

Abstract
Statins are a class of drugs widely used worldwide to manage hypercholesterolemia and the prevention of secondary heart attacks. Currently, available statins vary in terms of their pharmacokinetic and pharmacodynamic profiles. Although the primary target of statins is the inhibition of HMG-CoA reductase (HMGR), the rate-limiting enzyme in cholesterol biosynthesis, statins exhibit many pleiotropic effects downstream of the mevalonate pathway. These pleiotropic effects include the ability to reduce myocardial fibrosis, pathologic cardiac disease states, hypertension, promote bone differentiation, anti-inflammatory, and antitumor effects through multiple mechanisms. Although these pleiotropic effects of statins may be a cause for enthusiasm, there are many adverse effects that, for the most part, are unappreciated and need to be highlighted. These adverse effects include myopathy, new-onset type 2 diabetes, renal and hepatic dysfunction. Although these adverse effects may be relatively uncommon, considering the number of people worldwide who use statins daily, the actual number of people affected becomes quite large. Also, co-administration of statins with several other medications, herbal agents, and foods, which interact through common enzymatic pathways, can have untoward clinical consequences. In this review, we address these concerns.
Keywords: Statins, Pleiotropy, Myopathy, Diabetes, Drug Interactions
Graphical Abstract

1. Introduction
Statins are currently the most effective medications at lowering cholesterol. They competitively inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme of the cholesterol biosynthetic pathway. Stains lower total serum cholesterol, triglycerides (TG), and in particular, serum low-density lipoprotein cholesterol (LDL-C), which is the main culprit in the development of atherosclerosis and cardiovascular disease. Statins also upregulate hepatic low-density lipoprotein (LDL) receptors, which also aid in removing and hence decreasing circulating cholesterol levels. Currently available statins include atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, and simvastatin. These differ in their physicochemical, pharmacokinetic, and pharmacodynamic properties, influencing their potential/ultimate clinical indications.
Although the primary action of statins is the lowering of cholesterol through inhibition of HMG-CoA reductase, they have many other indirect pleiotropic effects. These include the induction of the endothelial nitric oxide synthase (eNOS), an enzyme that produces nitric oxide (NO), inhibition of arterial smooth muscle cell (SMC) proliferation, anti-inflammatory, anticancer, and immunomodulatory properties, amongst others. In this overview, we first discuss the properties of the statins that are currently available in clinical practice, then elucidate their pleiotropic mechanisms of action, and then hone on their potential adverse effects. Finally, we also discuss their dietary and drug-drug interactions, which are of clinical consequence.
2. Physio-chemical properties of statin
Functionally, all statins have the same mechanism of action; however, structural differences account for their various pharmacodynamic properties. Statins can be divided into two types with a unique HMG-like moiety that is either a lactone (type 1, e.g., lovastatin, simvastatin, mevastatin) or a carboxylic acid (type 2, e.g., fluvastatin, atorvastatin, pitavastatin, rosuvastatin) (Figure 1). These moieties, which represent the pharmacophore of the drugs, interact with the active site of the HMG-CoA Reductase (HMGR) and are larger and more hydrophobic than HMG-CoA (Istvan, 2003; Istvan, 2002). Of note, it is the carboxylic acid form that binds to the enzyme; thus, the lactone moiety must first be converted to the acid form before binding. Therefore, “lactone statins” may be viewed as prodrugs that need to be activated before they can inhibit the enzyme (Istvan, 2002). The rest of the molecules impart various degrees of lipophilicity, allowing maximum contact with the hydrophobic pocket on the enzyme (Istvan, 2002). In this regard, we can also look at the hydrophobic ring structure of these drugs, which also divides them into two groups. Type 1 statins have a decalin ring structure, along with a butyryl group (Istvan, 2003). Type 2 statins have central ring structures larger than the decalin ring, along with common fluorophenyl and methyl ethyl groups replacing the butyryl group of type 1 statins (Istvan, 2003). These structural features can influence the biological function and the binding interactions at the active site (Istvan, 2003). Lovastatin and pravastatin (type 1) are both fully natural statins that are derived from fungi, while simvastatin is a semi-synthetic statin derived from lovastatin [2]; all other statins (type 2) are synthetic.
Figure 1.
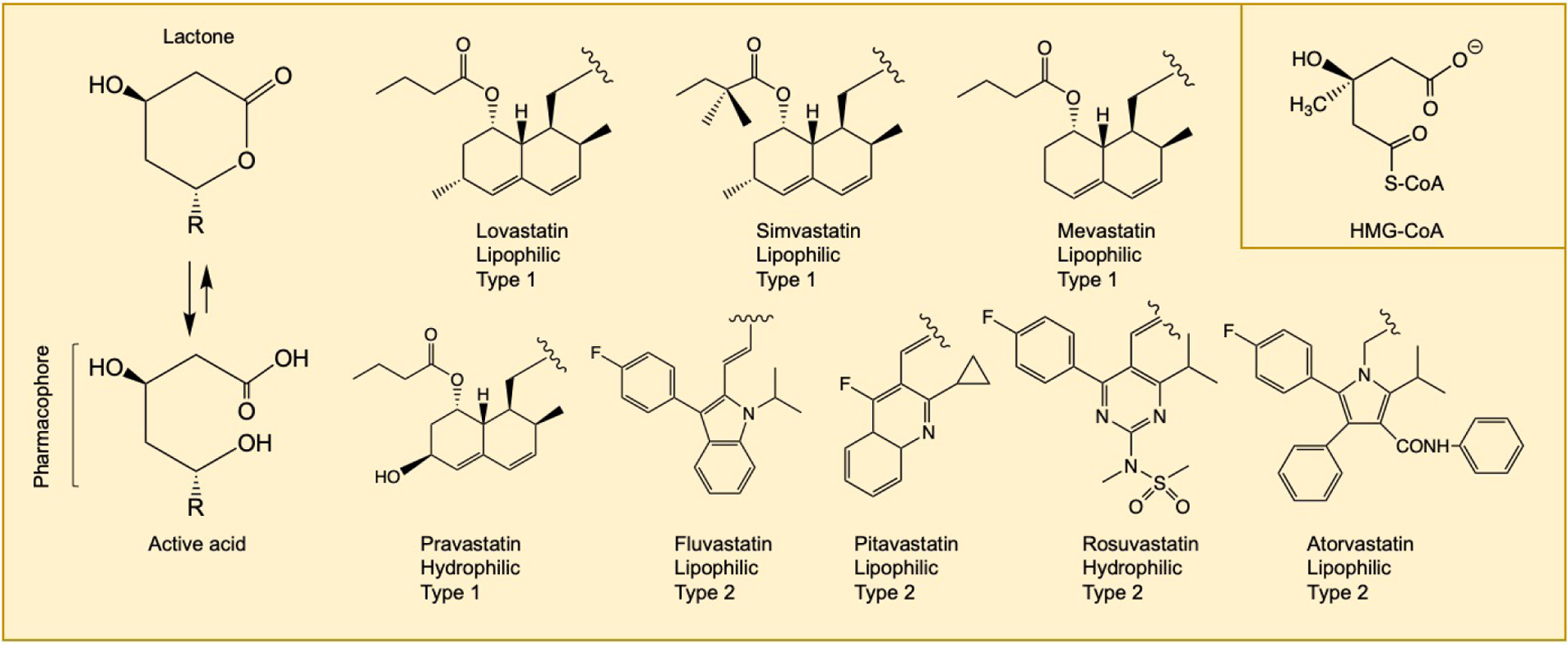
Chemical structures of statin medications. Statins exist in an equilibrium between their inactive lactone forms and their active dihydroxy-heptanoic acid forms, with equilibrium favoring the active form. Lovastatin, simvastatin, and mevastatin are administered in the lactone form, whereas all other statins are administered in their active acid form. The open acid form represents its pharmacophore and competitively inhibits HMGR. The R group determines lipophilicity, hepatic/myocyte selectivity. Type 1 statins have a decalin ring (naphthalene) structure. Type 2 statins have central ring structures larger than the decalin ring, along with common fluorophenyl and methyl ethyl groups replacing the butyryl group of type 1 statins. The structure of HMG-CoA is shown in the inset.
Hydrophilic statins may exhibit more hepatoselectivity than lipophilic ones, whereas lipophilic statins can undergo passive diffusion through both hepatic and non-hepatic tissues; hydrophilic statins undergo carrier-mediated uptake (Schachter, 2005). The increased hepatoselectivity may confer some advantages to hydrophilic statins, as evidenced by an observational cohort analysis, which found that hydrophilic statins were associated with a modest risk reduction in heart failure incidents compared to lipophilic statins (Imran et al., 2017).
2.1. Statin metabolism and excretion
Statins are active in their open-ring dihydroxy heptanoic acid form; they are considered inactive in their closed-ring lactone form. However, these two isoforms are in equilibrium with the equilibria favoring the hydroxy-acid form (Murphy et al., 2020) (Figure 1). The hydroxy-acid form, which is more stable and therefore favored in the human body, can be converted into the lactone form via glucuronidation by UGT enzymes, specifically by UGT1A1, UGT1A3 and UGT2B7, found primarily in the endoplasmic reticulum of cells of the liver, kidneys and intestines (Meech et al., 2019; Schirris et al., 2015); this can also be achieved by CoASH-dependent metabolism, potentially mediated by microsomal long-chain acyl-CoA synthetase (Li et al., 2006; Murphy et al., 2020). Conversely, the lactone form can be converted into the hydroxy-acid form by hydrolysis mediated by carboxylesterases, specifically by hCE1, found in the cytoplasm and endoplasmic reticulum of hepatocytes in the liver (Casey Laizure et al., 2013; Murphy et al., 2020). hCE1 mediated conversion is vital for lovastatin and simvastatin, which are administered in the lactone form (Casey Laizure et al., 2013). Statin uptake by the hepatocytes is crucial for their metabolism and eventual clearance. For many statins, this process is largely mediated by organic anion transporting polypeptides (OATP), specifically OATP1B1; statins that are substrates of OATP1B1 include atorvastatin, rosuvastatin, pravastatin, pitavastatin, and cerivastatin (Murphy et al., 2020). In addition, although the lipophilic nature of lovastatin and simvastatin allow for their uptake primarily through passive diffusion, their uptake is also partially mediated by OATP1B1. Statins are metabolized by the cytochrome P450 (CYP450) enzyme system found primarily in hepatocytes’ endoplasmic reticulum; the exception is pravastatin, which is not a substrate of any of the CYP450s, discussed below. Atorvastatin, lovastatin, and simvastatin are metabolized by CYP3A4 (Causevic-Ramosevac and Semiz, 2013). Fluvastatin is metabolized primarily by CYP2C9 (Causevic-Ramosevac and Semiz, 2013) but also metabolized in part by CYP3A4 and CYP2C8, although in a minor role. Both rosuvastatin and pitavastatin undergo negligible metabolism by the CYP450s (Murphy et al., 2020). Although pitavastatin metabolism is mainly accomplished by glucuronidation, CYP2C9 plays a minor role in its metabolism. Pravastatin is metabolized by sulfation in cytosolic hepatocytes. Statins are excreted primarily through the bile by active transport using multiple ATP-binding cassette (ABC) transporters, specifically multidrug resistant-associated protein (MRP2), breast cancer resistance protein (BCRP), and P-glycoprotein (Pg-P) (Murphy et al., 2020). Polymorphisms or drug-interactions involving any of these metabolic or excretory pathways can influence active drug concentration, leading to potential toxic effects or decreased efficacy.
3. Targets of statins beyond HMG-CoA reductase
Statins have pleiotropic effects beyond decreasing serum cholesterol, LDL-C, and triglyceride levels. These effects are downstream of the mevalonate pathway. These pleiotropic effects are discussed here.
3.1. Protein prenylation
One of the major mechanisms by which statins exert pleiotropic effects is by decreasing protein prenylation due to reduced production of the isoprenoid molecules farnesyl pyrophosphate and geranylgeranyl pyrophosphate (Figure 2). In the process of prenylation, the enzymes farnesyltransferase (FTase) and geranylgeranyltransferases I and II (GGTase I, GGTase II) catalyze the transfer of farnesyl or geranylgeranyl units from each of these molecules to post-translational proteins (Xu et al., 2015). Proteins with the CAAX motif on their C-terminal are targeted for prenylation by either FTase or GGTase I. The amino acid at the X position determines whether the protein is specifically targeted for farnesylation geranylgeranylation or both (Xu et al., 2015). The primary targets of protein prenylation are small GTPases. Proteins prenylated by FTase include members of the Ras family, including K-Ras, H-Ras, N-Ras, RhoE, Rheb, and RhoB. GGTase I prenylates other GTPases, including Rac1, Rac2, Ra1A, Rap1B, RhoA, RhoB, RhoC, Cdc42, Rab8, Rab11 and Rab13. Finally, GGTase II recognizes several specific motifs and prenylated proteins of the Rab family (Xu et al., 2015). Small GTPases require prenylation as a means of increasing their hydrophobic nature, allowing them to interact with cellular membranes; this is the site at which they can become activated and ultimately play a role in signal transduction pathways in many systems (Xu et al., 2015). The ability of statins to reduce the production of these isoprenoid molecules, which ultimately mediate this prenylation process, allows them to exert many pleiotropic effects.
Figure 2.
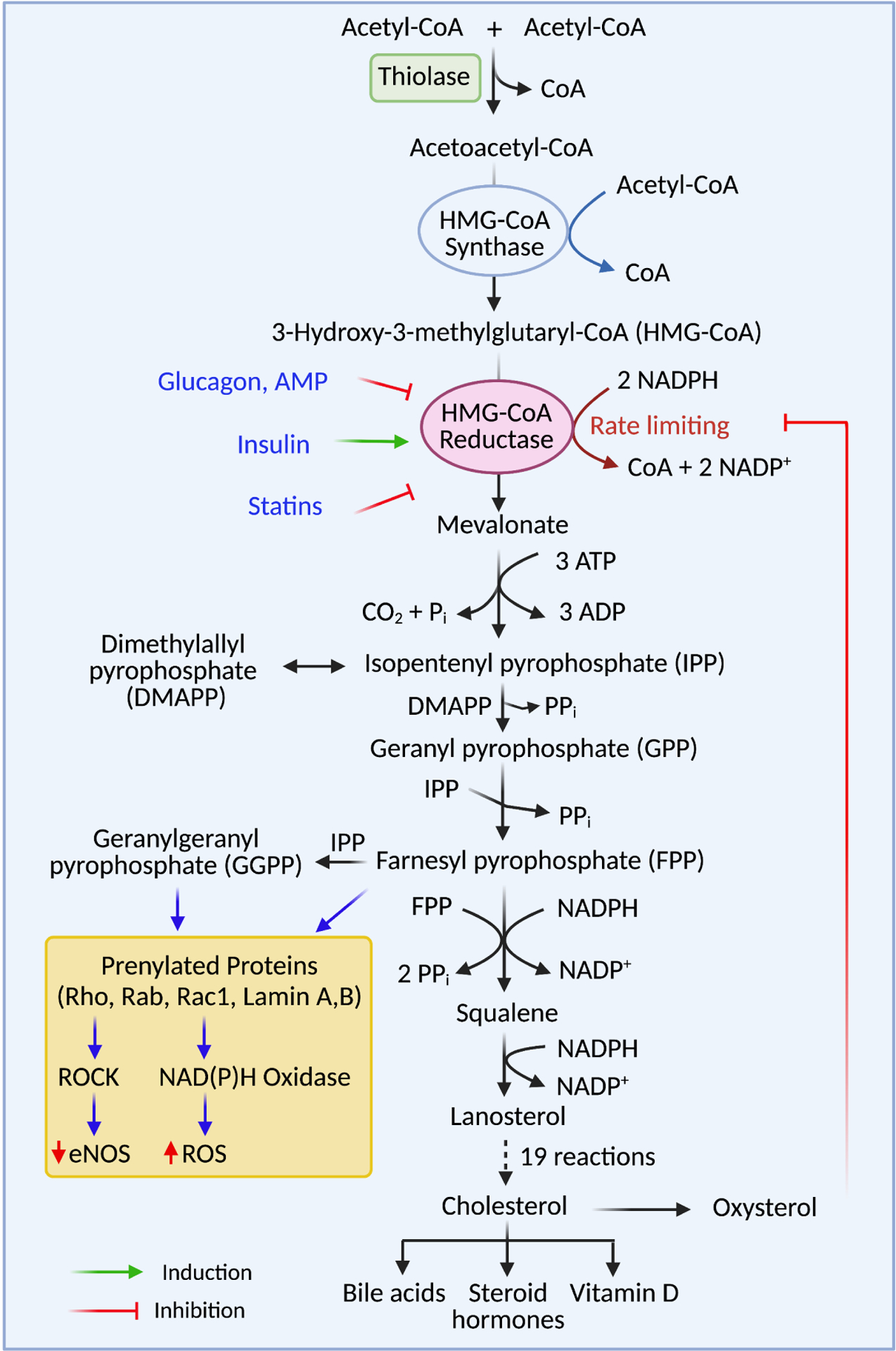
The cholesterol synthesis pathway. Cholesterol synthesis begins with two molecules of acetyl-CoA, which are converted to one molecule of acetoacetyl-CoA by the enzyme thiolase. Acetoacetyl-CoA and another molecule of acetyl-CoA are used to synthesize HMG-CoA by the enzyme HMG-CoA synthase. The rate-limiting enzyme of the pathway is HMGR. HMGR activity is stimulated or activated by several endogenous molecules, including insulin, AMP, and glucagon. Statins competitively inhibit HMGR, resulting in decreased concentrations of all downstream products. Protein prenylation is mediated by intermediate products of the cholesterol synthesis pathway, FPP and GGPP. Decreased protein prenylation decreases the activity of downstream signaling pathways such as ROCK and NADPH oxidases, accounting for many of the pleiotropic effects of statins.
