

Abstract
Activated group 2 innate lymphoid cells (ILC2s) accumulate and promote inflammatory resolution and tissue repair in host defense against acute respiratory viral infections. However, the heterogeneity of ILC2s in the lung and the mechanisms by which ILC2 cells contribute to tissue repair remain elusive. Using single-cell RNA-sequencing (scRNA-seq), we identify a transcriptionally distinct ILC2 subset that showed enrichment for wound healing signature genes and the transcription factor BATF. Notably, BATF promotes the proliferation and function of ILC2s and restricts their plasticity during infection with influenza virus. In the absence of BATF, ILC2s lose their immune protective properties and acquire pathogenic ILC3-like functions, leading to persistent neutrophil infiltration, tissue damage, and respiratory failure. Mechanistically, BATF directly binds to the cis-regulatory elements of wound healing genes, maintains their chromatin accessibility, and promotes their expression. Lastly, BATF plays an important role in an IL-33-ST2 feed-forward loop that supports ILC2 cell identity and function. Collectively, our findings shed light on a BATF-dependent ILC2 program, thereby providing a potential therapeutic target for terminating detrimental inflammation during acute viral infection.
One-sentence summary:
BATF regulates ILC2-mediated tissue repair and inflammation resolution during acute respiratory virus infection.
Introduction
Uncontrolled excessive inflammation in the lung, such as inflammatory cytokine storm, is widely appreciated as a unifying component resulting in lethality in many acute respiratory microbial infections, including influenza virus (1) and SARS-CoV-2 (2). Numerous studies have highlighted critical roles for group 2 innate lymphoid cells (ILC2s) in maintaining barrier tissue homeostasis by regulating immunity, resolution of inflammation, tissue repair and remodeling in the lung during acute respiratory virus infection (3–5). At the resolution phase, influenza infection augments the production of alarmin molecules, such as interleukin 33 (IL-33), which acts as a strong activator of the ILC2 response (4, 6). Activated ILC2s express a large number of genes associated with anti-inflammation and tissue repair, including cytokines (such as Il5, Il9, and Il13), extracellular matrix proteins (such as decorin, asporin, and dermatopontin), and epidermal growth factor family members (such as amphiregulin) (3). In ILC2s, either insufficient or excessive activation will result in adverse consequences such as impaired tissue repair, asthma, fibrosis and chronic obstructive pulmonary disease (COPD) (7–10). Yet, the intrinsic factors that regulate ILC2 activation and function after acute viral infection to control inflammatory resolution and tissue repair, a process vital to restoring health, remain elusive.
The transcription factor BATF contains a basic leucine zipper (bZip) domain, forms heterodimeric transcriptional complexes with other members of the AP-1 family, and regulates gene expression in various lymphocyte lineages (11–15). BATF has been shown to directly promote chromatin accessibility in T cells (16). In Th17 cells, loss of BATF is known to cause diminished chromatin accessibility and transcription factor recruitment at lineage-specifying loci (13, 17–19). Moreover, BATF modulates the Th2 locus control region (Rad50) in Th2 cells (20), and drives IL-33 receptor ST2 expression in non-lymphoid tissue Treg cells (21). Importantly, chromatin accessibility assays have revealed that the BATF binding motif is specifically enriched in group 2, but not group 1 and 3 ILCs (22). Recently, BATF was reported to regulate ILC hematopoiesis, periphery homeostasis and functions (23). In a pulmonary type 2 immune response, BATF appeared to be critical for the function of IL-25-responsive inflammatory ILC2 cells (iILC2s) (24). However, the precise mechanisms through which BATF functions to guard the fate of tissue resident ILC2s and how BATF regulates the activation and function of ILC2s, especially during the resolution phase of an acute respiratory virus infection, are largely unknown.
In this study, we characterize a BATF-dependent wound healing program in ILC2s that is required to terminate the inflammatory response in the lung. We show that there is heightened expression of BATF in an ILC2 subset that is specialized to promote wound healing. Furthermore, we show that BATF directly binds to ILC2 signature and wound healing-associated genes, thereby promoting chromatin accessibility and gene expression. In contrast, BATF-deficient ILC2s exhibit attenuated proliferation, impaired wound healing functions, and enhanced pro-inflammatory features due to altered transcriptional profiles and epigenetic landscapes after activation. Together, these findings define a BATF-dependent, IL-33-ST2 feed-forward loop that is critical to the support of ILC2 cell identity and function.
Results
scRNA-seq reveals enriched expression of BATF in a wound healing ILC2 subset during influenza virus infection
Innate lymphoid cells (ILCs), such as ILC1 and ILC2, play critical and distinct roles at different phases of influenza virus infection (3, 25). To further examine whether potential heterogeneity exists within ILC subsets in an unbiased way, we performed single-cell RNA-sequencing (scRNA-seq) on Lin−CD90.2+ ILCs sorted from the lungs of C57BL/6 mice at days 5 and 10 after influenza A (H1N1/PR8) sublethal infection (Fig. 1A and fig. S1A). In order to assess dynamic changes at the transcriptional and cellular levels in ILCs after infection, we integrated a published scRNA-seq dataset of ILCs from the lungs of PBS-treated mice and used it as a day 0 control sample (26). By using Uniform Manifold Approximation and Projection (UMAP) analysis in the R package Seurat, we detected four distinct clusters based on their transcriptomes (Fig. 1B and fig. S1B). Cells in cluster 1 exhibited high expression of Ncr1 and Tbx21 and are referred to here as ILC1s (Fig. 1B and fig. S1C). Clusters 2, 3 and 4 were identified as ILC2s based on their high expression of ILC2 signature genes, such as Gata3, Il1rl1 (encodes ST2), and Klrg1 (Fig. 1B and fig. S1C). Compared with cluster 2 (ILC2a) and cluster 3 (ILC2b), cluster 4 (ILC2c) was enriched in genes related to ILC2 activation and function, including Areg (encodes amphiregulin), Il5, Il13, Klrg1 and Pdcd1, thus reflecting the presence of activated ILC2s (Fig. 1, C and D). Of note, cluster 4 scored highest in a module score of wound healing genes and most probably corresponds to a wound healing subset (ILC2c) (Fig. 1E). The proportion of ILC2c was increased after influenza virus infection (Fig. 1F) and this subset demonstrated the highest proliferation module score as well (fig. S1D), suggesting that the induction and function of ILC2s play an essential role in promoting the resolution phase. It is apparent that Influenza virus infection leads to the generation of distinct ILC2 subsets (fig. S2A) that preferentially upregulated genes associated with ILC2 activation, including Il5, Il13, Pdcd1, Arg1, Klrg1, and Areg, as well as wound healing module scores at the resolution phase (fig. S2, B to D). Intriguingly, we found that transcription factor BATF was progressively expressed and enriched in the wound healing ILC2c subset (fig. S2, B and C). This finding suggests that BATF is likely to play an important role in supporting ILC2 proliferation, differentiation and function following virus-induced activation.
Fig.1. scRNA-seq analysis of pulmonary innate lymphoid cells responding to influenza viral infection.
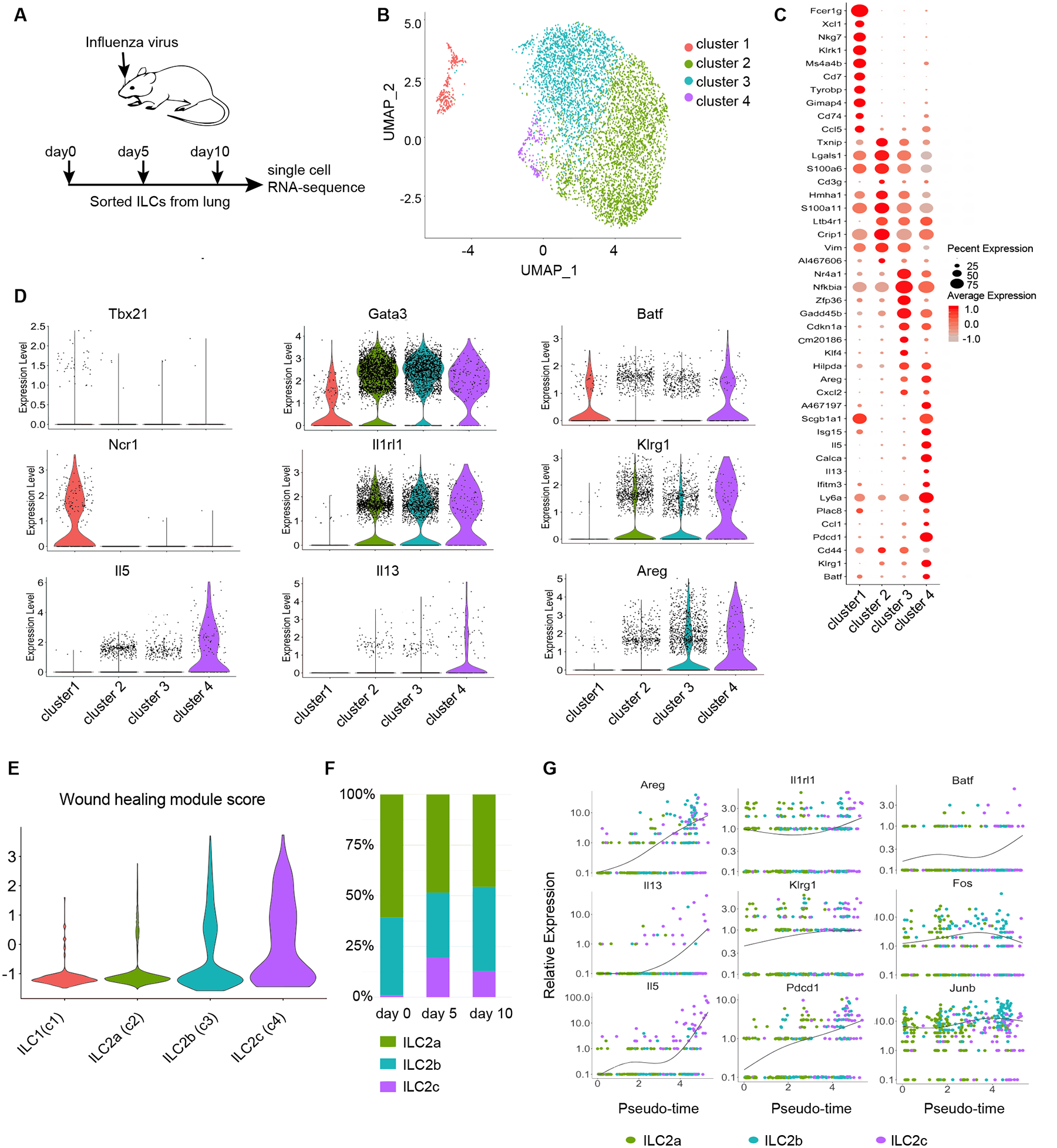
(A) Experimental design for single-cell RNA-sequencing of ILCs sorted from lungs of influenza virus infected mice. (B) UMAP plot of scRNA-seq data of lung-derived ILCs from three timepoints of influenza virus infection. Cells are colored by cluster. Day 0 Data from GEO: GSE102299 (26). (C) Representative differentially expressed genes between clusters. Dot size represents the percent of cells in a cluster expressing the gene and color shows the average relative expression of the gene. (D) Violin plots displaying distinct patterns of expression across clusters of key differentially expressed genes, including transcription factors, cell surface markers, and function genes across clusters. (E) Violin plots showing distribution of wound healing scores by cluster. The gene sets were taken from supplemental table 1 of a previously published study (3). (F) Proportions of ILC2 subsets at different timepoints of influenza virus infection. (G) Pseudotime analysis of expression of prominent genes. Cells are colored by cluster.
To further validate that BATF induction was associated with ILC2 subset formation, we explored the developmental relationship between ILC2s by employing pseudotime trajectory analysis (Fig. 1G). Indeed, Batf, as well as the activated ILC2-associated genes Il1rl1, Areg, Il5, Il13, Klrg1 and Pdcd1 exhibited progressive up-regulation along the wound healing ILC2c differentiation trajectory, whereas the expression trends of other bZIP family TFs, such as Fos and Junb remained largely unchanged (Fig. 1G). In addition, RNA Velocity analysis further indicated the activated ILC2s subsets were potentially derived from resting ILC2s following influenza virus infection (fig. S2E). Together, our single cell transcriptomics, pseudotime trajectory and RNA Velocity analyses suggest that during influenza virus infection, BATF expression is required for ILC2 activation and for the acquisition of wound healing function.
