

ABSTRACT
The retroviral surface envelope protein subunit (SU) mediates receptor binding and triggers membrane fusion by the transmembrane (TM) subunit. SU evolves rapidly under strong selective conditions, resulting in seemingly unrelated SU structures in highly divergent retroviruses. Structural modeling of the SUs of several retroviruses and related endogenous retroviral elements with AlphaFold 2 identifies a TM-proximal SU β-sandwich structure that has been conserved in the orthoretroviruses for at least 110 million years. The SU of orthoretroviruses diversified by the differential expansion of the β-sandwich core to form domains involved in virus-host interactions. The β-sandwich domain is also conserved in the SU equivalent GP1 of Ebola virus although with a significantly different orientation in the trimeric envelope protein structure relative to the β-sandwich of human immunodeficiency virus type 1 gp120, with significant evidence for divergent rather than convergent evolution. The unified structural view of orthoretroviral SU and filoviral GP1 identifies an ancient, structurally conserved, and evolvable domain underlying the structural diversity of orthoretroviral SU and filoviral GP1.
IMPORTANCE The structural relationships of SUs of retroviral groups are obscured by the high rate of sequence change of SU and the deep-time divergence of retroviral lineages. Previous data showed no structural or functional relationships between the SUs of type C gammaretroviruses and lentiviruses. A deeper understanding of structural relationships between the SUs of different retroviral lineages would allow the generalization of critical processes mediated by these proteins in host cell infection. Modeling of SUs with AlphaFold 2 reveals a conserved core domain underlying the structural diversity of orthoretroviral SUs. Definition of the conserved SU structural core allowed the identification of a homologue structure in the SU equivalent GP1 of filoviruses that most likely shares an origin, unifying the SU of orthoretroviruses and GP1 of filoviruses into a single protein family. These findings will allow an understanding of the structural basis for receptor-mediated membrane fusion mechanisms in a broad range of biomedically important retroviruses.
KEYWORDS: HIV-1, gp120, MLV, FELV, ALV, RBD, gammaretrovirus, alpharetrovirus, betaretrovirus, lentivirus, syncytin, EBOV, GP1, foamy virus, spumaretrovirus
INTRODUCTION
The retroviruses are an ancient group of viruses with wide genetic diversity. Of the three canonical retroviral genes, the env gene encoding the surface (SU) and transmembrane (TM) envelope protein subunits mediating receptor binding and membrane fusion during infection is the most diverse. TM is the more conserved of the two envelope protein subunits due to its role in membrane fusion during host cell infection. SU evolves more rapidly as it adapts to different hosts and receptors and during immune evasion. A long-standing question is if the SUs of widely divergent retroviruses share any structural similarities or whether deep-time evolution resulted in structurally distinct SUs without any remaining conserved structural elements.
The most biomedically important group of retroviruses is the orthoretroviruses. These include the alpharetroviruses, betaretroviruses, gammaretroviruses, deltaretroviruses, epsilonretroviruses, and lentiviruses, which induce a variety of pathologies, including oncogenesis and inflammatory and immunodeficiency syndromes in humans, mammals, and avian species. Endogenous elements related to the orthoretroviruses are widespread in vertebrate genomes (1). Most have large deletions and heavy mutational loads that often lead to defective envelope proteins. However, orthoretroviral env genes integrated into vertebrate genomes can evolve under purifying selection and be maintained in an intact and sometimes functional form for millions of years. Among these are the syncytins, retroviral env genes coopted for key roles in placentation (1, 2).
Excluding the epsilonretroviruses, the envelope proteins of orthoretroviruses have been classified as beta, gamma, and avian gamma types (3, 4). The beta-type SU and TM envelope protein subunits of betaretroviruses and lentiviruses are noncovalently associated, and the TM subunit has 2 conserved cysteine residues (3, 4). The gamma-type envelope proteins of gammaretroviruses and deltaretroviruses have SU and TM subunits that are covalently associated, with a third conserved cysteine residue in TM mediating that association (3). The avian gamma-type envelope protein is unique to the alpharetroviruses and is considered a variant of the gamma-type envelope (3). The transmembrane subunits of orthoretroviruses are class I fusion proteins forming α-helical coiled coils in the postfusion state (5). Other viral families with class I fusion proteins are the orthomyxoviruses, paramyxoviruses, coronaviruses, arenaviruses, and filoviruses (5). Limited but significant sequence and structural similarities between the TM subunit of retroviruses and the GP2 transmembrane envelope protein subunit of filoviruses in their postfusion conformation have been described (6, 7). However, similarities in the receptor-binding envelope protein subunits of retroviruses and filoviruses are limited to analogous structures anchoring the human immunodeficiency virus type 1 (HIV-1) gp120 and the Ebola virus (EBOV) GP1 surface envelope subunits to the transmembrane subunits through a pair of antiparallel β-strands (8) but without any apparent sequence or more extensive structural similarity.
The structural diversity of the SUs of orthoretroviruses is also apparent from the variety of domain organizations that are observed in different viral lineages. The SUs of gammaretroviruses have a modular organization with an amino-terminal receptor-binding domain (RBD) followed by a proline-rich region (PRR) linking the RBD to a more conserved carboxy-terminal C-domain of unknown structure (9–13). In contrast, HIV-1 gp120 does not have an independently folding RBD analogous to that of the gammaretroviruses (14). However, structural similarities, including fragmentary sequence similarities, are shared between the surface envelope proteins of lentiviruses and those of betaretroviruses (15, 16). The regions of structural similarity correspond to a β-sandwich structure in the TM-proximal region of the inner domain of HIV-1 g120. Sections of the HIV-1 gp120 β-sandwich participate in intersubunit interactions with the gp41 transmembrane subunit (8, 17). The conserved β-sandwich structure is expanded differentially in the betaretroviruses and lentiviruses to form structurally distinct but topologically equivalent domains (18). The apical regions of the betaretroviral SU models are topologically equivalent to the HIV-1 gp120 distal inner domain layer 2 that interacts with the coreceptor (19). In contrast, the region topologically equivalent to the HIV-1 gp120 inner domain layer 3 and outer domain (19) forms a short loop in the β-sandwich of the SU of betaretroviruses (18). Thus, the beta-type SU proteins evolved by the differential expansion of a conserved TM-proximal β-sandwich domain to form diverse structures mediating virus-host interactions.
It is not clear if the gamma-type SU variants, with their markedly different domain organizations, including an independent RBD in some lineages, also share these or any other aspects of structural conservation with the betaretroviruses or lentiviruses. The recent release of AlphaFold 2, a deep-learning-based structural prediction tool that can yield structures of high quality comparable to experimentally determined structures (20, 21), provides an opportunity to address the structural conservation in the SUs of retroviruses more broadly. Here, it is shown that the structural diversity of orthoretroviral SUs is based on the differential expansion of an ancient and structurally conserved TM-proximal β-sandwich domain. In addition, this β-sandwich is also conserved in a structurally equivalent region of the SU equivalent GP1 of filoviruses, unifying a wide range of seemingly unrelated orthoretroviral and filoviral surface envelope proteins into a single protein family.
RESULTS
Modeling of orthoretroviral SU.
A publicly available simplified version of AlphaFold 2 (22) accessible as a Colab notebook (see Materials and Methods) was used for structural modeling. AlphaFold 2 is not based on traditional threading structural modeling techniques but rather is based on deep-learning techniques (22). Briefly, the input sequence is used to identify similar sequences in databases, creating a multiple-sequence alignment (MSA) that defines variable regions. Following MSA generation, the relevant information is extracted from the MSA by a neural network in a series of iterative steps to identify likely residue interactions. This information is then passed to the structural prediction module to generate models based on these residue interactions, generating three-dimensional models. Matches with known structures in the MSA can be used as the templates for the predictions, although this is not necessary for predictions. AlphaFold yields a single structure (22) and therefore does not provide alternative conformations for flexible regions that may occur in viral proteins. In addition, low sequence coverage in the MSA, as may occur for certain highly divergent regions of viral proteins, may lead to low-reliability models.
