

SUMMARY
Background:
Ketogenic diet is a potential means of augmenting cancer therapy. Here, we explore ketone body metabolism and its interplay with chemotherapy in pancreatic cancer.
Methods:
Metabolism and therapeutic responses of murine pancreatic cancer were studied using KPC primary tumors and tumor chunk allografts. Mice on standard high-carbohydrate diet or ketogenic diet were treated with cytotoxic chemotherapy (nab-paclitaxel, gemcitabine, cisplatin). Metabolic activity was monitored with metabolomics and isotope tracing, including 2H- and 13C-tracers, liquid chromatography-mass spectrometry, and imaging mass spectrometry.
Findings:
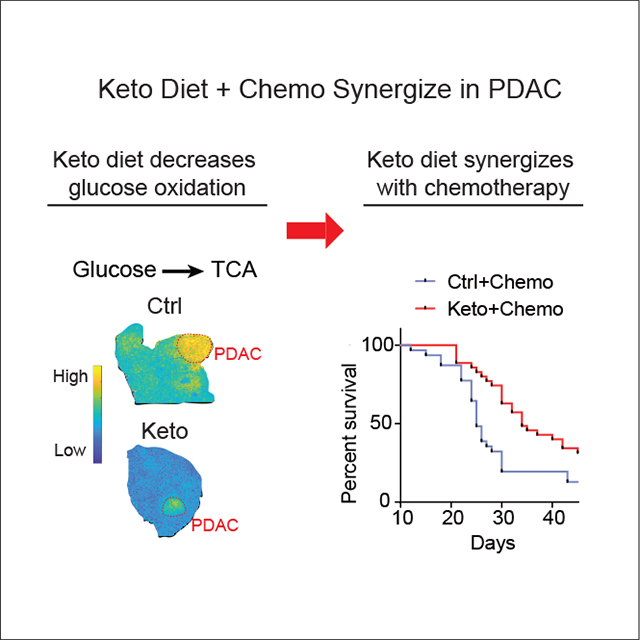
Ketone bodies are unidirectionally oxidized to make NADH. This stands in contrast to the carbohydrate-derived carboxylic acids lactate and pyruvate, which rapidly interconvert, buffering NADH/NAD. In murine pancreatic tumors, ketogenic diet decreases glucose’s concentration and tricarboxylic acid cycle contribution, enhances 3-hydroxybutyrate’s concentration and tricarboxylic acid contribution, and modestly elevates NADH, but does not impact tumor growth. In contrast, the combination of ketogenic diet and cytotoxic chemotherapy substantially raises tumor NADH and synergistically suppresses tumor growth, tripling the survival benefits of chemotherapy alone. Chemotherapy and ketogenic diet also synergize in immune-deficient mice, although long-term growth suppression was only observed in mice with an intact immune system.
Conclusions:
Ketogenic diet sensitizes murine pancreatic cancer tumors to cytotoxic chemotherapy. Based on these data, we have initiated a randomized clinical trial of chemotherapy with standard versus ketogenic diet for patients with metastatic pancreatic cancer (NCT04631445).
Graphical Abstract
Using murine KPC pancreatic cancer tumors, Yang et al. demonstrate synergy between ketogenic diet and classical cytotoxic chemotherapy (gemcitabine, nab-paclitaxel, cisplatin). Ketogenic diet decreases tumor glucose and glycolytic intermediates, enhances 3-hydroxybutyrate, and boosts reactive oxygen species. This metabolic rewiring results in markedly stronger and more durable benefits from chemotherapy.
INTRODUCTION
Cells require chemical energy in the form of ATP to maintain their function. Metabolism generates ATP both anaerobically, through glycolysis, and aerobically, through NADH and oxidative phosphorylation.
Cells have a finite respiratory capacity, determined by the number of healthy mitochondria and the availability of oxygen. In other words, there is a limit to how much NADH cells can handle. When this limit is approached, NADH and reduced respiratory chain complexes accumulate, leading to reactive oxygen species (ROS).1 This redox stress can be catastrophic in patients with genetic mitochondrial disorders, and may also occur in cells where respiration is impaired for other reasons—for example, oxygen deprivation as often occurs in tumors.2–5
Cells employ a variety of mechanisms to avoid excessive NADH accumulation. One is lactate excretion, with ATP generated through glycolysis. This requires an adequate supply of glucose, either from internal stores (glycogen) or the microenvironment, which is often glucose depleted in tumors.6,7 Another is feedback inhibition of mitochondrial NADH-generating enzymes, such as pyruvate dehydrogenase.8–10
Using these approaches, cultured cells with impaired electron transport chain function can survive but cannot proliferate in standard medium, in part because NADH buildup in mitochondria blocks the synthesis of aspartate and pyrimidines.8,9,11 Growth can be restored by providing pyruvate, whose reduction to lactate clears extra NADH. Within the body, pyruvate is available in the circulation. Rapid exchange between circulating lactate/pyruvate and tissue NADH/NAD has been hypothesized to buffer local redox perturbations, and whole-body reductive stress can be mitigated by injection of an engineered enzyme, LOXCAT, which converts circulating lactate into pyruvate.12
Pancreatic ductal adenocarcinoma (PDAC) tumors are characterized by stromal fibrosis that limits blood flow. They are hypoxic, nutrient-deprived, and have lower glucose and higher lactate levels than surrounding pancreatic tissue.13,14 Somehow, despite this harsh metabolic environment, PDAC tumors grow irrepressibly. Diagnosis typically occurs after metastasis with associated poor prognosis. Recently, augmentation of the most common chemotherapy regimen of gemcitabine and albumin-bound nanoparticle paclitaxel (nab-paclitaxel) with cisplatin led to median survival of 16 months, a step forward but still terribly short.15–17 Accordingly, strategies to augment PDAC treatment are desperately needed.
One possibility is dietary manipulation. Diets lacking in serine/glycine or methionine have shown efficacy in murine colorectal cancer models.18,19 Fasting or fasting-mimicking diet enhanced therapeutic activity of endocrine therapeutics in hormone-receptor-positive breast tumor models20 and, in a phase II study, a fasting-mimicking diet showed favorable pathological and radiographic trends when combined with cytotoxic chemotherapy for breast cancer.21
A ketogenic diet—very low carbohydrate, low protein, and high fat—mimics several aspects of fasting without calorie restriction. Even in the fed state, glucose and insulin are low and ketone bodies (3-hydroxybutyrate [3HB], acetoacetate, and acetone) are high. The ketogenic diet has attracted attention as a potential means of enhancing cancer treatment.22–25 Proposed mechanisms include suppression of pro-tumorigenic insulin signaling, depletion of glucose as a preferred tumor fuel, imbalances between saturated and unsaturated fatty acids, and antitumor effects of ketone bodies, which may potentially include reductive stress or histone deacetylation inhibition.26–28
Although phosphatidylinositol 3-kinase (PI3K) inhibitors have not shown utility in pancreatic cancer, Hopkins et al. showed that these inhibitors are effective in pancreatic cancer mouse models when combined with the ketogenic diet.29 In this context, the ketogenic diet prevents the PI3K inhibitor-induced hyperglycemia and associated hyperinsulinemia which otherwise tend to override the pharmacological PI3K inhibition, resulting in effective blockade of both PI3K signaling and tumor growth.
Here, we test the impact of a ketogenic diet (with roughly balanced saturated versus unsaturated fatty acid composition) on the growth of murine pancreatic KPC tumors (spontaneous in pancreas or passaged as flank allograft). While ineffective on its own, when combined with the current standard-of-care chemotherapy for PDAC, the ketogenic diet impairs tumor growth and significantly prolongs survival. The tumor growth impairment is associated with glucose depletion, altered tricarboxylic acid (TCA) substrate usage, and NADH elevation.
RESULTS
Syngeneic subcutaneous KPC tumor chunk model
Human pancreatic cancer tumors have extensive desmoplastic stroma. Such stroma is absent in typical subcutaneous allografts but recapitulated in genetically engineered pancreatic cancer models including the KPC model (LSL-KrasG12D, p53R172H, Pdx-Cre). If KPC cells are passaged in vitro and then implanted subcutaneously, however, the resulting tumors are mainly epithelial. To address this limitation, while benefiting from the ease of implanting and monitoring subcutaneous tumors, we established a syngeneic subcutaneous KPC tumor chunk model in which tumors are implanted subcutaneously without ever being passaged in vitro. In this model, pancreatic tumors are harvested from KPC mice, optionally frozen (enabling easy sharing between labs), minced with surgical scissors into small pieces, suspended in Dulbecco’s modified Eagle’s medium, mixed with Matrigel, and injected subcutaneously into the mouse flank. The passaging process can be repeated: subcutaneous tumors can be used to seed new subcutaneous tumors, with a maximum of two subcutaneous passages used here. These tumors retain various cell types observed in pancreatic cancer, including both epithelial cancer cells and cancer-associated fibroblasts (Figure S1A).
Redox buffering by lactate-pyruvate but not 3-hydroxybutyrate-acetoacetate
Before diving into pancreatic cancer metabolism, we started by investigating the metabolism of the classical ketone bodies 3HB and acetoacetate in healthy mice. Like lactate and pyruvate, 3HB and acetoacetate chemically differ by one hydride: 3HB is a hydride donor and acetoacetate a hydride acceptor (Figure 1A). The recognition that lactate is a major circulating fuel, with rapid exchange between pyruvate made via glycolysis and circulating lactate, gave rise to the idea that circulating lactate and pyruvate may collectively act as a “redox buffer”—that is, these metabolites flow back and forth between cells and the circulation, donating or accepting hydrides as needed such that local NADH/NAD ratios are maintained at healthy levels.2,30–33
Figure 1. Circulating 3-hydroxybutyrate, unlike circulating lactate, unidirectionally delivers hydride to tissues.

(A) Illustration of exchange between circulatory lactate with tissue pyruvate and circulatory 3-hydroxybutyrate with tissue acetoacetate. 2H-tracers report on the exchange flux shown in the lactate schematic (and not in the 3HB schematic, because such exchange does not occur). Experimentally, whole-body exchange flux is the difference between 2H-tracer turnover flux and 13C-tracer turnover flux. Red, 2H (deuterium); blue, 13C.
