution Conformations Trapped as Specific Protonation States during the Process of Electrospray Ionization?
There are many reports stating that ESI appears to capture solution-like conformations in the gas phase.26,31–37 The evaporative-cooling phenomenon associated with droplet shrinkage appears to “freeze out” a number of structures that are in equilibrium in solution, as illustrated in Figure 1.36,38–41
Figure 1.

Hypothetical drawing of the process of nanoelectrospraying proteins into the gas phase (right) and its implications on the protein free-energy landscape (left). In solution, rapid interconversion of protein structures [native (N), intermediate (I), denatured (D)] is mediated by solvent. During early states of n-ESI, the droplet initiates with solution populations in equilibrium with rapidly exchanging protons (bottom). As the droplet dries, the solution equilibrium is quenched due to evaporative cooling (middle). Just before entering the MS instrument, protein structures are kinetically trapped on a new, gas-phase free energy landscape associated with the solvent-free ion (top).
Protein structures in solution resemble rugged free-energy landscapes where rapid interconversion of structures is mediated by solvent. During n-ESI, the droplets containing protein solutions undergo rapid cooling to below room temperature, resulting from desolvation. The temperature drop over this short time scale is likely to be substantial;39,42–44 however, it is difficult to estimate an exact temperature due to various factors including solvent behavior, ambient collisions that warm the droplet, and instrumental conditions. Nonetheless, within the lower temperature droplet environment, the energy barriers between structures on the free-energy landscape become more difficult and take longer to overcome, resulting in the “freezing out” of protein structures in the absence of a lubricating solvent. Changes to the free energy of the system during n-ESI are likely to be small; recent thermochemical results (ΔG, ΔH, ΔS, and ΔCp) derived from solution temperature-dependent IMS data agree with literature values provided from experiments done directly in solution with calorimetry.45
As distinct solution conformations emerge into the gas phase as desolvated ions, different numbers of protons (and other excess ions) are retained according to the types of residues exposed, leading to the observed distribution of protonation states in ESI-MS (e.g., [M + nH]n+, [M + (n + 1)H]n+1, [M + (n + 2)H](n+2)+, etc.).46–49 The outcome is that individual charge states may be specific to protein structure in a manner that allows access to even subtle differences in solution conformations. Below, we revisit prior published results27,45,50 and find them to be consistent with the notions presented here. It is likely that this analogy has not been drawn previously because little is known about the structures of non-native protein conformations and the interconversions among them. Equilibrium ensembles and folding intermediates51–54 often have short lifetimes, and their fleeting existence leaves them difficult to characterize.55 Because of this, we begin by considering a simple system with large energetic barriers between well-characterized structures such that structural transitions are slow and obvious. Such a situation is often found for peptide bonds involving a proline residue, which can rotate between cis ↔ trans forms, inducing large structural changes.56,57 Folding studies reveal that these transitions are often rate limiting.58
EXAMPLES
Charge-State Populations Controlled by a Conformational Change: The PPI to PPII Helix.
The simple model peptide polyproline-7 (Pro7) provides an excellent starting point for understanding conformational regulation of proton transfer.27 When dissolved in a nonpolar solvent such as 1-propanol, Pro-7 favors the right-handed PPI helical structure in which each of the six peptide bonds adopts a cis configuration. In aqueous solutions, the all-trans PPII left-handed helix is observed. The PPI helix possesses a macrodipole in the direction of the N-terminal residue that is stabilized upon protonation. As shown in Figure 2a, when PPI is electro-sprayed from a 1-propanol solution, a single peak in the mass spectrum, having a size (collision cross section) consistent with the singly protonated ion [PPI + H]+, is observed. Upon dilution of the Pro7 PPI helix into a more aqueous environment (e.g., 2.0 min after dilution as shown in Figure 2a), the singly protonated PPI helical species persists as the only ion formed upon electrospray. At longer times, doubly protonated PPII is formed by the reaction: PPI+PrOH + H3O+ → PPII2+PrOH/aq + H2O.
Figure 2.

(a) Collision cross section (CCS) distributions for PPIPrOH → PPIIaq transition for Pro7. Bottom CCS distributions obtained by integrating the ions with different m/z values show the formation of the PPI helix in propanol and the formation of the PPII helix in water. Kinetic studies from 2 to 246 min at 296 K in 40:59.5:0.5 1-propanol/H2O/HOAc (v/v/v) show the transition from PPI to PPII. Structures derived from molecular dynamic simulations are shown for each CCS peak. (b) Relative abundance of different charge states and conformers as a function of transition time. See text and ref 27 for details. Adapted ref 27. Copyright 2015 American Chemical Society.
The long time scale associated with the formation of the PPII2+ ion is remarkable as it is the slowest proton-transfer reaction ever reported due to the slow interconversion of structures. The kinetics associated with this reaction are shown in Figure 2b. The structure of PPI must not be able to instantaneously accommodate an additional charge. Instead, transfer of the proton from solution to form the doubly protonated state is regulated by a slow structural transition, in this case one in which every peptide bond flips to a trans configuration, leading to the PPII helix. Ultimately, the final location of the proton depends upon the structure of Pro7.
