

Abstract
Innate pattern recognition receptors, including toll-like receptors (TLRs), can alter the tumor microenvironment and prime adaptive anti-tumor immunity. However, TLR agonists have not been well-tolerated due to toxicities associated with widespread immune activation following systemic administration. To design a therapeutic that is suitable for systemic delivery and capable of eliciting a tumor-targeted response, we developed a novel class of immune-stimulating antibody conjugates (ISACs) comprising a TLR7/8 agonist conjugated to tumor-targeting antibodies. Systemically administered anti-HER2 ISACs were well-tolerated in vivo and elicited robust activation of intratumoral myeloid cells, resulting in tumor clearance and subsequent immunological memory. In vitro potency and in vivo efficacy required tandem activity driven by intact Fc functionality and TLR agonism to enable phagocytosis and T cell-mediated anti-tumor immunity. ISAC-mediated immunological memory was not limited to HER2, as ISAC-treated mice were protected from rechallenge with a HER2-negative tumor. These results provide strong rationale for the clinical development of ISACs and show that synergy between FcγR and TLR7/8 signaling contributes to the mechanism of ISAC-mediated anti-tumor immunity.
Introduction:
Prior to the end of the 20th century, cancer patients generally had three treatment modalities available: surgical resection, radiation, and chemotherapy. The emergence of cancer immunotherapy added a powerful fourth modality that greatly expanded the oncology repertoire beginning with the approval of rituximab for non-Hodgkin’s lymphoma and extending to the widespread adoption of checkpoint inhibitors 1,2. However, any one therapy, including immunotherapy, is temporally limited unless it engenders durable biologic and immunologic responses that overcome tumor heterogeneity and mutational burden 3–5.
Checkpoint inhibitors such as anti-PD-1 and PD-L1 antibodies dramatically improve outcomes in several indications, including in patients with high tumor mutational burden or microsatellite instability, and one determinant of tumor susceptibility to checkpoint immunotherapy is the extent of pre-existing T cell infiltration. Patients with “cold” tumors that have a sparse T cell infiltrate are less responsive to checkpoint blockade and have poorer prognoses compared to patients with “hot” tumors where immunosuppressed tumor-reactive T cells are present within the tumor microenvironment (TME) 6,7. Tumor biopsy and immunohistochemistry indicate that professional antigen-presenting cells (APCs) such as dendritic cells (DCs) are often more frequent within the TME than T cells, albeit still susceptible to immunosuppressive mechanisms 8,9. Given the presence of myeloid cells in the TME and their role in promoting T cell activation, there has been an increasing effort to develop therapies that activate tumor-associated APCs to elicit widespread T cell immunity and an effective anti-tumoral immune response.
It is now well-documented that local delivery of immunostimulatory agents in preclinical models can reprogram suppressed tumor resident APCs into activated cells capable of processing and presenting tumor associated antigens, including neoantigens, to T cells that subsequently mediate anti-tumor immunity 10–13. One such strategy pioneered by Carmi et al demonstrated that intra-tumoral delivery of allogeneic tumor-targeting antibody with a Toll-like receptor (TLR)-3 agonist or with TNFα and CD40L functionally rescued tumor-associated APCs leading to enhanced antigen uptake and subsequent induction of anti-tumor T cell immunity. This combination strategy, which leveraged antibody effector function together with TLR agonism, led to the eradication of primary tumors and metastatic lesions, and protected the host from tumor recurrence 11,14. Similarly, agonistic anti-OX40 antibody in combination with a TLR9 agonist, CpG, activated APCs and T cells within the tumor, resulting in impressive efficacy against multiple tumor types in mice 13.
The effective translation of APC activating strategies into the clinic is being investigated in multiple indications with clinical data in patients with melanoma suggesting that intra-tumoral delivery of TLR9 agonists in combination with anti-PD-1 antibody can lead to tumor shrinkage and abscopal responses in non-injected lesions 15. Similarly, several groups are pursuing TLR7/8 agonists for tumor immunotherapy as the expression pattern of TLR7/8 in human APCs more closely mirrors the expression pattern of TLR7 and TLR9 in murine APCs16–18. Ultimately, the adoption of intra-tumoral delivery strategies, if efficacious, will be limited by tumor accessibility as systemic delivery of immune agonists targeting APCs has been restricted by toxicity, short half-lives, and sub-optimal delivery to target sites.
Our goal was to design an immune-stimulating antibody conjugate (ISAC) comprising a tumor-targeting monoclonal antibody conjugated to an immune agonist as a novel therapeutic approach to elicit durable anti-tumor immunity. Systemic administration of the ISAC elicited localized APC activation in the TME that was dependent on Fc effector function and TLR activation while circumventing typical toxicities associated with systemic delivery of TLR agonists. Moreover, systemic administration of these conjugates to tumor-bearing mice enabled antigen processing and presentation to T cells leading to sustained anti-tumor immunity in multiple models that are resistant to the unconjugated tumor-targeting antibody.
Results:
ISAC Design and Characterization
To enable conjugation of a TLR agonist to an antibody, molecular docking tools were employed to design a novel TLR7/8 agonist (T785) with a butyl-amine that served as a solvent-accessible point of attachment to the antibody (Figure 1A, Supplemental Figure 1A–C). T785 engaged human TLR7 with an EC50 of 0.643 μM and human TLR8 with an EC50 of 1.60 μM in HEK293 cell lines that co-express an NF-κB-inducible secreted alkaline phosphatase reporter and either TLR7 or TLR8 (Figure 1B). Specificity of T785 for TLR7/8 was confirmed by the inability of T785 to induce NF-κB activity in HEK293 reporter cells that lack TLR7/8 expression (Supplemental Figure 2A). To confirm agonistic activity in primary cells, freshly isolated monocytes and DCs, termed myeloid APCs (~96% monocytes, 4% DC; Supplemental Figure 3A), which express both TLR7 and TLR8, were stimulated overnight with 1 μM T785 or R848, a well-established and characterized TLR7/8 agonist 19. T785 and R848 stimulated myeloid APCs to a similar extent (Figure 1C) 20,21. Activity of T785 was also confirmed in freshly isolated human plasmacytoid DCs (pDCs), which express TLR7 but not TLR8 (Supplemental Figure 2B).
Figure 1: ISAC Design and Characterization.
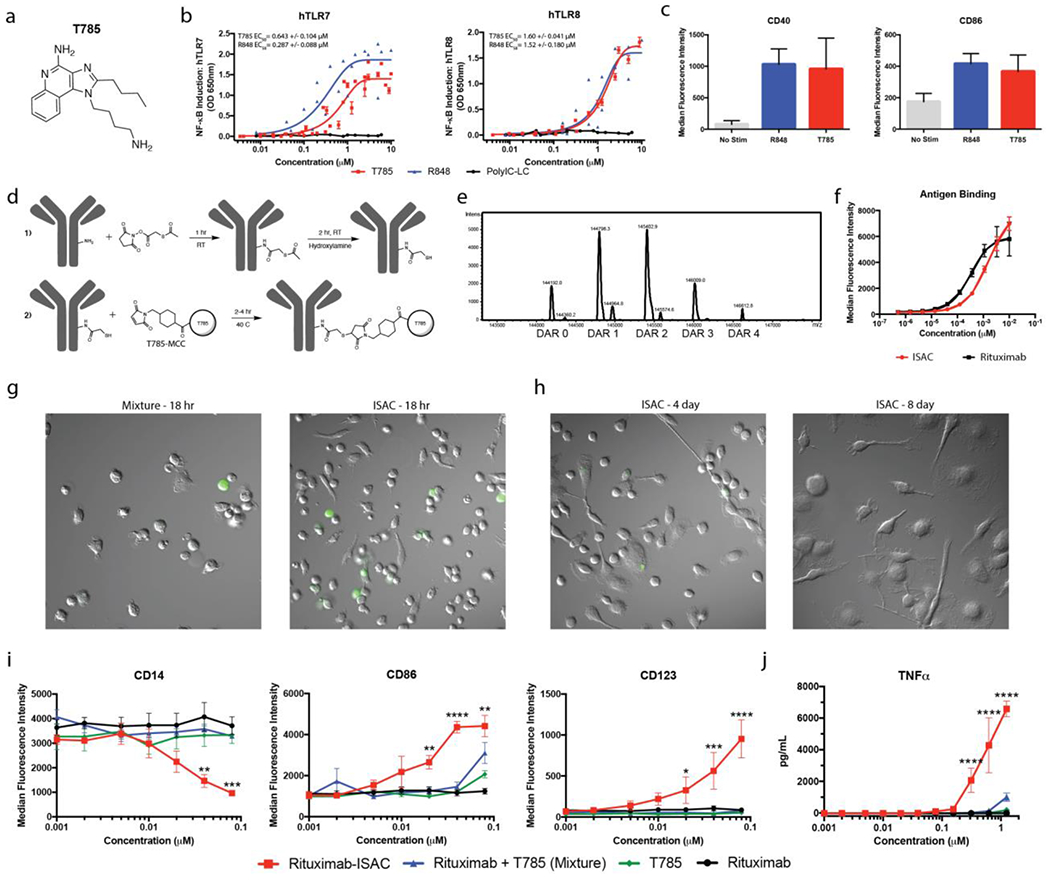
(a) Chemical structure of T785 (b) HEK-Blue-TLR7 or TLR8 reporter cells were cultured for 18 hours in the presence of T785, R848 or Poly-ICLC prior to assessment of NF-κB-induced SEAP activity. Data shown are from 8 experiments and EC50 values are calculated as mean with SEM. (c) Freshly isolated human myeloid APCs were stimulated with 1 μM of T785 or R848 for 18 hours prior to assessment of myeloid activation by flow cytometry. Data are from 1 experiment with 3 donors and are representative of >9 donors. (d) Rituximab was reacted with SATA via lysine residues to convert free amines into protected sulfhydryl groups prior to deacetylation with hydroxylamine and subsequent reaction with T785-MCC to yield the rituximab-ISAC. (e) LC-MS analysis of the rituximab-ISAC following PNGase F treatment. DAR was calculated based upon the linker-agonist mass addition of 606 g/mol. (f) Fluorescently labeled rituximab or rituximab-ISAC was incubated with CD20+ Toledo tumor cells at 4°C for 2 hours prior to analysis via flow cytometry. Data are from 1 experiment with triplicate samples and are representative of 2 experiments. (g-h) Freshly isolated human myeloid APCs were cultured with rituximab, T785, rituximab and T785 or the rituximab-ISAC in the presence of CFSE-labeled CD20+ Toledo tumor cells at a 3:1 effector to target ratio. The rituximab concentration is depicted on the X-axis with the concentration of T785 in these assays being consistent with the amount of T785 conjugated to the rituximab-ISAC. (g) Hoffman modulation contrast microscopy shown at 40X magnification after (g) 18 hours following stimulation with 80 nM of rituximab-ISAC. Data are representative of >10 donors. (h) Myeloid APCs were analyzed via flow cytometry 18 hours after stimulation. Data shown are from 3 donors and are representative of >10 donors (mean and SEM); *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To investigate the effects of ISACs in human in vitro cultures, an ISAC was generated with rituximab (Figure 1D), a clinically validated anti-CD20 monoclonal antibody used for the treatment of B cell malignancies, by conjugation to T785 in a multi-step reaction with compatible heterobifunctional crosslinkers 22. T785-ISAC conjugation efficiency was measured by LC-MS following deglycosylation with PNGase F, resulting in an average drug to antibody ratio (DAR) of 1.71 (Figure 1E). We confirmed that the ISAC retained comparable binding to CD20 following flow cytometry experiments with the CD20-expressing Toledo tumor cell line, a well-characterized cell line derived from a patient with diffuse large cell lymphoma (Figure 1F) 23. T785-ISAC binding to the human Fc receptors (FCGR3A, FCGR2A, FCGR2B, FCGR1) was assessed by Biacore and was comparable to rituximab (Supplemental Table 1).