BATF deficiency in ILCs results in sustained inflammation and impaired tissue repair in the lung after Influenza virus infection
It is known that Zbtb16 (which encodes PLZF) is highly expressed in common helper innate lymphoid progenitors (CHILP) (27). To test whether BATF plays an intrinsic role in ILC development, we crossed mice carrying floxed Batf alleles (Batffl/fl) to mice expressing a Cre recombinase/green fluorescent protein (eGFP) fusion protein from the Zbtb16 locus (PLZF-Cre), thereby generating mice with a conditional knockout of BATF (cKO, Batffl/fl PLZF-Cre) in ILCs. This strategy should result in the loss of BATF in all mature ILC subsets, including ILC1s, ILC2s and ILC3s. Intracellular staining confirmed efficient ILC-specific (including ILC2-specific) deletion of BATF in the lungs of these mice (fig. S3A). Notably, when compared to Batffl/fl (hereafter referred to as wild type, WT) littermate controls, cKO mice had unaltered numbers of common lymphoid progenitors (CLP), CHILP, ILC2 progenitors (ILC2P), as well as ILC2s in bone marrow, but reduced total numbers of ILC2s in the lung (fig. S3, B to D). Furthermore, we found that BATF expression positively correlated with ST2 and GATA3 expression in Lin−CD90.2+ cells in the lung (fig. S3, E to H), indicating that BATF may positively maintain ILC2 lineage identity in the lung. Next, we found that the total numbers of ILC2s in the small intestine lamina propria (SI) and large intestine lamina propria (LI) were also significantly decreased in the cKO mice, whereas, with the exception of reduced number of lymphoid tissue inducer (LTi) cells in the SI, all other ILC populations in the intestine (e.g., ILC1s, ILC3s and LTi cells) were not affected (fig. S3, I to L). Moreover, the percentage and cellular number of other lymphocytes, including T cells, B cells, NK cells, NKT cells and γδ T cells, were not greatly affected in cKO mice (fig. S4, A to F). Together, these data indicated that BATF does not affect the cellularity of ILC2 progenitors in the bone marrow of BATF cKO mice, but is selectively critical for the homeostasis of ILC2s in peripheral tissues.
To further examine whether BATF deficiency affects ILC2 responses after exposure to influenza virus infection, Batffl/fl and cKO mice were infected with a sublethal dose of influenza PR8 virus (50 PFU) and monitored for weight loss and survival over 14 days. Compared to the transient weight loss in Batffl/fl mice, cKO mice exhibited continuous weight loss even after day 9, and close to 80% of the animals succumbed to infection within 14 days (Fig. 2, A and B). The increased susceptibility of cKO mice to viral infection at the resolution phase may be due to their compromised capacity for tissue repair. As expected, loss of BATF in ILCs significantly impaired epithelial integrity as measured by elevated red blood cell leakage in the bronchoalveolar lavage fluid (BAL), and by increased myeloid cell infiltration at day 14 post infection (Fig. 2, C to E). In agreement with these observations, histological examination demonstrated severe lung inflammation in cKO mice when compared to Batffl/fl mice, which was characterized by extensive tissue damage and lymphocyte infiltration at both day 9 and day 14 (Fig. 2, F to G). Specifically, cKO mice displayed a persistent increase in the percentage of neutrophils (Neu), inflammatory monocytes (iMon) and inflammatory macrophages (iMac) over time (fig. S5, A to C) and a decrease in the percentage of cells associated with the resolution of inflammation and tissue repair, including alveolar macrophages (aMac) (28) and eosinophils (Eos) (29) (fig. S5, A to C). Moreover, cKO mice exhibited a slight delay in virus clearance, but had comparable numbers of tetramer-positive CD8+ T cells in the lung to Batffl/fl mice (Fig. 2, H and I). These observations promoted us to assess if alterations in tissue repair genes could be detected in the lungs of Batffl/fl and cKO mice. It is well established that, upon IL-33 stimulation, ILC2s secrete amphiregulin for tissue repair during the resolution phase of influenza infection (3). Indeed, we found that Il33 was upregulated in the lungs of both Batffl/fl and cKO mice. However, the expression of Areg was markedly decreased in cKO mice compared with their Batffl/fl counterparts (Fig. 2J), suggesting that the susceptibility of cKO mice to tissue damage may be corelated with the impaired tissue repair function of ILC2s. Collectively, these data demonstrate that BATF deficiency in ILCs causes prolonged inflammation, impaired tissue repair in the lung, and increased susceptibility to influenza virus infection.
Fig. 2. BATF deficiency in ILCs results in susceptibility to Influenza virus Infection.
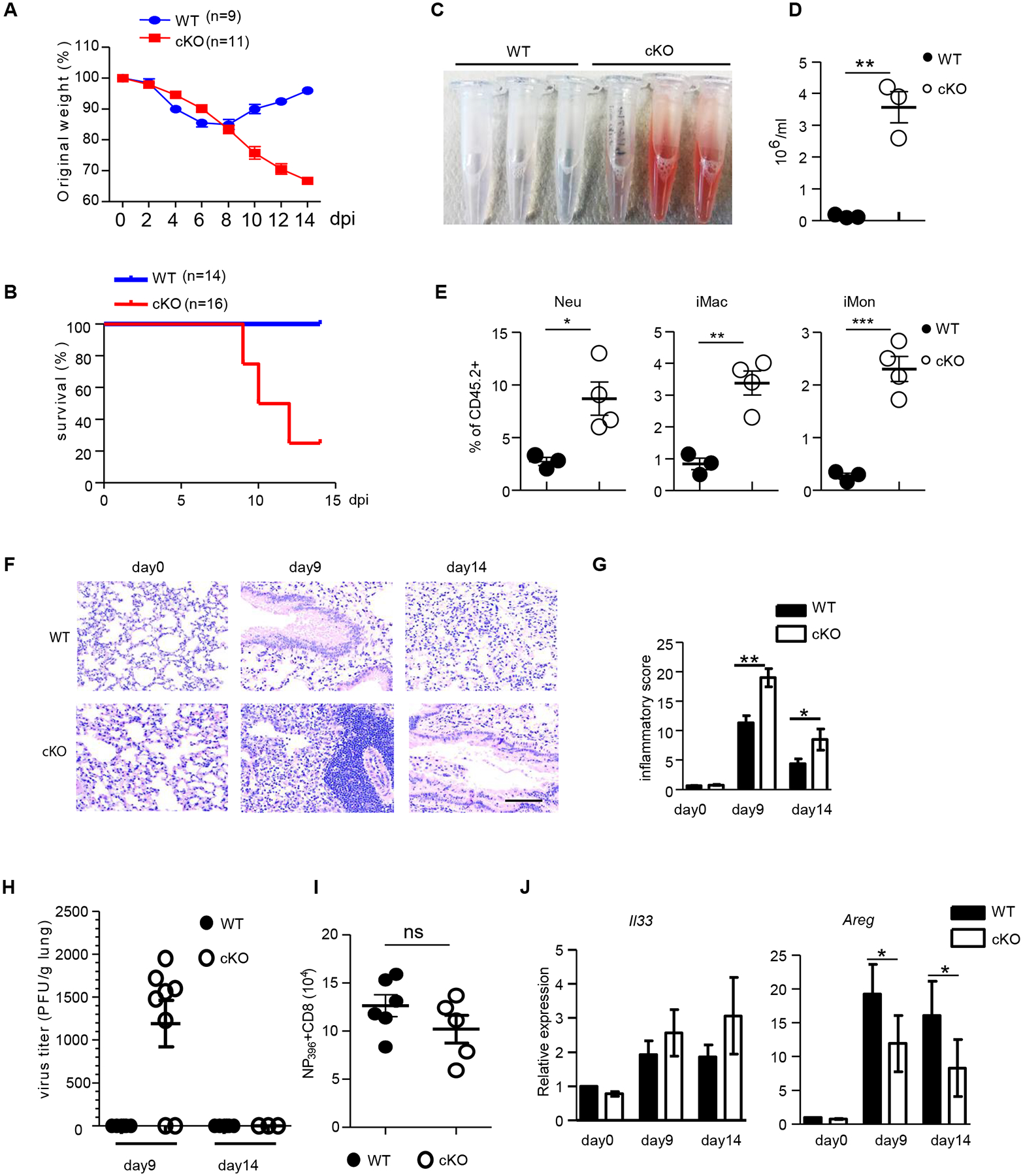
(A and B) Batffl/fl (WT) and Batffl/fl PLZF-cre (cKO) mice were infected with sublethal PR8 influenza virus (50 PFU) and monitored for survival (A) and body weight changes (B) for two weeks after infection. (C-D) BAL was harvested at day 14 from lungs of Batffl/fl (WT) and BATF cKO mice given sublethal PR8 influenza infection. Visual appearance (C) and numbers of red blood cells (D) in BAL fluid are shown. (E) The frequency of neutrophils (CD45.2+CD11b+Ly6G+), inflammatory macrophages (CD45.2+CD11b+CD11c−F4/80+Ly6C+), and inflammatory monocytes (CD45.2+CD11b+F4/80−Ly6C+) in the BAL fluids of WT and cKO mice infected with sublethal PR8 virus infection at day 14 (n=3–4 mice per group). (F and G) Lung histology of Batffl/fl (WT) and cKO mice infected with sublethal PR8 influenza at the indicated days (F) and inflammatory score of the lungs (G). Scale bar, 100 μm. (H) Tissue virus titers in the lungs of Batffl/fl (WT) and BATF cKO mice infected with sublethal PR8 influenza. (I) Quantified total cell number of NP396 tetramer positive CD8+ T cells in the lung of Batffl/fl (WT) and cKO mice at day 9 after influenza virus infection. (J) qRT-PCR analysis of mRNA expression of Il33 and Areg in the lung homogenates after sublethal PR8 virus infection at the indicated days. Data shown as the mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001 (two-tailed unpaired t-test). Each dot represents one mouse. Data are pooled from three (A) or five (B) or two (I) independent experiments or are representative of at least two independent experiments.
Lung ILC2 expansion and function are impaired in influenza-infected BATF cKO mice
We next sought to investigate whether the prolonged inflammation and delayed pulmonary restoration and tissue repair after influenza virus infection in BATF cKO mice were associated with diminished numbers and/or a functional defect of pulmonary ILC2s. Utilizing fluorescent antibody labeling in vivo, we observed very few ILC2s were labeled when compared with influenza virus-specific, tetramer-positive CD8+ T cells (fig. S6A). Moreover, ILC2s, not iILC2s, predominated in the lung (fig. S6B). All these data indicate that most ILC2s are resident in the lung after influenza virus infection, which is not the case for circulating iILC2s, as published previously (24, 30). Furthermore, there was a significant reduction in the frequency and overall number of ILC2s in the lung and mediastinal lymph nodes of cKO mice when compared to littermate controls at both day 9 and day 14 post influenza virus infection (Fig. 3, A and B, and fig. S6, C and D). Conversely, the total cell number, but not frequency of ILC1s, was increased modestly in cKO mice (fig. S6, E and F), suggesting that BATF is selectively required for the differentiation or maintenance of ILC2s, but not ILC1s, during influenza infection. Furthermore, ILC2s from cKO mice exhibited significantly reduced expression of the cell proliferation marker Ki-67 (Fig. 3, C and D) as well as IL-5, IL-13 and amphiregulin production (Fig.3, E to H). Accordingly, the protein levels of IL-5, IL-13 and amphiregulin in BAL were all significantly decreased in cKO mice (Fig. 3I). Moreover, compared to Batffl/fl ILC2s, cKO ILC2s exhibited impaired IL-33-mediated upregulation of amphiregulin expression in a dose-dependent manner in vitro (fig. S6, G and H), supporting the notion that IL-33-induced amphiregulin expression in ILC2s is BATF-dependent.
Fig. 3. Impaired lung ILC2 expansion and wound-healing function in virus-infected BATF cKO mice.
