

Abstract
This review aims to provide a comprehensive overview of condensation-based methods for the C−H bond functionalization of amines that feature azomethine ylides as key intermediates. These transformations are typically redox-neutral and share common attributes with classic name reactions such as the Strecker, Mannich, Friedel−Crafts, Pictet−Spengler, and Kabachnik−Fields reaction, while incorporating a redox-isomerization step. This approach provides an ideal platform to rapidly transform simple starting materials into complex amines.
Keywords: C−H bond functionalization, amines, heterocycles, redox-neutral, redox-isomerization, condensation
Graphical Abstract
1. Introduction
1.1. General Remarks
Fully and partially saturated cyclic amines are ubiquitous core structures of natural products, pharmaceutical drugs, and other molecules of interest.1 While numerous methods have been devised that enable the synthesis of such azacycles from acyclic precursors, the preparation of ring-substituted cyclic amines directly from their readily available parent heterocycles by means of C−H bond functionalization holds a special appeal. A broad range of mechanistically diverse approaches have emerged, including but not limited to methods involving deprotonation with strong bases, oxidation, transition metal catalyzed C–H insertion, C–H insertion with carbenes or metal carbenoids, intra- and intermolecular hydride transfer, α-amino radical formation, hydroaminoalkylation, hydrogen borrowing, electrochemistry, and photoredox catalysis.2 Although not necessarily representing a limiting factor, the vast majority of individual methods that fall within these categories are restricted to tertiary or protected amines, while others require special types of substrates (most commonly, tertiary N-aryl amines). Methods for the direct C−H bond functionalization of unprotected primary and secondary amines remain comparatively rare.
1.2. Overview
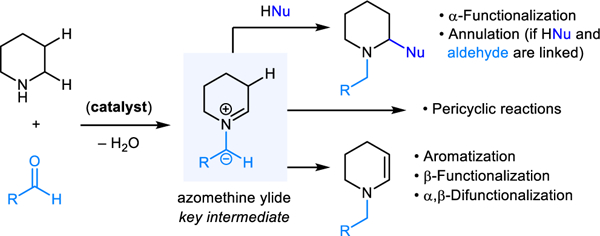
Transformations involving the condensation of primary and secondary amines with aldehydes/ketones enjoy a rich history in organic chemistry and continue to be used widely. Iminium ions such as 1 are typically invoked as intermediates on the path to products 2 (Scheme 1). These transformations include well-known name reactions (e.g., Strecker, Mannich, Friedel−Crafts, Pictet−Spengler, Kabachnik−Fields) among many others. The reactions that are the topic of this review share many features with these classic transformations as they are based on closely related if not identical starting materials (Scheme 1). Water is the only byproduct in both types of reactions. In contrast to classic transformations however, condensation reactions resulting in C–H bond functionalization incorporate a redox-isomerization step. In essence, these transformations merge reductive N-alkylation with oxidative C–H bond functionalization in an overall redox-neutral event.3 As revealed by computational and experimental studies, azomethine ylides (e.g., 4) are key intermediates in this process. While other pathways to 4 play a role in certain variants, the most common precursor to 4 appears to be an N,O-acetal such as 3. The latter can expel one equivalent of carboxylic acid via intramolecular proton abstraction/elimination to form azomethine ylide 4. This species can reengage carboxylic acid to form N,O-acetal 5, a regioisomer of 3. Reaction of a nucleophile with 3 provides α-functionalized products 6, which represent regioisomers of the classic products 2. Because 3 and 5 exist in equilibrium (via 4), and 3 can engage nucleophiles to form classic condensation products 2, the classic and the redox-isomerization pathways tend to compete (vide infra). Other reaction outcomes are also possible. For instance, 5 can undergo elimination of carboxylic acid to form enamine 7, providing the basis for numerous subsequent transformations including aromatization with concurrent substitution of multiple ring positions, β-functionalization, and α,β-difunctionalization. Azomethine ylides 4 derived from N,O-acetals 3 also participate in pericyclic reactions such as (3+2) cycloadditions and 1,5-electrocyclizations. While this type of reactivity is what azomethine ylides are best known for,4 their intermediacy in redox-isomerization reactions predates their first known participation in (3+2) cycloadditions by many decades, although this was not recognized at the time.
Scheme 1.
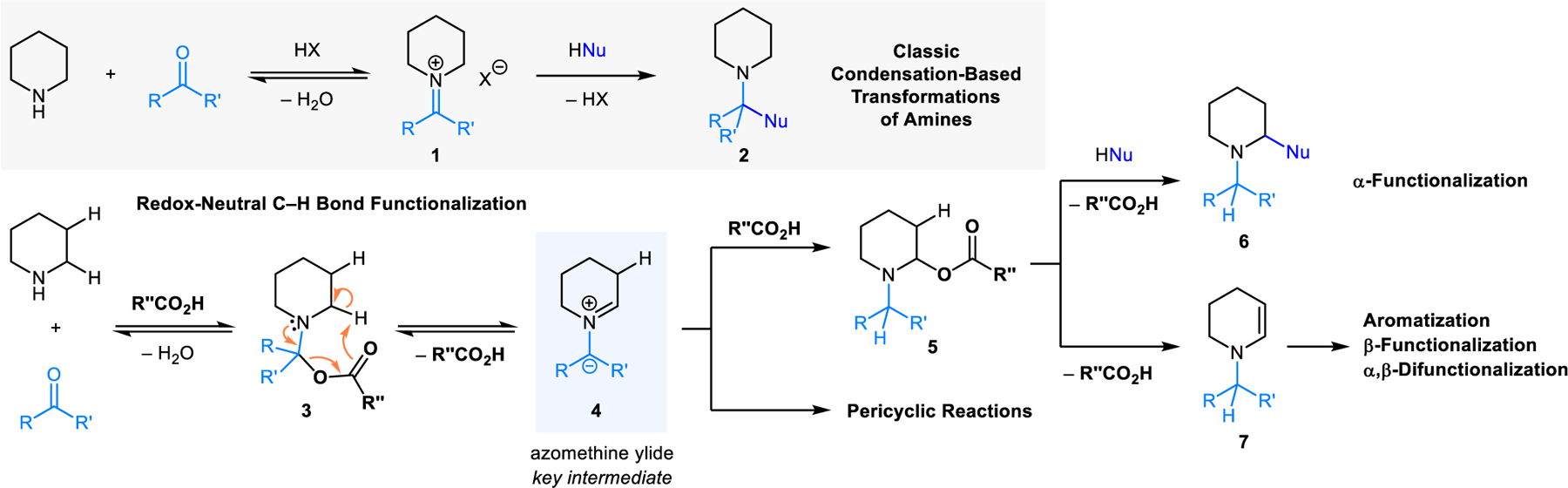
Classic transformations vs. condensation-based C−H functionalization using piperidine as a prototypical example.
1.3. Scope of This Review
Aspects of this topic were previously covered in the form of Accounts by us in 2015,5 and by the Jana group in 2016.6 This Review aims to provide a more comprehensive coverage, including early contributions that have not been reviewed in detail elsewhere, in addition to the many studies that have appeared since the publication of the above mentioned Accounts. The coverage of this Review extends to the end of 2020. While most of the material relates to reactions of cyclic amines, related reactions involving acyclic amines are also discussed as appropriate. Transformations are categorized according to the diverging reaction pathways that follow the formation of the key azomethine ylide intermediate. Outside the scope of this Review are redox-neutral reactions involving intramolecular hydride transfer. While some of these transformations also incorporate a condensation step, the amine undergoing C–H bond functionalization is typically not involved in this directly.2h,7 Also not discussed are reactions that are related to those covered but that are based on the decarboxylative transformation of α-amino acids rather than on C–H bond functionalization.8 Further excluded are reactions of amine substrates possessing activating electron-withdrawing groups in the α-position (e.g., esters of amino acids).
2. Aromatization of Cyclic Amines
2.1. Pyridines from Piperidine
The formation of 3,5-disubstituted pyridines from piperidine and aromatic aldehydes most likely is the earliest known example of a condensation-based C−H bond functionalization of an amine (Scheme 2). This transformation was reported by Rügheimer in 1891 and involves heating of N-benzoyl piperidine and benzaldehyde in neat form at 240−250 °C.9 The product obtained from this reaction was determined to be 3,5-dibenzylpyridine (8a) by comparing its potassium permanganate oxidation products with 3,5-dibenzoylpyridine and pyridine-3,5-dicarboxylic acid. Skraup and Böhm in 1926,10 as well as Parker and Furst in 1958,11 reported on reactions of piperidine and aromatic aldehydes catalyzed by piperidinium acetate in refluxing toluene. The formation of unidentified products was noted. Inspired by the work of Parker and Furst, a few years later, Poirier12 and Burrows13,14 independently studied the reaction of piperidinium acetate with benzaldehyde. The structures of the thus obtained products are identical to the product of Rügheimer’s reaction. This was verified by NMR and comparison with an authentic sample of 3,5-dibenzylpyridine (8a) (prepared independently from pyridine-3,5-dicarboxylic acid). Rügheimer’s reaction is also believed to occur between piperidine and benzaldehyde, with the former being obtained in situ from the hydrolysis of N-benzoyl piperidine. A mechanism for this remarkable transformation was proposed by Poirier and Burrows, who envisioned the isomerization of the initially formed exocyclic iminium ion 9 to endocyclic iminium ion 10, followed by the formation of the enamine intermediate 11. However, omitted from their discussions was the detailed mechanism of the proposed isomerization, in addition to several other key steps.
Scheme 2.
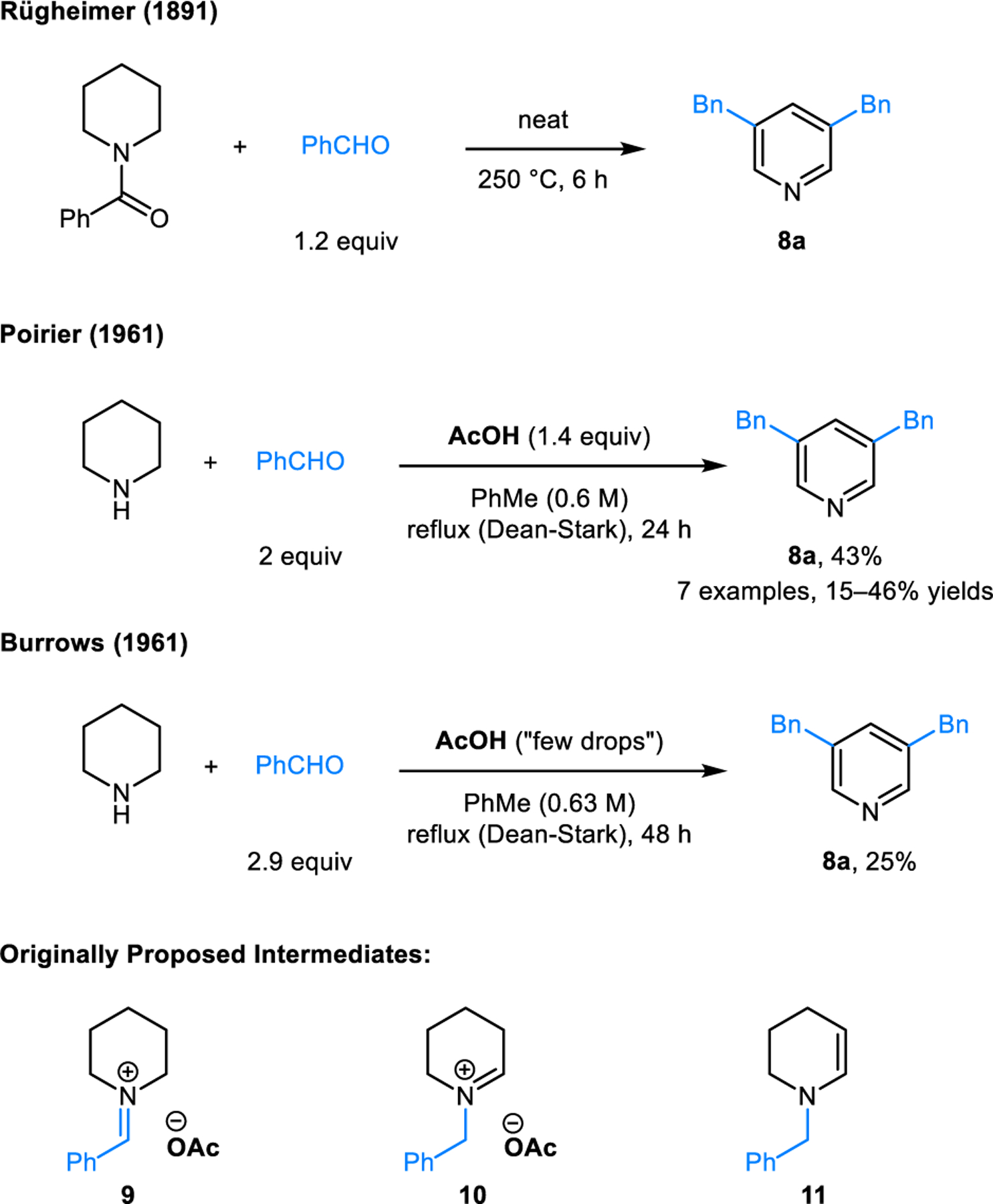
Early examples of 3,5-dibenzylpyridine formation from piperidine and benzaldehyde (Rügheimer-Burrows reaction) and proposed intermediates.
Somewhat surprisingly, only sporadic examples of the Rügheimer-Burrows reaction were reported in the following decades. Kameswari and coworkers reported the synthesis of 3-picoline (12), which was obtained as the major product upon passing a mixture of piperidine and trioxane over γ-Al2O3 (Scheme 3).15 Compared to reactions performed in the presence of acetic acid, which usually proceed in refluxing toluene, this reaction under basic conditions requires over 400 °C. The corresponding reaction performed at 250 °C mainly yields 3,5-dimethylpyridine. Neves and Lodeiro applied the Rügheimer-Burrows reaction to aldehyde 13, enabling the formation of 3,5-bisporphyrinylpyridine 14 which functions as a fluorescent ratiometric probe for zinc ions (Scheme 4).16 Catalytic amounts of lanthanum triflate were found to improve the yield of this reaction. Our group developed modified conditions for conducting Rügheimer-Burrows reactions that are applicable to a range of arylaldehydes.17 Employing benzoic acid as an additive under microwave irradiation greatly shortens the reaction time and improves the yields of pyridines 8 (Scheme 5). Chang and coworkers identified Amberlyst-15 as a catalyst for Rügheimer-Burrows reactions performed in refluxing toluene.18
Scheme 3.
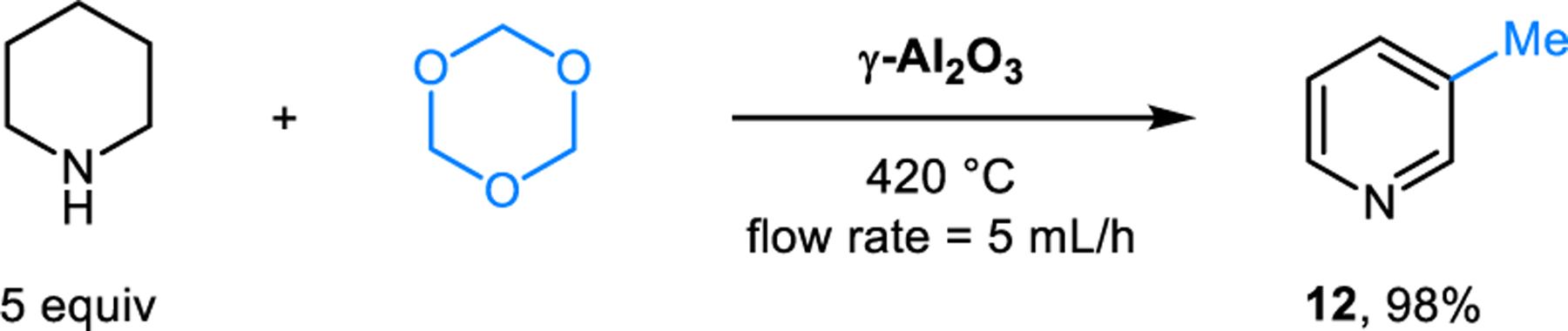
Synthesis of 3-picoline from piperidine and trioxane under basic reaction conditions.
Scheme 4.
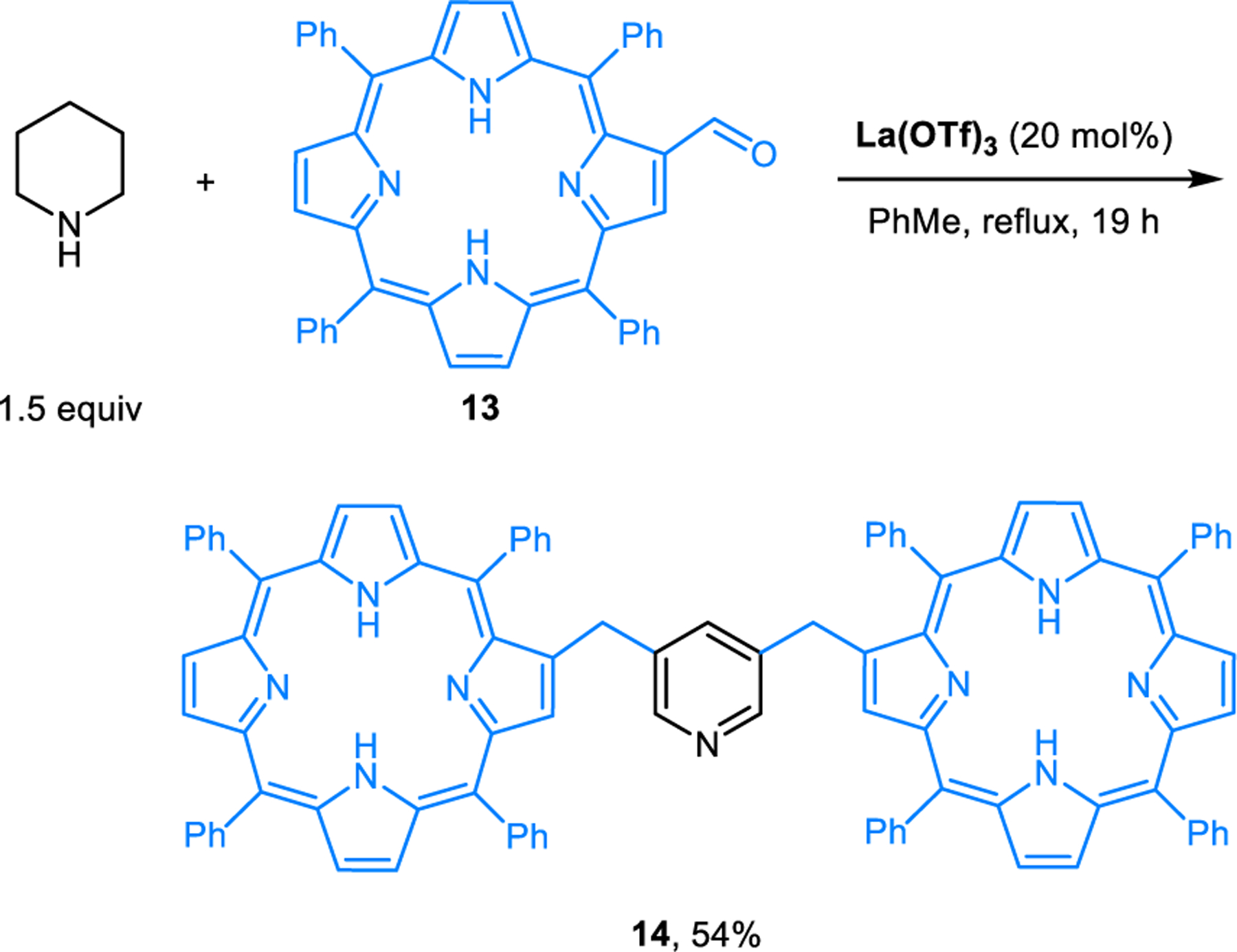
La(OTf)3-catalyzed synthesis of 3,5-bisporphyrinylpyridine via a Rügheimer-Burrows reaction.
Scheme 5.
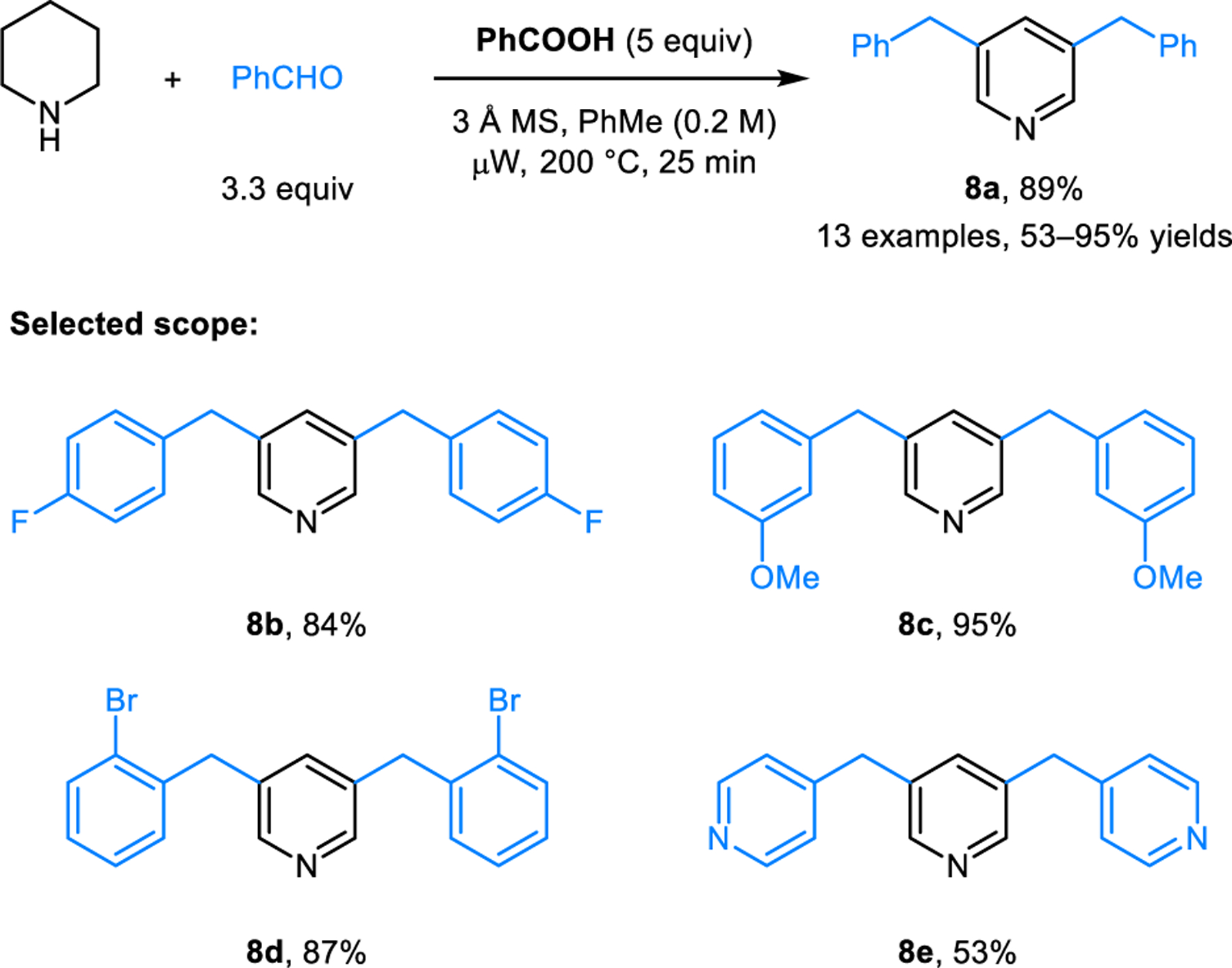
Modified Rügheimer-Burrows reaction under microwave conditions.
2.2. Isoquinolines from Tetrahydroisoquinolines and Quinolines from Tetrahydroquinolines
The Rügheimer-Burrows reaction is also applicable to the synthesis of 4-benzylisoquinoline (15a) from 1,2,3,4-tetrahydroisoquinoline (THIQ), as well as 3-benzylquinolines from substituted 1,2,3,4-tetrahydroquinolines (THQ) such as 3-benzyl-6-chloroquinoline (16) from 6-chloro-THQ (Scheme 6). Burrows first reported these reactions in 1963.14 It is noteworthy that parent THQ lacking the chlorine atom at the 6-position fails to form the desired product. Instead, two equivalents of THQ combine with benzaldehyde in a Friedel-Crafts-type reaction producing 6,6’-(phenylmethylene)bis-THIQ (not shown). The modified reaction conditions developed by our group for the preparation of 3,5-disubstituted pyridines are also applicable to the synthesis of isoquinolines 15 (Scheme 7) and quinolines (e.g., 17, Scheme 8).17
Scheme 6.
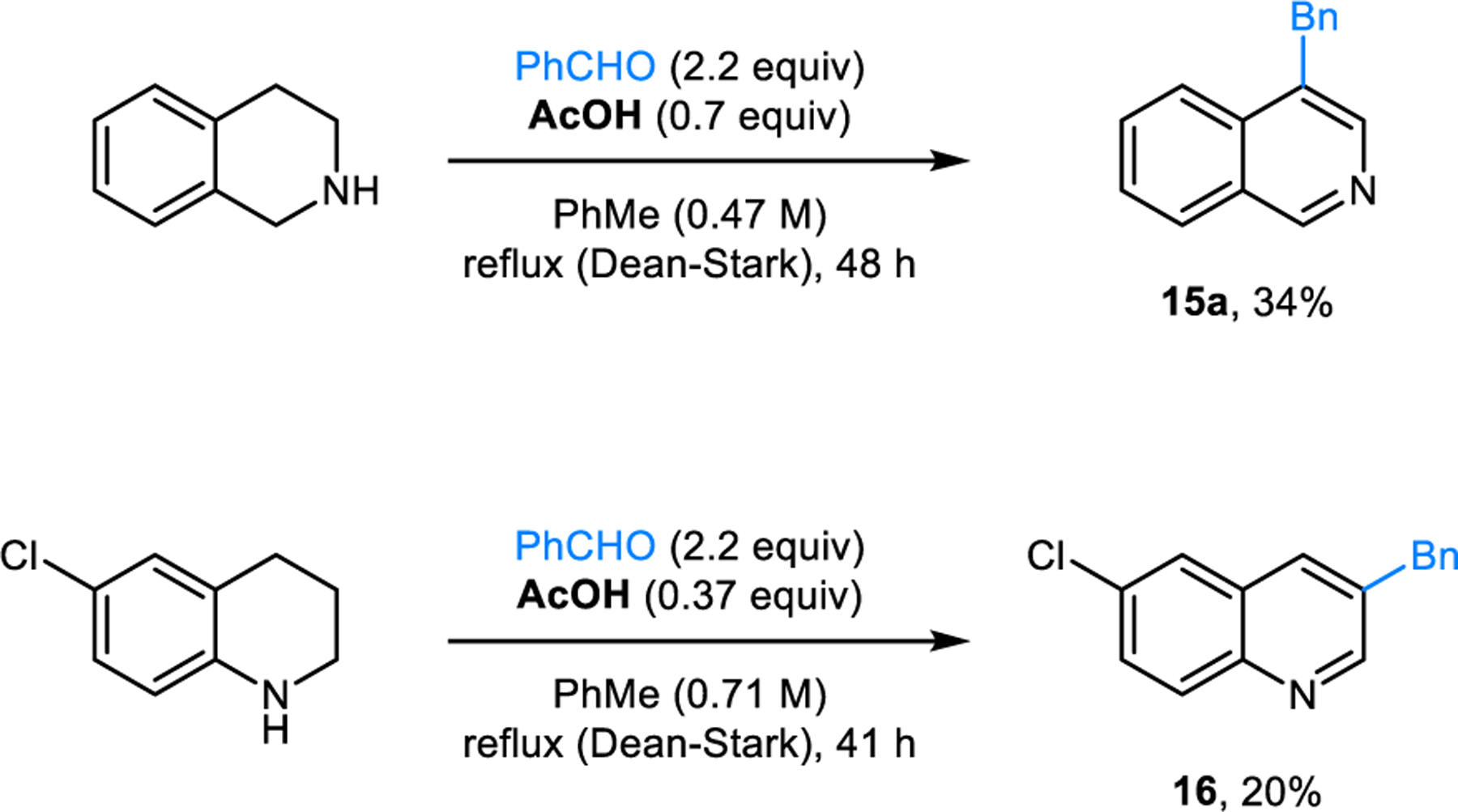
Rügheimer-Burrows reactions with other azacycles.
Scheme 7.
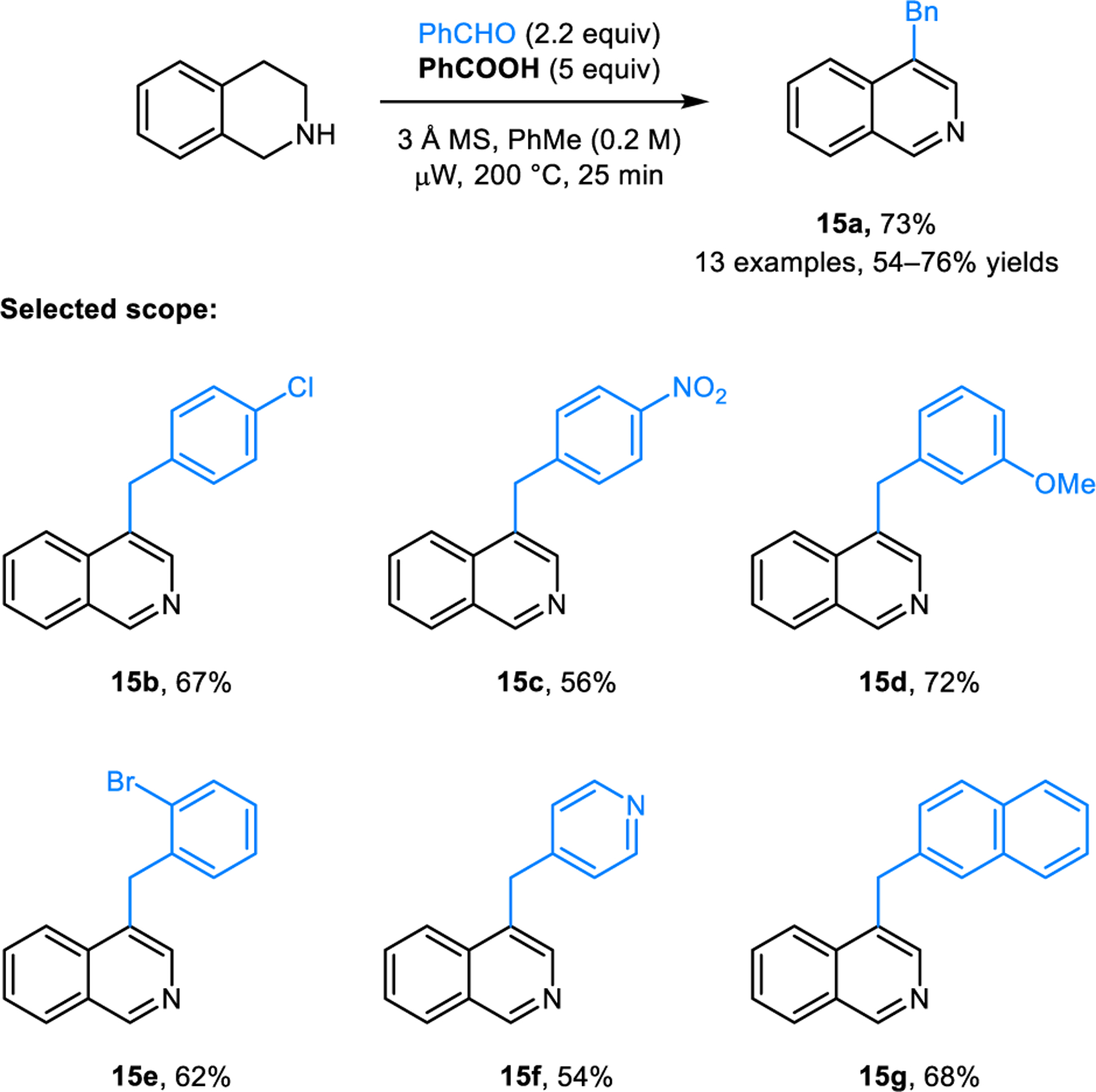
Synthesis of 3-benzylisoquinolines.
Scheme 8.
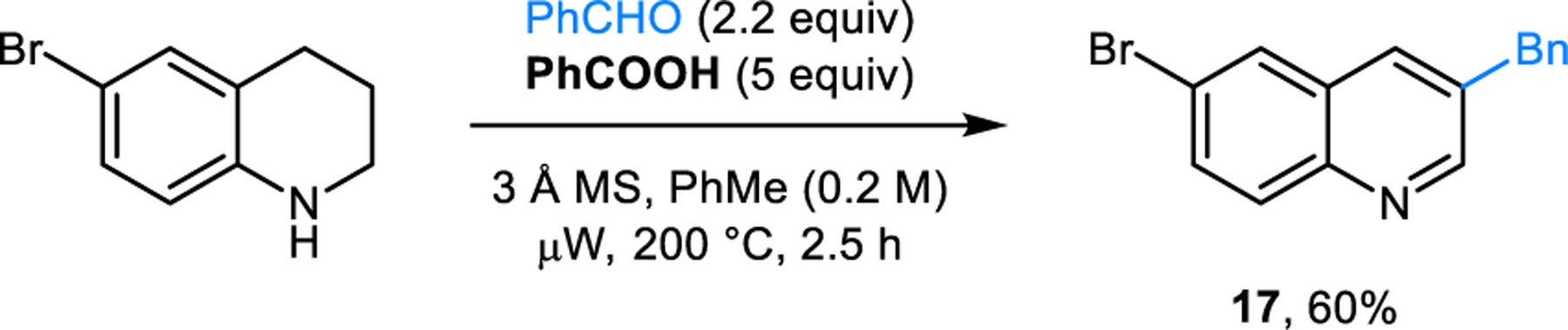
Synthesis of a 3-benzyl-substituted quinoline.
A plausible mechanistic scenario for the Rügheimer-Burrows reaction that is based on previous reports by others, along with our more recent exploration of related transformations (vide infra), is outlined in Scheme 9. Condensation of THIQ and benzaldehyde in the presence of benzoic acid first results in the formation of N,O-acetal 18. This species undergoes isomerization to N,O-acetal 20 via azomethine ylide intermediate 19, consistent with the general mechanism outlined in Scheme 1. N,O-acetal 20 is then thought to undergo further isomerization to N,O-acetal 22 via the intermediacy of azomethine ylide 21. Elimination of acetic acid then results in the formation of enamine 23. Reaction of 23 with benzaldehyde and benzoic acid provides 24 which subsequently generates azomethine ylide 25 upon loss of benzoic acid. Protonation of 25 by benzoic acid leads to aromatization and formation of 26 which then undergoes debenzylation to yield final product 15a. The intermediacy of enamine 23 was established by Sainsbury19 and Dannhardt.20 In addition, indirect evidence for the existence of azomethine ylide 19 was obtained by Dannhardt by trapping this intermediate in a (3+2) dipolar cycloaddition with benzaldehyde.21 An alternative scenario involving azomethine ylide 27 cannot be ruled out but appears less likely in light of computational studies on related systems (vide infra).
Scheme 9.
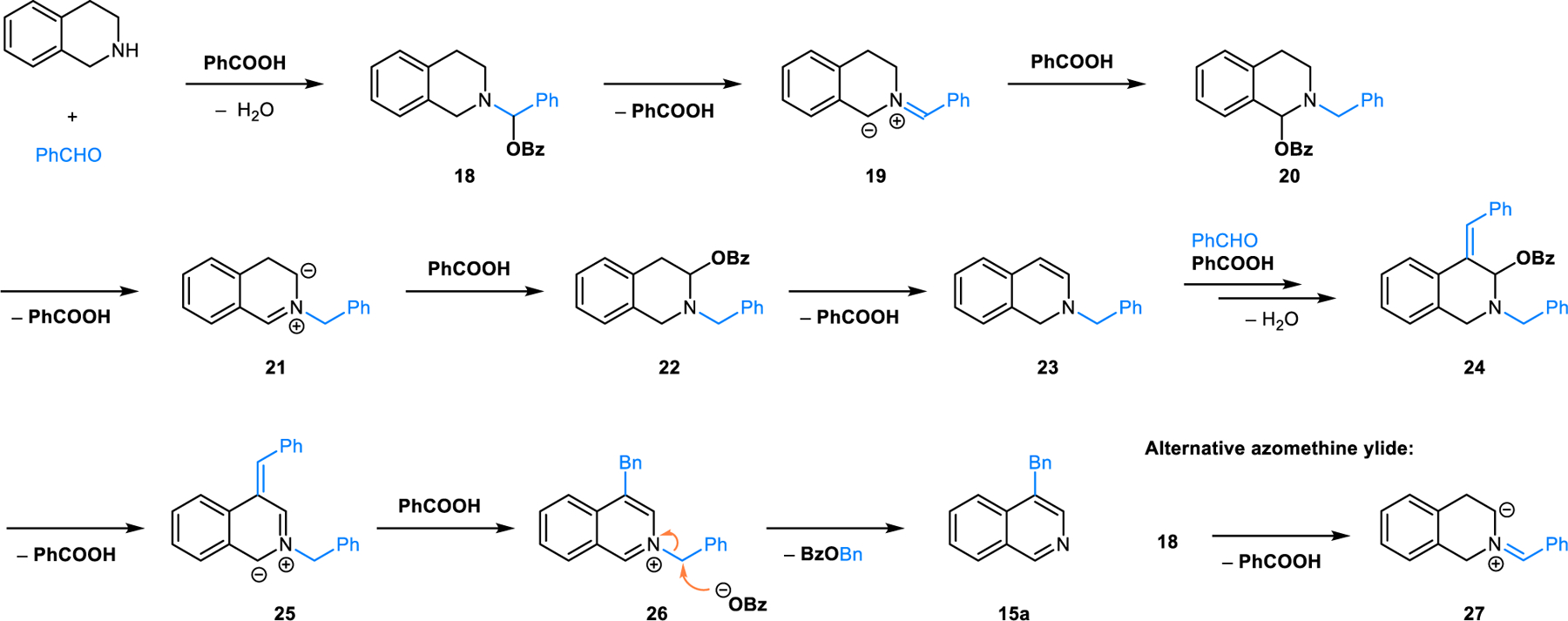
Proposed mechanism for the redox-neutral synthesis of 3-benzylisoquinoline from THIQ and benzaldehyde with benzoic acid.
2.3. Pyrrole from 3-Pyrroline or Pyrrolidine
In 1996, Sayre and coworkers reported on the ability of 3-pyrroline to act as an irreversible inactivator of quinone-dependent amine oxidase.22 The proposed mechanism of inactivation involves the condensation of 3-pyrroline with a p-quinone-type compound, followed by aromatization via the isomerization of an iminium ion intermediate to yield an N-arylpyrrole product (not shown). Cook and coworkers subsequently reported the synthesis of N-cyclohexylpyrrole (28) by heating 3-pyrroline and cyclohexanone under reflux in xylenes, with water being removed by a Dean-Stark apparatus (Scheme 10).23 In addition to 3-pyrroline, redox-neutral pyrrole formation is also feasible with pyrrolidine as the starting material, as reported by Oda and coworkers (Scheme 11).24 In this case, N-alkylation/aromatization is accompanied by alkylation of the 3-position. For instance, heating a mixture of pyrrolidine and isobutyraldehyde in toluene at 180 °C for 20 hours results in the formation of pyrrole 29a in 79% yield. Analogous reactions involving aromatic aldehydes generally provide the corresponding products in lower yields. Toma and coworkers modified Oda’s reaction by employing microwave irradiation conditions.25 While reaction temperatures are still high, significantly shorter reaction times and improved yields were achieved. It is noteworthy that both Cook and Oda proposed iminium ion intermediates and a 1,3-hydride transfer mechanism to account for the overall transformation. However, based on orbital symmetry considerations, 1,3-hydride shifts would need to occur in antarafacial fashion, rendering them essentially geometry-forbidden processes.
Scheme 10.
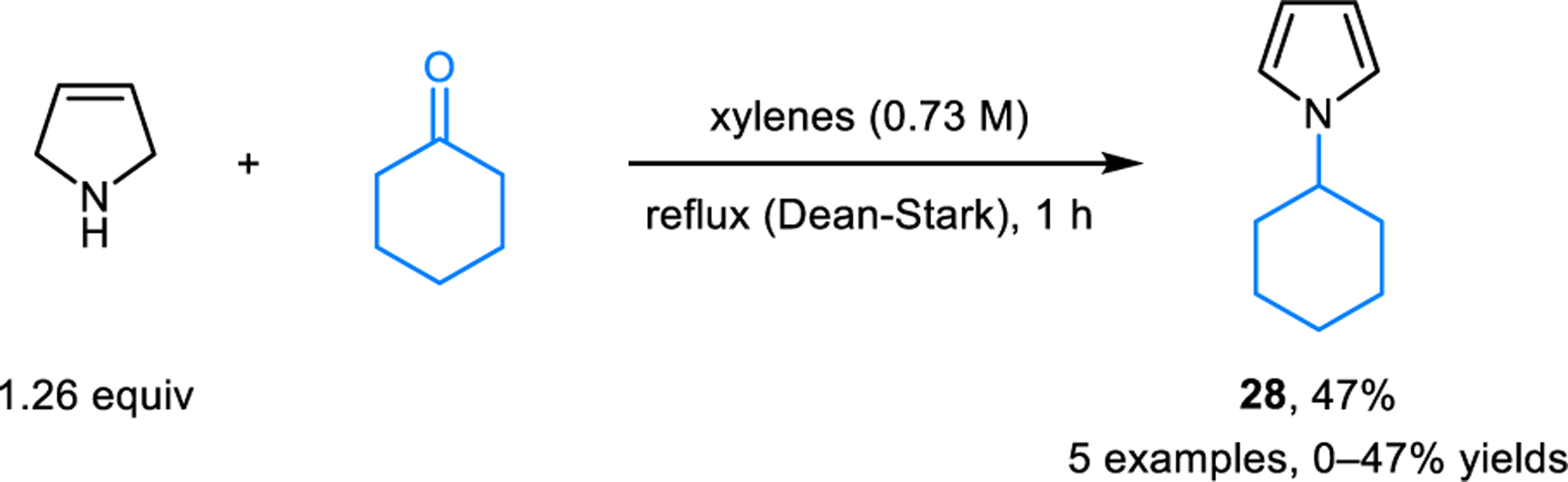
Synthesis of N-cyclohexylpyrrole from cyclohexanone and 3-pyrroline.
Scheme 11.
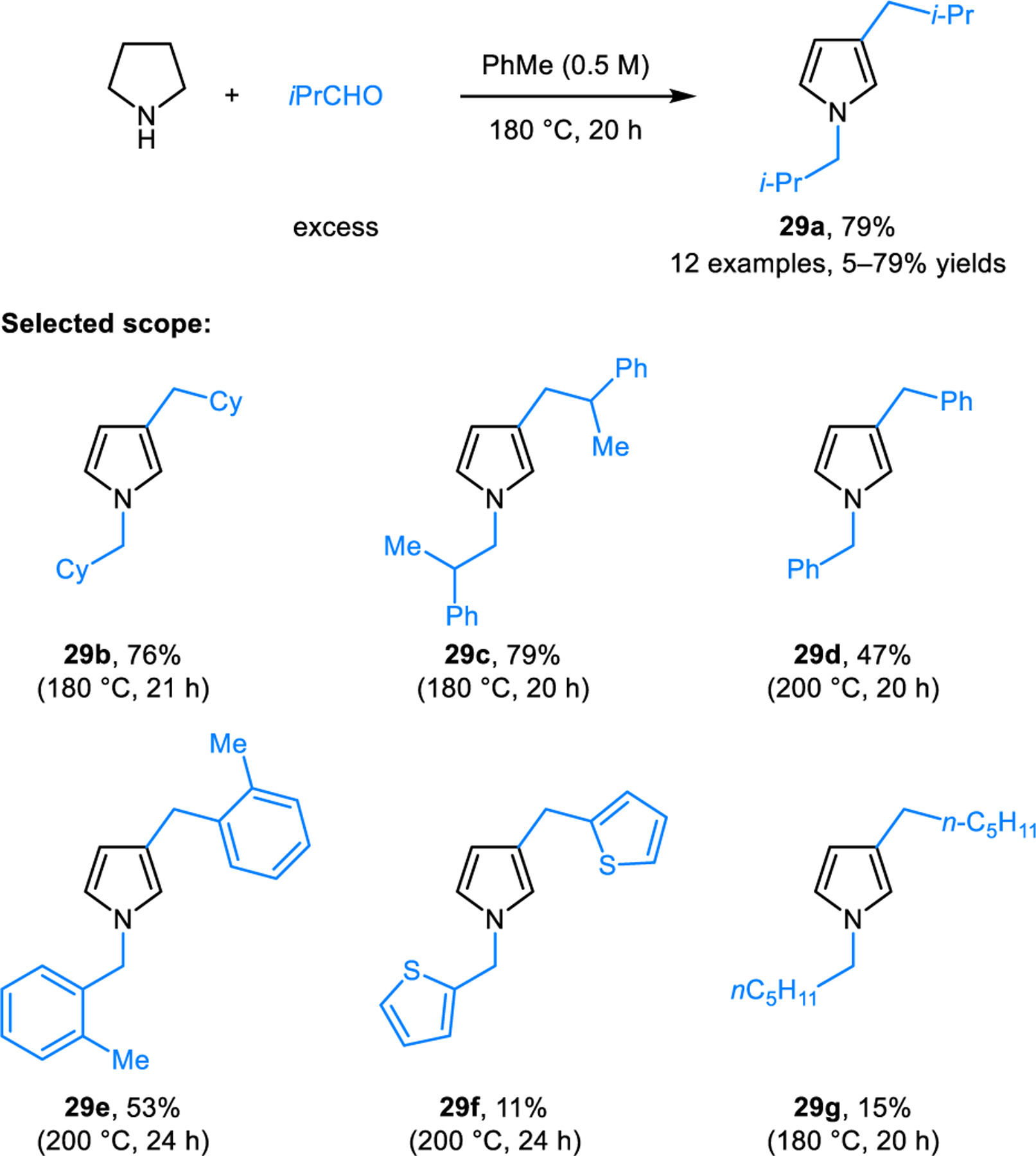
Synthesis of 1,3-dialkylpyrrole from pyrrolidine and aldehydes.
A significantly improved procedure for the formation of N-alkyl pyrroles 30 and 31 from 3-pyrrolines was reported by Tunge and coworkers (Scheme 12).26 Application of benzoic acid as the catalyst allows for these reactions to be performed under milder conditions (reflux in toluene). This transformation exhibits a broad substrate scope and provides N-alkyl pyrroles in good to excellent yields. The mechanism of this reaction was investigated computationally by Yu and coworkers who first proposed the formation of azomethine ylides from carboxylic-acid-derived N,O-acetals in these types of transformations.27 The mechanism for pyrrole formation is related to that shown in Scheme 9.
Scheme 12.
