

Abstract
BACKGROUND:
Central histamine (HA) signaling modulates diverse cortical and subcortical circuits throughout the brain, including the nucleus accumbens (NAc). The NAc, a key striatal subregion directing reward-related behavior, expresses diverse HA receptor subtypes that elicit cellular and synaptic plasticity. However, the neuromodulatory capacity of HA within interneuron microcircuits in the NAc remains unknown.
METHODS:
We combined electrophysiology, pharmacology, voltammetry, and optogenetics in male transgenic reporter mice to determine how HA influences microcircuit motifs controlled by parvalbumin-expressing fast-spiking interneurons (PV-INs) and tonically active cholinergic interneurons (CINs) in the NAc shell.
RESULTS:
HA enhanced CIN output through an H2 receptor (H2R)-dependent effector pathway requiring Ca2+-activated small-conductance K1 channels, with a small but discernible contribution from H1Rs and synaptic H3Rs. While PV-IN excitability was unaffected by HA, presynaptic H3Rs decreased feedforward drive onto PV-INs via AC-cAMP-PKA (adenylyl cyclase-cyclic adenosine monophosphate-protein kinase A) signaling. H3R-dependent plasticity was differentially expressed at mediodorsal thalamus and prefrontal cortex synapses onto PV-INs, with mediodorsal thalamus synapses undergoing HA-induced long-term depression. These effects triggered downstream shifts in PV-IN- and CIN-controlled microcircuits, including near-complete collapse of mediodorsal thalamus-evoked feedforward inhibition and increased mesoaccumbens dopamine release.
CONCLUSIONS:
HA targets H-|R, H2R, and H3Rs in the NAc shell to engage synapse- and cell type-specific mechanisms that bidirectionally regulate PV-IN and CIN microcircuit activity. These findings extend the current conceptual framework of HA signaling and offer critical insight into the modulatory potential of HA in the brain.
The nucleus accumbens (NAc) integrates information encoded by diverse neurotransmitter and monoamine systems to drive adaptive behavior. Although extensively innervated by canonical monoamine centers, such as the ventral tegmental area and raphe nuclei, the NAc shell (NAcSh) also receives dense histaminergic afferents from the tuberomammillary nucleus of the posterior hypothalamus (1,2). The NAcSh expresses multiple histamine (HA) heteroreceptor subtypes, including Gi/o-coupled H3 receptors (H3Rs), Gαq-coupled H1Rs, and Gαs-coupled H2Rs (3,4). Extrahypothalamic HA signaling is implicated in NAc-dependent affective states, including addiction, depression, and stress (5–7). Furthermore, stress-associated H3R signaling elicits cell type- and synapse-specific effects on glutamatergic transmission in the NAc (7,8). However, the full functional repertoire of HA in the NAc, particularly within HA receptor-expressing microcircuit motifs, remains unknown.
NAcSh microcircuits are established by distinct interneuron subpopulations, including parvalbumin-expressing fast-spiking interneurons (PV-INs) and tonically active cholinergic interneurons (CINs) (9–12). Despite comprising only approximately 5% of cells in the NAc, PV-INs and CINs perform specialized network operations that are indispensable for adaptive and pathological learning (13–16). Whereas PV-INs gate medium spiny neuron (MSN) output through feedforward inhibition (FFI), CINs are uniquely coupled to mesoaccumbens dopamine (DA) release (17–2Q). CINs use regenerative pacemaker mechanisms to provide background acetylcholine (ACh) tone that engages nicotinic ACh receptors (nAChRs) and muscarinic ACh receptors (mAChRs) throughout the NAc (21,22). Unlike CINs, PV-IN output requires glutamatergic drive from the same corticolimbic structures that innervate MSNs, such as the prefrontal cortex (PFC) and mediodorsal thalamus (MDT) (23,24). The coincident activation of PV-INs by these regions allows PV-INs to exert precise temporal control over synaptically driven MSN action potential (AP) firing (24). It remains unexplored whether these microcircuits are regulated by the HA system.
We used pharmacology, electrophysiology, voltammetry, and optogenetics in transgenic reporter mice to dissect histaminergic effects on PV-IN and CIN function in the NAcSh. Whereas PV-IN excitability was unaffected by HA signaling, CIN output was potently enhanced through an H2R-predominant pathway requiring small conductance Ca2+-activated K+ (SK) channels and H3R-sensitive GABAergic (gamma-aminobutyric acidergic) synapses. In contrast, presynaptic AC-cAMP-PKA (adenylyl cyclase-cyclic adenosine monophosphate-protein kinase A)-coupled H3Rs triggered a robust decrease in feedforward drive onto PV-INs that was differentially expressed at thalamocortical synapses onto PV-IN synapses. These effects were accompanied by downstream microcircuit alterations, including near-complete MSN escape from thalamically evoked FFI and increased DA release via β2-containing nAChRs. Our findings indicate that HA signaling via multiple receptor subtypes engages distinct interneuron microcircuits to redistribute NAcSh microcircuit activity.
METHODS AND MATERIALS
Forafull description of Methods and Materials, see Supplemental Methods and Materials.
Pharmacology
Picrotoxin was purchased from Sigma-Aldrich. All other drugs were purchased from Tocris Bioscience.
Statistics and Data Analysis
Electrophysiological experiments were analyzed using Clampfit 10.4 (Molecular Devices, LLC) and GraphPad Prism 8.4 (GraphPad Software). Demon Voltammetry and Analysis Software (Wake Forest Innovations) was used for all analyses of fast-scan cyclic voltammetry data. Data were modeled via analysis of peak and decay kinetics to determine the peak. Paired or unpaired t tests were used to analyze statistical differences between data sets. Sidak’s post hoc analyses were used for analyses of variance requiring multiple comparisons. Power analyses were performed with preliminary data during the acquisition of each new data set. The sample size obtained from each power analysis calculation was then compared with sample sizes reported in the literature for similar experiments. For all analyses, α was set as 0.05, with p < α indicating a statistically significant difference. See Supplemental Methods and Materials for additional details.
RESULTS
HA Differentially Modulates CIN and PV-IN Excitability in NAcSh
To interrogate histaminergic actions on discrete interneuron microcircuits in the NAcSh, we focused our analysis on PV-INs and CINs, as these interneuron subtypes exert profound but distinct control over NAc circuit dynamics. Whole-cell patchclamp recordings of terminal deoxynucleotidyl transferase (tdT)-positive cells in slices from PVtdT mice revealed fast-spiking APs, no hyperpolarization-induced voltage sag (VSAG), and absent spontaneous AP (sAP) firing, consistent with the properties of PV-INs in the NAc (Figure 1A, B). To determine if HA modulates PV-IN excitability and resting membrane potential (VRMP), sensitive to membrane-delimited HA signaling in most neuronal populations, current-clamp recordings were performed in slices containing HA (10 μM) or artificial cerebrospinal fluid (ACSF) alone. Neither VRMP- nor somatic current injection-elicited high-frequency AP firing was changed by HA (Figure 1C–F). Similarly, the H3R agonist RAMH [R-(–)-α-methyl-HA] (1 μM), H2R agonist dimaprit (10 μM), and H-|R agonist 2-PyEA (2-pyridylethylamine) (10 μM) failed to shift Iholding (Figure 1G, H). These data suggest that HA does not acutely regulate the intrinsic excitability of PV-INs in the NAcSh.
Figure 1.