ISACs activate human myeloid APCs
The immunostimulatory potential of the rituximab T785-ISAC was assessed following coculture of freshly isolated human myeloid APCs with CFSE-labeled CD20+ Toledo tumor cells for 18 hours. Myeloid APCs cultured with rituximab T785-ISAC underwent rapid morphologic changes consistent with morphology of DCs, whereas DC morphology was not observed in cocultures incubated with rituximab, T785 or an equimolar mixture of rituximab and T785 (Figure 1G). APCs stimulated with rituximab T785-ISAC remained viable and dendritic in morphology for up to eight days whereas APCs stimulated with the mixture were mostly non-viable by day eight (Figure 1H). Multicolor flow cytometry analysis after 18 hours indicated that APCs stimulated with rituximab T785-ISAC downregulated CD14 and CD16 and concomitantly upregulated CD123, a cell surface marker shown to be up-regulated on activated myeloid DCs 24. Conversely, APCs stimulated with rituximab, T785 or the mixture did not modulate expression of these markers (Figure 1I, Supplemental Figure 4A, gating strategy described in Supplemental Figures 3B–C).
Human APCs stimulated with rituximab T785-ISAC also upregulated costimulatory molecule CD86 in a dose-dependent manner, with similar trends observed with CD40 and HLA-DR upregulation consistent with the phenotype of activated APCs (Figure 1I & Supplemental Figure 4B). Dose-dependent changes in surface marker expression were consistent whether assessed as median fluorescence intensity (MFI, Figure 1) or as the percent of the total population of cells that is positive for the respective marker (Supplemental Figure 5). Subsequent analysis of cell-free supernatants demonstrated that rituximab T785-ISAC-stimulated APCs secreted higher quantities of TNFα and IL-1β (Figure 2J and Supplementary Figure 4C). The immunostimulatory potential of T785-ISACs was not restricted to rituximab, as an T785-ISAC generated with trastuzumab also elicited enhanced myeloid activation (Supplemental Figure 4D).
Figure 2: ISACS elicit distinct signaling patterns in myeloid APCs.
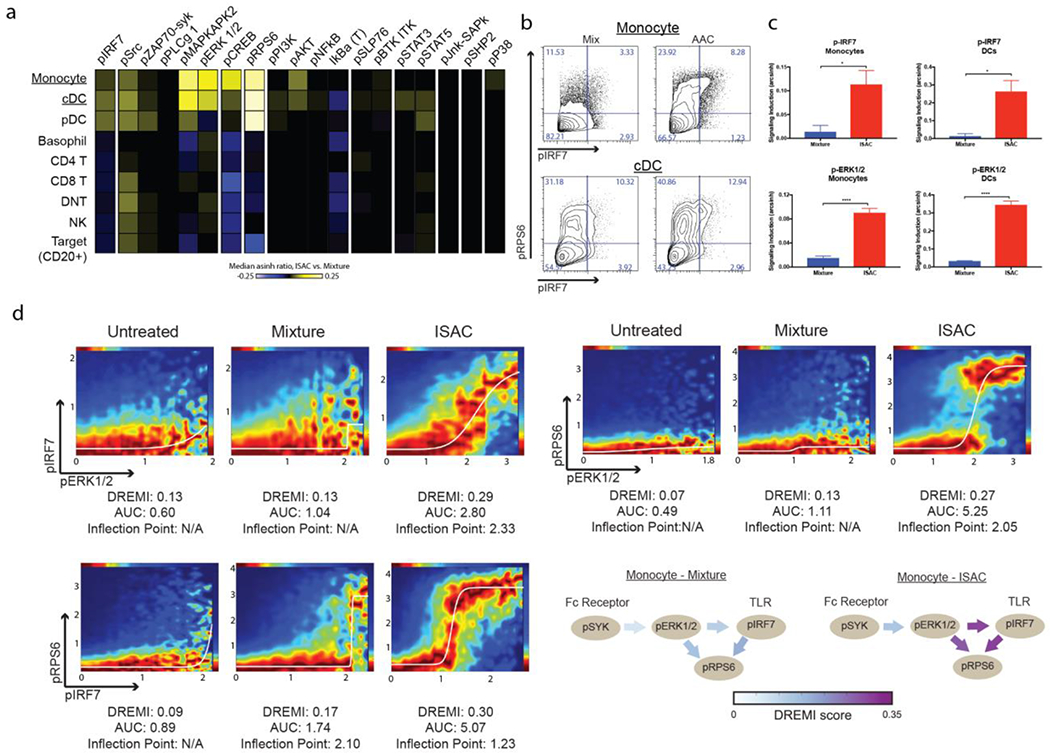
(a-d) Freshly isolated human PBMC were stimulated with 1 μM of the rituximab-ISAC or an equimolar equivalent of the mixture in the presence of CD20+ Toledo tumor cells at a 1:1 ratio for 15 minutes. (a) The median arcsinh ratio of ISAC over signaling measured following mixture stimulation for various phosphoproteins as depicted by a heat map, with yellow indicative of increased signaling and blue indicative of reduced signal. (b) Representative flow cytometry plots for pIRF7 and pRPS6 in monocytes and cDCs. (c) Fold change of signaling induced was computed as the arcsinh ratio over the unstimulated control. IRF7 and ERK1/2 phosphorylation was measured in monocytes and cDCs following ISAC or mixture stimulation. Data are from one experiment with six donors (mean and SEM); *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. (d) Representative DREVI plots following DREMI analysis with the area under the curve (AUC) and inflection point (IP) denoted. DREVI plots were visually inspected for curve fit and signal-to-noise, and inflection points were calculated for those with proper sigmoidal curve fits (N/A indicates no curve fit).
The capacity of ISAC-stimulated APCs to elicit antigen-presentation and cross-priming of CD8+ T cells was assessed using the ovalbumin (OVA) model antigen system 25,26. Splenic CD11c+ DCs isolated from wildtype mice were cultured overnight with immune complexes formed with OVA and either the anti-OVA antibody or ISAC. OVA-loaded DCs were quantified and incubated with OT-I CD8+ T cells bearing TCR specificity for the OVA peptide, SIINFEKL, complexed with MHC-I. Anti-OVA ISAC mediated enhanced cross-presentation as measured by the amount of MHC-I bound SIINFEKL peptide and enhanced cross-priming as measured by CD8+ T cell proliferation (Supplemental Figures 6A & 6B).
ISACs elicit amplified TLR and FcγR-associated intracellular signaling in human myeloid APCs
Activation of FcγRs and TLRs induce a complex phospho-signaling cascade, and the magnitude and characteristics of these responses differ among various immune cell subsets 27. We investigated the downstream intracellular signaling pathways engaged in various cell types within PBMC-tumor cocultures stimulated with the rituximab T785-ISAC compared to equimolar mixture controls using mass cytometry (CyTOF) 28. Leukocyte subgroups (i.e. monocytes, CD4+ T cells) were identified via SPADE clustering using lineage markers with target cells encompassing both CD20+ Toledo tumor cells and healthy B cells 29. Phosphorylation events associated with TLR (pIRF7) and FcγR (pERK1/2) signaling were increased 15 minutes after stimulation with the T785-ISAC in monocytes and cDCs, as compared to the individual components mixture 30,31. Intracellular signaling was largely restricted to cells of myeloid origin (monocytes and cDCs) as well as pDCs, which all express the requisite FcγRs and TLR7 and/or TLR8. In monocytes and cDCs, the stimulation induced by the ISAC also resulted in significantly higher phosphorylation of MAPKAPK2, p38, CREB, and ribosomal protein S6 (RPS6), a requirement for initiating downstream protein synthesis (Figure 2A–C & Supplemental Figure 7A). CD141+ cDCs and CD1c+ cDCs showed comparable levels of pIRF7 across both cDC subsets, while levels of RPS6 and pERK1/2 trended higher in CD141+ cDCs, which excel in CD8+ T cell priming and antigen cross-presentation, as compared to CD1c+ cDCs (Supplemental Figure 7B). The kinetics of T785-ISAC stimulation were consistent with expected signaling dynamics: ERK1/2 phosphorylation peaked after 5 minutes, while it’s downstream partner RPS6 didn’t reach peak phosphorylation until the 15-minute timepoint (Supplemental Figure 7C). These kinetics support expected dynamics as signaling propagates over time, and suggests that there is a sustained and amplified signaling response to ISAC stimulation of APCs.
Given the amplified response observed following ISAC stimulation associated with FcγR and TLR signaling pathways, we next utilized the conditional-Density Resampled Estimate of Mutual Information (DREMI) and conditional-Density Rescaled Visualization (DREVI) algorithms to quantify the strength of pairwise relationships within the FcγR and TLR signaling pathways 32. These algorithms generate DREVI plots for visualization, where the area under the curve (AUC) is an indicator of overall signal intensity, inflection point (IP) is a measure of the activation threshold, and the DREMI score quantifies the pairwise dependence of the two phosphoproteins in question. This analysis identified increased DREMI scores indicative of enhanced pairwise dependence between proteins associated with FcγR and TLR signaling pathways following T785-ISAC stimulation as compared to the equimolar mixture controls (Figure 2D). A strong interdependence between pERK1/2 and pIRF7 was uniquely observed following stimulation with T785-ISAC, but not mixture, as indicated by an increased DREMI score, amplified signal intensity and reduced activation threshold required for IRF7 phosphorylation (Figure 2D). This analysis also indicated a reduced activation threshold required to initiate protein translation (RPS6 phosphorylation) following ISAC stimulation as quantified by increased DREMI scores and reduced IPs for the pairwise combinations of pERK1/2-pRPS6 and pIRF7-pRPS6. Furthermore, the level of RPS6 phosphorylation was amplified as a result of ERK1/2 or IRF7 phosphorylation as determined by increased pairwise AUCs (Figure 2D). These combined results not only confirm signaling of the ISAC through the expected FcγR and TLR pathways, but furthermore, support a synergy between the two pathways resulting in reduced activation thresholds and amplified signal intensities.
ISACs require Fc effector function to mediate activation of myeloid APCs
The contribution of Fc activity to ISAC function was investigated following ablation of downstream FcγR signaling with R406, a small molecule kinase inhibitor of Syk that prevents signal transduction downstream of the activating FcγRs 33,34. Addition of R406 abrogated T785-ISAC-mediated myeloid activation as evidenced by the lack of upregulation of the costimulatory molecule CD86 but did not alter the cellular response to TLR7/8 activation by T785 (Figure 3A & Supplemental Figure 8). Syk inhibition with R406 also abrogated signaling observed in monocytes following T785-ISAC stimulation, with significantly reduced levels of phosphorylated MAPKAPK-2, ERK1/2 and IRF7 measured (Supplemental Figure 7D).
Figure 3: ISACs require Fc effector function and TLR agonism to mediate myeloid activation.