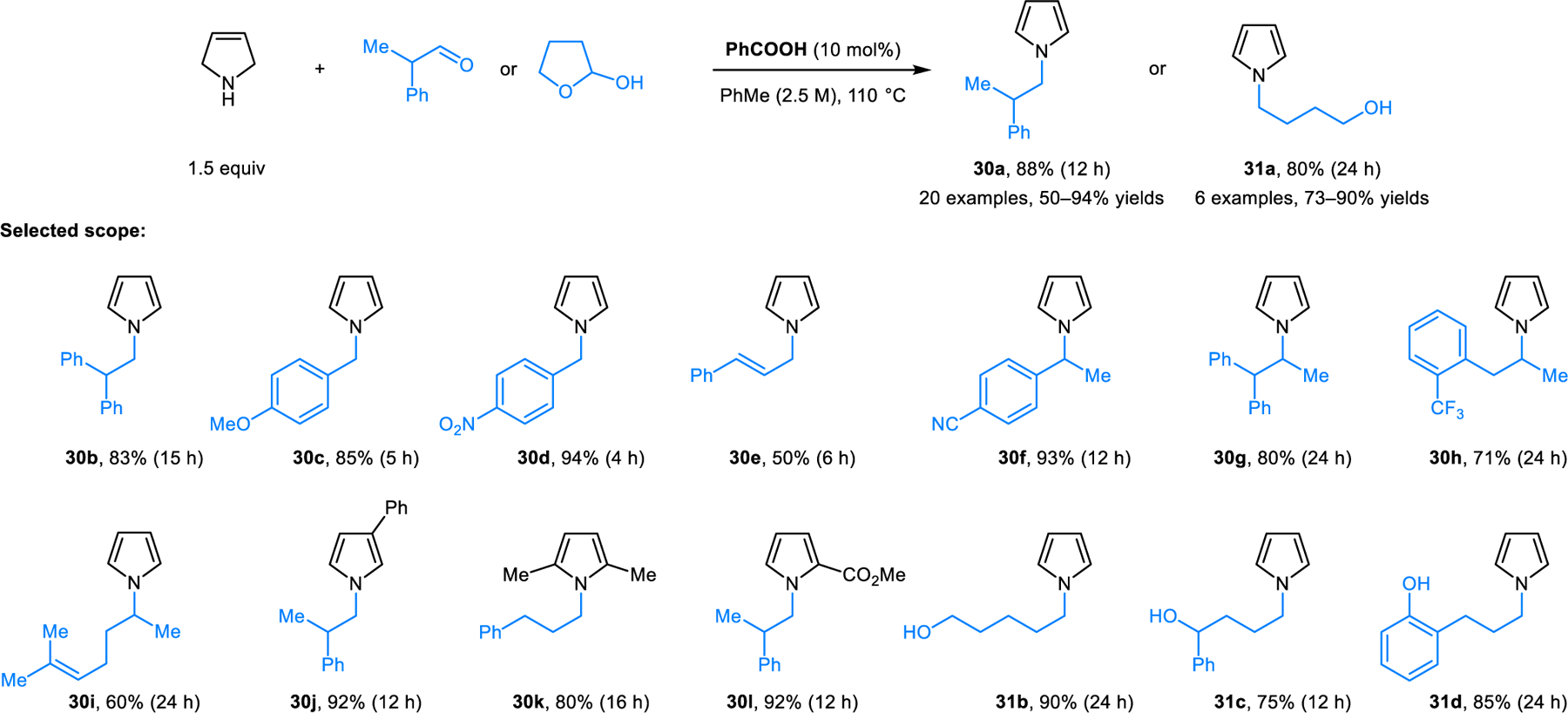
Benzoic acid-catalyzed redox-neutral synthesis of N-alkylpyrroles from 3-pyrrolines and carbonyl compounds.
2.4. Indoles from Indolines
Condensation-based aromatization has also been achieved in the synthesis of N-benzylindoles 32 from indoline and aldehydes. As reported by Pan and coworkers, products 32 are obtained by heating a mixture of indoline and an aromatic aldehyde in toluene at 125 °C in the presence of catalytic amounts of benzoic acid (Scheme 13).28 To account for the overall redox-isomerization, the authors invoked a mechanism involving a 1,3-hydride shift leading to the isomerization of an iminium ion intermediate. Interestingly, under identical reaction conditions, indoline and salicylaldehyde combine to form N-benzylindoline (33) instead of N-benzylindole (32h) (Scheme 14). This divergent reactivity was rationalized by the stabilization of intermediate 34 via intramolecular hydrogen bonding. Ultimately, acid promoted loss of water ensues and intermediate 35 is reduced by intermolecular hydride transfer from an additional equivalent of indoline. Consistent with this pathway, indole is formed as a byproduct. An alternative mechanism involves elimination of indole from expected product 32h which may in fact be the primary product. Conjugate addition of indoline to the resulting ortho-quinone methide 36 then provides 33. This latter mechanism is consistent with what has been observed in redox-annulations of salicylaldehydes with secondary amines (vide infra).
Scheme 13.
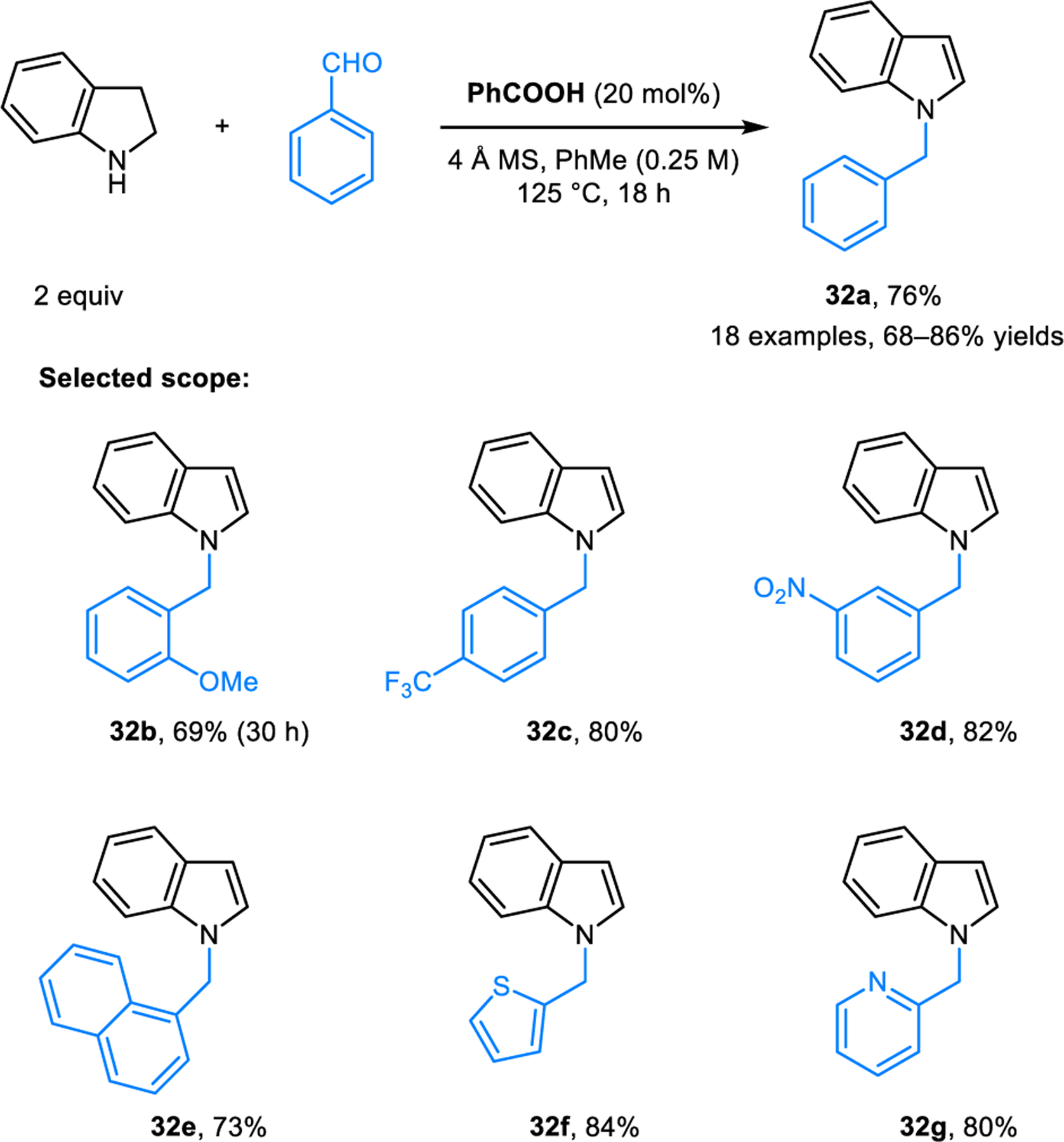
Benzoic acid-catalyzed redox-neutral synthesis of N-alkylindoles from indoline and aromatic aldehydes.
Scheme 14.
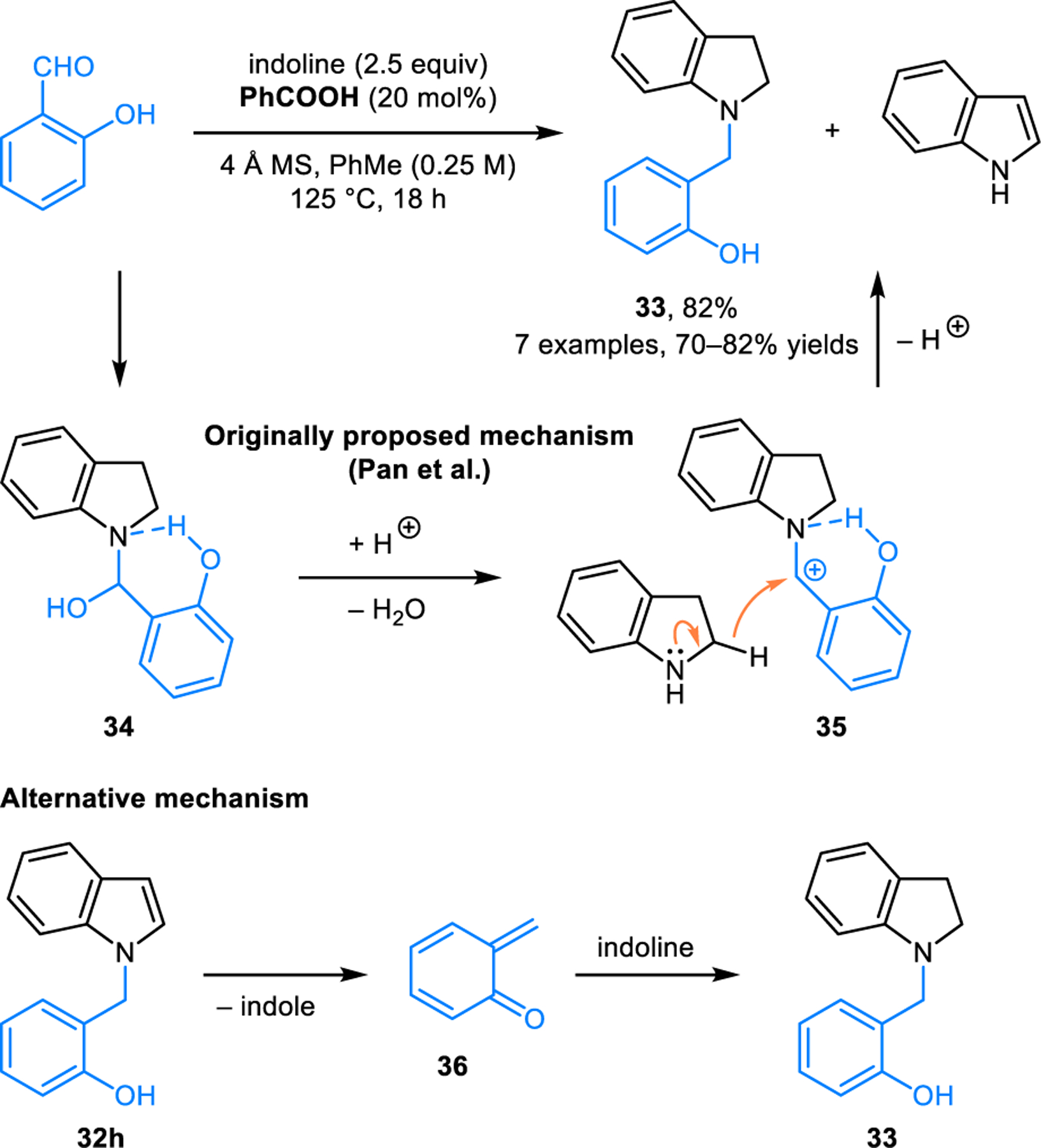
Divergent reactivity with salicylaldehyde.
The synthesis of N-benzylindoles 32 from indolines and aldehydes was independently developed by our group (Scheme 15).29 Under reaction conditions that involve microwave irradiation, N-benzylindole 32h is still the major product when salicylaldehyde is employed as the starting material, with only minor amounts of N-benzylindoline 33 being isolated. Interestingly, the reaction of 2-methylindoline with 4-chlorobenzaldehyde also provides the corresponding N-benzyl-2-methylindoline in 20% yield (not shown). Here, reduction of an intermediate via intermolecular hydride transfer appears likely. Strong support for the intermediacy of an azomethine ylide was obtained in reactions of indolines with an aromatic aldehyde containing a pendant dipolarophile in the ortho-position (Scheme 16). In the case of parent indoline, similar amounts of the (3+2) cycloaddition product 37a and the aromatized product 38a were obtained. With 5-bromoindoline, only the (3+2) cycloaddition product 37b was isolated.
Scheme 15.
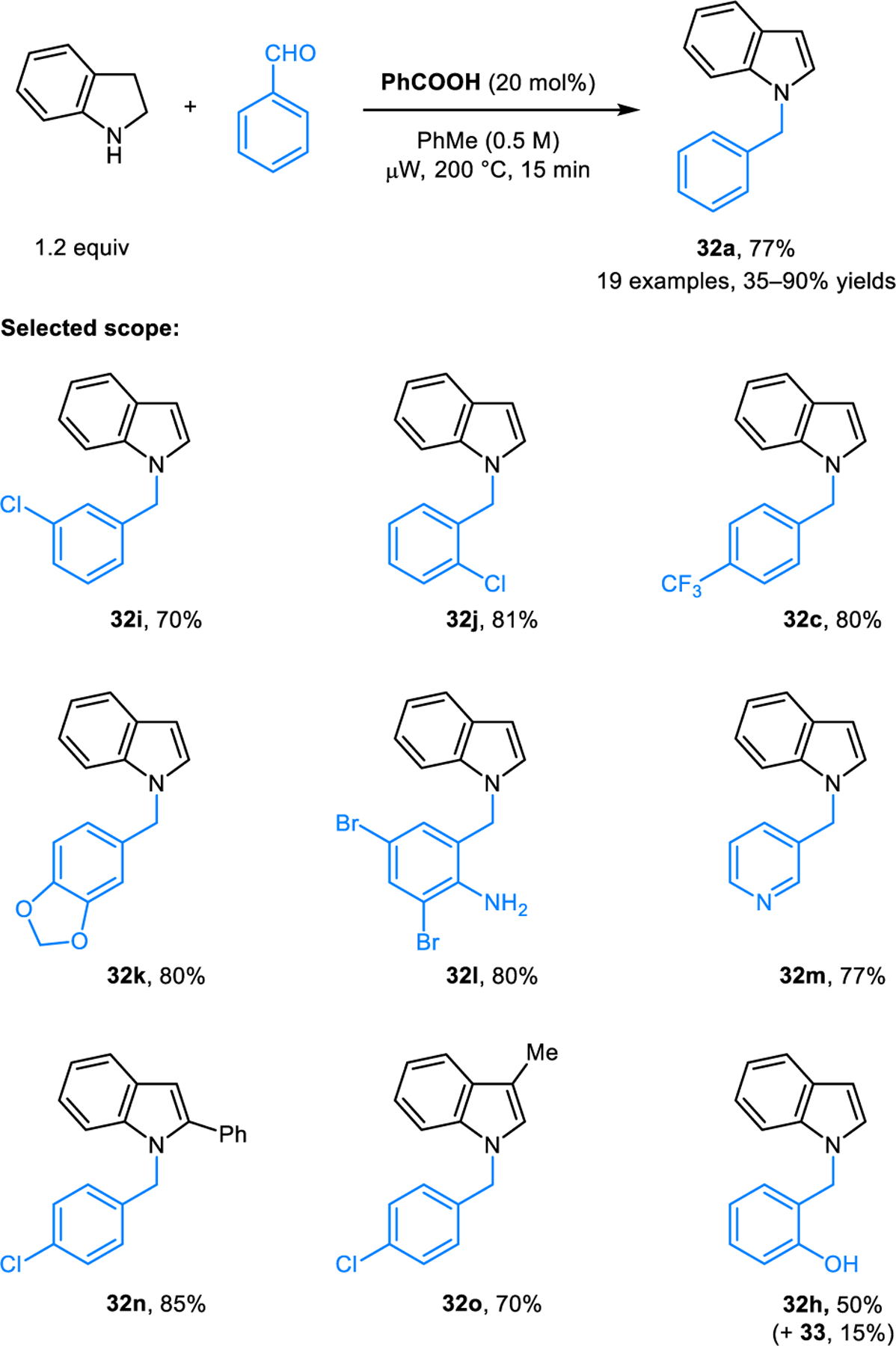
Benzoic acid-catalyzed redox-amination of indoline with aromatic aldehydes under microwave conditions.
Scheme 16.
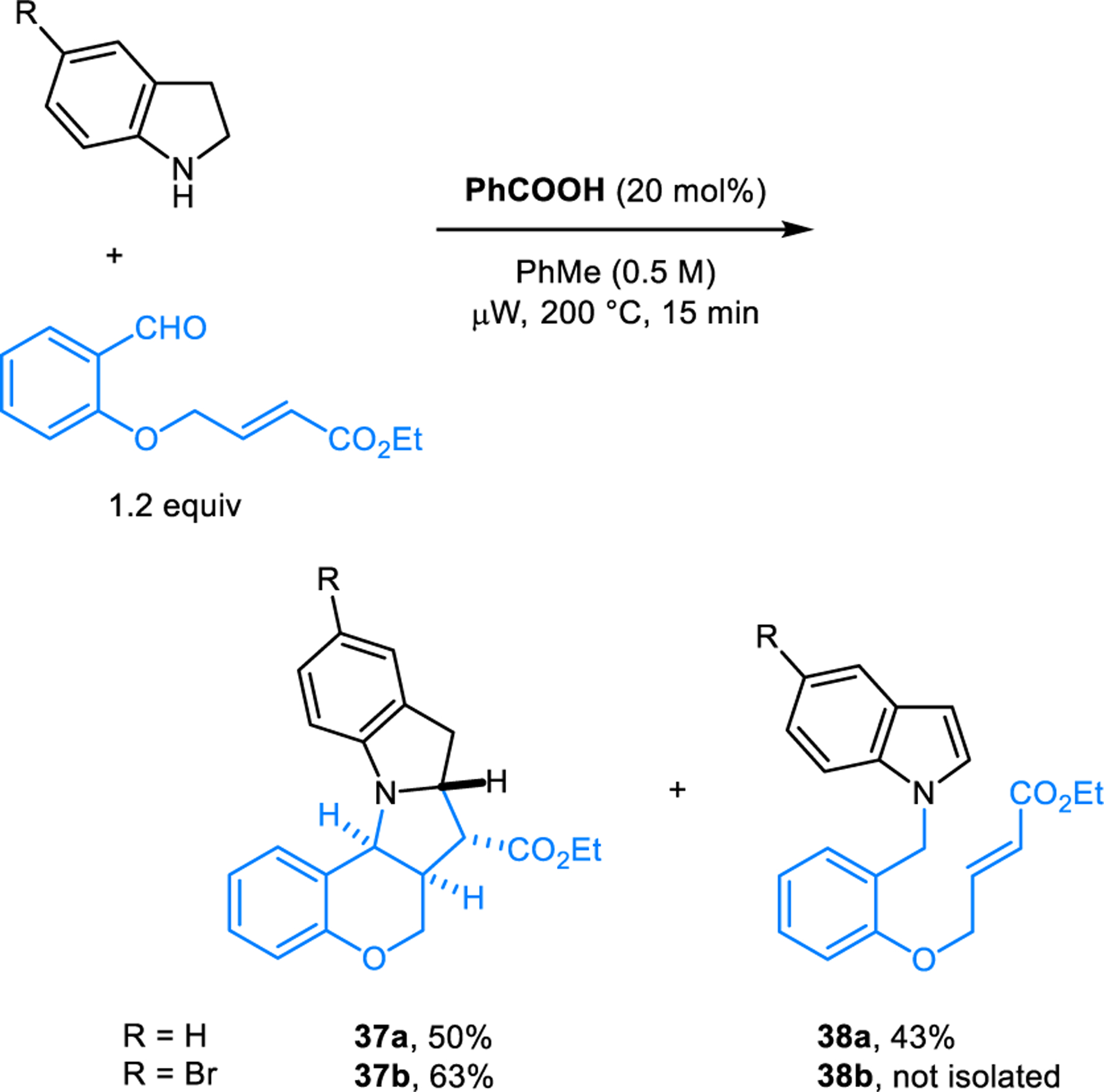
Trapping of azomethine ylide intermediates by intramolecular [3+2] dipolar cycloaddition.
Saracoglu and coworkers later realized the synthesis of N-alkylindoles from indoline and aliphatic ketones.30 While no reaction takes place with Brønsted acids such as benzoic acid or trifluoroacetic acid, bismuth nitrate pentahydrate was identified as a viable catalyst. In addition to the desired N-alkylindole products formed by the redox-amination, secondary byproducts from alkylation of the aromatic ring were also obtained. Indoline was proposed to act as the hydride donor in the formation of these byproducts. A similar reaction between indoline and benzylic ketones not only provides the corresponding N-alkylindoles, but also yields N-alkylindolines.31
Tunge and coworkers reported a redox-amination reaction forming N-aryl-1-aminoindoles based on the condensation of indolines and nitrosobenzenes (Scheme 17).32 The reaction is performed by adding nitrosobenzene slowly over a period of one hour to a mixture of indoline and 30 mol% of benzoic acid catalyst in toluene at 110 °C. A variety of functional groups are tolerated, and N-aryl-1-aminoindoles 39 are obtained in generally good to excellent yields. The authors also developed a one-pot procedure for the redox-amination starting from aniline (not shown).
Scheme 17.
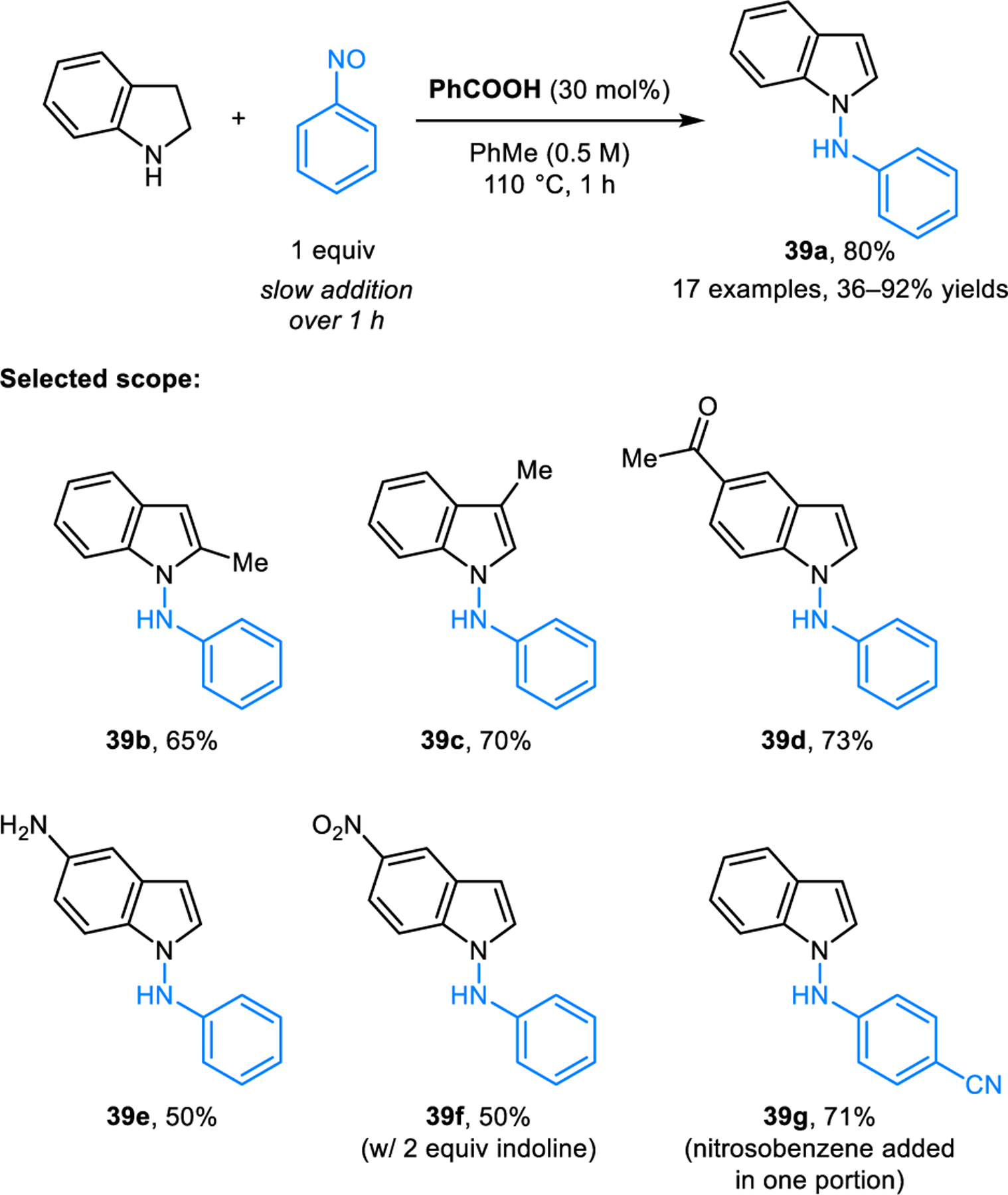
Synthesis of N-aryl-1-aminoindoles via redox-amination between indolines and nitrosobenzenes.
3. Pericyclic Reactions
3.1. (3+2) Dipolar Cycloaddition
Azomethine ylides generated via the condensation-based approach readily engage in pericyclic reactions, the most common of which are (3+2) dipolar cycloadditions with dipolarophiles such as methyl acrylate and N-methylmaleimide. Dependent on the nature of the substrates involved, different mechanisms account for the generation of the azomethine ylide intermediates. In 1986, Grigg et al. reported the first examples of (3+2) cycloadditions involving azomethine ylides formed via condensation of secondary cyclic amines and 1,2-dicarbonyl compounds, such as isatin and phenyl glyoxaldehyde (Scheme 18).33 These 1,2-dicarbonyl compounds exhibit bifunctional character. In the sequence proposed by Grigg and coworkers, one carbonyl group is involved in iminium ion formation while the vicinal carbonyl group participates in a 1,5-proton shift, resulting in the formation of a resonance-stabilized azomethine ylide 40/40’.34 Such reactions are stereospecific in that the anti-dipole of the azomethine ylide 40’ is formed due to the geometry required for the 1,5-proton shift.35 Grigg et al. later extended the scope to carbonyl compounds in which a 1,5-proton shift mechanism is not feasible.35 For instance THIQ and benzaldehyde was found to be a viable combination. Here, azomethine ylide generation was proposed to stem from deprotonation of an iminium ion intermediate. Under these conditions, a mixture of four diastereomeric cycloadducts is obtained, with the major product diastereomers being derived from the anti-dipole.
Scheme 18.
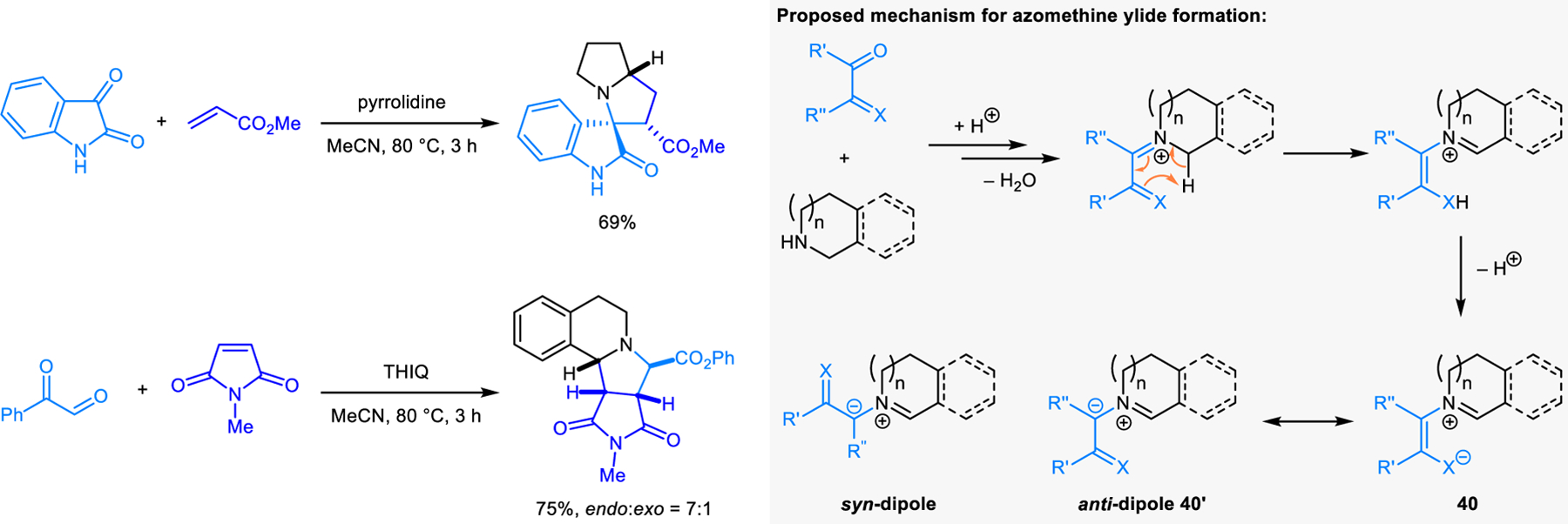
[3+2] Dipolar cycloaddition involving azomethine ylides formed by the condensation of cyclic amines and 1,2-dicarbonyl compounds and the proposed mechanism of azomethine ylide formation.
In reactions of aldehyde 41 possessing a pendent dipolarophile in the ortho-position, the diastereoselectivity of the reaction is dependent on the electronic nature of the dipolarophile. When N-methylmaleimide is used as an external dipolarophile, due to its high reactivity, it reacts preferentially with the kinetically-favored anti-dipole, and the anti-syn dipole equilibrium is not reached. Only two diastereomers (42 and 43) derived from the anti-dipole are then obtained in a ratio of 2:1, isolated as a mixture also containing 44, a side product arising from the conjugate addition of THIQ to N-methylmaleimide (Scheme 19). In comparison, the intramolecular cycloaddition between THIQ and 41 in refluxing xylene results in both anti-dipole adduct 45 and syn-dipole adduct 46, with the former being the dominant product (Scheme 20). In this case, it was proposed that cycloaddition is slower than anti-syn dipole equilibration, and thus is rate-determining. In contrast, aldehyde 47a, containing an α,β-unsaturated ester as a significantly more reactive dipolarophile, leads to formation of 48a as a single diastereomer (exclusive reaction of the anti-dipole).
Scheme 19.
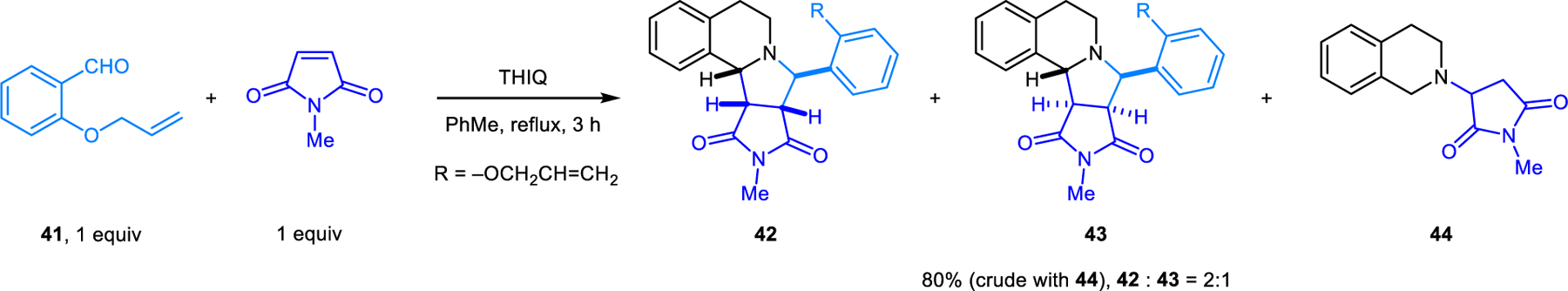
Stereospecific intermolecular [3+2] dipolar cycloaddition of azomethine ylide anti-dipole using N-methylmaleimide as dipolarophile.
Scheme 20.
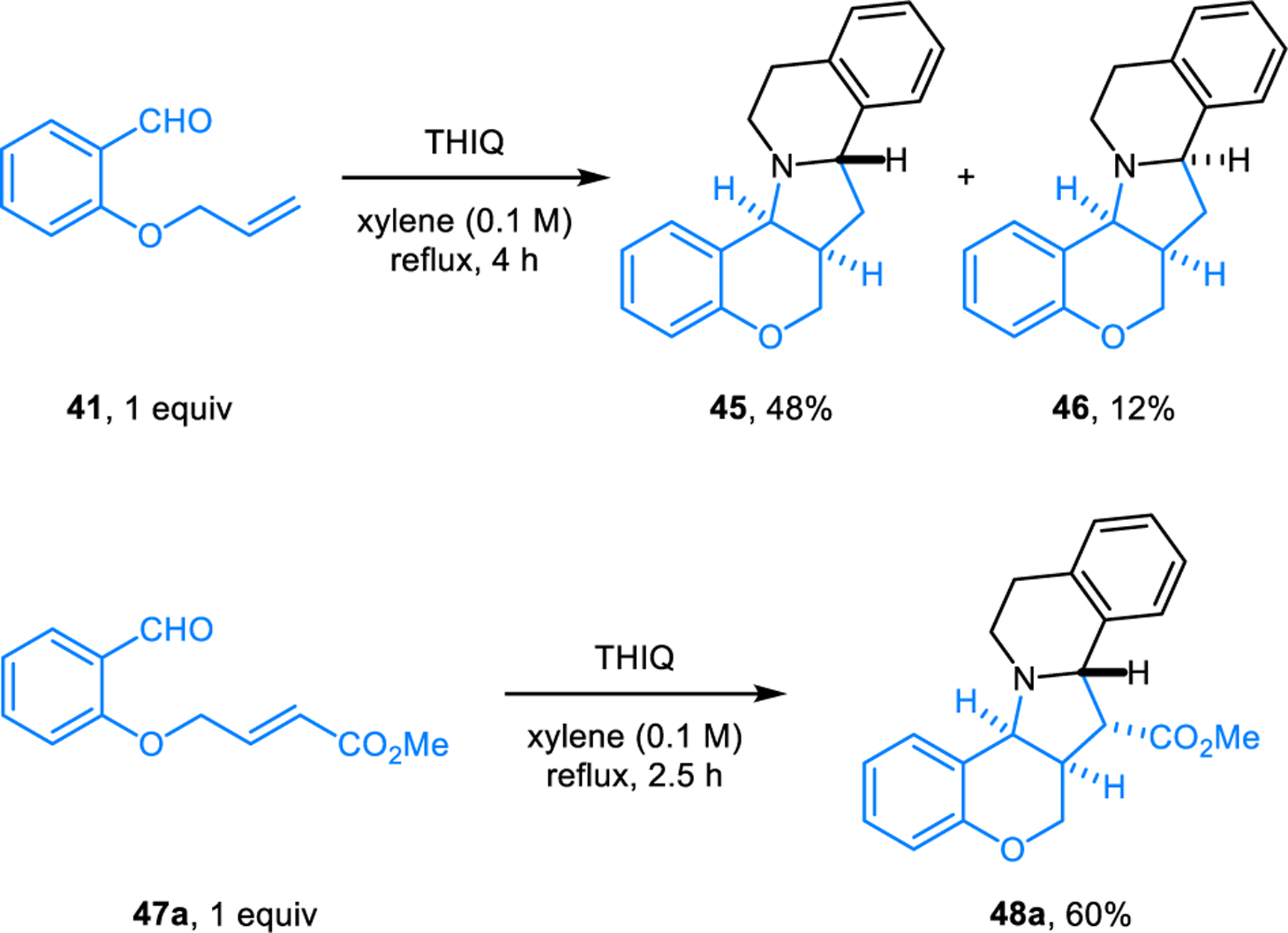
Dipolarophile-dependent stereoselective and stereospecific intramolecular [3+2] dipolar cycloaddition.
Risch and coworkers showed that acyclic amines also participate in azomethine ylide formation followed by (3+2) cycloaddition (Scheme 21).36 For instance, heating of a mixture of benzaldehyde and dibenzylamine in toluene with the removal of water using a Dean-Stark apparatus, results in the formation of oxazolidine 49 in good yield and excellent diastereoselectivity. In this case, two equiv of aldehyde are required, with one equiv acting as the dipolarophile. However, the dipolarophile is not limited to benzaldehyde: methyl arylate, phenyl vinyl sulfone, dimethyl fumarate and N-methyl maleimide are viable substrates as well (not shown).
Scheme 21.
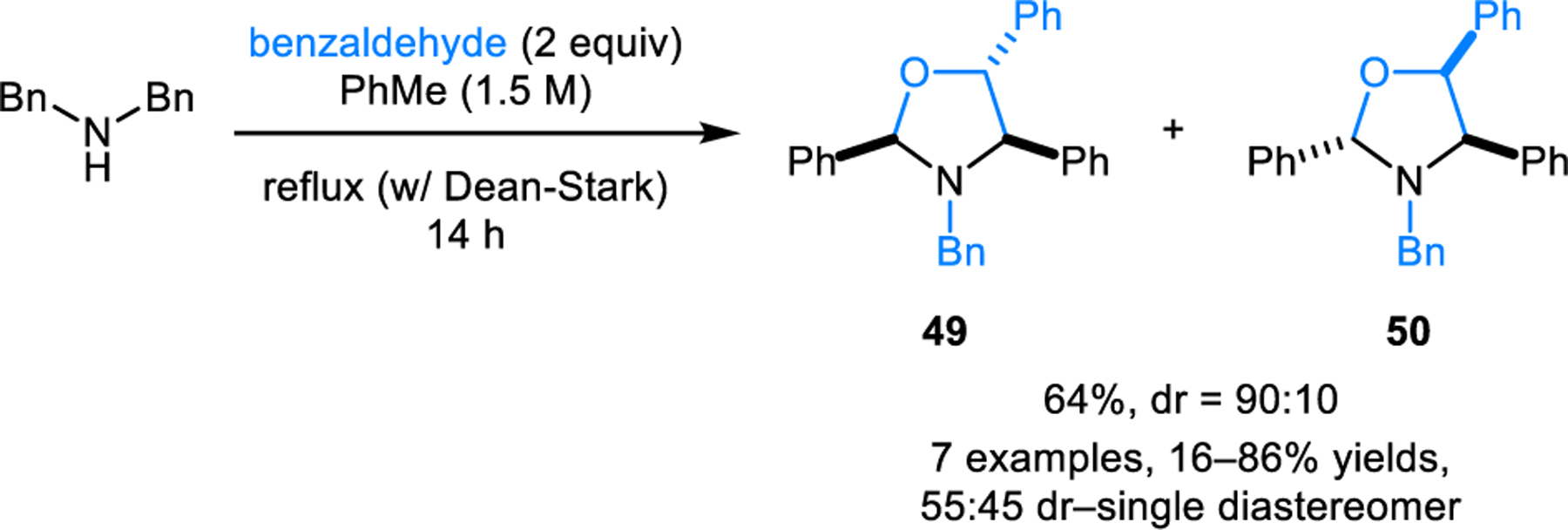
Formation of oxazalidines via redox-neutral [3+2] dipolar cycloaddition of azomethine ylide and benzaldehyde.
Dang, Bai and coworkers reported an interesting two-component reaction of aldehydes with acyclic amines containing a pendant dipolarophile (Scheme 22).37 Specifically, diastereoselective intramolecular (3+2) cycloadditions are achieved with aromatic aldehydes and pyrimidine derivatives such as 51 to provide the corresponding products 52 in good to excellent yields. Dramatically diminished yields or no products are obtained with aliphatic aldehydes (e.g. 52f). Product 52h does not form from the corresponding substrate containing an E-methyl group at the terminal alkene, possibly due to unfavorable interactions between the methyl group and the phenyl group in the cycloaddition step. On the other hand, substrate 51i provides the corresponding cycloaddition product 52i in moderate yield. Favorable π-π interactions stabilizing the transition-state were invoked in the formation of 52i.
Scheme 22.
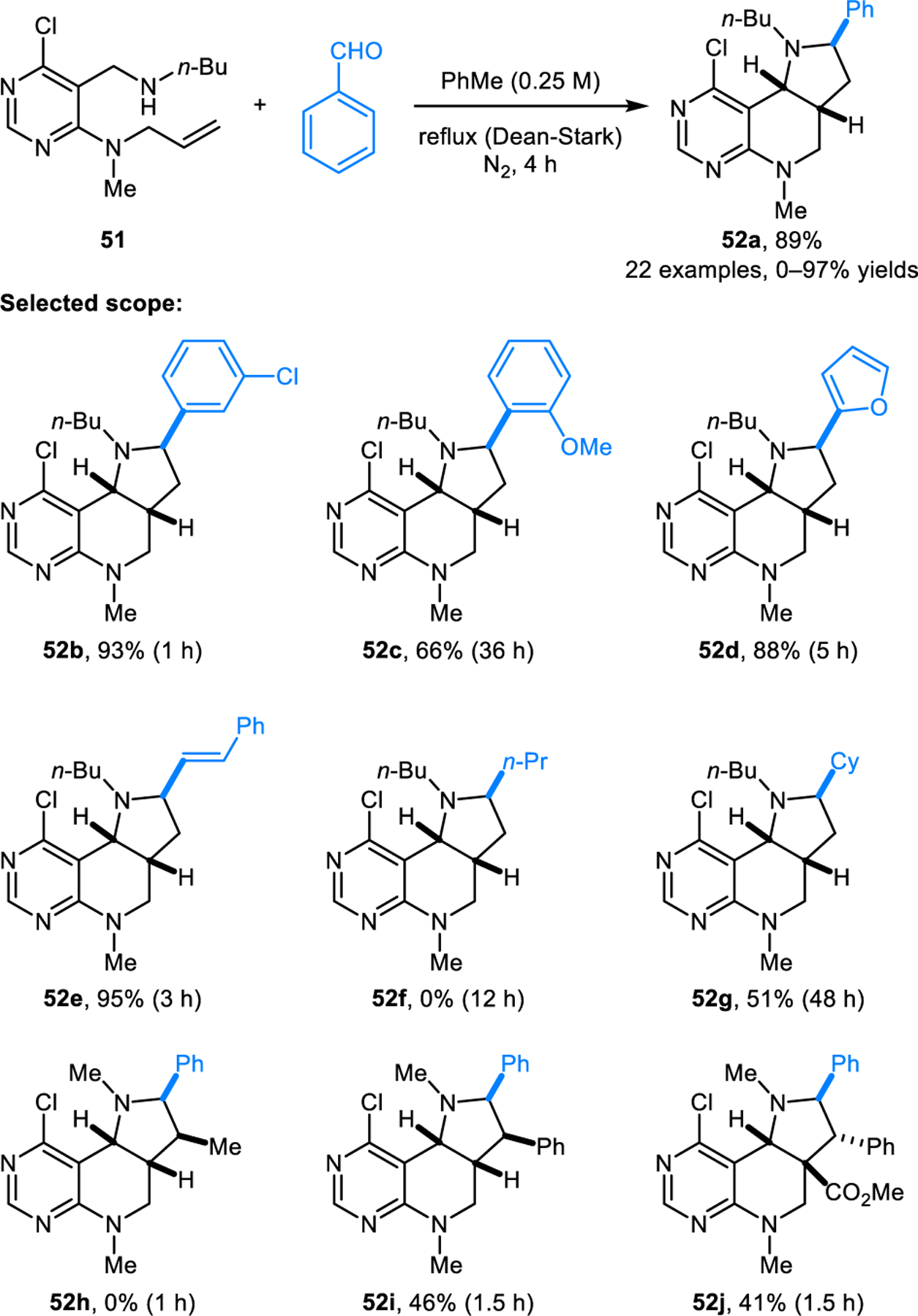
Diastereoselective intramolecular [3+2] dipolar cycloaddition involving pyrimidine-based aliphatic amines.
To determine whether the carboxylic acid catalyzed pathway to azomethine ylides is compatible with pericyclic chemistry, our group investigated Grigg’s classic intramolecular (3+2) cycloaddition reaction of aldehydes such as 53 with benzoic acid as the catalyst (Schemes 23 and 24).38 In the presence of molecular sieves, THIQ and other amines with activated benzylic C–H bonds readily participate in the reaction at temperatures as low as room temperature, while providing products (e.g., 54a) in high yields and diastereoselectivities. A wide range of non-benzylic cyclic amines are also viable substrates at elevated temperatures under microwave irradiation conditions. Reactions involving non-symmetrical cyclic amines such as 2-methylpyrrolidine and 2-phenylpyrrolidine exhibit substrate-dependent regio- and diastereoselectivities. 2-Methylpyrrolidine is functionalized at the sterically more accessible position to provide a mixture of two diastereomeric products. In contrast, 2-phenylpyrrolidine produces both regioisomers while still favoring the sterically more approachable α-position. In this case, both products are formed in diastereoselective fashion.
Scheme 23.
