

Abstract
Background:
Inflammatory cascades following traumatic brain injury (TBI) can have both beneficial and detrimental effects on recovery. Single biomarker studies do not adequately reflect the major arms of immunity and their relationships to long-term outcomes. Thus, we applied treelet transform (TT) analysis to identify clusters of interrelated inflammatory markers reflecting major components of systemic immune function for which substantial variation exists among individuals with moderate-to-severe TBI.
Methods:
Serial blood samples from 221 adults with moderate-to-severe TBI were collected over 1–6 months post-injury (n = 607 samples). Samples were assayed for 33 inflammatory markers using Millipore multiplex technology. TT was applied to standardized mean biomarker values generated to identify latent patterns of correlated markers. Treelet clusters (TC) were characterized by biomarkers related to adaptive immunity (TC1), innate immunity (TC2), soluble molecules (TC3), allergy immunity (TC4), and chemokines (TC5). For each TC, a score was generated as the linear combination of standardized biomarker concentrations and cluster load for each individual in the cohort. Ordinal logistic or linear regression was used to test associations between TC scores and 6- and 12-month Glasgow Outcome Scale (GOS), Disability Rating Scale (DRS), and covariates.
Results:
When adjusting for clinical covariates, TC5 was significantly associated with 6-month GOS (odds ratio, OR = 1.44; p-value, p = 0.025) and 6-month DRS scores (OR = 1.46; p = 0.013). TC5 relationships were attenuated when including all TC scores in the model (GOS: OR = 1.29, p = 0.163; DRS: OR = 1.33, p = 0.100). When adjusting for all TC scores and covariates, only TC3 was associated with 6- and 12-month GOS (OR = 1.32, p = 0.041; OR = 1.39, p = 0.002) and also 6- and 12-month DRS (OR = 1.38, p = 0.016; OR = 1.58, p = 0.0002). When applying TT to inflammation markers significantly associated with 6-month GOS, multivariate modeling confirmed that TC3 remained significantly associated with GOS. Biomarker cluster membership remained consistent between the GOS-specific dendrogram and overall dendrogram.
Conclusions:
TT effectively characterized chronic, systemic immunity among a cohort of individuals with moderate-to-severe TBI. We posit that chronic chemokine levels are effector molecules propagating cellular immune dysfunction, while chronic soluble receptors are inflammatory damage readouts perpetuated, in part, by persistent dysfunctional cellular immunity to impact neuro-recovery.
Keywords: Adaptive immunity, Biomarker, Chemokine, Cytokine, Glasgow outcome scale, Inflammation, Innate immunity, Soluble receptor, Traumatic brain injury, Treelet transform analysis
1. Introduction
Traumatic brain injury (TBI) results in both a systemic and neuroinflammatory response that impacts global recovery and increases risk for secondary conditions (Kochanek et al., 2008; Kumar et al., 2016, 2015). The inflammatory cascades following neurological injury have both beneficial and detrimental effects on recovery (Correale and Villa, 2004; Ziebell and Morganti-Kossmann, 2010). Several experimental models have assessed anti-inflammatory drugs, including ibuprofen and minocycline, for their ability to mitigate cellular immune damage, producing inconclusive results (Browne et al., 2006; Bye et al., 2007), as cellular immunity was minimally reduced with treatment. Clinical studies often attribute perturbations of a single inflammatory marker as contributing to outcomes or secondary conditions resulting from the TBI (Stein et al., 2011; Hergenroeder et al., 2010; Hayakata et al., 2004; Kirchhoff et al., 2008; Singhal et al., 2002). However, these studies do not consider inflammatory signaling pathways or contextualize the contributions of variable arms of immunity to post-injury sequelae.
Previous studies have linked acute neuroinflammation to TBI severity (Singhal et al., 2002; Hensler et al., 2002). The central nervous system’s (CNS) innate immune response primarily involves microglia, which release inflammatory mediators in response to blood–brain barrier (BBB) disruption. Microglial activation and polarization shape functional states and responses based on local environment cues. Classic responses include pro-inflammatory signaling, chemokine-mediated cell trafficking, phagocytic activity, and repair and remodeling processes (Ebert et al., 2005; Loane and Kumar, 2016). Cellular responses to inflammatory signaling have been modeled with respect to tissue integrity and immune resolution (Vaughan et al., 2018). Reactive astrocytes also release damage-associated molecular patterns (Thelin et al., 2017; Wang et al., 2018), in addition to interleukin (IL)-6, which contribute to cerebrovascular instability (Hariri et al., 1994; Lau and Yu, 2001). CNS cell mitochondrial dysfunction also activates neuroinflammation, including inflammasome activation (Suliman et al., 2016; Gong et al., 2018), in response to cellular injury and the innate immune responses that trigger neuronal death (Mortezaee et al., 2018).
BBB disruption occurs rapidly post-TBI and results in systemic exposure to infiltrating CNS antigens (Chodobski et al., 2011; Morganti-Kossmann et al., 2007). Recent identification of the astrocyte-regulated glymphatic system (Rasmussen et al., 2018) suggests that, along with BBB breach, antigenic exposure from CNS-derived debris likely promotes an early systemic immune response to TBI (Louveau et al., 2015; Yang et al., 2017). Systemic inflammation is perpetuated by both the sympathetic and parasympathetic nervous systems (Elenkov et al., 2000), via cytokine- and chemokine-driven communication networks, which further activate and mobilize cellular and humoral immunity (Hazeldine et al., 2015).
Injury-induced systemic immune system activation is followed by immuno-suppression occurring, in part, via catecholamine activity and nicotinic α7 mediated cholinergic activity (Hazeldine et al., 2015). Hypothalamic-pituitary adrenal (HPA) axis mediated glucocorticoid release may further influence immunosuppression. Our previous work shows acute cerebrospinal fluid (CSF) cortisol effects on neuroinflammation may have a divergent detrimental role on outcome, by exacerbating inflammation in some and contributing to immunoparalysis in others (Santarsieri et al., 2014).
The systemic exposure to CNS antigens that occurs after a post-injury BBB breach (Chodobski et al., 2011) also initiates and regulates the adaptive immune response. After spinal cord injury (SCI) (Riegger et al., 2009), adaptive immune response is suppressed, and many individuals experience lymphopenia following the aseptic, acute inflammatory response (Jeong et al., 2013). Similarly, profound systemic immune system deficits, such as impaired T-cell function and exacerbated inflammatory and reactive oxygen species production, contribute to persistent immunological disruption and hinder homeostatic restoration and tissue recovery after TBI and stroke (Hazeldine et al., 2015; Shim and Wong, 2016).
Our previous research indicates that lymphopenia also occurs with TBI and is linked to longer hospital length-of-stay and worse long-term outcomes (Calcagno et al., 2018). Suppressed immune responses post-injury can facilitate infections and impair wound healing, negatively impacting neurological recovery (Riegger et al., 2009; Calcagno et al., 2018). Recent knowledge suggests a complex humoral triad of CNS and systemic signaling, both cellular and humoral, that communicates via sympathetic nervous system and HPA axis signaling. This triad influences and is influenced by inflammatory feedback loops and other humoral signaling markers, including hormones and neurotrophins, in the context of TBI (Wagner and Kumar, 2019). Central to this point is evidence for central-peripheral immune system crosstalk leading to a post-acute immune-related prospectus rooted in cellular signaling regulation (Thelin et al., 2017).
Our group has done seminal work in a moderate-to-severe TBI cohort in characterizing chronic serum inflammatory markers elevations and their association with global outcome (Kumar et al., 2015). We demonstrated how inflammatory load scores can broadly measure outcome-sensitive, systemic inflammation. We also identified acute CSF inflammatory markers using principal component analysis (PCA) followed by cluster analysis to distinguish individual inflammatory profiles among those with unfavorable outcomes (Kumar et al., 2016). PCA allowed us to create numeric representations of correlated variable patterns (components) (Bryant and Yarnold, 1995), which were then used to cluster individuals.
Based on this literature and our own work, we hypothesized that serum inflammatory markers serve as quantifiable readouts of systemic immunity dynamics, BBB integrity and ongoing CNS antigenic exposure, and neuroinflammatory effects on systemic immunity during chronic recovery. Also, investigating signaling molecules requires contextualization with respect to their membrane-bound or soluble receptors, target cells, or cellular byproducts, to determine the multiplicity of roles associated with inflammatory markers. For example, Tumor Necrosis Factor (TNF)-α is a multifaceted inflammatory marker best understood for its interactions with TNF receptors 1 (TNFR1, p55) and 2 (TNFR2, p75). Both soluble forms of TNFRs are truncated extracellular domains of the surface receptors (Cope et al., 1995) formed via proteolytic cleavage upon activation by TNFα (Suvannavejh et al., 2000); therefore, soluble receptors in circulation arise as byproducts of their respective signaling pathways. We also hypothesized that persistent innate immune activation and adaptive immune system suppression contribute to dysfunctional chronic inflammatory states post-injury that affect outcome.
We applied treelet transform (TT) analysis, which leverages both PCA and cluster analysis, to aggregate and quantify inflammatory data into distinct clusters of correlated biomarkers that contribute to biomarker variation in our cohort. TT derived clusters were used to delineate how TBI affected various arms of immunity during the first 6 months post-injury. We used multivariable regression to assess associations of treelet cluster (TC) scores with age, injury and clinical factors, as well as global outcomes. This report features TT as a primary analytic tool for its dimension reduction and clustering capacity in order to make inferences about domains of immunity and their impacts on individuals with moderate-to-severe TBI during the early-chronic recovery phase. We demonstrate how TT was used to identify distinct and interpretable clusters of inflammatory markers relevant to TBI outcomes.
2. Methods
2.1. Study design, population description and sample collection
The University of Pittsburgh Institutional Review Board (IRB) approved this study. Informed consent was provided by next-of-kin for participants with moderate-to-severe TBI who survived their acute injury. Participants whose cognitive status improved sufficiently over the course of the study were given the opportunity to self-consent. Uninjured, control volunteers self-consented to provide blood samples to derive reference values for biomarkers measured. This prospective cohort study included 221 individuals, recruited from either acute admission or inpatient rehabilitation at the University of Pittsburgh Medical Center (UPMC) as a part of a larger study evaluating biomarkers after TBI. Fig. 1 summarizes data availability within this study cohort. We included men and women with moderate-to-severe TBI, aged 17–78 years, from whom blood samples (n = 607) were collected monthly over the first six months post-injury. Samples were collected on a monthly schedule (within a 2-week window) to the extent possible. Our clinical coordinators conducted home follow-up visits within a 2-week window of each monthly timepoint for each participant to collect a blood sample. These samples were drawn at each monthly visit unless participants refused blood draw, were unable to provide a sample, were lost to follow-up, or died. A portion of patients were also enrolled after the first month blood draw window (n = 61), thus missing that data point. After blood collection, samples were rested for ~ 30 min, stored at 4 degrees C for transport and then centrifuged at 2500 RPM for ten minutes at room temperature. Supernatant was aliquoted into 0.5 mL tubes, and immediately stored at −80 degreesC until batch analysis. On average, 3.23 sample timepoints were collected per study participant.
Fig. 1. Study CONSORT Diagram.
Month 1–6 inflammatory input data was available for the 33 markers of interest in a cohort of n = 185 individuals with moderate to severe TBI. Treelet transform analysis was conducted with these individuals’ inflammatory data. A subset of these individuals were survivors at six months available and assessed for long-term recovery assessments (GOS and DRS).
2.2. Demographic and clinical variables
Relevant demographic and clinical variables were collected through personal interview and medical record review. Variables collected include: age, sex, body mass index (BMI), Glasgow Coma Scale (GCS) score, Injury Severity Scale (ISS) score (both head and non-head regions), mechanism of injury, hospital length-of-stay, and acute care computed tomography (CT) findings. Injury types from acute care CT reports were obtained via medical chart review for multiple, pre-specified lesion types. Intra-axial lesions included intra-ventricular hemorrhage (IVH), intra-parenchymal hemorrhage (IPH), and contusions. Extra-axial lesions included subdural hemorrhage (SDH), subarachnoid hemorrhage (SAH), and epidural hemorrhage (EDH). Data was also abstracted on diffuse axonal injury (DAI) and midline shift. The number of lesions identified on CT scan for the three sub-categories of intra-axial hemorrhages was not mutually exclusive, as an individual could have more than one lesion type. ISS assesses overall anatomical trauma severity (Baker et al., 1974) and is calculated based on the Abbreviated Injury Scale (AIS), an established scoring system sub-divided into six body regions: head/neck, face, chest, abdomen, extremities/pelvis, and external. The ISS is the sum of the squared, highest AIS score of the three most severely injured regions—ranging from 0 to 75 (Baker et al., 1974). We report ISS non-head scores, which are calculated from the top three most severely injured regions, excluding the head/neck (Baker et al., 1974; Copes et al., 1988). The GCS is a commonly used neurological injury severity scale in the clinical setting and is routinely used as an inclusion criterion for TBI clinical trials (Robertson et al., 2014; Wright et al., 2014; Meythaler et al., 2002) and for clinical cohort biomarker and genomics (Santarsieri et al.., 2014; Munoz et al., 2017; Garringer et al., 2013; Diamond et al., 2015; Kumar et al., 2018) studies inclusion criterion. The measure objectively assesses consciousness post-TBI via verbal, eye opening and motor response (Teasdale et al., 1979). The best GCS score collected within the first 24 h of admission was used for analysis.
2.3. Inflammatory marker Luminex Bead assay
Thirty-three inflammatory biomarkers were measured in (n = 607 samples) using a Luminex™ bead array assay (Millipore, Billerica, Massachusetts), requiring a total of ~ 35 μl serum to run. These multiplex assays used microsphere technology where assay beads were tagged with various fluorescent-labeled markers. The protein binding onto the multiplex bead was analyzed with a fluorescence detection laser optic system. The Human High Sensitivity T-cell Magnetic Bead Panel included IL-10, IL-12p70, IL-13, IL17A, IL-1β, IL-2, IL-21, IL-4, IL-23, IL-5, IL-6, IL-7, IL-8, Macrophage Inflammatory Protein (MIP)-1α, MIP-1β, TNF-α, Fractalkine, Granulocyte Macrophage Colony Stimulating Factor (GM-CSF), Interferon-inducible T-cell alpha chemoattractant (ITAC), and Interferon (IFN)-γ. The intra-assay coefficient of variance (CV) was < 5%. The inter-assay CV was < 20%. The Human Neurodegenerative Disease Magnetic Bead included soluble Intracellular Adhesion Molecule (sICAM)-1, Regulated upon Activation, Normal T-cell Expressed and Secreted (RANTES), Neural Cell Adhesion Molecule (NCAM), and soluble Vascular Adhesion Molecule (sVCAM)-1. The intra-assay CV was < 6%. The inter-assay CV was < 13%. The Human Soluble Cytokine Receptor Magnetic Bead Panel included soluble (s)CD30, soluble glycoprotein (sgp)130, soluble interleukin-1 receptor (sIL-1R)-I, sIL-1RII, sIL-2α, sIL-4R, sIL-6R, soluble tumor necrosis factor receptor 1 (sTNFRI), and sTNFRII. The intra-assay CV was < 10% while inter-assay CV was < 15%. Of note, sIL-1RI assayed poorly for 68% of samples. Thus, this marker was omitted from analysis. All other inflammatory markers assayed were quantifiable in n = 185 individuals for ~ 3.23 sample timepoints per individual over the first 6 months post-injury.