The SU sequences of 18 extant exogenous orthoretroviruses and endogenous retroviral elements were used for modeling (Fig. 1 and 2). For simplicity, endogenous elements are classified here according to the orthoretroviral lineages from which they derive. Within the alpharetroviruses, the SU of avian leukemia virus (ALV) and the lizard endogenous elements Mab-Env3 and Mab-Env4 (23) were modeled. The SUs of a wide range of the more diverse gammaretroviruses and related endogenous elements were also modeled. These include representatives of the type C and D gammaretroviruses (Fig. 1A). The type C gammaretroviruses include murine (MLV) and feline (FELV) leukemia viruses and porcine (PERV) (24) and python (PyERV) (25) endogenous retroviruses. The type D gammaretroviruses include avian reticuloendotheliosis virus (REV) and endogenous baboon (BaEV) and feline RD114 endogenous retroviruses. Mason-Pfizer monkey virus (MPMV) is a betaretrovirus that acquired a gamma-type env gene by recombination with a virus related to, REV (26) and is classified here as a type D gammaretrovirus based on its envelope protein sequence for simplicity (Fig. 1A). In addition, models were also obtained for the SU of human syncytin-1 and -2 (2, 27), which cluster by TM sequence with the type D gammaretroviruses; the unclassified gammaretrovirus-related mammalian syncytin-Mar1 (28), syncytin-Car1 (29), and syncytin-Rum1 (30); and the SU encoded by endogenous gammaretroviral elements from spiny-rayed fish (31). The latter include the SUs encoded by the mudskipper percomORF retroviral env endogenous element and a related retroviral endogenous element from the Japanese eel, named here Env-Psc and Env-Aja respectively. Modeling of the SUs of additional endogenous gammaretroviral elements, deltaretroviruses, and epsilonretroviruses was unsuccessful, yielding mostly unfolded models that were not further analyzed.
FIG 1.

Modeling of orthoretroviral SU. (A) Relationship of orthoretroviral TM and filoviral GP2 transmembrane subunit ectodomain sequences. A neighbor-joining cladogram was generated with Geneious Prime using Ebola virus GP2 to root the cladogram. Orthoretroviral and filoviral groups are color-coded as indicated in the cladogram except for unclassified gammaretroviruses, which are shown in black. Numbers indicate node support (percent) from 1,000 bootstrap runs. (B to I) pLDDT scores of SU models. pLDDT scores along the SU sequence are indicated for each model, grouping similar models in the same panels.
FIG 2.
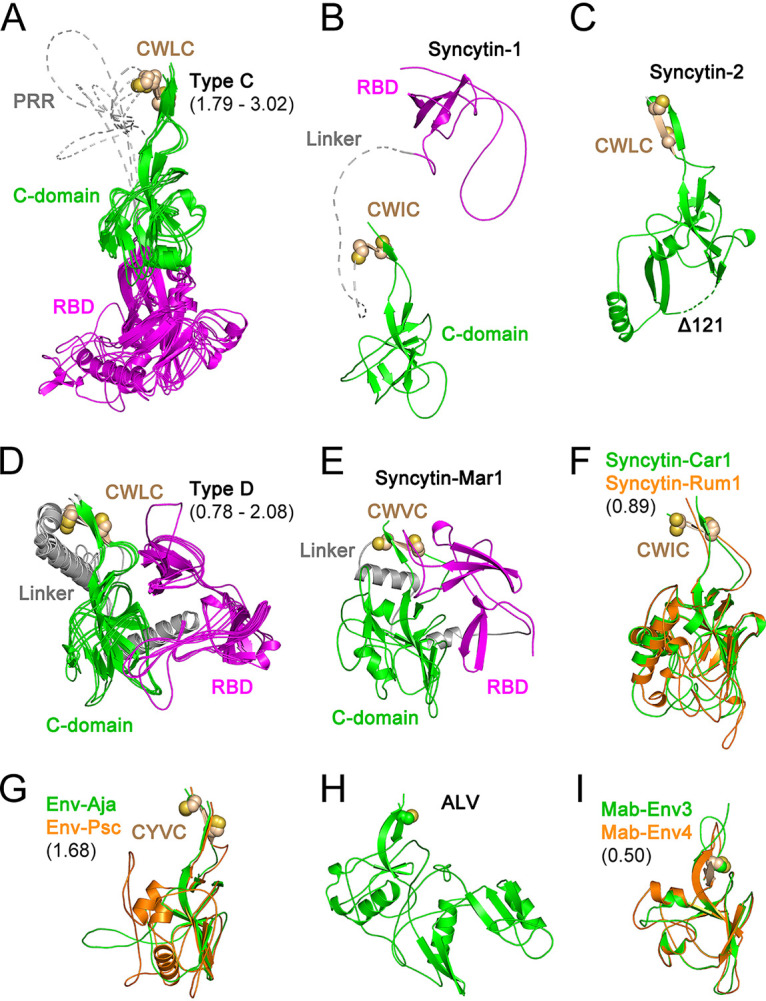
Orthoretroviral SU structural models. Shown are models of the SUs of type C gammaretroviruses (A), syncytin-1 (B), syncytin-2 (C), type D gammaretroviruses (D), syncytin-Mar1 (E), syncytin-Car1 and Rum1 (F), Env-Aja and Env-Psc (G), ALV (H), and Mab-Env3 and Mab-Env4 (I). Panels A, D, F, G, and I show superpositions of structurally similar models, with minimum and maximum root mean square deviations of aligned models in angstroms shown in parentheses. The models in panels A, B, D, and E show the RBD in magenta and the linker regions joining the RBD to the C-domain in gray. The models in panels F, G, and I are shown in colors as indicated in each panel. The disordered PRR and linker regions are shown as dotted lines in panels A and B. The conserved cysteine residues in the gammaretroviral CWLC consensus motifs in panels A to G or preceding the first β-strand in the alpharetrovirus SU models in panels H and I are shown as spheres. The deletion in the syncytin-2 SU model (Δ122) is shown as a dotted line.
Although template-based modeling was enabled in the predictions, none of the input sequences identified suitable templates of known structure among the 12,662 (4,016 unique) sequence hits in all the MSAs except for the RBD regions of MLV and FELV for the type C gammaretroviral SU sequences (see Materials and Methods for data availability). In addition, relevant in this case, no hits to HIV-1 gp120 (UniProt accession number P05478) or EBOV GP1 (UniProt accession number Q05320) were identified in the MSAs. Thus, the structural models produced here were predicted de novo without the input of templates except for the known structures of the MLV and FELV RBDs. In essence, outside the RBD regions of a subset of models, the models presented here emerged from the statistical properties of sequence variation and residue covariation within the MSAs processed by neural networks and not by modification of known structures by simple threading.
A high diversity of modeled SU structures was observed, with SU models clustering according to TM sequence similarities in most cases (Fig. 1 and 2). SU models had predicted local distance difference test (pLDDT) scores of at least 70 and often above 90, which indicate high-confidence main chain and side chain modeling, respectively (20) (Fig. 1B and Fig. 3). Scores of between 50 and 70 indicate moderate main chain modeling confidence, and pLDDT scores of below 50 indicate poor modeling confidence (20). Many of the poor-confidence regions (pLDDT < 50) correspond to terminal regions and internal regions that correspond to long linkers between domains (Fig. 1B and Fig. 3). For the syncytin-2 SU, a high-scoring model similar to SU models of gammaretroviruses was obtained only after deleting 122 residues from its midsection (Fig. 2C). This deletion was determined empirically and is analogous to the empirical deletion of flexible regions from proteins for structural determination except that, in this case, it removes regions that cannot be modeled and interfere with the modeling of other protein regions that can otherwise be modeled with high confidence if the interfering region is deleted. In the case of syncytin-2, the deleted region corresponds to a relatively long loop that is variable among the retroviral SU structural models (see below). Thus, the syncytin-2 SU structure modeled here is significantly different from the syncytin-2 SU model in the AlphaFold protein structure database (20) without the deletion (UniProt accession number P60508), which has generally lower-quality pLDDT scores in the SU region. The quality of the SU models obtained allowed the comparison of SU domain organizations and overall secondary and tertiary structural elements among a wide range of orthoretroviruses. The low-confidence regions do not impact these analyses as they are often located in terminal or loop regions that are not critical to compare overall models and structures.
FIG 3.

pLDDT scores mapped on orthoretroviral SU models. pLDDT scores are mapped on cartoon representations of type C gammaretroviruses (A), syncytin-1 (B), syncytin-2 (C), type D gammaretroviruses (D), syncytin-Mar1 (E), syncytin-Car1 and Rum1 (F), Env-Aja and Env-Psc (G), ALV (H), and Mab-Env3 and Mab-Env4 (I). Models are shown in the same order and orientation as in Fig. 2. The pLDDT score scale is shown next to panel A. Low-confidence model regions with scores below 50 are shown in red. Unstructured PRR and linker regions are shown as dotted lines. The RBD, C-domains, PRR, and linker regions of type C, type D, syncytin-1, and syncytin-Mar1 are indicated. The deletion in the syncytin-2 model is indicated as dotted lines in panel C.