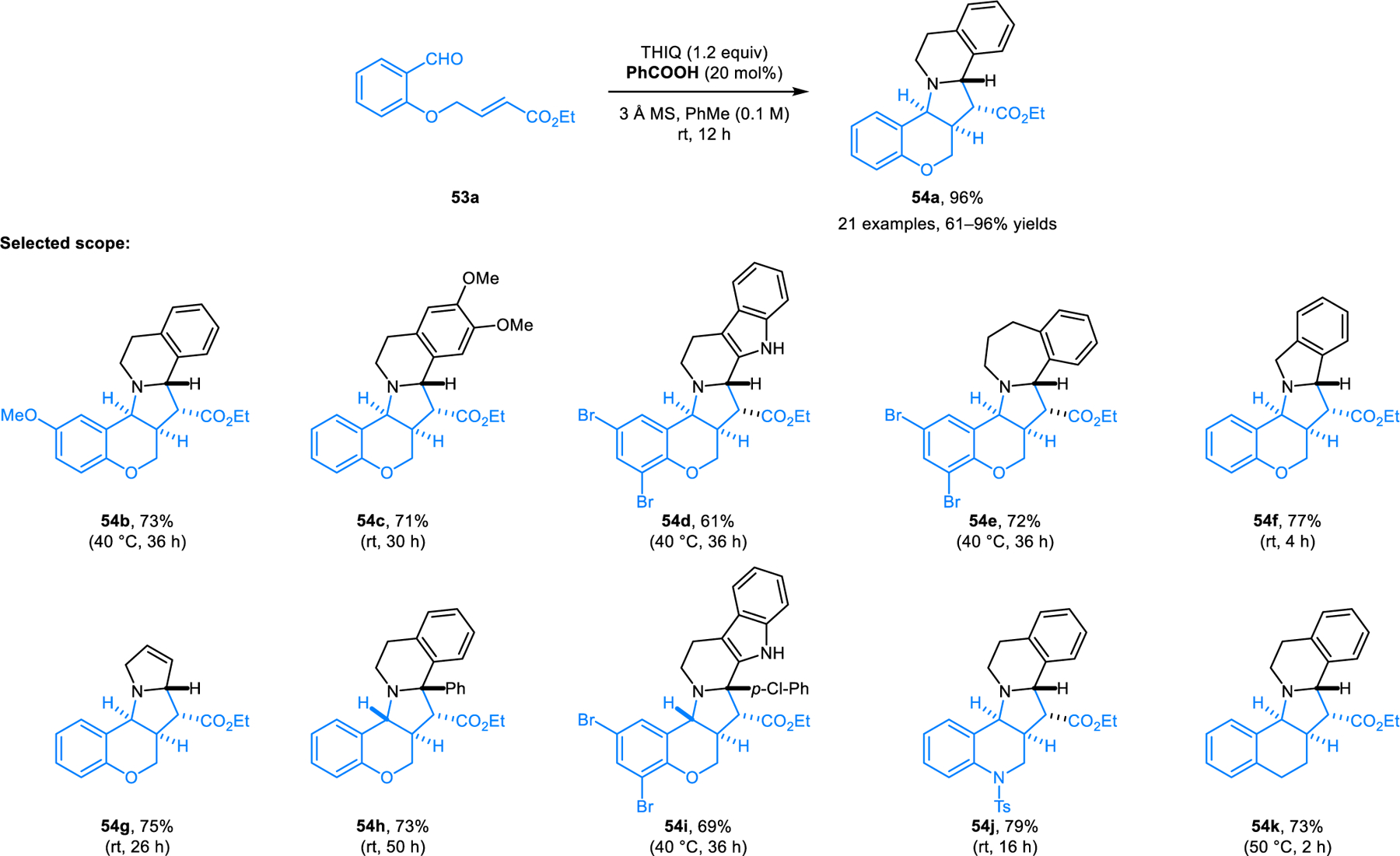
Benzoic acid-catalyzed intramolecular [3+2] dipolar cycloaddition of azomethine ylides derived from benzylic cyclic amines.
Scheme 24.
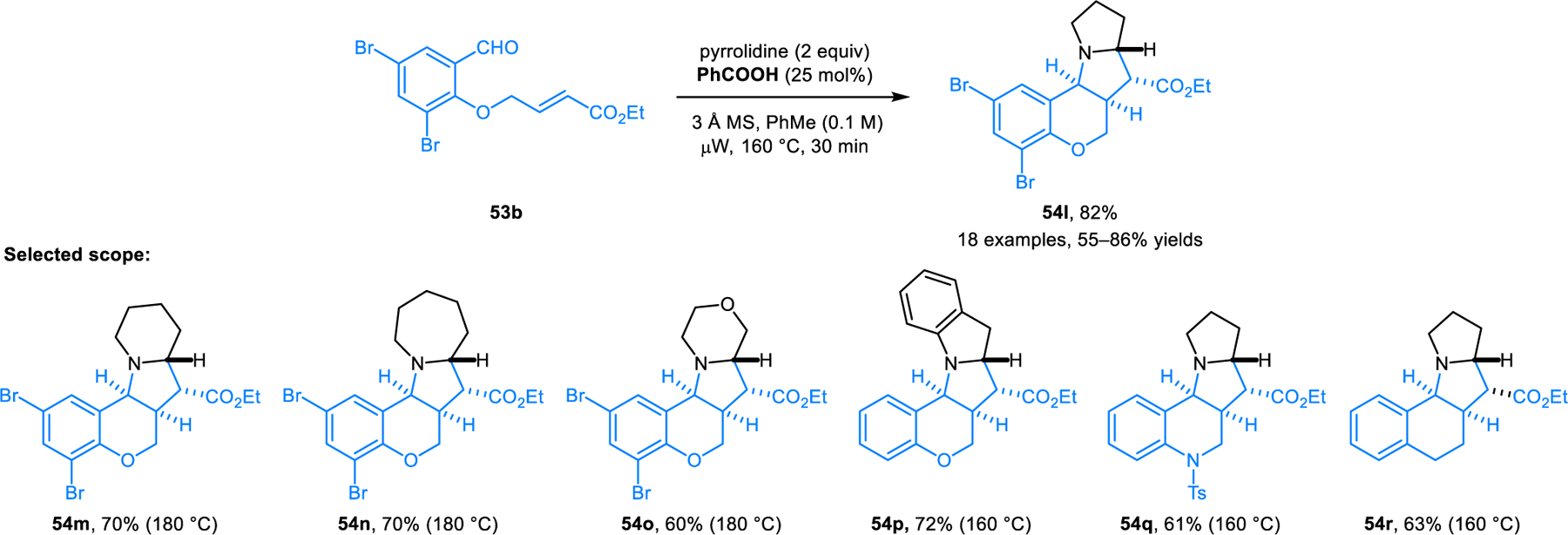
Benzoic acid-catalyzed intramolecular [3+2] dipolar cycloaddition of azomethine ylides derived from non-benzylic cyclic amines.
Related intramolecular (3+2) cycloaddition reactions of alkene dipolarophiles and azomethine ylides derived from the condensation of secondary amines and aromatic aldehydes were also reported by Parma et al.39 and Mantelingu, Rangappa, and coworkers.40 Notably, the latter study includes the use of 1-substituted THIQs as substrates, which provide cycloaddition products containing tetra-substituted carbon centers. Zanoni, Protti and coworkers developed a variant of this reaction which is catalyzed by photo-excited 2-naphthol (Scheme 25).41 The reaction involving aldehyde 47 and THIQ in the presence of 50 mol% of 2-naphthol and 5 mol% of Ru[bpy]3Cl2 under 410 nm LED light irradiation gives (3+2) cycloaddition product 48a and 48b in 65% and 25% yield respectively at room temperature. The effect of 2-naphthol on catalyzing this reaction is attributed to its dramatically increased acidity upon photo-excitation. The pKa of photo-excited 2-naphthol was found to be more than six orders of magnitude lower than its ground-state pKa in water.42
Scheme 25.
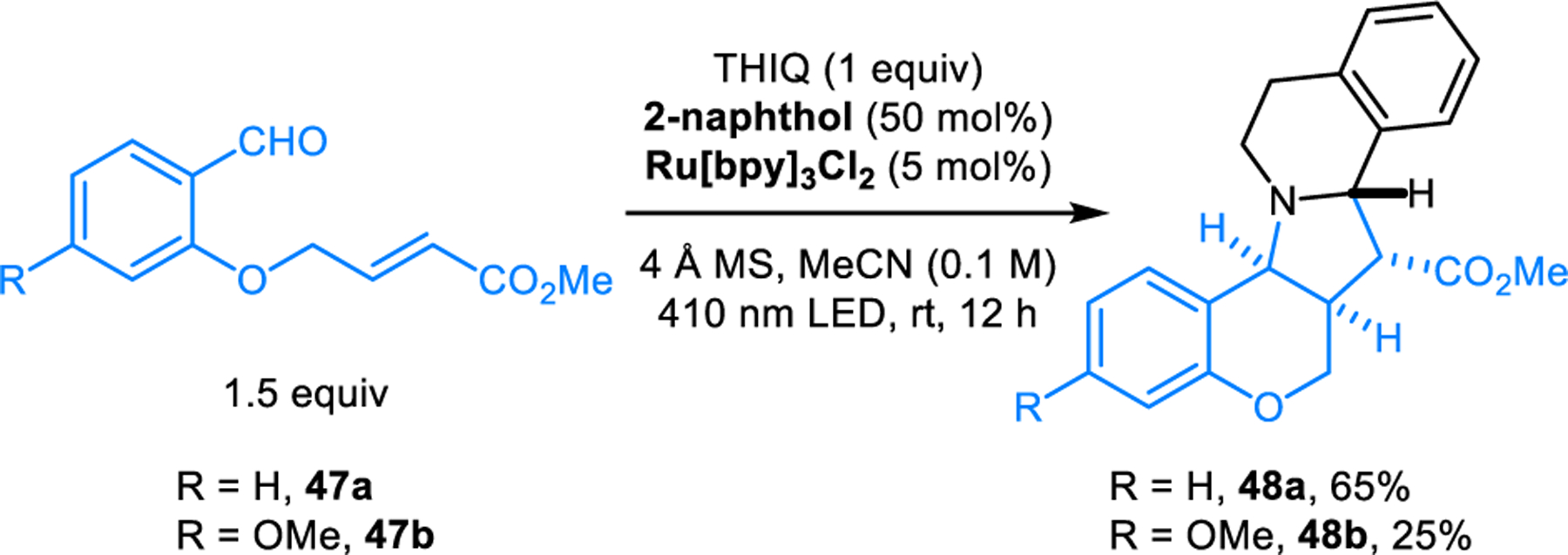
Intramolecular [3+2] dipolar cycloaddition of azomethine ylide derived from THIQ catalyzed by photo-excited 2-naphthol.
Aromatic aldehydes act as dipolarophiles in (3+2) cycloaddition reactions with azomethine ylides derived from pyrrolidine or piperidine and another equivalent of aldehyde, as reported by Hajra and coworkers (Scheme 26).43 Reactions are performed under microwave conditions and are dramatically accelerated by catalytic amounts of potassium acetate. Products 55 are obtained as single diastereomers. The scope of this transformation was later extended by Mantelingu, Rangappa and coworkers to include cyclic amines with activated benzylic C–H bonds to form oxazolidines 56 (Scheme 27).44 These reactions are catalyzed by benzoic acid and operate under milder conditions. Recently, Wu, Li and coworkers reported related reactions involving E-cinnamaldehydes to access products 57 (Scheme 28).45 Here, highly reactive ethyl ketomalonate acts as the dipolarophile. Reactions are performed under mild conditions. The method exhibits broad functional group tolerance on the cinnamaldehyde component. Several research groups reported intermolecular (3+2) dipolar cycloadditions of electron-deficient alkenes and azomethine ylides derived from isatins and cyclic amines.46–49 In addition to α,β-unsaturated alkenes and aldehydes, fullerenes50–52 and α,β-unsaturated alkynes53 are also viable dipolarophiles in (3+2) cycloadditions with azomethine ylides derived from the condensation of cyclic amines and aldehydes.
Scheme 26.
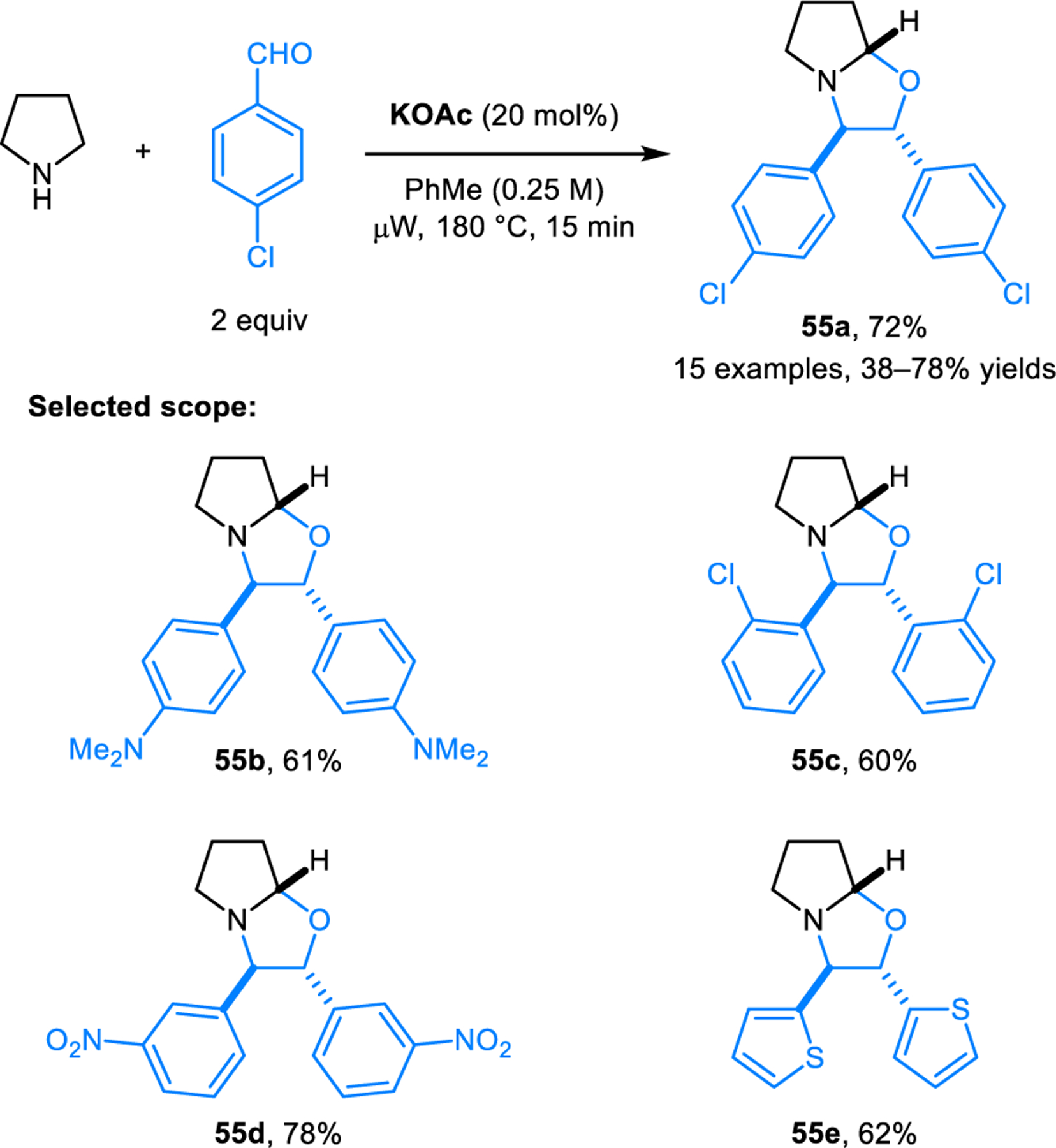
Synthesis of ring-fused oxazolines via [3+2] dipolar cycloaddition of aromatic aldehydes and azomethine ylides derived from alicyclic amines.
Scheme 27.
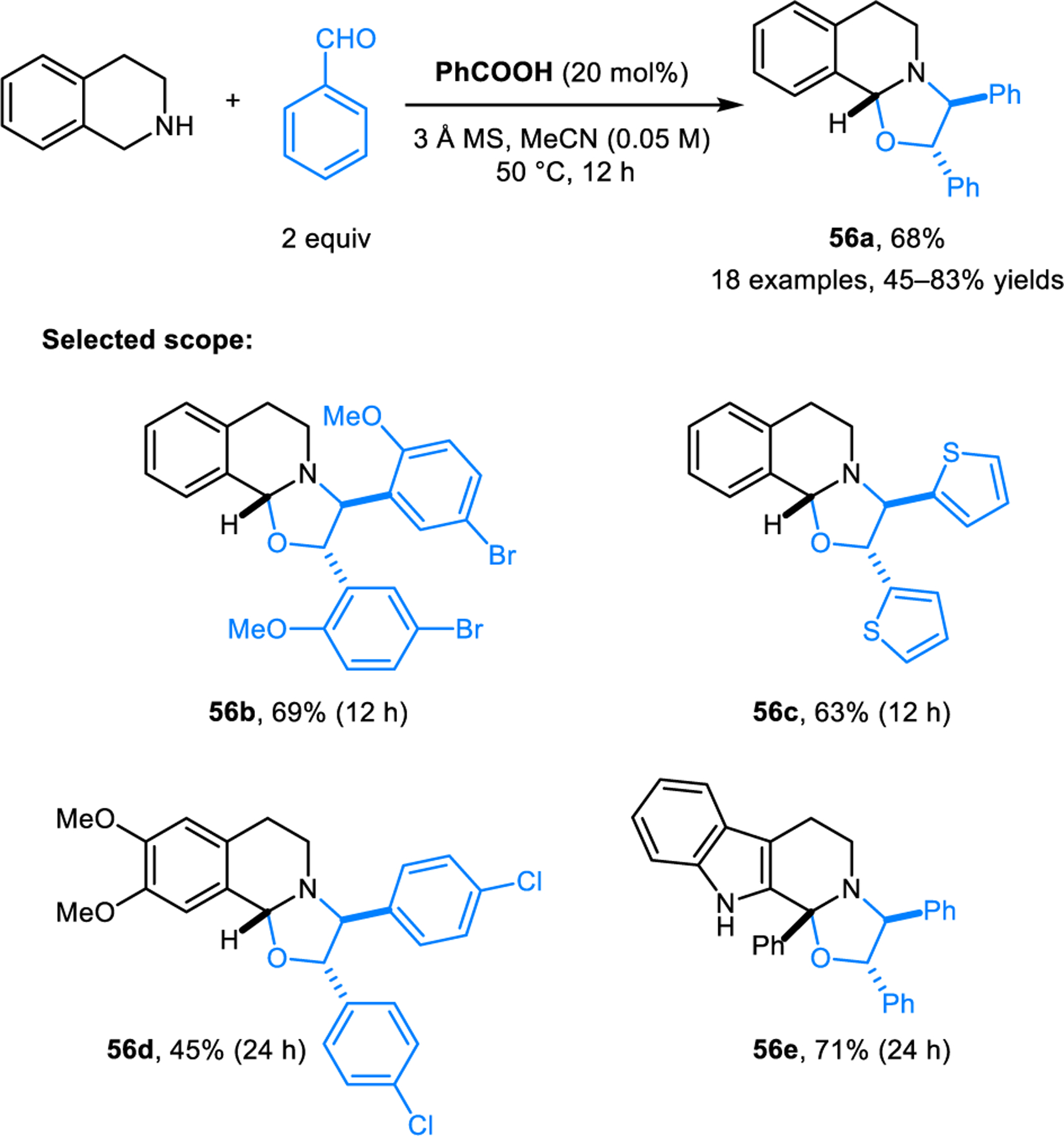
Synthesis of ring-fused oxazolines via [3+2] dipolar cycloaddition of aromatic aldehydes and azomethine ylides derived from benzylic cyclic amines.
Scheme 28.
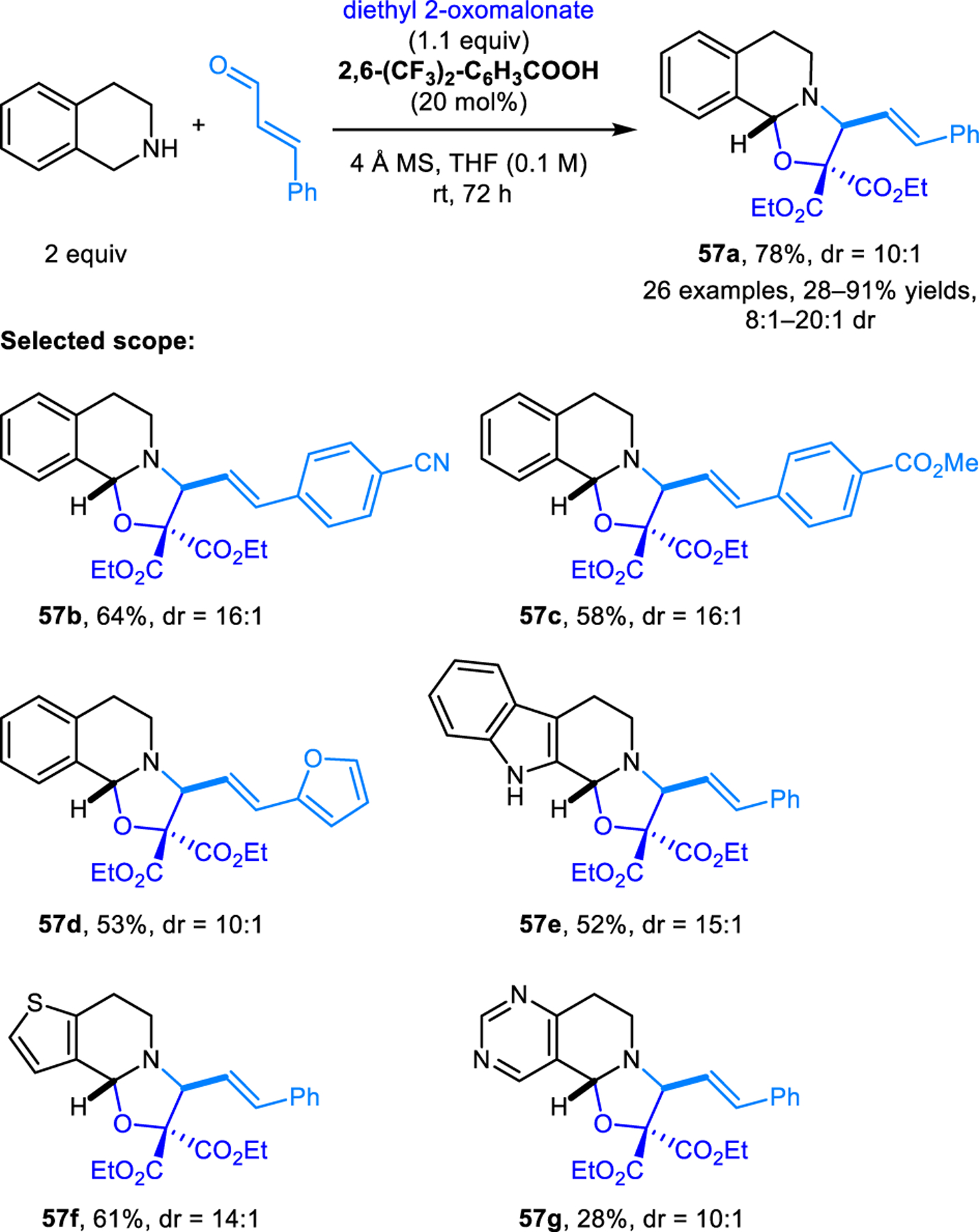
Synthesis of ring-fused oxazolines via [3+2] dipolar cycloaddition of diethyl 2-oxomalonate and azomethine ylides derived from benzylic cyclic amines and E-cinnamaldehydes.
Intermolecular (3+2) cycloaddition reactions of (E)-β-nitrostyrenes and azomethine ylides, derived from the condensation of DHIQ and phenylglyoxal monohydrate or phenylglyoxalate, were reported by the Wu group (Scheme 29).54 Here, the cycloaddition step is followed by aromatization, triggered by elimination of the nitro group and oxidation, providing pyrroloisoquinoline 58 as products. This reaction was applied to the synthesis of lamellarin G trimethyl ether.
Scheme 29.
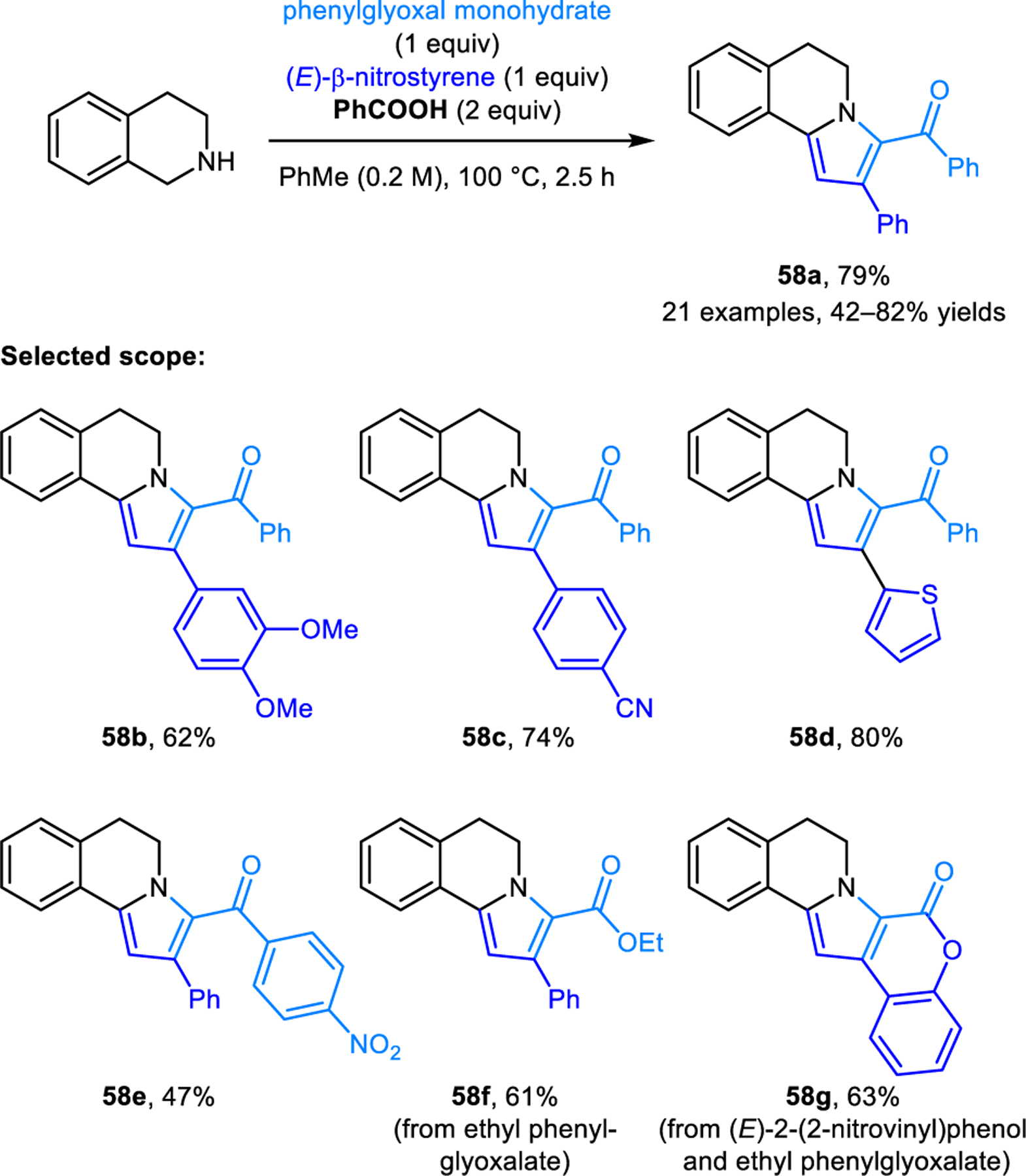
Intermolecular [3+2] dipolar cycloaddition between (E)-β-nitrostyrene and azomethine ylides derived from the condensation of DHIQ and phenylglyoxal or phenylglyoxalate.
3.2. 6π-Electrocyclizations
The first 6π-electrocyclization in the context of amine/aldehyde condensation was reported by Grigg and coworkers (Scheme 30).55 1-Vinyl-THIQ was found to engage with aldehydes to form dihydropyrroloisoquinolines such as 59. Following the formation of azomethine ylide intermediate 60, 1,5-electrocyclization provides enamine 61. This intermediate subsequently reacts with a second equivalent of aldehyde, followed by dehydration (presumably via 62) and finally tautomerization/aromatization.
Scheme 30.
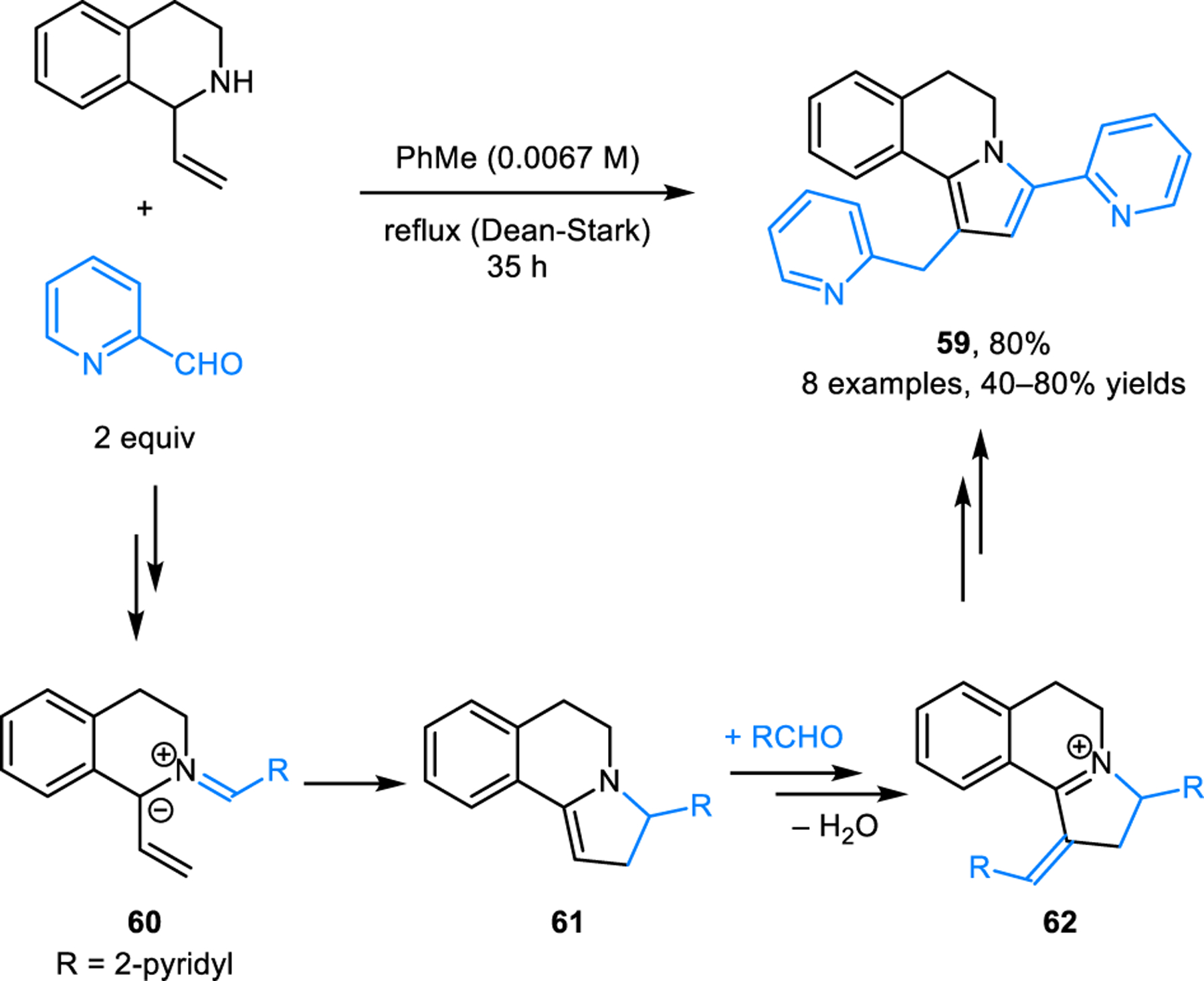
1,5-Electrocyclization of azomethine ylide derived from the condensation of 1-vinyl-THIQ and pyridine 2-carboxyaldehyde.
Based on a single earlier example,56a our group developed the redox-neutral formation of pyrroles (e.g., 63) from cyclic amines and 1,3-diketones (Scheme 31).56b Heating a mixture of dibenzoylmethane and pyrrolidine without additive in toluene at 280 °C under microwave irradiation was found to yield N-benzoylpyrrolidine (65) as the major product, the result of a retro-Claisen reaction (not shown). However, in the presence of p-toluenesulfonic acid (p-TSA), the retro-Claisen pathway is suppressed, albeit not completely, and bicyclic pyrrole 63a is formed as the major product. The reaction pathway likely involves enaminone 64 (also obtained as a side product), which is envisioned to undergo 1,6-proton transfer, followed by 6π-electrocyclization of the resulting azomethine ylide intermediate. Finally, dehydration provides 63a.
Scheme 31.
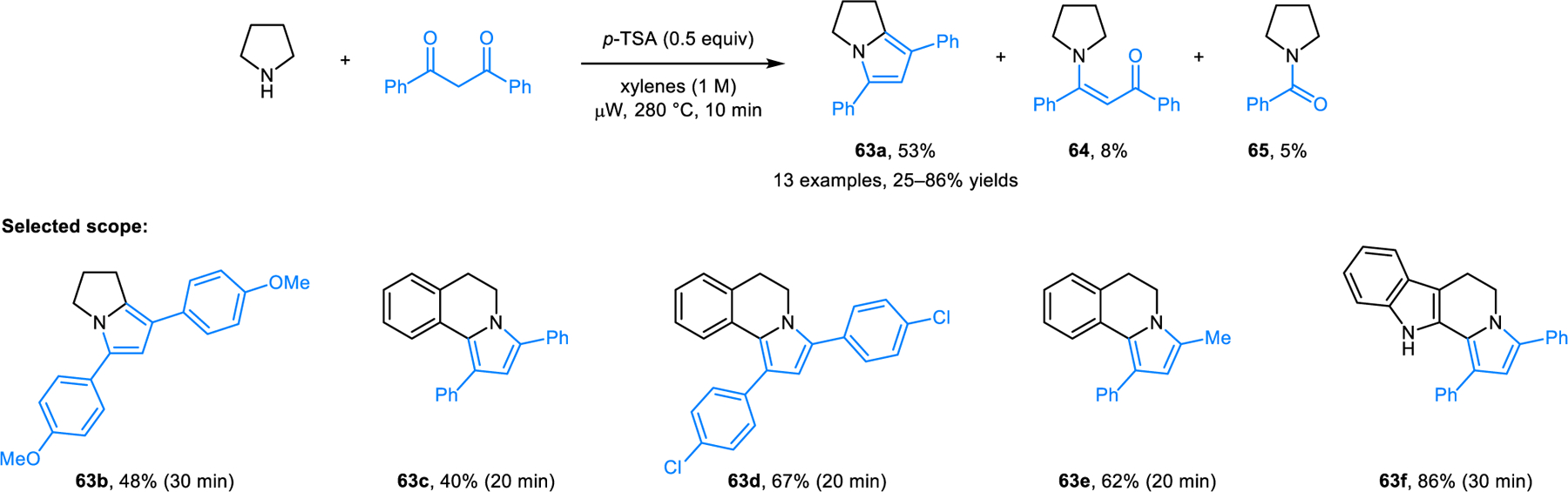
Synthesis of pyrroles from cyclic amines and 1,3-diketones via 1,5-electrocyclization.
Annulation reactions of cyclic amines and α,β-unsaturated ketones were reported by our group (Scheme 32).57 Polycyclic products such as 66 are readily obtained by heating the amine (e.g., pyrrolidine or THIQ) and an α,β-unsaturated ketone in toluene in the presence of stoichiometric amounts of benzoic acid. Interestingly, reactions involving THIQ provide 66f–j as sole products instead of the expected products such as 67. Apparently, isomerization of 67 to the thermodynamically more favored enamine product 66f is facile under the reaction conditions. Reactions are thought to proceed via the condensation of cyclic amines with α,β-unsaturated ketones, followed by formation of azomethine ylide intermediates such as 68. 6π-Electrocyclization of 68 leads to the final product.
Scheme 32.
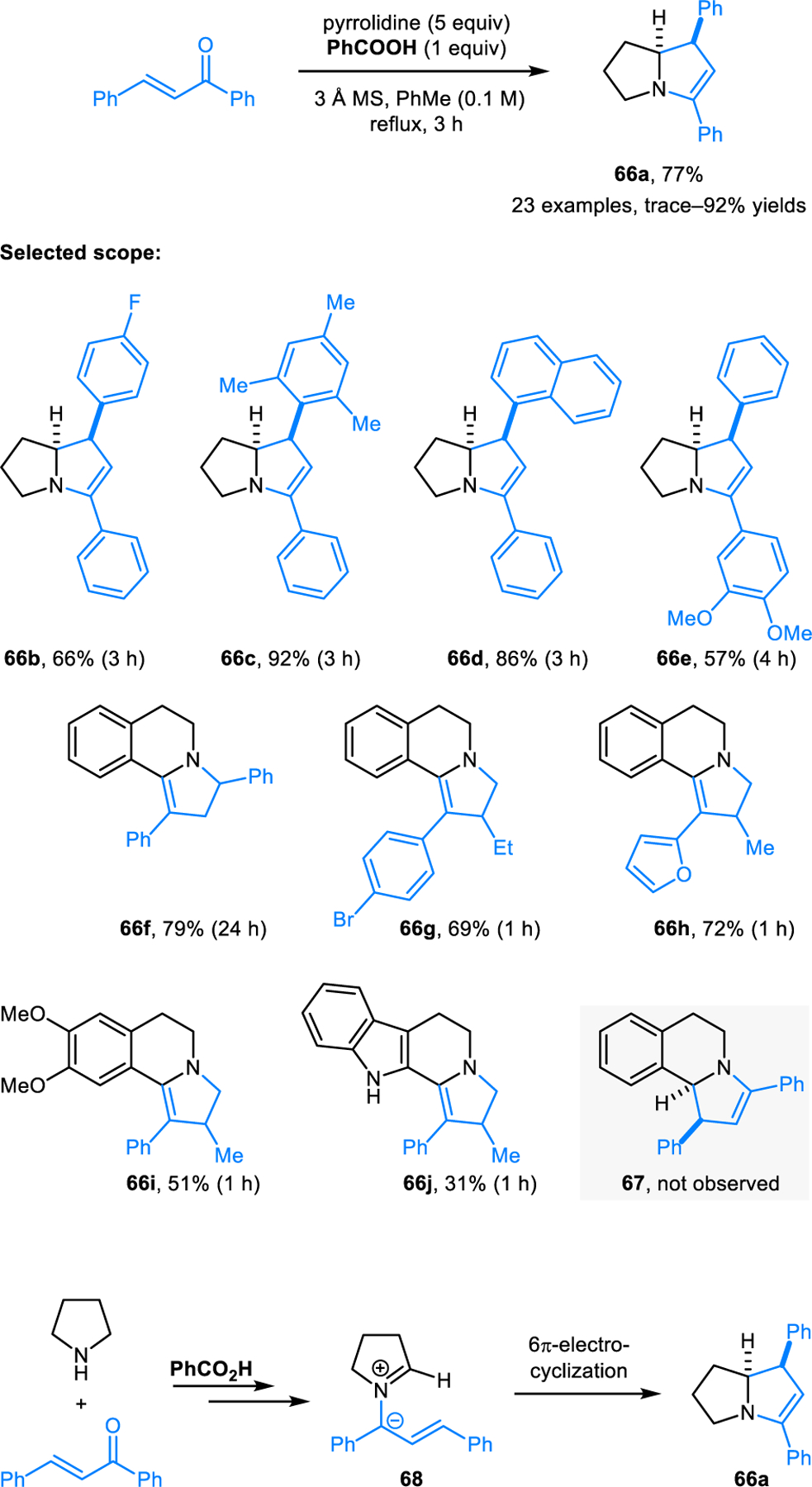
Redox-annulation of cyclic amines and α,β-unsaturated ketones via 1,5-Electrocyclization.
Duan, Lin and coworkers reported the synthesis of pyrroloisoquinolines 69 from 3-chloro E-cinnamaldehydes and DHIQs, via a base-promoted 6π-electrocyclization reaction pathway (Scheme 33).58 Reactions are performed at 120 °C in dimethylformamide with 2.5 equiv of triethylamine, resulting in moderate to high yields of the desired products. The reaction sequence is proposed to initiate via the conjugate addition of DHIQ to 3-chloro E-cinnamaldehyde, followed by the elimination of chloride to form 70. The latter intermediate then condenses with a second equivalent of DHIQ generating a conjugated enamino iminium ion 71, which is deprotonated by triethylamine to provide the zwitterionic intermediate 72 needed for the subsequent 6π-electrocyclization. Aromatization provides the final product.
Scheme 33.
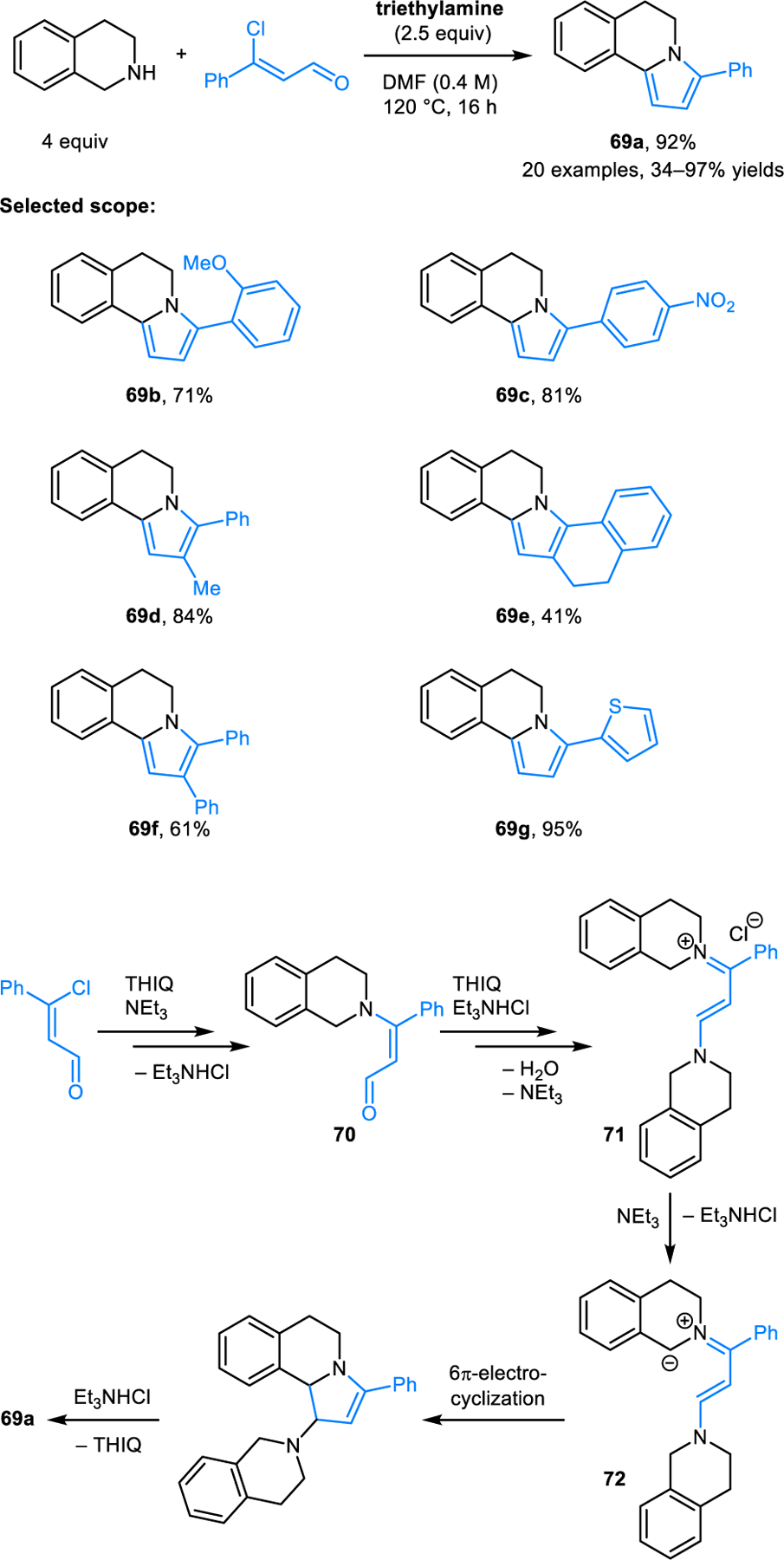
Synthesis of pyrroloisoquinolines via 6π-electrocyclization based on the condensation of DHIQ and E-cinnamaldehydes.
Ou, Yang and coworkers reported a unique (3+3) cycloaddition/dimerization of azomethine ylides derived from N-alkyl isatin and cyclic amines (Scheme 34).59 Reactions are promoted by Brønsted acids such as trifluoroacetic acid and 3,5-dinitrobenzoic acid and provide dispirooxindole-piperazines products such as 73a. The regioselectivity of the cycloaddition depends on the nature of the amine. Reactions involving THIQs form products via a “head-to-tail” dimerization and in most cases provide a mixture of two diastereomers. On the other hand, reactions involving pyrrolidine and glycine methyl ester afford single product diastereomers via a “head-to-head” approach.
Scheme 34.
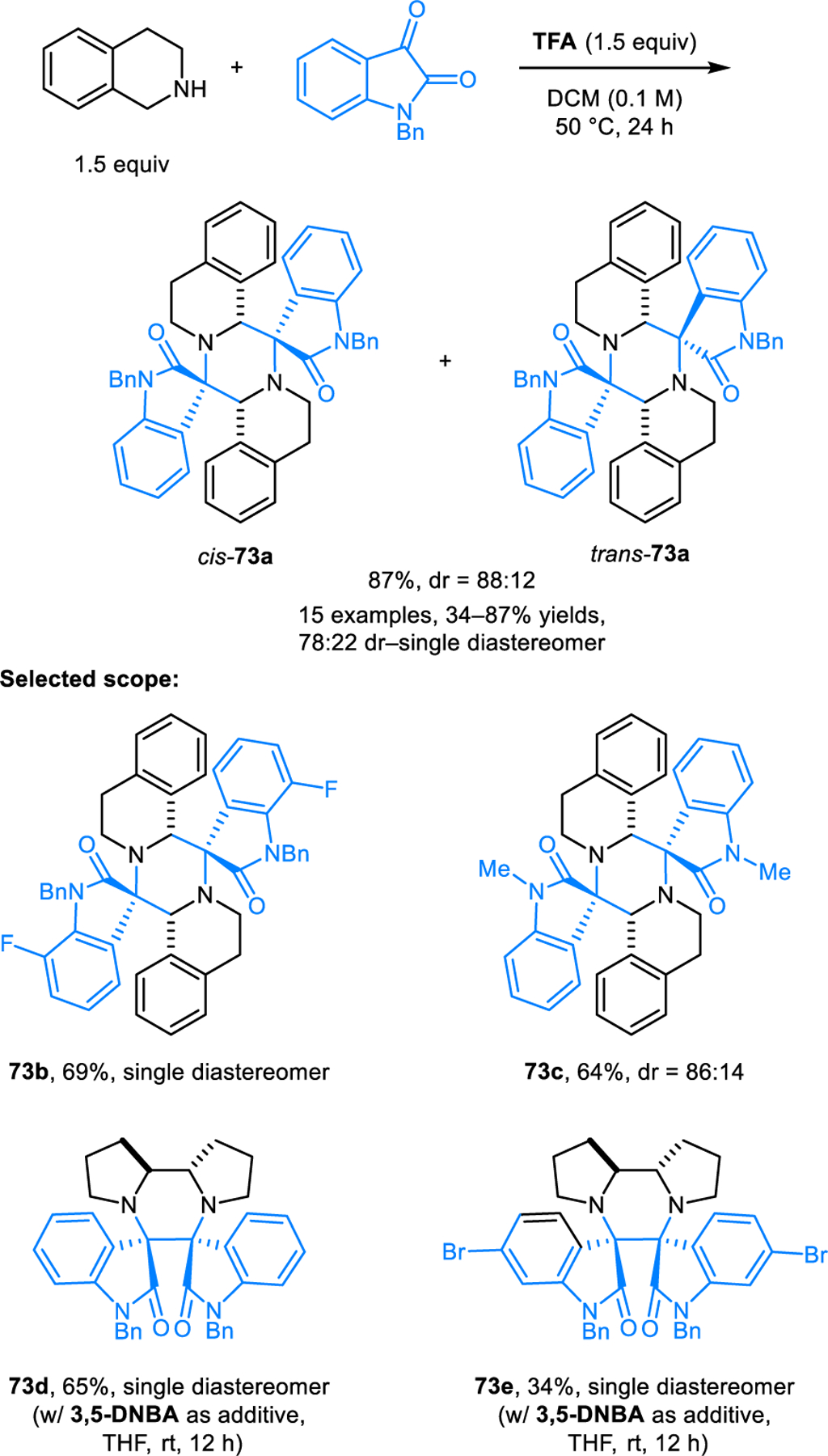
Regioselectivity-tunable 1,3-dipolar (3+3) dimerizations of azomethine ylides derived from the condensation of N-alkyl isatins and cyclic amines.
