

ABSTRACT
The aim of the HERITAGE Family Study was to investigate individual differences in response to a standardized endurance exercise program, the role of familial aggregation, and the genetics of response levels of cardiorespiratory fitness and cardiovascular disease and diabetes risk factors. Here we summarize the findings and their potential implications for cardiometabolic health and cardiorespiratory fitness. It begins with overviews of background and planning, recruitment, testing and exercise program protocol, quality control measures, and other relevant organizational issues. A summary of findings is then provided on cardiorespiratory fitness, exercise hemodynamics, insulin and glucose metabolism, lipid and lipoprotein profiles, adiposity and abdominal visceral fat, blood levels of steroids and other hormones, markers of oxidative stress, skeletal muscle morphology and metabolic indicators, and resting metabolic rate. These summaries document the extent of the individual differences in response to a standardized and fully monitored endurance exercise program and document the importance of familial aggregation and heritability level for exercise response traits. Findings from genomic markers, muscle gene expression studies, and proteomic and metabolomics explorations are reviewed, along with lessons learned from a bioinformatics-driven analysis pipeline. The new opportunities being pursued in integrative -omics and physiology have extended considerably the expected life of HERITAGE and are being discussed in relation to the original conceptual model of the study.
Key Words: EXERCISE GENOMICS, HERITABILITY, EXERCISE TRAINING, CARDIOMETABOLIC RISK FACTORS, EXERCISE AND GENE EXPRESSION, EXERCISE AND PROTEOMICS, EXERCISE AND METABOLOMICS, EXERCISE AND BIOINFORMATICS
The aim of this article is to summarize the findings of the HERITAGE (HEalth, RIsk factors, exercise Training And GEnetics) Family Study and their potential implications for cardiometabolic health and exercise performance. HERITAGE has contributed substantially to our understanding of adaptation to exercise and endurance training over the last 25 yr. It started as a collaborative endeavor among scientists from five research institutions in the United States and Canada. Over the years, investigators from institutions around the world have joined the effort to use HERITAGE data to address novel questions of scientific interest.
Scientists who have been involved in the early phases of the study or are currently leveraging the biobank and data of HERITAGE to address underlying biological mechanisms have undertaken the present review. The review covers foundational topics such as background and planning, recruitment, testing and exercise program protocol, panel of quality control measures, and other relevant issues. This is followed by a review of the findings on cardiometabolic traits, including adiposity and specific fat depots, cardiorespiratory fitness, exercise hemodynamics, insulin and glucose metabolism, lipid and lipoprotein profile, blood levels of steroids and other hormones, markers of oxidative stress, skeletal muscle morphology and metabolic indicators, and resting metabolic rate (RMR). Subsequently, reviews of findings arising from genomic markers, muscle gene expression studies, and proteomic and metabolomic explorations are provided, along with lessons learned from a bioinformatics pipeline. Finally, an overview of the strengths and limitations of the study, and considerations on new opportunities offered by advances in -omics and bioinformatics are offered.
Background
In a series of experiments performed in the laboratory of C. Bouchard (CB) at Laval University, Quebec, Canada, during the 1980s, two important conclusions were reached. The first was that there were large interindividual differences in the response level to a standardized exercise program at a given dose (1–6). The second was that there was strong indication of a genetic component to the variation in human trainability (4,6,7). Based on the aforementioned observations, it became evident that a major exercise training study encompassing a substantial number of subjects from nuclear families was necessary to test the genetic hypothesis more thoroughly. To this end, CB established a team of seasoned investigators who agreed to work together in the context of a research consortium. They included Arthur S. Leon (ASL) (University of Minnesota), D. C. Rao (DCR) (Washington University), James S. Skinner (JSS) (initially at Arizona State University and then at Indiana University), and the late Jack H. Wilmore (JHW) (University of Texas at Austin).
Historical notes
The team began planning the study in 1988 and a series of meetings were held to prepare an application to the National Institutes of Health. The National Heart, Lung and Blood Institute (NHLBI) expressed an interest in the proposed research, and Dr. Millicent Higgins from the Division of Epidemiology and Clinical Applications came forward to provide guidance for a multicenter grant application involving American and Canadian investigators. Our grant application was reviewed in June 1990, and a revision went to a Special Review Committee in May 1991; the latter received an excellent score and was recommended for funding. Dr. Claude Lenfant, Director of NHLBI, and Dr. Higgins met with CB to discuss the project funding and to ask for an expansion of the research to a substantial sample of African American families. The budget was adjusted accordingly, and the project was approved for funding (five coordinated R01 grants to CB, ASL, DCR, JHW, and JSS) with a start date of September 1992 (phase 1). Data collection began in February 1993 and was completed in the spring of 1997. The five grants were renewed from 1997 to 2000 (phase 2) and from 2001 to 2003 (phase 3). The grants for the two sites (CB and DCR) with expertise in genetics were renewed from 2006 to 2010 (phase 4).
Aim of HERITAGE
The aim of the HERITAGE research consortium was to investigate the magnitude of the individual differences in response to a standardized endurance exercise program, the importance of familial aggregation, and the genetics of the response levels of cardiorespiratory fitness and cardiovascular disease and diabetes risk factors. Figure 1 depicts a schematic of the initial conceptual model underlying HERITAGE.
FIGURE 1.
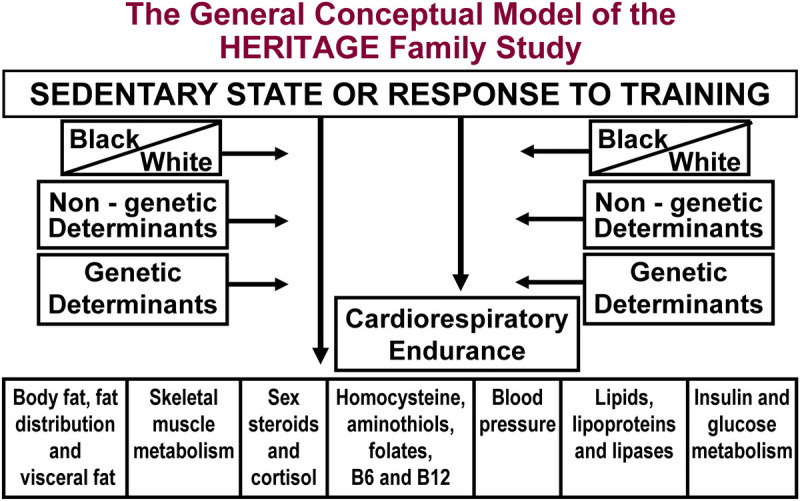
The 1992 model integrating the various components of HERITAGE. Subjects were measured in the sedentary state and after the exercise program intervention. Cardiorespiratory fitness, exercise hemodynamics, and subsets of cardiometabolic and diabetic risk factors were monitored. Differences between Blacks and Whites were investigated. Studies on genetic and nongenetic determinants of human variability in the sedentary state and in response to endurance exercise training were pursued.
Organization of the research consortium
The HERITAGE Family Study was led by a Steering Committee composed of the five principal investigators (CB (Chair), DCR, ASL, JHW, and JSS) and the Project Director (Jacques Gagnon from 1992 to 1999 and Tuomo Rankinen from 1999 to 2012). An administrator (Jean-Paul Albert) supported the work of the Steering Committee from 1992 to 1999 (Supplementary Table S1, Supplemental Digital Content, Appendix, http://links.lww.com/MSS/C482). An Advisory Board supported and advised the Steering Committee (Supplementary Table S2, Supplemental Digital Content, Appendix, http://links.lww.com/MSS/C482). During phase 1, the most demanding period of the project, the research was organized around a Consortium Coordinating Center (Laval University), a Data Coordinating Center (Washington University), four Clinical Centers responsible for recruitment, testing, and training of subjects (Arizona State University and then Indiana University, Laval University, University of Minnesota, and University of Texas at Austin), and four core laboratories (lipids, steroid hormones, glucose-insulin–related traits, and cell lines) located at Laval University. The leadership of these HERITAGE units and their personnel is identified in Supplementary Table S3 (Supplemental Digital Content, Appendix, http://links.lww.com/MSS/C482). By 2021, more than 200 peer-reviewed articles have been published from the HERITAGE Family Study. Most of these articles were based solely on HERITAGE data. In other publications, HERITAGE contributed data along with multiple other cohorts mainly in the context of multicenter genome-wide association studies (GWAS). A list of all HERITAGE-based publications and other general information regarding the study can be found at www.HeritageFamilyStudy.com.
PROTOCOL AND QUALITY CONTROL MEASURES
The HERITAGE Family Study enrolled a total of 855 individuals from 101 two-generation nuclear families of European ancestry (hereafter Whites), each with both biological parents and at least two offspring, and from 117 families of African ancestry (hereafter Blacks) (Table 1). The final sample at baseline included a total of 529 White and 326 Black individuals. Participants were within 17–65 yr of age, “sedentary” for at least 3 months before the study, and otherwise in good health. They were evaluated before and after a 20-wk standardized training program, providing an opportunity to assess familial aggregation of responsiveness to regular exercise (8).
TABLE 1.
Number of families, men, women, and total subjects at baseline and posttraining in the HERITAGE Family Study.
| Ancestry | Baseline | Posttraininga | ||||||
|---|---|---|---|---|---|---|---|---|
| Families | Men | Women | Total | Families | Men | Women | Total | |
| Whites | 101 | 262 | 267 | 529 | 99 | 233 | 250 | 483 |
| Blacks | 117 | 117 | 209 | 326 | 105 | 87 | 172 | 259 |
| Total | 218 | 379 | 476 | 855 | 204 | 322 | 422 | 742 |
aPosttraining numbers include only the subjects who were classified as completers having completed at least 57 (95%) of the 60 exercise training sessions.
Families were recruited from media (newspapers, TV, radio), posted advertisements, personal contacts, and institutional communication services. Telephone and mailed questionnaires were used for initial eligibility screening. All potential families underwent an orientation session to explain the study and answers questions. Participants were compensated up to $1000 in incremental payments for successful completion of the study (i.e., after completing the 20-wk training program, after completing baseline and posttraining battery of tests). The study was approved by each participating institution’s institutional review board. Written informed consent was obtained from each study participant, including permission to use individual cell lines, DNA, and collected specimens for the initial project and future collaborative studies.
The recruitment went smoothly for the families of Whites, as more than 90% of the families included both parents and three or more offspring. Recruiting similar types of families for participants who self-identified as Black was challenging. This led to a revision of the recruitment requirements for the latter families, which was approved by the HERITAGE Steering Committee, the HERITAGE Advisory Board, and NHLBI. The priority shifted to the recruitment of trios and pairs of first degree relatives by descent in Black communities at all HERITAGE sites. This revised approach worked well in the end, as we were able to enroll 326 Black adults from 117 families (Table 1). It has been brought to our attention that the description 27 yr ago of the challenge we were facing in the recruitment of families of African ancestry in the first HERITAGE paper devoted to aims, design, and measurement protocols of the study (8) was offensive to some. We agree, and in retrospect, we should have used a more sensitive description of the situation.
The initial screening included a health history, physical examination, a resting electrocardiogram (ECG), and an exercise test with ECG monitoring. At baseline, participants completed a health questionnaire, including habits relating to smoking and alcohol consumption; the ARICBaecke Physical Activity Questionnaire (9,10); the Willett Food Frequency Questionnaire to assess usual food nutrient patterns (11); the Minnesota Eating Pattern Assessment Tool (EPAT) to evaluate dietary fat sources (12,13); a menstrual history; and a detailed family history questionnaire (Table 2). The health habit and EPAT questionnaires were repeated during midtraining (week 10). Participants were instructed at the beginning and at midtraining to not change their baseline health habits. The health habit and EPAT questionnaires were administered again at the end of training (week 20).
TABLE 2.
List of the concomitant and behavioral variables available in the HERITAGE Family Study.
| Phenotype | Baseline | Midtraining | Posttraining |
|---|---|---|---|
| Age, sex, and ethnicity | x | ||
| Medical history: previous diseases and physical examination | x | ||
| Personal history: medications, smoking, alcohol consumption, level of education, occupation, sleeping habits | x | x | x |
| Family history: myocardial infarction, other cardiovascular disease, stroke, hypertension, hypercholesterolemia, obesity, diabetes, cancer | x | ||
| Menstrual cycle: age at menarche, menstrual status (premenopausal and postmenopausal), number of pregnancies, babies over 9 lb, estrogen, and PROG replacement | x | x | |
| Physical activity habits: leisure time index, work index, sports index | x | ||
| EPAT: Score section I and section II | x | x | x |
| Food frequency questionnaire | x | ||
| Anxiety levels | x | x |
Panel of Tests and Measurements
Anthropometric and body composition measurements were made before and after training, as described elsewhere (14). Measurements included standing height, body mass, skinfold thicknesses at eight different sites, and waist circumference (see Ref. [8]). Underwater weighing (the gold standard measure of body composition at the time) was performed in the postabsorptive state before and after training to determine body density, fat mass (FM), fat-free mass, and relative body fat using sex- and ethnic-specific equations (15). Abdominal visceral and subcutaneous fat were quantified before and after training by computed tomography (CT) (16). Scans were taken between the fourth and fifth lumbar vertebrae (L4–L5 space) (14). Subjects were examined in the supine position, with arms stretched above their heads. Total and visceral (AVF) abdominal fat areas (in cm2) were calculated by delineating those areas with an electronic graph pen and then computing the adipose tissue surfaces by using an attenuation range of −30 to −190 Hounsfield units. The abdominal subcutaneous fat area was calculated as the difference between the total and visceral fat areas.
Resting blood pressure (BP) measurements were made twice on separate days before the start of exercise training and at 24- and 72 h after training. Subjects were tested before 11:00 a.m. in the postabsorptive state with no caffeine-containing beverages and tobacco products for at least 2 h before measurements. BP was determined using a properly fitted cuff connected to a Colin STBP-780 automated unit (17).
To measure plasma lipid and lipoprotein levels, blood samples were collected in the morning after a 12-h fast, kept on ice until centrifugation, and then stored at 4°C until analyzed within 10 d (18). Total cholesterol (TC) and triglycerides (TG) levels were determined in plasma and lipoprotein fractions (VLDL, HDL, LDL) by enzymatic methods using the Technicon RA-1000 analyzer plasma. Apolipoprotein (Apo) B and ApoA-I concentrations were measured, and those of LDL cholesterol (LDL-C), LDL-TG, and VLDL-ApoB were calculated. The cholesterol content of HDL2 and HDL3 subfractions was also determined. ApoE phenotype was measured by an isoelectrofocusing method. Lipoprotein lipase (LPL) and hepatic-TG lipase (HL) activities were measured in plasma obtained from 12-h fasted subjects, 10 min after intravenous injection of heparin (60 IU·kg−1 of body weight).
An intravenous glucose tolerance test (IVGTT) was performed in the morning after an overnight fast. Blood samples were collected at 16 time points over 3 h to determine plasma glucose, insulin, and connecting peptide (C-peptide) concentrations. The updated MINMOD model was used to quantify the acute response to insulin, insulin sensitivity index (Si), disposition index, and other parameters (19).
The plasma steroid hormone profile was assayed, including androstenedione (DELTA4), testosterone (TESTO), dihydrotestosterone (DHT), androsterone glucuronide (ADTG), androstane-3α, 17β-diol glucuronide, pregnenolone (PREGE) fatty acid esters, dehydroepiandrosterone and its fatty acid, progesterone (PROG), 17-hydroxyprogesterone, cortisol (CORT), aldosterone (ALDO), estradiol (E2), and dehydroepiandrosterone sulfate. Table 3 summarizes the variables measured and the posttraining timing of measurements. Three exercise cardiorespiratory fitness tests all on cycle ergometers were performed before and after the exercise program. The first test was a ramp protocol with increasing workload every 2 min leading to exhaustion and the first estimate of V̇O2max. The second test was a submaximal test with workloads of 50 W and then 60% of the power output (PO) at V̇O2max, each lasting 8 min in to obtain physiological data in steady state. The third test repeated the conditions of the preceding submaximal tests, then proceeded to a PO corresponding to 80% of V̇O2max, followed by a progressively increasing workload until exhaustion and the second estimate of V̇O2max. Resting and exercise BP, fasting insulin, glucose and C-peptide, lipid and lipoprotein profile, and steroid hormone profile measurements were performed twice before the exercise training program, as well as twice posttraining (24 and 72 h after the last exercise session), as specified in Table 3. The anthropometry, body composition, CT assessment of abdominal fat, IVGTT, postheparin LPL and HL activities, and the plasma levels of various substrates during exercise were obtained only once before and once post training. Additional traits measured in subsamples of the HERITAGE cohort were performed once before and once after the exercise program.
TABLE 3.
List of phenotypes available in the HERITAGE Family Study.
| Phenotype | Baseline | Posttraining | ||
|---|---|---|---|---|
| Test 1 | Test 2 | Test 3 | Test 4 | |
| Body fat and body composition | ||||
| Weight, height, and BMI | x | x | ||
| FM and fat-free mass (underwater weighing) | x | x | ||
| Regional fat distribution and visceral fat | ||||
| CT total, subcutaneous, and visceral fat | x | x | ||
| Sum of 8 skinfolds | x | x | ||
| Waist and hip girths | x | x | ||
| BP | ||||
| Resting | x | x | x (24 h) | x (72 h) |
| Exercise at 50 W and at 60% and 80% V̇O2max | x | x | x | x |
| Insulin and glucose metabolism | ||||
| IVGTT: Glucose, insulin, C-peptide | x | x | ||
| Fasting: Glucose, insulin, C-peptide | x | x | x | x |
| Lipids, lipoproteins, and lipases | ||||
| Postheparin lipolytic activities: LPL and HL | x | x (72 h) | ||
| Fasting lipid, lipoprotein, apolipoprotein profile | x | x | x (24 h) | x (72 h) |
| Steroids | ||||
| Panel of androgens, estrogens plus SHBG | x | x | x (24 h) | x (72 h) |
| Cardiorespiratory endurance | ||||
| Submaximal and maximal indicators of cardiorespiratory fitness | x | x | x | x |
| SV, cardiac output, and other hemodynamic indicators during exercise | x | x | x | x |
| Plasma protein, glucose, FFA, lactate at 50 W, 60% V̇O2max and 80% V̇O2max, and max | x | x | ||
| Additional phenotypes on subsamples | ||||
| Leptin (resting and exercise) | x | x | ||
| Insulin-like growth factors 1 and 2 | x | x | ||
| Homocysteine, cysteine, glutathione, cysteinyl glycine, folates, vitamins B6, and B12 | x | |||
| LDL oxidation, TBARS, hemolysis, GSH-Px, α-tocopherol, β-carotene, retinol, phospholipid fatty acids, Lp(a) | x | x | ||
| Skeletal muscle fiber types, capillary density, enzymes | x | x (96 h) | ||
| RMR | x | x | ||
24 h, 72 h, and 96 h refer to tests performed 24, 72, and 96 h after the last training session, respectively.
Three exercise tests were performed before and after training. First, a maximal cardiorespiratory fitness test was performed. It was a cycle ergometer test with increasing workload leading to exhaustion and VO2max. The data from this test were used to compute the specific HR and PO needed for the exercise prescription of each participant. The second test was submaximal and included exercising for 8 min at 50W followed by 8 min at the PO associated with 60% of VO2max. The last test repeated the conditions of the second test, followed by a workload corresponding to 80% of VO2max, and then increasing workloads until exhaustion was reached to provide a second estimate of VO2max. In the post-training period, these tests were performed at 24, 48, and 72 h following the last exercise training bout.
Inclusion and Exclusion Criteria
Age
Participants outside 17–65 yr old were excluded to reduce potential complications from maturation at the low end and aging at the high end.
Activity level
All participants were required to be “sedentary” at baseline. Sedentary was defined as no regular physical activity (occurring no more than once a week) involving an energy expenditure of ≥7 METs for subjects 50 yr and older and ≥8 METs for those younger than 50 yr over at least the previous 3 months.
Body mass index
Body mass index (BMI) was <40 kg·m−2 because of metabolic abnormalities and difficulty in exercising that is associated with extreme obesity. However, a few subjects with a BMI of 40 kg·m−2 or more were included because it was determined that they could perform the exercise and the testing program.
BP and medications
Resting BP was required to be <160/100 mm Hg for two out of three readings. Individuals on diuretic or antihypertensive drugs at the initial interview were permitted to enter the study if they were free of hypertensive complications, their personal physician permitted them to discontinue their medication(s), and their BP level met the aforementioned criteria after at least 3 months off medication. Subjects unable to be removed from their antihypertensive treatment were excluded from the study.
Absence of significant medical conditions and diseases
A detailed medical history and physical examination were conducted by a physician or a nurse practitioner under the supervision of a physician after screening. For subjects with suspicious symptoms or suggestive medical histories, their personal physicians were asked to provide additional medical information, test results, and hospital records before inclusion or exclusion. A past history and/or physical or laboratory finding of certain medical conditions required exclusion from the study (8).
Exercise Testing Protocol
Three exercise tests were administered both before and after training: maximal, steady-state submaximal, and submaximal–maximal. All tests were performed on the same cycle ergometer (Ergo-Metrics 800S from SensorMedics; different from the cycle ergometer used for the training program) in the sitting position at approximately the same time of day, with at least 48 h difference between tests at baseline. The first test was to establish the participant’s V̇O2max, as well as to verify the normality of the exercise ECG at baseline. The maximal test started at an initial intensity of 50 W for 3 min and increased by 25 W every 2 min until volitional exhaustion. For older, smaller, or less fit individuals, the test started at 40 W, with increases of 10–20 W each 2 min thereafter. This was done in to increase the number of stages they could complete before reaching maximum because the results were used to select the PO for the subsequent exercise tests (20). During the second test, participants exercised at 50 W and at 60% V̇O2max, determined from the initial maximal test, for 8 min at each PO to measure steady-state ventilation (VE), V̇O2, VCO2, respiratory exchange ratio (RER), systolic (SBP) and diastolic (DBP) BP, heart rate (HR), cardiac output (˙Q), and stroke volume (SV). During the third and final test, each participant repeated the submaximal steady-state exercise at 50 W and 60% V̇O2max of the second test, after which the PO was increased to 80% V̇O2max and then continued to increase until the subject reached volitional exhaustion. In addition, a venous catheter was inserted in the left arm to obtain blood samples at rest, during exercise at 50 W and at 60% and 80% V̇O2max, and immediately upon completion of the maximal test. Blood samples were analyzed for glucose, free fatty acids, lactate, and total proteins. Metabolic measurements and ˙Q (CO2 rebreathing technique) were determined using a SensorMedics 2900 metabolic cart, BP with a Colin STBP-780 automated BP monitor, and HR from the ECG. SV was derived from HR and ˙Q.
Exercise Training Protocol
The training protocol required a total of 60 training sessions, three per week for 20 wk. All training sessions were held on-site at the participating clinical centers. Each participant trained on a cycle ergometer (Universal Aerobicycles, Cedar Rapids, IA) using the same standardized training protocol at the four clinical centers. Training intensity was adjusted for individual differences in V̇O2max and began at the HR associated with 55% of baseline V̇O2max (derived from the first, maximal exercise test) for 30 min for the first 2 wk. The intensity or duration of the training sessions was increased every 2 wk, until subjects were working at the HR associated with 75% of baseline V̇O2max for 50 min during the last 6 wk (Table 4). The PO of the cycle ergometer was adjusted to maintain each subject’s HR at the prescribed level; this was controlled by direct HR monitoring at all times during all training sessions using the Universal Gym Mednet (Cedar Rapids, IA) computerized system. All training sessions were supervised on-site by trained personnel to ensure that the equipment was working properly, and the participants were compliant with the protocol. A total of 742 participants completed at least 95% of the training sessions (57 sessions) and were defined as “completers.”
TABLE 4.
Overview of the 20-wk training program in HERITAGE.
| Weeks | Frequency (Sessions per Week) |
Intensity (% V̇O2max) |
Duration (Minutes per Session)a |
|---|---|---|---|
| 2 | 3 | 55 | 30 |
| 2 | 3 | 55 | 35 |
| 2 | 3 | 65 | 35 |
| 2 | 3 | 65 | 40 |
| 2 | 3 | 70 | 40 |
| 2 | 3 | 70 | 45 |
| 2 | 3 | 75 | 45 |
| 6 | 3 | 75 | 50 |
aDoes not include 5-min warm-up or 3-min cool-down.
Subjects were not allowed to exercise more than four sessions per week or less than one per week and could not get ahead or fall behind by more than two sessions. Adherence was monitored several times per week. If subjects were falling behind, a plan was developed to bring them back on schedule as soon as possible. In premenopausal women, tests and measurements were scheduled during the follicular phase of the menstrual cycle. This required that some women exercised a few or several more sessions until they had reached the proper phase of their menstrual cycle.
Core Laboratories, Cell Lines, Biobank
Four core laboratories were established at Laval University in Quebec, Canada, for assaying lipids, sex steroid hormones, and glucose-insulin–related traits, and to establish cell lines. Lymphoblastoid cell lines were established for each participant to ensure a continuous source of DNA. Cell lines were obtained by transformation of human lymphocytes with the Epstein–Barr virus. Such cell lines grow well in culture, have an infinite life span, and present chromosomal stability over years (21). The procedure requires isolation of monocyte cells, transformation with the virus, and cryopreservation of transformed cell lines.
Training of Personnel and Data Quality
Study personnel were centrally trained on recruitment, measurement, exercise testing, exercise training, and data entry using a detailed Manual of Procedures. Retraining of staff on measurement protocols was implemented as needed at each clinical center under the Project Director on a yearly basis. Appropriate training of new personnel was provided when necessary. These actions were designed to maximize quality of all data acquired at each clinical center and to minimize potential unwarranted data heterogeneity.
Reproducibility, Clinical Center Differences, and Drift over Time
As part of the quality control program of HERITAGE, plans were made to quantify assay and measurement variability, within-subject variability, potential clinical site differences, and drift over time in protocol at each clinical center. As a control group was not deemed necessary by the investigators and external reviewers given the primary aim of HERITAGE, the quality control measures became particularly important and useful. The metrics of these quality control measures have been addressed in several publications (18,22–25), and they are commented upon hereinafter in the various sections of this article. Briefly, four approaches were used to assess assay and measurement reproducibility and within-subject variability. First, for all traits assayed from blood samples, the assay was repeated on split samples from the same blood draw. In addition, the assays were performed twice on 5% of all samples selected at random. Second, for all variables that were measured days apart, twice before and twice after the exercise program, reliability coefficients (intraclass correlations), technical errors (within-subject SD), and coefficients of variation (CV) (100 × SD/mean) could be computed across all subjects. Third, an additional 15 subjects meeting the inclusion criteria of the study were recruited at each clinical center for the sole purpose of being tested three times over 3 wk for most of the HERITAGE panel of tests. Fourth, four subjects (a total of eight individuals over the period) traveled to all four clinical centers in a random order across three yearly cycles under the supervision of the Project Director to take the full battery of tests and measurements at each clinical center.
Table 5 summarizes the test–retest data at baseline plus the quality control study data on subjects measured three times over 3 wk. Intraclass correlations, technical errors, and CV are presented for a subset of variables to illustrate the various measurement domains of HERITAGE. Under both designs, the three indicators of reproducibility can be considered highly satisfactory and well within values reported in the literature for these types of measurements.
TABLE 5.
Reproducibility and technical error for selected traits as derived from the test–retest of the HERITAGE protocol at baseline and from a quality control substudy performed on subjects measured three times within 3 wk across the four clinical centers.
| Heritage Family Subjects (n Varies from 279 to 745) | Quality Control Substudy (n Varies from 55 to 60) | |||||
|---|---|---|---|---|---|---|
| Intraclass Correlation | Technical Error | Coefficient of Variation (%) | Intraclass Correlation | Technical Error | Coefficient of Variation (%) | |
| Body weight (kg) | 1.00 | 0.1 | 0.2 | 1.0 | 0.7 | 0.2 |
| HR 50 W (bpm) | 0.89 | 6.1 | 4.4 | 0.79 | 6.5 | 6.1 |
| V̇O2 at 60% max (mL O2·min−1) | 0.99 | 53.4 | 3.6 | 0.98 | 62.2 | 3.5 |
| Cardiac output at 60% max (L·min−1) | 0.93 | 0.8 | 5.9 | 0.95 | 0.7 | 4.5 |
| Max HR (bpm) | 0.88 | 5.4 | 2.9 | 0.88 | 3.7 | 2.0 |
| V̇O2 max (mL·min−1) | 0.95 | 119.9 | 5.1 | 0.96 | 136.9 | 4.7 |
| Cholesterol (mmol·L−1) | 0.95 | 0.21 | 4.7 | 0.93 | 0.24 | 6.0 |
| HDL-Chol (mmol·L−1) | 0.94 | 0.06 | 6.2 | 0.94 | 0.06 | 6.0 |
| LPL (nmol·min−1·mL−1) | 0.95 | 8.1 | 15.4 | 0.95 | 7.6 | 14.7 |
| TESTO (nmol·L−1) | ||||||
| Male | 0.95 | 1.9 | 13 | 0.95 | 1.8 | 11 |
| Female | 0.90 | 0.3 | 23 | 0.92 | 0.2 | 13 |
| DHEAS (nmol·L−1) | ||||||
| Male | 0.96 | 955 | 16 | 0.94 | 868 | 13 |
| Female | 0.97 | 673 | 18 | 0.92 | 801 | 18 |
| SHBG (nmol·L−1) | ||||||
| Male | 0.97 | 4.1 | 11 | 0.98 | 3.6 | 9 |
| Female | 0.97 | 11.7 | 15 | 0.99 | 11.9 | 12 |
One aspect of the Quality Control Program was to verify periodically whether there were differences in measurements across clinical sites (24–27). No substantive differences among the four clinical centers were found. Likewise, there were no substantive differences between Blacks and Whites (23). Finally, data were interrogated across six time periods of the data collection phase to verify whether there was any evidence of drift over time (22). None was detected.
General Overview of Statistical Analyses
In general, standard statistical methods (e.g., correlations, linear regressions, general linear models, t-tests) were used to assess the associations within and between phenotypes at baseline and in response to exercise training. The specific procedures used were specific to the research questions and tests being performed. Models were run in the total sample or stratified by ethnic, sex, or age group, or post hoc analysis was used to determine differences between subgroups. In most HERITAGE individual studies, statistical significance was established at P < 0.05. Thus, for any text hereafter referring to results from previously published HERITAGE studies, particularly results related to clinical phenotypes (i.e., non-omics variables), the term “significance” refers to P < 0.05, unless otherwise noted. Moreover, if text states that a value “increased” or “decreased” without also using the word “significant,” it should be assumed the increase/decrease was significant unless otherwise noted.
EXERCISE, ADIPOSITY, AND FAT DEPOTS
The reproducibility of anthropometric and body composition measures in HERITAGE was determined in 60 men and women who completed the entire anthropometric and body composition test battery on three different days within a period of 3 wk (26). A high degree of reproducibility was observed across all traits measured and at each of the clinical centers. For instance, intraclass correlations ranged from 0.95 to 0.99 for anthropometric measures and from 0.97 to 1.00 for body composition measures, all with low technical error variance; this suggests that it would be possible to detect small changes in adiposity phenotypes in response to the endurance training program, with a view to investigate the genetic basis of these changes.
Baseline observations and effects of training
HERITAGE participants were slightly overweight, with average baseline BMI values ranging from 24.9 ± 13.9 kg·m−2 in White women to 27.9 ± 6.0 kg·m−2 in Black women (29). Significant ethnic differences were found for most adiposity phenotypes in women (but not in men), with higher values in Black women than in White women. At baseline, higher values of adiposity (sum of eight skinfolds, percent body fat, and FM) were observed in men than in women. Both before and after training, men had more AVF than women, Whites had more than Blacks, and parents had about twice as much adiposity as their offspring (14).
In response to training, there was only a marginal but significant decrease (delta values ± SE) in body weight (−0.2 ± 0.1 kg). Decreases were also observed for the sum of skinfolds (−6.2 ± 0.7 mm), waist girth (−0.9 ± 0.1 cm), percent body fat (−0.8 ± 0.1), FM (−0.7 ± 0.1 kg), abdominal subcutaneous fat (8.9 ± 1.1 cm2), AVF (−4.6 ± 0.6 cm2), and abdominal total fat (−13.5 ± 1.4 cm2) in the total sample. The greatest changes were observed for total FM and percent body fat (>3%) and AVF (6%). There were several differences in training response by sex and ethnicity but not by generation (parents vs children). For example, reductions in FM and AVF were significantly higher in men than in women. For the sum of eight skinfolds, reductions were higher in Whites than in Blacks (14). After controlling for reductions in both BMI and waist girth, there were significant reductions in FM, percent body fat, and all CT abdominal fat measurements in all sex–ethnic groups (30).
Familial aggregation studies
The topic of familial aggregation in the amount and distribution of fat was addressed (31,32). The heritability estimates for baseline AVF and FM in 86 White families reached 47% and 55%, respectively, with no sex or generation differences in the familial correlations for these measures (32). Figure 2 displays the CT AVF level adjusted for age, sex, and FM by family ranked on the basis of familial mean value. For a given level of total adiposity, there are families with low levels and others with high levels of visceral fat. The heritability of AVF adjusted for age, sex, and FM was 47% (32).
FIGURE 2.
