

Abstract
Hundreds of genetic loci increasing risk for neuropsychiatric disorders have recently been identified. This success, perhaps paradoxically, has posed challenges for therapeutic development, which are amplified by the highly polygenic and pleiotropic nature of these genetic contributions. Success requires understanding the biological impact of single genetic variants and predicting their effects within an individual. Comprehensive functional genomic annotation of risk loci provides a framework for interpretation of neurobiological impact, requiring experimental validation with in vivo or in vitro model systems. Systems-level, integrative pathway analyses are beginning to elucidate the additive, polygenic contributions of risk variants on specific cellular, molecular, developmental, or circuit-level processes. Although most neuropsychiatric disease modeling has focused on genes disrupted by rare, large-effect-size mutations, common smaller-effect-size variants may also provide solid therapeutic targets to inform precision medicine approaches. Here we enumerate the promise and challenges of a genomics-driven approach to uncovering neuropsychiatric disease mechanisms and facilitating therapeutic development.
The high heritability of neuropsychiatric disorders (46.3% as a class)1 is a tantalizing clue that genetics will finally provide a rigorous neurobiological framework for comprehending conditions that have evaded biological understanding for decades2. Heritability estimates indicate that inherited genetic variants contribute substantially to disease liability, often more so than early environmental influences or noninherited, de novo mutations, but clearly gene and environment usually contribute together (Fig. 1)2–6. Initial linkage and candidate gene studies of psychiatric disease often yielded inconsistent findings, as a result of limited power and difficulty accounting for systematic biases such as population stratification. When interpreting results from large-scale genomics studies, it is important to take statistical power into consideration7. So, in contrast to candidate gene studies, results from more recent, large-scale genome-wide studies have yielded much more robust results3.
Figure 1.
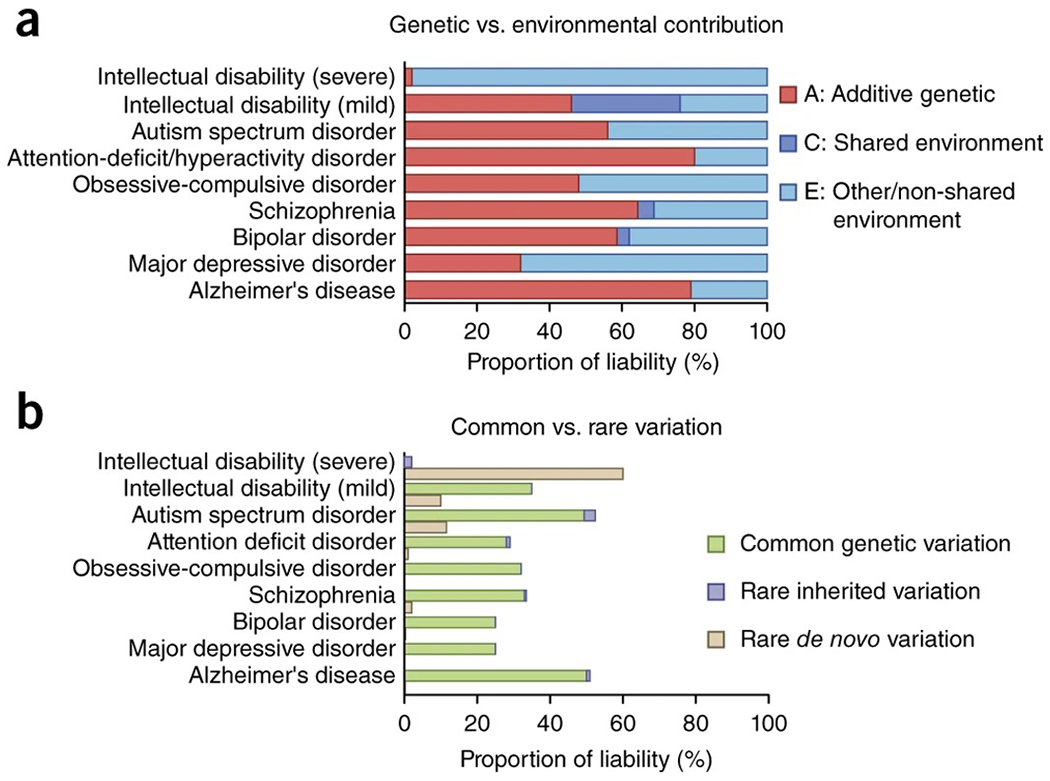
Genetic and environmental contribution to liability for neuropsychiatric disease. (a) ACE model liability estimates (see Box 2) are compiled for various neuropsychiatric disorders derived from large-scale twin and/or population-based studies. (b) Genetic contributions can be further partitioned by variant classes, including common, rare inherited, and rare de novo mutations. The contribution of de novo variants to disease liability is lower than their overall frequency in cases due to incomplete penetrance. Data are compiled from refs. 2,3,8–10,13,29,31,123,133–138.
The genetic architecture of psychiatric disease has received much attention and is the subject of several recent reviews2–6,8–10. Genetic variants associated with neuropsychiatric disease take several forms based on detection methodology and study design (Box 1 and Table 1)4. They can also be classified by effect size, which can be inferred from population genetics models that predict an inverse relationship between variant frequency and effect size11.
Box 1. Large-scale genetic investigation of complex traits, such as neuropsychiatric disease.
Technological advances now enable cost-effective, genome-wide interrogation of genetic variation in large cohorts, but they necessitate careful power analysis and study design to maximize variant discovery (Table 1)7. Microarray-based platforms can detect structural anomalies such as CNVs or genomic rearrangements. SNP microarrays provide a cost-effective platform for common trait GWAS. A genome-wide SNP backbone coupled with imputation to an ancestry-matched reference panel enables efficient genomic coverage. Population-specific platforms have been developed, such as the PsychChip, which has higher density in regions associated with psychiatric disease, including rare CNVs and exome variants. Despite this, coverage remains incomplete and generally limited to common or previously identified rare variants. Massively parallel, high-throughput sequencing platforms identify variants with single-base-pair resolution and can theoretically capture the full range of allele frequencies (for example, common, rare, private) and variant types (SNVs, indels, CNVs). In WES, the ~1% protein-coding portion of the genome is captured and then sequenced, to reduce cost and bolster interpretability of identified variants. WGS surveys the entire genomic space, although coverage is still often incomplete because of difficulties mapping repeat-dense regions. Sufficient depth is critical to overcoming potential sequencing errors and capturing heterozygous SNVs. Sanger sequencing is often performed as a confirmatory test.
Study design is an important factor when considering large-scale genetic studies. Case/control is a standard design that compares allele frequencies across a diverse set of cases and controls en masse. However, subtle biases (for example, population stratification) must be rigorously accounted for and inheritance patterns cannot be determined. Family designs that include a proband and both parents (‘trio’) can account for population stratification and identify inheritance patterns but are more difficult and expensive to collect. Filtering for de novo variants in a proband with unaffected parents can facilitate interpretation of pathogenicity. However, ‘unaffected’ parents may harbor incompletely penetrant mutations, especially for complex traits.
Table 1.