3.3. 1,5-Proton shift
Quinoenzymes catalyze the oxidation of amines to imines.60,61 The mechanism of this oxidation involves the initial condensation of the amine with quinone, a step often followed by transamination via a (formal) 1,5-proton shift generating an imine intermediate, related to the process proposed in Grigg’s (3+2) dipolar cycloaddition (Scheme 18). While the step breaking the amine α-C−H bond is redox-neutral, the overall reaction often requires external oxidant to convert phenolic substrate or pre-catalyst to quinone. This quinone-mediated, process is attributed to several bioinspired reactions, mostly involving amine dehydrogenation to form imines, a topic which has been extensively reviewed.62–64 Other reactions that fit within the topic of this review involve the condensation of stoichiometric amounts of quinones with amines in overall redox-neutral α-C–H bond functionalizations. Qu and coworkers reported a redox-neutral approach to ring-fused N,O-acetals (e.g., 74, Scheme 35).65 Pyrrolidine undergoes a reaction with 3,5-di-tert-butyl o-quinone at room temperature in 2,2,2-trifluoroethanol (TFE), giving the resulting N,O-acetal product in high yield. Significantly, excellent regioselectivity and diastereoselectivity is observed with 2-substituted pyrrolidines, as exemplified by the reaction of 2-methylpyrrolidine providing 74. The latter product is isolated as the kinetically more favored trans-isomer. Ring-fused N,O-acetals readily undergo ring-opening reactions with organometallic nucleophiles such as Grignard and organolithium reagents, allowing for the facile construction of disubstituted products such as 75. Deprotection under oxidative conditions furnishes trans-2,5-disubstituted pyrrolidines (e.g., 76). The proposed mechanism of this transformation involves a 1,5-proton shift, resulting in the isomerization of exocyclic iminium ion 77 to endocyclic iminium ion 78. Qu and coworkers later developed modified reaction conditions in which products such as 74 are further oxidized by excess 3,5-di-tert-butyl o-quinone to form ring-fused iminium ions, followed by hydrolysis to provide N-aryl-pyrrolidin-2-ones. The latter products provide γ-lactams upon deprotection. This reaction was applied to the synthesis of (S)-Vigabatrin and analogues.66
Scheme 35.
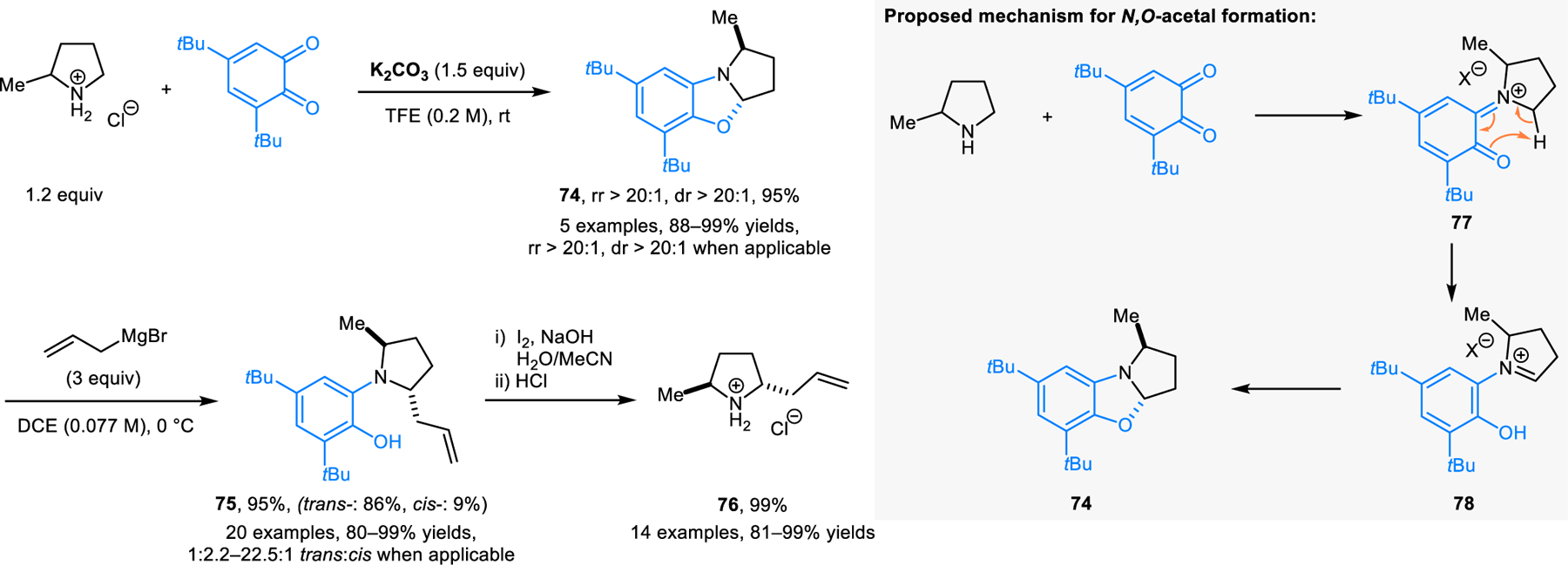
Synthesis of trans-2,5-disubstituted pyrrolidines via quinone-mediated redox-neutral ring-fused N,O-acetal formation.
Quinone-mediated amine α-C−H bond functionalization reactions of primary, α-tertiary amines were developed by Dixon and coworkers (Scheme 36).67 Condensation of a primary amine with 3,5-di-tert-butyl o-quinone provides an N-o-hydroxyaryl imine intermediate via a 1,5-proton shift. These N-o-hydroxyaryl imines are susceptible to the direct addition of various nucleophiles, such as organometallic reagents and cyanide sources to form products such as 79a and 79b. Alternatively, N-o-hydroxyaryl imines can also be converted to α-amino radicals via proton-coupled electron transfer (PCET), catalyzed by a iridium(III) photocatalyst under visible-light in the presence of Hantzsch ester as the proton source. These α-Amino radicals can engage in polarity-reversed reactions with electrophilic radical acceptors, such as (tert-butyl-2-((phenylsulfonyl)methyl)acrylate), yielding α-functionalized products (e.g. 79c). Oxidative deprotection of the o-hydroxyaryl group eventually affords primary α-tertiary amines. This strategy was later extended by the same group to the synthesis of benzo[1,4]oxazines.68 Here, imine intermediates are converted to enamines under basic conditions, followed by the oxidation with iodine to provide α-iodo imines. Finally, displacement of the iodide by the ortho-hydroxyl group provides benzo[1,4]oxazines.
Scheme 36.
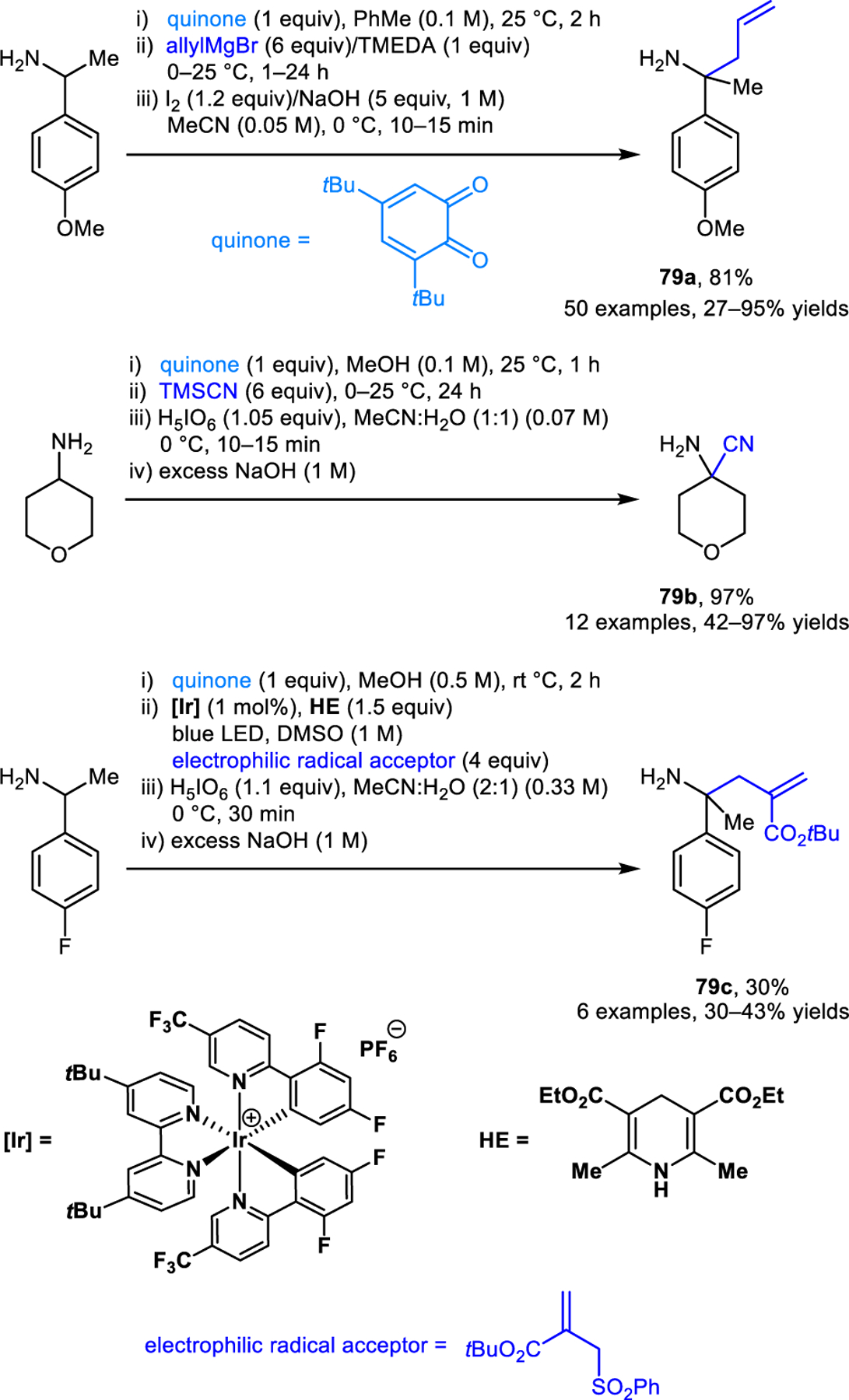
Synthesis of primary α-tertiary amines via quinone-mediated amine α-C−H bond functionalization.
4. Redox-Variants of Classic Transformations incorporating a C–H Bond Functionalization Step
4.1. α-Cyanation
In 2012, our group reported a method for the redox-neutral α-cyanation of cyclic amines. This transformation appears to represent the first example of a new type of intermolecular non-pericyclic reaction pathway for azomethine ylide intermediates derived from the condensation of secondary amines and aldehydes (Scheme 37).69 α-Aminonitrile 80a, the product of a classic Strecker reaction, readily isomerizes to its ring-substituted regioisomer 81a under catalysis by simple carboxylic acids. α-Aminonitriles 80a and 81a equilibrate under microwave heating via the intermediacy of N,O-acetal 83 and azomethine ylide 84. Iminium-cyanide ion pair 82 might also be involved in the process. The 18:1 regioisomeric ratio (rr) of 81a and 80a is thought to represent the thermodynamic equilibrium ration. As a case in point, upon exposure of regioisomerically pure 81a to identical reaction conditions, the same 18:1 regioisomeric ratio of 81a and 80a is obtained. Computational studies by Li and coworkers suggest that the electron density at the benzylic position of azomethine ylide 84 is higher than at the ring-position, thus favoring exocyclic protonation.8e
Scheme 37.
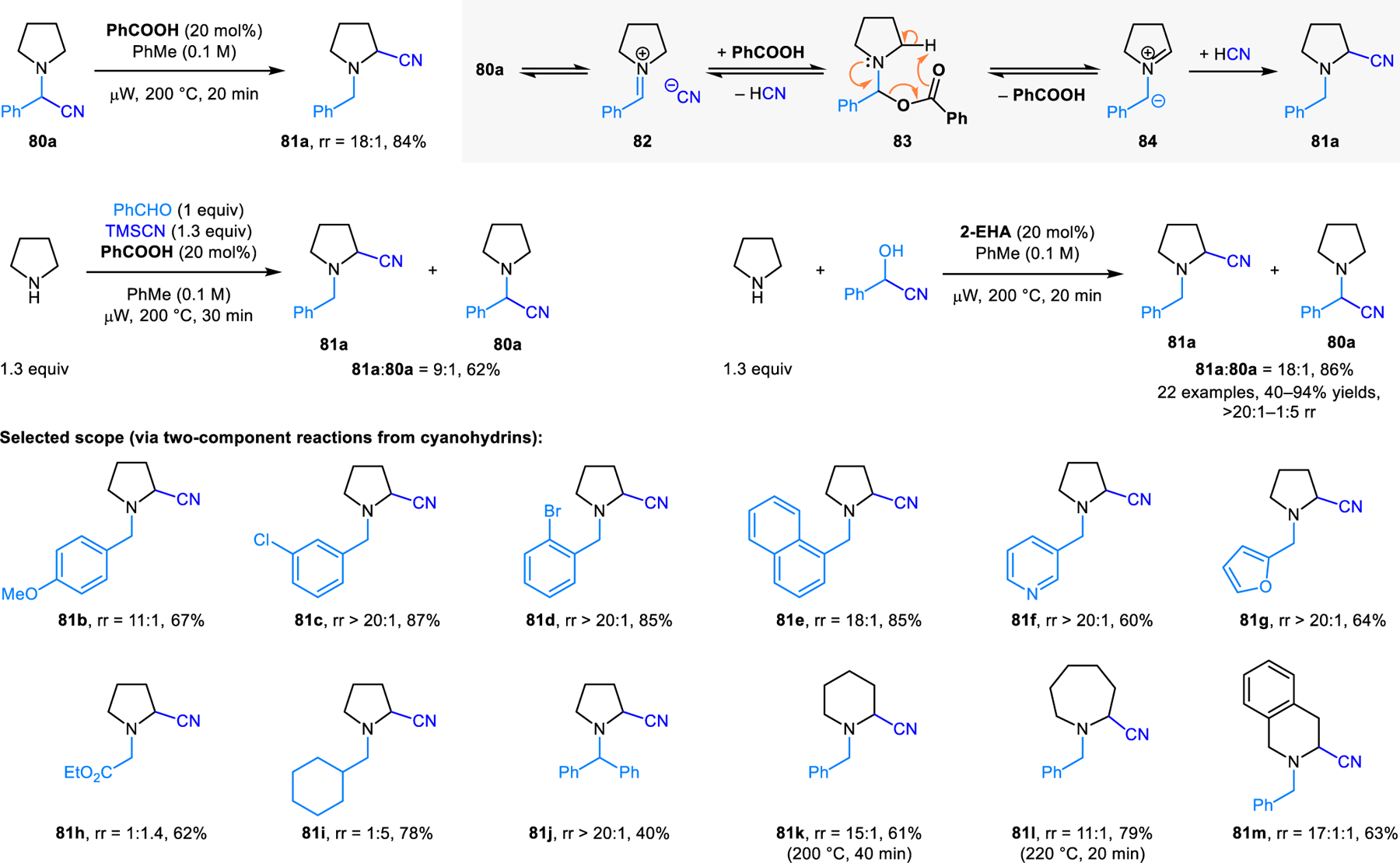
Redox-neutral α-cyanation of cyclic amines.
Aminonitrile 81a can be accessed directly via the three-component condensation of pyrrolidine, benzaldehyde and TMSCN, albeit in reduced regioisomeric ratio (Scheme 37). More favorable results are obtained in reactions of pyrrolidine with cyanohydrins, with the regioisomeric ratio of α-aminonitriles being strongly dependent on the nature of the N-substituent. In these transformations, 2-ethylhexanoic acid (2-EHA) provides more favorable results than benzoic acid. This two-component condensation with cyanohydrins is applicable to a range of amines. As a unique feature of the redox-Strecker reaction, reaction of THIQ with benzaldehyde-derived cyanohydrin predominantly yields the product derived from functionalization of the less activated, non-benzylic C3 position. This outcome is regio-complementary to well-established oxidative methods (typically employing N-aryl-THIQs) which lead to exclusive functionalization of the benzylic C1 position.70
4.2. α-Alkynylation
The regioselective α-alkynylation of cyclic amines was first reported by our group (Scheme 38).71,72 In contrast to the corresponding α-aminonitriles (e.g., 80a and 81a, Scheme 37),regioisomeric propargylic amines such as 85 and 86 do not readily engage in isomerization. Thus, reaction conditions had to be developed that directly furnish ring-functionalized 85 over the classic A3 reaction product 86. Redox-A3-reactions are successfully realized with 2,6-dichlorobenzaldehyde and Cu(II)-2-ethylhexanoate as the catalyst. Pyrrolidine provides excellent regioisomeric ratios favoring the redox-A3 product, while amines such as piperidine and azepane provide redox-A3 products in less favorable but still notable ratios. The regular A3 product is the major regioisomer in reactions with morpholine. The scope of the redox-A3 reaction was subsequently expanded by Yu and coworkers to include cyclic amines containing α-benzylic C–H bonds such as THIQ and tryptoline to generate products such as 87 selectively over 88 (Scheme 39).73 Given the more activated nature of the substrates, reactions proceed under milder reaction conditions (50 °C). Importantly, copper(I) iodide was found to be a uniquely effective catalyst. Regioselectivities are excellent for most aromatic aldehydes and dramatically diminished for aliphatic aldehydes. Interestingly, under otherwise identical conditions, other copper(I) salts such as copper(I) chloride and copper(I) bromide preferentially afford undesired regioisomers.
Scheme 38.
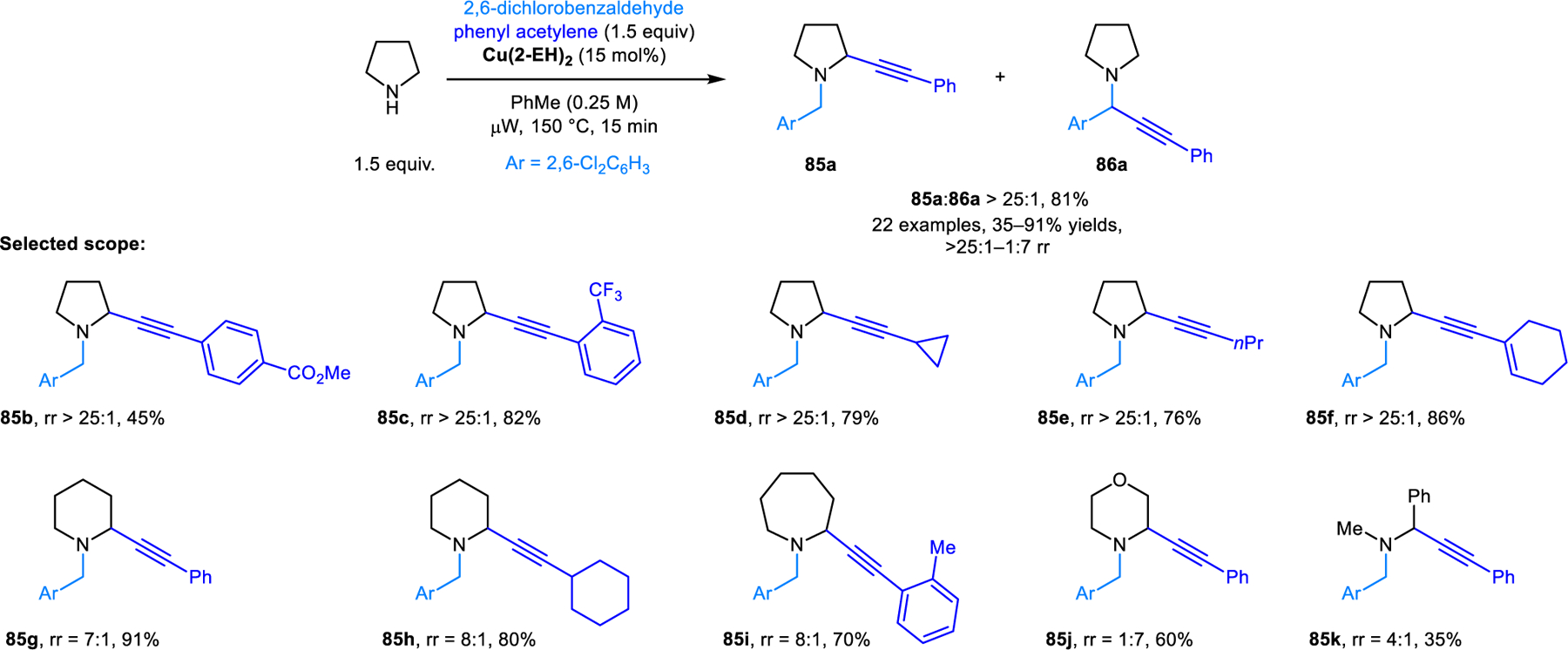
Redox-neutral α-alkynylation of alicyclic amines.
Scheme 39.
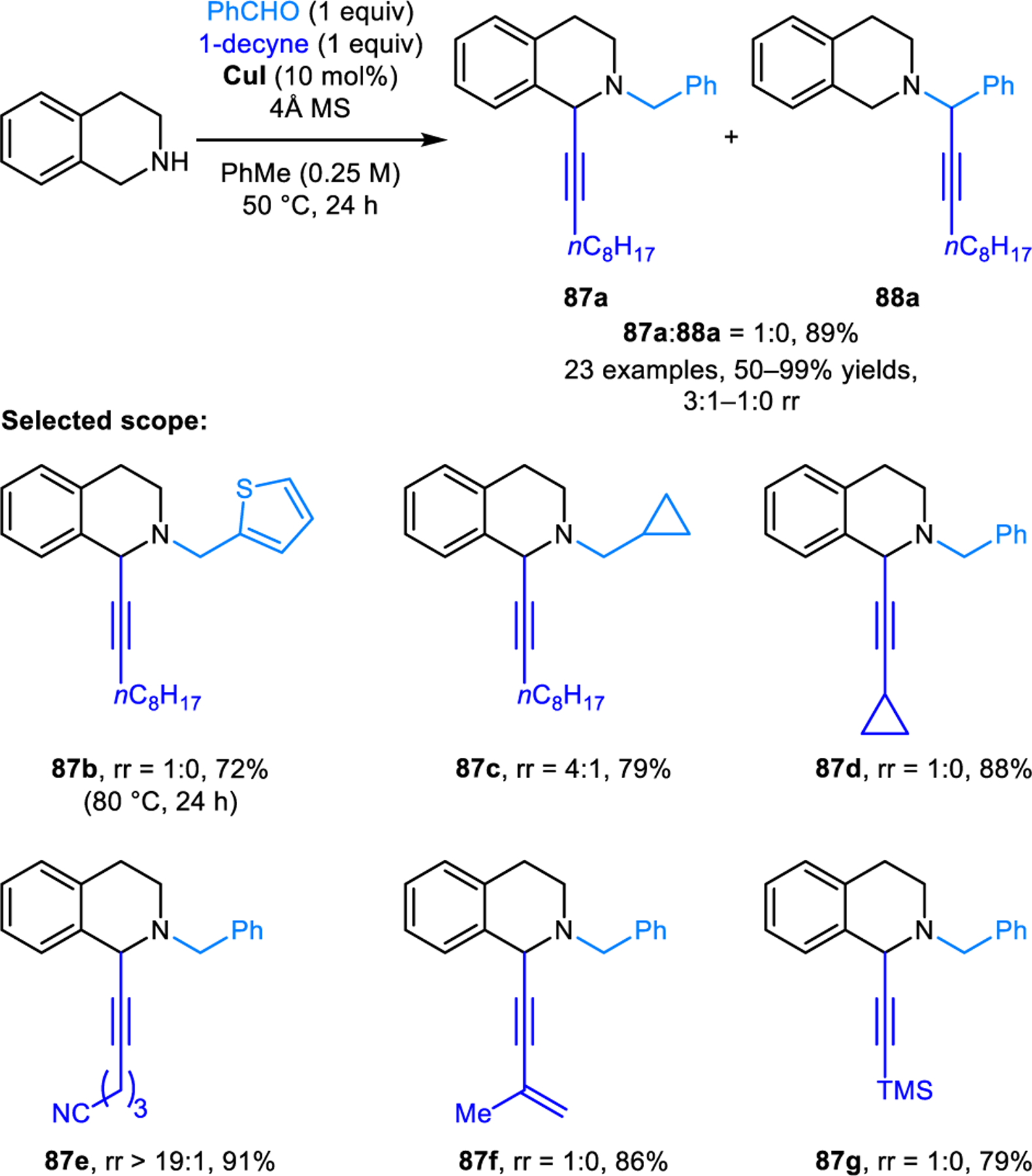
Redox-neutral α-alkynylation of THIQ.
Following the initial reports on redox-A3 reactions, an important advance was published by the Ma group.74 These researchers discovered that low catalysts loadings and highly favorable regioisomeric ratios in redox-A3 reactions of THIQ can be realized with copper(I) bromide, when used in combination with triphenylphosphine. The authors further developed a catalytic enantioselective version of this reaction, utilizing catalytic amounts of copper(I) iodide, a (R,R)-N-Pinap ligand, and benzoic acid (Scheme 40). Excellent enantioselectivities were obtained for a range of products 87. This transformation represents the first and remains one of the few known catalytic enantioselective reactions that follow the azomethine ylide path to redox-neutral α-C−H bond functionalization. Enantioselective α-alkynylations reaction were later applied by the Ma group to the facile synthesis of the natural products (+)-dysoxyline, (+)-crispine A, (−)-trolline, and (+)-oleracein E.75,76 In addition, Tong and coworkers also utilized this reaction in the total synthesis of numerous protoberberine and aporhoeadane alkaloids.77–79
Scheme 40.
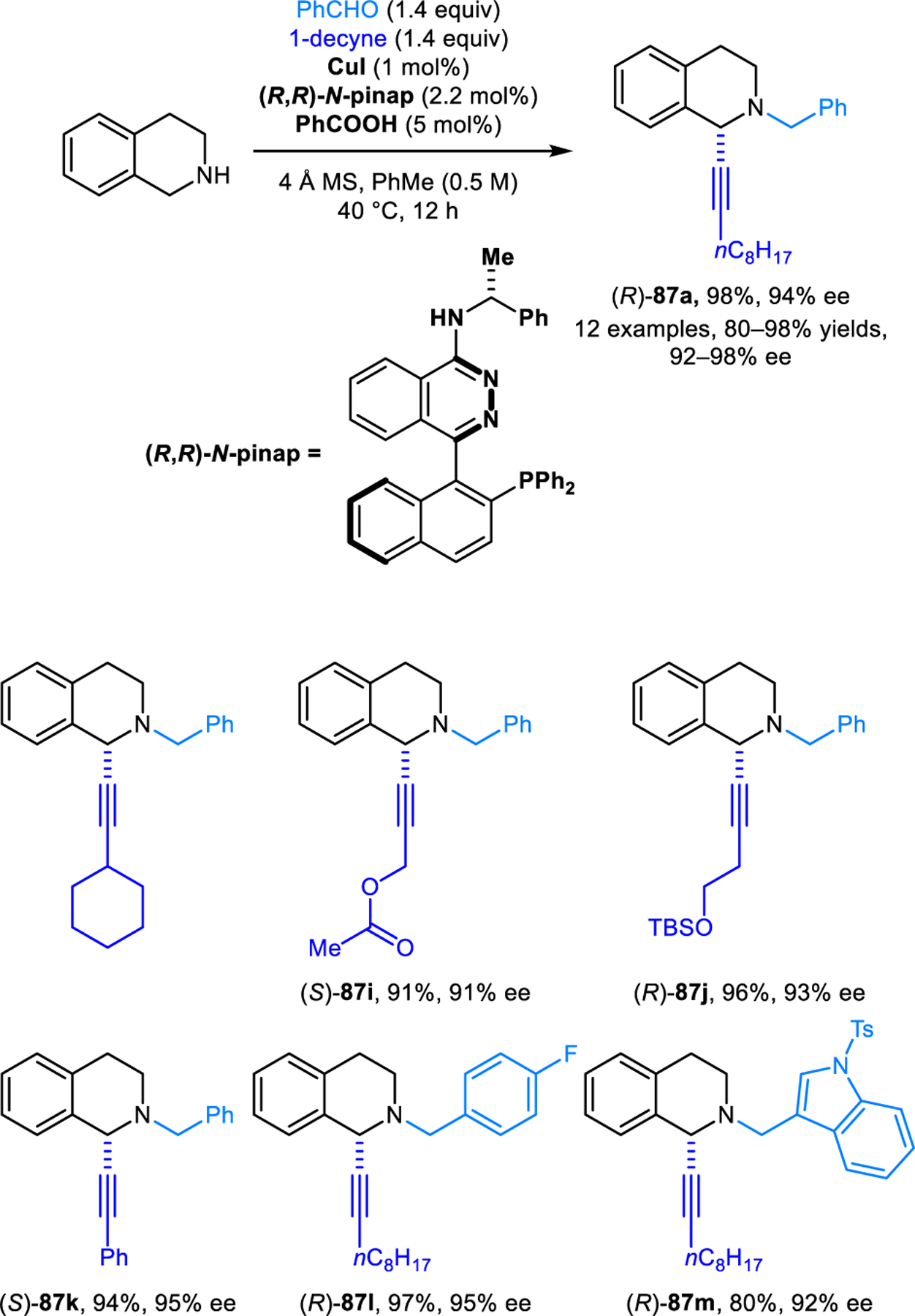
Catalytic enantioselective redox-A3 reaction of THIQ.
Further developments on redox-A3 reactions have appeared over the years. Transition metals other than copper have been explored as catalysts. For instance, Liu and coworkers identified gold(III) bromide as an efficient catalyst in redox-A3 reactions of cyclic amines and N-protected indole-2-carboxaldehyde, providing excellent regioisomeric ratios even with the challenging substrate morpholine (e.g, 88b) (Scheme 41).80 Shao, Cao and coworkers established the ability of silver acetate to catalyze redox-A3 reactions involving THIQ and aromatic aldehydes to yield products including 89 (Scheme 42).81 Kouznetsov and coworkers compared the catalytic performance of an iridium(II) visible-light photocatalyst, silver nitrate, and copper(I) bromide/PPh3, and found that Cu(I)-based systems provide superior results in the redox-A3 reaction of THIQs.82 Heterogenous catalytic systems have also been developed to facilitate catalyst recovery and product isolation. Such catalysts include copper(I) iodide immobilized on a 3-(2-aminoethylamino)propyl-functionalized mesoporous MCM-41 material developed by the Cai group,83 a hematite (Fe2O3)-supported copper oxide nano particle developed by Rawat and coworkers,84 as well as a Cu/C3N4 system developed by the Yao group.85 These heterogenous catalyst systems can be recycled by filtration or the use of a magnet. Reuse of recovered catalysts in multiple cycles without the loss of catalytic activity was demonstrated. A related redox-neutral α-alkynylation reaction was published by Liu and coworkers (Scheme 43).86 Instead of using aldehydes as carbonyl donors, α-alkynylation of cyclic amines is achieved with p-quinol 90 to provide N-aryl propargylic amines such as 91 in good yields. According to the proposed mechanism, the isomerization of the initially formed iminium ion is facilitated by the departure of the trimethylsiloxy group with concurrent aromatization of the quinol ring.
Scheme 41.
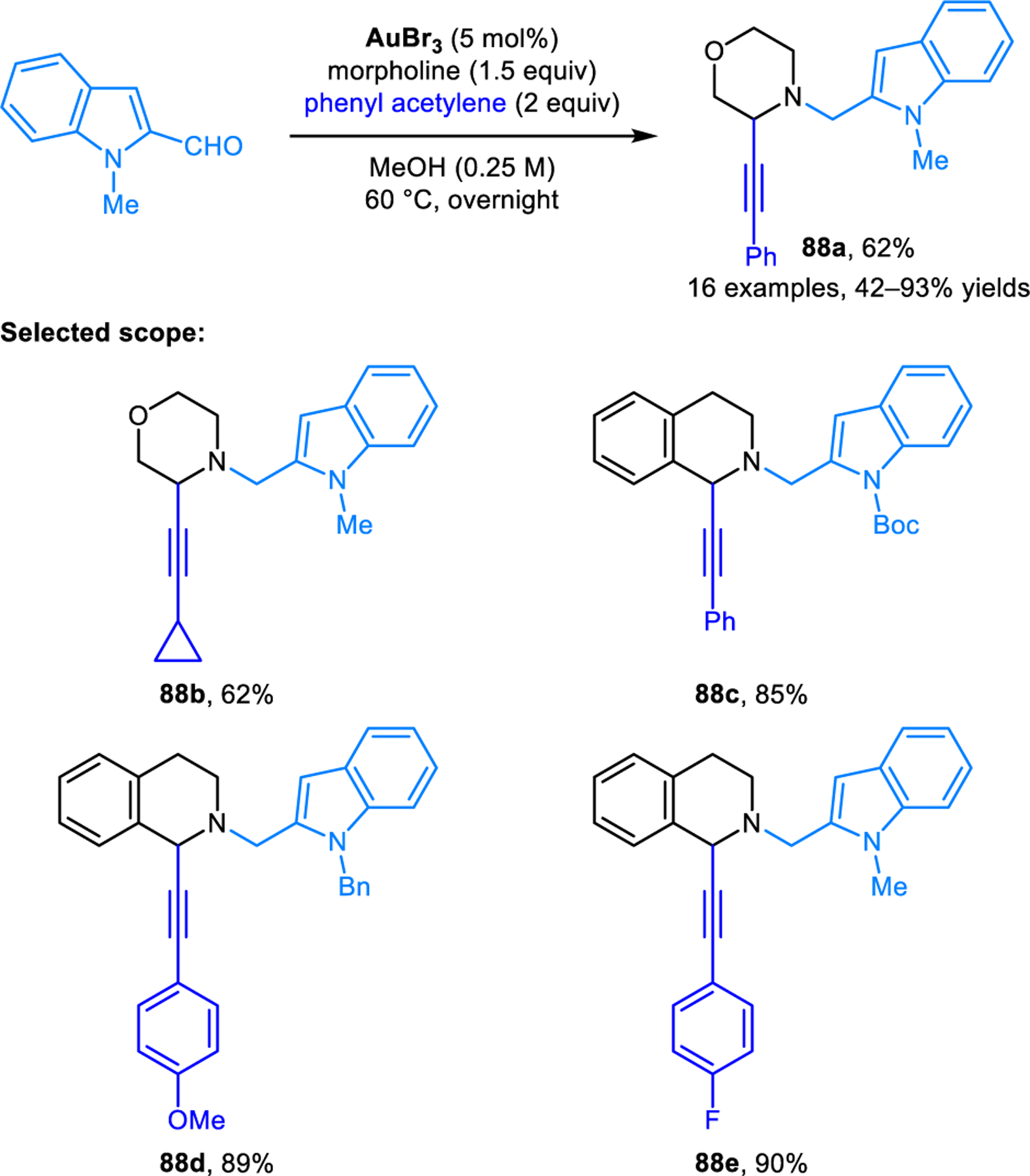
AuBr3-catalyzed redox-neutral α-alkynylation of cyclic amines involving N-methylindole-2-carboxaldehyde.
Scheme 42.
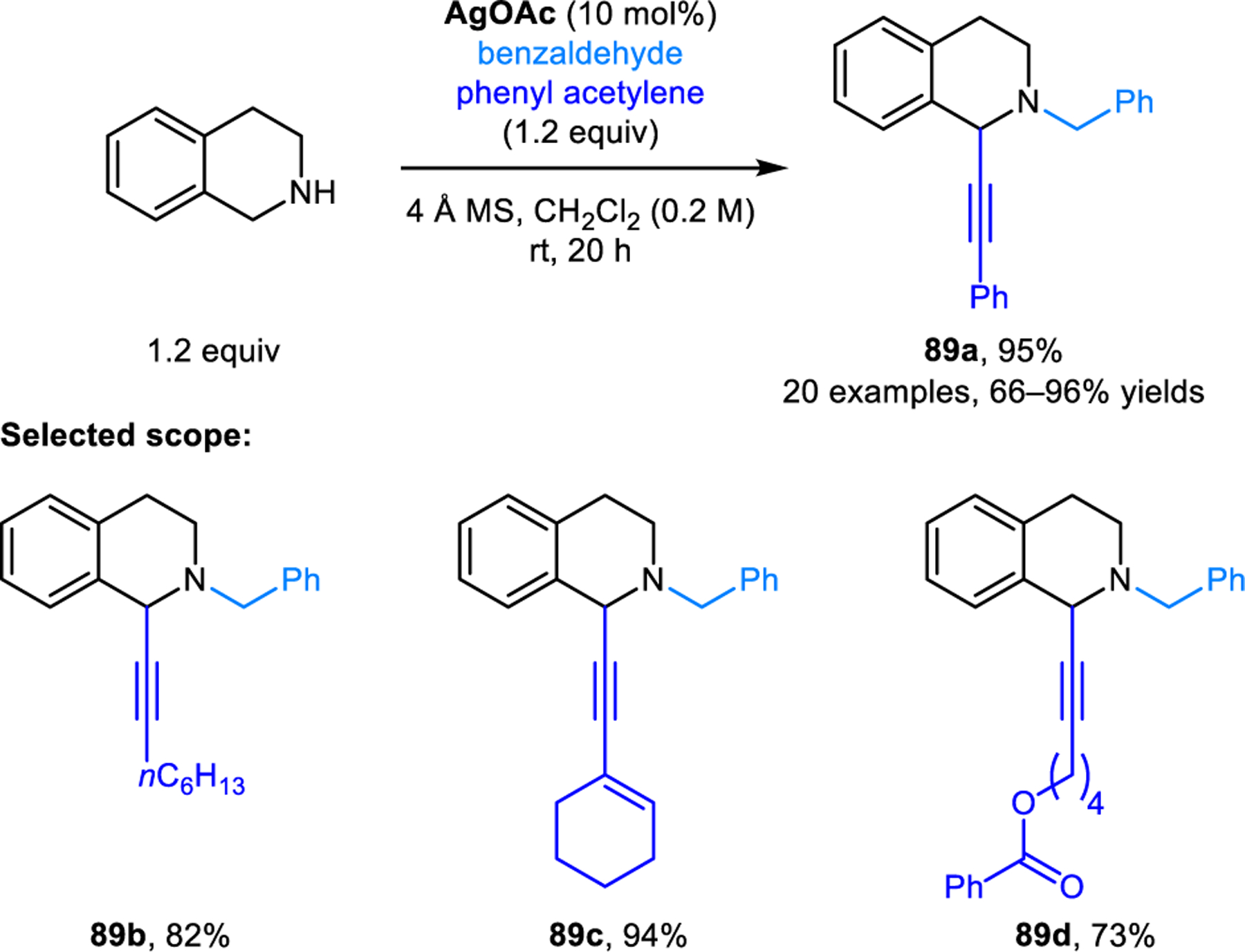
AgOAc-catalysed redox-neutral α-alkynylation of THIQ.
Scheme 43.
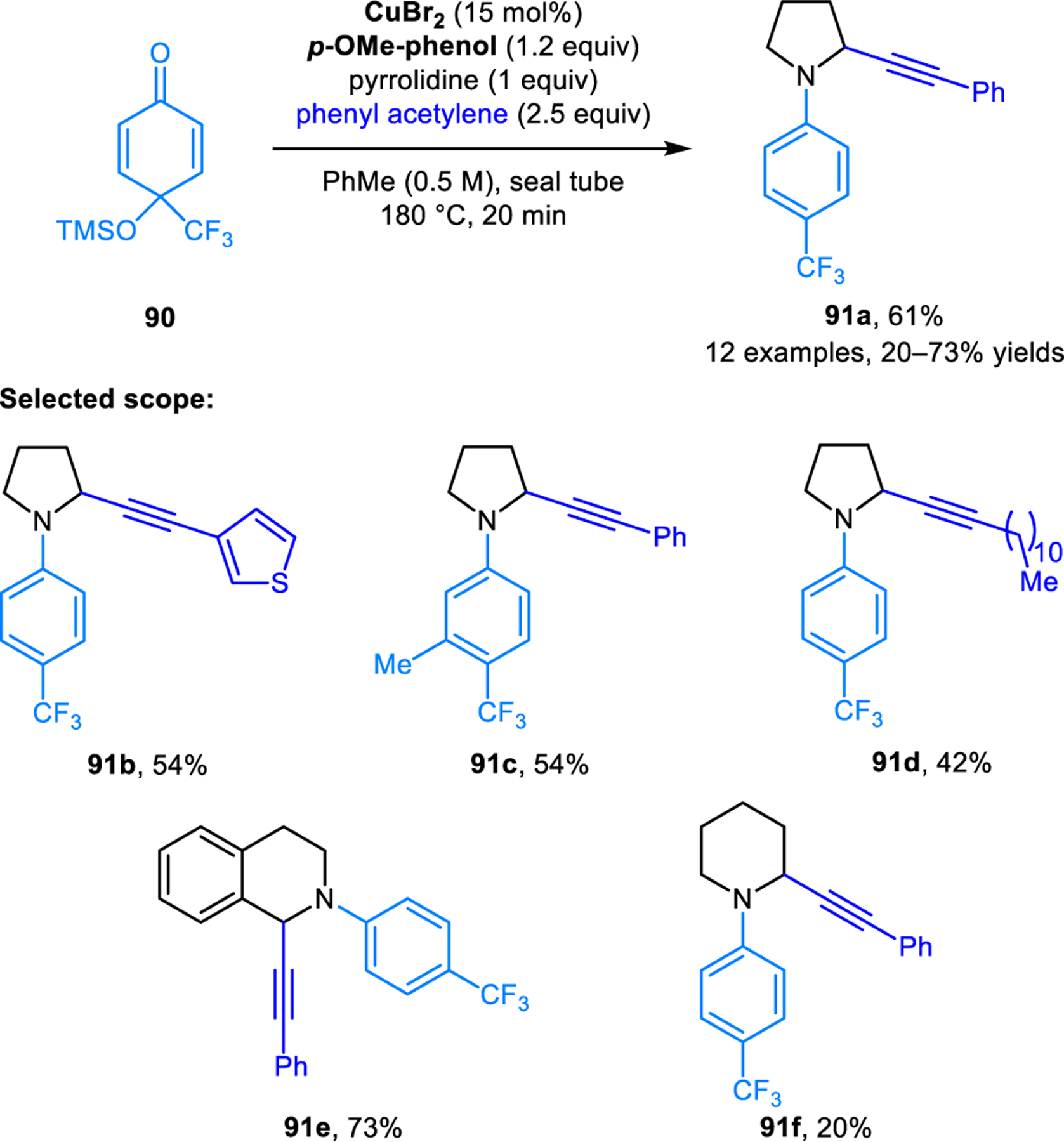
Redox-neutral α-alkynylation of pyrrolidine based on condensation with p-quinols.
4.3. α-Phosphonation
The redox-neutral α-phosphonation of pyrrolidine using phosphine oxides as nucleophiles was reported by our group (Scheme 44).87 The reaction provides the desired redox-Kabachnik-Fields product in high selectivity when 2,6-dichlorobenzaldeyde is employed, even without added carboxylic acid promoters. However, with benzaldehyde, catalytic amounts of benzoic acid are essential to maintain good regioselectivity. As was observed in the corresponding redox-Strecker reaction (Scheme 37), benzoic acid catalyzes the isomerization of the regular Kabachnik-Fields product 93a to the desired product 92a. Unfortunately, the desired regioselectivity is only obtained in reactions involving pyrrolidine and aromatic aldehydes or ketones. Reactions with aliphatic aldehydes and/or other non-benzylic cyclic amines all exhibit significantly decreased regioselectivities in the presence or absence of carboxylic acid additives. Gao and coworkers reported silver acetate catalyzed redox-Kabachnik-Fields reactions involving THIQs.88 Regioselectivities are dependent on the nature of the nucleophile. Dialkyl phosphonates exclusively provide the desired ring-functionalized products, while diphenylphosphine oxide only forms undesired regioisomers. Aliphatic aldehydes afford lower regioselectivity. However, redox-Kabachnik-Fields products are still obtained as the major regioisomers.88
Scheme 44.
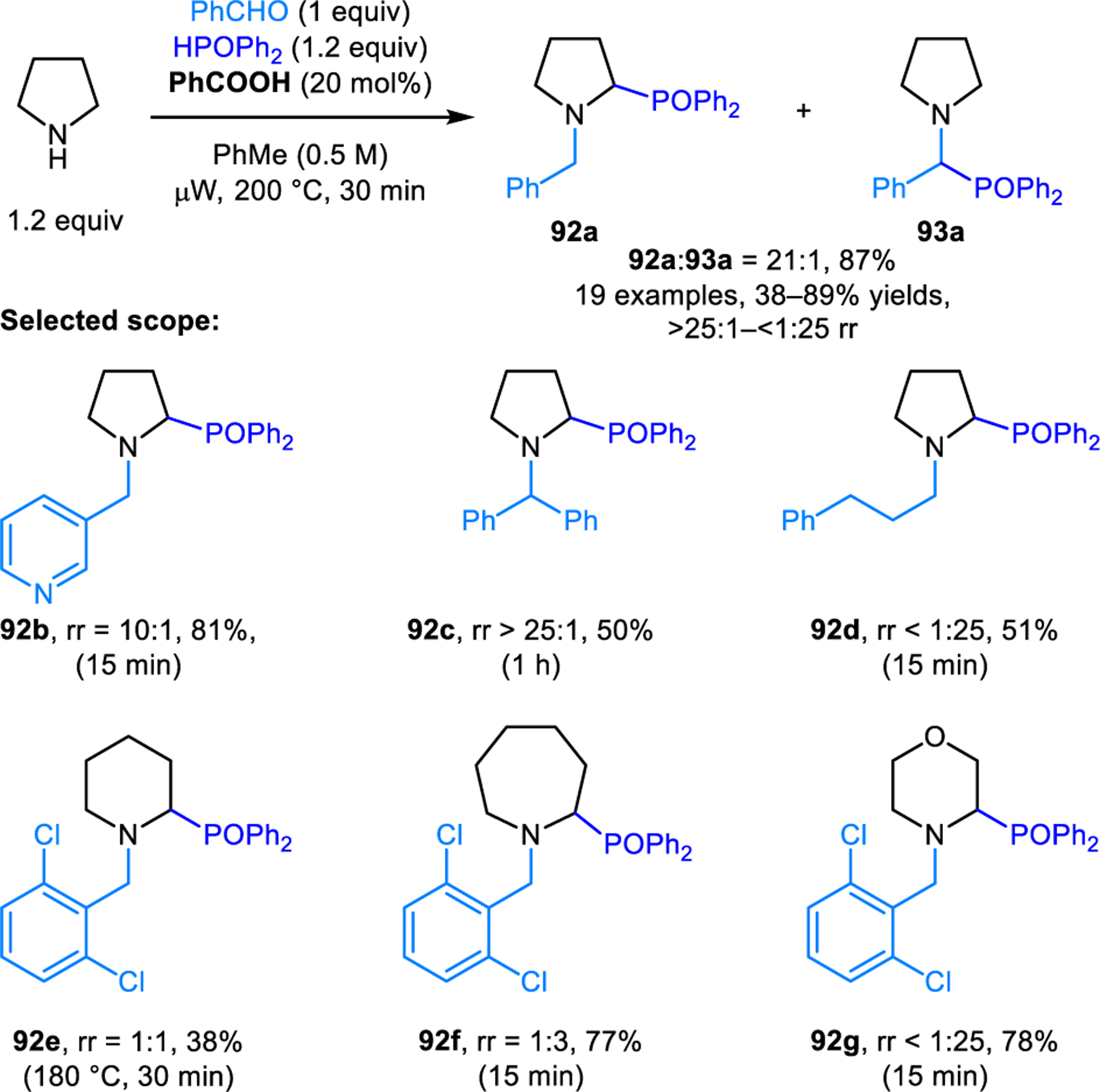
Redox-neutral α-phosphonation of alicyclic amines.
