

Abstract
Purpose:
Although androgen deprivation therapy (ADT) and androgen receptor signaling inhibitors (ARSI) are effective in metastatic prostate cancer (PC), resistance occurs in most patients. This phase I/II trial assessed the safety, pharmacokinetic impact, and efficacy of the glucocorticoid receptor (GR) antagonist mifepristone (Mif) in combination with enzalutamide (Enz) for castration-resistant PC (CRPC).
Patients and Methods:
106 patients with CRPC were accrued. Phase I subjects were treated with Enz monotherapy at 160 mg per day for 28 days to allow steady-state accumulation. Patients then entered the dose escalation combination portion of the study. In phase II, patients were randomized 1:1 to either receive Enz alone or Enz plus Mif. The primary endpoint was PSA progression free survival (PFS), with radiographic PFS, and PSA response rate (RR) as key secondary endpoints. Circulating tumor cells were collected before randomization for exploratory translational biomarker studies.
Results:
We determined a 25% dose reduction in Enz, when added to Mif resulted in equivalent drug levels compared to full dose Enz and was well tolerated. However, the addition of Mif to Enz following a 12-week Enz lead-in did not delay time to PSA, radiographic or clinical PFS. The trial was terminated early due to futility.
Conclusion:
This is the first prospective trial of dual AR-GR antagonism in CRPC. Enz combined with Mif was safe and well tolerated but did not meet its primary endpoint. The development of more specific GR antagonists combined with AR antagonists, potentially studied in an earlier disease state, should be explored.
Introduction
Although androgen deprivation therapy (ADT) initially controls metastatic prostate cancer (PC), failure of ADT and progression to castration-resistance occurs in the vast majority of patients.1 This transition to castrate-resistant prostate cancer (CRPC) is an important clinical landmark that correlates with an increased risk of morbidity and death.2 Despite CRPC development, androgen receptor (AR) signaling remains a key component driving CRPC progression.3,4 Therapeutics that more potently block AR signaling, such as the highly selective AR antagonist enzalutamide (Enz) and the androgen synthesis inhibitor abiraterone acetate are established standards of care for metastatic CRPC.5-9 More recently, AR signaling inhibition (ARSI) has been shown to improve outcomes earlier in the PC continuum and such inhibitors are used heavily in combination with ADT in the first line castration-sensitive setting.10-12
Depending on therapeutic context, the duration of ARSI efficacy varies before resistance emerges; however, resistance is a near universal eventuality.13-19 Beyond potent ARSI, therapeutic options are limited; targeting specific ARSI-resistance pathways is vital to reduce death from prostate cancer.
Multiple mechanisms may explain CRPC progression, including AR splice variants, AR mutations, and ligand-independent AR activation.15-19 There are also nuclear-hormone signaling independent resistance mechanisms, with some evidence supporting the hypothesis that alternative nuclear hormone signaling pathways, such as glucocorticoid receptor (GR) signaling, may compensate for AR signaling to enable PC cell survival despite potent ARSI. The GR and AR are in the same nuclear hormone receptor family and share target DNA sequence binding homology.20,21 Although in primary PC specimens GR expression is relatively low, recent studies demonstrated that GR expression significantly increases after ARSI.22-24 Subsequent to ARSI, GR activation promotes PC cell survival and proliferation, and GR activation conferred protection from Enz-associated growth suppression. 25-27 In a subset of CRPC patients, those who developed tumors with high GR expression correlated with a poor response to Enz.27 In preclinical models, GR antagonism with mifepristone (Mif), a non-selective steroidal nuclear hormone antagonist, or other more selective GR modulators delay CRPC progression in combination with ARSI.26,27 These data suggest increased GR expression compensates for diminished AR activity in PC cells treated with ARSI and may represent a therapeutic target for progressive CRPC.
However, since Mif is an inhibitor of CYP2C8 and CYP3A4 (responsible for Enz metabolism) it can increase Enz plasma exposure when given concurrently. The safety of this combination is unknown and requires further inquiry.
We hypothesized that after potent ARSI with Enz, increased GR expression and function compensates for diminished AR signaling in CRPC, facilitating cell survival and castrate resistant progression. The safe co-administration of Mif, a potent GR inhibitor FDA approved for Cushing’s Syndrome, with Enz would block this pathway and improve patient outcomes. We thus conducted a phase I/II open label trial (NCT:02012296) of study of Enz combined with Mif to assess the feasibility and impact on disease progression with dual AR and GR antagonism. The study started with a phase I portion focused on safety and pharmacokinetics (PK), followed by a randomized phase II portion.
Materials and Methods
Patient Selection
Eligible patients had histologically confirmed CRPC defined according to PC working group (PCWG) criteria.28 Any prior systemic therapy for PC was acceptable except CYP17 antagonists or inhibitors that block androgen production (such as abiraterone) or prior ARSIs. Eligibility included an Eastern Cooperative Oncology Group (ECOG) performance status of ≤2, acceptable bone marrow, hepatic, and renal function, and adequate baseline blood pressure and electrolytes.
Study Design and End Points
Phase I
The phase I portion assessed the safety of the two-drug combination along with the PK impact of Mif on Enz exposure. The primary objective was to determine the recommended phase II dose (RP2D) of Enz combined with Mif. Patients were treated with single agent Enz at 160 mg/day for 28 days. Baseline steady-state plasma levels of Enz and its M2 metabolite (N-desmethyl enzalutamide) were determined (Ctrough). Subjects then entered the combination portion of the study, at 300 mg/day of Mif combined with dose reduced Enz at 40 mg/day, chosen as a conservative starting dose due to Enz-Mif drug-drug interactions (decreased Enz clearance).
Interpatient dose escalation of Enz in patient cohorts of (at least) 6 patients was based on safety and PK parameters performed by InVentiv/Medivation, utilizing a constant Mif dose. The R2PD was determined as the Mif dose combined Enz such that ≤33% of patients (2/6 patients per cohort) experienced dose limiting toxicities (DLTs) and the ratio of day (57/29) Enz plus active metabolites was ≥ 0.75 and ≤ 1.5, along with a doubling of serum cortisol, providing support that GR was systemically antagonized. DLTs were defined as grade 3 or 4 toxicities that were potentially therapy-related. An independent safety monitor oversaw the study conduct specifically with regards to safety.
Phase II
This was a multicenter randomized open-label study conducted at five sites within the Department of Defense supported Prostate Cancer Clinical Trials Consortium (PCCTC). As GR expression was reported to increase with Enz,27 and to enrich for acquired Enz-resistance (as opposed to de novo resistance), patients in the phase II portion began treatment with Enz 160 mg/day as a single agent for 12 weeks. This was followed by randomization, in a one-to-one ratio, to receive either Enz 160 mg/day or Enz+Mif at the RP2D. To randomize, subjects needed stable disease or better at 12 weeks of single agent Enz, defined by PSA≤1.25 times the PSA at the start of Enz, lack of radiographic progression as defined by PCWG criteria,28 clinical stability (by treating physician), and toleration of Enz 160 mg/day.