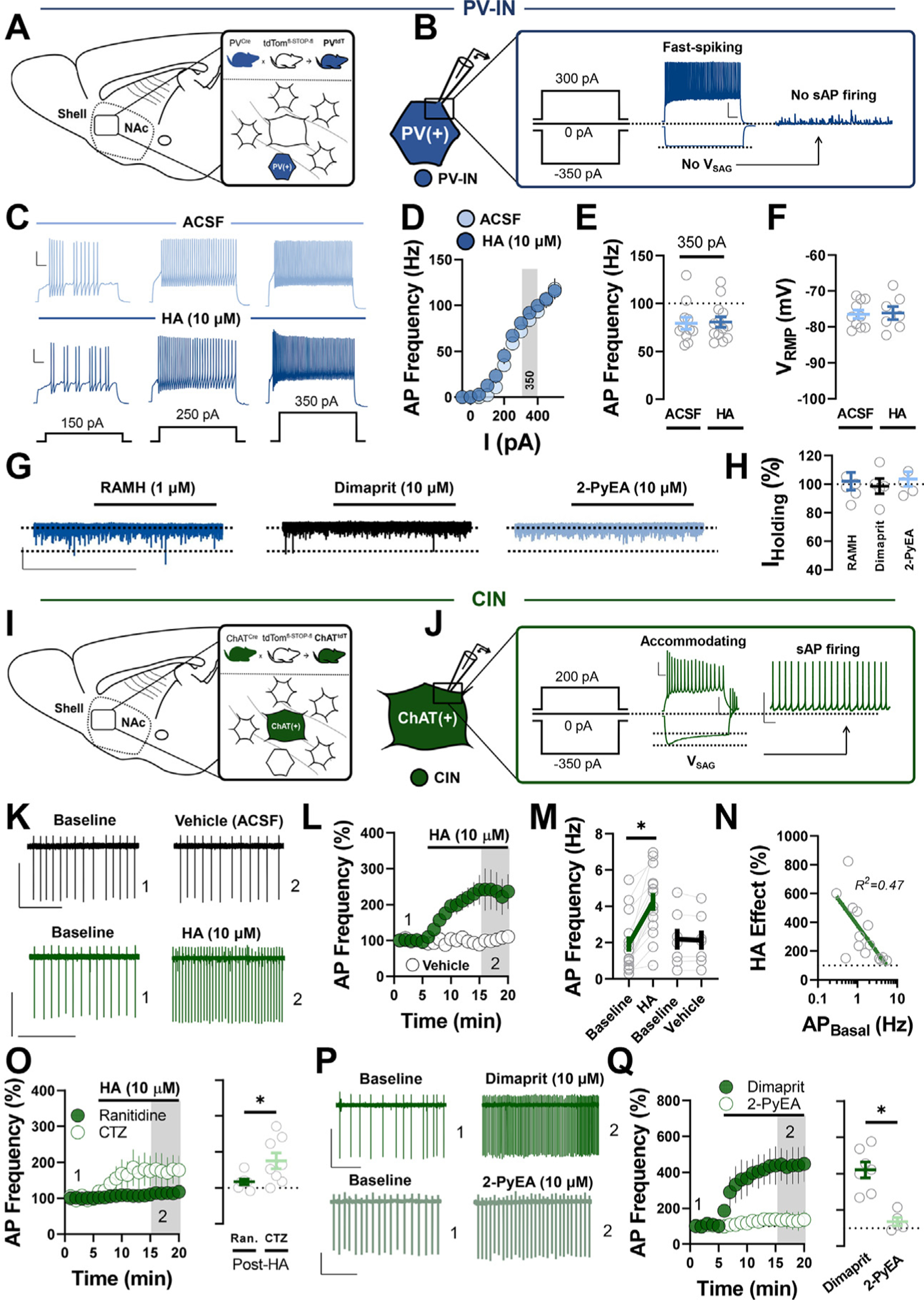
HA differentially regulates PV-IN and CIN excitability in the NAc shell. (A) Schematic depicting transgenic reporter strategy labeling tdT-positive PV-INs in the NAc shell of PVtdT mice. (B) Representative traces of PV-IN AP firing (left), membrane hyperpolarization (middle), and absent sAP firing (right) following +300 pA, −350 pA, and 0 pA current injection, respectively (scale bar = 20 mV/100 ms). (C) Representative traces of PV-IN AP firing evoked via +150 pA, +250 pA, and +350 pA current injection in ACSF (top, light blue) and HA (bottom, dark blue) (scale bar = 20 mV/100 ms). (D) Input-output curve of PV-IN AP firing in ACSF and HA. (E) Quantification of AP frequency in ACSF or HA following 350 pA current injection (AP350-pA ACSF: 80.58 ± 5.43 Hz, n = 11; AP350-pA HA: 79.31 ± 6.36 Hz, n =13, p = .880). (F) VRMP of PV-INs in ACSF vs. HA (VRMP ACSF: −76.50 ± 1.18 mV, n = 10; VRMP HA: −76.11 ± 1.81 mV, n = 7, p = .853). (G) Representative traces of IHolding in PV-INs exposed to RAMH (dark blue), dimaprit (black), or 2-PyEA (light blue) (scale bar = 30 pA/4 min). (H) Postdrug IHolding normalized within-cell to baseline (ACSF) IHolding RAMH: 102.01% ± 6.23%, n = 6; folding dimaprit: 98.60% ± 6.28%, n = 5; IHding 2-PyEA: 103.60% ± 5.08%, n = 5; one-way analysis of variance, F2,13 = 0.19, p = .829). (I) Schematic depicting transgenic reporter strategy labeling tdT-positive CINs in the NAc shell of ChATtdT mice. (J) Representative traces of CIN accommodating APs (scale bar = 20 mV/100 ms), hyperpolarization-activated Vsag (scale bar = 50 mV/100 ms), and tonic sAP firing (scale bar = 50 mV/500 ms), following +200 pA, −350 pA, and 0 pA current injection, respectively. (K) Representative traces of CIN sAP firing in pre- and post-vehicle (ACSF) (black) or HA (green) (scale bars = 50 pA/3 s). (L) Normalized time-course summary of sAP firing frequency at t(gray) following HA or vehicle application. (M) Average sAP frequency before and after HA or ACSF at t(gray) (baseline: 1.93 ± 0.42 Hz, HA: 4.256 ± 0.49 Hz, n = 14, p < .0001; baseline: 2.185 ± 0.58 Hz, vehicle: 2.14 ± 0.53 Hz, n = 7, p = .511). (N) Semilogarithmic plot of the HA-induced increase in sAP firing (HA effect) against basal sAP firing rate (HA effect vs. AP Basal R2 = 0.471). (O) Normalized time-course summary (left) and quantification (right) of sAP firing frequency during HA application in cetirizine or ranitidine (HA in ranitidine: 116.6% ± 7.67%, n = 7; HA inCTZ: 175.1% ± 22.08%, n = 8, p = .034). (P) Representative traces of CIN sAP firing in dimaprit or 2-PyEA (scale bars = 50 pA/3s). (Q) Normalized time-course summary (left) and quantification (right) of sAP firing frequency during dimaprit or 2-PyEA application (dimaprit: 420.8% ± 43.76%, n = 7; 2-PyEA: 135.7% ± 22.60%, n = 5, p = .005). Error bars indicate SEM. *p < .05. ACSF, artificial cerebrospinal fluid; AP, action potential; CIN, cholinergic interneuron; CTZ, cetirizine; HA, histamine; NAc, nucleus accumbens; PV-IN, parvalbumin-expressing fast-spiking interneuron; Ran., ranitidine; RMP, resting membrane potential; sAP, spontaneous AP; tdT, terminal deoxynucleotidyl transferase.