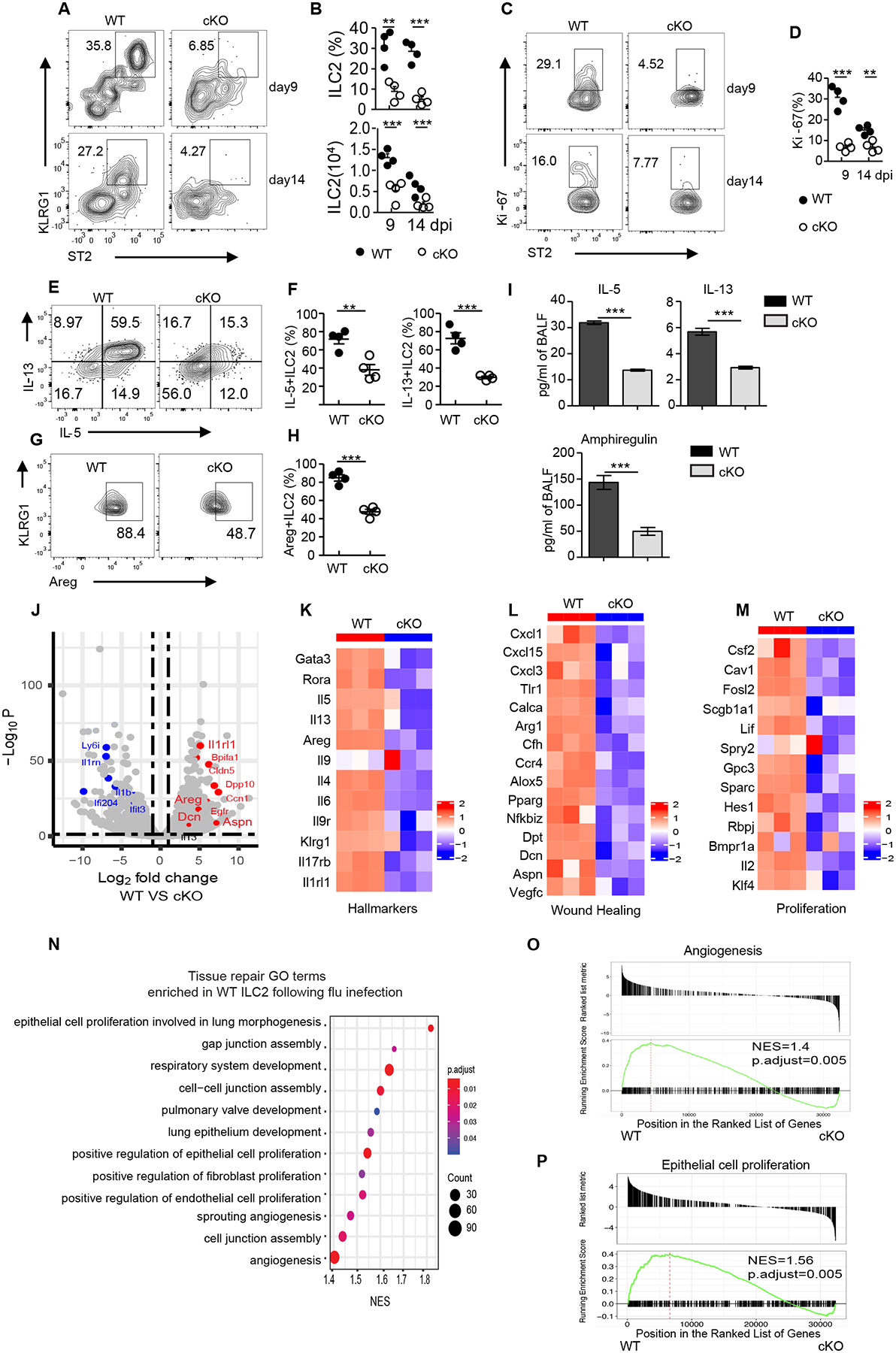
(A) Representative flow cytometry plot of ILC2s in the lungs of Batffl/fl (WT) or BATF cKO mice given sublethal PR8 virus infection at the indicated days, data pre-gated on Lin−CD90.2+. (B) Frequency and numbers of lung ILC2s in (A). (C) Representative flow cytometry plot showing Ki-67 expression in lung ILC2s as assessed in (A), numbers represent percentage of Ki-67+ cells in each gate. (D) Frequency of Ki-67+ ILC2s in (C). (E) Representative flow plots of IL-5- and IL-13-expressing ILC2s (pre-gated on Lin−CD90.2+ KLRG1+) isolated from lung after influenza infection at day 9. (F) Frequency of IL-5+ (left) and IL-13+ (right) ILC2s in (E). (G) Amphiregulin expression of lung ILC2s (pre-gated on Lin−CD90.2+ KLRG1+) after stimulation with PMA plus ionomycin ex vivo. (H) Frequency of Areg+ ILC2s in (G). (I) Protein level of IL-5, IL-13 and amphiregulin in BAL from Batffl/fl (WT) (n=6) and cKO (n=6) after influenza virus infection at day 9. Data are combined from two independent experiments. (J) Volcano plot comparing differentially expressed genes from Batffl/fl (WT) versus cKO ILC2s sorted from lungs after influenza infection on day 9 and subjected to bulk RNA sequencing. Red and blue dots represent upregulated and downregulated genes, respectively, in WT mice. (K-M) Heatmaps of ILC2 hallmark genes (K), wound healing genes (L), and proliferation-related genes (M) from Batffl/fl (WT) versus cKO ILC2s. (O) Selective list of tissue repair Gene Ontology pathways enriched in Batffl/fl (WT) ILC2s. The pathways presented in the plot are significantly enriched (Benjamini-Hochberg adjusted p < 0.05), with the size and color of each dot indicating the number of genes in a pathway and the adjusted p-value, respectively. (O and P) GSEA of selected pathways enriched in Batffl/fl (WT) ILC2. (A-H) Data shown as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001 (two-tailed unpaired t-test). Each dot represents one mouse (n=4 per group). Data are representative of at least two independent experiments.
To further assess how BATF deficiency affects ILC2 gene expression, bulk RNA sequencing analyses were performed on ILC2s sorted from the lungs of Batffl/fl and cKO mice at day 9 after influenza virus infection. Loss of BATF significantly altered gene expression (1998 upregulated and 3211 downregulated) in cKO ILC2s (log2 fold change > 1.5; false discovery rate test, p < 0.01) (Fig. 3J). Consistent with our flow cytometry results, ablation of BATF in ILC2s also led to substantially reduced expression of ILC2 hallmark genes, including Gata3, Rora, Il1rl1, Klrg1, Il5, Il13, Il4, Il9r and Areg (Fig. 3K). Moreover, a survey of genes known to be involved in wound healing and cell proliferation programs, most of which have been previously reported to be upregulated in ILC2s responding to influenza infection (3) showed that wound healing genes (Areg, Cxcl3, Calca and Pparg), tissue remodeling genes (Dpt, Dcn and Aspn), and known proliferation genes (such as Csf2, Cav1 and Fosl2) were all dramatically down-regulated in the absence of BATF (Fig. 3, J, L and M). As expected, Gene Ontology (GO) pathway and gene set enrichment analysis (GSEA) analysis indicated that Batffl/fl ILC2s were enriched for genes implicated in tissue repair processes, such as angiogenesis, extracellular matrix organization, cell-cell junction assembly, and epithelial cell proliferation (Fig. 3, N to P). Next, we examined the molecular pathways using KEGG and GSEA analysis. As anticipated, genes involved in the PI3K-Akt signaling pathway that is associated with cell proliferation, as well as genes involved in the Wnt, Hippo and TGFβ pathways (31–33) that play positive roles in tissue repair and wound healing, were enriched in Batffl/fl ILC2s. By contrast, inflammatory related pathways, such as NF-κB and TNF signaling were significantly enriched in cKO ILC2s (fig. S6, I and J). Taken together, these data indicate that BATF stabilizes ILC2 identity by promoting cell proliferation and maintaining the wound healing function of these cells during influenza virus infection.
Amphiregulin fails to rescue BATF cKO mice following influenza virus infection
A previous study demonstrated that administration of recombinant amphiregulin (rAreg), but not rIL-13, is sufficient to restore lung function in ILC-depleted Rag1−/− mice after influenza virus infection (3). This prompted us to test whether delivery of rAreg could play similar role in influenza virus-infected BATF cKO mice (fig. S7A). After sublethal influenza virus infection, cKO mice receiving rAreg exhibited similar rates of weight loss and survival compared to a PBS-treated control group (fig. S7, B and C), suggesting that replenishment of rAreg is not sufficient to restore lung tissue homeostasis in cKO mice following influenza virus infection.
Batf−/− ILC2s produce IL-17A and promote an inflammatory response in the lungs during influenza virus infection
Given that administration of rAreg did not substantially restore lung tissue homeostasis in cKO mice and that Batf−/− ILC2s displayed loss of expression of ILC2 signature genes in influenza infected-cKO mice, we hypothesized that BATF might control ILC2 plasticity in response to influenza infection. In support of this, various genes associated with the IL-17 signaling pathway, including Il17a, Il23r, Il12rb, Il6ra and Il1b, were found to be upregulated in Batf−/− ILC2s (Fig. 4A). Consistently, the IL-17 signaling pathway was significantly enriched in Batf−/− ILC2s (Fig. 4B). Given that IL-17A-producing ILC2s have been reported by several groups (34–36), we sought to determine if BATF affects the level of IL-17A production in ILC2s during influenza virus infection. To do this, ILC2s were sorted from the lungs of Batffl/fl and cKO mice on day 9 after influenza infection and IL-17A expression was assessed by intracellular staining. As expected, IL-17A was rarely detected in Batffl/fl ILC2s, but was found to be robustly expressed in BATF cKO ILC2s (Fig. 4, C to E). Of note, IL-17A+ ILC2s from BATF cKO mice co-expressed IFNγ, but not amphiregulin or IL-5 (Fig. 4, C and D). Taken together, these results suggest that Batf−/− ILC2s display a pathogenic ILC3-like phenotype, as IL-17A is known to promote neutrophil migration under inflammatory conditions. Indeed, the cKO mice exhibited higher neutrophil infiltration in both the BAL fluid and lung parenchyma at various time points after influenza infection (Fig. 2E and fig. S5C). Furthermore, numerous inflammatory responses GO terms were found enriched in Batf−/− ILC2s following influenza infection, including those associated with positive regulation of T cell mediated immunity, positive regulation of neutrophil migration and acute inflammatory response (Fig. 4F). Besides increased IL-17A production, the cKO ILC2s also broadly upregulated chemokines promoting monocyte and neutrophil migration (37, 38), such as Ccl2, Ccl22, Ccl24, Cxcl10 and Cxcl16 (Fig. 4G), and exhibited a significantly enriched neutrophil chemotaxis pathway (Fig. 4H). These results collectively demonstrate that ablation of BATF in ILC2s causes the increased formation of pathogenic ILC2s with elevated inflammatory cytokine production, which in turn promotes neutrophils infiltration and exacerbates the tissue damage associated with influenza virus infection.
Fig. 4. Batf −/− ILC2s display IL-17A producing inflammatory ILC3-like phenotype after influenza infection.
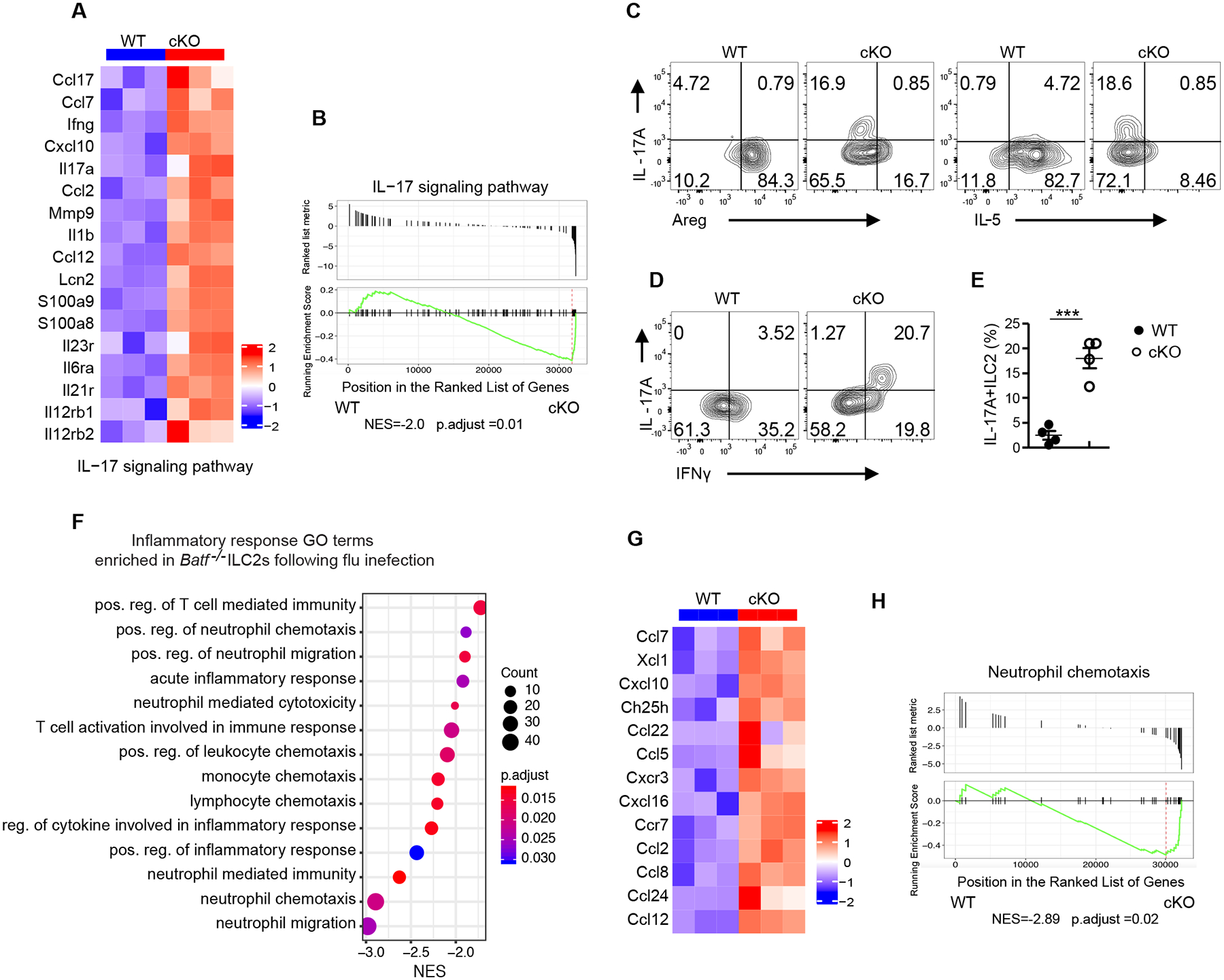
(A) Heatmap of genes encoding for the IL-17 signaling pathway from WT versus cKO ILC2s sorted from lung after influenza infection at day 9. (B) GSEA plot of the IL-17 pathway, which was enriched in BATF cKO ILC2s. (C-D) Representative flow plot of intracellular staining of IL-17A vs Areg, IL17A vs IL-5 (C), and IL-17A vs IFNγ in ILC2s (pre-gated on Lin−CD90.2+ KLRG1+) (D). (E) Quantification of plots in (D). (F) Selected list of inflammatory response Gene Ontology pathways enriched in cKO ILC2s. The pathways presented in the plot are significantly enriched (Benjamini-Hochberg adjusted p < 0.05), with the size and color of each dot indicating the number of genes in a pathway and the adjusted p-value, respectively. (G) Heatmaps of genes involved in the GO positive regulation of neutrophil migration. (H) GSEA plot of the positive regulation of neutrophil chemotaxis pathway, which was enriched in cKO ILC2s. (C-E) Data shown as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001 (two-tailed unpaired t-test). Each dot represents one mouse (n=4 per group). Data are representative of at two independent experiments.