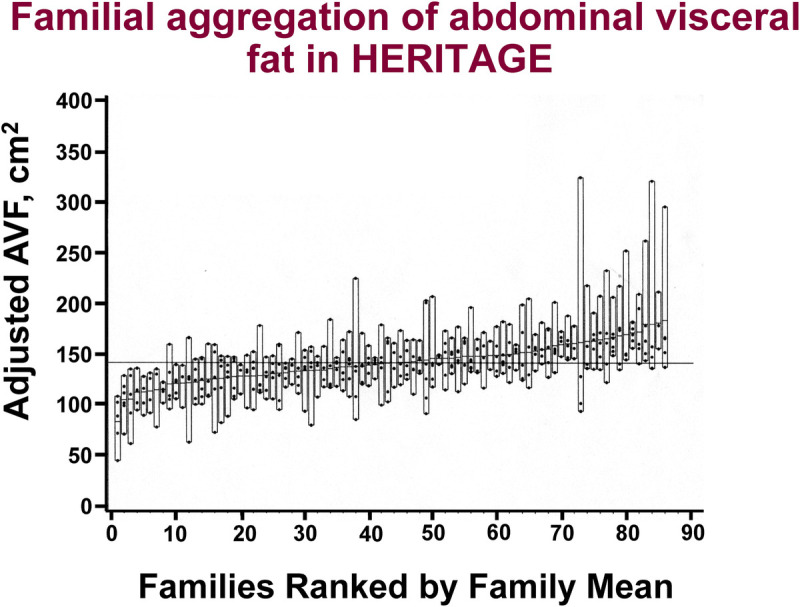
CT measured AVF level at baseline adjusted for age, sex, and FM plotted by family rank among the families of European descent in HERITAGE. Dots within each bar represent individual values for each member of the given family. The dash within each bar is the mean AVF level of each family. For a given level of total adiposity, some families exhibit a mean adjusted AVF level of about 70 to 90 cm2, whereas others have AVF levels in the range of 120 to 150 cm2. Families are ranked by the mean-adjusted AVF level. A strong familial aggregation is observed with a heritability estimate of 47%. Adapted with permission from Rice et al. (32).
Familial aggregation of the amount and distribution of subcutaneous fat (skinfold measures of trunk fat, extremity fat, subcutaneous fat, and trunk to extremity fat ratio; waist circumference) at baseline and in response to training was examined in 483 White subjects from 99 families. Significant familial aggregation was found for all phenotypes measured at baseline and for training-induced changes in trunk fat and waist circumference (31). The heritability estimates for baseline skinfold measures of fat ranged from 31% (total subcutaneous fat) to 51% (waist circumference), whereas heritability estimates for the response phenotypes were lower (range, 14%–21%) and not significant for the change in waist circumference (31).
CARDIORESPIRATORY FITNESS
This section deals with indicators of maximal and submaximal exercise capacity, including V̇O2max, maximal and submaximal PO (at 60% and 80% V̇O2max), and V̇O2 at the ventilatory threshold (VO2VT). Because exercise testing was done at four clinical centers, it was deemed important to determine the reproducibility of submaximal and maximal cycle ergometer exercise tests (as described previously). The within-subject SD for repeated V̇O2max measures was 114 mL before training, 108 mL after training in >700 subjects, 137 mL for the 60 subjects tested three times in a period of 3 wk, and 115 mL in 8 traveling subjects tested four times, once at each clinical center, within a period of 2 wk. The CV for repeated measures ranged from 4.1% to 5.0% and the intraclass correlations were >0.96 (Refs. [20,27,33,34] and unpublished data). VO2VT was also deemed satisfactorily reproducible with a test–retest correlation of 0.83 (35).
In a study on the accuracy of commonly used age-based prediction equations for maximal HR (HRmax), Sarzynski et al. (36) compared intrinsic (baseline or pretraining) HRmax obtained in 762 subjects who underwent two separate maximal exercise tests and where V̇O2max was directly measured, and compared them with the Fox (220 − age) (37) and Tanaka (208 − 0.7 × age) (38) formulas. It was observed that the measured HRmax was not highly correlated with estimated HRmax using these formulas; the standard error of the estimate for the formulas was ~11–12 bpm and largely consistent across sex and ethnicity. These findings highlight the limitations of using age to estimate HRmax.
Baseline observations
HERITAGE participants were quite representative of sedentary populations of relevant age groups. As depicted in Figure 3, V̇O2max (in milliliters of O2 per kilogram of body mass) averaged 33 in the sample of 493 Whites and 27 in 245 Blacks, with SDs ranging from about 7 to 9. A wide range of cardiorespiratory fitness in the sedentary state was observed in each ethnic group, with a range of V̇O2max values extending from 15 to 55.
FIGURE 3.
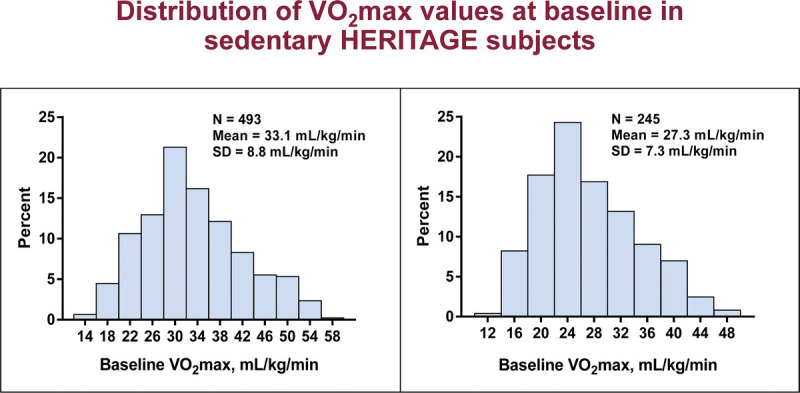
Distribution of V̇O2max values at baseline in sedentary HERITAGE subjects.
Training response
The steady-state HR and V̇O2 were measured during cycle ergometer exercise tests before and after training (39). The HR associated with 55%, 65%, 70%, and 75% of each subject’s pretraining V̇O2max was used to prescribe exercise intensity across the 20 wk of training. Using the linear relationship between HR, V̇O2, and PO, PO was predicted for each of the 60 training sessions at the respective programmed HR. The average ratio of the actual training HR to programmed HR was 0.99; that is, subjects typically trained within 1–2 bpm of their prescribed HR. One could ask whether minute-by-minute differences in the ratio of actual/predicted PO influenced the V̇O2max training response. Secondary analysis showed that the training program significantly increased mean V̇O2max across all actual/predicted PO classes (39). The minute-by-minute difference between actual and predicted PO accounted for 6% of the variance in gain in V̇O2max expressed in milliliters of O2. (33).
The training program produced significant improvements in V̇O2max (17%) and PO at 60% V̇O2max (26 W or 28%) (40). There was wide interindividual variation in V̇O2max response, with values ranging from −4.7% to +47.8% or −110 to +1100 mL O2·min−1, as illustrated in Figure 4A.
FIGURE 4.
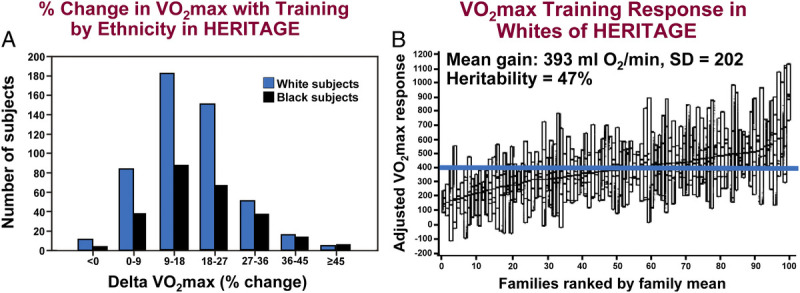
A, Distribution of the gains in V̇O2max expressed as a percentage of baseline V̇O2max level in the samples of Blacks and Whites. B, Familial aggregation of the age and sex adjusted V̇O2max changes with the exercise program in the sample of Whites of European descent. Families are ordered by the mean gain for all family members. The F ratio from the ANOVA shows that there are 2.5 times more variance between families than within families (P = 0.0001). The horizontal blue line represents the mean gain for the whole group. The heritability of the V̇O2max training response reached 47%. Adapted with permission from Bouchard et al. (41).
The effects of exercise training intensity relative to the VT on changes in PO (in watts) and VO2VT, as well as changes in V̇O2max were evaluated (35). VT could be determined at baseline and after training in 432 sedentary White and Black men (n = 224) and women (n = 208). While training started at a fixed intensity according to their V̇O2max (individual’s HR corresponding to 55% of baseline V̇O2max), there was substantial variability in the training intensity relative to VT. The range of participant’s V̇O2 at VT to V̇O2max (VT%V̇O2max = VO2VT/V̇O2max) was 34% to 83%, reflecting differences in intrinsic fitness among participants despite everyone being sedentary for at least 3 months before the study. In analyses that related baseline VT%V̇O2max to the changes in VO2VT and V̇O2max after training, individuals who trained at higher intensities (relative to VT) had greater improvements in VO2VT after training. Overall, training intensity relative to VT accounted for ~26% of the training effect on VO2VT, whereas the absolute training intensity (measured in watts) accounted for ~56% of the training effect. In contrast, there was no effect in the training intensity relative to VT on V̇O2max changes (35).
In a separate investigation, the changes in HR at a given percent of V̇O2max with training were investigated across the full HERITAGE cohort and by age, sex, and ethnicity (42). Although HR decreased at the same absolute intensity (PO in watts) with training, HR at the same relative intensity (%V̇O2max) remained constant across all subgroups. These findings in a large and diverse cohort were the first to show that frequent testing and recalibration of HR relative to %V̇O2max during a training program is unnecessary.
Age, sex, and ethnicity
The influence of age, sex, ethnicity, and initial fitness on the response of V̇O2max to training was examined in 435 Whites and 198 Blacks (287 men and 346 women) (43). As expected, the average V̇O2max in milliliters of O2 per minute increased significantly for all sex and ethnic groups, but responses in men and women and Blacks and Whites of all ages were slightly heterogeneous (Table 6). However, there were no mean differences in the gains in V̇O2max expressed as a percent of baseline levels across sex, ethnicity, and age classes. The mean gain was 17% ± 9% (43). Blacks began with lower values but had similar responses to those of Whites. Older subjects had a lower absolute change but a similar percent gain as younger subjects. There were low, average, and high responders at all ages, both sexes, both ethnic groups, and all levels of initial V̇O2max. In the aggregate, age, sex, ethnicity, and initial fitness had limited influence on V̇O2max response to standardized training in a large heterogeneous sample of sedentary Black and White men and women. It has been estimated that age, sex, ethnicity, and baseline V̇O2max explain about 11% of the variance in the response of V̇O2max to training, with age, sex, and ethnicity, each explaining about 3% and baseline level about 2% (33,44).
TABLE 6.
Maximal HR and V̇O2max training responses across HERITAGE categories of participants.
| Group | n | Post Max HR (bpm) | ΔVO2max (mL O2) | ΔVO2max (%) |
|---|---|---|---|---|
| All | 633 | 184 ± 13 | 389 ± 204 | 17 ± 9 |
| Men | 287 | 183 ± 13 | 444 ± 226 | 16 ± 8 |
| Women | 346 | 185 ± 12 | 344 ± 172 | 19 ± 10 |
| Blacks | 198 | 184 ± 13 | 366 ± 172 | 18 ± 10 |
| Whites | 435 | 185 ± 13 | 399 ± 217 | 17 ± 9 |
| 17–29 yr | 315 | 191 ± 9 | 397 ± 216 | 17 ± 9 |
| 30–49 yr | 200 | 181 ± 11 | 402 ± 189 | 19 ± 9 |
| 50–65 yr | 118 | 172 ± 12 | 244 ± 192 | 18 ± 10 |
The 633 subjects retained for this table were completers with no missing data. Adapted with permission from Skinner et al. (43).
Familial aggregation
We reported that there was familial resemblance of intrinsic V̇O2max levels (i.e., V̇O2max in the sedentary state), with 2.7 times more variance between families than within families based on 86 White families, and a heritability level of 51% for V̇O2max (in milliliters per minute) adjusted for age, sex, body mass, FM, and fat-free mass (45). A significant spousal correlation in intrinsic V̇O2max (0.14 ≤ r ≤ 0.26) was observed, consistent with results from prior studies (46,47). Importantly, the variability in V̇O2max in the sedentary state was best explained by a model in which maternal inheritance accounted for about 60% of the heritability level instead of the expected 50% (45).
Familial aggregation of three different measures of submaximal V̇O2 (V̇O2 at 60% and 80% V̇O2max) and submaximal PO (PO at 60% and 80% V̇O2max) was investigated in White participants at baseline (48). In analyses that adjusted for age, sex, and body mass, there were two to five times more variance between than within families for these baseline traits, with significant heritability estimates ranging from 48% to 74%, reflecting contributions from both genetic and shared environmental influences. These heritability estimates, as well as the individual patterns of familial resemblance (e.g., spouse, sibling, and parent–offspring correlations), were consistent with results from prior studies of submaximal performance measures from the Quebec Family Study (49) and family participants in the Canadian Fitness Survey (50).
The familial aggregation of the response of V̇O2max to training was examined in 481 adults from 98 White families. There was 2.5 times more variance between families than within families for V̇O2max response to training (41). This pattern of familial clustering of V̇O2max trainability is illustrated in Figure 4B. The most parsimonious familial correlation model yielded a maximal heritability estimate of 47% for V̇O2max response, with a maternal transmission accounting for slightly more than half of the estimate (about 28%). Significant familial aggregation was observed for VT traits, with heritability levels >50% for baseline measurements but more moderate levels for training responses (heritability ranging from 22% to 51%) (51). Heritability values ranged from 23% to 57% for the training response of other indicators of submaximal cardiorespiratory fitness (48). These findings are supported by findings from monozygotic twin studies showing significant within-pair correlations in training gains in submaximal exercise V̇O2 and PO after training (7,52).
EXERCISE AND HEMODYNAMICS
The data on exercise hemodynamics are derived from two maximal exercise tests performed at baseline, two maximal tests at 24 and 72 h after training, and submaximal exercise at 50 W and 60% V̇O2max before and after the exercise program. Measurements included VE, V̇O2, VCO2, RER, SBP, DBP, HR, ˙Q, and SV, plus plasma glucose, free fatty acids, lactate, and total proteins. Cardiovascular, respiratory, and metabolic adaptations to submaximal exercise were moderately to highly reproducible, with CV values less than 9% and intraclass correlations greater than 0.80 (20,27). Reproducibility was generally better at higher PO. Except for SBP, DBP, and RER, cardiovascular, respiratory, and metabolic responses were also highly reproducible during maximal exercise, with intraclass correlations greater than 0.86 (20,27). Day-to-day variations were modest, and reproducibility of submaximal and maximal exercise values was comparable across the four clinical centers.
Baseline familial aggregation studies
A series of studies reported familial aggregation for intrinsic (baseline) cardiovascular variables and cardiorespiratory adaptation to training. The heritability of resting SBP and DBP among 482 White participants from 99 HERITAGE families was quantified (53). Heritability estimates for baseline SBP and DBP were 54% and 41%, respectively, and are comparable to those from prior studies (50,54,55) despite having lower CV values reflecting a more uniform (i.e., sedentary) HERITAGE sample combined with repeated baseline measurements. A subsequent investigation (56) showed substantially higher maximal heritability estimates for intrinsic resting SBP among African American families (68%), a population with limited existing data (57). Familial aggregation of exercise SV and ˙Q at 60% V̇O2max was significant with moderate heritability levels (46% for both) (58).
Effects of the exercise program
Resting HR and BP responses to exercise training were investigated according to age, sex, and ethnicity. Small but consistent reductions in resting HR (−2.7 to −4.6 bpm) across subgroups were found (17). Prior studies have produced conflicting data about the effect of training on resting HR (59). In an ancillary study of HERITAGE, home-based HR monitoring was used to confirm the accuracy of resting HR recorded in the laboratory (17). Resting HR was measured on different days to determine intraindividual variability. Furthermore, and distinct from results of prior studies (60), there was no significant change in resting BP after training, which is likely explained by the lower baseline BP (118/68 mm Hg) of the participants. In contrast, SBP, DBP, and mean BP during submaximal exercise (at an absolute PO of 50 W) were generally lowered with training.
The response to training of SV, ˙Q, and arteriovenous oxygen difference (a-V̇O2 diff) during submaximal and maximal exercise was also evaluated (61) (Table 7). Steady-state V̇O2 at 50 W decreased only slightly (~3%) with training, likely the result of improved ergometer cycling efficiency. As expected, V̇O2 improved considerably at maximal PO (17.4%) and at 60% V̇O2 max (15.3%) with training. A significant decrease in HR was observed during exercise at 50 W (mean reduction of 11 bpm), but only a minor reduction of 4 bpm at 60% V̇O2max, with no change in HRmax. Slight increases in SV (4%) and a-V̇O2 diff (2%) were found at 50 W, which in combination with the lower HR led to a slight decrease (−5%) in ˙Q (−5%). During exercise at 60% V̇O2max, there were substantial increases in SV (11%), ˙Q (7%), and a-V̇O2 diff (6%) with training. Whereas HR at maximal exercise was unchanged, there were substantial gains in ˙Q (10%) and V̇O2max (17%). These observations are largely consistent with those from prior but smaller studies (62–65).
TABLE 7.
Changes with the exercise program in hemodynamic variables during submaximal and maximal exercise in HERITAGE participants.
| Variable | Pretraining | Posttraining | Change | % Change |
|---|---|---|---|---|
| V̇O2 at 50 W (mL·min−1) | 1029.7 | 994.9 | −34.8 | −3.2 |
| HR 50 W (bpm) | 120.2 | 108.8 | −11.4 | −9.5 |
| SV 50 W (mL per beat) | 95.9 | 99.8 | 3.9 | 4.1 |
| Cardiac output 50 W (L·min−1) | 11.3 | 10.7 | −0.6 | −5.31 |
| a-V̇O2 diff 50 W (mL per 100 mL per beat) | 9.2 | 9.4 | 0.2 | 2.2 |
| V̇O2 at 60% V̇O2max (mL·min−1) | 1423.3 | 1625.9 | 202.6 | 15.3 |
| HR 60% (bpm) | 140.1 | 135.8 | −4.3 | −3.1 |
| SV 60% (mL per beat) | 98.6 | 109.2 | 10.6 | 10.8 |
| Cardiac output 60% (L·min−1) | 13.7 | 14.7 | 1.0 | 7.3 |
| a-V̇O2 diff 60% (mL per 100 mL per beat) | 10.3 | 10.9 | 0.6 | 5.8 |
| V̇O2max (mL·min−1) | 2343.4 | 2725.9 | 382.6 | 17.4 |
| Max HR (bpm) | 184.9 | 184.3 | −0.6 | −0.3 |
| Max cardiac output (L·min−1) | 18.2 | 20.1 | 1.9 | 10.4 |
Based on >631 subjects with complete data. All training effects are significant at P < 0.05. Adapted from Wilmore et al. (61) and unpublished data.
The role of menopause and hormone replacement on cardiovascular physiological traits was also examined (66). The response of submaximal and maximal exercise cardiovascular traits in premenopausal (n = 338) women, postmenopausal women on hormone replacement therapy (HRT; n = 28), and postmenopausal women not on HRT (n = 29) were explored. In a novel finding, among all cardiovascular phenotypes examined, only baseline SBP during submaximal exercise (50 W) was significantly lower among premenopausal women than in postmenopausal women after adjustment for age. SBP during maximal but not submaximal exercise was significantly lower among postmenopausal women on HRT than those not taking HRT, a finding consistent with prior investigations (67,68). This may be due to estrogen’s vasodilatory effects at high exercise intensity. However, regular exercise-induced improvements in exercise hemodynamic traits among women are independent of age and menopausal or hormone replacement status.
Familial aggregation of response to the exercise program
No familial aggregation was observed for resting BP or HR in response to training in the full HERITAGE cohort (53). However, comparing families who had elevated BP (>135 and/or 80 mm Hg, SBP or DBP, respectively) in this largely normotensive population with those who had normal BP, there was a genetic component (35%–44%) to the variance in training-induced resting HR and SBP changes. These findings are consistent with prior works (69) and further supported by a subsequent HERITAGE investigation (70), which found that SBP and HR responses to training were characterized by genetic contributions, but DBP responses seemed to be largely independent of family lines.
The familial aggregation of training-induced changes in cardiorespiratory hemodynamic traits was considered in a series of studies. Heritability estimates for the training-induced changes in submaximal exercise V̇O2 and PO (60% or 80% V̇O2max) were significant (range of 23% to 57%), but tended to be lower than those for the baseline measures (48%–74%), as detailed previously (48). In contrast to baseline measurements, there was minimal correlation among spouses in training responses, suggesting that familial aggregation among these traits was largely due to genetic influences. The effects of the exercise program on HR and BP during submaximal exercise among 99 White families (n = 481 participants) were investigated, and heritability estimates of HR at 50 W and 60% V̇O2max were 34% and 29%, respectively, and 22% for SBP at 50 W (71). These findings were independent of the effects of age, sex, BMI, tobacco use, and baseline V̇O2max. In contrast, minimal heritability existed among DBP responses, SBP at 60% V̇O2max, and all of the cardiovascular traits among Black participants (all families with a total of 257 participants). These findings were the first published analyses of familial aggregation of submaximal exercise BP and HR responses to an endurance exercise training program but have not been replicated yet.
The association of cardiorespiratory fitness with risk of future coronary heart disease (CHD) was examined using a revised Framingham Heart Study algorithm (72). Compared with the highest tertile, the lowest tertile of V̇O2max had a 62% greater risk of future CHD. Another study found that those with V̇O2max above the group median had lower levels of FM, AVF, and total abdominal fat than those below for a given BMI (30). These data suggest that lower total adiposity and abdominal fat is a mean by which higher V̇O2max attenuates the health risks attributable to obesity.
EXERCISE, INSULIN, AND GLUCOSE METABOLISM
Fasting plasma glucose and insulin were assayed in the fasted state at baseline and 24 to 36 h after the last exercise training session. An IVGTT was performed before and 24 h after training with blood samples collected at 16 time points over 3 h to determine plasma glucose, insulin, and connecting peptide (C-peptide) concentrations, as described earlier (73). The protocol did not include an injection of insulin or tolbutamide. From the MINMOD Millenium software (19), the following traits were derived: Si, AIRg, Di, Kg, and Sg. The Si measures the ability of an increment in plasma insulin to enhance the net disappearance of glucose from plasma and is used as a measure of insulin sensitivity. The acute insulin response to glucose (AIRg) is defined as the integrated area under the insulin curve between 0 and 10 min of the IVGTT and represents a measurement of insulin response. The disposition index (Di) was calculated as Si multiplied by AIRg and measures the ability of the pancreatic beta cells to compensate for changes in insulin sensitivity. Kg is an estimate of the disappearance rate of plasma glucose calculated as the slope of glucose from 10 to 60 min postintravenous glucose injection. Glucose effectiveness (Sg) measures the ability of glucose, independent of change in plasma insulin, to increase glucose disposal and to suppress endogenous glucose output.
At baseline, mean Si was 61% higher in Whites than in Blacks and 12% higher in women than in men (74) (Table 8). Slight but clinically nonsignificant mean differences were seen in fasting glucose. Mean fasting insulin was comparable between men and women but was higher in Blacks than in Whites. The baseline AIRg was more than twice as high in Blacks as in Whites. A positive correlation between Kg and glucose area below fasting glucose concentration was observed at baseline (r = 0.49, P < 0.001). Age was significantly correlated with baseline Si, Sg, AIRg, and Kg (r = −0.11, −0.13, −0.24, and −0.33, respectively; all P < 0.01).
TABLE 8.
Measures of insulin and glucose homeostasis at baseline and posttraining in HERITAGE by sex and ethnicity.
| Variables | Total Participants (n ≥ 610) | Women (n ≥ 321) | Men (n ≥ 289) | Blacks (n ≥ 178) | Whites (n ≥ 432) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Before | After | Before | After | Before | After | Before | After | Before | After | |
| Fasting glucose (mmol·L−1) | 5.1 ± 0.6 | 5.2 ± 0.6* | 5.0 ± 0.6 | 5.1 ± 0.6* | 5.2 ± 0.6 | 5.3 ± 0.6 | 5.1 ± 0.6 | 5.3 ± 0.7* | 5.1 ± 0.6 | 5.1 ± 0.6** |
| Fasting insulin (pmol·L−1) | 70.7 ± 50.4 | 64.2 ± 39.3* | 68.5 ± 50.3 | 61.5 ± 36.0* | 73.2 ± 50.5 | 67.1 ± 42.6* | 81.9 ± 67.7 | 70.5 ± 43.3* | 66.1 ± 40.5 | 61.6 ± 37.4* |
| Si (10−4 min−1·[μU/mL]−1)a | 3.9 ± 2.9 | 4.1 ± 2.7* | 4.1 ± 2.9 | 4.2 ± 2.6 | 3.6 ± 2.8 | 4.1 ± 2.7* | 2.8 ± 2.3 | 3.1 ± 2.1* | 4.3 ± 2.9 | 4.6 ± 2.8* |
| AIRg (pmol·L−1 × 10 min) | 956 ± 1042 | 903 ± 933* | 980 ± 1143 | 948 ± 1021 | 931 ± 924 | 855 ± 827* | 1660 ± 1524 | 1570 ± 1348* | 660 ± 524 | 623 ± 457* |
| Sg (min−1) | 0.02 ± 0.01 | 0.02 ± 0.01* | 0.02 ± 0.01 | 0.02 ± 0.01* | 0.02 ± 0.009 | 0.02 ± 0.01* | 0.02 ± 0.01 | 0.02 ± 0.01* | 0.02 ± 0.008 | 0.02 ± 0.01* |
| Dib | 2713 ± 2411 | 2902 ± 2473* | 2903 ± 2571 | 3053 ± 2714 | 2510 ± 2216 | 2741 ± 2177 | 3622 ± 3398 | 4015 ± 3348 | 2329 ± 1709 | 2435 ± 1804 |
| Kg (%·min−1) | 1.70 ± 0.6 | 1.75 ± 0.6* | 1.80 ± 0.6 | 1.84 ± 0.6 | 1.58 ± 0.6 | 1.65 ± 0.6* | 1.85 ± 0.7 | 1.95 ± 0.7* | 1.63 ± 0.6 | 1.66 ± 0.6 |
Values shown as mean ± SD.
aUnits are taken from the MINMOD program. To convert value to SI units (10−4 min−1 [pmol/mL]−1), multiply by 0.167.
bDi is calculated as Si generated from the MINMOD program (10−4 min−1 [μU/mL]−1) multiplied by AIRg (pmol·L−1 × 10 min).
*P < 0.05 for difference between baseline and posttraining mean values.
**P < 0.05 for difference in mean training response value between ethnic groups.
Si, insulin sensitivity index; AIRg, acute insulin response to glucose; Sg, glucose effectiveness; Di, disposition index; Kg, glucose disappearance rate
Adapted from Boule et al. (74) and unpublished data.
Effects of the exercise program
Mean Si increased by 10% (P < 0.006) after the training program, along with a 3% (P < 0.001) decrease in AIRg, a nonsignificant 7% increase in Di, an index of glucose homeostasis and ability of beta cell to compensate for insulin resistance, a 11% (P < 0.002) increase in Sg, a 3% (P < 0.02) increase in Kg, and a 7% (P < 0.02) decrease in glucose area below fasting glucose concentration (74). Fasting plasma glucose was unchanged with training. However, among 55 subjects with impaired fasting glucose (5.6–7.0 mmol·L−1 or 100–125 mg·dL−1) at baseline, 24 became normoglycemic in response to the exercise program.
Interindividual variations in response to the exercise intervention were observed in IVGTT-derived measures (74). For instance, AIRg increased 7% in the quartile of subjects displaying the lowest baseline glucose tolerance, whereas it decreased 14% in the upper quartile. Such a diverging insulin response between the extreme quartiles of glucose tolerance warrants further investigation regarding its underlying biology. Another novel result was the significant reduction in area under the glucose curve below fasting levels (74). This suggests that training improves glucose regulation, not only by decreasing hyperglycemia but also by attenuating the risk of hypoglycemia.
In a secondary analysis, 38 HERITAGE Whites and 17 Blacks were found to have an increase in fasting insulin in response to the exercise program that was defined as adverse; that is, the increase was 24 pmol·L−1 or more, which is twice the technical error for fasting insulin (75). Using the same definition of a fasting insulin adverse response, it was found across six exercise intervention studies, encompassing 1687 subjects, that the prevalence of an adverse fasting insulin response was 8.3%. Adverse responders were found in all six studies. This is a topic that has not received sufficient attention thus far and warrants further research because of its potential implications for public health and type 2 diabetes management.
A significant interaction (P = 0.02) with sex was found for the Si training response, with the mean increase in Si larger in men (15%) than in women (5%). The mean increase in Si was larger in Blacks (16%) than Whites (8%) and smaller in normal weight (6%) than in overweight (14%) and obese (15%) participants, but these differences were not statistically significant. Similarly, there was no difference in the Si response between 272 premenopausal and 40 postmenopausal women (74). No difference between Blacks and Whites was found for the AIRg response to training. At baseline, the IVGTT area under the glucose curve below fasting levels was inversely correlated with changes in Kg (r = −0.30, P < 0.001), suggesting that those who had the lowest glucose at the end of the IVGTT had smaller improvements in glucose tolerance. Changes in AIRg were weakly but positively associated (r = 0.09–0.13, P < 0.05), whereas changes in Si were not associated with changes in body mass, waist circumference, cardiorespiratory fitness, and plasma LDL-C, HDL cholesterol (HDL-C), or TG. These findings suggest that participants who had more weight loss had a greater reduction in insulin secretion, despite not having larger changes in insulin sensitivity (74).
After the 20-wk training program, fasting plasma insulin and glucose were measured 1 and 3 d after the last exercise bout (74). Fasting glucose in 547 participants slightly increased between 24 and 72 h after the last exercise bout, a response that was larger in Blacks than in Whites. Fasting insulin was decreased by 8% 24 h after the last exercise bout, which was comparable to the mean 10% increase in Si estimated from IVGTT. Moreover, fasting insulin returned to baseline levels after 72 h. This finding is consistent with those of previous studies, suggesting that improvements in insulin sensitivity and glucose tolerance were short-lived and became undetectable 60–72 h after the last exercise session (76–78).
Familial aggregation of IVGTT-derived traits
Significant familial resemblance and heritability coefficients were observed for the baseline levels of the IVGTT-derived variables in both Blacks and Whites (79). After adjustment for age, sex, and BMI, the heritability estimates in Blacks and Whites, respectively, were 48% and 25% for Kg, 44% and 46% for AIRg, 38% and 44% for Si, and 32% and 24% for Di. Interestingly, Blacks had significantly higher heritability for overall glucose tolerance than Whites, but there was no ethnic difference in heritability estimates for insulin sensitivity or insulin secretion. No sex or generation differences were found in the familial correlations for any of the MINMOD traits (79). In the FUSION Study (a cohort of offspring and spouses of adults with type 2 diabetes), the reported heritability estimates for Di (23%) and AIRg (35%) were comparable to those from HERITAGE, whereas the heritability estimates for Si (28%), Sg (18%), and AIRg (35%) were somewhat lower in FUSION (80).
Changes in fasting insulin, Si, and Sg were characterized by significant familial aggregation in their response patterns. Specifically, familial aggregation analyses based on general linear models with adjusted data in MERLIN (81) showed that heritability estimates reached 8% for fasting insulin, 14% for Sg, and 16% for Si responses to exercise training (unpublished data). Changes induced by the exercise program in AIRg were not characterized by significant familial resemblance.
In summary, the large sample of participants in a well-controlled exercise regimen in HERITAGE provided a unique experimental context to study individual variations in the response of glucose and insulin metabolism to training. On average, training led to beneficial changes in fasting insulin levels, insulin sensitivity, and other glucose-insulin–related traits derived from an IVGTT. Fasting glucose and insulin measurements made 24 and 72 h after training indicate that, on average, the favorable changes are greatly attenuated 3 d after the last exercise session. A more beneficial response pattern was observed among participants who had the least favorable glucose-insulin profile at baseline.
TRAINING, LIPID, LIPOPROTEINS, AND LIPASES
Plasma lipid and lipoprotein levels were assayed from blood samples collected 2 d apart in the morning after a 12-h fast at baseline and, for most variables, 24 and 72 h after the last training session. Concentration values were adjusted for potential plasma volume changes resulting from the training program based on plasma total protein concentration difference between baseline and posttraining measurements. Plasma TC and TG levels were determined, and the following lipoprotein fractions were assayed by enzymatic methods: VLDL, HDL, and LDL. ApoB and ApoA-I concentrations were measured, and those of LDL-C, LDL-TG, and VLDL-ApoB were calculated. LPL and HL activities were measured in plasma obtained from 12-h fasted subjects, 10 min after intravenous injection of heparin (60 IU·kg−1 of body mass). The procedures and assays used have been described in several publications (18,82–85). Reproducibility of assays and the day-to-day variation in plasma levels have been reported in detail (18). Briefly, the day-to-day variation in lipid and lipoprotein fractions was estimated to be small, with intraclass coefficients >0.87 for repeated measurements in 379 participants for all traits except HDL3-C (0.79). Similar levels of stability were observed based on the assays performed on three samples obtained within 3 wk in 60 subjects. Again, HDL3-C had the lowest coefficient of 0.71. CV values were 5% to 8% for all traits except TG (about 20%) and VLDL-C (about 30%). However, when the assays were repeated on split samples from the same blood draw, the intraclass coefficients were >0.93, with CV values <5%, except for HDL2-C (10%). Plasma post-HL and LPL were also reliably measured with split-sample repeatability coefficients >0.95.