3.1.1. Statins and Rho-associated kinases (ROCK)
Rho kinases (ROCK), of which there are two isoforms, are serine/threonine kinases found downstream in the Rho GTPase signal transduction pathway (Hartmann et al., 2015). The two isoforms are termed ROCK1 and ROCK2, they have a similar biological function, share 65% identity in their amino acid sequences and 92% identity in their kinase domain, but they have different subcellular distributions and binding preferences to cellular lipids (Hartmann et al., 2015; Nakamura et al., 1999; Yoneda et al., 2005). ROCK activation is modulated by RhoA GTPase, which in turn is activated through upstream signaling by a diverse group of proteins including angiotensin II, endothelin-1, fibroblast growth factor (FGF), and transforming growth factor-beta (TGF-β), amongst others (Seccia et al., 2020). ROCKs have regulatory properties in multiple tissues, including the vascular smooth muscle cells, endothelial cells, inflammatory cells, fibroblasts, and cardiac myocytes (Oesterle et al., 2017). ROCK1 has roles in inducing apoptosis in cardiomyocytes (Chang et al., 2006; Del Re et al., 2007; Seccia et al., 2020), but it may also induce cardiac fibrosis (Zhang et al., 2006) or impaired contractile function and subsequent cardiac dilation (Shi et al., 2008) in situations of pathologic hypertrophy, as demonstrated by in vivo mice experiments. In addition, ROCK1 plays a role in neointima formation by recruiting leukocytes following vascular injury (Noma et al., 2008). ROCK2, meanwhile, promotes cardiomyocyte hypertrophy along with fibrosis in pathologic conditions (Shimizu and Liao, 2016).
In endothelial cells, ROCK phosphorylation of phosphatase and tensin homology (PTEN) inhibits the pro-survival PI3K/Akt pathway and decreases eNOS expression. NO bioavailability is also decreased by ROCK through induction of NADPH oxidase, which generates reactive oxidative species (ROS) (Seccia et al., 2020). Furthermore, the Rho/ROCK pathway promotes thrombogenesis through various mechanisms; these include increases in intercellular adhesion molecule-1 (ICAM-1), tissue factor, and endothelial plasminogen activator inhibitor-1 (Eto et al., 2002; Nakakuki et al., 2005). In smooth muscle cells, ROCK activity leads to phosphorylation and inactivation of myosin light-chain phosphatase (MLCP), which causes sensitization of smooth muscle cells to calcium and contraction (Seccia et al., 2020). ROCK also promotes smooth muscle cell proliferation and migration at the genetic level via activation of the myocyte enhancer factor-2 (MEF-2) (Pagiatakis et al., 2012; Seccia et al., 2020). Altogether, leukocyte ROCK activity has been demonstrated to be elevated in patients with cardiovascular diseases such as hypertension, metabolic syndrome, dyslipidemia, coronary artery diseases, and atherosclerosis (Hata et al., 2011; Liu et al., 2007; Nohria et al., 2006; Nohria et al., 2009), as reviewed in (Shimizu and Liao, 2016). Statins can decrease ROCK activation and its downstream effects by inhibiting the formation of FTase, thereby interfering with the prenylation and activation of Rho proteins. In multiple studies, statins have been shown to decrease ROCK activity in patients with cardiac diseases, such as atherosclerosis, congestive heart failure, hypertension, coronary artery disease, and others (Liu et al., 2009; Nohria et al., 2009; Rawlings et al., 2009). Of note, this pleiotropic effect is not correlated with decreases in serum LDL cholesterol levels (Rawlings et al., 2009), thus indicating cholesterol-independent effects. In addition, this decreased ROCK activation can manifest multiple cellular effects, including reduced intimal smooth muscle cell thickening and cellular proliferation, as well as reduced myocardial fibrosis and left ventricular hypertrophy (Oesterle et al., 2017).
Statin can also improve cardiac function by inducing eNOS expression and activity (Oesterle et al., 2017). eNOS induction is due to increases in eNOS mRNA half-life, which is reversed through GGPP addition; this explains how statins increase NO production (Rikitake and Liao, 2005). Thus, RhoA/Rock inhibition is generally thought to be the driving mechanism behind eNOS induction (Margaritis et al., 2014). This mechanism also decreases miRNA-155 expression, a micro-RNA that inhibits eNOS expression (Margaritis et al., 2014).
3.1.2. Statins and Rac1
Rac1 is another Rho GTPase protein that statins can target. This GTPase protein has been implicated in cardiac disease, specifically cardiac hypertrophy. Mechanistically, Rac1 activates the enzyme NADPH oxidase. During its activation and migration to the membrane, Rac1 translocates to the membrane alongside the rest of the enzyme complex and binds guanosine triphosphate (GTP) (Mital and Liao, 2004). Once again, inhibition of isoprenylation by statins can inhibit the activation of Rac1 and thus improve cardiac hypertrophy (Mital and Liao, 2004; Oesterle et al., 2017). As mentioned earlier, inhibition of NADPH oxidase can also increase the availability of NO.
3.2. The role of statins in osteogenesis
Statins may also promote bone differentiation and protect against bone resorption through multiple mechanisms, demonstrated in many animal studies and clinical trials (Chamani et al., 2021; Chuengsamarn et al., 2010; Duan and Bonewald, 2016; Mundy et al., 1999; Sugiyama et al., 2000). In a clinical trial that compared any statin use to control patients with hyperlipidemia and osteopenia, patients in the statin arm demonstrated increased neck bone mineral density (Chamani et al., 2021; Chuengsamarn et al., 2010). In in vitro studies using mice calvarium and in vivo rat studies, simvastatin and lovastatin increased bone formation and bone volume, respectively (Mundy et al., 1999). Simvastatin, at doses of 1–200 nM, increased the expression of osteogenic transcription factors such as Runx2 and osterix (Osx) using mouse embryonal stem cells (Chamani et al., 2021; Qiao et al., 2011). Runx2 is a key gene for bone formation, increasing protein expression of osteocalcin, type I collagen, and osteopontin, to prime the differentiation of osteochondroprogenitor cells into precursor osteoblasts (Jang et al., 2012; Sinha and Zhou, 2013). Osx is subsequently important in completing the differentiation of these cells into mature osteoblasts (Sinha and Zhou, 2013). This is likely induced by the induction of the bone morphogenetic protein 2 (BMP-2) gene, upstream of these transcription factors. Statins such as simvastatin, mevastatin, and fluvastatin can promote bone formation by inducing BMP-2, which is responsible for promoting osteoblast differentiation; this effect was not observed with pravastatin at equivalent doses (Mundy et al., 1999; Sugiyama et al., 2000). Elimination of this effect upon supplementing mevalonate, FPP and GGPP suggests that this effect is downstream of HMG-CoA reductase, by decreased expressions of FPP and GGPP (Chamani et al., 2021; Ohnaka et al., 2001; Sugiyama et al., 2000; Weivoda and Hohl, 2011; Yoshida et al., 2006). Specifically, these osteogenic effects of statins may be induced by upstream activation of the Wnt/ß-catenin pathway (Chamani et al., 2021). ß-catenin is an essential protein for bone formation and allows pluripotent mesenchymal stem cells to differentiate into either osteoblasts or chondrocytes; therapy that activates the ß-catenin pathway has improved bone formation and healing (Duan and Bonewald, 2016).
In addition, because osteoblasts and adipocytes are both derived from the same mesenchymal stem cells, the administration of different agents can favor the formation of one over the other (Chamani et al., 2021). In the case of statins, both simvastatin (0.1–2.0 μM) and lovastatin (0.5–10 μM) have demonstrated the ability to inhibit differentiation into adipocytes and shunt mesenchymal formation into osteoblasts by decreasing the expression of pro-adipocyte transcription factor (PPAR-γ2) and increasing the expression of pro-osteoblast transcription factors (runx2 and osteocalcin promoter) (Li et al., 2003; Song et al., 2003). These effects may also occur secondary to statin inhibition of the Rho-kinase pathways, potentially negatively regulating bone formation (Ohnaka et al., 2001).
Statins simultaneously protect against bone resorption by preventing differentiation and activation of osteoclasts (Figure 3). In animal models, statin-induced the production of osteoprotegerin, an anti-osteoclastic protein (Kaji et al., 2005). Although the production of RANKL, an osteoclast activating ligand, was also increased by statins, they also inhibited downstream activation of NF-κB required for osteoclast activation (Kaji et al., 2005). In addition, Simvastatin induced the estrogen receptor ɑ; estrogen has a protective role in bone resorption by inhibiting osteoclast activity (Li et al., 2011).
Figure 3.
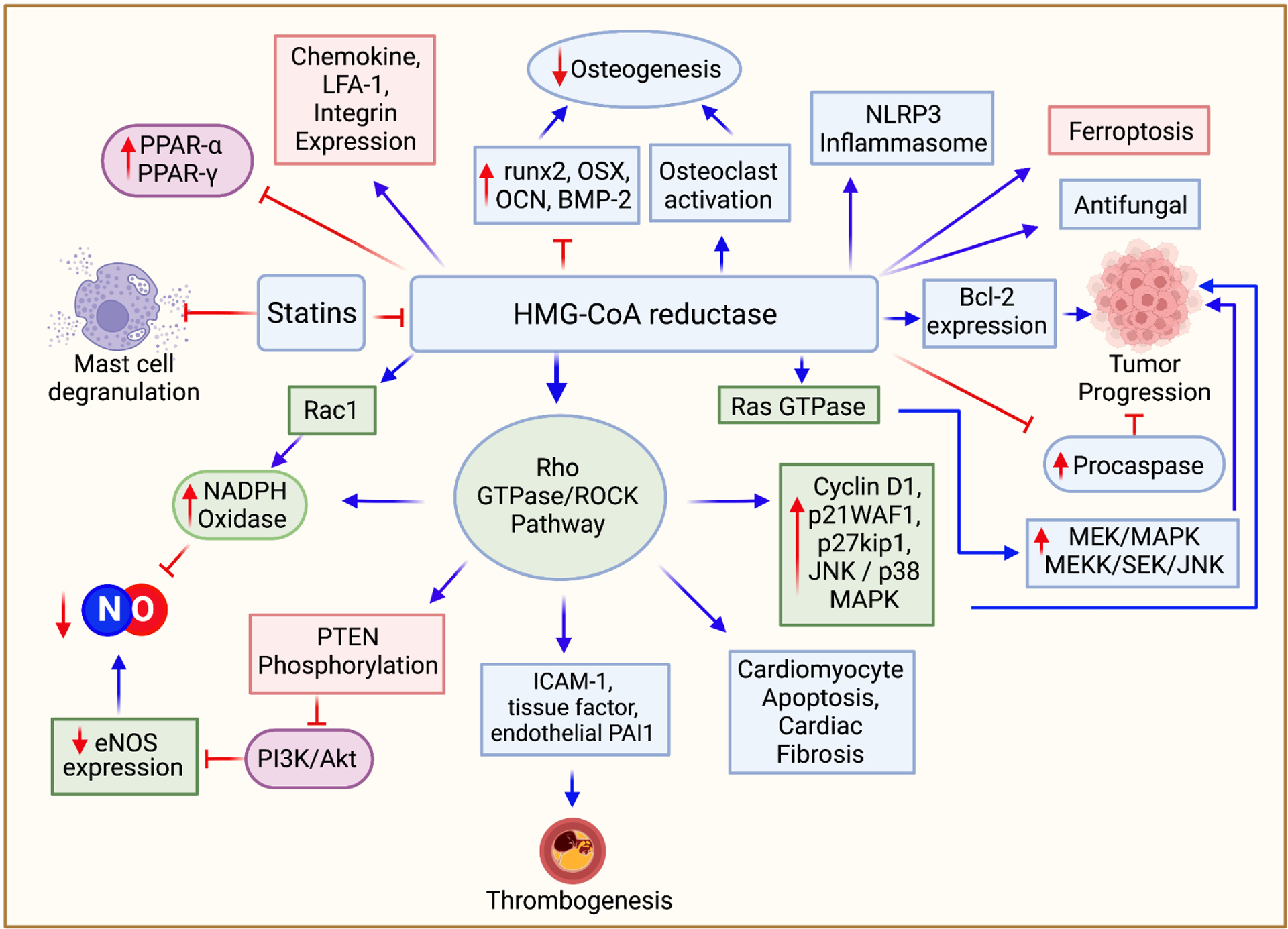
Pleiotropic effects of statins. Statins’ impact extends beyond cholesterol reduction. Inhibition of FPP and GGPP mediated protein prenylation can be the root of many pleiotropic effects. Inhibition of the Rho/ROCK pathway is responsible for many of these effects. Statin use is associated with increased induction of eNOS, increased bioavailability of NO, decreased induction of NADPH oxidase, reduced incidence of thrombogenesis, cardioprotective effects arising from decreased cardiomyocyte apoptosis, and cardiac fibrosis, and anticancer effects. Statins can induce ferroptosis by decreasing CoQ10 levels. Anti-inflammatory mechanisms of statins include inhibition of the NLRP3 inflammasome, induction of PPA receptor-α and PPA receptor-γ, inhibition of mast cell degranulation, and inhibition of chemokine and integrin expression. Statins also have potential implications in osteogenesis, inducing the differentiation of osteoblasts while simultaneously inhibiting osteoclast activation. Statins also have antifungal properties secondary to FPP inhibition.
As observed with simvastatin and atorvastatin (at doses of 10−9-10−6 M), bone cells are dose-dependently affected by statins, which inhibited osteoblast proliferation in MG-63 cells (Ruiz-Gaspa et al., 2007). Furthermore, a micromolar dose of simvastatin enhanced the mRNA expression of osteocalcin – a protein hormone that primarily modulates glucose homeostasis and has a minor effect on bone mineralization (Figure 3) (Baek et al., 2005). However, simvastatin has also been shown to reduce the proliferation of human bone marrow stromal cells at micromolar to sub-nanomolar concentrations (Baek et al., 2005).
3.3. Statins and the immune response
3.3.1. Statins target several molecules of the inflammatory response
The anti-inflammatory effects of statins have been well documented (Altaf et al., 2015; Davaro et al., 2014; Koushki et al., 2021; Lim and Staudt, 2013; Pinal-Fernandez et al., 2018). In addition, there is a good correlation between statin use and decreased levels of patients’ serum inflammatory markers such as C-reactive protein (CRP) and serum amyloid A (SAA), which are often elevated in inflammatory states (Pinal-Fernandez et al., 2018). Below we discuss the anti-inflammatory mechanisms through which statins act.
Statins may influence the inflammatory response by either inhibiting or stimulating the Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome or toll-like receptors 2 and 4 (TL receptor 2, TL receptor 4) (Koushki et al., 2021). Both NLRP3 and TL receptors are components of the innate immune response; these receptors detect ligands that are released during inflammatory states as byproducts of cellular stress or tissue damage (Koushki et al., 2021). TL receptors are activated by damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs); activation of these TL receptors can lead to downstream activation of several different signaling pathways, including NF-κB and MAPK (Lim and Staudt, 2013). NLRP3 activation occurs in two steps: (i) activation of the NF-κB pathway either through TL receptors or other receptors; this leads to the transcription of NLRP3 as well as pro-IL-1β; (ii) activated NLRP3 oligomerizes and creates an inflammasome which can lead to the proteolytic activation of inflammatory cytokines such as IL-1β and IL-18, this process is also aided by caspase-1 (Koushki et al., 2021).
Statins may act on multiple targets in the inflammasome activation pathway to exert anti-inflammatory effects. First, statins may be able to decrease the expression or the downstream signaling pathways of DAMP and PAMP molecules which would trigger NLRP3 activation; these molecules include oxidized LDL (ox-LDL) or TNF-α via the induction of pregnane X receptors (Wang et al., 2017), a nuclear receptor which acts as a transcription factor for the drug-inducible expression of several genes, and the high mobility group box 1 (HMGB1) protein. This chromatin-associated protein has been associated with several hematologic malignancies (Lv et al., 2017). Second, statins may also decrease the expression of TL receptor 2 and TL receptor 4, thereby decreasing activation of the inflammasome (Moutzouri et al., 2012; Yang et al., 2012). This has been demonstrated both in vitro studies using the THP-1 cell line treated with atorvastatin (0.1–20 μM), as well as in randomized blind endpoint clinical studies in hypercholesterolemia patients using simvastatin (40 mg) or simvastatin/ezetimibe (10/10 mg) (Moutzouri et al., 2012; Yang et al., 2012). The mechanisms of these reductions are not well described; however, the findings of in vitro studies suggest that these effects are not mediated simply by reduced cholesterol levels. Finally, statins can also inhibit NF-κB, which is responsible for transcription of the inflammasome (Dichtl et al., 2003; Hernández-Presa et al., 2002; Ortego et al., 1999). This process is through increased production of nitric oxide (NO), by previously discussed mechanisms, causing the induction and stabilization of the NF-κB inhibiting protein, IκB-α (Figure 3) (Peng et al., 1995). In addition, PX receptor activation by statins inhibits activation of NLRP3 by NF-κB (Koushki et al., 2021). However, the effect of statins on the activation of the NLRP3 is controversial since some studies suggest that statins inhibit NLRP3 (Altaf et al., 2015; Davaro et al., 2014; Koushki et al., 2021), while others suggest activation (Henriksbo et al., 2014).