Platforms for large-scale, genome-wide interrogation of genetic variation
| Technology | Outcome measure in individual | Outcome measure in population | Challenges to interpretation |
|---|---|---|---|
| Chromosomal microarray | CNV | Recurrence | (1) Pleiotropy (2) Incomplete penetrance (3) Pathogenic gene(s) not directly identified |
| Genome-wide SNP microarray | SNP Polygenic score | Genome-wide significant index SNP or haplotype | (1) Identifying causal variant (2) Identifying functional effect of variant (3) Function of noncoding regions often not well established, especially in CNS |
| Whole exome sequencing | (De novo) SNVs | Gene burden test | (1) Pathogenicity often difficult to establish unless multiple instances observed (2) Functional significance often unclear, especially for missense mutations |
| Whole genome sequencing | SNPs (De novo) SNVs, indels | Gene burden test or Recurrence | (1) Pathogenicity often difficult to establish unless multiple instances observed (2) Functional significance often unclear, especially for missense mutations (3) Function of noncoding regions often not well established, especially in CNS |
Hundreds of causal genetic variants with varying effect sizes have been robustly associated with neuropsychiatric disorders, with thousands more likely involved3,12–16. An essential next step is deciphering the biological impact of these variants. Here we discuss biological interpretation of genetic variation, focusing on rare variants of moderate to large effect and common variants with small effect. This genetics-driven approach has several advantages. First, genetics accounts for a majority of disease liability for many neuropsychiatric disorders and is therefore expected to be a high-yield area of investigation (Box 2 and Fig. 1). Second, genetic variants indicate biological causality. Third, human genetics is grounded in human biology, which is especially important for neuropsychiatric phenotypes that may not be fully conserved across species. Finally, next-generation sequencing technology provides a near-complete survey of the genetic search space in an unbiased fashion at genome-wide scale, circumventing many of the limitations in reproducibility that undermined earlier genetic approaches (Table 1).
Box 2. Genetic architecture of neuropsychiatric disease.
A fundamental question for any complex human trait is the degree to which genetic or environmental factors influence phenotypic variance. Heritability (h2) refers to the proportion of phenotypic variance due to genetic factors and in the narrow sense is also referred to as additive genetic variance (A). Environmental factors can be partitioned into the common, shared environment (C) and the residual, nonshared environmental variance (E). While the common, shared environment can be difficult to precisely pinpoint, it is often interpreted as in utero and early childhood factors. Classically, twin studies have been used to estimate these various components, although more sophisticated statistical methods have been developed (for example, generalized linear mixed models)139. Importantly, de novo genetic variation, which can contribute substantially to disorders such as ASD or intellectual disability, is generally not captured in heritability estimates. Disease-associated genetic variation can be further partitioned by allele frequency and inheritance patterns. Common variants (minor allele frequency >0.5%) generally have small effect sizes with odds ratios <1.3. Rare variants, including CNVs, have much larger effect sizes (odds ratios typically 2–60), and yet penetrance for specific clinically defined disorders can vary widely. Mutations of larger effect size have been constrained by natural selection because of negative effects on reproductive fitness and therefore tend to be both rare and de novo. The contribution of common genetic variation to overall disease liability (for example, SNP heritability) can be estimated using genome-wide complex trait analysis (GCTA)140 or methods that partition heritability, such as LD-score regression (Fig. 1)57. Except for severe intellectual disability (IQ <50), current estimates indicate that rare variants contribute an order of magnitude less to overall disease liability than do common variants, although this varies across conditions.
Interpreting rare genetic variation
An early clue of the genetic contribution to major psychiatric conditions was their association with rare Mendelian syndromes, such as DiGeorge, Rett, or fragile X, each with characteristic morphologic, cognitive, and neuropsychiatric phenotypes. The advent of chromosomal microarrays enabled the detection of copy number variation (CNV), submicroscopic deletions or duplications in DNA. More recently, whole exome sequencing (WES) and whole genome sequencing (WGS) have enabled the large-scale detection of rare, unique and private single nucleotide variants (SNVs), small chromosomal rearrangements (<50 kb), indels, and inversions. Chromosomal microarrays and WES have such a high yield in identifying genetic variants underlying neurodevelopmental disorders that they are becoming the standard of care for children with autism spectrum disorder (ASD)17.
Detection, association, and interpretation of disease-causing genetic variants have many challenges, largely driven by the relatively high number of potentially disease-causing rare variants in every genome18,19. Sequencing studies are rarely sufficiently powered to detect disease association at a variant level, given the vast size of the genomic search space and potential number of ultra-rare or even private variants. To improve power, gene-based approaches are often applied, in which association testing is performed after variants are aggregated at the gene level7. Formal statistical significance should be assessed at genome-wide thresholds and statistical evidence of association should not be superseded by biological plausibility or ‘functionality’.20 The genome of a random individual will have on average 100 loss-of-function or likely gene-disrupting variants (nonsense, frameshift, and splice-site mutations), approximately one of which will be de novo. Furthermore, every individual carries on average 20 completely inactivated genes19. Synonymous variants are far more common and are therefore usually set aside, although there is now evidence that synonymous variation can have gene regulatory functions and can contribute to disease risk21. Individual rare genetic variants must therefore be interpreted in the context of the specific locus’s or gene’s tolerance for mutations, evolutionary or selective constraint18, and population allele frequency20. Several bioinformatic tools exist that predict the deleteriousness of a given variant or tolerance for mutation at a gene level18,22, although this remains an area of active development.
Even after taking into account inheritance and the predicted functional severity of a mutation, causal ambiguity often still exists even in cases of de novo protein truncating mutations, resulting in the assignment of ‘variant of unknown significance’. Robustly identifying the most likely causal rare variants requires more complete genomic annotations (including the noncoding part of the genome) and extensive population allele frequency databases from several populations (Fig. 2). A rare allele in one population may actually be common in another and without strong phenotypic consequences, substantially changing the interpretation of pathogenicity20. To confront this, several large-scale efforts have been made to aggregate population-level genomic variation into searchable databases, including among others ExAC23, DGV, and ClinVar. As an example, variants that are not found in the ExAC database, which includes WES results from over 60,000 unrelated adults without history of severe pediatric disease, are more likely to be deleterious23,24. Finally, the noncoding genome plays important regulatory roles, but is excluded from WES and has not been analyzed in the majority of published WGS papers. Having more comprehensive annotation of the noncoding genome in neural tissues is therefore a pressing goal of current research25. Standard pathway analyses should be applied only once variants have statistical support to avoid risk of false-positive results due to potential biases in these analyses, as well as inherent sensitivity to inclusion of spurious genes and population stratification (Box 3)7,26,27.
Figure 2.