BATF supports chromatin accessibility of wound healing genes in ILC2s
A previous study demonstrated that the binding motif for BATF was enriched in chromatin regions uniquely accessible in pulmonary ILC2s (22). Thus, we hypothesized that BATF may specifically promote chromatin remodeling in ILC2s upon activation. To test this, we compared chromatin accessibility in Batffl/fl and Batf−/− ILC2s sorted from lungs at day 9 after influenza virus infection using assay for transposase-accessible chromatin using sequencing (ATAC-seq) (fig. S8A). Although we observed that over 50% of the open regions (6627 peaks) were shared, 6390 and 4877 peaks were distinctly open in WT and Batf−/− ILC2s respectively (Fig. 5A). Moreover, the distribution of open chromatin regions across the genome was largely unchanged between Batffl/fl (WT) and Batf−/− ILC2s (Fig. 5B). Next, we compared the differentially open peaks located in the enhancer and promoter regions, respectively, and observed that Batffl/fl (WT) ILC2s exhibited more chromatin accessibility of wound healing related genes, including Il1rl1, Areg, and Il13 (Fig. 5C and fig. S8B). On the other hand, chromatin of the ILC3 signature genes, Il23r and Il12rb2, exhibited accessibility exclusively in Batf−/− ILC2s (Fig. 5C and fig. S8B). These results agree with our RNA-seq analysis, and further support a pioneer function of BATF in promoting the accessibility of chromatin in ILC2 signature genes loci after infection with influenza virus. Since we observed key differences in the epigenetic landscape induced by viral infection in Batffl/fl (WT) versus Batf−/− ILC2s, we next sought to investigate the potential binding sites open to transcriptional regulators that might contribute to these differences. HOMER de novo motif analysis identified BATF as the top enriched motif in Batffl/fl (WT) ILC2s, supporting that BATF is directly responsible for chromatin accessibility (Fig. 5, D and E). Moreover, known motif analysis revealed regions more accessible in Batffl/fl (WT) compared to cKO ILC2s that were enriched for consensus bZIP (71%) binding motifs, confirming the established role of bZIP transcription factors (e.g., BATF, Fra1, JunB and AP-1) in ILC2 activation and effector function (fig. S8, C to E). By contrast, de novo motif enrichment in cKO ILC2s ranked highest for Ets1 (Fig. 5, F and G), which has been shown to promote pro-inflammatory immunity mediated by nature killer (NK) cells and iILC2s (39, 40). Remarkably, Genomic Regions Enrichment of Annotation Tools (GREAT) analysis showed that Batffl/fl (WT) and cKO ILC2s distinctly enriched differential GO (Gene Ontology) biological processes pathways. Cell-cell adhesion is a key process employed by macrophages (41) and mucosal-associated invariant T (MAIT) cells (42) to promote tissue repair. Consistently, positive regulation of cell adhesion pathways was observed in Batffl/fl (WT) ILC2s, whereas Batf−/− ILC2s displayed negative regulation of cell-cell adhesion at enhancer regions (Fig. 5H) and negative regulation of wound healing at promoter regions (fig. S8F).
Fig. 5. BATF supports chromatin accessibility of wound-healing gene loci in ILC2s.
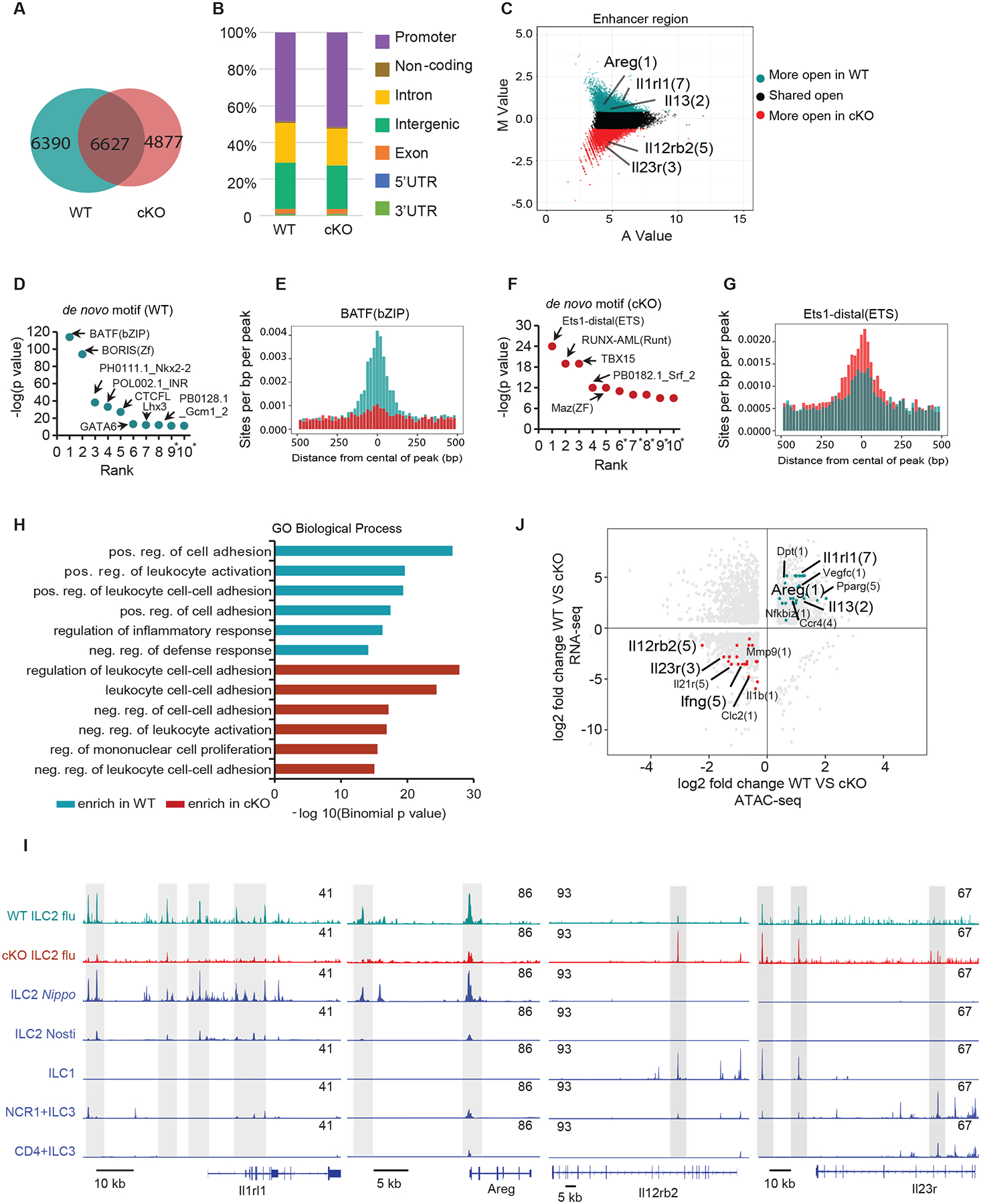
(A) Venn diagrams showing overlap of ATAC-seq peaks identified in ILC2s sorted from lungs of Batffl/fl (WT) (dark green) and BATF cKO (dark red) mice after influenza infection at day 9. (B) Distribution of ATAC-seq peaks across the genome in WT and cKO ILC2s. (C) Scatter plot (MA plot) showing M value (log2(read density in WT ÷ read density in cKO)) vs A value (0.5 × log2(read density in WT × read density in cKO)) of the merged set of Batffl/fl (WT) and cKO enhancer ATAC-seq peaks after normalization. The top 5,000 peaks are highlighted for Batffl/fl (WT) (dark green) and cKO (dark red) ILC2 cells. Selected genes are labeled and with the number of enhancer peaks in parentheses. (D) Transcription factor binding motifs significantly enriched (top 10, p < 10−10) in WT ILC2 cells and plotted based on their rank in the top 5,000 Batffl/fl (WT) specific enhancers. (E) The distributions of BATF binding motifs in the top 5,000 WT- and cKO-specific enhancers (dark green and dark red, respectively). Regions shown are +/− 500 bp from the center of each enhancer. (F) Transcription factor binding motifs significantly enriched (top 10, p < 10−10) in cKO ILC2 cells and plotted based on their rank in the top 5,000 cKO specific enhancers, * represent possible false positive. (G) The distributions of Ets1 binding motifs in the top 5,000 WT- and cKO-specific enhancers (dark green and dark red, respectively). Regions shown are +/− 500 bp from the center of each enhancer. (H) Gene ontology pathways of genes with differential chromatin accessibility between Batffl/fl (WT) (dark green) and cKO (dark red) was performed using the GREAT tool. The -log10(binomial p-value) is shown for the top 6 Molecular Signature Database pathways enriched in Batffl/fl (WT) and cKO-specific enhancers, with pathways of interest highlighted based on sample enrichment. (I) Accessibility of indicated loci in Batffl/fl (WT) and cKO ILC2s and other ILCs as determined by ATAC-seq. Data in blue from (22), GEO: GSE77695 and (43), GEO: GSE131996. (J) Relationship between differential accessibility and differential expression genes in Batffl/fl (WT) verses cKO ILC2s. The data represent the log2 ratio of normalized ATAC-seq signal compared to the log2 ratio of RNA expression for WT and cKO ILC2s. Only the top 5,000 WT- and cKO-specific non- promoter ATAC-seq peaks that are associated with genes that are significantly differently expressed between Batffl/fl (WT) and cKO ILC2s are included. Some notable genes with higher ATAC-seq signal and RNA expression in Batffl/fl (WT) ILC2s (dark green) or cKO ILC2s (dark red) are highlighted, with the number of enhancers in parentheses.
To obtain additional insight into the effects of influenza infection on ILC2, we compared our ATAC-seq peaks with previously published data on ILCs (22, 43) to evaluate the accessibility of some key signature genes. As expected, ILC2 hallmark genes, such as Il1rl1, Areg, Il15 and Il13, gained accessibility in activated Batffl/fl (WT), but not in Batf−/− ILC2s following influenza or ILC2s following Nippostrongylus brasiliensis (Nippo) parasite infection (Fig. 5I and fig. S8G). The IL-9-IL-9R axis has been reported to act as a unique and autocrine amplifier of ILC2 function to promote tissue repair in the recovery phase after helminth-induced lung inflammation (44), and we found that the accessibility of both of these genes was decreased in BATF deficient ILC2s (fig. S8G). In contrast, the inflammatory genes, such as Il23r, Il12rb2 and Ifng, acquired more accessibility in Batf−/− ILC2s than their Batffl/fl (WT) counterparts (Fig. 5I and fig. S8G). Moreover, BATF-dependent gains and losses in ATAC-seq signals were consistent with the differentially expressed (DE) genes in RNA-seq. Notably, among these DE genes, wound healing genes (such as Il1rl1, Areg, Pparg, Dpt and Vegfc) gained more accessibilities in WT ILC2s, whereas IL-17A inflammatory pathways genes (Il23r, Il12rb2, Il21r and Ifng) showed more open regions in Batf−/− ILC2s (Fig. 5J). Taken together, these data indicate that Batf-deficient ILC2s display a hybrid effector function as characterized by decreased chromatin accessibility in tissue repair related genes and increased accessibility for inflammatory genes, this profile strongly supports the notion that BATF maintains ILC2 function and lineage stability by directly regulating chromatin accessibility.
BATF promotes expression of tissue repair genes and represses inflammatory genes
To investigate BATF binding sites in ILC2s, we subjected lung ILC2s to chromatin sequencing using Cleavage Under Targets and Tagmentation (CUT&Tag). Strong enrichment for occupancy by BATF, but not control IgG, was noted within 1 kb of the central peaks observed across the whole genome of ILC2s (Fig. 6A). Inspection of genome-wide peak distribution revealed that the majority of BATF binding sites were enriched at intergenic regions, exons and introns (Fig. 6B). In addition, HOMER de novo motif analysis identified BATF as the top binding motif, which further validated the specificity of CUT&Tag sequencing (Fig. 6C). Next, we asked how many ILC2-relevant genes could be directly regulated by BATF. To quantify this, we combined CUT&Tag data with the differentially expressed genes (DEs) of RNA-seq datasets and found that 34% (1811 of 3408) of DE genes contained BATF binding sites, suggesting that these genes were directly regulated by BATF in ILC2 cells (Fig. 6D). These BATF-targeted genes included key ILC2 signature genes, wound healing genes and tissue remodeling associated genes, such as Il1rl1 and Areg, which were positively regulated by BATF, as well as the inflammatory genes, such as Il17a and Il21r, which were negatively regulated by BATF (Fig. 6E). As expected, the Il1rl1, Areg and Il17a loci were directly bound by BATF, but not IgG, further supporting that BATF exerts a direct effect on modulation of ILC2s signature genes expression (Fig. 6F). Since BATF has a dual role in regulating genes expression, we wondered if BATF co-bound distinct transcription factors to exert its effect on target genes regulation. To this end, we employed HOMER motif analysis and found that both the positively and negatively regulated genetic regions bound by BATF were enriched for bZIP family transcription factors (Fig. 6G). These data suggested that BATF preferentially heterodimerizes with other bZIP TFs to form activating, or suppressive transcription factor complexes in the overall context of ILC2s activation. Strikingly, the positively regulated genes were enriched for genes involved in cell projection morphogenesis, regulation of cell projection organization and positive regulation of epithelial cell proliferation, all of which are pathways involved in wound healing and tissue repair as reported previously (42, 45, 46). In contrast, genes negatively regulated by BATF were enriched for genes involved in the inflammatory response, myeloid leukocyte activation and neutrophil migration (Fig. 6H). Combining ATAC-seq with BATF-specific CUT&Tag analysis, we assessed the status of unique open enhancer peaks in WT or Batf−/− ILC2s. Over 24% (1576 of 6391) of peaks that lose accessibility in Batf−/− ILC2s (e.g., Il1rl1, Areg and Il13) showed BATF binding in the CUT&Tag-seq dataset, whereas 12% (594 of 4878) of peaks that gain accessibility in Batf−/− ILC2s (e.g., Il12rb2 and Ifnb) were bound by BATF (Fig. 6I). Overall, these results provide compelling evidence that BATF works as an epigenetic switch to govern ILC2 lineage stability and tissue repair function.