(B) Circulatory turnover flux (units of nmol/min/g body weight) for 2H- versus 13C-lactate in healthy mice fed standard diet. Greater 2H than 13C flux indicates reversible hydride exchange. Mean ± SEM, n = 5 for 2H-lactate, n = 10 for 13C-lactate. *p < 0.05 by two-tailed Student’s t test.
(C) Circulatory turnover flux for 2H- versus 13C-3HB in healthy mice fed standard diet. Equal turnover flux indicates unidirectional catabolism into acetoacetate. Mean ± SEM, n = 5 for 2H-3HB, n = 7 for 13C-3HB.
(D) Mass isotope distribution of KPC tumor chunk allograft lactate and pyruvate following [U-13C]lactate infusion for mice fed standard diet. Identical labeling patterns are consistent with rapid interconversion of these metabolites. Mean ± SEM, n = 6.
(E) Mass isotope distribution of tumor 3HB and acetoacetate following [U-13C]3HB infusion for mice fed standard diet. Lack of acetoacetate labeling reflects its production from sources other than circulating 3HB. Mean ± SEM, n = 6.
See also Figure S1
To confirm this idea and to test whether the same might be true of circulating 3HB and acetoacetate, we devised a set of isotope-tracer infusion experiments. The circulatory turnover flux (Fcirc) of a metabolite refers to the rate at which tissues collectively take up the metabolite from arterial circulation; alternatively, Fcirc can be defined as the rate at which tissues collectively excrete the metabolite into the venous circulation, as these two rates are equal at steady state for minimally pertur-bative tracer infusions. Importantly, production and consumption that do not alter isotope labeling (for example, removal of an unlabeled hydride followed by addition of a different unlabeled hydride in its place) do not register as circulatory turnover flux. By comparing the Fcirc values of 13C-labeled metabolites (for which oxidation and reduction reactions do not register as Fcirc) and 2H-labeled metabolites (for which they do), one can evaluate the extent to which different circulating metabolites undergo rapid reversible redox reactions.
When we infused uniformly 13C-labeled lactate into fasted mice, the measured lactate turnover flux was 380 nmol/g/min, consistent with our previous published measurements, and very fast (about 3-fold faster than glucose Fcirc on a molar basis, or 1.5-fold greater than glucose flux on a carbon atom basis).32 When 2H-labeled lactate (labeled only at the active hydride) was infused instead, the measured turnover flux increased to 550 nmol/g/min (Figures 1B, S1B, and S1C). This increase implies that lactate frequently enters tissues, delivers hydrides to NAD, and is then excreted as pyruvate or again reduced to lactate.
In contrast, the 3HB turnover flux as measured with uniformly 13C-labeled 3HB versus 2H-labeled 3HB was indistinguishable (~40 nmol/g/min) (Figures 1C, S1D, and S1E). Furthermore, from infused [3,4,4,4-2H]3HB (M+4), we did not observe substantial product arising from oxidation to acetoacetate followed by re-reduction to 3HB ([4,4,4-2H]3HB, M+3) (Figure S1F). Thus, unlike lactate-pyruvate, the 3HB-acetoacetate pair does not participate in reversible cellular redox reactions, and instead is unidirectionally catabolized. In short, while lactate-pyruvate acts as a redox buffer, 3HB is a unidirectional hydride donor.
Next, we infused [U-13C]lactate and [U-13C]3HB, separately, into mice bearing subcutaneous KPC tumor chunk allografts and fed them a regular carbohydrate-rich diet. After infusions reached steady state, tumors were extracted and metabolites analyzed by liquid chromatography-mass spectrometry. With 13C-lactate infusion, the tumor pyruvate and lactate isotope labeling patterns were indistinguishable, consistent with their rapid interconversion (Figure 1D). In contrast, when 13C-3HB was infused, acetoacetate labeling remained minimal (7-fold lower than 3HB), indicating that, in mice on the control diet, tumor acetoacetate neither comes from nor exchanges with 3HB (Figure 1E).
The ketogenic diet is a physiological means of dramatically increasing circulating 3HB and acetoacetate. To check whether the ketogenic diet induces hydride exchange between these ketone bodies, we infused [3,4,4,4-2H]3HB to mice (without tumors) fed the ketogenic diet. In the ketogenic organ of liver, most 3HB and acetoacetate were unlabeled, coming from local synthesis. Relative to [3,4,4,4-2H]3HB, [4,4,4-2H]3HB (M+3) and acetoacetate (M+3) were substantial, indicating that 3HB and acetoacetate exchange locally in liver (Figure S1G). Nevertheless, circulating [4,4,4-2H]3HB (M+3) and acetoacetate (M+3) were minimal, implying limited reversible flux between the ketone bodies at the whole-body level (Figure S1G). Thus, irrespective of dietary status, 3HB acts largely as a unidirectional hydride donor rather than a reversible redox exchanger such as lactate.
Ketogenic diet suppresses tumor glucose utilization and induces 3-hydroxybutyrate burning
To explore the metabolic effects of the ketogenic diet in mice bearing tumors, after KPC tumor chunk allograft implantation the mice were fasted overnight and then were either maintained on a control diet or switched to a ketogenic diet. The ketogenic diet suppressed circulating glucose concentration and glucose and lactate Fcirc. Bulk tumor glucose and lactate concentrations also decreased (Figure 2A, S2A, and S2B). Intratumoral glucose fell to 1.3 mM, sufficiently low to potentially impair tumor glycolysis (Figure 2A). Circulating insulin, c-peptide, and insulin-like growth factor-1 were also suppressed, while 3HB levels and Fcirc were dramatically increased (Figures 2A, 2B, S2B, and S2C).
Figure 2. Ketogenic diet suppresses glucose metabolism and induces ketone body oxidation in KPC tumors (A–G, tumor chunk allograft; H, primary).

(A) Metabolite concentrations of serum and bulk KPC tumors in mice fed with control or ketogenic diet. Mean ± SEM, n ≥ 5.
(B) Serum insulin and c-peptide level in mice fed with control or ketogenic diet. Mean ± SEM, n = 4 for insulin, n = 6 for c-peptide.
(C) Intratumor redox pairs in mice fed with control or ketogenic diet. Mean ± SEM, n = 17.
(D) Tumor versus circulating lactate labeling from [U-13C]glucose and [U-13C]lactate infusion. In ketogenic diet, tumor lactate comes more from circulating lactate, as opposed to from intratumoral glycolysis. Mean ± SEM, n = 3.
(E) Tumor metabolite labeling from [U-13C]3HB infusion. Tumor metabolite enrichment was normalized to the enrichment of circulatory [U-13C]3HB. Mean ± SEM, n = 3 for control diet, n = 4 for ketogenic diet.
(F) Direct contributions of circulating nutrients to tumor acetoacetate. Mean ± SD.
(G) Direct contributions of circulating nutrients to tumor TCA cycle (malate as representative metabolite). Mean ± SD.
(H) MALDI-imaging mass spectrometry of malate labeling from 13C-glucose (top) and 13C-3HB (bottom) in pancreas from KPC mice. Fractional enrichment indicates 13C enrichment normalized to the 13C enrichment of circulatory infusates. T, tumor; F, peritumoral fibrosis.
*p < 0.05, **p < 0.01, ***p < 0.001 by two-tailed Student’s t test. n.s., not significant. See also Figure S2.
We also monitored how the ratios of metabolic redox pairs changed on the ketogenic diet. While the bulk tumor lactate/pyruvate ratio, which can reflect cytoplasmic NADH/NAD, remained roughly the same in mice on the ketogenic diet, the overall NADH/NAD ratio increased by ~40%, suggesting mitochondrial NADH buildup (Figure 2C).
To probe tumor glycolysis, we infused [U-13C]glucose and harvested tumors at labeling pseudo-steady state (2.5 h). Tumor lactate was labeled by glucose, to a yet greater extent than was circulating lactate, indicating active tumor glycolysis at a rate sufficient to outpace lactate exchange with the circulation and drive tumor lactate accumulation, as observed also in human breast cancer tumors.34 Faster glycolysis than lactate exchange was further proven by [U-13C]lactate infusion, whereby more than 60% of intratumor lactate pool was diluted by unlabeled sources such as glycolysis (Figures 2D, S2D, and S2E). In addition to lowering the intratumoral lactate concentration, the ketogenic diet decreased tumor lactate and pyruvate enrichment from infused glucose and increased the contribution from circulating lactate (Figures 2D, S2D, and S2E). These data are consistent with the ketogenic diet suppressing tumor glycolysis.
To track the fate of circulating 3HB in tumors, we infused [U-13C]3HB. The ketogenic diet dramatically enhanced the fraction of tumor acetoacetate labeled from circulating 3HB, indicating induction of intratumoral 3HB catabolism. Moreover, 3HB extensively labeled TCA intermediates and glutamate (which rapidly exchanges with α-ketoglutarate) selectively in mice on the ketogenic diet (Figure 2E).
To understand more comprehensively which nutrients are oxidized in pancreatic tumors, on both the control and ketogenic diets we performed a series of 13C-tracer infusion experiments in tumor-bearing mice with 13C-labeled glucose, lactate, oleate (C18:1, the most abundant monounsaturated fatty acid), palmitate (C16:0, the most abundant saturated fatty acid), acetate, glutamine, and a mixture of branched-chain amino acids. On control diet, tumor acetoacetate came more from fat than 3HB. In contrast, on the ketogenic diet, circulating 3HB was the dominant tumor acetoacetate source (Figure 2F). In both dietary conditions, glutamine was the largest contributor to tumor TCA cycle intermediates. Both glucose and lactate contributed directly to the tumor TCA cycle, but these contributions fell in the ketogenic diet, being replaced by increased contributions from oleate and 3HB (Figures 2G and S2F). The 3HB contribution to the tumor TCA far exceeded that in normal pancreas (Figure 2G).
Heterogeneity in tumor nutrient use probed by imaging mass spectrometry
To explore tumor heterogeneity with respect to nutrient oxidation, we infused [U-13C]glucose or [U-13C]3HB to KPC mice with spontaneous pancreatic tumors and analyzed the spatial distribution of downstream TCA cycle labeling using matrix-assisted laser desorption ionization (MALDI)-imaging mass spectrometry. We observed higher glucose contribution to the TCA cycle of tumors compared with the surrounding pancreas, with the greatest contribution in regions rich in epithelial cancer cells. On the ketogenic diet, although PDAC tumors continued to show greater glucose contribution to TCA relative to surrounding pancreas, the overall glucose TCA contribution was dramatically suppressed (Figures 2H and S2G).