To assess whether HA modulates CIN excitability, we first confirmed that tdT-positive cells in the NAcSh of ChATtdT mice exhibited characteristic properties of CINs, including large, morphologically distinct somata, significant spike accommodation, Cs+-sensitive VSAG, and sAP firing at VRMP (Figure 1I, J). Taking advantage of the fact that CINs exhibit pacemaker activity, we examined the effects of HA on CIN excitability in cell-attached configuration in drug-free ACSF. HA application resulted in a pronounced increase in sAP firing relative to time-matched ACSF controls (Figure 1K–M). The HA-induced increase in sAP firing inversely correlated with basal firing rate, indicating that CINs with lower firing rates undergo greater HA-induced elevations in sAP firing (Figure 1N). Thus, unlike PV-INs, HA positively regulates CIN excitability in the NAcSh.
We next asked which HA receptor subtype mediates the excitatory actions of HA on CINs, focusing first on H1 and H2 Gαq- and Gαs-coupled receptors with established excitatory effects on excitability, respectively (4,25). Bath application of HA in the continuous presence of the H1R antagonist cetirizine (1 μM) only modestly reduced the HA-induced increase in sAP firing. However, the H2R antagonist ranitidine (1 μM) significantly attenuated the excitatory effects of HA (Figure 1O). Further discernment of the contribution of H1Rs and H2Rs can be found in Figure S1A, B. Additionally, the H2R agonist dimaprit triggered a marked increase in sAP firing in CINs relative to the H1R agonist 2-PyEA (Figure 1P, Q). These data indicate that HA potentiates CIN output by targeting H2Rs with a small contribution from H1Rs.
HA Selectively Reduces GABAergic Transmission Onto CINs to Increase Excitatory-Inhibitory Synaptic Balance
To address whether glutamatergic and GABAergic transmission contributes to the HA-induced increase in sAP firing in CINs, we quantified synaptic influences on basal sAP frequency (Figure 2A). Blockade of GABAA/B receptors with picrotoxin (50 μM) and SCH 50911 (5 μM) increased sAP firing in CINs, whereas blockade of AMPA and NMDA receptors with NBQX (5 μM) and APV (50 μM) decreased sAP firing, indicating a bidirectional influence on basal CIN output (Figure 2B–D). However, while blockade of ionotropic glutamate receptors, on average, had no effect on the HA-induced increase in sAP firing, GABA antagonists reduced the magnitude of the HA-induced increase (Figure 2E–I). Therefore, although both GABAergic and glutamatergic drive influence basal CIN sAP firing, GABAergic transmission, in part, contributes to the excitatory effects of HA on CIN output.
Figure 2.
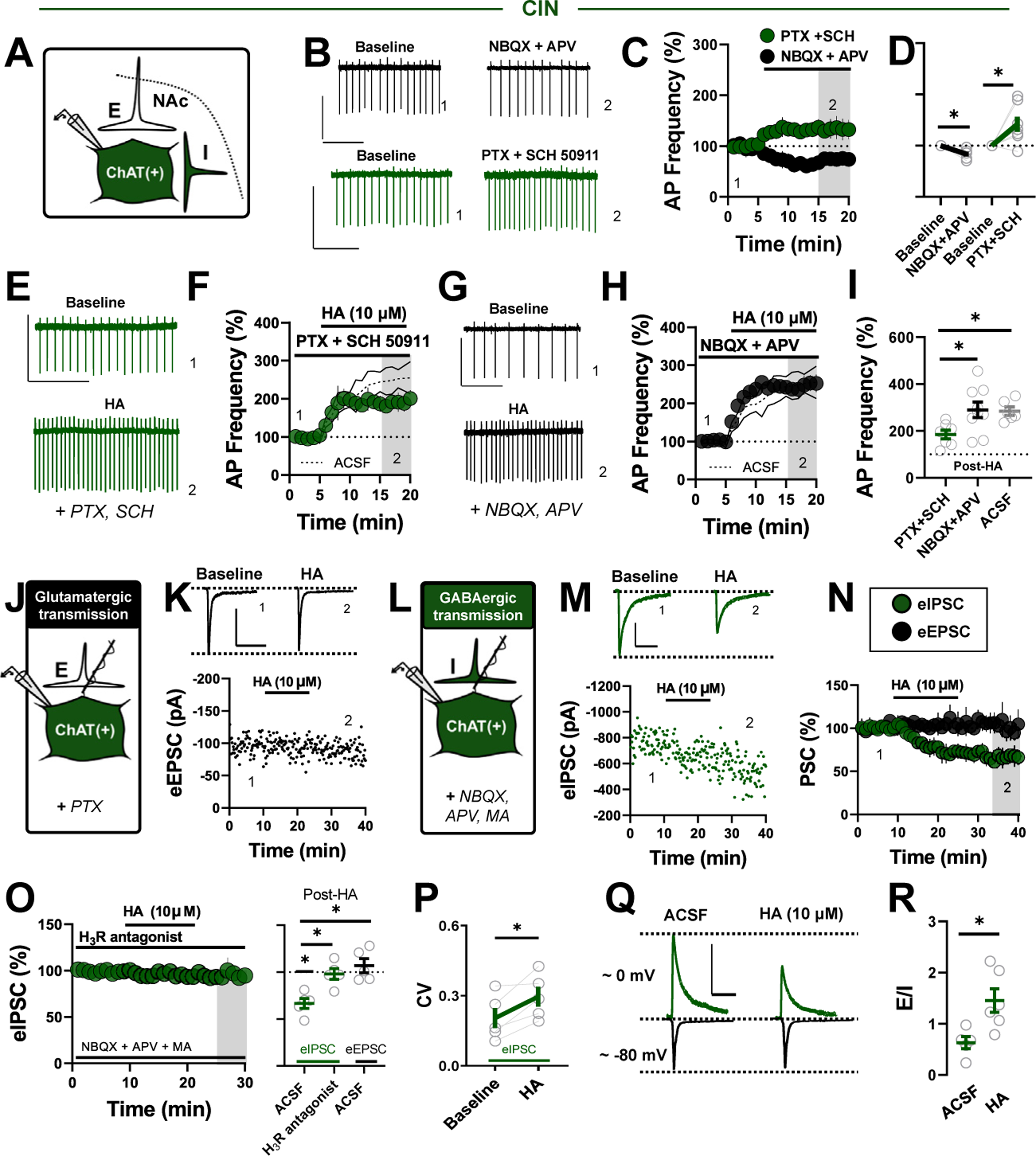
HA engages presynaptic H3Rs to increase E/I synaptic balance onto CINs. (A) Schematic depicting local excitatory (E) and inhibitory (I) synapses onto CINs. (B) Representative traces of CIN sAP firing before and after a cocktail of glutamatergic (NBQX+APV) (black) or GABAergic synaptic blockers (SCH 50911+PTX) (green) (scale bars = 50 pA/2 s). (C) Normalized time-course summary of sAP firing frequency during NBQX/APV application or SCH and PTX application. (D) Quantification of average sAP firing at t(gray) following the application of each synaptic blocker cocktail (NBQX/APV: 85.63% ± 4.78%, n = 5, p = .026; PTX/SCH: 141.60% ± 14.67%, n = 7, p = .029). (E) Representative traces of CIN sAP firing before and after HA application in GABAergic synaptic blockers (scale bar = 50 pA/2 s). (F) Normalized time-course summary of sAP firing frequency during HA application in SCH/PTX superimposed on ghosted ACSF replicate. (G) Representative traces of CIN sAP firing before and after HA application in glutamatergic synaptic blockers (scale bar = 50 pA/2 s). (H) Normalized time-course summary of sAP firing frequency during HA application in NBQX/APV superimposed on ghosted ACSF replicate. (I) Quantification of average sAP firing at t(gray) following HA in each synaptic blocker cocktail (HA in PTX/SCH: 184.7% ± 18.62%, n = 7; HA in NBQX/APV: 290.90% ± 32.91%, n = 9, GABA vs. glutamate receptor blockers, p = .038; HA in ACSF [replicated]: 284.6% ± 25.41%, n = 6, ACSF vs. GABA blockers, p = .024; one-way analysis of variance, F2,19 = 1.739, Sidak’s post hoc, p = .0214). (J) Schematic depicting whole-cell recording strategy examining local glutamatergic (E) transmission onto CINs. (K) Representative traces superimposed on an experiment of eEPSCs recorded before and after HA application (scale bar = 100 pA/50 ms). (L) Schematic depicting whole-cell recording