2.4. Six- and twelve-month outcome assessment
Individuals in this cohort were interviewed at 6 and 12 months post-injury to assess global recovery using the Glasgow Outcome Scale (GOS) and Disability Rating Scale (DRS). GOS and DRS scores are commonly used study endpoints for both TBI clinical trials (Giacino et al., 2012; Meythaler et al., 2002; Robertson et al., 2014; Wright et al., 2014) and biomarker and genomics cohort (Wagner et al., 2011; Goyal et al., 2013; Myrga et al., 2016; Barton et al., 2016) research. GOS measures global, neurological recovery using a semi-structured interview and ranges from 1 to 5. The scores on this scale are defined as: 1) dead, 2) vegetative state, 3) severe disability, 4) moderate disability, and 5) good recovery (Jennett and Bond, 1975). For analyses, we grouped individuals into two categories: 1) unfavorable outcome (GOS = 2–3) and 2) favorable outcome (GOS = 4–5). The DRS similarly measures global recovery, via items assessing basic neurological function, cognitive capacity to direct activities of daily living, and capacity for more complex independent activities of daily living (e.g. employment). DRS scores range from 0 to 30, with lower scores corresponding to little/no disability, higher scores corresponding to more disability, and a score of 30 indicating death (Rappaport et al., 1982). DRS scores were categorized for analyses by severity: 0–4 (partial to no disability), 4–14 (moderate or severe disability), 15–29 (extreme severe disability, vegetative state). One participant, deceased at 6 months, was excluded from analysis.
2.5. Statistical analysis
Descriptive statistics, including median, inter-quartile range (IQR), mean, and standard error of the mean (SE), were computed for continuous variables. Univariate binary logistic, ordinal logistic, or linear regression models were used, as applicable, to assess associations to demographic and clinical factors. Pearson correlations measured strength and direction of associations between two continuous variables. Frequencies and percentages were calculated for categorical variables, and the Chi-square test (or Fisher’s exact test as appropriate) was used to identify group differences between these variables. Average values were computed for each biomarker assessed in samples collected 1–6 months post-injury; these values were then standardized to a mean of 0 and standard deviation of 1 prior to TT analysis. Biomarker patterns were analyzed in two stages: 1) TT and 2) bivariate comparisons between the TC scores derived from stage 1 and other demographic, clinical, injury, and outcome variables. Multivariable binary or ordinal logistic regressions were then performed, as applicable, to assess the association of TC scores with outcome. Statistical analyses were performed using SAS 9.4 (Cary, North Carolina) and Stata 15.0 (College Station, Texas).
2.5.1. Treelet transform methods
TT was applied to standardized inflammatory biomarker data to identify latent patterns of correlated markers. TT was conducted using the tt add-on for Stata. The TT process and methods have been described previously (Gorst-Rasmussen, 2012; Gorst-Rasmussen et al., 2011). TT is a linear dimension-reduction methodology that conducts PCA within a hierarchal cluster analysis (Gorst-Rasmussen, 2012) and requires complete data for all input variables (i.e. month 1–6 means for all 33 biomarkers).
TT produces clusters of variables based on their correlation structure. TT joins the two most highly correlated variables by local PCA and replaces these variables with the resulting sum variable (the first principal component). This process is repeated for all remaining and local PCA-based sum variables until all have joined, or branched, into a single sum variable. TT requires a selection of 1) the number of components (≥2 input variables preferred to retain a component) and 2) a cut-level at which components cluster. The ideal cut-level maximized should explain variance across 5 Monte Carlo (MC) simulations of 10-fold cross-validation over 100 bootstrap samples for the previously determined number of components. The defined cut-level maximizes variance capture of components while maintaining interpretability and sparseness of cluster membership. Biomarkers in the branches left of the cut-level are given a cluster assignment and a loading value, while TT uniquely assigns loadings of 0 to markers beyond the cluster. TT results are summarized in a dendrogram, with the highest correlated variables branching first. Mean cross-validation scores across 100 bootstrap samples in the 5-component treelet were plotted by cut-level to identify a range of viable values (Fig. 2). A cut-level of 16 was used for the final model.
Fig. 2. Cross-Validation Score Across 100 Bootstrap Iterations of Treelet Cross-Validation.
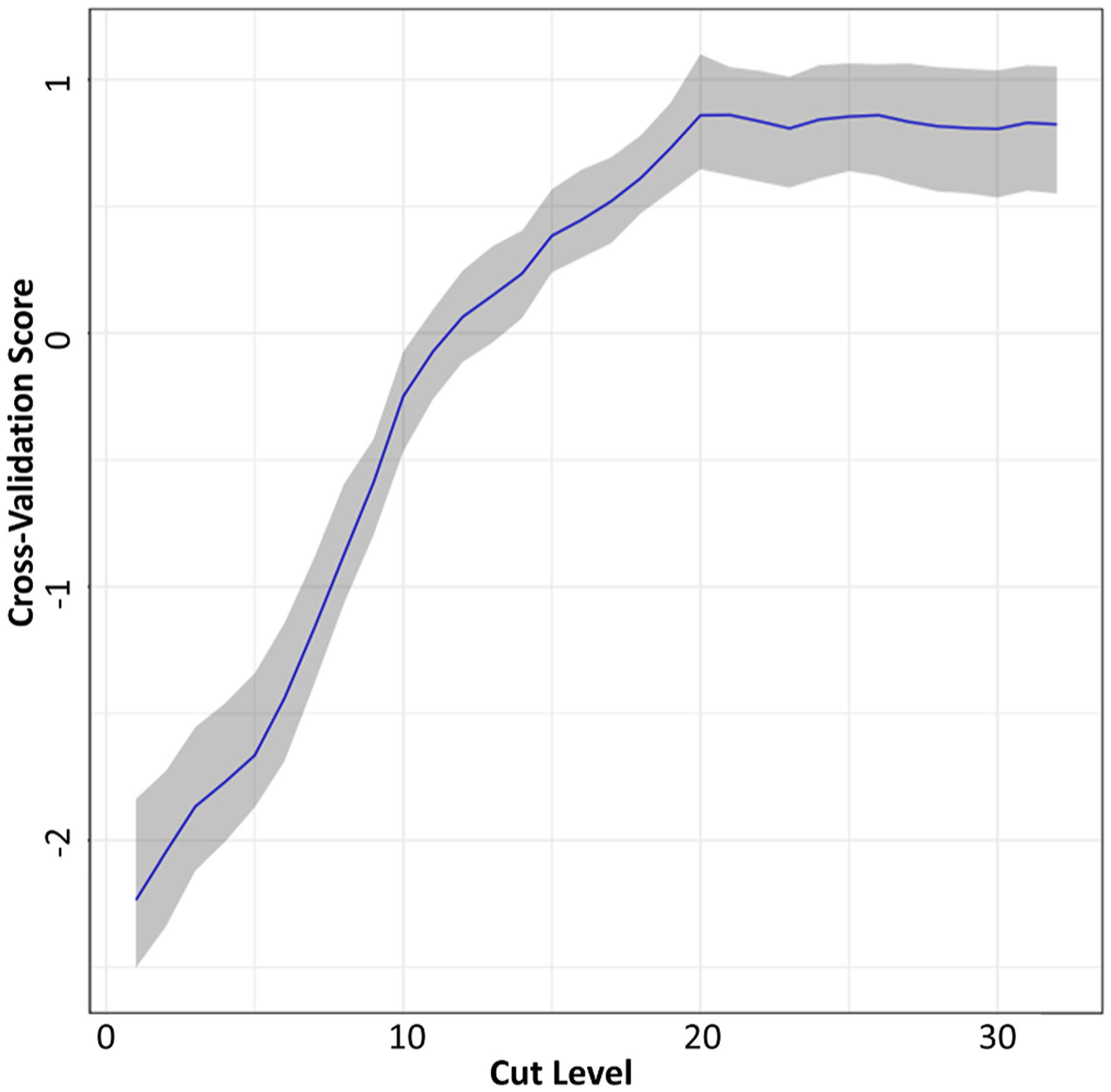
Treelet transform cross-validation was carried out for 5 component treelet transform, in 100 bootstrap samples of the analytic cohort (80% sampled with replacement). Cross-validation scores within each iteration were standardized, and the mean cross-validation score (across 100 bootstrap iterations) plotted across cut-off levels. The blue line represents the standardized mean score by cut-level and gray shaded area indicates the 95% confidence interval. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
TT assigns each clustered marker a quantitative contribution or loading to its respective cluster. For each retained cluster, or TC, a score was generated as the linear combination of standardized inflammatory marker concentration and the markers’ respective loading value. TC scores represent an aggregate of inflammatory levels for markers in each TC, with higher TC scores indicating higher levels of a TC’s inflammatory markers and adherence to a particular cluster pattern. In simulations, TT was more suitable than PCA for sparse data with small sample sizes, where the algorithm correctly joined together the subsets of variables with a sample size as little as n = 100 (Lee et al., 2008). While we did not test power or minimal sample size, we do provide results that assess the stability of the dimension reduction through another bootstrap resampling to observe cluster stability across TT iterations. We then integrated these aggregate measures of immunity into our analyses to explore TC scores (i.e. immunity domain) associations with clinical and injury severity scores (GCS, ISS, and ISS non-head), demographic characteristics, and global outcome measures (GOS and DRS) to determine how chronic immune dysfunction impacts TBI recovery.
As TT analysis is not designed for repeated measures data, we averaged inflammatory marker levels over the 6-month sampling period for each subject prior to analysis. Prior to averaging inflammatory data values for samples collected over the first six months post injury for the purposes of conducting treelet transformation analyses, average values for each marker measured were graphed to evaluate temporal dynamics over the 6-month sampling period. We observed minimal temporal dynamics for each inflammatory marker at the cohort level, particularly after the first month post-TBI. In fact, markers closely followed a zero-order pattern over the time-course averaged. This preliminary work (data not shown) provided justification for averaging values six-month time period.
2.5.2. Bivariate and multivariable analyses
Bivariate analyses were conducted to identify relationships among and between TC scores and demographic, injury, and global outcome variables. Pearson correlation coefficients and univariate linear regression models were assessed between TC scores and continuous variables including age, sex, GCS score, ISS, and ISS non-head scores. Univariate binary logistic regressions were conducted to observe associations between each TC score and 6- and 12-month dichotomized GOS outcome groups. Univariate ordinal logistic regressions were conducted to observe associations between each TC score and 6- and 12-month categorized DRS. We also performed age, sex, and GCS adjusted multivariable models with and without other TC scores for all outcomes. We checked for possible multicollinearity using the variance inflation factor (VIF) for each model. A VIF < 10 was deemed as normal range (Kutner and Nachtsheim, 2005). The proportional odds assumption was also investigated for all ordinal logistic regression models (data not shown).
3. Results
3.1. Demographic and clinical characteristics
Table 1 presents demographic information describing our cohort. The median age was 31 years, and the majority of the population included Caucasian men. Motor vehicle and motorcycle accidents and falls accounted for the majority of TBI causes. Of the 185 participants with inflammatory data, CT data was also available for 165 patients. One hundred thirty-two individuals had CT intra-axial hemorrhages, including IVH, IPH, and contusions; and, 150 individuals had extra-axial hemorrhages, including SDH, SAH, and EDH.
Table 1.
Study Cohort Demographic and Clinical Characteristics (n = 185).
| Variable | Median | IQR |
|---|---|---|
| Age (Years) | 31 | 23–48 |
| BMI (kg/m2) | 25.85 | 22.8–29.3 |
| GCS score (Best in 24 h) | 7 | 6–10 |
| ISS score (Head/neck) | 29 | 22–36 |
| ISS score (Non-head) | 9 | 4–18 |
| Length of Stay in Hospital (Days) | 19 | 12–28 |
| n | % | |
| Sex, Male | 144 | 77.8 |
| Race, Caucasian | 166 | 89.7 |
| Mechanism of Injury | n | % |
| MVA | 85 | 45.9 |
| Motorcycle | 34 | 18.4 |
| Fall | 41 | 22.2 |
| Violent/Gunshot Wound | 4 | 2.1 |
| Sports-related | 7 | 3.8 |
| Other | 6 | 3.2 |
| Unknown | 8 | 4.3 |
| CT Injury Type | n | % |
| Intra-axial Hemorrhage | 132 | 71.4 |
| IVH | 53 | 28.6 |
| IPH | 87 | 47.0 |
| Contusion | 101 | 54.6 |
| Extra-axial Hemorrhage | 150 | 81.1 |
| SDH | 116 | 62.7 |
| SAH | 117 | 63.2 |
| EDH | 29 | 15.7 |
| Diffuse Axonal Injury | 20 | 10.8 |
| Midline Shift | 55 | 29.7 |
| Unknown | 20 | 10.8 |
3.2. Treelet transform dendrogram
The TT analysis was applied to 33 inflammatory biomarkers from n = 185 individuals. The cut-level was set by examining cross-validation scores, which compared multiple possible cut-levels that could be adopted (Fig. 2 and Fig. 3).
Fig. 3. Treelet Dendrogram.
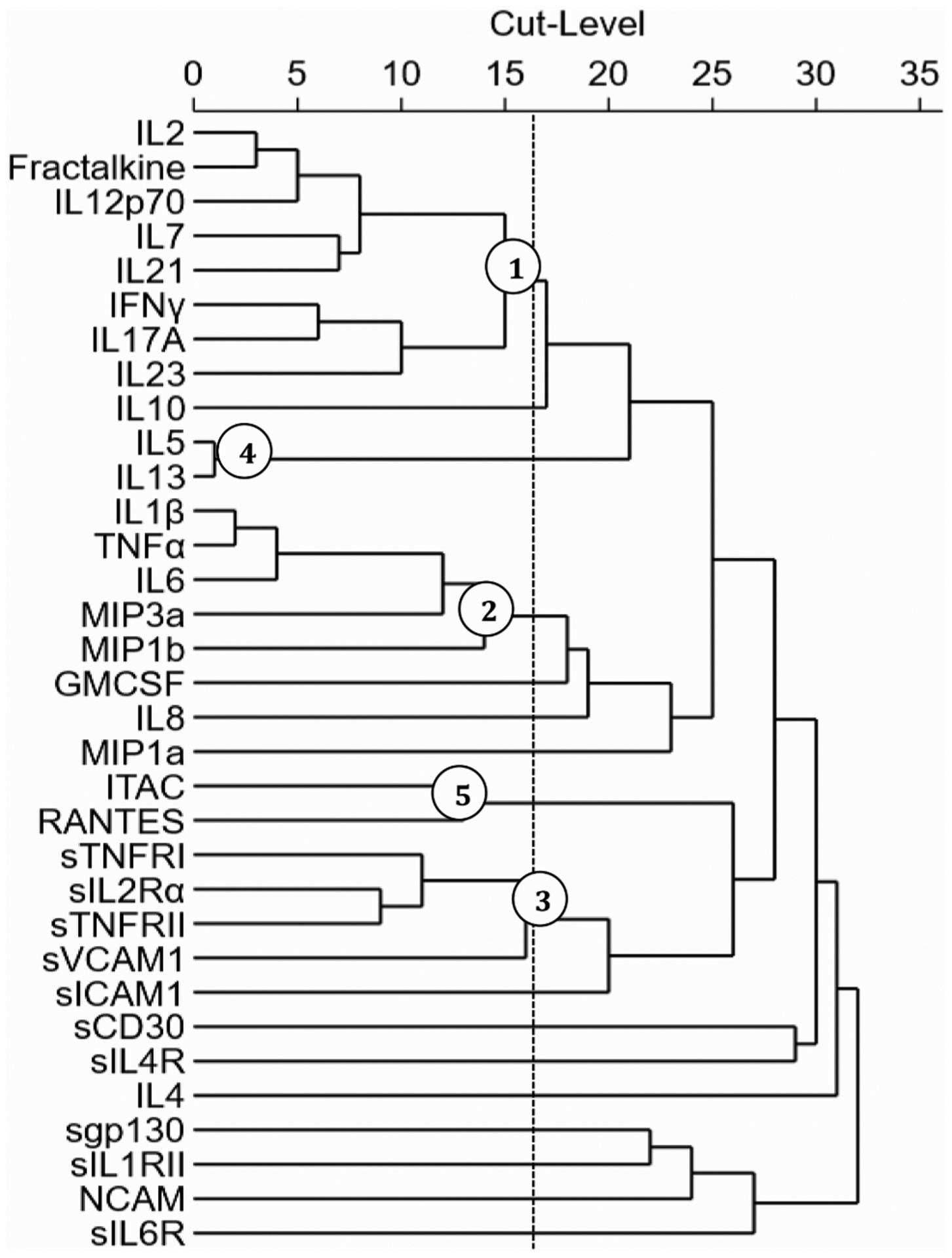
The treelet cluster analysis created a Dendrogram for 33 inflammatory markers. The dotted vertical line depicts the cut-level used for the analysis (16), in which the last branch before the line contains a significant cluster (designated by circled node) of inflammatory markers.