Fig. 6. BATF promotes expression of tissue repair genes and represses inflammatory genes.
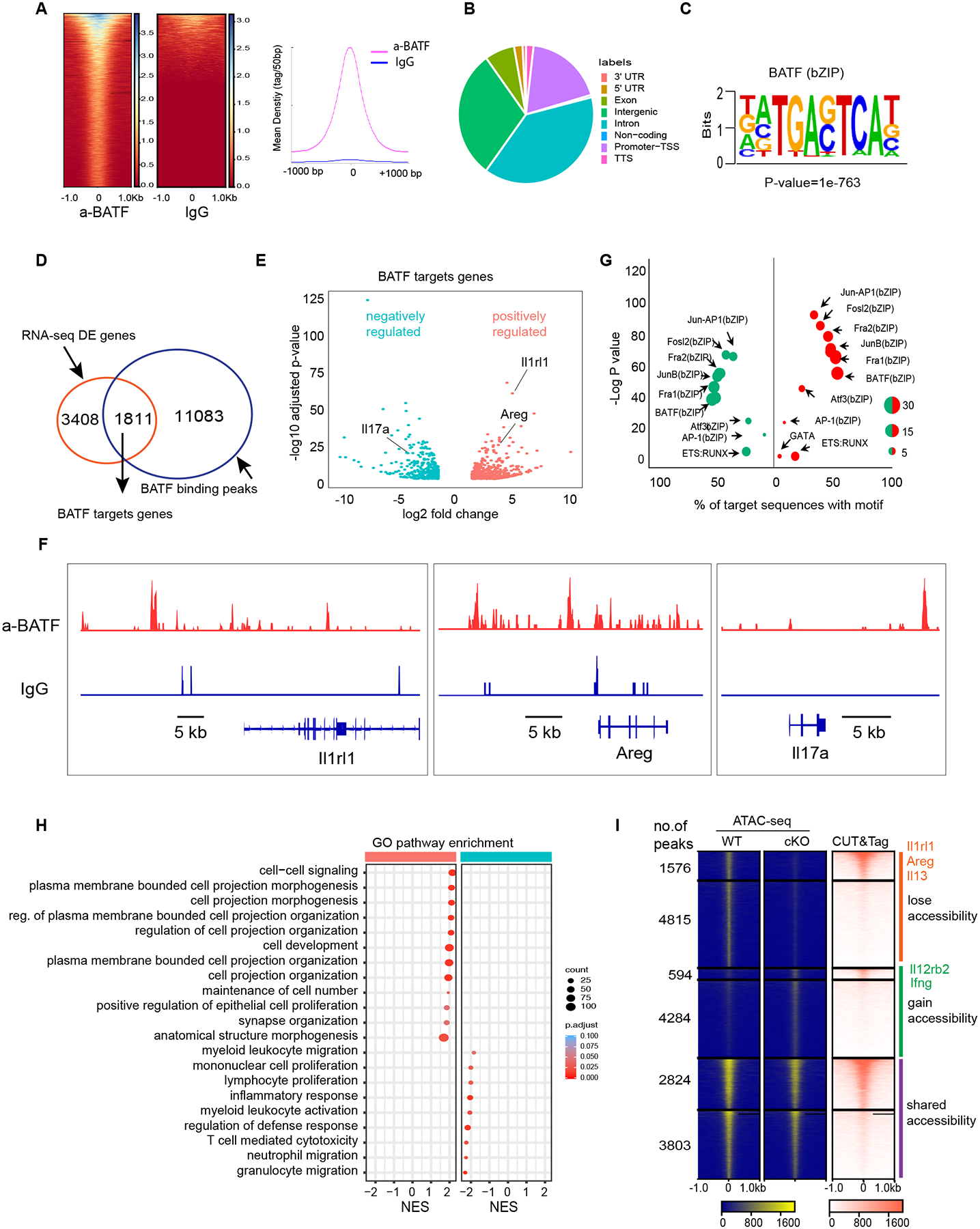
(A) Heatmap for genome-wide distribution of anti-BATF and rabbit control IgG binding signals at peak centers in ILC2s sorted from the lung of WT mice using CUT&Tag sequencing. (B) Distribution of BATF binding peaks across the genome of ILC2s. (C) HOMER de novo motif analysis of TF binding motif scores highest in BATF binding regions. (D) Venn diagram of BATF-regulated genes and BATF-bound genes. (E) Volcano plots of expression levels of BATF target genes in WT versus cKO ILC2s sorted from lungs after influenza infection at day 9. Red, increased expression in WT ILC2 cells. Green, reduced expression in WT, the represented genes are highlighted. (F) Representative alignments of CUT&Tag ChIP-seq data of BATF and IgG binding to the indicated loci in ILC2s. (G) List of top HOMER known motifs enriched in BATF binding peaks, size of dot indicate the percentage of no target sequence with motif, color of dot indicate BATF positively (red) or negatively (green) regulated as in (E). (H) Selective list of Gene Ontology pathways enriched in WT and cKO ILC2s based on (E). The pathways presented in the plot are significantly enriched (Benjamini-Hochberg adjusted p < 0.05), with the size and color of each dot indicating the number of genes in a pathway and the adjusted p-value, respectively. (I) Heatmap of all atlas peaks of ATAC-seq of enhancers in WT and cKO ILC2s, including peaks that gain and lose accessibility between WT and cKO ILC2s and those in common between the two (left and middle panels), the right panel shows heatmap of BATF binding peaks according to atlas peaks in the left and middle panels.
BATF promotes ILC2 function via IL-33-BATF-ST2 feed-forward loop
The IL-33-ST2 axis plays a major role in ILC2 activation and our finding that BATF binds directly to the Il1rl1 locus (encodes ST2) (Fig. 6F) prompted us to test whether IL-33 affects the expression of BATF in ILC2s. To do this, we sorted ILC2s from lung and cultured them in the presence of different doses of recombinant IL-33. Consistent with a previous report demonstrating that IL-33 upregulated BATF expression in gut ILC2s (26), we found that BATF expression in pulmonary ILC2s was increased upon IL-33 stimulation in a dose-dependent manner (Fig. 7A). In addition, we examined the expression of BATF in ILC2s following exposure to IL-33 in vivo. To do this, IL-33 was administered to mice via intraperitoneal injection for 3 continuous days. As expected, IL-33 induced ILC2 development in the lungs and bone marrow when compared with a control, PBS injection group. However, BATF deficiency significantly attenuated the expansion of ILC2s in response to IL-33 stimulation (Fig. 7, B and C, and fig. S9, A and B). Of note, BATF, KLRG1 and ST2 expression in WT (Batffl/fl) ILC2s increased in response to IL-33 but this was not observed in cKO mice (Fig. 7, D and E, and fig. S9, C and D). The expression of the ILC2 activation marker amphiregulin was reduced in the cKO mice compared to control mice (Fig. 7, D and E). Moreover, the ability of IL-5 and IL-13 cytokine secretion of lung ILC2s in cKO mice was impaired (Fig. 7, F and H). In addition, we observed a reduction of proliferation marker Ki-67 positive ILC2s in cKO mice compared with control mice (Fig. 7, I and J, and fig. S9, E and F). To further validate if the observed IL-33-mediated proliferation of ILC2s is BATF-dependent and cell intrinsic, lung ILC2s were sorted, stained with the proliferation dye CellTrace Violet (CTV), and cultured in vitro for 2 days. As expected, when levels of CTV staining were compared, a high proportion of the ILC2s from WT (Batffl/fl) mice showed a significantly lower level of CTV intensity (Fig. 7K), indicating proliferation and expansion of ILC2s is BATF-dependent.
Fig. 7. BATF promotes ILC2 function via the IL-33-BATF-ST2 axis.
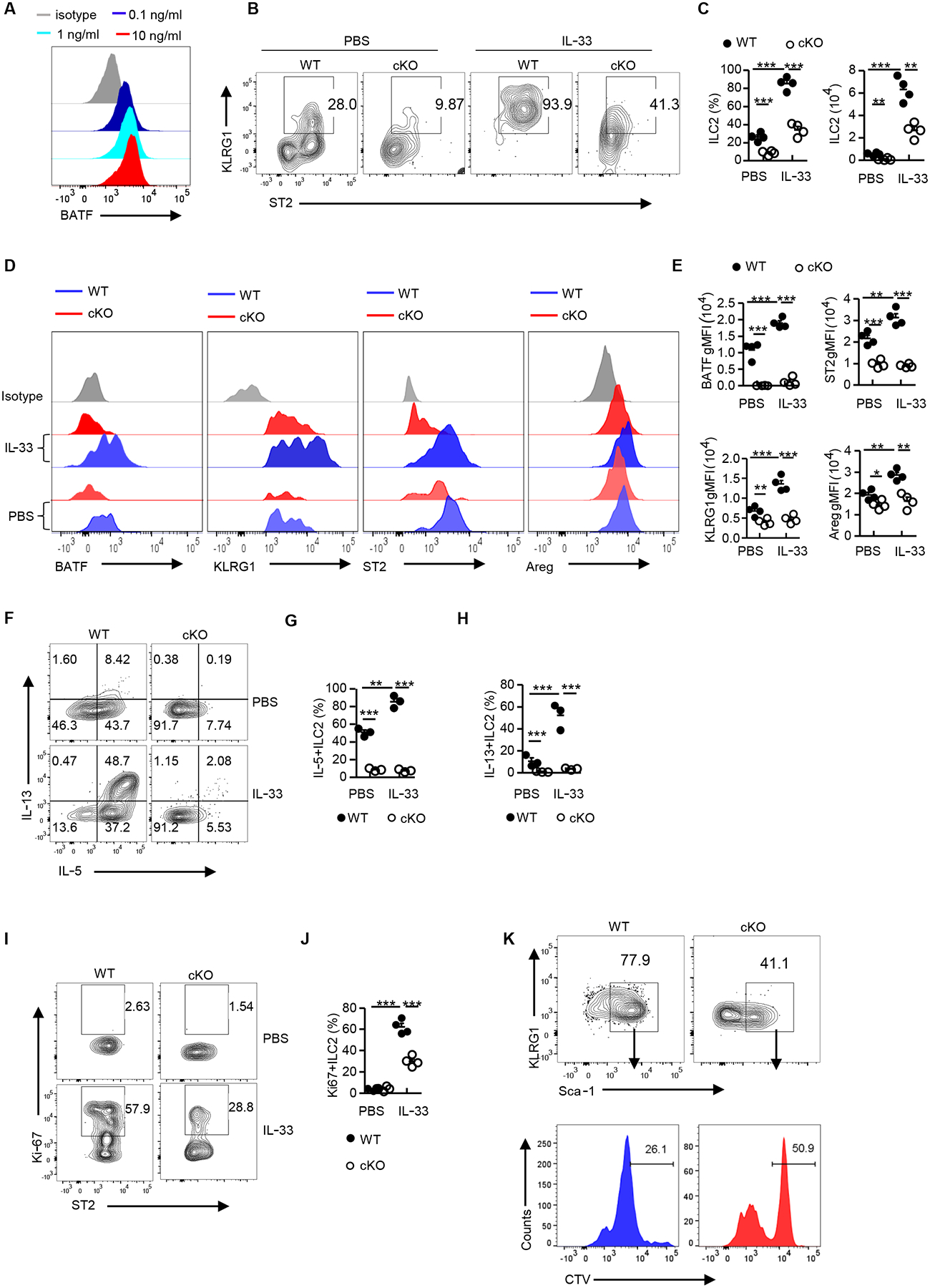
(A) Flow cytometry analysis of BATF expression in ILC2s (pre-gated on Lin−CD90.2+ST2+) sorted from Batffl/fl (WT) mouse lungs and cultured in vitro with different doses of IL-33 for 2–3 days. Histograms are representative of two independent experiments. (B) Representative flow plot showing lung ILC2s in Batffl/fl (WT) and cKO treated with PBS or IL-33, pre-gated on Lin−CD90.2+ cells. (C) Frequency and number of lung ILC2s in (C) (n=4 per group). (D) Flow cytometry analysis of BATF, KLRG1, ST2 and Areg expression in ILC2s (pre-gated on Lin−CD90.2+ST2+) as assessed in (B). (E) Quantification of BATF, KLRG1, ST2, and Areg gMFI in (D) (n=4 per group). (F) Flow cytometry analyzing intracellular levels of IL-5 and IL-13 from lung mILC2s (pre-gated on Lin−CD90.2+ KLRG1+) as in (B) but following stimulation with PMA plus ionomycin ex vivo. (G-H) Frequency of IL-5+ (G) or IL-13+ (H) lung ILC2s in (F) (n=4 per group). (I) Flow cytometry analysis of Ki-67 expression in lung ILC2s as in (B). Numbers in flow plots represent the percentage of Ki-67+ cells in each gate. (J) Frequency of Ki-67 positive ILC2s in (I) (n=4 per group). (K) FACS analysis of lung ILC2s sorted from Batffl/fl (WT) and cKO mice as indicated in (A), labeled with CTV, and cultured in vitro for 2–3 days. Data shown as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001 (two-tailed unpaired t-test). Each dot represents one mouse. Data are representative of at least two independent experiments.