strategy examining local GABAergic (I) transmission onto CINs. (M) Representative traces superimposed on an experiment of eIPSCs recorded before and after HA application (scale bar = 300 pA/50 ms). (N) Normalized time course summary of eIPSCs and eEPSCs during HA application (eEPSCs HA: 106.90% ± 7.44%, n = 5, p = .891; eIPSCs HA: 66.55% ± 5.37%, n = 5, p < .001; one-way analysis of variance, F2,12 = 11.52, Sidak’s post hoc, p = .0016; eIPSCs HA vs. eEPSCs HA, p = .002). (O) Normalized time-course summary of eIPSCs during HA application in ACSF containing H3R antagonist thioperamide (left) and quantification ofaverage eIPSC and eEPSC amplitude at t(gray) following HAor HA+H3R antagonist (eIPSCs HA in H3R: 98.11 % ± 5.74%, n = 5; eIPSCs HA vs. eIPSCs HA in H3R, p = .010). (P) CV of eIPSCs before and after HA (baseline CV: 0.20 ± 0.04, HA CV: 0.296 ± 0.05, n = 5, p = .028). (Q) Representative traces ofelectrochemically isolated eIPSCs and eEPSCs within cell before and after HA (scale bar = 200 pA/50 ms). (R) Quantification of average E/I ratio in ACSF vs. HA (E/I ACSF: 0.63 ± 0.12, n = 5; E/I HA: 1.45 ± 0.23, n = 6, p = .016). Error bars indicate SEM. *p < .05. ACSF, artificial cerebrospinal fluid; AP, action potential; CIN, cholinergic interneuron; CV, coefficient of variation; eEPSC, electrically evoked excitatory PSC; E/I, excitatory-inhibitory; eIPSC, electrically evoked inhibitory PSC; GABAergic, gamma-aminobutyric acidergic; H3R, H3 receptor; HA, histamine; MA, mecamylamine; NAc, nucleus accumbens; PSC, postsynaptic current; PTX, picrotoxin; sAP, spontaneous action potential; SCH, SCH 50911.
We assessed synapse-specific effects of HA more directly by first recording electrically evoked excitatory postsynaptic currents (eEPSCs) from CINs at −70 mV in picrotoxin-containing ACSF. In contrast to glutamatergic synapses onto MSNs, which undergo HA-induced long-term depression (LTD), HA had no effect on eEPSC amplitude in CINs (Figure 2J, N, O). Next, we recorded electrically evoked inhibitory postsynaptic currents (eIPSCs) in CINs at −70 mV in ACSF containing NBQX, APV, and the broad-spectrum nAChR antagonist mecamylamine (1 μM) to prevent recurrent polysynaptic IPSCs (Figure S2) (26). Interestingly, bath application of HA resulted in a significant decrease in monosynaptic eIPSC amplitude that was blocked by the H3R antagonist thioperamide (10 μM) and accompanied by an increase in the coefficient of variation, indicating that HA decreases GABAergic transmission onto CINs via presynaptic inhibitory H3Rs (Figure 2J–P). A reduction in GABAergic, but not glutamatergic, synaptic transmission onto CINs suggests that HA increases excitatory-inhibitory (E/I) balance onto CINs. Thus, HA-induced shifts in E/I balance were assessed within cells by electrochemically isolating eIPSCs and eEPSCs at approximately 0 mV and −80 mV, respectively. E/I balance was significantly increased by HA relative to ACSF alone, indicating that, in addition to the primary H2R-dependent excitation of CIN sAP firing, HA increases E/I balance onto CINs by targeting presynaptic H3Rs at GABAergic synapses (Figure 2Q, R).
Decreased SK Channel Function Contributes to the Activity-Dependent Excitatory Effects of H2Rs on CIN Output
Hyperpolarization-activated, cyclic nucleotide-gated cation (HCN) channels are regulators of pacemaker rhythms in NAc CINs (27). Given that H2Rs are Gαs protein-coupled receptors coupled to cAMP production, we hypothesized that H2Rs increase sAP firing by targeting HCN-regulated pacemaker activity. We quantified the HCN-dependent VSAG in current-clamp following somatic current injection between −400 pA and −200 pA (Figure 3A). Neither HA nor dimaprit affected VSAG magnitude (Figure 3B, C). Moreover, the HCN channel blocker ZD7288 (1 μM) failed to prevent the dimaprit-induced increase in sAP firing, indicating that the mechanism engaged by H2Rs is likely HCN channel independent (note: prolonged or high-dose application of ZD7288 abolished CIN firing) (Figure 3D, G). To determine if H2R effector function proceeds independently of cAMP/PKA signaling, we superfused dimaprit in the presence of the cell-permeant, phosphodiesterase-resistant cAMP analog, dibutyryl cAMP (db-cAMP) (500 μM) or PKA inhibitor H89 (1 μM). The dimaprit-induced increase in sAP firing in CINs was also unaffected by db-cAMP or H89 application. Taken together with basal effects of each compound on sAP firing (Figure S3A), these data suggest that the H2R-induced increase in CIN output is cAMP/PKA independent (Figure 3E–G).
Figure 3.