Water Stabilizes Charge and Conformation.
If an understanding of how many different conformations contribute to the charge state distribution in an electrospray mass spectrum is inhibited by the fleeting nature of “non-native” states, then one might imagine that appreciating the role of solvent will be even more challenging. The competition for protons is defined not only by the accessible basic sites on each conformation but also the willingness of the solvent to give them up. That is, it is the interactions of the system, consisting of the molecule and the solvent, that lead to the protonation state. Of the many examples of this, consider the key role of hydration illustrated by recent studies of 4-aminobenzoic acid (4-ABA).50 The protonation of the amine group of 4-ABAH+ is favored in protic solvents (N-protomer), whereas protonation of the carboxylic acid group is favored in the gas phase and in aprotic solvents (O-protomer). A series of hydrated 4-ABAH+ ions are observed upon electrospraying the analyte into a cryogenic IMS-MS with a range of n = 0 to ∼50 water molecules [4-ABAH+(H2O)n]. The proton transfer of hydrated 4-ABAH+(H2O)n from the N- to O-protomer occurs at n = 6, which is mediated by a stable water bridge between the polar groups. The head-to-tail arrangement of this water bridge strongly implies proton hopping between water molecules, i.e., the Grotthuss mechanism.1 In the presence of acetonitrile, the proton migration is inhibited owing to favorable solvent interactions with the N-protomer. We note that there are results that do not appear to be consistent with a Grotthuss-type mechanism. For example, in nonaqueous solvents, this mechanism would not work well. But it does appear that such a mechanism is reasonable under aqueous conditions.59 Similar to this example, several recent studies have found evidence for specific conformations of isotopomers.60,61 It is important to note that during the final stages of the dehydration process, transporting protons away from their solution phase locations can fundamentally alter the structural integrity of polar molecules.
Evidence That Specific Non-native Conformations Formed upon Denaturation Favor Specific Protonation States.
The configurational regulation of protons in the small Pro7 and 4-amino benzoic acid systems is also apparent in larger systems. Consider, for example, protein denaturation, which can be induced by many types of perturbations, including introduction of chaotropes,62 variation of pH,63 and thermal heating.64 The ESI charge state distribution is extremely sensitive to the structure of proteins in solution. A narrow distribution of low charge states is often observed for “native” electrospray; a broader distribution of m/z peaks corresponding to higher charge states is observed from denatured solutions. But, do specific solution conformations contribute uniquely to specific charge states in these larger systems?
An example of this is found upon analyzing the 64 amino acid protein chymotrypsin inhibitor 2 (CI-2). The solution transition from native to denatured states of CI-2 has been studied extensively by traditional calorimetric techniques.65–67 Protein denaturation is generally viewed as a cooperative two-state process in which loss of the native structure gives rise to the denatured state a distribution of random, amorphous forms.65 However, it is postulated that different types of intermediate structures should be populated near the phase transition between the native and denatured forms.68 Figure 3a shows the analysis of CI-2 with variable-temperature ESI to measure the cooperative two-state transition by means of an average charge state derived from temperature-dependent MS data. Fitting these data to a two-state sigmoidal transition yields a melting temperature (Tm) which provides an ensemble signature of stability for the native fold. Tm values measured by this analysis have been in agreement with values measured with traditional techniques.26,69,70
Figure 3.

Variable-temperature ESI-IMS-MS studies of CI-2 in water at pH 2.6. (a) Weighted average charge state as a function of solution temperature for CI-2 with a Tm = 48.5 ± 0.3 °C; inset shows the mass spectra at various solution temperatures. (b) Relative abundance as a function of solution temperature for each of the six individual charge states of CI-2. (c) Free-energy landscape depicting ΔG as a function of cross section (for each charge state) showing 41 unique solution structures. Relative abundance of CCS distributions for each charge state are shown at the bottom of each landscape. Each black line represents the free energy of a structure populated in solution, arbitrarily referenced to product conformation 1 (P1) shown as a red line at ΔG = 0 kJ·mol–1. At low solution temperature (26 °C, bottom), structures present within the +6 and +7 distributions are favored with low ΔG values; at the highest temperatures (top), denatured structures become the favored signals while the folded structures in the +6 and +7 charge states become unfavorable. See text and ref 45 for details. Adapted from ref 45. Copyright 2020, American Chemical Society.