The review boards of all participating institutions approved the study which was conducted according to the Declaration of Helsinki and Good Clinical Practice guidelines of the International Conference on Harmonization. All patients signed a written informed consent before the conduct of any study procedures and after a full explanation of the study to the patient by the study investigator.
Phase II Endpoints
The primary phase II endpoint was whether Mif combined with Enz prolonged time to PSA progression compared to Enz alone. PSA progression was defined according to PCWG criteria28,29 as a PSA that is ≥1.25 times (25% increase) the PSA at randomization (week 12). Time to PSA progression was used as a pharmacodynamic biomarker of GR antagonism within CRPC tumors as activation of both the GR and the AR can drive PSA expression in PC.21,30 Secondary objectives included evaluating the effect of Mif on endocrine biomarkers such as cortisol, thyrotropin, and testosterone. Additional secondary objectives included PSA RR (≥ 50% reduction in PSA after 12 weeks of therapy), and time to radiographic and clinical progression, all according to PCWG criteria.28,29
Exploratory circulating tumor cell (CTC) studies were performed on CTCs collected once after 12 weeks of Enz monotherapy.
On Study Evaluations
Visits occurred every two weeks for the first 8 weeks, then monthly. Standard blood counts, chemistries, PSA, endocrine markers, and plasma samples were regularly collected.
Baseline and on-study EKG were obtained to monitor for QTc prolongation. Disease burden at baseline and every 12 weeks was assessed with standard nuclear medicine bone scans and abdominal/pelvic cross-sectional imaging.
Enz or Enz+Mif continued until progression of disease was noted by PCWG criteria.28,29 PSA was measured monthly, but did not determine study drug termination.
CTC Evaluation
All CTC studies were performed centrally by Epic Sciences (Supplementary Figure 1A). CTC identification was performed using Epic Sciences’ CTC-specific platform for GR expression.31 Blood collected in Streck™ tubes was shipped overnight to Epic Sciences. Nucleated cells from the blood were plated onto glass microscopy slides, fixed, and bio-banked at −80°C until analysis. Biomarker expression studies were performed on four slides per patient; 2 slides for GR. Two slides correspond to the analysis of 6 million nucleated cells within the blood draw. Each assay respectively stained for pan-cytokeratin (CK), CD45, and DAPI in addition to GR. A CTC is defined as any CK+, CD45−, DAPI+ cell. After staining the slide, each nucleated cell was imaged using high-throughput florescence microscopy, and CTCs were identified using Epic Sciences’ proprietary digital pathology algorithms. Candidate CTCs were then confirmed by trained human technicians. The final cell counts in each sub-group (GR+/−) were then tabulated.
For GR evaluation (Supplementary Figure 1A), the monoclonal antibody specific to the GR (D6H2L, Cell Signaling Technology, Rabbit IgG, Cat #12041) C-terminal domain was utilized. If a patient had greater than 0 detectable CTC per milliliter of tested blood, they were classified in a binary fashion as “CTC positive.” The threshold for a CTC to be positive for GR is not formally known, and it is not known to what extent GR expression correlates with activity, therefore cell-line cells spiked into healthy donor blood from DU145 (GR positive, RRID: CVCL_0105) and LNCaP (GR negative) were analyzed in parallel. For each cell detected on the slide the mean fluorescence intensity from the GR antibody detection was recorded and classified cells as GR positive (GR+) if it had GR expression detectable above background fluorescence within identified CTCs. Otherwise, cells were classified GR negative (GR−).
Formulation
Phase II Enz was supplied by Astellas and Medivation (now Pfizer) as 40 mg capsules, 120 capsules per bottle. Mif was provided by Corcept Therapeutics as 300 mg tablets, 30 tablets/bottle. Enz and Mif were taken concurrently once daily.
Statistical Analysis
In phase I, steady-state Ctrough for Enz and its metabolite were determined after 28 days of Enz alone and after an additional 28 days of combination therapy. The mean of the day 57/29 ratio of trough concentrations for Enz and its metabolite were calculated for each dosing cohort and used for dose adjustment determinations in the following cohort. Standard deviation and range of the Ctrough and ratio were calculated.
Randomization for phase II was done at the University of Chicago in a 1:1 block fashion, using block sizes of 4, 6, and 8. There was no specific stratification as the 12-week standard of care Enz lead-in was hypothesized to add homogeneity to the population. For the randomized phase II study, the primary endpoint was PSA PFS post randomization, defined as time to PSA progression or death, whichever came first. We assumed median time to PSA progression in the control (Enz alone) arm would be 6 months. This was based on the phase III Enz trial AFFIRM,13 in which ~10% of treated patients had PSA progression prior to 12 weeks, the PSA PFS was approximately 42% at 9 months, corresponding to a 6-month rate, conditional on no event at 12 weeks, of 0.42/0.9=0.47. Thus, our enrichment strategy was projected to lead to a patient population with a median PFS of 6 months (24 weeks) in the control arm post randomization. To detect a hazard ratio (HR) of 0.60, corresponding to an increase in the median from 24 to 40 weeks, with 80% power, a sample size of 84 patients (42/arm) was required, using a one-sided test at the α=0.10 significance level. This assumed a two-year accrual period and a subsequent one-year follow-up period. Since it was anticipated that ~10% of enrolled patients in phase II would not be randomized due to progression or intolerance, 92 patients were to be enrolled. An interim futility analysis was conducted after 50% of PSA progression events were observed. A conditional power for the primary endpoint of <25% at this analysis would lead to study termination for futility. As shown in Results, using the protocol-defined PSA criteria above, futility was reached and accrual to the trial was terminated early. For subsequent analyses, the PSA progression endpoint was modified to conform with PCWG criteria to require both a 25% increase from baseline or nadir and an absolute 5ng/mL increase. As will be seen, the conclusions were unaltered.
For time to PSA progression, Kaplan-Meier (KM)32-35 curves were generated for the two treatment arms and compared using a log-rank test. Median time-to-event in each group was estimated along with 90% confidence intervals using the method of Brookmeyer and Crowley.34 Radiographic progression-free survival was analyzed with KM curves. For patients without progression, PFS was censored at the date of the patient’s last assessment.
Relative PSA change from baseline within each arm was also reported using a waterfall plot.28 Adverse events were summarized by grade and type. Group comparisons used the chi-square or Fisher’s exact tests.
We also compared endocrine PD marker differences between the Enz alone versus the Enz+Mif treatment arms. In previous placebo-controlled trials with Mif, serum cortisol levels reliably doubled from a baseline of approximately 15 μg/dL to >30 μg/dL.38,39 Based on the reported interquartile ranges and assuming normality of the distribution, the coefficient of variation was estimated at 50%. At time of interim analysis, the difference in cortisol levels was analyzed. A lack of biomarker effect could provide justification for closing the trial. The PK data was obtained as above and were summarized using standard descriptive methods (means, standard deviations, medians, and ranges).