Baseline Observations
The baseline plasma lipid, lipoprotein, and lipase activity profiles were compared between men and women and Blacks and Whites and associated with cardiometabolic risk factors. Sex differences were examined in a series of publications in Whites (18,82,86), whereas ethnic and sex differences were examined in two other articles (29,83). In general, men had less favorable profiles than women (18,83,86) and Blacks more favorable profiles than Whites (83) (Table 9). The ethnic differences in plasma HL and LPL activities are concordant with results from previous studies (88–90). The differences in plasma lipid profile between men and women were largely explained by differences in visceral adiposity and plasma LPL activity, whereas ethnicity had only a minor contribution (29,83). Total body adiposity accounted for 26% to 36% of the variance in baseline blood lipids (29). Higher LPL activity was associated with a more favorable lipid–lipoprotein profile, whereas higher HL activity was associated with an unfavorable profile in both men and women (86). Fasting insulin was the only significant predictor of LPL activity in men (negative association), whereas menstrual status and fasting insulin (negative associations) and plasma sex hormone–binding globulin (SHBG) levels (positive association) were all independent predictors of LPL activity in women (86).
TABLE 9.
Plasma lipids, lipoproteins, and postheparin lipases in response to the HERITAGE exercise program by sex and ethnicity.
| Variables | Total Participants (n = 675) | Women (n = 376) | Men (n = 299) | Blacks (n = 250) | Whites (n = 481) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Before | After | Before | After | Before | After | Before | After | Before | After | |
| Total Ca | 171.0 ± 36.1 | 172.5 ± 35.8* | 168.0 ± 33.6 | 169.0 ± 33.4 | 174.7 ± 39.9 | 173.9 ± 39.4** | 165.9 ± 33.6 | 167.5 ± 35.7 | 174.3 ± 36.9 | 175.6 ± 36.1 |
| TGa | 110.1 ± 63.2 | 110.2 ± 67.2* | 95.9 ± 48.7 | 95.3 ± 52.3 | 127.4 ± 74.1 | 121.5 ± 70.9*,** | 92.5 ± 55.0 | 90.3 ± 58.5 | 122.1 ± 69.1 | 120.4 ± 69.9 |
| LDL-Ca | 114.4 ± 31.5 | 114.6 ± 30.8 | 115.5 ± 29.4 | 111.1 ± 28.7 | 118.0 ± 33.7 | 117.1 ± 34.0 | 111.2 ± 29.4 | 111.8 ± 29.6 | 116.2 ± 31.8 | 116.2 ± 31.3 |
| VLDL-Ca | 15.5 ± 12.6 | 15.4 ± 12.9 | 12.2 ± 8.2 | 12.1 ± 9.3 | 19.5 ± 15.6 | 18.4 ± 14.4 | 12.0 ± 11.1 | 11.6 ± 10.9 | 17.6 ± 13.2 | 17.2 ± 13.2 |
| VLDL-TGa | 69.5 ± 56.6 | 65.8 ± 56.0* | 53.5 ± 39.0 | 52.2 ± 43.0 | 88.5 ± 67.5 | 82.0 ± 64.8** | 47.8 ± 40.3 | 43.4 ± 37.7* | 78.7 ± 59.0 | 76.2 ± 59.3 |
| HDL-Ca | 41.1 ± 10.6 | 42.2 ± 11.0* | 44.3 ± 10.2 | 45.7 ± 10.7* | 37.1 ± 9.7 | 38.2 ± 10.2* | 42.3 ± 12.3 | 43.4 ± 12.9* | 40.3 ± 9.9 | 42.0 ± 10.5* |
| HDL2-Ca | 13.9 ± 7.6 | 14.7 ± 8.0* | 16.4 ± 7.6 | 17.9 ± 8.0* | 10.8 ± 6.3 | 11.5 ± 6.7*,** | 14.3 ± 9.0 | 15.9 ± 9.7* | 13.5 ± 6.9 | 14.4 ± 7.5*,*** |
| HDL3-Ca | 27.2 ± 5.2 | 27.5 ± 5.0* | 28.1 ± 5.5 | 27.8 ± 4.9 | 26.3 ± 5.0 | 26.7 ± 5.6* | 28.0 ± 5.7 | 27.6 ± 5.3 | 26.8 ± 5.1 | 27.6 ± 5.0*,*** |
| TC/HDL-Ca | 4.4 ± 1.3 | 4.3 ± 1.4* | 4.0 ± 1.1 | 3.9 ± 1.1* | 5.0 ± 1.6 | 4.8 ± 1.5*,** | 4.2 ± 1.4 | 4.1 ± 1.4* | 4.6 ± 1.5 | 4.4 ± 1.4* |
| ApoA-1a | 117.5 ± 16.7 | 119.8 ± 16.1* | 119.7 ± 17.1 | 121.8 ± 16.8* | 114.3 ± 15.8 | 117.0 ± 15.2* | 115.5 ± 16.9 | 116.8 ± 16.0 | 118.2 ± 17.0 | 121.9 ± 16.4*,*** |
| ApoBa | 84.4 ± 23.7 | 85.1 ± 23.6 | 80.4 ± 21.8 | 81.2 ± 21.8 | 88.5 ± 25.2 | 88.1 ± 25.8** | 80.9 ± 22.5 | 81.8 ± 22.7 | 86.3 ± 24.1 | 86.8 ± 23.8 |
| LDL-ApoBa | 76.7 ± 21.4 | 77.5 ± 21.4* | 73.8 ± 20.1 | 74.3 ± 19.7 | 79.8 ± 22.8 | 79.7 ± 23.5 | 74.6 ± 20.7 | 75.4 ± 21.1 | 78.0 ± 21.6 | 78.5 ± 21.5 |
| LPLb | 61.3 ± 32.2 | 68.2 ± 29.0* | 67.4 ± 33.7 | 72.2 ± 31.1* | 54.2 ± 28.8 | 63.5 ± 25.4*,** | 71.8 ± 34.2 | 76.5 ± 29.1* | 56.6 ± 30.1 | 64.3 ± 28.1* |
| HLb | 190.0 ± 74.0 | 179.4 ± 71.4* | 158.2 ± 63.9 | 150.6 ± 62.5* | 228.2 ± 68.1 | 213.4 ± 66.2*,** | 152.8 ± 65.1 | 143.4 ± 63.9* | 208.9 ± 70.7 | 196.5 ± 68.4* |
| HL·L−1PLb | 5.3 ± 8.8 | 3.6 ± 3.4* | 3.5 ± 3.9 | 2.8 ± 2.5* | 7.5 ± 12.0 | 4.5 ± 4.1*,** | 3.1 ± 3.3 | 2.3 ± 1.7* | 6.4 ± 10.2 | 4.2 ± 3.9*,*** |
To convert mg·dL−1 to mmol·L−1, divide cholesterol values by 38.7 and TG by 88.54.
amg·dL−1.
bnmol·mL−1·min−1.
*P < 0.05 for difference between baseline and posttraining mean values.
**P < 0.05 for difference in mean training response value between sex groups.
***P < 0.05 for difference in mean training response value between ethnic groups.
The distribution of the apolipoprotein composition of HDL, specifically the concentration of HDL containing apolipoprotein A-I only (LpAI) versus HDL with both apoA-I and apoA-II (LpAI/AII), was examined (82). Women had higher LpAI and lower LpAI/AII levels than men. As expected, LpAI was correlated with HDL2-C, whereas LpAI/AII was more closely associated with HDL3-C levels in both sexes. In both sexes, higher LpAI levels were associated with higher HDL-C and apoA-I and lower TC/HDL-C ratio, whereas LpAI/AII was positively correlated with TC, apoA-I, apoB, and insulin. LpAI levels were negatively correlated with body FM, waist circumference, and fasting levels of TG, apoB, and insulin in women only.
Familial Aggregation Studies at Baseline
Familial aggregation of baseline plasma lipid, lipoprotein, and lipase traits was assessed in 437 sedentary, White adults (171 parents, 266 offspring) from 86 families (85). Significant familial aggregation was observed for all age-adjusted phenotypes (TC, HDL-C, LDL-C, TG, LPL, HL), with heritability estimates of 62%, 83%, and 50% for TC, HDL-C, and LDL-C, respectively. For plasma TG levels and HL activity, the heritability estimates were 55% and 40%, respectively, but significant spouse correlations were found, suggesting common familial environment contributed to familial resemblance in addition to genetic factors. For plasma LPL activity, sex differences were found, with higher heritability in female pairs (76%) than in male pairs (30%) and opposite-sex pairs (44%).
Effects of the Training Program
The training responses of the lipid, lipoprotein, and lipase profile are summarized in Table 9. A 3.6% increase in plasma HDL-C was found in the total sample, mostly due to an increase in HDL2-C, with an associated increase of apoA-I (84). Significant reductions in plasma TG and VLDL-TG levels were observed in the 24-h samples only, reflecting a response to the last bout of exercise and not a training effect, per se. No changes were observed in TC, LDL-C, VLDL-C, and apoB. The findings for HDL-C are concordant with those from a review of 100 training studies that found changes in HDL-C ranged from 4% to 22% (91), as well as from a meta-analysis that found a modest net increase in HDL-C of 2.53 mg·dL−1 across 25 randomized clinical trials (92).
Effects of training on the lipoprotein subclass profile
The investigation of the effects of training on lipids–lipoproteins was expanded by examining changes in the lipoprotein subclass profile measured by NMR (LabCorp, Morrisville, NC) (93). Overall, the concentration of large HDL (HDL-P) and large LDL (LDL-P) particles increased with training, while the concentration of small LDL-P and total, large, medium, and small VLDL-P and mean VLDL-P size decreased with training. These changes mirrored those found in a previous HERITAGE report, where the increase in HDL-C was mostly driven by increases in the larger HDL2-C subclass and not the smaller HDL3-C subclass (84). These results were expanded in a meta-analysis of 10 exercise interventions from six studies (including HERITAGE), which found significant changes in the following lipoprotein subclasses (independent of age, sex, ethnicity, baseline BMI, and baseline trait value): decreases in the concentration of large VLDL-P, small LDL-P, and medium HDL-P and mean VLDL-P size, with significant increases in the concentration of large LDL-P and large HDL-P and mean LDL-P size (93). These results highlight that despite several studies showing no changes in some of the traditional measures of lipids–lipoproteins (e.g., TG and LDL-C did not change in HERITAGE), NMR subclass profiling revealed significant and favorable changes in several lipoprotein subclasses in response to training.
Association of plasma lipid, lipoprotein, and lipase activity responses to training, with concomitant changes in cardiometabolic risk factors
A study categorized men into four groups based on baseline TG (low/high) and HDL-C (low/high) concentrations and found that endurance training may be particularly beneficial in men with low HDL-C, elevated TG, and abdominal obesity (94). Greater fat loss, characterized by loss of FM, AVF, and abdominal subcutaneous fat, was associated with a better lipid response to training, characterized by an increase in HDL-C and decreases in TG, LDL-C, TC, and TC/HDL-C (29). In fact, a composite variate of change in body fat accounted for 21% of the variance in a composite variate of change in blood lipids in Blacks and 7–10% in Whites. Change in AVF was not as strongly associated with changes in lipids in Blacks as in Whites. Changes in lipid traits were not related to concomitant changes in V̇O2max (95). Weak but significant correlations were also found between change in submaximal HR at 50 W with change in HDL2-C (r = −0.17; significant in men, women, Whites) and LDL-C (r = 0.09, significant in men and Blacks) (40).
Although the predictors of lipoprotein subclass trait responses to training are largely unknown, a recent study (96) found that baseline levels of a circulating biomarker of liver fat called dimethylguanidino valeric acid (DMGV) were positively associated with changes in small HDL-P (β = 0.14) and inversely associated with changes in medium and total HDL-P (β = −0.15 and −0.19, respectively) and apoA-I (β = −0.14).
Ethnic and sex differences in plasma lipid, lipoprotein, and lipase activity responses to training
No differences in the HDL-C response were noted by age, sex, or ethnicity (84,87). However, Blacks experienced greater increases in HDL2-C. Relative to apoA-I, there were greater increases in women than in men, in Blacks than in Whites, and in offspring than in parents (84). At the univariate level, absolute change in FM was correlated with change in HDL-C (r = −0.23), HDL2-C (r = −0.17), and TC/HDL-C (r = 0.24) in men and with change in LDL-C (r = 0.22), TC (r = 0.19), and TC/HDL-C (r = 0.15) in women (95). Ethnicity was a significant predictor of changes in TG in both sexes.
Significant reductions in plasma HL activity and increases of LPL activity were observed in all sex and ethnic subgroups, except that the increase in LPL activity was not significant in Black men (97). Change in LPL activity was correlated with change in AVF (r = −0.17 for Whites and −0.26 for Blacks). In Whites, change in LPL activity was correlated with change in waist circumference (r = −0.18) and subcutaneous fat (r = −0.23), as well as change in TG (r = −0.20) and HDL-C and HDL2-C (r = 0.21 and 0.23). The finding of increased LPL activity associated with a decrease in TG reflects the hypothesis of an increased ability to clear TG-rich lipoproteins with regular exercise.
Variability in plasma lipid and lipoprotein responses to training
Although the mean increase in HDL-C was 3.6%, there was wide variability in responsiveness to training, ranging from a mean decrease of 9.3% in quartile 1 (mean reduction of 0.11 mmol·L−1) to a mean increase of 18% in quartile 4 (mean increase of 0.18 mmol·L−1) of the HDL-C response distribution (87). Parallel changes in HDL2-C and HDL3-C, apoA-I levels, and LPL activity were seen across quartiles of HDL-C response. A multivariate regression analysis that included baseline variables accounted for 15.5% of the variability in HDL-C response to training, with percent change in TG accounting for 8.8%, baseline HDL-C for 1.5%, baseline LPL activity for 1.2%, and baseline insulin for 1.0% (87).
The variability in lipid and lipoprotein responses to training was also highlighted in the previously mentioned study examining the prevalence of potentially unfavorable responses to training across six studies (75). Adverse responses were defined as a decrease in HDL-C ≤0.12 mmol·L−1 or an increase of TG ≥0.42 mmol·L−1. Figure 5 depicts the changes in HDL-C and TG for all HERITAGE subjects, with the individual values highlighted in red representing those with unfavorable responses. The prevalence of adverse response for change in HDL-C was 6% in Whites and 8% in Blacks and 8% in both Blacks and Whites for change in TG. Across all six studies (n = 1687), the prevalence of adverse response was 13.3% for HDL-C and 10.3% for TG. The prevalence of unfavorable responses seemed to be similar across variable doses of training.
FIGURE 5.
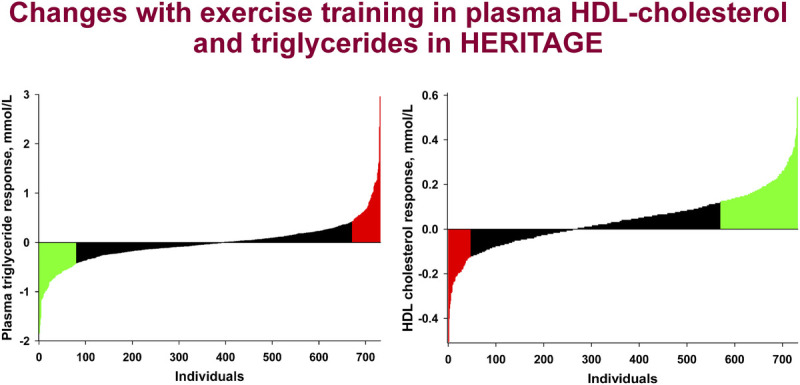
Distribution of the response of HDL-C (left panel) and TG (right panel) to the HERITAGE exercise program with potential adverse responders highlighted in red, and the most favorable responders shown in green, both defined as plus or minus 2 × the technical error for each trait. To convert mmol·L−1 of HDL-C to mg·dL−1, divide by 0.02586. To convert mmol·L−1 of TG to mg·dL−1, divide by 0.01129. Adapted with permission from Bouchard et al. (75).
Familial Aggregation Studies of Training Response
Familial aggregation was the strongest predictor of interindividual variation in plasma lipid responsiveness to training. For example, there was 1.8 times more variance between families than within families for HDL-C changes (44). Familial correlations for the training responses were significant for most lipid–lipoprotein variables, with heritability estimates ranging from 25% to 38% (98). Exceptions were for heritability levels near 60% for changes in apoB in Blacks and HDL2-C in Whites and a lack of heritability for change in LDL-C in Blacks. Heritability estimates ranged from 26% (Blacks) to 29% (Whites) for change in HDL-C and from 29% (Whites) to 32% (Blacks) for change in TG. In Whites, the heritability estimate for training-induced changes in LPL activity was 15% (99).
TRAINING, STEROIDS, AND OTHER HORMONES
Although mass spectrometry measurements are nowadays considered the gold standard for the quantification of plasma steroid levels (100), the assays used in HERITAGE were antibody based. However, the approach included prior extraction of steroids as well as high-pressure liquid chromatography separation before antibody measurements, and each step of the process was adequately controlled through calculation of recovery rates. Moreover, two blood samples were obtained at least 24 h apart at baseline, as well as two samples after training, one 24 and the other 72 h after the last exercise session. A large panel of steroid hormones or their derivatives were assayed (see Ukkola et al. (101) and He et al. (28)), including commonly measured hormones such as TESTO, E2, PROG, ALDO, and CORT. Steroid precursors such as DELTA4, PREGE, or the free, fatty-acid ester, and sulfated moieties of dehydroepiandrosterone (DHEA, DHEAE, DHEAS) were also examined, in addition to steroid metabolites such as DHT, ADTG, and androstenediol glucuronide (DIOLG). Circulating levels of the peptide steroid transporter SHBG were also examined.
Extensive quality control analyses were used to quantify the reproducibility of the steroid assays and document the day-to-day variability of hormone levels in the HERITAGE population. Moreover, blood samples were obtained in the early follicular phase of the menstrual cycle in premenopausal women before and after training. In brief, the reliability of repeated assays from split samples was highly significant, with intraclass coefficients generally >0.9 in 35 males and 25 females (28). Test–retest on 2 d apart in the whole cohort yielded coefficients of >0.8 in men and women, except for CORT (0.52) and E2 in males (0.59). All assays performed three times over 3 wk had intraclass coefficients of ≥0.70, except for ALDO in women (0.40), CORT in men (0.55), and PREGE in men (0.69).
Baseline observations
The baseline levels of all steroids and their response to training for the sample of men by ethnicity are displayed in Table 10. The steroid levels before and after training are shown in Table 11 for premenopausal women taking or not taking oral contraceptives, and for postmenopausal women with and without HRT.
TABLE 10.
Steroid hormone levels at baseline and in response to the HERITAGE exercise program in men.
| Variables | Men (n = 267) | Blacks (n = 67) | Whites (n = 200) | |||
|---|---|---|---|---|---|---|
| Before | After | Before | After | Before | After | |
| ADTG | 156 ± 85 | 158 ± 91 | 160 ± 70 | 157 ± 67 | 155 ± 89 | 158 ± 98 |
| ALDO | 0.26 ± 0.1 | 0.28 ± 0.2 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.3 ± 0.2*** | 0.3 ± 0.2 |
| CORT | 375 ± 111 | 394 ± 122* | 349 ± 99 | 364 ± 113 | 383 ± 113*** | 404 ± 123 |
| DELTA4 | 2.8 ± 1.4 | 2.7 ± 1.3 | 2.7 ± 1.1 | 2.4 ± 1 | 2.8 ± 1.5 | 2.9 ± 1.4** |
| DHEA | 15.6 ± 10.0 | 14.5 ± 9.1* | 14.7 ± 6.6 | 12.9 ± 5.7 | 15.9 ± 10.9 | 15 ± 10 |
| DHEAE | 9.3 ± 5.7 | 8.7 ± 5.1 | 10.1 ± 4.7 | 8.1 ± 3.6 | 9 ± 6 | 8.9 ± 5.5** |
| DHEAS | 5652 ± 3091 | 5593 ± 3032 | 5469 ± 2586 | 5065 ± 2247 | 5715 ± 3247 | 5770 ± 3238** |
| DHT | 2.7 ± 1.2 | 2.7 ± 1.1 | 3.2 ± 1.3 | 3.0 ± 1.3 | 2.6 ± 1.2*** | 2.6 ± 1.0 |
| DIOLG | 28.5 ± 13.7 | 29.9 ± 20.8 | 29.5 ± 12.6 | 30.8 ± 13.5 | 28.2 ± 14.1 | 29.6 ± 22.8 |
| E2 | 71.3 ± 44.1 | 72.3 ± 43.5 | 79.3 ± 39.8 | 82.2 ± 50.8 | 68.6 ± 45.2 | 69.0 ± 40.3 |
| OHPROG | 5.5 ± 2.8 | 5.5 ± 2.7 | 4.3 ± 2.2 | 4.5 ± 2.1 | 5.8 ± 2.9*** | 5.9 ± 2.9 |
| PREGE | 16.6 ± 9.5 | 15.3 ± 8.8* | 18.8 ± 9.2 | 14.9 ± 6.8 | 15.8 ± 9.6 | 15.5 ± 9.4** |
| PROG | 1.66 ± 0.8 | 1.63 ± 0.9 | 1.80 ± 0.8 | 1.70 ± 0.8 | 1.60 ± 0.8 | 1.60 ± 0.9 |
| SHBG | 39.2 ± 16.5 | 39.2 ± 16.2 | 37.6 ± 15.7 | 38.5 ± 15.4 | 39.8 ± 16.7 | 39.4 ± 16.5 |
| TESTO | 15.0 ± 6.0 | 15.1 ± 5.6 | 16.2 ± 6 | 16.2 ± 5.9 | 14.7 ± 6 | 14.7 ± 5.5 |
Units are nmol·L−1, except for E2 (pmol·L−1).
*P < 0.05 for difference between baseline and posttraining mean values.
**P < 0.05 for difference in mean training response value between ethnic groups.
***P < 0.05 for differences between ethnic groups at baseline.
TABLE 11.
Steroid hormone levels at baseline and in response to the HERITAGE exercise program in women by menopause and hormonal drug status.
| Variables | Premenopausal, No OC (n = 167) | Premenopausal, OC (n = 88) | Postmenopausal, No HRT (n = 24) | Postmenopausal, HRT (n = 22) | ||||
|---|---|---|---|---|---|---|---|---|
| Before | After | Before | After | Before | After | Before | After | |
| ADTG | 101 ± 55 | 98 ± 55 | 82 ± 41 | 84 ± 43 | 66 ± 37 | 68 ± 41 | 61 ± 45 | 58 ± 42 |
| ALDO | 0.2 ± 0.2 | 0.2 ± 0.2 | 0.3 ± 0.2 | 0.3 ± 0.2 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.2 | 0.2 ± 0.2 |
| CORT | 358 ± 134 | 414 ± 174 | 662 ± 275 | 683 ± 305 | 368 ± 129 | 401 ± 137 | 369 ± 129 | 427 ± 165 |
| DELTA4 | 2.6 ± 1.5 | 2.6 ± 1.5 | 2.5 ± 1.6 | 2.8 ± 1.3 | 1.5 ± 0.8 | 1.5 ± 0.8 | 1.3 ± 0.6 | 1.3 ± 0.7 |
| DHEA | 14.8 ± 9.6 | 14.5 ± 10.2 | 16.3 ± 11.0 | 17.0 ± 9.7 | 7.9 ± 4.8 | 8.5 ± 7.0 | 6.8 ± 4.0 | 7.2 ± 5.5 |
| DHEAE | 8.4 ± 5.4 | 7.5 ± 4.8* | 7.9 ± 5.5 | 8.2 ± 5.4 | 5.6 ± 3.7 | 5.6 ± 3.5 | 3.8 ± 2.0 | 4.5 ± 2.5 |
| DHEAS | 4023 ± 2212 | 3823 ± 2185* | 4132 ± 2271 | 4087 ± 2186 | 1882 ± 1019 | 1930 ± 1061 | 1677 ± 830 | 1709 ± 1002 |
| DHT | 0.5 ± 0.2 | 0.6 ± 0.4 | 0.5 ± 0.3 | 0.5 ± 0.3 | 0.5 ± 0.3 | 0.5 ± 0.2 | 0.4 ± 0.1 | 0.4 ± 0.2 |
| DIOLG | 15.6 ± 8.1 | 16.1 ± 9.1 | 12.5 ± 5.8 | 13.4 ± 7.2* | 12.1 ± 6.5 | 11.8 ± 5.5 | 9.4 ± 5.8 | 9.2 ± 5.8 |
| E2 | 130 ± 106 | 137.1 ± 126 | 125 ± 221 | 105 ± 127 | 45 ± 55 | 36 ± 51 | 333 ± 389 | 325 ± 415 |
| OHPRG | 2.3 ± 1.7 | 2.7 ± 3.4 | 2.1 ± 1.4 | 2.1 ± 1.3 | 1.7 ± 0.9 | 1.6 ± 0.9 | 1.2 ± 0.8 | 1.2 ± 0.8 |
| PREGE | 15.2 ± 9.0 | 14.5 ± 8.7 | 13.4 ± 9.4 | 14.3 ± 10.7 | 11.2 ± 6.3 | 10.9 ± 7.3 | 6.7 ± 3.0 | 7.3 ± 4.1 |
| PROG | 1.6 ± 0.8 | 1.5 ± 0.8 | 1.4 ± 0.7 | 1.4 ± 0.7 | 0.9 ± 0.6 | 0.8 ± 0.5 | 0.7 ± 0.5 | 0.7 ± 0.4 |
| SHBG | 66.1 ± 27.0 | 68.4 ± 34.3 | 113.3 ± 58.2 | 109 ± 55.2 | 65.9 ± 24.4 | 62.7 ± 25.6 | 118.6 ± 61.8 | 113.2 ± 59.0 |
| TESTO | 1.3 ± 0.5 | 1.3 ± 0.5 | 1.4 ± 0.6 | 1.4 ± 0.6 | 1.4 ± 1.2 | 1.1 ± 0.4 | 1.1 ± 0.5 | 1.0 ± 0.5 |
Units for all variables are nmol·L−1, except E2 (pmol·L−1).
*P < 0.05 for difference between baseline and posttraining mean values.
OC, oral contraceptives.
Adapted from He et al. (28) and unpublished data.
Baseline levels of plasma steroids were examined in several publications (101,102,104). As summarized by Ukkola and colleagues (101), age was a predictor for most steroid hormones, with the strongest relationship observed for DHEAS (r = −0.39), whereas BMI was also associated with the variability of several measurements, including SHBG (r = 0.20) and TESTO (r = 0.12). When age and BMI were taken into account, ethnicity still contributed significantly to variations in CORT and DHT (for men and women) and in PROG for women. However, ethnicity had a much lower influence on plasma steroid levels than age and BMI. In addition, caloric and nutrient intakes, and smoking influenced steroid hormone variability, but their contributions were minor once age, BMI, and ethnicity were taken into account (101).
The well-documented decline in androgen levels with age observed in males (105,106) was also observed in men of HERITAGE. Interestingly, the concomitant age-related increase in total body FM explained an important part of the variation in steroid levels in men, particularly for TESTO levels (104). These results are consistent with those from studies showing an important contribution of adiposity indices to variations in steroid hormone levels in men (107), women (108) and in men and women of HERITAGE (102). In another HERITAGE report, baseline sex hormone and SHBG levels were examined in men and women with an emphasis on postheparin lipolytic enzyme activities (86). Activities of LPL and HL are known to be sensitive to androgens and estrogens, which likely explains part of the sex differences in blood lipids, particularly for HDL-C (109). TESTO was found to be an independent predictor of HL activity (r = 0.13), consistent with the known effects of androgens in decreasing HDL-C levels. In women, SHBG levels, which are regulated in part by androgens and estrogens, were correlated with HL activity (r = −0.39), independent of adiposity. This study showed that the sensitivity of lipase activities to sex hormones was confirmed in a large sample of men and women (86).
At baseline, increasing V̇O2max was related to higher androgen levels (TESTO and DHT), as well as higher SHBG levels after adjustment for age and ethnicity (28). In women, no significant hormonal variations were observed as a function of V̇O2max, except for the free androgen index (TESTO/SHBG × 100) which was lower across increasing baseline V̇O2max quartiles. However, the overall strength of the associations between baseline hormones and V̇O2max was relatively low.
Familial aggregation at baseline
Baseline hormone and SHBG levels were examined as a function of familial aggregation (110–114). The factors examined in these analyses were DHEA, DHEAE and DHEAS (111,113), SHBG (110,112), and androgens and glucuronide conjugates (114). Regarding DHEAS, Whites showed a heritability estimate of 58% at baseline. Segregation analyses suggested a major Mendelian locus affecting baseline DHEAS accounting for about half of the trait variance (111). Ethnicity-related differences in familial aggregation patterns were also examined for DHEAS, and a comparable level of heritability of 66% was found (113).
Baseline levels of SHBG differed by sex, with higher levels in women, whereas SHBG levels were higher in fathers than in sons (111). Familial aggregation analyses of SHBG levels are consistent with the idea that multiple factors regulate the secretion of this hepatokine (115). A significant amount of the variance in SHBG was accounted for by TESTO, E2, age, BMI, and insulin. Nevertheless, the heritability estimate was 64%, independent of these factors. Analyses in Blacks revealed similar heritability levels (110).
CORT levels were significantly different between sex and generation groups at baseline in Whites, with higher values in females and offspring (116). In Blacks, lower baseline CORT levels were found in mothers than in fathers and higher levels in offspring and in daughters than in sons. The heritability of CORT was lower than for other steroids examined (116). Interestingly, familial cross-trait correlations and bivariate segregation analyses generated consistent results, suggesting common familial components underlying CORT and body fat covariation caused by common polygenic determinants (117). Such genetic pleiotropy was not found for the association of SHBG and adiposity indices (118). Evidence of genetic pleiotropy between CORT levels and adiposity phenotypes agrees with the well-known coregulation of adiposity, body fat distribution, and glucocorticoid homeostasis (119,120).
Finally, familial resemblance of circulating androgens and androgen metabolites at baseline was also examined (114). There were sex and generation differences at baseline, with parents having lower levels than offspring and males higher levels than females in both generations and ethnicities. This analysis again revealed high heritability estimates for most of the androgens and metabolites examined. For example, heritability levels in Whites reached 69% and 87% for TESTO and DHT, respectively, which were similar in Blacks (70% and 73%, respectively). The androgen metabolite DIOLG has been the focus of many studies in the field of obesity and body fat distribution (121,122). The relatively high heritability estimates obtained for this steroid (74% and 62% in Whites and Blacks, respectively) need to be considered in light of results from more recent twin studies indicating that differences in circulating DHT between lean and heavier co-twins are related to increased expression of androgen-metabolizing enzyme aldo-ketoreductase 1C2 in adipose tissue (123,124). Genome-wide linkage scans were performed in HERITAGE to identify loci influencing the various moieties of DHEA (125), as well as other steroids and SHBG (126). The results from these studies are of particular interest in the context of the growing body of evidence supporting the existence of an elaborate set of steroid-converting enzymes in human adipose tissues (122).
Effects of the exercise program
The training program had only marginal effects on fasting plasma levels of steroid hormones and SHBG, as evidenced by data in Tables 10 and 11. In males, only CORT had a significant increase with training, whereas DHEA and PREGE showed a decrease (Table 10). In premenopausal women not taking oral contraceptives, DHEAS decreased significantly with training, whereas DIOLG increased in those taking oral contraceptives (Table 11). No significant change in any steroid was observed in postmenopausal women with or without HRT, a finding that may have been impacted by the relatively small sample size of women in each group.
The impact of baseline steroid levels on changes in cardiorespiratory fitness and body composition in response to training was examined in HERITAGE (28,102,103). Sex hormones were not related to the change in V̇O2max, suggesting that high baseline endogenous androgen levels do not predict higher responses to endurance exercise training in previously sedentary adults (28). No association was detected between baseline steroid hormone levels and changes in adiposity traits (102) or the improvements in resting and exercise BP (103). In another report, the impact of menopausal status and HRT on exercise hemodynamics during exercise was examined (66). Menopausal status was found to influence training adaptation of SBP, as menopausal women improved more. Age was a major covariate of variability in hemodynamic parameters (66). Postmenopausal women showed less improvement in SBP during submaximal exercise with the training program when taking HRT (66). Among other findings, no sex, generation, or ethnic differences were observed for mean change in SHBG with training (110). In Blacks, no differences between sexes and generations were observed for CORT training response.
Familial aggregation of responses to the training program
An analysis based on White families showed that changes in plasma DHEAS was characterized by a nonsignificant heritability level of 30% (113). The training response of SHBG level was also characterized by very low heritability estimates (110,112). Segregation analyses based on the White families provided evidence for a major locus effect influencing the CORT response to training; this was not detected in Blacks, perhaps because of the smaller sample size (116).
TRAINING AND OXIDATIVE STRESS
Excess reactive oxygen species production is thought to contribute to the development of atherosclerosis and cardiovascular disease. Oxidative alterations of LDL particles are thought to play a critical role in atherosclerosis and in inflammatory processes. Hemolysis induced by reactive oxygen species is an indicator of the overall antioxidant capacity. An ancillary study investigated these areas on a HERITAGE subsample at baseline and in response to training, with an emphasis on total homocysteine (tHcy), LDL oxidation, erythrocyte glutathione peroxidase activity (GSH-Px), retinol, and erythrocyte resistance to hemolysis (127).