Analogous MALDI-imaging tracing studies with 3HB confirmed minimal 3HB contribution to normal or pancreatic tumor TCA cycle in the standard diet condition. In mice fed the ketogenic diet, however, tumors oxidized circulating 3HB as a source of high-energy electrons, with particularly intense TCA isotope labeling observed in the regions of peritumoral fibrosis (Figures 2H and S2G). Thus, the ketogenic diet suppresses tumor glucose oxidation and induces tumor 3HB burning.
Ketogenic diet synergizes with cytotoxic chemotherapy
Motivated by the ketogenic diet-induced changes in tumor nutrient use, we looked for downstream effects. We observed no impact on nucleotides, energy charge, or growth rate of subcutaneous KPC tumor chunk allografts, or on the survival of tumor-bearing animals (Figures 3A, 3C, and S3A).
Figure 3. Ketogenic diet synergizes with cytotoxic chemotherapy.

(A) Chemotherapy regimen (triple chemotherapy: nab-paclitaxel, gemcitabine, cisplatin) with doses administered on day 4 and day 7 after diet switch in C57BL/6 mice with KPC tumor chunk allografts.
(B): Tumor volumes for diet only without chemotherapy (batch #2, mean ± SEM, n = 8 mice per group).
(C) Kaplan-Meier curve for diet only without chemotherapy (n = 14 mice per group).
(D) Allograft tumor volumes in mice treated with control diet plus chemotherapy or ketogenic diet plus chemotherapy (batch #2, mean ± SEM, n = 11 mice per group; for other batches, see Figure S3B).
(E) Kaplan-Meier curve for mice treated with control diet plus chemotherapy or ketogenic diet plus chemotherapy (n = 31 mice for Ctrl + Chemo and n = 35 mice for Keto + Chemo).
(F) Representative H&E staining, cleaved caspase-3 staining, and Ki67 staining from tumor tissue treated with control diet plus chemotherapy or ketogenic diet plus chemotherapy. Scale bar, 50 μm.
(G) Quantification of cleaved caspase-3-positive cells and Ki67-positive cells within tumor tissues from (F) (n = 10 images for Ctrl + Chemo and n = 10 images for Keto + Chemo group).
(H) Primary tumor volumes in KPC genetically engineered mouse model (GEMM) (each line represents one mouse).
(I) Kaplan-Meier curve for KPC GEMM (n = 5 for Ctrl + Chemo group, n = 4 for Keto + Chemo group).
*p < 0.05, ***p < 0.001. Two-way ANOVA in (B); log-rank (Mantel-Cox) test in (C), (E), and (I); two-tailed Student’s t test in (G). n.s., not significant. See also Figure S3.
Given the lack of direct antitumor activity of the ketogenic diet, we tested its impact in combination with cytotoxic chemotherapy (gemcitabine, nab-paclitaxel, cisplatin).15–17 Mice with palpable KPC allograft tumors were treated with the triple chemotherapy 4 and 7 days after switching permanently to the ketogenic diet (compared with remaining on the control diet) (Figure 3A). This experiment was repeated four times, for a total of 66 mice randomized to either ketogenic or control diet over the course of 2 years. The combination of ketogenic diet and chemotherapy synergistically suppressed tumor growth and prolonged survival (Figures 3D and 3E). The benefits were evident in each individual experiment (Figures S3B and S3C). Overall, the average survival gain due to chemotherapy alone was 5.8 days, and this increased to 16 days for mice on the ketogenic diet (Figure 3E). These survival benefits were associated with increased tumor necrosis (but not apoptosis) and modestly decreased Ki67 staining, reflecting lower tumor cell proliferation (Figures 3F and 3G).
We conducted similar studies in genetically engineered tumors arising spontaneously within the pancreas of KPC mice,35,36 again finding significant survival benefits for the triple chemotherapy-ketogenic diet combination (Figures 3H, 3I, and S3D). Thus, the ketogenic diet robustly augments the effectiveness of cytotoxic chemotherapy for KPC murine pancreatic cancer.
Ketogenic diet and classical chemotherapy disrupt redox homeostasis
To investigate potential mechanisms underlying the effectiveness of the triple chemotherapy-ketogenic diet combination, we examined histone acetylation, as 3HB is a known histone deacetylase inhibitor.28 We did not, however, observe increased acetylation in the ketogenic diet tumors, arguing against histone deacetylation inhibition as the key ketogenic diet mechanism (Figures S4A and S4C).
We next examined metabolic effects of the ketogenic diet-chemotherapy combination, carrying out metabolomics 24 h after the second dose of chemotherapy. Surprisingly, triple chemotherapy alone increased the tumor NADH/NAD ratio, perhaps indicating chemotherapy-induced damage to tumor oxygen supply or mitochondrial function (Figure 4A). The ketogenic diet also increased the tumor NADH/NAD ratio (Figure 4A). When we analyzed tumors in all four treatment groups together, we observed a significant negative correlation between NADH/NAD ratio and tumor growth: Tumors with the highest NADH/NAD ratio tended to have regressed in the week prior to harvest, while those with low ratios tended to grow (Figure 4B).
Figure 4. Ketogenic diet and classical chemotherapy disrupt redox homeostasis.

(A) NADH/NAD ratio in KPC allografttumors (mean ± SEM, n = 17 for Ctrl group, n = 12 for Keto group, n = 17 for Ctrl + Chemo group, n = 14 for Keto + Chemo group).
(B) Correlation of intratumor NADH/NAD ratio with tumor growth suppression. Mice were sacrificed on day 8, and NADH/NAD measured and plotted versus tumor growth (or, for values <1, regression) over the preceding 7 days.
(C) Tumor glycolytic metabolite levels. Mean ± SEM, n = 13 for Ctrl group, n = 13 for Keto group, n = 10 for Ctrl + Chemo group, n = 13 for Keto + Chemo group.
(D) Tumor nucleotide levels.
(E) Tumor NADPH, NADP, and their ratios.
(F) Tumor GSH, oxidized glutathione (GSSG), and their ratios. Mean ± SEM, n = 13 for Ctrl group, n = 13 for Keto group, n = 10 for Ctrl + Chemo group, n = 13 for Keto + Chemo group.
(G) Tumor protein 4-HNE adduct levels (mean ± SEM, n = 4 for Ctrl + Chemo group, n = 6 for Keto + Chemo group).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. One-way ANOVA with Tukey’s test in (A), (C) to (F); Pearson correlation in (B); two-tailed Student’s t test in (G).
Cells with impaired respiration can maintain ATP production by inducing glycolysis. In mice on the control diet, chemotherapy-treated tumors showed significant increases of glycolytic intermediates including fructose bisphosphate and 3-phospho-glycerate (Figure 4C). This apparent shift toward more glycolytic metabolism was associated with elevated nucleotide levels, perhaps reflecting decreased nucleotide consumption for nucleic acid synthesis in the chemotherapy-treated tumors (Figure 4D). It was further accompanied by higher NADPH and reduced glutathione (GSH) (Figures 4E and 4F). These adaptive metabolic responses to chemotherapy were ablated by the ketogenic diet, with intratumoral glycolytic intermediate, nucleotide, and GSH levels lower in the triple chemotherapy plus ketogenic diet condition compared with triple chemotherapy plus standard diet (Figures 4C and 4F).
Consistent with the ketogenic diet in combination with chemotherapy augmenting tumor redox stress, the lipid peroxidation marker 4-hydroxylnonenal (measured as its protein adduct) increased significantly (Figures 4G and S4D). Thus, for chemotherapy-treated tumors, the ketogenic diet depletes glucose, nucleotides, and GSH while increasing NADH (indicative of tumor electron overloading) and lipid peroxidation.
Ketogenic diet induces inflammatory gene expression in the tumor
To further explore potential mechanisms by which the ketogenic diet may sensitize tumors to chemotherapy, we extracted RNA from tumors 7 days after mice were switched to either the control or ketogenic diet. Overall, the tumor gene expression signature was only modestly changed. The few significantly upregulated gene sets revolved around inflammation: IFN Response, IL6/JAK/STAT3 Signaling, and Inflammatory Response (Figures 5A and 5B). In addition, selected genes related to fibrosis, including collagen Col12A1, were increased. Circulating cytokine analysis revealed elevated FGF21 (as expected based on the ketogenic diet being low in protein) and S100a9, which is produced by and may regulate myeloid cells.37,38 Bulk tumor cytokine protein analysis did not show significant differences dependent on dietary condition (Figures S5A and S5B). Consistent with the increased collagen gene expression, in the ketogenic diet condition, tumor fibrosis as measured by Sirius red and α-smooth muscle actin staining increased (Figure S5C).
Figure 5. Ketogenic diet induces inflammatory gene expression in the tumor.

(A) Gene expression in allograft KPC tumors of mice fed control or ketogenic diet. Ketogenic diet increases expression of genes related to the immune response and reduces expression of genes related to cholesterol synthesis.
(B) Gene set enrichment analysis highlights increased pro-inflammatory genes in tumors of mice fed ketogenic diet. FDR, false discovery rate.
See also Figure S5.
Ketogenic diet-chemotherapy synergy does not require the adaptive immune system
Motivated by the gene expression evidence for increased intratumor inflammation, we attempted studies combining ketogenic diet and triple chemotherapy in immunodeficient NSG mice, but NSG mice did not tolerate the triple chemotherapy. Accordingly, we administered the standard-of-care therapy of gemcitabine and nab-paclitaxel, without cisplatin.17 Despite the lack of an adaptive immune system, in combination with chemotherapy the ketogenic diet suppressed tumor growth and augmented the lifespan, indicating direct antitumor activity (Figures 6A, 6C, S6A, and S6B). Among chemotherapy-treated tumors, in the NSG mice we again observed depletion of glycolytic intermediates, nucleotide triphosphates, NADPH, and glutathione in the ketogenic diet condition (Figures 6D and 6G). The improvement in overall survival attributable to the ketogenic diet was, however, less than in the immunocompetent mice, with no durable tumor control, suggesting that the durable therapeutic benefits in immunocompetent mice on the ketogenic diet involve a component of immune surveillance.
Figure 6. Ketogenic diet-chemotherapy synergy does not require the adaptive immune system.