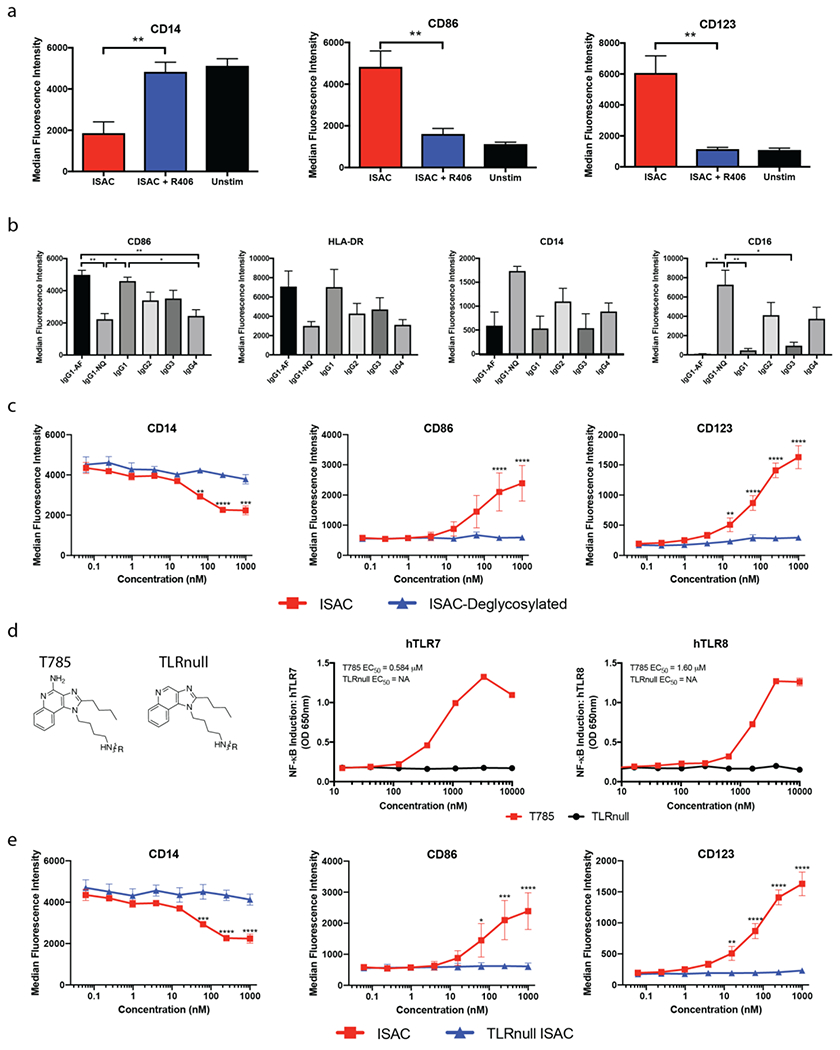
Freshly isolated human myeloid APCs were cultured for 18 hours in the presence of CD20+ Toledo tumor cells at a 3:1 effector to target ratio and (a) 80 nM of the rituximab-ISAC with or without 1 μM R406, (b) rituximab-ISACs on the indicated antibody isotype, (c) rituximab-ISAC or deglycosylated rituximab-ISAC (ISAC-deglycosylated). (a-c) Cells were analyzed by flow cytometry and data is reported as median fluorescence intensity. (d) HEK-Blue-TLR7 or TLR8 reporter cells were cultured for 18 hours in the presence of T785 or TLRnull prior to assessment of NF-κB-induced SEAP activity. Data shown are from 1 experiment and representative of at least 3 experiments. (e) Freshly isolated human myeloid APCs were cultured for 18 hours in the presence of CD20+ Toledo tumor cells and the indicated rituximab-ISACs and analyzed by flow cytometry (data shown as median fluorescence intensity). (a-e) Data shown are representative of 1-4 additional experiments and ≥3 donors for each experiment (mean and SEM). Statistical significance is defined as *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Given these data, rituximab T785-ISACs were formulated with comparable DARs on each of the four naturally occurring isotypes to define the extent to which T785-ISAC activity varies with known FcγR binding affinities 35,36. Aglycosylated and afucosylated rituximab ISACs were also investigated, as aglycosylation ablates FcγR binding while afucosylation increases the binding affinity for FCGR3A 37. The immunostimulatory capacity of the rituximab T785-ISAC correlated with the FcγR affinity of the antibody isotype (Figure 3B). Conversely, the afucosylated IgG1 (IgG1-AF), wildtype IgG1 and IgG3 T785-ISACs elicited myeloid activation, with the IgG1-AF T785-ISAC inducing the highest levels of CD86 upregulation (Figure 3B). Deglycosylation of the wildtype rituximab T785-ISAC with PNGase F, which reduces the Fc-FcγR binding affinity resulted in diminished ISAC immune stimulatory activity in vitro, supporting the importance of a functionally active Fc region (Figure 3C).
ISACs require TLR agonism to mediate myeloid activation
The contribution of TLR activity on ISAC function was investigated following ablation of TLR agonism by removing the 2-amino pyridine from T785 that is believed to form a salt bridge with critical aspartate residues in the binding pockets of TLR7 and TLR8 38 (TLRnull, Figure 3D). The TLRnull compound was conjugated to rituximab (TLRnull ISAC) and assessed for myeloid activation potential in the presence of CD20+ tumor cells in vitro. The TLRnull ISAC did not induce upregulation of costimulatory molecule CD86 or modulate the expression of CD14 or CD123, indicating that TLR agonism is needed for the activation phenotype observed with T785-ISACs (Figure 3E). Furthermore, the TLRnull ISAC did not induce phosphorylation of IRF-7 in monocytes, consistent with its inability to engage TLR7/8 (Supplementary Figure 7E).
Systemically administered trastuzumab T785-ISAC eliminates tumors in the absence of B, T or NK cell activity in human xenograft tumor models
The ability of ISACs to induce myeloid- and neutrophil-mediated destruction of solid tumors in the absence of B, T and NK cell activity was assessed using human xenograft models 39,40. Trastuzumab, a clinically validated HER2-targeting antibody, was selected for investigation to determine whether the trastuzumab T785-ISAC could overcome tumor resistance to trastuzumab in HER2+ solid tumors models (Figure 4 and Supplemental Figure 9) 41,42. First, systemic administration of trastuzumab T785-ISAC was compared to the intra-tumoral delivery of a mixture of trastuzumab and T785. The trastuzumab T785-ISAC provoked tumor regression and clearance whereas delivery of an equimolar mixture of components provided minimal therapeutic benefit compared to trastuzumab alone (Figure 4A). Subsequent studies comparing tumor-targeted ISACs with their appropriate isotype controls confirmed that anti-tumor activity required tumor-targeting (Figure 4B and Supplemental Figure 10). Importantly, systemic administration of the trastuzumab T785-ISAC and its isotype control were well-tolerated, with minimal impact on body weight (Supplemental Figure 11).
Figure 4: T785-ISACs elicit robust anti-tumor immunity in trastuzumab-resistant tumor xenograft models.
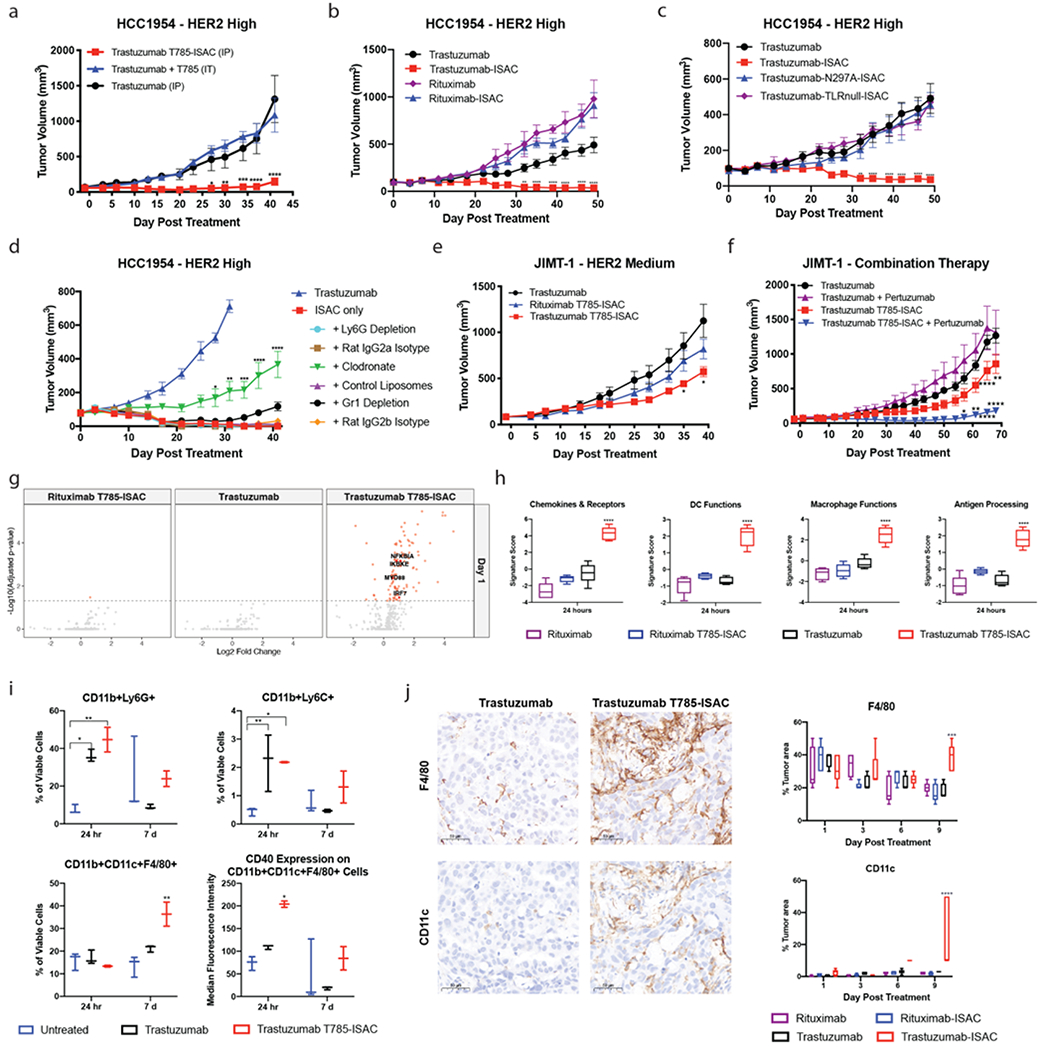
(a-d) SCID/Beige or NSG mice were implanted with the HCC1954 tumor cell line and were randomized when the tumor volume reached 50 – 75 mm3. (a) Mice were treated once by intraperitoneal (IP) with 5 mg/kg trastuzumab T785-ISAC or trastuzumab, or with molar equivalent dosing with a mixture of trastuzumab and T785 to the ISAC via intra-tumoral (IT) injection. Data are from one experiment with n=3-5 mice per group. (b) Mice were treated via intraperitoneal injection with 5 mg/kg of trastuzumab, trastuzumab-ISAC, rituximab, or rituximab-ISAC Q5DX6 (n= 5 mice per group). (c) Mice were treated via intraperitoneal injection with 5 mg/kg of trastuzumab, trastuzumab T785-ISAC, trastuzumab N297A-ISAC, or trastuzumab TLRnull-ISAC (n=5 mice per group). (d) HCC1954 tumor cells were implanted into SCID/Beige mice, and cells of interest were depleted prior to and during trastuzumab T785-ISAC treatment (5 mg/kg, Q5DX3) using anti-Ly6G antibody (rat IgG2a control), clodronate liposomes (control liposomes as control) or anti-Gr1 antibody (rat IgG2b control). Data are from one experiment with n=4-6 mice per group. (e, f) NSG or Rag2/IL2rg double knockout mice were implanted with the JIMT-1 tumor cell line and randomized when the tumor volume reached 50 – 75 mm3. Mice were subsequently treated with the indicated treatments at 5 mg/kg intraperitoneally with a frequency of (e) Q5DX6 or (f) Q5DX5 (n=3-6 mice per group). (b-f) Data shown are representative of at least two experiments. (g-j) HCC-1954 tumors were harvested from cohorts of mice at the indicated timepoints after treatment with a single dose of 5 mg/kg ISAC or control antibodies. Tumors were processed for flow cytometry analysis, NanoString mRNA quantification or formalin fixed and paraffin embedded for immunohistochemistry. (g) Volcano plots depict log2 fold change of gene expression in treated vs isotype control tumors measured by NanoString at 24 hours (n=5 mice per group). Changes with an adjusted p value of < 0.05 are shown in red. (h) Gene signature scores quantified by nSolver Advanced Analysis Pathway Score for tumors analyzed 24 hours after treatment (n=5 mice per group). (i) Flow cytometry analysis of tumoral cellular composition 24 hours and 7 days following treatment (n=2-4 mice per group). Data are representative of at least 2 experiments. (j) Representative images as well as quantification of F4/80 and CD11c IHC of tumors harvested 9 days after each indicated treatment. Scale bars are 50 μm. (a-j) Data are shown as mean with SEM and statistics are shown with *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To determine whether the mechanism of action contributing to ISAC efficacy in vivo is similar to that described in vitro, Fc-incompetent (trastuzumab N297A-ISAC) and trastuzumab TLRnull-ISACs were tested in the same breast tumor xenograft model in parallel with a wildtype trastuzumab T785-ISAC. While the trastuzumab T785-ISAC led to tumor regression and clearance, both the Fc-incompetent and TLRnull ISACs failed to elicit anti-tumor activity (Figure 4C). Together these data indicate a requirement for tumor targeting, and both FcγR and TLR-engagement for ISAC anti-tumor activity.
The differential contribution of neutrophils versus other myeloid cells in mediating anti-tumor effects was assessed by cell -specific depletions. Neutrophils were depleted using a Ly6G antibody, while depletion of both neutrophils and immature APCs (monocytes) was achieved with an anti-Gr1 antibody. Phagocytic cells were depleted using clodronate-loaded liposomes. We found that phagocytic cells, presumably macrophages, contributed significantly to mediating anti-tumor activity following ISAC treatment (Figure 4D). However, neutrophil depletion with anti-Ly6G had no impact on ISAC efficacy. Gr1 depletion did not immediately impact ISAC-mediated efficacy but reduced the duration of tumor control (Figure 4D).