The primary objective of the CTC correlative study was to assess intra- and inter-patient variability in GR within CTCs from patients with progressive CRPC. The secondary objective of this aim was to explore the correlation between baseline GR expression and PSA PFS in patients treated with Enz ± Mif. The components of variability were estimated using analysis of variance. Finally, Cox33 regression models for time to PSA progression were fit using GR expression as a binary covariate and incorporating treatment-by-marker interaction terms to determine the marker’s prognostic and/or predictive value.
Phase I Dose Escalation Methods
Six patient dose cohorts escalated through the pre-planned dose combinations of Mif and Enz (see Figure 1). During the phase I portion of the study, patients could potentially receive Mif doses of either 300 mg daily or 300 mg every other day combined with Enz at either 40, 80, 120, or 160 mg daily. Per protocol guidelines, the starting dose for the first cohort was 40 mg of enzalutamide and 300 mg of mifepristone daily. PK analyses were then performed by InVentiv/Medivation. The first PK levels of Enz and its main M2 metabolite were then taken after 28 days of Enz at standard dosing to reach a steady state with less variability of Enz drug levels, after which time combination dosing with Mif occurred to find dose escalation cohorts. After 28 days of combination, another trough PK was taken of both Enz and its active metabolite (M2). A ratio of trough drug concentration after 28 days of combined dosing (day 57 of the study) divided by trough drug concentrations after 28 days of Enz alone dosing (day 29 of the study) was then performed, defined as ri. An average of ri for all patients within a dose cohort (i.e. mean of 6 or 12 patients) was then calculated, defined as ř. For ř >1.5, enzalutamide was then decreased by one level (e.g. 80 mg daily to 40 mg daily) and the patient re-entered the dose escalation algorithm. If ř <0.75, Enz was then increased by one level (e.g. 40 mg daily to 80 mg daily) as below and the patient re-entered the dose escalation algorithm. If ř ≤1.5, ≥ 0.75, then Mif dose changes were determined by the rate of dose limiting toxicity (DLT). DLT was defined as any grade 3 or 4 toxicity that was potentially related to the therapy. There was a 28-day DLT monitoring period after combination treatment during the phase 1 study in which safety was examined prior to dose escalation. A 28-day DLT period was chosen as the majority of dose limiting toxicity seen with Enz, including rare seizures, were seen in previous phase I/II studies within 6 weeks of beginning Enz (patients had been on Enz for four weeks prior to combination dosing already). The DLT period therefore began at day 29, upon the onset of concurrent Mif and Enz drug daily administration, and ended at day 57, four weeks later. If ř ≤1.5 and ≥ 0.75 Mif was escalated one level and the patient re-entered the dose escalation algorithm provided the dose limited toxicity rate (DLT) was less than 33%. The decision to escalate Mif was made by investigators and an independent safety monitor after evaluation of toxicity data from the current dose level. If the DLT rate was greater than 33% (>2 in 6 patients per cohort), then it was concluded the maximum tolerated dose (MTD) of mifepristone was exceeded and the prior dose level was determined to be the MTD which was then expanded to 12 patients. If the MTD was exceeded with Mif 300 mg every day, the dose was reduced to 300 mg every other day. The R2PD was defined as the highest dose tested where at most two of the six patients developed DLT during the first cycle of treatment and ř ≤1.5.
Figure 1: Consort Diagram.
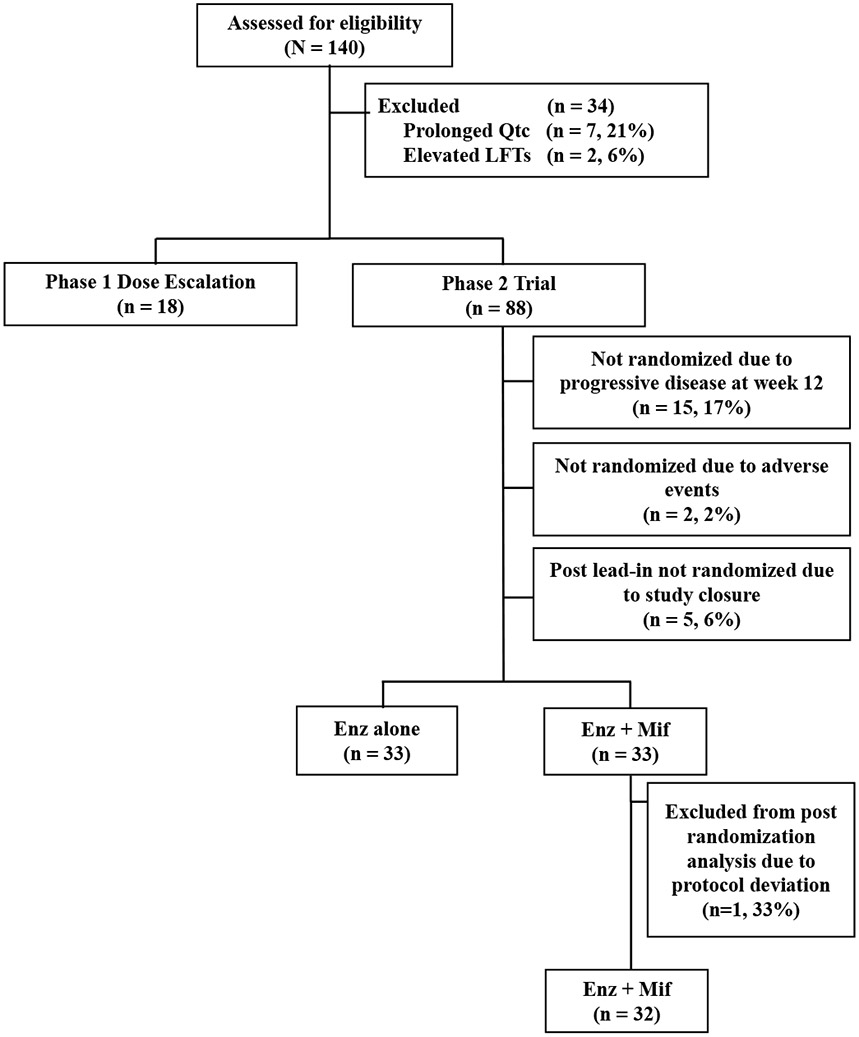
Enz, enzalutamide; Enz alone, Enz 160mg daily after 12-week enzalutamide monotherapy lead in; Enz+Mif, Enz 120mg and Mif 300 mg daily after 12-week Enz lead-in; LFTs, liver function tests; Mif, mifepristone.
The data generated in this study are available upon request from the corresponding author.