(A) Chemotherapy regimen (nab-paclitaxel, gemcitabine) for immunocompromised NSG mice with KPC tumor allografts.
(B) Tumor volumes in NSG mice (mean ± SEM, n = 12 for control diet + nab-paclitaxel + gemcitabine, n = 11 for ketogenic diet + nab-paclitaxel + gemcitabine).
(C) Kaplan-Meier curve for NSG mice (n = 17 mice per group).
(D) Tumor glycolytic metabolite levels.
(E) Tumor nucleotide levels.
(F) Tumor NADPH, NADP, and their ratios.
(G) Tumor GSH, GSSG, and their ratios.
In (D) to (G), n = 16 for control diet + nab-paclitaxel + gemcitabine, n = 11 for ketogenic diet + nab-paclitaxel + gemcitabine. *p < 0.05, **p < 0.01, ***p < 0.001. Two-way ANOVA in (B); log-rank (Mantel-Cox) test in (C); two-tailed Student’s t test in (D) to (G). See also Figure S6.
DISCUSSION
Aerobic glycolysis was the first molecular phenotype assigned to cancer and underlies tumor diagnosis by fluorodeoxyglucose positron emission tomography imaging.39–43 However, efforts to pharmacologically target tumor glycolysis, or other central metabolic pathways, have yet to succeed clinically. An alternative way to target tumor metabolism is via diet. A ketogenic diet—mainly fat and very low carbohydrate—suppresses the normal feeding-induced insulin spike and produces persistently elevated circulating ketone bodies, mimicking a chronic fasted state.44–47 It has shown promise, alone or in combination with chemotherapy agents, in various xenograft tumor models.23–25,48,49 Recently, Cantley and colleagues showed that the ketogenic diet rendered PI3K inhibitors, which are normally inactive against pancreatic cancer, effective in KPC tumors.29 Here we show that, in KPC tumors, the ketogenic diet synergizes with the clinically active cytotoxic chemotherapy regimen of gemcitabine, nab-paclitaxel, and cisplatin. Strikingly, while having no effect on tumor growth in its own, the ketogenic diet triples the survival benefits of triple chemotherapy.
What is the metabolic effect of the ketogenic diet in tumors? It has been suggested that tumors do not burn ketone bodies,50 and indeed on the control diet this is the case for pancreatic cancer. However, on the ketogenic diet KPC pancreatic cancer tumors avidly burn ketones, with the ketone contribution to TCA more than double that in regular pancreas. Isotope tracing reveals that this ketone burning involves unidirectional conversion of 3HB into acetoacetate, with minimal flux in the opposite direction. Based on localization of the responsible dehydrogenase (3HBDH), this conversion presumably occurs in mitochondria. As such, it generates mitochondrial NADH and, via acetoacetate, acetyl-coenzyme A, which under physiological conditions are valuable fuels. Imaging mass spectrometry analysis revealed that, within primary KPC tumors, the strongest contribution of 3HB to TCA metabolism occurred in regions of peritumoral fibrosis. Consistent with such regions being avid ketone users, the ketogenic diet also increased overall tumor fibrosis.
One likely driver of the avid tumor ketone body uptake is high monocarboxylate transporter (MCT) expression.51 Commonly thought of as lactate exporters, in typical carbohydrate-rich diets MCTs can enhance tumor growth by promoting glycolysis and redox homeostasis.30,33,52,53 However, MCTs are not specific lactate exporters. Depending on intracellular and environmental levels and pH, MCTs can catalyze uptake or excretion of lactate, pyruvate, 3HB, or acetoacetate.51,54 In the ketogenic diet, high 3HB hijacks tumor MCTs, driving tumor ketone catabolism.
Rapid ketone catabolism generates mitochondrial NADH which, in the context of a poorly perfused tumor such as pancreatic cancer, can lead to accumulation of reduced respiratory chain complexes resulting in oxygen radical generation, lipid peroxidation, and glutathione depletion.55 This contrasts with previous reports where 3HB has been reported to directly scavenge ROS or induce antioxidant genes,22,28,56–59 and may reflect the net impact of ketosis on oxidative stress being context dependent, tending to promote ROS generation when oxygen is scarce.
Unexpectedly, in addition to the ketogenic diet increasing tumor NADH, so did triple chemotherapy. Whether this reflects the chemotherapy impacting tumor vascularization, mitochondria, or another factor remains an open question. The rise in NADH/NAD ratio induced by therapy correlated, across different allograft tumors on control or ketogenic diet, with therapeutic response, with high NADH associated with tumor regression.
In addition to modulating tumor NADH, the ketogenic diet may augment sensitivity to chemotherapy by suppressing histone deacetylation, insulin signaling, or tumor glycolysis.28,29,60–62 We observed that the ketogenic diet suppressed tumor glucose levels and associated TCA intermediate labeling. In the standard diet, triple chemotherapy elevated glycolytic intermediate levels. This response was blocked by the ketogenic diet, and bulk tumor glucose levels were lowest in the combined chemotherapy-ketogenic diet condition. Thus, glycolysis may be particularly critical for tumor cells to survive chemotherapy, and, by limiting glucose availability, the ketogenic diet may promote chemotherapy efficacy.
Moreover, the ketogenic diet may impact antitumor immunity. In immunocompetent mice, the ketogenic diet induces pro-inflammatory tumor gene expression.63 This is not essential for the chemotherapy-ketogenic diet synergy, as the therapeutic benefits occur also in mice lacking an adaptive immune system. However, the immune system likely contributes to long-term tumor control.
To explore the possibility for such tumor control to translate to the clinic, we have initiated clinical trial NCT04631445. This randomized trial tests standard versus ketogenic diet in combination with triple chemotherapy for metastatic pancreatic cancer. Patient dietary choices are supported by continuous remote medical care delivered over the internet.64 Initial compliance is encouraging and further patient recruitment ongoing.
Limitations of the study
The present paper examines a genetically engineered mouse model of pancreatic cancer, primarily using early-passage flank tumor chunk allografts. The therapeutic benefits of combining cytotoxic chemotherapy and ketogenic diet are validated in genetically engineered mice that spontaneously develop intrapancreatic tumors. An inherent deficiency of these models is lack of the full spectrum of mutations found in human pancreatic cancer.
In terms of the mechanism of the chemotherapy-ketogenic diet synergy, analyses were largely limited to descriptive studies in the syngeneic flank allografts. Unfortunately, the tumor chunk allograft model, while advantageously involving the full complement of tumor cell types and avoiding any passaging in vitro, is not readily genetically manipulable. Accordingly, we did not directly test whether the observed chemotherapy-diet synergy is mechanistically driven by NADH elevation, ROS, or lipid peroxidation. Tools such as LbNOX can be used for such purposes in the future. We also did not definitively determine whether the long-term survival in the ketogenic diet-triple chemotherapy condition is due to the immune system or to the inclusion of cisplatin (which was not tolerated in the immune-deficient NSG mice).
The ketogenic diet itself is a complex manipulation, which alters dietary protein and fat in addition to carbohydrate. Effects of insulin are difficult to dissociate from those of ketone bodies, and we did not independently experimentally manipulate these herein.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Joshua D. Rabinowitz (joshr@princeton.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-seq data and other data in this manuscript are deposited in Mendeley. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cleaved Caspase-3 | Cell Signaling | Cat#9661, RRID:AB_2341188 |
| Ki67 | Abcam | Cat#ab15580, RRID:AB_443209 |
| Anti-Histone H3(acetyl K37) | Abcam | Cat#ab4729, RRID:AB_2118291 |
| Anti-Histone H3 | Abcam | Cat#ab1791, RRID:AB_302613 |
| Anti-Histone H4 | Abcam | Cat#ab10158, RRID:AB_296888 |
| Histone H3ac (pan-acetyl) | Active Motif | Cat#39040, RRID:AB_2687871 |
| Histone H4ac (pan-acetyl) | Active Motif | Cat#39026, RRID:AB_2687872 |
| Acetyl-lysine | Cell Signaling | Cat#9441, RRID:AB_331805 |
| α-Smooth muscle actin | Santa Cruz | Cat# sc-32251, RRID:AB_262054 |
| 4 Hydroxynonenal | Abcam | Cat#ab46545, RRID:AB_722490 |
| β-actn | Cell Signaling | Cat#4970, RRID:AB_2223172 |
| Biotinylated Goat Anti-Rabbit IgG | Vector | Cat#BA-1000, RRID:AB_2313606 |
| Cytokine multiplex antibodies | R&D | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| [U-13C] glucose | Cambridge isotope laboratory | Cat#CLM-1396 |
| [U-13C] lactate | Cambridge isotope laboratory | Cat#CLM-1579 |
| [U-13C] oleate | Cambridge isotope laboratory | Cat#CLM-8763 |
| [U-13C] palmitate | Cambridge isotope laboratory | Cat#CLM-6059 |
| [U-13C] 3-HB | Cambridge isotope laboratory | Cat#CLM-3853 |
| [U-13C] glutamine | Cambridge isotope laboratory | Cat#CLM-1822 |
| [U-13C] leucine | Cambridge isotope laboratory | Cat#CLM-2262 |
| [U-13C] valine | Cambridge isotope laboratory | Cat#CLM-2249 |
| [U-13C] isoleucine | Cambridge isotope laboratory | Cat#CLM-2248 |
| [U-13C] acetate | Cambridge isotope laboratory | Cat#CLM-440 |
| [2-2H] lactate | CDN Isotopes | Cat#D-3837 |
| [3,4,4,4-2H] 3-HB | Cayman Chemicals | Cat#14158 |
| Abraxane | Abraxis Bioscience | Cat#NDC code: 68817-134-50 |
| Gemcitabine | Hospira | Cat#NDC code: 0409-0181-01 |
| Cisplatin | Sigma | Cat#PHR1624 |
| 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide | Sigma | Cat#03449 |
| 3-Nitrophenylhydrazine | Sigma | Cat#N21804 |
| pyridine | Sigma | Cat#270970 |
| DMEM | Corning | Cat# 10-017-CV |
| Matrigel | Corning | Cat# 354234 |