To probe the ability of T785-ISACs to control tumor growth in models with lower HER2 expression, we next modeled efficacy in the trastuzumab resistant, HER2-medium expressing tumor model, JIMT-1. The JIMT-1 cancer cell line is estimated to have approximately 6.5 x105 copies of surface HER2, and expresses a reduced level of HER2 as compared to the HCC1954 tumor cell line by flow cytometry 43. While the trastuzumab T785-ISAC significantly slowed tumor growth compared to the isotype ISAC or trastuzumab controls, no tumor regression or clearance was observed (Figure 4E). We hypothesized that pertuzumab, which binds a HER2 epitope distinct from trastuzumab’s binding site, would increase Fc-clustering, and subsequently enhance ADCP and efficacy of the ISAC in the JIMT-1 model. The combination of the trastuzumab T785-ISAC with pertuzumab led to significantly enhanced efficacy and significant tumor regression (Figure 4F). Without the immune stimulation of the trastuzumab T785-ISAC, the combination of pertuzumab with trastuzumab provided no benefit.
We next investigated the molecular and cellular events triggered by trastuzumab T785-ISAC that ultimately result in anti-tumor efficacy. Early gene expression changes in tumors were measured with the NanoString mouse pan-cancer immune profiling panel, revealing a significant upregulation of 13.7% of the genes within 24 hours of treatment including genes associated with TLR7 signaling and activation (IRF-7, NF-kB associated genes) as compared to 0-0.1% following trastuzumab or isotype T785-ISAC treatment (Figure 4G) 44. Analysis of gene expression pathways assessed by nSolver Advanced Analysis Pathway Score revealed significant upregulation of key immune-related gene signatures (Macrophage Functions, DC Functions, Antigen Processing & Presentation) which were consistent with the in vitro and in vivo findings that Fc-FcγR interactions are critical for ISAC-mediated activity (Figure 4H and Supplemental Figure 12). Gene signature scores associated with chemokines and cytokines were highly elevated following trastuzumab T785-ISAC treatment, consistent with protein quantification performed measuring cytokine and chemokine secretion within the tumor 24 hours following administration (Figure 4H and Supplemental Figure 13). These findings, in congruence with the in vitro and in vivo efficacy data, indicate a robust TLR- and FcγR-mediated response is induced by the ISAC.
Flow cytometry and IHC were used to assess whether increased T785-ISAC-mediated activation and chemokine expression ultimately led to the recruitment of immune effector cells. Myeloid immune infiltrates were profiled by flow cytometry 24 hours and 7 days post administration of a single dose of trastuzumab T785-ISAC or trastuzumab and compared to untreated controls. Analysis of tumors 24 hours following treatment revealed an infiltration of CD11b+Ly6C+ monocytes and CD11b+Ly6G+ granulocytes in the tumors treated with trastuzumab or trastuzumab T785-ISAC compared to the untreated controls (Figure 4I). Seven days post treatment, CD11b+CD11c+F4/80+ positive myeloid APCs were significantly increased in the trastuzumab T785-ISAC treated group compared to trastuzumab or untreated controls (Figure 4I) 45,46. This increase in myeloid APCs may be attributed to the significant increase in production of myeloid-related chemokines following trastuzumab T785-ISAC treatment. While there was not a significant increase in frequency of myeloid APCs at the 24-hour timepoint, cells of this phenotype were activated after trastuzumab T785-ISAC treatment, as measured by an upregulation of CD40 on the cell surface, compared to the same population from trastuzumab or untreated controls (Figure 4I). This finding, along with the NanoString data, further demonstrates the trastuzumab T785-ISAC’s ability to engage and activate myeloid cells in the tumor microenvironment. Given the CD11b positivity of the myeloid APCs observed one week following treatment, it is possible that the early infiltrate of monocytes are precursors to the myeloid APCs population that matures in the ISAC-treated tumors but not in trastuzumab treated tumors. However, it remains to be determined how the pro-inflammatory proximal events measured 24 hours following ISAC stimulation by NanoString and MSD ultimately influenced distal events such as maturation of myeloid cells in the transforming tumor microenvironment at the later 6-day timepoint, and ultimately, influenced tumor regression and clearance.
Finally, IHC was performed to assess the presence and location of key immune cell populations in the tumor following treatment with the trastuzumab T785-ISAC as compared to its controls. Consistent with the day 7 flow cytometry, F4/80 expressing cells in the tumor and peritumoral region were elevated at day 9 after a single treatment with trastuzumab T785-ISAC (Figure 4J). Furthermore, targeted ISAC treatment resulted in a dramatic increase in CD11c+ cells in the peritumoral region that was not seen after rHER2 antibody treatment or treatment with isotype or isotype-ISAC (Figure 4J). The elevation in CD11c+ cells by both flow cytometry and IHC was also evidenced in NanoString data, in which mRNA expression of ITGAX was significantly elevated at days 6 and 9 following trastuzumab T785-ISAC administration (Supplemental Figure 14).
Tumor-targeted T785-ISACs elicit tumor regression in the syngeneic MMC tumor model
To assess the capacity of ISACs to mediate anti-tumor efficacy in the presence of B, T and NK cells, we used a syngeneic rat HER2 (rHER2)-expressing mouse mammary carcinoma (MMC) cell line that was derived from a spontaneous mammary carcinoma in rHER2-expressing FVB mice. To minimize cross-species immunogenicity associated with rHER2 expression in MMC, transgenic mice that endogenously express rat Her2 under the control of the MMTV promotor were used as the host (Supplemental Figure 15) 47. Leveraging the presence of a fully intact immune system, we aimed to assess the ISAC’s ability to combat tumors with a large tumor burden of 500 mm3, as larger tumors are generally more difficult to treat in pre-clinical models 48. Systemic administration of all the treatments, including the rHER2 T785-ISAC was well-tolerated, with minimal impact on body weight (Supplemental Figure 16A). Only the rHER2 T785-ISAC, however, was able to cure mice bearing large, established tumors (Figure 5A). As was seen in the xenograft model, cell depletion studies showed that significant anti-tumor activity was lost when phagocytic cells were depleted with clodronate-loaded liposomes (Figure 5B). While the ISAC is not expected to act directly on T cells, mice pre-treated with a CD8-depleting antibody lost the ability to clear tumors after two administrations of rHER2 T785-ISAC (Figure 5C). This suggests tumor-targeted ISACs can act indirectly through T cells, following presentation of tumor-associated antigens. Consistent with the presence of immunological memory, mice cured of MMC tumors following rHER2 T785-ISAC treatment were protected against tumor rechallenge (Figure 5D).
Figure 5: T785-ISACs elicit robust anti-tumor effects in MMC syngeneic tumor model.
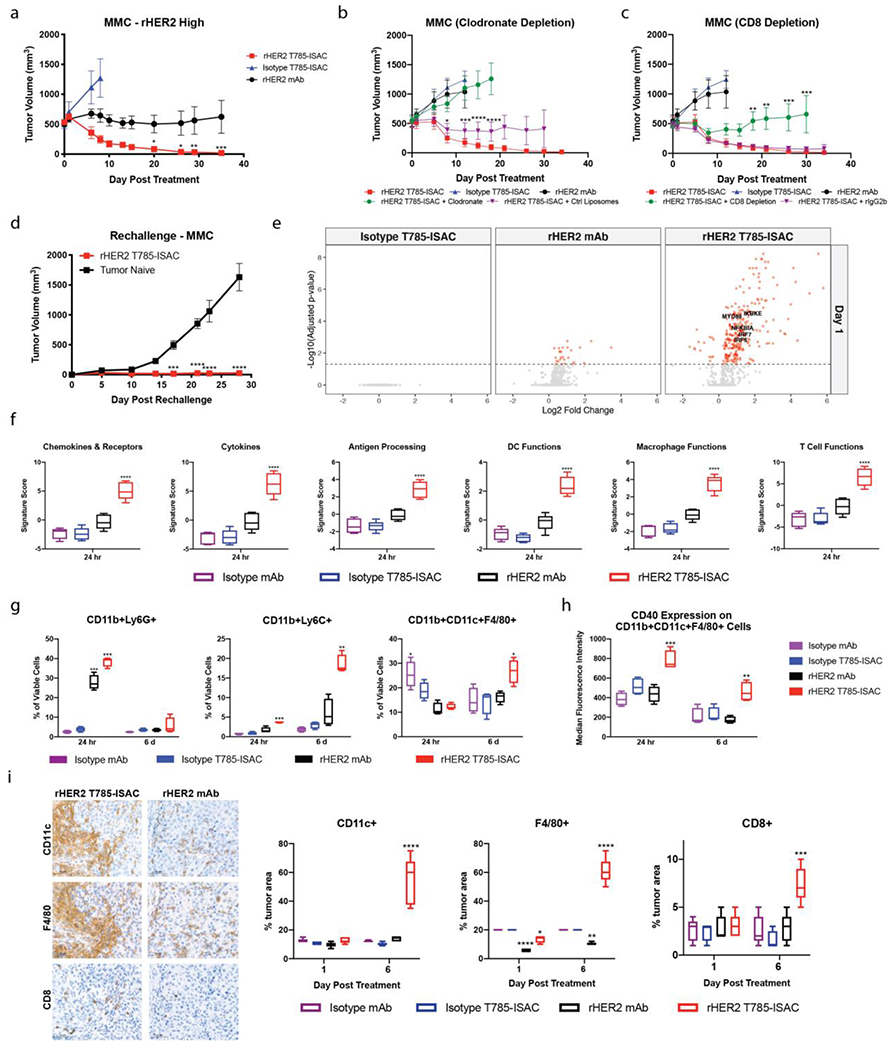
(a-h) FVB/N-TgN (MMTV-Erbb2) mice were implanted with the MMC tumor cell line. (a) Female mice were randomized when the tumor volume reached about 500 mm3. Mice were then treated via intraperitoneal injection with 5 mg/kg q5d x 2 of mouse anti-rat HER2 antibody, mouse anti-rat HER2 T785-ISAC, or isotype T785-ISAC (TA99) (n=4-7 mice per arm). (b, c) Male mice were randomized when the tumor volume reached about 500 mm3 and were pre-treated starting one day before the initial treatment date with clodronate-loaded or control liposomes for phagocyte depletion (6 cycles, through Day 15) or anti-CD8 depleting antibody (rIgG2b control) for CD8 T cell depletion (8 cycles, through Day 21). Animals were treated with 5 mg/kg rHER2 T785-ISAC q5d x 2 (n=6 mice per group) intraperitoneally. For phagocyte depletion, statistics reflect comparison of T785-ISAC treatment with clodronate or control liposomes, while for CD8 T cell depletion, statistics reflect comparison of T785-ISAC with T785-ISAC + CD8 Depletion or T785-ISAC + rat isotype IgG2b. (d) Anti-rHER2 T785-ISAC treated mice that experienced complete tumor regression for >60 days after their last treatment were challenged with the MMC tumor cell line. Tumor naïve mice served as implantation controls (n=5 mice per group). (a-d) Data are representative of at least 2 experiments. Mice were humanely euthanized once tumors reached 2,000 mm3. Data are shown for those groups in which no tumors had reached 2,000 mm3.(e-i) Cohorts of mice were randomized when MMC tumors tumor volumes reached about 500 mm3 and treated with 5 mg/kg ISAC or control antibodies dosed at day 0 and day 5. Tumors were processed for cytokine measurement by MSD, flow cytometry analysis, NanoString mRNA quantification or formalin fixed and paraffin embedded for immunohistochemistry at the times shown. (e) Volcano plots depict log2 fold change of gene expression in treated vs isotype control tumors measured by NanoString at 24 hours (n=5 mice per group). Changes with an adjusted p value of < 0.05 are shown in red. (f) Gene signature scores quantified by nSolver Advanced Analysis Pathway Score for tumors analyzed 24 hours after treatment (n=5 mice per group). (g, h) Flow cytometry analysis of tumoral cellular composition and activation state 24 hours and 6 days following initiation of treatment (n=4 mice per group). Data are representative of at least 2 experiments. (i) Representative images as well as quantification of F4/80, CD11c and CD8 IHC of tumors harvested 6 days after each indicated treatment. Scale bars are 50 μm. (a-i) Data are reported as mean and SEM; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To further probe the mechanism underlying ISAC efficacy in the syngeneic model, we next investigated the cellular, transcriptional and cytokine changes in the tumor microenvironment following ISAC dosing. Two cohorts of mice bearing large MMC tumors were treated with rHER2 T785-ISAC or controls. Tumors from the first cohort were harvested 24 hours after dosing (Day 1), when all the tumors were of similar size (Supplemental Figure 16B). The other cohort was given a second dose 5 days after the first, then harvested 24 hours later (Day 6), at which point tumors in all the treatment groups had grown measurably except those treated with T785-ISAC (Supplemental Figure 16B). Tumors were analyzed by IHC, mRNA expression and for cytokine secretion in protein lysates.