A recent study has found that BATF is also critical for ILC development (23). To further analyze the role of BATF specifically in mature ILC2s (mILC2s), we generated Batffl/fl ERT2CreRosamTmG mice by crossing Batffl/fl with ERT2CreRosamTmG color tracing mice. In this model, administration of tamoxifen (TAM) leads to nuclear accumulation of Cre recombinase and concomitant deletion of the floxed Batf locus as well as the floxed reporter locus. This causes Batf-sufficient cells to gain green fluorescent protein (GFP) expression once Cre recombinase is activated and the cells become Batf-deficient. We sorted pulmonary mILC2s from Batffl/fl ERT2CreRosamTmG mice and cultured then with TAM (4-Hydroxytamoxifen, 4-OHT) to induce BATF deletion (fig. S10, A to C). After stimulating the cells with IL-33 for expansion, the expression of BATF in GFP+ ILC2s was dramatically decreased as compared to Batf-replete GFP− ILC2s (fig. S10D). Moreover, loss of BATF significantly reduced ST2 and amphiregulin expression (fig. S10, E and F), as well as the chromatin accessibility in the Il1rl1 (encode ST2) and Areg loci by ATAC-seq analysis (fig. S10, G and H). Collectively, these data indicate that IL-33-induced BATF expression in ILC2s maintains ST2 levels for ILC2 activation and proliferation via an IL-33-BATF-ST2 forward feedback loop, which in turn further promotes ILC2 lineage stability and functionality.
Discussion
In this study, through scRNA-seq, we discovered that a subset of activated ILC2s in the lung expresses high levels of genes associated with wound healing after influenza virus infection. This subset is also characterized by high expression of the transcription factor BATF. Furthermore, we discovered a novel role for BATF in pulmonary ILC2s following acute respiratory virus infection. Mechanistically, we have demonstrated that BATF acts as a critical node in governing the transcriptional, epigenetic and functional programs of pulmonary resident ILC2s.
Previous scRNA-seq data showed the presence of BATF in both IL-33- and IL-25-responsive pulmonary ILC2 subsets (26). These two subsets represent natural ILC2s (nILC2s), which respond strongly to IL-33, and inflammatory ILC2s (iILC2s), which migrate between mucosal sites and respond primarily to IL-25. Consistently, it has recently been reported that BATF is required for a normal ILC2 response to IL-25 and IL-33 (23) and high BATF expression was also found in iILC2s, where it is essential for iILC2-mediated anti-helminth immunity (24). Our data show that BATF is enriched in the pulmonary wound healing ILC2 subset after influenza virus infection. Notably, we demonstrated that the vast majority of pulmonary ILC2s are tissue resident and have a distinct transcriptional profile. These data support the idea that wound healing ILC2s are not the same subset as iILC2s identified during helminth infection (24). Consistent with a previous study (23), we found that BATF was essential for ILC2 peripheral homeostasis. However, a study from another group did not find a significantly lower number of lung-resident nILC2s in naïve total BATF−/− mice as compared to Batffl/fl mice (24). In addition, BATF has been reported to regulate ILC hematopoiesis (23). However, we did not observe any significant defect in the number of ILC2 progenitors in the BM of cKO mice. The similarities and discrepancies of our observations with previous studies are most likely due to the use of different mouse models and/or different gating strategies. However, whether BATF deficiency does not affect the cell number but instead alters the developmental features of ILC2 progenitors in cKO mice requires further investigation. Therefore, utilization of a mature ILC2-specific BATF deletion model might be a better choice to separate ILC developmental and effector functions.
Our results demonstrate that BATF broadly and directly controls core functional gene expression of pulmonary ILC2s during influenza virus infection, such as Il5, Il6, Il9, Il13 and Areg. Numerous studies have shown that all these cytokines play beneficial roles in wound healing. For example, ILC2-derived IL-5 is important for eosinophil homeostasis (47), which helps to reduce inflammation and promote tissue homeostasis (48). In our study, the reduction of IL-5 expression in cKO ILC2s likely explains the decreased eosinophil infiltration noted at later stages following influenza infection. IL-13 can promote goblet cell hyperplasia in the lung (49), and IL-6 has been shown to control levels of adhesion molecules and growth factors that are essential to the wound healing process (50). Additionally, ILC2s exhibit high expression of IL-9R and IL-9 to promote an autocrine mechanism of epithelial cell maintenance in the lung (44, 51). Notably, our study demonstrates that BATF binds to a core set of wound healing genes, including Areg, and promotes chromatin accessibility and gene expression. Notably, replenishment of amphiregulin failed to rescue the recovery of cKO mice, suggesting that BATF deficiency shifts the properties of ILC2s from tissue repair to proinflammatory in the context of influenza virus infection, which may cause increased morbidity and mortality in cKO mice. It has been observed that in certain ILC2s, plasticity exists to allow them to respond to different challenges and to adapt to the microenvironment (52, 53). Indeed, we show that BATF directly regulates ILC2 identity during influenza virus infection. During the resolution phase, IL-33 upregulates BATF expression in ILC2s, allowing BATF to bind directly to the locus of Il1rl1 (encodes ST2) in order to maintain, or promote, ST2 levels for ILC2 activation and proliferation via an IL-33-BATF-ST2 feed forward loop. Of note, Il1rl1 is one of many genes that are defective in expression following Batf ablation, but this may not be the only mechanism responsible for defective ILC responses. Moreover, in humans, BATFhi ILC2s exhibit more resistance to plasticity than BATFneg ILC2s, suggesting that BATF guards ILC2 identity in a graded manner (54). Our study revealed the amounts of BATF in pulmonary ILC2s were strongly and positively correlated with the expression of ILC2 hallmarks, such as GATA3. Given that GATA3 is known to regulate ST2 expression in ILC2s (55, 56), it remains to be established if the role of BATF as a regulator of the IL-33-ST2 axis depends on GATA3.
In the absence of BATF, ILC2 signature genes are globally downregulated and a proinflammatory IL-17A signature is upregulated. IL-23 binding to IL-23R is generally considered as an essential initiator to drive pathogenic Th17 cell formation (57). Our data suggest that the loss of BATF in ILC2s increases Il23r expression, which probably initiates the formation of pathogenic IL-17A-producing ILC2s upon influenza virus infection. In addition, previous evidence has shown that IL-25-responsive ILC2s (iILC2s) can produce IL-17A, but maintain high GATA3 expression (30). Considering that the reduction of GATA3, as well as ST2 (IL-33R) and Il17rb (encoding a subunit IL-25R) are most likely to make Batf−/− ILC2s fail to respond to IL-33 and IL-25, we conclude that the phenotype of Batf−/− ILC2s in our study is distinct from iILC2s. Similar phenotypes have been noted for Bcl11b−/− ILC2s (58) and Gfi1−/− ILC2s (59). Additionally, the mere reduction of GATA3 in Batf−/− ILC2s could be sufficient for IL-17A production, which is supported by earlier studies showing that ILC2s can only produce IL-17A upon downregulation of GATA3 (10, 60).
Notably, it has been shown that BATF functions as an activator to induce IL-17A expression in Th17 cells (18) and iNKT cells (61). We have observed the opposite in ILC2s, but have provided data to explain this conundrum. First, BATF maintains chromatin accessibility the Il17a locus in Th17 cells (13, 17, 18) but this effect is not seen in ILC2s. Moreover, it is possible that BATF forms transcription complexes with different factors in these two cell types. In this regard, BATF and RORγt co-bind the Il17a promoter site (−155 to −187) to synergistically promote IL-17A expression in Th17 cells (18, 62). However, we observed very low BATF binding peaks at this promoter region in ILC2s, probably due to lack of RORγt in ILC2s. Additionally, one of the IL-17A positive regulator motifs, AP-1-IRF composite element (AICE), was enriched in BATF binding peaks in Th17 cells (13, 63); however, bZIP motifs (such as Jun-AP1, Fosl2, Fra2 and JunB) rather than AICE were significantly enriched in peaks bound by BATF in ILC2s. These data indicate BATF may preferentially heterodimerize with AP-1 family members to suppress IL-17A expression in ILC2s, which is supported by the notion that BATF, which lacks a transcriptional activation domain, initially functions as an inhibitor of AP-1 driven transcription (11). Overall, a dual role for BATF in gene regulatory events was reported in Th17 cells (17), CD8+ T cells (64) and type 1 regulatory cells (Tr1s) (65). Our data demonstrate that BATF plays distinct roles in maintaining ILC2 identity and subsequently promoting ILC2-mediated tissue repair during influenza virus infection.
Another mechanism by which ILC2s promote tissue homeostasis is through cell-cell crosstalk. During influenza virus infection, the typical inflammatory process is characterized by decreased aMac and increased monocyte recruitment (66). Importantly, aMac numbers must be quickly re-established to resolve infection and repair the tissue (67, 68). In our study, we observed that the typical inflammatory process persists in the lungs of BATF cKO mice. Given that aMacs are a major contributor to amphiregulin expression for tissue repair during parasite infection (69), the impaired recovery of aMacs and the persistent higher infiltration of iMons and iMacs in cKO mice exacerbate the shortage of amphiregulin and prolong the inflammatory possess. It will be important to further investigate the mechanism by which ILC2s regulate the balance of aMacs and iMons in the lung.
In summary, from a molecular and cellular perspective, our study strongly demonstrates that the induction of BATF allows ILC2s to recognize tissue damage and induce the expression of wound healing genes, allowing for efficient tissue repair and for the functional restoration of the vascular barrier. Importantly, the BATF-dependent expression of wound healing genes in ILC2s in our model establishes ILC2s as a major player in limiting inflammation-induced tissue damage in lung epithelium. We conclude that targeting ILC2s represents a promising intervention to promote tissue repair and control inflammation caused by pathogenic acute viral infection.
MATERIALS AND METHODS
Study design
The purpose of this study was to understand the intrinsic role of BATF in promoting pulmonary resident ILC2-mediated tissue repair during acute respiratory virus infection. ScRNA-seq, bulk RNA-seq, ATAC-seq, CUT&Tag-seq and flow cytometry were used to analyze the leukocyte milieu in the lung of WT (Batffl/fl) or Batf cKO mice. Influenza virus infections were ended upon mouse sacrifice at indicated days after infection. In general, experiments were performed at least twice unless indicated otherwise. Data were not excluded from analysis. Experimenters were not blinded to intervention groups for flow cytometry analysis.
Mice and Influenza Virus infection
Six to eight-week-old female C57BL/6 mice were obtained through the National Cancer Institute grantees program (Frederick, MD). Mice were bred and maintained in a closed breeding facility, and mouse handling conformed to the requirements of the Institutional Animal Care and Use Guidelines of Medical College of Wisconsin. Batffl/fl mice was previously described (70) and PLZF-Cre recombinase mice (stock #:024529) was purchased from Jackson and crossed to one another to generate either Batffl/flPLZF-Cre– or Batffl/flPLZF-Cre+ mice. Mice were infected with influenza A/PR8/34 (~50 PFU/mouse) to establish acute sublethal infection. Infection were performed by intranasal (i.n.) under anesthesia as described before (71). R26-CreERT2 mice (stock #: 008463) and RosamTmG reporter mice (stock #: 007576) were purchased from Jackson laboratory. Batffl/fl mice were crossed with ERT2CreRosamTmG mice to generate Batffl/fl ERT2CreRosamTmG mice.
Isolation of ILC cells
ILC cells were isolated from tissues including lung, bone marrow (BM), small intestine, and large intestine. For the lung, mice were euthanized and perfused with 10 ml cold DPBS (Corning) via the right heart ventricle. Removed lung lobes were cut into fine pieces and digested at 37°C with Collagenase IV (1 mg/ml; Worthington) for 40 minutes. The tissue pieces were further grounded and homogenized in a 70-μm cell strainer. After spinning down, red blood cells were removed by addition of ACK buffer (Lonza) for 10 minutes on ice. Subsequently, the single-cell suspension was washed with DPBS containing 0.5% bovine serum albumin (Fisher Scientific) and 2mM EDTA (Lonza). For bone marrow, euthanized mice were cut down the right femur and tibia and BMs were mechanically dispersed into a single-cell suspension. For the gut, intestines were cut open longitudinally and fit tissue and Peyer’s patches were removed. Next, intestines were washed with cold PBS and cut into pieces. Epithelial layers were removed by shaking incubation in 5 mM EDTA Ca2+ and Mg2+ free 1640 medium (Life Technologies) for 30 min each at 37°C. Then, intestines were cut into fine pieces and digested twice for 40 min each at 37°C with Collagenase II and III (1 mg/ml; Worthington), and DNase I (200 μg/ml; Roche). Lymphoid cells were isolated with 30%–60% Percoll gradient, and washed twice with cold DPBS.