Table 2 indicates the proportion of variance explained by each cluster and loadings, which signify contribution by each inflammatory marker to the given component. The five clusters capture a cumulative 43.64% of total variability with inflammation levels among individuals. The variance captured by each cluster are as follows: TC1 (14.66%), TC2 (10.45%), TC3 (7.84%), TC4 (5.92%), and TC5 (4.77%). The greatest contributors to cluster formation were: IL-2 and Fractalkine (TC1), IL-1β and TNF-α (TC2), sIL-2Rα and sTNFRII (TC3), while the markers of TC4 (IL-5, IL-13) and TC5 (ITAC, RANTES) had equal load contribution. Fig. 3 provides a correlation matrix dendrogram for the biologically justifiable cut-level of 16 established with five hierarchical components of the markers ranked by the proportion of variance contributed. The clusters represent the highest branching nodes to the left of the optimal cut-level.
Table 2.
Percent Variance Captured of Inflammatory Markers to Respective Treelet Cluster.
| Adaptive Immunity (14.66%) | Innate Immunity (10.45%) | Soluble Molecules (7.84%) | Allergy Immunity (5.92%) | Chemokines (4.77%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| TC1 | Load | TC2 | Load | TC3 | Load | TC4 | Load | TC5 | Load |
| IL-2 | 0.3818 | IL-1β | 0.4923 | sTNFRI | 0.4953 | IL-5 | 0.7071 | ITAC | 0.7071 |
| Fractalkine | 0.3818 | TNF-α | 0.4923 | sIL-2Rα | 0.5313 | IL-13 | 0.7071 | RANTES | 0.7071 |
| IL-12p70 | 0.3689 | IL-6 | 0.4678 | sTNFRII | 0.5313 | ||||
| IL-7 | 0.3581 | MIP3α | 0.3947 | sVCAM-1 | 0.4361 | ||||
| IL-21 | 0.3581 | MIP1β | 0.3750 | ||||||
| IFN-γ | 0.3318 | ||||||||
| IL-17A | 0.3318 | ||||||||
| IL-23 | 0.3105 | ||||||||
3.2.1. Biological contextualization of clusters
Based on these results, and the established literature regarding the inflammatory markers that clustered (Table 3), we adopted the following cluster nomenclature to reference TCs as representing specific domains of immunity. TC1 represents markers associated with adaptive immunity including IL-2, Fractalkine, IL-12p70, IL-7, IL-21, IFNγ, IL-23, and IL-17A (Feghali and Wright, 1997; Lundström et al., 2012; Needham et al., 2019; Noack and Miossec, 2014; Rancan et al., 2004; Schroder et al., 2004). TC2 is reflective of innate immunity and includes markers IL-1β, TNFα, IL-6, MIP-1β, and MIP-3α (Ziebell and Morganti-Kossmann, 2010; Feghali and Wright, 1997; Needham et al., 2019; Keane et al.., 2006; McKee and Lukens, 2016; Woodcock and Morganti-Kossmann, 2013). Soluble molecules, including sTNFRI, sTNFRII, sIL-2Rα and sVCAM-1, clustered into TC3 (Feghali and Wright, 1997; McKee and Lukens, 2016; Sedger and McDermott, 2014). The allergy cluster (TC4) included IL-5 and IL-13 (Feghali and Wright, 1997; Noack and Miossec, 2014; Galli et al., 2008). Lastly, the chemokine cluster (TC5) included ITAC and RANTES (Appay and Rowland-Jones, 2001; Huang et al., 2000; Mohan et al., 2002; Springer, 1994).
Table 3.
Biological Contextualization of Clusters.
| Treelet Cluster | Biological Context | References |
|---|---|---|
|
Adaptive Immunity (TC1)
L-2, Fractalkine, IL-12p70, IL-7, IL-21, IFNγ, IL-23, IL- 17A |
|
(Feghali and Wright, 1997; Lundström et al., 2012; Needham et al., 2019; Noack and Miossec, 2014; Rancan et al., 2004; Schroder et al., 2004) |
|
Innate Immunity (TC2) IL-1β, TNF-α, IL-6, MIP-1β, and MIP-3α |
|
(Ziebell and Morganti-Kossmann, 2010; Feghali and Wright, 1997; Needham et al., 2019; Keane et al.:1–5., 2006; McKee and Lukens, 2016; Woodcock and Morganti-Kossmann, 2013) |
|
Soluble Molecules (TC3) sTNFRI, sTNFRII, sIL2Rα, sVCAM-1 |
|
(Feghali and Wright, 1997; McKee and Lukens, 2016; Sedger and McDermott, 2014) |
|
Allergy Immunity (TC4) IL-5, IL-13 |
|
(Feghali and Wright, 1997; Noack and Miossec, 2014; Galli et al., 2008) |
|
Chemokines (TC5) ITAC, RANTES |
|
(Appay and Rowland-Jones, 2001; Huang et al., 2000; Mohan et al., 2002; Springer, 1994) |
3.2.2. Treelet stability assessment
A bootstrapping method was used as a sensitivity analysis to assess the stability of TC membership with respect to the biomarkers included in each TC. The results for this bootstrapping method are presented in Table 4. These results justify the exclusions in identified TCs of adjacent inflammatory markers that appear less frequently (<20% of iterations) due to the clear drop off in clustering percentages among markers. Clustered inflammatory markers appeared in at least 63% of the total iterations, with the exception of sVCAM-1, which had a clustering rate of 41.7%. Due to biological relevance to other inflammatory markers in TC3 in representing soluble molecule biology, we concluded that sVCAM-1 inclusion was justifiable at this cut-level. These relatively high inflammatory marker inclusion rates, evident from the bootstrapping technique, support adequate cluster stability for use in subsequent analyses.
Table 4.
Treelet Cluster Stability Assessment.
| Adaptive Immunity | Innate Immunity | Soluble Molecules | Allergy Immunity | Chemokines | |||||
|---|---|---|---|---|---|---|---|---|---|
| TC1 | % | TC2 | % | TC3 | % | TC4 | % | TC5 | % |
| IL-2 | 83.0 | IL-1β | 85.2 | sTNFRI | 94.0 | IL-5 | 96.1 | ITAC | 80.6 |
| Fractalkine | 83.0 | TNFα | 85.2 | sIL-2Rα | 94.0 | IL-13 | 96.1 | RANTES | 80.6 |
| IL-12p70 | 83.0 | IL-6 | 85.2 | sTNFRII | 94.0 | ||||
| IL-7 | 83.0 | MIP1β | 78.6 | sVCAM-1 | 41.7 | ||||
| IL-21 | 83.0 | MIP3α | 77.3 | ||||||
| IFNγ | 70.2 | ||||||||
| IL-23 | 70.2 | ||||||||
| IL-17A | 63.7 | ||||||||
| IL-10 | 15.4 | GM-CSF | 16.0 | - | - | IFN-γ | 10.2 | IL-8 | 7.2 |
| IL-4 | 13.6 | IL-8 | 5.7 | - | - | IL-17A | 10.2 | - | - |
| - | - | - | - | - | - | IL-23 | 10.2 | - | - |
A 1000-fold iterative bootstrap of treelet transform performed with 5 clusters and a cut-level of 16 yielded sufficiently stable cluster formation. That is all markers contained in the final model appeared at least 60% of all bootstrapping iterations, with the exception of sVCAM-1 (41.7%) for which inclusion was justified based on biological relevance to other cluster variables. Markers below the solid line were excluded from the final treelet model due to low clustering percentages. Event rate for which an inflammatory marker was contained by a particular cluster is denoted by the clustering percentage (%).
3.3. Pearson correlations between TC scores
Fig. 4 depicts all significant relationships based on correlations between the TC scores. We visually depicted how the different arms of immunity rendered by the TT are linked or related. TC1 had a moderately large association and significant relationship with TC2 (r = 0.33, p < 0.001) and TC4 (r = 0.37, p < 0.001). TC1 also had a small but significant association with TC3 (p = 0.029) and TC5 (p = 0.036). Further, TC2 had a moderately strong correlation coefficient that was associated with TC5 (r = 0.29, p < 0.001). TC2 also had a small but significant association with TC3 (p = 0.037). Lastly, TC3 had a moderately strong correlation with TC5 (r = 0.39, p < 0.001). These results indicate how certain components of immunity post-injury are biologically interrelated, and that they may have at least some degree of statistical collinearity.
Fig. 4. Pearson Correlations Between TC Scores.
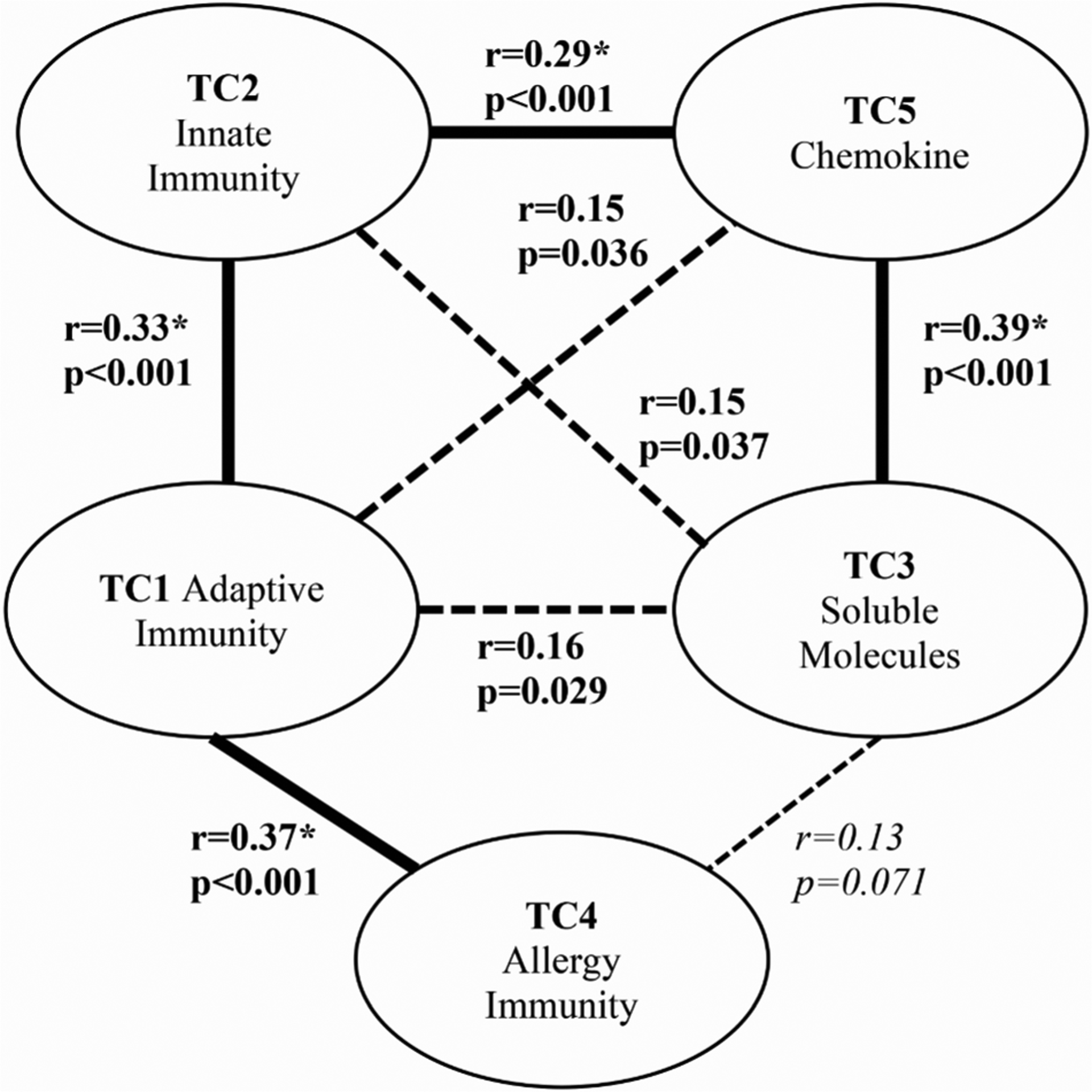
Correlations significant at the 10% level (p < 0.1) between TCs are shown. Lines between the cluster domains of immunity, are bolded if the relationship is moderately strong (r > 0.25) or dashed if the relationship is expressed to a weaker degree (p ≤ 0.1; r < 0.25). Non-significant relationships are not shown.
3.4. TC score associations with demographic and injury variables
Univariate linear regression models illustrate associations between TC scores and demographic variables (Table 5).
Table 5.
Univariate Linear Regression-Based Associations Between TC Scores and Demographic and Injury Characteristics.
| Beta (SE) p-value | TC1 Score Adaptive | TC2 Score Innate | TC3 Score Soluble Receptors | TC4 Score Allergy | TC5 Score Chemokine |
|---|---|---|---|---|---|
| Age | −0.012 (0.009) | 0.004 (0.008) | 0.029 (0.007) | −0.007 (0.001) | −0.001 (0.001) |
| n = 185 | 0.236 | 0.631 | <0.001 | 0.304 | 0.831 |
| Sex | −0.496 (0.389) | −0.026 (0.330) | −0.156 (0.285) | 0.215 (0.248) | −0.148 (0.223) |
| n = 185 | 0.204 | 0.937 | 0.585 | 0.387 | 0.506 |
| GCS Score | −0.096 (0.048) | −0.101 (0.042) | 0.034 (0.037) | −0.007 (0.031) | −0.056 (0.028) |
| n = 174 | 0.050 | 0.018 | 0.361 | 0.810 | 0.049 |
| ISS Score | 0.015 (0.015) | 0.031 (0.015) | 0.011 (0.012) | −0.017 (0.009) | 0.038 (0.009) |
| n = 147 | 0.314 | 0.037 | 0.355 | 0.079 | <0.001 |
| ISS Non-Head Score | −0.011 (0.021) | 0.001 (0.020) | 0.012 (0.017) | −0.0156 (0.013) | 0.052 (0.013) |
| n = 119 | 0.597 | 0.977 | 0.496 | 0.254 | <0.001 |
Significant associations (p < 0.05) are bolded and trend level associations (p < 0.1) italicized.