Regulation of the anti-inflammatory effects of statins may also occur through induction of the peroxisome proliferator-activated receptor alpha (PPA receptor-α). Statins can increase PPA receptor-α and PPA receptor-γ activity by increasing the production of endogenous ligands such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PJ2); these ligands are produced downstream of cyclooxygenase-2 (COX-2) by the ERK/MAPK pathways (Yano et al., 2007). Statins can also induce the expression of the PPA receptor-α gene (Landrier et al., 2004). Statin administration in HUVEC cell culture decreased IL-1β and IL-6 mRNA and protein expression via PPA receptor-α and PPA receptor-γ (Inoue et al., 2000).
The endothelial adhesion and transendothelial migration of leukocytes are early events in the inflammatory process. The most comprehensive model of leukocyte adhesion and transmigration at sites of inflammation involves five steps. The first step in this process involves the reversible adhesion of leukocytes to the vascular endothelium by interactions between E-selectin and P-selectin on the endothelial vasculature to one of several ligands on leukocytes (Leick et al., 2014). This reversible adhesion, also termed “leukocyte rolling,” is subsequently strengthened by the secretion of chemokines by the endothelium, which induces the expression of the integrins VLA4, LFA-1, and Mac-1 on endothelial vasculature (Leick et al., 2014). These integrins can bind to the molecules intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) on leukocytes, causing a stronger adhesion, termed “leukocyte arrest” (Leick et al., 2014). Statins, except for pravastatin, interfere with leukocyte adhesion by allosteric inhibition of the LFA-1 integrin, independent of their inhibition of HMG-CoA reductase, lovastatin and simvastatin are the most potent in this effect. Statins can also prevent leukocyte adhesion by decreasing the expression of both chemokine molecules and receptors via inhibition of the geranylgeranyl isoprenoid pathway (Veillard et al., 2006). Inhibition of the Rho GTPase signaling pathways by statins may also play a role in blocking leukocyte adhesion (Ghittoni et al., 2007). Rho GTPases appear to have roles in inducing E-selectin expression and localization; these effects have been characterized in clinical studies (Cernuda-Morollón and Ridley, 2006; Romano et al., 2000; van Haelst et al., 2001).
The anti-inflammatory effects of statins appear to allow them to exert beneficial effects in treatment of atherosclerosis, independent of their lipid-lowering properties. In vitro studies in non-human primates, with treatment arms consisting of pravastatin (40 mg/kg/day) or simvastatin (20 mg/kg/day) demonstrated decreased recruitment of macrophages to sites of atherosclerotic lesions, with accordant decreases in the expression of active matrix metalloproteinases (MMPs) (Sukhova et al., 2002). This was also observed in cell cultures of mouse and human macrophages, using fluvastatin (5–100 μmol/L), which led to a dose-dependent reduction of up to 40% in MMP-9 expression (Bellosta et al., 1998). The overexpression of MMPs is correlated with decreased plaque stability, with increased risk of fatal events, due to decreased integrity of the collagenous extracellular matrix of the plaque’s fibrous cap (Libby et al., 1996). A proposed mechanism by which statins may influence macrophage accumulation is by reducing expression of histone mRNA, a marker of cell proliferation, on macrophages as determined by an in vivo rabbit study using cerivastatin (0.6 mg/kg/day) (Aikawa et al., 2001). Pravastatin treatment demonstrated the added benefit of decreasing expression of the pro-coagulant tissue factor (TF) (Sukhova et al., 2002). These findings suggest statins influence outcomes in hypercholesterolemic patients independent of their ability to reduce serum cholesterol levels.
3.3.2. Statins impair mast cell degranulation
Inhibition of mast cell activity by statins also contributes to their anti-inflammatory properties. Mast cells are components of the innate immune system which have a role in wound healing, inflammation, tumor growth, and parasitic infection (Bachelet et al., 2006). Upon binding IgE to the FcεRI receptor, mast cells are degranulated to release several inflammatory mediators, including histamine. Statins, particularly fluvastatin, have shown to be effective in inhibiting mast cell degranulation (Figure 3) (Kolawole et al., 2016). Furthermore, lovastatin (20 μM), atorvastatin (1–50 μM), and cerivastatin (1–50 μM) have all demonstrated the ability to inhibit histamine release in vivo in human HMC-1 and rat peritoneal mast cells (Krauth et al., 2006; Roche et al., 1995). Statins also have variable effects on the release of pro-inflammatory cytokines; atorvastatin and lovastatin decrease TNF-α and IL-6 production from mast cells, whereas fluvastatin increases their production (Kouhpeikar et al., 2020).
3.3.3. Statins interfere with T-lymphocyte migration
T-lymphocytes have a vital role in the immune response as effectors of the adaptive immune system. T-lymphocyte activation begins with presenting foreign antigens as peptides on the heterodimer major histocompatibility complexes (MHC) located on the plasma membrane of antigen-presenting cells (APCs). The recognition of a foreign antigen allows T-cells to proliferate and mature to either CD4+ helper T-lymphocytes or CD8+ cytotoxic T-lymphocytes, acquiring additional functions which will enable them to exact one of several immune responses (Fabbri et al., 2003).
Statins can impact the migration of T-lymphocytes to sites of inflammation by decreasing the expression of T-lymphocyte attractant chemokines. For example, atorvastatin inhibits the expression of IL-8, a chemokine produced by the endothelial cells, which attracts T-lymphocytes towards sites of infection (Ghittoni et al., 2005; Tunon and Egido, 2004; Weitz-Schmidt, 2002). Simvastatin can also block both chemokine and chemokine receptor expression in human vascular endothelial cells and macrophages without affecting its capacity to inhibit cholesterol production (Weitz-Schmidt, 2002). The effects of statins in preventing leukocyte adhesion and transmigration, described in section 3.3.1, also apply to T-cell migration processes. In particular, inhibition of the LFA-1 integrin by statins is vital, as it normally provides a co-stimulatory signal for T-cell activation (Weitz-Schmidt et al., 2001).
Statins can impact T-lymphocyte activation and proliferation by exerting an inhibitory influence over APCs and MHCs, as demonstrated in cell culture studies as well as clinical studies. While statins have not been shown to affect mature MHCs, simvastatin (20 μM) inhibits the upregulation of IFN-γ-inducible MHC II molecules on APCs, thus reducing antigen presentation to CD4 T-lymphocytes (Ghittoni et al., 2006). This effect is likely achieved due to the downstream impacts of Rho GTPase inhibition (Weitz-Schmidt, 2002). Statin treatment can also inhibit the maturation of cytokine-stimulated dendritic cells, leading to reduced T-lymphocyte proliferation (Yilmaz et al., 2004). In addition, statins also impede the antigen processing and presentation functions of APCs. Simvastatin was found to significantly diminish the processing and presentation of tetanus toxoid to CD4 T-lymphocytes by preventing protein antigen uptake via receptor-mediated endocytosis and macropinocytosis, again secondary to defective prenylation by Rab and Rho GTPases (Ghittoni et al., 2006). Statins can also impact intrinsic T-cell activation processes. Simvastatin was shown to block the expression of markers of T-cell activation, CD69, and CD25. This was mediated by interruption of the T-cell receptor (TCR) signaling cascade at steps involving Ras-like GTPases (Ghittoni et al., 2005).
3.4. Statins and cancer
Multiple studies have reported that statins can be used to prevent various types of cancers, including that of the colon (Jung et al., 2016; Pikoulis et al., 2016; Poynter et al., 2005), breast (Ji et al., 2016; Koyuturk et al., 2007; Muck et al., 2004), ovarian, pancreatic, lung, and lymphomas, reviewed in (Kashfi, 2018; Vallianou et al., 2014). Statins such as simvastatin, fluvastatin, lovastatin, and pravastatin inhibit cancer growth by inhibiting the Rho and Ras GTPases, both of which activate downstream signaling pathways that are deregulated in tumors (Figure 3). Rho GTPases upregulate the JNK and p38 MAPK pathways, which regulate cell proliferation, differentiation, survival, and migration (Ahmadi et al., 2017; Wagner and Nebreda, 2009). In addition, Rho GTPase activation upregulates the expression of several proteins in the cell cycle, including Cyclin D1, p21WAF1, and p27kip1 (Ahmadi et al., 2017). Furthermore, Ras GTPases activate the oncogenic pathways PI3K/Akt, MEK/MAPK, and MEKK/SEK/JNK in a broad spectrum of cancers (Ahmadi et al., 2017; Hoxhaj and Manning, 2020). Most statins inhibit proliferation and induce apoptosis in cell culture and animal studies. The pro-apoptotic effects are mediated through activation of caspase 3, caspase 8, caspase 9, and Bax (Ahmadi et al., 2017; Yanae et al., 2011), as well as downregulation of the anti-apoptotic Bcl-2 protein (Kato and Honda, 2020; Spampanato et al., 2012). Statins may also cause the inactivation of the mTOR protein and induce AMP-activated protein kinase (Gorabi et al., 2019; Pisanti et al., 2014; Sanli et al., 2011). Statins inhibit tumor growth at clinically relevant doses and diminish metastasis in animal models (Kusama et al., 2002; Paragh et al., 2003). Statins can also lead to the degradation of mutant p53 enzymes in cancer cells while preserving normal p53 expression in non-cancer cells, likely via influencing the stability of mutant p53; this effect is dependent on the inhibition of the mevalonate pathway (Freed-Pastor et al., 2012; Parrales et al., 2016).
The ability of statins to induce eNOS activation and otherwise increase NO can be useful in cancer management. eNOS expression may be induced by inhibiting the RhoA/ROCK GTPase signaling pathway, which primarily leads to induced stability of eNOS mRNA (Margaritis et al., 2014; Rikitake and Liao, 2005). This induction is also potentially aided by the PI3K/Akt pathway, which activates eNOS post-translationally (Margaritis et al., 2014); however, there are also suggestions that statins may inhibit the PI3K/Akt pathway (Beckwitt et al., 2018; Yanae et al., 2011). NO has a dichotomous role in cancer biology, with some reports suggesting that NO possesses antitumor and anti-inflammatory properties, while others implicate NO in tumor promotion and inflammation (Kashfi, 2018; Kashfi and Vannini, 2019). These paradoxes have been explained in terms of a dual or biphasic effect dependent on the local flux of NO (Kashfi, 2018; Kashfi and Vannini, 2019; Ridnour et al., 2006). Statins dose-dependently can either increase angiogenesis or have angiostatic properties (Urbich et al., 2002). These effects may be due to levels of eNOS expression, with low concentrations favoring angiogenesis and higher levels having inhibitory effects.
Statins may also trigger cancer cell death by inducing ferroptosis. Ferroptosis is a form of cell death which occurs due to accumulation of lipid peroxidation products (Stockwell et al., 2017). One mechanism by which this accumulation occurs is through inhibition of endogenous antioxidant systems such as glutathione peroxidase 4 (GPX4), and ferroptosis suppressor protein 1 (FSP1), which degrade peroxidation products (Stockwell et al., 2017). FSP1 acts to inhibit ferroptosis through reduction of coenzyme Q10 (CoQ10) which is NADH-dependent; the reduced CoQ10 acts as a radical-trapping antioxidant, preventing accumulation of these peroxidation products (Bersuker et al., 2019). Statins can likely induce ferroptosis by depleting CoQ10, a derivative of the mevalonate pathway (Zhu et al., 2021). Another mechanism by which statins may contribute to ferroptosis is inhibition of the selenoprotein enzyme GPX4, limiting selenocysteine production (Stockwell et al., 2017; Viswanathan et al., 2017). This effect is indirect; statins inhibit selenocysteine synthesis by inhibition of isopentenylation of selenocysteyl tRNA (Warner et al., 2000). Induction of ferroptosis has been investigated as a novel target for cancer therapy, although ferroptosis inducing therapy has not yet been approved clinically, due to in vivo cytotoxicity (Viswanathan et al., 2017; Ye et al., 2021). There is a lack of current literature exploring the effects of statins on cancer cell death from the perspective of ferroptosis, although these effects may be significant.
The lipophilicity of statins may impact their efficacy in treating extrahepatic tumors. Hydrophilic statins may be less effective as cancer therapeutics because of their inability to cross extrahepatic cellular membranes (Ahmadi et al., 2017; Beckwitt et al., 2018; Kato et al., 2010). Some reports also suggest that hydrophilic statins may promote tumor progression due to total reductions in cholesterol burden, which has an anti-oxidant role. As a result, there may be compensatory increases in the mevalonate pathway within the extrahepatic tissues, leading to increases in the local milieu of cholesterol, which could promote carcinogenesis (Ahmadi et al., 2017).
The activation of the Wnt/β-catenin signaling pathway has been implicated in several types of tumors and hematologic malignancies (Zhang and Wang, 2020). As mentioned in section 3.2., statins’ positive effects in osteogenesis are mediated by activation of the Wnt/β-catenin signaling pathway leading to increased differentiation of mesenchymal stem cells to osteoblasts. It appears that statins have the opposite effect on the Wnt/β-catenin signaling pathway using tumor cell lines. Simvastatin and atorvastatin administration in lung cancer (Calu-1) and melanoma (A375) cell lines both led to decreased β-catenin phosphorylation in a dose-dependent manner (up to 10 μM) (Lim et al., 2021). Other in vitro studies using gastric cancer cells and uterine meiolyomas corroborate these effects (El Sabeh et al., 2021; Liu et al., 2020).
3.5. Antifungal effects of statins
Fungi are a significant cause of global mortality, responsible for 1.5 million deaths each year (Tavakkoli et al., 2020). In addition, fungi are developing resistance to antimicrobial medication at an alarming rate, necessitating novel therapies (Fisher et al., 2018). Statins can be added to the small repertoire of antifungal agents. Statins express antifungal activity against Candida spp., Aspergillus spp., and zygomycetes, in addition to a myriad of other pathogenic yeasts and molds (Lefebvre et al., 2010). The primary mechanism by which statins are thought to exert anti-fungal effects is by inhibiting the production of ergosterol (Tavakkoli et al., 2020), the most abundant sterol in fungal cell membranes. Analogous to cholesterol in eukaryotic membranes, ergosterol mediates cell membrane fluidity and permeability in fungi and is the end product of the mevalonate pathway in fungi, not cholesterol. Therefore HMG-CoA reductase inhibition can also decrease ergosterol production in fungi (Tavakkoli et al., 2020). Many of the other mechanisms of statins’ antifungal effects stem from inhibition of FPP production. Inhibition of Ras GTPases mediated by FPP inhibition allows statins to induce apoptosis and impair fungal cell morphogenesis. Inhibition of heme A synthesis as another downstream product of the mevalonate pathway also causes reductions in cytochrome C levels, impairing mitochondrial oxidative phosphorylation (Tavakkoli et al., 2020). While it is possible that all statins possess antifungal activity, cerivastatin and pitavastatin do not have documented antifungal activity (Tavakkoli et al., 2020). The synthetic statins, atorvastatin, fluvastatin, and rosuvastatin, have been reported as more potent anti-fungal agents than the fungal-derived statins, lovastatin, and pravastatin. These synthetic statins are effective against a broader range of species and are effective at lower concentrations than fungal-derived statins (Natesan et al., 2008; Nyilasi et al., 2009; Tavakkoli et al., 2020). Specifically, fluvastatin and simvastatin appeared to be the most potent antifungal statin, while pravastatin exhibited the least anti-fungal properties (Cabral et al., 2013; Nyilasi et al., 2014; Tavakkoli et al., 2020). Statins may also have a role as adjuvant therapy to current antifungal medications. Fluvastatin, lovastatin, atorvastatin, rosuvastatin, and atorvastatin potentiated the efficacy of azole, polyene, allylamine, and echinocandin antifungals in a synergistic manner (Cabral et al., 2013; Campoy and Adrio, 2017; Chin et al., 1997; Nyilasi et al., 2010a; Nyilasi et al., 2014; Nyilasi et al., 2010b; Nyilasi et al., 2009; Tavakkoli et al., 2020; Venturini et al., 2011). Fluvastatin also displayed lower MIC than several designated antifungal agents, which bodes well for translation of antifungal effects to clinical practice (Tavakkoli et al., 2020).
4. Adverse events of statins: Not all that glitter is gold
While statins are significantly used in managing hypercholesterolemia and have many pleiotropic effects, they also have many dose-dependent adverse effects that necessitate caution in their widespread use. These include muscle myopathies, new-onset type 2 diabetes, cognitive, renal, and hepatic dysfunctions. There are also drug-drug interactions that one must consider when using statins, as this is an underappreciated concern for most part. In the following sections, we aim to underscore these potential effects, some of which can be life-threatening.