| Lactate dehydrogenase | Sigma | Cat#59747 |
| hydroxybutyrate dehydrogenase | Sigma | Cat#10127833001 |
| TRIzol | Invitrogen | Cat#15596026 |
| NAD | Sigma | Cat#N0632 |
| RIPA buffer (containing | Millipore | Cat#20-188 |
| proteinase inhibitor | Roche | Cat#4693159001 |
| goat serum | Fisher Scientific | Cat#16210064 |
| horseradish peroxidase streptavidin | Vector Laboratories | Cat#SA-5704-100, |
| DAB | Agilent/Dako | Cat#K346811-2 |
| hematoxylin | Sigma | Cat#GHS216 |
| Cytoseal 60 mounting medium | Thermo Scientific | Cat#23-244256 |
| Sirus Red | Sigma | Cat#365548 |
| SignalStain® Boost IHC Detection Reagent (HRP, Rabbit) | Cell Signaling | Cat#8114, RRID:AB_10544930 |
| Immunohistochemistry Application Solutions Kit for rabbit | Cell Signaling | Cat#13079 |
| Critical commercial assays | ||
| insulin | crystal chem | Cat#90080 |
| c-peptide | crystal chem | Cat#90050 |
| IGF1 | R&D systems | Cat#MG100 |
| 4-HNE | Cell Biolabs | Cat#STA-838 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River Laboratories | Cat# 027; RRID:IMSR_CRL:027 |
| Mouse: NSG | Jackson Laboratory | Cat# 005557; RRID:IMSR_JAX: 005557 |
| Mouse: KPC (KrasLSL-G12D/+Trp53R172H/+Pdx-1-Cre) mice | From Haiyonghan’s lab | N/A |
| Mouse: KrasLSL-G12D/+Trp53−/− Pdx-1-Cre mice | From Shawn Davidson’s lab | N/A |
| Software and Algorithms | ||
| El-Maven | Elucidata | https://resources.elucidata.io/elmaven |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
| Isocope | In house developed | |
| Image J | Image J | https://imagej.nih.gov/ij/ |
| Deposited Data | Mendeley data | https://data.mendeley.com/datasets/chm3xr2j7v/1 |
| Others: | ||
| Ketogenic diet | Bio-serv | Cat#S3666 |
| Control diet | Lab diet | Cat#PicoLab 5053 |
| LC-MS Q-Exactive plus | Thermofisher | Cat# IQLAAEGAAPFALGMBDK |
| Biotek Synergy HT | Biotek | N/A |
| XBridge BEH Amide XP column | Waters | Cat# 186006725 |
| Acquity UPLC BEH C18 column | Waters | Cat# 186002350 |
| Mouse Button | Instech | Cat# VABM1B/25 |
| Mouse Jugular Vein Catheter | Instech | Cat# C20PU-MJV1301 |
| Mouse Double Catheter Button | Instech | Cat# VABM2B/25R25 |
| Mouse Carotid Artery Catheter | Instech | Cat# C10PU-MCA1459 |
| Surgery tools | Fine Science Tools | N/A |
| Cyromill | Retsch | Cat# 20.749.0001 |
| Beads | Inframat | Cat# 4039GM-S005 |
| microvette CB300 Z | Sarstedt | Cat# 16.440 |
| SolariX XR MS | Bruker | https://www.bruker.com/products/mass-spectrometry-and-separations/mrms/solarix.html |
| Just infusion Syringe Pump | Just infusion | Cat# NE-300 |
| Visualsonics Vevo 770 system | Fujifilm VisualSonics | N/A |
| cryomicrotome | Leica | Cat#CM3050S |
| HTX TM-Sprayer | HTX Technologies | N/A |
| ChemiDoc MP system | Bio-Rad | N/A |
| Nikon Eclipse 80i microscope | Nikon | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse husbandry and PDAC tumor models
All mouse work was approved by the Institute Animal Care and Use Committees (IA-CUCs) at Princeton University or the University of Arizona. Wild type C57BL/6 mice were obtained at 8-weeks of age from Charles River Laboratories and NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were obtained at 8-weeks of age from Jackson Laboratory. The KPC (KrasLSL-G12D/+Trp53R172H/+Pdx-1-Cre) mice were generated from three mouse parental strains (LSL-KrasG12D/+; LSL-Trp53R172H/+; and Pdx-1-Cre) obtained from National Cancer Institute (NCI) mouse repository and following established procedures described by Hingorani and colleagues.35 All mice were housed under normal light cycle (7:00–19:00) and fed a standard chow diet ad lib (PicoLab Rodent 5053 laboratory Diet St. Louis, MO). NSG mice were provided with acidic drinking water (pH 3.0), which was refreshed weekly. Syngeneic PDAC allograft tumors were established by harvesting tumors from KPC mice, mincing the tissue with surgical scissors into small particles able to pass through a 16G needle, suspending in DMEM medium, mixing with Matrigel (Corning Cat #354234) at a 1:1 ratio (v/v), and then injecting 200 microliters of the mixture subcutaneously into the mouse flank. Allograft tumors were passaged up to two times in syngeneic recipient mice before implantation for use in experiments. Further passaging diminished ketogenic diet responsiveness. Tumor growth was monitored by measuring the tumor dimensions (length, width, and height) twice per week using a caliper. Tumor volume was calculated as 0.5 × (Length × Width × Height).
METHOD DETAILS
Tumor monitoring and chemotherapy
KPC tumor tissue was implanted into flanks of C57BL/6 or NSG mice. Once the tumors were palpable, mice were fasted overnight (16 h) and randomly assigned to standard chow or ketogenic (Bioserv, S3666) diet groups. The ketogenic diet was served in a petri dish and replaced every other day. For studies with PDAC GEMM, KPC mice were monitored biweekly for tumor development using high resolution ultrasound imaging (Visualsonics Vevo 770 system, Fujifilm VisualSonics, Ontario, Canada) and upon tumor sizes reaching 150 mm3, mice were randomly assigned to treatment groups.
The triple chemotherapy regimen included nab-paclitaxel (NDC code: 68817–134-50) dosed at 50 mg/kg via retro-orbital injection, gemcitabine (NDC code: 0409–0181-01) at 70 mg/kg via intraperitoneal injection and cisplatin (sigma PHR1624) at 4 mg/kg via intraperitoneal injection. This triple chemotherapy regimen was administrated three days and six days (day 4 and 7) post-dietary group assignment, with cisplatin omitted for the NSG mice due to poor tolerability. Mouse weight and tumor sizes were monitored until humane endpoint was reached (20% weight loss or tumor size of 1,000 mm3 or tumor length of 20 mm).
Isotope tracer infusion
For lactate circulatory turnover flux measurements, 10–14 weeks old C57BL/6 mice were catheterized via both left carotid artery and right jugular vein, as non-perturbative arterial blood sampling is required for precise measurement of circulating lactate labeling. Mice were allowed to recover for 5 days. Mice were fasted from ZT2 to ZT6.5. Sodium [2-2H] or [U-13C]lactate solution was diluted to 5% w/w in water, and infused into the double catheterized mice from ZT6.5 to ZT9 to pseudo steady state. Arterial blood was sampled to calculate the turnover rate for 2H or 13C lactate.
For other tracing experiments, 10–14 weeks old C57BL6/J mice or mice bearing tumors (KPC flank allografts or KrasLSL-G12D/+Trp53−/− Pdx-1-Cre GEMM, tumor sizes ~ 150 mm3) were catheterized via right jugular vein. Mice were allowed to recover from surgery for 3–5 days. For 3HB circulatory turnover flux experiments, mice were fasted from ZT2 to ZT6.5, and [3,4,4,4-2H] or [U-13C]3HB were infused to catheterized mice from ZT6.5 to ZT9. Tail blood was sampled to calculate the turnover rate for 2H or13C3HB.
To compare the turnover rates of nutrients for mice in different diets, catheterized mice were fasted overnight (16 hours) and then fed a standard chow or ketogenic diet for 1 week. Isotope tracers were infused into the jugular vein catheter at a steady rate for 2.5 hours (ZT14-ZT16.5) followed by tail blood collection, euthanasia by cervical dislocation and tissue (tumor, pancreas) harvesting.
For metabolomics studies, tail blood and tissues were collected from ad lib fed mice at ZT16 on day 7 post-dietary group assignment (one day after second dosage of chemotherapy in those treatment groups).
Blood samples were cooled on ice to coagulate and then serum was collected following centrifugation at 16,000 × g for 10 minutes at 4°C. Tissues samples were collected by clamping in aluminum foil with a pre-cooled Wollenberger like clamp and immediately transferred into liquid nitrogen. All serum and tissues samples were stored at −80°C until further analysis.
LC-MS sample preparation
Tissue samples were dissected into small pieces, transferred into 2 ml Eppendorf tubes, and pulverized using a Cryomill (Retsch). 10–20 mg of the resulting tissue powder was weighed into a pre-cooled Eppendorf tube for metabolite extraction. Water soluble metabolites were extracted with 40:40:20 methanol: acetonitrile: water with 0.5% formic acid pre-cooled to −20°C. Extraction solvent was added at 40X the sample quantity (400 μl of extraction buffer per 10 μg of tissue powder and 120 μl of extraction buffer per 3 μl serum). After vortexing for 10s and keeping samples in ice for 10 min, samples were neutralized by adding 15% ammonium bicarbonate (NH4HCO3) aqueous solution (8.75% v/v of extraction buffer). Samples were then again mixed by vortexing for 10s, centrifuged at 16,000× g for 30 min at 4 °C, and then supernatant was transferred to LC-MS vials for analysis. In Figure 2A, internal standards of glucose,lactate, and 3-HB were spiked into solution, and we consider that 1mg of tissue sample equals to 1ml.
LC-MS method
Water soluble metabolite measurements were obtained by running samples on the Q Exactive PLUS hybrid quadrupole-orbitrap mass spectrometer (Thermo Scientific) coupled with hydrophilic interaction chromatography (HILIC). An XBridge BEH Amide column (150 mm × 2.1 mm, 2.5 μM particle size, Waters, Milford, MA) was used. The gradient was solvent A (95%:5% H2O:acetonitrile with 20 mM ammonium acetate, 20 mM ammonium hydroxide, pH 9.4) and solvent B (100% acetonitrile) 0 min, 90% B; 2 min, 90% B; 3 min, 75%; 7 min, 75% B; 8 min, 70%, 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 14 min, 25% B; 16 min, 0% B, 20.5 min, 0% B; 21 min, 90% B; 25 min, 90% B. The flow rate was 1 50 μL/min with an injection volume of 5 μL and a column temperature of 25°C. The MS scans were in negative ion mode with a resolution of 140,000 at m/z 200. The automatic gain control (AGC) target was 1 × 106 and the scan range was m/z 75–1000. All data from isotope labeling experiments were analyzed by El-MAVEN with natural abundance correction.