This interpretation does not preclude a more complex process involving many structures that are hidden within each charge state. Figure 3b shows that each charge state varies uniquely with solution temperature, requiring that multiple solution structures are contributing differently to each of the charge states within the MS distribution. Inspection of the mobilities within each charge state shows multiple peaks that behave differently with solution temperature. A Gaussian model of the temperature-dependent cross section distributions is used to elucidate 41 unique structures behaving differently with solution temperature. Figure 3c shows an example of the conformational free energy landscape of CI-2 consisting of 41 structures at various solution temperatures. Because of the broad dynamic range of IMS-MS techniques, the 41 structures can be seen across many temperature ranges even if they are in very low abundance. We might think of this as a transition that is taking place similar to a more flexible, membrane-like surface; as the temperature changes, the free energy surface evolves to favor some new species (unfolded structures with high collision cross sections) and disfavor others (folded structures with low collision cross sections). In this way, a conformation that we would consider energetically unfavored may be preferred at elevated temperatures. Of course, depending upon the nature of the system, new non-native structures may be formed by lowering the temperature as well (i.e., cold denaturation).
Are These Ideas Consistent with Other Protein Mass Spectrometry Data?
Many clues about the relationship of conformations and charge-state distribution come from early electrospray studies. Since Chowdhury, Katta, and Chait’s classic 1990 paper describing how the populations of the +8 to +18 charge states of cytochrome c change upon varying the pH of aqueous solutions (as shown in Figure 4),46 many reports of variations in protonation states with changing solution environment have been made.71 Implicit in the interpretation of these results is the impression that conformation states are distinct from one another, but each is assumed to give rise to multiple charge states in the electrospray distribution. This behavior is common for many proteins and changes in charge states can be induced by other means of protein denaturation, e.g., variation in solvent composition72 or solvent temperature.73–75 The results described above suggest that far more detail about many conformations is likely to be accessible from the MS distribution.
Figure 4.

Classic examples of changes in charge state and conformation of cytochrome c measured by MS depicted from ref 46 and adapted from refs 76 (copyright 1998 American Chemical Society), and 77 (copyright 1997 American Chemical Society). Left: changes in MS charge-state distribution of cytochrome c at different solvent pH: 2.6 (bottom), 3.0 (middle), 5.2 (top). Top right: isotopic 1H/2H exchange profiles of different charge states of cytochrome c in the gas phase formed by ESI (circles), IR laser heating (squares), collisional heating (open triangles), and charge stripping (closed triangles). Bottom right: cross sections of cytochrome c populated in each charge state at high (filled circles) and low (open circles) injection energies measured by IMS-MS.
The idea that protons are far more mobile than conformational states would suggest that composites of multiple distinct species are hidden within a charge state distribution. In their 1993 paper, McLafferty and co-workers rigorously tested this hypothesis by reacting gaseous cytochrome c ions with deuterated solvents and measuring the isotopic 1H/2H exchange profiles of different charge states as shown in Figure 4.78 They interpreted the varying exchange levels for different charge states as evidence for families of different gas-phase conformations, that in some cases appeared to be correlated to unique structures in solution. Subsequent studies showed that unique conformations could be resolved based on differences in their mobilities through a buffer gas on millisecond time scales (Figure 4).25,77 The first remarkable aspect of these results is that each requires multiple, well-defined conformations that can coexist in the absence of solvent. Additionally, these measurements reveal that unlike the rapid interconversion of conformations in solution, when solvent is removed, specific structures can be stable for long time periods (at least milliseconds in the case of mobility studies to many minutes, or even hours, as revealed by 1H/2H exchange).79 The large differences imply that the solution acts as a lubricant, allowing structures that are long-lived in the gas-phase to be highly flexural in solution. Several years later, Breuker and McLafferty discussed the time scales of the formation of specific types of intramolecular interactions in the gas phase.29 Their estimates suggest that gentle instrument conditions can preserve solution structures into the gas phase and the ESI dehydration process may even stabilize aspects of the solution fold.
Mobile Protons in the Gas Phase.
In the absence of a conducting medium (in nonactivating, gas-phase conditions), protons are solvated by intramolecular interactions with the peptide backbone groups and side chains. For example, carbonyl (CO) and amide (NH) groups along the backbone have lone-pair electrons and thus high gas-phase basicities that stabilize protons80, which has the effect of fixing their positions within the gas-phase conformation. Highly charged gas-phase protein structures may be influenced by nearby protons and undergo Coulomb-induced unfolding; however, it is anticipated that protons that are farther than ∼10 Å apart are not influenced by one another.81–84 In general, the maximum charge deposited onto a protein structure in the gas-phase is usually no greater than the total number of basic residues and N-termini in the sequence.72,80,85 A previous model by Williams and collaborators has been used to elucidate the influence of Coulomb repulsion on the gas-phase basicities of common proteins, showing that the low gas-phase basicities of highly charged proteins causes solvent ions to be protonated instead of basic protein sites.86 Such Coulomb forces can also be exploited to stabilize local rearrangements of protein solution structures into the gas-phase by use of counterions.37,87,88
Upon activation of peptides or proteins by collisions, photons, temperature, etc., protons that were originally sequestered become mobile. A mobile proton is hungry for an electron and thus weakens nearby electron-dense regions associated with covalent bonding interactions, which facilitates dissociation. Wysocki, Gaskell, Harrison, and others describe this as the mobile proton model that results in peptide fragments that are valuable for determining oligomer sequences.89 We note that in order to take advantage of the sensitivity of ESI to monitor protein conformations in solution, careful attention to instrument tuning is necessary. Efforts toward developing instrumental protocols to reduce the amount of ion heating associated with measurements of protein solution conformations are have been presented and are ongoing.90
CONCLUSIONS
We have explored the Grotthuss mechanism and its implications on protonation of specific macromolecular structures in solution. This mechanism would predict that small changes in structure are nearly instantaneously accompanied by changes in protonation configuration/state. Because of the differences in time scales of proton migration and protein structural changes, changes in the equilibrium distribution of structures in solution may appear as changes in the ESI charge state distribution. One outcome of these considerations is that ESI may be exquisitely sensitive to protein and peptide conformations in a manner that has not been appreciated previously. As such, theoretical treatments of proteins in solution should consider the rapid changes in protonation states that accompany small changes in structure, an idea that is currently not practical in molecular dynamic simulations.