Baseline characteristics
Significant sex and ethnic differences in tHcy were observed before training in the full cohort (n = 816). tHcy was highest in Black men (11.0 ± 7.2 μmol·L−1), followed by White men (9.4 ± 3.7 μmol·L−1), Black women (8.5 ± 4.2 μmol·L−1), and White women (7.7 ± 2.7 μmol·L−1) (128). Although results from cross-sectional studies corroborate the finding of greater tHcy in males, evidence for ethnic differences is mixed, with some studies reporting White males having the highest tHcy levels (129) and others reporting no significant differences by ethnicity (130). Sex differences did not persist in measures of oxidative stress other than tHcy.
Other measures of oxidative stress were examined in a subset of 146 subjects from 28 two-generation, White, nuclear families from the Quebec clinical center (Table 12). A significant proportion (31.6%) of the variation in LDL oxidation lag time was accounted for by LDL-C, plasma α-tocopherol, serum uric acid, and plasma phospholipid oleic acid (127). Baseline plasma α-tocopherol, β-carotene, and retinol levels, as well as GSH-Px, were all significantly higher in parents than in children of either sex, whereas plasma glutathione was higher in children than in parents (127). The relationship between oxidative stress and aging is well established, particularly mitochondria oxidative stress commonly considered as one of the drivers of the aging processes (131). Thus, the observed differences in baseline markers of oxidative stress across generations were expected and support an association between aging and elevated oxidative stress.
TABLE 12.
Baseline, training response, and familial aggregation of measures of oxidative stress in Whites of the HERITAGE Family Study.
| Oxidative Stress Trait | Baseline | Training Response | ||||||
|---|---|---|---|---|---|---|---|---|
| Familial Aggregation | Familial Aggregation | |||||||
| n | Mean (SD) | F | H 2 | Mean (SD) | Training Effect (P Value) | F | H 2 | |
| Plasma glutathione (μmol·L−1) | 142 | 4.5 (1.5) | 2.56 | 0.44 | 0.04 (1.8) | NS | 3.20 | 0.46 |
| Erythrocyte antioxidant enzyme GSH-Px (U·g−1 hemoglobin erythrocyte) | 109 | 57.2 (15.4) | 6.09 | 1.00 | −0.5 | NS | NS | 0.002 |
| Resistance to hemolysis (C50-AAPH; mmol·L−1) |
106 | 75.9 (14.0) | 3.06 | 0.36 | 3.6 (16.1) | 0.02 | 2.08 | 0.35 |
| LDL oxidation lag phase (min) | 71 | 58.4 (23.3) | 3.12 | 0.38 | 6.2 (38.9) | NS | 2.97 | 0.84 |
| LDL oxidation propagation rate (nmol·mg−1 LDL·min−1) | 71 | 8.9 (2.6) | 2.02 | 0.31 | 0.3 (4.0) | NS | 2.12 | 0.44 |
| Plasma TBARS (nmol MDA·mL−1) | 138 | 1.9 (0.7) | 3.19 | 0.43 | −0.04 (0.9) | NS | 3.15 | 0.51 |
Mean values and training response P values have not been adjusted for any covariates. F ratio compares the between-family with the within-family variances and thus provides an estimate of familial resemblance. H2 is the heritability coefficient.
NS, not significant (P > 0.05).
Adapted with permission from Blache et al. (127) and unpublished data.
Effects of the training program
tHcy did not change in response to training. However, a subgroup analysis revealed differential responses by baseline tHcy levels and ethnicity. Exercise training resulted in a significant reduction of tHcy (−3.5 ± 7.0 μmol·L−1) in subjects with elevated baseline tHcy (≥15 μmol·L−1), whereas a significant increase (+0.3 ± 1.5 μmol·L−1) was observed in subjects with normal baseline levels of tHcy (<15 μmol·L−1) (128). Furthermore, training resulted in a nominal decrease in tHcy in Black males and a nominal increase in White males (P = 0.04 between Blacks and Whites) (128). In the ancillary study of Whites from the Quebec center, the only measure of oxidative stress that significantly changed with training was erythrocyte resistance to hemolysis, with no change in the other markers (Table 12). Interestingly, smoking status was a significant modifier of the beneficial erythrocyte response to training. Instead of beneficial effects of training, erythrocytes of smokers had a greater susceptibility to hemolysis after training (127). In sex-specific analyses, the percent increase in total glutathione was significant in both men and women, whereas erythrocyte resistance to hemolysis and plasma α-tocopherol was improved with training in women only (127).
Familial aggregation profile
There was significant familial aggregation of measures of oxidative stress at baseline and in response to training (Table 12). Specifically, LDL oxidation lag time and propagation rate, erythrocyte resistance to hemolysis, plasma thiobarbituric acid–reacting substance (TBARS), and plasma glutathione all exhibited significant familial aggregation at baseline and after training. Erythrocyte GSH-Px showed significant familial aggregation at baseline, but not in response to training (127). The baseline traits showed two to six times more variance between families than within families with maximal heritability estimates ranging from 31% to >90%, whereas maximal heritability estimates for training response traits ranged from 35% to 84%.
To conclude, subjects with elevated tHcy at baseline had significantly reduced tHcy levels with training. Although the clinical utility of tHcy for assessing CHD risk has been called into question (132), some evidence suggests that the observed reductions in tHcy may be enough to reduce CHD risk in those with hyperhomocysteinemia (133). The other beneficial effect of training was increased resistance to hemolysis of erythrocytes in women, which was significantly attenuated by smoking. Recently, distinct hematological patterns were found in smokers, and the differences were associated with greater risk of CHD in smokers (134). Similar results were reported in a cohort of over 100,000 Danish individuals where smoking was causally associated with increased blood leukocytes, neutrophils, lymphocytes, and monocytes. In addition, they suggest that smoking results in decreased erythrocyte life span due to increased hemolysis (135). Thus, cessation of smoking is likely necessary to improve erythrocyte fragility through training.
Overall, training only modestly improved markers of oxidative stress. These results were somewhat surprising, as regular exercise has been previously shown to decrease systemic oxidative stress and attenuate oxidative damage throughout the body (136,137). A potential reason for the apparent disagreement between the HERITAGE findings and those from other studies could be use of animal models in many studies examining training and oxidative stress that may not be generalizable to humans. Furthermore, training may reduce oxidative stress in older individuals only (136); thus, the HERITAGE cohort with its age inclusion limited to 65 yr may not represent the ideal target population for exercise interventions aimed at improving antioxidant defenses. Further research in diverse populations is needed to elucidate the impact of training on markers of oxidative stress.
EXERCISE AND SKELETAL MUSCLE BIOLOGY
Biopsies of the vastus lateralis muscle were obtained before and after training in a subset of 78 subjects from 19 White families (138). This subsample had a mean age of 33 yr and a mean BMI of 25 kg·m−2. Fiber type distribution, fiber areas, capillarity, and enzyme activities were assayed as described previously (139–141). This HERITAGE ancillary study was led by Dr. Jean-Aimé Simoneau, who passed away shortly after the completion of data collection.
Baseline observations
Means and SDs for the proportion of type 1, 2A, and 2B fibers were close to the values previously reported from the same laboratory in samples of 203 adult females and 215 adult males ranging in age from 16 to 33 yr (142). The proportions of type 1 fibers were 43.2% (SD, 11.8) and 20.0% for type 2B (138). Mean fiber type areas were ~20% larger in the HERITAGE subsample than in the 10-yr younger cohort of 418 adults reported by Simoneau and Bouchard (142). Muscle enzyme activities were characterized by considerable heterogeneity, with CV values of 20% to 25% for creatine kinase (CK), phosphorylase (PHOS), hexokinase (HK), phosphofructokinase (PFK), glyceraldehyde phosphate dehydrogenase (GAPDH), carnitine palmitoyl transferase (CPT), 3-beta-hydroxyacyl CoA dehydrogenase (HADH), citrate synthase (CS), and cytochrome c oxidase (COX) (143). Variability of the enzyme activities in the HERITAGE sample was slightly lower than in the larger cohort of younger sedentary adults (142). The more glycolytic type 2B fibers were characterized by a lower capillary density than in muscle type 1 and 2A fibers.
Effects of the training program
As depicted in Table 13, there were increases in the proportion of type 1 fibers (about 3%) and decreases in type 2B fibers with no change in type 2A (138). The significance of these changes is unclear, as they are well within the confidence intervals of the fiber type assessment procedures (139). There were no changes in the surface areas of the various fiber types. The number of capillaries surrounding each fiber type increased with training, but the ratio of capillaries to fiber type areas decreased for all fiber types. Muscle enzyme activities were substantially and significantly increased after training, except for GAPDH and LPL.
TABLE 13.
Effects of the endurance exercise program on vastus lateralis muscle fiber type, capillarity per fiber, and enzyme activities, and the familial aggregation of the training response.
| Training Effect | Familial Aggregation of Training Response | ||||
|---|---|---|---|---|---|
| Muscle Trait | Pretraining | Training Response | P | F Ratio | P |
| % type 1 | 43.2 (11.8) | 3.5 (9.4) | 0.003 | 0.62 | NS |
| % type 2 A | 36.8 (8.9) | 1.8 (9.9) | NS | 1.81 | 0.057 |
| % type 2B | 20.0 (11.2) | −5.4 (10.3) | 0.0001 | 1.13 | NS |
| Type 1 caps | 4.58 (0.81) | 0.76 (0.72) | 0.0001 | 1.65 | 0.09 |
| Type 2A caps | 4.40 (1.00) | 0.63 (0.77) | 0.0001 | 1.75 | 0.07 |
| Type 2B caps | 3.47 (1.00) | 0.55 (0.68) | 0.0001 | 0.99 | NS |
| CK | 389 (68) | 27 (69) | 0.0009 | 3.97 | 0.0001 |
| PHOS | 19.9 (4.9) | 1.6 (3.3) | 0.0001 | 2.14 | 0.017 |
| HK | 2.5 (0.5) | 0.4 (0.5) | 0.0001 | 4.01 | 0.0001 |
| PFK | 57.3 (13.9) | 4.9 (10.6) | 0.0002 | 1.85 | 0.043 |
| GAPDH | 422 (94) | 7 (66) | NS | 2.39 | 0.007 |
| LPL | 0.87 (0.27) | −0.01 (0.28) | NS | 1.35 | NS |
| CPT | 0.12 (0.02) | 0.05 (0.02) | 0.0001 | 2.47 | 0.006 |
| HADH | 16.2 (3.0) | 4.6 (3.0) | 0.0001 | 2.13 | 0.017 |
| CS | 11.5 (2.1) | 5.7 (2.5) | 0.0001 | 2.73 | 0.002 |
| COX | 7.0 (1.7) | 2.9 (2.5) | 0.0001 | 2.07 | 0.021 |
Mean and (SD). F ratio of the between- to within-family variance components. Number of subjects ranged from 60 to 78 depending on the muscle trait. The Vmax of enzymes was expressed in micromoles of substrate per gram of wet weight tissue per minute. Total muscle LPL was expressed as nanomicromoles of FFA transformed per gram of wet weight tissue per minute.
NS, not significant (P > 0.05); caps, number of capillaries per fiber; CK, creatine kinase; PHOS, phosphorylase; HK, hexokinase; PFK, phosphofructokinase; GAPDH, glyceraldehyde phosphate dehydrogenase; LPL, lipoprotein lipase; CPT, carnitine palmitoyl transferase; HADH, 3-beta-hydroxyacyl CoA dehydrogenase; CS, citrate synthase; COX, cytochrome c oxidase.
Familial aggregation profile
The muscle biopsy data came from 19 nuclear families. Considering that there is a dearth of data on the genetic epidemiology of skeletal muscle phenotypes, HERITAGE offered an opportunity to test whether there was familial aggregation in the sedentary state and in response to training (138). There was no consistent evidence for familial aggregation of fiber type distribution or fiber type areas in the sedentary state, with the exception of type 1 fibers. However, significant familial resemblance was observed for the number of capillaries and the number of capillaries per surface area around fiber types 1 and 2A. No evidence of family lines was detected in the observed variability of fiber type distributions, surface areas, and capillarity in response to training, although there were trends of familial aggregation for number of capillaries surrounding types 1 and 2A fibers. In contrast, strong familial aggregations for muscle enzyme activities (CK, PHOS, HK, PFK, CPT, HADH, CS, and COX) were observed at baseline (except GAPDH and LPL) and in response to the exercise program, with the exception of total muscle LPL (143). Studies with much larger sample sizes would be needed to provide more definitive quantification of the variability within and between families, along with relevant heritability estimates.
TRAINING AND RESTING METABOLIC RATE
Research is equivocal on the effects of endurance training on RMR (144–152). Seventy-seven participants from the Texas clinical center (40 men) age 17 to 63 yr completed two RMR measurements by indirect calorimetry before training and one measurement each 24 and 72 h after the last exercise bout. Identical RMR testing procedures were used for each test (153). Reliability was high (intraclass correlation ≥0.90) between the two pretests, including resting V̇O2, resting energy expenditure, HRsleep, and HRRMR.
There was no change in RMR due to training at 24 or 72 h after training. Adjusting RMR values for age, sex, fat-free mass, and pretraining V̇O2max did not affect the results (Table 14). There was a large pretraining to posttraining increase in V̇O2max (17.9%) and small but significant differences in HRsleep, (61.0–59.3 bpm), HRRMR (60.7–58.7 bpm), percent body fat (29.2%–28.2%), FM (23.1–22.5 kg), and fat-free mass (54.3–55.0 kg) in this subsample.
TABLE 14.
RMR before and after the HERITAGE exercise training program.
| Resting Metabolic Rate | Pretraining | Posttraining | ||||
|---|---|---|---|---|---|---|
| Trial 1 | Trial 2 | Mean | Trial 3: 24 h | Trial 4: 72 h | Mean | |
| V̇O2 (mL·min−1) | ||||||
| Men | 253 ± 34 | 249 ± 32 | 252 ± 33 | 255 ± 34 | 253 ± 34 | 255 ± 33 |
| Women | 194 ± 29 | 196 ± 29 | 196 ± 31 | 198 ± 33 | 200 ± 32 | 199 ± 31 |
| Total | 226 ± 43 | 224 ± 42 | 225 ± 42 | 227 ± 44 | 228 ± 42 | 228 ± 42 |
| MJ·d−1 | ||||||
| Men | 7.32 ± 0.97 | 7.25 ± 0.94 | 7.31 ± 0.95 | 7.37 ± 0.98 | 7.35 ± 0.98 | 7.38 ± 0.97 |
| Women | 5.62 ± 0.83 | 5.69 ± 0.91 | 5.70 ± 0.89 | 5.74 ± 0.95 | 5.80 ± 0.92 | 5.78 ± 0.89 |
| Total | 6.56 ± 1.24 | 6.51 ± 1.21 | 6.54 ± 1.22 | 6.56 ± 1.26 | 6.61 ± 1.23 | 6.61 ± 1.23 |
| kJ·kg −1 FFM·d−1 | ||||||
| Men | 118.0 ± 11.7 | 117.6 ± 14.2 | 118.0 ± 12.6 | 117.1 ± 12.1 | 116.7 ± 14.6 | 117.2 ± 13.0 |
| Women | 125.9 ± 13.4 | 126.8 ± 14.2 | 127.2 ± 13.4 | 125.5 ± 13.4 | 128.9 ± 13.4 | 127.2 ± 12.1 |
| Total | 121.3 ± 13.0 | 121.3 ± 14.6 | 121.8 ± 13.8 | 121.3 ± 13.4 | 121.8 ± 15.1 | 121.8 ± 13.4 |
Mean ± SD. Maximum number of subjects: 40 men and 37 women. No significant effect of the training program was found on RMR expressed various ways.
Reproduced with permission from Wilmore et al. (153).
The effect of aerobic training has continued to be a topic of interest, with most studies since the 1998 HERITAGE publication showing that aerobic training does not affect RMR (154–166). One study showed that White women did not experience a change in RMR after 6 months of training, whereas Black women saw a decrease (167). Another study showed an increase at 7 wk, but a return to pretraining levels after 14 wk of training (168). Three studies have shown an increase in RMR with aerobic training (169–171). There is no clear explanation for these apparent differences.
CANDIDATE GENE AND GWAS STUDIES
Genetic studies of exercise-related traits at the genome level in HERITAGE began in 1997 and have been characterized by several phases that closely matched advances in genomic technologies. The first wave of studies focused on candidate genes. It was followed by a series of reports based on an unbiased search of the genome using panels of microsatellite markers and linkage analytical strategies, which led to positional cloning and fine mapping of regions of interest. In a next phase, sets of candidate genes were uncovered by probing the variability in skeletal muscle gene expression, which were then submitted to further investigation. Finally, in more recent times, millions of single nucleotide polymorphisms (SNPs) covering the whole genome are being used in association studies of HERITAGE traits. A brief summary of the findings across these approaches follows.
Candidate gene studies
A total of 37 studies published between 1997 and 2010 tested associations between 34 candidate genes and a wide variety of traits measured at baseline and in response to training. The phenotypes investigated included cardiorespiratory fitness traits (maximal and submaximal V̇O2 and PO), hemodynamic-related traits (resting and submaximal exercise HR, SBP and DBP, and submaximal SV and ˙Q), adiposity-related traits (BMI, sum of eight skinfold thicknesses, FM, percent body fat, and abdominal fat), plasma lipid and lipoprotein traits (TC, TG, LDL-C, HDL-C, and apolipoproteins), and insulin and glucose metabolism traits (fasting glucose, insulin and C-peptide levels, Si, AIRg, and Sg). In most studies, the association with baseline and response traits was tested in Black and White subjects, and the associations tended to be ethnic specific. Table 15 summarizes the findings by indicating whether an association was reported at baseline (B) or in response to training (R) for any given phenotype in each trait category.
TABLE 15.
Summary of candidate gene studies with baseline (B) and response (R) traits in HERITAGE.
| Gene Symbol | Gene Name | CRF | Hemodynamic | Adiposity | Lipids and Lipoproteins | Insulin and Glucose | Other | References |
|---|---|---|---|---|---|---|---|---|
| ACE | Angiotensin I converting enzyme | B and R | R | (172,173) | ||||
| ADIPOQ | Adiponectin, C1Q and collagen domain containing | B | B | B | (174) | |||
| ADRA2A | Adrenoreceptor alpha 2A | B | (175) | |||||
| ADRB2 | Adrenoreceptor beta 2 | B and R | (176,177) | |||||
| ADRB3 | Adrenoreceptor beta 3 | R | (177) | |||||
| AGRP | Agouti related neuropeptide | B | Dietary intake | (178–180) | ||||
| AGT | Angiotensinogen | B and R | B | (172,181) | ||||
| AMPD1 | Adenosine monophosphate deaminase 1 | B and R | B and R | (182) | ||||
| ANG | Angiogenin | B | (183) | |||||
| APOE | Apolipoprotein E | B and R | (184,185) | |||||
| ATP1A2 | ATPase Na+/K+ transporting subunit alpha 2 | R | (186) | |||||
| BCHE | Butyrylcholinesterase | Anxiety | (187) | |||||
| CETP | Cholesteryl ester transfer protein | B and R | (185) | |||||
| CKM | Creatine kinase, M type | B and R | (188) | |||||
| DRD2 | Dopamine receptor D2 | PA level | (189) | |||||
| EDN1 | Endothelin 1 | R | (190) | |||||
| FHL1 | Four and a half LIM domains 1 | B and R | (191) | |||||
| FTO | FTO alpha-ketoglutarate dependent dioxygenase | B and R | (192) | |||||
| GHRL | Ghrelin and obestatin prepropeptide | B | B | (193) | ||||
| GNB3 | G protein subunit beta 3 | R | R | (194) | ||||
| LEP | Leptin | R | (195) | |||||
| LEPR | Leptin receptor | B | R | (195,196) | ||||
| LIPC | Lipase C, hepatic type | B | R | (197) | ||||
| LIPE | Hormone-sensitive lipase | B | (198) | |||||
| LPL | Lipoprotein lipase | B | (199) | |||||
| MC4R | Melanocortin 4 receptor | (200) | ||||||
| NOS3 | Nitric oxide synthase 3 | R | (201) | |||||
| PON1 | Paraoxonase 1 | Anxiety | (187) | |||||
| PPARG | Peroxisome proliferator–activated receptor gamma | R | B and R | R | (202,203) | |||
| PTPN1 | Protein tyrosine phosphatase nonreceptor type 1 | B | B | (204) | ||||
| SLC4A5 | Solute carrier family 4 member 5 | B | B | (205) | ||||
| TGFB1 | Transforming growth factor beta 1 | B | (206) | |||||
| TNF | Tumor necrosis factor | CRP B and R | (207) | |||||
| UCP3 | Uncoupling protein 3 | R | (208) |
An entry indicates that at least one trait showed significant (P < 0.05) evidence of association with a variant in the gene locus. Gene symbols and names are from the National Center for Biotechnology Information.
CRF, cardiorespiratory fitness; PA, physical activity.
For cardiorespiratory fitness and hemodynamic-related traits, ACE, AGT, AMPD1, ANG, ATP1A2, CKM, GHRL, GNB3, NOS3, PPARG, and SLC4A5 showed evidence of association, with at least one phenotype measured at baseline or in response to training. For example, one of the first candidate gene studies showed that muscle-specific creatine kinase (CKM), a gene previously related to intrinsic cardiorespiratory fitness, was associated with response of V̇O2max to training adjusted for age, sex, BMI, and pretraining V̇O2max, but not baseline levels of V̇O2max in White subjects (188). A later study found that, in Black subjects, minor allele homozygotes (T/T) at PPARG rs2016520 showed smaller training-induced improvements in V̇O2max than C-allele carriers (202). Several candidate genes, ADIPOQ, ADRA2A, ADRB2, AGRP, AGT, FTO, GHR, GNB3, LEPR, LIPE, PTPN1, and UCP3, showed evidence of association with at least one adiposity-related trait. However, no association was found between polymorphisms in the ADRB3 and MC4R genes and adiposity. Minor allele homozygotes (A/A) at FTO rs8050136 were heavier and fatter in White men but not White women, whereas C allele carriers showed three times greater reductions in FM and percent fat than A/A homozygotes across all White subjects (192). These findings suggest that resistance to training-induced reductions in adiposity is one mechanism by which FTO is related to obesity.
For lipid and lipoprotein traits, evidence of association was found with these candidate genes: ADIPOQ, APOE, CETP, LIPC, LPL, and PPARG. The APOE polymorphism was associated with baseline lipoprotein levels, with the E2/3 genotype having the lowest and E3/4 and E4/4 genotypes the highest LDL-C levels in men and women of both ethnic groups. White female carriers of E3 had higher baseline HDL-C levels, and White female carriers of E2/3 and E3/3 showed larger increases in HDL-C in response to training than E4/4 carriers (184). A subsequent study showed that CETP genotypes and haplotypes were associated with HDL phenotypes, including that CETP rs1800775 A/A homozygotes had higher baseline HDL-C levels in both Blacks and Whites and greater increases in HDL3-C and apoA-I with training in Whites only (185). Combined models found that the APOE and CETP rs1800775 genotypes accounted for 6%–9% of the variance in baseline and training-induced changes in apoA-I and HDL phenotypes. For traits related to insulin and glucose metabolism, evidence of association was observed with ADIPOQ, FHL1, LEP, LEPR, LIPC, PPARG, and PTPN1. For example, an association was observed between DNA sequence variation in the FHL1 gene and the response of fasting insulin and insulin sensitivity to training in White men (191). Evidence of a gene-training interaction was uncovered, with PPARG Ala carriers displaying greater improvements in glucose (Kg, Sg) and insulin (AIRg, Di) metabolism in response to training (203).
BP phenotypes were linked to several candidate genes (ACE, AGT, AMPD1, ANG, EDN1, GNB3, NOS3, SLC4A5, TGFB1). The relationship between variants in angiotensinogen (AGT M235T, rs699) and angiotensin-converting enzyme (ACE insertion/deletion) and training-induced changes in resting and exercise BP was studied in 476 Whites from 99 families. It was found that men with the AGT MM and MT genotypes demonstrated improved DBP responses to training, whereas men with the TT genotype had minimal responses and, moreover, had significantly higher DBP responses during acute maximal exercise (172). In a sample that included both ethnic groups of HERITAGE, an angiogenin (ANG) gene polymorphism was associated with both pretraining and posttraining resting and exercise SBP among Blacks, but not Whites (183). These findings extended previously known associations between ethnicity and resting BP to exercise BP responses. Subsequently, a polymorphism in the transforming growth factor-beta (TGF-β) gene was associated with SBP at rest and during acute exercise both before and after the exercise program among Whites, but not Blacks (206). Data from the Aerobics Center Longitudinal Study and HERITAGE were used to examine the association of endothelin 1 (EDN1) SNPs and BP phenotypes. In a case–control study design of incident hypertension, the investigators found that, although the allele and genotype frequencies of EDN1 tagging SNPs were not different between the two groups, there was a significant interaction between the EDN1 rs5370 genotype and baseline cardiorespiratory fitness levels on the future risk of hypertension. The EDN1 rs5370 risk allele (T) was also associated with attenuated responses in SBP and pulse pressure after training in HERITAGE White subjects, highlighting the capacity for physical activity and fitness status to potentially modify the influence of genes on cardiovascular outcomes (190).
Other studies found evidence of associations between genetic variation in BCHE and PON1 genes and anxiety scores (187), DRD2 gene and physical activity level (189), TNF gene and C-reactive protein (CRP) levels (207), and AGRP with total dietary and macronutrient intake (178).
Genome-wide linkage studies
Before the advent of GWAS to identify loci associated with complex traits, genome-wide linkage analysis (GWLA) was a common unbiased method used to identify regions of the genome (known as quantitative trait loci or QTLs) harboring genes that could influence complex traits. In HERITAGE, 23 GWLAs were undertaken to identify QTLs associated with traits measured at baseline or in response to training. For these linkage scans, up to 700 markers covering the 22 autosomes with a mean spacing of 4.1 Mb on the physical map were used. In these studies, the sibpair linkage method was used to test for linkage between the phenotype of interest and microsatellites, a class of highly polymorphic genetic markers consisting of variable copy number of repetitive DNA sequences found in the noncoding regions of the genome. Typically, multipoint linkage analysis was performed using a regression-based model, as implemented in MERLIN (81). Briefly, siblings who share a greater proportion of alleles identical-by-descent at the marker locus will also show a greater resemblance in the phenotype. The phenotypic resemblance of the siblings is linearly regressed on the estimated proportion of alleles that the sibling pair shares identical-by-descent at each marker locus (209).
The first HERITAGE GWLA undertaken on baseline V̇O2max and its response to training led to the identification of four QTLs for baseline V̇O2max and five QTLs for V̇O2max response (210). Several other QTLs for traits related to cardiorespiratory fitness (211,212), hemodynamic phenotypes (213–218), adiposity (219,220), plasma lipids and lipoproteins (221,222), sex hormones and steroids (125,126), insulin and glucose metabolism (223–225), energy and macronutrient intake (226), CRP levels (227), and metabolic syndrome (228) have been identified. Two bivariate GWLAs were also undertaken to identify pleiotropic QTLs affecting adiposity and TG levels (229) and TG and HDL-C levels (230). Results suggested that one or more QTLs in the 19q13 region jointly influence TG levels and adiposity, with the APOE and LIPE (hormone-sensitive lipase) genes being strong candidates for the source of the pleiotropic QTL (229). Dense mapping of three QTLs associated with the changes in submaximal exercise (50 W) SV (2q31-q32 and 10p11.2) and HR (2q33.q34) led, respectively, to the identification of the titin (231), kinesin family member 5B (232), and cAMP responsive element binding protein 1 (233) genes as strong candidate genes for hemodynamic responses to training. A QTL for submaximal exercise capacity (V̇O2 at 60% V̇O2max) on 13q12 was followed by dense mapping with more than 1800 SNPs over a 7.9-Mb region (211). Association analyses narrowed the target region to loci encoding MIPEP and SGCG.
Genome-wide association studies
To date, five GWAS based on HERITAGE data have been published. The first GWAS examined the association of 324,611 SNPs with V̇O2max response to endurance training and identified a panel of 21 SNPs explaining about half of the variance in the trainability of V̇O2max (234). A second GWAS revealed that nine SNPs accounted for the genetic variance of the submaximal exercise HR response to training (235). A third study performed genome-wide and transcriptome-wide profiling to identify genes associated with the TG response to training (236). The GWAS identified four SNPs accounting for the genetic variance of the TG response, whereas a molecular signature based on the baseline expression of 11 genes predicted 27% of TG changes in response to training. Association analyses of SNPs in or near these 11 predictor genes revealed that SNPs from only four genes were associated with TG changes. A composite score based on the top four SNPs, each from the genomic and transcriptomic analyses, was found to be the strongest predictor of the TG response to training in a model including over 50 clinical predictor variables (236). A fourth GWAS based on data from three different cohorts including HERITAGE identified 52 SNPs from 7 loci showing suggestive evidence of association (P < 1.0 × 10−6) with AVF and subcutaneous fat assessed by CT scan (237). A fifth GWAS revealed associations between SNPs and insulin sensitivity (Si). More specifically, the GWAS analysis identified one SNP significantly associated with baseline Si (P = 3.79 × 10−8) plus 7 SNPs with suggestive association (P < 10−5), and 10 SNPs with suggestive association (P < 10−5) with Si response to training. Interestingly, mouse knockouts in four of the genes associated with these SNPs showed phenotypes compatible with alterations with glucose metabolism (238).
HERITAGE involvement in large GWAS consortia
HERITAGE joined the GIANT (Genetic Investigation of ANthropometric Traits) and CHARGE consortia, international collaborative efforts aimed at identifying genetic loci associated with human body size, obesity, and behavioral and physiological traits. The primary focus of these large-scale projects has been meta-analysis of GWAS data. The GWAS data from HERITAGE Whites (n ~ 483) have been included in GWAS meta-analyses of obesity (239), height (240), BMI (241,242), and traits related to body shape/composition (waist circumference, waist-to-hip ratio, body fat percentage) (243–246). HERITAGE also contributed to GWAS meta-analyses of HR (247), plasma leptin (248), dietary macronutrient intake (249), and principal components of anthropometric traits (250).
These meta-analyses involved up to hundreds of thousands of individuals from dozens of studies, with genotype data on tens of thousands to millions of SNPs. For example, in one of the largest GWAS meta-analyses of BMI, 339,224 individuals were included from 80 studies in stage 1 and another 34 studies in stage 2 and found 97 BMI-associated loci (P < 5 × 10−8) (241). Conversely, a study of the influence of age and sex on genetic associations with body size and shape examined up to ~2.8 M SNPs in over 320,000 individuals (246). A number of these efforts have dealt with potential interactions with smoking, sleep, or educational attainment. One report investigated genome-wide lipid loci stratified by sleep duration (251), whereas another searched for loci related to resting BP and sleep (252). The smoking by genotype interactions on the risk of type 2 diabetes and fasting plasma glucose was considered in a recent study (253). Finally, the potential interactions with level of education were investigated for genome-wide loci associated with resting BP (254). This series of gene–environment interaction reports has successfully identified new genomic loci relevant to plasma lipids, blood glucose, and BP.
These GWAS meta-analyses are among the most cited articles in the fields of genetic epidemiology and obesity genetics and have contributed greatly to our understanding of the genetic architecture of anthropometric traits in adults.
Genome-wide interaction studies
Understanding the genetic and environmental factors and their interactions (G × E) underlying complex disease traits may provide insights into more effective intervention and therapeutic strategies. More recently, a major initiative was launched to pursue genome-wide interaction studies (GWIS) (255). Several GWIS reports identified important gene–lifestyle interactions for several cardiometabolic traits (243,246,256–262). The HERITAGE study investigators participated in most of these GWIS investigations, which contributed novel findings.
For example, several loci showing strong evidence for regulatory features supporting shared pathophysiology between cardiometabolic and addiction traits, such as smoking, were identified (262). GWIS reports have also highlighted a role in BP regulation for biological candidates such as modulators of vascular structure and function (CDKN1B, BCAR1-CFDP1, PXDN, EEA1), ciliopathies (SDCCAG8, RPGRIP1L), telomere maintenance (TNKS, PINX1, AKTIP), and central dopaminergic signaling (MSRA, EBF2). Similarly, analyses of gene–smoking interactions in lipids have identified several new loci associated with lipids, some of which were detected only in smokers, thereby highlighting the importance of gene–behavior interactions (256). Despite some interesting findings, it is still unclear how prevalent such interactions are and what their full impact may be on the genetic and environmental architecture of complex traits. Only comprehensive and properly powered GWIS studies can address these uncertainties.
GENE EXPRESSION STUDIES
A subset of HERITAGE participants from the Quebec clinical center underwent vastus lateralis skeletal muscle biopsies to examine the association between the muscle gene expression profile and cardiometabolic phenotypes at baseline and in response to training.
A first study examined muscle gene expression in 16 subjects discordant for the response of insulin sensitivity (Si) to the training program: 8 showing no changes (low responders) and 8 showing marked improvements (high responders; response ~4 times higher than that of low responders) (263). Differences in gene expression before and after training were tested for 703 and 1166 transcripts, respectively, using pooled RNA from each group. A total of 38 transcripts were differentially expressed between response groups at both baseline and posttraining, with 22 having known functions: upregulated genes were related to cell adhesion/growth, signal transduction, and electron and adenine transport, whereas downregulated genes were involved with oxidative phosphorylation, transcript regulation, and protein biosynthesis. Differential expression at either baseline or posttraining was confirmed by real time quantitative reverse polymerase chain reaction in four of the five genes selected for validation (Fig. 6) (263). In a follow-up report on three SNPs at the FHL1 locus, one of these four genes, it was found that allelic variants were associated with changes in fasting insulin, Si, Kg, and Di response to training in White men (191). Further analysis of the relationship between gene expression and Si was examined in a study where a transcriptional signature linked to the transcription factor MEF2A was found to be predictive of the response of Si both in HERITAGE and in an independent cohort (238).