Acetate and ketone bodies measurements
5 μL serum or 10mg pulverized tumor tissue was added to 100 μL of 12 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), 15 mM 3-Nitrophenylhy-drazine and pyridine (2% v/v) mixture in methanol, incubated at 4°C for 1 h, centrifuged for 10 min at 16,000 × g, and then the supernatant quenched with 0.5 mM beta-mercaptothion and 0.1% formic acid in water. The resulting samples were used for LC-MS analysis with negative mode electrospray ionization scanning m/z from 100 to 300 at 1 Hz with 140,000 resolution on Q Exactive. LC separation was reversed phase C18 on an Acquity UPLC BEH C18 column (2.1 mm × 100 mm, 1.7 5 μm particle size, 130 Å pore size; Waters, Milford, MA) using a gradient of solvent A (water), solvent B (methanol): 0 min, 10% B; 1 min, 10% B; 5 min, 30% B; 7 min, 100% B; 11 min, 100% B; 11.5 min, 10% B; 14 min, 10% B. Flow rate was 200 μL/min and column temperature was 60°C with an injection volume of 10 μL.
RNA preparation and RNA-seq analysis
Total RNA was extracted using TRIzol (Invitrogen, 15596026) from snap-frozen tumor samples. RNA quality was confirmed using the Agilent 2100 Bioanalyzer. Libraries were prepared from 100–500 ng of total RNA with barcoded multiplexing. Single-end sequencing was carried out on an Illumina HiSeq 2500 with a 100-bp read length. For image analysis and base calling, Illumina CASAVA-1.8.2 were utilized. After the read quality was assessed with fastqc, reads were mapped against the reference genome and transcript annotation (GRCm38.p6) using STAR65 with the default setting plus the “–quantMode TranscriptomeSAM” and “–outSAMtype BAM” options. Gene expression was quantified from the BAM files by RSEM66 using the “rsem-calculate-expression” function with the “–estimate-rspd” and “–append-names” options. Differential expression gene analysis was performed with DESeq2.67 Briefly, the results of RSEM were imported by the “DESeqDataSetFromTximpor” function in the tximport package with the following options: “type=‘rsem’, txIn = FALSE, txOut=FASLE”. After the establishment of DESeq data set “estimateSizeFactors”, counts were normalized with the “counts” function. Differential expression analysis between Keto-Diet and Chow-Diet was performed with the “DESeq” command, and the results exported by the “results” command.
Cytokine multiplex sample preparation and analysis
Luminex discovery assay was performed using mouse premixed multi-analyte kit (R&D systems). Analyte-specific antibodies pre-coated on magnetic microparticles are embedded with fluorophores at a set ratio for each unique bead region. Samples, standards, and microparticles are mixed in a well of the microtiter plate. Analyte of interest binds to the immobilized antibodies. A wash step is performed to remove any unbound substances. A biotinylated antibody cocktail specific to the analyte of interest is added to each well. After a wash to remove unbound biotinylated antibody, streptavidin-phycoerythrin conjugate (Streptavidin-PE) is added to each well that binds to the biotinylated antibody. Final washes remove unbound Streptavidin-PE, the microparticles are resuspended in buffer and read using Luminex 200. Analysis with the Luminex 200 uses one laserto excite the dyes inside each bead to identify the bead region and the second laser to excite the PE to measure the amount of analyte bound to the bead.
MALDI imaging mass spectrometry
At time of harvesting, tumor samples were snap-frozen on dry ice for 30 min and stored at −80°C until sectioning. 10 μm-thick tumor sections were collected on a cryomicrotome (Leica CM3050S, Wetzlar, Germany). Then the tissues were thaw-mounted on indium tin oxide (ITO)-coated glass (Bruker Daltonics, Bremen, Germany) and kept in desiccator under vacuum for 20 mins. Serial sections were collected on standard glass slides for H&E staining.
Tissue sections after desiccation were coated with matrix by spraying 10 mg/ml N-(1-naphthyl) ethylenediamine dihydrochloride (NEDC) in 70% MeOH using an HTXTM-Sprayer (HTX Technologies, LLC). The sprayer temperature was set to 80°C with a flow rate of 0.1 ml/min, a velocity of 1000 mm/min, track spacing of 2 mm and pressure of 10 psi. Ten passes of matrix were applied to each slide with 30 s of drying time between each pass. The MALDI-FTICR measurements were obtained using a solarix XR FTICR mass spectrometer equipped with 9.4 T magnet and parcel (Bruker Daltonics, Bremen, Germany). The resolving power of the parcel was R=120,000 at m/z 500. The x-y raster width was set to 50 μm using Smartbeam-II laser optics. A spectrum was accumulated from 300 laser shots at 1200 Hz. All images and isotope data were examined and analyzed using the open-source in-house software IsoScope.
Kinetic isotope effect (KIE) measurements
We carried out in vitro enzyme assays to assess the KIE for lactate dehydrogenase and 3HB dehydrogenase. 20 mM [U-13C]sodium lactate, [2-2H]sodium lactate, [U-13C]sodium 3HB, and [3,4,4,4-2H]sodium 3HB substrate solutions were prepared in advance. 200 mM Tris pH 8.0 and 25 mM NAD (Sigma N0632) solutions were prepared on day of experiment. Lactate dehydrogenase (Sigma 59747), and hydroxybutyrate dehydrogenase (Sigma 10127833001) enzymes were diluted to 0.5U/ml. Solution A, consisting of 12.5 μl of Tris buffer, 1 μl of NAD solution, 24.5 μl of water, and 2 μl of enzyme solution, was prepared as a mixture and added directly in half-area 96 well plates (Corning 3881). 10 μl of each substrate solution was added into solution A and NADH absorbance was measured at 340 nm. For each enzyme and substrate combination, the NADH absorbance was plotted over time to determine the slope and the kinetic isotope effect was calculated from the ratio of the 13C-substrate slope to the 2H-substrate slope.
Western blotting for histone acetylation
Pulverized tumor tissue was weighed (10mg) and proteins were extracted with RIPA buffer (Millipore:20–188) containing proteinase inhibitor (Roche: 4693159001) for 1 hour, followed by centrifuging at 16,000g for 10 mins to get supernatant. Protein concentrations were determined using Pierce BCA protein assay kit (ThermoFisher#23225). Around 20 μg of protein was loaded (mixed with loading buffer) into 4–20% gel (Bio-Rad: 4568095), transferred onto membrane, incubated with primary antibodies against β-actin (Cell Signaling #4970), total H3(ab1791), total H4 (ab10158), Pan-H3ac (Active Motif 39040), Pan-H4ac (Active Motif 39026), Pan-acetyllysine (Cell Signaling 9441), 4-HNE(ab46545), washed with TBST, incubated with secondary antibody and visualized using the Bio-Rad ChemiDoc MP system.
ELISA
Serum samples for insulin (crystal chem 90080), c-peptide (crystal chem 90050) and IGF1 (R&D systems MG100) were analyzed according to ELISA kit instruction. Tissue samples for protein-4HNE adduct were analyzed according to Cell Biolabs OxiSelect HNE adduct competitive ELISA kit (STA-838). Basically, pulverized tissues were lysed in RIPA buffer sufficiently and centrifuged to get supernatant. 50 μl of supernatant and standard solutions were added into 4-HNE conjugated plates for 10 mins Incubation. 50 μl of 4-HNE antibody was added for 1 hour incubation with sufficient wash afterwards. 100 μl of secondary antibody-HRP conjugates was added each well for 1 hour incubation with sufficient wash afterwards. Absorbance values were read after suggested substrate solution and stop solution addition. Results were calculated according to the standard curve.
Immunohistochemical staining
Tumor specimens from the mouse models were fixed by immersing in 10% buffered formalin overnight at 4°C and maintained in 70% ethanol for paraffin embedding. Immunohistochemical staining was performed with antibodies against H3K27ac (1:1000; Abcam, ab4729) and alpha-SMA (1:1000; Santa Cruz, ab32251), using Immunohistochemistry Application Solutions Kit for rabbit (Cell Signaling, 13079) or mouse antibody (Cell Signaling, 8114) following the manufacturer’s protocols. Antigen retrieval was performed in a pressure cooker for 15 min with citric acid-based buffer, pH 6.0 (Vector Labs, H-3300). Sirius Red staining was performed with 0.1% Direct Red 80 (Sigma, 365548) in 1.3% picric acid (Sigma, P6744) after hematoxylin staining. Slides were scanned using a VS-100 scanning microscope (Olympus), and quantifications was performed with ImageJ.
For Ki67 and cleaved caspase-3 staining, paraffin sections were deparaffinized, rehydrated, and heated for 25 min at 95°C in 10 mM (pH 6) citrate buffer (K035, Diagnostic Biosystems). After that, paraffin sections were incubated with 3% hydrogen peroxide (715333, Walgreens) for 10 min and blocked in 10% goat serum (16210064, Fisher Scientific) for 1 h at room temperature and then incubated overnight at 4°C with primary antibody against Ki67 (1:2000 dilution, ab15580, Abcam), Cleaved Caspase-3 (1:150 dilution, 9661, Cell Signaling). The following day, paraffin sections were incubated with biotin-conjugated secondary antibody for 30 min (BA-1000, Vector), horseradish peroxidase streptavidin for 10 min (SA-5704–100, Vector Laboratories) and developed by DAB (K346811–2, Agilent/Dako) followed by hematoxylin (GHS216, Sigma) staining. Sections were then dehydrated, mounted in Cytoseal 60 mounting medium (23–244256, Thermo Scientific) and analyzed using a Nikon Eclipse 80i microscope. For quantification of IHC on Ki67, active caspase-3 were analyzed by quantifying at least 10 images at 20× magnification using ImageJ.
Metabolic flux quantitation
Circulatory turnover flux, Fcirc was calculated as previously described32, using arterial serum for lactate due to its substantial arterial-venous labeling gradient and tail vein serum for other nutrients:
R is the infusion rate of the labeled tracer and L is the fractional enrichment of infused labeled form of the metabolite tracer. Fcirc values of 2H-labeled metabolites were corrected for kinetic isotope effect (KIE, determined from the enzymatic assays) by multiplying the observed Fcirc by KIE value of 2H tracer.