4.1. Myopathy
Myopathy is a common dose-dependent side effect associated with statins. It is characterized by stiffness, cramps, weakness, or loss of strength during exertion (Istvan, 2003; Rosenbaum et al., 2013). Myopathy can be classified as myalgia, myositis, or rhabdomyolysis, depending on the presenting symptoms and levels of creatine kinase (CK). Symptoms such as muscle pain and stiffness are the most common side effects of statin, but rhabdomyolysis is the most severe and rarest (Bitzur et al., 2013; Ramkumar et al., 2016). Asymptomatic CK elevation up to 4 times the upper limit of normal (ULN) is observed in up to 26% of patients (Turner and Pirmohamed, 2019). Myalgias are events in which patients experience muscle pain, tenderness, or weakness with normal or slightly elevated CK levels; these occur in an estimated 2–7% of patients (Tomaszewski et al., 2011; Turner and Pirmohamed, 2019). Myopathy is defined as symptomatic muscle pain in which CK is elevated to greater than 4 times the ULN, while severe myopathy is classified as muscle symptoms with CK between 10–50 times ULN. Muscle-related symptoms are generally lower (1.5–5%) in randomized controlled trials than in clinical practice. It demonstrates that statin use on its own is associated with an insignificantly greater risk of developing muscle toxicity (Tomaszewski et al., 2011). This can reflect the significant risk factors of myopathies, which can include oversight of drug-drug interactions of statins in clinical practice or external factors such as heavy exercise. Rhabdomyolysis is the most feared complication of statin use and is associated with renal failure and a high mortality rate reported as 0.3 per 100,000 person-years. However, the risk of statin-induced rhabdomyolysis increases in those taking other medications such as fibrates (Tomaszewski et al., 2011). Of note, in 2001, cerivastatin was withdrawn from the market due to a much higher incidence of rhabdomyolysis compared to any other statin. Statins metabolized by the CYP3A4 enzyme are much more likely to cause myotoxicity and/or rhabdomyolysis due to greater chances of drug-drug interactions (Tomaszewski et al., 2011). Other risk factors that increase the likelihood and severity of statin-induced myotoxicity include female gender, age over 80, other underlying neuromuscular diseases, and alcohol abuse (Thompson et al., 2016). In many patients, mild muscle symptoms and slight rises in creatinine kinase are observed even with low doses of statins. Unfortunately, in some cases, the myopathy is not relieved by the termination of statin therapy, which alludes to the possibility of statin-induced necrotizing autoimmune myopathy (SINAM) (Thompson et al., 2016).
There are several theories regarding the mechanisms behind statin-induced muscle symptoms (Bitzur et al., 2013). Statins can reduce levels of coQ10, altering calcium channel function in skeletal muscles, leading to cell injury and apoptosis (Figure 4)(Yokote et al., 2011). CoQ10 is also a cofactor in the mitochondrial electron transport chain; thus, deficiencies in CoQ10 may impair mitochondrial oxidative phosphorylation (Crane, 2001; Fragaki et al., 2016). Meta-analysis of 12 studies looking at patients with statin-associated myopathy indicated that CoQ10 supplementation ameliorated statin-associated muscle symptoms (Qu et al., 2018). This strongly suggests that Co-Q10 supplementation may be a complementary approach in managing statin-induced myopathy (Qu et al., 2018). Lactone forms of statin can inhibit complex III of the mitochondrial electron transport chain by binding to an allosteric site, the Q0 of the complex (Figure 4). Some statins may impact the electron transport chain at other sites as well; simvastatin in its lactone form, for example, has been shown to impair mitochondrial complex I. Any mitochondrial impairment can lead to ATP deficiency of the muscle, which could play a role in inducing apoptosis (Turner and Pirmohamed, 2019). Calcium channel defects may also have a role in statin myopathy, as statin treatment has been shown to reduce the stability of the ryanodine receptor 1 channel by inducing dissociation of the FKBP12 stabilizing protein; this can cause excess calcium release from the sarcoplasmic reticulum (Figure 4). Increased calcium release induces excess muscle contraction, which may trigger myopathy (Turner and Pirmohamed, 2019). Calcium release may also lead to the upregulation of pro-apoptotic proteins such as caspase 3 (Turner and Pirmohamed, 2019). Mutations in ryanodine receptor 1 have been linked to statin-associated myopathy (Isackson et al., 2018), although this alone is not likely to trigger myopathy. Another mechanism by which statins can induce myopathy is disrupting the protein prenylation and activation of small GTPases. Specifically, statin-mediated PI3K/Akt inhibition may play a role in myopathy, inducing muscle atrophy via the forkhead box class O (FOXO) activation. FOXO activates the two ubiquitin ligase molecules, atrogin-1, and muscle-specific ring finger-1, which degrade protein molecules. The resultant protein deficient state can potentially lead to muscular atrophy (Hoffman and Nader, 2004; Thompson et al., 2016). Akt phosphorylation typically causes phosphorylation of FOXO, preventing it from entering the nucleus. However, with statin therapy inhibiting the phosphorylation of the Akt molecule, unphosphorylated FOXO can enter the nucleus and cause muscular atrophy (Cao et al., 2009). In the nucleus, FOXO can also activate pyruvate dehydrogenase kinase (PDK) transcription; increased activity of PDK inactivates muscle pyruvate dehydrogenase complex (Mallinson et al., 2009). The resultant inhibition of the tricarboxylic acid cycle leading to decreased ATP production could also be a mechanism of statin-induced myopathy.
Figure 4.
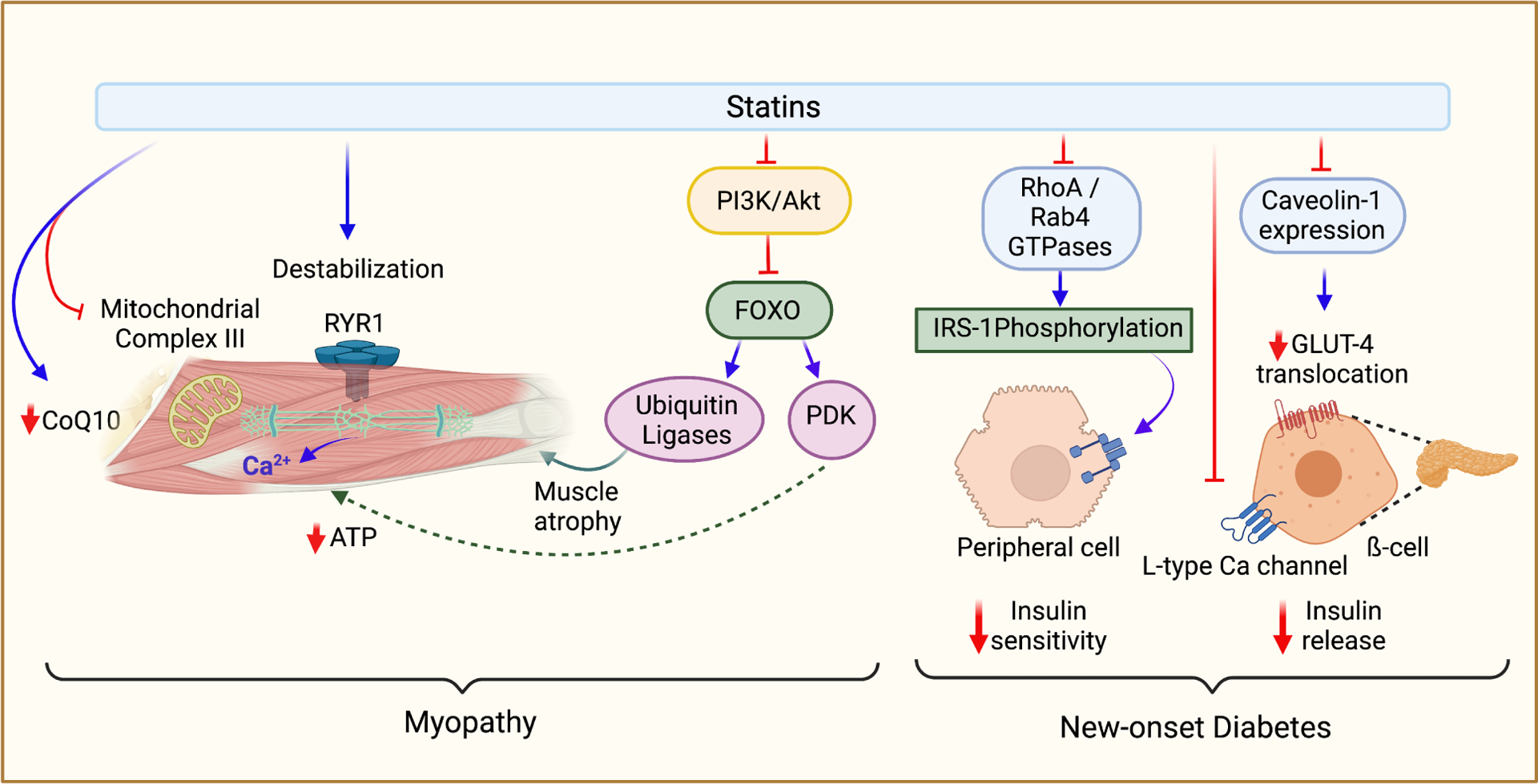
Adverse effects of statins. The two most common adverse effects associated with statins are myopathy and new-onset type 2 diabetes; the mechanisms of this pathology are described above. Myopathy is primarily due to reduced coQ10 production following statin treatment. Reduced levels of coQ10 can decrease mitochondrial respiration, leading to less ATP production in skeletal muscle cells. In a similar mechanism, lactone statins have also been shown to inhibit complex III of the electron transport chain by offsite binding to the Q0 site. Myopathy can also be triggered by destabilized activation of RYR1, leading to excess calcium release in a pathology similar to malignant hyperthermia. PI3K/Akt inhibition by statins leads to activation of the FOXO protein, which can then increase the nuclear expression of ubiquitin ligases and PDK, both of which can lead to muscle atrophy. There are multiple mechanisms of statin-induced diabetes. Insulin release from pancreatic ß-cells may be impaired by statin-mediated downregulation of GLUT-2 transporter. Inhibition of L-type calcium channels in pancreatic ß-cells (due to cholesterol depletion-associated membrane alteration) can lead to impaired insulin release from the pancreas. Several mechanisms may also lead to insulin resistance. Inhibition of prenylation of RhoA and Rab4 GTPases can lead to inhibition of IRS-1 phosphorylation and thus inhibition of downstream insulin signaling pathways. Downregulation of caveolin-1 expression can lead to decreased GLUT4 translocation to cellular membranes.
4.2. Diabetes
The first indication that statins may precipitate new-onset diabetes was reported in 2008 (JUPITER study) when the beneficial effects of rosuvastatin were evaluated in people with elevated high-sensitivity C-reactive protein levels but without hyperlipidemia (Ridker et al., 2008). This study indicated that rosuvastatin did not cause a significant increase in myopathy or cancer but did cause a higher incidence of diabetes (Ridker et al., 2008). Meta-analyses of 13 clinical trials evaluating 91,140 participants using statins for high cholesterol management over a mean period of 4 years indicated that statins dose-dependently increased the risk of new-onset diabetes by 9% (Sattar et al., 2010). This analysis did not distinguish between type 1 and type 2 diabetes. Despite this, statins are still widely prescribed because they offer a significant reduction in overall cardiovascular risk (Robinson, 2015). There are two general mechanisms by which statins may cause type 2 diabetes, (i) pancreatic ß-cell dysfunction and (ii) insulin resistance in peripheral tissue (Mach et al., 2018). By suppressing GLUT-2 transporter expression on the pancreatic ß cells, statins can impair insulin release. GLUT-2 transporters on the ß cells allow for glucose influx which then sets off a cascade of cellular changes culminating in the release of insulin (Fu et al., 2013). Simvastatin, atorvastatin, and pravastatin have all been shown to inhibit GLUT-2 expression in a dose-dependent manner (up to 100 μM), in vivo in human pancreatic β cells and in the mouse MIN6 cell line (Zhao and Zhao, 2015; Zhou et al., 2014a). Another mechanism by which statins may impair insulin release is by direct inhibition of calcium channels of the pancreatic ß islet cells. Insulin release from these cells is directly stimulated by rising concentrations of cytoplasmic calcium (Fu et al., 2013). As such, inhibition of these calcium channels can induce impaired insulin release. In an animal model, simvastatin disrupted membrane lipid rafts which caused a conformation change in L-type calcium channels leading to its inhibition (Paseban et al., 2019; Xia et al., 2008). Statins can also upregulate low-density lipoprotein receptors (LDLR) on the pancreatic ß cells, leading to cholesterol accumulation intracellularly (Perego et al., 2019). This can lead to reduced ATP production by the mitochondria following glucose metabolism and reduced trafficking of secretory insulin granules (Perego et al., 2019). Finally, high-dose statin treatment can induce ß-cell apoptosis and reduce the ß-cell survival rate (Zhao and Zhao, 2015).
Apart from impaired insulin release, insulin resistance can also account for the diabetogenic effects of statins. The two mechanisms by which statins may increase insulin resistance are via downregulating GLUT4 transporter translocation and inhibiting insulin receptor substrate (IRS-1), diminishing the downstream signaling pathways (Robinson, 2015; Takaguri et al., 2008). Caveolins are cholesterol-enriched proteins that can aggregate to form microdomains called caveolae; these caveolae can serve as signaling platforms for many signaling cascades. Insulin signaling causes translocation of GLUT4 transporters to caveolin platforms (Cohen et al., 2003), allowing these GLUT4 transporters to facilitate glucose influx. In animal models, atorvastatin and lovastatin caused reductions in GLUT4 translocation (Robinson, 2015). Atorvastatin also decreased levels of caveolin-1 and simvastatin demonstrated inhibition of caveolar vesicle docking (Figure 4) (Khan et al., 2009; Nakata et al., 2006). Furthermore, in the insulin receptor cascade, insulin binding to its receptor causes phosphorylation of IRS-1, leading to further downstream signaling (Mardilovich et al., 2009), which is in part is modulated by the GTPases RhoA and Rab4 (Paseban et al., 2019). Statin treatment can thus induce insulin resistance by inhibiting the activity of these GTPases, as seen by atorvastatin (Takaguri et al., 2008).
Though statin use does have an association with an increased risk of type 2 diabetes, not all statins display this effect. For example, pitavastatin and pravastatin do not affect glucose levels in patients with and without diabetes (Mach et al., 2018; Yokote et al., 2011).
4.3. Cognitive function
The effects of statins on cognitive function are controversial. Although there is evidence supporting the neuroprotective effects of statins, there are also reports suggesting that statins may adversely affect cognitive function. Several epidemiological studies and meta-analyses have reported a lower risk of dementia in statin users (Jick et al., 2000; Wolozin et al., 2000; Wong et al., 2013). Other case studies have also documented reports of reversible cognitive impairment, manifesting in restlessness, mental confusion, and short-term memory loss after beginning statin therapy or increasing the dosage (Elias et al., 2005; Mach et al., 2018; Wagstaff et al., 2003). Decreased serum cholesterol has also been associated with poor cognitive function (Elias et al., 2005). Adverse effects are thought to result from CNS exposure to higher doses of lipophilic statins that can effectively cross the blood-brain barrier (BBB) and lower cholesterol levels in the CNS. In this context, cholesterol has many important functions in the brain, including myelin sheath formation, neuro signaling processes, and mitochondrial function (Schultz et al., 2018). However, several systematic reviews have shown that the effects of statins on cognitive dysfunction are negligible (Ott et al., 2015; Song et al., 2013; Suraweera et al., 2016). In perspective, the FDA has put a neurological side effect warning label on statins due to the possibility of increased dementia, mild cognitive impairment, or cognitive performance decline (Vahabzadeh and Mohammadpour, 2015). The cognitive effects of statins have thus not been fully elucidated.
4.4. Renal Function
Statins have generally been recognized for their renal protective functions in patients with chronic kidney disease (CKD) and are recommended in non-dialysis patients with stage 3 CKD (Esmeijer et al., 2019). A network meta-analysis of 43 RCTs revealed that statins slowed the progressive decline of eGFR and reduced proteinuria in patients (Esmeijer et al., 2019). Although the effects of statins on renal function in these patients are generally positive, there are some adverse effects with regards to the kidneys to keep in mind. Rhabdomyolysis can classically induce renal failure due to the release of myoglobin from muscle tissues. Myoglobin can cause renal dysfunction due to induction of renal vasoconstriction, the formation of intratubular casts, and toxic effects on tubular cells mediated by reactive oxygen species (Petejova and Martinek, 2014). This adverse effect, however, is exceedingly uncommon in the general population. There is also an increased risk of hospital admission with acute kidney injury in patients taking high-intensity statins compared to low-intensity statins (Dormuth et al., 2013; Tonelli et al., 2019), with the strongest effect observed within the first 4 months of starting statin therapy (Dormuth et al., 2013). There are also reports indicating that high-dose statin therapy is associated with proteinuria (Agarwal, 2004). This may suggest a dose-dependent injurious effect of statin therapy on renal function.