Two major SU groups with different amino-terminal RBDs were identified within the gammaretroviruses (Fig. 2A, B, D, and E). One group includes the SU of type C gammaretroviruses with an RBD similar to those in the previously described MLV and FELV RBD crystal structures (12, 13) (Fig. 2A). The other group includes the type D gammaretroviruses as well as the SUs of human syncytin-1 and rodent syncytin-Mar1 (Fig. 2B, D, and E). In neither group, the position of the RBD relative to other domains of the models may be reliable due to the long and apparently flexible linkers between domains. The type D gammaretroviruses have an amino-terminal RBD that consists of two topologically similar β-sheets (Fig. 4A and B). The syncytin-1 and syncytin-Mar1 SU models have RBDs structurally similar to the carboxy-terminal subdomain of the REV RBD model despite little to no sequence similarity (Fig. 4B to D). No significant structural similarities were observed between the RBDs of type C and D gammaretroviruses.
FIG 4.
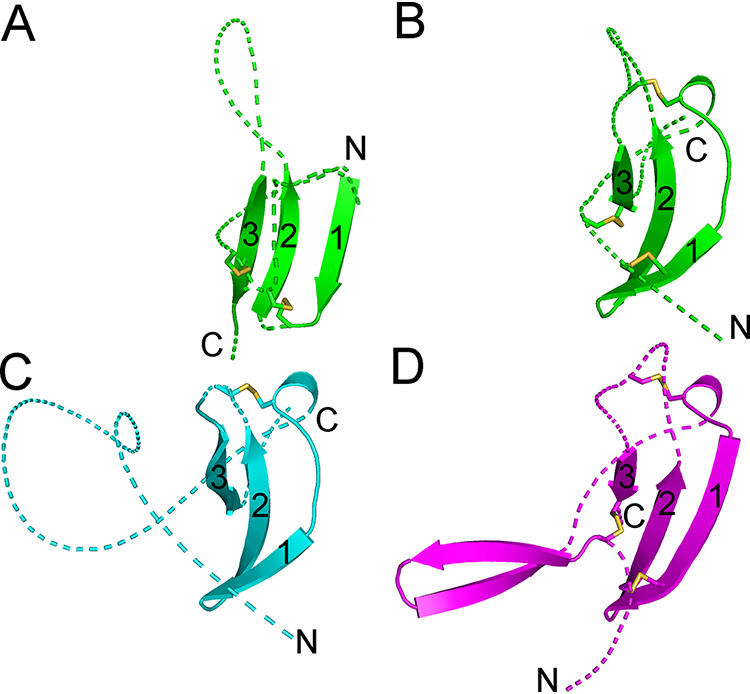
RBDs of type D gammaretroviruses, syncytin-1, and syncytin-Mar1. (A) First, amino-terminal β-sheet of the type D REV RBD. (B) Second, carboxy-terminal REV RBD β-sheet. (C) Syncytin-1 RBD. (D) Syncytin-Mar1 RBD. The β-sheets are shown in the same orientation in all panels with the three sequential β-strands indicated by numbers. Loops linking β-strands and flanking the β-sheet region are shown as dotted lines for clarity. Disulfides are shown as sticks. The amino and carboxy termini of each section are indicated.
An independently folding carboxy-terminal domain in the SU of type C and D gammaretroviruses, syncytin-1, and syncytin-Mar1 corresponds to the entire SU C-domain. This domain starts with a β-strand including the conserved gammaretroviral CWLC consensus motif that mediates intersubunit covalent interactions (32, 33) (Fig. 2A to G). Surprisingly, the SU models of a subset of unclassified gammaretroviral elements are comprised of a C-domain structure without an independent RBD, including the SU of the type D-like gammaretroviral syncytin-2 (Fig. 2C, F, and G). The CWLC-like consensus motif β-strand in these models is located near the SU amino terminus. Several of these gammaretroviral syncytins are fusogenic (27–30), indicating that receptor binding is mediated by the C-domain equivalents of these envelope proteins. The models for the SUs of alpharetroviruses do not have an independent amino-terminal RBD (Fig. 2H and I). The alpharetroviruses do not have a CWLC-like motif, but the first β-strand is preceded by a conserved cysteine residue (Fig. 2H and I). In the Mab-Env3 SU model, this cysteine forms a disulfide with an amino-terminal cysteine residue in an unstructured region outside the main domain, while in the ALV and Mab-Env4 SU models, these cysteines are unpaired.
Identification of a conserved orthoretroviral SU structure.
Despite the apparent structural diversity in the SU models of different retroviral groups, a highly conserved domain shared by all orthoretroviral lineages was identified (Fig. 5 and 6). This domain comprises the C-domain of type C and D gammaretroviruses and the entire SUs of unclassified gammaretroviruses without an independent RBD and alpharetroviruses. The conserved domain corresponds to the gp41-proximal HIV-1 gp120 β-sandwich (14, 19) (Fig. 6A), also conserved in the SUs of other lentiviruses and betaretroviruses (18). A structural alignment of the models excluding the amino-terminal RBD of gammaretroviruses defined 12 consensus β-strands, 10 of which have homologues in the HIV-1 gp120-proximal inner domain structure (Fig. 5). This domain thus represents a highly conserved TM-proximal domain (PD) of orthoretroviral SU. To harmonize the nomenclature of regions in the different models and structures, the consensus β-strands of the PD are numbered 1 to 12, and connecting regions are denoted sequentially from A to L (Fig. 5 and 6). The PD region homologous to the region that in HIV-1 gp120 faces the trimer axis is named “layer 1” according to its designation in HIV-1 gp120 (19). This region is structurally diverse and has lower pLDDT scores in several models. All cysteines in the PD except those in terminal β-strand 1 with the consensus CWLC motif in the gammaretroviruses are involved in disulfide bonds in the models but with some notable differences in the arrangement of disulfide bonds by structurally equivalent cysteines in different viral lineages (Fig. 5A and Fig. 6). Although distinct, all disulfides are compatible with the conserved PD β-sandwich fold. For reasons that are unclear, 4 cysteines in the type C gammaretroviral PD models form disulfides that are distinct from the experimentally determined disulfides in the SUs of MLV and the related mink cell focus-forming virus (34, 35). The experimentally determined disulfides would not be compatible with the conserved PD β-sandwich fold. However, in alpharetroviral SU, receptor binding is sufficient to disrupt a disulfide between the β-strand 8 and 11 homologues (36), suggesting that disulfide bonds may be relatively labile in that region. The conserved gammaretroviral CWLC consensus motif in β-strand 1 is expected to be located near the TM cysteine that covalently links with SU by analogy with the HIV-1 prefusion trimeric envelope crystal structure (8).
FIG 5.

Conserved orthoretroviral and filoviral PDs. (A) Orthoretroviral and filoviral PD sequences structurally aligned to the PD of Env-Aja. Note that the sequences are not aligned by sequence similarity but rather represent residue alignments in three-dimensional space to show the equivalent β-strand regions of the PD structure and the boundaries of connecting loops. The structural alignments were generated by the DALI server, except for the terminal β-strands 1 and 12. These two β-strands, which are not part of the core PD and are modeled in different orientations in different models, cannot be automatically aligned and were aligned manually. Insertions relative to the minimal Env-Aja SU model are not shown to highlight the β-strands shared by the models and structures, except for insertions in the major variable loop F and K regions, which are shown by the number of amino acid residues in parentheses. The lengths of all main loop regions within the PD are shown at the bottom of Fig. 7. Only one representative of each orthoretroviral and filoviral lineage is shown. The consensus β-strands are indicated by boxes and arrows. Cysteine residues are highlighted in yellow. Disulfide bonds in models and structures are shown below the structural alignment in matching lowercase letters, with the last line indicating the experimentally determined disulfide bonds in the MLV SU. The proximal domain is highlighted in green, except for the apical, layer 1, and loop K regions, which are highlighted in red, blue, and cyan. Terminal β-strands 1 and 12 are highlighted in orange. The boundaries of the HIV-1 gp120 and EBOV GP1 sequences correspond to residues labeled as aa 31 to 497 of chain A and aa 62 to 187 of chain I in the structures under PDB accession numbers 3JWD and 3CSY, respectively. (B) Schematic representation of the PD structures and major extended regions in different viral lineages. Regions are colored as described above for panel A, with the PD, apical region, layer 1, and K regions shown in green, red, blue, and cyan. The location of HIV-1 gp41 relative to the PD in the trimer structure is shown in gray. The asterisk in layer 1 indicates a β-strand that sometimes extends the β-sheet formed by extended β-strands 7 and 10. The conserved disulfide bond between β-strands 5 and 6 in the apical domain is shown as an orange line. The regions with major expansions in different lineages are indicated. MLD, mucin-like domain.