Results
Patient Characteristics
As above, the clinical trial had a two-part study design with phase I focused on safety and PK followed by phase II. Between January 2014 and January 2019, 140 total patients were assessed for trial eligibility; 106 patients were ultimately accrued (Figure 1). The most common reason for not meeting eligibility were (N, %) prolonged QTc (7, 21%) and elevated liver function tests (2, 6%).
Eighteen patients were enrolled in phase I dose escalation and treated in three combination dosing cohorts. These include (1) Enz 40 mg and Mif 300 mg, (2) Enz 80 mg and Mif 300 mg and (3) Enz 120mg and Mif 300 mg. 6 patients were in each dosing cohort. Demographics were consistent with a general metastatic PC population (Table 1).
Table 1:
Baseline Characteristics
| Overall (n=106) |
Phase I (n=18) |
Phase II (n=88) | |||
|---|---|---|---|---|---|
| Enz (n=33) |
Enz + Mif (n=33) |
NR (n=22) |
|||
| Age, years | |||||
| Median (range) | 69 (52-58) | 70 (55-84) | 71 (52-83) | 71 (57-85) | 68 (52-58) |
| >75, No. (%) | 34 (32) | 5 (28) | 10 (30) | 14 (42) | 5 (23) |
| ECOG PS, No (%) | |||||
| 0 or 1 | 46 (43) | 10 (56) | 13 (39) | 16 (49) | 7 (32) |
| 2 | 60 (57) | 8 (44) | 20 (61) | 17 (52) | 15 (68) |
| Race, No. (%) | |||||
| White | 76 (72) | 12 (67) | 20 (61) | 26 (79) | 16 (73) |
| AA | 25 (24) | 6 (33) | 9 (27) | 7 (21) | 3 (14) |
| Asian | 2 (2) | 0 | 2 (6) | 0 | 0 |
| Other | 3 (3) | 0 | 2 (6) | 0 | 1 (5) |
| Prior Docetaxel No. (%) | 36 (34) | 7 (39) | 9 (27) | 6 (19) | 14 (64) |
| Disease Location, No. (%) | |||||
| Bones | 73 (69) | 16 (89) | 22 (67) | 19 (58) | 16 (73) |
| Lymph Nodes | 57 (54) | 11 (61) | 17 (52) | 18 (55) | 11 (50) |
| Viscera | 30 (28) | 6 (33) | 9 (27) | 6 (18) | 9 (41) |
| PSA (ng/mL), median (range) | 11.0 (0.1-616) | 7.1 (1.5-616) | 14.4 (2.2-342) | 12.3 (0.2-77.7) | 10.4 (0.1-380) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; NR, not randomized; PS, performance status
88 patients were enrolled in phase II (Figure 1). After the 12-week Enz lead in, 66 patients ultimately underwent randomization. 33 patients continued to receive Enz alone (160mg/day), while 33 patients were randomized to receive the RP2D of 120 mg/day Enz and 300 mg/day Mif (Enz+Mif). Fifteen (17%) patients were not randomized due to progressive disease prior to 12 weeks, 2 (2%) patients not randomized due to adverse events, and 5 (6%) not randomized due to study closure during their 12-week Enz lead in. One patient was excluded from post-randomization analysis due to a protocol deviation of combined treatment initiation. Patient characteristics were well balanced between the two phase 2 groups (Table 1). Despite the study being written anticipating post-docetaxel Enz, most patients received treatment in the pre-docetaxel setting (34% of patients received docetaxel previously).
Patients who failed to randomize after the 12-week Enz lead-in had a higher proportion of prior docetaxel therapy and a higher percentage of visceral disease relative to those randomized.
Phase I
Enz PK
The main PK outcomes are summarized in Figure 2. The Day 57 / Day 29 PK trough ratio of Enz and metabolites concentration (Figure 2) for cohort 1, Enz 40mg and Mif 300mg, was 0.6 (standard deviation 0.2, range 0.4-0.8). Per protocol, the dose of Enz was then escalated in the second cohort to Enz 80mg, Mif 300 mg. Day 57/29 mean PK ratios were still suboptimal per protocol at 0.7 (standard deviation 0.1, range 0.5-0.8). Cohort 3 was then dosed Enz 120 mg, Mif 300 mg and had a mean Day 57/29 PK ratio of 1.0 (standard deviation 0.2, range 0.8-1.2), reaching the protocol goal of 0.75-1.5 as being acceptable for phase 2.
Figure 2: Enzalutamide steady-state drug levels.

Boxplot showing median, interquartile range, and full range of day 57/29 Enz and metabolite drug levels by dose escalation cohort. The Enz drug level trough drug concentration (Ctrough) achieved equivalent PK levels at a dosing of Enz 120 mg, Mif 300 mg with ratio of 1.0 for cohort 3, which became the recommended phase 2 dose. “X” denotes mean within each group.
Abbreviations: Ctrough, concentration of Enz and metabolite on given day; Enz, enzalutamide; Mif, mifepristone.
Endocrine Pharmacodynamic Effects
To ensure adequate GR antagonism, we assessed change in cortisol after 4 weeks of Mif treatment. Serum cortisol levels were measured prior to Enz initiation, after 28 days of Enz monotherapy, and then at day 57 after randomization to combination dosing with Mif. Cortisol routinely doubled as expected after combination dosing with Mif for each cohort (Supplementary Figure 2).
Phase 1 Safety
Study-drug-attributable adverse events in at least 15% of patients in any trial phase is seen in Table 2. Overall, the combination of Mif+Enz was generally well-tolerated. There were no DLTs in any cohort and no study-drug-attributable grade 4 or 5 adverse events. In phase I the most common all-grade side effects were fatigue (72%), anorexia (28%) and diarrhea (28%). The only notable study-drug-attributable grade 3 side effects were fatigue (6%), cognitive disturbance (6%), and hypertension (6%). There were no significant differences in adverse events when comparing the various Enz dosing cohorts.