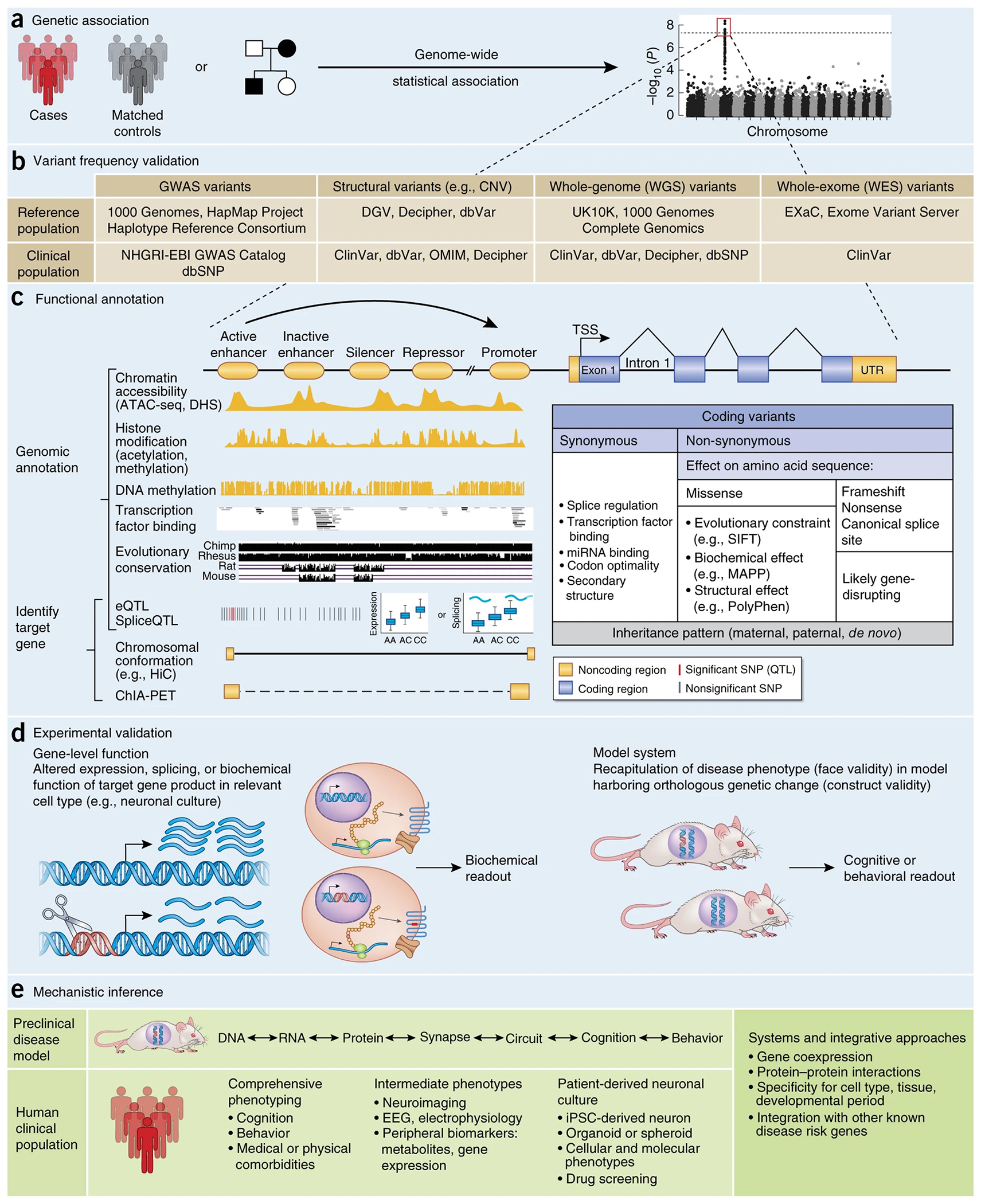
Neurobiological framework for interpretation of individual disease-associated variants. (a) When considering a neurobiological framework for interpretation of disease-associated genetic variation, it is most important to begin with variants that meet genome-wide significance thresholds20. (b) Independent replication is also critical, which can be supported by prior reported associations in a clinical genetic database (for example, ClinVar) and by an appropriate observed frequency in large population reference databases (for example, ExAC). (c) Functional annotation differs for coding and noncoding variants, although some general principles apply to both (for example, inheritance, evolutionary conservation). For coding variants, the target gene is known and annotation is initially based on impact to the amino acid sequence. Synonymous mutations, often interpreted as neutral, can contribute to human disease risk by changing transcription factor or microRNA binding or by altering mRNA stability or secondary structure21. Nonsense, frameshift, and canonical splice site mutations are generally placed in the most deleterious, likely gene disrupting category, although their disease association must still be statistically supported. Interpretation of missense mutations is more difficult, relying typically on evolutionary constraint or by inferred disruption of protein structure or biochemical function22. Functional annotation of noncoding variants is a rapidly evolving area, but can be broadly conceptualized as (top) predicting a regulatory effect and (bottom) identifying target gene(s). Computational methods can predict the likelihood that noncoding regions act as enhancers, repressors, or insulators within a given tissue or cell line on the basis of epigenetic annotations49. Gene targets can be inferred through statistical frameworks such as eQTL or by mapping intrachromosomal physical binding interactions through chromosome conformation capture methods. (d) Predictions of the potential impact of a variant on the target gene should be experimentally validated. Gene-level disruption can be confirmed in a cell-based experimental system, as long as genomic and epigenetic context are considered. Model organisms with construct validity may also be useful. (e) Once the proximal biological effect of a disease-associated variant is determined, disease mechanisms can begin to be inferred through follow up investigation in preclinical or clinical settings. Performing comprehensive clinical and medical phenotyping of individuals harboring specific, known disease-associated variants will be especially important for mechanistic insight as well as future ‘genotype-first’ precision medicine approaches35. NHGRI, National Human Genome Research Institute; EBI, European Bioinformatics Institute; ATAC-seq, assay for transposase-accessible chromatin with sequencing; DHS, DNase I hypersensitivity sites; ChIA-PET, chromatin interaction analysis by paired-end tag sequencing; TSS, transcription start site; SIFT, sorting intolerant from tolerant; MAPP, multivariate analysis of protein polymorphism.
Box 3. Lessons in reproducibility.
Psychiatric genetics is susceptible to false positive results, a problem amplified by frequent comorbidities, overlapping symptoms and limited biomarkers. The candidate gene era was fraught with false positives, which have been limited by genome-wide analyses3. However, in the era of whole exome and genome sequencing, nonpathogenic rare and private variants will be identified in every individual genome, so extra care must be taken to avoid overinterpretation of results7,20.
Replication is critical; genotypes and phenotypes between discovery and replication sets should be comparable. For example, an early genetic finding in schizophrenia was a linkage peak including the DTNBP1 locus. Replication studies measured different markers around DTNBP1 without imputation to a common reference, each defining a different haplotype as the risk allele, with no concordance of findings141. And indeed, the largest schizophrenia GWAS to date has failed to find any association near the DTNBP1 locus.
In case/control studies it is critical to account for all potential biological (for example, age, sex) and technical confounds, especially those related to experimental design, such as batch effects. For example, a study profiling gene expression in cell lines derived from subjects of European and Asian ancestry reported that 25% of genes were differentially expressed across ethnicities, which was claimed to reflect common genetic variation142. However, these results disappeared after accounting for a strong group × batch confound143. Similarly, a recent high-profile GWAS of longevity reported 33 genome-wide significant SNPs, which were able to predict lifespan in an independent cohort with a remarkable 77% accuracy144. This study was later retracted after it was determined that a batch effect likely accounted for the signal.
Subtle differences in allele frequencies between subpopulations within case and control groups (termed “population stratification”) or (cryptic) relatedness among subjects can also introduce significant bias. A recent study claimed to predict a diagnosis of ASD with a remarkable ~70% accuracy using only 237 common SNPs, but did not properly account for population stratification, as claimed145. Rather, these SNPs were strongly associated with ethnicity differences between subjects, and did not predict ASD status146. Similarly, a recent paper claimed to identify eight genetically defined subtypes of schizophrenia in 4,196 patients and 3,827 controls, but did not account for population stratification147. One should be concerned that, without explicit correction, these results are driven by ancestry or other hidden confounds.
Finally, rare variants are present in every genome, can have a predicted functional effect without actually being pathogenic, can segregate with traits owing to hidden factors (for example, linkage disequilibrium), or can aggregate by chance in affected family members. A recent paper reported a new Mendelian form of multiple sclerosis caused by a rare mutation in NR1H3, identified in two multiplex families with a severe form of the disease148. The authors also show that the purported disease variant causes transcriptional dysregulation of NR1H3 and its target genes. However, a study with 13-fold larger sample size found no such association. Rather, the results can be accounted for by a previously identified genome-wide significant common multiple sclerosis variant in moderate LD ~400 kb away149. Potential pathogenic variants should be assessed in large population-scale databases whenever possible, and evidence of a biological effect in a model system does not provide evidence for genetic association.