FIG 6.
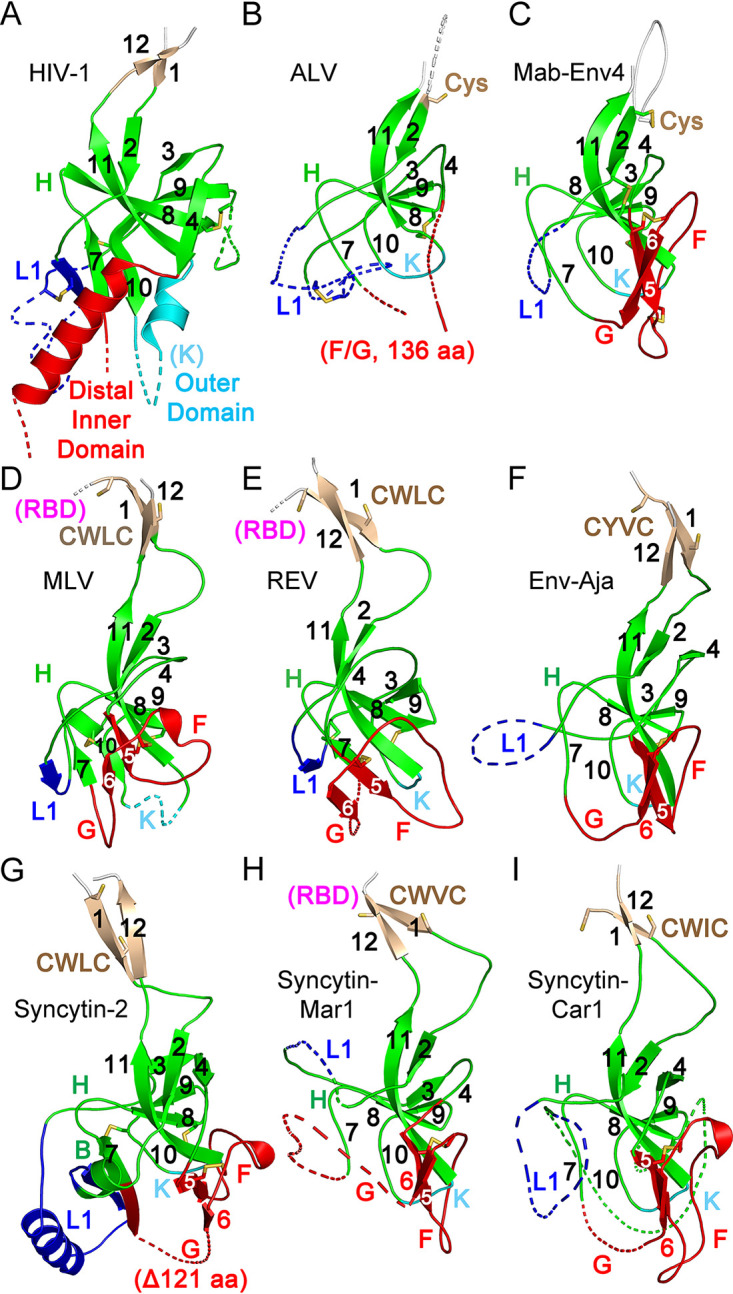
Conserved orthoretroviral SU proximal domain β-sandwich. (A) Structure of the HIV-1 gp120 PD region (PDB accession number 3JWD). (B to I) PD regions of the ALV (B), Mab-Env4 (C), MLV (D), REV (E), Env-Aja (F), syncytin-2 (G), syncytin-Mar1 (H), and syncytin-Car1 (I) SU models. Parts of layer 1 (L1) and regions F and G and the HIV-1 gp120 distal inner and outer domains are shown as dotted lines for clarity. The PD, apical domain, and layer 1 regions are shown in green, red, and blue. Selected loop and β-strand regions are labeled in each panel. Cysteine residues are shown as sticks. The locations of the conserved CWLC consensus motifs of gammaretroviral SU in β-strand 1 are indicated in panels D to I. The connections to the RBD in the models in panels D, E, and H are shown in magenta. All structures and models are shown in the same orientation as those in Fig. 2 and 3.
Structural conservation of the PD β-sandwich is observed despite the absence of any significant sequence similarity between orthoretroviral groups within the region (Fig. 5A). The structural conservation includes the complex topological arrangement of β-strands and their connections within the β-sandwich (Fig. 5B and Fig. 6). In all models, the chain between β-strands 2 and 4 circles around the conserved domain in the same clockwise direction as that observed from the virion surface (Fig. 5B and Fig. 6). The β-sheet formed by antiparallel strands 8, 9, and 11 have β-strand 8 centrally located between the other two β-strands. The two β-strands that are observed in the PDs of all SU models but not in HIV-1 gp120 are parallel β-strands 5 and 6, which extend the β-sheet formed by antiparallel β-strands 9, 8, and 11 (Fig. 5B and Fig. 6). These two β-strands are joined in a right-handed configuration by disulfide-constrained loop F, which wraps around the beginning of β-strand 11 in most models except in the SUs of type C gammaretroviruses, which have a shorter loop F. Together with loop G, these form an apical region in the conserved PD. Analysis of the previously described betaretroviral SU models (18) identified β-strand 5 and 6 homologues, including the conserved disulfide, and region F and G homologues. Thus, the SU of lentiviruses is unique in that the region corresponding to the apical region of the orthoretroviral PD forms an α-helix that projects toward the distal end of the inner domain (14, 18) rather than a conserved loop.
Structural alignments with significant DALI similarity Z-scores (37) were obtained between most SU models (Fig. 7). The exceptions are the HIV-1 gp120 crystal structure and the betaretroviral mouse mammary tumor virus (MMTV) SU model, which had the lowest overall similarity scores and failed to automatically align with some SU models. The highest average structural similarity scores were observed for the SU models of human syncytin-2 and the unclassified gammaretroviral Env-Aja endogenous element. These SU models have PD structures that are the closest to the consensus of all models. The SU models with the highest DALI Z-scores when aligned with the syncytin-2 SU model were derived from Env-Aja followed by the type D gammaretroviruses, gammaretroviral syncytins, the Env-Psc percomORF endogenous element, ALV and the related alpharetroviral lizard syncytins Mab-Env3 and Mab-Env4, type C gammaretroviruses and betaretroviruses, and, finally, the crystal structure of HIV-1 gp120 (Fig. 7). The conservation of very specific structural features indicates a common origin for the PDs of different orthoretroviral lineages.
FIG 7.

Orthoretroviral SU and filoviral GP1 DALI structural similarity Z-score matrix. The viral lineages and the scale for DALI Z-scores are shown on the right. The lengths in amino acids of selected PD regions of different viruses and endogenous elements are shown below the matrix. Color-coding indicates lengths below (blue tones), above (red tones), or equal to the median for each region.
Differential expansion of the PD in the orthoretroviruses.
The SU of most alpha- and gammaretroviruses is formed by expansions and extensions of the PD, as has been observed for the betaretroviruses and lentiviruses but with notable differences among and within lineages (Fig. 6). The PD of the type C and D gammaretroviruses does not have long internal expansions analogous to the betaretroviral and lentiviral PD expansions (Fig. 6D and E). Instead, the major expansion of the PD of type C and D gammaretroviruses, syncytin-1, and syncytin-Mar1 is the amino-terminal linker and RBD (Fig. 2A, B, D, and E and Fig. 6D, E, and H). As mentioned above, the SUs of the unclassified gammaretroviral endogenous elements Env-Aja, syncytin-2, syncytin-Car1, syncytin-Rum1, and Env-Psc do not have a domain comparable to the type C and D gammaretroviral RBDs (Fig. 2C, F, and G). Instead, the syncytin-2 SU has a long, unmodeled expansion of loop G that is topologically equivalent to the betaretroviral SU apical region (Fig. 6G). The syncytin-Car1, syncytin-Rum1, and Env-Psc SUs are more compact, with smaller expansions of different subsets of layer 1, the section from regions C to D, and apical loop G (Fig. 2F and G and Fig. 7). The Env-Aja SU is comprised of a PD with no significant extensions or expansions relative to other models (Fig. 2G and Fig. 6F). The minimalistic SU domain organization of Env-Aja combined with its near-consensus PD structure indicates that it is closely related structurally to a basal orthoretroviral SU.