4.5. Hepatic function
Current guidelines recommend determining liver function and creatine levels at baseline, and then 6–8 weeks after starting or adjusting the medication/dose, and after that annually or as needed (Lowe et al., 2013). Statins can cause transaminitis, at an incidence of 0.5% to 3%, generally within the first 3 months of starting statin therapy, which is rarely significant at the clinical level (Bhardwaj and Chalasani, 2007; Jose, 2016). These elevations are dose-dependent and generally short-term; they subside with continued statin use or can be managed by reducing the dose (Bitzur et al., 2013; Chitturi and George, 2002; Mach et al., 2018; Yokote et al., 2011). There is no distinction between statin type and the incidence of transaminitis. Transaminitis may be related to changes in the hepatocyte cellular membrane (Calderon et al., 2010). When the hepatocyte cellular membrane’s lipid concentration decreases, the membrane subsequently becomes more permeable and allows lipophilic enzymes to leak (Dujovne, 2002; Tolman, 2000).
Autoimmune hepatitis is the most common statin-induced liver injury; statins can initiate idiopathic inflammatory myositis or immune-mediated necrotizing myopathy by stimulating the production of autoantibodies against HMG-CoA reductase (Mammen et al., 2011). The mechanisms underlying the statin-induced liver injury can vary depending on the type of statin. Atorvastatin-associated liver injury has decreased biliary flow from the liver to the duodenum, whereas simvastatin has been associated with hepatocellular injury (Björnsson et al., 2012). Combining statin therapy with other hepatotoxic substances such as alcohol, calcium channel blockers, and fibrates may increase the tendency of statin-induced hepatotoxicity (Mancini et al., 2011; Tolman, 2002).
5. Xenobiotic interactions of statins
Several CYP450s metabolize statins; thus, other xenobiotics that are also metabolized by the same enzyme systems could potentially have profound clinical consequences. In this respect, dietary components and over-the-counter medications should also be considered when statins are used. These interactions are discussed in the following sections.
5.1. Dietary interactions
5.1.1. Grapefruit
The current guidelines for statin use are to refrain from consuming any form of grapefruit during statin therapy. Two prominent furanocoumarins in the juice, 6′,7′-dihydroxybergamottin (DHB) and bergamottin (BG), are important contributors to grapefruit juice-drug interactions (He et al., 1998; Paine et al., 2005). Small intestinal CYP3A4 is responsible for metabolizing atorvastatin, lovastatin, and simvastatin (Kolars et al., 1992); this enzyme is inhibited by BG and DHB, causing an increase in the bioavailability of these statins.
Not all interactions between grapefruit and statins have adverse effects. For example, grapefruit also contains hydroxy methyl glutaryl flavonoids (HMGF), which can inhibit HMG-CoA reductase and acyl-CoA-cholesterol acyltransferase (ACAT) (Bok et al., 1999). Inhibition of these two enzymes reduces the formation of cholesterol esters (Wilcox et al., 2001), thus affecting lipoprotein formation.
In addition, naringin, another naturally occurring flavonoid present in grapefruit, prevents cholesterol from oxidizing while also activating AMPK, which decreases cholesterol levels (Li et al., 2019). Furthermore, naringin increases fecal cholesterol excretion, reducing its absorption from the small intestine (Jeon et al., 2007; Jeon et al., 2004).
5.1.2. St. John’s Wort
Hypericum perforartum (St. John’s Wort) is a flowering plant primarily used to treat mental health conditions such as obsessive-compulsive disorder and somatic symptom disorder, especially in Europe (Linde, 2009). In the US, it is available over-the-counter, and because it has not received FDA approval, it is marketed as a dietary supplement. St. John’s Wort may induce the activation of the enzymes P-glycoprotein/MDR1, which excretes statins into the bile (Murphy et al., 2020), and CYP3A4 (Markowitz et al., 2003). Concomitant use of St. John’s Wort with statins will elicit a decrease in statin bioavailability. In a study of 24 hypercholesterolemic patients on simvastatin who also used a St. John’s Wort product, total cholesterol levels and LDL-cholesterol were significantly increased, without any changes in HDL-cholesterol levels (Eggertsen et al., 2007).
5.2. Drug-drug interactions
All statins, except pravastatin, are metabolized by the CYP450 superfamily of enzymes, primarily by either CYP3A4 or CYP2C9. Therefore, any xenobiotic which inhibits these enzymes could significantly increase statin plasma concentrations. On the other hand, any xenobiotic which induces these enzymes could significantly decrease statin plasma levels (Causevic-Ramosevac and Semiz, 2013). Many medications inhibit the CYP3A4 system, thus interfering with simvastatin, lovastatin, and atorvastatin metabolism. For example, desethylamiodarone a hepatic metabolite of amiodarone, is a key noncompetitive inhibitor of CYP3A4 (Causevic-Ramosevac and Semiz, 2013). Concomitant administration of amiodarone and lovastatin leads to a 1.8-fold increase in lovastatin levels, with the same holding true for simvastatin (Wiggins et al., 2016), Table 1. Other key drug-drug interactions involving inhibition of CYP3A4 are with the dihydropyridine calcium channel blockers (benidipine, azelnidipine, nifedipine), the benzene acetonitrile calcium channel blocker (verapamil), and the benzodiazepine calcium channel blocker (diltiazem) (Causevic-Ramosevac and Semiz, 2013), Table 1. Other drug classes which inhibit the CYP3A4 system include the protease inhibitors (lopinavir, ritonavir), azole antifungals (itraconazole, ketoconazole), and macrolide antibiotics (erythromycin, clarithromycin, troleandomycin, telithromycin) (Causevic-Ramosevac and Semiz, 2013), Table 1. The primary consequence of CYP3A4 inhibition is statin toxicity, leading to myopathies.
Table 1:
Interactions of statins with other medications.
| Target | Medication | Statin | Effect on statin levels | Ref |
|---|---|---|---|---|
| ↓ CYP3A4 | Amiodarone | Lovastatin | 1.8 × AUC | (Wiggins et al., 2016) |
| Simvastatin | 1.8 × AUC | |||
| ↓ CYP3A4 | Amlodipine | Simvastatin | 1.8 × AUC | (Zhou et al., 2014b) |
| Lovastatin | Minor magnitude | |||
| Azelnidipine | Simvastatin | 1.9 × AUC | ||
| Lercanidipine | Simvastatin | 56% Increased bioavailability | ||
| Lercanidipine | Fluvastatin | 1.13 × AUC | ||
| ↓ CYP3A4 | Diltiazem | Lovastatin | 4.6 × AUC | (Wiggins et al., 2016) |
| Simvastatin | 3.6 × AUC | |||
| Atorvastatin | 1.51 × AUC | |||
| ↓ CYP3A4 | Verapamil | Lovastatin | 3.6 × AUC | (Wiggins et al., 2016) |
| Simvastatin | 2.5 × AUC | |||
| ↓ OATP1B1 | Cyclosporine | Lovastatin | 5–20 × AUC | (Wiggins et al., 2016) |
| Atorvastatin | 6–15 × AUC | |||
| Pravastatin | 5–10 × AUC | |||
| Simvastatin | 6–8 × AUC | |||
| Rosuvastatin | 7 × AUC | |||
| Pitavastatin | 5 × AUC | |||
| Fluvastatin | 2–4 × AUC | |||
| ↓ OATP1B1 | Gemfibrozil | Lovastatin | 2–3 × AUC | (Wiggins et al., 2016) |
| Simvastatin | 2–3 × AUC | |||
| Pravastatin | 2 × AUC | |||
| Rosuvastatin | 1.6–1.9 × AUC | |||
| Pitavastatin | 1.5 × AUC | |||
| Atorvastatin | 1.4 × AUC | |||
| No known interaction | Fenofibrate/Fenofebric Acid | Fluvastatin | Magnitude likely to be minor | (Wiggins et al., 2016) |
| Lovastatin | ||||
| Atorvastatin | 1.0 × AUC | |||
| Rosuvastatin | 1.1 × AUC | |||
| Simvastatin | 1.1 × AUC | |||
| Pitavastatin | 1.2 × AUC | |||
| ↓ OATP1B1, ↓ p-gp and ↓ CYP3A4 | Clarithromycin | Simvastatin | 10–11 × AUC | (Abu Mellal et al., 2019; Eljaaly and Alshehri, 2017) |
| Atorvastatin | 3.2–4.4 × AUC | (Eljaaly and Alshehri, 2017) | ||
| Pravastatin | 2 × AUC | |||
| Erithromycin | Pitavastatin | 2.8 × AUC | (Kellick et al., 2014) | |
| Telithromycin | Simvastatin | 4.0 × AUC | ||
| ↓ CYP3A4 | Itroconazole | Simvastatin | 19 × AUC | (Eljaaly and Alshehri, 2017) |
| Lovastatin | 15 × AUC | |||
| Atorvastatin | 2.5–3.3 × AUC | |||
| Fluconazole | Fluvastatin | 1.7 × AUC | (Kellick et al., 2014) | |
| Posaconazole | Simvastatin | 4.0 × AUC | ||
| ↑ CYP3A4 | Rifampin | Simvastatin | 0.13 × AUC | (Kyrklund et al., 2000) |
| ↓ OAT1B1, ↓ CYP3A4 | Lopinavir/Ritonavir | Atorvastatin | 2.9 × AUC | (Kellick et al., 2014) |
| Rosuvastatin | 2.1 × AUC | |||
| Tipranavir/Ritonavir | Atorvastatin | 9.4 × AUC | ||
| Telepravir | 7.88 × AUC | |||
| Saquinavir/Ritonavir | 3.9 × AUC | |||
| Darunavir/Ritonavir | 3.9 × AUC | |||
| Fosamprenavir/Ritonavir | 2.53 × AUC | |||
| Nelfinavir | 1.74 × AUC | |||
| Atazanavir/Ritonavir | Rosuvastatin | 3.1 × AUC | ||
| Not fully understood | Digoxin | Atorvastatin | 1.2 × AUC | (Wiggins et al., 2016) |
| ↓ p-gp | Dronedarone | Simvastatin | 3.9 × AUC | (Wiggins et al., 2016) |
| Lovastatin | Unknown, likely similar to simvastatin | |||
| ↓ CYP3A4, ↓ p-gp | Ranolazine | Simvastatin | 1.9 × AUC | |
| Lovastatin | Unknown, likely similar to simvastatin | |||
| ↓ CYP3A4, ↓ p-gp | Conivaptan | Lovastatin | 3 × AUC | (Wiggins et al., 2016) |
| Simvastatin | 3 × AUC | |||
| ↓ CYP3A4, ↓ p-gp | Ticagrelor | Simvastatin | 2–3 × AUC | (Wiggins et al., 2016) |
| Lovastatin | Unknown, likely similar to simvastatin | |||
| Atorvastatin | 1.4 × AUC | |||
| ↓ CYP2C9 | Warfarin | Simvastatin | Increase in simvastatin exposure is unlikely to be significant; effects may include increase in INR due to increased warfarin exposure | (Wiggins et al., 2016) |
| Fluvastatin | ||||
| Lovastatin | ||||
| Rosuvastatin |
Another target of drug-drug interactions is the OATP transporter proteins which regulate the hepatic uptake of statins and other medications. Inhibitors of the OATP1B1 transporter can also lead to statin toxicity (Shitara, 2011). Cyclosporine and gemfibrozil are the key medications that inhibit the OATP1B1 transporter (Causevic-Ramosevac and Semiz, 2013). Macrolide antibiotics also increase the concentrations of statin drugs by inhibiting the P-gp efflux pump and OATP1B1, in addition to CYP3A4 (Causevic-Ramosevac and Semiz, 2013).
Induction of CYP3A4 by medications would decrease statin concentrations and thus efficacy at a given dose; rifampicin and phenytoin are strong inducers of the CYP3A4 system (Causevic-Ramosevac and Semiz, 2013).
6. Conclusion and perspectives
Statins have many pleiotropic effects which extend beyond their ability to lower cholesterol synthesis. Many of these effects relate to decreased production intermediates and downstream products of the cholesterol synthesis pathway. For example, many pleiotropic effects result from lower amounts of FPP and GGPP, which have a major role in post-translational modification of proteins by prenylation. Decreases in protein prenylation allow statins to exert cardioprotective, anti-oxidant, anti-inflammatory, and anticancer effects. In addition, statins increase NO production by inducting eNOS expression and activity through RhoA/ROCK GTPase inhibition (Rikitake and Liao, 2005). Other pleiotropic effects of statins include stimulation of osteogenesis and potential anti-fungal effects through several mechanisms. Although statins have demonstrated the ability to exert many positive pleiotropic effects, not all that glitters is gold. For example, statin therapy is associated with several adverse events that can be chronic or even fatal. The most recognized adverse event related to statins is myopathy, which ranges from myalgia to rhabdomyolysis. Statin-induced myopathic effects appear to stem from other pleiotropic effects, including inhibition of the mitochondrial electron transport chain. These effects are also exacerbated by many other risk factors; interactions with other xenobiotics, including many different medications. Another notable adverse event is a potential increase in new-onset type 2 diabetes. Thus, careful monitoring of patients is needed when statins are prescribed, as they are not necessarily without adverse effects themselves.
There are several potential applications of statins that are secondary to their use as cholesterol-lowering agents. Perhaps some of the pleiotropic properties of statins can be made use of in combination with other agents. However, in this regard, one has to be aware of potential adverse effects.
In general, a current limitation of mechanistic analyses of statin use are that the hydrophilicity of different statins is not generally considered when claiming the pleiotropic effects that the class of drugs may exert. Lipid solubility plays an important role in allowing diffusion to several tissues and likely plays a significant role in determining the efficacy of each individual statin in exerting its pleiotropic effects. Lipophilic statins may be able to exert stronger pleiotropic effects due to their increased ability to diffuse extrahepatic tissues. However, these characteristics should be directly assessed in future experimental designs when comparing the effects of hydrophilic and lipophilic statins. In addition, it is important to consider the doses of statins that can be safely administered therapeutically when designing in vitro and animal model studies (Björkhem-Bergman et al., 2011). Although many of the papers cited in this review used therapeutically feasible concentrations of statins, there were several studies which used statin dosages that were above those of clinical relevance. Although these pleiotropic effects may still exist, as evidenced by results of clinical trials, caution should be exercised when in vitro and animal models are being designed. It is important to use clinically achievable dosages in order to assure the validity of any derived conclusions.
Acknowledgments
KK supported in part by the National Institutes of Health [R24 DA018055; R01GM123508] and the Professional Staff Congress-City University of New York (PSC-CUNY) [TRADB-49-271].