The results indicate that older age was associated with higher TC3 scores (β = 0.029, p < 0.001). Lower GCS scores were associated with higher TC1 (β = −0.096, p = 0.05), TC2 (β = −0.101, p = 0.018), and TC5 (β = −0.056, p = 0.049) scores. These findings indicate that the worse (lower) GCS score, the higher the adaptive, innate, and chemokine burden. Higher ISS scores were also associated with higher TC2 (innate) (β = 0.031, p = 0.037) and TC5 (chemokine) (β = 0.038, p < 0.001), while ISS non-head scores were only associated with higher TC5 (chemokine) scores (β = 0.052, p = <0.001).
3.5. TC score associations to GOS scores
We performed binary logistic regressions to model the relationships between each TC score and GOS scores, at 6 and 12 months post-injury (Table 6). Results indicate that for every unit increase in TC5 score, there were 56.9% higher odds for worse 6-month outcomes (p = 0.006). For each unit increase in TC2 (p = 0.051) and TC3 (p = 0.062), results indicate trend level associations wherein there were 23% and 22% increased odds of unfavorable GOS outcomes respectively. At 12 months post-injury, each unit increase in TC2 and TC3 was associated with 22.7% and 24.3% higher TC2 (p = 0.034) and TC3 (p = 0.046) scores respectively.
Table 6.
Associations between TC Scores and 6- and 12-month GOS Scores.
| Treelet Cluster Score | Unfavorable 6-month GOS Score | Unfavorable 12-month GOS Score | ||
|---|---|---|---|---|
| OR [95% CI] | p-value | OR [95% CI] | p-value | |
| TC1: Adaptive | 1.07 [0.92, 1.23] | 0.411 | 1.02 [0.87, 1.19] | 0.843 |
| TC2: Innate | 1.23 [1.0, 1.48] | 0.051 | 1.23 [1.02, 1.48] | 0.034 |
| TC3: Soluble Molecules | 1.22 [0.99, 1.51] | 0.062 | 1.24 [1.0, 1.54] | 0.046 |
| TC4: Allergy | 0.85 [0.64, 1.14] | 0.2806 | 0.92 [0.70, 1.21] | 0.554 |
| TC5: Chemokines | 1.57 [1.14, 2.16] | 0.006 | 1.19 [0.92, 1.54] | 0.195 |
Each line represents a single univariate logistic regression model of unfavorable dichotomized GOS score (2,3). Significant associations (p < 0.05) are bolded and trend level associations italicized (p < 0.1).
3.6. TC score associations to DRS scores
Similarly, individual ordinal regressions were performed to model the associations of TC scores and categorized DRS at 6 and 12 months post-TBI (Table 7). One unit increase in the TC3 (p = 0.034) and TC5 (p = 0.002) scores was associated with a 25% and 60% increase, respectively, in the odds of being in the higher DRS category (worse outcome) at 6 months post-TBI. Higher TC2 scores showed trend level associations (p = 0.072) with higher (worse) DRS categories at 6 months. Proportional odds assumptions were met for all 6-month models. At month 12, only TC3 was associated (p = 0.024) with worse DRS; indicating that for every unit increase in TC3 scores, there is a 27% increased odds of a higher DRS score. Proportional odds assumptions were also met for all 12-month models.
Table 7.
Associations between TC Scores and 6- and 12-month DRS Scores.
| Treelet Cluster Score | 6-month DRS Score | 12-month DRS Score | ||
|---|---|---|---|---|
| OR [95% CI] | p-value | OR [95% CI] | p-value | |
| TC1: Adaptive | 1.01 [0.87, 1.18] | 0.891 | 1.01 [0.87, 1.18] | 0.861 |
| TC2: Innate | 1.14 [0.99, 1.31] | 0.072 | 1.11 [0.97, 1.28] | 0.131 |
| TC3: Soluble Molecules | 1.25 [1.02, 1.54] | 0.034 | 1.27 [1.03, 1.57] | 0.024 |
| TC4: Allergy | 0.87 [0.65, 1.16] | 0.345 | 1.01 [0.8, 1.28] | 0.923 |
| TC5: Chemokines | 1.60 [1.19, 2.15] | 0.002 | 1.11 [0.85, 1.44] | 0.441 |
Each line represents a single univariate ordinal logistic regression model of categorized DRS: 0–3 (partial to no disability), 4–14 (moderate or severe disability), 15–29 (extreme severe disability, vegetative state). Significant associations (p < 0.05) are bolded and trend level associations italicized (p < 0.1).
3.7. Multivariable models of outcome metrics
TC score correlations (Fig. 4), and the association of these scores with different demographic and clinical characteristics (Table 5), indicate the potential for confounding and interaction effects among variables. Thus 6- and 12-month binary GOS and 6- and 12-month categorized DRS regressions were developed using age, sex, and GCS with one TC score taken at a time. All TC scores were considered together in a full model (Tables 8–11). We tested for all possible 2-way interactions (data not shown) for each outcome, and none were significant in the full model. Proportional odds assumptions were met for all multivariable models.
Table 8.
Multivariable models for associations between TC Scores and 6-month GOS Scores.
| Variable OR p-value | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | Model 6 |
|---|---|---|---|---|---|---|
| Age | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 0.99 |
| 0.831 | 0.952 | 0.689 | 0.928 | 0.938 | 0.451 | |
| Gender | 0.70 | 0.69 | 0.75 | 0.70 | 0.75 | 0.76 |
| Male vs. Female | 0.432 | 0.403 | 0.515 | 0.427 | 0.533 | 0.562 |
| GCS Score | 0.77 | 0.78 | 0.76 | 0.77 | 0.78 | 0.77 |
| Best in 24 h | p < 0.001 | 0.001 | p < 0.001 | p < 0.001 | 0.001 | 0.001 |
| TC1: Adaptive | 1.0 | 0.98 | ||||
| 0.987 | 0.837 | |||||
| TC2: Innate | 1.12 | 1.03 | ||||
| 0.264 | 0.787 | |||||
| TC3: Soluble Molecules | 1.33 | 1.32 | ||||
| 0.022 | 0.041 | |||||
| TC4: Allergy | 0.85 | 0.80 | ||||
| 0.31 | 0.251 | |||||
| TC5: Chemokines | 1.44 | 1.29 | ||||
| 0.025 | 0.163 |
Each model represents a multivariable logistic regression of unfavorable dichotomized GOS score (2,3) adjusting for all covariates. Significant associations (p < 0.05) are bolded.
Table 11.
Multivariable models for associations between TC Scores and 12-month DRS Scores.
| Variable OR p-value | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | Model 6 |
|---|---|---|---|---|---|---|
| Age | 0.99 | 0.99 | 0.98 | 0.99 | 0.99 | 0.97 |
| 0.321 | 0.284 | 0.108 | 0.336 | 0.313 | 0.061 | |
| Gender | 0.71 | 0.69 | 0.72 | 0.71 | 0.71 | 0.71 |
| Male vs. Female | 0.423 | 0.394 | 0.45 | 0.419 | 0.429 | 0.442 |
| GCS Score | 0.84 | 0.86 | 0.83 | 0.85 | 0.85 | 0.82 |
| Best in 24 h | 0.014 | 0.027 | 0.01 | 0.016 | 0.017 | 0.01 |
| TC1: Adaptive | 0.98 | 0.89 | ||||
| 0.755 | 0.282 | |||||
| TC2: Innate | 1.08 | 1.10 | ||||
| 0.302 | 0.252 | |||||
| TC3: Soluble Molecules | 1.46 | 1.58 | ||||
| 0.003 | 0.002 | |||||
| TC4: Allergy | 1.02 | 1.0 | ||||
| 0.858 | 0.987 | |||||
| TC5: Chemokines | 1.061 | 0.872 | ||||
| 0.676 | 0.402 |
Each model represents a multivariable ordinal logistic regression of categorized DRS: 0–4 (partial to no disability), 4–14 (moderate or severe disability), 15–29 (extreme severe disability, vegetative state), adjusting for all covariates. Significant associations (p < 0.05) are bolded.
3.7.1. GOS score
After adjusting for covariates, higher TC3 (Model 3, Table 8) and TC5 (Model 5, Table 8) were associated with 6-month unfavorable GOS. One unit increase in TC3 was associated (p = 0.022) with 32.7% and one unit increase in TC5 was associated (p = 0.025) with 44.4% increased odds of 6-month unfavorable GOS. However, in the fully adjusted model (Model 6, Table 8) only TC3 remained significantly associated (p = 0.041) with GOS. Although TC scores and covariates were somewhat correlated, the VIFs were within normal range and all < 2 (Age: 1.22, GCS: 1.17, TC1: 1.38, TC2: 1.27, TC3: 1.27, TC4: 1.23, TC5: 1.21), suggesting that multi-collinearity is not present to a substantial degree in this model.
Higher TC2 scores (Model 2, Table 9) had a marginal association (OR = 1.17, p = 0.096) with 12-month unfavorable GOS scores. However, one unit increase in TC3 scores (Model 3, Table 9) was associated with 34.7% (OR = 1.35, p = 0.015) higher odds of 12-month unfavorable GOS. In the fully adjusted model (Model 6, Table 9), only TC3 was significant (p = 0.017); one unit increase in TC3 was associated with 38.5% higher odds of 12-month unfavorable GOS. GCS was significantly associated with both 6- and 12-month GOS in all models.
Table 9.
Multivariable models for associations between TC Scores and 12-month GOS Scores.
| Variable OR p-value | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | Model 6 |
|---|---|---|---|---|---|---|
| Age | 1.0 | 1.0 | 0.99 | 1.0 | 1.0 | 0.90 |
| 0.884 | 0.742 | 0.543 | 0.872 | 0.826 | 0.36 | |
| Gender | 0.59 | 0.57 | 0.60 | 0.60 | 0.60 | 0.57 |
| Male vs. Female | 0.226 | 0.204 | 0.26 | 0.239 | 0.24 | 0.215 |
| GCS Score | 0.83 | 0.85 | 0.82 | 0.83 | 0.84 | 0.83 |
| Best in 24 h | 0.008 | 0.024 | 0.006 | 0.008 | 0.011 | 0.014 |
| TC1: Adaptive | 0.98 | 0.91 | ||||
| 0.815 | 0.374 | |||||
| TC2: Innate | 1.17 | 1.18 | ||||
| 0.096 | 0.122 | |||||
| TC3: Soluble Molecules | 1.347 | 1.39 | ||||
| 0.015 | 0.017 | |||||
| TC4: Allergy | 0.94 | 0.94 | ||||
| 0.643 | 0.713 | |||||
| TC5: Chemokines | 1.139 | 0.964 | ||||
| 0.361 | 0.813 |
Each model represents a multivariable logistic regression of unfavorable dichotomized GOS score (2,3) adjusting for all covariates. Significant associations (p < 0.05) are bolded and trend level associations italicized (p < 0.1).
3.7.2. DRS score
After adjusting for covariates, one unit increase in TC3 (Model 3, Table 10) and TC5 (Model 5, Table 10) showed a 37.4% (p = 0.008) and 45.9% (p = 0.013) higher odds respectively of worse 6-month DRS scores. In the fully adjusted model (Model 6, Table 10), only TC3 was significant (p = 0.016); one unit increase in TC3 was associated with 37.2% higher odds of worse 6-month DRS scores. TC5 was marginally significant (p = 0.1); one unit increase in TC5 was associated with 32.6% higher odds of having worse 6-month DRS scores. At 12 months, only TC3 (Model 3, Table 11) was associated with DRS (OR = 1.46, p = 0.003), alone and in the fully adjusted model (Model 6, Table 11). One unit increase in TC3 was associated (p = 0.002) with 57.5% higher odds of having worse 12-month DRS scores. Proportional odds assumptions were met for all models presented in Tables 10 and 11. GCS was significantly associated with both 6- and 12-month DRS in all models.
Table 10.
Multivariable models for associations between TC Scores and 6-month DRS Scores.
| Variable OR p-value | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | Model 6 |
|---|---|---|---|---|---|---|
| Age | 1.01 | 1.0 | 1.0 | 1.0 | 1.0 | 0.99 |
| 0.688 | 0.755 | 0.775 | 0.76 | 0.917 | 0.502 | |
| Gender | 0.72 | 0.72 | 0.77 | 0.74 | 0.84 | 0.80 |
| Male vs. Female | 0.448 | 0.45 | 0.544 | 0.482 | 0.687 | 0.619 |
| GCS Score | 0.76 | 0.77 | 0.75 | 0.76 | 0.77 | 0.76 |
| Best in 24 h | p < 0.001 | 0.001 | p < 0.001 | p < 0.001 | 0.001 | p < 0.001 |
| TC1: Adaptive | 0.96 | 0.91 | ||||
| 0.587 | 0.347 | |||||
| TC2: Innate | 1.07 | 1.01 | ||||
| 0.362 | 0.875 | |||||
| TC3: Soluble Molecules | 1.37 | 1.37 | ||||
| 0.008 | 0.016 | |||||
| TC4: Allergy | 0.88 | 0.87 | ||||
| 0.402 | 0.446 | |||||
| TC5: Chemokines | 1.459 | 1.326 | ||||
| 0.013 | 0.100 |
Each model represents a multivariable ordinal logistic regression of categorized DRS: 0–4 (partial to no disability), 4–14 (moderate or severe disability), 15–29 (extreme severe disability, vegetative state), adjusting for all covariates. Significant associations (p < 0.05) are bolded and trend level associations italicized (p < 0.1).
3.7.3. Post-hoc assessment of age and GCS score interaction
Associations presented in Table 5 indicate that TC3 was associated with age (p < 0.001) but not GCS (p = 0.361). When age and GCS were taken together, there was a trend level significant interaction (p = 0.058) with TC3 (Table 12a). No similar effects were found with TC5. The increasing slopes of TC3 versus age at higher GCS levels (Table 12b) suggests that at older ages and higher GCS scores, the rates of change in TC3 levels were higher than the TC5 level rate of change. This relationship explains why TC3 levels had a stronger association with 6-month outcome than TC5. TC3 was also correlated with TC5 (r = 0.39, Fig. 4) and TC3 possibly had a confounding effect on TC5 when both variables were included in this model, despite VIFs < 2.
Table 12a.
Effects of age and GCS score on TC3 (soluble molecules) and TC5 (chemokines).
| Variable | Outcome: TC3 | Outcome: TC5 | ||
|---|---|---|---|---|
| Beta (SE) | p-value | Beta (SE) | p-value | |
| Age | −0.006 (0.020) | 0.746 | 0.018 (0.016) | 0.263 |
| GCS | −0.159 (0.087) | 0.070 | 0.004 (0.071) | 0.953 |
| Age × GCS | 0.004 (0.002) | 0.058 | −0.002 (0.002) | 0.301 |
Table 12b.