The avian gamma-type alpharetroviral ALV SU model has a domain organization that is analogous to those of the SUs of betaretroviruses and syncytin-2 (Fig. 6B). Similarities include a relatively long expansion of apical regions F and G but not region K. The alpharetrovirus-like syncytins Mab-Env3 and Mab-Env4 have PD structures that most closely resemble the PD of the ALV SU but without any extensions of region G or any other region relative to the ALV SU (Fig. 2I, Fig. 6C, and Fig. 7). These two endogenous elements, like the Env-Aja SU, also have a minimal SU comprised of a PD without significant extensions except for a slightly longer loop F and layer 1 than those of Env-Aja (Fig. 7).
Conservation of the PD in filoviral GP1.
The structural similarities among the SUs of distantly related orthoretroviruses can be observed despite any obvious similarities in the underlying sequences. Given the sequence and structural similarities between the TM subunit of retroviruses and the transmembrane envelope protein GP2 of filoviruses, it is possible, although it has not been reported, that these structural similarities extend to filoviral GP1. Visual inspection of EBOV trimeric envelope protein crystal structures (38, 39) revealed that the virion-proximal region of the GP1 subunit, including the base and head subdomains, has a fold similar to the conserved orthoretroviral PD β-sandwich (Fig. 8A). The topology of the chain in the filoviral PD homologue is the same as that for the orthoretroviral PD. Structural similarities include the clockwise turn of the chain around the domain between the β-strand 2 and 4 homologues and the relative positions of the antiparallel β-strand 8, 9, and 11 homologues. In addition, the apical region formed by the β-strand 5 and 6 and loop F homologues of EBOV GP1 is structurally similar to the apical region of orthoretroviral SU models, including the conserved disulfide (Fig. 5A and Fig. 8A). Differences include a slightly expanded region B; a slightly longer loop F than those of the gammaretroviral SU, Mab-Env3, and Mab-Env4; and continuous β-strands 1/2 and 11/12 in the proximal region in EBOV GP1. Structural alignments between EBOV GP1 and the SUs of orthoretroviruses were significant in all cases except for the SUs of MMTV and HIV-1 gp120, with the highest similarity scores for the alignments with the consensus Env-Aja and syncytin-2 model structures (Fig. 7). The glycan cap and mucin-like domains that are cleaved prior to the binding of GP1 to the receptor in the endosome (38, 40) are thus carboxy-terminal extensions of the conserved β-sandwich PD structure of GP1 (Fig. 5B).
FIG 8.

Conservation and orientation of the PD in EBOV GP1. (A) EBOV GP1 base and head subdomains (PDB accession number 3CSY) comprising the PD, shown in an orientation similar to that of the PD of orthoretroviruses in Fig. 6. The β-strand and selected loop regions are labeled and colored as described in the legends of Fig. 5 and 6. Disulfides in the apical region are shown as sticks. (B and C) EBOV (PDB accession number 5HJ3) (B) and HIV-1 (PDB accession number 4TVP) (C) trimeric envelope protein crystal structures shown from the top, distal side. The apical domain in the GP1 protomers in panel B and the distal region of the inner domains and the outer domains of gp120 in panel C are omitted for clarity. The β-sandwich, terminal β-strand, and layer 1 homologues are shown in green, wheat, and blue. Region H and β-strand 3 homologues are shown in red and light blue and highlighted with red and light blue asterisks. The arrows in panel B indicate the clockwise direction of the major 120° rotation of GP1 relative to gp120 protomers in the trimeric structures. (D and E) The same structures as the ones in panels B and C, rotated 90°, with the virion-proximal side facing up.
The most remarkable difference between the PDs of HIV-1 gp120 and EBOV GP1 is in their orientations within envelope protein trimers. The EBOV GP1 PD is rotated clockwise by approximately 120° around the axis perpendicular to the viral membrane compared to the HIV-1 PD as seen from the top of the trimer (Fig. 8B and C). This results in different sets of PD regions occluded in the envelope protein trimers of HIV-1 gp120 and EBOV GP1. For instance, whereas region H and β-strand 3 in the gp120 protomers of the HIV-1 trimeric envelope protein crystal structure face the trimer axis and surface, respectively, the opposite is true for the homologous regions of GP1 protomers in the EBOV trimeric envelope protein crystal structure (Fig. 8B to E). As a consequence, none of the intersubunit contacts mediated by residues in the PD are structurally equivalent in these two viruses.
Orientation of the PD of gammaretroviruses in trimeric envelope proteins.
The discrepancy between lentiviral and filoviral PD orientations in the trimeric envelope protein structures raises the question of whether the orientation of the PD in envelope protein trimers of other orthoretroviruses is more similar to the orientation of the PD in filoviruses or lentiviruses. This was addressed by analysis of glycosylation patterns in the PD of gammaretroviruses. Glycosylation sites would not be expected in PD regions closely associated with the TM subunit. In fact, the HIV-1 PD surface that interacts with the trimer axis, including region H, is devoid of glycosylation sites (14, 17). In contrast, region H of the PD of some extant type C and D gammaretroviruses has a glycosylation site (see Fig. 10), suggesting that the PD orientation in envelope protein trimers differs between the gammaretroviruses and lentiviruses. The gammaretroviral PD surface equivalent to the EBOV PD surface interacting with GP2 is mostly devoid of glycosylation sites (Fig. 9D and I), suggesting that the gammaretroviral PD orientation in trimeric structures is similar to that of the EBOV PD or intermediate between those of the filoviruses and lentiviruses.
FIG 10.
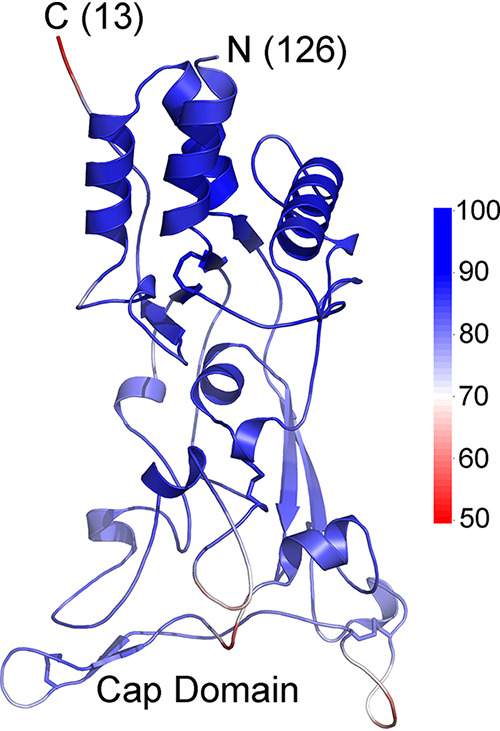
African green monkey simian foamy retrovirus SU model. The modeled structure is shown with colors indicating pLDDT scores along the chain, with the scale shown to the right. The amino and carboxy termini of the model are indicated with the number of residues not modeled on each end. The amino-terminal section outside the model shown has helical regions that do not pack with the rest of the SU and are not structurally similar to the SU of orthoretroviruses. Disulfide bonds are indicated by sticks. All cysteine residues in the modeled region participate in disulfide bonds. A cap minidomain located distally relative to the chain termini is indicated.
FIG 9.

Glycosylation sites of gammaretroviral PD regions. (A) Cartoon representation of the FELV PD model. (B) Space-filling representation of the same model as the one in panel A. (C to E) Sequential 90° rotations of the model in panel B. (F) Cartoon representation of the MPMV PD model. (G) Space-filling representation of the same model as the one in panel F. (H to J) Sequential 90° rotations of the model in panel G. Regions are colored as described in the legend of Fig. 6, with the PD, apical region, layer 1, and K regions shown in green, red, blue, and cyan. Asparagine residues in potential glycosylation sites are shown in yellow. The H region that in HIV-1 is closely associated with gp41 is indicated with an asterisk. Note the glycosylation site centrally located in region H in panels A to C and F to H. Regions prior to and beyond β-strands 1 and 12 are not shown. The FELV and MPMV PD surfaces homologous to the EBOV GP1 surface facing GP2 in the trimeric envelope protein structure are shown in panels D and I.