While these simple considerations of the motions of protons and proteins is useful when analyzing electrospray charge states and conformations, we note that there are several factors that influence the preservation of native-like protein structures into the gas-phase environment that we have not discussed in detail here and are still poorly understood. Instrumental conditions, such as applied electric field, distance from MS inlet, capillary temperature, and numerous other factors influence the desolvation and emergence of protein ions into the gas-phase. Solvent effects, such as using a high-salt buffer or organic solvent, will also influence solution structures and MS charge states.
It is instructive to close this critical insight by considering the influence of the Grotthuss mechanism in understanding the velocities of electrons.10 As J. J. Thomson was “discovering the electron”, he used an analogy with Grotthuss chains in 1889 to explain the very high velocities of electrons through gases.91 That is, he considered the existence of a polarized chainlike structure within the gas as a means of conducting electrons. This extension of the ideas of Grotthuss also applies as we consider electronic motion and nuclear motion in small molecules. Born and Oppenheimer realized that nuclear motion in molecules was so much slower than electronic motion that in theory the two are separable. Like protons in proteins, for small molecules we would say that electrons are fast and smart; nuclei are slow and dumb. The irony of this is while the Grotthuss mechanism for protons has essentially been manifested in quantum chemical theories for calculating molecular structure (i.e., Born’s and Oppenheimer’s approximation which allows electronic structure to be treated separately in calculations from nuclear motion), the implications have not yet been realized for theoretical treatment of macromolecules in the solution environment for which it was first proposed.
ACKNOWLEDGMENTS
This work is supported in part by funds from National Institutes of Health Grant Nos. 5R01GM121751 (D.E.C., D.H.R., A.L.) and P41GM128577 (D.H.R.) and partial support from Grant Nos. DP2GM123486 (A.L.), and R01GM114494 (S.V.); National Science Foundation Grant Nos. 2031083 [provided from the Center for Biological Metrology (DEC)] and CHE-1707675 (DHR); and the Robert and Marjorie Mann Graduate Research Fellowship from the Indiana University College of Arts and Sciences (SAR).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/jasms.1c00100
The authors declare no competing financial interest.
Contributor Information
Shannon A. Raab, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States
Tarick J. El-Baba, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States
Arthur Laganowsky, Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States.
David H. Russell, Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States
Stephen J. Valentine, Department of Chemistry, West Virginia University, Morgantown, West Virginia 26506, United States.
David E. Clemmer, Department of Chemistry, Indiana University, Bloomington, Indiana 47405, United States.
REFERENCES
- (1).Grotthuss C. J. T. d. Collection of C. Cole. Méoire sur la décomposition de l’eau: et des corps qu’ elle tient en dissolution àl’aide de l’éĺectricitégalvanique. [publisher not identified]: Rome, 1805. [Google Scholar]
- (2).DeGregorio N; Iyengar SS Challenges in constructing accurate methods for hydrogen transfer reactions in large biological assemblies: rare events sampling for mechanistic discovery and tensor networks for quantum nuclear effects. Faraday Discuss. 2020, 221 (0), 379–405. [DOI] [PubMed] [Google Scholar]
- (3).Hassanali A; Giberti F; Cuny J; Kühne, T. D.; Parrinello, M. Proton transfer through the water gossamer. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (34), 13723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Avogadro A. Essay on a manner of determining the relative masses of the elementary molecules of bodies, and the proportions in which they enter into these compounds. Journal de physique 1811, 73, 58–76. [Google Scholar]