4.4. α-Arylation
Our group developed a redox-variant of the Friedel–Crafts alkylation process, namely the α-arylation of cyclic amines with electron-rich aromatic nucleophiles including 2-naphthols, phenols, indoles and pyrroles (Scheme 45).89 As revealed in the initial development of this reaction, direct heating of a mixture of pyrrolidine, 2,6-dichlorbenzaldehyde and 2-naphthol results in only small amounts of the desired redox-Friedel–Crafts product 94a, and none of the classic condensation product 97. Considerable amounts of side products 95 and 96 are obtained, resulting from competing reaction pathways involving enamine intermediates. Desired product 94a is obtained in excellent yields by keeping the concentration of the aldehyde low throughout the reaction, which is achieved by slow addition. Interestingly, reactions without carboxylic acid additive provide higher yields, indicating that 2-naphthol itself may be sufficiently acidic to catalyze the reaction. In contrast, when indoles and pyrroles are used, stoichiometric 2-EHA is required as an additive to achieve excellent regioselectivities. Regioselectivities once again depend on the nature of the aldehyde. Only redox-Friedel–Crafts products form for both 2-naphthol and indole when 2,6-dichlorobenzaldehyde or mesitylaldehyde is used, while undesired regioisomers predominate with benzaldehyde. Nearly concurrently to our report, the Jana group also disclosed a similar reaction where 9-fluorenone is used in place of 2,6-dichlorobenzaldehyde, achieving excellent regioselectivities in redox-Friedel–Crafts reactions.90 These researchers later utilized this strategy in the diastereoselective and enantiospecific α’-arylation of optically pure prolinol and 2-methylpyrrolidine (Scheme 46).91 Products 98 were obtained in good yields. 9H-fluoren-9-imine outperforms 9-fluorenone in these reactions. Jana et al. also reported related α-arylations of THIQs to form products such as 99a (Scheme 47).92 Yi, Zhang and coworkers developed redox-arylations of THIQs using indoles as nucleophiles, catalyzed by a combination of copper(I) bromide/triphenylphosphine, and acetic acid (Scheme 48).93 Side products derived from Friedel-Crafts reactions between indole and aromatic aldehydes are formed in the absence of triphenylphosphine and inferior yields of products 100 are obtained with either no or higher amounts of acetic acid. Luo, Lv and coworkers later reported very similar reactions catalyzed by a Lewis acidic tritylium salt.94 In addition, redox-neutral α-arylation reactions involving THIQs were also reported by Deb, Baruah, and coworkers using L-proline as the catalyst.95 The Rawat group realized closely related transformations under catalyst-free conditions in a biodegradable PEG solvent.96a Catalyst-free α-arylations of THIQ with indole and salicylaldehyde were reported by Kumar and coworkers.96b
Scheme 45.
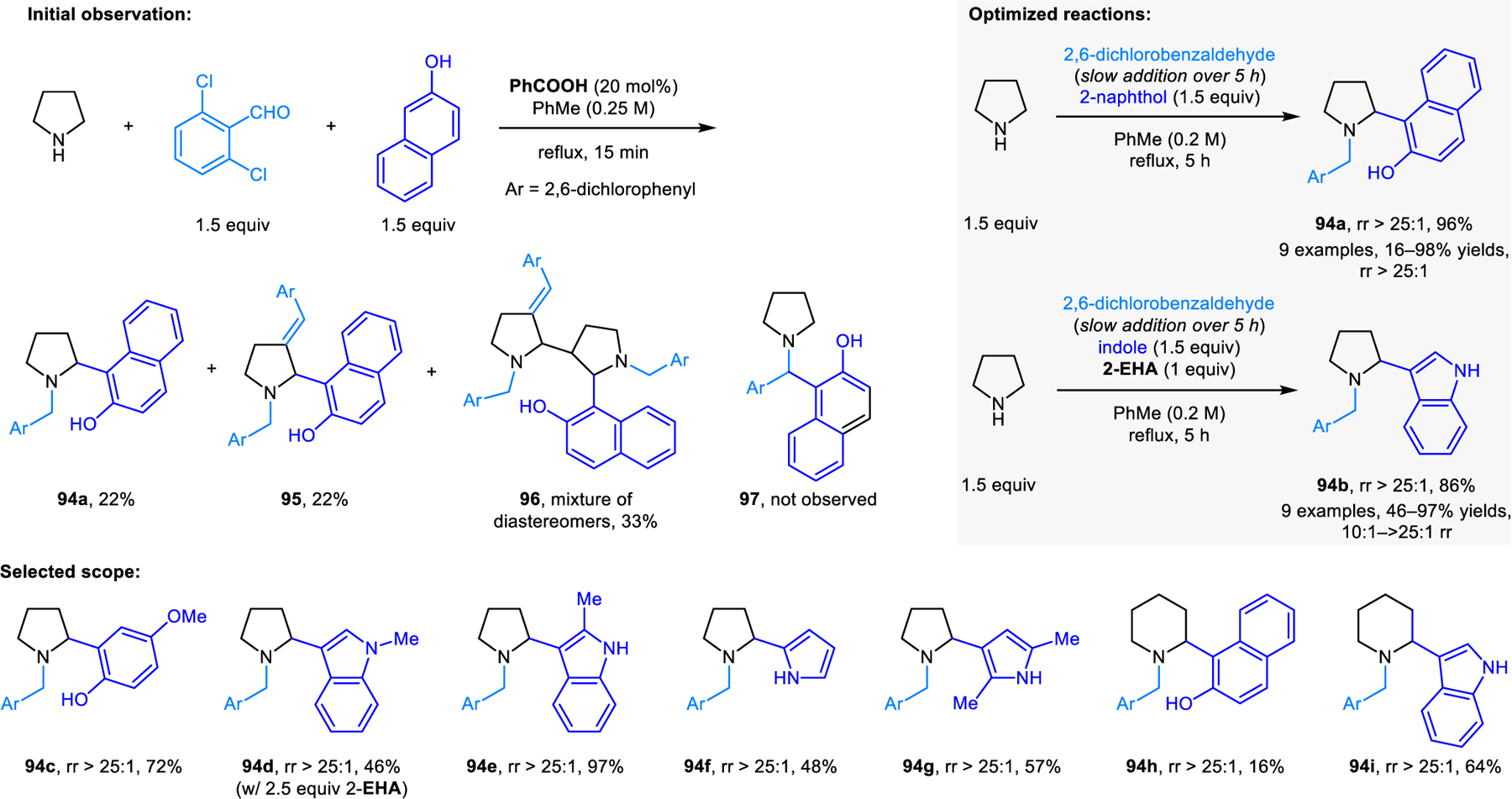
Redox-neutral α-arylation of pyrrolidine.
Scheme 46.
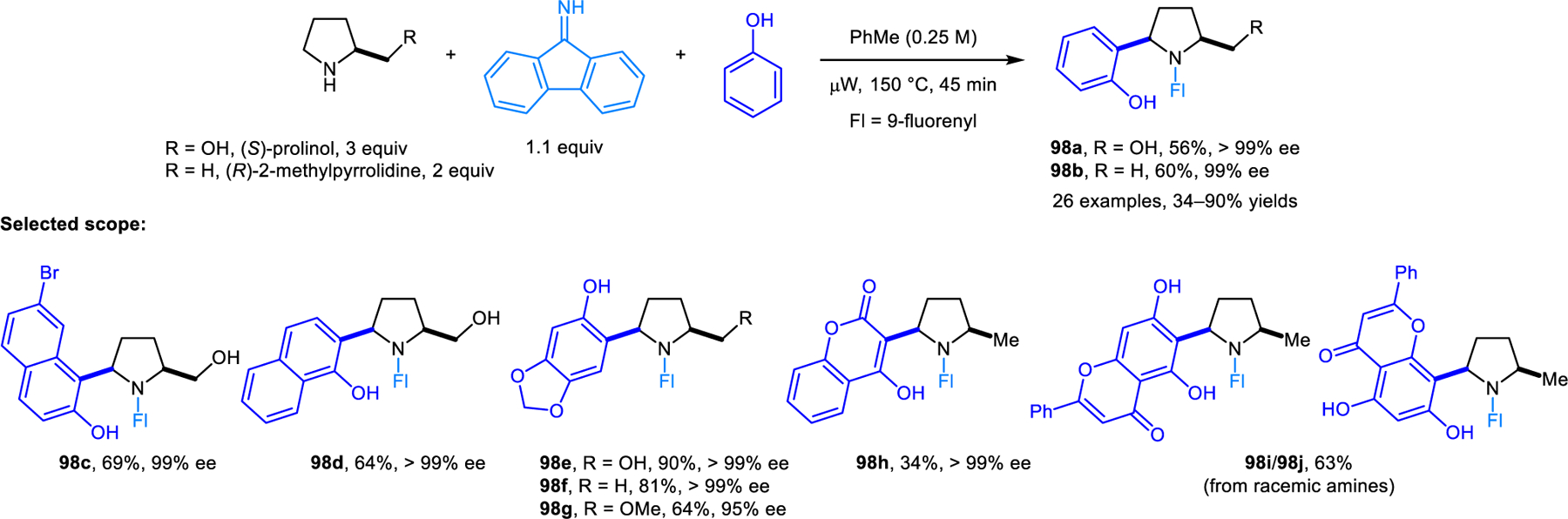
Diastereoselective and enantiospecific redox-neutral α’-arylation of enantioenriched prolinol and 2-methylpyrrolidine.
Scheme 47.
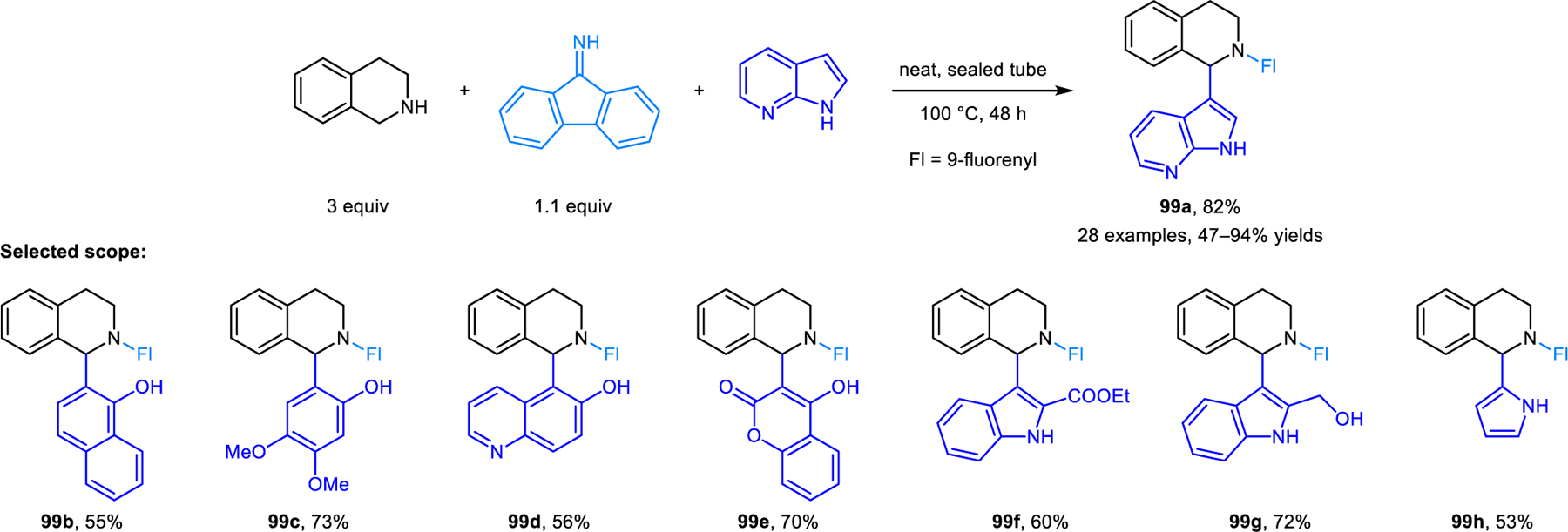
Redox-neutral α-arylation of THIQ with various nucleophiles.
Scheme 48.
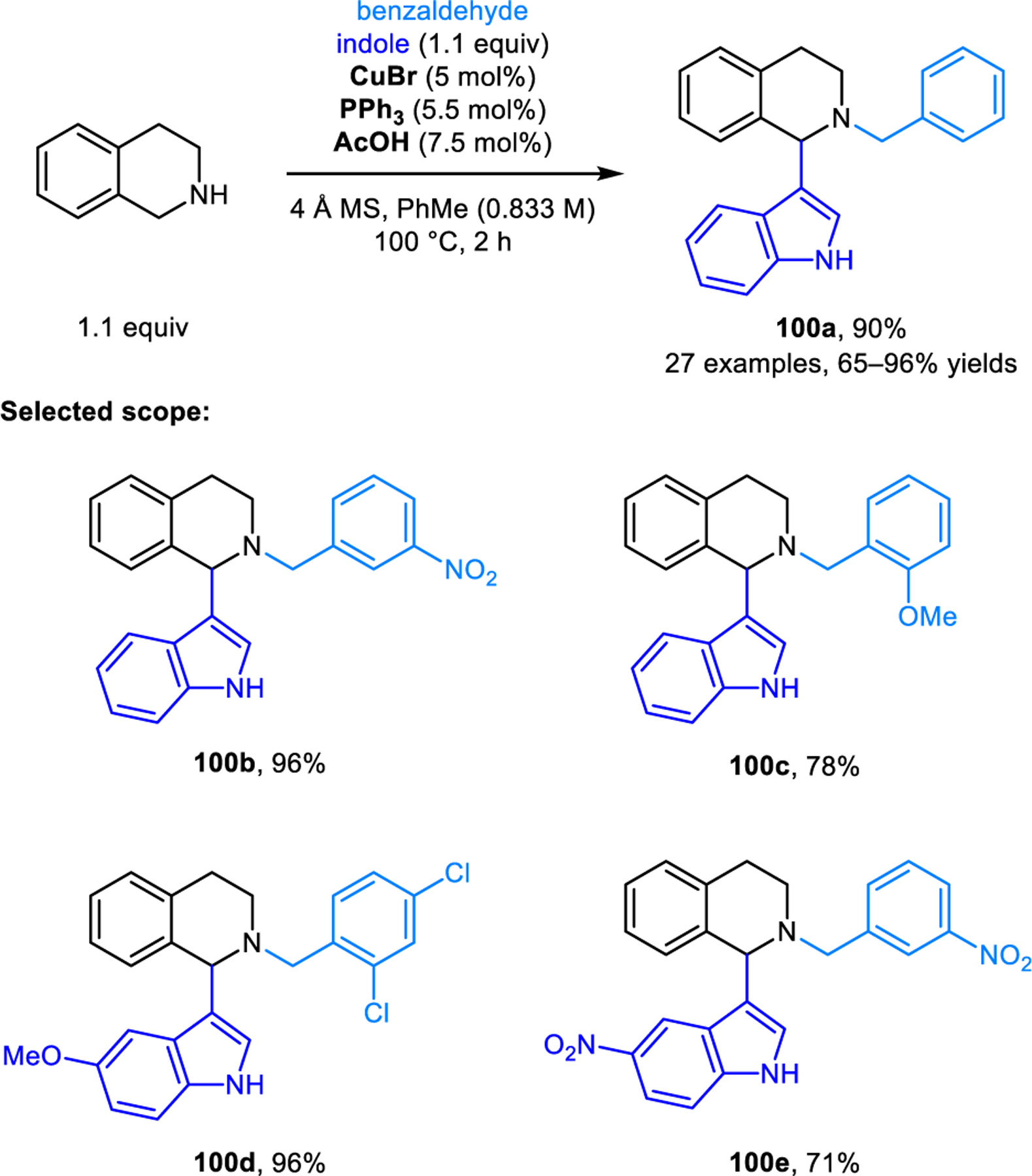
Copper and acetic acid co-catalyzed redox-arylation of DHIQ involving indoles as nucleophiles.
4.5. α-Alkylation with Ketones
Redox-Mannich reactions of cyclic amines were first reported by our group, employing enolizable ketones and nitroalkanes as nucleophiles (Scheme 49 and 50).97 In the case of pyrrolidine, 2,6-dichlorobenzaldehyde and slow addition conditions are critical to obtaining acceptable yields of products 101. Reactions involving THIQ can be performed under simplified conditions at 50 °C. Interestingly, no classic Mannich reaction products are observed in these reactions, likely due to the facile formation of chalcones via amine elimination.
Scheme 49.
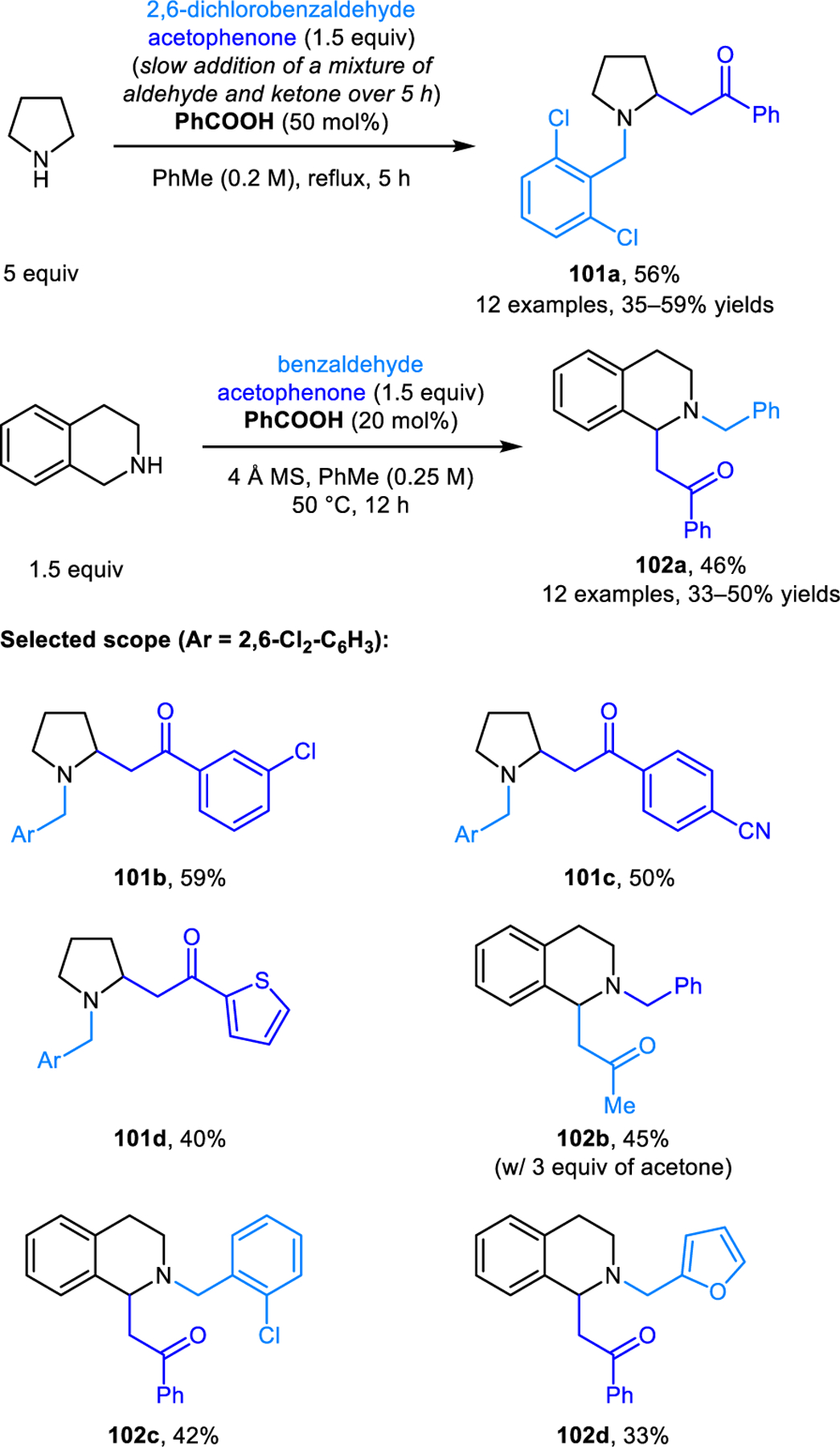
Redox-Mannich reactions of pyrrolidine and THIQ involving ketones as nucleophiles.
Scheme 50.
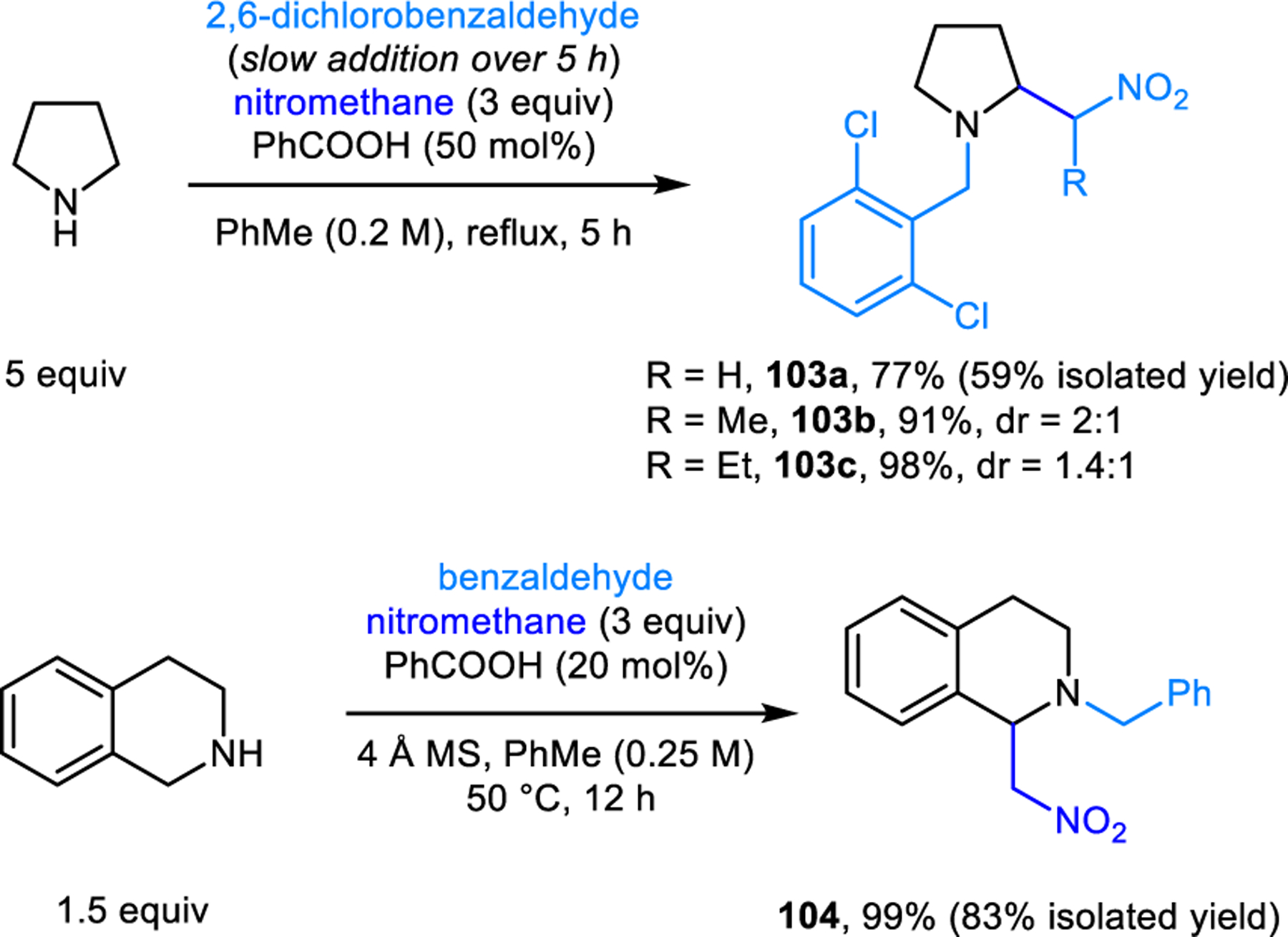
Redox-Mannich reactions with nitromethane.
4.6. Redox-Ugi Reactions
A new variant of the classic Ugi reaction involving the α-C−H bond functionalization of cyclic amines was reported by our group (Scheme 51).98 Amines readily react with aromatic aldehydes/ketones and isocyanides under reflux in toluene. These redox-Ugi reactions are promoted by acetic acid and lack the prototypical Mumm rearrangement. Instead, nitrilium ion intermediates are directly hydrolyzed to provide N-alkyl α-amino amide products such as 105. Pyrrolidine and THIQ are viable substrates. As in several examples mentioned previously, reactions involving pyrrolidine are limited to carbonyl reaction partners such as fluorenone, mesitaldehyde, and 2,6-dichlorobenzalehyde. THIQ exhibits a broader scope regarding aldehydes. In most cases, water is added to improve the yields. A publication by Feng and coworkers details closely related transformations in which imides (e.g., 106) are isolated as the final products (Scheme 52).99 In the presence of molecular sieves, nitrilium ion intermediates react further with carboxylic acids. Aromatic carboxylic acids perform well while acetic acid results in lower yields. Only hydrolyzed products are obtained with formic acid. Not surprisingly, the regioselectivity is highly dependent on the reaction conditions. Desired ring-functionalized products of reactions involving THIQ are obtained at high temperatures in toluene, while classic Ugi products form at room temperature in acetonitrile. Jana and coworkers developed a redox-Ugi-azide four-component reaction (Scheme 53).100 These reactions involve trapping of the nitrilium ion intermediates by TMSN3, generating α-tetrazolyl amines (e.g., 107).
Scheme 51.
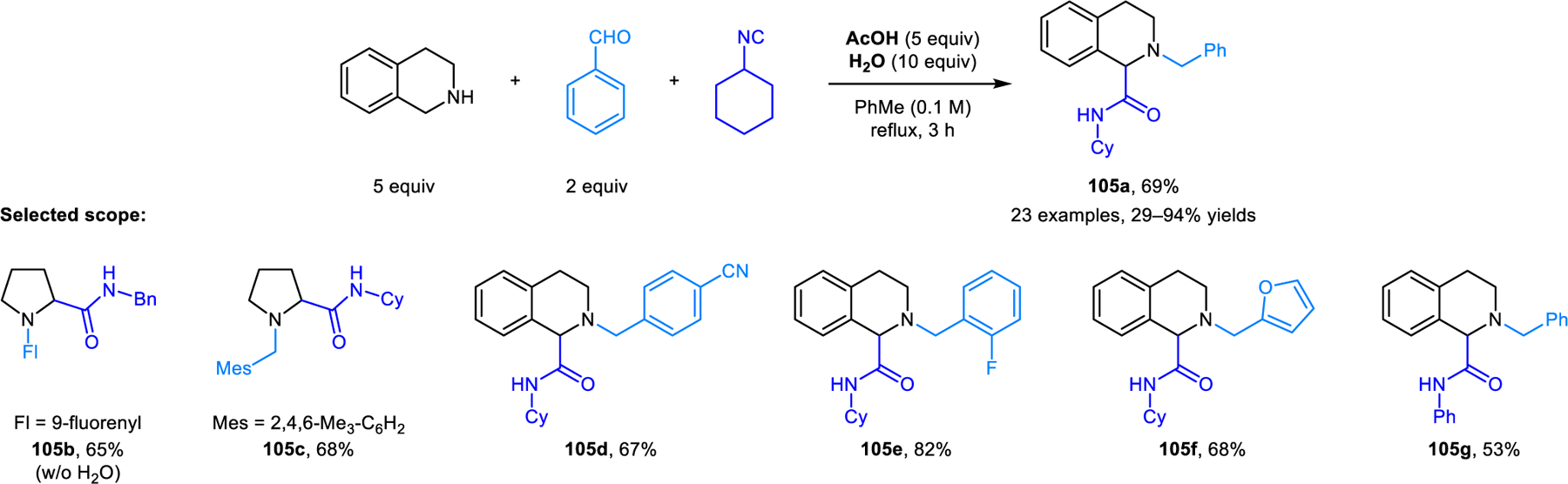
Redox-Ugi reactions involving hydrolysis of nitrilium ion.
Scheme 52.
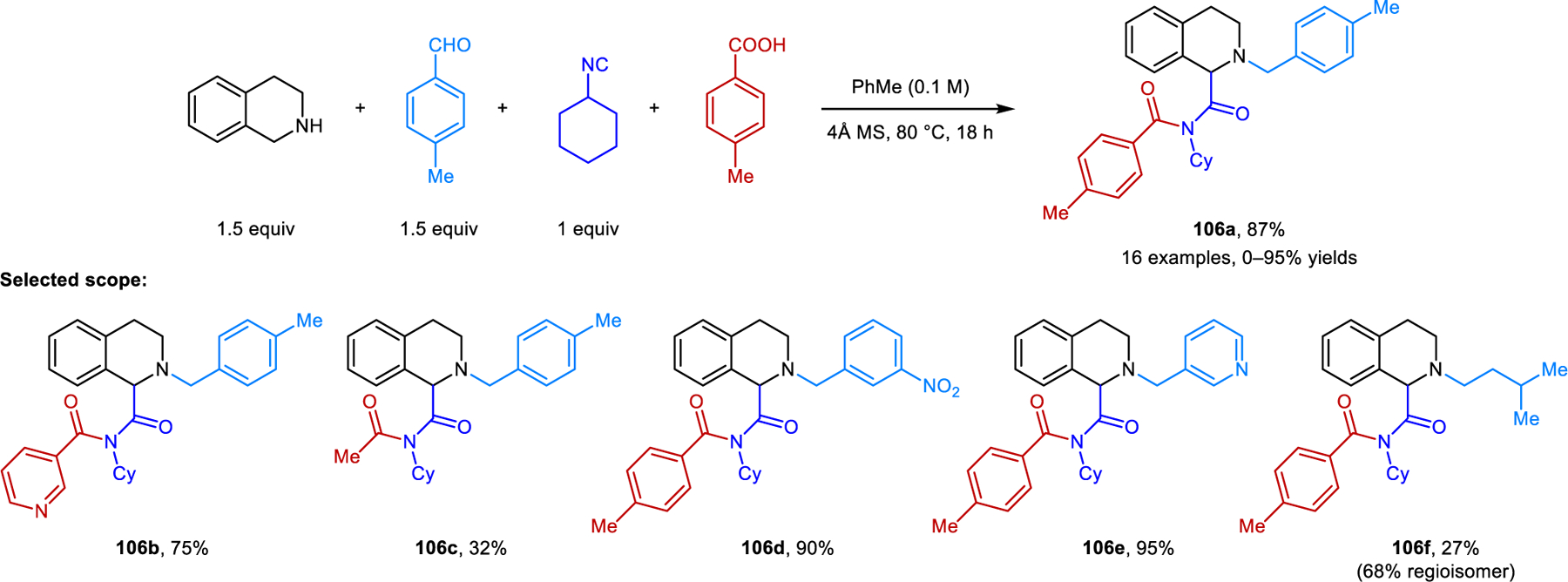
Redox-Ugi reactions involving carboxylic acid addition to nitrilium ion.
Scheme 53.
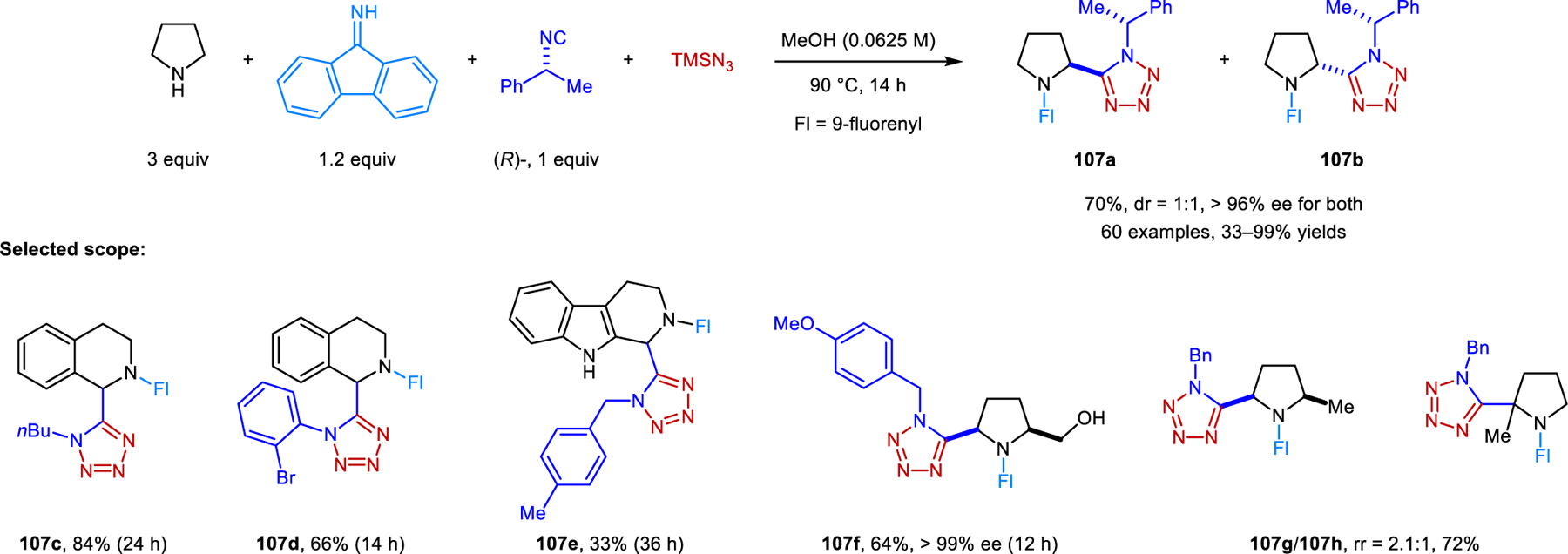
Redox-Ugi-azide reaction forming α-tatrazolyl amines.
4.7. Miscellaneous Intermolecular Reactions
The redox-neutral benzylic C−H bond functionalization of THIQ employing diazo carbonyl compounds as nucleophiles was achieved by Zhou and coworkers (Scheme 54).101 Reactions providing products such as 108 are facilitated by catalytic amounts of silver acetate or stoichiometric benzoic acid. With silver acetate, the regioselectivity of this reaction is highly dependent on the aldehyde used. 2,6-Dimethyl and 2,6-dichlorobenzaldehyde provide excellent selectivities while sterically less demanding aromatic aldehydes produce undesired regioisomers in significant amounts. The formation of undesired regioisomers is completely suppressed when silver acetate is replaced with one equiv of benzoic acid, albeit at the expense of product yields.
Scheme 54.
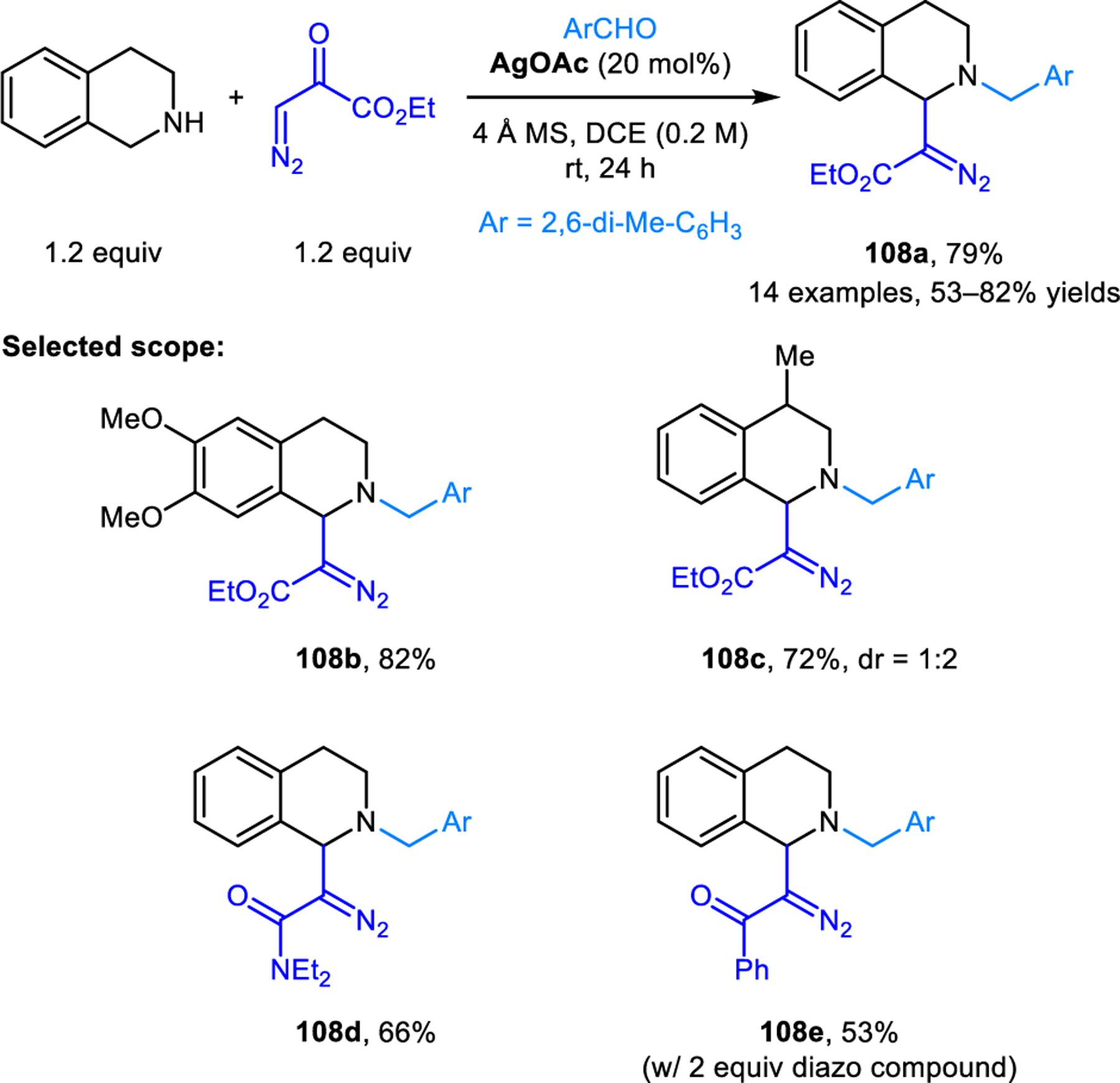
Redox-neutral α-functionalization of THIQs involving diazo compounds as nucleophiles.
The first redox-Petasis reaction was reported by Qu and coworkers (Scheme 55).102 Vinyl and aryl boronic acids or borates are viable nucleophiles for the α-C−H bond functionalization of 3-pyrrolidinol, based on the condensation of the latter with the mono-dimethylketal of p-quinone. The hydroxyl group of 3-pyrrolidinol is critical as it acts as a directing group in the formation of products 109. This strategy was applied to a 4-step enantioselective total synthesis of 8-epi-(−)-lentiginosine.
Scheme 55.
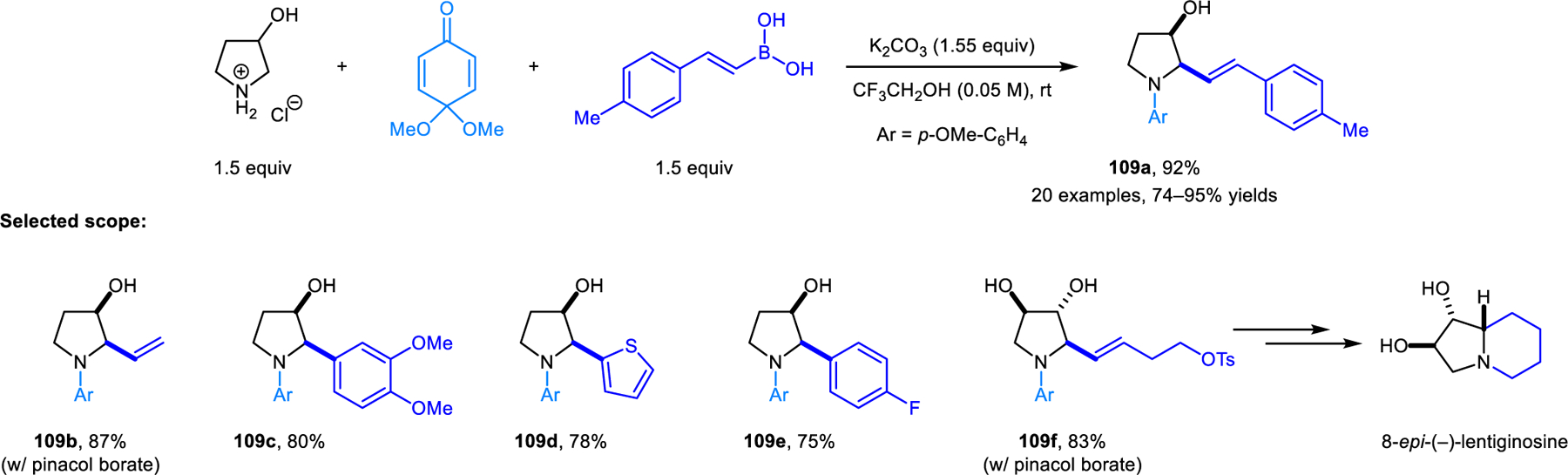
Redox-Petasis reaction of 3-pyrrolidinol.
Jana and coworkers reported an unusual N-amination of amines that proceeds with concurrent α-oxidation (Scheme 56).103 Specifically, pyrrolidine engages nitrosobenzenes in the presence of 2,4-dichlorobenzoic acid (DCBA) to provide cyclic hydrazides such as 110. THIQ is also a viable substrate. This transformation is related to the redox-amination/aromatization reported by Tunge and coworkers (c.f. Scheme 17) but involves a subsequent oxidative step.32
Scheme 56.
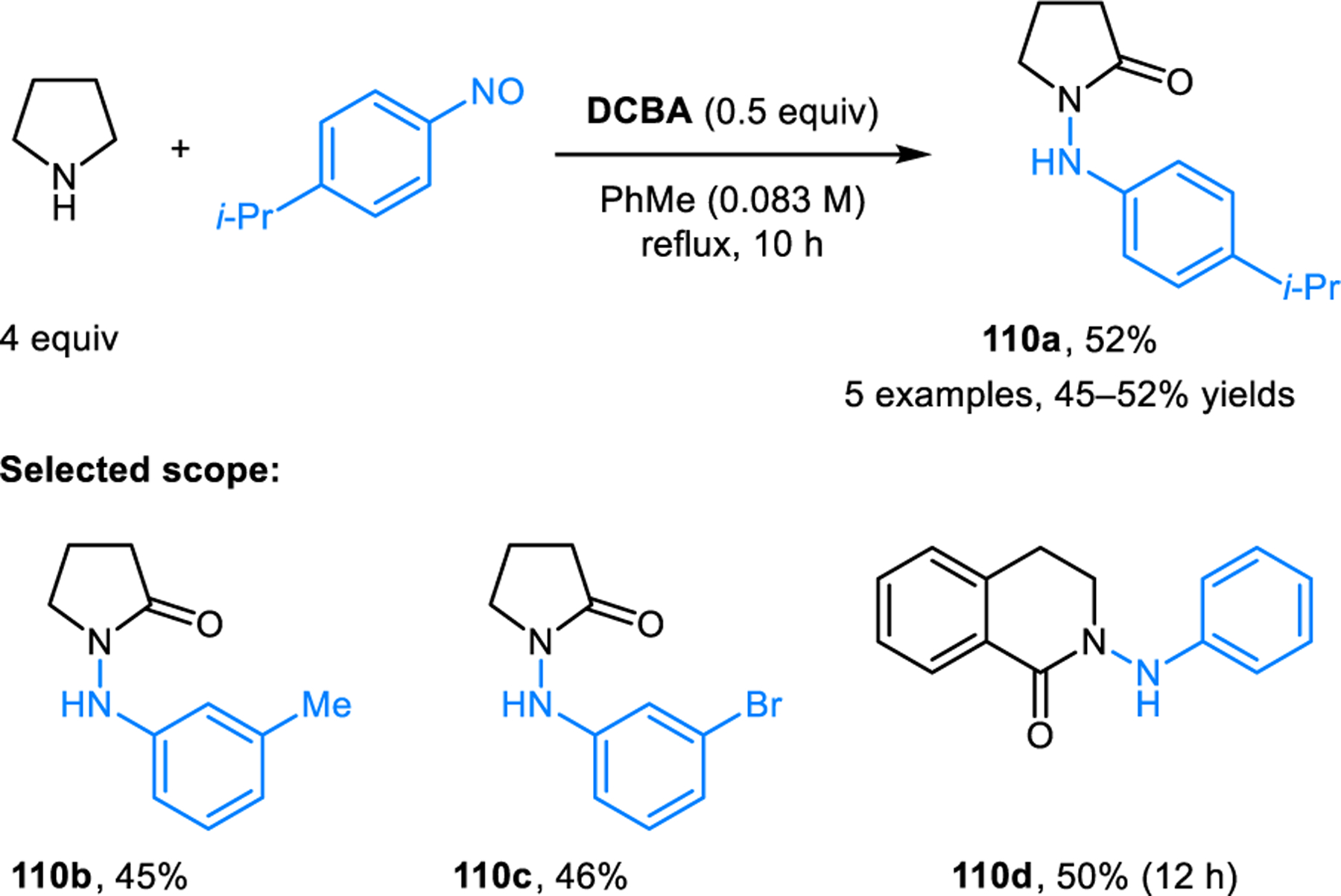
N-amination of cyclic amines with nitrosobenzene.
Very recently, Wang and coworkers reported an N-heterocyclic carbene (NHC)-catalyzed redox-aza-benzoin condensation of THIQs (Scheme 57).104 Sodium benzoate is used as the base in the formation of the active NHC catalyst. Products such as 111a are formed in good to excellent yields.
Scheme 57.