DISCUSSION
The structural models presented here provide a unified view of the structure of the SU of most biomedically important orthoretroviruses and related endogenous elements that play critical roles in placentation. This is likely to extend to the SUs of deltaretroviruses and all lentiviruses for which structures or reliable models have not yet been obtained. Furthermore, the same structural type is also observed in the GP1 protein of filoviruses, thus unifying the envelope proteins of these different viral families, including the receptor-binding subunits, into a single structural protein family. AlphaFold consistently yields models with similar features despite the absence of sequence similarity between different viral lineages in the regions modeled. The number of models converging to similar structures and the relatively high pLDDT reliability scores in the core PD regions indicate robust and reliable models, especially in the PD region. Importantly, none of the retroviral SU structural models were derived by using either HIV-1 gp120, EBOV GP1, or any other known PD-like structures as templates, and thus, the similarities of the PD models to these proteins arise not due to starting from a gp120- or GP1-like structure modified by threading-like techniques but rather from the principles embedded in AlphaFold’s neural networks, not informed about expected structural features of the models. Essentially, the models were generated de novo and not based on preexisting structures but nonetheless converged into structures similar to those of the gp120 inner-proximal domain and the EBOV GP1 base and cap subdomains. In fact, the consensus of all models is neither HIV-1 gp120 nor EBOV GP1 that would be expected for threading-like modeling but rather the model for the SU of a fish endogenous element. In addition, modeling of the SU of spumaretroviruses, the sister group of the orthoretroviruses within the Retroviridae, with AlphaFold 2 using the same parameters results in high-reliability models that are structurally distinct from the orthoretroviral SU models, with no PD structure equivalent and unrelated to any known structure in the Protein Data Bank (Fig. 10). This indicates that AlphaFold does not default to PD-like structures when modeling retroviral SU structures and that the PD structure is restricted to the orthoretroviruses within the Retroviridae. Some regions are poorly modeled, for example, the apical regions of the syncytin-2 and ALV SUs and, perhaps, the type D gammaretroviral RBDs. The overall domain organization of the SUs of different orthoretroviruses can nonetheless be gleaned from the models to identify the main shared structural features and the structural evolution of these proteins.
The major insight that emerges from the unified structural view is that at the center of the structurally diverse and rapidly evolving receptor-interacting SU of orthoretroviruses and filoviruses sits a structurally conserved but evolvable domain, the PD β-sandwich. Extreme sequence variation is tolerated within the PD β-sandwich while retaining its basic structure. At the same time, the PD allows large expansions that emanate from several loops within the domain or its termini, resulting in a diversity of SU and GP1 structures with important functions in receptor interaction and immune evasion in different viral lineages.
A second insight emerging from this unified structural view is the age of the conserved SU PD β-sandwich structure. The oldest known members of the orthoretroviral family with the conserved PD structure are the spiny-rayed fish percomORF endogenous elements. The percomORF element genomic integration event was dated to at least 110 million years ago (31), setting a lower-bound estimate for the age of the conserved orthoretroviral PD structure. The retroviral element encoding Env-Aja has not been dated, but its env gene seems to be basal to the percomORF elements by TM protein sequence analysis (31) and may therefore derive from an even older orthoretroviral lineage. This estimate indicates the age of the PD within the orthoretroviral lineage but not in the filoviruses. The ultimate origin of the orthoretroviral env gene with its conserved SU is unresolved. It is possible that orthoretroviruses acquired an env gene from another viral lineage early in their evolution, similar to env gene acquisition by long terminal repeat (LTR) retrotransposable elements of invertebrates (41–43). One possible source is an envelope protein gene from a filovirus-like or unknown or extinct viral lineage. However, due to the antiquity of orthoretroviral SUs within the orthoretroviruses, it is possible that a filovirus-like lineage directly or indirectly acquired its envelope protein gene from an orthoretrovirus with an SU structurally similar to that of Env-Aja or an unknown or extinct orthoretroviral lineage with a C-terminal extension in the PD. Deep-time divergent evolution has since erased most or all SU sequence similarities while retaining the PD structural fold. That is, evolutionary relationships in that region can now be assessed only by structural analysis rather than by traditional sequence-based phylogenetic analyses. A similar transfer based on sequence similarities between the orthoretroviral TM and GP2 was previously suggested (7). The definition of the basic shared structural elements of this conserved domain as well as the patterns of sequence variation in the structural models described here and in endogenous elements in genomic data from multiple vertebrate species may allow the identification of more distantly related members of this protein family.
An alternative to be considered is that the PD arose independently and convergently in the filoviruses and orthoretroviruses, perhaps as a structure necessary for the attachment of the terminal β-strands to the transmembrane subunits mediating fusion or fusion mechanisms. The complete lack of sequence similarity and the lack of disulfide conservation outside the apical region may suggest convergence, although a similar lack of sequence similarity and differences in disulfide bonding patterns are also observed within the orthoretroviruses (Fig. 5A). However, several considerations argue strongly against convergence of the PD in the orthoretroviruses and filoviruses. The PD fold is unique, not found in any other known protein structure outside the orthoretroviruses and filoviruses, arguing against independent emergence due to an inherent simplicity of the fold. This is in contrast to the relatively simple coiled-coil postfusion configuration of the transmembrane subunits that could have emerged independently in several viral groups. In fact, the complex structure and topology of the PD fold and the richness of details shared between the PDs of orthoretroviruses and filoviruses are unlikely to have arisen independently more than once. The details include the strict conservation in the order of 10 β-strands in the primary sequence, which is not necessary for the β-sandwich fold; the same spatial distribution of the β-strands and their relative parallel and antiparallel configurations; the long clockwise turn of the chain around the domain linking β-strands 2 and 4, which are otherwise located proximally to each other and could be joined by a simpler, shorter loop; the lack of any clear commonalities in intersubunit or terminal strand interactions mediated by the long-chain section around the domain in HIV-1 and EBOV or a clear structural necessity for this loop around the domain for the β-sandwich fold or stability; the presence of a conserved parallel β-strand apical structure involved in receptor binding in several, if not all, of these viruses (see below); and the conservation of tertiary structure configurations, such as β-strands 7 and 10, that are removed from close contact with the terminal strands and do not form equivalent intersubunit interactions in HIV-1 and EBOV. No structural principles dictate the necessity of any of these structural details for terminal strand interaction with the transmembrane subunits or β-sandwich fold and stability. In addition, the different orientations of the PDs in HIV-1 and EBOV envelope protein trimers and the unrelated contacts that the PD makes with the transmembrane subunits in these viruses argue against convergence due to intersubunit contacts. Finally, PD-like structures are not required for terminal β-strand attachment to transmembrane subunits. This is exemplified by the paramyxoviruses and orthomyxoviruses, which also have class I fusion machineries and similar β-strand attachments in the fusion protein or subunit (8) but lack any obvious structures related to the PD fold.
Convergence or divergence of the envelope proteins of retroviruses and filoviruses should be considered in the totality of these proteins, beyond the specifics of the PD structure. The envelope proteins of alpharetroviruses, gammaretroviruses, and filoviruses can be described in very similar terms, all of which could be attributed in principle to convergent evolution. In a roughly decreasing order of likelihood of convergence are quaternary structure and furin-mediated subunit processing typical of class I fusion machineries. Not strictly required for class I fusion mechanisms and less likely to be the product of convergence are covalent interactions between receptor-binding and fusion subunits through a Cys residue embedded in a conserved CX6CC motif in the latter that is not present in any other viral class I fusion machinery; significant sequence similarities between the transmembrane subunits of retroviruses and filoviruses, also not shared with any other class I fusion proteins (6, 7); and a structurally complex PD in the surface subunit. That is, both the surface and transmembrane subunits of the envelope proteins of retroviruses and filoviruses share specific signatures that are not shared with any other class I envelope proteins or strictly required for class I fusion mechanisms. Importantly, the CX6CC motif in the transmembrane subunit, a rather specific signature as interpreted within the alpha- and gammaretroviruses and shared in the same position in the filoviruses (6, 7), is not a requisite for envelope proteins with a PD structure as the lentiviruses and betaretroviruses have a corresponding CX7C motif without a covalent intersubunit interaction, which thus has no direct functional linkage with the SU PD. Therefore, the linkage of a PD in the surface subunit to a CX6CC motif in the transmembrane subunit of filoviruses and orthoretroviruses is unlikely to be a product of convergent evolution. Although any of these individual features could in principle be attributed to convergent evolution with higher or lower confidence, the compounding of several independent putative convergent events in both subunits for the evolution of similar orthoretroviral and filoviral envelope proteins, some of which are rather unlikely even in isolation, makes convergence significantly less likely than the more parsimonious interpretation of divergent evolution after horizontal gene transfer. An analogous reasoning has been used to support a single origin with divergent rather than convergent evolution of capsid proteins with the jelly-roll fold in different RNA and DNA virus families (44).