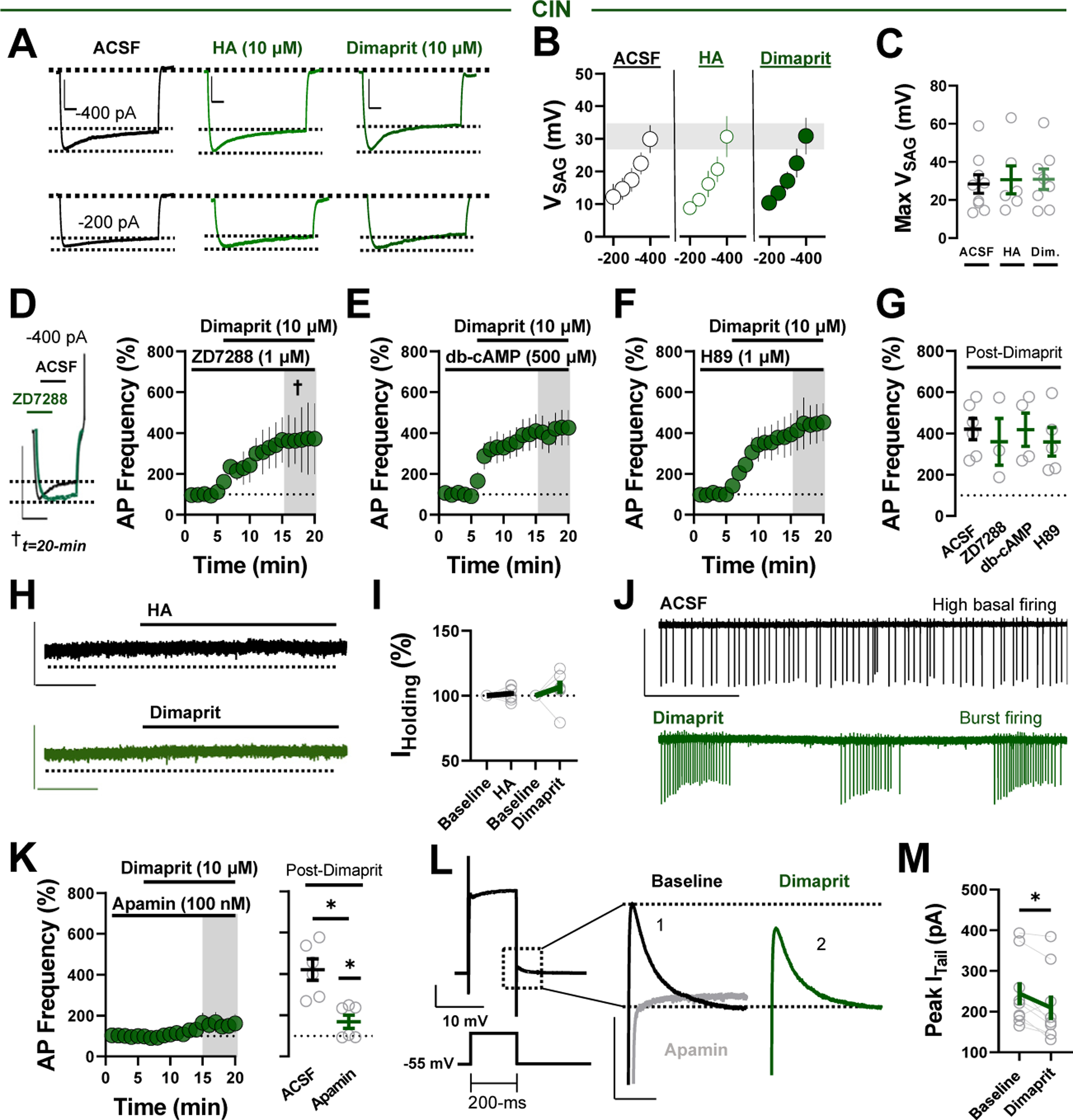
Decreased small conductance Ca2+-activated K+ channel function contributes to excitatory H2 receptor signaling in CINs. (A) Representative traces of VSAG in CINs following −200 pA and −400 pA current injection in ACSF (black), HA (light green), and dimaprit (dark green). (B) Average VSAG in ACSF, HA, or dimaprit following hyperpolarizing current steps (scale bars = 50 mV/100 ms). (C) Quantification of the maximum VSAG (at current injection = −400 pA) in ACSF or following each pharmacological manipulation (VSAG ACSF: 28.36 ± 4.87 mV, n = 9; VSAG HA: 30.63 ± 7.34 mV, n = 6; VSAG dimaprit: 30.92 ± 5.41 mV, n = 8; one-way analysis of variance, ACSF vs. drug, F2,20 = 0.66, Sidak’s post hoc, p = .979). (D) Representative traces (left) of VSAG before and after ZD7288 at approximately t = 20 minutes (†) obtained from separate CIN in Iclamp following −400 pA current step. Note absence of hyperpolarization-activated, cyclic nucleotide-gated cation-mediated VSAG. Normalized time-course summary (right) of sAP firing frequency during dimaprit application in the presence of ZD7288. (E) Normalized time-course summary of sAP firing frequency during dimaprit application in the presence of db-cAMP. (F) Normalized time-course summary of sAP firing frequency during dimaprit application in the presence of H89. (G) Quantification of average sAP firing at t(gray) following dimaprit in each pharmacological manipulation relative to ACSF (re-depicted from Figure 1L) (dimaprit in ZD7288: 359.30% ± 113.3%, n = 3, p = .580; dimaprit in db-cAMP: 417.7% ± 8.98%, n = 4, p = .974; dimaprit in H89: 359.11% ± 69.03%, n = 5, p = .483). (H) Representative traces of IHolding (VM = −70 mV) at baseline (ACSF) and in HA (black) or dimaprit (green) (scale bars = 40 pA/5 min). (I) Average IHolding before and after HA or dimaprit (HA IHolding: 101.70% ± 1.85%, n = 8, p = .542; dimaprit IHolding: 106.61% ± 75.14%, n = 8, p = .248). (J) Representative traces of sAPs in CINs exhibiting a high basal firing rate in ACSF (black) and the resulting dimaprit-induced transition to burst firing (green) (scale bars = 50 pA/3 s). (K) Normalized time-course summary (left) and quantification (right) of sAP firing frequency during dimaprit application in the presence of apamin (dimaprit in apamin: 168.40% ± 32.09%, n = 6, p = .002) (ACSF re-depicted from Figure 1L). (L) Representative traces of the voltage step-induced current response (scale bar = 500 pA/200 ms) and ITail at baseline (ACSF) (black) and following dimaprit (green) or apamin (gray) (ITail scale bars = 200 pA/50 ms). (M) Quantification of maximum ITail before and after dimaprit (peak ITail before dimaprit: 244.38 ± 28.07 pA; peak ITail after dimaprit: 209.6 ± 29.66 pA, n = 9, p = .017). Error bars indicate SEM. *p < .05. ACSF, artificial cerebrospinal fluid; AP, action potential; CIN, cholinergic interneuron; db-cAMP, dibutyryl cyclic adenosine monophosphate; Dim., dimaprit; HA, histamine; Max, maximum; sAP, spontaneous AP.
To define effector mechanisms targeted by H2Rs, we tested whether HA or dimaprit evokes a postsynaptic conductance in CINs. Iholding was recorded from CINs in voltage-clamp configuration at a hyperpolarized command voltage to prevent sAP firing. After obtaining an Iholding baseline, HA or dimaprit was incorporated into the ACSF bath for ≥10 minutes (Figure 3H). Neither manipulation was accompanied by a change in Iholding, an intriguing finding given that excitatory HA signaling often modulates membrane properties (Figure 3H, I). Although several biophysical possibilities may explain this outcome, we postulated that the system targeted by H2Rs in CINs operates minimally in the absence of sAP firing. Furthermore, we noted periodic dimaprit-induced conversions to burst firing in cells displaying high basal firing rates (e.g., ≥6 Hz) (Figure 3J). An activity-dependent, nondepolarizing effector mechanism qualitatively associated with burst firing may point to the involvement of SK channels (22,28).
To determine if SK channels mediate the excitatory effects of H2Rs on CIN sAP firing, we recorded sAP firing in CINs in the presence of the SK channel antagonist apamin (50 nM). Apamin significantly reduced the dimaprit-induced increase in sAP firing (Figure 3K). We next examined whether H2R activity modulates SK channel-dependent medium afterhyperpolarizations in CINs by measuring the amplitude of SK channel-mediated (apamin-sensitive) tail currents (ITail) following a 200-ms voltage step to +10 mV from −55 mV (Figure 3L) (28,29). Using a 12- to 20-second ISI to minimize Goldman rectification, we found that dimaprit triggered a reduction in peak ITail amplitude relative to baseline, suggesting that H2Rs negatively regulate SK channel function in CINs and that the effects of apamin likely occluded those of dimaprit (Figure 3M). These findings indicate that H2Rs potentiate CIN output by inhibiting SK channels in an HCN-, cAMP-, and PKA-independent manner.