The fact that de novo loss-of-function variants are predicted to have high impact14,28 has made them attractive targets to study (see “Disease modeling” below). The vast majority of disease models have therefore been based on manipulation of genes harboring these alleles of large effect size29. However, most of these mutations are pleiotropic in nature, associated with variable but often severe abnormalities in multiple cognitive, medical, and behavioral domains. Understanding which molecular, anatomical, or physiological abnormalities relate to specific cognitive or behavioral phenotypes is difficult, and experiments that attempt to do so are rare. A notable recent example capitalized on an allelic series identified in SHANK3, in which two different loss-of-function variants have been associated with distinct clinical phenotypes in humans, namely schizophrenia and autism, albeit in only a few individuals30. Comparison of mice harboring orthologous mutations identified distinct neurobiological effects of the different variants, correlated with distinctive changes in prefrontal and striatal circuitry between models, a remarkable dichotomy30. Studying other alleles on different genetic backgrounds, and different genes showing similar phenotypic divergence in humans, as well as larger human cohorts with variable phenotypes associated with different alleles, will be necessary for appreciating the generalizability of these findings in mice to the observed divergence in disease mechanisms in humans.
Many high-penetrance rare mutations predispose to multiple clinically distinct disorders, including intellectual disability, epilepsy, autism, schizophrenia28,31. For example, about one-third of individuals with 22q11.2 deletions will have ASD and one-third schizophrenia28,32. As such, mice carrying a deletion syntenic to the human 22q11.2 locus should be viewed as a general model of neurodevelopmental disease, rather than a single disorder. Disease-associated CNVs have also been occasionally observed in apparently healthy carriers, for example in mothers with dup15q11–13 who pass the duplication to their affected children33. There is evidence that both genetic background and the environment can potentially have a large impact on the phenotypic outcome in these cases32,34. To account for this, it is prudent to conduct experimental manipulations at these loci on at least two genetic backgrounds. Furthermore, comprehensive clinical phenotyping of individuals with rare variants in the same locus will be essential to help decipher underlying neurobiological mechanisms35. Indeed, large-scale cognitive assessment of individuals carrying major-effect CNVs in the Icelandic population found substantially reduced performance in specific cognitive domains, even in carriers without a psychiatric diagnosis36. Neuroimaging has begun to elucidate the neuroanatomic and circuit-level impact of these rare variants, highlighting the promise of this bottom-up approach to mapping gene–brain–behavior relationships37. Furthermore, studying such people harboring the same mutation, but with different clinical outcomes, is likely to be high yield36. Finally, measuring other forms of genetic variation within rare-variant carriers, such as polygenic risk, may provide a potential explanation for underlying pleiotropy, as recently shown in schizophrenia34.
Another approach to disentangling mechanisms is to study allelic series of variants with different effects on the phenotype in one locus30. Here one would expect to see concentration of phenotypes within specific subcategories of variants: for example, milder phenotypes in patients with heterozygous or missense mutations in genes known to cause severe recessive disorders. The application of WES and WGS in larger populations will enable us to answer this question in more detail and will be a boon to genotype–phenotype studies in humans.
Interpreting common genetic variation
Genome-wide association studies (GWAS) have successfully identified thousands of common genetic variants associated with complex diseases (http://www.ebi.ac.uk/gwas/), including several hundred loci for neuropsychiatric disorders3,12,13,16,38,39. Population-level screening for common genetic contributions to human phenotypes is on the near horizon. Despite these GWAS successes, the number of resolved psychiatric disease genes remains small due to the difficulty identifying the causal variant(s) and their functional impact.
GWAS does not identify a gene per se, but a region that is associated with disease status. When genome-wide significance is achieved (set at P < 5 × 10−8), the effective confidence interval surrounding a ‘lead’ or ‘index’ SNP (with the lowest P-value in a given locus) is set by the surrounding region of linkage disequilibrium (LD), which spans on average ~40 kb, but is highly variable throughout the genome. Identifying the underlying ‘causal’ variant(s) within a target region, and its biological effect, is typically an enormous challenge. In schizophrenia, for example, the strongest GWAS signal maps to the major histocompatibility complex (MHC) locus and spans several hundred genes12. Recent work elegantly dissects this locus to identify the likely causal variants within a few genes, including C4A (ref. 40), which we describe later in more detail.
A majority of common disease-associated genetic variation lies outside coding regions and is enriched in regulatory elements such as enhancers or promoters. Variants in these regulatory elements act to modulate the expression and splicing of distal gene targets, potentially with large effect. Regulatory elements also tend to act in a cell-type- and tissue-specific manner and can be inferred through evolutionary conservation, chromatin accessibility, and characteristic histone marks (Box 3)41–44. Projects such as ENCODE45, the NIH Epigenetics Roadmap46, PsychEncode25 and GTEX47 are building tissue-specific atlases of human gene regulation. However, these annotations are generally derived from only a few individuals and are far from complete, especially in neural tissues, directly limiting our ability to annotate genetic variants relevant to human brain disorders. There also is substantial evidence that gene regulation can occur at long intrachromosomal distances48. Consequently, identifying the gene targets of regulatory regions is a challenging problem and an area of active investigation using both computational49 and experimental approaches, such as HiC50. Gene targets can also be inferred statistically, relying on expression quantitative trait loci (eQTL; see “Integrative approaches” below), which identifies variants that are associated with changes in gene expression in a given cell type or tissue. Although most (~80% of) variants acting as eQTLs occur within 100 kb of their target gene, many loci act on genes hundreds of kilobases away47,48,51. Once a regulatory effect such as an eQTL or physical promoter–enhancer interaction is confirmed experimentally, further conclusive evidence can be derived from showing that such relationships exist in human brain and are altered in the disease-affected brain. Complementing such studies by investigating the effects of common disease-associated SNPs on human phenotypes, such as brain structure and function, can provide further insight into circuit mechanisms52.
Capturing polygenicity
The biological effect of individual common variants (or loci) in most cases will be very small4,53. Since individual common variants account for such a small proportion of disease liability, how can they be of use? One major insight came from the work of Visscher, Wray, and colleagues, who used quantitative genetic reasoning to demonstrate that one could capture the aggregate effect of genetic variants (polygenicity), many of which fail to meet highly conservative genome-wide significance thresholds but nonetheless contribute to disease liability54. In schizophrenia, there are predicted to be over 8,000 disease-associated common variants13. A similar level of polygenicity is expected for virtually every major common neuropsychiatric disorder3. How this plays out in an individual patient is not yet known, and environmental factors (such as smoking55 or cannabis use56) potentially contribute. Indeed, it is clear that genome-wide significant loci represent the tip of the iceberg in terms of the biological signal captured by GWAS4,53,54. New approaches such as LD score regression57 can quantify the aggregate ‘SNP heritability’ captured by common variants within a given study, which can then be used to calculate genetic correlations across disorders or with other traits of interest58, especially intermediate phenotypes59. An extension of this method can quantify the proportion of heritability attributed to SNPs within various functional categories (such as enhancers for specific cell types)60. Conceptually similar, polygenic risk scoring (PRS) quantifies within an individual the aggregate effect of common variants for a given trait, typically calculated as the sum of trait-associated alleles across the genome, weighted by effect size61,62. PRS can be used to identify high-risk individuals for closer clinical assessment, phenotyping before disorder onset to better understand disease trajectory, or to stratify for clinical trials, choosing or refining treatments on the basis of genetic signal. In addition, PRS provides a continuous, quantitative measure of genetic load that can be correlated with phenotypic or endophenotypic measures, such as structural or functional neuroimaging63. However, PRS is likely to be population-specific and is limited by the power of the initial GWAS. There is urgent need to expand such studies to more diverse populations of African, Hispanic, and Asian descent, so that individuals within these populations can benefit from the promise of genetic advances.