- (5).Clarke DT; Doig AJ; Stapley BJ; Jones GR The α-helix folds on the millisecond time scale. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (13), 7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lin MM; Mohammed OF; Jas GS; Zewail AH Speed limit of protein folding evidenced in secondary structure dynamics. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (40), 16622–16627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sekhar A; Vallurupalli P; Kay LE Defining a length scale for millisecond-timescale protein conformational exchange. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (28), 11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen EHL; Lu TTY; Hsu JCC; Tseng YJ; Lim TS; Chen RPY Directly monitor protein rearrangement on a nanosecond-to-millisecond time-scale. Sci. Rep. 2017, 7 (1), 8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dill KA; MacCallum JL The Protein-Folding Problem, 50 Years On. Science 2012, 338 (6110), 1042. [DOI] [PubMed] [Google Scholar]
- (10).Born M; Oppenheimer R. Zur Quantentheorie der Molekeln. Ann. Phys. 1927, 389 (20), 457–484. [Google Scholar]
- (11).Fenn JB; Mann M; Meng CK; Wong SF; Whitehouse CM Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246 (4926), 64–71. [DOI] [PubMed] [Google Scholar]
- (12).Konermann L; Ahadi E; Rodriguez AD; Vahidi S. Unraveling the Mechanism of Electrospray Ionization. Anal. Chem. 2013, 85 (1), 2–9. [DOI] [PubMed] [Google Scholar]
- (13).Iribarne J; Thomson B. On the evaporation of small ions from charged droplets. J. Chem. Phys. 1976, 64 (6), 2287–2294. [Google Scholar]
- (14).Daub CD; Cann NM How are completely desolvated ions produced in electrospray ionization: insights from molecular dynamics simulations. Anal. Chem. 2011, 83 (22), 8372–8376. [DOI] [PubMed] [Google Scholar]
- (15).Iavarone AT; Williams ER Mechanism of charging and supercharging molecules in electrospray ionization. J. Am. Chem. Soc. 2003, 125 (8), 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ahadi E; Konermann L. Modeling the behavior of coarse-grained polymer chains in charged water droplets: implications for the mechanism of electrospray ionization. J. Phys. Chem. B 2012, 116 (1), 104–112. [DOI] [PubMed] [Google Scholar]
- (17).Fenn JB Electrospray wings for molecular elephants (Nobel lecture). Angew. Chem., Int. Ed. 2003, 42 (33), 3871–3894. [DOI] [PubMed] [Google Scholar]
- (18).Kebarle P; Verkerk UH Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom. Rev. 2009, 28 (6), 898–917. [DOI] [PubMed] [Google Scholar]
- (19).Cech NB; Enke CG Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2001, 20 (6), 362–387. [DOI] [PubMed] [Google Scholar]
- (20).Liuni P; Wilson DJ Understanding and optimizing electrospray ionization techniques for proteomic analysis. Expert Rev. Proteomics 2011, 8 (2), 197–209. [DOI] [PubMed] [Google Scholar]
- (21).Rayleigh L; On XX the equilibrium of liquid conducting masses charged with electricity. London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science 1882, 14 (87), 184–186. [Google Scholar]
- (22).Wortmann A; Kistler-Momotova A; Zenobi R; Heine MC; Wilhelm O; Pratsinis SE Shrinking droplets in electrospray ionization and their influence on chemical equilibria. J. Am. Soc. Mass Spectrom. 2007, 18 (3), 385–393. [DOI] [PubMed] [Google Scholar]
- (23).El-Faramawy A; Siu KWM; Thomson BA Efficiency of Nano-Electrospray Ionization. J. Am. Soc. Mass Spectrom. 2005, 16 (10), 1702–1707. [DOI] [PubMed] [Google Scholar]
- (24).Karas M; Bahr U; Dülcks, T. Nano-electrospray ionization mass spectrometry: addressing analytical problems beyond routine. Fresenius’ J. Anal. Chem. 2000, 366 (6), 669–676. [DOI] [PubMed] [Google Scholar]
- (25).Clemmer DE; Hudgins RR; Jarrold MF Naked protein conformations: cytochrome c in the gas phase. J. Am. Chem. Soc. 1995, 117 (40), 10141–10142. [Google Scholar]
- (26).El-Baba TJ; Woodall DW; Raab SA; Fuller DR; Laganowsky A; Russell DH; Clemmer DE Melting Proteins: Evidence for Multiple Stable Structures upon Thermal Denaturation of Native Ubiquitin from Ion Mobility Spectrometry-Mass Spectrometry Measurements. J. Am. Chem. Soc. 2017, 139 (18), 6306–6309. [DOI] [PubMed] [Google Scholar]
- (27).Shi L; Holliday AE; Khanal N; Russell DH; Clemmer DE Configurationally-Coupled Protonation of Polyproline-7. J. Am. Chem. Soc. 2015, 137 (27), 8680–8683. [DOI] [PubMed] [Google Scholar]
- (28).Xia Z; Williams ER Effect of droplet lifetime on where ions are formed in electrospray ionization. Analyst 2019, 144 (1), 237–248. [DOI] [PubMed] [Google Scholar]
- (29).Breuker K; McLafferty FW Stepwise evolution of protein native structure with electrospray into the gas phase, 10–12 to 102 s. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (47), 18145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rovelli G; Jacobs MI; Willis MD; Rapf RJ; Prophet AM; Wilson KR A critical analysis of electrospray techniques for the determination of accelerated rates and mechanisms of chemical reactions in droplets. Chemical Science 2020, 11 (48), 13026–13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ruotolo BT; Robinson CV Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol. 2006, 10 (5), 402–408. [DOI] [PubMed] [Google Scholar]