Abbreviations:
- ABC
ATP-binding cassette
- ACAT
acyl-CoA:cholesterol acyltransferase
- APC
antigen presenting cell
- AUC
area under curve
- BCRP
breast cancer resistance protein
- BMP-2
bone-morphogenic protein 2
- CK
creatine kinase
- CKD
chronic kidney disease
- CoQ10
co-enzyme Q10
- CYP450
cytochrome P450
- DAMP
damage associated molecular pattern
- DHB
6′,7′-dihydroxybergamottin
- eNOS
endothelial nitric oxide synthase
- FOXO
forkhead box class O
- FPP
farnesyl pyrophosphate
- FTase
farnesyltransferase
- GGPP
geranylgeranyl pyrophosphate
- GGTase I
gernylgernayltransferase I
- GGTase II
geranylgeranyltransferase II
- GTP
guanosine triphosphate
- HMGB1
high mobility group box 1
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- HMGF
hydroxy methyl glutaryl flavonoids
- HMGR
HMG-CoA reductase
- ICAM-1
intracellular adhesion molecule-1
- IRS-1
insulin receptor substrate-1
- LDL
low-density lipoprotein
- LDL-C
low-density lipoprotein cholesterol
- LFA-1
lymphocyte function-associated antigen 1
- Mac-1
macrophage 1
- MEF-2
myocyte enhancer factor-2
- MHC
major histocompatibility complex
- MLCP
myosin light-chain phosphate
- MMP
matrix metalloproteinase
- MRP2
multidrug resistant-associated protein
- NO
nitric oxide
- NLRP3
Nod-like receptor family pyrin domain containing 3
- SMC
smooth muscle cell
- OATP
organic anion transporting polypeptides
- OSX
osterix
- OCN
osteocalcin
- ox-LDL
oxidized LDL
- PAMP
pathogen associated molecular pattern
- PDK
pyruvate dehydrogenase kinase
- PI3K/Akt
phosphatidylinositol-3-kinase/Akt
- Pg-P
P-glycoprotein
- PPA receptor-ɑ
peroxisome proliferator-activated receptor alpha
- PPA receptor-γ2
peroxisome proliferator-activated receptor γ2
- PTEN
phosphate and tensin homology
- ROCK
Rho kinase
- FGF
fibroblast growth factor
- ROS
reactive oxidative species
- SINAM
statin-induced necrotizing autoimmune myopathy
- TF
tissue factor
- TG
triglycerides
- TGF-β
transforming growth factor-beta
- TL receptor
toll-like receptor
- ULN
upper limit of normal
- VLA4
very late antigen 4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
References
- Abu Mellal A, Hussain N, Said AS, 2019. The clinical significance of statins-macrolides interaction: comprehensive review of in vivo studies, case reports, and population studies. Ther Clin Risk Manag 15, 921–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal R, 2004. Statin Induced Proteinuria: Renal Injury or Renoprotection? Journal of the American Society of Nephrology 15, 2502. [DOI] [PubMed] [Google Scholar]
- Ahmadi Y, Ghorbanihaghjo A, Argani H, 2017. The balance between induction and inhibition of mevalonate pathway regulates cancer suppression by statins: A review of molecular mechanisms. Chem Biol Interact 273, 273–285. [DOI] [PubMed] [Google Scholar]
- Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y, Shiomi M, Schoen FJ, Libby P, 2001. An HMG-CoA Reductase Inhibitor, Cerivastatin, Suppresses Growth of Macrophages Expressing Matrix Metalloproteinases and Tissue Factor In Vivo and In Vitro. Circulation 103, 276–283. [DOI] [PubMed] [Google Scholar]
- Altaf A, Qu P, Zhao Y, Wang H, Lou D, Niu N, 2015. NLRP3 inflammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coronary Artery Disease 26. [DOI] [PubMed] [Google Scholar]
- Bachelet I, Levi-Schaffer F, Mekori YA, 2006. Mast Cells: Not Only in Allergy. Immunology and Allergy Clinics of North America 26, 407–425. [DOI] [PubMed] [Google Scholar]
- Baek K, Lee WY, Oh K, Tae H, Lee JM, Lee E, Han J-H, Kang M, Cha BY, Lee K, Son H, Kang S, 2005. The Effect of Simvastatin on the Proliferation and Differentiation of Human Bone Marrow Stromal Cells. Journal of Korean medical science 20, 438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwitt CH, Shiraha K, Wells A, 2018. Lipophilic statins limit cancer cell growth and survival, via involvement of Akt signaling. PLOS ONE 13, e0197422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellosta S, Via D, Canavesi M, Pfister P, Fumagalli R, Paoletti R, Bernini F, 1998. HMG-CoA Reductase Inhibitors Reduce MMP-9 Secretion by Macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology 18, 1671–1678. [DOI] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, Bassik MC, Nomura DK, Dixon SJ, Olzmann JA, 2019. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj SS, Chalasani N, 2007. Lipid-Lowering Agents That Cause Drug-Induced Hepatotoxicity. Clinics in Liver Disease 11, 597–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitzur R, Cohen H, Kamari Y, Harats D, 2013. Intolerance to statins: mechanisms and management. Diabetes Care 36 Suppl 2, S325–S330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkhem-Bergman L, Lindh JD, Bergman P, 2011. What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br J Clin Pharmacol 72, 164–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björnsson E, Jacobsen EI, Kalaitzakis E, 2012. Hepatotoxicity associated with statins: Reports of idiosyncratic liver injury post-marketing. Journal of Hepatology 56, 374–380. [DOI] [PubMed] [Google Scholar]
- Bok SH, Lee SH, Park YB, Bae KH, Son KH, Jeong TS, Choi MS, 1999. Plasma and hepatic cholesterol and hepatic activities of 3-hydroxy-3-methyl-glutaryl-CoA reductase and acyl CoA: cholesterol transferase are lower in rats fed citrus peel extract or a mixture of citrus bioflavonoids. J Nutr 129, 1182–1185. [DOI] [PubMed] [Google Scholar]
- Cabral ME, Figueroa LIC, Fariña JI, 2013. Synergistic antifungal activity of statin–azole associations as witnessed by Saccharomyces cerevisiae- and Candida utilis-bioassays and ergosterol quantification. Revista Iberoamericana de Micología 30, 31–38. [DOI] [PubMed] [Google Scholar]
- Calderon R, Cubeddu L, Goldberg R, Schiff E, 2010. Statins in the Treatment of Dyslipidemia in the Presence of Elevated Liver Aminotransferase Levels: A Therapeutic Dilemma. Mayo Clinic proceedings. Mayo Clinic; 85, 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campoy S, Adrio JL, 2017. Antifungals. Biochemical pharmacology 133, 86–96. [DOI] [PubMed] [Google Scholar]
- Cao P, Hanai J. i., Tanksale P, Imamura S, Sukhatme VP, Lecker SH, 2009. Statin-induced muscle damage and atrogin-1 induction is the result of a geranylgeranylation defect. The FASEB Journal 23, 2844–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey Laizure S, Herring V, Hu Z, Witbrodt K, Parker RB, 2013. The Role of Human Carboxylesterases in Drug Metabolism: Have We Overlooked Their Importance? Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 33, 210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causevic-Ramosevac A, Semiz S, 2013. Drug interactions with statins. Acta Pharmaceutica 63, 277–293. [DOI] [PubMed] [Google Scholar]
- Cernuda-Morollón E, Ridley AJ, 2006. Rho GTPases and Leukocyte Adhesion Receptor Expression and Function in Endothelial Cells. Circ Res 98, 757–767. [DOI] [PubMed] [Google Scholar]
- Chamani S, Liberale L, Mobasheri L, Montecucco F, Al-Rasadi K, Jamialahmadi T, Sahebkar A, 2021. The role of statins in the differentiation and function of bone cells. European Journal of Clinical Investigation 51, e13534. [DOI] [PubMed] [Google Scholar]
- Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, Schwartz RJ, 2006. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci U S A 103, 14495–14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin NX, Weitzman I, Della-Latta P, 1997. In vitro activity of fluvastatin, a cholesterol-lowering agent, and synergy with flucanazole and itraconazole against Candida species and Cryptococcus neoformans. Antimicrob Agents Chemother 41, 850–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitturi S, George J, 2002. Hepatotoxicity of Commonly Used Drugs: Nonsteroidal Anti-Inflammatory Drugs, Antihypertensives, Antidiabetic Agents, Anticonvulsants, Lipid-Lowering Agents, Psychotropic Drugs. Seminars in liver disease 22, 169–183. [DOI] [PubMed] [Google Scholar]
- Chuengsamarn S, Rattanamongkoulgul S, Suwanwalaikorn S, Wattanasirichaigoon S, Kaufman L, 2010. Effects of statins vs. non-statin lipid-lowering therapy on bone formation and bone mineral density biomarkers in patients with hyperlipidemia. Bone 46, 1011–1015. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Combs TP, Scherer PE, Lisanti MP, 2003. Role of caveolin and caveolae in insulin signaling and diabetes. American Journal of Physiology-Endocrinology and Metabolism 285, E1151–E1160. [DOI] [PubMed] [Google Scholar]
- Crane FL, 2001. Biochemical functions of coenzyme Q10. J Am Coll Nutr 20, 591–598. [DOI] [PubMed] [Google Scholar]
- Davaro F, Forde SD, Garfield M, Jiang Z, Halmen K, Tamburro ND, Kurt-Jones E, Fitzgerald KA, Golenbock DT, Wang D, 2014. 3-Hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor (statin)-induced 28-kDa interleukin-1β interferes with mature IL-1β signaling. J Biol Chem 289, 16214–16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Re DP, Miyamoto S, Brown JH, 2007. RhoA/Rho Kinase Up-regulate Bax to Activate a Mitochondrial Death Pathway and Induce Cardiomyocyte Apoptosis *. Journal of Biological Chemistry 282, 8069–8078. [DOI] [PubMed] [Google Scholar]
- Dichtl W, Dulak J, Frick M, Alber HF, Schwarzacher SP, Ares MPS, Nilsson J, Pachinger O, Weidinger F, 2003. HMG-CoA Reductase Inhibitors Regulate Inflammatory Transcription Factors in Human Endothelial and Vascular Smooth Muscle Cells. Arteriosclerosis, Thrombosis, and Vascular Biology 23, 58–63. [DOI] [PubMed] [Google Scholar]
- Dormuth C, Hemmelgarn B, Paterson M, James M, Teare G, Raymond C, Lafrance J-P, Levy A, Garg A, Ernst P, 2013. Use of high potency statins and rates of admission for acute kidney injury: multicenter, retrospective observational analysis of administrative databases. BMJ (Clinical research ed.) 346, f880. [DOI] [PubMed] [Google Scholar]
- Duan P, Bonewald LF, 2016. The role of the wnt/β-catenin signaling pathway in formation and maintenance of bone and teeth. Int J Biochem Cell Biol 77, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujovne CA, 2002. Side effects of statins: hepatitis versus “transaminitis”—myositis versus “CPKitis”. Am J Cardiol 89, 1411–1413. [DOI] [PubMed] [Google Scholar]
- Eggertsen R, Andreasson A, Andrén L, 2007. Effects of treatment with a commercially available St John’s Wort product (Movina) on cholesterol levels in patients with hypercholesterolemia treated with simvastatin. Scand J Prim Health Care 25, 154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Sabeh M, Saha SK, Afrin S, Borahay MA, 2021. Simvastatin Inhibits Wnt/β-Catenin Pathway in Uterine Leiomyoma. Endocrinology 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias PK, Elias MF, D’Agostino RB, Sullivan LM, Wolf PA, 2005. Serum cholesterol and cognitive performance in the Framingham Heart Study. Psychosom Med 67, 24–30. [DOI] [PubMed] [Google Scholar]
- Eljaaly K, Alshehri S, 2017. An updated review of interactions of statins with antibacterial and antifungal agents. Journal of Translational Science 3. [Google Scholar]
- Esmeijer K, Dekkers O, de Fijter J, Dekker F, Hoogeveen E, 2019. Effect of different types of statins on kidney function decline and proteinuria: a network meta-analysis. Scientific Reports 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Kozai T, Cosentino F, Joch H, Lüscher TF, 2002. Statin Prevents Tissue Factor Expression in Human Endothelial Cells. Circulation 105, 1756–1759. [DOI] [PubMed] [Google Scholar]
- Fabbri M, Smart C, Pardi R, 2003. T lymphocytes. Int J Biochem Cell Biol 35, 1004–1008. [DOI] [PubMed] [Google Scholar]
- Fisher M, Hawkins N, Sanglard D, Gurr S, 2018. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 360, 739–742. [DOI] [PubMed] [Google Scholar]
- Fragaki K, Chaussenot A, Benoist J-F, Ait-El-Mkadem S, Bannwarth S, Rouzier C, Cochaud C, Paquis-Flucklinger V, 2016. Coenzyme Q10 defects may be associated with a deficiency of Q10-independent mitochondrial respiratory chain complexes. Biol Res 49, 4–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon S-H, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell MJ, Osborne TF, Tian B, Lowe SW, Silva JM, Børresen-Dale A-L, Levine AJ, Bargonetti J, Prives C, 2012. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Gilbert ER, Liu D, 2013. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr Diabetes Rev 9, 25–53. [PMC free article] [PubMed] [Google Scholar]
- Ghittoni R, Lazzerini P, Laghi-Pasini or Pasini FL,F, Baldari C, 2007. T Lymphocytes as Targets of Statins: Molecular Mechanisms and Therapeutic Perspectives. Inflammation & allergy drug targets 6, 3–16. [DOI] [PubMed] [Google Scholar]
- Ghittoni R, Napolitani G, Benati D, Ulivieri C, Uliveri C, Patrussi L, Laghi-Pasini or Pasini FL,F, Lanzavecchia A, Baldari C, 2006. Simvastatin inhibits the MHC class II pathway of antigen presentation by impairing Ras superfamily GTPases. European journal of immunology 36, 2885–2893. [DOI] [PubMed] [Google Scholar]
- Ghittoni R, Patrussi L, Pirozzi K, Pellegrini M, Lazzerini PE, Capecchi PL, Pasini FL, Baldari CT, 2005. Simvastatin inhibits T-cell activation by selectively impairing the function of Ras superfamily GTPases. The FASEB Journal 19, 1–24. [DOI] [PubMed] [Google Scholar]
- Gorabi AM, Kiaie N, Hajighasemi S, Banach M, Penson PE, Jamialahmadi T, Sahebkar A, 2019. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J Clin Med 8, 2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann S, Ridley AJ, Lutz S, 2015. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front Pharmacol 6, 276–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata T, Soga J, Hidaka T, Idei N, Fujii Y, Fujimura N, Mikami S, Maruhashi T, Kihara Y, Chayama K, Kato H, Noma K, Liao JK, Higashi Y, Group R.S., 2011. Calcium channel blocker and Rho-associated kinase activity in patients with hypertension. J Hypertens 29, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Iyer KR, Hayes RN, Sinz MW, Woolf TF, Hollenberg PF, 1998. Inactivation of Cytochrome P450 3A4 by Bergamottin, a Component of Grapefruit Juice. Chemical Research in Toxicology 11, 252–259. [DOI] [PubMed] [Google Scholar]
- Henriksbo BD, Lau TC, Cavallari JF, Denou E, Chi W, Lally JS, Crane JD, Duggan BM, Foley KP, Fullerton MD, Tarnopolsky MA, Steinberg GR, Schertzer JD, 2014. Fluvastatin Causes NLRP3 Inflammasome-Mediated Adipose Insulin Resistance. Diabetes 63, 3742. [DOI] [PubMed] [Google Scholar]
- Hernández-Presa MA, Martín-Ventura JL, Ortego M, Gómez-Hernández A, Tuñón J, Hernández-Vargas P, Blanco-Colio LM, Mas S, Aparicio C, Ortega L, Vivanco F, Gerique JG, Dıaz C, Hernández G, Egido J, 2002. Atorvastatin reduces the expression of cyclooxygenase-2 in a rabbit model of atherosclerosis and in cultured vascular smooth muscle cells. Atherosclerosis 160, 49–58. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Nader GA, 2004. Balancing muscle hypertrophy and atrophy. Nature Medicine 10, 584–585. [DOI] [PubMed] [Google Scholar]
- Hoxhaj G, Manning BD, 2020. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nature Reviews Cancer 20, 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imran TF, Wong A, Schneeweiss S, Desai R, 2017. Abstract 17590: Hydrophilic Statins Are Associated With a Lower Risk of Incident Heart Failure Than Lipophilic Statins in a Large National Database. Circulation 136, A17590–A17590. [Google Scholar]
- Inoue I, Goto S. i., Mizotani K, Awata T, Mastunaga T, Kawai S. i., Nakajima T, Hokari S, Komoda T, Katayama S, 2000. Lipophilic HMG-CoA reductase inhibitor has an anti-inflammatory effect: Reduction of MRNA levels for interleukin-1β, interleukin-6, cyclooxygenase-2, and p22phox by regulation of peroxisome proliferator-activated receptor α (PPARα) in primary endothelial cells. Life Sciences 67, 863–876. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Wang J, Zia M, Spurgeon P, Levesque A, Bard J, James S, Nowak N, Lee TK, Vladutiu GD, 2018. RYR1 and CACNA1S genetic variants identified with statin-associated muscle symptoms. Pharmacogenomics 19, 1235–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istvan E, 2003. Statin inhibition of HMG-CoA reductase: a 3-dimensional view. Atherosclerosis Supplements 4, 3–8. [DOI] [PubMed] [Google Scholar]
- Istvan ES, 2002. Structural mechanism for statin inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase. American Heart Journal 144, S27–S32. [DOI] [PubMed] [Google Scholar]
- Jang W-G, Kim E-J, Kim D-K, Ryoo H-M, Lee K-B, Kim S-H, Choi H-S, Koh J-T, 2012. BMP2 protein regulates osteocalcin expression via Runx2-mediated Atf6 gene transcription. J Biol Chem 287, 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Kim HK, Kim HJ, Do GM, Jeong TS, Park YB, Choi MS, 2007. Hypocholesterolemic and antioxidative effects of naringenin and its two metabolites in high-cholesterol fed rats. Transl Res 149, 15–21. [DOI] [PubMed] [Google Scholar]