Table 2:
Adverse Events
| Phase I: No., % (n= 18) |
Phase II: Enz Alone No., % (n = 33) |
Phase II: Enz + Mif No., % (n= 33) |
||||
|---|---|---|---|---|---|---|
| All Grade |
Grade 3 | All Grade |
Grade 3 | All Grade |
Grade 3 | |
| Total Adverse Events (No. of patients, %) | 16 (89) | 3 (22) | 29 (88) | 5 (15) | 30 (91) | 6 (18) |
| Adverse Event | All Grade |
Grade 3 | All Grade |
Grade 3 | All Grade |
Grade 3 |
| Fatigue | 13 (72) | 1 (6) | 25 (76) | 1 (3) | 25 (76) | 4 (12) |
| Anorexia | 5 (28) | 0 (0) | 4 (12) | 0 (0) | 11 (33) | 0 (0) |
| Diarrhea | 5 (28) | 0 (0) | 6 (18) | 0 (0) | 4 (12) | 0 (0) |
| Hot Flashes | 5 (28) | 0 (0) | 15 (45) | 0 (0) | 15 (45) | 0 (0) |
| Nausea | 5 (28) | 0 (0) | 3 (9) | 0 (0) | 3 (9) | 0 (0) |
| Pain | 5 (28) | 0 (0) | 1 (3) | 0 (0) | 1 (3) | 0 (0) |
| Amnesia | 4 (22) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Edema | 3 (17) | 0 (0) | 1 (3) | 0 (0) | 3 (9) | 0 (0) |
| Generalized Muscle Weakness | 0 (0) | 0 (0) | 2 (6) | 0 (0) | 8 (24) | 0 (0) |
| Dizziness | 2 (11) | 0 (0) | 1 (3) | 0 (0) | 5 (15) | 0 (0) |
Enz, Enzalutamide; Enz alone, Enz 160mg daily after 12-week enzalutamide monotherapy lead in; Enz+Mif, Enz 120mg and Mif 300 mg daily after 12 week Enz lead-in; Mif, Mifepristone
Phase II Results
Phase II Safety
In the phase II study, the combination of Mif+Enz was well tolerated with minimal significant differences associated with the addition of Mif (study-drug-attributable adverse events listed in Table 2). When comparing the Enz alone arm to Enz+Mif arm, the most common any-grade side effects were fatigue (76% vs. 76%), hot flashes (45% vs. 45%), anorexia (12% vs. 33%, p>0.05), and generalized muscle weakness (6% vs. 24%, p>0.05). Grade 3 fatigue was higher in the Enz+Mif arm (12%) as compared to the Enz alone group (3%). Like phase I, there were no grade 4 or 5 attributable adverse events. There were very few treatment discontinuations due to adverse events (2 patients in the Enz arm, 1 patient in the Enz+Mif arm).
PSA Endpoints
PSA PFS was the primary endpoint. Per protocol, an interim futility analysis was performed after 50% of the planned PSA-progression events (35 events). At this analysis the hazard ratio (Enz+Mif/Enz alone) was 1.34 in favor of the control arm and the conditional power for finding a benefit to combination therapy if the trial continued was only 0.12. Based on lack of efficacy at interim analysis, the study was closed to accrual.
As described under Methods, additional analysis using a more stringent definition of PSA progression was performed. In this analysis, which included 26 events, median PSA PFS after randomization was 20.8 months in the Enz-alone arm compared with 16.5 months in the Enz+Mif arm. Cox regression analysis yielded a hazard ratio of 1.09 when comparing the two arms (logrank p=0.83, Figure 3A).
Figure 3: PSA and radiographic progression free survival.

A. Kaplan-Meier plot of time to PSA progression while receiving Enz alone compared with Enz+Mif after combination dosing began at 12 weeks, showing a median progression-free survival hazard ratio of 1.09 comparing both arms (log rank p=0.83).
B. Kaplan-Meier plot of time to radiographic progression. After a 12-week Enz lead-in, patients were randomized to receive either Enz alone or Enz+Mif, showing a median progression-free survival of NR in Enz alone arm and 16.5m in Enz+Mif arm (p=0.22).
Abbreviations: Enz, Enzalutamide; Enz alone, Enz 160mg daily after 12-week enzalutamide monotherapy lead in; Enz+Mif, Enz 120mg and Mif 300 mg daily after 12 week Enz lead-in; Mif, Mifepristone; PSA, prostate specific antigen
With respect to PSA response, the bulk of the PSA change occurred within the first 12 weeks of the initial Enz lead-in (Supplementary Figure 3A). The pre-randomization PSA RR (defined as PSA decline > 50%) was 81% in those subsequently randomized to Enz-alone group and 72% in the Enz+Mif group. Very few patients eligible for randomization experienced primary PSA progression (post-treatment PSA increase as best response during the lead-in: 2 patients in the Enz+Mif group, 1 in the Enz alone).
The maximum percentage decrease in PSA that patients achieved after randomization to either Enz-alone or Enz+Mif at week 12 was similar comparing the two treatment arms (Supplementary Figure 3B). The mean post-randomization decrease in PSA change to best response was −30.3% in the Enz arm and −28.8% in Enz+Mif arm (p=0.86). Nine patients (28%) in each arm had a post-randomization PSA decline > 50%. With respect to radiographic response, 4/33 patients in each arm had documented RECIST response.
In sum, these data suggest that the addition to Mif to Enz did not improve PSA-PFS or response.
Radiographic PFS
Time to radiographic progression was a secondary endpoint of phase II. Patients that came off study for either physician discretion, PSA progression, toxicity or data lock were censored at the time taken off study. There were no significant differences in time to radiographic progression between the two arms (Figure 3B). Median radiographic PFS was 16.5 months in the Enz+Mif group vs. not reached in the Enz only group (HR 1.70, p=0.22).
CTC Analyses
The goal of the CTC analysis was to test whether the presence of GR positive CTCs would predict benefit from the addition of Mif to Enz. Week 12 blood samples drawn immediately prior to randomization were available for CTC analyses on 24/33 pts in the Enz+Mif group (73%), 29/33 in the Enz-alone group (88%) and 6/17 (35%) of patients who did not proceed to randomization after the initial Enz lead-in. The majority (68%) of randomized patients were noted to have detectable CTCs compared with 6/6 (100%) of patients in the group that did not proceed to randomization (p=0.17). A higher percentage of patients in the Enz group (79%) had detectable CTCs at the time of randomization, compared with (54%) of those randomized to Enz+Mif arm (p=0.08). Adjusting the treatment arm comparison of PSA progression for the presence of CTCs, the hazard ratio decreased from 1.09 to 0.93, but remained non-statistically significant (p=0.87). Consistent with prior data,31 patients with detectable CTC at time of randomization were less likely to have had a PSA response (Supplementary Figure 1B) and more likely to have worse PSA PFS (Supplementary Figure 1C).
Amongst the entire study population, 30/42 (71%) of patients with detectable CTCs had GR positive (+) CTCs at week 12. A numerically higher percentage of patients who were not eligible to randomize had GR+ CTCs (5/6, 83%) compared to 25/36 (69%) of randomized patients (Supplementary Figure 1D). The incidence of GR+ CTC was similar in the two arms (74% Enz alone, 62% Enz+Mif, as expected from randomization) and the presence of GR+ CTC was not, contrary to the study hypothesis, a predictor of prolonged PSA-PFS (HR 1.15, p=0.84, Supplementary Figure 1E). In the converse analysis of PSA-PFS in patients who, at week 12 had GR negative (−) CTCs or lacked CTCs, there was no difference between arms (HR=0.69, p=0.59).