- (32).Pierson NA; Chen L; Valentine SJ; Russell DH; Clemmer DE Number of solution states of bradykinin from ion mobility and mass spectrometry measurements. J. Am. Chem. Soc. 2011, 133 (35), 13810–13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wyttenbach T; Pierson NA; Clemmer DE; Bowers MT Ion mobility analysis of molecular dynamics. Annu. Rev. Phys. Chem. 2014, 65, 175–196. [DOI] [PubMed] [Google Scholar]
- (34).Lanucara F; Holman SW; Gray CJ; Eyers CE The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6 (4), 281–294. [DOI] [PubMed] [Google Scholar]
- (35).McAllister RG; Metwally H; Sun Y; Konermann L. Release of native-like gaseous proteins from electrospray droplets via the charged residue mechanism: insights from molecular dynamics simulations. J. Am. Chem. Soc. 2015, 137 (39), 12667–12676. [DOI] [PubMed] [Google Scholar]
- (36).Silveira JA; Fort KL; Kim D; Servage KA; Pierson NA; Clemmer DE; Russell DH From Solution to the Gas Phase: Stepwise Dehydration and Kinetic Trapping of Substance P Reveals the Origin of Peptide Conformations. J. Am. Chem. Soc. 2013, 135 (51), 19147–19153. [DOI] [PubMed] [Google Scholar]
- (37).Khristenko N; Amato J; Livet S; Pagano B; Randazzo A; Gabelica V. Native Ion Mobility Mass Spectrometry: When Gas-Phase Ion Structures Depend on the Electrospray Charging Process. J. Am. Soc. Mass Spectrom. 2019, 30 (6), 1069–1081. [DOI] [PubMed] [Google Scholar]
- (38).Kim D; Wagner N; Wooding K; Clemmer DE; Russell DH Ions from solution to the gas phase: a molecular dynamics simulation of the structural evolution of substance P during desolvation of charged nanodroplets generated by electrospray ionization. J. Am. Chem. Soc. 2017, 139 (8), 2981–2988. [DOI] [PubMed] [Google Scholar]
- (39).Lee S-W; Freivogel P; Schindler T; Beauchamp JL Freeze-Dried Biomolecules: FT-ICR Studies of the Specific Solvation of Functional Groups and Clathrate Formation Observed by the Slow Evaporation of Water from Hydrated Peptides and Model Compounds in the Gas Phase. J. Am. Chem. Soc. 1998, 120 (45), 11758–11765. [Google Scholar]
- (40).Rodriguez-Cruz SE; Klassen JS; Williams ER Hydration of gas-phase gramicidin S (M+ 2H) 2+ ions formed by electrospray: the transition from solution to gas-phase structure. J. Am. Soc. Mass Spectrom. 1997, 8 (5), 565–568. [Google Scholar]
- (41).Clemmer DE; Russell DH; Williams ER Characterizing the Conformationome: Toward a Structural Understanding of the Proteome. Acc. Chem. Res. 2017, 50 (3), 556–560. [DOI] [PubMed] [Google Scholar]
- (42).Ando K; Arakawa M; Terasaki A. Freezing of micrometersized liquid droplets of pure water evaporatively cooled in a vacuum. Phys. Chem. Chem. Phys. 2018, 20 (45), 28435–28444. [DOI] [PubMed] [Google Scholar]
- (43).Breuker K; McLafferty FW Stepwise evolution of protein native structure with electrospray into the gas phase, 10–12 to 102 s. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (47), 18145–18152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kebarle P; Tang L. From ions in solution to ions in the gas phase-the mechanism of electrospray mass spectrometry. Anal. Chem. 1993, 65 (22), 972A–986A. [Google Scholar]
- (45).Raab SA; El-Baba TJ; Woodall DW; Liu W; Liu Y; Baird Z; Hales DA; Laganowsky A; Russell DH; Clemmer DE Evidence for Many Unique Solution Structures for Chymotrypsin Inhibitor 2: A Thermodynamic Perspective Derived from vT-ESI-IMS-MS Measurements. J. Am. Chem. Soc. 2020, 142 (41), 17372–17383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Chowdhury SK; Katta V; Chait BT Probing conformational changes in proteins by mass spectrometry. J. Am. Chem. Soc. 1990, 112 (24), 9012–9013. [Google Scholar]
- (47).Mohimen A; Dobo A; Hoerner JK; Kaltashov IA A chemometric approach to detection and characterization of multiple protein conformers in solution using electrospray ionization mass spectrometry. Anal. Chem. 2003, 75 (16), 4139–4147. [DOI] [PubMed] [Google Scholar]
- (48).Fort KL; Silveira JA; Pierson NA; Servage KA; Clemmer DE; Russell DH From solution to the gas phase: factors that influence kinetic trapping of Substance P in the gas phase. J. Phys. Chem. B 2014, 118 (49), 14336–14344. [DOI] [PubMed] [Google Scholar]