FIGURE 6.
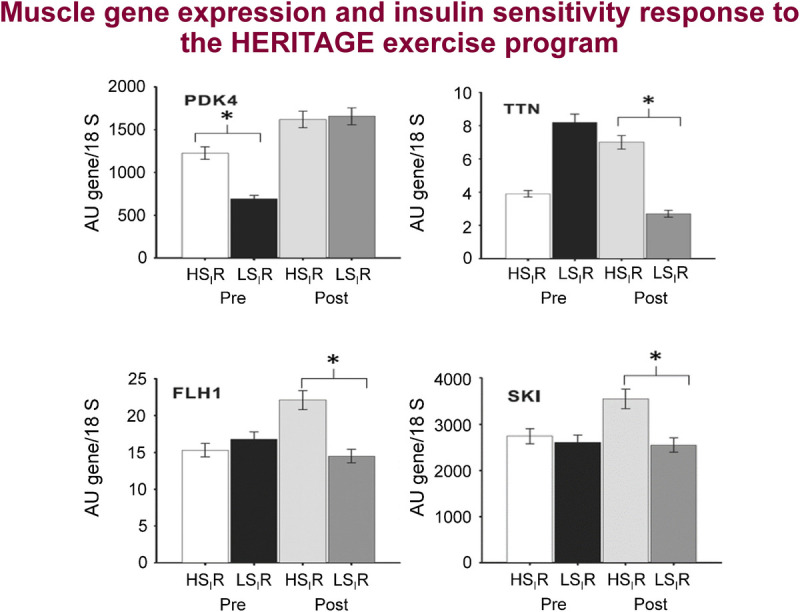
Real time quantitative reverse polymerase chain reaction validation of gene expression levels for insulin sensitivity (Si) response to training identified in microarray experiments. HSIR, high Si responder; LSIR, low Si responder; PDK4, pyruvate dehydrogenase kinase 4; TTN, titin; SKI, V-Ski avian sarcoma viral oncogene homolog; FHL1, four-and-a-half LIM domain 1. Levels normalized over the corresponding 18S rRNA value. *P < 0.05 for difference in mean value between response groups. Adapted with permission from Teran-Garcia et al. (263).
HERITAGE gene expression data were also used in a study that identified an age-related gene expression signature in human muscle (264). Using a total of 97 Affymetrix U133 + 2 gene chips from two independent studies, including 52 from HERITAGE, 580 genes were identified whose expression levels were related to a continuum of age (18–78 yr). The muscle age-related genes were related to serotonin, DNA topoisomerase antagonism, and RXR activation.
In one study, a combination of genome-wide muscle gene expression with DNA sequence variation screening was used to identify genes associated with changes in V̇O2max due to endurance exercise training (265). A regression analysis combining SNPs associated with 29 skeletal muscle transcripts predicting changes in V̇O2max and SNPs previously associated with the trainability of V̇O2max led to the identification of a set of 11 genetic variants explaining 23% of the variance in V̇O2max training response. Subsequently, regulatory molecules coordinating the complex network of ~800 genes found to be differentially regulated in response to training (i.e., training-responsive transcriptome) were examined by contrasting subjects who demonstrated the least versus the greatest improvement in V̇O2max (low vs high responders) resulting in the identification of 86 genes (266). A panel of 3400 SNPs near these 86 genes were genotyped, and 24 were nominally associated with responder status (P < 0.01) but not statistically significant after accounting for multiple testing: 8 of them belonged to the HLA cluster of genes on chromosome 6, whereas other significant SNPs were in loci coding for proteins largely involved in cell proliferation or tumor suppression (ASNS, DLC1, FABP5, NID1, PDZD2, SASH1, SPARC, and TRAM1 genes).
A data-driven, reverse-engineering approach was used to develop a genome-wide network model of muscle homeostasis in response to training (267). The analysis initially focused on identifying pairs of genes that were differentially coexpressed between pretraining and posttraining skeletal muscle in HERITAGE (n = 52). The analysis identified a differential coexpression network, which the algorithm organized into 25 interconnected gene clusters. Further analysis of the inferred network revealed that genes found within core metabolic and translational pathways are a predominant feature of these networks. Interestingly, the eukaryotic translation initiation factor Eif6 was the most connected ribosome associated factor in the network leading to the hypothesis that Eif6 could be a part of an mTor-independent mechanism regulating metabolic adaptation. Analysis of the Eif6+/− haploinsufficient mouse (50% reduction in eIF6 expression) found different transcriptional profiles compared with wild-type mice (genes differentially expressed in the gastrocnemius: 2651 upregulated, 829 downregulated; soleus: 2482 upregulated, 1846 downregulated; tibialis anterior: 1361 upregulated, 958 downregulated; false discovery rate < 10%) and that the profile of the gastrocnemius muscle mirrored the predicted Eif6 signature found in HERITAGE both in functional profile and in direction of change. Further experiments of mitochondrial functionality and muscle performance in Eif6 heterozygote mice revealed alterations in electron transport chain dynamics, superoxide generation, and reduced exercise capacity, further supporting the role of this factor in regulating muscle energy homeostasis (267).
Finally, a study that utilized transcriptome-wide profiling to identify a baseline muscle gene expression signature of the response of TG to training was described previously (236). Briefly, after transcriptome-wide association analysis, a molecular signature based on the baseline expression of 11 genes predicted 27% of TG changes in response to exercise training (Fig. 7).
FIGURE 7.
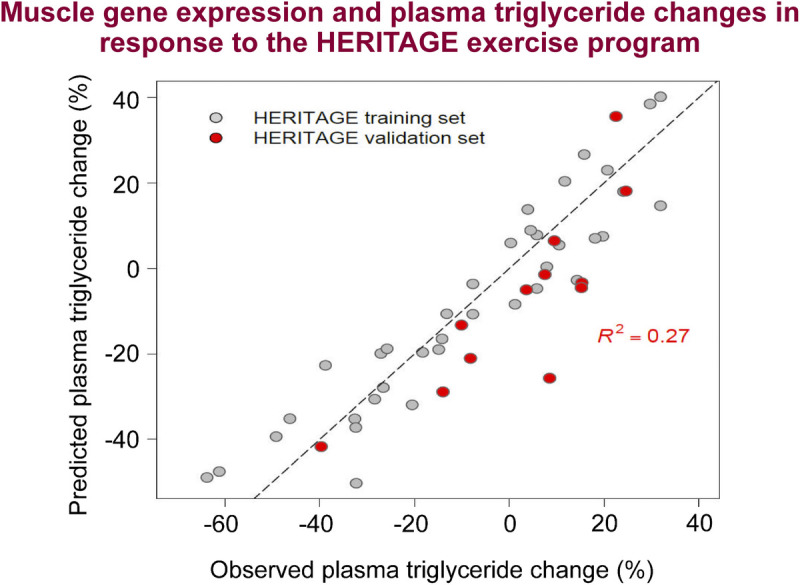
Performance of the vastus lateralis gene expression profile regression model derived from a training set (n = 37, gray dots) and a validation set (n = 12, red dots) for the prediction of exercise training-induced changes in TG in HERITAGE. Reproduced with permission from Sarzynski et al. (236).
METABOLOMIC STUDIES
Targeted, liquid chromatography tandem mass spectrometry–based metabolomics was performed to profile 186 plasma metabolites in 441 White subjects before and after training. DMGV, a recently identified circulating biomarker of liver fat that predicts incident type 2 diabetes and decreases in body weight after weight loss surgery (268), was investigated to examine its relationship to cardiometabolic changes with training (96). In age- and sex-adjusted analyses, baseline DMGV levels were positively correlated with adverse body mass and composition (including visceral fat), lipid and lipoproteins, and glucose and insulin homeostasis traits (β coefficient ranges for associations meeting Bonferroni statistical significance: 0.17–0.46). After adjustment for both BMI and AVF, associations of DMGV levels with all glucose and insulin traits and 16 of 22 lipid metabolism traits remained highly significant (96). DMGV levels decreased with training (median change. −9.4%), and changes in DMGV were positively correlated with changes of several lipid and glucose and insulin traits (e.g., total, medium, and large VLDL-P; VLDL-P size; TG; total and medium HDL-P; small LDL-P, TC; fasting insulin and glucose).
Baseline levels of DMGV were associated with attenuated improvements in HDL traits (inverse association with total and medium HDL particle concentration and apoA-I; positive association with small HDL particles) after training, even after adjusting for age, sex, baseline level of each outcome variable, and changes in BMI and visceral fat (96) (Fig. 8). Baseline DMGV levels were also inversely associated with changes in Si (β = −0.13; P = 3.0 × 10−3) and positively with changes in AVF (β = 0.11; P = 0.03) and body mass (β = 0.11; P = 0.04) in minimally adjusted models, but the associations were not significant in fully adjusted models. In summary, DMGV identified adverse metabolic traits in healthy adults of European descent in HERITAGE and was associated with attenuated improvements in lipid traits and insulin sensitivity with training.
FIGURE 8.
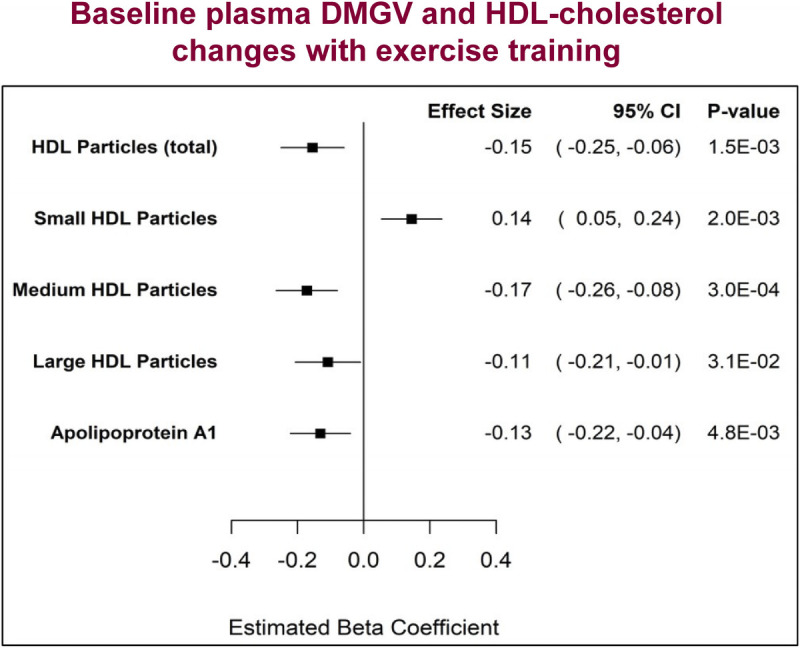
Associations between baseline DMGV levels and HDL traits responsiveness to exercise training in HERITAGE Whites. Reproduced with permission from Robbins et al. (96).
PROTEOMIC STUDIES
The most recent molecular profiling platform applied to HERITAGE has been proteomics. Specifically, to measure large-scale circulating proteomic changes after endurance exercise training, baseline and posttraining plasma samples were analyzed using the Slow-Off rate Modified Aptamer (SOMAmer)-based platform, SomaScan® (SomaLogic, Inc., Boulder, CO). This platform utilizes affinity binding to quantify approximately 5000 distinct protein targets in circulation (269,270).
In a novel, proof-of-concept study utilizing SomaLogic proteomics data, the association of plasma proteins with cross-sectional V̇O2max was analyzed (271). An unpaired crossover sampling method (with 50% of samples from subjects at baseline and 50% at posttraining) was used to avoid correlation from pairs and to increase the observed range of V̇O2max values in the data set. An elastic net linear regression model was derived, refined, and validated on 80%, 10%, and 10% of participants, respectively. The derivation model (80%, or 523 subjects) identified 115 proteins that were highly correlated with measured V̇O2max, with an r2 of 0.80; this was replicated in the validation data set (10%, or n = 62), with an r2 of 0.71 (Fig. 9). The top 3 proteins with the largest quantitative contribution to V̇O2max were leptin, C1QR1 (complement component C1Q receptor, part of immune system), and GGH (gamma glutaryl hydrolase, regulation of intracellular folate) (271).
FIGURE 9.
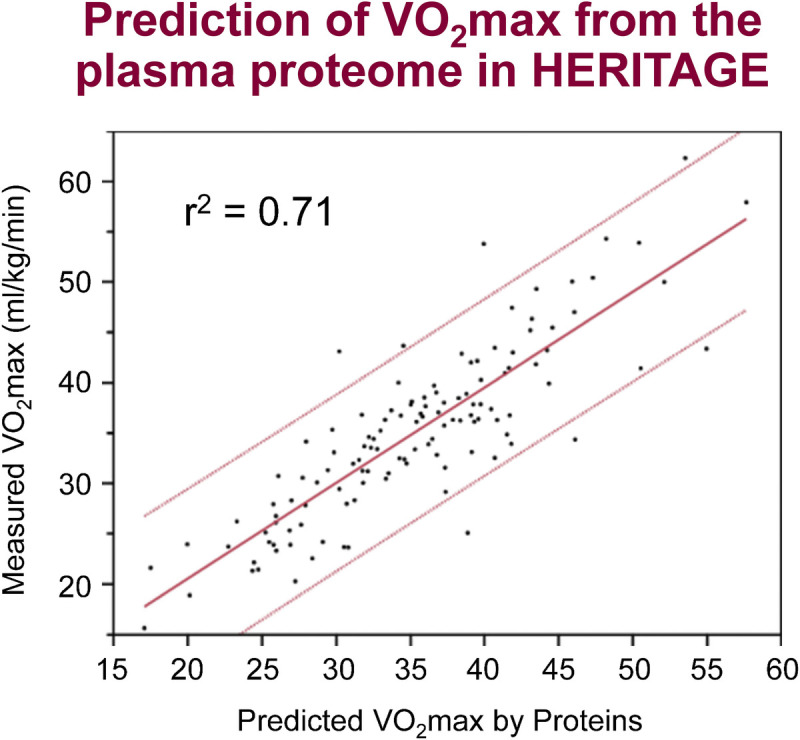
Prediction of measured V̇O2max by 124 plasma protein levels in the validation data set of HERITAGE. Reproduced with permission from Williams et al. (271).
The plasma proteomic profile of V̇O2max was further refined in analyses that characterized proteins associated with baseline V̇O2max and changes in V̇O2max in response to training in more than 650 subjects (272). After adjustment for age, sex, BMI, and ethnicity, 147 circulating proteins were associated with baseline V̇O2max (Fig. 10, left panel), including 85 proteins that were positively associated and 62 proteins negatively associated. Proteins positively associated with baseline V̇O2max spanned specific organ systems and biologic processes including angiogenesis (e.g., extracellular matrix protein 1, anthrax toxin receptor 2), coagulation and hematopoiesis (e.g., complement decay-accelerating factor, tetranectin), and lipid metabolism (e.g., apolipoprotein F, lipase member K). Several well-known markers of metabolic dysregulation known to be positively associated with adiposity were inversely associated with baseline V̇O2max, including leptin, CRP, and insulin.
FIGURE 10.
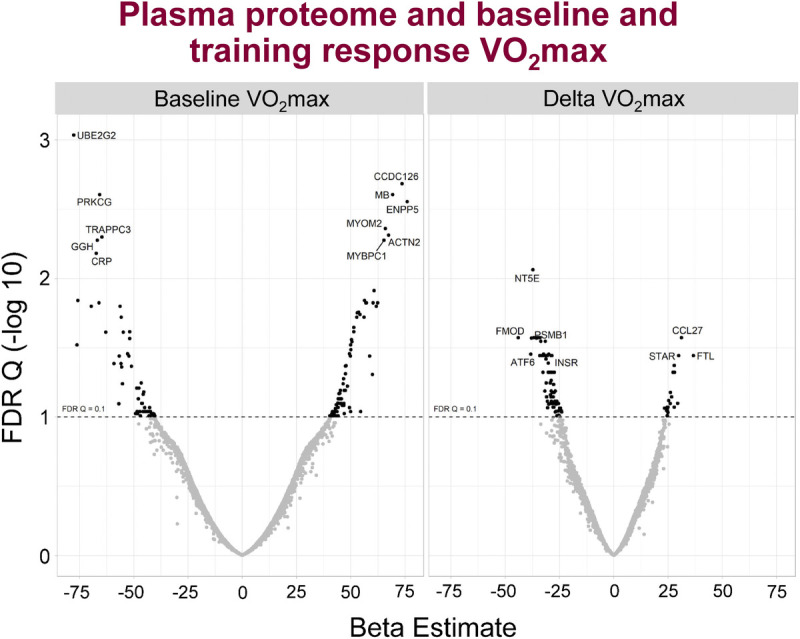
Plasma proteins positively and negatively associated with baseline V̇O2max (left panel) and change in V̇O2max (right panel) adjusted for age, sex, ethnicity, and BMI in HERITAGE. Reproduced with permission from Robbins et al. (272).
In addition, 102 baseline proteins were associated with changes in V̇O2max with training in a linear regression model adjusted for age, sex, BMI, ethnicity, and the baseline level of V̇O2max (Fig. 10, right panel). Minimal overlap was found between the proteins associated with baseline V̇O2max versus change in V̇O2max, as only five proteins (T132B, ATF6A, CO9A1, insulin, and PIANP) were associated with both traits, proteomic findings that are consistent with clinical findings in HERITAGE demonstrating a lack of correlation between these two V̇O2max traits (see the Cardiorespiratory Fitness section). Subsequently, analyses were aimed at determining the ability of plasma proteins to identify V̇O2max gains (>15% change from baseline) to exercise training in comparison to clinical traits alone. A model based solely on clinical traits (age, sex, ethnicity, BMI) yielded an area under the curve of 0.62, whereas the addition of 56 proteins to the clinical trait model increased the area under the curve to 0.84, a substantial improvement in predicting who is likely to be a higher V̇O2max responder. Lastly, the association of V̇O2max-related proteins with mortality was examined in the Framingham Offspring Study. It was found that 12 proteins associated with baseline V̇O2max and 9 proteins associated with change in V̇O2max were associated with incident all-cause mortality (false discovery rate q < 0.1) (272).
BIOINFORMATICS EXPLORATIONS OF CARDIORESPIRATORY FITNESS TRAITS
Here, the main findings from integrative bioinformatics explorations of two cardiorespiratory fitness traits representing intrinsic V̇O2max and trainability of V̇O2max are summarized. These two studies are separated in time by about 6 yr, and their methodologies reflect the evolving trends and capabilities of the bioinformatics field in general.
In 2013, an analysis examining the contributions of genes and biological pathways to variations in trainability of V̇O2max (hereafter called delta V̇O2max) was published (273). Using GWAS SNP associations to delta V̇O2max, significant contributions at the “gene set” or pathway level instead of individual SNP level were investigated. By considering association scores from all gene members of a gene set, pathways related to the broad categories of immune function, cardiomyopathy, extracellular matrix regulation, and metabolism (PPAR signaling, pantothenate, and CoA biosynthesis) were found to be significantly associated with V̇O2max trainability. Examining pathway overrepresentation among genes above a threshold of genetic association to delta V̇O2max further identified a network of 35 genes, of which 31 were associated with delta V̇O2max at P ≤ 0.005. These network genes could be partitioned into pathways related to calcium signaling, nitric oxide signaling, and protein kinase A signaling.
A hypothesis-free gene prioritization tool was used to rank genes moderately associated with delta V̇O2max in GWAS analysis. One group of genes (PINX1, CD44, PARK2, RYR2, ADCY5, and SHANK2) demonstrated consistently strong scores across all analyses, whereas a second group (KCNQ5, GRIK4, RPTOR, ACVR1C, and ACSL1) were strongly associated only in a subset. Several of the genes identified (CAMTA1, RYR2, ACSL1, CD44, BIRC7/YTHDF1) have been further replicated (274). This bioinformatics exploration of the biology underlying V̇O2max adaptation suggests that pathways related to calcium signaling, energy sensing and partitioning, mitochondrial biogenesis, angiogenesis, immune functions, and regulation of autophagy and apoptosis are some key mechanisms through which the physiological responses of V̇O2max to training are mediated.
The bioinformatics explorations of the genetics and biology underlying V̇O2max in the sedentary state (275) differed extensively in complexity and depth compared with the 2013 analysis of V̇O2max trainability. In the 6-yr interim, there had been a profound expansion in databases and data analysis tools, especially access to large public data sets and the ability to formulate hypotheses about the function of noncoding SNPs, thereby greatly expanding the scope of understanding the putative roles of genetic variants. Using summary statistics from intrinsic V̇O2max GWAS as a starting point, the focus was first on the “discovery” of candidate genes and pathways, followed by a “validation” phase of candidate genes identified in the discovery phase in which knockout phenotypes and exercise cardiovascular and muscle physiology phenotypes were interrogated (275).
Intrinsic V̇O2max bioinformatics: Discovery phase
A pipeline consisting of seven steps was used to generate gene targets and hypotheses regarding the biology underlying V̇O2max.
Quantifying gene-level association to intrinsic V̇O2max, based on SNP data. The most likely causal genes at associated loci were prioritized. This exercise led to the identification of genes such as PRADC1, TPM2, IGFN1, and CASQ2.
Effects of noncoding SNPs on promoter and enhancer binding sites in key tissues. A statistically significant enrichment for enhancer-associated SNPs was observed in tissues related to cardiorespiratory functions (aorta, right ventricle, skeletal muscle, myoblasts, and satellite cells). The analysis further revealed an increased overlap with active enhancers (H3K4me1 and H3K27ac marks) and active promoters (H3K4me3 and H3K9ac marks) in skeletal muscle-related samples, and increased levels of active enhancer associated H3K4me1-site overlaps in aorta. These findings provided insights into some possible roles of regulatory SNPs in influencing intrinsic V̇O2max.
Effects of noncoding SNPs on nearby gene expression (cis-eQTLs) in relevant tissues. A total of 216 SNPs was found to be significantly associated with cis-gene expression (eQTL P ≤ 1 × 10−05). The top 3 most significant eQTL associations were observed for SNP rs2838815 regulating ADARB1 gene expression in brain (P ≤ 1 × 10−18), a cluster of highly linked SNPs (clustered at ATE1 gene intron) regulating NSMCE4A gene expression in pancreas (P < 1 × 10−14), and another cluster of tightly linked SNPs affecting expression of the PRADC1 gene (P ≤ 1 × 10−12) in adipose tissue, as well as the heart and skeletal muscle. Analysis for joint effects, that is, genomic loci overlapping for both eQTL and histone binding sites, demonstrated the strongest overlaps in heart tissue, followed by the pancreas, skeletal muscle, and adipose tissue.
Enrichment of intrinsic V̇O2max-associated candidate genes in tissue-specific functional networks. The functional connectivity of tissue-expressed genes to the set of GWAS-significant genes was examined. The top 5 tissues with high network connectivity included the placenta, heart, skeletal muscle, pancreas, and liver. Analysis of skeletal muscle specific subnetworks demonstrated a high degree of connectivity for the genes SSB, EED, PICALM, and TIMM8B.
Examining skeletal muscle gene expression profiles. Whole-genome expression profiling data were analyzed to identify genes that transcriptionally correlated with intrinsic V̇O2max levels. Forty-seven Affymetrix probes were significant. Among these, three genes (CASQ2, COX7A2L, PRADC1) were also identified in our other bioinformatic analyses.
Enrichment of intrinsic V̇O2max-associated genes in biological pathways. Pathway enrichment analysis identified 34 pathways as significant. Hierarchical clustering of the significant pathways identified groups of pathways with shared genes, for example, pathways involved in calcium and inositol 1,4,5-trisphosphate signaling, and G-protein signaling. Additional pathway analysis further identified pathways related to insulin and PI3 signaling as significant.
Weighted ranking of candidate intrinsic V̇O2max-associated genes identified in prior steps. To generate a priority list of candidate genes based on the findings from individual bioinformatics analysis, a weighted-ranking algorithm was developed. The top 10 genes identified through this exercise included ADARB1, PRADC1, TPM2, FBXO41, IGFN1, NOTO, CASQ2, ARL6IP5, RAB11FIP5, and TMF1.
Intrinsic V̇O2max bioinformatics: Validation phase
The prioritized genes were filtered against the evidence available from knockout mouse studies and from new data on intermediate cardiorespiratory fitness endophenotypes available in HERITAGE.
Examining the phenotypic effects of candidate gene knockouts in mouse models. Knockout mouse phenotype data from Mouse Genome Informatics were available for 21 of the 38 candidate genes identified through the bioinformatics analyses. Notably, knockout of the Casq2 gene affected all endophenotype categories (cardiovascular, hematopoietic, skeletal muscle, metabolism), with most observed phenotypes related to the cardiovascular system. Knockout of Picalm affected mostly hematopoietic traits. Sgcg knockout produced effects on cardiovascular and muscle phenotypes, whereas Pradc1 knockout led to increased circulating glucose.
Examining SNPs in candidate genes for associations to cardiovascular and muscle intermediate endophenotypes of intrinsic V̇O2max. Based on the results from the bioinformatics analyses, gene expression studies, and knockout mouse phenotypes, the next step was to investigate whether candidate genetic loci were associated with physiological and metabolic traits known to be relevant to intrinsic cardiorespiratory fitness and measured in HERITAGE. SNPs in genes such as ADRB1, ATE1, CASQ2, DMRT2, NOTO, and SGCG were found to be associated with exercise-related cardiovascular traits such as HR at 60%V̇O2max, SV at 60%V̇O2max, and SBP pressure at maximal exercise. Multiple SNPs of SGCG, DMRT2, ADARB1, and CASQ2 genes were nominally associated with muscle-related traits, including percent fiber type, capillary area per fiber, and muscle enzyme activities. As an example, CASQ2 SNP rs7523715 was associated with ˙Q and HR during submaximal and maximal exercise, respectively, whereas CASQ2 SNP rs2999460 was associated with the percentage of type 1 and type 2B muscle fibers. CASQ2 gene expression was also positively associated with enzyme activities related to aerobic cellular respiration (cytochrome oxidase, CS, and HADH) and negatively associated with the rate-limiting enzyme of glycolysis (PFK).
Several candidate genes from the aforementioned integrative analyses provided new insights into the genetic regulation of intrinsic V̇O2max. Of these, two genes—CASQ2 and SGCG—are highlighted hereinafter for illustrative purposes. The CASQ2 gene, encoding calsequestrin, a calcium-binding protein localized in the sarcoplasmic reticulum in cardiac and skeletal muscle cells, was highly ranked in the bioinformatics analysis, positively correlated in gene expression to intrinsic V̇O2max, harbored SNPs positively associated with several muscle phenotype data from HERITAGE (e.g., greater proportion of type I and lower proportion of type IIb fibers), while also displaying knockout mouse phenotypes in all four classes of endophenotypes. The most notable effects were either cardiovascular (cardiac muscle contractility, increased heart mass, cardiac hypertrophy, sinus arrhythmia, etc.) or muscle-related (abnormal sarcoplasmic reticulum morphology, reduced muscle contractility). These findings in HERITAGE corroborate those from a study of high- and low-active mice in which CASQ1, the muscle form of CASQ2, was overexpressed in the soleus (primarily comprising type I fibers) of high-active mice compared with low-active (276). Moreover, transient knockdown of CASQ1 protein in high-active mice resulted in a significant decrease in physical activity. Rare variant mutations in CASQ2 (rs121434549, rs786205106, and rs121434550) have been implicated in catecholaminergic polymorphic ventricular tachycardia 2 (CPVT2) (277–279). These findings would suggest that common variants in CASQ2 also influence cardiac and skeletal muscle function relevant to intrinsic V̇O2max. Although CASQ2 was predominantly associated with cardiovascular phenotypes, the largest number of muscle-related phenotypes was observed in sarcoglycan gamma (SGCG) knockout mouse models. Notably, SGCG is a sarcolemmal transmembrane glycoprotein and a component of the sarcoglycan complex linking F-actin cytoskeleton and the extracellular matrix in muscle cells. Impairments in SGCG function result in early-onset recessive muscular dystrophy, especially limb–girdle muscular dystrophy, in humans (280,281). This study suggests a role for common variations in the SGCG gene in the regulation of intrinsic V̇O2max.
This effort based on a bioinformatics pipeline applied to V̇O2max data in the sedentary state suggests four gene loci related to cardiovascular physiology (ATE1, CASQ2, NOTO, and SGCG), four loci related to hematopoiesis (PICALM, SSB, CASQ2, and CA9), four loci related to skeletal muscle function (SGCG, DMRT2, ADARB1, and CASQ2), and eight loci related to metabolism (ATE1, PICALM, RAB11FIP5, GBA2, SGCG, PRADC1, ARL6IP5, and CASQ2) as candidates for human variability in cardiorespiratory fitness.
EPILOGUE
The HERITAGE Family Study has been a remarkable story of scientific collaboration among multiple sites and a large number of scientists, fellows, and research associates over a period of 30 yr. As with all studies, HERITAGE has strengths and limitations. Strengths of the study include the relatively large sample size for an exercise training study, the inclusion of family members as opposed to singletons, and substantial samples of both sexes, different ages, and Blacks and Whites. An extensive panel of quality control measures were implemented to standardize testing and training procedures, staff training, and minute-by-minute monitoring of each training session for each participant with a level of compliance reaching >95%. These efforts included substudies to evaluate within-person variability over time in sedentary people, and whether there were between-site differences and drift in testing protocol over time by clinical center sites. There were extensive panels of behavioral, physiological, and metabolic phenotypes measured before and after training, with most traits being measured twice before and then 24 and 72 h after training, all complemented by a comprehensive data management system and an extensive biobanking resource.
There are also limitations to the study, the most obvious of which being the lack of a control group. As mentioned previously, it was agreed 30 yr ago that a control group was not necessary to address the main aims of HERITAGE. Indeed, the consensus was that the funding available would be better devoted to reaching a larger sample size of families and participants in both ethnic groups than to add a control group. The lack of a control group became a challenge when the HERITAGE resource was used in secondary analyses. However, a substantial panel of quality control measures were added to the protocol in to quantify the assay reproducibility (for blood-based assays), the reproducibility and technical error of physiological and metabolic measurements, and the within-person variability over time of these traits, plus estimates of between clinical center differences and drift over time in data, with the goal of compensating for the lack of a control group in secondary analyses. Other limitations include that only one standardized dose of exercise was tested, children and older adults were not incorporated into the study, and skeletal muscle samples were available on only a subsample of participants.
What we have learned has been reviewed in the preceding sections. The original aims of HERITAGE were to investigate the magnitude of the individual differences for exercise-related traits and their response levels with exposure to endurance training, to evaluate whether there was familial aggregation and compute the heritability level for each trait and their response to training, and to explore a set of candidate genes and unbiased genomic microsatellite markers associated with exercise traits and their trainability. As detailed previously, these aims have been thoroughly covered across the various phases of the HERITAGE research program. The most novel and impactful findings of the study are summarized in Supplementary Table S4 (Supplemental Digital Content, Appendix, http://links.lww.com/MSS/C482). An illustration of the general trends in the reported findings is provided here with an emphasis on cardiorespiratory fitness, as evaluated by V̇O2max.
Individual differences in V̇O2max among sedentary adults were found to be large. An appreciation of their magnitude can be found in Figure 11. The left panel depicts the distribution of V̇O2max per kilogram of body weight per minute, based on two tests, within 1 wk, in a group of 174 young adult males age 17 to 35 yr who were all physically inactive (mean of 40.6 mL O2·kg−1·min−1, SD, 8.0). There is a 3-fold range between the low and high values of “intrinsic” cardiorespiratory fitness. The same observation was made for women, older subjects, and both ethnic groups. The right panel illustrates the V̇O2max training response in the same homogenous sample of young adult males. The mean gain reached 15.7%, with SD of 8.7%. Even though most tended to improve by about 10% to 20%, the range of responses is wide, with some experiencing no gain and others improving by 40% to 60%, as illustrated by the skewed distribution. Comparable variability in baseline levels and responses to training was observed for other traits, including submaximal exercise capacity, exercise HR and BP, visceral adiposity, insulin sensitivity, HDL-C, lipoprotein and hepatic lipases, and other exercise-related traits as was summarized in previous sections of this review.
FIGURE 11.
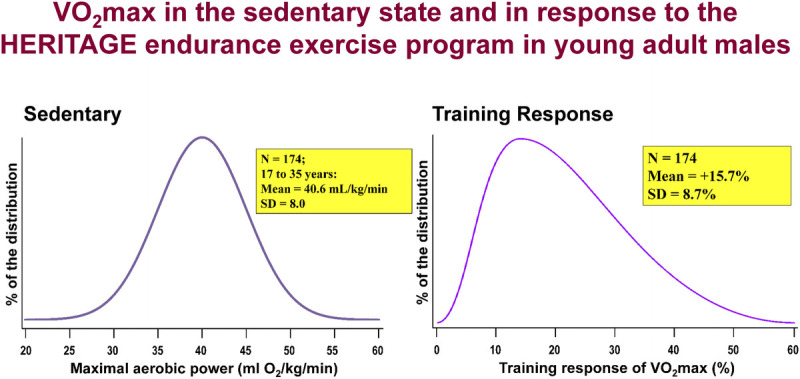
Individual differences in intrinsic V̇O2max (left panel) and its response to the standardized training program (right panel) in HERITAGE young adult males age 17 to 35 yr. Intrinsic V̇O2max is derived from two tests at baseline in the sedentary state. The gain in V̇O2max was calculated using the mean of two post training values minus the baseline level.