Endocrinologic Pharmacodynamic Effects
Cortisol
As observed in phase I, serum cortisol was expected to increase by as much as double after treatment with Mif.36,37 The ratio of week 16 to week 12 cortisol was 1.45 (0.47-2.43, Enz alone) vs. 2.40 (1.90-2.89, Enz+Mif) indicating cortisol nearly doubled after introducing Mif, as expected (p=0.06). Given positive skewness, a logarithmic comparison of week 16 to week 12 cortisol demonstrated strong statistical significance (p=0.0002).
Testosterone
Prior studies demonstrated that Mif markedly increases serum androgen levels in castrate patients.32 This increase was thought secondary to Mif’s inhibition of GR resulting in an increase in adrenocorticotropic hormone leading to an increase in adrenal androgen production. As shown in Figure 4, testosterone increased slightly after 12 weeks of Enz lead-in, but significantly increased with Mif after randomization. The average post-randomization testosterone was 16.0 ng/dL in the Enz alone group vs. 38.8 ng/dL in the Enz+Mif group (p=0.0012). Therefore, Mif led to a nearly 2.5x increase in androgen production for the Enz+Mif population.
Figure 4: Effect of treatment on serum testosterone levels.

Combination dosing with Enz+Mif did not begin until after week 12 (which represents at least 12 weeks from baseline). In situations in which cycle 1 day 1 data was not available for baseline, the date of the pre-study PSA was used for dating testosterone baseline if patients were screened and started within a 5-day window. Post randomization data represents first testosterone value at least 4 weeks after week 12 testosterone level. Baseline testosterone levels were in the castrate range and did not significantly increase between baseline and Week 12. Combination dosing with Enz+Mif did not start until after week 12. Testosterone increased markedly after the addition of Mif in the Enz+Mif arm due to increased adrenal testosterone production. Testosterone did not substantially change for the group continuing to receive Enz alone. Star (*) represents p<0.01 difference between Enz and Enz+Mif at post-randomization time point.
Abbreviations: Enz, Enzalutamide; Enz alone, Enz 160mg daily after 12-week enzalutamide monotherapy lead in; Enz+Mif, Enz 120mg and Mif 300 mg daily after 12 week Enz lead-in; Mif, Mifepristone
Discussion
This was the first reported randomized clinical trial to test the hypothesis that continuous GR pathway inhibition would be safe and improve outcomes in combination with ARSI for mCRPC. We found that a 25% dose reduction in Enz, when added to Mif, resulted in equivalent drug levels compared to full dose Enz. Although generally well-tolerated, the addition of Mif to Enz following a 12-week Enz lead-in did not delay time to PSA or radiographic PFS.
Of note, this study demonstrated a median PSA PFS after randomization of 20.8 months in the Enz-alone arm and 16.5 months in the Enz+Mif arm. This implies a total median PSA PFS of 23.8 months with Enz, considerably longer than the median time to PSA progression seen in prior studies.13,38 However, our study did not allow patients who progressed on Enz monotherapy in the first 12 weeks to proceed to randomization. By focusing on such “non-progressors” we were selecting for patients with significantly above-average PSA PFS from Enz.
This trial had several strengths. While there is significant preclinical evidence that the GR contributes to Enz resistance,23,26,27,39,40 this is the first ever prospective randomized study to study combined GR and AR antagonism in a CRPC clinical population. In addition, our study was multi-institutional, involving over 100 CRPC patients across four cancer centers throughout the United States. Our study provides some of the first clinical evidence of the therapeutic combination of GR modulators with AR antagonism in prostate cancer. Furthermore, our phase I study was able to demonstrate how safe, pharmacologically guided dosing of Enz combined with a CYP3A4, CYP3C8/9 inhibitor could achieve equivalent therapeutic effect. Given Enz is associated with increased risk of seizures at higher plasma concentrations and may be less effective at lower blood doses, careful attention to PK is of paramount importance.41,42 This study can be used to suggest how to safely combine Enz with other agents that have PK properties like Mif. Furthermore, this study was the first to show that long-term GR blockade, at a pharmacologically active dose, along with ARSI would be safe or tolerable.43
A unique feature of this trial was that it was the first ever to utilize the interrogation of CTCs to analyze the GR as a biomarker in a prospective CRPC trial. The goal of this analysis was to determine if GR expression could be a predictive biomarker for Mif efficacy. Our study did show that patients with CTCs had a less robust response and a shorter PSA PFS from Enz regardless of the treatment they ultimately received, consistent with prior data validating pre-treatment CTCs as a prognostic biomarker in men starting either abiraterone or Enz.31 However, our study did not demonstrate that the presence of GR+ CTCs was predictive of a specific lack of response to Enz, nor that individuals with GR+ CTCs benefited from GR directed therapy with Mif.
Our study had several shortcomings. First, the phase II portion was open-label and lacked a placebo-controlled arm. Secondly, the primary endpoint of phase II was PSA PFS, chosen since PSA is a pharmacodynamic biomarker of nuclear hormone activity. Instead, radiographic PFS may have better reflected clinical status. Finally, the definition of GR positivity in CTCs is not well established. We took a binary approach of classifying CTCs as GR+ should they exhibit any expression beyond background. There are potential pitfalls to this approach. GR expression is likely on a spectrum. Focusing instead on only high GR expression (e.g. highest decile) rather than a binary approach may better reflect GR positivity. GR expression may also not correspond with downstream pathway activation. Future studies could use paired biopsies as more accurate correlative markers of GR activation. A more nuanced view of GR expression within progressive mCRPC may better support GR’s utility as a therapeutic biomarker. Additionally, our trial collected CTCs only at randomization with the hypothesis that GR+ CTC would predict benefit of dual antagonism. CTCs at baseline, randomization, and end of study could determine if Mif helped clear GR+ CTCs. Future studies could collect CTCs at more time points.
Although Mif did not improve PSA PFS, this trial does not refute the hypothesis that GR antagonists have a benefit in combination with ARSI in PC. The exact mechanisms of castrate resistance and subsequent resistance mutations is an active area of investigation. We may have randomized patients to Mif after too long a period of Enz exposure. Our trial design originally posited that GR expression would be enriched after 12 weeks of Enz monotherapy. Emerging evidence suggests that GR enrichment may occur much earlier in PC.44 A recent study of androgen biosynthesis inhibition in the pre-operative setting demonstrated that GR enrichment may occur as early as the neo-adjuvant castrate-sensitive stage.44 Conducting this study in the CRPC setting after an additional 12 weeks of Enz may have inadvertently enriched for intra-patient pleotropic ARSI-resistance mechanisms beyond GR. Prior to becoming castrate-resistant, PC disease biology is likely more homogenous. Introducing GR inhibition in an earlier clinical stage before the activation of adaptive cell survival mechanisms (such as p53 mutations or Rb loss) may have therapeutic merit.
Androgen receptor mutations (such as AR-amplification, AR splice variant expression, and aberrant AR co-regulator activities) have been implicated in castrate resistance and could have also contributed to the study’s failure.45,46 Despite randomization, the trial may have had an imbalance in such other AR mutations, (e.g. AR-v7) between the two arms.