In schizophrenia, PRS can currently capture ~7% of variance in disease liability in independent populations of European ancestry12. While far from complete, this translates into odds ratios of 8–20 when comparing the highest vs. lowest decile groups, depending on population12. This finding was recently replicated in an independent UK population, in which PRS was found to account for 5.4% of variance in disease liability translating to an odds ratio of 7.7 between the highest and lowest deciles. As such, PRS is among the most strongly reproducible biological disease predictors to date64.
PRS can also be a powerful tool for identifying patient subgroups. For example, polygenic risk for bipolar disorder predicts manic symptoms in schizophrenia, but not other clinical symptoms, suggesting a distinct mechanistic underpinning for this symptom domain65. A similar approach was recently taken in inflammatory bowel disease, in which PRS can distinguish ulcerative colitis from Crohn’s disease and identify distinct subtypes of Crohn’s disease66. In ASD, LD score regression was recently used to demonstrate that genetic risk for deficits in social function fall along a continuous, bell-shaped distribution within the general population24, as previously predicted67. These studies demonstrate that quantification of polygenic risk coupled with systemic phenotypic assessment can facilitate new insights into disease biology.
However, a major challenge that remains is to understand the mechanisms by which multiple genetic risk factors of low individual effect size actually coalesce to increase disease risk. We emphasize the view that systems biology and integrative approaches as described below are a necessary step in prioritizing potential disease mechanisms and drug targets for therapeutic development3,68. Such approaches provide platforms on which to understand convergence in disease and protective mechanisms from human population genetic data3,68.
Systems genetics
Some of the same technological advances that have enabled large-scale genetic investigation of complex diseases have also enabled systematic characterization of epigenetic, molecular, cellular, and circuit-level landscapes of the human brain across typical development25,69,70. These resources now enable comprehensive pathway-based, systems-level approaches to articulating the neurobiological context in which genetic variation may exert its effects, as recently reviewed71. Perhaps most relevant for CNS disorders, disease relevant gene sets can be investigated for temporal, spatial, and cell-type specificity using large reference data sets. The BrainSpan69 and BrainCloud72 projects profiled gene expression in hundreds of human brain samples across the lifespan, beginning with early fetal timepoints. Spatial patterns are captured in exquisite anatomic detail in adult73 and fetal74 human brain samples, as well as primate75, by the Allen Brain Institute. CNS cell-type-specific transcriptomes have been defined using single-cell RNA sequencing (RNA-seq) or cell sorting methods in primate76, mouse77, and now human78. Overlapping the growing list of reproduced genetic hits in psychiatric disease with more refined cell-type-specific profiles is likely to provide key circuit-level insight into disease79.
Using these approaches, common genetic variation for schizophrenia, bipolar disorder, and depression has been suggested to converge on pathways for histone methylation, immune signaling, and neuronal signaling, although this must be viewed as preliminary owing to the small number of known loci in this analysis80. Gene coexpression networks can identify modules of genes with predicted functional relationships at specific spatiotemporal timepoints in brain. Intersecting these modules with risk genes can yield insights into disease biology68,71,81,82. Clustering genes on the basis of experimentally defined physical properties, such as protein–protein interactions, can identify sub-networks of convergent biological processes, such as chromatin remodeling and histone regulation in ASD83–85. Combining protein–protein interaction, gene expression, and other data into truly integrated networks reflecting CNS function will be critical to understanding pathway convergence of manifold genetic risk variants in these disorders.
Integrative approaches
Allele-specific expression and eQTL studies link genetic variation with altered transcript expression. Sample size and tissue specificity are critical limiting factors, as 10–45% and ~70% of eQTLs are predicted to be tissue and cell-type specific, respectively47,86. This has prompted several consortium-level efforts to generate eQTL databases of human brain, including GTEx47, UKBEC87, and CommonMind88, among others. As current human brain eQTL studies contain at most a few hundred samples, they remain vastly underpowered given a large statistical search space relating a dense map of genetic variation to expression of ~20,000 genes. Furthermore, as eQTLs are often highly cell-type specific86, tissue-level profiling of brain tissue homogenate likely obscures contributions from underlying individual cell types.
Nevertheless, psychiatric GWAS studies have found enrichment of brain-specific eQTL among disease-implicated SNPs as a class, suggesting that intersection with these regulatory data sets may provide important biological insights39. Critical steps moving forward will be to intersect GWAS-implicated disease variants with large-scale eQTL studies, followed by verification of the significance (and directionality) of predicted functional relationships through case-control transcriptome profiling. Recent innovative studies have begun to directly integrate GWAS and eQTL data to perform transcriptome-wide association studies, which have the potential to provide powerful genecentric insights into disease mechanisms89,90. On balance, however, we note that overlap of eQTL and disease association peaks does not provide evidence of a causal relationship to disease, since linkage disequilibrium acts on both signals and some degree of overlap is expected by chance alone. Furthermore, eQTL studies may be less statistically conservative in correcting for multiple comparisons than GWAS, leading to a higher propensity for false positive results91.
Similar approaches exist for defining the landscape of epigenetic regulation of gene expression, which represents an additional layer of biological complexity44. Major psychiatric risk genes include CHD8, which encodes a chromatin remodeling enzyme associated with ASD and macrocephaly35, and SETD1A, which encodes a histone methyltransferase and was recently associated with schizophrenia, developmental delay, intellectual disability, and epilepsy92. Common genetic variants for schizophrenia and bipolar disorder have also been linked to histone methylation, albeit less directly80. Recent, in-depth characterization of the spatial and developmental trajectory of methylation in human brain demonstrated that schizophrenia-associated variants strongly overlap with fetal brain methylation-QTL signals70,93. Similar approaches are being undertaken for histone acetylation QTL94, for example, as part of PsychEncode25.
Partitioning the GWAS SNP heritability from schizophrenia and bipolar disorder on the basis of functional categories defined by these epigenetic signatures identified strong CNS enrichment for common genetic variation in both disorders and fetal brain, specifically in schizophrenia60,95. Concordantly, genetic variants conferring risk for schizophrenia so far seem enriched in fetal prefrontal cortex gene coexpression networks81,82. These results suggest that fetal brain development represents one critical window during which genetic risk factors for certain specific neuropsychiatric disorders exert their effects.
Finally, the most powerful approaches will integrate multiple orthogonal data sets to assess differing levels of genetic, epigenetic, and neurobiological regulation. An exemplary recent example of this type of approach was the investigation of the top genome-wide significant locus in schizophrenia, spanning the highly complex MHC region40. This work combined fine mapping of this locus in schizophrenia with a newly generated reference of structural haplotypes to predict that disease-associated variants function by increasing expression of the complement component 4A gene (C4A) in brain. The role of C4A was verified using gene expression profiling in schizophrenia brain samples, and the C4 protein was shown to regulate synaptic pruning in a rodent model, identifying one of the causal neurobiological mechanisms contributing to disease risk. Integrative approaches have also been undertaken to characterize other GWAS loci in schizophrenia—for example, identifying risk variants that function as eQTL and map to enhancer regions encoding the L-type calcium channel CACNA1C95.
Disease modeling
Many powerful basic research tools now exist that can guide mechanistic insight into disease-associated genetic variation, ranging from in vivo animal models to in vitro culture systems of human fetal neuron progenitor cells, adult induced pluripotent stem cell–derived neurons, and cerebral organoids96,97, each with advantages and limitations29. Caution is always warranted, as insights from behavioral and circuit-level analyses related to human higher cognition and behavior are limited by evolutionary divergence. Even at a molecular level, some genes and signaling pathways are not well conserved between humans and rodent models98,99. In addition, genetic risk alleles for psychiatric disease may converge on human-specific transcriptional processes, or pathways that are not well preserved in lower organisms100–102.