The topic of familial aggregation and heritability has been addressed throughout this review. After adjustment for age, sex, body mass, and other appropriate concomitants, familial aggregation of maximal and submaximal indicators of cardiorespiratory fitness remained significant, with approximately 2.5 times more variance between families relative to within families. Familial aggregation for response traits persisted after adjustment for multiple covariates for the same traits. Heritability levels commonly reported in the scientific literature or traits such as body mass and body composition, fat distribution, visceral fat, BP, insulin and glucose traits, lipids and lipoproteins were generally replicated in HERITAGE sedentary state measurements. Intrinsic V̇O2max was characterized by a heritability level of the order of 50% after adjustment for age, sex, body mass, and body composition. Globally, the heritability of the training response traits tends to be lower than the heritability in the sedentary state. One notable exception to this trend pertains to the trainability of V̇O2max, whose heritability level reaches 45% to 50% when assessed in both the untrained and trained states.
Human heterogeneity in the response to a given dose of exercise over a given time period is deeply rooted in the biology of human adaptability, with genomic differences playing an important role. Using the changes in V̇O2max in the HERITAGE cohort, we have attempted to quantify the correlates and apparent sources of response variability. Our findings have been summarized earlier and are depicted in Figure 12 (33). Age, sex, ethnicity, baseline body weight, and baseline V̇O2max were all weakly associated with the gains in V̇O2max, each accounting for 2% to 3% of the variance. The minute-by-minute fluctuations in achieved HR and PO compared with the prescribed HR and PO during each exercise session for each subject were computed. These differences between achieved versus prescribed were found to account for about 6% of the V̇O2max training response. We estimated as discussed previously that the measurement error and the day-to-day variation in V̇O2max explained about 20% of the variability in training response. Finally, as described earlier (Fig. 4), the genetic component of V̇O2max trainability accounted for 47% of the variance, a heritability level compatible with estimates obtained in selection experiments performed in rats (282). Thus, in the aggregate, the sources of variation or correlates of V̇O2max trainability explain 87% of the variance, as detailed in Figure 12 (33).
FIGURE 12.
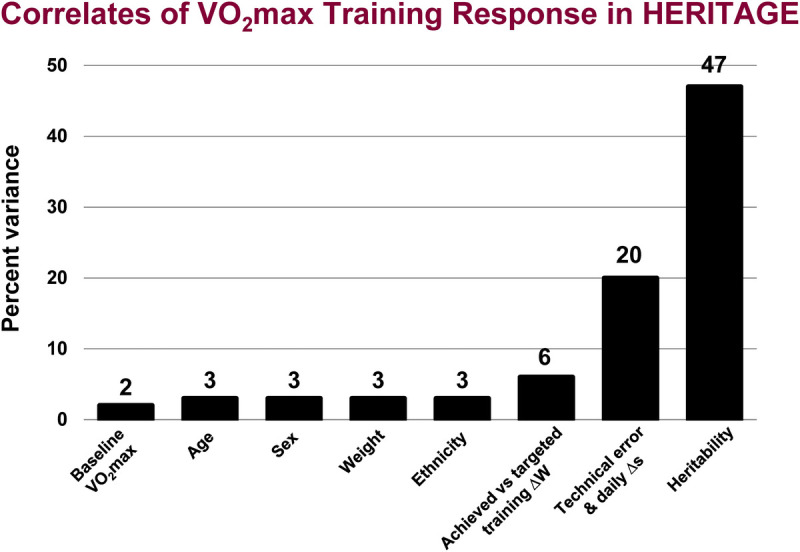
A summary of the correlates of the gains in V̇O2max in response to the standardized and fully monitored 20-wk exercise program of HERITAGE. A maximum of 482 Whites and 259 Blacks were available for these calculations. Age ranged from 17 to 65 yr. The mean gain in V̇O2max was 384 mL O2 (SD, 202). Reproduced with permission from Sarzynski et al. (33).
One important question is whether the HERITAGE findings on V̇O2max trainability apply to the training-induced changes in other traits of interest. Individual differences in response level have been observed for a panel of seven cardiometabolic and cardiorespiratory fitness traits (44,283). Changes in V̇O2max were shown to be independent of the changes in adiposity, visceral adipose tissue, insulin, lipid and lipoprotein, and inflammatory phenotype changes (Table 16). For instance, the correlation coefficients between the gains in V̇O2max and the changes in cardiometabolic traits are less than 0.10. Similarly, the coefficients among several cardiometabolic trait changes are generally about 0.20 or less, except for the changes between percent body fat and AVF, which reached 0.35 (283). Thus, the response levels across traits do not aggregate in any given individual, indicating that there is a high degree of trait specificity in responsiveness to an endurance training program. One implication of these observations is that the profile of correlates and determinants for the training-induced gains in V̇O2max is unlikely to be applicable to other response traits. It is also unlikely that the underlying biology of training responsiveness (as defined by molecular drivers, transcription factors, activators and repressors, small and large molecules, gene networks, pathways, and systems) will be the same for all response traits. Thus, each response trait or family of traits (e.g., a set of adiposity-related variables) may have its unique biological and molecular signature.
TABLE 16.
Correlation coefficients of exercise training responses among cardiometabolic traits in the HERITAGE Family Study.
| % Fat | AVF | Insulin | HDL-C | Small LDL-P | GlycA | |
|---|---|---|---|---|---|---|
| V̇O2max | −0.09 | −0.05 | 0.03 | 0.06 | −0.07 | −0.05 |
| % body fat | 0.35* | 0.16* | −0.07 | 0.08 | 0.03 | |
| AVF | 0.23* | −0.07 | 0.10 | 0.08 | ||
| Fasting insulin | −0.09 | 0.05 | 0.13* | |||
| HDL-C | −0.27* | −0.11 | ||||
| Small LDL-P | 0.20* |
All listed traits represent the change in trait value in response to training (i.e., delta trait).
Values are Pearson correlation coefficients and (P value). Correlations adjusted for age, sex, ethnicity, and baseline value of the response traits. Multiple testing corrected threshold for statistical significance P < 0.007. Based on 564 subjects with complete data.
*P < 0.007 for statistical significance.
Fasting insulin, fasting plasma insulin; GlycA, plasma inflammatory marker; AVF, abdominal visceral fat; fasting plasma insulin; LDL-P, low-density lipoprotein particle, plasma inflammatory marker GlycA.
Adapted with permission from Barber et al. (283).
HERITAGE and the early training experiments performed with numerous pairs of sedentary monozygotic twins have successfully established that there are considerable individual differences in the response level to a given dose of exercise of cardiorespiratory fitness, physiological and cardiometabolic traits, and that genetic components account for substantial fractions of the response variability. Subsequently, HERITAGE was the first study to embark on a systematic search for the genomic variants associated with training response variability. Before the GWAS and omics era, multiple candidate genes were explored, and panels of microsatellite markers were used to perform unbiased screens of the genome taking advantage of the pairs of siblings and nuclear family structure of HERITAGE participants. One of the major lessons learned from these early genetic explorations was that some of the prevailing views during the first 15 yr or so of the HERITAGE research program were later found to be inadequate. The genetic component of complex, multifactorial traits, such as V̇O2max trainability or the training response of any physiological and metabolic trait, could not be accounted for by a relatively small number of genes and genomic variants. Advances in technologies and high-throughput systems have allowed the recognition that such complex traits are commonly associated with large numbers of genomic variants, with each of them contributing only a tiny or small fraction of the genetic variance. Since then, HERITAGE resources have been mobilized to incorporate genome-wide and molecular-wide (e.g., proteome and metabolome) screening approaches based on panels of millions of measured and imputed genomic markers, and thousands of muscle and plasma molecular markers.
This has made it possible to extend the original intent of the HERITAGE project to complex and multi-omics exploration of the biology of cardiovascular and diabetes related traits in the sedentary state and in response to endurance training. Displayed in Figure 13 is the contrast between the original HERITAGE plan versus the new and expanded effort currently in progress based on the contributions of several laboratories. The interest in both baseline exercise-related phenotypes in a sedentary cohort and in the variability of the response to training persists. However, the goal of trying to account for the biology of human variation based primarily on demographics, physiology, metabolism, behavior, and genetics has been greatly augmented. Because of the availability of the HERITAGE biobank, it has become possible to deploy other omics, including more extensive panels of genomic variants (about 18 million measured and imputed SNPs), muscle transcriptomic (in the subset with muscle biopsies), and plasma proteomic (a panel of 5000 proteins), metabolomic (a panel of about 3000 targeted and untargeted metabolites), and lipidomic profiles, as well as to increase our capability to define the biology and molecular foundation of exercise-related phenotypes. This whole new effort is supported by additional resources in computational biology and bioinformatics.
FIGURE 13.
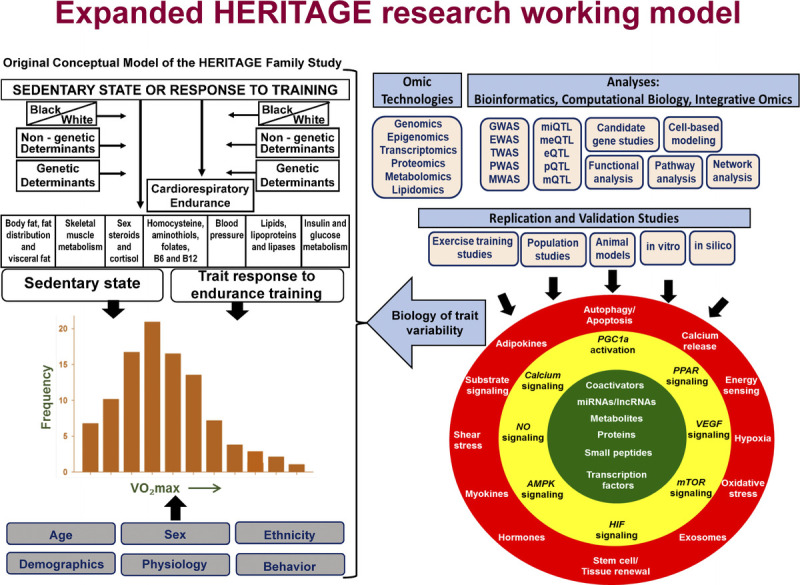
Schematic display of the original HERITAGE working model (left side of the figure) and the expanded research strategy deployed to investigate the biology and molecular determinants of exercise-related traits in the sedentary state and their responsiveness to an endurance training program meeting the requirements of the Physical Activity Guidelines.
Physical activity level and cardiorespiratory fitness are both related to risk profiles, common morbidities, the risk of cardiovascular disease, diabetes and obesity, and all-cause and cardiovascular mortality. Physical inactivity has been shown to have an impact on all-cause mortality comparable to that of smoking and obesity, and is estimated to be responsible for more than 5 million deaths worldwide (284). Forty years ago, it was shown for the first time that cardiorespiratory fitness was associated with mortality (285). Since then, many studies have confirmed that low cardiorespiratory fitness level was associated with a higher rate of all-cause and cardiovascular mortality, even after adjustment for other known risk factors (286). However, in spite of a large number of observational studies, it is still not clear whether the observed favorable effect of cardiorespiratory fitness depends on the intrinsic level observed in the sedentary state or if it is mainly driven by the low to moderate improvements commonly seen with exposures to physical activity (287). The expanded research program of HERITAGE cannot provide direct evidence in favor of or against either hypothesis, but it does provide highly relevant information on the molecular transducers and highly complex biology of cardiorespiratory fitness and common cardiometabolic traits in the causal paths linking physical activity, fitness, and health.
Supplementary Material
Acknowledgments
Thanks are expressed to the HERITAGE families who participated in the study. The authors also want to express our gratitude to all colleagues, research collaborators, and technical personnel who were involved in the planning and execution of the study. They are listed in Supplementary Table S3 (http://links.lww.com/MSS/C482). The authors thank Melanie Peterson for her support in the preparation and editing of the manuscript.
The HERITAGE Family Study was initially funded by the National Heart, Lung and Blood Institute (NHLBI) through the following grants: HL45670 (C. Bouchard, PI) from 1992 to 2010, HL47317 (D. C. Rao, PI) from 1992 to 2010, HL47321 (J.H. Wilmore, PI, deceased) from 1992 to 2003, HL47323 (A. S. Leon, PI) from 1992 to 2003, and HL 47327 (J.S. Skinner, PI) from 1992 to 2003. Current funding for HERITAGE is provided to Dr. Sarzynski (R01HL146462) and to Drs. Gerszten and Sarzynski (R01NR019628). Dr. Bouchard is supported by the John W. Barton Sr. Chair in Genetics and Nutrition and by National Institutes of Health (NIH) P30GM118430. Dr. Clish is supported by NIH U24DK112340. Dr. Després is the Scientific Director of the International Chair on Cardiometabolic Risk supported by the Fondation de l’Université Laval, and he is supported by the Canadian Institutes of Health Research (FDN-167278). Dr. Gerszten is supported by NIH U54GM115428, R01DK081572, R01NR019628, and U24DK112340. Dr. Ghosh is supported by NIGMS U54GM104940 andP20GM103528 and the National Medical Research Council, Ministry of Health Singapore (WBS R913200076263). Dr. Robbins is supported by NHLBI K23HL150327-02 and John S. LaDue Memorial Fellowship in Cardiology at Harvard Medical School. Dr. Sarzynski is supported by NIH R01HL146462, R01NR019628, R01DK128057, and SC INBRE P20GM103499. Dr. Tchernof is Co-director of the Research Chair in Bariatric and Metabolic Surgery at Laval University. Dr. Tremblay is the holder of the Canada Research Chair in Environment and Energy Balance funded by the Natural Sciences and Engineering Research Council of Canada. Mr. Barber is supported by American Heart Association Predoctoral Fellowship Award 833917 and NIH SC INBRE P20 GM103499.
HERITAGE website: A website containing general information on the HERITAGE Family Study, including a listing of all associated publications, can be found at www.HeritageFamilyStudy.com.
APPENDIX HERITAGE Family Study Review Paper
SUPPLEMENTARY TABLE S1.
HERITAGE Consortium Steering Committee.
| Claude Bouchard, PhD; Chairperson | Laval University (until 1999); later Pennington Biomedical Research Center |
| Jacques Gagnon, PhD; Project Director (1992–1999) | Laval University |
| Jean Paul Albert, MBA; Administrator (1992–1999) Tuomo Rankinen, PhD; Project Director (1999–2012) |
Laval University Pennington Biomedical Research Center |
| Arthur S. Leon, MD | University of Minnesota |
| D. C. Rao, PhD | Washington University |
| James S. Skinner, PhD | Arizona State University (until 1995); Indiana University (1996+) |
| Jack H. Wilmore, PhD | The University of Texas at Austin (until 1997), later Texas A&M University, then back to The University of Texas at Austin after 2003 |
SUPPLEMENTARY TABLE S2.
HERITAGE Consortium Advisory Board (1992–2008).
| Elizabeth Barrett-Connor, MD | University of California, San Diego |
| Jean Davignon, MD | Clinical Research Institute of Montreal |
| E. Randy Eichner, MD (1992–2002) | University of Oklahoma |
| Robert C. Elston, PhD | Louisiana State University and Case Western Reserve University |
| William L. Haskell, PhD | Stanford University |
| Rudy Leibel, MD (2002–2008) | Columbia University |
| Eric Ravussin, PhD (2002–2008) | Pennington Biomedical Research Center |
SUPPLEMENTARY TABLE S3.
HERITAGE coordinating centers, clinical centers, and core laboratories: list of personnel.
| Laval University Consortium Coordinating Center and Clinical Center | |
| PI: Claude Bouchard, PhD Local Project Coordinator: Marcelle Lareau, MSc | |
| Jean-Paul Albert, MBA | Benoit Lamarche, PhD |
| Manon Baril, BSc | Claude Leblanc, MSc |
| Jean Bergeron, MD | Gilles Lortie, MD, PhD |
| Come S. Bouchard, MSc | Isabel Mercier, MSc |
| Anne-Marie Bricault, MSc | François Michaud, BSc |
| Yvon Chagnon, PhD | Chantal Paré, BSc |
| Jean-Pierre Despres, PhD | Louis Pérusse, PhD |
| Diane Drolet, MSc | Denis Prud’homme, MD, MSc |
| Jacques Gagnon, PhD | Jean-Aime Simoneau, PhD |
| My-Anh Ho-Kim, MSc | Germain Thériault, MD |
| Karen Horth, | Angelo Tremblay, PhD |
| Louise Laberge, | John Weisnagel, MD, PhD |
| Washington University Data Coordinating Center | |
| PI: D. C. Rao, PhD | |
| Ping An, MD | Zhaohai Li, PhD |
| Ingrid B. Borecki, PhD | Stephen Mendel, PhD |
| Harry Cheng, MSc | Laura E. Mitchell, PhD |
| Gu Chi, PhD | Michael A. Province, PhD |
| Warwick Daw, PhD | Treva Rice, PhD |
| Habib El-Moalem, PhD | Kathryn A. Schallert, BSc |
| John O. Holloszy, MD | Kenneth B. Schechtman, PhD |
| Yuling Hong, MD | George P. Vogler, PhD |
| David J. Lerner, BSc | |
| Laval University Core Laboratories | |
| Diabetes Research Unit | Lipid Research Center |
| André Nadeau, MD, PhD; Director | Paul Lupien, MD, PhD; Director |
| Yolande Montreuil, RN | Jean-Pierre Després, PhD |
| Gilles Tancrède, MSc | Sital Moorjani, PhD |
| DNA and Cell Line Unit | Steroid Laboratory |
| France T. Dionne, PhD; Director | Alain Bélanger, PhD; Director |
| Monique Chagnon, ART | Simon Caron, MSc |
| Caroline Noel, BSc | Fernand Labrie, MD, PhD |
| Marie-Christine Thibault, PhD | Line Lavoie, MSc |
| University of Minnesota Clinical Center | |
| PI: Arthur S. Leon, MD Local Project Coordinator: Ava J. Walker, PhD | |
| Marilyn Borkon, | Marcella Meyers, PhD |
| Fernando S. Branco, MD | James P. Norton, MSc |
| Erik J. Ekstrom, RD | Robert C. Serfass, PhD |
| Steve Gaskill, PhD | M. Katie Schmitz, MSc |
| William V. Mendoza, MD | |
|
Arizona State University Clinical Center (Until 1995) Indiana University Clinical Center (1996+) | |
| PI: James S. Skinner, PhD Local Project Coordinator in Arizona: Kristine M. Wilmore, MA Local Project Coordinator in Indianapolis: Joanne Krasnoff, PhD | |
| Kelly Enders, RN | |
| Anna A. Jaskolska, PhD | |
| Artur J. Jaskolski, PhD | |
| University of Texas at Austin Clinical Center | |
| PI: Jack H. Wilmore, PhD Local Project Coordinator: Philip R. Stanforth, MSc | |
| Melissa Domenick, MA | |
| David W. Hayes, MD | |
| Paul J. Roach, MD | |
SUPPLEMENTARY TABLE S4.
Listing of some of the most novel or impactful findings from the HERITAGE Family Study from 1995 to 2021.
| Novel or Impactful Findings | Trait(s) | Details | Citation(s) |
|---|---|---|---|
| Heterogeneity of training responses | |||
| Large interindividual differences in phenotype responses to endurance training | V̇O2max | V̇O2max responses ranged from no gain to >1 L·min−1 (−5% to +48%). | (43,44,74) |
| IVGTT traits | The proportion of subjects who had no change or decreases in IVGTT traits ranged from 42% to 55%. | ||
| No correlation between baseline V̇O2max and its changes with training | V̇O2max | The correlation between baseline and delta V̇O2max was 0.08, with baseline levels after adjustments explaining only 2% of the variance in delta V̇O2max. | (33,43) |
| No aggregation of training responsiveness in individuals or subgroups | V̇O2max | There were low, average, and high V̇O2max responses across ages, both sexes, both ethnic groups, and levels of initial V̇O2max. | (43) |
| V̇O2max, % fat, AVF, insulin, HDL-C, small LDL, GlycA | Almost half of the cohort had at least one high response and one low response across seven cardiometabolic traits. There is a high degree of trait specificity in training responsiveness. |
(283) | |
| Some unfavorable responses to endurance training | Fasting insulin; HDL-C; TG; SBP |
Prevalence of potentially “adverse” response in HERITAGE ranged from 6% to 9% for the four traits. | (75) |
| Familial aggregation of baseline phenotype levels | |||
| Within-subject variability | V̇O2max | Within-subject SD from measures repeated days and weeks apart range from 108 to 137 mL O2·min−1 with CV of 4.1% to 5.0%. | (20,27,33,34), plus unpublished data |
| Significant familial resemblance for intrinsic V̇O2max | V̇O2max | 2.7 times more variance between families than within families. Heritability of intrinsic V̇O2max was 51%, with significant maternal heritability (29%). | (45) |
| Significant familial aggregation for intrinsic submaximal V̇O2 | V˙O2 at 50 W, 60%, 80% | Maximal heritability estimates ranged from 48% to 70% and 29% to 48% for maternal heritability. | (48) |
| Significant familial aggregation for intrinsic submaximal Q and SV | SV and Q at 50 W and 60% V̇O2max | Maximal heritability estimates of 46% for SV and Q at 60% V̇O2max and 41% and 42% at 50 W | (58) |
| Significant familial aggregation for visceral fat | AVF | Heritability of 47% to 48%, independent of total body fat | (32) |
| Higher heritability of resting BP in Black subjects | Resting SBP and DBP | Heritabilities of SBP and DBP were 68% and 56% in Black subjects vs 43% and 24% in White subjects. | (56) |
| Significant familial aggregation for muscle enzyme activities | CK, PHOS, HK, PFK, GAPDH, LPL, CPT, HADH, CS, COX | Strong familial aggregation for activities of baseline muscle enzymes related to PCr, glycolytic, and oxidative metabolism. | (138,143) |
| Significant familial aggregation for markers of oxidative stress | LDL-ox, C50-AAPH, TBARS, glutathione | Heritability for oxidative stress traits ranged from 31% to 44%. | (127) |
| Familial aggregation of training response | |||
| Significant familial aggregation for V̇O2max training response | V̇O2max | Heritability was 47%, with maternal inheritance accounting for 28%. | (41) |
| Significant familial aggregation for submaximal V̇O2 training response | V̇O2 at 50 W, 60%, 80% | Heritability values ranged from 23% to 57% for the training response of submaximal measures of V̇O2. | (48) |
| Significant familial aggregation of submaximal exercise BP and HR training responses | HR50, HR60%, SBP50 | Heritabilities for HR50, HR60%, and SBP50 responses were 34%, 29%, and 22%. Heritability for DBP traits and all HR and BP traits were lower in Black subjects. | (71) |
| Significant familial aggregation for submaximal Q & SV training responses | SV & Q at 50 W and 60% V̇O2max | Heritabilities ranged from 24% to 38% for SV and Q at 50 W and 60% V̇O2max. | (58) |
| Significant familial aggregation of interindividual variation in plasma lipid responsiveness to training | TC, TG, LDL-C, apoB, HDL-C, HDL2-C, HDL3-C, apoA-I | Heritability ranged from 25% to 38% for lipid response traits. Exceptions were for heritability levels near 60% for changes in apoB in Blacks and HDL2-C in Whites and a lack of heritability for change in LDL-C in Black subjects. | (98) |
| Significant familial aggregation for training response of markers of oxidative stress | LDL-ox, C50-AAPH, TBARS, glutathione | Heritability for oxidative stress training response traits ranged from 35% to 84%. | (127) |
| Significant familial aggregation for training response of muscle enzyme activities | CK, PHOS, HK, PFK, GAPDH, CPT, HADH, CS, COX | Strong familial aggregation was found for training response of muscle enzymes related to PCr, glycolytic, and oxidative metabolism. | (138,143) |
| Ethnic and sex differences in responses to training | |||
| Training responses of submaxmial exercise measures of hemodynamic traits differed by sex and ethnic groups | HR, SBP, DBP at 50 W | Submaximal HR, SBP, and DBP decreased with training, with greater reductions in women compared with men and in Black and older subjects compared with White and younger subjects. | (17), pp. 10–116 |
| a-V̇O2 diff, SV, Q, and V̇O2 at 50 W | Black men did not increase a-V̇O2 diff at 50 W. Thus, on average, Black men had greater increases in SV50 and smaller decreases in Q50 compared with White men to achieve similar VO250. | (61), MSSE, pp. 99–106 | |
| Significant sex interactions for insulin sensitivity response to training | Si | Insulin sensitivity increased by 10% in the total cohort, with the increase larger in men than women (16% vs 5%). | (74) |
| Significant ethnic and sex differences in lipid, lipoprotein, and lipase activity responses to training | ApoA-I, HDL2-C | ApoA-I increased more in women than men, in Black than White subjects, and in offspring than in parents. Black subjects experienced greater increases in HDL2-C compared with White subjects. | (84) |
| LPL activity | LPL activity increased in all subgroups except Black men. | (97) | |
| Response of other traits to training | |||
| Little influence of endurance training on markers of oxidative stress | LDL-ox, C50-AAPH, TBARS, antioxidants, and aminothiols | Only erythrocyte resistance to hemolysis significantly changed with training, which interacted with smoking status (smokers did not experience beneficial effects of training on erythrocytes), and was significant in women only. | (127) |
| No change in RMR with training. | RMR via indirect calorimetry | Sample size (N = 77) was larger than previous studies. There was no change in RMR at 24 or 72 h after training. | (153) |
| Favorable changes in clinically relevant lipoprotein subfractions in response to training | NMR-based lipoprotein subfractions | Large HDL-P and LDL-P increased, whereas small LDL-P and all VLDL subfractions and VLDL-P size decreased with training. These findings were not captured with traditional lipid profiling (i.e., TG and LDL-C did not change in total sample). | (93) |
| Genome-wide linkage studies of baseline and response phenotypes | |||
| Identification of QTLs for baseline V̇O2max and for V̇O2max response | V̇O2max | QTLs on 4q, 8q, 11p, and 14q were reported for baseline V̇O2max. QTLs on 1p, 2p, 4q, 6p, and 11p were identified for change in V̇O2max. | (210) |
| Dense mapping of 4 QTLs identified strong candidate genes for submaximal exercise capacity and hemodynamic responses to training | SV50, HR50 | Titin, kinesin family member 5B, cAMP responsive element binding protein 1, MIPEP, and SGCG genes were identified as strong candidates for changes in submaximal SV and HR and submaximal exercise capacity. | (211,231,233) |
| First GWAS of exercise response traits | |||
| GWAS of V̇O2max response to endurance training | V̇O2max | 39 SNPs were associated at P < 1.5 × 10−4, with a panel of 21 SNPs accounting for 49% of the variance in V̇O2max trainability. | (234) |
| GWAS of submaximal HR response to training | HR50 | 40 SNPs associated at P < 9.9 × 10–5, with top hit 8 × 10−7. Nine SNPs accounted for the genetic variance of the submaximal exercise HR response to training. | (235) |
| Molecular signatures of exercise response derived from integrative analyses of genomic and transcriptomic profiles | |||
| Muscle gene expression and SNP signatures predict V̇O2max response to endurance training | V̇O2max | Genome-wide baseline muscle gene expression and validation identified a 29-RNA signature that predicted changes in V̇O2max. Candidate genes from this predictor and the literature led to a 11-SNP signature that explained 23% of the variance in V̇O2max trainability. | (265) |
| Combined genome-wide and transcriptome-wide analysis identifies SNPs associated with TG response to training | TG | GWAS identified 4 SNPs accounting for the genetic variance of TG response, whereas molecular signature based on the baseline expression of 11 genes predicted 27% of TG changes in response to training. An 8-SNP score comprising 4 SNPs each from transcriptomics and GWAS was the strongest predictor of TG training response. | (236) |
| GWAS and transcriptional signature of insulin sensitivity response to training | Si | Integrative analysis of functional genomic and transcriptomic profiles identified combined variation in genes linked to cholinergic, calcium, and chemokine signaling associated with Si training response. MEF2A transcription factor was the most significant candidate driving the transcriptional signature associated with ∆Si, strengthening the relevance of calcium signaling in exercise training-mediated Si response. | (238) |
| Proteomic signatures of V̇O2max and its trainability | |||
| Plasma protein signature of cardiorespiratory fitness level | V̇O2max | Elastic net regression identified 115 proteins highly correlated (r2 = 0.80) with measured V̇O2max, which was replicated in the validation data set, with an r2 of 0.71 (Fig. 9). | (271) |
| Plasma proteins associated with intrinsic V̇O2max and its trainability | V̇O2max | 147 proteins were associated with baseline V̇O2max, whereas 102 baseline proteins were associated with changes in V̇O2max, with minimal overlap (only 5 proteins) between protein sets. A baseline 56-protein signature improved prediction of V̇O2max response (AUC 0.84) compared with a model of only clinical variables (AUC 0.62). | (272) |
| Bioinformatics explorations of intrinsic V̇O2max and its trainability | |||
| Integrative pathway analysis of V̇O2max response to training GWAS | V̇O2max | Using GWAS results followed by candidate gene prioritization and pathway analysis, pathways related to calcium signaling, energy sensing and partitioning, mitochondrial biogenesis, angiogenesis, immune functions, and regulation of autophagy and apoptosis were identified as key mechanisms through which the physiological responses of V̇O2max to training are mediated. | (273) |
| Genetics and biology underlying intrinsic V̇O2max | V̇O2max | A bioinformatics pipeline applied to V̇O2max data in the sedentary state suggests 4 loci related to cardiovascular physiology (ATE1, CASQ2, NOTO, and SGCG), four loci related to hematopoiesis (PICALM, SSB, CASQ2, and CA9), 4 loci related to skeletal muscle function (SGCG, DMRT2, ADARB1, and CASQ2), and 8 loci related to metabolism (ATE1, PICALM, RAB11FIP5, GBA2, SGCG, PRADC1, ARL6IP5, and CASQ2) as candidates for human variability in cardiorespiratory fitness among sedentary adults. | (275) |
| Metabolomic biomarker of training responsiveness | |||
| DMGV is a biomarker of metabolic response to endurance training | Fasting glucose, insulin, and lipids | Baseline levels of plasma DMGV associated with lack of improvement in HDL traits. DMGV levels decreased with training and were positively correlated with several lipid, glucose, and insulin traits | (96) |
Footnotes
This review is dedicated to the late Jack H. Wilmore, PhD, and to the HERITAGE Project Directors: Jacques Gagnon, PhD (from 1992 to 1999), and Tuomo Rankinen, PhD (from 1999 to 2012).
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.acsm-msse.org).
Contributor Information
MARK A. SARZYNSKI, Email: SARZ@mailbox.sc.edu.
TREVA K. RICE, Email: treva@wustl.edu.
JEAN-PIERRE DESPRÉS, Email: Jean-Pierre.Despres@criucpq.ulaval.ca.
LOUIS PÉRUSSE, Email: Louis.Perusse@kin.ulaval.ca.
ANGELO TREMBLAY, Email: angelo.tremblay@kin.ulaval.ca.
PHILIP R. STANFORTH, Email: p.stanforth@austin.utexas.edu.
ANDRÉ TCHERNOF, Email: andre.tchernof@criucpq.ulaval.ca.
JACOB L. BARBER, Email: barberj6@email.sc.edu.
FRANCESCO FALCIANI, Email: F.Falciani@liverpool.ac.uk.
CLARY CLISH, Email: clary@broadinstitute.org.
JEREMY M. ROBBINS, Email: jrobbin1@bidmc.harvard.edu.
SUJOY GHOSH, Email: sg45653@gmail.com.
ROBERT E. GERSZTEN, Email: rgerszte@bidmc.harvard.edu.
ARTHUR S. LEON, Email: leonx003@umn.edu.
JAMES S. SKINNER, Email: jimskinnrphd@gmail.com.