Bronchoalveolar lavage fluid collection
Mice were anesthetized by injection of ketamine and xylazine in PBS. After exposure the trachea, bronchoalveolar lavage fluid was collected by lavage twice with 0.5 ml ice cold PBS into the lung. The fluid was spun down at 500 g for 5 minutes. The pellets were washed with ice cold PBS and then counted blood cells or stained with indicated antibodies for flow cytometry analysis. The supernatant was stored at −80°C for further analysis.
ELISA
BAL fluids were collected from WT and cKO mice at day 9 post-influenza virus infection (A/PR8/34, 50 PFU/mouse). Measurement of protein level of IL-5, IL-13, and amphiregulin were performed followed by instructions as provided by commercial ELISA kits (ThermoFisher).
Flow cytometry and cell sorting
Filtered single-cell suspensions were isolated from the above-mentioned tissues or ILC2s from vitro cultured system were then stained with fluorophore-conjugated antibodies against cell surface antigens for 30–60 minutes at 4°C. For intracellular staining, cells were incubated in complete cell culture medium with 50 ng/mL phorbol 12-myristate 13-acetate (Sigma-Aldrich) and 1 μM ionomycin (Sigma-Aldrich) for 30 minutes at 37°C. Brefeldin A (2 mg/mL, Biolegend) was then added for 4.5 h. The fixation and staining were done using the BD Fixation/Permeabilization Solution Kit (BD Biosciences). For nuclear transcription factor staining, cells were fixed and permeabilized with the Foxp3 staining buffer kit (eBioscience). Live and dead cells were discriminated by Live and Dead violet viability kit (Invitrogen). All flow cytometry data were acquired on an LSRII cytometer (BD Biosciences) and analyzed by FlowJo (TreeStar, OR). For innate lymphoid cells sorting, ILCs were isolated from the lungs of indicted mice, then stained with lineage markers including biotin-labeled antibodies, including CD3, CD8α, TCRβ, TCRγ/δ, CD11b, CD11c, B220, Gr1, NK1.1 and TER119. After incubation on ice for 15 minutes, the cells were washed with cold PBS. Lineage negative cells were then purified through Streptavidin-RapidSpheresTM 50001 (STEMCELL) incubation and magnet for column-free immunomagnetic separation. The purified ILCs were stained for 15 minutes on ice with antibodies against CD90.2 and ST2, live/dead cell staining and streptavidin. Stained cells were washed and sorted using a FACSAria III cytometer (BD Biosciences). All antibodies used for flow cytometry are listed in Table S1.
Single-cell RNA sequencing and Analysis
Pulmonary ILCs (Lin−CD90.2+) were FACS-sorted from four influenza-infected mice on each timepoint day 5 and day 10 post infection and were loaded on the Chromium Controller (10x Genomics). Single-cell RNA-seq libraries were prepared using the Chromium Single Cell 3’ v2 Reagent Kit (10x Genomics) according to manufacturer’s protocol. Libraries were loaded onto an Illumina NextSeq with the NextSeq 500/550 High Output Kit v2 (150 cycles) (FC-404-2002, Illumina) with the following conditions: 26 cycles for read 1, 98 cycles for read 2, and 8 cycles for i7 index. Python Run Downloader (Illumina) was used to download raw sequencing data. Cell Ranger (10x Genomics) functions mkfastq and count were used to demultiplex the sequencing data and generate gene-barcode matrices, respectively. As a control, a single cell RNA-sequencing (scRNA-seq) dataset of pulmonary ILCs obtained from uninfected C57BL/6 mice was obtained from GEO (GSE102299 - GSM2733478_PBS_Rep1) (26). Data were imported into R (version 3.6.1) using the Seurat package (version 3.1.1) (72). Integrated analysis was used to combine the samples. Uniform manifold approximation and projection (UMAP) was used to visualize the cells, which formed 9 separate clusters based on the Louvain algorithm. We removed contaminating cells and kept ILCs based on high expression of Ncr1 and Il1rl1 (encodes ST2). Reclustering the cells provided 4 separate clusters. Cells were analyzed for gene expression while grouped by both cluster and timepoint. The R package Monocle (v 2.12.0) was used to determine gene expression in pseudotime (73). RNA Velocity was used to project predicted differentiation trajectories onto UMAP plots of the cells (74).
Bulk RNA-sequencing and Analysis
2000 ILC2s (Lin−CD90.2+ST2+) from the lungs of WT and cKO mice after influenza virus infection at day 9 were directly sorted into lysis buffer containing Protector RNase Inhibitor (Roche). Three replicates from each group were subsequently sequenced. 2 ng of purified RNA were used as input for a modified SMART-Seq2 protocol entailing RNA secondary structure denaturation (72°C for three minutes), reverse transcription with Maxima Reverse Transcriptase (Life Technologies), and whole transcriptome amplification (WTA) with KAPA HiFi HotStart ReadyMix 2X (Kapa Biosystems) for 12 cycles. WTA products were purified with AMPure XP beads (Beckman Coulter) and quality assessed with a High Sensitivity DNA Chip run on a Bioanalyzer 2100 system (Agilent). 0.2 ng of purified WTA product was used as input for the Nextera XT DNA Library Preparation Kit (Illumina). Uniquely barcoded libraries were pooled and sequenced with a NextSeq 500 sequencer using a high output V2 75 cycle kit (Illumina) and 2×38 paired end reads. Bulk RNA-seq data were aligned to the Mus musculus mm10 genome and quality control was performed using Nextflow pipeline (nf-core/rnaseq 1.4.2) (DOI:10.5281/zenodo.1400710) (75). Gene expression was quantified at the gene level using Salmon. RNA-seq libraries were then normalized and genes were tested for differential expression between BATF deficient and wild-type ILC2s with DESeq2 v1.24.0 (76). DESeq2 Wald tests were used to determine whether fold changes were significantly different from zero. For visualization, data were transformed using the regularized logarithmic transformation (76). Pre-ranked gene set enrichment analyses were conducted using shrunken fold-changes and clusterProfiler v3.12.0 (77). KEGG (78), Reactome (79), and GO (80) databases were used for GSEA. The Benjamini-Hochberg method was used to adjust p-values for false discovery in both differential expression and GSEA analyses.
ATAC-sequencing and Analysis
ATAC-seq was performed according to a published protocol (81) with minor modification. 50–80,000 ILC2 cells were sorted from the lungs of WT and cKO mice after influenza virus infection at day 9 or from mILC2s cultured in vitro (GFP−ILC2 or GFP+ILC2s). The sorted ILC2s were spin down and washed with 50μl cold PBS, followed by treatment with 50μl lysis buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). After pelleting the nuclei by centrifuging at 500 g for 10 minutes, the pellets were resuspended in a 40 μl transposition reaction with 2 μl Tn5 transposase (FC-121-1030; Illumina) to tag and fragmentalize accessible chromatin. The reaction was incubated at 37°C with for 30 minutes. The tagmented DNAs were then purified using a QIAGEN MinElute kit and amplified with 12 cycles of PCR based on the amplification curve. Once the libraries were purified using a QIAGEN PCR cleanup kit. The quantified libraries were sequenced using NextSeq 500/550 kit v2 with 75 cycles and 400 million reads with paired end sequencing. Reads were aligned to mouse reference genome mm9 using Bowtie2 (82), and then filtered using SAMtools (-F 1796) (83). Duplicates were marked with Picard and removed. Finally, reads aligning to the mitochondrial genome were removed. Peaks were then called using MACS2 (--no model --shift −100 --ext size 200 -q 0.01) (version 2.1.1) (84). Data from different subsets was normalized and subset-specific peaks were identified using MAnorm (version 0.1.0) (85). Motif analysis was performed using HOMER (-h to calculate enrichment using cumulative hypergeometric distribution) (version 4.10.4) (86). Gene ontology analysis of subset-specific enhancers was done using GREAT (version 4.0.4) (87).
ILC2s In vitro Culture
ILC2s were sorted from the lungs and cultured in vitro with RPMI-1640 medium (Invitrogen) with 10% heat-inactivated FBS (HyClone Fisher), 100 U/ml penicillin and 0.1 mg/ml streptomycin (Thermo Fisher Scientific), 50 μM 2-mercaptoethanol (Sigma-Aldrich), 10 ng/mL of IL-2 (Peprotech), 10 ng/mL of IL-7 (Peprotech), and 10 ng/mL of IL-33 (Peprotech) to expand the cells.
CUT&Tag ChIP-sequencing and Analysis
ILC2s were sorted from the lungs of C57BL/6 mice and cultured for 2 weeks to expand the cells. 100,000 ILC2s were processed and library construction according to the CUT&Tag ChIP-Seq approach mentioned (version 2) (88) (stepwise protocol can be found at https://www.protocols.io/view/bench-top-cut-amp-tag-z6hf9b6). Rabbit normal serum IgG1 was used as a control. 37 cycles of paired-end sequencing were performed on a NextSeq 550 and about 5 to 10 million reads were generated for each sample. For data analysis, first of all, each dataset was first downsampled to the same read depth. For standardization between experiments, Escherichia. coli DNA derived from transposase protein production was used to normalize sample read counts based on the recommendation of the CUT&TAG protocol. Reads were aligned to mouse reference genome mm10 and E. coli (strain K12) using Bowtie2 (version 2.2.5) with options: --local --very- sensitive-local --no-unal --no-mixed --no-discordant --phred33 -I 10 -X 700. Peaks were called using SEACR (version1.1) with options: 0.01 non-stringent. Peaks were annotated with HOMER (version 4.9.1) and visualizations were created using deep Tools (version 3.3.0) and IGV (version 2.8.2). k-means clustering of BATF ChIP-seq data was performed using seqMINER (version 1.3.3).
Bulk RNA-seq and CUT&Tag ChIP -seq comparison
To compare bulk RNA-seq and CUT&Tag ChIP-seq, peak gene symbols and significant RNA-seq gene symbols were intersected in order to find the overlaps of genes. Visualizations were created using ggplot (v3.2.1) and EnhancedVolcano (v1.2.0). Pre-ranked GSEA was conducted using the shrunken log2 fold changes from the bulk RNA-seq data using the genes also present (overlapping) with CUT&Tag peaks. For CUT&Tag ChIP-seq, peaks were compared. IDRfilter from ChIPpeakAnno v3.18.2 (89) was used to assess the consistency of replicate experiments and obtain a high-confidence single set of peaks. Peaks overlapping and binding pattern were analyzed and visualized with ChIPpeakAnno.
RNA isolation and quantitative real-time PCR
After influenza virus infection at the indicated days, fractions of lung were snap-frozen on dry ice and subsequently stored in a −80°C freezer. Samples were stabilized in RNAlater-ICE (ThermoFisher) before proceeding with tissue homogenization and RNA isolation using the RNAqueous-Micro Kit (ThermoFisher) according to the manufacturer’s instructions. Equal quantities of RNA from each sample were reverse transcribed into cDNA using the SuperScript II Reverse Transcriptase and oligo dT(18) primers. Quantitative real-time PCR was performed using the Maxima SYBR Green/ROX qPCR Master Mix. Expression of target genes was normalized to Gapdh expression. All primers are listed in table S1.
Histological analysis
Lungs were fixed for at least 24 h with 10% formalin and were embedded in paraffin. 4 μm lung sections were stained with hematoxylin and eosin (H&E). Pathological inflammation and damage scores were evaluated by the following parameters: bronchitis, interstitial inflammation, edema, endothelialitis, pleuritis and thrombus formation. Each parameter was graded on a scale of 0 to 4 (0: absent, 1: mild, 2: moderate, 3: severe, 4: very severe). The total histopathological score was expressed as the sum of the scores for the different parameters, the maximum being 24.
Amphiregulin administration
Recombinant murine Amphiregulin (R&D Systems) was administrated by intraperitoneal injection. Each dose of 5 μg was used every two days starting from influenza virus infection at day 0. Virus-infected WT and cKO mice treated with PBS were used as control group.
IL-33-mediated ILC2 expansion in vivo
Mice were anesthetized with isoflurane and treated intraperitoneal injection (IP) with 200 μL of IL-33 (500 ng per mouse) in PBS for 4 consecutive days. PBS treated mice were used as controls.
CTV labeling and Proliferation assay
ILC2s were sorted from the lungs of WT and cKO mice and cultured in vitro as described above, followed by labeling with CellTrace Violet (Thermo Fisher Scientific) according to manufacturer’s instructions. After 2-day culture the cells were stained with indicated antibodies and analyzed by flow cytometry on a BD LSRFortessa cytometer (BD Biosciences).
mILC2 culture and induced deletion of Batf in vitro
mILC2s were sorted from the lungs of Batffl/fl ERT2CreRosamTmG mice and cultured in vitro in RPMI-1640 medium (Invitrogen) with 10% heat-inactivated FBS, 100 U/mL penicillin, 0.1 mg/mL streptomycin, 50 μM 2-mercaptoethanol, 10 ng/mL of IL-2, 10 ng/mL of IL-7, and 0.04 ng/mL of 4-Hydroxytamoxifen (4-OHT) for conditional inactivation of Batf. Two days after 4-OHT treatment, IL-33 (10 ng/mL) was added for expansion and cells were cultured continuously in the presence of 4-OHT. After 6–7 days expansion, cells were collected and stained with the indicated antibodies for flow cytometry analysis, or GFP− and GFP+ ILC2s were FACS sorted to perform ATAC-sequence analysis.
Fluorophore-conjugated antibody labeling in vivo
For direct labeling of circulation leukocytes in vivo, the fluorophore-conjugated antibody FITC-CD45.2 was diluted with PBS and intravenously into BALB/c mice after influenza infection at day 9 (5 μg/mouse). After 5 minutes, mice were scarified and lungs were processed for flow cytometry detection. The PBS injected mice were used as a control.