TC3-age relationship at different values of GCS.
| GCS level | dTC3/dAge | Delta-method SE | p-value |
|---|---|---|---|
| 3 | 0.005 | 0.014 | 0.706 |
| 6 | 0.017 | 0.010 | 0.075 |
| 9 | 0.029 | 0.008 | <0.001 |
| 12 | 0.041 | 0.010 | <0.001 |
| 15 | 0.053 | 0.015 | <0.001 |
Each line represents the slopes, standard error (using Delta method), and p-value of TC3 vs. age, at GCS = 3, 6, 9, 12, 15. Significant slopes (p < 0.05) are bolded and trend level significant slopes italicized (p < 0.1).
3.8. An exemplar: treelet transform analysis of GOS-specific markers
Cluster scores provide a robust approach from which to investigate aggregated inflammatory relationships to outcome metrics. However, markers that do not cluster may still individually track to the outcome of interest and are therefore not captured in this analysis. Also, markers that cluster but do not individually track to the outcome of interest, may attenuate the strength of outcome relationships at the cluster level. To this end, we produced a GOS-specific treelet exemplar to address these caveats.
The treelet dendrogram presented in Section 3.2 represents the correlation structure of all 33 inflammatory markers studied and the associated contribution to overall variance by each significant cluster. However, 12 inflammatory markers are associated (p < 0.1) with 6-month GOS scores (Table 13).
Table 13.
Associations Between Inflammatory 1–6 Month Standardized Means and Dichotomized 6-month GOS group Using Univariate Logistic Regression.
| Standardized Month 1–6 Mean | Unfavorable 6-month GOS Score | Full Treelet (33 markers) Cluster Membership | |
|---|---|---|---|
| OR | p-value | ||
| IL-7 | 1.54 | 0.0146 | TC1: Adaptive |
| IL-21 | 1.49 | 0.0181 | TC1: Adaptive |
| IL-1b | 1.32 | 0.0908 | TC2: Innate |
| MIP-3a | 1.71 | 0.0355 | TC2: Innate |
| MIP-1b | 1.75 | 0.0326 | TC2: Innate |
| sIL-2Ra | 1.45 | 0.0500 | TC3: Soluble Molecules |
| sTNFRII | 1.64 | 0.0111 | TC3: Soluble Molecules |
| ITAC | 1.63 | 0.0277 | TC5: Chemokines |
| RANTES | 1.71 | 0.0089 | TC5: Chemokines |
| MIP-1a | 2.15 | 0.0022 | DNC |
| sICAM-1 | 1.54 | 0.0187 | DNC |
| sIL-4R | 0.60 | 0.0150 | DNC |
| *DNC: Did not cluster | |||
Only markers indicating a relationship to GOS at a threshold of p < 0.1 are reported. All markers, except sIL-4R, show increased odds of unfavorable outcome with higher expression. TC membership, based on the TT dendrogram generated from all 33 markers, is reported for reference in order to assess independent contribution of markers from each cluster in outcome distinction.
We used this subset of variables (IL-7, IL-21, IL-1β, MIP-3α, MIP-1β, ITAC, RANTES, sIL-2Rα, sTNFRII, MIP-1α, sICAM-1, sIL-4R) to generate a GOS-specific treelet dendrogram (Fig. 5).
Fig. 5. Treelet Dendrogram of Inflammatory Markers Specific to 6-month GOS Score.
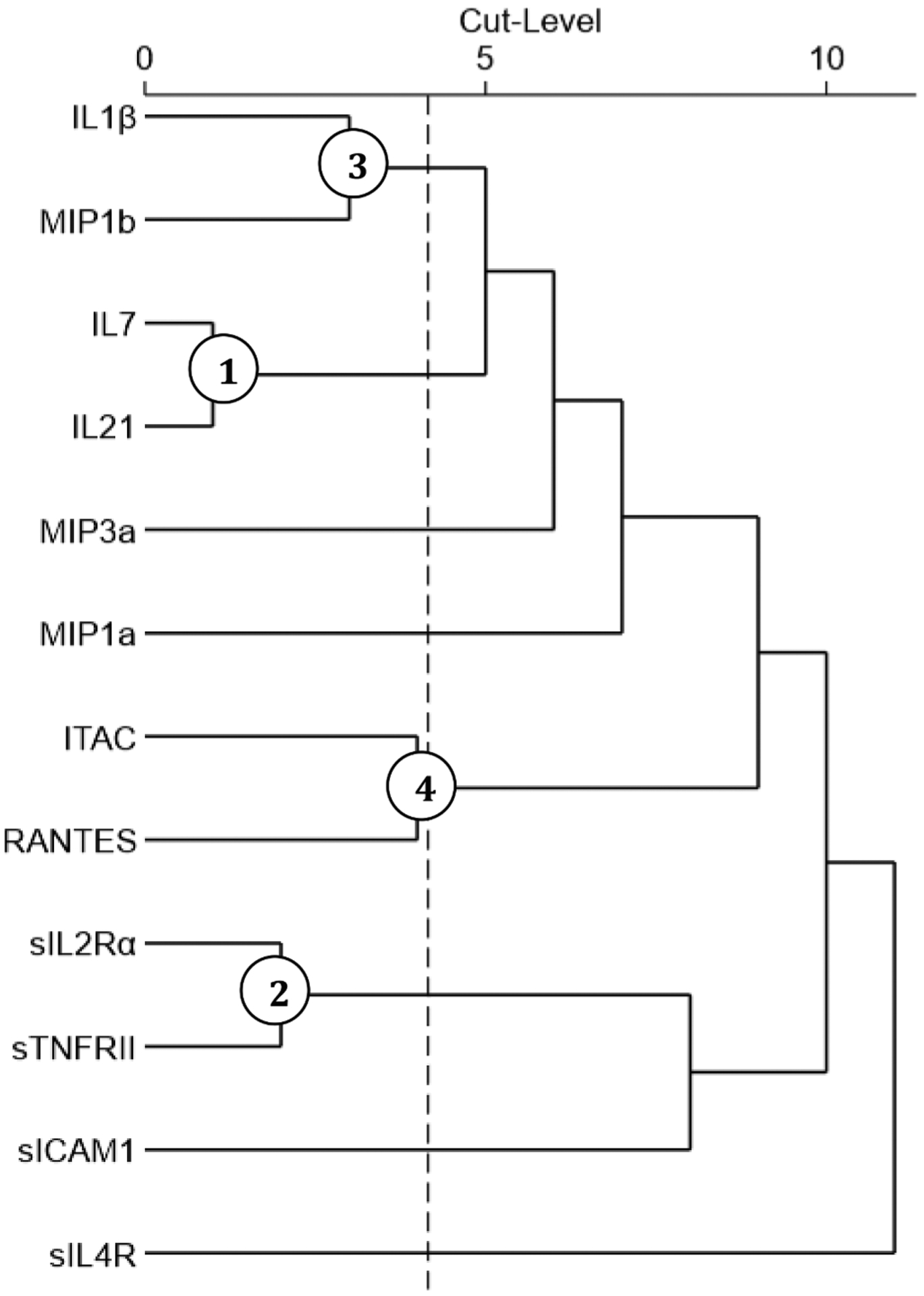
TT yielded the following correlation matrix for the subset of 12 inflammatory marker means associated (p < 0.1) with GOS outcome group. The dotted line depicts the optimal cut-level (4) and circled nodes represent significant clusters ranked by contribution to overall variance capture.
As described in Section 3.2, a 100-iteration cross validation process was used to determine the chosen cut point by assessing the frequency ofoptimal cut point ranges. The cut-level chosen was 4, and this TT yielded the following clusters and their respective contribution to percent variance captured: TC1GOS: IL-7 and IL-21 (14.69%), TC2GOS: sIL-2Rα and sTNFRII (14.05%), TC3GOS: IL-1β and MIP-1β (13.85%), and TC4GOS: ITAC and RANTES (13.13%). Together these clusters explained a total 55.73% of the variability among individuals’ GOS-related inflammatory marker profiles. All markers contribute equally to their respective clusters with loading values of 0.7071. TC1GOS was associated with GCS and ISS (extracranial), TC2GOS with age, TC3GOS with GCS, and TC4GOS with GCS, ISS (extracranial), and ISS (non-head) (Table 14). Individual logistic regressions showed a significant association of TC1GOS (OR = 1.36, p = 0.0129), TC2GOS (OR = 1.42, p = 0.0153), TC3GOS (OR = 1.35, p = 0.0390), and TC4GOS (OR = 1.57, p = 0.0058) with 6-month GOS (Table 15).
Table 14.
Univariate Linear Regression-Based Associations Between 6-month GOS-Specific TC Scores (TCGOS) and Demographic and Injury Characteristics.
| Beta (SE) p-value | TC1GOS Score Adaptive IL-7, IL-21 | TC2GOS Score Soluble Molecules sIL-2Ra, sTNFRII | TC3GOS Score Innate IL-1b, MIP-1b | TC4GOS Score Chemokine ITAC, RANTES |
|---|---|---|---|---|
| Age | −0.0036 (0.01) | 0.0202 (0.01) | −0.0016 (0.01) | −0.0012 (0.01) |
| n = 185 | 0.5532 | 0.0005 | 0.7828 | 0.8309 |
| Sex | −0.1973 (0.24) | −0.2112 (0.23) | 0.0482 (0.23) | −0.1484 (0.22) |
| n = 185 | 0.4026 | 0.3596 | 0.8334 | 0.5058 |
| GCS Score | −0.080 (0.03) | 0.0181 (0.03) | −0.0804 (0.03) | −0.0561 (0.03) |
| n = 174 | 0.0071 | 0.5399 | 0.0065 | 0.0494 |
| ISS Score | 0.0216 (0.01) | 0.0109 (0.01) | 0.0182 (0.01) | 0.0376 (0.01) |
| n = 147 | 0.0288 | 0.2705 | 0.0745 | <0.0001 |
| ISS Non-Head Score | 0.0033 (0.01) | 0.0115 (0.01) | −0.0011 (0.01) | 0.0520 (0.01) |
| n = 119 | 0.8113 | 0.4078 | 0.9382 | 0.0001 |
Significant associations (p < 0.05) are bolded and trend level associations (p < 0.1) italicized.
Table 15.
Associations between GOS-Specific TC Scores (TCGOS) and 6-month GOS Scores.
| Treelet Cluster Score | Unfavorable 6-month GOS Score | |
|---|---|---|
| OR [95% CI] | p-value | |
| TC1GOS: Adaptive IL-7, IL-21 | 1.36 [1.07–1.74] | 0.0129 |
| TC2GOS: Soluble Molecules sIL-2Ra, sTNFRII | 1.42 [1.07–1.88] | 0.0153 |
| TC3GOS: Innate IL-1b, MIP-1b | 1.35 [1.02–1.8] | 0.0390 |
| TC4GOS: Chemokines ITAC, RANTES | 1.57 [1.14–2.16] | 0.0058 |
Each line represents a single univariate logistic regression model of unfavorable dichotomized GOS score (2,3). Significant associations (p < 0.05) are bolded and trend level associations are italicized (p < 0.1).
In the adjusted models (Table 16), the main effect of TC4GOS (OR = 1.44, p = 0.0245), and the interaction effects TC2GOS × GCS and TC3GOS × GCS, were significant (both p = 0.046) (Model 2, 3, and 4). That is, the effects on global outcome of TC2GOS (soluble molecules) and TC3GOS (innate) differ in the context of different GCS scores demonstrating how injury severity contributes to inflammatory pathophysiology post-TBI. In the fully adjusted model with all covariates and TCGOS scores (Model 6, Table 16), only the interaction TC2GOS × GCS was significant (p = 0.048); one unit increase in TC2GOS was associated with 4.41 × 0.89GCS times higher odds of 6-month unfavorable GOS. The VIFs were within the normal range (Age: 1.17, GCS: 1.18, TC1GOS: 1.62, TC2GOS: 1.21, TC3GOS: 1.5, TC4GOS: 1.25). GCS was associated with 6-month GOS in all models.
Table 16.
Multivariable models for associations between GOS-specific TC Scores and 6-month GOS Scores.
| Variable OR p-value | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 |
|---|---|---|---|---|---|
| Age | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| 0.916 | 0.798 | 0.979 | 0.938 | 0.73 | |
| Gender | 0.72 | 0.67 | 0.67 | 0.75 | 0.68 |
| Male vs. Female | 0.463 | 0.398 | 0.378 | 0.533 | 0.419 |
| GCS Score | 0.76 | 0.76 | 0.82 | 0.78 | 0.80 |
| Best in 24 h | 0.0009 | 0.001 | 0.016 | 0.0006 | 0.007 |
| TC1GOS: Adaptive IL-7, IL-21 | 1.22 | 0.98 | |||
| 0.135 | 0.896 | ||||
| TC2 GOS : Soluble Molecules sIL-2Ra, sTNFRII | 4.73 | 4.41 | |||
| 0.007 | 0.013 | ||||
| TC3GOS: Innate IL-1b, MIP-1b | 0.40 | 0.40 | |||
| 0.116 | 0.208 | ||||
| TC4 GOS : Chemokines ITAC, RANTES | 1.44 | 1.29 | |||
| 0.0245 | 0.204 | ||||
| TC2GOS × GCS | 0.89 | 0.89 | |||
| 0.046 | 0.048 | ||||
| TC3GOS × GCS | 1.21 | 1.18 | |||
| 0.046 | 0.158 |
Each model represents a multivariable logistic regression of unfavorable dichotomized GOS score (2,3) adjusting for all covariates. Significant associations (p < 0.05) are bolded and trend level associations are italicized (p < 0.1).
Performing TT on a subset of the marker panel (GOS-associated markers) allowed us to test reproducibility and stability of marker clusters from the original treelet dendrogram. We observed a similar correlation structure and an understanding about adjacent cluster pairs (i.e. adaptive and innate immunity related markers) from the larger treelet, which may have commonalities that jointly influence outcome.
4. Discussion
Our understanding of inflammation hinges on a holistic evaluation of the immune system, including its cellular functions and humoral signaling after TBI. Despite the frequency of single marker studies, an understanding of the overarching immune response to TBI and impact on recovery remains elusive. As such, attempts to reach a consensus characterization of possible “beneficial” and “detrimental” aspects of post-TBI inflammation at the individual marker level are challenging due to 1) the heterogeneous nature of the injury itself, 2) variable time windows of marker measurement, 3) timing for potential treatment intervention, and 4) population heterogeneity. This realization has motivated recent work delineating temporal and correlational patterns among inflammatory and cellular mediators to achieve a more complete characterization of the overall state of immunity post-TBI (Kumar et al., 2016; Vaughan et al., 2018; Helmy et al., 2012). We leveraged the dimension reduction technique, TT analysis to study early chronic inflammation post-TBI, to extract interpretable and related clusters of biomarkers based on co-expression patterns. This work represents the broadest immune profile characterization of systemic inflammation carried out in the field to date, and TT analysis effectively identified relevant biological function and interactions between the inflammatory biomarkers measured, which were then evaluated in the context of global recovery metrics.
4.1. Novelty of treelet transform in studying inflammation
TT has been used to explore dietary patterns and myocardial infarction risk, and more recently, metabolite profiles and prostate cancer risk (Gorst-Rasmussen et al., 2011; Schmidt et al., 2020). We hypothesized that TT would be useful in characterizing distinct inflammatory patterns associated with demographic and clinical factors, as well as TBI outcomes. TT incorporates both PCA and cluster analysis to identify underlying data patterns. TT also streamlines the interpretation of clustered biomarkers by assigning a numeric biomarker loading based on their overall contribution to a given cluster (Gorst-Rasmussen, 2012). One consideration with TT is the discretion of choosing a cut point to produce the viable clusters used in later analyses. However, Fig. 2 shows the utility of cross validation in choosing cut levels that maximize variance captured by the clusters, while retaining interpretability of clustering markers.