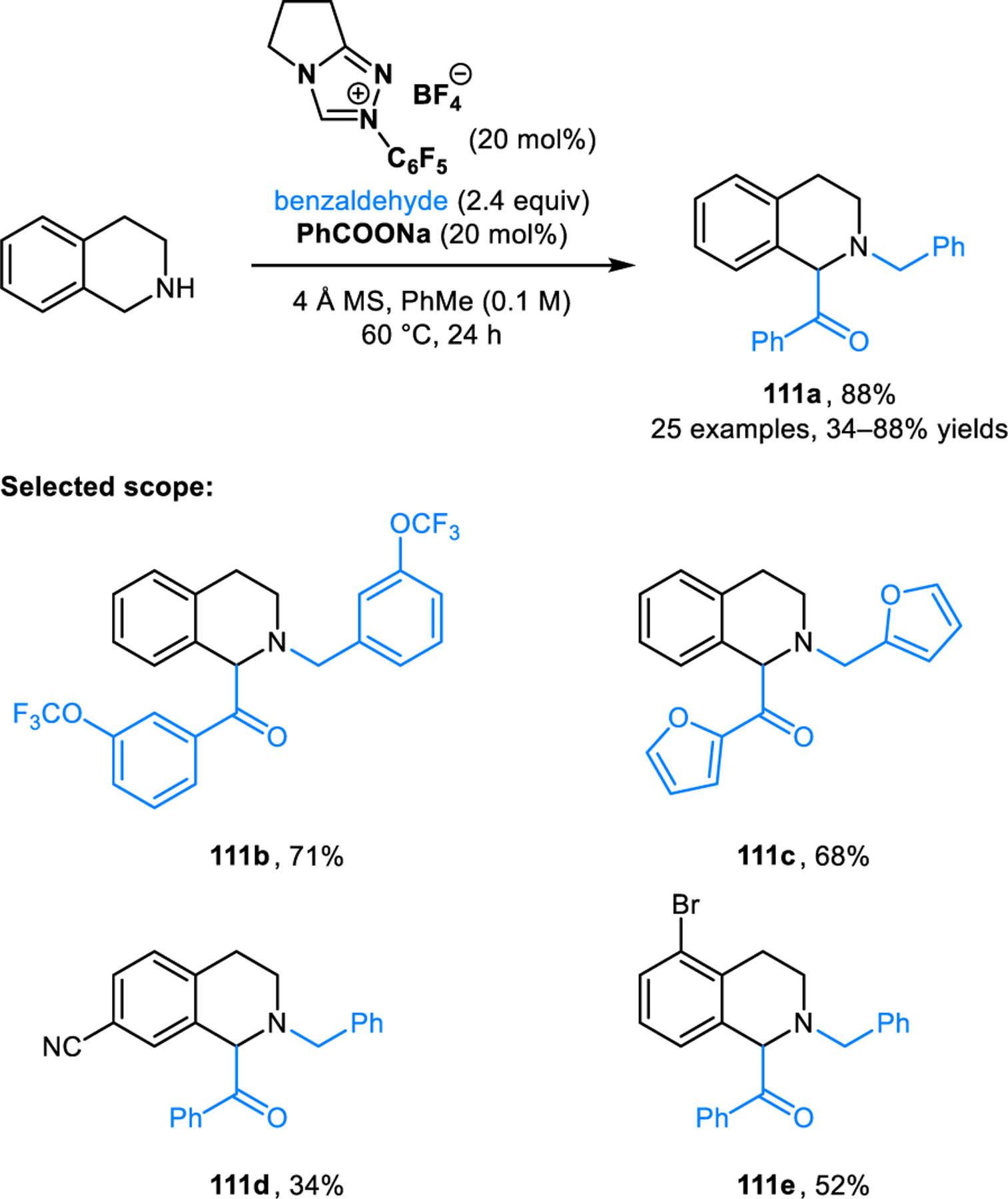
NHC-catalyzed redox-aza-benzoin reaction of THIQs.
5. Redox-Annulations
Redox-annulations represent intramolecular variants of some of the reactions discussed in the previous sections. Redox-neutral annulations of amines with concurrent C–H and C–N bond formation are achieved by covalently linking the aldehyde/ketone reaction partner to the (pro)nucleophile. These transformations are powerful in that they facilitate the rapid formation of polycyclic azacycles from simple starting materials. A number of redox-annulations precede the development of the first intermolecular variants. The first redox-annulation was discovered serendipitously by our group. Ortho-aminobenzaldehydes were found to readily engage in redox-annulations with amines (e.g., pyrrolidine) to provide ring-fused aminal products 112 (Scheme 58).105 Good yields of aminals are typically obtained simply by heating a mixture of an ortho-aminobenzaldehyde and an amine in an alcoholic solvent. The scope of both components is relatively broad. While amines such as pyrrolidine and THIQ undergo aminal formation at moderate temperatures (reflux in EtOH), other substrates such as piperidine require elevated temperatures. Cyclic amines with an existing α-substituent (e.g., 2-methylpyrrolidine) provide mixtures of regioisomeric products, favoring functionalization of the sterically more hindered α-position. Aminoketones represent challenging substrates, but engage pyrrolidine under microwave irradiation at 250 °C, providing products as a mixture of diastereomers in low to moderate yields.106 The mechanism of this reaction was later elucidated based on extensive experimental and computational studies.107 Accordingly, pyrrolidine and ortho-aminobenzaldehyde exist in equilibrium with hemiaminal 113. The latter undergoes transformation to azaquinone methide 114 upon elimination of water via a six-membered ring transition state. According to charge-density calculations, 114 is best thought of as a zwitterion, as illustrated by the resonance structure 114’. Rate-determining 1,6-proton transfer on 114’ produces azomethine ylide 115.
Scheme 58.
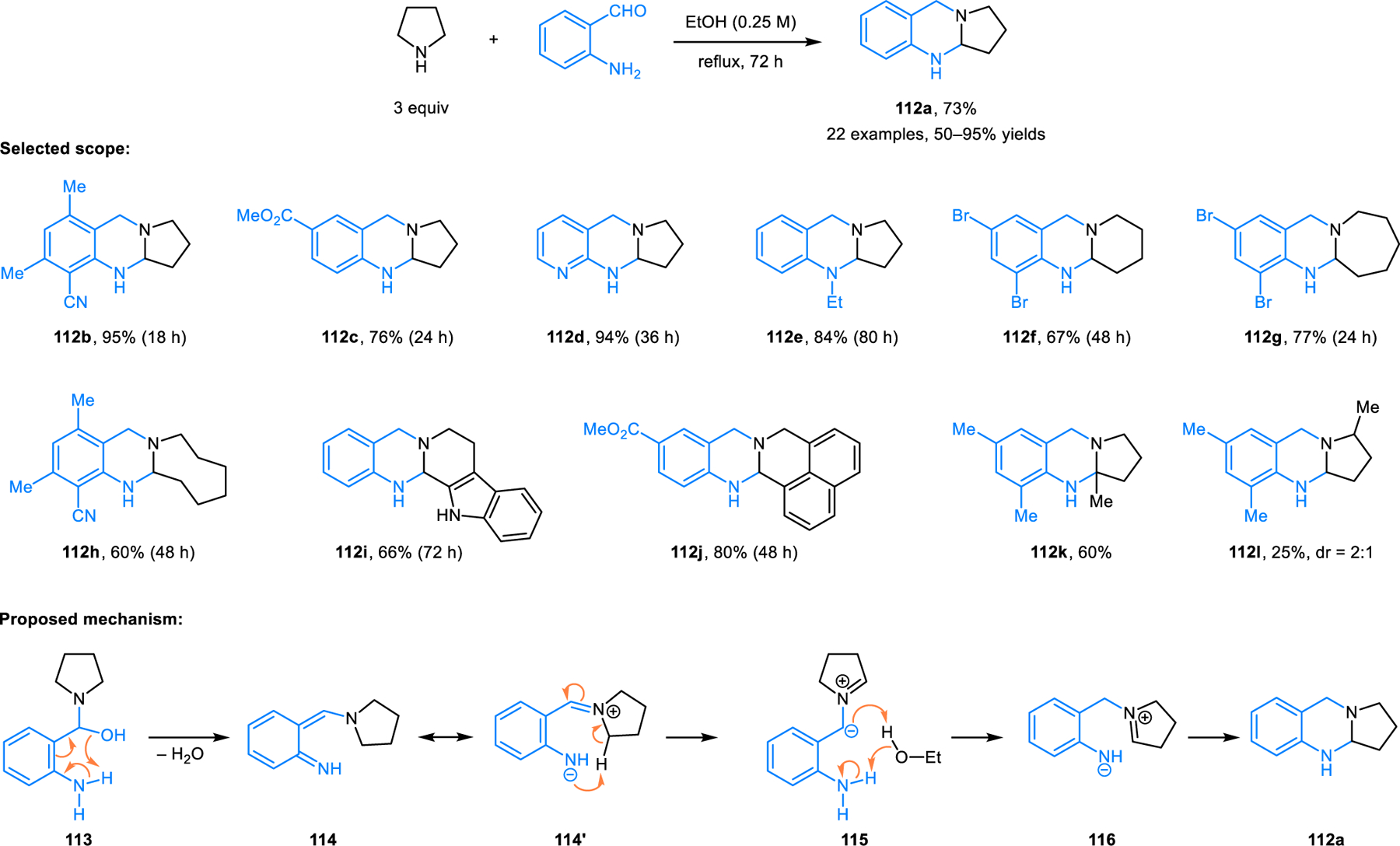
Ring-fused aminals from the redox-annulation of cyclic amines and ortho-aminobenzaldehydes.
Ethanol-mediated proton transfer then results in the formation of zwitterion 116 which subsequently undergoes ring closure to provide the final product 112a. Remarkably, no intermediates with overall formal charges are involved in the entire reaction sequence – such species were found to be prohibitively high in energy. Experimental support for proposed intermediates 114 and 115 comes from trapping these species via cycloaddition reactions. The aminal formation was applied to the facile synthesis of deoxyvasicinone, rutaecarpine and related natural products.105,106,108 Nearly simultaneously to our initial report on annulative amine α-amination, Dang, Bai and coworkers independently reported a closely related transformation. While the bulk of their study dealt with decarboxylative annulations, these researchers also showed that aminals such as 117a form in condensations of an aminopyrimidine carboxaldehyde and dibenzylamine or N-methylbenzylamine (Scheme 59).109 An interesting, alternative pathway to aminals and the corresponding amidines was recently reported by Chusov and coworkers.110 Ortho-nitrobenzaldehydes serve as starting materials in place of ortho-aminobenzaldehydes. Reactions are promoted by iron pentacarbonyl, enabling the formation of ring-fused aminals and amidines under mild conditions. Deb, Baruah and coworkers reported three-component annulation reactions of THIQ, benzaldehyde, and aminouracil.111 These reactions involve an oxidation step and furnish amidines rather than aminals.
Scheme 59.
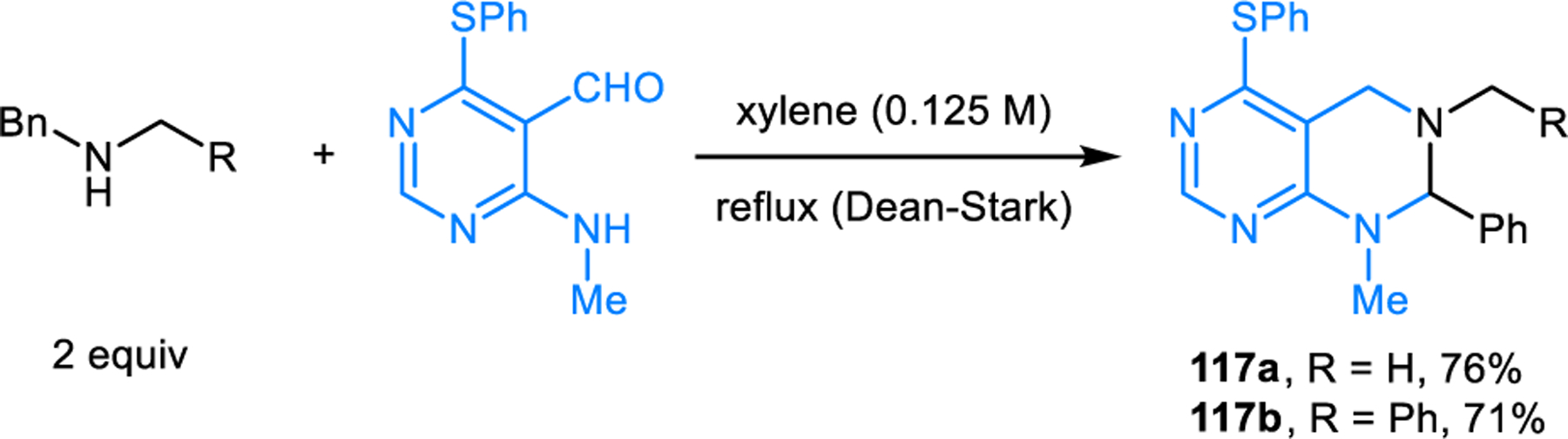
Aminals from the redox-annulation of benzylic amines and aminopyrimidine carboxaldehydes.
In analogy to the synthesis of ring-fused aminals from ortho-aminobenzaldehydes, salicylaldehydes and thiosalicyl-aldehydes engage in the formation of the corresponding N,O-acetals (e.g., 118, Scheme 60)112 and N,S-acetals (e.g., 121, Scheme 61).113 Despite the apparent similarity to aminal formation, the synthesis of N,O- and N,S-acetals requires substantially different reaction conditions. For efficient N,O-acetal formation, carboxylic acid additives such as acetic acid are needed. According to computational studies, they key role of acetic acid is not only in its participation in C–H bond functionalization. Rather, acetic acid significantly lowers the energy barrier of a subsequent key step by acting as a proton shuttle in the generation of zwitterion 120 from azomethine ylide 119. The computational profile for N,S-acetal formation mirrors that of N,O-acetal formation, with overall lower energy barriers. This is consistent with the experimental observation that lower reaction temperatures are required in the synthesis of N,S-acetals. It is noteworthy that N,O-acetals such as 118 exist in equilibrium with zwitterionic species (e.g., 120), as shown by NMR studies. The corresponding equilibrium also exists for ring-fused N,S-acetals, as revealed in detailed studies by Roberts and coworkers.114 Other approaches to ring-fused N,O-acetal have also emerged. Jana et al. reported the synthesis of N,O-acetals such as 122 from cyclic amines and 2-hydroxy 1-naphtholaldehydes and ketones (Scheme 62).115 Reactions are performed under microwave irradiation without any additive. Interestingly, sterically more demanding 2-hydroxy 1-naphtholketone provides product in a higher yield than the corresponding aldehyde, while requiring slightly higher reaction temperatures.
Scheme 60.
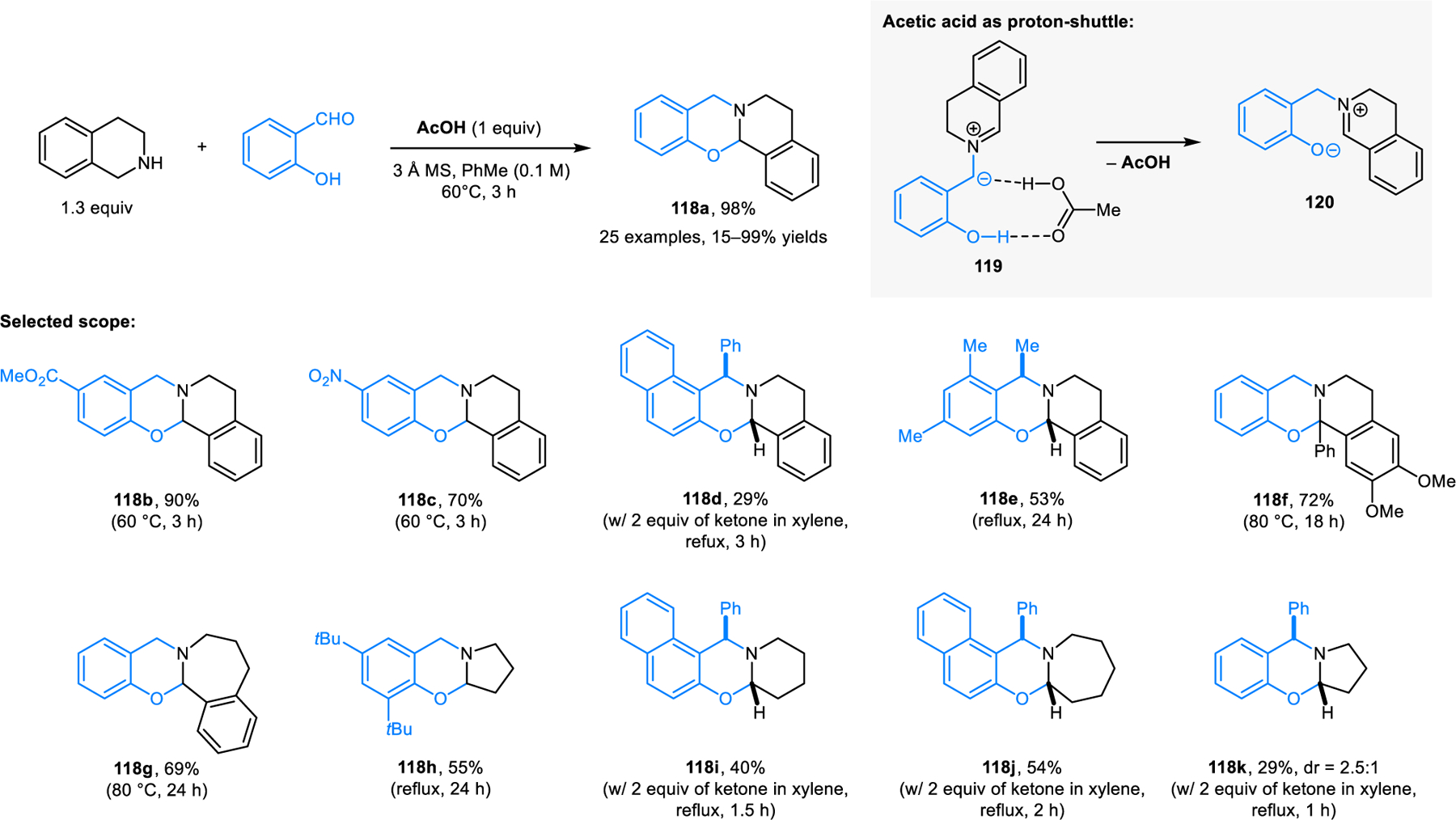
Redox-neutral synthesis of ring-fused N,O-acetal.
Scheme 61.
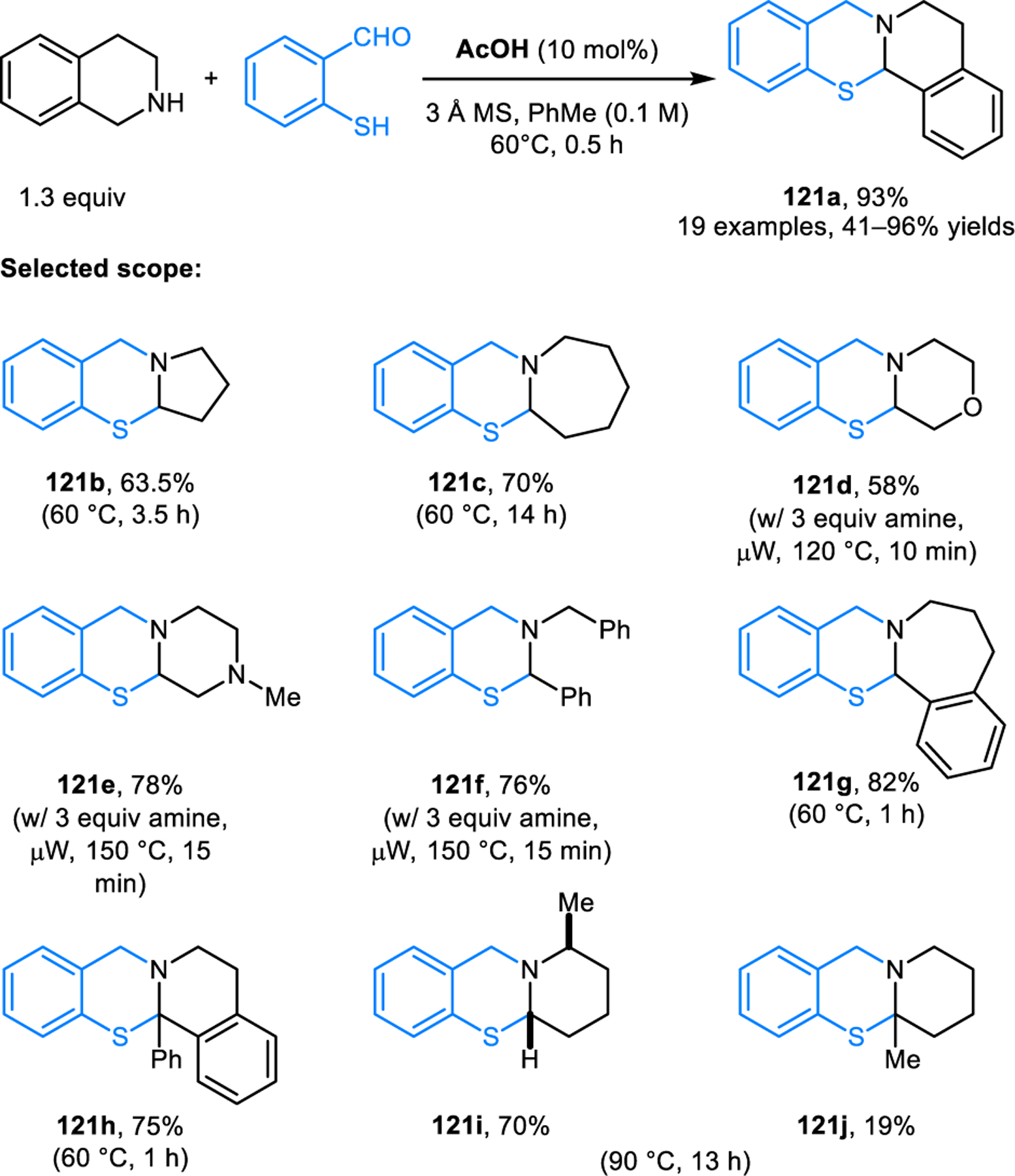
Redox-neutral synthesis of ring-fused N,S-acetal.
Scheme 62.
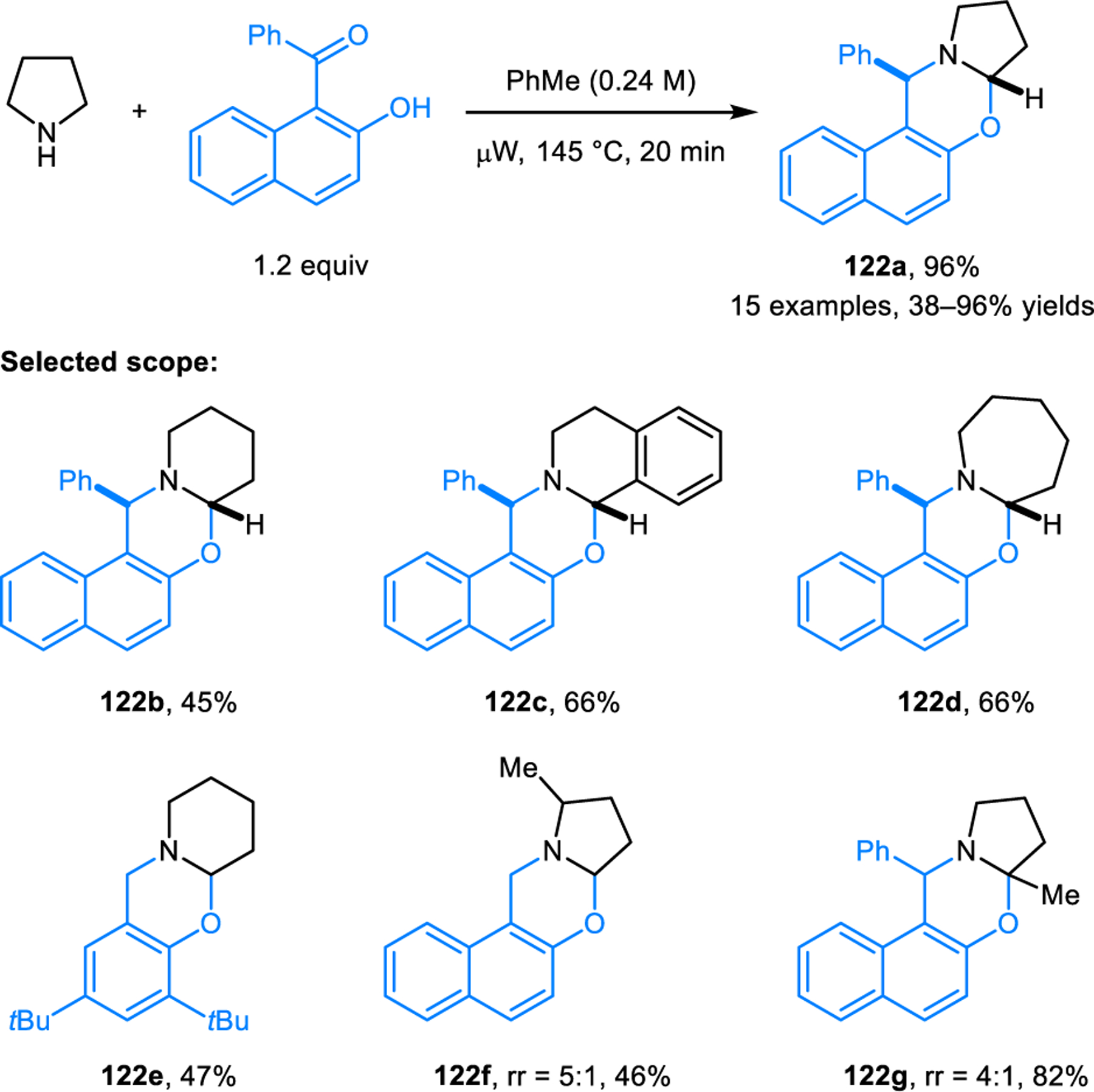
Redox-neutral synthesis of ring-fused N,O-acetal without additive.
Redox-annulations involving C−C bond formation have been explored extensively. The first examples, reported by our group, represent redox-variants of Pictet–Spengler reactions (Scheme 63).116 Indoles and naphthols containing an aldehyde functionality condense with amines at elevated temperatures to form annulation products such as 123. In some cases, slow addition of the aldehyde leads to higher yields whereas other substrate combinations perform best under microwave conditions.
Scheme 63.
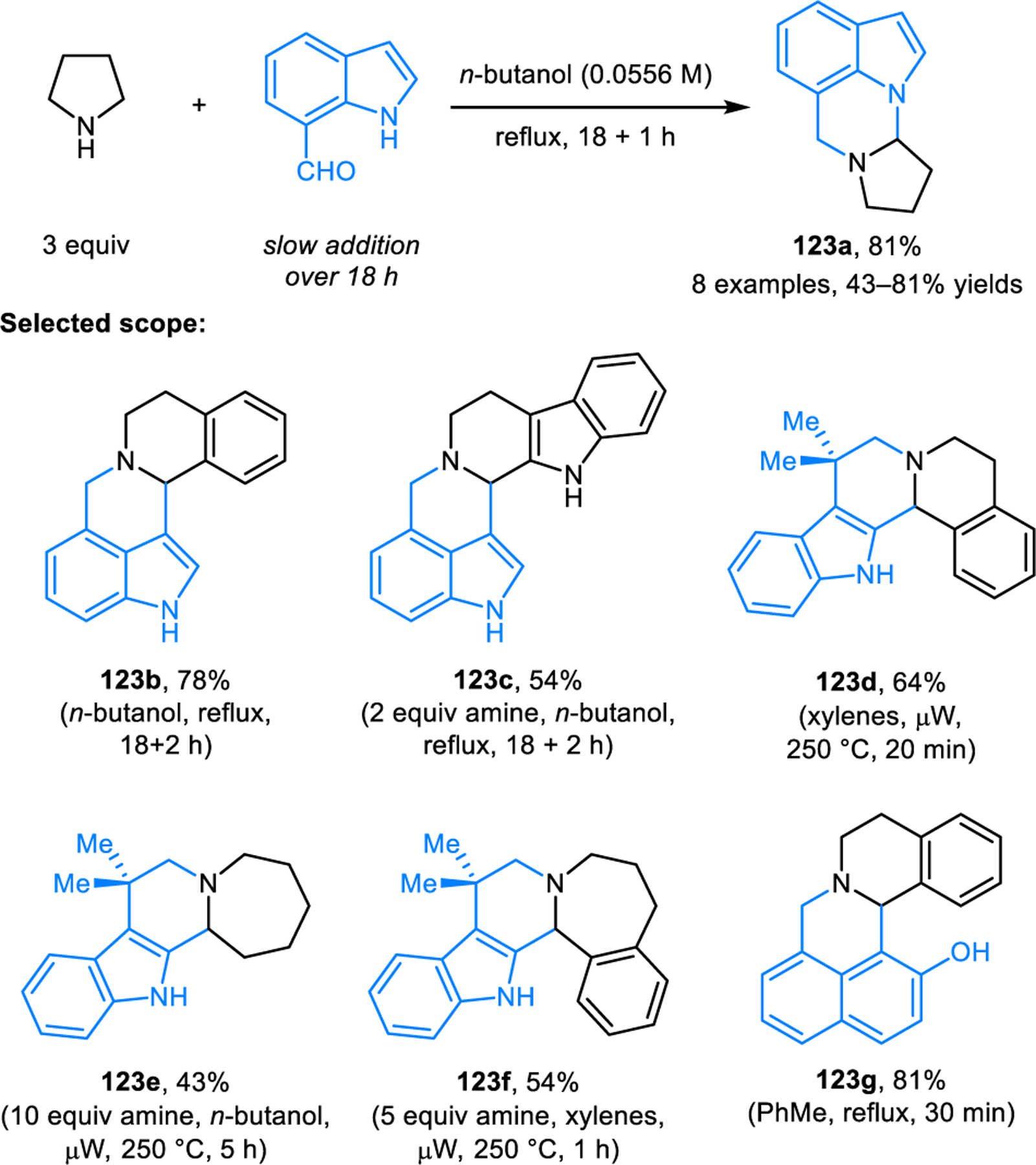
Redox-annulation of cyclic amines with aromatic compounds bearing aldehyde moieties.
Redox-annulation of THIQs with 2-formylaryl malonates provide facile access to the core structures of natural products in the tetrahydroprotoberberine family (Scheme 64).117 In the initial development of this reaction with pyrrolidine to form polycyclic amine 124a, dealkoxycarbonylated side product 125 was frequently observed. Compound 126 containing an exocyclic double bond in the β-position was also identified as an additional side product. Under optimal conditions (heating in toluene in the presence of 10 equiv of 2-EHA), the formation of these side products is suppressed almost entirely. This reaction was utilized in the synthesis (±)-thalictricavine and its epimer.
Scheme 64.
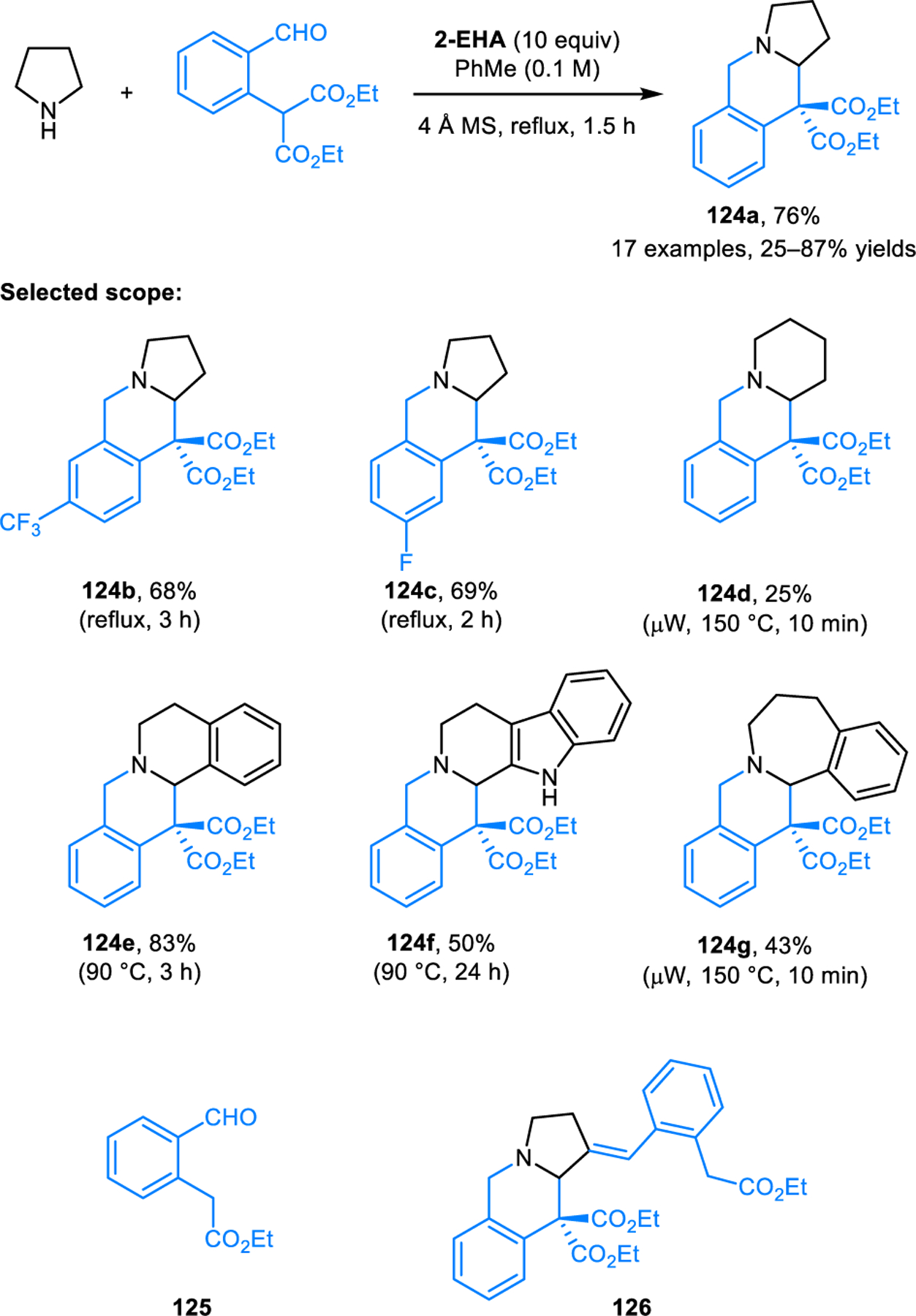
Redox-annulation of cyclic amines with 2-formylaryl malonates.
Redox-annulations with ortho-tolualdehydes possessing a single activating group have also been developed. Our group reported annulation reactions of cyclic amines with ortho-cyanomethylbenzaldehydes (Scheme 65).118 These reactions are promoted by 20 equiv of acetic acid, providing products 127 in good yields and moderate to good diastereomeric ratios. Epimerization of the products occurs under the reaction conditions; thus, the diastereomeric ratios correspond to the thermodynamic equilibrium ratios. While cyclic amines with benzylic α-C–H bonds readily participate in annulations with ortho-cyanomethylbenzaldehydes, less activated substrates such as pyrrolidine and piperidine fail to undergo annulation. However, the desired annulation products can be obtained with proline and pipecolic acid via a decarboxylative pathway. In related work, we reported redox-annulations with ortho-(nitromethyl)benzaldehydes (Scheme 66).119 While no diastereoselectivity is observed for product 128d (thermodynamic equilibrium ratio is 1:1), products such as 128a are obtained essentially as single diastereomers. The nitro group in the products can be removed, effectively rendering the annulation process traceless.
Scheme 65.
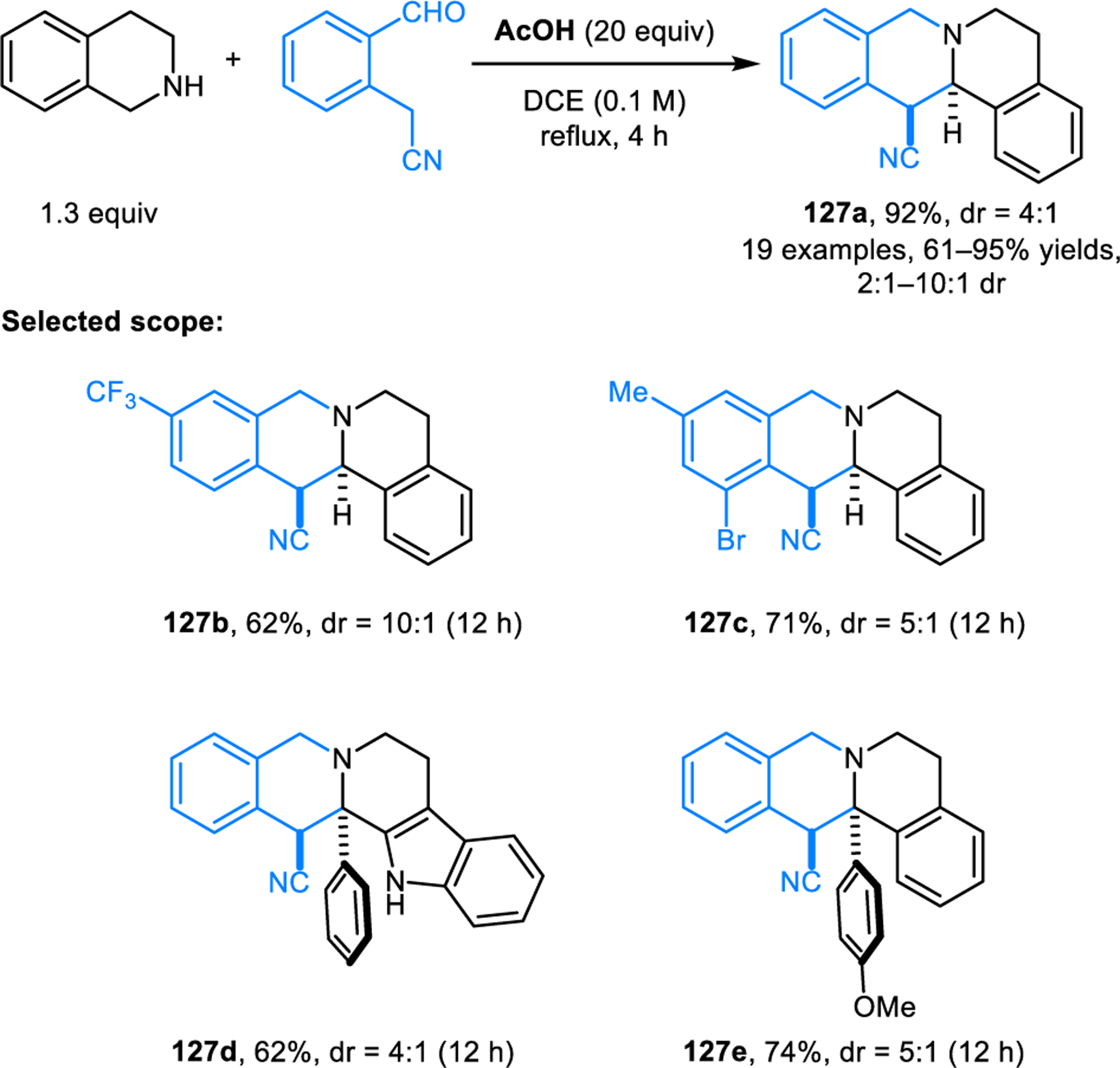
Redox-annulation of THIQs and ortho-cyanomethylbenzaldehydes.
Scheme 66.
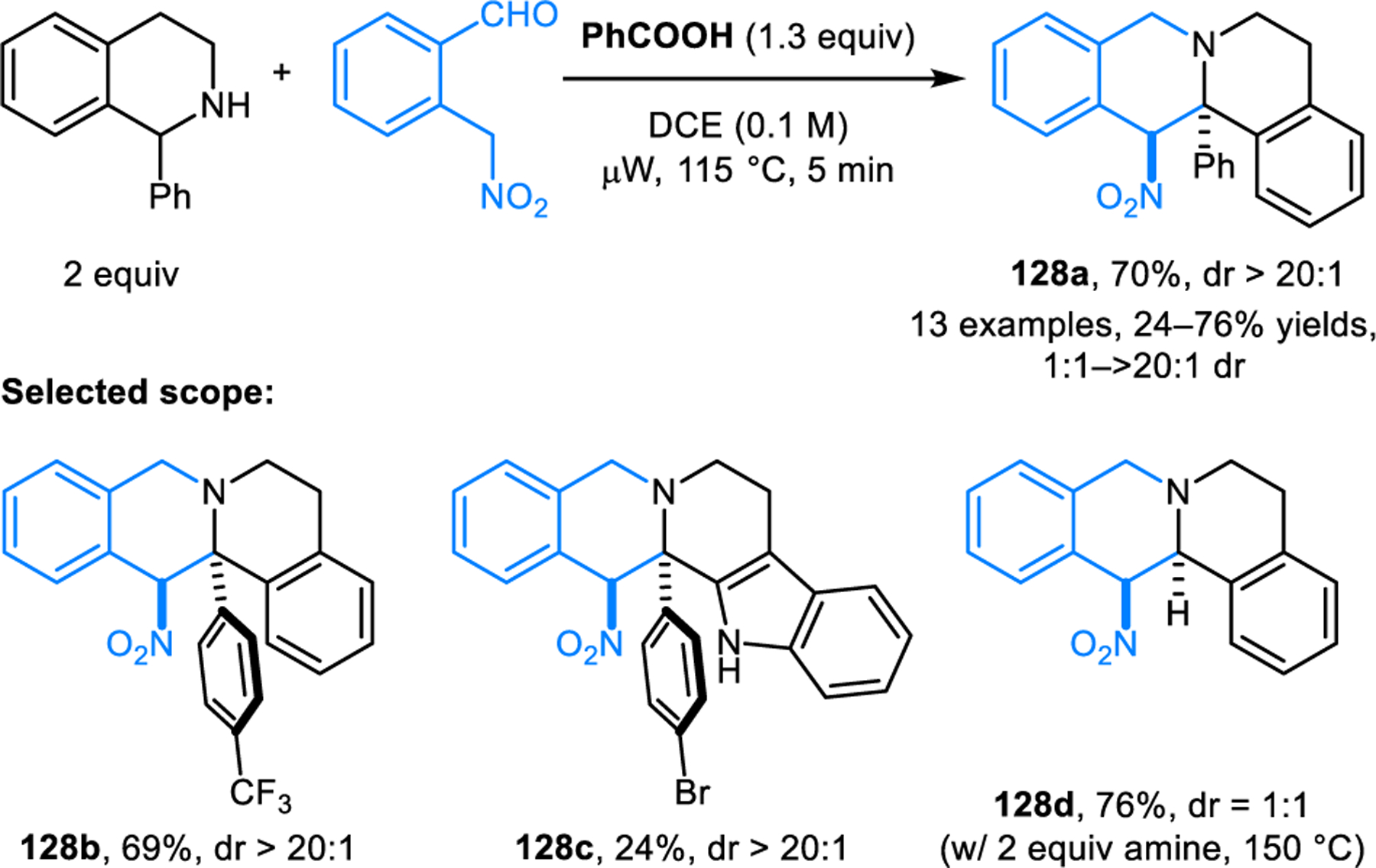
Redox-annulation of benzylic cyclic amines with ortho-(nitromethyl)benzaldehydes.
As an important step towards expanding the scope of these transformations, redox-annulations have also been achieved with enolizable aldehydes, enabling asymmetric variants. Enantioenriched 4-nitrobutyraldehydes (e.g., 129), substrates that are easily obtained in highly enantioenriched form from aldehydes and nitroalkenes by well-established organocatalytic enantioselective methods, undergo enantiospecific redox-annulations with activated cyclic amines such as THIQ (Scheme 67).120 The addition of 10 equiv of acetic acid is critical in these reactions. The level of diastereoselectivity is dependent on the aldehyde used. Diastereoselectivity is usually low with α-branched 4-nitrobutyraldehydes, a result of reversible enamine formation during the reaction (e.g., products 130a/131a). Moderate to good diastereoselectivities are observed with 4-nitrobutyraldehydes lacking an α-substituent.
Scheme 67.
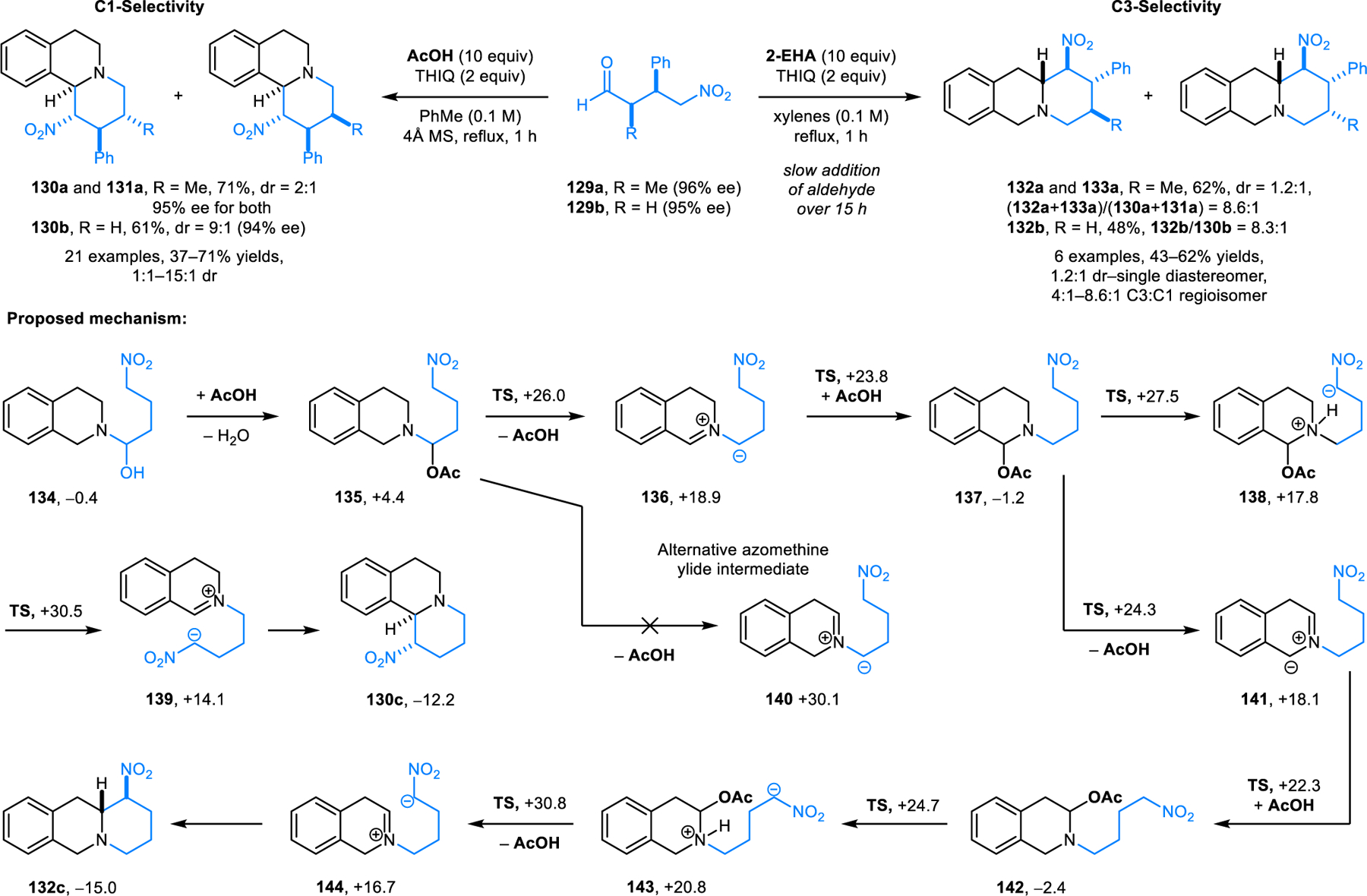
Asymmetric redox-annulation of cyclic amines with 4-nitrobutyraldehyde and calculated mechanism (free energies in kcal/mol).