D. C. RAO, Email: rao@wustl.edu.
REFERENCES
- 1.Despres JP, Bouchard C, Savard R, Prud’homme D, Bukowiecki L, Theriault G. Adaptive changes to training in adipose tissue lipolysis are genotype dependent. Int J Obes (Lond). 1984;8(1):87–95. [PubMed] [Google Scholar]
- 2.Savard R, Després JP, Marcotte M, Bouchard C. Endurance training and glucose conversion into triglycerides in human fat cells. J Appl Physiol (1985). 1985;58(1):230–5. [DOI] [PubMed] [Google Scholar]
- 3.Despres JP Moorjani S Tremblay A, et al. Heredity and changes in plasma lipids and lipoproteins after short-term exercise training in men. Arteriosclerosis. 1988;8(4):402–9. [DOI] [PubMed] [Google Scholar]
- 4.Simoneau JA, Lortie G, Boulay MR, Marcotte M, Thibault MC, Bouchard C. Inheritance of human skeletal muscle and anaerobic capacity adaptation to high-intensity intermittent training. Int J Sports Med. 1986;7(3):167–71. [DOI] [PubMed] [Google Scholar]
- 5.Lortie G, Simoneau JA, Hamel P, Boulay MR, Landry F, Bouchard C. Responses of maximal aerobic power and capacity to aerobic training. Int J Sports Med. 1984;5(5):232–6. [DOI] [PubMed] [Google Scholar]
- 6.Prud’homme D, Bouchard C, Leblanc C, Landry F, Fontaine E. Sensitivity of maximal aerobic power to training is genotype-dependent. Med Sci Sports Exerc. 1984;16(5):489–93. [DOI] [PubMed] [Google Scholar]
- 7.Hamel P, Simoneau JA, Lortie G, Boulay MR, Bouchard C. Heredity and muscle adaptation to endurance training. Med Sci Sports Exerc. 1986;18(6):690–6. [PubMed] [Google Scholar]
- 8.Bouchard C, Leon AS, Rao DC, Skinner JS, Wilmore JH, Gagnon J. The HERITAGE Family Study. Aims, design, and measurement protocol. Med Sci Sports Exerc. 1995;27(5):721–9. [PubMed] [Google Scholar]
- 9.Baecke JA, Burema J, Frijters JE. A short questionnaire for the measurement of habitual physical activity in epidemiological studies. Am J Clin Nutr. 1982;36(5):936–42. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs DR, Jr, Ainsworth BE, Hartman TJ, Leon AS. A simultaneous evaluation of 10 commonly used physical activity questionnaires. Med Sci Sports Exerc. 1993;25(1):81–91. [DOI] [PubMed] [Google Scholar]
- 11.Willett WC Sampson L Stampfer MJ, et al. Reproducibility and validity of a semiquantitative food frequency questionnaire. Am J Epidemiol. 1985;122(1):51–65. [DOI] [PubMed] [Google Scholar]
- 12.Hunninghake DB, Mullis RM, Brekke ML, Peters JR, Quiter ES. A simple tool for the assessment of dietary fat intake in clinical populations. Circulation. 1987;76(IV Suppl):35. [Google Scholar]
- 13.Peters JR Quiter ES Brekke ML, et al. The Eating Pattern Assessment Tool: a simple instrument for assessing dietary fat and cholesterol intake. J Am Diet Assoc. 1994;94(9):1008–13. [DOI] [PubMed] [Google Scholar]
- 14.Wilmore JH Despres JP Stanforth PR, et al. Alterations in body weight and composition consequent to 20 wk of endurance training: the HERITAGE Family Study. Am J Clin Nutr. 1999;70(3):346–52. [DOI] [PubMed] [Google Scholar]
- 15.Behnke AR, Wilmore JH. Evaluation and Regulation of Body Build and Composition. Englewood Cliffs (NJ).: Prentice-Hall; 1974, xiv. 236 p. [Google Scholar]
- 16.Sjostrom L, Kvist H, Cederblad A, Tylen U. Determination of total adipose tissue and body fat in women by computed tomography, 40K, and tritium. Am J Physiol. 1986;250(6 Pt 1):E736–45. [DOI] [PubMed] [Google Scholar]
- 17.Wilmore JH Stanforth PR Gagnon J, et al. Heart rate and blood pressure changes with endurance training: the HERITAGE Family Study. Med Sci Sports Exerc. 2001;33(1):107–16. [DOI] [PubMed] [Google Scholar]
- 18.Després JP Gagnon J Bergeron J, et al. Plasma post-heparin lipase activities in the HERITAGE Family Study: the reproducibility, gender differences, and associations with lipoprotein levels. HEalth, RIsk factors, exercise Training and GEnetics. Clin Biochem. 1999;32(3):157–65. [DOI] [PubMed] [Google Scholar]
- 19.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther. 2003;5(6):1003–15. [DOI] [PubMed] [Google Scholar]
- 20.Skinner JS Wilmore KM Jaskolska A, et al. Reproducibility of maximal exercise test data in the HERITAGE Family Study. Med Sci Sports Exerc. 1999;31(11):1623–8. [DOI] [PubMed] [Google Scholar]
- 21.Nilsson K. Establishment of permanent human lymphblastoid cell lines in vitro. In: Bloom BR, David JR, editors. In Vitro Methods in Cell-Mediated and Tumor Immunity. New York (NY): Academic Press; 1976. pp. 713–35. [Google Scholar]
- 22.Daw EW Province MA Gagnon J, et al. Reproducibility of the HERITAGE Family Study intervention protocol: drift over time. Ann Epidemiol. 1997;7(7):452–62. [DOI] [PubMed] [Google Scholar]
- 23.El-Moalem HE Gagnon J Province M, et al. Race differences in reproducibilities: the HERITAGE Family Study. Am J Hum Biol. 1997;9(4):415–24. [DOI] [PubMed] [Google Scholar]
- 24.Gagnon J Province MA Bouchard C, et al. The HERITAGE Family Study: quality assurance and quality control. Ann Epidemiol. 1996;6(6):520–9. [DOI] [PubMed] [Google Scholar]
- 25.Stanforth PR Gagnon J Rice T, et al. Reproducibility of resting blood pressure and heart rate measurements. The HERITAGE Family Study. Ann Epidemiol. 2000;10(5):271–7. [DOI] [PubMed] [Google Scholar]
- 26.Wilmore JH Stanforth PR Domenick MA, et al. Reproducibility of anthropometric and body composition measurements: the HERITAGE Family Study. Int J Obes Relat Metab Disord. 1997;21(4):297–303. [DOI] [PubMed] [Google Scholar]
- 27.Wilmore JH Stanforth PR Turley KR, et al. Reproducibility of cardiovascular, respiratory, and metabolic responses to submaximal exercise: the HERITAGE Family Study. Med Sci Sports Exerc. 1998;30(2):259–65. [DOI] [PubMed] [Google Scholar]
- 28.He Z, Rankinen T, Leon AS, Skinner JS, Tchernof A, Bouchard C. Plasma steroids and cardiorespiratory fitness response to regular exercise. In: Spiegelman B, editor. Hormones, Metabolism and the Benefits of Exercise. Cham (Switzerland); 2017. pp. 25–42. [PubMed] [Google Scholar]
- 29.Ardern CI Katzmarzyk PT Janssen I, et al. Race and sex similarities in exercise-induced changes in blood lipids and fatness. Med Sci Sports Exerc. 2004;36(9):1610–5. [DOI] [PubMed] [Google Scholar]
- 30.Janssen I Katzmarzyk PT Ross R, et al. Fitness alters the associations of BMI and waist circumference with total and abdominal fat. Obes Res. 2004;12(3):525–37. [DOI] [PubMed] [Google Scholar]
- 31.Perusse L Rice T Province MA, et al. Familial aggregation of amount and distribution of subcutaneous fat and their responses to exercise training in the HERITAGE Family Study. Obes Res. 2000;8(2):140–50. [DOI] [PubMed] [Google Scholar]
- 32.Rice T Despres JP Daw EW, et al. Familial resemblance for abdominal visceral fat: the HERITAGE Family Study. Int J Obes Relat Metab Disord. 1997;21(11):1024–31. [DOI] [PubMed] [Google Scholar]
- 33.Sarzynski MA, Ghosh S, Bouchard C. Genomic and transcriptomic predictors of response levels to endurance exercise training. J Physiol. 2017;595(9):2931–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shephard RJ, Rankinen T, Bouchard C. Test–retest errors and the apparent heterogeneity of training response. Eur J Appl Physiol. 2004;91(2–3):199–203. [DOI] [PubMed] [Google Scholar]
- 35.Gaskill SE Walker AJ Serfass RA, et al. Changes in ventilatory threshold with exercise training in a sedentary population: the HERITAGE Family Study. Int J Sports Med. 2001;22(8):586–92. [DOI] [PubMed] [Google Scholar]
- 36.Sarzynski MA Rankinen T Earnest CP, et al. Measured maximal heart rates compared to commonly used age-based prediction equations in the HERITAGE Family Study. Am J Hum Biol. 2013;25(5):695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fox SM, 3rd, Naughton JP, Haskell WL. Physical activity and the prevention of coronary heart disease. Ann Clin Res. 1971;3(6):404–32. [PubMed] [Google Scholar]
- 38.Tanaka H, Monahan KD, Seals DR. Age-predicted maximal heart rate revisited. J Am Coll Cardiol. 2001;37(1):153–6. [DOI] [PubMed] [Google Scholar]
- 39.Skinner JS Wilmore KM Krasnoff JB, et al. Adaptation to a standardized training program and changes in fitness in a large, heterogeneous population: the HERITAGE Family Study. Med Sci Sports Exerc. 2000;32(1):157–61. [DOI] [PubMed] [Google Scholar]
- 40.Wilmore JH Green JS Stanforth PR, et al. Relationship of changes in maximal and submaximal aerobic fitness to changes in cardiovascular disease and non-insulin-dependent diabetes mellitus risk factors with endurance training: the HERITAGE Family Study. Metabolism. 2001;50(11):1255–63. [DOI] [PubMed] [Google Scholar]
- 41.Bouchard C An P Rice T, et al. Familial aggregation of VO(2max) response to exercise training: results from the HERITAGE Family Study. J Appl Physiol (1985). 1999;87(3):1003–8. [DOI] [PubMed] [Google Scholar]
- 42.Skinner JS Gaskill SE Rankinen T, et al. Heart rate versus %VO2max: age, sex, race, initial fitness, and training response—HERITAGE. Med Sci Sports Exerc. 2003;35(11):1908–13. [DOI] [PubMed] [Google Scholar]
- 43.Skinner JS Jaskolski A Jaskolska A, et al. HERITAGE Family Study . Age, sex, race, initial fitness, and response to training: the HERITAGE Family Study. J Appl Physiol (1985). 2001;90(5):1770–6. [DOI] [PubMed] [Google Scholar]
- 44.Bouchard C, Rankinen T. Individual differences in response to regular physical activity. Med Sci Sports Exerc. 2001;33(6 Suppl):S446–51; discussion S52-3. [DOI] [PubMed] [Google Scholar]
- 45.Bouchard C Daw EW Rice T, et al. Familial resemblance for VO2max in the sedentary state: the HERITAGE Family Study. Med Sci Sports Exerc. 1998;30(2):252–8. [DOI] [PubMed] [Google Scholar]
- 46.Lesage R, Simoneau JA, Jobin J, Leblanc J, Bouchard C. Familial resemblance in maximal heart rate, blood lactate and aerobic power. Hum Hered. 1985;35(3):182–9. [DOI] [PubMed] [Google Scholar]
- 47.Montoye HJ, Gayle R. Familial relationships in maximal oxygen uptake. Hum Biol. 1978;50(3):241–9. [PubMed] [Google Scholar]
- 48.Pérusse L Gagnon J Province MA, et al. Familial aggregation of submaximal aerobic performance in the HERITAGE Family Study. Med Sci Sports Exerc. 2001;33(4):597–604. [DOI] [PubMed] [Google Scholar]
- 49.Pérusse L Leblanc C Tremblay A, et al. Familial aggregation in physical fitness, coronary heart disease risk factors, and pulmonary function measurements. Prev Med. 1987;16(5):607–15. [DOI] [PubMed] [Google Scholar]
- 50.Pérusse L, Leblanc C, Bouchard C. Inter-generation transmission of physical fitness in the Canadian population. Can J Sport Sci. 1988;13(1):8–14. [PubMed] [Google Scholar]
- 51.Gaskill SE Rice T Bouchard C, et al. Familial resemblance in ventilatory threshold: the HERITAGE Family Study. Med Sci Sports Exerc. 2001;33(11):1832–40. [DOI] [PubMed] [Google Scholar]
- 52.Bouchard C Tremblay A Després JP, et al. The response to exercise with constant energy intake in identical twins. Obes Res. 1994;2(5):400–10. [DOI] [PubMed] [Google Scholar]
- 53.An P Rice T Perusse L, et al. Complex segregation analysis of blood pressure and heart rate measured before and after a 20-week endurance exercise training program: the HERITAGE Family Study. Am J Hypertens. 2000;13(5 Pt 1):488–97. [DOI] [PubMed] [Google Scholar]
- 54.Carter CL, Kannel WB. Evidence of a rare gene for low systolic blood pressure in the Framingham heart study. Hum Hered. 1990;40(4):235–41. [DOI] [PubMed] [Google Scholar]
- 55.Rice T, Province M, Perusse L, Bouchard C, Rao DC. Cross-trait familial resemblance for body fat and blood pressure: familial correlations in the Quebec Family Study. Am J Hum Genet. 1994;55(5):1019–29. [PMC free article] [PubMed] [Google Scholar]
- 56.Gu C Borecki I Gagnon J, et al. Familial resemblance for resting blood pressure with particular reference to racial differences: preliminary analyses from the HERITAGE Family Study. Hum Biol. 1998;70(1):77–90. [PubMed] [Google Scholar]
- 57.Moll PP, Harburg E, Burns TL, Schork MA, Ozgoren F. Heredity, stress and blood pressure, a family set approach: the Detroit Project Revisited. J Chronic Dis. 1983;36(4):317–28. [DOI] [PubMed] [Google Scholar]
- 58.An P Rice T Gagnon J, et al. Familial aggregation of stroke volume and cardiac output during submaximal exercise: the HERITAGE Family Study. Int J Sports Med. 2000;21(8):566–72. [DOI] [PubMed] [Google Scholar]
- 59.Wilmore JH Stanforth PR Gagnon J, et al. Endurance exercise training has a minimal effect on resting heart rate: the HERITAGE study. Med Sci Sports Exerc. 1996;28(7):829–35. [DOI] [PubMed] [Google Scholar]
- 60.Kelley G, Tran ZV. Aerobic exercise and normotensive adults: a meta-analysis. Med Sci Sports Exerc. 1995;27(10):1371–7. [PubMed] [Google Scholar]
- 61.Wilmore JH Stanforth PR Gagnon J, et al. Cardiac output and stroke volume changes with endurance training: the HERITAGE Family Study. Med Sci Sports Exerc. 2001;33(1):99–106. [DOI] [PubMed] [Google Scholar]
- 62.Ehsani AA, Ogawa T, Miller TR, Spina RJ, Jilka SM. Exercise training improves left ventricular systolic function in older men. Circulation. 1991;83(1):96–103. [DOI] [PubMed] [Google Scholar]
- 63.Mier CM, Domenick MA, Turner NS, Wilmore JH. Changes in stroke volume and maximal aerobic capacity with increased blood volume in men women. J Appl Physiol (1985). 1996;80(4):1180–6. [DOI] [PubMed] [Google Scholar]
- 64.Seals DR, Hagberg JM, Spina RJ, Rogers MA, Schechtman KB, Ehsani AA. Enhanced left ventricular performance in endurance trained older men. Circulation. 1994;89(1):198–205. [DOI] [PubMed] [Google Scholar]
- 65.Spina RJ, Ogawa T, Martin WH, 3rd, Coggan AR, Holloszy JO, Ehsani AA. Exercise training prevents decline in stroke volume during exercise in young healthy subjects. J Appl Physiol (1985). 1992;72(6):2458–62. [DOI] [PubMed] [Google Scholar]
- 66.Green JS Stanforth PR Gagnon J, et al. Menopause, estrogen, and training effects on exercise hemodynamics: the HERITAGE study. Med Sci Sports Exerc. 2002;34(1):74–82. [DOI] [PubMed] [Google Scholar]
- 67.Lewandowski J Pruszczyk P Elaffi M, et al. Blood pressure, plasma NPY and catecholamines during physical exercise in relation to menstrual cycle, ovariectomy, and estrogen replacement. Regul Pept. 1998;75–76:239–45. [DOI] [PubMed] [Google Scholar]
- 68.Pines A Fisman EZ Shapira I, et al. Exercise echocardiography in postmenopausal hormone users with mild systemic hypertension. Am J Cardiol. 1996;78(12):1385–9. [DOI] [PubMed] [Google Scholar]
- 69.Hagberg JM, Ferrell RE, Dengel DR, Wilund KR. Exercise training-induced blood pressure and plasma lipid improvements in hypertensives may be genotype dependent. Hypertension. 1999;34(1):18–23. [DOI] [PubMed] [Google Scholar]
- 70.Rice T An P Gagnon J, et al. Heritability of HR and BP response to exercise training in the HERITAGE Family Study. Med Sci Sports Exerc. 2002;34(6):972–9. [DOI] [PubMed] [Google Scholar]
- 71.An P Pérusse L Rankinen T, et al. Familial aggregation of exercise heart rate and blood pressure in response to 20 weeks of endurance training: the HERITAGE Family Study. Int J Sports Med. 2003;24(1):57–62. [DOI] [PubMed] [Google Scholar]
- 72.Katzmarzyk PT Gagnon J Leon AS, et al. Fitness, fatness, and estimated coronary heart disease risk: the HERITAGE Family Study. Med Sci Sports Exerc. 2001;33(4):585–90. [DOI] [PubMed] [Google Scholar]
- 73.Walton C, Godsland IF, Proudler AJ, Felton C, Wynn V. Evaluation of four mathematical models of glucose and insulin dynamics with analysis of effects of age and obesity. Am J Physiol. 1992;262(5 Pt 1):E755–62. [DOI] [PubMed] [Google Scholar]
- 74.Boule NG Weisnagel SJ Lakka TA, et al. Effects of exercise training on glucose homeostasis: the HERITAGE Family Study. Diabetes Care. 2005;28(1):108–14. [DOI] [PubMed] [Google Scholar]
- 75.Bouchard C Blair SN Church TS, et al. Adverse metabolic response to regular exercise: is it a rare or common occurrence? PLoS One. 2012;7(5):e37887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burstein R, Polychronakos C, Toews CJ, MacDougall JD, Guyda HJ, Posner BI. Acute reversal of the enhanced insulin action in trained athletes. Association with insulin receptor changes. Diabetes. 1985;34(8):756–60. [DOI] [PubMed] [Google Scholar]
- 77.Heath GW, Gavin JR, 3rd, Hinderliter JM, Hagberg JM, Bloomfield SA, Holloszy JO. Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. J Appl Physiol Respir Environ Exerc Physiol. 1983;55(2):512–7. [DOI] [PubMed] [Google Scholar]
- 78.Schneider SH, Amorosa LF, Khachadurian AK, Ruderman NB. Studies on the mechanism of improved glucose control during regular exercise in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1984;26(5):355–60. [DOI] [PubMed] [Google Scholar]
- 79.Hong Y Weisnagel SJ Rice T, et al. Familial resemblance for glucose and insulin metabolism indices derived from an intravenous glucose tolerance test in Blacks and Whites of the HERITAGE Family Study. Clin Genet. 2001;60(1):22–30. [DOI] [PubMed] [Google Scholar]
- 80.Watanabe RM Valle T Hauser ER, et al. Familiality of quantitative metabolic traits in Finnish families with non-insulin-dependent diabetes mellitus. Finland–United States Investigation of NIDDM Genetics (FUSION) study investigators. Hum Hered. 1999;49(3):159–68. [DOI] [PubMed] [Google Scholar]
- 81.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101. [DOI] [PubMed] [Google Scholar]
- 82.Couillard C Bergeron J Despres JP, et al. Apolipoprotein AI- and AI:AII-containing lipoproteins in White men and women of the HERITAGE Family Study: associations with metabolic risk profile variables. Metabolism. 2003;52(12):1530–6. [DOI] [PubMed] [Google Scholar]
- 83.Despres JP Couillard C Gagnon J, et al. Race, visceral adipose tissue, plasma lipids, and lipoprotein lipase activity in men and women: the Health, Risk Factors, Exercise Training, and Genetics (HERITAGE) family study. Arterioscler Thromb Vasc Biol. 2000;20(8):1932–8. [DOI] [PubMed] [Google Scholar]
- 84.Leon AS Rice T Mandel S, et al. Blood lipid response to 20 weeks of supervised exercise in a large biracial population: the HERITAGE Family Study. Metabolism. 2000;49(4):513–20. [DOI] [PubMed] [Google Scholar]
- 85.Perusse L Rice T Despres JP, et al. Familial resemblance of plasma lipids, lipoproteins and postheparin lipoprotein and hepatic lipases in the HERITAGE Family Study. Arterioscler Thromb Vasc Biol. 1997;17(11):3263–9. [DOI] [PubMed] [Google Scholar]
- 86.Desmeules A Couillard C Tchernof A, et al. Post-heparin lipolytic enzyme activities, sex hormones and sex hormone–binding globulin (SHBG) in men and women: the HERITAGE Family Study. Atherosclerosis. 2003;171(2):343–50. [DOI] [PubMed] [Google Scholar]
- 87.Leon AS Gaskill SE Rice T, et al. Variability in the response of HDL cholesterol to exercise training in the HERITAGE Family Study. Int J Sports Med. 2002;23(1):1–9. [DOI] [PubMed] [Google Scholar]
- 88.Carr MC, Brunzell JD, Deeb SS. Ethnic differences in hepatic lipase and HDL in Japanese, Black, and White Americans: role of central obesity and LIPC polymorphisms. J Lipid Res. 2004;45(3):466–73. [DOI] [PubMed] [Google Scholar]
- 89.Friday KE Srinivasan SR Elkasabany A, et al. Black–White differences in postprandial triglyceride response and postheparin lipoprotein lipase and hepatic triglyceride lipase among young men. Metabolism. 1999;48(6):749–54. [DOI] [PubMed] [Google Scholar]
- 90.Vega GL, Clark LT, Tang A, Marcovina S, Grundy SM, Cohen JC. Hepatic lipase activity is lower in African American men than in White American men: effects of 5′ flanking polymorphism in the hepatic lipase gene (LIPC). J Lipid Res. 1998;39(1):228–32. [PubMed] [Google Scholar]
- 91.Durstine JL, Grandjean PW, Davis PG, Ferguson MA, Alderson NL, DuBose KD. Blood lipid and lipoprotein adaptations to exercise: a quantitative analysis. Sports Med. 2001;31(15):1033–62. [DOI] [PubMed] [Google Scholar]
- 92.Kodama S Tanaka S Saito K, et al. Effect of aerobic exercise training on serum levels of high-density lipoprotein cholesterol: a meta-analysis. Arch Intern Med. 2007;167(10):999–1008. [DOI] [PubMed] [Google Scholar]
- 93.Sarzynski MA Burton J Rankinen T, et al. The effects of exercise on the lipoprotein subclass profile: a meta-analysis of 10 interventions. Atherosclerosis. 2015;243(2):364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Couillard C Despres JP Lamarche B, et al. Effects of endurance exercise training on plasma HDL cholesterol levels depend on levels of triglycerides: evidence from men of the Health, Risk Factors, Exercise Training and Genetics (HERITAGE) Family Study. Arterioscler Thromb Vasc Biol. 2001;21(7):1226–32. [DOI] [PubMed] [Google Scholar]
- 95.Katzmarzyk PT Leon AS Rankinen T, et al. Changes in blood lipids consequent to aerobic exercise training related to changes in body fatness and aerobic fitness. Metabolism. 2001;50(7):841–8. [DOI] [PubMed] [Google Scholar]
- 96.Robbins JM Herzig M Morningstar J, et al. Association of dimethylguanidino valeric acid with partial resistance to metabolic health benefits of regular exercise. JAMA Cardiol. 2019;4(7):636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bergeron J Couillard C Despres JP, et al. Race differences in the response of postheparin plasma lipoprotein lipase and hepatic lipase activities to endurance exercise training in men: results from the HERITAGE Family Study. Atherosclerosis. 2001;159(2):399–406. [DOI] [PubMed] [Google Scholar]
- 98.Rice T Després JP Pérusse L, et al. Familial aggregation of blood lipid response to exercise training in the Health, Risk Factors, Exercise Training, and Genetics (HERITAGE) Family Study. Circulation. 2002;105(16):1904–8. [DOI] [PubMed] [Google Scholar]
- 99.Hong Y Rice T Gagnon J, et al. Familiality of triglyceride and LPL response to exercise training: the HERITAGE study. Med Sci Sports Exerc. 2000;32(8):1438–44. [DOI] [PubMed] [Google Scholar]
- 100.Handelsman DJ, Wartofsky L. Requirement for mass spectrometry sex steroid assays in the Journal of Clinical Endocrinology and Metabolism. J Clin Endocrinol Metab. 2013;98(10):3971–3. [DOI] [PubMed] [Google Scholar]
- 101.Ukkola O Gagnon J Rankinen T, et al. Age, body mass index, race and other determinants of steroid hormone variability: the HERITAGE Family Study. Eur J Endocrinol. 2001;145(1):1–9. [DOI] [PubMed] [Google Scholar]
- 102.He Z, Rankinen T, Leon AS, Skinner JS, Tchernof A, Bouchard C. Plasma steroids, body composition, and fat distribution: effects of age, sex, and exercise training. Int J Obes (Lond). 2018;42(7):1366–77. [DOI] [PubMed] [Google Scholar]
- 103.He Z, Rankinen T, Leon AS, Skinner J, Tchernof A, Bouchard C. Plasma steroids are not associated with resting and exercise blood pressure. Int J Sports Med. 2018;39(13):967–71. [DOI] [PubMed] [Google Scholar]
- 104.Couillard C Gagnon J Bergeron J, et al. Contribution of body fatness and adipose tissue distribution to the age variation in plasma steroid hormone concentrations in men: the HERITAGE Family Study. J Clin Endocrinol Metab. 2000;85(3):1026–31. [DOI] [PubMed] [Google Scholar]
- 105.Harman SM Metter EJ Tobin JD Pearson J Blackman MR, Baltimore Longitudinal Study of Aging . Longitudinal effects of aging on serum total and free testosterone levels in healthy men. Baltimore Longitudinal Study of Aging. J Clin Endocrinol Metab. 2001;86(2):724–31. [DOI] [PubMed] [Google Scholar]
- 106.Labrie F, Belanger A, Cusan L, Gomez JL, Candas B. Marked decline in serum concentrations of adrenal C19 sex steroid precursors and conjugated androgen metabolites during aging. J Clin Endocrinol Metab. 1997;82(8):2396–402. [DOI] [PubMed] [Google Scholar]
- 107.Blouin K Despres JP Couillard C, et al. Contribution of age and declining androgen levels to features of the metabolic syndrome in men. Metabolism. 2005;54(8):1034–40. [DOI] [PubMed] [Google Scholar]
- 108.Marchand GB Carreau AM Weisnagel SJ, et al. Increased body fat mass explains the positive association between circulating estradiol and insulin resistance in postmenopausal women. Am J Physiol Endocrinol Metab. 2018;314(5):E448–56. [DOI] [PubMed] [Google Scholar]
- 109.Perret B, Mabile L, Martinez L, Terce F, Barbaras R, Collet X. Hepatic lipase: structure/function relationship, synthesis, and regulation. J Lipid Res. 2002;43(8):1163–9. [PubMed] [Google Scholar]
- 110.An P Rice T Gagnon J, et al. Population differences in the pattern of familial aggregation for sex hormone–binding globulin and its response to exercise training: the HERITAGE Family Study. Am J Hum Biol. 2001;13(6):832–7. [DOI] [PubMed] [Google Scholar]
- 111.An P Rice T Gagnon J, et al. A genetic study of dehydroepiandrosterone sulfate measured before and after a 20-wk endurance exercise training program: the HERITAGE Family Study. Metabolism. 2000;49(3):298–304. [DOI] [PubMed] [Google Scholar]
- 112.An P Rice T Gagnon J, et al. A genetic study of sex hormone–binding globulin measured before and after a 20-week endurance exercise training program: the HERITAGE Family Study. Metabolism. 2000;49(8):1014–20. [DOI] [PubMed] [Google Scholar]
- 113.An P Rice T Gagnon J, et al. Race differences in the pattern of familial aggregation for dehydroepiandrosterone sulfate and its responsiveness to training in the HERITAGE Family Study. Metabolism. 2001;50(8):916–20. [DOI] [PubMed] [Google Scholar]
- 114.Hong Y Gagnon J Rice T, et al. Familial resemblance for free androgens and androgen glucuronides in sedentary Black and White individuals: the HERITAGE Family Study. Health, Risk Factors, Exercise Training and Genetics. J Endocrinol. 2001;170(2):485–92. [DOI] [PubMed] [Google Scholar]
- 115.Hammond GL. Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. J Endocrinol. 2016;230(1):R13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Feitosa MF Rice T Rosmond R, et al. A genetic study of cortisol measured before and after endurance training: the HERITAGE Family Study. Metabolism. 2002;51(3):360–5. [DOI] [PubMed] [Google Scholar]
- 117.Feitosa MF Rice T Rosmond R, et al. Pleiotropic relationships between cortisol levels and adiposity: the HERITAGE Family Study. Obes Res. 2002;10(12):1222–31. [DOI] [PubMed] [Google Scholar]
- 118.Feitosa MF Borecki IB Rankinen T, et al. Lack of pleiotropic genetic effects between adiposity and sex hormone–binding globulin concentrations before and after 20 weeks of exercise training: the HERITAGE Family Study. Metabolism. 2003;52(1):35–41. [DOI] [PubMed] [Google Scholar]
- 119.Chapman K, Holmes M, Seckl J. 11beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93(3):1139–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Incollingo Rodriguez AC, Epel ES, White ML, Standen EC, Seckl JR, Tomiyama AJ. Hypothalamic-pituitary-adrenal axis dysregulation and cortisol activity in obesity: a systematic review. Psychoneuroendocrinology. 2015;62:301–18. [DOI] [PubMed] [Google Scholar]
- 121.Tchernof A Brochu D Maltais-Payette I, et al. Androgens and the regulation of adiposity and body fat distribution in humans. Compr Physiol. 2018;8(4):1253–90. [DOI] [PubMed] [Google Scholar]
- 122.Tchernof A, Mansour MF, Pelletier M, Boulet MM, Nadeau M, Luu-The V. Updated survey of the steroid-converting enzymes in human adipose tissues. J Steroid Biochem Mol Biol. 2015;147:56–69. [DOI] [PubMed] [Google Scholar]
- 123.Vihma V Heinonen S Naukkarinen J, et al. Increased body fat mass and androgen metabolism—a twin study in healthy young women. Steroids. 2018;140:24–31. [DOI] [PubMed] [Google Scholar]
- 124.Vihma V Naukkarinen J Turpeinen U, et al. Metabolism of sex steroids is influenced by acquired adiposity—a study of young adult male monozygotic twin pairs. J Steroid Biochem Mol Biol. 2017;172:98–105. [DOI] [PubMed] [Google Scholar]
- 125.An P Rosmond R Borecki IB, et al. Genome-wide linkage scan to detect loci influencing levels of dehydroepiandrosterones in the HERITAGE Family Study. Metabolism. 2001;50(11):1315–22. [DOI] [PubMed] [Google Scholar]
- 126.Ukkola O Rankinen T Gagnon J, et al. A genome-wide linkage scan for steroids and SHBG levels in Black and White families: the HERITAGE Family Study. J Clin Endocrinol Metab. 2002;87(8):3708–20. [DOI] [PubMed] [Google Scholar]
- 127.Blache D Lussier-Cacan S Gagnon J, et al. Effect of exercise training on in vitro LDL oxidation and free radical-induced hemolysis: the HERITAGE Family Study. Antioxid Redox Signal. 2007;9(1):123–30. [DOI] [PubMed] [Google Scholar]
- 128.Okura T Rankinen T Gagnon J, et al. Effect of regular exercise on homocysteine concentrations: the HERITAGE Family Study. Eur J Appl Physiol. 2006;98(4):394–401. [DOI] [PubMed] [Google Scholar]
- 129.Estrada DA, Billett HH. Racial variation in fasting and random homocysteine levels. Am J Hematol. 2001;66(4):252–6. [DOI] [PubMed] [Google Scholar]
- 130.Ganji V, Kafai MR. Population reference values for plasma total homocysteine concentrations in US adults after the fortification of cereals with folic acid. Am J Clin Nutr. 2006;84(5):989–94. [DOI] [PubMed] [Google Scholar]
- 131.Romano AD, Serviddio G, de Matthaeis A, Bellanti F, Vendemiale G. Oxidative stress and aging. J Nephrol. 2010;23(15 Suppl):S29–36. [PubMed] [Google Scholar]
- 132.Clarke R Bennett DA Parish S, et al. Homocysteine and coronary heart disease: meta-analysis of MTHFR case–control studies, avoiding publication bias. PLoS Med. 2012;9(2):e1001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schnyder G Roffi M Pin R, et al. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med. 2001;345(22):1593–600. [DOI] [PubMed] [Google Scholar]
- 134.Malenica M Prnjavorac B Bego T, et al. Effect of cigarette smoking on haematological parameters in healthy population. Med Arch. 2017;71(2):132–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pedersen KM, Colak Y, Ellervik C, Hasselbalch HC, Bojesen SE, Nordestgaard BG. Smoking and increased White and red blood cells. Arterioscler Thromb Vasc Biol. 2019;39(5):965–77. [DOI] [PubMed] [Google Scholar]
- 136.Franzoni F Ghiadoni L Galetta F, et al. Physical activity, plasma antioxidant capacity, and endothelium-dependent vasodilation in young and older men. Am J Hypertens. 2005;18(4 Pt 1):510–6. [DOI] [PubMed] [Google Scholar]
- 137.Sallam N, Laher I. Exercise modulates oxidative stress and inflammation in aging and cardiovascular diseases. Oxid Med Cell Longev. 2016;2016:7239639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rico-Sanz J Rankinen T Joanisse DR, et al. Familial resemblance for muscle phenotypes in the HERITAGE Family Study. Med Sci Sports Exerc. 2003;35(8):1360–6. [DOI] [PubMed] [Google Scholar]
- 139.Simoneau JA, Bouchard C. Genetic determinism of fiber type proportion in human skeletal muscle. FASEB J. 1995;9(11):1091–5. [DOI] [PubMed] [Google Scholar]
- 140.Simoneau JA, Lortie G, Boulay MR, Thibault MC, Bouchard C. Repeatability of fibre type and enzyme activity measurements in human skeletal muscle. Clin Physiol. 1986;6(4):347–56. [DOI] [PubMed] [Google Scholar]
- 141.Simoneau JA, Lortie G, Boulay MR, Thibault MC, Theriault G, Bouchard C. Skeletal muscle histochemical and biochemical characteristics in sedentary male and female subjects. Can J Physiol Pharmacol. 1985;63(1):30–5. [DOI] [PubMed] [Google Scholar]
- 142.Simoneau JA, Bouchard C. Human variation in skeletal muscle fiber-type proportion and enzyme activities. Am J Physiol. 1989;257(4 Pt 1):E567–72. [DOI] [PubMed] [Google Scholar]
- 143.Rankinen T, Bouchard C, Rao DC. Familial resemblance for muscle phenotypes: the HERITAGE Family Study. Corrigendum. Med Sci Sports Exerc. 2005;37(11):2017. [DOI] [PubMed] [Google Scholar]
- 144.Tremblay A, Fontaine E, Poehlman ET, Mitchell D, Perron L, Bouchard C. The effect of exercise-training on resting metabolic rate in lean and moderately obese individuals. Int J Obes (Lond). 1986;10(6):511–7. [PubMed] [Google Scholar]
- 145.Poehlman ET, Danforth E, Jr. Endurance training increases metabolic rate and norepinephrine appearance rate in older individuals. Am J Physiol. 1991;261(2):E233–9. [DOI] [PubMed] [Google Scholar]