To calculate the average carbon atom labeling of metabolites, the following formula was used:
N is the number of carbon atoms (including both 12C and 13C) in the metabolite, i is the number of 13C atoms in the metabolite, and M is the measured abundance of the given isotopic form (when M is reported in fractional abundance, as is common, then the denominator is 1 and can be omitted). The normalized carbon labeling of downstream metabolite (Y) from a tracer (X) is then calculated by dividing the average carbon labeling of the metabolite of interest (either in serum or tissues) to the average carbon labeling of the tracer in serum.
The direct contribution of circulating nutrients to the metabolites of interest is calculated using the logic described previously.32,68 Basically, every 13C-labeled carbon in each metabolite of interest can derived from two ways: directly from the tracer or indirectly from another circulating nutrient labeled by the tracer. By performing independent infusions of each circulating nutrient that labels the metabolite of interest, we can solve for the direct contribution of each circulating nutrient to a given metabolite pool. For example, to calculate the direct contribution of glucose and lactate to the TCA metabolite malate within a KPC tumor, we can write two equations:
Where Ltumoral←glc and Lserumlac←glc represent normalized carbon labeling of tumor malate or serum lactate from U-13C glucose tracer, Ltumormal←lac and Lserumglc←lac represent normalized labeling of tumor malate or serum glucose from U-13C lactate tracer, and fmal←glucose and fmal←lac represent the direct contribution of malate from circulating glucose or lactate. As all the L values are empirically measured, we can determine the f values using linear algebra (2 equations with 2 unknowns). We can expand this logic to multiple tracers (1..n), and write the algebra matrix in the following format to solve for each f value (n equations with n unknowns):
This same approach was used to calculate intratumoral pyruvate/lactate sources from [U-13C]glucose and [U-13C]lactate infusions. Optimization were performed in MATLAB function “fmincon”. Errors of the output f were obtained by Monte Carlo simulations. Each measured element L (in both the matrix and the vector) were randomly sampled multiple times (50), assuming a normal distribution with the experimentally measured standard deviation.69
QUANTIFICATION AND STATISTICAL ANALYSIS
Samples sizes are defined in each figure legend. Unless otherwise specified, results for biological replicates are presented as mean ± SEM. Statistical significance was calculated using unpaired Student’s t test when comparing two groups, one-way ANOVA followed by Tukey test when comparing three or more, two-way ANOVA followed by Tukey test when comparing tumor growth in different groups, and log-rank by Mantel-Cox test for Kaplan-Meier curve.
Supplementary Material
Highlights.
Ketogenic diet synergizes with classical chemotherapy to treat murine pancreas cancer
In response to ketosis, tumors suppress glucose use and induce 3-hydroxybutyrate use
Therapeutic benefit correlates with elevated tumor NADH/NAD ratio
Survival gain from chemotherapy is roughly tripled in the ketogenic diet condition
Context and significance.
Pancreatic ductal adenocarcinoma is one of most deadly cancers. Despite improvements in chemotherapy, survival remains terribly short. Dietary manipulation is an understudied strategy to improve cancer therapy. Ketogenic diet involves eating mainly fat with almost no carbohydrates. Using a mouse cancer model, we show that ketogenic diet changes pancreatic cancer metabolism and its response to chemotherapy, decreasing insulin and glucose use while increasing use of 3-hydroxybutyrate (a ketone body) and causing oxidative stress in cancer cells. This diet-driven change results in increased tumor sensitivity to chemotherapy, with ketogenic diet roughly tripling the survival benefits of chemotherapy alone. A randomized clinical trial testing whether these benefits translate to patients with metastatic pancreatic cancer is open and currently enrolling patients.
ACKNOWLEDGMENTS
This work was supported by funding to J.D.R. from the US National Institutes of Health (NIH) (R01CA163591 and DP1DK113643), Stand Up to Cancer (SU2-CAACR-DT-20-16), and Ludwig Cancer Research. L.Y. was supported by a postdoctoral fellowship from the New Jersey Commission on Cancer Research. W.L. was supported by NIH grant R50CA211437. J.Y.G. is supported by NIH grant R01CA237347-01A1, ACS grant 134036-RSG-19-165-01-TBG, Rutgers Busch Biomedical Grant, and the New Jersey Health Foundation. R.E. is a March of Dimes Chair in Molecular and Developmental Biology fellow at the Salk Institute and a Nomis Distinguished Scholar, and is supported by NIH grant #DK057978 the Lustgarten Foundation, the Don and Lorraine Freeberg Foundation, and a gift from the David C. Copley Foundation.
Funding:
NIH R01CA163591, R50CA211437, R01CA237347-01A1, R01DK057978; NJCCR; NJHF; SU2C-AACR-DT-20-16; ACS134036-RSG-19-165-01-TBG; Rutgers Busch Biomedical Grant; Freeberg Foundation; Copley Foundation; Ludwig Cancer Research.
Footnotes
DECLARATION OF INTERESTS
J.D.R. is an adviser and stockholder in Kadmon Pharmaceuticals, Colorado Research Partners, L.E.A.F. Pharmaceuticals, Bantam Pharmaceuticals, Barer Institute, and Rafael Pharmaceuticals; a paid consultant of Pfizer; a founder, director, and stockholder of Farber Partners, Serien Therapeutics, and Sofro Pharmaceuticals; a founder and stockholder in Toran Therapeutics; inventor of patents and patent applications held by Princeton University, including a patent application related to ketogenic diet for cancer therapy.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.medj.2021.12.008.
REFERENCES
- 1.Korge P, Calmettes G, and Weiss JN (2015). Increased reactive oxygen species production during reductive stress: the roles of mitochondrial glutathione and thioredoxin reductases. Biochim Biophys Acta 1847, 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodman RP, et al. (2020). Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 583, 122–126. 10.1038/s41586-020-2337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hompland T, et al. (2018). Combined MR imaging of oxygen consumption and supply reveals tumor hypoxia and aggressiveness in prostate cancer patients. Cancer Res 78, 4774–4785. [DOI] [PubMed] [Google Scholar]
- 4.Koong AC, et al. (2000). Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol 48, 919–922. [DOI] [PubMed] [Google Scholar]
- 5.Titov DV, et al. (2016). Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Favaro E, et al. (2012). Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab 16, 751–764. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan MR, et al. (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birsoy K, et al. (2015). An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan LB, et al. (2015). Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, et al. (2020). Serine catabolism feeds NADH when respiration is impaired. Cell Metab 31, 809–821.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martínez-Reyes I, et al. (2020). Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292. 10.1038/s41586-020-2475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patgiri A, et al. (2020). An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD+ imbalance. Nat Biotechnol 38, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erkan M, et al. (2012). The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol 9, 454–467. [DOI] [PubMed] [Google Scholar]
- 14.Kamphorst JJ, et al. (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci 110, 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jameson GS, et al. (2017). A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer. J Clin Oncol 35, 341. [Google Scholar]
- 16.Jameson GS, et al. (2020). Response rate following albumin-bound paclitaxel plus gemcitabine plus cisplatin treatment among patients with advanced pancreatic cancer: a phase 1b/2 pilot clinical trial. JAMA Oncol 6, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Von Hoff DD, et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao X, et al. (2019). Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572, 397–401. 10.1038/s41586-019-1437-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddocks ODK, et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. [DOI] [PubMed] [Google Scholar]
- 20.Caffa I, et al. (2020). Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 583, 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Groot S, et al. (2020). Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat Commun 11, 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shukla SK, et al. (2014). Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weber DD, Aminazdeh-Gohari S, and Kofler B (2018). Ketogenic diet in cancer therapy. Aging 10, 164–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber DD, et al. (2020). Ketogenic diet in the treatment of cancer—where do we stand? Mol Metab 33, 102–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zahra A, et al. (2017). Consuming a ketogenic diet while receiving radiation and chemotherapy for locally advanced lung cancer and pancreatic cancer: the university of Iowa experience of two phase 1 clinical trials. Radiat. Res. 187, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lien EC, et al. (2021). Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 5997, 302–307. 10.1038/s41586-021-04049-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poff A, Ari C, Arnold P, Seyfried T, and D’Agostino D (2014). Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int. J. Cancer J. Int. Cancer 135, 1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimazu T, et al. (2013). Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hopkins BD, et al. (2018). Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks GA (2018). The science and translation of lactate shuttle theory. Cell Metab. 27, 757–785. [DOI] [PubMed] [Google Scholar]
- 31.Faubert B, et al. (2017). Lactate metabolism in human lung tumors. Cell 171, 358–371.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hui S, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rabinowitz JD, and Enerbäck S (2020). Lactate: the ugly duckling of energy metabolism. Nat. Metab. 2, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghergurovich JM, et al. (2021). Local production of lactate, ribose phosphate, and amino acids by human triple-negative breast cancer. Med 2, 736–754.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hingorani SR, et al. (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- 36.Olive KP, et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogl T, et al. (2007). Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 13, 1042–1049. [DOI] [PubMed] [Google Scholar]