A remarkable feature of this reaction is that the regioselectivity of the C–H bond functionalization can be changed by adjusting reaction conditions. The kinetically more favored benzylic (C1) C−H bond functionalization products (e.g., 130/131) are obtained by directly heating the mixture of starting materials with acetic acid in refluxing toluene for one hour. Alternatively, the thermodynamically more-favored non-benzylic (C3) C−H bond functionalization products (e.g., 132/133) are obtained as major products upon heating of the amine with 2-EHA in refluxing xylenes, with slow addition of the 4-nitrobutyraldehyde over 15 hours. Detailed computational studies were conducted to shed light on the regiodivergent behavior of this annulation. Accordingly, N,O-acetal 135 is first generated from hemiaminal 134. Isomerization of 135 to 137 occurs via the intermediacy of azomethine ylide 136. N,O-acetal 137 then undergoes intramolecular proton transfer to form zwitterion 138. The latter undergoes rate-determining elimination of acetic acid to form zwitterion 139. Finally, ring closure of 139 affords the annulation product 130c. Regarding the formation of C3-functionalized product, it was found that direct access to this position via formation of azomethine ylide from N,O-acetal 135 is not feasible due to the prohibitively high energy of 140. Instead, the reaction pathway diverges from intermediate 137, which can undergo a second intramolecular fragmentation involving a C3-proton of the THIQ ring, generating endocyclic azomethine ylide 141. Identical subsequent steps from 141 lead to the regioisomeric annulation product 132c. As can be seen from the calculated energy profiles, the energy barrier of the rate-determining step providing 139 is slightly lower than that for forming 144. On the other hand, 132c is thermodynamically more stable than 130c. This picture is consistent with the observation that C3-functionalized annulation products form at higher reaction temperatures and require longer reaction times. This redox-annulation was applied to a 7-step total synthesis of the natural product (−)-protoemetinol.
β-Ketoaldehydes have also been shown to undergo redox-annulations with activated amines (Scheme 68).121 High yields of products such as 145a are obtained for non-enolizable β-ketoaldehydes simply by heating with the appropriate amine and 10 equiv of acetic acid in toluene. However, slow addition of the aldehyde is usually required for reactions involving more challenging enolizable β-ketoaldehydes, presumably to avoid competing aldol reactions. This method is applicable to the synthesis of bioactive compounds such as (±)-tetrabenazine. As is true for related products, the diastereomeric ratios obtained from reactions involving enolizable β-ketoaldehydes likely represent the thermodynamic equilibrium ratios of the two diastereomers, since the interconversion between such annulation products and the ring-opened isoquinolinium ions are known to take place under acidic conditions.122 Indeed, the same diastereomeric ratio obtained in the annulation reaction is reached by exposure of either diastereomerically pure isomer to the reaction conditions.
Scheme 68.
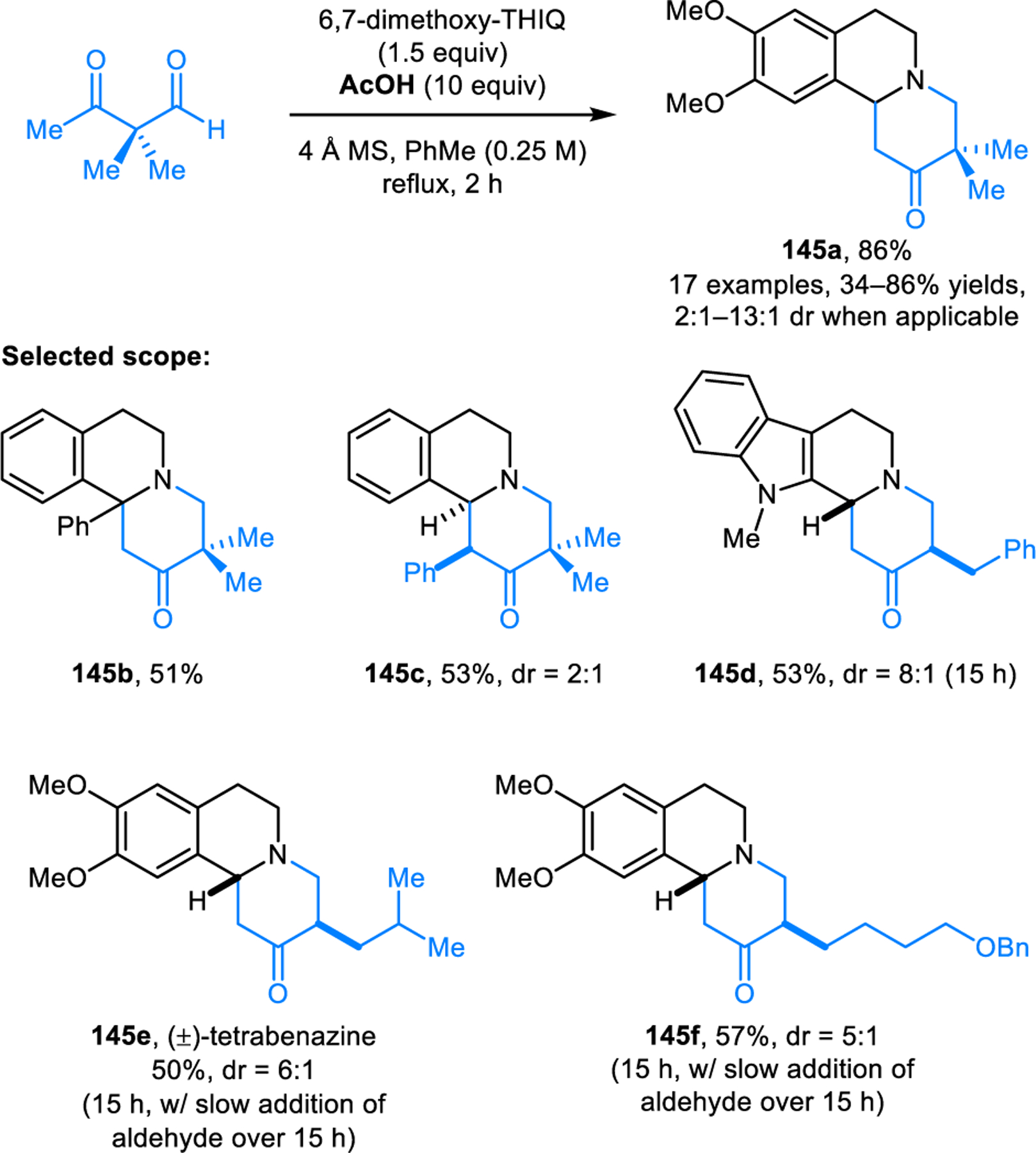
Redox-annulation of cyclic amines with β-ketoaldehydes.
The scope of redox-annulations has also been expanded to ortho-methyl heteroaryl aldehydes such as 2-alkylquinoline-3-carbaldehydes. A Lewis-acid catalyzed variant of this reaction was first reported by Wang and coworkers (Scheme 69).123 2-Methylquinoline-3-carbaldehyde undergoes redox-annulations with THIQ or tryptoline in the presence of aluminium triflate, providing the corresponding products 146 in moderate to high yields. The role of Al(OTf)3 was proposed to include coordination to the quinoline nitrogen, an interaction that is thought to facilitate the deprotonation of the methyl group which in turn would assist in the C–H functionalization step. The same group later achieved this transformation in catalytic enantioselective form using a chiral disulfonimide (DSI) Brønsted acid catalyst (Scheme 70).124 It was proposed that the enantioselectivity in the formation of products such as (S)-146e is controlled via the chiral ion pair 147. While only moderate enantioselectivities were obtained, it is important to note that this challenging transformation currently remains as the only catalytic enantioselective variant of an amine redox-annulation. Shortly after Wang’s initial disclosure of their racemic variant, our group independently reported a different approach to the same transformation (Scheme 71).125 Redox-annulation products are obtained by heating a mixture of an ortho-methyl heteroaryl aldehyde and a cyclic amine in a 1:1 mixture of toluene and acetic acid under reflux. While likely not amenable to an asymmetric variant, this approach results in an expanded scope for both reaction components. In addition to quinoline carbaldehydes, pyridine carbaldehydes are viable substrates. The 2-methyl group can be replaced with ethyl or benzyl groups while still maintaining excellent reactivities. THIQs and tryptolines containing substituents at the benzylic position readily participate in the reactions as well. Moreover, redox-annulations are readily achieved with 4-methylazarene carbaldehydes, providing products such as146m in good yields. Pyrrolidine performs poorly under standard reaction conditions but provides the corresponding annulation product in moderate yield under microwave irradiation at 200 °C.
Scheme 69.
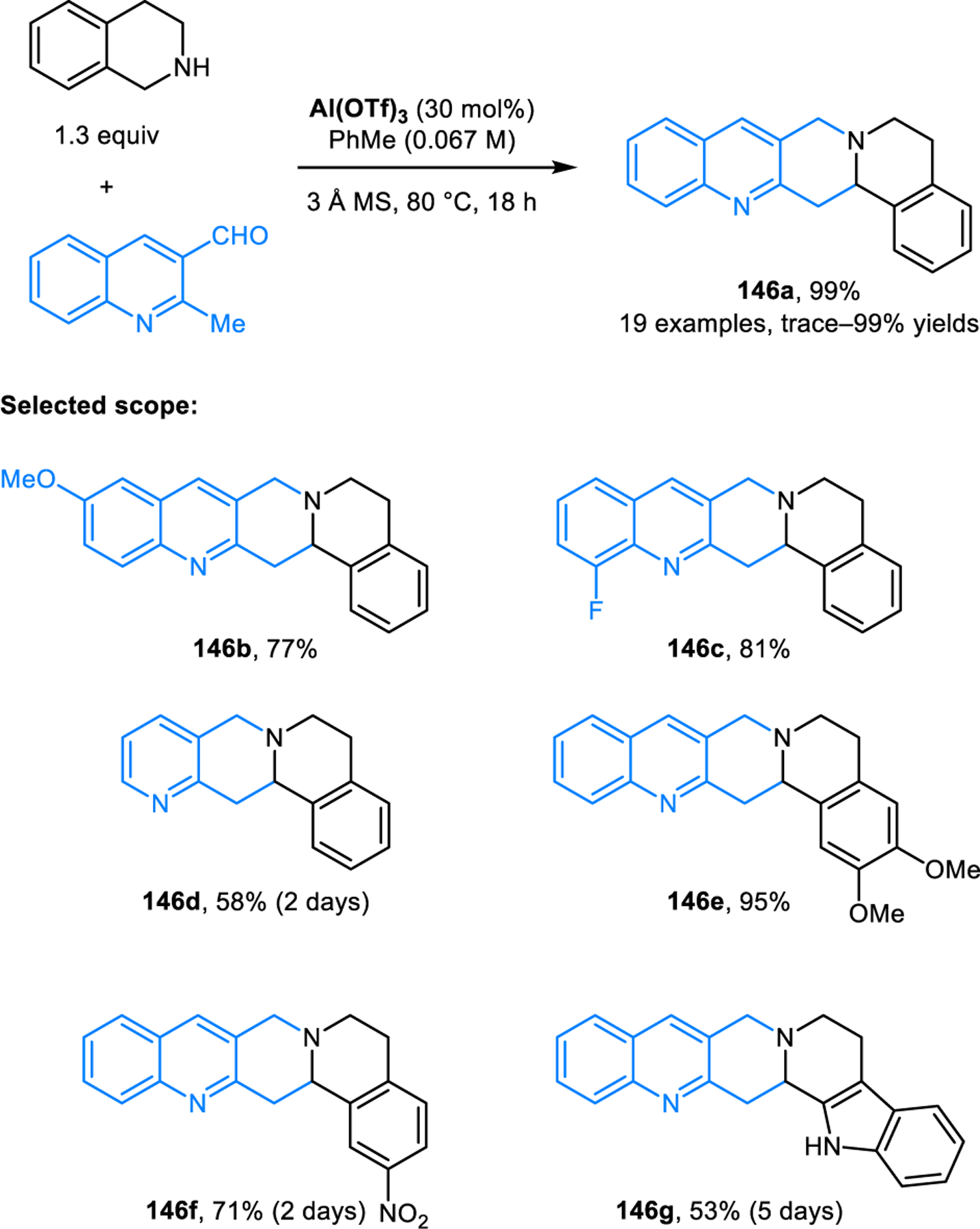
Lewis-acid-catalyzed redox-annulation of cyclic amines with ortho-methylazarene carbaldehydes.
Scheme 70.
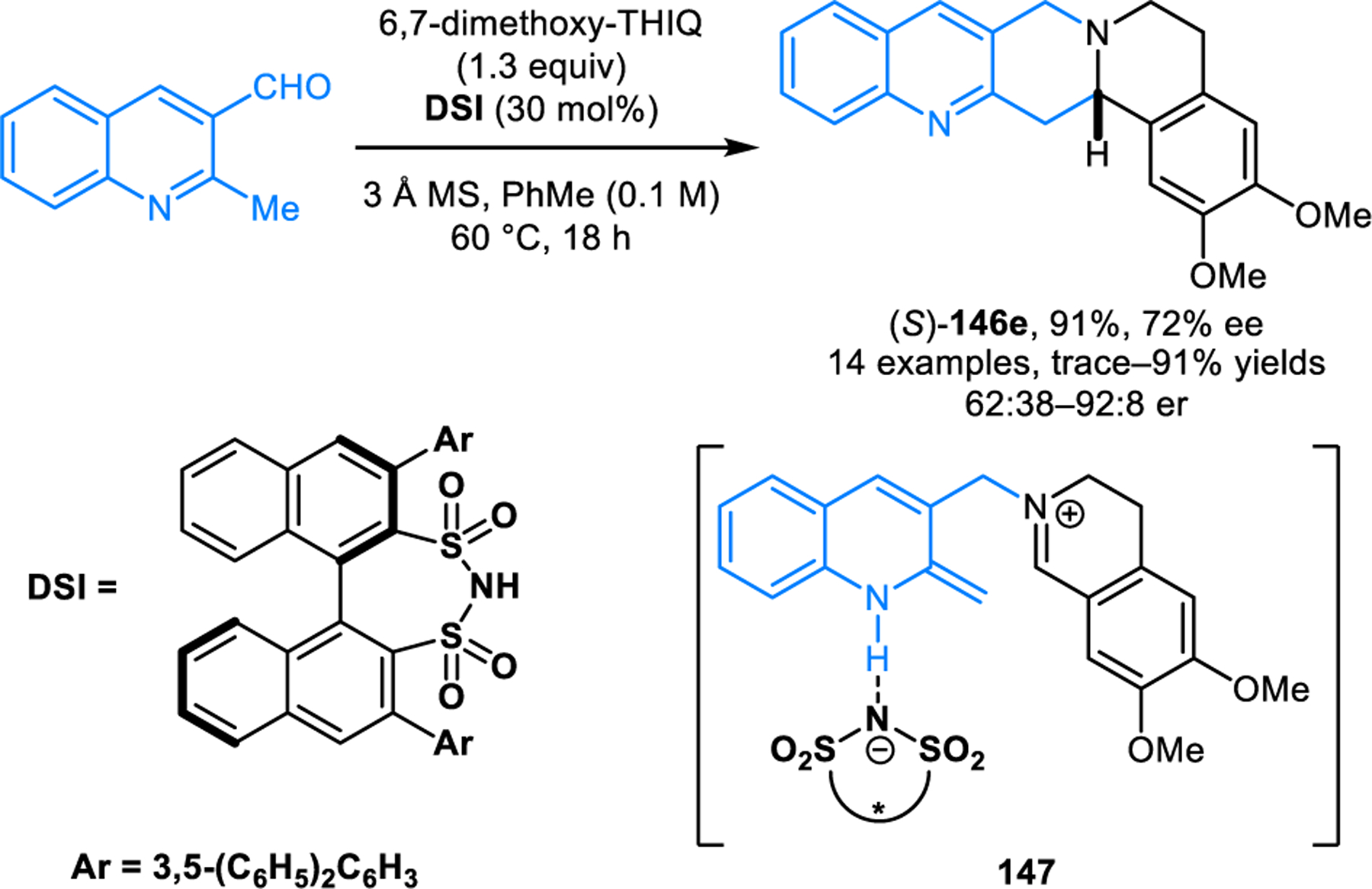
Catalytic enantioselective redox-annulation of cyclic amines with 2-methylquinoline-3-carbaldehyde.
Scheme 71.
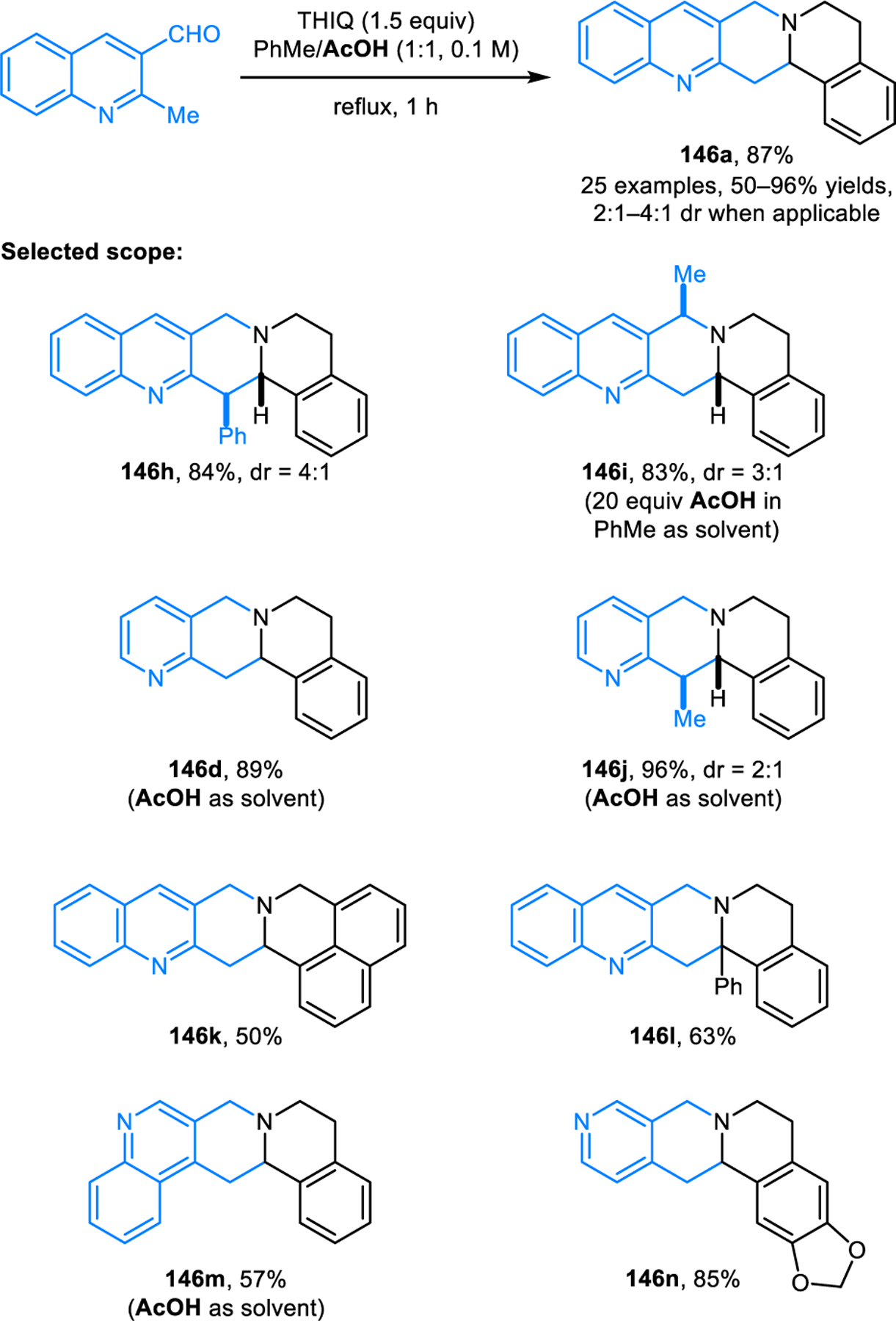
Redox-annulation of cyclic amines with alkylazarene carbaldehydes promoted by acetic acid.
As alluded to earlier, simple o-tolualdehyde is insufficiently activated to participate in redox annulations. However, highly electron-deficient o-tolualdehydes undergo redox-annulations with amines such as THIQ to form the corresponding products 148 (Scheme 72).126 The presence of two activating nitro groups on the aryl aldehyde was found to be a strict requirement.
Scheme 72.
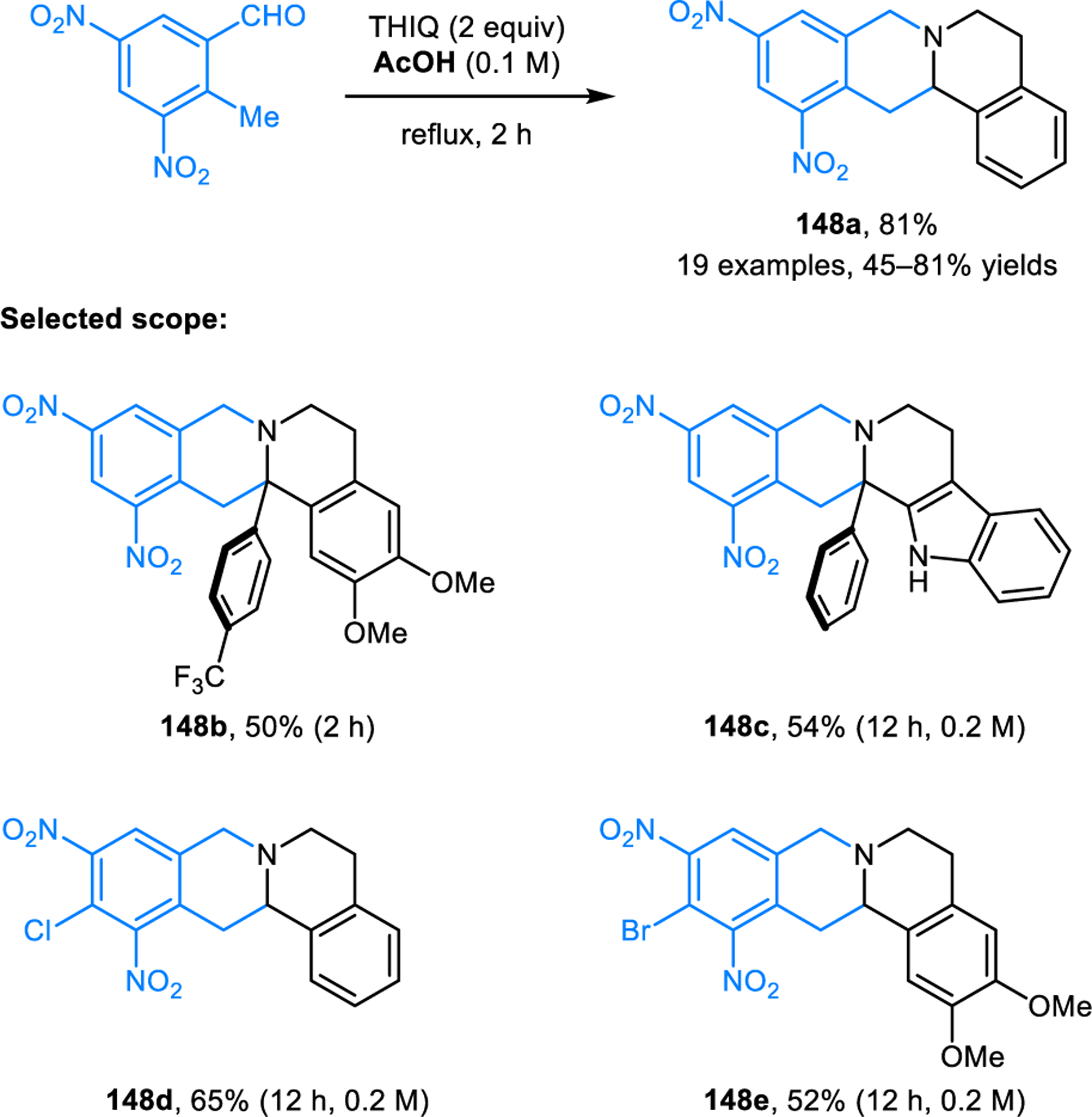
Redox-annulation of cyclic amines with electron-deficient o-tolualdehydes.
While most known redox-annulations involve the formation of six-membered rings, reactions that form five-membered rings have also been developed. Our group reported the formation of imidazolidinones via redox-neutral annulations of cyclic amines with α-ketoamides (Scheme 73).127 This reaction utilizes benzoic acid as the catalyst. Amines such as THIQ, pyrrolidine and azepane give good yields and in most cases excellent diastereoselectivities in reactions conducted in toluene solution under reflux. For instance, product 150a was obtained from pyrrolidine and α-ketoamide 149 as a single observable diastereomer in excellent yield. Less reactive amines such as piperidine, morpholine and thiomorpholine also undergo the corresponding annulations but require higher reaction temperatures and microwave irradiation, providing products with typically low levels of diastereoselectivity. As in redox-annulations involving β-ketoaldehydes and ortho-cyanomethylbenzaldehydes, the diastereomeric ratios obtained in these reactions likely correspond to thermodynamic equilibrium ratios, due to imidazolidinones undergoing reversable ring-opening under the reaction conditions. Shortly after the publication of our study, the Wu group independently reported a closely related reaction, albeit mostly without the use of any catalyst.128 It should be noted that annulations of amines with α-ketoamides, while possibly proceeding in mechanistic pathways similar to those involving the formation of six-membered rings, could also plausible involve a 1,5-electrocyclization step and thus represent another example of a pericyclic reaction.
Scheme 73.
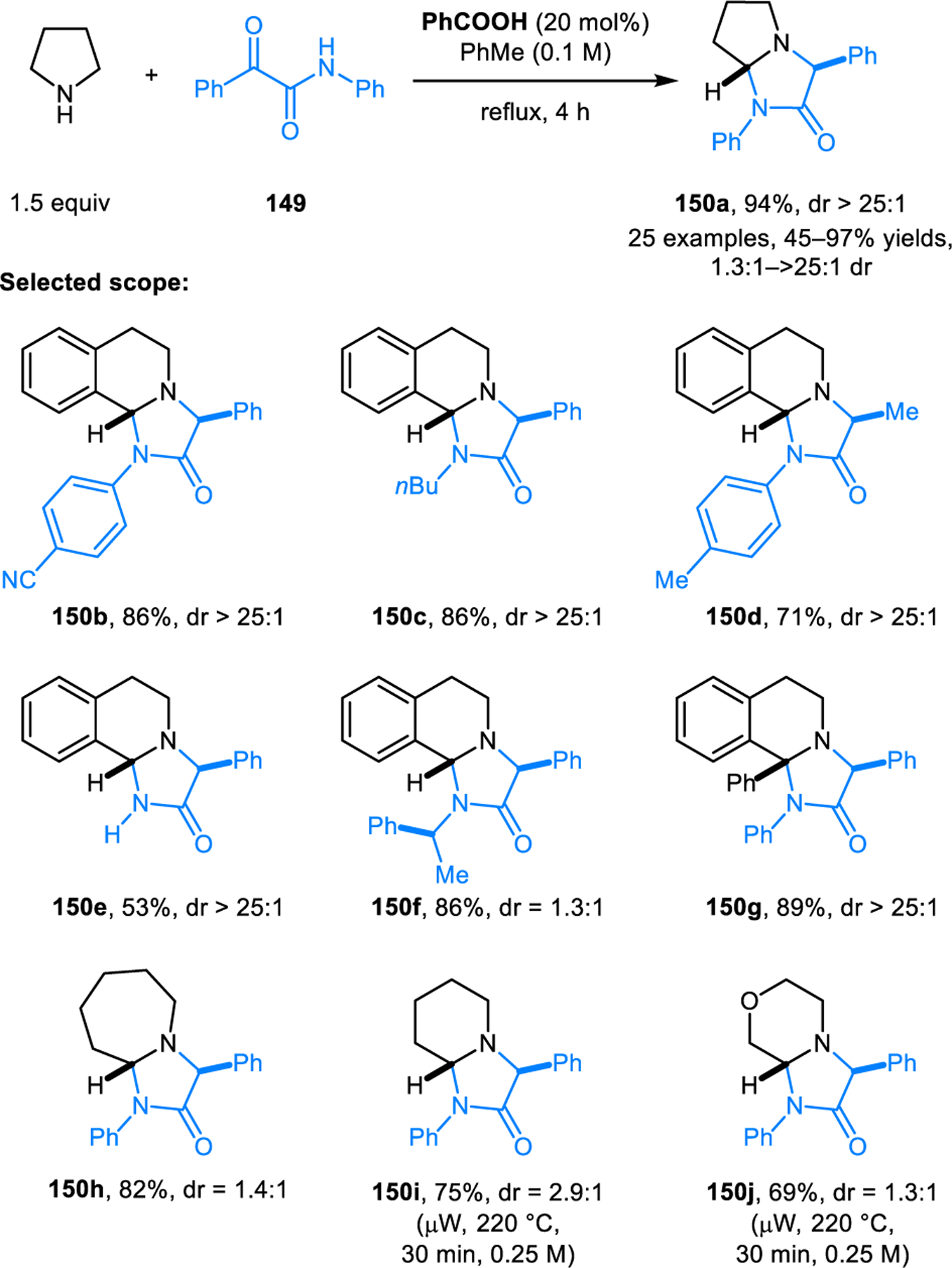
Redox-annulation of cyclic amines with α-ketoamides forming imidazolidinones.
Our group later reported another annulation process forming five-membered ring containing products 151, employing 2-(2-oxoethyl)malonates as reaction partners for cyclic amines (Scheme 74).129 Unlike in reactions leading to imidazolidinones, a pericyclic pathway can be ruled out. The reaction is limited to relatively activated amines, although products formally derived from pyrroline or piperidine can be obtained via a decarboxylative approach. Interestingly, benzoazepane does not form the expected C1-functionalized product. Instead, regioselective annulation occurs at the non-benzylic C3-position.
Scheme 74.
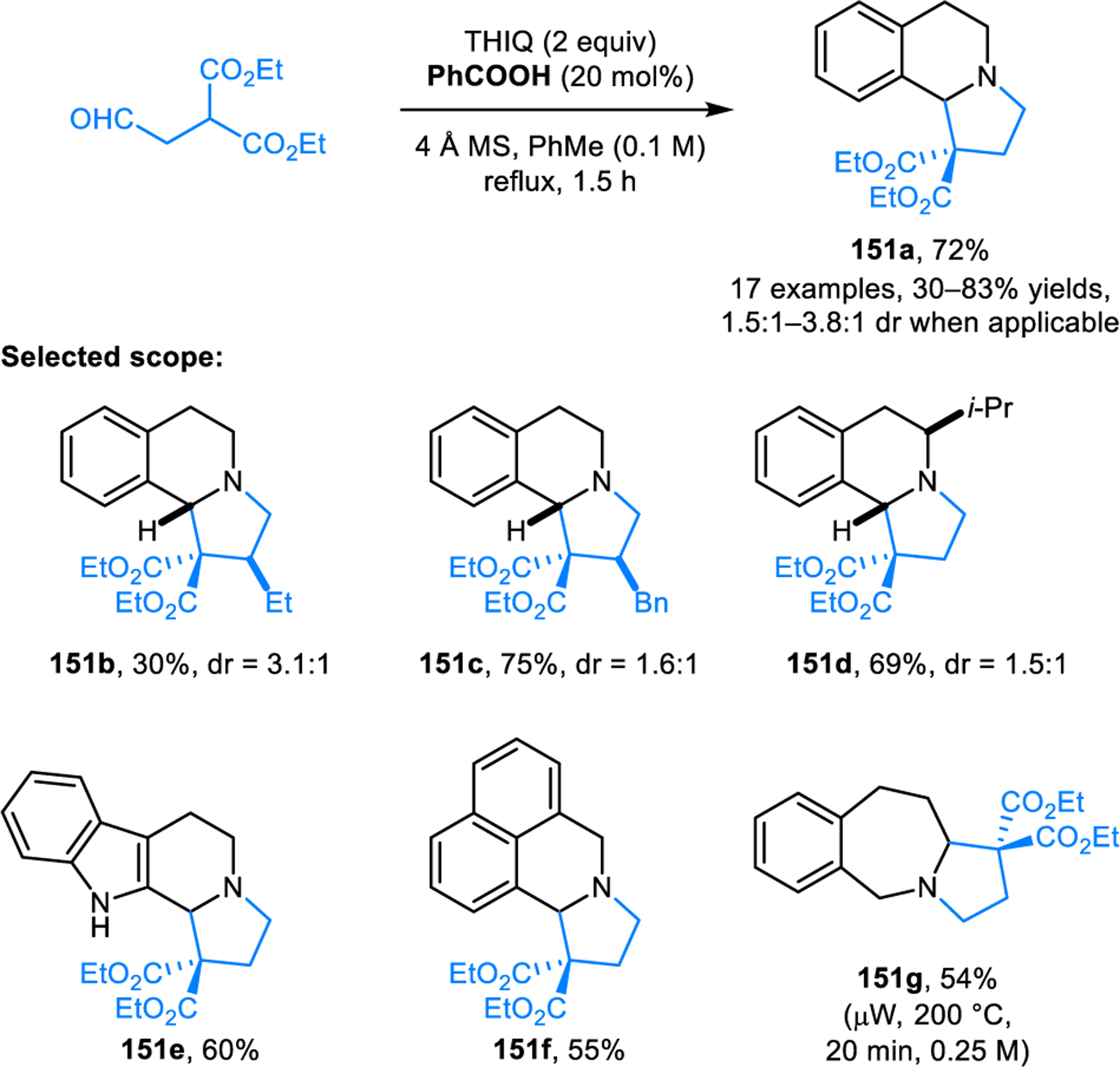
Redox-annulation of cyclic amines with 2-(2-oxoethyl)malonates.
6. Reactions Involving β-C−H Bond Functionalization
Other than reactions that lead to amine aromatization with concurrent β-C−H bond functionalization as discussed in section 2, the condensation-based functionalization of β-C−H bonds in cyclic amines is much less explored than α-C−H bond functionalization. All studies reported thus far invariably involve enamines as intermediates and often lead to concurrent amine α-functionalization. As shown in Scheme 1, enamines such as 7 can be derived from the corresponding N,O-acetals 5 by elimination of carboxylic acid. Alternatively, enamines can form via deprotonation of appropriate endocyclic iminium ion precursors. Although β-functionalized amines were occasionally isolated as byproducts in studies focusing on amine α-C−H bond functionalization (see, for instance, Scheme 45) the first systematic study on amine β-C−H bond functionalization was reported by our group in 2014 (Scheme 75).130 It was found that readily available 1-(aminomethyl)-β-naphthols, compounds which are also known as Betti bases, react with aromatic aldehydes in the absence of any additives to provide polycyclic N,O-acetals 153 in diastereoselective fashion. The optimized procedure involves heating under microwave irradiation. For example, the reaction of substrate 152 with benzaldehyde provides N,O-acetal 153a. The complementary reaction of 4-chlorobenzaldehyde and 1-(aminomethyl)-β-naphthol derived from benzaldehyde provides regioisomeric product 153b. These observations suggest the following mechanistic scenario: 1-(aminomethyl)-β-naphthol first undergoes fragmentation to an ortho-quinone methide and free amine. The latter then engages the aldehyde to form an enamine which then reengages the ortho-quinone methide in a formal [4+2] cycloaddition via proposed transition state 154. A reported X-ray structure of a 1-(aminomethyl)-β-naphthol compound supports this notion.131 Hydrogen bonding is observed between the hydroxyl group and the amine nitrogen, which is thought to facilitate the fragmentation step. This transformation exhibits a relatively broad scope regarding both reaction partners. In addition to 1-(aminomethyl)-β-napthols, 2-(aminomethyl)-phenols are also viable substrates. While the standard procedure employs Betti bases as substrates, a one-pot, two-step reaction from pyrrolidine, benzaldehyde and β-naphthol is equally efficient, providing the corresponding product in high yields on gram scale.
Scheme 75.
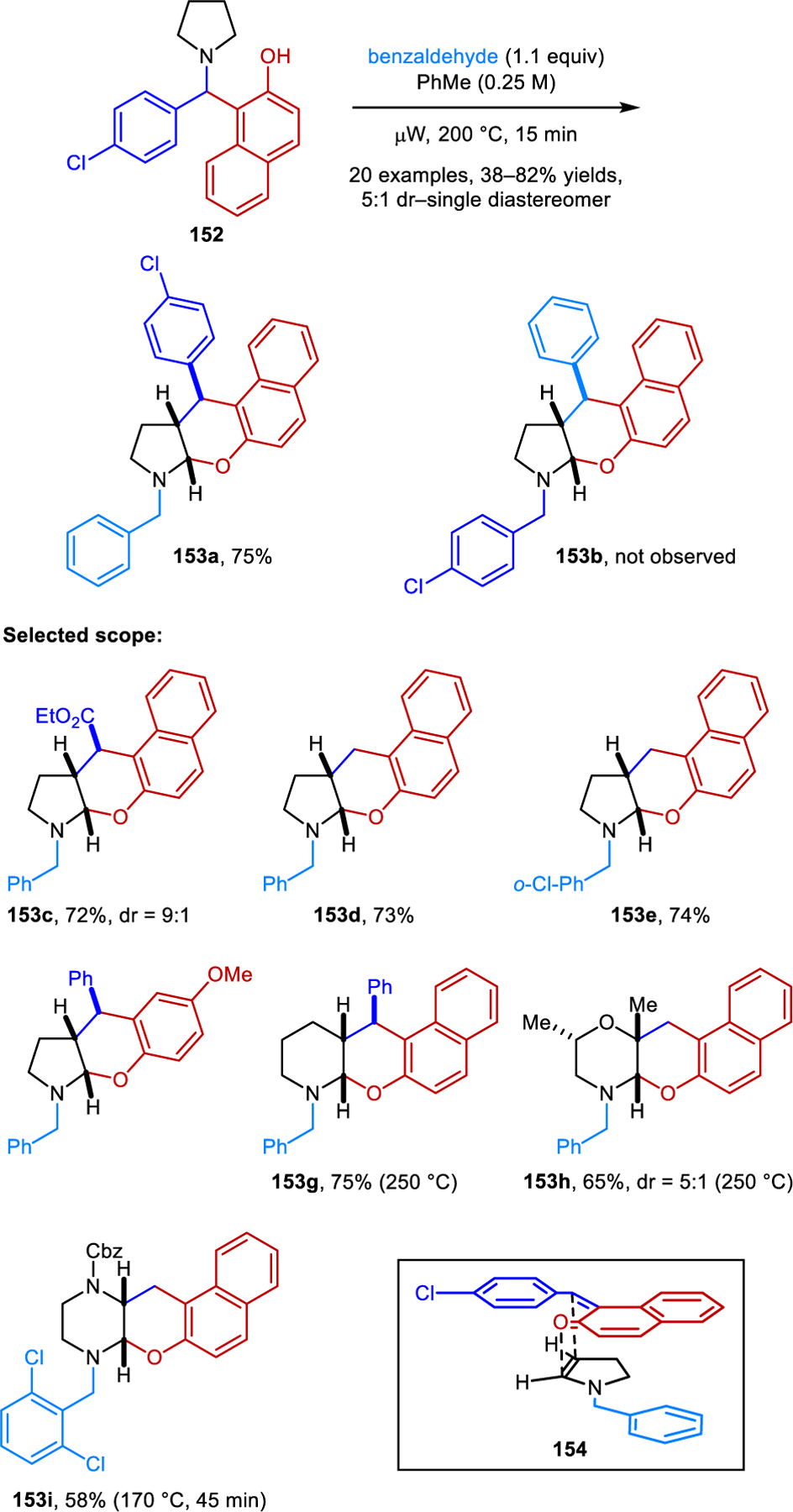
Redox-neutral α,β-difunctionalization of cyclic amines forming polycyclic N,O-acetals.
As part of detailed mechanistic studies conducted by our group, it was found that a mixture of the diastereomeric (3+2) dipolar cycloaddition products 156–159 is obtained upon heating a mixture of pyrrolidine, benzaldehyde and N-methylmaleimide with stoichiometric amounts of benzoic acid (Scheme 76).132 Products 156–159 contain a β-exocyclic double bond and are all derived from conjugated azomethine ylide 164. The ratio of the isomers is dependent on the amounts of benzoic acid and benzaldehyde. Simple (3+2) cycloaddition product 155 is also obtained when the amount of pyrrolidine exceeds that of benzaldehyde. Conjugate addition product 160 is observed as a side product under certain conditions. Based on computational studies, conjugated azomethine ylide 164 is derived from N,O-acetal 163, which in turn comes from enamine 162. Intermediate 162 is obtained by the reaction of enamine 161 with benzaldehyde and benzoic acid, followed by dehydration. During our study, it was also discovered that enamine dimers 165 readily form upon exposure of 2,6-dichlorobenzadehyde to excess amine in the presence of 20 mol% benzoic acid (Scheme 77). In polar protic solvents such as methanol, these enamine dimers exist in equilibrium with their corresponding enamine monomers. Enamines thus generated engage in conjugate additions with β-nitrostyrenes. Yan and coworkers later reported transformations resembling reactions shown in Scheme 76, with the modification that N-methylmaleimide is replaced by other electron-deficient alkenes.133,134
Scheme 76.
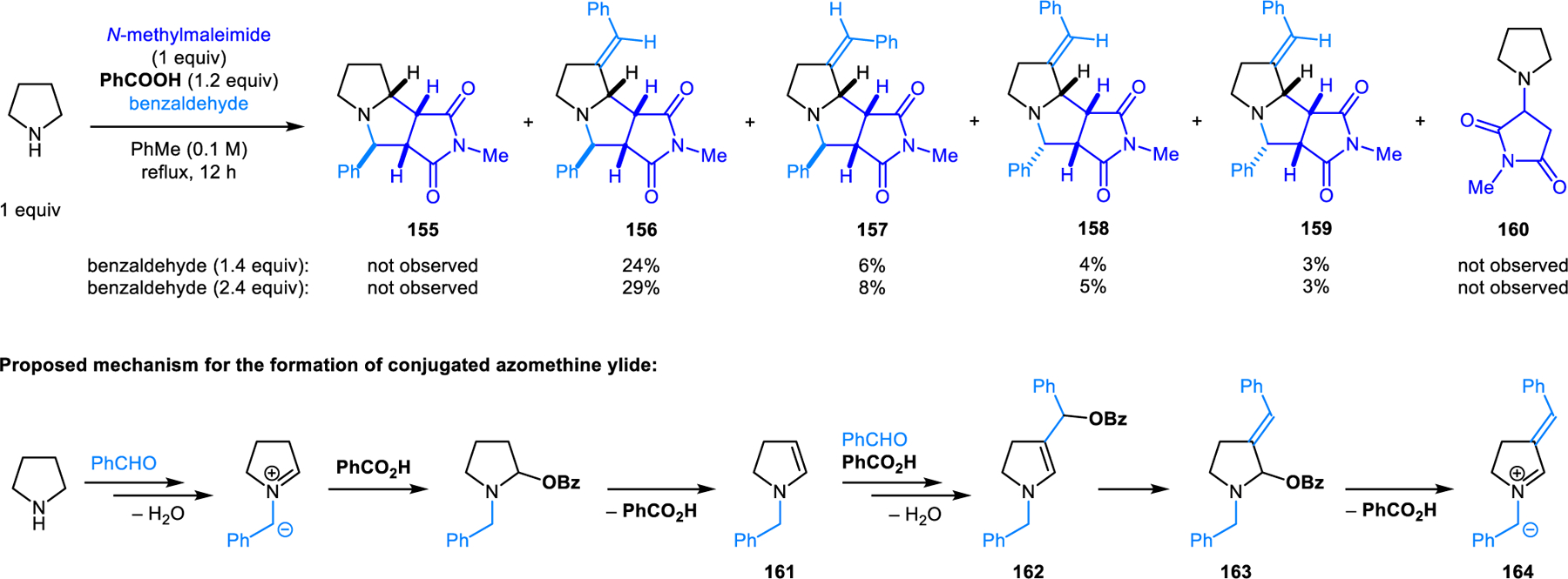
Benzoic acid-promoted (3+2) dipolar cycloaddition involving conjugated azomethine ylides derived from pyrrolidine and benzaldehyde.
Scheme 77.
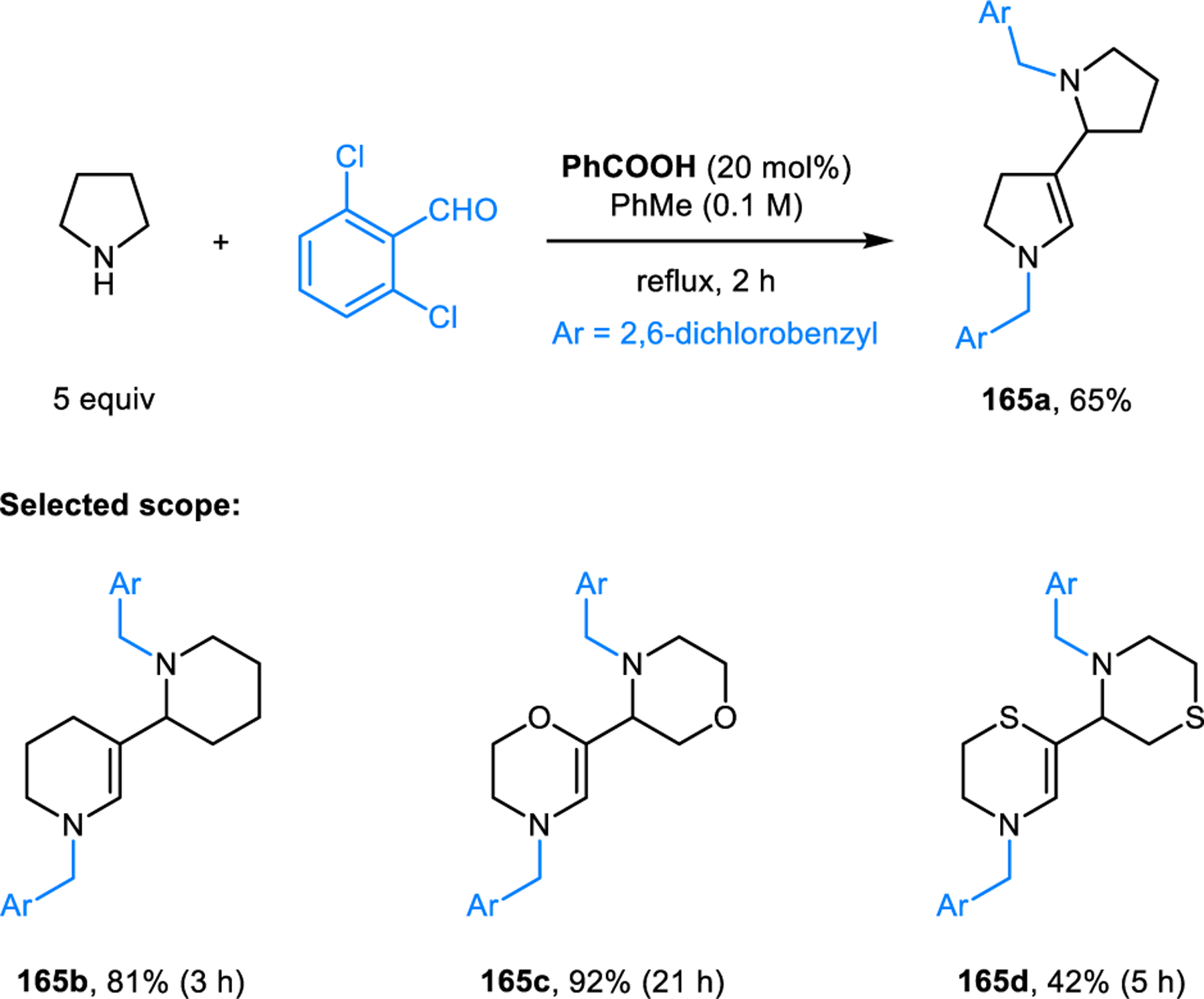
Redox-neutral formation of enamine dimers.