- Jeon SM, Park YB, Choi MS, 2004. Antihypercholesterolemic property of naringin alters plasma and tissue lipids, cholesterol-regulating enzymes, fecal sterol and tissue morphology in rabbits. Clin Nutr 23, 1025–1034. [DOI] [PubMed] [Google Scholar]
- Ji Y, Rounds T, Crocker A, Sussman B, Hovey RC, Kingsley F, Muss HB, Garber JE, Wood ME, 2016. The Effect of Atorvastatin on Breast Cancer Biomarkers in High-Risk Women. Cancer prevention research (Philadelphia, Pa.) 9, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA, 2000. Statins and the risk of dementia. The Lancet 356, 1627–1631. [DOI] [PubMed] [Google Scholar]
- Jose J, 2016. Statins and its hepatic effects: Newer data, implications, and changing recommendations. J Pharm Bioallied Sci 8, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YS, Park CH, Eun CS, Park DI, Han DS, 2016. Statin use and the risk of colorectal adenoma: A meta-analysis. Journal of gastroenterology and hepatology 31, 1823–1830. [DOI] [PubMed] [Google Scholar]
- Kaji H, Kanatani M, Sugimoto T, Chihara K, 2005. Statins modulate the levels of osteoprotegerin/receptor activator of NFkappaB ligand mRNA in mouse bone-cell cultures. Horm Metab Res 37, 589–592. [DOI] [PubMed] [Google Scholar]
- Kashfi K, 2018. The dichotomous role of H2S in cancer cell biology? Deja vu all over again. Biochemical pharmacology 149, 205–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashfi K, Vannini F, 2019. Nitric Oxide and Cancer: To Inhibit or To Induce iNOS: That Is the Question?, in: Bonavida L.M.a.B. (Ed.), Therapeutic Application of Nitric Oxide in Cancer and Inflammatory Disorders. Academic Press, Oxford, United Kingdom, pp. 93–111. [Google Scholar]
- Kato S, Honda K, 2020. Use of Biomarkers and Imaging for Early Detection of Pancreatic Cancer. Cancers (Basel) 12, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Smalley S, Sadarangani A, Chen-Lin K, Oliva B, Brañes J, Carvajal J, Gejman R, Owen GI, Cuello M, 2010. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. J Cell Mol Med 14, 1180–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellick KA, Bottorff M, Toth PP, 2014. A clinician’s guide to statin drug-drug interactions. Journal of Clinical Lipidology 8, S30–S46. [DOI] [PubMed] [Google Scholar]
- Khan T, Hamilton MP, Mundy DI, Chua SC, Scherer PE, 2009. Impact of simvastatin on adipose tissue: pleiotropic effects in vivo. Endocrinology 150, 5262–5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB, 1992. Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. The Journal of Clinical Investigation 90, 1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolawole EM, McLeod JJA, Ndaw V, Abebayehu D, Barnstein BO, Faber T, Spence AJ, Taruselli M, Paranjape A, Haque TT, Qayum AA, Kazmi QA, Wijesinghe DS, Sturgill JL, Chalfant CE, Straus DB, Oskeritzian CA, Ryan JJ, 2016. Fluvastatin Suppresses Mast Cell and Basophil IgE Responses: Genotype-Dependent Effects. J Immunol 196, 1461–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouhpeikar H, Delbari Z, Sathyapalan T, Simental-Mendía LE, Jamialahmadi T, Sahebkar A, 2020. The Effect of Statins through Mast Cells in the Pathophysiology of Atherosclerosis: a Review. Current Atherosclerosis Reports 22, 19. [DOI] [PubMed] [Google Scholar]
- Koushki K, Shahbaz SK, Mashayekhi K, Sadeghi M, Zayeri ZD, Taba MY, Banach M, Al-Rasadi K, Johnston TP, Sahebkar A, 2021. Anti-inflammatory Action of Statins in Cardiovascular Disease: the Role of Inflammasome and Toll-Like Receptor Pathways. Clin Rev Allergy Immunol 60, 175–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuturk M, Ersoz M, Altiok N, 2007. Simvastatin induces apoptosis in human breast cancer cells: p53 and estrogen receptor independent pathway requiring signalling through JNK. Cancer letters 250, 220–228. [DOI] [PubMed] [Google Scholar]
- Krauth MT, Majlesi Y, Sonneck K, Samorapoompichit P, Ghannadan M, Hauswirth AW, Baghestanian M, Schernthaner GH, Worda C, Müller MR, Sperr WR, Valent P, 2006. Effects of various statins on cytokine-dependent growth and IgE-dependent release of histamine in human mast cells. Allergy 61, 281–288. [DOI] [PubMed] [Google Scholar]
- Kusama T, Mukai M, Iwasaki T, Tatsuta M, Matsumoto Y, Akedo H, Inoue M, Nakamura H, 2002. 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors reduce human pancreatic cancer cell invasion and metastasis. Gastroenterology 122, 308–317. [DOI] [PubMed] [Google Scholar]
- Kyrklund C, Backman JT, Kivistö KT, Neuvonen M, Laitila J, Neuvonen PJ, 2000. Rifampin greatly reduces plasma simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther 68, 592–597. [DOI] [PubMed] [Google Scholar]
- Landrier J-F, Thomas C, Grober J, Duez H, Percevault F, Souidi M, Linard C, Staels B, Besnard P, 2004. Statin Induction of Liver Fatty Acid-Binding Protein (L-FABP) Gene Expression Is Peroxisome Proliferator-activated Receptor-α-dependent*. Journal of Biological Chemistry 279, 45512–45518. [DOI] [PubMed] [Google Scholar]
- Lefebvre M, Alshawa K, Dupont B, 2010. L’activité antifongique des statines. Journal De Mycologie Medicale - J MYCOLOGIE MEDICALE 20, 212–217. [Google Scholar]
- Leick M, Azcutia V, Newton G, Luscinskas FW, 2014. Leukocyte recruitment in inflammation: basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell Tissue Res 355, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Subramanian R, Yu S, Prueksaritanont T, 2006. Acyl-coenzyme a formation of simvastatin in mouse liver preparations. Drug Metab Dispos 34, 102–110. [DOI] [PubMed] [Google Scholar]
- Li S, Zhang Y, Sun Y, Zhang G, Bai J, Guo J, Su X, Du H, Cao X, Yang J, Wang T, 2019. Naringenin improves insulin sensitivity in gestational diabetes mellitus mice through AMPK. Nutr Diabetes 9, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cui Q, Kao C, Wang G-J, Balian G, 2003. Lovastatin inhibits adipogenic and stimulates osteogenic differentiation by suppressing PPARγ2 and increasing Cbfa1/Runx2 expression in bone marrow mesenchymal cell cultures. Bone 33, 652–659. [DOI] [PubMed] [Google Scholar]
- Li X, Song Q-S, Wang J-Y, Leng H. j., Chen Z-Q, Liu Z-J, Dang G-T, Song C-L, 2011. Simvastatin induces estrogen receptor-alpha expression in bone, restores bone loss, and decreases ERα expression and uterine wet weight in ovariectomized rats. Journal of Bone and Mineral Metabolism 29, 396–403. [DOI] [PubMed] [Google Scholar]
- Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT, 1996. Macrophages and atherosclerotic plaque stability. Current Opinion in Lipidology 7. [DOI] [PubMed] [Google Scholar]
- Lim K-H, Staudt LM, 2013. Toll-like receptor signaling. Cold Spring Harb Perspect Biol 5, a011247–a011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim W-J, Lee M, Oh Y, Fang X-Q, Lee S, Lim C-H, Park J, Lim J-H, 2021. Statins Decrease Programmed Death-Ligand 1 (PD-L1) by Inhibiting AKT and β-Catenin Signaling. Cells 10, 2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde K, 2009. St. John’s Wort – an Overview. Complementary Medicine Research 16, 146–155. [DOI] [PubMed] [Google Scholar]
- Liu P-Y, Chen J-H, Lin L-J, Liao JK, 2007. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J Am Coll Cardiol 49, 1619–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P-Y, Liu Y-W, Lin L-J, Chen J-H, Liao JK, 2009. Evidence for statin pleiotropy in humans: differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation 119, 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Xia H, Zhou S, Tang Q, Zhou J, Ren M, Bi F, 2020. Simvastatin Inhibits the Malignant Behaviors of Gastric Cancer Cells by Simultaneously Suppressing YAP and β-Catenin Signaling. Onco Targets Ther 13, 2057–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe RN, Marrs JC, Saseen JJ, 2013. Patterns of serum laboratory monitoring for safety and efficacy in patients on chronic statin therapy. Ther Adv Drug Saf 4, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z-H, Phuong TA, Jin S-J, Li X-X, Xu M, 2017. Protection by simvastatin on hyperglycemia-induced endothelial dysfunction through inhibiting NLRP3 inflammasomes. Oncotarget 8, 91291–91305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach F, Ray KK, Wiklund O, Corsini A, Catapano AL, Bruckert E, De Backer G, Hegele RA, Hovingh GK, Jacobson TA, Krauss RM, Laufs U, Leiter LA, März W, Nordestgaard BG, Raal FJ, Roden M, Santos RD, Stein EA, Stroes ES, Thompson PD, Tokgözoglu L, Vladutiu GD, Gencer B, Stock JK, Ginsberg HN, Chapman MJ, European Atherosclerosis Society Consensus, P., 2018. Adverse effects of statin therapy: perception vs. the evidence - focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur Heart J 39, 2526–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallinson JE, Constantin-Teodosiu D, Sidaway J, Westwood FR, Greenhaff PL, 2009. Blunted Akt/FOXO signalling and activation of genes controlling atrophy and fuel use in statin myopathy. The Journal of Physiology 587, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, Casciola-Rosen LA, 2011. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 63, 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini GBJ, Baker S, Bergeron J, Fitchett D, Frohlich J, Genest J, Gupta M, Hegele RA, Ng D, Pope J, 2011. Diagnosis, Prevention, and Management of Statin Adverse Effects and Intolerance: Proceedings of a Canadian Working Group Consensus Conference. Canadian Journal of Cardiology 27, 635–662. [DOI] [PubMed] [Google Scholar]
- Mardilovich K, Pankratz SL, Shaw LM, 2009. Expression and function of the insulin receptor substrate proteins in cancer. Cell Communication and Signaling 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margaritis M, Channon KM, Antoniades C, 2014. Statins as regulators of redox state in the vascular endothelium: beyond lipid lowering. Antioxid Redox Signal 20, 1198–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz JS, Donovan JL, DeVane CL, Taylor RM, Ruan Y, Wang J-S, Chavin KD, 2003. Effect of St John’s Wort on Drug Metabolism by Induction of Cytochrome P450 3A4 Enzyme. JAMA 290, 1500–1504. [DOI] [PubMed] [Google Scholar]
- Meech R, Hu DG, McKinnon RA, Mubarokah SN, Haines AZ, Nair PC, Rowland A, Mackenzie PI, 2019. The UDP-Glycosyltransferase (UGT) Superfamily: New Members, New Functions, and Novel Paradigms. Physiological Reviews 99, 1153–1222. [DOI] [PubMed] [Google Scholar]
- Mital S, Liao JK, 2004. Statins and the myocardium. Semin Vasc Med 4, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutzouri E, Tellis CC, Rousouli K, Liberopoulos EN, Milionis HJ, Elisaf MS, Tselepis AD, 2012. Effect of simvastatin or its combination with ezetimibe on Toll-like receptor expression and lipopolysaccharide – Induced cytokine production in monocytes of hypercholesterolemic patients. Atherosclerosis 225, 381–387. [DOI] [PubMed] [Google Scholar]
- Muck AO, Seeger H, Wallwiener D, 2004. Inhibitory effect of statins on the proliferation of human breast cancer cells. International journal of clinical pharmacology and therapeutics 42, 695–700. [DOI] [PubMed] [Google Scholar]
- Mundy G, Garrett R, Harris S, Chan J, Chen D, Rossini G, Boyce B, Zhao M, Gutierrez G, 1999. Stimulation of Bone Formation in Vitro and in Rodents by Statins. Science 286, 1946. [DOI] [PubMed] [Google Scholar]
- Murphy C, Deplazes E, Cranfield CG, Garcia A, 2020. The Role of Structure and Biophysical Properties in the Pleiotropic Effects of Statins. International Journal of Molecular Sciences 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakakuki T, Ito M, Iwasaki H, Kureishi Y, Okamoto R, Moriki N, Kongo M, Kato S, Yamada N, Isaka N, Nakano T, 2005. Rho/Rho-Kinase Pathway Contributes to C-Reactive Protein–Induced Plasminogen Activator Inhibitor-1 Expression in Endothelial Cells. Arteriosclerosis, Thrombosis, and Vascular Biology 25, 2088–2093. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Moss AJ, Brown MW, Kinoshita M, Kawai C, 1999. Long-term nitrate use may be deleterious in ischemic heart disease: A study using the databases from two large-scale postinfarction studies. American Heart Journal 138, 577–585. [DOI] [PubMed] [Google Scholar]
- Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T, 2006. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia 49, 1881–1892. [DOI] [PubMed] [Google Scholar]
- Natesan SK, Chandrasekar PH, Alangaden GJ, Manavathu EK, 2008. Fluvastatin potentiates the activity of caspofungin against Aspergillus fumigatus in vitro. Diagnostic Microbiology and Infectious Disease 60, 369–373. [DOI] [PubMed] [Google Scholar]
- Nohria A, Grunert ME, Rikitake Y, Noma K, Prsic A, Ganz P, Liao JK, Creager MA, 2006. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ Res 99, 1426–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohria A, Prsic A, Liu P-Y, Okamoto R, Creager MA, Selwyn A, Liao JK, Ganz P, 2009. Statins inhibit Rho kinase activity in patients with atherosclerosis. Atherosclerosis 205, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, Hiroi Y, Shimizu K, Luscinskas FW, Sun J, Liao JK, 2008. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest 118, 1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyilasi I, Kocsubé S, Galgoczy L, Papp T, Pesti M, Vágvölgyi C, 2010a. Effect of different statins on the antifungal activity of polyene antimycotic. Acta Biologica Szegediensis 54, 33–36. [Google Scholar]
- Nyilasi I, Kocsubé S, Krizsán K, Galgóczy L, Papp T, Pesti M, Nagy K, Vágvölgyi C, 2014. Susceptibility of clinically important dermatophytes against statins and different statin-antifungal combinations. Med Mycol 52, 140–148. [DOI] [PubMed] [Google Scholar]
- Nyilasi I, Kocsubé S, Krizsán K, Galgóczy L, Pesti M, Papp T, Vágvölgyi C, 2010b. In vitro synergistic interactions of the effects of various statins and azoles against some clinically important fungi. FEMS Microbiol Lett 307, 175–184. [DOI] [PubMed] [Google Scholar]
- Nyilasi I, Kocsubé S, Pesti M, Lukacs G, Papp T, Vágvölgyi C, 2009. In vitro interactions between primycin and different statins in their effects against some clinically important fungi. Journal of medical microbiology 59, 200–205. [DOI] [PubMed] [Google Scholar]
- Oesterle A, Laufs U, Liao JK, 2017. Pleiotropic Effects of Statins on the Cardiovascular System. Circ Res 120, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnaka K, Shimoda S, Nawata H, Shimokawa H, Kaibuchi K, Iwamoto Y, Takayanagi R, 2001. Pitavastatin Enhanced BMP-2 and Osteocalcin Expression by Inhibition of Rho-Associated Kinase in Human Osteoblasts. Biochemical and Biophysical Research Communications 287, 337–342. [DOI] [PubMed] [Google Scholar]
- Ortego M, Bustos C, Hernández-Presa MA, Tuñón J, Dıaz C, Hernández G, Egido J, 1999. Atorvastatin reduces NF-κB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis 147, 253–261. [DOI] [PubMed] [Google Scholar]
- Ott BR, Daiello LA, Dahabreh IJ, Springate BA, Bixby K, Murali M, Trikalinos TA, 2015. Do statins impair cognition? A systematic review and meta-analysis of randomized controlled trials. J Gen Intern Med 30, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagiatakis C, Gordon JW, Ehyai S, McDermott JC, 2012. A novel RhoA/ROCK-CPI-17-MEF2C signaling pathway regulates vascular smooth muscle cell gene expression. J Biol Chem 287, 8361–8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine MF, Criss AB, Watkins PB, 2005. Two major grapefruit juice components differ in time to onset of intestinal CYP3A4 inhibition. J Pharmacol Exp Ther 312, 1151–1160. [DOI] [PubMed] [Google Scholar]
- Paragh G, Kertai P, Kovacs P, Paragh G Jr., Fulop P, Foris G, 2003. HMG CoA reductase inhibitor fluvastatin arrests the development of implanted hepatocarcinoma in rats. Anticancer research 23, 3949–3954. [PubMed] [Google Scholar]
- Parrales A, Ranjan A, Iyer SV, Padhye S, Weir SJ, Roy A, Iwakuma T, 2016. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol 18, 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paseban M, Butler AE, Sahebkar A, 2019. Mechanisms of statin-induced new-onset diabetes. Journal of Cellular Physiology 234, 12551–12561. [DOI] [PubMed] [Google Scholar]
- Peng H-B, Libby P, Liao JK, 1995. Induction and Stabilization of IκBα by Nitric Oxide Mediates Inhibition of NF-κB *. Journal of Biological Chemistry 270, 14214–14219. [DOI] [PubMed] [Google Scholar]
- Perego C, Da Dalt L, Pirillo A, Galli A, Catapano AL, Norata GD, 2019. Cholesterol metabolism, pancreatic β-cell function and diabetes. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1865, 2149–2156. [DOI] [PubMed] [Google Scholar]
- Petejova N, Martinek A, 2014. Acute kidney injury due to rhabdomyolysis and renal replacement therapy: a critical review. Crit Care 18, 224–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikoulis E, Margonis GA, Angelou A, Zografos GC, Antoniou E, 2016. Statins in the chemoprevention of colorectal cancer in established animal models of sporadic and colitis-associated cancer. European journal of cancer prevention : the official journal of the European Cancer Prevention Organisation (ECP) 25, 102–108. [DOI] [PubMed] [Google Scholar]