- (49).Shi H; Clemmer DE Evidence for two new solution states of ubiquitin by IMS–MS analysis. J. Phys. Chem. B 2014, 118 (13), 3498–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hebert MJ; Russell DH Tracking the Structural Evolution of 4-Aminobenzoic Acid in the Transition from Solution to the Gas Phase. J. Phys. Chem. B 2020, 124 (11), 2081–2087. [DOI] [PubMed] [Google Scholar]
- (51).Daggett V; Fersht A. The present view of the mechanism of protein folding. Nat. Rev. Mol. Cell Biol. 2003, 4 (6), 497–502. [DOI] [PubMed] [Google Scholar]
- (52).Merchant KA; Best RB; Louis JM; Gopich IV; Eaton WA Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (5), 1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Neudecker P; Lundström P; Kay LE Relaxation dispersion NMR spectroscopy as a tool for detailed studies of protein folding. Biophys. J. 2009, 96 (6), 2045–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Englander SW; Mayne L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (45), 15873–15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Motlagh HN; Toptygin D; Kaiser CM; Hilser VJ Single-molecule chemo-mechanical spectroscopy provides structural identity of folding intermediates. Biophys. J. 2016, 110 (6), 1280–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Schimmel PR; Flory PJ Conformational energy and configurational statistics of poly-L-proline. Proc. Natl. Acad. Sci. U. S. A. 1967, 58 (1), 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Warnke S; Baldauf C; Bowers MT; Pagel K; von Helden G. Photodissociation of Conformer-Selected Ubiquitin Ions Reveals Site-Specific Cis/Trans Isomerization of Proline Peptide Bonds. J. Am. Chem. Soc. 2014, 136 (29), 10308–10314. [DOI] [PubMed] [Google Scholar]
- (58).Chen Y; Ding F; Nie H; Serohijos AW; Sharma S; Wilcox KC; Yin S; Dokholyan NV Protein folding: then and now. Arch. Biochem. Biophys. 2008, 469 (1), 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Oomens J; Moore DT; Meijer G; von Helden G. Infrared multiple photon dynamics and spectroscopy of cationic PABA and its dehydroxylated fragment ion. Phys. Chem. Chem. Phys. 2004, 6 (4), 710–718. [Google Scholar]
- (60).Li J; Pacheco A; Raghavachari K; Iyengar S. A Grotthuss-like proton shuttle in the anomalous C2H3+ Carbocation: Energetic and vibrational properties for isotopologues. Phys. Chem. Chem. Phys. 2016, 18, 29395. [DOI] [PubMed] [Google Scholar]
- (61).Kaszycki JL; Bowman AP; Shvartsburg AA Ion Mobility Separation of Peptide Isotopomers. J. Am. Soc. Mass Spectrom. 2016, 27 (5), 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Myers JK; Nick Pace C; Martin Scholtz J. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4 (10), 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Anderson DE; Becktel WJ; Dahlquist FW pH-Induced denaturation of proteins: a single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 1990, 29 (9), 2403–2408. [DOI] [PubMed] [Google Scholar]
- (64).Wintrode PL; Makhatadze GI; Privalov PL Thermodynamics of ubiquitin unfolding. Proteins: Struct., Funct., Genet. 1994, 18 (3), 246–53. [DOI] [PubMed] [Google Scholar]
- (65).Jackson SE; Fersht AR Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 1991, 30 (43), 10428–10435. [DOI] [PubMed] [Google Scholar]
- (66).Kazmirski SL; Wong KB; Freund SM; Tan YJ; Fersht AR; Daggett V. Protein folding from a highly disordered denatured state: the folding pathway of chymotrypsin inhibitor 2 at atomic resolution. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (8), 4349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Itzhaki LS; Otzen DE; Fersht AR The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: evidence for a nucleation-condensation mechanism for protein folding. J. Mol. Biol. 1995, 254 (2), 260–88. [DOI] [PubMed] [Google Scholar]
- (68).Baldwin RL The Search for Folding Intermediates and the Mechanism of Protein Folding. Annu. Rev. Biophys. 2008, 37 (1), 1–21. [DOI] [PubMed] [Google Scholar]
- (69).Woodall DW; El-Baba TJ; Fuller DR; Liu W; Brown CJ; Laganowsky A; Russell DH; Clemmer DE Variable-Temperature ESI-IMS-MS Analysis of Myohemerythrin Reveals Ligand Losses, Unfolding, and a Non-Native Disulfide Bond. Anal. Chem. 2019, 91 (10), 6808–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Woodall DW; Henderson LW; Raab SA; Honma K; Clemmer DE Understanding the Thermal Denaturation of Myoglobin with IMS-MS: Evidence for Multiple Stable Structures and Trapped Pre-equilibrium States. J. Am. Soc. Mass Spectrom. 2021, 32 (1), 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Cassou CA; Sterling HJ; Susa AC; Williams ER Electrothermal supercharging in mass spectrometry and tandem mass spectrometry of native proteins. Anal. Chem. 2013, 85 (1), 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Loo JA; Loo RRO; Udseth HR; Edmonds CG; Smith RD Solvent-induced conformational changes of polypeptides probed by electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1991, 5 (3), 101–105. [DOI] [PubMed] [Google Scholar]