Further supporting our observations in the xenograft models that tumor targeting is required for ISAC activity, mRNA analysis of tumors 24 hours after rHER2 T785-ISAC treatment showed dramatic upregulation of 34.1% of the genes in the NanoString mouse pan-cancer immune profiling panel compared to isotype-treated tumors. The anti-rHER2 antibody alone had only a slight impact on gene expression (3.5% of the genes analyzed) whereas the non-targeted isotype T785-ISAC did not affect gene expression (Figure 5E). As seen in the HCC1954 xenograft model, genes associated with TLR7-mediated signaling and activation were significantly upregulated by the tumor-targeting rHER2 T785-ISAC, indicative of a robust TLR-mediated response (Figure 5E). Gene expression pathway analysis again revealed significant upregulation of key immune-related gene signatures, including signatures for gene expression pathway analysis revealed significant upregulation of key immune-related gene signatures following rHER2 T785-ISAC treatment, including those associated with chemokines and cytokines (Figure 5F). Protein quantification was performed on the same tumors as analyzed by NanoString, and consistent with the gene signature findings, significantly increased levels of pro-inflammatory cytokines and chemokines were measured at 24 hours following rHER2 T785-ISAC treatment (Supplemental Figure 17). Gene signature scores were also significantly elevated for Antigen Processing & Presentation, Dendritic Cell Function, and Macrophage Function, further supporting an Fc-FcγR mediated and phagocytosis-driven response 24 hours following rHER2 T785-ISAC treatment (Figure 5F and Supplemental Figure 18). In addition to significantly elevated gene signature scores affiliated with a robust TLR- and FcγR-mediated response, the gene signature score for T cell Functions was also significantly increased within 24 hours of rHER2 T785-ISAC treatment, further supporting the finding that T cells are required for ISAC-mediated efficacy (Figure 5F and Supplemental Figure 18).
To characterize the immune cells that may contribute to the early increase in proinflammatory cytokine and chemokine production, tumors were analyzed by flow cytometry 24 hours following the first dose of rHER2 T785-ISAC or the controls and 6 days following initiation of treatment, 24 hours after a second dose. Analysis of tumors at Day 1 revealed an infiltration of CD11b+Ly6C+ monocytes in the tumors treated with rHER2 T785-ISAC, and CD11b+Ly6G+ granulocytes in the tumors treated with rHER2 mAb or rHER2 T785-ISAC, but not the isotype ISAC or isotype antibody controls (Figure 5G). Levels of monocytes were even further increased in rHER2 T785-ISAC treated tumors following 6 days. Similar to what we observed in the HCC1954 xenograft model, myeloid APCs (CD11b+CD11c+F4/80+) were significantly elevated at day 6 following rHER2 T785-ISAC treatment compared to the controls (Figure 5G and Supplemental Figure 19). Furthermore, myeloid APCs were found to be activated specifically in the rHER2 T785-ISAC treated group at both timepoints, as measured by an upregulation of CD40 on the cell surface (Figure 5H). Importantly, an increase in tumoral myeloid APCs was not measured following phagocyte depletion using clodronate-loaded liposomes (Supplemental Figure 20).
Finally, IHC was performed to assess the presence and location of key immune cell populations in the MMC model following treatment with the rHER2 T785-ISAC. As was seen in the HCC1954 xenograft model, treatment with the rHER2 T785-ISAC resulted in a dramatic increase in CD11c+ and F4/80+ cells in the peritumoral region that was not seen after rHER2 antibody treatment or treatment with isotype or isotype T785-ISAC (Figure 5I). Consistent with the early expression of the T cell chemokines and the increased gene signature score for T Cell Function, significant infiltration of CD8+ cells was seen by IHC within the tumors of rHER2 T785-ISAC treated mice at Day 6, whereas CD8 staining in mice treated with anti-rHER2 antibody or isotype controls remained low (Figure 5I). Increased CD8+ IHC staining, together with amplified gene expression of T cell function-associated genes and CD8-dependent tumor clearance, support the potential of the T785-ISAC to turn a cold tumor hot.
CL264-ISACs elicit tumor regression in HER2-medium expressing xenograft tumor model
To demonstrate that ISACs are a robust platform across multiple immune-stimulating payloads, we developed an ISAC using a well-known TLR7 agonist, CL264 with a DAR comparable to the trastuzumab T785-ISAC (Figure 6A and Supplemental Figure 21). The capacity of trastuzumab ISACs produced with CL264 or T785 to mediate intra-tumoral myeloid activation was assessed in the HCC1954 xenograft model 24 hours after a single administration of trastuzumab, trastuzumab CL264-ISAC or trastuzumab T785-ISAC. Enhanced myeloid activation was observed with trastuzumab CL264-ISAC as evidenced by increased CD40 expression on tumor infiltrating CD11b+CD11c+F4/80+ myeloid APCs as compared to trastuzumab or trastuzumab T785-ISAC (Figure 6B).
Figure 6: CL264-containing ISACs elicit tumor clearance in HER2 medium expressing xenografts and T cell mediated tumor clearance, immunologic memory, and epitope spreading in a syngeneic tumor model.
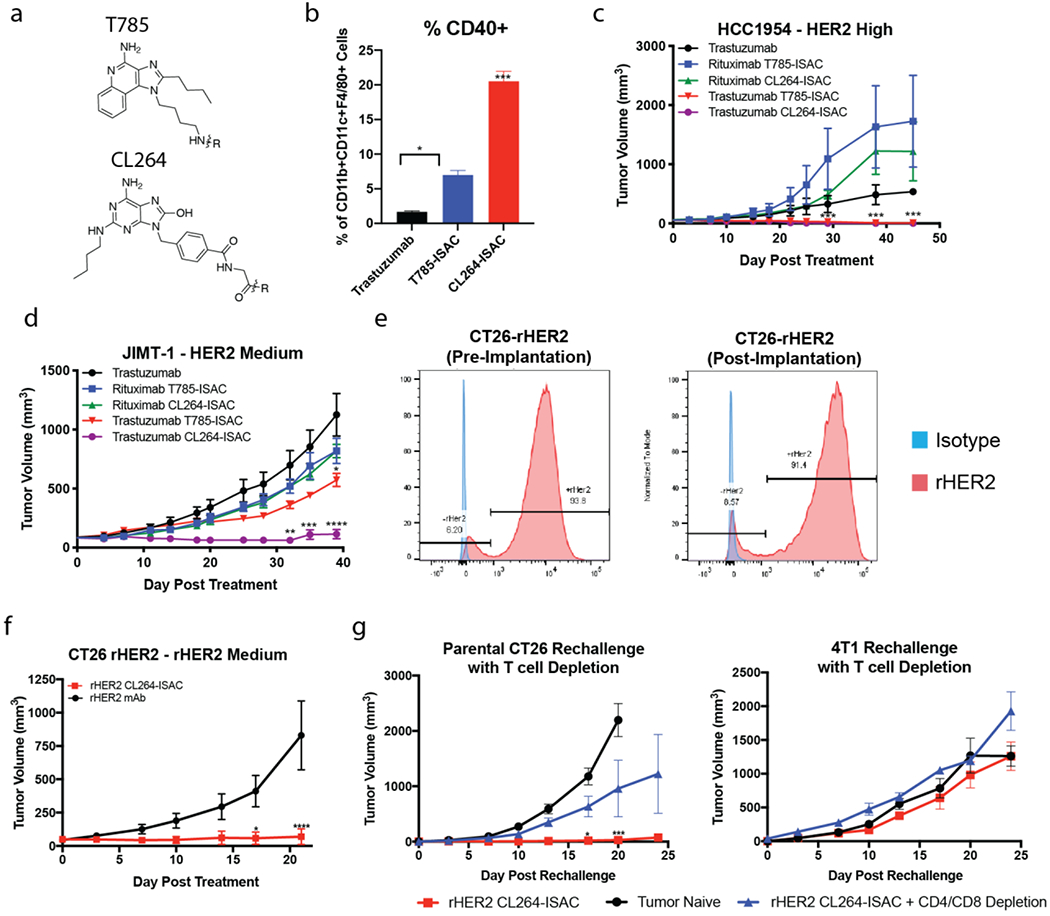
(a) Chemical structures of T785 and CL264 adjuvants used for ISAC generation. (b) NSG mice implanted with the HCC1954 tumor cell line were randomized when the tumor volume reached 50 – 75 mm3. Mice were treated with once via intraperitoneal injection of 5 mg/kg trastuzumab, trastuzumab T785-ISAC or trastuzumab CL264-ISAC. Tumors were harvested 20 hours post administration, processed to a single cell suspension, and analyzed by flow cytometry to assess activation of tumor-infiltrating myeloid APCs. (c-d) NSG or Rag2/IL2rg double knockout mice were implanted with the indicated human tumor cell line and randomized when the tumor volume reached 50 – 75 mm3 (HCC1954) or 75 – 150 mm3 (JIMT-1). Mice were treated via intraperitoneal injection with 5 mg/kg of rituximab, trastuzumab, trastuzumab T785-ISAC, trastuzumab CL264-ISAC or the respective isotype-ISACs, every 5 days for a total of 6 treatments. (e) rHER2 expression was measured on CT26-rHER2 tumor cells in culture prior to implantation (left flow plot) and in tumors nine days post-implantation (right flow plot) by flow cytometry with fluorescently-conjugated anti-rHER2 antibody (red) or an isotype control (blue). (f) Balb/c mice were implanted with the CT26-rHER2 tumor cell line and were randomized when the tumor volume reached 50 mm3. Mice were then treated via intraperitoneal injection with 10 mg/kg of mouse anti-rat HER2 or mouse anti-rat HER2 CL264-ISAC every 5 days for a total of 6 treatments. Data shown are from 1 experiment with 8 mice per arm and are representative of 3 experiments. (g) Anti-rat HER2 CL264-ISAC treated mice that experienced complete tumor regression for >21 days after their last treatment were challenged with parental CT26 (left flank) and 4T1 cell lines (right flank), with or without CD4 and CD8 T cell depletion (n =3 each). Tumor naïve mice (n = 6) challenged with the parental CT26 and 4T1 cell lines were included as controls. (a-i) Data are shown from individual experiments with 3-6 mice per arm and are representative of 1-4 experiments with a minimum of 3 mice per arm in each experiment. Data are shown as mean with SEM and statistics are shown with *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To determine if the enhanced myeloid activation capacity of the CL264-ISAC translated to greater anti-tumor activity in vivo, the trastuzumab CL264-ISAC was compared to the T785-ISAC in the HER2High HCC1954 and the HER2Med JIMT-1 tumor models. While both trastuzumab ISACs mediated complete tumor regression in the HCC1954 HER2High model, the CL264-ISAC demonstrated increased anti-tumor activity in the JIMT-1 HER2Med model suggesting that ISAC potency and the ability to target lower antigen density tumors may be linked (Figure 6C–D).
Both ISACs were well tolerated, but we observed transient body weight loss of 5-10% in animals treated with either the trastuzumab CL264-ISAC or rituximab CL264-ISAC, with body weight recovering to or above baseline by the third dose (Supplemental Figure 16A). Systemic cytokine secretion was measured following treatment with T785-ISAC or CL264-ISAC. Low levels of TNFα were measured in the serum of animals four hours after administration of the T785-ISAC, while both the targeted and isotype CL264-ISACs elicited higher levels of TNFα secretion systemically (Supplemental Figure 11). It is unlikely that these effects between T785 and CL264 ISACs were due to a direct impact of the ISAC on tumor cell growth, as the ISAC did not inhibit cancer cell proliferation in vitro (Supplemental Figure 9).
CL264-ISACs elicit tumor clearance and immunologic memory in syngeneic tumor models
Given the increased potency and efficacy observed with the CL264-ISAC in human tumor xenograft models with varied HER2 antigen density, we next characterized the activity of CL264-ISACs in syngeneic tumor models. Similar to the T785-ISAC in the MMC model, the CL264-ISAC led to tumor regression and clearance in a T cell dependent manner, leading to durable immunologic memory (Supplemental Figure 22A).