HA Decreases Feedforward Glutamatergic Drive Onto PV-INs via the AC-cAMP-PKA Signaling Pathway
Unlike CINs, PV-INs lack intrinsic pacemaker activity and rely on glutamatergic drive to reach AP threshold (23). Thus, we speculated that HA targets glutamatergic inputs onto PV-Ins to influence synaptic efficacy through the feedforward microcircuit. To test this, we recorded eEPSCs from PV-INs at −70 mV (Figure 4A). Superfusion of HA persistently decreased eEPSC amplitude and increased paired-pulse ratio (PPR) and coefficient of variation, suggestive of a decrease in glutamate release probability (Figure 4B–D). Consistent with presynaptically expressed inhibitory H3Rs, the HA-induced decrease in EPSC amplitude was completely blocked by the H3R antagonist thioperamide and recapitulated by RAMH (Figure 4C, E-G). Thus, HA signaling via H3Rs decreases glutamatergic drive onto PV-INs.
Figure 4.
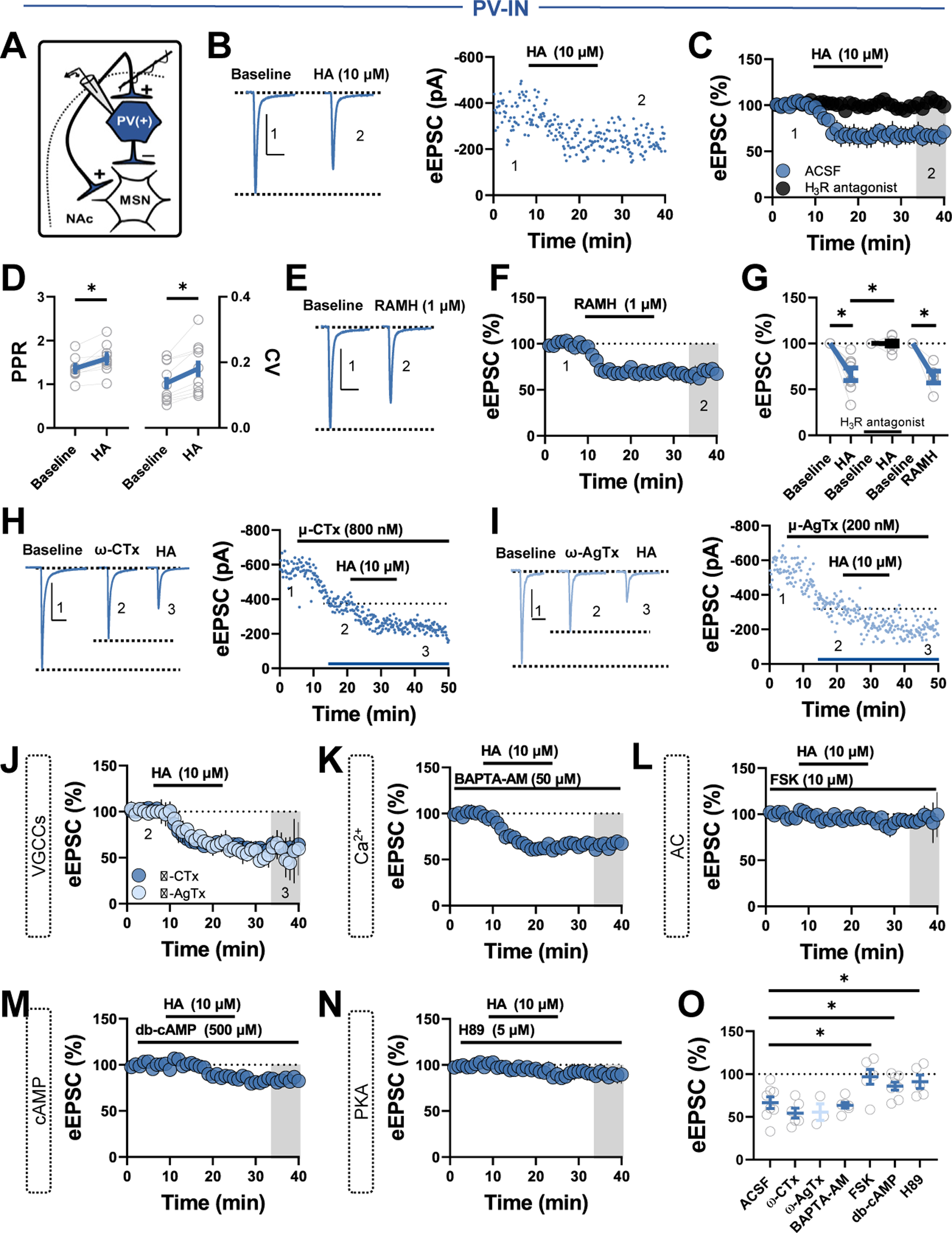
HA decreases feedforward glutamatergic drive onto PV-INs via AC-cAMP-PKA signaling. (A) Schematic depicting whole-cell recording strategy of local glutamatergic inputs onto PV-INs within feedforward inhibition microcircuits. (B) Representative traces (left) and experiment (right) of eEPSCs in PV-INs depicting the effects of HA. (C) Normalized time-course summary of eEPSCs following HA in ACSF or H3R antagonist thioperamide (HA: 66.50% ± 6.79%, n = 8, p = .002). (D) PPR (left) and CV (right) before and after HA (baseline PPR: 1.36 ± 0.08, HA PPR: 1.59 ± 0.10, n = 10, p = .001; baseline CV: 0.14 ± 0.02, HA CV: 0.18 ± 0.25, n = 10, p = .001). (E) Representative traces of eEPSCs depicting the effects of RAMH. (F) Normalized time-course summary of eEPSCs following RAMH. (G) Quantification of average eEPSC amplitude at t(gray) following each pharmacological manipulation (RAMH: 63.45% ± 6.41 %, n = 5, p = .005; HA in H3R antagonist: 99.95% ± 2.59%, n = 8, p = .981). (H) Representative occlusion experiment (left) and traces (right) depicting the effects of HA in ω-CTx. Representative occlusion experiment (left) and traces (right) depicting the effects of HA in ω-AgTx. (J) Normalized time-course summary of eEPSCs following HA in ACSF containing ω-CTx or ω-AgTx (HA in CTx: 54.31 % ± 5.371%, n = 6, p = .370; HA in AgTx: 55.44% ± 9.67%, n = 3, p = .591). (K) Normalized time-course summary of eEPSCs following HA in slices incubated for 1 hour in BAPTA-AM (HA in BAPTA-AM: 63.41% ± 3.39%, n = 6, p = .721). (L) Normalized time-course summary of eEPSCs following HA in slices incubated for at least 2 hours in FSK (HA in FSK: 96.82% ± 8.65%, n = 6, p = .016). (M) Normalized time-course summary of eEPSCs following HA application in db-cAMP (HA in db-cAMP: 85.88% ± 4.59%, n = 8, p = .032). (N) Normalized time-course summary of eEPSCs following HA in H89 (HA in H89: 91.01% ± 7.84%, n = 5, p = .041). (O) Quantification of average eEPSC amplitude at t(gray) following each pharmacological manipulation. All scale bars = 300 pA/50 ms. Error bars indicate SEM. *p < .05. AC, adenylyl cyclase; ACSF, artificial cerebrospinal fluid; cAMP, cyclic adenosine monophosphate; CV, coefficient of variation; db-cAMP, dibutyryl cAMP; eEPSC, electrically evoked excitatory postsynaptic current; FSK, forskolin; H3R, H3 receptor; HA, histamine; MSN, medium spiny neuron; NAc, nucleus accumbens; ω-AgTx, ω-agatoxin TK; ω-CTx; ω-conotoxin GVIA; PKA, protein kinase A; PPR, paired-pulse ratio; PV-IN, parvalbumin-expressing fast-spiking interneuron; Ran., ranitidine; VGCC, voltage-gated Ca2+ channel.