Among the significant TCs that emerged, they ranked in contribution to overall variance capture as follows: adaptive immunity, innate immunity, soluble molecules, allergy immunity, and chemokines. These findings highlight the importance of systemic inflammation after neurotrauma and identify biomarker groups that differ most among the TBI population and contribute to heterogeneous injury responses. Notably, the adaptive immunity cluster contributes most to the population variance.
4.2. Role for systemic immune readouts in understanding chronic neuroinflammation
We characterized peripheral inflammatory patterns as a reflective indicator of chronic TBI-induced neuroinflammation due to supporting evidence in other clinical models for systemic immune marker readouts reflecting CNS pathology and degree of functional and emotional impairment. For example, Frodl and Amico review work in major depression disorder, elderly cognitive impairment, and Alzheimer’s disease (AD) that link magnetic resonance imaging (MRI)-detected and region-specific structural brain changes, and the subsequent emotional and cognitive deficits, to peripheral markers of brain inflammation (Frodl and Amico, 2014). Both clinical and experimental models of aging and AD suggest that neural activation and functional brain connectivity are vulnerable to peripheral inflammatory-induced abnormalities (Labrenz et al., 2016; Walker et al., 2020).
There is increasing attention on the CNS-systemic interface and joint involvement in mounting and coordinating an immune response to brain injury. Recent literature suggests that modulation of the CNS and systemic inflammatory systems is regulated by the autonomic nervous system (Kenney and Ganta, 2014); however, in a state of neurological physiological disruption like brain injury, homeostasis becomes difficult to maintain. This is evidenced by the chronic manifestation of serum inflammatory derangements for months after injury (Kokiko-Cochran and Godbout, 2018; Kumar et al., 2015) and TBI-induced inflammatory burden associations with long-term sequelae like cognitive dysfunction and depression (Bodnar et al., 2018; Walker and Tesco, 2013).
We hypothesized that TCs captured groups of interrelated inflammatory molecules that reflect different arms of immunity. Our results indicated that the generated TCs were biologically relevant and mapped well to the domains of adaptive immunity, innate immunity, allergy immunity, soluble signaling molecules/receptors, and chemokines. We leveraged TC scores to inform imbalanced inflammatory states after TBI, rather than narrowly assess perturbations of a single inflammatory marker. While TC score associations with demographic and injury factors (Table 5) were diverse, variation in global outcome and disability long-term were consistently attributed to elevated innate immunity, soluble molecule, and chemokine burden (Tables 6 and 7).
4.3. Innate immunity
The innate immune response is dominated by chronic expression of IL-1β, TNF-α, and IL-6 and macrophage inducible proteins that act to propagate inflammatory activity well after the injury has occurred. We show that worse anatomical and neurological injury (i.e. higher ISS and lower GCS scores) sustains innate immunity elevations and, in turn, increases odds of unfavorable global outcome over the first-year post-injury (Table 6). This finding suggests that initial injury severity impacts acute pathophysiology and exacerbates maladaptive chronic innate immune response patterns, providing evidence that systemic inflammatory dysfunction is a common secondary condition post-TBI. Prolonged inflammation post-injury can activate the HPA axis, which when unregulated, may contribute to poor recovery, chronic stress, and depression (Bodnar et al., 2018). The innate immunity cluster was strongly correlated with the adaptive and chemokine clusters (Fig. 4), demonstrating how this non-specific component of the immune response can elicit a more specific, targeted injury repair response over time and a continued cellular immune response. The correlative nature between innate immunity and chemokines suggests early innate pathway propagation of chemokine production and unfavorable outcomes post-TBI (Ziebell and Morganti-Kossmann, 2010). The correlation between the innate and soluble molecules supports the idea that the innate immune cluster has an active role in perpetuating the overall immune response, including cellular immunity.
4.4. Chemokines and soluble receptor signaling
Once initiated, the immune response is intricately controlled by expression of cell signaling molecules that guide cell migration, proliferation, and communication via other cellular molecules. This biology was captured across two chronic inflammation TCs consisting of ITAC and RANTES (chemokines) and sTNFRI, sTNFRII, sIL-2Rα, and sVCAM-1 (soluble receptors/adhesion molecules). The correlational results show the innate cluster as associated with the chemokine cluster, which itself is associated with the soluble molecule cluster (Fig. 4). These relationships suggest a biologically relevant chain of events related to cell trafficking, adhesion, and activation that support the perpetuation of proinflammatory cascades.
Soluble receptors have a unique capacity for outcome discrimination with respect to age-related pathophysiology (Table 5 and 6), as expression increases with age and increases odds of unfavorable outcome and greater disability post-injury. This finding suggests that inflammatory marker families be studied in aggregate to assess degree to which circulating cytokines signal via membrane-bound versus soluble receptors. The study of soluble molecule biology, particularly the conditions that facilitate proteolytic cleavage, may further delineate potential mechanistic underpinnings for parent cytokine contributions to adverse outcomes (Levine, 2008).
The soluble receptor cluster was specifically associated with aging post-TBI. TNF molecules, including sTNFRI and sTNFRII, contribute to and are a product of early lymphopenia (Calcagno et al., 2018; Wagner et al., 2019), a phenomenon that may underlie impaired adaptive immune function mediated tissue repair (Suvannavejh et al., 2000). The literature suggests that harmful versus beneficial aspects of systemic immunity depend on timing post-injury and underlying demographics pre-injury (Chodobski et al., 2011; Timaru-Kast et al., 2012). Here, age-related associations with chronic soluble receptor burden pose another unique contributor to age-related risk for poor post-TBI recovery (Timaru-Kast et al., 2012).
As a post-hoc assessment of shared variance between age, GCS score, and soluble molecule expression, an interaction term between age and GCS score was included in the multivariable models (Section 3.7.3). The degree of TC3 changes with respect to age (slope: δTC3/δAge) increases with milder neurological injury severity (higher GCS score). That is, in the context of milder injuries associated with a tempered TNFα response (Woodcock and Morganti-Kossmann, 2013), age is a greater contributor to TNF soluble receptor activity (Patel and Brewer, 2008) and contributes to detrimental outcomes.
Interestingly, chronic chemokine production was associated with both initial injury severity and long-term global outcome and disability (Tables 5–7). Chemokines were the only cluster to track with non-head ISS, which may implicate their role in peripheral injury severity and repair, healing, and/or infection more so than clusters that track with neurological severity (Table 5). RANTES supports chemotaxis and T-cell expression (Mikolajczyk et al., 2016), while ITAC has potent chemoattractant properties regulated by IFNγ to recruit IL-2 activated T-cells (Cole et al., 1998). Together, this persistent chemo-attractant influence may render the chemokine TC particularly valuable in outcome discrimination capacity.
4.5. Adaptive and allergy immunity
The adaptive immunity cluster is important for prolonged immune responses involved in neuro-repair. In this context, some individuals may be better equipped to immunologically generate a lympho-reparative response after TBI than those with reduced adaptive immune function due to factors like aging and stress (Gregory et al., 2007; Lundström et al., 2012). The adaptive cluster (TC1) explains the most variance captured in the TT analysis. Yet no significant associations were observed between TC1, DRS, and GOS. This finding may be due to other inflammatory responses having a larger (and perhaps overlapping) role in terms of pathophysiology and discriminating outcome. Alternatively, adaptive immune impacts may be more demonstrable with the development of specific secondary conditions and less sensitive to broad multidimensional outcomes like GOS and DRS. Notably, the adaptive immune cluster was correlated with the innate and allergy clusters (Fig. 4).
The literature describes links between CNS-derived autoantibody production in traumatic SCI (Arevalo-Martin et al., 2018; Hergenroeder et al., 2016), which occurs, in part, due to the adaptive immune system. In particular, immunoglobulin M (IgM) antibodies exhibit protective roles: recognize self-antigen, enhance phagocytosis of damaged cells, promote tissue homeostasis, and initiate adaptive immune responses by moderating humoral cell activity and autoantibody production (Grönwall et al., 2012). Adaptive immune molecules like IL-7 have been linked to lymphoproliferative mechanisms, specifically in producing IgM autoantibodies. Also, IL-21 enhances innate immunity features like apoptosis and phagocytosis, and it influences adaptive elements like auto-regulation and differentiation of T-cells and B-cells as well as Ig production (Schluns et al., 2000; Spolski and Leonard, 2014). Our recent work shows systemic IgM autoantibody production specific to the pituitary and hypothalamus as potentially reparative, associated with multiple adaptive immune markers, and associated with reduced neuroendocrine dysfunction in men with moderate-to-severe TBI (Vijapur et al., 2020). Together, these data support the need to further study adaptive immunity and how it may facilitate neurorecovery and repair via IgM related autoimmune mechanisms.
4.6. Integrating immune domains
Based on our data, we hypothesized that failure to resolve immune system homeostasis contributes significantly to susceptibility to complications and secondary conditions associated with TBI. We proposed that these systemic inflammatory profiles provide biomarker readouts reflective of ongoing neuro-dysfunction after TBI and persistent lack of inflammatory regulation over the first 6 months post-injury. Perturbations in inflammatory signaling networks likely also impact neurotrophic and endocrine functioning in their contributions to TBI deficits (Wagner and Kumar, 2019).
Our results add to the discussion regarding the continued state of systemic inflammatory dysregulation after TBI. The literature shows ongoing microglial activation years after TBI (Norden et al., 2015) and examples of individual inflammatory marker elevations in both civilian and military populations with repetitive and moderate/severe TBI (Devoto et al., 2017; Rodney et al., 2020; Rusiecki et al., 2020). Chemokines are linked to both initial injury severity and 6-month long-term outcomes suggesting a key role for these markers on the longitudinal impacts of immune dysfunction. Soluble receptors, however, map to both 6- and 12-month recovery scores (Tables 8–11). These results may indicate that chemokines are effector molecules centered on propagating and regulating immune function, while soluble receptors are inflammatory readouts of damage that has occurred via persistent dysfunctional cellular immunity pathways.
4.7. GOS-specific treelet
The 33-marker treelet dendrogram provided a comprehensive characterization of post-TBI chronic inflammation and identified significant clusters contributing to variance in our TBI population irrespective of outcome. A post-hoc exemplar treelet was then generated for the subset of 12 inflammatory markers individually related to 6-month GOS. Similar cluster membership and associations between all TCs and global outcome were retained. However, covariate-adjusted multivariable models suggest that the chemokine cluster has an independent capacity for outcome discrimination that holds after demographic and injury variable adjustment (Table 16). After adjusting for all GOS-specific TC scores, the interaction term between soluble molecule load and GCS score (TC2xGCS) remained the only cluster-related variable significantly associated with unfavorable outcome. This suggests that initial neurological injury is a persistent contributor to systemic inflammation, particularly soluble molecule expression (sIL-2Rα, sTNFRII).
4.8. Strategies for therapeutic intervention
Assessing TC score profiles characteristic of both 1) demographic and injury groups and 2) longitudinal outcome groups provides information related to immune domains in dysfunction across the TBI recovery continuum. In addition to their descriptive and mechanistic value in characterizing early chronic immunity patterns after TBI, TC scores may support the selection of immunomodulatory agents to utilize as post-acute treatment strategies. Aside from direct therapeutic immune-agents, rehabilitation modalities like exercise could be assessed for their impact on these immune domains (Griesbach, 2011; Piao et al., 2013). When understanding how markers within these TCs covary after injury, the manipulation of one marker can be contextualized for its impact on the expression of another, an approach that may outperform single biomarkers to guide therapeutic intervention.
4.9. Limitations & future directions
This study is not without limitations. The main inflammatory kit utilized was a 21-plex T-Cell High Sensitivity array. The clusters presented here were limited to the markers present in our three multiplex assays. Therefore, T-cell related markers were well characterized and incorporated into this treelet dendrogram while other immune domains may benefit from additional marker inclusion. Furthermore, a future direction might include creating a treelet capturing immune function beyond the first 6 months post-injury and characterizing their associations with outcome. We can validate our findings by replicating this treelet methodology in an independent cohort. Utilizing this treelet methodology in another population would allow us to evaluate if/how the cluster memberships might vary and be impacted by the underlying population.
Our data suggest that neurological injury severity, as measured by the GCS in this cohort with moderate-to-severe TBI, has a strong influence on inflammatory levels across TC domains for all multivariate analyses. Our descriptive analyses suggest that those with a “best in 24-hour” GCS in the 13–15 (or milder injury range) have lower adaptive, innate, and chemokine TC scores, suggesting that future directions should explore inflammatory patterns among cohorts with true mild TBI or concussion. Among those particularly interesting to study would be cohorts of individuals who go on to have persistent chronic symptoms versus those who recover without ongoing protracted symptomatic sequelae. Also, larger cohorts with TBI of all severities may be useful for sex or age stratified treelet transformation analyses evaluating unique inflammatory patterns and subsequent associations with TBI outcomes. Survivors with moderate-to-severe TBI, individuals often have multiple injury types, making inflammatory attributions by specific injury or lesion type challenging. However, larger cohorts may have a subset of individuals with isolated injury types for which such subanalyses might be possible.
Future work should also focus on markers that did not cluster because their expression patterns are not correlated with other serum inflammatory markers in our panel. They still hold capacity for outcome discrimination (Table 13) as the markers MIP-1α, sICAM-1, and sIL-4R did not associate with GOS score at the cluster level but did associate in a bivariate comparison. There is a potential masking effect within identified clusters for dominant markers (higher cluster load) related to a particular outcome. For example, IL-7 is related to GOS score in a bivariate comparison, but the adaptive immunity cluster itself is not. Perhaps when evaluating other survivor-based outcomes (cognition, depression, etc.), we may further our understanding of cluster and non-clustered marker relationships to recovery.
Another future direction would be to examine how inflammatory clusters can help understand the underlying pathophysiology associated with secondary conditions, including persistent hypogonadotropic hypogonadism (PHH) (Vijapur et al., 2020), cognition (Milleville et al., 2020), depression (Schulz et al., 2018), epilepsy (Diamond et al., 2014), and others. Also, it would be beneficial to consider how personal biology, injury factors (GCS in particular), and acute care hospitalization characteristics (lymphopenia, acute infection status) impact these immune domains chronically.
As noted in the methods, TT analysis does not lend itself well to studying temporal biomarker dynamics, and future work may consider how to employ latent growth curve analyses (Felt et al., 2017) or group-based trajectory analyses (Kumar et al., 2015; Niyonkuru et al., 2013) to evaluate important latent individual and subgroup temporal biomarker dynamics not appreciated when graphing markers at the cohort level. Future work may include a neural network-based approach for extracting models from dynamic (temporal) data using ordinary and partial differential equations (Sun et al., 2020). In particular, given the spatio-temporal nature of the data (biomarkers sampled over different time intervals), we can identify a model that takes into account the intrinsic differential structure. Resulting models may be parameterized by using both (shallow) multilayer perceptrons and nonlinear differential terms, in order to incorporate relevant correlations between biomarkers collected at varying timepoints. Finally, methods like agent-based modeling (ABM) may also be an appropriate extension to this work to attribute inflammatory marker profiles to activated cellular immune patterns during the chronic phases of TBI recovery. ABM would utilize the temporal cytokine signaling data and logical rules grounded in scientific literature to simulate interactive networks between agents (immune cells) generating and receiving inflammatory signals and inform states changes of healing, damage, and overall recovery (Anderson and Vadigepalli, 2016; Folcik et al., 2011).