To assess ISAC efficacy in a syngeneic tumor model with reduced antigen expression, we developed a CT26 cell line that stably expresses rat HER2 (CT26-rHER2). Importantly, about 8.5% of the CT26 cells do not express rHER2 after tumor implantation (Figure 6E). Wildtype Balb/c mice treated with the rHER2 CL264-ISAC displayed substantial anti-tumor immunity in that 75% (6 out of 8) of the mice cleared their tumors and remained tumor free for the duration of the experiment, whereas none of the mice treated with the anti-rHER2 antibody had complete regression (Figure 6F). Transient weight loss of about 10% body weight following rHER2 CL264-ISAC treatment was observed, with body weight recovering by eight days post initiation of the study (Supplemental Figure 22B). We assessed whether the rHER2 CL264-ISAC mediated immunologic memory against a tumor lacking expression of the targeted antigen, rHER2, by challenging mice previously cured of CT26-rHER2 tumors with the parental CT26 cell line that lacked rHER2 expression. Mice previously cured of CT26-rHER2 tumors with rHER2 ISAC were fully protected from challenge with the parental CT26 cell line (Figure 6I). Depletion of CD4 and CD8 T cells prior to the tumor rechallenge showed that T cells are required for protection. Finally, we tested whether the immunological memory developed in mice previously cured with the ISAC was specific to tumor-associated antigens of the primary tumor treated. During the parental CT26 tumor rechallenge, mice were simultaneously challenged with a different tumor, 4T1, implanted in the contralateral flank. The data show that development of immunological memory was specific to CT26 tumor-associated antigens distinct from rHER2, as tumor growth of 4T1 tumors was not impacted (Figure 6G).
Discussion:
The results presented here provide compelling evidence supporting ISACs as a novel technology for tumor immunotherapy. In vitro modeling of ISACs demonstrated their ability to activate primary human myeloid APCs in an FcγR and TLR-dependent manner and to enhance antigen cross-presentation using the murine OVA model system. Activation through upregulation of costimulatory molecule CD40 was not only observed in vitro but was also seen on tumor-residing myeloid APCs in both xenograft and syngeneic tumor models.
One of the striking findings of this study was that covalent attachment of a TLR agonist to a tumor-targeted monoclonal antibody in the form of an ISAC altered the immunostimulatory outcome and overall efficacy that is typically anticipated with TLR agonists. A comparison of local delivery of trastuzumab combined with T785 to systemic delivery of the T785-ISAC demonstrated that only the ISAC was successful in reducing tumor burden. Local delivery of the mixture provided no benefit in the HCC1954 tumor xenograft model, with comparable effects to trastuzumab alone. This finding supports that not only the local delivery, but also the simultaneous delivery of the antibody and TLR agonist, is essential for the therapeutic efficacy observed with the ISAC.
Mechanistic data generated in the HCC1954 tumor xenograft model demonstrated the in vivo requirement for FcγR engagement and TLR engagement for efficacy, as both the TLRnull-ISAC and Fc-inactive ISAC failed to control tumor growth. Furthermore, systemic administration of ISACs resulted in sustained anti-tumor efficacy in rat HER2+ syngeneic tumor models and human HER2+ breast cancer xenografts that were resistant to treatment with the unconjugated antibody. HER2 was selected as the target antigen for preclinical in vivo studies because neoadjuvant, adjuvant and first line therapy for patients with locally advanced or metastatic breast cancer are dominated by HER2-targeted antibodies that are not designed to stimulate the immune system, and approved T cell checkpoint inhibitor blocking antibodies have been minimally effective in these patients. These patients could benefit from ISACs that retain the functionality of the parent antibodies and add significant immunostimulatory potential. As with any immunotherapeutic, the possibility to generate or exacerbate anti-drug antibodies exists and, in the case of the ISAC, may ultimately depend on the immunogenicity of the parent antibody. These assessments are best performed clinically and trastuzumab may be an ideally suited for investigation as the reported immunogenicity rate of Herceptin™ in patients is less than 1%.
In vivo data demonstrating the requirement for simultaneous delivery of the antibody and adjuvant is further supported by work detailing the intracellular signaling governing ISAC activity through simultaneous FcγR and TLR signaling. While additional studies may further elucidate the intracellular ISAC signaling dynamics, the studies described here highlight the potential signaling advantages of conjugating a TLR agonist to an antibody, as is typically observed when antibodies engage pathogens during protective immunity. CyTOF-based analysis of intracellular signaling in human PBMCs was employed to further understand and differentiate the activity of the mixture and ISAC. The results revealed an amplified signature following ISAC stimulation compared to that elicited by the mixture of individual components, further supporting the potential for enhancement of activity through chemical conjugation of the antibody and TLR agonist. While TLR7 and TLR8-specific potency of ISACs cannot be measured using HEK293 reporter cells, the demonstration of ISAC-induced phosphorylation of IRF-7 suggests active TLR7/8 agonism, as IRF-7 is known to be downstream of TLR7 and TLR8 MyD88-driven signaling 30. The data indicate that the ISAC not only elicits increased signaling of canonical pathways associated with TLR and FcγR pathways, but also reduces the pairwise activation threshold required to trigger phosphorylation of downstream signaling proteins such as ribosomal protein S6, a key regulator of protein translation in the cell. High DREMI scores, particularly measured in monocytes as well as in DCs, support the strong conditional dependence induced by stimulation with the ISAC that is not seen with the mixture. This phenomenon may be facilitated by intracellular changes in abundance, localization, and/or recruitment of receptors within the endosome, but further investigation is required to confirm an exact mechanism. Importantly, DREMI analysis identified relationships consistent with known signaling partners as a result of ISAC stimulation. In accordance with its known inhibitory role in canonical NF-κB signaling 49, we observed reduced dependency (i.e. DREMI score) between pIRF7 and inhibitor of kappa B (IκB) when myeloid cells were stimulated with ISAC. Collectively, these results support an amplified and sustained intracellular response via relevant signal transduction pathway molecules downstream of ISAC stimulation in addition to synergy between the TLR and FcγR-related signaling pathways.
Our results revealed that functional Fc-FcγR interactions are essential for ISAC-mediated activation of myeloid APCs. Native IgG1 Fc, which has the highest affinity for the activating FcγRs of the naturally existing IgG isotypes, was superior to other IgG isotypes with lower affinities for the activating FcγRs. Further enhancement of the Fc-FcγR interaction through afucosylation enhanced potency while diminution of the Fc-FcγR interaction through aglycosylation reduced potency. As expected, Syk signaling downstream of FcγR engagement in addition to subsequent TLR agonism were required for ISACs to elicit maximal stimulation. These finding are supported by studies demonstrating a dramatic enhancement of myeloid stimulation following co-stimulation with plate-bound IgG and the TLR agonist PAM3CSK 50. They also suggest that more extensive Fc crosslinking, on the target cell-surface or within the in vitro plate setting, may occur, or differential signaling networks are engaged, following ISAC stimulation as opposed to the mixture of components.
Translation of the ISAC platform in vivo was first assessed with human HER2+ tumor cell lines in mice that lack fully functional B, T and NK cells. Remarkably, the trastuzumab ISAC was significantly more effective than trastuzumab across multiple tumors with varying levels of HER2 expression. Furthermore, the data demonstrated the dependence on tumor-targeting for anti-tumor effects of the ISAC, as the isotype-ISAC was unable to induce tumor regression and clearance. While in vitro modeling demonstrated a necessity for TLR and FcγR activity to enable ISAC potency, such models were not successful in measuring the dependence on ISAC antigen-targeting. No differences were measured between targeted and isotype T785-ISACs in plate-based assays with healthy human myeloid APCs co-cultured with tumor cells (data not shown). The lack of target-dependence measured in vitro may be attributed to multiple factors: myeloid APCs are found to upregulate FcγRs on the cell surface following in vitro culturing, which may enhance the ability for soluble IgG to bind to FcγR, leading to an amplified signal from the isotype ISAC. Additionally, the cultured system lacks endogenous IgG and additional serum factors present in vivo that may compete for FcγR in vivo and dampen effects seen with an isotype ISAC. Nonetheless, the in vivo models clearly demonstrated the ISAC’s reliance on tumor-targeting for anti-tumor efficacy. Not only did isotype ISACs show little impact on tumor growth in efficacy studies, but they were also found to induce no impact on the genetic level in tumors, as shown in xenograft and syngeneic tumor models. In addition to demonstrating target-dependent efficacy in vivo, the T785-ISAC was tolerated by animals and induced minimal weight loss, demonstrating little sign of off-target effects and adverse events in the mouse. These data further support the ability of a systemically-delivered tumor-targeting ISAC to travel to the tumor and induce a localized pro-inflammatory anti-tumor response.
The importance of myeloid cell-mediated effector functions was demonstrated in cell depleting studies, in which clodronate depletion of phagocytes ablated the ISAC’s ability to induce tumor clearance. Interestingly, although tumors initially regressed after depletion of Gr1 positive cells, the tumors subsequently grew out. This phenomenon may have occurred as a result of depletion of phagocyte precursors such as monocytes, which may have later differentiated into functional and mature phagocytic cells 51. Further molecular and cellular insights were obtained upon quantifying mRNA levels and changes following ISAC treatment. These data were further corroborated by immunohistochemistry as well as protein quantification, indicating an infiltration of myeloid cells into the tumor along with production of a pro-inflammatory cytokine TNFα and myeloid chemoattractants CCL2 (MCP-1) and CCL4 (Mip1b).
The receptors for CCL2 and CCL4, CCR2 and CCR5 are expressed on monocytes and macrophages, respectively, suggesting these cell types could be recruited to ISAC-treated tumors. Furthermore, Cxcl9 and Cxcl11 mRNA, which encode T cell-specific chemokines and have been shown to be expressed by DCs in the TME, were also upregulated by trastuzumab ISAC treatment (Supplemental Figure 23) 52,53.
A trastuzumab ISAC with increased potency, using the agonist CL264, exhibited enhanced efficacy in tumor models expressing less HER2. This is important given that patients whose tumors express low HER2 have more limited treatment options and are not eligible for trastuzumab or other trastuzumab-containing therapies. Transient weight loss of about 5-10% and increased systemic secretion of TNFα were observed in animals treated with both the targeted and isotype CL264-ISACs, indicative of possible off-target effects in addition to the robust anti-tumor activity observed.
The collective data presented in the xenograft models suggest that ISAC-mediated phagocytosis is an effective mechanism to eliminate tumors, but this mechanism might be enhanced following antigen presentation of tumor neoantigens to T cells in immunocompetent hosts. Toward this end, the ability of ISACs to promote anti-tumor immunity in wildtype mice was investigated with anti-rHER2 ISACs in two different rHER2-expressing syngeneic models (MMC and CT26-rHER2). Both T785 and CL264 containing ISACs were assessed in the syngeneic MMC model given the high antigen expression of rHER2, as both the lower and higher potency ISACs were postulated to be efficacious with high antigen density on the tumor cell. Both rHER2 ISACs led to complete tumor clearance in animals bearing large tumors (350-950 mm3) in the MMC model whereas the unconjugated antibody failed to control tumor growth. Analysis of mRNA transcript levels in the MMC model revealed a dramatic shift in regulation of many genes at 24 hours following treatment with the targeted rHER2 T785-ISAC relative to the rHER2 antibody or isotype ISAC, including within genes involved in myeloid cell biology, TLR biology, and adaptive immune responses. Increased infiltrates of CD11c- and F4/80-expressing cells were seen by immunohistochemistry, similar to the trend seen in the HCC1954 xenograft tumor model. Furthermore, CD8 T cell infiltration was observed by immunohistochemistry, supporting the generation of an adaptive immune response following ISAC treatment.
In addition to the demonstrated importance of phagocytes in the xenograft model, depletion studies in the MMC syngeneic tumor model revealed a strong dependence on phagocytes, as depletion using clodronate-loaded liposomes halted ISAC-mediated anti-tumor effects. Moreover, the syngeneic studies demonstrated a critical role for T cells in ISAC efficacy. ISAC-driven tumor clearance was heavily dependent on CD8 T cell activity, as depletion of CD8 T cells inhibited anti-tumor efficacy. The data suggest that T cell activity is driven through TLR mediated activation of ISAC-stimulated myeloid cells. Such requirement for T cells, presumably related to presentation of tumor associated antigens by ISAC-stimulated myeloid cells, supports that ISACs likely mediate their anti-tumor activity through at least two mechanisms: phagocytosis and antigen presentation. While phagocytes provide an early anti-tumor response following ISAC treatment, antigen priming of T cells ultimately provides the tumor clearance and durable effects measured in fully immunocompetent models. This combination would be highly desirable for cancer patients as ISAC treatment could lead to robust and durable anti-tumor immunity.