Classic model organisms used for molecular genetics have predominantly consisted of fruit fly (Drosophila melanogaster), zebrafish (Danio rerio), and mouse (Mus musculus), owing to the relative ease of genetic manipulation and potential for high throughput investigation. Recent advances in genome engineering have facilitated the creation of transgenic rat103 and primate104 models of neuropsychiatric disease, limiting throughput but enabling investigation of more complex neural circuitry105. Model organisms have historically been used to investigate the effect of rare, deleterious variants or Mendelian syndromes associated with neuropsychiatric disease. Common genetic variants are much more difficult to model in animals as most lie in regulatory regions poorly conserved across species. Transgenic mice have been used to model major effect forms of autism (including mutations in FMR1, TSC1, TSC2, CNTNAP2, and MeCP2), as well as copy-number variation (16p11.2, 22q11.2, and dup15q11), as recently reviewed29. Adult rescue of phenotypic deficits has been demonstrated in major gene mouse models of neurodevelopmental disorders, such as fragile X syndrome, tuberous sclerosis and Rett syndrome, providing hope for treatment. However, analogous treatments in the human clinical populations have largely failed, for largely unknown reasons106. Similar models of rare variants have been investigated in flies, including loss-of-function mutations in the FMR1 homolog dmfr1 (ref. 107), and zebrafish, such as cntnap2 mutants108. Notwithstanding the above caveats, major advantages of in vivo models include the ability to directly interrogate complex circuit-level alterations, to assess basic cognitive phenotypes, to measure and manipulate neurodevelopmental processes, and to perform large-scale genetic or pharmacologic screens, among others. Modeling of 16p11.2 deletion syndrome in mice, for example, has enabled circuit-level phenotypic dissection, identifying a number of abnormalities in the physiology and function of the basal ganglia109. Molecular genetic dissection of this locus in zebrafish implicated a single gene in this region, KCTD13, as mediating the underlying neuroanatomic phenotype110. However, the region is complex and it is likely that other genes in this region contribute to the broader cognitive and behavioral phenotypes.
Recent developments in stem cell biology have enabled the in vitro generation of human neurons, providing a greatly needed experimental platform for phenotypic characterization and drug screening97. Much of the excitement centers on the potential for creating patient-derived ‘virtual biopsies’ for a tissue is inaccessible to direct investigation. Characterizing neurons derived from human induced pluripotent stem cells from subjects with known penetrant mutations111 and those without established genetic causes of disease both have value. In the latter, the likely causal heterogeneity requires higher numbers than are typically studied to yield generalizable results112. Advantages of this approach include the ability to capture polygenicity, incorporation of genetic background, ability to investigate human-specific biological processes, and potential for high throughput assays113. Pharmacologic screening is thereby possible for patient-derived mutations114, presaging future precision medicine approaches. One limitation is that until we are able to develop mechanistic knowledge based on our genetic findings, it is not clear what relevant cellular or molecular phenotypes should be screened for in vitro29. Systematic approaches, such as gene expression profiling, are likely a good starting point and, critically, can be used to quantify the relative maturity, variability, cellular, and regional identity more rigorously than individual markers115. Other technical hurdles include line-to-line heterogeneity, a limited number of neuronal cell types that can be differentiated, and an inability to form complex circuits. More sophisticated approaches have recently been undertaken to address some of these limitations, including the development of cerebral organoids112 and human cortical spheroids96, which exhibit a cytoarchitectural structure with cortical lamination, incorporate neuronal and glial cell types, form functional synapses, and display spontaneous electrical activity. Considering genetic background effects, a final critical factor is sample size, which can be partially mitigated using either unaffected family members as controls or isogenic lines in which the genetic risk alleles have been corrected.
Pathways to precision health
Moving forward, how can we translate genetic hits into mechanistic insight to reinvigorate a stalled CNS drug development pipeline116? The genomics era has instilled much optimism in this regard117, having recently identified new causal pathways in schizophrenia40, new genetic predictors of treatment response in bipolar disorder118, and genetic risk factors for serious side effects of psychotropic medication119, among others. It is notable that most of these advances are the product of large-scale collaborative approaches120.
A related question that remains is how to prioritize genetically identified biological targets for development of new medicines. To date, such efforts have disproportionately focused on the mutations with the largest effect sizes, which are easier to identify, interpret, and model in preclinical settings. However, there is evidence that small effect-size (typically common, inherited, polygenic) and large-effect size (typically rare, noninherited) variants converge on distinct biological processes. In ASD, for example, inherited variants converge largely on postnatal synaptic processes, whereas de novo loss-of-function variants are enriched for developmental regulation and chromatin modification pathways15,71. A potential interpretation is that more highly penetrant mutations disproportionately disrupt the robustness of the neurodevelopmental trajectory to an environmental or genetic perturbation (‘canalization’)121. This would explain the association of rare variation with more severe and pleiotropic syndromes including intellectual disability, epilepsy, and ASD. This would also predict that clinical disease specificity is guided by distinct factors, such as environmental or common variants, in accordance with recent evidence34.
We propose that genes and pathways affected by common variants may be at least equally, if not more, amenable to therapeutic intervention than those disrupted by high penetrance mutations (Box 4). First, the small effect size of common variants suggests that disease risk is inherently modifiable and that ‘protective’ environmental exposures in the form of biological intervention could prevent disease or reduce risk. Second, common variation by definition is present in a larger proportion of the population and therefore is likely more generalizable. Third, for most neuropsychiatric disorders, common variation is predicted to contribute more substantially to disease liability than highly penetrant mutations, often by an order of magnitude31,122,123. Finally, in other complex disorders, successful new drug targets can often be retrospectively substantiated by genome-wide significant variants (Table 2)124. In hyperlipidemia, for example, targets of statins (HMGCR) and the new class of lipid-lowering PCSK9-inhibitors (PCSK9) are among the top GWAS-identified risk variants125, although these targets were discovered before the GWAS era. There are enormous challenges to targeting common variants using traditional methods. First, we need to better characterize composite genetic risk in individuals—what common and/or rare risk variants are necessary and sufficient to cause disease in an individual. Individual genetic subtypes of a disorder could be identified on the basis of convergent risk profiles defined by population scale WGS, thus stratifying patients by their underlying biology65,66.
Box 4. FDA-approved medications supported by common-variant association.
Nearly all classes of medications currently used to treat neuropsychiatric disease were discovered by serendipity and target the same molecular pathways as their prototypes, developed decades ago116. Novel therapeutic targets are greatly needed and genetics provides an avenue for their identification117. Preclinical drug development has historically favored targets based on rare, moderately penetrant genetic variants, which are easier to identify, interpret, and investigate in model organisms. Although this has been successful in some cases, the recent dismal approval rate of candidate drugs entering clinical trials for neuropsychiatric disorders suggests that alternative approaches may be needed106. We argue that pathways enriched for common genetic variation should receive more attention for drug development. In support of this, we have surveyed the literature for examples of FDA-approved medications that are supported by GWAS-identified targets (Table 2). While most of these drugs were developed before the GWAS-era, their targets can be retrospectively validated by genome-wide significant loci associated with disease risk. One can extrapolate from these successes to predict that additional pathways enriched for common variation from disease GWAS can identify future efficacious drug targets131. We note that this is neither prospective nor a formal statistical analysis assessing enrichment of approved drugs acting on GWAS-identified targets. However, others have estimated that genetic evidence as a whole could double the success rate of clinical drug development124.
Table 2.