The unified structural view also provides information about the structural evolution of SUs in different orthoretroviral lineages. The structural models indicate that the divergence of alpha- and gammaretroviruses occurred before or shortly after the start of structural diversification by PD expansion. Both the alpha- and gammaretroviruses include endogenous elements encoding SU proteins consisting of minimal or almost minimal PD structures without an amino-terminal RBD or other major expansions. The most parsimonious interpretation of the data is that the minimal SUs of Env-Aja, Mab-Env3, and Mab-Env4 are closely related structurally to a basal orthoretroviral SU and that most or all of the structural diversification by terminal and internal expansions of the PD occurred independently and in parallel after the divergence of these lineages. For the beta-type envelope proteins, it is not clear if SU structural diversification occurred before or after the divergence of the betaretroviruses from the lentiviruses as no beta-type minimal SU structures have been identified. Finally, the SU of lentiviruses is the most divergent structurally within the orthoretroviruses and filoviruses, with one or more outer domains derived from region K and a structurally unique distal inner domain in place of the PD apical domain (14, 18).
An unexpected source of structural variation in this protein family is the orientation of the PD in oligomeric envelope protein structures. This results in different faces of the PD interacting with the transmembrane subunits in HIV-1 and EBOV trimeric envelope proteins. The only structurally equivalent interactions of the PD of HIV-1 and EBOV with TM and GP2 are the terminal regions of the domain outside the β-sandwich (8). Not only is the PD oriented differently in these viruses, but TM and GP2 also fold differently and make contacts with the PD that are not structurally analogous in the respective prefusion trimeric envelope protein structures. Therefore, the intersubunit interface seems to be evolutionarily malleable, with the terminal strand attachment of the PD to the transmembrane subunit being the only interaction that remained structurally conserved but with some degree of structural variation. The differences in PD orientation and intersubunit interactions mediated by the β-sandwich structure of HIV-1 gp120 and EBOV GP1 suggest that the deep-time conservation of this structure is not primarily due to the preservation of specific intersubunit interactions. Other functional activities or structural roles of the PD probably account for its deep-time structural conservation.
The conserved PD apical region structure may be a critical element in the mechanism of receptor-induced membrane fusion. Direct or indirect interaction of the PD apical region with a receptor appears to be a shared functional event for triggering membrane fusion in this protein family. This has been shown for the filoviruses, alpharetroviruses, and even the type C gammaretroviruses, which depend on sequences that include the PD apical region to induce fusion after receptor binding by the RBD (9–12, 40, 45, 46). HIV-1 gp120 interacts with its coreceptor through a structurally distinct but topologically equivalent region, the distal region of the inner domain, that connects with the rest of the PD in a manner similar to that of the apical region of other viral lineages (14). Incidentally, the structural, and, therefore, functional, equivalence between the apical regions of the PDs of alpharetroviruses, gammaretroviruses, and filoviruses, which is not predicted based on sequence analyses, emerges in the models without any functional information input, providing further support for the models. The minimal orthoretroviral SU variants identified here may serve as simple and structurally tractable systems to elucidate the basic functions of the conserved PD structure without the layers of structural complexity used by extant orthoretroviruses and filoviruses for receptor binding and immune evasion.
MATERIALS AND METHODS
Structural modeling.
The mature orthoretroviral SU sequences were used as the input for structural modeling with AlphaFold 2 (20). The GenBank accession numbers and boundaries of the sequences used for modeling were as follows: P03391 for FELV (Gardner-Arnstein strain), amino acids (aa) 28 to 465; NP_057935 for MLV (Moloney strain), aa 33 to 469; ACD35952 for PERV, aa 38 to 459; AAN77282 for PyERV, aa 26 to 416; GQ375848 for REV (strain HLJ07I), aa 37 to 398; NP_056893 for MPMV, aa 23 to 394; YP_001497149 for RD114, aa 19 to 378; YP_009109691 for BaEV, aa 19 to 376; AAF28334 for syncytin-1, aa 22 to 317; NM_001305587 for syncytin-Mar1, aa 20 to 370; NM_207582 for syncytin-2, aa 16 to 350; NM_001305592 for syncytin-Car1, aa 21 to 276; NM_001305454 for syncytin-Rum1, aa 25 to 276; KI305800 for Env-Aja, polypeptide encoded by nucleotides 10308 to 10850, antisense; JACK01022596 for Env-Psc, polypeptide encoded by nucleotides 2606 to 3280, antisense; NC_015116 for ALV subgroup J (isolate SCDY1), aa 69 to 374; MG254890 for Env-Mab3, aa 20 to 213; MG254891 for Env-Mab4, aa 16 to 215; and MF582544 for African green monkey simian foamy virus 3 (strain SFVcae_FV2014), aa 219 to 558. A version of AlphaFold 2 run on the ColabFold server (22), with graphical processing unit support accessible at https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb (September 2021), was used for modeling with templates, without amber relaxation and homooligomer set to 1. For each SU, the top model with the highest average pLDDT score was selected for analysis. A 122-amino-acid region in loop G of the syncytin-2 SU (aa 158 to 279 from the initiation codon) was removed for modeling. Initial modeling of the full-length syncytin-2 SU showed a distorted structure with parallel β-strands 5 and 6 being significantly displaced. The deleted region in the final model was determined by empirically testing different deletions in modeling to identify the shortest deletion yielding models with the β-strand 5 and 6 homologues in the apical region in the expected location of the PD. Atomic coordinates and pLDDT scores in PDB format of the retroviral SU structural models and MSA sequence hits are available in the Dryad database at https://doi.org/10.5061/dryad.m63xsj435.
Model analyses.
SU models were analyzed with PyMOL version 2.5.1 (Schrödinger, LLC). The pLDDT scores were extracted from the column corresponding to B-factors (column 11) in the PDB files with atomic coordinates of the models. Secondary structures and disulfides were automatically determined by PyMOL. Structural alignments and similarities between models and with other structures in the Protein Data Bank were determined with the DALI server at http://ekhidna2.biocenter.helsinki.fi (37). DALI similarity Z-scores above 2 were considered significant (37). The RBD and linker sequences in the SU models of gammaretroviruses, syncytin-1, and syncytin-Mar1 up to 1 residue prior to the CWLC consensus sequence were removed before running structural alignments to avoid alignments of this gammaretrovirus-specific domain.
Contributor Information
Isidro Hötzel, Email: ihotzel@gene.com.
Frank Kirchhoff, Ulm University Medical Center.