Several other research groups have reported condensation-based reactions involving the β-C−H bond functionalization of cyclic amines. Wu and coworkers reported a process in which a mixture of pyrrolidine, an α-ketoaldehyde, and an acrylate undergo a reaction that leads to simultaneous α- and β-C−H bond functionalization of the amine to furnish bicyclic 2,3-dihydro-1H-pyrrolizines 166 (Scheme 78).135 The use of acetic acid as a cosolvent in combination with toluene is crucial to obtaining high yields of products. The products were proposed to form via a (3+2) dipolar cycloaddition of a β-conjugated azomethine ylide with acrylate, followed byoxidation of the resulting pyrrolizidine to a 2,3-dihydro-1H-pyrrolizine. Products lacking β-substitution are obtained as side products.
Scheme 78.
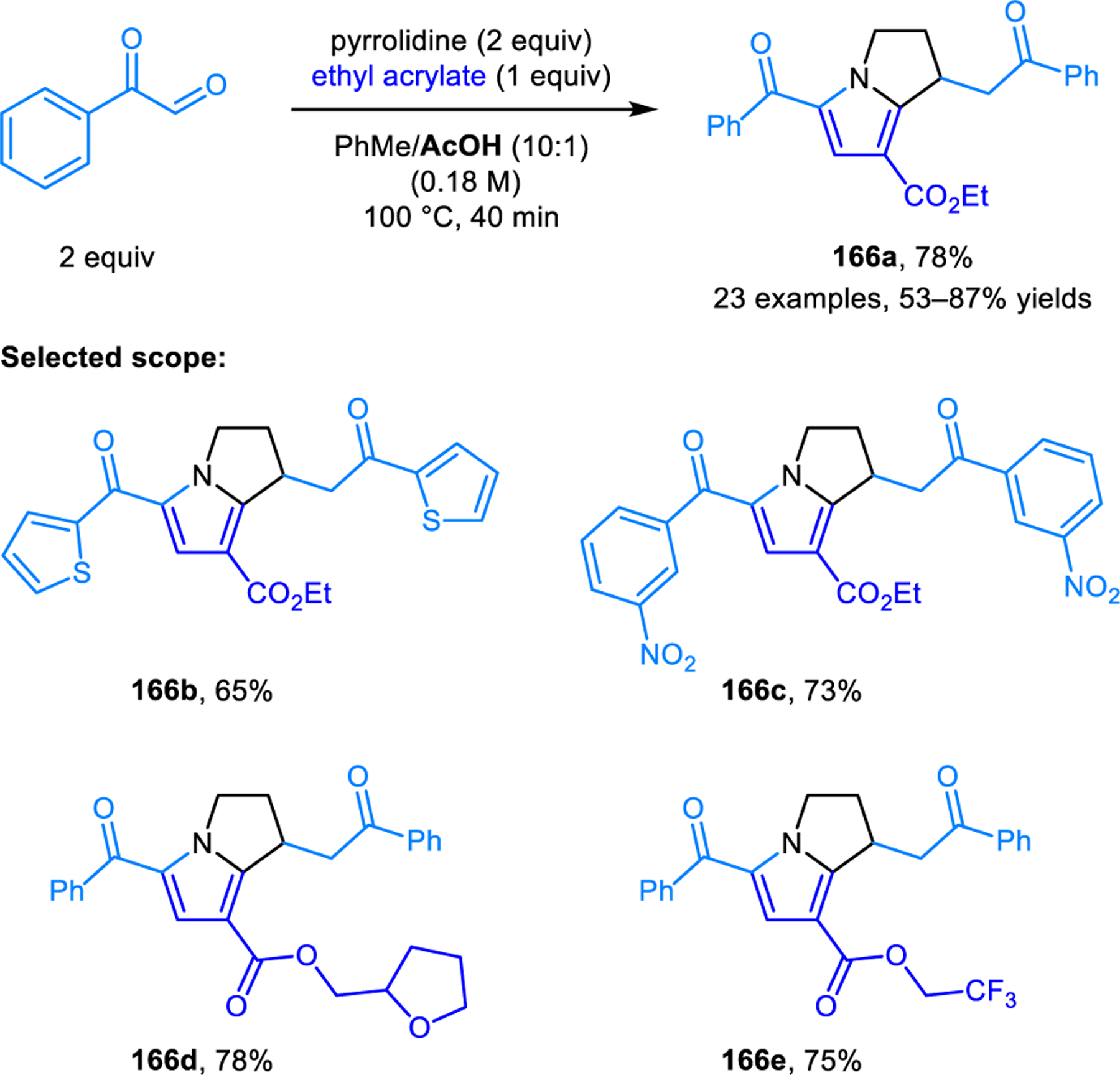
α,β-Difunctionalization of pyrrolidine forming 2,3-dihydro-1H-pyrrolizine derivatives.
A mechanistically related reaction involving piperidine, N-protected isatin, and alkenyl disulfone 167 was reported by Yu, Meng and coworkers (Scheme 79).136 The products 168 are the result of α,α,β,β-tetrafunctionalization. Product yields in this transformation are enhanced by utilizing 1 equiv of 4-(trifluoromethyl)benzoic acid (4-TFBA) as an additive.
Scheme 79.
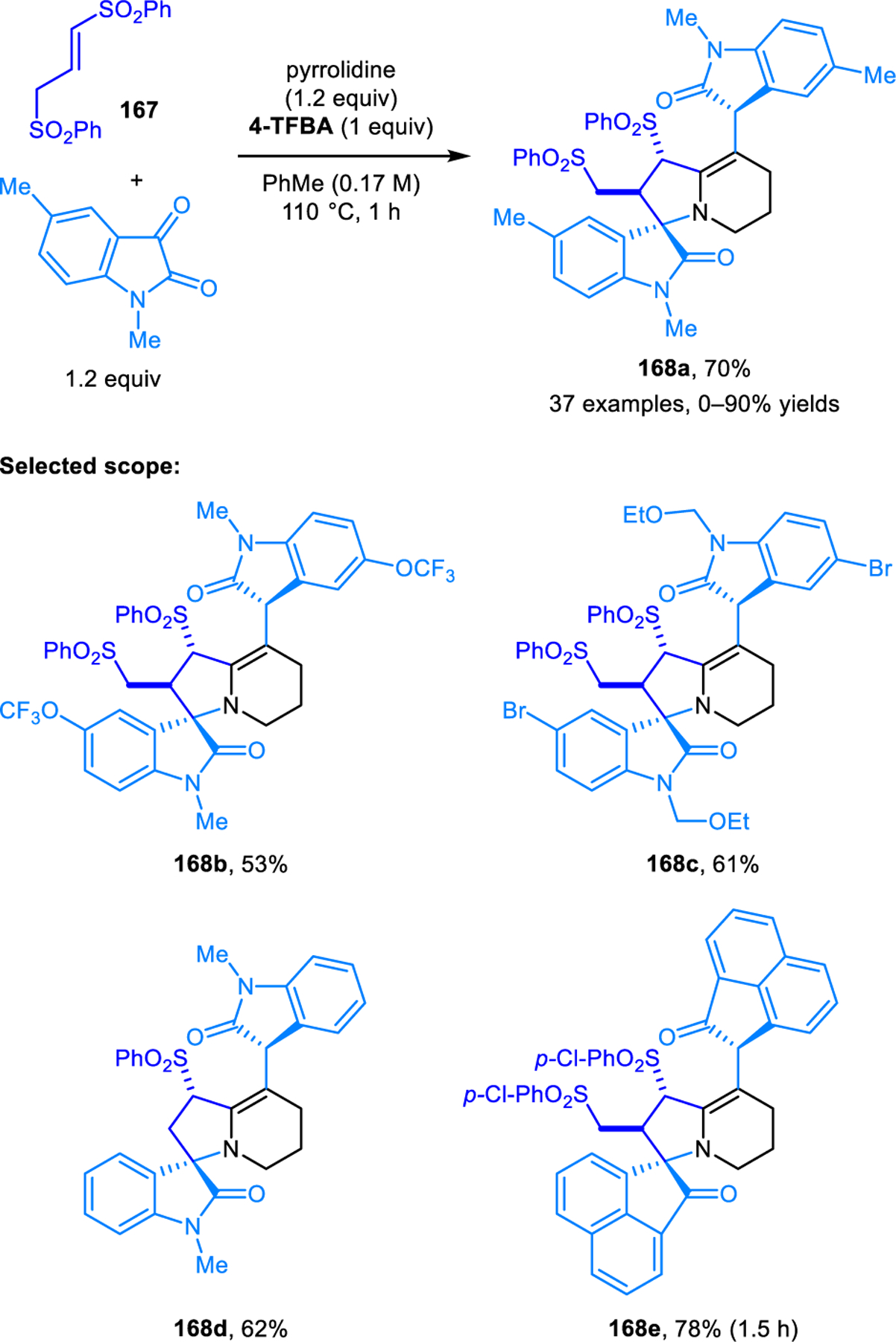
α,α,β,β-Tetrafunctionalization of piperidine forming spirooxindoles.
Jana and coworkers reported a set of divergent redox-neutral transformations based on the condensation of cyclic amines and aromatic aldehydes in the presence of another reaction partner (Scheme 80).137 Polycyclic N,O-acetals such as 169 are obtained by heating a mixture of cyclic amine, aromatic aldehyde, and 4-hydroxycoumarine (or 1,3-cyclohexanedione) in toluene with 2,4-dichlorobenzoic acid (DCBA) as the promoter. While N,O-acetals 169 resemble compounds 153 (c.f. Scheme 75), the authors proposed a different mechanism to account for their formation, based in part on control experiments. Accordingly, β-conjugated iminium ion intermediate 172 is formed from the condensation of pyrrolidine, carboxylic acid, and two equiv of aldehyde. Iminium ion 172 engages in conjugate addition with 4-hydroxycoumarine to form enamine 173. Intramolecular proton transfer followed by diastereoselective ring-closure provides the final product. A different reaction outcome is observed when 4-hydroxycoumarine is replaced with cinnamic acid. Bicyclic pyrrolizidine derivatives 170 containing β-exocyclic double bonds are obtained via (3+2) cycloaddition of the corresponding β-conjugated azomethine ylide and cinnamic acid. Interestingly, in reactions employing 9-fluorenone imine instead of aldehyde, the carboxylic acid group present in the initial cycloaddition product further reacts with the β-exocyclic double bond, resulting in the highly diastereoselective formation of fused tricyclic bowl-shaped compound 171.
Scheme 80.
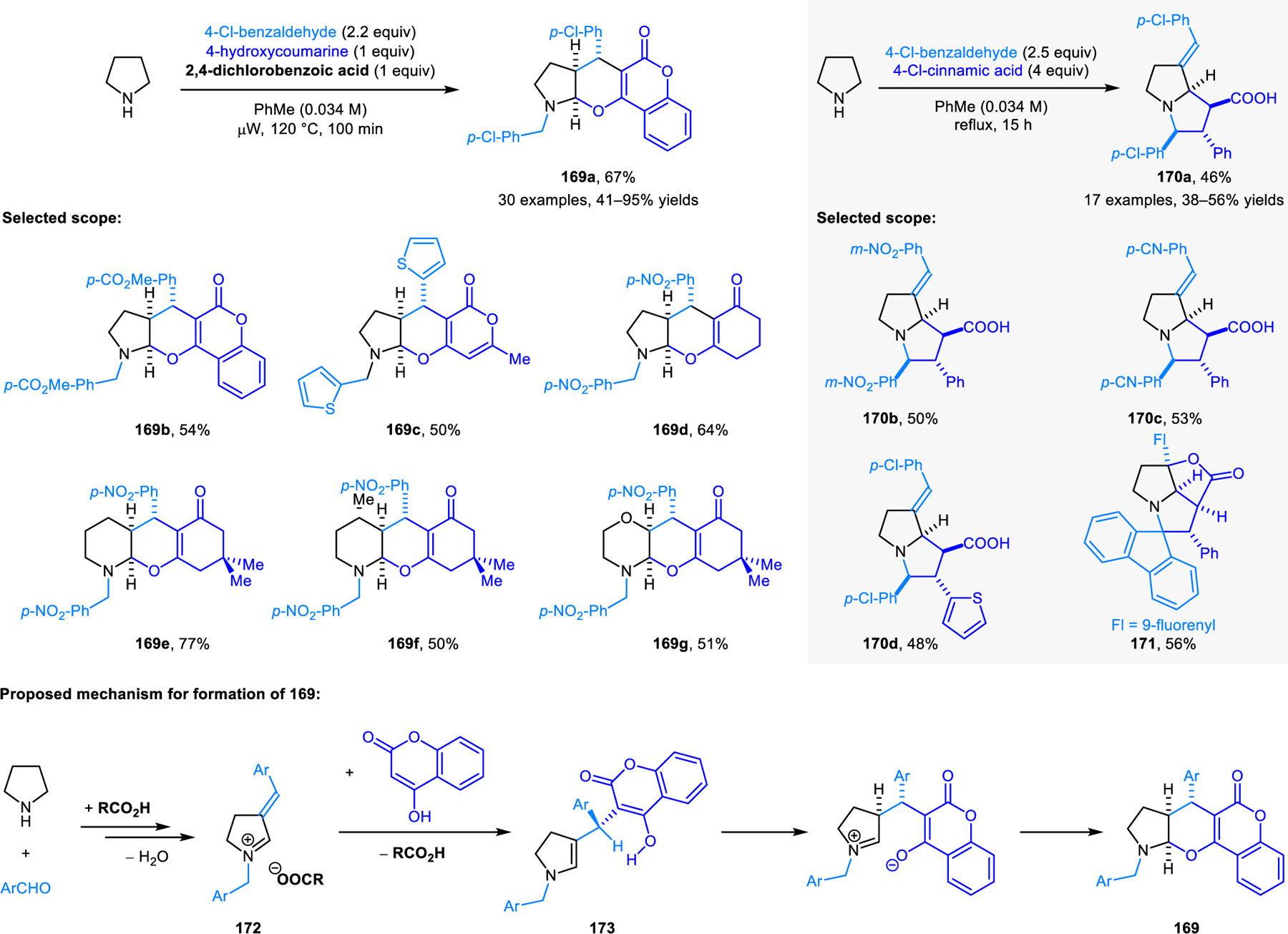
Redox-neutral synthesis of polycyclic heterocycles involving β-C−H bond functionalization of cyclic amines.
Chang and coworkers reported a reaction that results in α,β-annulation of cyclic amines via condensation with 2-formylazarenes (Scheme 81).138 Reactions are conducted under reflux in toluene in the presence of acetic acid, providing polycyclic pyrrolo azaarenes 174 in good to excellent yields. Reactions harness the reactivity of an enamine intermediate. Following a formal (3+2) cycloaddition with a second equivalent of 2-formylazarene, dehydration/aromatization results in the formation of the final product.
Scheme 81.
Redox-neutral synthesis of polycyclic pyrrolo azaarenes.
Indole derivatives such as 175 and 176 can be prepared from secondary N-aryl amines and nitrosobenzenes, as described in a study by Jana and coworkers (Scheme 82).139 The proposed reaction mechanism involves the formation of N-aryl-1-aminoiminium ions 177 followed by redox-isomerization to 178 and subsequent formation of enamine 179. The latter then undergoes a [3,3]-sigmatropic rearrangement providing β-aryl imine 180. Intramolecular addition/elimination provides the final 3-substituted indole product. Reactions employing N-alkyl anilines or aminopyridines are accompanied by the loss of the N-aryl group during the reaction course (e.g., formation of 176a).
Scheme 82.
Synthesis of indole derivatives via an acid-mediated coupling of secondary amines and nitrosobenzene.
A unique example of amine β-C−H bond functionalization, namely the formation of β-conjugated cyclic imines such as 181 from the corresponding amines, was reported by the Jana group (Scheme 83).140 This reaction is promoted by 3,5-dinitrobenzoic acid (DNBA) and, just like other reactions discussed in this section, is initiated by the condensation of a cyclic amine with an aldehyde. However, the overall transformation is oxidative in nature. Reactions involving pyrrolidine and aromatic aldehydes bearing various substituents give products 181 in high yields. Piperidine is also a viable substrate but provides slightly lower yields. The formation of the products was proposed to result from a reaction between aldehydes and enamines such as 2-pyrroline or 2-piperideine. These enamines are thought to be derived from the corresponding imines which in turn result from the hydrolysis or aminolysis of endocyclic N-benzyl iminium ions. According to this scenario, the formation of β-conjugated cyclic imines requires the consumption of two equivalents of aldehyde. Considering the substrate stoichiometry, thermal aerial oxidation of benzyl alcohols (resulting from the hydrolysis of N-benzyl iminium ions) to benzaldehydes, as well as oxidation of N-benzyl amines (resulting from the aminolysis of N-benzyl iminium ions) and cyclic amines to imines may occur under the reaction conditions. In fact, reactions involving pyrrolidine and either benzyl alcohol or N-benzyl pyrrolidine under otherwise identical conditions also provide the desired product, albeit in low yields. Yield dramatically decrease in reactions performed under an argon atmosphere.
Scheme 83.
Synthesis of α,β-unsaturated imines based on the condensation of alicyclic amines and aldehydes.
7. Outlook
Condensation-based methods for the C−H bond functionalization of secondary amines proceeding via the intermediacy of azomethine ylides are typically redox-neutral in nature. They thus provide access to functionalized amines via a strategy that is complementary to other approaches which are still dominated by oxidative methods. Much progress has been made since the early days of serendipitous discovery, but significant challenges remain. While the application of carboxylic acids as catalysts/promoters has enabled the development of previously inconceivable transformations by lowering reactions barriers, high reaction temperatures are still required in many if not most transformations. Future efforts thus must target new strategies for further lowering energy barriers for the key C–H bond functionalization step, particularly in amine substrates lacking benzylic or otherwise activated α-C−H bonds. This in turn should help facilitate the development of catalytic enantioselective variants of which there are only two so far.
Acknowledgment
We are grateful to the current and former members of the Seidel research group who have contributed to the development of the research summarized in this review.
Funding Information
Financial support from the NIH–NIGMS (grant no. R01GM101389) is gratefully acknowledged.
References
- (1).(a) Taylor RD; MacCoss M; Lawson ADG, J. Med. Chem 2014, 57, 5845; [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT, J. Med. Chem 2014, 57, 10257; [DOI] [PubMed] [Google Scholar]; (c) Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A, Nat. Chem 2018, 10, 383. [DOI] [PubMed] [Google Scholar]
- (2).(a) For a general overview on amine C–H bond functionalization, see: Dutta S; Li B; Rickertsen DRL; Valles DA; Seidel D, SynOpen 2021, 05, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Other selected reviews: Campos KR, Chem. Soc. Rev 2007, 36, 1069; [DOI] [PubMed] [Google Scholar]; (c) Jazzar R; Hitce J; Renaudat A; Sofack-Kreutzer J; Baudoin O, Chem. Eur. J 2010, 16, 2654; [DOI] [PubMed] [Google Scholar]; (d) Yeung CS; Dong VM, Chem. Rev 2011, 111, 1215; [DOI] [PubMed] [Google Scholar]; (e) Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes BUW, Chem. Eur. J 2012, 18, 10092; [DOI] [PubMed] [Google Scholar]; (f) Jones KM; Klussmann M, Synlett 2012, 23, 159; [Google Scholar]; (g) Peng B; Maulide N, Chem. Eur. J 2013, 19, 13274; [DOI] [PubMed] [Google Scholar]; (h) Girard SA; Knauber T; Li C-J, Angew. Chem. Int. Ed 2014, 53, 74; [DOI] [PubMed] [Google Scholar]; (i) Haibach MC; Seidel D, Angew. Chem. Int. Ed 2014, 53, 5010; [DOI] [PubMed] [Google Scholar]; (j) Wang L; Xiao J, Adv. Synth. Catal 2014, 356, 1137; [Google Scholar]; (k) Vo C-VT; Bode JW, J. Org. Chem 2014, 79, 2809; [DOI] [PubMed] [Google Scholar]; (l) Qin Y; Lv J; Luo S, Tetrahedron Lett 2014, 55, 551; [Google Scholar]; (m) Beatty JW; Stephenson CRJ, Acc. Chem. Res 2015, 48, 1474; [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Qin Y; Zhu L; Luo S, Chem. Rev 2017, 117, 9433; [DOI] [PubMed] [Google Scholar]; (o) Cheng M-X; Yang S-D, Synlett 2017, 28, 159; [Google Scholar]; (p) Chu JCK; Rovis T, Angew. Chem. Int. Ed 2018, 57, 62; [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Stateman LM; Nakafuku KM; Nagib DA, Synthesis 2018, 50, 1569; [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Edwards PM; Schafer LL, Chem. Commun 2018, 54, 12543; [DOI] [PubMed] [Google Scholar]; (s) Gonnard L; Guérinot A; Cossy J, Tetrahedron 2019, 75, 145; [Google Scholar]; (t) Liu S; Zhao Z; Wang Y, Chem. Eur. J 2019, 25, 2423; [DOI] [PubMed] [Google Scholar]; (u) Antermite D; Bull JA, Synthesis 2019, 51, 3171; [Google Scholar]; (v) Zhang T; Wu Y-H; Wang N-X; Xing Y, Synthesis 2019, 51, 4531; [Google Scholar]; (w) Trowbridge A; Walton SM; Gaunt MJ, Chem. Rev 2020, 120, 2613; [DOI] [PubMed] [Google Scholar]; (x) Kapoor M; Singh A; Sharma K; Hua Hsu M, Adv. Synth. Catal 2020, 362, 4513; [Google Scholar]; (y) An X-D; Xiao J, Org. Chem. Front 2021, 8, 1364; [Google Scholar]; (z) Basak S; Winfrey L; Kustiana BA; Melen RL; Morrill LC; Pulis AP, Chem. Soc. Rev 2021, 50, 3720; [DOI] [PubMed] [Google Scholar]; (aa) Caplin MJ; Foley DJ, Chem. Sci 2021, 12, 4646; [DOI] [PMC free article] [PubMed] [Google Scholar]; (ab) Ohno S; Miyoshi M; Murai K; Arisawa M, Synthesis.
- (3).(a) Selected reviews on various types of redox-neutral transformations: Burns NZ; Baran PS; Hoffmann RW, Angew. Chem. Int. Ed 2009, 48, 2854; [DOI] [PubMed] [Google Scholar]; (b) Mahatthananchai J; Bode JW, Acc. Chem. Res 2014, 47, 696; [DOI] [PubMed] [Google Scholar]; (c) Ketcham JM; Shin I; Montgomery TP; Krische MJ, Angew. Chem. Int. Ed 2014, 53, 9142; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Huang H; Ji X; Wu W; Jiang H, Chem. Soc. Rev 2015, 44, 1155. [DOI] [PubMed] [Google Scholar]
- (4).(a) Selected reviews on the chemistry of azomethine ylides: Padwa A, Wiley: New York, N. Y, 1984;; (b) Padwa A; Editor, Wiley: New York, N. Y, 1984;; (c) Padwa A; Pearson WH, Wiley: Chichester, U. K, 2002;; (d) Najera C; Sansano JM, Curr. Org. Chem 2003, 7, 1105; [Google Scholar]; (e) Coldham I; Hufton R, Chem. Rev 2005, 105, 2765; [DOI] [PubMed] [Google Scholar]; (f) Pandey G; Banerjee P; Gadre SR, Chem. Rev 2006, 106, 4484; [DOI] [PubMed] [Google Scholar]; (g) Pinho e Melo TMVD, Eur. J. Org. Chem 2006, 2873; [Google Scholar]; (h) Bonin M; Chauveau A; Micouin L, Synlett 2006, 2349; [Google Scholar]; (i) Nair V; Suja TD, Tetrahedron 2007, 63, 12247; [Google Scholar]; (j) Najera C; Sansano JM, Top. Heterocycl. Chem 2008, 12, 117; [Google Scholar]; (k) Stanley LM; Sibi MP, Chem. Rev 2008, 108, 2887; [DOI] [PubMed] [Google Scholar]; (l) Nyerges M; Toth J; Groundwater PW, Synlett 2008, 1269; [Google Scholar]; (m) Pineiro M; Pinho e Melo TMVD, Eur. J. Org. Chem 2009, 5287; [Google Scholar]; (n) Burrell AJM; Coldham I, Curr. Org. Synth 2010, 7, 312; [Google Scholar]; (o) Anac O; Gungor FS, Tetrahedron 2010, 66, 5931; [Google Scholar]; (p) Adrio J; Carretero JC, Chem. Commun 2011, 47, 6784; [DOI] [PubMed] [Google Scholar]; (q) Adrio J; Carretero JC, Chem. Commun 2014, 50, 12434; [DOI] [PubMed] [Google Scholar]; (r) Hashimoto T; Maruoka K, Chem. Rev 2015, 115, 5366; [DOI] [PubMed] [Google Scholar]; (s) Meyer A; Ryan J, Molecules 2016, 21, 935; [Google Scholar]; (t) Otero-Fraga J; Montesinos-Magraner M; Mendoza A, Synthesis 2017, 49, 802; [DOI] [PubMed] [Google Scholar]; (u) Döndas HA; de Gracia Retamosa M; Sansano JM, Synthesis 2017, 49, 2819; [Google Scholar]; (v) Tang S; Zhang X; Sun J; Niu D; Chruma JJ, Chem. Rev 2018, 118, 10393; [DOI] [PubMed] [Google Scholar]; (w) Wei L; Chang X; Wang C-J, Acc. Chem. Res 2020, 53, 1084. [DOI] [PubMed] [Google Scholar]
- (5).Seidel D Acc. Chem. Res 2015, 48, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mahato S; Jana CK Chem. Rec 2016, 16, 1477. [DOI] [PubMed] [Google Scholar]
- (7).(a) Examples of hydride transfer-based methods involving a condensation step: Zhang C; Murarka S; Seidel D, J. Org. Chem 2009, 74, 419; [DOI] [PubMed] [Google Scholar]; (b) Mori K; Ohshima Y; Ehara K; Akiyama T, Chem. Lett 2009, 38, 524; [Google Scholar]; (c) Haibach MC; Deb I; De CK; Seidel D, J. Am. Chem. Soc 2011, 133, 2100; [DOI] [PubMed] [Google Scholar]; (d) Bai G; Dong F; Xu L; Liu Y; Wang L; Li S-S, Org. Lett 2019, 21, 6225; [DOI] [PubMed] [Google Scholar]; (e) Wang S; Shen Y-B; Li L-F; Qiu B; Yu L; Liu Q; Xiao J, Org. Lett 2019, 21, 8904. [DOI] [PubMed] [Google Scholar]
- (8).(a) For a review, see: Rahman M; Mukherjee A; Kovalev IS; Kopchuk DS; Zyryanov GV; Tsurkan MV; Majee A; Ranu BC; Charushin VN; Chupakhin ON; Santra S, Adv. Synth. Catal 2019, 361, 2161. [Google Scholar]; (b) Selected examples: Cohen N; Blount JF; Lopresti RJ; Trullinger DP, J. Org. Chem 1979, 44, 4005; [Google Scholar]; (c) Bi H-P; Zhao L; Liang Y-M; Li C-J, Angew. Chem. Int. Ed 2009, 48, 792; [DOI] [PubMed] [Google Scholar]; (d) Zhang C; Seidel D, J. Am. Chem. Soc 2010, 132, 1798; [DOI] [PubMed] [Google Scholar]; (e) Bi H-P; Teng Q; Guan M; Chen W-W; Liang Y-M; Yao X; Li C-J, J. Org. Chem 2010, 75, 783; [DOI] [PubMed] [Google Scholar]; (f) Das D; Richers MT; Ma L; Seidel D, Org. Lett 2011, 13, 6584; [DOI] [PubMed] [Google Scholar]; (g) Dighe SU; K. S, A. K.; Srivastava S; Shukla P; Singh S; Dikshit M; Batra S, J. Org. Chem 2015, 80, 99; [DOI] [PubMed] [Google Scholar]; (h) Kang Y; Seidel D, Org. Lett 2016, 18, 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Rügheimer L Ber 1891, 24, 2186.; [Google Scholar]; (b) Rügheimer L Ber 1892, 25, 2421. [Google Scholar]
- (10).Skraup S; Böhm K Ber 1926, 59, 1015. [Google Scholar]
- (11).Parker ED; Furst A J. Org. Chem 1958, 23, 201. [Google Scholar]
- (12).Poirier RH; Morin RD; McKim AM; Bearse AE J. Org. Chem 1961, 26, 4275. [Google Scholar]
- (13).Burrows EP; Hutton RF; Burrows WD J. Org. Chem 1962, 27, 316. [Google Scholar]
- (14).Burrows WD; Burrows EP J. Org. Chem 1963, 28, 1180. [Google Scholar]
- (15).Kameswari U; Pillai CN Catal. Lett 1996, 38, 53. [Google Scholar]
- (16).Moura NMM; Núñez C; Santos SM; Faustino MAF; Cavaleiro JAS; Paz FAA; Neves MGPMS; Capelo JL; Lodeiro C Chem. Eur. J 2014, 20, 6684.24782336 [Google Scholar]
- (17).Platonova AY; Seidel D Tetrahedron Lett 2015, 56, 3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chang M-Y; Tsai Y-L J. Org. Chem 2020, 85, 5651. [DOI] [PubMed] [Google Scholar]
- (19).Sainsbury M; Dyke SF; Brown DW; Lugton WGD Tetrahedron 1968, 24, 427. [Google Scholar]
- (20).Dannhardt G; Mayer KK; Obergrusberger I; Roelcke J Arch. Pharm. (Weinheim, Ger.) 1986, 319, 977. [Google Scholar]
- (21).Dannhardt G; Roelcke J Arch. Pharm. (Weinheim, Ger.) 1992, 325, 671. [Google Scholar]
- (22).Lee Y; Huang H; Sayer LM J. Am. Chem. Soc 1996, 118, 7241. [Google Scholar]
- (23).Cook AG; Switek KA; Cutler KA; Witt AM Lett. Org. Chem 2004, 1, 1. [Google Scholar]
- (24).Oda M; Fukuchi Y; Ito S; Thanh NC; Kuroda S Tetrahedron Lett 2007, 48, 9159. [Google Scholar]
- (25).Poláčková V; Veverková E; Toma S; Bogdal D Synth. Commun 2009, 39, 1871. [Google Scholar]
- (26).Pahadi NK; Paley M; Jana R; Waetzig SR; Tunge JA J. Am. Chem. Soc 2009, 131, 16626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xue X; Yu A; Cai Y; Cheng J-P Org. Lett 2011, 13, 6054. [DOI] [PubMed] [Google Scholar]
- (28).Mao H; Xu R; Wan J; Jiang Z; Sun C; Pan Y Chem. Eur. J 2010, 16, 13352. [DOI] [PubMed] [Google Scholar]
- (29).Deb I; Das D Seidel D Org. Lett 2011, 13, 812. [DOI] [PubMed] [Google Scholar]
- (30).Bayindir S; Erdogan E; Kilic H; Aydin O; Saracoglu N J. Heterocyclic Chem 2015, 52, 1589. [Google Scholar]
- (31).Aydin O; Kilic H; Bayindir S; Erdogan E; Saracoglu N J. Heterocyclic Chem 2015, 52, 1540. [Google Scholar]
- (32).Ramakumar K; Tunge JA Chem. Commun 2014, 50, 13056. [DOI] [PubMed] [Google Scholar]
- (33).Ardill H; Grigg R; Sridharan V; Surendrakumar S; Thianpatanagul S; Kanajun S J. Chem. Soc., Chem. Commun 1986, 602. [Google Scholar]
- (34).Ardill H; Dorrity MJR; Grigg R; Leon-Ling M-S; Malone JF; Sridharan V; Thianpatanagul S Tetrahedron 1990, 46, 6433. [Google Scholar]
- (35).Ardill H; Fontaine XLR; Grigg R; Henderson D; Montgomery J; Sridharan V; Surendrakumar S Tetrahedron 1990, 46, 6449. [Google Scholar]
- (36).Wittland C; Arend M; Risch N Synthesis 1996, 367. [Google Scholar]
- (37).Xie H; Xiang J; Dang Q; Bai X Synlett, 2012, 23, 585. [Google Scholar]
- (38).Mantelingu K; Lin Y; Seidel D Org. Lett 2014, 16, 5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sutariya TR; Labana BM; Parmar NJ; Kant R; Gupta VK; Plata GB; Padrόn JM New. J. Chem 2015, 39, 2657. [Google Scholar]
- (40).Kumar CSP; Harsha KB; Sandhya NC; Ramesha AB; Mantelingu K; Rangappa KS New J. Chem 2015, 39, 8397. [Google Scholar]
- (41).Strada A; Fredditori M; Zanoni G; Protti S Molecules 2019, 24, 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tolbert LM; Haubrich JE J. Am. Chem. Soc 1994, 116, 10593. [Google Scholar]
- (43).Rahman M; Bagdi AK; Mishra S; Hajra A Chem. Commun 2014, 50, 2951. [DOI] [PubMed] [Google Scholar]
- (44).Kumar CSP; Harsha KB; Mantelingu K; Rangappa KS RSC Adv 2015, 5, 61664. [Google Scholar]
- (45).Wu X; Zhu Z-H; He H; Ren L; Zhu C-F; Li Y-G J. Org. Chem 2020, 85, 6216. [DOI] [PubMed] [Google Scholar]
- (46).Zhang X; Liu M; Qiu W; Evans J; Kaur M; Jasinski JP; Zhang W ACS Sustainable Chem. Eng 2018, 6, 5574. [Google Scholar]
- (47).Guo J; Zhao Y; Fang D; Wang Q; Bu Z Org. Biomol. Chem 2018, 16, 6025. [DOI] [PubMed] [Google Scholar]
- (48).Huang Y; Huang Y-X; Sun J; Yan C-G RSC Adv 2018, 8, 23990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Boudriga S; Haddad S; Askri M; Soldera A; Knorr M; Strohmann C; Golz C RSC Adv 2019, 9, 11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Safaei-Ghomi J; Masoomi R RSC Adv 2015, 5, 15591. [Google Scholar]
- (51).Yang H-T; Tan Y-C; Ge J; Wu H; Li J-X; Yang Y; Sun X-Q; Miao C-B J. Org. Chem 2016, 81, 11201. [DOI] [PubMed] [Google Scholar]
- (52).Shi J-L; Zhang X-F; Wang H-J; Li F-B; Zhong X-X; Liu C-X; Liu L; Liu C-Y; Qin H-M; Huang Y-S J. Org. Chem 2016, 81, 7662. [DOI] [PubMed] [Google Scholar]
- (53).Zheng K; Zhuang S; You M; Shu W; Wu A; Wu Y ChemistrySelect 2017, 2, 10762. [Google Scholar]
- (54).Zheng K-L; You M-Q; Shu W-M; Wu Y-D; Wu A-X Org. Lett 2017, 19, 2262. [DOI] [PubMed] [Google Scholar]
- (55).Grigg R; Gunaratne HQN; Henderson D; Sridharan V Tetrahedron 1990, 46, 1599. [Google Scholar]
- (56).(a) Soeder RW; Bowers K; Pegram LD; Cartaya-Marin CP, Synth. Commun 1992, 22, 2737; [Google Scholar]; (b) Deb I; Seidel D, Tetrahedron Lett 2010, 51, 2945. [Google Scholar]
- (57).Kang Y; Richers MT; Sawicki CH; Seidel D Chem. Commun 2015, 51, 10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Yang Z; Lu N; Wei Z; Cao J; Liang D; Duan H; Lin Y J. Org. Chem 2016, 81, 11950. [DOI] [PubMed] [Google Scholar]
- (59).Xia P-J; Sun Y-H; Xiao J-A; Zhou Z-F; Wen S-S; Xiong Y; Ou G-C; Chen X-Q; Yang H J. Org. Chem 2015, 80, 11573. [DOI] [PubMed] [Google Scholar]
- (60).Mure M; Mills SA; Klinman JP Biochemistry 2002, 41, 9269. [DOI] [PubMed] [Google Scholar]
- (61).Mure M Acc. Chem. Res 2004, 37, 131. [DOI] [PubMed] [Google Scholar]
- (62).Largeron M; Fleury M-B Science 2013, 339, 43. [DOI] [PubMed] [Google Scholar]
- (63).Wendlandt AE; Stahl SS Angew. Chem. Int. Ed 2015, 54, 14638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Largeron M Org. Biomol. Chem 2017, 15, 4722. [DOI] [PubMed] [Google Scholar]
- (65).Cheng Y-F; Rong H-J; Yi C-B; Yao J-J; Qu J Org. Lett 2015, 17, 4758. [DOI] [PubMed] [Google Scholar]
- (66).Rong H-J; Cheng Y-F; Liu F-F; Ren S-J; Qu J J. Org Chem 2017, 82, 532. [DOI] [PubMed] [Google Scholar]
- (67).Vasu D; de Arriba ALF; Leitch JA; de Gombert A; Dixon DJ Chem. Sci 2019, 10, 3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Vasu D; Leitch JA; Dixon DJ Tetrahedron 2019, 75, 130726. [Google Scholar]
- (69).Ma L; Chen W; Seidel D J. Am. Chem. Soc 2012, 134, 15305. [DOI] [PubMed] [Google Scholar]
- (70).Murahashi S-I; Komiya N; Terai H; Nakae T J. Am. Chem.Soc 2003, 125, 15312. [DOI] [PubMed] [Google Scholar]
- (71).Das D; Sun AX; Seidel D Angew. Chem. Int. Ed 2013, 52, 3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Seidel D Org. Chem. Front 2014, 1. 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Zheng Q; Meng W; Jiang G; Yu Z Org. Lett 2013, 15, 5928. [DOI] [PubMed] [Google Scholar]
- (74).Lin W; Cao T; Fan W; Han Y; Kuang J; Luo H; Miao B; Tang X; Yu Q; Yuan W; Zhang J; Zhu C; Ma S Angew. Chem. Int. Ed 2014, 53, 277. [DOI] [PubMed] [Google Scholar]
- (75).Lin W; Ma S Org. Chem. Front 2014, 1, 338. [Google Scholar]
- (76).Lin W; Ma S Org. Chem. Front 2017, 4, 958. [Google Scholar]
- (77).Zhou S; Tong R Chem. Eur. J 2016, 22, 7084. [DOI] [PubMed] [Google Scholar]
- (78).Zhou S; Tong R Org. Lett 2017, 19, 1594. [DOI] [PubMed] [Google Scholar]
- (79).Yu J; Zhang Z; Zhou S; Zhang W Tong R Org. Chem. Front 2018, 5, 242. [Google Scholar]
- (80).Li J; Wang H; Sun J; Yang Y; Liu L Org. Biomol. Chem 2014, 12, 2523. [DOI] [PubMed] [Google Scholar]
- (81).Shao G; He Y; Xu Y; Chen J; Yu H; Cao R Eur. J. Org. Chem 2015, 4615. [Google Scholar]
- (82).Villamizar MCO; Galvis CEP; Kouznetsov VV Synthesis, 2021, 53, 547. [Google Scholar]
- (83).Zhao H; He W; Wei L; Cai M Catal. Sci. Technol 2016, 6, 1488. [Google Scholar]
- (84).Gulati U; Rawat S; Rajesh UC; Rawat DS New J. Chem 2017, 41, 8341. [Google Scholar]
- (85).Xu H; Wang J; Wang P; Niu X; Luo Y; Zhu L; Yao X RSC Adv 2018, 8, 32942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Shi L; Wang M; Pan L; Li Y; Liu Q Chem. Commun 2018, 54, 8721. [DOI] [PubMed] [Google Scholar]
- (87).Das D; Seidel D Org. Lett 2013, 15, 4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Hu G; Chen W; Ma D; Zhang Y; Xu P; Gao Y; Zhao Y J. Org. Chem 2016, 81, 1704. [DOI] [PubMed] [Google Scholar]
- (89).Chen W; Wilde RG; Seidel D Org. Lett 2014, 16, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Haldar S; Mahato S; Jana CK Asian J. Org. Chem 2014, 3, 44. [Google Scholar]
- (91).Haldar S; Roy SK; Maity B; Koley D; Jana CK Chem. Eur. J 2015, 21, 15290. [DOI] [PubMed] [Google Scholar]
- (92).Haldar S; Jana CK Org. Biomol. Chem 2019, 17, 1800. [DOI] [PubMed] [Google Scholar]
- (93).Yi F; Su J; Zhang S; Yi W; Zhang L Eur. J. Org. Chem 2015, 7360. [Google Scholar]
- (94).Zhang Q; Lv J,; Luo S Acta Chim. Sinica 2016, 74, 61. [Google Scholar]
- (95).Rahman I; Deka B; Thakuria R; Deb ML; Baruah PK Org. Biomol. Chem 2020, 18, 6514. [DOI] [PubMed] [Google Scholar]
- (96).(a) Gulati U; Rawat S; Rawat DS, Tetrahedron Lett 2020, 61, 152304; [Google Scholar]; (b) Kaur P; Gurjar KK; Kumar V; Gohit S; Gupta V; Kumar R ChemistrySelect 2020, 5, 12514. [Google Scholar]
- (97).Chen W; Seidel D Org. Lett 2014, 16, 3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Zhu Z; Seidel D Org. Lett 2016, 18, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Yan J; Bai Q; Xu C; Feng G Synthesis 2016, 48, 3730. [Google Scholar]
- (100).Haldar S; Saha S; Mandal S; Jana CK Green Chem 2018, 20, 3463. [Google Scholar]
- (101).Huang J; Li L; Xiao T; Mao Z; Zhou L Asian J. Org. Chem 2016, 5, 1204. [Google Scholar]
- (102).Yi C-B; She Z-Y; Cheng Y-F; Qu J Org. Lett 2018, 20, 668. [DOI] [PubMed] [Google Scholar]
- (103).Purkait A; Jana CK Synthesis 2019, 51, 2687. [Google Scholar]
- (104).Jiang D, Wu Z and Wang J, Chin. J. Chem 2020, 38, 135–138. [Google Scholar]
- (105).Zhang C; De CK; Mal R; Seidel D J. Am. Chem. Soc 2008, 130, 416. [DOI] [PubMed] [Google Scholar]
- (106).Richers MT; Deb I; Platonova AY; Zhang C; Seidel D Synthesis 2013, 45, 1730. [PMC free article] [PubMed] [Google Scholar]
- (107).Dieckmann A; Richers MT; Platonova AY; Zhang C; Seidel D; Houk KN J. Org. Chem 2013, 78, 4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Richers MT; Zhao C; Seidel D Beilstein J. Org. Chem 2013, 9, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Zheng L; Yang F; Dang Q; Bai X Org. Lett 2008, 10, 889. [DOI] [PubMed] [Google Scholar]
- (110).Afanasyev OI; Podyacheva E; Rudenko A; Tsygankov AA; Makarova M; Chusov D J. Org. Chem 2020, 85, 9347. [DOI] [PubMed] [Google Scholar]
- (111).Deb ML; Borpatra PJ; Baruah PK Green Chem 2019, 21, 69. [Google Scholar]
- (112).Richers MT; Breugst M; Platonova AY; Ullrich A; Dieckmann A; Houk KN; Seidel D J. Am. Chem. Soc 2014, 136, 6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Jarvis CL; Richers MT; Breugst M; Houk KN; Seidel D Org. Lett 2014, 16, 3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Kirkeby EK; Roberts AG Chem. Commun 2020, 56, 9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Mahato S; Haque MA; Dwari S; Jana CK RSC Adv 2014, 4, 46214. [Google Scholar]
- (116).Zhang C; Das D; Seidel D Chem. Sci 2011, 2, 233. [Google Scholar]
- (117).Ma L; Seidel D Chem. Eur. J 2015, 21, 12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Paul A; Chandak HS; Ma L; Seidel D Org. Lett 2020, 22, 976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Rickertsen DRL; Ma L; Paul A; Abboud KA; Seidel D SynOpen 2020, 4, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Kang Y; Chen W; Breugst M; Seidel D J. Org. Chem 2015, 80, 9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Chen W; Seidel D Org. Lett 2016, 18, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Openshaw HT; Whittaker NJ J. Chem. Soc 1963, 1461. [Google Scholar]
- (123).Li J; Qin C; Yu Y; Fan H; Fu Y; Li H; Wang W Adv. Synth. Catal 2017, 359, 2191. [Google Scholar]
- (124).Li J; Fu Y; Qin C; Yu Y; Li H; Wang W Org. Biomol. Chem 2017, 15, 6474. [DOI] [PubMed] [Google Scholar]
- (125).Zhu Z; Seidel D Org. Lett 2017, 19, 2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Paul A; Adili A; Seidel D Org. Lett 2019, 21, 1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Zhu Z; Lv X; Anesini J, J. E.; Seidel D Org. Lett 2017, 19, 6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Liu Y; Wu J; Jin Z; Jiang H Synlett 2018, 29, 1061. [Google Scholar]
- (129).Zhu Z; Chandak HS; Seidel D Org. Lett 2018, 20, 4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Chen W; Kang Y; Wilde RG; Seidel D Angew. Chem. Int. Ed 2014, 53, 5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Mukhopadhyay C; Rana S; Butcher RJ Synth. Commun 2012, 42, 3077. [Google Scholar]
- (132).Ma L; Paul A; Breugst M; Seidel D Chem. Eur. J 2016, 22, 18179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Huang Y; Fang H-L; Huang Y-X; Sun J; Yan C-G J. Org. Chem 2019, 84, 12437. [DOI] [PubMed] [Google Scholar]
- (134).Fang H-L; Han Y; Sun J; Yan C-G ChemistrySelect 2020, 5, 14086. [Google Scholar]
- (135).Zheng K-L; Shu W-M; Ma J-R; Wu Y-D; Wu A-X Org. Lett 2016, 18, 3526. [DOI] [PubMed] [Google Scholar]
- (136).Du Y; Yu A; Jia J; Zhang Y; Meng X Chem. Commun 2017, 53, 1684. [DOI] [PubMed] [Google Scholar]
- (137).Mandal S; Dwari S; Jana CK J. Org. Chem 2018, 83, 8874. [DOI] [PubMed] [Google Scholar]
- (138).Chang M-Y; Wu Y-S J. Org. Chem 2019, 84, 3638. [DOI] [PubMed] [Google Scholar]
- (139).Roy SK; Purkait A; Aziz SMT; Jana CK Chem. Commun 2020, 56, 3167. [DOI] [PubMed] [Google Scholar]
- (140).Mandal S; Mahato S; Jana CK Org. Lett 2015, 17, 3762. [DOI] [PubMed] [Google Scholar]