To begin to identify the mechanism recruited by H3Rs at PV-IN synapses, we examined the purported contribution of N- and P/Q-type voltage-gated Ca2+ channels (VGCCs) (30,31). Bath application of the N-type VGCC blocker ω-conotoxin GVIA (600 nM) or P/Q-type VGCC blocker ω-agatoxin TK (200 nM) reduced eEPSC amplitude, confirming that N-type and P/Q-type VGCCs facilitate glutamatergic transmission onto PV-INs (Figure 4H, I). However, subsequent application of HA decreased eEPSC amplitude comparably to control conditions, suggesting that H3R function at this synapse does not require N-type or P/Q-type VGCCs (Figure 4H–J, O). Furthermore, the cell-permeant Ca2+ chelator BAPTA-AM (5 μM) had no effect on the HA-induced decrease in eEPSCs, reducing the likelihood of indiscriminate actions at either VGCC subtype (Figure 4K, O).
We next examined the contribution of the AC-cAMP-PKA signaling pathway modulated by Gi/o-coupled receptors, including H3Rs (32). Preincubating slices for 2 hours in the AC activator forskolin (1 μM) blocked the HA-induced decrease in eEPSC amplitude (Figure 4L, O). To determine if HA triggers AC-induced shifts in cAMP levels, we used a bath application of HA containing db-cAMP. Indicative of an AC-stimulated effector pathway, db-cAMP attenuated the HA-induced depression (Figure 4M, O). If H3R function at this synapse proceeds via AC-cAMP-PKA signaling, inhibiting PKA activity should disrupt the HA-induced decrease in eEPSC amplitude. Indeed, bath application of HA in H89 attenuated the depression in eEPSC amplitude (Figure 4N, O). Interpreted alongside basal effects of each compound on eEPSCs (Figure 3B), these findings suggest that H3Rs decrease feedforward glutamatergic drive onto PV-INs via AC-cAMP-PKA signaling.
Thalamocortical Synapses Onto PV-INs Are Differentially Regulated by HA Signaling
We previously reported that HA shifts thalamocortical glutamatergic drive onto D1-MSNs in the NAc, with inputs originating from the PFC undergoing robust LTD (8). To determine if these inputs onto PV-INs exhibit similar sensitivity to HA, we expressed channelrhodopsin-2 in the PFC or MDT of PVtdT mice (Figure 5A). Optically evoked EPSCs (oEPSCs) from the PFC or MDT in PV-INs were abolished by the voltage-gated Na+ channel blocker tetrodotoxin (500 nM) and restored by the voltage-gated K+ channel blocker 4-AP (4-aminopyridine) (1 mM), confirming a monosynaptic connection to NAcSh PV-INs. Subsequent application of NBQX abolished oEPSCs in PV-INs from the PFC or MDT, indicating that monosynaptic PFC and MDT-to-PV-IN synapses are glutamatergic (Figure 5B, C).
Figure 5.
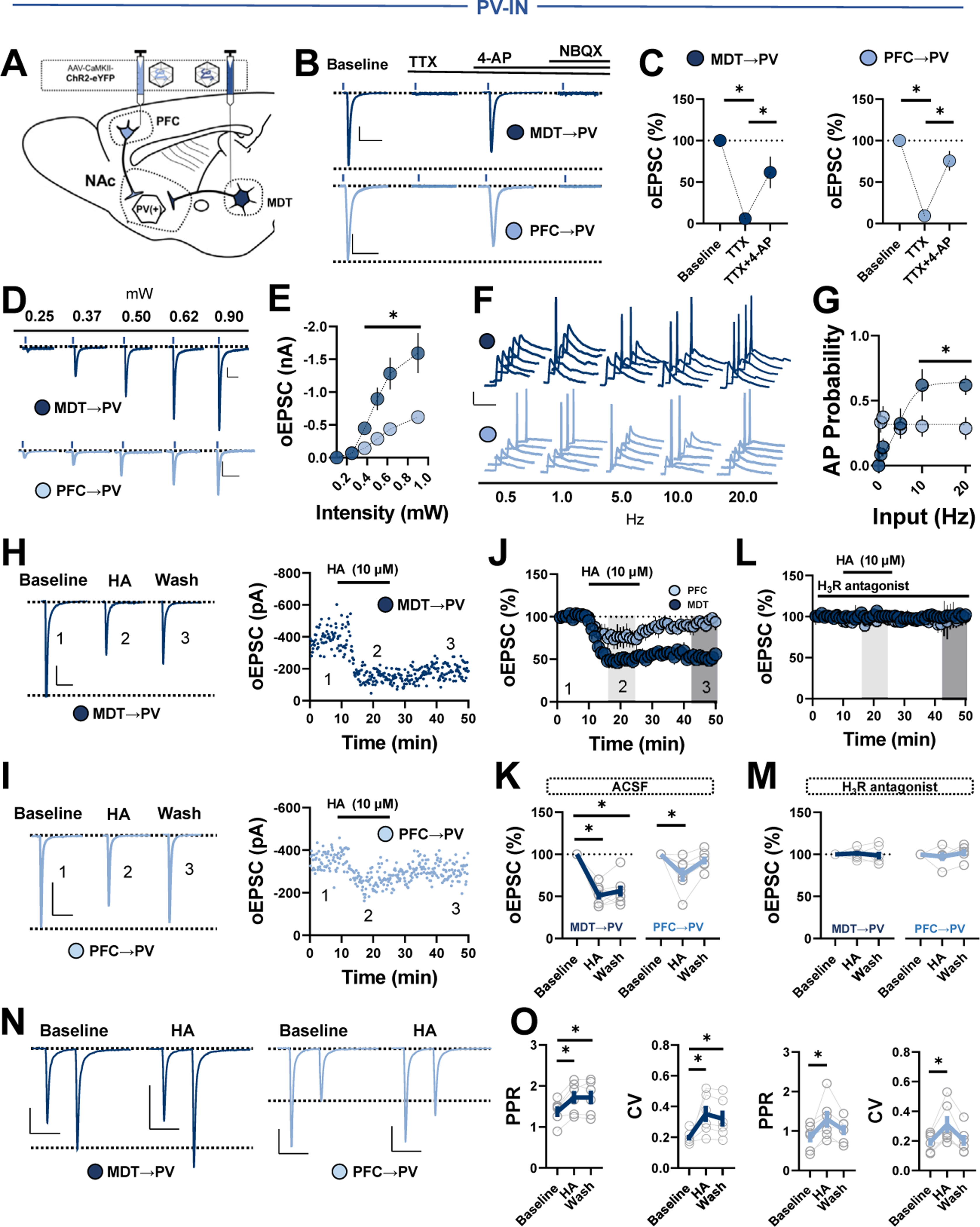
Thalamocortical transmission differentially engages PV-INs in an HA-biased microcircuit. (A) Schematic depicting stereotaxic delivery of channelrhodopsin-2 into the PFC or MDT of PVtdT mice. (B) Representative traces of MDT-evoked (dark blue) or PFC-evoked (light blue) oEPSCs in PV-INs at baseline and following the sequential addition of TTX, APV, and NBQX (scale bars = 100 pA/50 ms). (C) Average oEPSC amplitude following TTX and TTX+4-AP at MDT-to-PV-IN (left) or PFC-to-PV-IN (right) synapses (TTX MDT: 11.20% ± 6.93%, n = 3, p = .006; TTX+4-AP MDT: 61.68% ± 18.90%, n = 3, p = .039; TTX PFC: 9.21% ± 1.95%, n = 3, p < .001; TTX+4-AP PFC: 75.59% ± 11.74%, n = 3, p = .033). (D) Representative traces of MDT- and PFC-evoked oEPSCs at increasing light stimulus intensities (scale bars = 100 pA/50 ms). (E) Quantification of average oEPSC amplitude at MDT vs. PFC inputs across stimulus intensities (MDT0.25: −0.065 ± 0.021 nA, MDT0.375: −0.447 ± 0.084 nA, MDT0.5: −0.897 ± 0.169 nA, MDT0.625: −1.28 ± 0.239 nA, MDT0.9: −1.59 ± 0.300 nA, n = 6; PFC0.25: −0.070 ± 0.018 nA, PFC0.375: −0.144 ± 0.027 nA, PFC0.5: −0.288 ± 0.054 nA, PFC0.625: −0.437 ± 0.064 nA, PFC0.9: −0.618 ± 0.0691 nA, n = 11; one-way repeated measures ANOVA, MDT vs. PFC, Sidak’s post hoc, p < .0001). (F) Representative traces of MDT- and PFC-evoked APs at increasing stimulus frequencies (scale bars = 10 mV/50 ms). (G) AP probability at increasing stimulus frequencies at MDT-to-PV-IN or PFC-to-PV-IN synapses (MDT0.5: 0.086 ± 0.054, MDT1 0.143 ± 0.066, MDT5: 0.324 ± 0.116, MDT10: 0.619 ± 0.124, MDT20: 0.619 ± 0.074, n = 8; PFC0.5: 0.333 ± 0.079, PFC1 0.373 ± 0.083, PFC5: 0.286 ± 0.073, PFC10: 0.303 ± 0.819, PFC20: 0.286 ± 0.086, n = 20; one-way repeated measures ANOVA, MDT20 vs. PFC20, p = .0341). (H) Representative traces (left) and experiment (right) of MDT-evoked eEPSCs in PV-INs depicting the effects of HA (scale bars = 100 pA/50 ms). (I) Representative traces (left) and experiment (right) of PFC-evoked eEPSCs in PV-INs depicting the effects