- 146.Poehlman ET, Gardner AW, Arciero PJ, Goran MI, Calles-Escandon J. Effects of endurance training on total fat oxidation in elderly persons. J Appl Physiol. 1994;76(6):2281–7. [DOI] [PubMed] [Google Scholar]
- 147.Broeder CE, Burrhus KA, Svanevik LS, Wilmore JH. The effects of either high-intensity resistance or endurance training on resting metabolic rate. Am J Clin Nutr. 1992;55(4):802–10. [DOI] [PubMed] [Google Scholar]
- 148.Bingham SA, Goldberg GR, Coward WA, Prentice AM, Cummings JH. The effect of exercise and improved physical fitness on basal metabolic rate. Br J Nutr. 2007;61(2):155–73. [DOI] [PubMed] [Google Scholar]
- 149.Meijer GA, Westerterp KR, Seyts GH, Janssen GM, Saris WH, ten Hoor F. Body composition and sleeping metabolic rate in response to a 5-month endurance-training programme in adults. Eur J Appl Physiol Occup Physiol. 1991;62(1):18–21. [DOI] [PubMed] [Google Scholar]
- 150.Frey-Hewitt B, Vranizan KM, Dreon DM, Wood PD. The effect of weight loss by dieting or exercise on resting metabolic rate in overweight men. Int J Obes (Lond). 1990;14(4):327–34. [PubMed] [Google Scholar]
- 151.Meredith CN Frontera WR Fisher EC, et al. Peripheral effects of endurance training in young and old subjects. J Appl Physiol (1985). 1989;66(6):2844–9. [DOI] [PubMed] [Google Scholar]
- 152.Westerterp KR, Meijer GA, Schoffelen P, Janssen EM. Body mass, body composition and sleeping metabolic rate before, during and after endurance training. Eur J Appl Physiol Occup Physiol. 1994;69(3):203–8. [DOI] [PubMed] [Google Scholar]
- 153.Wilmore JH Stanforth PR Hudspeth LA, et al. Alterations in resting metabolic rate as a consequence of 20 wk of endurance training: the HERITAGE Family Study. Am J Clin Nutr. 1998;68(1):66–71. [DOI] [PubMed] [Google Scholar]
- 154.Herrmann SD, Willis EA, Honas JJ, Lee J, Washburn RA, Donnelly JE. Energy intake, nonexercise physical activity, and weight loss in responders and nonresponders: the Midwest Exercise Trial 2. Obesity (Silver Spring). 2015;23(8):1539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jennings A, Alberga A, Sigal R, Jay O, Boule N, Kenny G. The effect of exercise training on resting metabolic rate in type 2 diabetes mellitus. Med Sci Sports Exerc. 2009;41(8):1558–65. [DOI] [PubMed] [Google Scholar]
- 156.Keytel LR, Lambert MI, Johnson J, Noakes TD, Lambert EV. Free living energy expenditure in post menopausal women before and after exercise training. Int J Sport Nutr Exerc Metab. 2001;11(2):226. [DOI] [PubMed] [Google Scholar]
- 157.Karstoft K, Brinkløv CF, Thorsen IK, Nielsen JS, Ried-Larsen M. Resting metabolic rate does not change in response to different types of training in subjects with type 2 diabetes. Front Endocrinol (Lausanne). 2017;8:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kraemer WJ Volek JS Clark KL, et al. Physiological adaptations to a weight-loss dietary regimen and exercise programs in women. J Appl Physiol (1985). 1997;83(1):270–9. [DOI] [PubMed] [Google Scholar]
- 159.Lee M-G, Sedlock DA, Flynn MG, Kamirmori GH. Resting metabolic rate after endurance exercise training. Med Sci Sports Exerc. 2009;41(7):1444–51. [DOI] [PubMed] [Google Scholar]
- 160.Poehlman ET Denino WF Beckett T, et al. Effects of endurance and resistance training on total daily energy expenditure in young women: a controlled randomized trial. J Clin Endocrinol Metab. 2002;87(3):1004–9. [DOI] [PubMed] [Google Scholar]
- 161.Mourier A Gautier JF De Kerviler E, et al. Mobilization of visceral adipose tissue related to the improvement in insulin sensitivity in response to physical training in NIDDM. Effects of branched-chain amino acid supplements. Diabetes Care. 1997;20(3):385–91. [DOI] [PubMed] [Google Scholar]
- 162.Sawczyn S Mishchenk V Moska W, et al. Strength and aerobic training in overweight females in Gdansk, Poland. Open Med (Wars). 2015;10(1):152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Scharhag-Rosenberger F, Meyer T, Walitzek S, Kindermann W. Effects of one year aerobic endurance training on resting metabolic rate and exercise fat oxidation in previously untrained men and women. Metabolic endurance training adaptations. Int J Sports Med. 2010;31(7):498–504. [DOI] [PubMed] [Google Scholar]
- 164.Short KR, Vittone JL, Bigelow ML, Proctor DN, Nair KS. Age and aerobic exercise training effects on whole body and muscle protein metabolism. Am J Physiol Endocrinol Metab. 2004;286(1):E92–101. [DOI] [PubMed] [Google Scholar]
- 165.van Aggel-Leijssen DP, Saris WH, Hul GB, van Baak MA. Long-term effects of low-intensity exercise training on fat metabolism in weight-reduced obese men. Metabolism. 2002;51(8):1003–10. [DOI] [PubMed] [Google Scholar]
- 166.Dolezal BA, Potteiger JA. Concurrent resistance and endurance training influence basal metabolic rate in nondieting individuals. J Appl Physiol (1985). 1998;85(2):695–700. [DOI] [PubMed] [Google Scholar]
- 167.Santa-Clara H, Szymanski L, Ordille T, Fernhall B. Effects of exercise training on resting metabolic rate in postmenopausal African American and Caucasian women. Metabolism. 2006;55(10):1358–64. [DOI] [PubMed] [Google Scholar]
- 168.Morio B Montaurier C Pickering G, et al. Effects of 14 weeks of progressive endurance training on energy expenditure in elderly people. Br J Nutr. 2007;80(6):511–9. [DOI] [PubMed] [Google Scholar]
- 169.Araiza P, Hewes H, Gashetewa C, Vella CA, Burge MR. Efficacy of a pedometer-based physical activity program on parameters of diabetes control in type 2 diabetes mellitus. Metabolism. 2006;55(10):1382–7. [DOI] [PubMed] [Google Scholar]
- 170.Christensen B Nellemann B Larsen MS, et al. Whole body metabolic effects of prolonged endurance training in combination with erythropoietin treatment in humans: a randomized placebo controlled trial. Am J Physiol Endocrinol Metab. 2013;305(7):E879–89. [DOI] [PubMed] [Google Scholar]
- 171.Potteiger JA, Kirk EP, Jacobsen DJ, Donnelly JE. Changes in resting metabolic rate and substrate oxidation after 16 months of exercise training in overweight adults. Am J Physiol Endocrinol Metab. 2008;18(1):79–95. [DOI] [PubMed] [Google Scholar]
- 172.Rankinen T Gagnon J Pérusse L, et al. AGT M235T and ACE ID polymorphisms and exercise blood pressure in the HERITAGE Family Study. Am J Physiol Heart Circ Physiol. 2000;279(1):H368–74. [DOI] [PubMed] [Google Scholar]
- 173.Rankinen T Pérusse L Gagnon J, et al. Angiotensin-converting enzyme ID polymorphism and fitness phenotype in the HERITAGE Family Study. J Appl Physiol (1985). 2000;88(3):1029–35. [DOI] [PubMed] [Google Scholar]
- 174.Ukkola O Santaniemi M Rankinen T, et al. Adiponectin polymorphisms, adiposity and insulin metabolism: HERITAGE Family Study and Oulu diabetic study. Ann Med. 2005;37(2):141–50. [DOI] [PubMed] [Google Scholar]
- 175.Garenc C Perusse L Chagnon YC, et al. The alpha 2-adrenergic receptor gene and body fat content and distribution: the HERITAGE Family Study. Mol Med. 2002;8(2):88–94. [PMC free article] [PubMed] [Google Scholar]
- 176.Garenc C Perusse L Chagnon YC, et al. Effects of beta2-adrenergic receptor gene variants on adiposity: the HERITAGE Family Study. Obes Res. 2003;11(5):612–8. [DOI] [PubMed] [Google Scholar]
- 177.Ukkola O Rankinen T Rice T, et al. Interactions among the beta2- and beta3-adrenergic receptor genes and total body fat and abdominal fat level in the HERITAGE Family Study. Int J Obes Relat Metab Disord. 2003;27(3):389–93. [DOI] [PubMed] [Google Scholar]
- 178.Loos RJ Rankinen T Rice T, et al. Two ethnic-specific polymorphisms in the human agouti-related protein gene are associated with macronutrient intake. Am J Clin Nutr. 2005;82(5):1097–101. [DOI] [PubMed] [Google Scholar]
- 179.Argyropoulos G Rankinen T Bai F, et al. The agouti-related protein and body fatness in humans. Int J Obes Relat Metab Disord. 2003;27(2):276–80. [DOI] [PubMed] [Google Scholar]
- 180.Argyropoulos G Rankinen T Neufeld DR, et al. A polymorphism in the human agouti-related protein is associated with late-onset obesity. J Clin Endocrinol Metab. 2002;87(9):4198–202. [DOI] [PubMed] [Google Scholar]
- 181.Rankinen T Gagnon J Perusse L, et al. Body fat, resting and exercise blood pressure and the angiotensinogen M235T polymorphism: the HERITAGE Family Study. Obes Res. 1999;7(5):423–30. [DOI] [PubMed] [Google Scholar]
- 182.Rico-Sanz J Rankinen T Joanisse DR, et al. Associations between cardiorespiratory responses to exercise and the C34T AMPD1 gene polymorphism in the HERITAGE Family Study. Physiol Genomics. 2003;14(2):161–6. [DOI] [PubMed] [Google Scholar]
- 183.Rivera MA Echegaray M Rankinen T, et al. Angiogenin gene-race interaction for resting and exercise BP phenotypes: the HERITAGE Family Study. J Appl Physiol (1985). 2001;90(4):1232–8. [DOI] [PubMed] [Google Scholar]
- 184.Leon AS Togashi K Rankinen T, et al. Association of apolipoprotein E polymorphism with blood lipids and maximal oxygen uptake in the sedentary state and after exercise training in the HERITAGE Family Study. Metabolism. 2004;53(1):108–16. [DOI] [PubMed] [Google Scholar]
- 185.Spielmann N Leon AS Rao DC, et al. CETP genotypes and HDL-cholesterol phenotypes in the HERITAGE Family Study. Physiol Genomics. 2007;31(1):25–31. [DOI] [PubMed] [Google Scholar]
- 186.Rankinen T Perusse L Borecki I, et al. The Na(+)-K(+)-ATPase alpha2 gene and trainability of cardiorespiratory endurance: the HERITAGE Family Study. J Appl Physiol (1985). 2000;88(1):346–51. [DOI] [PubMed] [Google Scholar]
- 187.Sklan EH Lowenthal A Korner M, et al. Acetylcholinesterase/paraoxonase genotype and expression predict anxiety scores in Health, Risk Factors, Exercise Training, and Genetics study. Proc Natl Acad Sci U S A. 2004;101(15):5512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rivera MA Dionne FT Simoneau JA, et al. Muscle-specific creatine kinase gene polymorphism and VO2max in the HERITAGE Family Study. Med Sci Sports Exerc. 1997;29(10):1311–7. [DOI] [PubMed] [Google Scholar]
- 189.Simonen RL Rankinen T Perusse L, et al. A dopamine D2 receptor gene polymorphism and physical activity in two family studies. Physiol Behav. 2003;78(4–5):751–7. [DOI] [PubMed] [Google Scholar]
- 190.Rankinen T Church T Rice T, et al. Effect of endothelin 1 genotype on blood pressure is dependent on physical activity or fitness levels. Hypertension. 2007;50(6):1120–5. [DOI] [PubMed] [Google Scholar]
- 191.Teran-Garcia M Rankinen T Rice T, et al. Variations in the four and a half LIM domains 1 gene (FHL1) are associated with fasting insulin and insulin sensitivity responses to regular exercise. Diabetologia. 2007;50(9):1858–66. [DOI] [PubMed] [Google Scholar]
- 192.Rankinen T, Rice T, Teran-Garcia M, Rao DC, Bouchard C. FTO genotype is associated with exercise training-induced changes in body composition. Obesity (Silver Spring). 2010;18(2):322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ukkola O Ravussin E Jacobson P, et al. Role of ghrelin polymorphisms in obesity based on three different studies. Obes Res. 2002;10(8):782–91. [DOI] [PubMed] [Google Scholar]
- 194.Rankinen T Rice T Leon AS, et al. G protein beta 3 polymorphism and hemodynamic and body composition phenotypes in the HERITAGE Family Study. Physiol Genomics. 2002;8(2):151–7. [DOI] [PubMed] [Google Scholar]
- 195.Lakka TA Rankinen T Weisnagel SJ, et al. Leptin and leptin receptor gene polymorphisms and changes in glucose homeostasis in response to regular exercise in nondiabetic individuals: the HERITAGE Family Study. Diabetes. 2004;53(6):1603–8. [DOI] [PubMed] [Google Scholar]
- 196.Chagnon YC Wilmore JH Borecki IB, et al. Associations between the leptin receptor gene and adiposity in middle-age Caucasian males from the HERITAGE Family Study. J Clin Endocrinol Metab. 2000;85(1):29–34. [DOI] [PubMed] [Google Scholar]
- 197.Teran-Garcia M Santoro N Rankinen T, et al. Hepatic lipase gene variant −514C > T is associated with lipoprotein and insulin sensitivity response to regular exercise: the HERITAGE Family Study. Diabetes. 2005;54(7):2251–5. [DOI] [PubMed] [Google Scholar]
- 198.Garenc C Perusse L Chagnon YC, et al. The hormone-sensitive lipase gene and body composition: the HERITAGE Family Study. Int J Obes Relat Metab Disord. 2002;26(2):220–7. [DOI] [PubMed] [Google Scholar]
- 199.Garenc C Pérusse L Bergeron J, et al. Evidence of LPL gene-exercise interaction for body fat and LPL activity: the HERITAGE Family Study. J Appl Physiol (1985). 2001;91(3):1334–40. [DOI] [PubMed] [Google Scholar]
- 200.Jacobson P Ukkola O Rankinen T, et al. Melanocortin 4 receptor sequence variations are seldom a cause of human obesity: the Swedish obese subjects, the HERITAGE Family Study, and a Memphis cohort. J Clin Endocrinol Metab. 2002;87(10):4442–6. [DOI] [PubMed] [Google Scholar]
- 201.Rankinen T Rice T Perusse L, et al. NOS3 Glu298Asp genotype and blood pressure response to endurance training: the HERITAGE Family Study. Hypertension. 2000;36(5):885–9. [DOI] [PubMed] [Google Scholar]
- 202.Hautala AJ, Leon AS, Skinner JS, Rao DC, Bouchard C, Rankinen T. Peroxisome proliferator-activated receptor-delta polymorphisms are associated with physical performance and plasma lipids: the HERITAGE Family Study. Am J Physiol Heart Circ Physiol. 2007;292(5):H2498–505. [DOI] [PubMed] [Google Scholar]
- 203.Ruchat SM Rankinen T Weisnagel SJ, et al. Improvements in glucose homeostasis in response to regular exercise are influenced by the PPARG Pro12Ala variant: results from the HERITAGE Family Study. Diabetologia. 2010;53(4):679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ukkola O Rankinen T Lakka T, et al. Protein tyrosine phosphatase 1B variant associated with fat distribution and insulin metabolism. Obes Res. 2005;13(5):829–34. [DOI] [PubMed] [Google Scholar]
- 205.Stutz AM, Teran-Garcia M, Rao DC, Rice T, Bouchard C, Rankinen T. Functional identification of the promoter of SLC4A5, a gene associated with cardiovascular and metabolic phenotypes in the HERITAGE Family Study. Eur J Hum Genet. 2009;17(11):1481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Rivera MA Echegaray M Rankinen T, et al. TGF-beta(1) gene-race interactions for resting and exercise blood pressure in the HERITAGE Family Study. J Appl Physiol (1985). 2001;91(4):1808–13. [DOI] [PubMed] [Google Scholar]
- 207.Lakka HM Lakka TA Rankinen T, et al. The TNF-alpha G-308A polymorphism is associated with C-reactive protein levels: the HERITAGE Family Study. Vascul Pharmacol. 2006;44(5):377–83. [DOI] [PubMed] [Google Scholar]
- 208.Lanouette CM Chagnon YC Rice T, et al. Uncoupling protein 3 gene is associated with body composition changes with training in HERITAGE study. J Appl Physiol (1985). 2002;92(3):1111–8. [DOI] [PubMed] [Google Scholar]
- 209.Haseman JK, Elston RC. The investigation of linkage between a quantitative trait and a marker locus. Behav Genet. 1972;2(1):3–19. [DOI] [PubMed] [Google Scholar]
- 210.Bouchard C Rankinen T Chagnon YC, et al. Genomic scan for maximal oxygen uptake and its response to training in the HERITAGE Family Study. J Appl Physiol (1985). 2000;88(2):551–9. [DOI] [PubMed] [Google Scholar]
- 211.Rice TK Sarzynski MA Sung YJ, et al. Fine mapping of a QTL on chromosome 13 for submaximal exercise capacity training response: the HERITAGE Family Study. Eur J Appl Physiol. 2012;112(8):2969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Rico-Sanz J Rankinen T Rice T, et al. Quantitative trait loci for maximal exercise capacity phenotypes and their responses to training in the HERITAGE Family Study. Physiol Genomics. 2004;16(2):256–60. [DOI] [PubMed] [Google Scholar]
- 213.An P Rice T Rankinen T, et al. Genome-wide scan to identify quantitative trait loci for baseline resting heart rate and its response to endurance exercise training: the HERITAGE Family Study. Int J Sports Med. 2006;27(1):31–6. [DOI] [PubMed] [Google Scholar]
- 214.Rankinen T An P Perusse L, et al. Genome-wide linkage scan for exercise stroke volume and cardiac output in the HERITAGE Family Study. Physiol Genomics. 2002;10(2):57–62. [DOI] [PubMed] [Google Scholar]
- 215.Rankinen T An P Rice T, et al. Genomic scan for exercise blood pressure in the Health, Risk Factors, Exercise Training and Genetics (HERITAGE) Family Study. Hypertension. 2001;38(1):30–7. [DOI] [PubMed] [Google Scholar]
- 216.Rice T Cooper RS Wu X, et al. Meta-analysis of genome-wide scans for blood pressure in African American and Nigerian samples. The National Heart, Lung, and Blood Institute GeneLink Project. Am J Hypertens. 2006;19(3):270–4. [DOI] [PubMed] [Google Scholar]
- 217.Rice T Rankinen T Chagnon YC, et al. Genomewide linkage scan of resting blood pressure: HERITAGE Family Study. Health, Risk Factors, Exercise Training, and Genetics. Hypertension. 2002;39(6):1037–43. [DOI] [PubMed] [Google Scholar]
- 218.Spielmann N Leon AS Rao DC, et al. Genome-wide linkage scan for submaximal exercise heart rate in the HERITAGE Family Study. Am J Physiol Heart Circ Physiol. 2007;293(6):H3366–71. [DOI] [PubMed] [Google Scholar]
- 219.Chagnon YC Rice T Perusse L, et al. Genomic scan for genes affecting body composition before and after training in Caucasians from HERITAGE. J Appl Physiol (1985). 2001;90(5):1777–87. [DOI] [PubMed] [Google Scholar]
- 220.Rice T Chagnon YC Perusse L, et al. A genomewide linkage scan for abdominal subcutaneous and visceral fat in Black and White families: the HERITAGE Family Study. Diabetes. 2002;51(3):848–55. [DOI] [PubMed] [Google Scholar]
- 221.Feitosa MF Borecki IB Rankinen T, et al. Evidence of QTLs on chromosomes 1q42 and 8q24 for LDL-cholesterol and apoB levels in the HERITAGE Family Study. J Lipid Res. 2005;46(2):281–6. [DOI] [PubMed] [Google Scholar]
- 222.Feitosa MF Rice T Rankinen T, et al. Evidence of QTLs on chromosomes 13q and 14q for triglycerides before and after 20 weeks of exercise training: the HERITAGE Family Study. Atherosclerosis. 2005;182(2):349–60. [DOI] [PubMed] [Google Scholar]
- 223.An P Hong Y Weisnagel SJ, et al. Genomic scan of glucose and insulin metabolism phenotypes: the HERITAGE Family Study. Metabolism. 2003;52(2):246–53. [DOI] [PubMed] [Google Scholar]
- 224.An P Teran-Garcia M Rice T, et al. Genome-wide linkage scans for prediabetes phenotypes in response to 20 weeks of endurance exercise training in non-diabetic Whites and Blacks: the HERITAGE Family Study. Diabetologia. 2005;48(6):1142–9. [DOI] [PubMed] [Google Scholar]
- 225.Lakka TA Rankinen T Weisnagel SJ, et al. A quantitative trait locus on 7q31 for the changes in plasma insulin in response to exercise training: the HERITAGE Family Study. Diabetes. 2003;52(6):1583–7. [DOI] [PubMed] [Google Scholar]
- 226.Collaku A Rankinen T Rice T, et al. A genome-wide linkage scan for dietary energy and nutrient intakes: the Health, Risk Factors, Exercise Training, and Genetics (HERITAGE) Family Study. Am J Clin Nutr. 2004;79(5):881–6. [DOI] [PubMed] [Google Scholar]
- 227.Lakka TA Rankinen T Rice T, et al. Quantitative trait locus on chromosome 20q13 for plasma levels of C-reactive protein in healthy Whites: the HERITAGE Family Study. Physiol Genomics. 2006;27(2):103–7. [DOI] [PubMed] [Google Scholar]
- 228.Loos RJ Katzmarzyk PT Rao DC, et al. Genome-wide linkage scan for the metabolic syndrome in the HERITAGE Family Study. J Clin Endocrinol Metab. 2003;88(12):5935–43. [DOI] [PubMed] [Google Scholar]
- 229.Feitosa MF Rice T North KE, et al. Pleiotropic QTL on chromosome 19q13 for triglycerides and adiposity: the HERITAGE Family Study. Atherosclerosis. 2006;185(2):426–32. [DOI] [PubMed] [Google Scholar]
- 230.Feitosa MF Rice T Borecki IB, et al. Pleiotropic QTL on chromosome 12q23–q24 influences triglyceride and high-density lipoprotein cholesterol levels: the HERITAGE Family Study. Hum Biol. 2006;78(3):317–27. [DOI] [PubMed] [Google Scholar]
- 231.Rankinen T Rice T Boudreau A, et al. Titin is a candidate gene for stroke volume response to endurance training: the HERITAGE Family Study. Physiol Genomics. 2003;15(1):27–33. [DOI] [PubMed] [Google Scholar]
- 232.Argyropoulos G Stutz AM Ilnytska O, et al. KIF5B gene sequence variation and response of cardiac stroke volume to regular exercise. Physiol Genomics. 2009;36(2):79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Rankinen T, Argyropoulos G, Rice T, Rao DC, Bouchard C. CREB1 is a strong genetic predictor of the variation in exercise heart rate response to regular exercise: the HERITAGE Family Study. Circ Cardiovasc Genet. 2010;3(3):294–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bouchard C Sarzynski MA Rice TK, et al. Genomic predictors of the maximal O2 uptake response to standardized exercise training programs. J Appl Physiol (1985). 2011;110(5):1160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Rankinen T, Sung YJ, Sarzynski MA, Rice TK, Rao DC, Bouchard C. Heritability of submaximal exercise heart rate response to exercise training is accounted for by nine SNPs. J Appl Physiol (1985). 2012;112(5):892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sarzynski MA Davidsen PK Sung YJ, et al. Genomic and transcriptomic predictors of triglyceride response to regular exercise. Br J Sports Med. 2015;49(23):1524–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sung YJ Perusse L Sarzynski MA, et al. Genome-wide association studies suggest sex-specific loci associated with abdominal and visceral fat. Int J Obes (Lond). 2016;40(4):662–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Takeshita L Davidsen PL Herbert JM, et al. Genomics and transcriptomics landscapes associated to changes in insulin sensitivity in response to endurance exercise. Sci Rep. 2021;11:23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Heid IM Huth C Loos RJ, et al. Meta-analysis of the INSIG2 association with obesity including 74,345 individuals: does heterogeneity of estimates relate to study design? PLoS Genet. 2009;5(10):e1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wood AR Esko T Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46(11):1173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Locke AE Kahali B Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Qi Q Kilpelainen TO Downer MK, et al. FTO genetic variants, dietary intake and body mass index: insights from 177,330 individuals. Hum Mol Genet. 2014;23(25):6961–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Justice AE Winkler TW Feitosa MF, et al. Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat Commun. 2017;8:14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lu Y Day FR Gustafsson S, et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun. 2016;7:10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Shungin D Winkler TW Croteau-Chonka DC, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518(7538):187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Winkler TW Justice AE Graff M, et al. The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLoS Genet. 2015;11(10):e1005378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.den Hoed M Eijgelsheim M Esko T, et al. Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet. 2013;45(6):621–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kilpelainen TO Carli JF Skowronski AA, et al. Genome-wide meta-analysis uncovers novel loci influencing circulating leptin levels. Nat Commun. 2016;7:10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Merino J Dashti HS Li SX, et al. Genome-wide meta-analysis of macronutrient intake of 91,114 European ancestry participants from the cohorts for heart and aging research in genomic epidemiology consortium. Mol Psychiatry. 2019;24(12):1920–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ried JS Jeff MJ Chu AY, et al. A principal component meta-analysis on multiple anthropometric traits identifies novel loci for body shape. Nat Commun. 2016;7:13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Noordam R Bos MM Wang H, et al. Multi-ancestry sleep-by-SNP interaction analysis in 126,926 individuals reveals lipid loci stratified by sleep duration. Nat Commun. 2019;10(1):5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Wang H Noordam R Cade BE, et al. Multi-ancestry genome-wide gene-sleep interactions identify novel loci for blood pressure. Mol Psychiatry. 2021;26(11):6293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wu P Rybin D Bielak LF, et al. Smoking-by-genotype interaction in type 2 diabetes risk and fasting glucose. PLoS One. 2020;15(5):e0230815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.de Las Fuentes L Sung YJ Noordam R, et al. Gene–educational attainment interactions in a multi-ancestry genome-wide meta-analysis identify novel blood pressure loci. Mol Psychiatry. 2021;26(6):2111–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Rao DC Sung YJ Winkler TW, et al. CHARGE Gene-Lifestyle Interactions Working Group* . Multiancestry study of gene–lifestyle interactions for cardiovascular traits in 610 475 individuals from 124 cohorts: design and rationale. Circ Cardiovasc Genet. 2017;10(3):e001649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Bentley AR Sung YJ Brown MR, et al. Multi-ancestry genome-wide gene–smoking interaction study of 387,272 individuals identifies new loci associated with serum lipids. Nat Genet. 2019;51(4):636–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.de Vries PS Brown MR Bentley AR, et al. Multiancestry genome-wide association study of lipid levels incorporating gene–alcohol interactions. Am J Epidemiol. 2019;188(6):1033–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Feitosa MF Kraja AT Chasman DI, et al. Novel genetic associations for blood pressure identified via gene–alcohol interaction in up to 570K individuals across multiple ancestries. PLoS One. 2018;13(6):e0198166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kilpelainen TO Bentley AR Noordam R, et al. Multi-ancestry study of blood lipid levels identifies four loci interacting with physical activity. Nat Commun. 2019;10(1):376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Manning AK Hivert MF Scott RA, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44(6):659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sung YJ de Las Fuentes L Winkler TW, et al. A multi-ancestry genome-wide study incorporating gene–smoking interactions identifies multiple new loci for pulse pressure and mean arterial pressure. Hum Mol Genet. 2019;28(15):2615–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sung YJ Winkler TW de Las Fuentes L, et al. A large-scale multi-ancestry genome-wide study accounting for smoking behavior identifies multiple significant loci for blood pressure. Am J Hum Genet. 2018;102(3):375–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Teran-Garcia M, Rankinen T, Koza RA, Rao DC, Bouchard C. Endurance training-induced changes in insulin sensitivity and gene expression. Am J Physiol Endocrinol Metab. 2005;288(6):E1168–78. [DOI] [PubMed] [Google Scholar]
- 264.Phillips BE Williams JP Gustafsson T, et al. Molecular networks of human muscle adaptation to exercise and age. PLoS Genet. 2013;9(3):e1003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Timmons JA Knudsen S Rankinen T, et al. Using molecular classification to predict gains in maximal aerobic capacity following endurance exercise training in humans. J Appl Physiol (1985). 2010;108(6):1487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Keller P Vollaard NBJ Gustafsson T, et al. A transcriptional map of the impact of endurance exercise training on skeletal muscle phenotype. J Appl Physiol (1985). 2011;110(1):46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Clarke K Ricciardi S Pearson T, et al. The role of Eif6 in skeletal muscle homeostasis revealed by endurance training co-expression networks. Cell Rep. 2017;21(6):1507–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.O’Sullivan JF Morningstar JE Yang Q, et al. Dimethylguanidino valeric acid is a marker of liver fat and predicts diabetes. J Clin Invest. 2017;127(12):4394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Jacob J Ngo D Finkel N, et al. Application of large-scale aptamer-based proteomic profiling to planned myocardial infarctions. Circulation. 2018;137(12):1270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ngo D Sinha S Shen D, et al. Aptamer-based proteomic profiling reveals novel candidate biomarkers and pathways in cardiovascular disease. Circulation. 2016;134(4):270–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Williams SA Kivimaki M Langenberg C, et al. Plasma protein patterns as comprehensive indicators of health. Nat Med. 2019;25(12):1851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Robbins JM Peterson B Schranner D, et al. Human plasma proteomic profiles indicative of cardiorespiratory fitness. Nat Metab. 2021;3(6):786–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ghosh S Vivar JC Sarzynski MA, et al. Integrative pathway analysis of a genome-wide association study of (V)O(2max) response to exercise training. J Appl Physiol (1985). 2013;115(9):1343–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Williams CJ Williams MG Eynon N, et al. Genes to predict VO2max trainability: a systematic review. BMC Genomics. 2017;18(8 Suppl):831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Ghosh S Hota M Chai X, et al. Exploring the underlying biology of intrinsic cardiorespiratory fitness through integrative analysis of genomic variants and muscle gene expression profiling. J Appl Physiol (1985). 2019;126(5):1292–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ferguson DP, Dangott LJ, Schmitt EE, Vellers HL, Lightfoot JT. Differential skeletal muscle proteome of high- and low-active mice. J Appl Physiol (1985). 2014;116(8):1057–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.di Barletta MR Viatchenko-Karpinski S Nori A, et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114(10):1012–9. [DOI] [PubMed] [Google Scholar]
- 278.Knollmann BC Chopra N Hlaing T, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116(9):2510–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Lahat H Pras E Olender T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69(6):1378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Georgieva B, Todorova A, Tournev I, Mitev V, Kremensky I. C283Y gamma-sarcoglycan gene mutation in the Bulgarian Roma (Gypsy) population: prevalence study and carrier screening in a high-risk community. Clin Genet. 2004;66(5):467–72. [DOI] [PubMed] [Google Scholar]
- 281.McNally EM Duggan D Gorospe JR, et al. Mutations that disrupt the carboxyl-terminus of gamma-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5(11):1841–7. [DOI] [PubMed] [Google Scholar]
- 282.Troxell ML, Britton SL, Koch LG. Selected contribution: variation and heritability for the adaptational response to exercise in genetically heterogeneous rats. J Appl Physiol (1985). 2003;94(4):1674–81. [DOI] [PubMed] [Google Scholar]
- 283.Barber JL Ruiz-Ramie JJ Robbins JM, et al. Regular exercise and patterns of response across multiple cardiometabolic traits: the HERITAGE Family Study. Br J Sports Med. 2022;56(2):95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Lee IM Shiroma EJ Lobelo F, et al. Effect of physical inactivity on major non-communicable diseases worldwide: an analysis of burden of disease and life expectancy. Lancet. 2012;380(9838):219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Wilhelmsen L Bjure J Ekstrom-Jodal B, et al. Nine years’ follow-up of a maximal exercise test in a random population sample of middle-age men. Cardiology. 1981;68(2 Suppl):1–8. [PubMed] [Google Scholar]
- 286.Kodama S Saito K Tanaka S, et al. Cardiorespiratory fitness as a quantitative predictor of all-cause mortality and cardiovascular events in healthy men and women: a meta-analysis. JAMA. 2009;301(19):2024–35. [DOI] [PubMed] [Google Scholar]
- 287.Bouchard C, Blair SN, Katzmarzyk PT. Less sitting, more physical activity, or higher fitness? Mayo Clin Proc. 2015;90(11):1533–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.