- 38.Xia C, et al. (2019). MRP14 enhances the ability of macrophage to recruit T cells and promotes obesity-induced insulin resistance. Int. J. Obes. 43, 2434–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antoch G, et al. (2004). Accuracy of whole-body dual-modality fluorine-18–2-fluoro-2-deoxy-D-glucose positron emission tomography and computed tomography (FDG-PET/CT) for tumor staging in solid tumors: comparison with CT and PET. J. Clin. Oncol. 22, 4357–368. [DOI] [PubMed] [Google Scholar]
- 40.Blodgett TM, Meltzer CC, and Townsend DW (2007). PET/CT: form and function. Radiology 242, 360–385. [DOI] [PubMed] [Google Scholar]
- 41.Juweid ME, and Cheson BD (2006). Positron-emission tomography and assessment of cancer therapy. N. Engl.J. Med. 354, 496–507. 10.1056/NEJMra050276. [DOI] [PubMed] [Google Scholar]
- 42.Lardinois D, et al. (2003). Staging of non-small-cell lung cancer with integrated positron-emission tomography and computed tomography. N. Engl. J. Med. 348, 2500–2507. [DOI] [PubMed] [Google Scholar]
- 43.Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464. [DOI] [PubMed] [Google Scholar]
- 44.Barborka CJ (1928). Ketogenic diet treatment of epilepsy in adults. J. Am. Med. Assoc. 91, 73. [Google Scholar]
- 45.Bates M (1971). Kinetics of ketone body metabolism in fasted and diabetic rats. Am. J. Physiol. 221, 984–991. [DOI] [PubMed] [Google Scholar]
- 46.McGarry JD, Guest MJ, and Foster DW (1970). Ketone body metabolism in the ketosis of starvation and alloxan diabetes. J. Biol. Chem. 245, 4382–4390. [PubMed] [Google Scholar]
- 47.Roberts MN, et al. (2017). A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 26, 539–546.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aminzadeh-Gohari S, et al. (2017). A ketogenic diet supplemented with medium-chain triglycerides enhances the anti-tumor and anti-angiogenic efficacy of chemotherapy on neuroblastoma xenografts in a CD1-nu mouse model. Oncotarget 8, 64728–64744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morscher RJ, et al. (2015). Inhibition of neuroblastoma tumor growth by ketogenic diet and/or calorie restriction in a CD1-nu mouse model. PLoS One 10, e0129802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanarek N, Petrova B, and Sabatini DM (2020). Dietary modifications for enhanced cancer therapy. Nature 579, 507–517. [DOI] [PubMed] [Google Scholar]
- 51.Pérez-Escuredo J, et al. (2016). Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 1863, 2481–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Draoui N, and Feron O (2011). Lactate shuttles at a glance: from physiological paradigms to anti-cancer treatments. Dis. Model. Mech. 4, 727–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanner LB, et al. (2018). Four key steps control glycolytic flux in mammalian cells. Cell. Syst. 7, 49–62.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.García-Cañaveras JC, Chen L, and Rabinowitz JD (2019). The tumor metabolic microenvironment: lessons from lactate. Cancer Res. 79, 3155–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luengo A, et al. (2021). Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 81, 691–707.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haces ML, et al. (2008). Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 211, 85–96. [DOI] [PubMed] [Google Scholar]
- 57.Jain SK, Kannan K, and Lim G (1998). Ketosis (acetoacetate) can generate oxygen radicals and cause increased lipid peroxidation and growth inhibition in human endothelial cells. Free Radic. Biol. Med. 25, 1083–1088. [DOI] [PubMed] [Google Scholar]
- 58.Maalouf M, Sullivan PG, Davis L, Kim DY, and Rho JM (2007). Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rojas-Morales P, Pedraza-Chaverri J, and Tapia E (2020). Ketone bodies, stress response, and redox homeostasis. Redox Biol. 29, 101395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Badgley MA, et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Puchalska P, and Crawford PA (2017). Multidimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab 25, 262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu GY, and Thompson JR (1988). The effect of ketone bodies on alanine and glutamine metabolism in isolated skeletal muscle from the fasted chick. Biochem. J. 255, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lussier DM, et al. (2016). Enhanced immunity in a mouse model of malignant glioma is mediated by a therapeutic ketogenic diet. BMC Cancer 16, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Athinarayanan SJ, et al. (2019). Long-term effects of a novel continuous remote care intervention including nutritional ketosis for the management of type 2 diabetes: a 2-year non-randomized clinical trial. Front. Endocrinol 10, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dobin A, et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hui S, et al. (2020). Quantitative fluxomics of circulating metabolites. Cell Metab. 32, 676–688.e4. https://doi.org/10.1016Zj.cmet.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.TeSlaa T, et al. (2021). The source of glycolytic intermediates in mammalian tissues. Cell Metab. 33, 367–378.e5. 10.1016/j.cmet.2020.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data and other data in this manuscript are deposited in Mendeley. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cleaved Caspase-3 | Cell Signaling | Cat#9661, RRID:AB_2341188 |
| Ki67 | Abcam | Cat#ab15580, RRID:AB_443209 |
| Anti-Histone H3(acetyl K37) | Abcam | Cat#ab4729, RRID:AB_2118291 |
| Anti-Histone H3 | Abcam | Cat#ab1791, RRID:AB_302613 |
| Anti-Histone H4 | Abcam | Cat#ab10158, RRID:AB_296888 |
| Histone H3ac (pan-acetyl) | Active Motif | Cat#39040, RRID:AB_2687871 |
| Histone H4ac (pan-acetyl) | Active Motif | Cat#39026, RRID:AB_2687872 |
| Acetyl-lysine | Cell Signaling | Cat#9441, RRID:AB_331805 |
| α-Smooth muscle actin | Santa Cruz | Cat# sc-32251, RRID:AB_262054 |
| 4 Hydroxynonenal | Abcam | Cat#ab46545, RRID:AB_722490 |
| β-actn | Cell Signaling | Cat#4970, RRID:AB_2223172 |
| Biotinylated Goat Anti-Rabbit IgG | Vector | Cat#BA-1000, RRID:AB_2313606 |
| Cytokine multiplex antibodies | R&D | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| [U-13C] glucose | Cambridge isotope laboratory | Cat#CLM-1396 |
| [U-13C] lactate | Cambridge isotope laboratory | Cat#CLM-1579 |
| [U-13C] oleate | Cambridge isotope laboratory | Cat#CLM-8763 |
| [U-13C] palmitate | Cambridge isotope laboratory | Cat#CLM-6059 |
| [U-13C] 3-HB | Cambridge isotope laboratory | Cat#CLM-3853 |
| [U-13C] glutamine | Cambridge isotope laboratory | Cat#CLM-1822 |
| [U-13C] leucine | Cambridge isotope laboratory | Cat#CLM-2262 |
| [U-13C] valine | Cambridge isotope laboratory | Cat#CLM-2249 |
| [U-13C] isoleucine | Cambridge isotope laboratory | Cat#CLM-2248 |
| [U-13C] acetate | Cambridge isotope laboratory | Cat#CLM-440 |
| [2-2H] lactate | CDN Isotopes | Cat#D-3837 |
| [3,4,4,4-2H] 3-HB | Cayman Chemicals | Cat#14158 |
| Abraxane | Abraxis Bioscience | Cat#NDC code: 68817-134-50 |
| Gemcitabine | Hospira | Cat#NDC code: 0409-0181-01 |
| Cisplatin | Sigma | Cat#PHR1624 |
| 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide | Sigma | Cat#03449 |
| 3-Nitrophenylhydrazine | Sigma | Cat#N21804 |
| pyridine | Sigma | Cat#270970 |
| DMEM | Corning | Cat# 10-017-CV |
| Matrigel | Corning | Cat# 354234 |
| Lactate dehydrogenase | Sigma | Cat#59747 |
| hydroxybutyrate dehydrogenase | Sigma | Cat#10127833001 |
| TRIzol | Invitrogen | Cat#15596026 |
| NAD | Sigma | Cat#N0632 |
| RIPA buffer (containing | Millipore | Cat#20-188 |
| proteinase inhibitor | Roche | Cat#4693159001 |
| goat serum | Fisher Scientific | Cat#16210064 |
| horseradish peroxidase streptavidin | Vector Laboratories | Cat#SA-5704-100, |
| DAB | Agilent/Dako | Cat#K346811-2 |
| hematoxylin | Sigma | Cat#GHS216 |
| Cytoseal 60 mounting medium | Thermo Scientific | Cat#23-244256 |
| Sirus Red | Sigma | Cat#365548 |
| SignalStain® Boost IHC Detection Reagent (HRP, Rabbit) | Cell Signaling | Cat#8114, RRID:AB_10544930 |
| Immunohistochemistry Application Solutions Kit for rabbit | Cell Signaling | Cat#13079 |
| Critical commercial assays | ||
| insulin | crystal chem | Cat#90080 |
| c-peptide | crystal chem | Cat#90050 |
| IGF1 | R&D systems | Cat#MG100 |
| 4-HNE | Cell Biolabs | Cat#STA-838 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River Laboratories | Cat# 027; RRID:IMSR_CRL:027 |
| Mouse: NSG | Jackson Laboratory | Cat# 005557; RRID:IMSR_JAX: 005557 |
| Mouse: KPC (KrasLSL-G12D/+Trp53R172H/+Pdx-1-Cre) mice | From Haiyonghan’s lab | N/A |
| Mouse: KrasLSL-G12D/+Trp53−/− Pdx-1-Cre mice | From Shawn Davidson’s lab | N/A |
| Software and Algorithms | ||
| El-Maven | Elucidata | https://resources.elucidata.io/elmaven |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
| Isocope | In house developed | |
| Image J | Image J | https://imagej.nih.gov/ij/ |
| Deposited Data | Mendeley data | https://data.mendeley.com/datasets/chm3xr2j7v/1 |
| Others: | ||
| Ketogenic diet | Bio-serv | Cat#S3666 |
| Control diet | Lab diet | Cat#PicoLab 5053 |
| LC-MS Q-Exactive plus | Thermofisher | Cat# IQLAAEGAAPFALGMBDK |
| Biotek Synergy HT | Biotek | N/A |
| XBridge BEH Amide XP column | Waters | Cat# 186006725 |
| Acquity UPLC BEH C18 column | Waters | Cat# 186002350 |
| Mouse Button | Instech | Cat# VABM1B/25 |
| Mouse Jugular Vein Catheter | Instech | Cat# C20PU-MJV1301 |
| Mouse Double Catheter Button | Instech | Cat# VABM2B/25R25 |
| Mouse Carotid Artery Catheter | Instech | Cat# C10PU-MCA1459 |
| Surgery tools | Fine Science Tools | N/A |
| Cyromill | Retsch | Cat# 20.749.0001 |
| Beads | Inframat | Cat# 4039GM-S005 |
| microvette CB300 Z | Sarstedt | Cat# 16.440 |
| SolariX XR MS | Bruker | https://www.bruker.com/products/mass-spectrometry-and-separations/mrms/solarix.html |
| Just infusion Syringe Pump | Just infusion | Cat# NE-300 |
| Visualsonics Vevo 770 system | Fujifilm VisualSonics | N/A |
| cryomicrotome | Leica | Cat#CM3050S |
| HTX TM-Sprayer | HTX Technologies | N/A |
| ChemiDoc MP system | Bio-Rad | N/A |
| Nikon Eclipse 80i microscope | Nikon | N/A |