- Pinal-Fernandez I, Casal-Dominguez M, Mammen AL, 2018. Statins: pros and cons. Med Clin (Barc) 150, 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanti S, Picardi P, Ciaglia E, D’Alessandro A, Bifulco M, 2014. Novel prospects of statins as therapeutic agents in cancer. Pharmacological Research 88, 84–98. [DOI] [PubMed] [Google Scholar]
- Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, Low M, Greenson JK, Rennert G, 2005. Statins and the risk of colorectal cancer. The New England journal of medicine 352, 2184–2192. [DOI] [PubMed] [Google Scholar]
- Qiao LJ, Kang KL, Heo JS, 2011. Simvastatin promotes osteogenic differentiation of mouse embryonic stem cells via canonical Wnt/β-catenin signaling. Mol Cells 32, 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H, Guo M, Chai H, Wang W.t., Gao Z.y., Shi D.z., 2018. Effects of Coenzyme Q10 on Statin-Induced Myopathy: An Updated Meta-Analysis of Randomized Controlled Trials. Journal of the American Heart Association 7, e009835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkumar S, Raghunath A, Raghunath S, 2016. Statin Therapy: Review of Safety and Potential Side Effects. Acta Cardiol Sin 32, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings R, Nohria A, Liu P-Y, Donnelly J, Creager MA, Ganz P, Selwyn A, Liao JK, 2009. Comparison of effects of rosuvastatin (10 mg) versus atorvastatin (40 mg) on rho kinase activity in caucasian men with a previous atherosclerotic event. Am J Cardiol 103, 437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ, 2008. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. New England Journal of Medicine 359, 2195–2207. [DOI] [PubMed] [Google Scholar]
- Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA, Isenberg JS, 2006. The biphasic nature of nitric oxide responses in tumor biology. Antioxid Redox Signal 8, 1329–1337. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Liao JK, 2005. Rho GTPases, statins, and nitric oxide. Circ Res 97, 1232–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JG, 2015. Statins and diabetes risk: how real is it and what are the mechanisms? Curr Opin Lipidol 26, 228–235. [DOI] [PubMed] [Google Scholar]
- Roche CM, Trimble ER, Ennis M, 1995. Effect of in vivo and in vitro Lovastatin Treatment on Mast Cell Activation. International Archives of Allergy and Immunology 108, 240–246. [DOI] [PubMed] [Google Scholar]
- Romano M, Mezzetti A, Marulli C, Ciabattoni G, Febo F, Di Ienno S, Roccaforte S, Vigneri S, Nubile G, Milani M, Davi G, 2000. Fluvastatin reduces soluble P-selectin and ICAM-1 levels in hypercholesterolemic patients: role of nitric oxide. Journal of Investigative Medicine 48. [PubMed] [Google Scholar]
- Rosenbaum D, Dallongeville J, Sabouret P, Bruckert E, 2013. Discontinuation of statin therapy due to muscular side effects: A survey in real life. Nutrition, Metabolism and Cardiovascular Diseases 23, 871–875. [DOI] [PubMed] [Google Scholar]
- Ruiz-Gaspa S, Nogues X, Enjuanes A, Monllau JC, Blanch J, Carreras R, Mellibovsky L, Grinberg D, Balcells S, Díez-Perez A, Pedro-Botet J, 2007. Simvastatin and atorvastatin enhance gene expression of collagen type 1 and osteocalcin in primary human osteoblasts and MG-63 cultures. Journal of Cellular Biochemistry 101, 1430–1438. [DOI] [PubMed] [Google Scholar]
- Sanli T, Liu C, Rashid A, Hopmans SN, Tsiani E, Schultz C, Farrell T, Singh G, Wright J, Tsakiridis T, 2011. Lovastatin Sensitizes Lung Cancer Cells to Ionizing Radiation: Modulation of Molecular Pathways of Radioresistance and Tumor Suppression. Journal of Thoracic Oncology 6, 439–450. [DOI] [PubMed] [Google Scholar]
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJM, Seshasai SRK, McMurray JJ, Freeman DJ, Jukema JW, Macfarlane PW, Packard CJ, Stott DJ, Westendorp RG, Shepherd J, Davis BR, Pressel SL, Marchioli R, Marfisi RM, Maggioni AP, Tavazzi L, Tognoni G, Kjekshus J, Pedersen TR, Cook TJ, Gotto AM, Clearfield MB, Downs JR, Nakamura H, Ohashi Y, Mizuno K, Ray KK, Ford I, 2010. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. The Lancet 375, 735–742. [DOI] [PubMed] [Google Scholar]
- Schachter M, 2005. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundamental & Clinical Pharmacology 19, 117–125. [DOI] [PubMed] [Google Scholar]
- Schirris TJ, Ritschel T, Bilos A, Smeitink JA, Russel FG, 2015. Statin Lactonization by Uridine 5’-Diphospho-glucuronosyltransferases (UGTs). Mol Pharm 12, 4048–4055. [DOI] [PubMed] [Google Scholar]
- Schultz BG, Patten DK, Berlau DJ, 2018. The role of statins in both cognitive impairment and protection against dementia: a tale of two mechanisms. Transl Neurodegener 7, 5–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seccia TM, Rigato M, Ravarotto V, Calò LA, 2020. ROCK (RhoA/Rho Kinase) in Cardiovascular-Renal Pathophysiology: A Review of New Advancements. J Clin Med 9, 1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhang Y-W, Summers LJ, Dorn GW 2nd, Wei L, 2008. Disruption of ROCK1 gene attenuates cardiac dilation and improves contractile function in pathological cardiac hypertrophy. J Mol Cell Cardiol 44, 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Liao JK, 2016. Rho Kinases and Cardiac Remodeling. Circ J 80, 1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitara Y, 2011. Clinical Importance of OATP1B1 and OATP1B3 in Drug–Drug Interactions. Drug Metabolism and Pharmacokinetics 26, 220–227. [DOI] [PubMed] [Google Scholar]
- Sinha KM, Zhou X, 2013. Genetic and molecular control of osterix in skeletal formation. J Cell Biochem 114, 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Guo Z, Ma Q, Chen Z, Liu Z, Jia H, Dang G, 2003. Simvastatin induces osteoblastic differentiation and inhibits adipocytic differentiation in mouse bone marrow stromal cells. Biochemical and Biophysical Research Communications 308, 458–462. [DOI] [PubMed] [Google Scholar]
- Song Y, Nie H, Xu Y, Zhang L, Wu Y, 2013. Association of statin use with risk of dementia: A meta-analysis of prospective cohort studies. Geriatrics & Gerontology International 13, 817–824. [DOI] [PubMed] [Google Scholar]
- Spampanato C, De Maria S, Sarnataro M, Giordano E, Zanfardino M, Baiano S, Cartenì M, Morelli F, 2012. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int J Oncol 40, 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD, 2017. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama M, Kodama T, Konishi K, Abe K, Asami S, Oikawa S, 2000. Compactin and Simvastatin, but Not Pravastatin, Induce Bone Morphogenetic Protein-2 in Human Osteosarcoma Cells. Biochemical and Biophysical Research Communications 271, 688–692. [DOI] [PubMed] [Google Scholar]
- Sukhova GK, Williams JK, Libby P, 2002. Statins Reduce Inflammation in Atheroma of Nonhuman Primates Independent of Effects on Serum Cholesterol. Arteriosclerosis, Thrombosis, and Vascular Biology 22, 1452–1458. [DOI] [PubMed] [Google Scholar]
- Suraweera C, de Silva V, Hanwella R, 2016. Simvastatin-induced cognitive dysfunction: two case reports. J Med Case Rep 10, 83–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaguri A, Satoh K, Itagaki M, Tokumitsu Y, Ichihara K, 2008. Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3T3L1 adipocytes. J Pharmacol Sci 107, 80–89. [DOI] [PubMed] [Google Scholar]
- Tavakkoli A, Johnston TP, Sahebkar A, 2020. Antifungal effects of statins. Pharmacology & Therapeutics 208, 107483. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Panza G, Zaleski A, Taylor B, 2016. Statin-Associated Side Effects. J Am Coll Cardiol 67, 2395–2410. [DOI] [PubMed] [Google Scholar]
- Tolman KG, 2000. Defining patient risks from expanded preventive therapies. Am J Cardiol 85, 15–19. [DOI] [PubMed] [Google Scholar]
- Tolman KG, 2002. The liver and lovastatin. Am J Cardiol 89, 1374–1380. [DOI] [PubMed] [Google Scholar]
- Tomaszewski M, Stępień KM, Tomaszewska J, Czuczwar SJ, 2011. Statin-induced myopathies. Pharmacol Rep 63, 859–866. [DOI] [PubMed] [Google Scholar]
- Tonelli M, Lloyd AM, Bello AK, James MT, Klarenbach SW, McAlister FA, Manns BJ, Tsuyuki RT, Hemmelgarn BR, for the Alberta Kidney Disease, N., 2019. Statin use and the risk of acute kidney injury in older adults. BMC Nephrology 20, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunon J, Egido J, 2004. [Endothelial dysfunction, inflammation and statins: new evidences]. Rev Esp Cardiol 57, 903–905. [PubMed] [Google Scholar]
- Turner RM, Pirmohamed M, 2019. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J Clin Med 9, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbich C, Dernbach E, Zeiher AM, Dimmeler S, 2002. Double-Edged Role of Statins in Angiogenesis Signaling. Circ Res 90, 737–744. [DOI] [PubMed] [Google Scholar]
- Vahabzadeh M, Mohammadpour A, 2015. Effect of Diabetes Mellitus on the Metabolism of Drugs and Toxins. Journal of Clinical Toxicology 05. [Google Scholar]
- Vallianou NG, Kostantinou A, Kougias M, Kazazis C, 2014. Statins and cancer. Anticancer agents in medicinal chemistry 14, 706–712. [DOI] [PubMed] [Google Scholar]
- van Haelst PL, van Doormaal JJ, May JF, Gans ROB, Crijns HJGM, Cohen Tervaert JW, 2001. Secondary prevention with fluvastatin decreases levels of adhesion molecules, neopterin and C-reactive protein. European Journal of Internal Medicine 12, 503–509. [DOI] [PubMed] [Google Scholar]
- Veillard NR, Braunersreuther V, Arnaud C, Burger F, Pelli G, Steffens S, Mach F, 2006. Simvastatin modulates chemokine and chemokine receptor expression by geranylgeranyl isoprenoid pathway in human endothelial cells and macrophages. Atherosclerosis 188, 51–58. [DOI] [PubMed] [Google Scholar]
- Venturini TP, Rossato L, Spader TB, Tronco-Alves GR, Azevedo MI, Weiler CB, Santurio JM, Alves SH, 2011. In vitro synergisms obtained by amphotericin B and voriconazole associated with non-antifungal agents against Fusarium spp. Diagn Microbiol Infect Dis 71, 126–130. [DOI] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, Viswanathan SR, Chattopadhyay S, Tamayo P, Yang WS, Rees MG, Chen S, Boskovic ZV, Javaid S, Huang C, Wu X, Tseng Y-Y, Roider EM, Gao D, Cleary JM, Wolpin BM, Mesirov JP, Haber DA, Engelman JA, Boehm JS, Kotz JD, Hon CS, Chen Y, Hahn WC, Levesque MP, Doench JG, Berens ME, Shamji AF, Clemons PA, Stockwell BR, Schreiber SL, 2017. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF, Nebreda ÁR, 2009. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature Reviews Cancer 9, 537–549. [DOI] [PubMed] [Google Scholar]
- Wagstaff LR, Mitton MW, Arvik BM, Doraiswamy PM, 2003. Statin-Associated Memory Loss: Analysis of 60 Case Reports and Review of the Literature. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 23, 871–880. [DOI] [PubMed] [Google Scholar]
- Wang S, Xie X, Lei T, Zhang K, Lai B, Zhang Z, Guan Y, Mao G, Xiao L, Wang N, 2017. Statins Attenuate Activation of the NLRP3 Inflammasome by Oxidized LDL or TNFα in Vascular Endothelial Cells through a PXR-Dependent Mechanism. Molecular Pharmacology 92, 256. [DOI] [PubMed] [Google Scholar]
- Warner GJ, Berry MJ, Moustafa ME, Carlson BA, Hatfield DL, Faust JR, 2000. Inhibition of Selenoprotein Synthesis by Selenocysteine tRNA[Ser]Sec Lacking Isopentenyladenosine *. Journal of Biological Chemistry 275, 28110–28119. [DOI] [PubMed] [Google Scholar]
- Weitz-Schmidt G, 2002. Statins as anti-inflammatory agents. Trends in pharmacological sciences 23, 482–486. [DOI] [PubMed] [Google Scholar]
- Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, Cottens S, Takada Y, Hommel U, 2001. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nature Medicine 7, 687–692. [DOI] [PubMed] [Google Scholar]
- Weivoda MM, Hohl RJ, 2011. Effects of Farnesyl Pyrophosphate Accumulation on Calvarial Osteoblast Differentiation. Endocrinology 152, 3113–3122. [DOI] [PubMed] [Google Scholar]
- Wiggins BS, Saseen JJ, Page RL 2nd, Reed BN, Sneed K, Kostis JB, Lanfear D, Virani S, Morris PB, 2016. Recommendations for Management of Clinically Significant Drug-Drug Interactions With Statins and Select Agents Used in Patients With Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 134, e468–e495. [DOI] [PubMed] [Google Scholar]
- Wilcox LJ, Borradaile NM, de Dreu LE, Huff MW, 2001. Secretion of hepatocyte apoB is inhibited by the flavonoids, naringenin and hesperetin, via reduced activity and expression of ACAT2 and MTP. J Lipid Res 42, 725–734. [PubMed] [Google Scholar]
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G, 2000. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 57, 1439–1443. [DOI] [PubMed] [Google Scholar]
- Wong WB, Lin VW, Boudreau D, Devine EB, 2013. Statins in the prevention of dementia and Alzheimer’s disease: A meta-analysis of observational studies and an assessment of confounding. Pharmacoepidemiology and Drug Safety 22, 345–358. [DOI] [PubMed] [Google Scholar]
- Xia F, Xie L, Mihic A, Gao X, Chen Y, Gaisano HY, Tsushima RG, 2008. Inhibition of Cholesterol Biosynthesis Impairs Insulin Secretion and Voltage-Gated Calcium Channel Function in Pancreatic β-Cells. Endocrinology 149, 5136–5145. [DOI] [PubMed] [Google Scholar]
- Xu N, Shen N, Wang X, Jiang S, Xue B, Li C, 2015. Protein prenylation and human diseases: a balance of protein farnesylation and geranylgeranylation. Science China Life Sciences 58, 328–335. [DOI] [PubMed] [Google Scholar]
- Yanae M, Tsubaki M, Satou T, Itoh T, Imano M, Yamazoe Y, Nishida S, 2011. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. Journal of Experimental & Clinical Cancer Research 30, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SS, Li R, Qu X, Fang W, Quan Z, 2012. Atorvastatin decreases Toll-like receptor 4 expression and downstream signaling in human monocytic leukemia cells. Cellular Immunology 279, 96–102. [DOI] [PubMed] [Google Scholar]
- Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, Motoshima H, Taguchi T, Sonoda K, Kukidome D, Takuwa Y, Kawada T, Brownlee M, Nishikawa T, Araki E, 2007. Statins Activate Peroxisome Proliferator-Activated Receptor γ Through Extracellular Signal-Regulated Kinase 1/2 and p38 Mitogen-Activated Protein Kinase–Dependent Cyclooxygenase-2 Expression in Macrophages. Circ Res 100, 1442–1451. [DOI] [PubMed] [Google Scholar]
- Ye L, Jin F, Kumar SK, Dai Y, 2021. The mechanisms and therapeutic targets of ferroptosis in cancer. Expert Opinion on Therapeutic Targets 25, 965–986. [DOI] [PubMed] [Google Scholar]
- Yilmaz A, Reiss C, Tantawi O, Weng A, Stumpf C, Raaz D, Ludwig J, Berger T, Steinkasserer A, Daniel WG, Garlichs CD, 2004. HMG-CoA reductase inhibitors suppress maturation of human dendritic cells: new implications for atherosclerosis. Atherosclerosis 172, 85–93. [DOI] [PubMed] [Google Scholar]
- Yokote K, Shimano H, Urashima M, Teramoto T, 2011. Efficacy and safety of pitavastatin in Japanese patients with hypercholesterolemia: LIVES study and subanalysis. Expert Review of Cardiovascular Therapy 9, 555–562. [DOI] [PubMed] [Google Scholar]
- Yoneda A, Multhaupt HAB, Couchman JR, 2005. The Rho kinases I and II regulate different aspects of myosin II activity. Journal of Cell Biology 170, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Asanuma M, Grossmann L, Fuse M, Shibata T, Yonekawa T, Tanaka T, Ueno K, Yasuda T, Saito Y, Tatsuno I, 2006. Geranylgeranyl-pyrophosphate (GGPP) synthase is down-regulated during differentiation of osteoblastic cell line MC3T3-E1. FEBS Letters 580, 5203–5207. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang X, 2020. Targeting the Wnt/β-catenin signaling pathway in cancer. J Hematol Oncol 13, 165–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-M, Bo J, Taffet GE, Chang J, Shi J, Reddy AK, Michael LH, Schneider MD, Entman ML, Schwartz RJ, Wei L, 2006. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. The FASEB Journal 20, 916–925. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhao S-P, 2015. Different effects of statins on induction of diabetes mellitus: an experimental study. Drug Des Devel Ther 9, 6211–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Li W, Xie Q, Hou Y, Zhan S, Yang X, Xu X, Cai J, Huang Z, 2014a. Effects of Simvastatin on Glucose Metabolism in Mouse MIN6 Cells. Journal of Diabetes Research 2014, 376570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y-T, Yu L-S, Zeng S, Huang Y-W, Xu H-M, Zhou Q, 2014b. Pharmacokinetic drug-drug interactions between 1,4-dihydropyridine calcium channel blockers and statins: factors determining interaction strength and relevant clinical risk management. Ther Clin Risk Manag 10, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P-F, Wang M-X, Chen Z-L, Yang L, 2021. Targeting the Tumor Microenvironment: A Literature Review of the Novel Anti-Tumor Mechanism of Statins. Frontiers in oncology 11, 761107–761107. [DOI] [PMC free article] [PubMed] [Google Scholar]