There is an emerging body of literature regarding CNS-derived exosomes that cross the BBB, serve as a source of systemic biomarkers, and may reflect a CNS environment facilitative of neural repair and/or complications after neurological injury (Manek et al., 2018; Mondello et al., 2020). In addition to inflammatory biomarker levels themselves, blood exosomes originating in the CNS may provide a more direct readout of CNS recovery state as they are expressed by brain cells directly and communicate with the periphery.
5. Conclusions
As the field shifts towards characterizing immune networks, TT analysis, as applied to biomarker data to capture inflammatory patterns, is novel. We utilized TT as a means of pattern analysis to extract inflammatory domains with respect to TBI pathology. Using TT, we uncovered underlying immune component associations with outcome and demographic factors in the form of biomarker clusters. Our results identify intersections between inflammatory patterns and long-term recovery post-TBI. Moving forward, we can use these findings as building blocks to understand immune networks post-TBI and the impact of immune modifying therapies on recovery.
Acknowledgments
This project used the UPCI Cancer Biomarkers Facility: Luminex Core Laboratory that is supported, in part, by the National Institute of Health [NIH P30CA047904]. A portion of clinical data was attained from the University of Pittsburgh Medical Center Trauma Registry. The authors would like to thank Jessa Darwin for her editorial support with this manuscript.
Funding
This work was supported, in part, by DoD W81XWH-071-0701, NIDILRR 90DP0041, & CDC R49 CCR 323155
References
- Anderson WD, Vadigepalli R, 2016. Modeling cytokine regulatory network dynamics driving neuroinflammation in central nervous system disorders. Drug Discov Today Dis Models. 19, 59–67. 10.1016/j.ddmod.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appay V, Rowland-Jones SL, 2001. RANTES: a versatile and controversial chemokine. Trends Immunol. 22 (2), 83–87. 10.1016/s1471-4906(00)01812-3. [DOI] [PubMed] [Google Scholar]
- Arevalo-Martin A, Grassner L, Garcia-Ovejero D, Paniagua-Torija B, Barroso-Garcia G, Arandilla AG, Mach O, Turrero A, Vargas E, Alcobendas M, Rosell C, Alcaraz MA, Ceruelo S, Casado R, Talavera F, Palazón R, Sanchez-Blanco N, Maier D, Esclarin A, Molina-Holgado E, 2018. Elevated autoantibodies in subacute human spinal cord injury are naturally occurring antibodies. Front. Immunol 9 10.3389/fimmu.2018.02365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker Susan.P., O’Neill Brian, Haddon William, Long William.B., 1974. The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. J. Trauma 14 (3), 187–196. [PubMed] [Google Scholar]
- Barton DJ, Kumar RG, McCullough EH, et al. , 2016. Persistent hypogonadotropic hypogonadism in men after severe traumatic brain injury: temporal hormone profiles and outcome prediction. J. Head Trauma Rehabil 10.1097/HTR.0000000000000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne KD, Iwata A, Putt ME, Smith DH, 2006. Chronic ibuprofen administration worsens cognitive outcome following traumatic brain injury in rats. Exp. Neurol 201 (2), 301–307. 10.1016/j.expneurol.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Bryant FB, Yarnold PR, 1995. Principal-components analysis and exploratory and confirmatory factor analysis. In: Reading and Understanding Multivariate Statistics. American Psychological Association, pp. 99–136. [Google Scholar]
- Bodnar CN, Morganti JM, Bachstetter AD, 2018. Depression following a traumatic brain injury: uncovering cytokine dysregulation as a pathogenic mechanism. Neural Regen. Res 13 (10), 1693–1704. 10.4103/1673-5374.238604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A, Kossmann T, Morganti-Kossmann MC, 2007. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp. Neurol 204 (1), 220–233. 10.1016/j.expneurol.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Calcagno J, Kumar R, Maza A, et al. , 2018. Lymphopenia after traumatic brain injury increases infections, hospital resource utilization, and worsens long-term outcomes. J. Neurotrauma 35. 10.1089/neu.2018.29013.abstracts. [DOI] [Google Scholar]
- Chodobski A, Zink BJ, Szmydynger-Chodobska J, 2011. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res 2 (4), 492–516. 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole KE, Strick CA, Paradis TJ, 1998. et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med 187 (12), 2009–2021. 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correale J, Villa A, 2004. The neuroprotective role of inflammation in nervous system injuries. J. Neurol 251 (11), 1304–1316. 10.1007/s00415-004-0649-z. [DOI] [PubMed] [Google Scholar]
- Cope AP, Aderka D, Wallach D, et al. , 1995. Soluble TNF receptor production by activated T lymphocytes: differential effects of acute and chronic exposure to TNF. Immunology 84 (1), 21–30. [PMC free article] [PubMed] [Google Scholar]
- Copes WS, Champion HR, Sacco WJ, Lawnick MM, Keast SL, Bain LW, 1988. The injury severity score revisited. J. Trauma 28 (1), 69–77. 10.1097/00005373-198801000-00010. [DOI] [PubMed] [Google Scholar]
- Devoto C, Arcurio L, Fetta J, Ley M, Rodney T, Kanefsky R, Gill J, 2017. Inflammation relates to chronic behavioral and neurological symptoms in military personnel with traumatic brain injuries. Cell Transplant 26 (7), 1169–1177. 10.1177/0963689717714098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond ML, Ritter AC, Failla MD, Boles JA, Conley YP, Kochanek PM, Wagner AK, 2014. IL-1β associations with posttraumatic epilepsy development: a genetics and biomarker cohort study. Epilepsia. 55 (7), 1109–1119. 10.1111/epi.12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond ML, Ritter AC, Jackson EK, Conley YP, Kochanek PM, Boison D, Wagner AK, 2015. Genetic variation in the adenosine regulatory cycle is associated with posttraumatic epilepsy development. Epilepsia. 56 (8), 1198–1206. 10.1111/epi.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert LM, Schaerli P, Moser B, 2005. Chemokine-mediated control of T cell traffic in lymphoid and peripheral tissues. Mol. Immunol 42 (7), 799–809. 10.1016/j.molimm.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES, 2000. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol. Rev 52 (4), 595–638. [PubMed] [Google Scholar]
- Feghali CA, Wright TM, 1997. Cytokines in acute and chronic inflammation. Front. Biosci 2, d12–26. 10.2741/a171. [DOI] [PubMed] [Google Scholar]
- Felt JM, Depaoli S, Tiemensma J, 2017. Latent growth curve models for biomarkers of the stress response. Front. Neurosci 11 10.3389/fnins.2017.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folcik VA, Broderick G, Mohan S, Block B, Ekbote C, Doolittle J, Khoury M, Davis L, Marsh CB, 2011. Using an agent-based model to analyze the dynamic communication network of the immune response. Theor. Biol. Med. Modell 8 (1) 10.1186/1742-4682-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodl T, Amico F, 2014. Is there an association between peripheral immune markers and structural/functional neuroimaging findings? Prog. Neuro-Psychopharmacol. Biol. Psychiatry 48, 295–303. 10.1016/j.pnpbp.2012.12.013. [DOI] [PubMed] [Google Scholar]
- Galli SJ, Tsai M, Piliponsky AM, 2008. The development of allergic inflammation. Nature 454 (7203), 445–454. 10.1038/nature07204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garringer JA, Niyonkuru C, McCullough EH, Loucks T, Dixon CE, Conley YP, Berga S, Wagner AK, 2013. Impact of aromatase genetic variation on hormone levels and global outcome after severe TBI. J. Neurotrauma 30 (16), 1415–1425. 10.1089/neu.2012.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacino JT, Whyte J, Bagiella E, Kalmar K, Childs N, Khademi A, Eifert B, Long D, Katz DI, Cho S, Yablon SA, Luther M, Hammond FM, Nordenbo A, Novak P, Mercer W, Maurer-Karattup P, Sherer M, 2012. Placebo-controlled trial of amantadine for severe traumatic brain injury. N. Engl. J. Med 366 (9), 819–826. 10.1056/NEJMoa1102609. [DOI] [PubMed] [Google Scholar]
- Gong Z, Pan J, Shen Q, Li M, Peng Y, 2018. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflammation. 15 (1), 242. 10.1186/s12974-018-1282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorst-Rasmussen A, 2012. tt: Treelet transform with Stata. Stata J. 12 (1), 130–146. [Google Scholar]
- Gorst-Rasmussen A, Dahm CC, Dethlefsen C, Scheike T, Overvad K, 2011. Exploring dietary patterns by using the treelet transform. Am. J. Epidemiol 173 (10), 1097–1104. 10.1093/aje/kwr060. [DOI] [PubMed] [Google Scholar]
- Goyal A, Failla MD, Niyonkuru C, Amin K, Fabio A, Berger RP, Wagner AK, 2013. S100b as a prognostic biomarker in outcome prediction for patients with severe traumatic brain injury. J. Neurotrauma 30 (11), 946–957. 10.1089/neu.2012.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory SG, Schmidt S, Seth P, et al. , 2007. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet 39 (9), 1083–1091. 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, 2011. Exercise after traumatic brain injury: is it a double-edged sword? PM R. 3 (6 Suppl 1), S64–72. 10.1016/j.pmrj.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Grönwall C, Vas J, Silverman GJ, 2012. Protective roles of natural IgM antibodies. Front. Immunol 3, 66. 10.3389/fimmu.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri RJ, Chang VA, Barie PS, Wang RS, Sharif SF, Ghajar JB, 1994. Traumatic injury induces interleukin-6 production by human astrocytes. Brain Res. 636 (1), 139–142. 10.1016/0006-8993(94)90188-0. [DOI] [PubMed] [Google Scholar]
- Hayakata T, Shiozaki T, Tasaki O, Ikegawa H, Inoue Y, Toshiyuki F, Hosotubo H, Kieko F, Yamashita T, Tanaka H, Shimazu T, Sugimoto H, 2004. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock 22 (2), 102–107. [DOI] [PubMed] [Google Scholar]
- Hazeldine J, Lord JM, Belli A, 2015. Traumatic brain injury and peripheral immune suppression: primer and prospectus. Front. Neurol 6, 235. 10.3389/fneur.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmy A, Antoniades CA, Guilfoyle MR, Carpenter KLH, Hutchinson PJ, Ikezu T, 2012. Principal component analysis of the cytokine and chemokine response to human traumatic brain injury. PLoS ONE 7 (6), e39677. 10.1371/journal.pone. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergenroeder GW, Moore AN, Schmitt KM, Redell JB, Dash PK, 2016. Identification of autoantibodies to glial fibrillary acidic protein in spinal cord injury patients. NeuroReport 27 (2), 90–93. 10.1097/WNR.0000000000000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensler T, Sauerland S, Bouillon B, Raum M, Rixen D, Helling H-J, Andermahr J, Neugebauer EAM, 2002. Association between injury pattern of patients with multiple injuries and circulating levels of soluble tumor necrosis factor receptors, interleukin-6 and interleukin-10, and polymorphonuclear neutrophil elastase. J. Trauma 52 (5), 962–970. [DOI] [PubMed] [Google Scholar]
- Hergenroeder GW, Moore AN, McCoy JP, Samsel L, Ward NH, Clifton GL, Dash PK, 2010. Serum IL-6: a candidate biomarker for intracranial pressure elevation following isolated traumatic brain injury. J Neuroinflammation. 7 (1) 10.1186/1742-2094-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Han Y, Rani MR, 2000. et al. Chemokines and chemokine receptors in inflammation of the nervous system: manifold roles and exquisite regulation. Immunol. Rev 177, 52–67. 10.1034/j.1600-065x.2000.17709.x. [DOI] [PubMed] [Google Scholar]
- Jennett B, Bond M, 1975. Assessment of outcome after severe brain damage. Lancet 1 (7905), 480–484. [DOI] [PubMed] [Google Scholar]
- Jeong H-K, Ji K, Min K, Joe E-H, 2013. Brain inflammation and microglia: facts and misconceptions. Exp. Neurobiol 22 (2), 59–67. 10.5607/en.2013.22.2.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane MP, Strieter RM, Belperio JA, 2006. Macrophage Inflammatory Protein. In: Laurent GJ, Shapiro SD (Eds.), Encyclopedia of Respiratory Medicine. Academic Press, pp. 1–5. 10.1016/B0-12-370879-6/00229-5. [DOI] [Google Scholar]
- Kenney MJ, Ganta CK, 2014. Autonomic nervous system and immune system interactions. In: Terjung R, (Ed.) Comprehensive Physiology. John Wiley & Sons, Inc.: 1177–1200. Accessed April 14, 2016. http://doi.wiley.com/10.1002/cphy.c130051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff C, Buhmann S, Bogner V, et al. , 2008. Cerebrospinal IL-10 concentration is elevated in non-survivors as compared to survivors after severe traumatic brain injury. Eur. J. Med. Res 13 (10), 464–468. [PubMed] [Google Scholar]
- Kochanek PM, Berger RP, Bayr H, Wagner AK, Jenkins LW, Clark RSB, 2008. Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: diagnosis, prognosis, probing mechanisms, and therapeutic decision making. CurrOpinCrit Care. 14 (2), 135–141. [DOI] [PubMed] [Google Scholar]
- Kokiko-Cochran ON, Godbout JP, 2018. The inflammatory continuum of traumatic brain injury and alzheimer’s disease. Front. Immunol 9 10.3389/fimmu.2018.00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RG, Rubin JE, Berger RP, Kochanek PM, Wagner AK, 2016. Principal components derived from CSF inflammatory profiles predict outcome in survivors after severe traumatic brain injury. Brain Behav. Immun 53, 183–193. 10.1016/j.bbi.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RG, Boles JA, Wagner AK, 2015. Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months postinjury. J Head Trauma Rehabil. 30 (6), 369–381. 10.1097/HTR.0000000000000067. [DOI] [PubMed] [Google Scholar]
- Kumar RG, Diamond ML, Boles JA, Berger RP, Tisherman SA, Kochanek PM, Wagner AK, 2015. Acute CSF interleukin-6 trajectories after TBI: Associations with neuroinflammation, polytrauma, and outcome. Brain Behav. Immun 45, 253–262. 10.1016/j.bbi.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Kumar RG, Breslin KB, Ritter AC, Conley YP, Wagner AK, 2018. Variability with astroglial glutamate transport genetics is associated with increased risk for post-traumatic seizures. J. Neurotrauma 10.1089/neu.2018.5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutner M, Nachtsheim C, 2005. Applied Linear Statistical Models. McGraw-Hill Irwin, Fifth. [Google Scholar]
- Lau LT, Yu AC, 2001. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J. Neurotrauma 18 (3), 351–359. 10.1089/08977150151071035. [DOI] [PubMed] [Google Scholar]
- Labrenz F, Wrede K, Forsting M, Engler H, Schedlowski M, Elsenbruch S, Benson S, 2016. Alterations in functional connectivity of resting state networks during experimental endotoxemia – an exploratory study in healthy men. Brain Behav. Immun 54, 17–26. 10.1016/j.bbi.2015.11.010. [DOI] [PubMed] [Google Scholar]