REFERENCES
- 1.Johnson WE. 2019. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol 17:355–370. 10.1038/s41579-019-0189-2. [DOI] [PubMed] [Google Scholar]
- 2.Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang X-Y, Edouard P, Howes S, Keith JC, McCoy JM. 2000. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403:785–789. 10.1038/35001608. [DOI] [PubMed] [Google Scholar]
- 3.Henzy JE, Johnson WE. 2013. Pushing the endogenous envelope. Philos Trans R Soc Lond B Biol Sci 368:20120506. 10.1098/rstb.2012.0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henzy JE, Coffin JM. 2013. Betaretroviral envelope subunits are noncovalently associated and restricted to the mammalian class. J Virol 87:1937–1946. 10.1128/JVI.01442-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallaher WR. 1996. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell 85:477–478. 10.1016/S0092-8674(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 7.Bénit L, Dessen P, Heidmann T. 2001. Identification, phylogeny, and evolution of retroviral elements based on their envelope genes. J Virol 75:11709–11719. 10.1128/JVI.75.23.11709-11719.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang G-Y, Ofek G, Stewart-Jones GBE, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. 2014. Structure and immune recognition of trimeric prefusion HIV-1 Env. Nature 514:455–461. 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavillette D, Boson B, Russell SJ, Cosset F-L. 2001. Activation of membrane fusion by murine leukemia viruses is controlled in cis or in trans by interactions between the receptor-binding domain and a conserved disulfide loop of the carboxy terminus of the surface glycoprotein. J Virol 75:3685–3695. 10.1128/JVI.75.8.3685-3695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnett AL, Davey RA, Cunningham JM. 2001. Modular organization of the Friend murine leukemia virus envelope protein underlies the mechanism of infection. Proc Natl Acad Sci USA 98:4113–4118. 10.1073/pnas.071432398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnett AL, Cunningham JM. 2001. Receptor binding transforms the surface subunit of the mammalian C-type retrovirus envelope protein from an inhibitor to an activator of fusion. J Virol 75:9096–9105. 10.1128/JVI.75.19.9096-9105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnett AL, Wensel DL, Li W, Fass D, Cunningham JM. 2003. Structure and mechanism of a coreceptor for infection by a pathogenic feline retrovirus. J Virol 77:2717–2729. 10.1128/jvi.77.4.2717-2729.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fass D, Davey RA, Hamson CA, Kim PS, Cunningham JM, Berger JM. 1997. Structure of a murine leukemia virus receptor-binding glycoprotein at 2.0 angstrom resolution. Science 277:1662–1666. 10.1126/science.277.5332.1662. [DOI] [PubMed] [Google Scholar]
- 14.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hötzel I, Cheevers WP. 2001. Conservation of human immunodeficiency virus type 1 gp120 inner-domain sequences in lentivirus and type A and B retrovirus envelope surface glycoproteins. J Virol 75:2014–2018. 10.1128/JVI.75.4.2014-2018.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hötzel I. 2008. Conservation of inner domain modules in the surface envelope glycoproteins of an ancient rabbit lentivirus and extant lentiviruses and betaretroviruses. Virology 372:201–207. 10.1016/j.virol.2007.10.038. [DOI] [PubMed] [Google Scholar]
- 17.Wyatt R, Desjardin E, Olshevsky U, Nixon C, Binley J, Olshevsky V, Sodroski J. 1997. Analysis of the interaction of the human immunodeficiency virus type 1 gp120 envelope glycoprotein with the gp41 transmembrane glycoprotein. J Virol 71:9722–9731. 10.1128/JVI.71.12.9722-9731.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hötzel I. 2022. Domain organization of lentiviral and betaretroviral surface envelope glycoproteins modeled with AlphaFold. J Virol 96:e01348-21. 10.1128/JVI.01348-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pancera M, Majeed S, Ban Y-EA, Chen L, Huang C, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci USA 107:1166–1171. 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, Bridgland A, Cowie A, Meyer C, Laydon A, Velankar S, Kleywegt GJ, Bateman A, Evans R, Pritzel A, Figurnov M, Ronneberger O, Bates R, Kohl SAA, Potapenko A, Ballard AJ, Romera-Paredes B, Nikolov S, Jain R, Clancy E, Reiman D, Petersen S, Senior AW, Kavukcuoglu K, Birney E, Kohli P, Jumper J, Hassabis D. 2021. Highly accurate protein structure prediction for the human proteome. Nature 596:590–596. 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mirdita M, Ovchinnikov S, Steinegger M. 2021. ColabFold—making protein folding accessible to all. bioRxiv 2021.08.15.456425. [DOI] [PMC free article] [PubMed]
- 23.Cornelis G, Funk M, Vernochet C, Leal F, Tarazona OA, Meurice G, Heidmann O, Dupressoir A, Miralles A, Ramirez-Pinilla MP, Heidmann T. 2017. An endogenous retroviral envelope syncytin and its cognate receptor identified in the viviparous placental Mabuya lizard. Proc Natl Acad Sci USA 114:E10991–E11000. 10.1073/pnas.1714590114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim NY, Lee D, Lee J, Park EW, Jung W-W, Yang JM, Kim YB. 2009. Characterization of the replication-competent porcine endogenous retrovirus class B molecular clone originated from Korean domestic pig. Virus Genes 39:210–216. 10.1007/s11262-009-0377-7. [DOI] [PubMed] [Google Scholar]
- 25.Huder JB, Böni J, Hatt J-M, Soldati G, Lutz H, Schüpbach J. 2002. Identification and characterization of two closely related unclassifiable endogenous retroviruses in pythons (Python molurus and Python curtus). J Virol 76:7607–7615. 10.1128/jvi.76.15.7607-7615.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonigo P, Barker C, Hunter E, Wain-Hobson S. 1986. Nucleotide sequence of Mason-Pfizer monkey virus: an immunosuppressive D-type retrovirus. Cell 45:375–385. 10.1016/0092-8674(86)90323-5. [DOI] [PubMed] [Google Scholar]
- 27.Blaise S, de Parseval N, Bénit L, Heidmann T. 2003. Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc Natl Acad Sci USA 100:13013–13018. 10.1073/pnas.2132646100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Redelsperger F, Cornelis G, Vernochet C, Tennant BC, Catzeflis F, Mulot B, Heidmann O, Heidmann T, Dupressoir A. 2014. Capture of syncytin-Mar1, a fusogenic endogenous retroviral envelope gene involved in placentation in the Rodentia squirrel-related clade. J Virol 88:7915–7928. 10.1128/JVI.00141-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cornelis G, Heidmann O, Bernard-Stoecklin S, Reynaud K, Véron G, Mulot B, Dupressoir A, Heidmann T. 2012. Ancestral capture of syncytin-Car1, a fusogenic endogenous retroviral envelope gene involved in placentation and conserved in Carnivora. Proc Natl Acad Sci USA 109:E432–E441. 10.1073/pnas.1115346109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornelis G, Heidmann O, Degrelle SA, Vernochet C, Lavialle C, Letzelter C, Bernard-Stoecklin S, Hassanin A, Mulot B, Guillomot M, Hue I, Heidmann T, Dupressoir A. 2013. Captured retroviral envelope syncytin gene associated with the unique placental structure of higher ruminants. Proc Natl Acad Sci USA 110:E828–E837. 10.1073/pnas.1215787110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henzy JE, Gifford RJ, Kenaley CP, Johnson WE. 2017. An intact retroviral gene conserved in spiny-rayed fishes for over 100 My. Mol Biol Evol 34:634–639. 10.1093/molbev/msw262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinter A, Kopelman R, Li Z, Kayman SC, Sanders DA. 1997. Localization of the labile disulfide bond between SU and TM of the murine leukemia virus envelope protein complex to a highly conserved CWLC motif in SU that resembles the active-site sequence of thiol-disulfide exchange enzymes. J Virol 71:8073–8077. 10.1128/JVI.71.10.8073-8077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim FJ, Manel N, Garrido EN, Valle C, Sitbon M, Battini J-L. 2004. HTLV-1 and -2 envelope SU subdomains and critical determinants in receptor binding. Retrovirology 1:41. 10.1186/1742-4690-1-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linder M, Linder D, Hahnen J, Schott H, Stirm S. 1992. Localization of the intrachain disulfide bonds of the envelope glycoprotein 71 from Friend murine leukemia virus. Eur J Biochem 203:65–73. 10.1111/j.1432-1033.1992.tb19828.x. [DOI] [PubMed] [Google Scholar]
- 35.Linder M, Wenzel V, Linder D, Stirm S. 1994. Structural elements in glycoprotein 70 from polytropic Friend mink cell focus-inducing virus and glycoprotein 71 from ecotropic Friend murine leukemia virus, as defined by disulfide-bonding pattern and limited proteolysis. J Virol 68:5133–5141. 10.1128/JVI.68.8.5133-5141.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith JG, Cunningham JM. 2007. Receptor-induced thiolate couples Env activation to retrovirus fusion and infection. PLoS Pathog 3:e198. 10.1371/journal.ppat.0030198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holm L. 2020. DALI and the persistence of protein shape. Protein Sci 29:128–140. 10.1002/pro.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454:177–182. 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bornholdt ZA, Ndungo E, Fusco ML, Bale S, Flyak AI, Crowe JE, Jr, Chandran K, Saphire EO. 2016. Host-primed Ebola virus GP exposes a hydrophobic NPC1 receptor-binding pocket, revealing a target for broadly neutralizing antibodies. mBio 7:e02154-15. 10.1128/mBio.02154-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, Shi Y, Song J, Qi J, Lu G, Yan J, Gao GF. 2016. Ebola viral glycoprotein bound to its endosomal receptor Niemann-Pick C1. Cell 164:258–268. 10.1016/j.cell.2015.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koonin EV, Dolja VV, Krupovic M. 2015. Origins and evolution of viruses of eukaryotes: the ultimate modularity. Virology 479–480:2–25. 10.1016/j.virol.2015.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayward A. 2017. Origin of the retroviruses: when, where, and how? Curr Opin Virol 25:23–27. 10.1016/j.coviro.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malik HS, Henikoff S, Eickbush TH. 2000. Poised for contagion: evolutionary origins of the infectious abilities of invertebrate retroviruses. Genome Res 10:1307–1318. 10.1101/gr.145000. [DOI] [PubMed] [Google Scholar]
- 44.Holmes EC. 2011. What does virus evolution tell us about virus origins? J Virol 85:5247–5251. 10.1128/JVI.02203-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmen SL, Federspiel MJ. 2000. Selection of a subgroup A avian leukosis virus [ALV (A)] envelope resistant to soluble ALV (A) surface glycoprotein. Virology 273:364–373. 10.1006/viro.2000.0424. [DOI] [PubMed] [Google Scholar]
- 46.Taplitz RA, Coffin JM. 1997. Selection of an avian retrovirus mutant with extended receptor usage. J Virol 71:7814–7819. 10.1128/JVI.71.10.7814-7819.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]