To further explore the importance of ISAC mediated T cell activation, mice cured of CT26-rHER2 tumors following rHER2 CL264-ISAC treatment were rechallenged, after 30 days of being tumor free, with the non-rHER2 expressing CT26 parental cell line. rHER2-CT26 cured animals were protected from the challenge with CT26 whereas as tumor naïve animals were unable to reject tumor growth. Additionally, to show that protection was specific to the CT26 tumor cell line, 4T1 was simultaneously implanted in the contralateral flank of parental CT26 challenged mice. Although CT26 tumors failed to grow in rHER-CT26 cured mice, 4T1 tumor growth was unencumbered and thus provides evidence that immunity is antigen-specific and dependent upon adaptive immune response. Furthermore, these studies suggest tumor targeted ISACs mediate immunological memory not only to the initial targeted tumor antigen but also to untargeted antigens. These data also suggest that myeloid cells may present tumor-associated antigens that drives additional CTL responses. The implications of these findings demonstrate that, even if the targeted tumor antigen is subsequently downregulated or unable to bind to the ISAC, immunological memory has already been invoked and is capable of sustaining a durable response. This response is not limited to the antigen being targeted by the ISAC but also to additional, unidentified, tumor expressed antigens.
Taken together, these data indicate that ISACs elicit qualitatively and quantitatively distinct biology relating to cellular activation and antigen presentation, T cell proliferation, and durable anti-tumor immunity.
Methods and Materials:
Antibody Conjugation & Characterization
For conjugation of adjuvants to the antibody, adjuvants were first synthesized to include a linker flanked by a reactive group. Conjugates produced using two step methods were first synthesized through the modification of mAb lysine residues with a heterobifunctional crosslinker SATA. Deprotection of the acetylated thiol exposed a reactive thiol which was then reacted at room temperature for 2-4 hours with adjuvant-linker flanked by a thio-reactive maleimide. For constructs produced using a single-step conjugation method, TFP esters were then conjugated to an IgG1 antibody. The TFP esters were dissolved in anhydrous DMSO to make a 20 mM stock solution and 5-10 molar equivalents (relative to the antibody) were added to the IgG antibody at 10 mg/mL in PBS. The conjugation reaction was performed at 4-40°C for 2-12 hours. The resulting immunoconjugates were buffer exchanged into PBS (pH 7.4) through desalting (Zeba Columns, Thermo Fisher Scientific) or dialysis to remove excess small molecular weight impurities. The final protein concentration was determined by measuring the absorbance at 280 nm on a Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific). The yields were >75% based on recovered protein. SEC analysis detected minimal aggregate present and DAR was determined by LC/MS analysis. The purified ISACs were filtered through a 0.2 μm sterile filter and stored at −20°C until use.
HEK Reporter Assay
HEK293 reporter cells expressing human TLR7 or human TLR8 were purchased from Invivogen (hkb-htlr7 or hkb-htlr8) and vendor protocols were followed for cellular propagation and experimentation. Briefly, cells were grown to 80-85% confluence at 5% CO2 in DMEM supplemented with 10% FBS, Zeocin, and Blasticidin. Cells were then seeded in 96-well flat plates at 4x104 cells/well with substrate containing HEK detection medium and immunostimulatory molecules. Activity was measured using a plate reader at 620-655 nm wavelength.
Biacore Analysis
His-tagged Fc Gamma Receptor (FCGR) proteins were obtained from R&D Systems (FCGR1 (CD64, cat# 1257-FC-050), FCGR2A (CD32a, cat# 1257-FC-050), FCGR2B (CD32b, cat# 1875-FC-050), FCGR3A (CD16a, cat# 4235-FC-050). Binding analysis was performed on a Biacore T200 instrument in HBS-EP+ buffer (GE BR100669) using a CM5 chip (GE BR100530) containing immobilized anti-HIS antibody (HIS Capture kit, GE 28995056). Rituximab or Rituximab-ISAC were injected over flow cells containing captured FCGRs or a reference surface anti-His antibody only. Data was double referenced with the anti-His surface and a buffer only injection. A kinetic titration method was used and surfaces were regenerated between cycles by injection of 10 mM glycine, pH 1.5. Data was fit using Biacore T200 evaluation software (V3.1) using kinetic fit (1:1) for FCGR1 and FCGR3A (average of 6 runs) and steady state affinity (1:1) for FCGR2A and FCGR2B (average of 3 runs).
Human Myeloid APC Isolation
Myeloid APCs, which predominantly consisted of monocytes (>95%), were isolated from whole blood of healthy donors (Stanford Blood Center) by density gradient centrifugation using a RosetteSep Human Monocyte Enrichment Cocktail (Stem Cell Technologies). Myeloid APCs were further isolated using a negative selection Human Monocyte Enrichment Kit without CD16 depletion (Stem Cell Technologies) to a final purity of >90% as determined by flow cytometry based on CD14, CD16, CD11c and HLA-DR expression.
Toledo Tumor Cell Preparation
Toledo cells (ATCC) were cultured in vendor recommended media and maintained at vendor recommended cell densities (ATCC). Cells were removed from culture, washed, and resuspended in PBS with 2% FBS and 2 mM EDTA at 1-10x106 cells/mL. In some cases, cells were subsequently labeled with 2 μM CFSE for 2 minutes and then washed once with RPMI-1640 (Lonza) supplemented with 10% FBS (Atlanta Biologicals) and 100 U/mL Pen/Strep (Lonza). Cells were then fixed in 2% paraformaldehyde and washed three times with PBS prior to further use.
Cellular Activation Assays
PBMCs or isolated monocytes were plated at 3x105 or 2x105 cells per well, respectively, in flat bottom 96-well plates in IMDM supplemented with 10% FBS (Atlanta Biologicals), 1 mM Sodium Pyruvate (Lonza), 100 μM non-essential amino acids (Lonza), and 100 U/mL Pen/Strep (Lonza). For monocyte and tumor co-culture experiments, monocytes were cultured with fixed allogeneic tumor cells at a 3 to 1 ratio. Cells were subsequently incubated with immunostimulatory agents or controls for 18-36 hours at 37°C with 10% CO2. Cellular activation was measured through cell surface expression of activation markers and cytokine production. Surface marker expression levels were measured by flow cytometry with antibodies against CD40 (5C3, BD), CD86 (IT2.2, BD), HLA-DR (L243, BD), CD16 (3G8, BD), CD14 (MΦP9, BD), and CD123 (7G3, BD). PBMCs were analyzed by flow cytometry with antibodies against the aforementioned activation markers as well as lineage markers CD19 (HIB19, BD), CD56 (B159, BD), CD3 (SK7, BD), CD4 (OKT4, BioLegend), CD8 (SK1, BD) and CD69 (FN50, BD). In all cases, cell culture supernatant was collected following 18 or 36 hour incubation and cytokine production was measured using either cytokine bead array kits (Human Inflammatory Cytokine Kit, BD Biosciences) and ELISA (TNFα Human ELISA kit, eBiosciences).
Mass Cytometry
PBMCs were isolated from healthy donor blood and stimulated with ISAC, a mixture of antibody and adjuvant, or no stimulus for 5-15 minutes at 37°C. Following stimulation, cells were fixed by adding 16% PFA (Electron microscopy sciences) to a final concentration of 1.6% and incubated for 10 minutes at room temperature (RT). Fixed cells were washed with cell staining medium (CSM; PBS with 0.5% BSA and 0.02% sodium azide (all Sigma) and barcoded using a palladium-based approach as previously described 54. Barcoded samples were combined into one composite sample for surface staining.
Surface staining was performed by adding antibodies (Supplemental Table 2) in CSM and incubating for 30 minutes at RT. Anti-human antibodies were either purchased pre-conjugated (Fluidigm) or conjugated to heavy metal isotopes using the MaxPar X8 antibody-labelling kit (Fluidigm) according to the manufacturer’s recommendations. Surface-stained cells were washed once in CSM and permeabilized with MeOH for 10 minutes on ice. Permeabilized cells were washed twice with CSM and antibodies against intracellular antigens were added for 30 minutes at RT. Cells were washed with CSM and finally resuspended in intercalation solution (1.6% PFA in PBS, 0.02% saponin (Sigma) and 0.5 μM iridium-intercalator (Fluidigm)) overnight at 4°C.
Before data acquisition, samples were washed once in CSM and twice in ddH2O. All samples were filtered through a cell strainer (Falcon) and resuspended at 1x106 cells/mL in ddH2O supplemented with 1x EQ four element calibration beads (Fluidigm) and data acquired on a CyTOF2 mass cytometer (Fluidigm). Signaling responses were evaluated using the SPADE implementation within cytobank 29. Analyses were run on all cells (no down-sampling), choosing the stated number of clusters and utilizing all relevant surface markers but no intracellular markers for clustering. Immune cell lineages were annotated based on their high-dimensional surface protein expression patterns.
Statistical analyses were conducted in R, an open source statistical software environment. Within each cell subset, treatment group and donor, average fold change was computed for the following twenty phospho-protein markers: p-Src, p-STAT5, p-cMET, p-AKT, p-MAPKAPK2, p-SHP2, p-SLP76, p-IRF-7, p-ZAP70/SYK, p-CREB, p-NFKB, p-PI3K, IKB (total), p-PLCG1, p-ERK1/2, p-p38, p-BTK/ITK, p-S6, p-STAT3 and p-JNK/SAPK. The average fold change was weighted by the total number of cells in each cluster, as defined following SPADE clustering. Paired t-tests were conducted to estimate the difference in average fold change, and to test the null hypothesis that there was no difference in average fold change of each marker across treatment and control conditions.
Xenograft Tumor Model Experimental Procedure
Tumor cell lines were purchased from ATCC (NCI-N87, HCC1954 and COLO205) or AddexBio (JIMT-1) and grown according to manufacturer’s guidelines. Cells were harvested when they reached 80-90% confluency by detaching with Accutase (Stemcell), washed with PBS, resuspended at 40 x 106 cells/mL in PBS and placed on ice for no longer than two hours. Immediately prior to implantation, suspended cells were mixed with an equal volume of Cultrex PathClear BME, Type 3 (R&D Systems), and 100 μL of the mixture (2 x 106 cells) were implanted subcutaneously into the right flank of 6-8-week-old female mice as follows: NCI-N87 and COLO205 cells were implanted in NSG mice (Jackson Laboratory), HCC1954 and JIMT-1 cells were implanted in Rag2/IL2rg double knockout (Taconic), and HCC1954 (for assessment with T785-ISAC) were implanted in NSG or SCID/beige mice (Jackson Laboratory or Taconic and Envigo). Tumor size was recorded twice a week and was estimated using the following formula: (length x width2)/2. Once tumors reached 50-100 mm3, usually within one week from tumor implantation, treatments were initiated. Mice were randomized by tumor size into treatment groups prior to initial treatment. Trastuzumab (EirGenix; EG12014), Trastuzumab ISAC, or Isotype ISAC was prepared in PBS at 1 mg/mL and administered at 5 mg/kg intraperitoneally every five days (Q5D) for a total of six doses. Mice whose tumors exceeded 2000 mm3 were euthanized.
HCC1954 and MMC Cellular Depletion Experimental Procedure
To deplete macrophages and other phagocytic cells, mice bearing HCC1954 or MMC tumors were treated via intraperitoneal injection two days prior to the initial ISAC treatment with clodronate liposomes or control liposomes not containing clodronate as a negative control (Encapsula, Catalog # CLD-8901) and then treated twice a week for a total of three weeks. To deplete neutrophils, mice were treated with depleting antibodies 1A8 to deplete Ly6G+ neutrophils only (BioXCell Catalog # BE0075-1) or RB6-8C5 to deplete Gr1+ cells, which includes Ly6G+ neutrophils and Ly6C+ cells (BioXCell Catalog # BE0075) or isotype controls 2A3 and LTF-2 (BioXCell Catalog # BE0089 and BE0090, respectively) two days before the initial ISAC treatment, then treated twice a week for three weeks. Depletion of cells in the blood was confirmed by flow cytometry (Supplemental Figure 24).