- Lee AB, Nadler B, Wasserman L, 2008. Treelets–An adaptive multi-scale basis for sparse unordered data. Ann. Appl. Stat 2 (2), 435–471. 10.1214/07-AOAS137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine SJ, 2008. Molecular mechanisms of soluble cytokine receptor generation. J. Biol. Chem 283 (21), 14177–14181. 10.1074/jbc.R700052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, 2016. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol 275 (Pt 3), 316–327. 10.1016/j.expneurol.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Harris TH, Kipnis J, 2015. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 36 (10), 569–577. 10.1016/j.it.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundström W, Fewkes NM, Mackall CL, 2012. IL-7 in human health and disease. Semin. Immunol 24 (3), 218–224. 10.1016/j.smim.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manek R, Moghieb A, Yang Z, Kumar D, Kobessiy F, Sarkis GA, Raghavan V, Wang KKW, 2018. Protein biomarkers and neuroproteomics characterization of microvesicles/exosomes from human cerebrospinal fluid following traumatic brain injury. Mol. Neurobiol 55 (7), 6112–6128. 10.1007/s12035-017-0821-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee CA, Lukens JR, 2016. Emerging roles for the immune system in traumatic brain injury. Front. Immunol 7 10.3389/fimmu.2016.00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meythaler JM, Brunner RC, Johnson A, Novack TA, 2002. Amantadine to improve neurorecovery in traumatic brain injury-associated diffuse axonal injury: a pilot double-blind randomized trial. JHead Trauma Rehabil. 17 (4), 300–313. [DOI] [PubMed] [Google Scholar]
- Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D, Sagan A, Wu J, Vinh A, Marvar PJ, Guzik B, Podolec J, Drummond G, Lob HE, Harrison DG, Guzik TJ, 2016. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 30 (5), 1987–1999. 10.1096/fsb2.v30.510.1096/fj.201500088R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milleville K, Awan N, DiSanto D, Kumar R, Wagner AK, 2020. Early chronic systemic inflammation and associations with cognitive performance, functional outcome and life quality after moderate to severe TBI. Brain Behav Immun-Health. 10.1016/j.bbih.2020.100185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan K, Ding Z, Hanly J, Issekutz TB, 2002. IFN-γ-inducible T cell α chemoattractant is a potent stimulator of normal human blood T lymphocyte transendothelial migration: differential regulation by IFN-γ and TNF-α. J. Immunol 168 (12), 6420–6428. 10.4049/jimmunol.168.12.6420. [DOI] [PubMed] [Google Scholar]
- Mondello S, Guedes VA, Lai C, Czeiter E, Amrein K, Kobeissy F, Mechref Y, Jeromin A, Mithani S, Martin C, Wagner CL, Czigler A, Tóth L, Fazekas B, Buki A, Gill J, 2020. Circulating brain injury exosomal proteins following moderate-to-severe traumatic brain injury: temporal profile, outcome prediction and therapy implications. Cells. 9 (4), 977. 10.3390/cells9040977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T, 2007. Modulation of immune response by head injury. Injury 38 (12), 1392–1400. 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Mortezaee K, Khanlarkhani N, Beyer C, Zendedel A, 2018. Inflammasome: its role in traumatic brain and spinal cord injury. J. Cell. Physiol 233 (7), 5160–5169. 10.1002/jcp.26287. [DOI] [PubMed] [Google Scholar]
- Munoz MJ, Kumar RG, Oh B-M, Conley YP, Wang Z, Failla MD, Wagner AK, 2017. Cerebrospinal fluid cortisol mediates brain-derived neurotrophic factor relationships to mortality after severe TBI: a prospective cohort study. Front. Mol. Neurosci 10 10.3389/fnmol.2017.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrga JM, Failla MD, Ricker JH, et al. , 2016. A dopamine pathway gene risk score for cognitive recovery following traumatic brain injury: methodological considerations, preliminary findings, and interactions with sex. J. Head Trauma Rehabil 31 (5), E15–E29. 10.1097/HTR.0000000000000199. [DOI] [PubMed] [Google Scholar]
- Niyonkuru C, Wagner AK, Ozawa H, Amin K, Goyal A, Fabio A, 2013. Group-based trajectory analysis applications for prognostic biomarker model development in severe TBI: a practical example. J. Neurotrauma 30 (11), 938–945. 10.1089/neu.2012.2578. [DOI] [PubMed] [Google Scholar]
- Norden DM, Muccigrosso MM, Godbout JP, 2015. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 96 (Pt A), 29–41. 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham EJ, Helmy A, Zanier ER, Jones JL, Coles AJ, Menon DK, 2019. The immunological response to traumatic brain injury. J. Neuroimmunol 332, 112–125. 10.1016/j.jneuroim.2019.04.005. [DOI] [PubMed] [Google Scholar]
- Noack M, Miossec P, 2014. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev 13 (6), 668–677. 10.1016/j.autrev.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Patel JR, Brewer GJ, 2008. Age-related changes to TNF receptors affect neuron survival in the presence of beta-amyloid. J. Neurosci. Res 86 (10), 2303–2313. 10.1002/jnr.21663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao C-S, Stoica BA, Wu J, Sabirzhanov B, Zhao Z, Cabatbat R, Loane DJ, Faden AI, 2013. Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiol. Dis 54, 252–263. 10.1016/j.nbd.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancan M, Bye N, Otto VI, Trentz O, Kossmann T, Frentzel S, Morganti-Kossmann MC, 2004. The chemokine fractalkine in patients with severe traumatic brain injury and a mouse model of closed head injury. J. Cereb. Blood Flow Metab 24 (10), 1110–1118. 10.1097/01.WCB.0000133470.91843.72. [DOI] [PubMed] [Google Scholar]
- Rappaport M, Hall K, Hopkins H, Belleza T, Cope D, 1982. Disability Rating Scale for severe head trauma: coma to community. Arch. Phys. Med. Rehabil 63 (3), 118–123. [PubMed] [Google Scholar]
- Rasmussen MK, Mestre H, Nedergaard M, 2018. The glymphatic pathway in neurological disorders. Lancet Neurol. 17 (11), 1016–1024. 10.1016/S1474-4422(18)30318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegger T, Conrad S, Schluesener HJ, Kaps H-P, Badke A, Baron C, Gerstein J, Dietz K, Abdizahdeh M, Schwab JM, 2009. Immune depression syndrome following human spinal cord injury (SCI): a pilot study. Neuroscience 158 (3), 1194–1199. 10.1016/j.neuroscience:2008.08.021. [DOI] [PubMed] [Google Scholar]
- Robertson CS, Hannay HJ, Yamal J-M, Gopinath S, Goodman JC, Tilley BC, Baldwin A, Rivera Lara L, Saucedo-Crespo H, Ahmed O, Sadasivan S, Ponce L, Cruz-Navarro J, Shahin H, Aisiku IP, Doshi P, Valadka A, Neipert L, Waguspack JM, Rubin ML, Benoit JS, Swank P, 2014. Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: a randomized clinical trial. JAMA 312 (1), 36. 10.1001/jama.2014.6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodney T, Taylor P, Dunbar K, Perrin N, Lai C, Roy M, Gill J, 2020. High IL-6 in military personnel relates to multiple traumatic brain injuries and post-traumatic stress disorder. Behav. Brain Res 392, 112715. 10.1016/j.bbr.2020.112715. [DOI] [PubMed] [Google Scholar]
- Rusiecki J, Levin LI, Wang L.i., Byrne C, Krishnamurthy J, Chen L, Galdzicki Z, French LM, 2020. Blast traumatic brain injury and serum inflammatory cytokines: a repeated measures case-control study among U.S. military service members. J Neuroinflammation. 17 (1) 10.1186/s12974-019-1624-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarsieri M, Niyonkuru C, McCullough EH, Dobos JA, Dixon CE, Berga SL, Wagner AK, 2014. Cerebrospinal fluid cortisol and progesterone profiles and outcomes prognostication after severe traumatic brain injury. J. Neurotrauma 31 (8), 699–712. 10.1089/neu.2013.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarsieri M, Kumar RG, Niyonkuru C, Kochanek PM, Wagner AK, 2014. Neuroendocrine-immune dysfunction in individuals with poor outcome after severe traumatic brain injury. J. Neurotrauma 31 (12). A-1-A–126. [Google Scholar]
- Schluns KS, Kieper WC, Jameson SC, Lefrançois L, 2000. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol 1 (5), 426–432. 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- Schmidt JA, Fensom GK, Rinaldi S, Scalbert A, Appleby PN, Achaintre D, Gicquiau A, Gunter MJ, Ferrari P, Kaaks R, Kühn T, Boeing H, Trichopoulou A, Karakatsani A, Peppa E, Palli D, Sieri S, Tumino R, Buenode-Mesquita B, Agudo A, Sánchez M-J, Chirlaque M-D, Ardanaz E, Larrañaga N, Perez-Cornago A, Assi N, Riboli E, Tsilidis KK, Key TJ, Travis RC, 2020. Patterns in metabolite profile are associated with risk of more aggressive prostate cancer: a prospective study of 3,057 matched case–control sets from EPIC. Int. J. Cancer 146 (3), 720–730. 10.1002/ijc.v146.310.1002/ijc.32314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA, 2004. Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol 75 (2), 163–189. 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Schulz J, Kumar RG, Awan N, Wagner AK, 2018. Poster 62: subacute systemic inflammation associated with depression at 12 months post-traumatic brain injury. PM&R. 10, S7. 10.1016/j.pmrj.2018.08.362. [DOI] [Google Scholar]
- Sedger LM, McDermott MF, 2014. TNF and TNF-receptors: from mediators of cell death and inflammation to therapeutic giants – past, present and future. Cytokine Growth Factor Rev. 25 (4), 453–472. 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Shim R, Wong C, 2016. Immunosuppression and infection-tackling the predicaments of post-stroke complications. Int. J. Mol. Sci 17 (1), 64. 10.3390/ijms17010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A, Baker AJ, Hare GMT, Reinders FX, Schlichter LC, Moulton RJ, 2002. Association between cerebrospinal fluid interleukin-6 concentrations and outcome after severe human traumatic brain injury. J. Neurotrauma 19 (8), 929–937. 10.1089/089771502320317087. [DOI] [PubMed] [Google Scholar]
- Spolski R, Leonard WJ, 2014. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov. 13 (5), 379–395. 10.1038/nrd4296. [DOI] [PubMed] [Google Scholar]
- Springer TA, 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 76 (2), 301–314. 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Stein DM, Lindell A, Murdock KR, Kufera JA, Menaker J, Keledjian K, Bochicchio GV, Aarabi B, Scalea TM, 2011. Relationship of serum and cerebrospinal fluid biomarkers with intracranial hypertension and cerebral hypoperfusion after severe traumatic brain injury. J. Trauma 70 (5), 1096–1103. 10.1097/TA.0b013e318216930d. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Piantadosi CA, Mattson MP, 2016. Mitochondrial quality control as a therapeutic target. Pharmacol. Rev 68 (1), 20–48. 10.1124/pr.115.011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhang L, Schaeffer H, 2020. NeuPDE: Neural Network Based Ordinary and Partial Differential Equations for Modeling Time-Dependent Data. Proceedings of Machine Learning Research 107, 352–372. [Google Scholar]
- Suvannavejh GC, Lee HO, Padilla J, Dal Canto MC, Barrett TA, Miller SD, 2000. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35–55)-induced experimental autoimmune encephalomyelitis. Cell. Immunol 205 (1), 24–33. 10.1006/cimm.2000.1706. [DOI] [PubMed] [Google Scholar]
- Teasdale G, Murray G, Parker L, Jennett B, 1979. Adding up the glasgow coma score. Acta Neurochir Suppl (Wien). 28 (1), 13–16. [DOI] [PubMed] [Google Scholar]
- Thelin EP, Nelson DW, Bellander B-M, 2017. A review of the clinical utility of serum S100B protein levels in the assessment of traumatic brain injury. Acta Neurochir (Wien). 159 (2), 209–225. 10.1007/s00701-016-3046-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelin EP, Tajsic T, Zeiler FA, Menon DK, Hutchinson PJA, Carpenter KLH, Morganti-Kossmann MC, Helmy A, 2017. Monitoring the neuroinflammatory response following acute brain injury. Front. Neurol 8 10.3389/fneur.2017.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timaru-Kast R, Luh C, Gotthardt P, Huang C, Schäfer MK, Engelhard K, Thal SC, Kleinschnitz C, 2012. Influence of age on brain edema formation, secondary brain damage and inflammatory response after brain trauma in mice. PLoS ONE 7 (8), e43829. 10.1371/journal.pone.0043829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan LE, Ranganathan PR, Kumar RG, Wagner AK, Rubin JE, 2018. A mathematical model of neuroinflammation in severe clinical traumatic brain injury. J Neuroinflammation. 15 (1), 345. 10.1186/s12974-018-1384-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijapur S, Yang Z, Barton D, et al. , 2020. Anti-pituitary and anti-hypothalamus autoantibody associations with inflammation and hypogonadotropic hypogonadism in men with traumatic brain injury. J. Neurotrauma 10.1089/neu.2019.6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Kumar RG, 2019. TBI rehabilomics research: conceptualizing a humoral triad for designing effective rehabilitation interventions. Neuropharmacology 145 (Pt B), 133–144. 10.1016/j.neuropharm.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Amin KB, Niyonkuru C, Postal BA, McCullough EH, Ozawa H, Dixon CE, Bayir H, Clark RS, Kochanek PM, Fabio A, 2011. CSF Bcl-2 and cytochrome C temporal profiles in outcome prediction for adults with severe TBI. J. Cereb. Blood Flow Metab 31 (9), 1886–1896. 10.1038/jcbfm.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A, Vaughan L, Wagner AK, 2019. Early neutrophil-to-lymphocyte ratio: associations with chronic systemic inflammation and long-term global outcome following TBI. J. Neurotrauma 36 (13). 10.1089/neu.2019.29100.abstracts. [DOI] [Google Scholar]
- Walker KR, Tesco G, 2013. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci 5 10.3389/fnagi.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker KA, Gross AL, Moghekar AR, Soldan A, Pettigrew C, Hou X, Lu H, Alfini AJ, Bilgel M, Miller MI, Albert MS, Walston J, 2020. Association of peripheral inflammatory markers with connectivity in large-scale functional brain networks of non-demented older adults. Brain Behav. Immun 87, 388–396. 10.1016/j.bbi.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KK, Yang Z, Zhu T, Shi Y, Rubenstein R, Tyndall JA, Manley GT, 2018. An update on diagnostic and prognostic biomarkers for traumatic brain injury. Expert Rev. Mol. Diagn 18 (2), 165–180. 10.1080/14737159.2018.1428089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, Goldstein FC, Caveney AF, Howlett-Smith H, Bengelink EM, Manley GT, Merck LH, Janis LS, Barsan WG, 2014. Very early administration of progesterone for acute traumatic brain injury. N. Engl. J. Med 371 (26), 2457–2466. 10.1056/NEJMoa1404304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock T, Morganti-Kossmann MC, 2013. The role of markers of inflammation in traumatic brain injury. Front. Neurol 4, 18. 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zhu T, Weissman AS, et al. , 2017. Autoimmunity and traumatic brain injury. Curr. Phys. Med. Rehabil. Rep In Press. [Google Scholar]
- Ziebell JM, Morganti-Kossmann MC, 2010. Involvement of pro-and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 7 (1), 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell JM, Morganti-Kossmann MC, 2010. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 7 (1), 22–30. 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]