Tissue processing and Viral Titration
Mouse lungs were collected in 2 mL ice cold PBS. Samples were weighed and transferred into M Tubes for dissociation in a gentleMACS instrument (Miltenyi Biotec). The gentleMACS Program RNA_01 was run to homogenize tissue. Samples were centrifuged at 600 g for 10 minutes. Supernatants were collected and stored at −80°C. For the virus titer assay, MDCK cells were cultured in a 96-well plate for 16–24 hours before virus incubation. Cells were washed with ice cold PBS and incubated with serially diluted virus for 90 minutes at 37°C, with each dilution having three replicates. Cells were washed and replaced with viral growth medium (DMEM, 0.2% final dilution of 7.5% BSA stock, 25 mM HEPES, 100 μg/ml pen/strep, and 2 μg/ml TPCK trypsin). After incubating at 37°C for 72 hours, wells were filled with 10% formalin and incubated for 10 minutes. Formalin was then removed. Monolayers were stained with 0.5% Crystal Violet in 20% methanol in deionized water. The stain was removed and wells washed with water until the water ran clear. Plates were read for plaque counting. Viral titers were determined by the Reed & Muench Calculator.
Statistical Analyses
Statistical tests were performed using GraphPad Prism software. p-values were calculated using two-way ANOVA analysis and two-tailed paired or unpaired Student’s t-tests. p-values < 0.05 were considered statistically significant.
Supplementary Material
Data file S1. Raw data file (Excel spreadsheet)
MDAR Reproducibility Checklist
Fig. S1. scRNA-seq analysis of pulmonary innate lymphoid cells responding to influenza viral infection.
Fig. S2. scRNA-seq analysis of pulmonary innate lymphoid cells after influenza viral infection.
Fig. S3. BATF is required for maintenance of peripheral ILC2s.
Fig. S4. cKO mice display normality of other major lymphocytes.
Fig. S5. BATF deficiency in ILCs results in susceptibility to Influenza virus Infection.
Fig. S6. Impaired lung ILC2 expansion and wound healing function in virus-infected cKO mice.
Fig. S7. BATF supports chromatin accessibility of wound healing gene loci in ILC2s.
Fig. S8. Amphiregulin failed to rescue cKO mice followed by influenza virus infection.
Fig. S9. BATF promotes IL-33-induced proliferation of ILC2s in the bone marrow.
Fig. S10. BATF regulates expression of ST2 and Areg on mILC2s in response to IL-33 stimulation.
Table S1. Key resources used in this study.
Acknowledgments
We thank Dr. S. Henikoff for providing 3XFlag-pA-Tn5-Fl plasmid and Dr. Nan Zhu for providing Protein A–Tn5 fusion protein.
Funding:
This work was supported by NIH grants AI125741 (W.C.), AI148403 (W.C.), AI112844 (J.S.), AI147394 (J.S.), AG047156 (J.S.), DK127526 (M.Y.K.), and AI153537 (R.Z.); an American Cancer Society Research Scholar Grant (W.C.); and an Advancing a Healthier Wisconsin Endowment (AHW) Grant (W.C). R.Z. is supported by the Cancer Research Institute Irvington Fellowship. M.Y.K. is a member of the Medical Scientist Training Program at the Medical College of Wisconsin, which is partially supported by a training grant from NIGMS (T32-GM080202). This research was completed in part with computational resources and technical support provided by the Research Computing Center at MCW.
Footnotes
Competing Interests: The authors declare no competing interests.
Data and Materials Availability:
The single-cell RNA-seq data are available in the GEO database with the accession code GSE149857. The bulk RNA-seq data are available in the GEO database with the accession code GSE149854. ATAC-seq data are available in the GEO database with the accession code GSE149847 and GSE188275. CUT&Tag ChIP-seq data are available in the GEO database with the accession code GSE149849. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Liu Q, Zhou YH, Yang ZQ, The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol 13, 3–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang C et al. , Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monticelli LA et al. , Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12, 1045–1054 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duerr CU et al. , Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol 17, 65–75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Califano D et al. , IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol 11, 209–219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Goffic R et al. , Infection with influenza virus induces IL-33 in murine lungs. Am J Respir Cell Mol Biol 45, 1125–1132 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Prefontaine D et al. , Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol 125, 752–754 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Lee JU et al. , Upregulation of interleukin-33 and thymic stromal lymphopoietin levels in the lungs of idiopathic pulmonary fibrosis. BMC Pulm Med 17, 39 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shim DH et al. , Pandemic influenza virus, pH1N1, induces asthmatic symptoms via activation of innate lymphoid cells. Pediatr Allergy Immunol 26, 780–788 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Silver JS et al. , Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol 17, 626–635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy TL, Tussiwand R, Murphy KM, Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol 13, 499–509 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Pham D et al. , Batf Pioneers the Reorganization of Chromatin in Developing Effector T Cells via Ets1-Dependent Recruitment of Ctcf. Cell Rep 29, 1203–1220 e1207 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li P et al. , BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Care MA et al. , SPIB and BATF provide alternate determinants of IRF4 occupancy in diffuse large B-cell lymphoma linked to disease heterogeneity. Nucleic Acids Res 42, 7591–7610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delacher M et al. , Precursors for Nonlymphoid-Tissue Treg Cells Reside in Secondary Lymphoid Organs and Are Programmed by the Transcription Factor BATF. Immunity 52, 295–312 e211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biddie SC et al. , Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43, 145–155 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciofani M et al. , A validated regulatory network for Th17 cell specification. Cell 151, 289–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schraml BU et al. , The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460, 405–409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitley SK et al. , IL-1R signaling promotes STAT3 and NF-kappaB factor recruitment to distal cis-regulatory elements that regulate Il17a/f transcription. J Biol Chem 293, 15790–15800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao K et al. , BATF Modulates the Th2 Locus Control Region and Regulates CD4+ T Cell Fate during Antihelminth Immunity. J Immunol 197, 4371–4381 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vasanthakumar A et al. , The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol 16, 276–285 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Shih HY et al. , Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 165, 1120–1133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, Kim MH, Friesen L, Kim CH, BATF regulates innate lymphoid cell hematopoiesis and homeostasis. Sci Immunol 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller MM et al. , BATF acts as an essential regulator of IL-25-responsive migratory ILC2 cell fate and function. Sci Immunol 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weizman OE et al. , ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 171, 795–808 e712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wallrapp A et al. , The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A, A committed precursor to innate lymphoid cells. Nature 508, 397–401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider C et al. , Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog 10, e1004053 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samarasinghe AE et al. , Eosinophils Promote Antiviral Immunity in Mice Infected with Influenza A Virus. J Immunol 198, 3214–3226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y et al. , IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol 16, 161–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bastakoty D, Young PP, Wnt/beta-catenin pathway in tissue injury: roles in pathology and therapeutic opportunities for regeneration. FASEB J 30, 3271–3284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Yu A, Yu FX, The Hippo pathway in tissue homeostasis and regeneration. Protein Cell 8, 349–359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan M et al. , Injury-activated transforming growth factor beta controls mobilization of mesenchymal stem cells for tissue remodeling. Stem Cells 30, 2498–2511 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai T et al. , IL-17-producing ST2(+) group 2 innate lymphoid cells play a pathogenic role in lung inflammation. J Allergy Clin Immunol 143, 229–244 e229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golebski K et al. , IL-1beta, IL-23, and TGF-beta drive plasticity of human ILC2s towards IL-17-producing ILCs in nasal inflammation. Nat Commun 10, 2162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernink JH et al. , c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nat Immunol 20, 992–1003 (2019). [DOI] [PubMed] [Google Scholar]
- 37.McDonald B, Kubes P, Chemokines: sirens of neutrophil recruitment-but is it just one song? Immunity 33, 148–149 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Struyf S et al. , Chemokines synergize in the recruitment of circulating neutrophils into inflamed tissue. Eur J Immunol 35, 1583–1591 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Ramirez K et al. , Gene deregulation and chronic activation in natural killer cells deficient in the transcription factor ETS1. Immunity 36, 921–932 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zook EC et al. , The ETS1 transcription factor is required for the development and cytokine-induced expansion of ILC2. J Exp Med 213, 687–696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu C et al. , Macrophages Mediate the Repair of Brain Vascular Rupture through Direct Physical Adhesion and Mechanical Traction. Immunity 44, 1162–1176 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Constantinides MG et al. , MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagashima H et al. , Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity 51, 682–695 e686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turner JE et al. , IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med 210, 2951–2965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakar MS et al. , Cellular forces and matrix assembly coordinate fibrous tissue repair. Nat Commun 7, 11036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franco JJ, Atieh Y, Bryan CD, Kwan KM, Eisenhoffer GT, Cellular crowding influences extrusion and proliferation to facilitate epithelial tissue repair. Mol Biol Cell 30, 1890–1899 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nussbaum JC et al. , Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugawara R et al. , Small intestinal eosinophils regulate Th17 cells by producing IL-1 receptor antagonist. J Exp Med 213, 555–567 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugita K et al. , Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL-13 in asthmatic patients. J Allergy Clin Immunol 141, 300–310 e311 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Lin ZQ, Kondo T, Ishida Y, Takayasu T, Mukaida N, Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J Leukoc Biol 73, 713–721 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Mohapatra A et al. , Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol 9, 275–286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krabbendam L, Bal SM, Spits H, Golebski K, New insights into the function, development, and plasticity of type 2 innate lymphoid cells. Immunol Rev 286, 74–85 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Colonna M, Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 48, 1104–1117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van der Ploeg EK et al. , Steroid-resistant human inflammatory ILC2s are marked by CD45RO and elevated in type 2 respiratory diseases. Sci Immunol 6, (2021). [DOI] [PubMed] [Google Scholar]
- 55.Zhu J, GATA3 Regulates the Development and Functions of Innate Lymphoid Cell Subsets at Multiple Stages. Front Immunol 8, 1571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoyler T et al. , The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 37, 634–648 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hasan Z et al. , JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat Commun 8, 15628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Califano D et al. , Transcription Factor Bcl11b Controls Identity and Function of Mature Type 2 Innate Lymphoid Cells. Immunity 43, 354–368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spooner CJ et al. , Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol 14, 1229–1236 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Ohne Y et al. , IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol 17, 646–655 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Jordan-Williams KL, Poston S, Taparowsky EJ, BATF regulates the development and function of IL-17 producing iNKT cells. BMC Immunol 14, 16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ichiyama K et al. , Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J Biol Chem 283, 17003–17008 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Glasmacher E et al. , A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338, 975–980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kurachi M et al. , The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol 15, 373–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karwacz K et al. , Critical role of IRF1 and BATF in forming chromatin landscape during type 1 regulatory cell differentiation. Nat Immunol 18, 412–421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hashimoto D et al. , Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang FF et al. , GM-CSF in the lung protects against lethal influenza infection. Am J Respir Crit Care Med 184, 259–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sever-Chroneos Z et al. , GM-CSF modulates pulmonary resistance to influenza A infection. Antiviral Res 92, 319–328 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allard B, Panariti A, Martin JG, Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Front Immunol 9, 1777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Betz BC et al. , Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med 207, 933–942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun J, Madan R, Karp CL, Braciale TJ, Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med 15, 277–284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Satija R, Farrell JA, Gennert D, Schier AF, Regev A, Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qiu X et al. , Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.La Manno G et al. , RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ewels PA et al. , The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol 38, 276–278 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu G, Wang LG, Han Y, He QY, clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kanehisa M, Goto S, KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fabregat A et al. , The Reactome Pathway Knowledgebase. Nucleic Acids Res 46, D649–D655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ashburner M et al. , Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ, ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–21 29 29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H et al. , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y et al. , Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shao Z, Zhang Y, Yuan GC, Orkin SH, Waxman DJ, MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13, R16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heinz S et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McLean CY et al. , GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaya-Okur HS et al. , CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu LJ et al. , ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 237 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Raw data file (Excel spreadsheet)
MDAR Reproducibility Checklist
Fig. S1. scRNA-seq analysis of pulmonary innate lymphoid cells responding to influenza viral infection.
Fig. S2. scRNA-seq analysis of pulmonary innate lymphoid cells after influenza viral infection.
Fig. S3. BATF is required for maintenance of peripheral ILC2s.
Fig. S4. cKO mice display normality of other major lymphocytes.
Fig. S5. BATF deficiency in ILCs results in susceptibility to Influenza virus Infection.
Fig. S6. Impaired lung ILC2 expansion and wound healing function in virus-infected cKO mice.
Fig. S7. BATF supports chromatin accessibility of wound healing gene loci in ILC2s.
Fig. S8. Amphiregulin failed to rescue cKO mice followed by influenza virus infection.
Fig. S9. BATF promotes IL-33-induced proliferation of ILC2s in the bone marrow.
Fig. S10. BATF regulates expression of ST2 and Areg on mILC2s in response to IL-33 stimulation.
Table S1. Key resources used in this study.
Data Availability Statement
The single-cell RNA-seq data are available in the GEO database with the accession code GSE149857. The bulk RNA-seq data are available in the GEO database with the accession code GSE149854. ATAC-seq data are available in the GEO database with the accession code GSE149847 and GSE188275. CUT&Tag ChIP-seq data are available in the GEO database with the accession code GSE149849. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.