Tumor Cytokine & mRNA Analysis Procedure
Tumors were implanted as previously described and were collected for analysis at the indicated time points post treatment. For immunohistochemistry and mRNA analysis, tumors were split, and mRNA analyzed with the NanoString mouse pan-cancer immune profiling panel. Data analysis was performed with nSolver Advanced Analysis Software according to manufacturer’s recommendations. Volcano plots were drawn with the ggplot2 package in R. To obtain tumor lysates for cytokine analysis, tumor fragments were kept in pre-made buffer (1 Pierce Protease Inhibitor Tablets (Thermo Fisher, Catalog #A32965) dissolved in 20 mL of T-PER Tissue Protein Extraction Reagent, Thermo Fisher, Catalog #78510) in GentleMACS M Tubes (Miltenyi, Catalog #130-093-236) immediately after necropsy. Tumor samples were then dissociated using a GentleMACS Octo Dissociator. All samples were spun down for 5 minutes at 300g after. Supernatants were then collected into microfuge tubes and were spun down again with a microcentrifuge. Cytokine analysis was performed on supernatants by MSD. Immunohistochemistry was performed using haemotoxylin and eosin to identify tissue architecture, as well as for the following markers where indicated in studies: CD11c (CST Catalog #87585) and CD8 (CST Catalog #98941).
Syngeneic Tumor Model Experimental Procedure
MMC cells were obtained from Dr. Disis (University of Washington) and were cultured in RPMI 1640 supplemented with 20% FBS, 1% penicillin-streptomycin, and L-glutamine at 37°C and 5% CO2 until 80-90% confluent. CT26 cells (ATCC) transfected to stably express rat HER2 protein were grown in RPMI 1640 supplemented with 10% FBS and 600 μg/mL G418 selective antibiotic (Thermo) at 37°C and 5% CO2 until 80-90% confluent. Once sufficiently confluent, cells were harvested by detaching with Accutase (Stemcell) and washing with PBS. Harvested cells were resuspended at 20 x 106 cells/mL (MMC) or 5 x 106 cells/mL (CT26-rHER2) in PBS and kept at 4°C/on ice until use. 100 μL of suspended cells were implanted subcutaneously into 6-8-week-old FVB/N-TgN(MMTV-Erbb2)NK1Mul/Jmice (MMC model, The Jackson Laboratory) or female BALB/c mice (CT26 model, The Jackson Laboratory). Tumor size was measured and recorded twice a week for the duration of the study. Tumor volume was estimated using the following formula: (length x width2)/2. Once tumors reached 200-500 mm3 (MMC) or 50-100 mm3 (CT26-rHER2), usually within one week from tumor implantation, treatments were initiated. Mice were randomized by tumor size into treatment groups prior to initial treatment. The anti-rHER2 mAb (BioXCell; 7.16.4) or anti-rHER2-ISAC were administered intraperitoneally (frequency as indicated in figure legends) for the duration of the study. Mice whose tumors exceeded 2000 mm3 were euthanized. T cell depletion was assessed in animals using CD4 (BioXCell; GK1.5) or CD8 (BioXCell; YTS 169.4) cell-depleting antibodies. Depletion was initiated prior to tumor implantation and continued with twice-weekly administration of 200 μg per antibody. For tumor rechallenge, CT26 and 4T1 cell lines were implanted at 5 x 106 cells per tumor line, and tumor naïve mice were challenged with both cell lines as controls.
Supplementary Material
Acknowledgements:
The authors wish to thank P. Basto, N. E. Reticker-Flynn, T. Prestwood, and B. Mallet for helpful discussion. We also thank the Stanford Blood Center Flow Cytometry Core, specifically L. Tolentino, O. Choi, and N.Wu. We also extend our appreciation to Dr. Mary L. (Nora) Disis and Denise Cecil from the University of Washington who provided the MMC tumor cell line.
Funding:
S. E. Ackerman was supported by a Stanford BioX Bowes Fellowship.
Footnotes
Competing Interests: All authors affiliated with Bolt Biotherapeutics, Inc are employees and hold stock options and/or equity in Bolt Biotherapeutics. E.G.E is a co-founder of Bolt Biotherapeutics. S.E.A, J.A.K, A.L, D.Y.J, E.G.E, and M.N.A are Stanford- and/or Bolt Biotherapeutics-affiliated inventors of PCT/US2017/041268.
References:
- 1.Davis TA et al. Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin’s lymphoma: safety and efficacy of re-treatment. J Clin Oncol 18, 3135–3143, doi: 10.1200/JCO.2000.18.17.3135 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Grillo-Lopez AJ et al. Rituximab: the first monoclonal antibody approved for the treatment of lymphoma. Curr Pharm Biotechnol 1, 1–9 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Lizotte PH et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight 1, e89014, doi: 10.1172/jci.insight.89014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spranger S et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A 113, E7759–E7768, doi: 10.1073/pnas.1609376113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beatty GL & Gladney WL Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 21, 687–692, doi: 10.1158/1078-0432.CCR-14-1860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridman WH, Pages F, Sautes-Fridman C & Galon J The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12, 298–306, doi: 10.1038/nrc3245 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Galon J & Bruni D Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 18, 197–218, doi: 10.1038/s41573-018-0007-y (2019). [DOI] [PubMed] [Google Scholar]
- 8.Gabrilovich DI, Ostrand-Rosenberg S & Bronte V Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12, 253–268, doi: 10.1038/nri3175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joyce JA & Fearon DT T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80, doi: 10.1126/science.aaa6204 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Carmi Y et al. Akt and SHP-1 are DC-intrinsic checkpoints for tumor immunity. JCI Insight 1, e89020, doi: 10.1172/jci.insight.89020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carmi Y et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature 521, 99–104, doi: 10.1038/nature14424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen BM et al. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat Med 26, 1125–1134, doi: 10.1038/s41591-020-0892-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sagiv-Barfi I et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 10, doi: 10.1126/scitranslmed.aan4488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spitzer MH et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 487–502 e415, doi: 10.1016/j.cell.2016.12.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milhem Mohammed M., Theresa Medina RG, Kirkwood John M., Buchbinder Elizabeth, Mehmi Inderjit, Niu Jiaxin, Shaheen Montaser, Weight Ryan, Margolin Kim, Luke Jason, Morris Aaron, Mauro David, Krieg Arthur M., Ribas Antoni CT144-Intratumoral toll-like receptor (TLR9) agonist, CMP-001, in combination with pembrolizumab can reverse resistance to PD-1 inhibition in a phase 1b trial in subjects with advanced melanoma (AACR Annual Meeting 2018, 2018). [Google Scholar]
- 16.Rook AH The beauty of TLR agonists for CTCL. Blood 119, 321–322, doi: 10.1182/blood-2011-11-391243 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Singh M et al. Effective innate and adaptive antimelanoma immunity through localized TLR7/8 activation. J Immunol 193, 4722–4731, doi: 10.4049/jimmunol.1401160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao BG, Vasilakos JP, Tross D, Smirnov D & Klinman DM Combination therapy targeting toll like receptors 7, 8 and 9 eliminates large established tumors. J Immunother Cancer 2, 12, doi: 10.1186/2051-1426-2-12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jurk M et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol 3, 499, doi: 10.1038/ni0602-499 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Chattergoon MA et al. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal Toll-like receptors without induction of type 1 interferon. PLoS Pathog 10, e1004082, doi: 10.1371/journal.ppat.1004082 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eigenbrod T, Pelka K, Latz E, Kreikemeyer B & Dalpke AH TLR8 Senses Bacterial RNA in Human Monocytes and Plays a Nonredundant Role for Recognition of Streptococcus pyogenes. J Immunol 195, 1092–1099, doi: 10.4049/jimmunol.1403173 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Mattson G et al. A practical approach to crosslinking. Mol Biol Rep 17, 167–183 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Gabay C, Ben-Bassat H, Schlesinger M & Laskov R Somatic mutations and intraclonal variations in the rearranged Vkappa genes of B-non-Hodgkin’s lymphoma cell lines. Eur J Haematol 63, 180–191, doi: 10.1111/j.1600-0609.1999.tb01766.x (1999). [DOI] [PubMed] [Google Scholar]
- 24.Alonso MN et al. T(H)1, T(H)2, and T(H)17 cells instruct monocytes to differentiate into specialized dendritic cell subsets. Blood 118, 3311–3320, doi: 10.1182/blood-2011-03-341065 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke SR et al. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol 78, 110–117, doi: 10.1046/j.1440-1711.2000.00889.x (2000). [DOI] [PubMed] [Google Scholar]
- 26.Zehn D, Lee SY & Bevan MJ Complete but curtailed T-cell response to very low-affinity antigen. Nature 458, 211–214, doi: 10.1038/nature07657 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondratova M et al. A multiscale signalling network map of innate immune response in cancer reveals cell heterogeneity signatures. Nat Commun 10, 4808, doi: 10.1038/s41467-019-12270-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bendall SC et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696, doi: 10.1126/science.1198704 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiu P et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol 29, 886–891, doi: 10.1038/nbt.1991 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawai T & Akira S TLR signaling. Cell Death Differ 13, 816–825, doi: 10.1038/sj.cdd.4401850 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Sanchez-Mejorada G & Rosales C Signal transduction by immunoglobulin Fc receptors. J Leukoc Biol 63, 521–533 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Krishnaswamy S et al. Systems biology. Conditional density-based analysis of T cell signaling in single-cell data. Science 346, 1250689, doi: 10.1126/science.1250689 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiefer F et al. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol Cell Biol 18, 4209–4220 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braselmann S et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther 319, 998–1008, doi: 10.1124/jpet.106.109058 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Bruhns P et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716–3725, doi: 10.1182/blood-2008-09-179754 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Nimmerjahn F & Ravetch JV Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 310, 1510–1512, doi: 10.1126/science.1118948 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Jefferis R Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci 30, 356–362, doi: 10.1016/j.tips.2009.04.007 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Tanji H, Ohto U, Shibata T, Miyake K & Shimizu T Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339, 1426–1429, doi: 10.1126/science.1229159 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Shultz LD et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174, 6477–6489, doi: 10.4049/jimmunol.174.10.6477 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Xia Z et al. Innate immune response to human bone marrow fibroblastic cell implantation in CB17 scid/beige mice. J Cell Biochem 98, 966–980, doi: 10.1002/jcb.20730 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Tanner M et al. Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol Cancer Ther 3, 1585–1592 (2004). [PubMed] [Google Scholar]
- 42.Luque-Cabal M, Garcia-Teijido P, Fernandez-Perez Y, Sanchez-Lorenzo L & Palacio-Vazquez I Mechanisms Behind the Resistance to Trastuzumab in HER2-Amplified Breast Cancer and Strategies to Overcome It. Clin Med Insights Oncol 10, 21–30, doi: 10.4137/CMO.S34537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li JY et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 29, 117–129, doi: 10.1016/j.ccell.2015.12.008 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Ning S, Pagano JS & Barber GN IRF7: activation, regulation, modification and function. Genes Immun 12, 399–414, doi: 10.1038/gene.2011.21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao Q et al. Renal F4/80+ CD11c+ mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy. J Am Soc Nephrol 26, 349–363, doi: 10.1681/ASN.2013121336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheng J et al. A Discrete Subset of Monocyte-Derived Cells among Typical Conventional Type 2 Dendritic Cells Can Efficiently Cross-Present. Cell Rep 21, 1203–1214, doi: 10.1016/j.celrep.2017.10.024 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Knutson KL, Almand B, Dang Y & Disis ML Neu antigen-negative variants can be generated after neu-specific antibody therapy in neu transgenic mice. Cancer Res 64, 1146–1151 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Moynihan KD et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med 22, 1402–1410, doi: 10.1038/nm.4200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siebenlist U, Franzoso G & Brown K Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol 10, 405–455, doi: 10.1146/annurev.cb.10.110194.002201 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Vogelpoel LT et al. FcgammaRIIa cross-talk with TLRs, IL-1R, and IFNgammaR selectively modulates cytokine production in human myeloid cells. Immunobiology 220, 193–199, doi: 10.1016/j.imbio.2014.07.016 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Daley JM, Thomay AA, Connolly MD, Reichner JS & Albina JE Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 83, 64–70, doi: 10.1189/jlb.0407247 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Broz ML et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26, 938, doi: 10.1016/j.ccell.2014.11.010 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Spranger S, Dai D, Horton B & Gajewski TF Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723 e714, doi: 10.1016/j.ccell.2017.04.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zunder ER et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat Protoc 10, 316–333, doi: 10.1038/nprot.2015.020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.