Although Mif is a potent GR antagonist it has several characteristics that may have made it a suboptimal GR antagonist for PC therapy. It is a non-selective steroidal nuclear hormone receptor antagonist that binds other NR3C family members including AR and the progesterone receptor. Modulation of these other receptors potentially led to unintended consequences. For example, in the setting of mutated AR ligand binding domains (LBD), other steroidal compounds can ligand and activate LBD-mutant AR. There are reports that Mif can activate AR (depending on AR LBD mutations and the Mif dose used)47,48 blunting our therapeutic approach. Future studies should study the evolution of LBD mutations in the context of GR antagonism.
Mif can also raise testosterone levels.32 Our study showed that testosterone levels significantly increased with Mif, potentially hindering Enz’s therapeutic effect in the Enz+Mif arm. Highly specific non-steroidal GR antagonists may not raise testosterone levels and thus may be more effective in PC.39,49 While Mif may have been a good starting point given its broad nuclear hormone receptor antagonism and clinical availability, phase I studies of more selective agents like relacorilant (NCT 03674814) or exicorilant (NCT 03437941) combined with Enz are ongoing, and can be used in future dual AR-GR antagonism PC studies.
While we achieved systemic cortisol receptor blockade with Mif as demonstrated by the increase in cortisol subsequent to Mif, other studies have shown that local tumor glucocorticoid levels stimulate the GR and contribute to Enz resistance.50 Given Mif binds and antagonizes GR it is unlikely that upstream glucocorticoid production would be sufficient to overcome Mif; however, it is possible that very high local glucocorticoid production outcompeted Mif for the GR. Future studies with imbedded tissue correlative studies can help answer whether this is a major contributing factor underlying insufficient GR antagonist activity.
In conclusion, this is the first prospective clinical trial reported of dual AR-GR antagonism in CRPC. Daily dosing of Enz combined with Mif was safe and well tolerated. The development of more specific GR antagonists, combined with AR antagonists, studied in an earlier stage more homogenous population may lead to more effective future therapeutic regimens. Additional work should clarify which biomarkers can identify patients who would benefit most from this approach.
Supplementary Material
Statement of Translational Relevance.
This is the first clinical trial to test the hypothesis that continuous glucocorticoid pathway inhibition would be safe and improve outcomes when combined with potent androgen receptor signaling inhibition for metastatic castrate resistant prostate cancer (CRPC). We found that a 25% dose reduction in enzalutamide, when added to mifepristone, resulted in equivalent drug levels compared to full dose enzalutamide. Although generally well tolerated, the addition of mifepristone to enzalutamide following a 12-week enzalutamide lead-in did not delay time to PSA progression. Similarly, the addition of mifepristone to enzalutamide did not prolong radiographic or clinical progression free survival. This provides some of the first clinical results regarding the safety and efficacy of combining androgen receptor targeted therapy with glucocorticoid receptor modulation in prostate cancer. In addition, this study was the first to incorporate glucocorticoid receptor evaluation within circulating tumor cells, an important biomarker for future clinical trials of this pathway.
Acknowledgements
Funding and Support (R. Szmulewitz)
Support for the clinical trial was provided by the Department of Defense CDMRP W81XWH-14-1-0021 and the CTC correlatives were supported by Prostate Cancer Foundation-Movember Challenge Award. Biostatistics and clinical trial office supported by University of Chicago NCI Cancer Center Support Grant (5P30CA014599-46).
Footnotes
Conflict of Interest Statements (Disclosures):
Anthony V. Serritella
No disclosures
Daniel Shevrin
No disclosures
Elisabeth I. Heath
Honoraria - Bayer; Dendreon; Sanofi
Consulting or Advisory Role - Agensys
Speakers' Bureau - Sanofi
Research Funding - Agensys (Inst); Celgene (Inst); Celldex (Inst); Dendreon (Inst); Genentech/Roche (Inst); Inovio Pharmaceuticals (Inst); Millennium (Inst); Seattle Genetics (Inst); Tokai Pharmaceuticals (Inst)
James L. Wade
No disclosures
Elia Martinez
No disclosures
Amanda Anderson, Joseph Schonhoft, Lincy Chu
Employees of Epic Sciences and received Epic stock options at the time of their work
Theodore Karrison
No disclosures
Walter M. Stadler
Consultant (DSMB) - AstraZeneca, Bayer, Eisai, Merck, Pfizer, Treadwell Therapeutics
Consultant (Other) - Caremark/CVS, EMA Wellness
CME providers Speakers Bureau (sponsorship unknown): Applied Clinical Education, Dava Oncology, Global Academy for Medical Education, OncLive, PeerView, Research to Practice, Vindico
Grant/Research Support (to institution) - Abbvie, Amgen, Astra-Zeneca, Astellas (Medivation), Bayer, Bristol-Myers-Squibb, Boehringer-Ingelheim, Calithera, Clovis, Corvus, Eisai, Exilixis, Genentech (Roche), Johnson & Johnson (Janssen), Merck, Novartis, Pfizer, Seattle Genetics, Tesaro, X4Pharmaceuticals
Expert Witness - Apotex, DRL, Mylan, Sandoz
Miscellaneous/Editorial- Cancer (ACS), Up-To-Date
Russell Z. Szmulewitz
Honoraria - Astellas Pharma
Consulting or Advisory Role - Abbvie; Amgen; Astellas Pharma; AstraZeneca; Exelixis; Janssen Oncology; Merck; Pfizer; Sanofi
Research Funding - Abbvie; Astellas Pharma; Incyte; Janssen Oncology; Macrogenics
Patents, Royalties, Other Intellectual Property - Patent licensed by University of Chicago of which I am co-inventor to Corcept Therapeutics for combination AR/GR inhibition in prostate cancer
Travel, Accommodations, Expenses - Corcept Therapeutics
References
- 1.Aragon-Ching JB, Dahut WL. Novel Androgen Deprivation Therapy (ADT) in the Treatment of Advanced Prostate Cancer. Drug Discov Today Ther Strateg. 2010;7(1-2):31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scher HI, Heller G. Clinical states in prostate cancer: toward a dynamic model of disease progression. Urology. 2000;55(3):323–327. [DOI] [PubMed] [Google Scholar]
- 3.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. [DOI] [PubMed] [Google Scholar]
- 4.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23(32):8253–8261. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. The lancet oncology. 2009;10(10):981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan CJ, Smith MR, Fizazi K, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16(2):152–160. [DOI] [PubMed] [Google Scholar]
- 9.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong AJ, Szmulewitz RZ, Petrylak DP, et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy With Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J Clin Oncol. 2019;37(32):2974–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis ID, Martin AJ, Stockler MR, et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. New England Journal of Medicine. 2019;381(2):121–131. [DOI] [PubMed] [Google Scholar]