- (73).Mirza UA; Cohen SL; Chait BT Heat-induced conformational changes in proteins studied by electrospray ionization mass spectrometry. Anal. Chem. 1993, 65 (1), 1–6. [DOI] [PubMed] [Google Scholar]
- (74).Benesch JL; Sobott F; Robinson CV Thermal dissociation of multimeric protein complexes by using nanoelectrospray mass spectrometry. Anal. Chem. 2003, 75 (10), 2208–14. [DOI] [PubMed] [Google Scholar]
- (75).Wang G; Abzalimov RR; Kaltashov IA Direct Monitoring of Heat-Stressed Biopolymers with Temperature-Controlled Electro-spray Ionization Mass Spectrometry. Anal. Chem. 2011, 83 (8), 2870–2876. [DOI] [PubMed] [Google Scholar]
- (76).McLafferty FW; Guan Z; Haupts U; Wood TD; Kelleher NL Gaseous conformational structures of cytochrome c. J. Am. Chem. Soc. 1998, 120 (19), 4732–4740. [Google Scholar]
- (77).Shelimov KB; Clemmer DE; Hudgins RR; Jarrold MF Protein structure in vacuo: gas-phase conformations of BPTI and cytochrome c. J. Am. Chem. Soc. 1997, 119 (9), 2240–2248. [Google Scholar]
- (78).Suckau D; Shi Y; Beu SC; Senko MW; Quinn JP; Wampler FM; McLafferty FW Coexisting stable conformations of gaseous protein ions. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (3), 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Freitas MA; Hendrickson CL; Emmett MR; Marshall AG Gas-phase bovine ubiquitin cation conformations resolved by gas-phase hydrogen/deuterium exchange rate and extent11Dedicated to Professor Michael T. Bowers on the occasion of his 60th birthday. Int. J. Mass Spectrom. 1999, 185–187, 565–575. [Google Scholar]
- (80).Hoaglund-Hyzer CS; Counterman AE; Clemmer DE Anhydrous Protein Ions. Chem. Rev. 1999, 99 (10), 3037–3080. [DOI] [PubMed] [Google Scholar]
- (81).Gronert S. Determining the gas-phase properties and reactivities of multiply charged ions. J. Mass Spectrom. 1999, 34 (8), 787–796. [DOI] [PubMed] [Google Scholar]
- (82).Douglass KA; Venter AR Predicting the highest intensity ion in multiple charging envelopes observed for denatured proteins during electrospray ionization mass spectrometry by inspection of the amino acid sequence. Anal. Chem. 2013, 85 (17), 8212–8218. [DOI] [PubMed] [Google Scholar]
- (83).Loo RRO; Loo JA Salt Bridge Rearrangement (SaBRe) Explains the Dissociation Behavior of Noncovalent Complexes. J. Am. Soc. Mass Spectrom. 2016, 27 (6), 975–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Stirnemann G; Kang S -g.; Zhou, R.; Berne, B. J. How force unfolding differs from chemical denaturation. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (9), 3413–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Covey TR; Bonner RF; Shushan BI; Henion J; Boyd R. The determination of protein, oligonucleotide and peptide molecular weights by ion-spray mass spectrometry. Rapid Commun. Mass Spectrom. 1988, 2 (11), 249–256. [DOI] [PubMed] [Google Scholar]
- (86).Schnier PD; Gross DS; Williams ER Electrostatic forces and dielectric polarizability of multiply protonated gas-phase cytochrome c ions probed by ion/molecule chemistry. J. Am. Chem. Soc. 1995, 117 (25), 6747–6757. [Google Scholar]
- (87).Merenbloom SI; Flick TG; Daly MP; Williams ER Effects of select anions from the Hofmeister series on the gas-phase conformations of protein ions measured with traveling-wave ion mobility spectrometry/mass spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22 (11), 1978–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Freeke J; Robinson CV; Ruotolo BT Residual counter ions can stabilise a large protein complex in the gas phase. Int. J. Mass Spectrom. 2010, 298 (1–3), 91–98. [Google Scholar]
- (89).Wysocki VH; Tsaprailis G; Smith LL; Breci LA Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 2000, 35 (12), 1399–1406. [DOI] [PubMed] [Google Scholar]
- (90).Ruotolo BT; Benesch JLP; Sandercock AM; Hyung S-J; Robinson CV Ion mobility–mass spectrometry analysis of large protein complexes. Nat. Protoc. 2008, 3 (7), 1139–1152. [DOI] [PubMed] [Google Scholar]
- (91).Falconer I. Corpuscles, Electrons and Cathode Rays JJ. Thomson and the ‘Discovery of the Electron’. Br. J. Hist. Sci. 1987, 20 (3), 241–276. [Google Scholar]