FDA-approved medications supported by GWAS variants
| Disease | Lead GWAS SNP | Genetic locus | FDA-approved medication Drug class |
|---|---|---|---|
| Psoriasis | rs9988642 | IL12R–IL23R | Ustekinumab Biologic |
| Hyperlipidemia | rs12916 | HMGCR | Many Statin |
| Hyperlipidemia | rs2479409 | PCSK9 | Alirocumab Biologic |
| Type 2 diabetes | rs1801282 | PPARG | Many Thiazolidinediones |
| Type 2 diabetes | rs5219 | KCNJ11 | Many Sulfonylurea |
| Osteoporosis | rs9533090 | TNFSF11 (RANKL) | Denosumab Biologic |
| Osteoporosis | rs7751941 | ESR1 | Many Selective estrogen receptor modulator |
| Schizophrenia | rs2514218 | DRD2 | Many Antipsychotic |
| Rheumatoid arthritis | rs2228145 | IL6R | Tocilizumab Biologic |
| Rheumatoid arthritis | rs3087243 | CTLA4 | Abatacept Biologic |
See Supplementary Table 1 for further details.
High-throughput precision health approaches are gaining traction and may provide an additional platform through which to validate potential drug targets. Phenome-wide association studies, which integrate clinical and genomic data to identify genotype–phenotype relationships on the basis of electronic medical records, offer great promise126,127 as evidenced by pharmacogenomic-based predictors of drug efficacy128. Other powerful new approaches include computational drug repositioning129,130, integrating, for example, a database of known drug targets with GWAS-implicated disease loci131 or with the transcriptomic profile of a drug from resources such as the Connectivity Map132. With large enough samples, the goal is that phenome-wide association studies will allow dissection of genetic contributions to specific phenotypes that, when combined, produce a specific clinical syndrome. Finally, the importance of environmental factors (such as gut microbiota) is becoming increasingly realized. Once genetic risk factors and pathways are accounted for, it will become possible to more systematically query the impact of the environment and its interaction with genetics. This approach has shown recent success in dissecting the role of smoking55 and cannabis56 use on risk of schizophrenia. As such, the knowledge imparted by understanding genetic contributions to disease risk can serve as a causal anchor, magnifying the power of follow-up studies and providing a strong foundation for finally unraveling the complex brain–behavior relationships underlying neuropsychiatric disease.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank A. Gordon, L. Perez-Cano, G. Ramaswami, E. Ruzzo, J. Rexach, G. Morris, and members of the Geschwind laboratory for discussions. This work was supported by US National Institute of Health (NIH) grants 5R01MH094714 (D.H.G.), 5P50MH106438 (D.H.G.), 1R01MH109912 (D.H.G.), and F30MH099886 (N.N.P.). This work was also supported by the Glenn/AFAR Postdoctoral Fellowship (20145357, H.W.), Basic Science Research Program through the National Research Foundation of Korea (2013024227, H.W.), and the UCLA Medical Scientist Training Program (N.N.P.). D.H.G. is a scientific advisor to Ovid Pharmaceuticals.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Polderman TJC et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet 47, 702–709 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Sullivan PF, Daly MJ & O’Donovan M Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet 13, 537–551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geschwind DH & Flint J Genetics and genomics of psychiatric disease. Science 349, 1489–1494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gratten J, Wray NR, Keller MC & Visscher PM Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat. Neurosc 17, 782–790 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarroll SA, Feng G & Hyman SE Genome-scale neurogenetics: methodology and meaning. Nat. Neurosci 17, 756–763 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kendler KS What psychiatric genetics has taught us about the nature of psychiatric illness and what is left to learn. Mol. Psychiatry 18, 1058–1066 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Sham PC & Purcell SM Statistical power and significance testing in large-scale genetic studies. Nat. Rev. Genet 15, 335–346 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Flint J & Kendler KS The genetics of major depression. Neuron 81, 484–503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawi Z et al. The molecular genetic architecture of attention deficit hyperactivity disorder. Mol. Psychiatry 20, 289–297 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Browne HA, Gair SL, Scharf JM & Grice DE Genetics of obsessive-compulsive disorder and related disorders. Psychiatr. Clin. North Am 37, 319–335 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manolio TA et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ripke S et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet 45, 1150–1159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanders SJ et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet 43, 977–983 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tammimies K et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. J. Am. Med. Assoc 314, 895–903 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacArthur DG et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacArthur DG et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sauna ZE & Kimchi-Sarfaty C Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet 12, 683–691 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Cooper GM & Shendure J Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet 12, 628–640 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson EB et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat. Genet 48, 552–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akbarian S et al. The PsychENCODE project. Nat. Neurosci 18, 1707–1712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen A et al. Evaluating methods for combining rare variant data in pathway-based tests of genetic association. BMC Proc. 5 (Suppl. 9), S48 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Leeuw CA, Neale BM, Heskes T & Posthuma D The statistical properties of gene-set analysis. Nat. Rev. Genet 17, 353–364 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Malhotra D & Sebat J CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de la Torre-Ubieta L, Won H, Stein JL & Geschwind DH Advancing the understanding of autism disease mechanisms through genetics. Nat. Med 22, 345–361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y et al. Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szatkiewicz JP et al. Copy number variation in schizophrenia in Sweden. Mol. Psychiatry 19, 762–773 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiroi N et al. Copy number variation at 22q11.2: from rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol. Psychiatry 18, 1153–1165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook EH Jr. et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet 60, 928–934 (1997). [PMC free article] [PubMed] [Google Scholar]
- 34.Tansey KE et al. Common alleles contribute to schizophrenia in CNV carriers. Mol. Psychiatry 21, 1085–1089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernier R et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stefansson H et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Maillard AM et al. The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol. Psychiatry 20, 140–147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sawcer S et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sekar A et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visel A, Rubin EM & Pennacchio LA Genomic views of distant-acting enhancers. Nature 461, 199–205 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaub MA, Boyle AP, Kundaje A, Batzoglou S & Snyder M Linking disease associations with regulatory information in the human genome. Genome Res. 22, 1748–1759 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nord AS, Pattabiraman K, Visel A & Rubenstein JLR Genomic perspectives of transcriptional regulation in forebrain development. Neuron 85, 27–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ernst J et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kundaje A et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanyal A, Lajoie BR, Jain G & Dekker J The long-range interaction landscape of gene promoters. Nature 489, 109–113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whalen S, Truty RM & Pollard KS Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet 48, 488–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y et al. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature 504, 306–310 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Esslinger C et al. Neural mechanisms of a genome-wide supported psychosis variant. Science 324, 605 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Price AL, Spencer CCA & Donnelly P Progress and promise in understanding the genetic basis of common diseases. Proc. Biol. Sci 282, 20151684 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wray NR, Goddard ME & Visscher PM Prediction of individual genetic risk of complex disease. Curr. Opin. Genet. Dev 18, 257–263 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Kendler KS, Lönn SL, Sundquist J & Sundquist K Smoking and schizophrenia in population cohorts of Swedish women and men: a prospective co-relative control study. Am. J. Psychiatry 172, 1092–1100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.French L et al. Early cannabis use, polygenic risk score for schizophrenia and brain maturation in adolescence. JAMA Psychiatry 72, 1002–1011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bulik-Sullivan BK et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flint J, Timpson N & Munafò M Assessing the utility of intermediate phenotypes for genetic mapping of psychiatric disease. Trends Neurosci. 37, 733–741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Euesden J, Lewis CM & O’Reilly PF PRSice: Polygenic Risk Score software. Bioinformatics 31, 1466–1468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pasaniuc B & Price AL Dissecting the genetics of complex traits using summary association statistics. Preprint at bioRxiv 10.1101/072934 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Franke B et al. Genetic influences on schizophrenia and subcortical brain volumes: large-scale proof of concept. Nat. Neurosci 19, 420–431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vassos E et al. An examination of polygenic score risk prediction in individuals with first episode psychosis. Biol. Psychiatry 10.1016/j.biopsych.2016.06.028 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Ruderfer DM et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol. Psychiatry 19, 1017–1024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cleynen I et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet 387, 156–167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Constantino JN & Todd RD Intergenerational transmission of subthreshold autistic traits in the general population. Biol. Psychiatry 57, 655–660 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Parikshak NN, Gandal MJ & Geschwind DH Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet 16, 441–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang HJ et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jaffe AE et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci 19, 40–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parikshak NN et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Colantuoni C et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hawrylycz MJ et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller JA et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bakken TE et al. A comprehensive transcriptional map of primate brain development. Nature 535, 367–375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bernard A et al. Transcriptional architecture of the primate neocortex. Neuron 73, 1083–1099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Darmanis S et al. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 112, 7285–7290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu X, Wells AB, O’Brien DR, Nehorai A & Dougherty JD Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci 34, 1420–1431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O’Dushlaine C et al. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci 18, 199–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gulsuner S et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gilman SR et al. Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat. Neurosci 15, 1723–1728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Roak BJ et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neale BM et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li J et al. Identification of human neuronal protein complexes reveals biochemical activities and convergent mechanisms of action in autism spectrum disorders. Cell Syst. 1, 361–374 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dimas AS et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325, 1246–1250 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ramasamy A et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci 17, 1418–1428 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fromer M et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Preprint at bioRxiv 10.1101/052209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gamazon IR et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet 47, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mackay TFC, Stone EA & Ayroles JF The genetics of quantitative traits: challenges and prospects. Nat. Rev. Genet 10, 565–577 (2009). [DOI] [PubMed] [Google Scholar]
- 92.Singh T et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci 19, 571–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hannon E et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat. Neurosci 19, 48–54 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.del Rosario RC-H et al. Sensitive detection of chromatin-altering polymorphisms reveals autoimmune disease mechanisms. Nat. Methods 12, 458–464 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Roussos P et al. A role for noncoding variation in schizophrenia. Cell Reports 9, 1417–1429 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pasca AM et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dolmetsch R & Geschwind DH The human brain in a dish: the promise of iPSC-derived neurons. Cell 145, 831–834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miller JA, Horvath S & Geschwind DH Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl. Acad. Sci. USA 107, 12698–12703 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seok J et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 110, 3507–3512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Konopka G et al. Human-specific transcriptional networks in the brain. Neuron 75, 601–617 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kwan KY et al. Species-dependent posttranscriptional regulation of NOS1 by FMRP in the developing cerebral cortex. Cell 149, 899–911 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Geschwind DH & Rakic P Cortical evolution: judge the brain by its cover. Neuron 80, 633–647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hamilton SM et al. Fmr1 and Nlgn3 knockout rats: novel tools for investigating autism spectrum disorders. Behav. Neurosci 128, 103–109 (2014). [DOI] [PubMed] [Google Scholar]
- 104.Liu Z et al. Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 530, 98–102 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Kaiser T & Feng G Modeling psychiatric disorders for developing effective treatments. Nat. Med 21, 979–988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jeste SS & Geschwind DH Clinical trials for neurodevelopmental disorders: at a therapeutic frontier. Sci. Transl. Med 8, 321fs1 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Dockendorff TC et al. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34, 973–984 (2002). [DOI] [PubMed] [Google Scholar]
- 108.Hoffman EJ et al. Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron 89, 725–733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Portmann T et al. Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Reports 7, 1077–1092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Golzio C et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485, 363–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Paşca SP et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med 17, 1657–1662 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mariani J et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162, 375–390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Patwardhan RP et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol 30, 265–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mei Y et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stein JL et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 83, 69–86 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hyman SE Revolution stalled. Sci. Transl. Med 4, 155cm11 (2012). [DOI] [PubMed] [Google Scholar]
- 117.Plenge RM, Scolnick EM & Altshuler D Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov 12, 581–594 (2013). [DOI] [PubMed] [Google Scholar]
- 118.Hou L et al. Genetic variants associated with response to lithium treatment in bipolar disorder: a genome-wide association study. Lancet 387, 1085–1093 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goldstein JI et al. Clozapine-induced agranulocytosis is associated with rare HLA-DQB1 and HLA-B alleles. Nat. Commun 5, 4757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lajonchere CM Changing the landscape of autism research: the autism genetic resource exchange. Neuron 68, 187–191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Félix M-A & Barkoulas M Pervasive robustness in biological systems. Nat. Rev. Genet 16, 483–496 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Klei L et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 3, 9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gaugler T et al. Most genetic risk for autism resides with common variation. Nat. Genet 46, 881–885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nelson MR et al. The support of human genetic evidence for approved drug indications. Nat. Genet 47, 856–860 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Teslovich TM et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bush WS, Oetjens MT & Crawford DC Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat. Rev. Genet 17, 129–145 (2016). [DOI] [PubMed] [Google Scholar]
- 127.Kohane IS Using electronic health records to drive discovery in disease genomics. Nat. Rev. Genet 12, 417–428 (2011). [DOI] [PubMed] [Google Scholar]
- 128.Nelson MR et al. The genetics of drug efficacy: opportunities and challenges. Nat. Rev. Genet 17, 197–206 (2016). [DOI] [PubMed] [Google Scholar]
- 129.Rastegar-Mojarad M, Ye Z, Kolesar JM, Hebbring SJ & Lin SM Opportunities for drug repositioning from phenome-wide association studies. Nat. Biotechnoi 33, 342–345 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Li J et al. A survey of current trends in computational drug repositioning. Brief. Bioinform 17, 2–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ruderfer DM et al. Polygenic overlap between schizophrenia risk and antipsychotic response: a genomic medicine approach. Lancet Psychiatry 3, 350–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lamb J et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 133.Lichtenstein P et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 373, 234–239 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lee SH et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet 45, 984–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Reichenberg A et al. Discontinuity in the genetic and environmental causes of the intellectual disability spectrum. Proc. Natl. Acad. Sci. USA 113, 1098–1103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gilissen C et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014). [DOI] [PubMed] [Google Scholar]
- 137.Plomin R, Haworth CM, Meaburn EL, Price TS & Davis OS Common DNA markers can account for more than half of the genetic influence on cognitive abilities. Psychol. Sci 24, 562–568 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ridge PG et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobioi. Aging 41, 200.e13–e20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tenesa A & Haley CS The heritability of human disease: estimation, uses and abuses. Nat. Rev. Genet 14, 139–149 (2013). [DOI] [PubMed] [Google Scholar]
- 140.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mutsuddi M et al. Analysis of high-resolution HapMap of DTNBP1 (dysbindin) suggests no consistency between reported common variant associations and schizophrenia. Am. J. Hum. Genet 79, 903–909 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Spielman RS et al. Common genetic variants account for differences in gene expression among ethnic groups. Nat. Genet 39, 226–231 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Akey JM, Biswas S, Leek JT & Storey JD On the design and analysis of gene expression studies in human populations. Nat. Genet 39, 807–808 author reply 808–809 (2007). [DOI] [PubMed] [Google Scholar]
- 144.Sebastiani P et al. Genetic signatures of exceptional longevity in humans. Science 2010, 1190532 (2010). [DOI] [PubMed] [Google Scholar]
- 145.Skafidas E et al. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol. Psychiatry 19, 504–510 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Belgard TG, Jankovic I, Lowe JK & Geschwind DH Population structure confounds autism genetic classifier. Mol. Psychiatry 19, 405–407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Arnedo J et al. Uncovering the hidden risk architecture of the schizophrenias: confirmation in three independent genome-wide association studies. Am. J. Psychiatry 172, 139–153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang Z et al. Nuclear receptor NR1H3 in familial multiple sclerosis. Neuron 90, 948–954 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cotsapas C NR1H3 p.Arg415Gln is not associated to multiple sclerosis risk. Preprint at bioRxiv 10.1101/061366 (2016). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.