- 12.Fizazi K, Tran N, Fein L, et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. New England Journal of Medicine. 2017;377(4):352–360. [DOI] [PubMed] [Google Scholar]
- 13.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–1197. [DOI] [PubMed] [Google Scholar]
- 14.Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 2015;4(3):365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadar MD. Small molecule inhibitors targeting the "achilles' heel" of androgen receptor activity. Cancer Res. 2011;71(4):1208–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai SJ, Ma A-H, Tepper CG, Chen H-W, Kung H-J. Inappropriate Activation of the Androgen Receptor by Nonsteroids: Involvement of the Src Kinase Pathway and Its Therapeutic Implications. Cancer Research. 2006;66(21):10449–10459. [DOI] [PubMed] [Google Scholar]
- 17.Sun S, Sprenger CC, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinkamp MP, O'Mahony OA, Brogley M, et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009;69(10):4434–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards J, Lim AC, Hay CW, et al. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res. 2012;72(9):2176–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Wang J, Yu G, Liu W, Pearce D. Androgen and glucocorticoid receptor heterodimer formation. A possible mechanism for mutual inhibition of transcriptional activity. J Biol Chem. 1997;272(22):14087–14092. [DOI] [PubMed] [Google Scholar]
- 21.Cleutjens CB, Steketee K, van Eekelen CC, van der Korput JA, Brinkmann AO, Trapman J. Both androgen receptor and glucocorticoid receptor are able to induce prostate-specific antigen expression, but differ in their growth-stimulating properties of LNCaP cells. Endocrinology. 1997;138(12):5293–5300. [DOI] [PubMed] [Google Scholar]
- 22.Mohler JL, Chen Y, Hamil K, et al. Androgen and glucocorticoid receptors in the stroma and epithelium of prostatic hyperplasia and carcinoma. Clin Cancer Res. 1996;2(5):889–895. [PubMed] [Google Scholar]
- 23.Szmulewitz RZ, Chung E, Al-Ahmadie H, et al. Serum/glucocorticoid-regulated kinase 1 expression in primary human prostate cancers. Prostate. 2012;72(2):157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yemelyanov A, Bhalla P, Yang X, et al. Differential targeting of androgen and glucocorticoid receptors induces ER stress and apoptosis in prostate cancer cells: a novel therapeutic modality. Cell Cycle. 2012;11(2):395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan TZ, Jin FS, Xie LP, Li LC. Relationship between glucocorticoid receptor signal pathway and androgen-independent prostate cancer. Urol Int. 2008;81(2):228–233. [DOI] [PubMed] [Google Scholar]
- 26.Isikbay M, Otto K, Kregel S, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer. 2014;5(2):72–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arora VK, Schenkein E, Murali R, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155(6):1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26(7):1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. [DOI] [PubMed] [Google Scholar]
- 30.Otto K, Griend DV, Conzen S, Szmulewitz R. Abstract 148: Glucocorticoid receptor-mediated cell survival following androgen receptor blockade in castrate-resistant prostate cancer. Cancer Research. 2012;72(8 Supplement):148. [Google Scholar]
- 31.Scher HI, Armstrong AJ, Schonhoft JD, et al. Development and validation of circulating tumour cell enumeration (Epic Sciences) as a prognostic biomarker in men with metastatic castration-resistant prostate cancer. European Journal of Cancer. 2021;150:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taplin ME, Manola J, Oh WK, et al. A phase II study of mifepristone (RU-486) in castration-resistant prostate cancer, with a correlative assessment of androgen-related hormones. BJU Int. 2008;101(9):1084–1089. [DOI] [PubMed] [Google Scholar]
- 33.Cox DR. Regression Models and Life-Tables. Journal of the Royal Statistical Society Series B (Methodological). 1972;34(2):187–220. [Google Scholar]
- 34.Brookmeyer R, Crowley J. A Confidence Interval for the Median Survival Time. Biometrics. 1982;38(1):29–41. [Google Scholar]
- 35.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. Journal of the American Statistical Association. 1958;53(282):457–481. [Google Scholar]
- 36.Page ST, Krauss RM, Gross C, et al. Impact of mifepristone, a glucocorticoid/progesterone antagonist, on HDL cholesterol, HDL particle concentration, and HDL function. J Clin Endocrinol Metab. 2012;97(5):1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pomara N, Hernando RT, de la Pena CB, Sidtis JJ, Cooper TB, Ferris S. The effect of mifepristone (RU 486) on plasma cortisol in Alzheimer's disease. Neurochemical research. 2006;31(5):585–588. [DOI] [PubMed] [Google Scholar]
- 38.Beer TM, Armstrong AJ, Rathkopf D, et al. Enzalutamide in Men with Chemotherapy-naive Metastatic Castration-resistant Prostate Cancer: Extended Analysis of the Phase 3 PREVAIL Study. Eur Urol. 2017;71(2):151–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kach J, Long TM, Selman P, et al. Selective Glucocorticoid Receptor Modulators (SGRMs) Delay Castrate-Resistant Prostate Cancer Growth. Mol Cancer Ther. 2017;16(8):1680–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puhr M, Hoefer J, Eigentler A, et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clinical Cancer Research. 2018;24(4):927–938. [DOI] [PubMed] [Google Scholar]
- 41.Foster WR, Car BD, Shi H, et al. Drug safety is a barrier to the discovery and development of new androgen receptor antagonists. Prostate. 2011;71(5):480–488. [DOI] [PubMed] [Google Scholar]
- 42.Scher HI, Beer TM, Higano CS, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375(9724):1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kach J, Conzen SD, Szmulewitz RZ. Targeting the glucocorticoid receptor in breast and prostate cancers. Science translational medicine. 2015;7(305):305ps319–305ps319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Efstathiou E, Davis JW, Pisters L, et al. Clinical and Biological Characterisation of Localised High-risk Prostate Cancer: Results of a Randomised Preoperative Study of a Luteinising Hormone-releasing Hormone Agonist with or Without Abiraterone Acetate plus Prednisone. Eur Urol. 2019;76(4):418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68(13):5469–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69(1):16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song L-N, Coghlan M, Gelmann EP. Antiandrogen Effects of Mifepristone on Coactivator and Corepressor Interactions with the Androgen Receptor. Molecular Endocrinology. 2004;18(1):70–85. [DOI] [PubMed] [Google Scholar]
- 48.Zhou H, Jachan N, Singh M, et al. Abstract 4172: Activation of AR signaling by mifepristone enhances prostate cancer growth and impairs enzalutamide response. Cancer Research. 2017;77(13 Supplement):4172–4172. [Google Scholar]
- 49.Benagiano G, Bastianelli C, Farris M, Brosens I. Selective progesterone receptor modulators: an update. Expert Opin Pharmacother. 2014;15(10):1403–1415. [DOI] [PubMed] [Google Scholar]
- 50.Li J, Alyamani M, Zhang A, et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.