of HA (scale bars = 100 pA/50 ms). (J) Normalized time-course summary of PFC- and MDT-evoked oEPSCs following HA. (K) Quantification of average oEPSC amplitude at t(light gray) following HA and during washout at t(dark gray) (MDT 51.18% ± 4.68%, p < .0001; wash: 56.38% ± 6.34%, p < .0001; F2,18 = 3.45, n = 7; PFC HA: 76.32% ± 5.57%, p = .0171; wash: 93.27% ± 4.57%, p = .641; F2,15 = 5.28, n = 6). (L) Normalized time-course summary PFC- and MDT-evoked oEPSCs following HA in JNJ 520785. (M) Quantification of MDT- and PFC-evoked oEPSCs following HA in JNJ 520785 (MDT HA in JNJ 520785: 101.2% ± 2.98%, p = .946; wash: 98.73% ± 4.55%, p = .943; F2,15 = 5.09, n = 6; PFC HA in JNJ 520785: 97.59% ± 5.24%, p = .871;wash: 102.2% ± 4.18%, p = .886; F2,12 = 1.67, n = 5). (N) Representative traces of 50-ms PPR at MDT- and PFC-to-PV-IN synapses before and after HA (scale bars = 100 pA/50 ms). (O) PPR (left) and CV (right) at baseline, in HA, and following washout (MDT baseline PPR: 1.37 ± 0.14, HA: 1.72 ± 0.16 [p = .008], wash: 1.71 ± 0.17[p = .032], n = 6, one-way ANOVA, F2,15 = 1.74; MDT baseline CV: 0.19 ± 0.17, HA: 0.35 ± 0.05 [p = .0439], wash: 0.32 ± 0.05 [p = .110], n = 6, one-way ANOVA, F2,15 = 3.53; PFC baseline PPR: 0.84 ± 0.14, HA: 1.30 ± 0.22 [p = .043], wash: 0.45 ± 0.16 [p = .451], n = 6, one-way ANOVA, F2,15 = 0.78; PFC baseline CV: 0.19 ± 0.03, HA: 0.31 ± 0.05 [p = .0439], wash: 0.32 ± 0.05 [p = .815], n = 6, one-way ANOVA, Sidak’s post hoc, F2,15 = 2.49). Error bars indicate SEM. *p < .05. ACSF, artificial cerebrospinal fluid; ANOVA, analysis of variance; AP, action potential; CV, coefficient of variation; HA, histamine; MDT, mediodorsal thalamus; NAc, nucleus accumbens; oEPSC, optically evoked excitatory postsynaptic current; PFC, prefrontal cortex; PPR, paired-pulse ratio; PV, parvalbumin; PV-IN, PV-expressing fast-spiking interneuron; TTX, tetrodotoxin.
To interpret consequences of HA on thalamocortical feedforward transmission, we assessed synaptic efficacy of each input onto PV-INs by first isolating the input-output relationship at increasing stimulus intensities (Figure 5D). Compared with PFC synapses, MDT synapses resulted in significantly larger oEPSCs in PV-INs at higher stimulus intensities (Figure 5E). Consistent with the input-output relationship, stimulation of MDT afferents sustained PV-IN AP firing probability at higher stimulus frequencies than PFC afferents (Figure 5F, G). Therefore, while glutamatergic inputs from both the MDT and the PFC evoke high-fidelity oEPSCs in PV-INs, MDT-to-PV-IN synapses more strongly engage PV-INs than PFC-to-PV-IN synapses in the NAcSh. These data provide a functional snapshot of the strength with which thalamocortical transmission can drive PV-IN-mediated feedforward inhibition in the NAcSh.
Next, we tested whether MDT-to-PV-IN synapses are modulated by HA signaling (Figure 5H). HA triggered a persistent decrease in oEPSC amplitude (Figure 5H–K) that was completely blocked by the H3R antagonist JNJ 5207852 (1 μM) (Figure 5L, M). Opposite of what we observed at PFC-to-NAc MSN synapses, HA triggered only a modest JNJ 5207852-sensitive decrease in PFC-oEPSC amplitude in PV-INs that returned to baseline (Figure 5I–M). Consistent with the presynaptic localization of H3Rs, the effects of HA at each input coincided with increases in paired-pulse ratio and coefficient of variation that reversed following washout only at PFC synapses (Figure 5N, O). These data suggest that presynaptic, H3R-dependent HA-LTD is expressed at thalamoaccumbens synapses onto PV-INs in the NAcSh.
Interneuron-Specific HA Signaling Shifts PV-IN and CIN-Embedded Microcircuit Output
Our findings indicate that HA differentially modulates PV-INs and CINs in the NAcSh. Thus, we examined the effects of HA on downstream microcircuit motifs controlled by PV-INs and CINs, such as FFI of MSNs and mesoaccumbens DA release (17,33). To accomplish this, we measured PFC- and MDT-evoked FFI of MSNs and electrically stimulated DA release, respectively. Afferent-evoked FFI of MSNs was assessed by stimulating PFC or MDT channelrhodopsin-2 and recording optically evoked IPSCs (oIPSCsFFI) in MSNs at approximately 0 to 5 mV (Figure 6A). HA triggered near-complete blockade of oIPSCsFFI in MSNs from the MDT with the residual oIPSC likely originating from another source of GABA in the NAcSh (Figure 6B–E). In contrast, HA decreased PFC-evoked oIPSCsFFI in MSNs to a significantly smaller degree, aligning with monosynaptic recordings obtained from PV-INs (Figure 6B–E). Thus, the robust inhibitory effects of HA at thalamoaccumbens synapses onto PV-INs translates into a near collapse of MDT-evoked FFI of MSNs.
Figure 6.
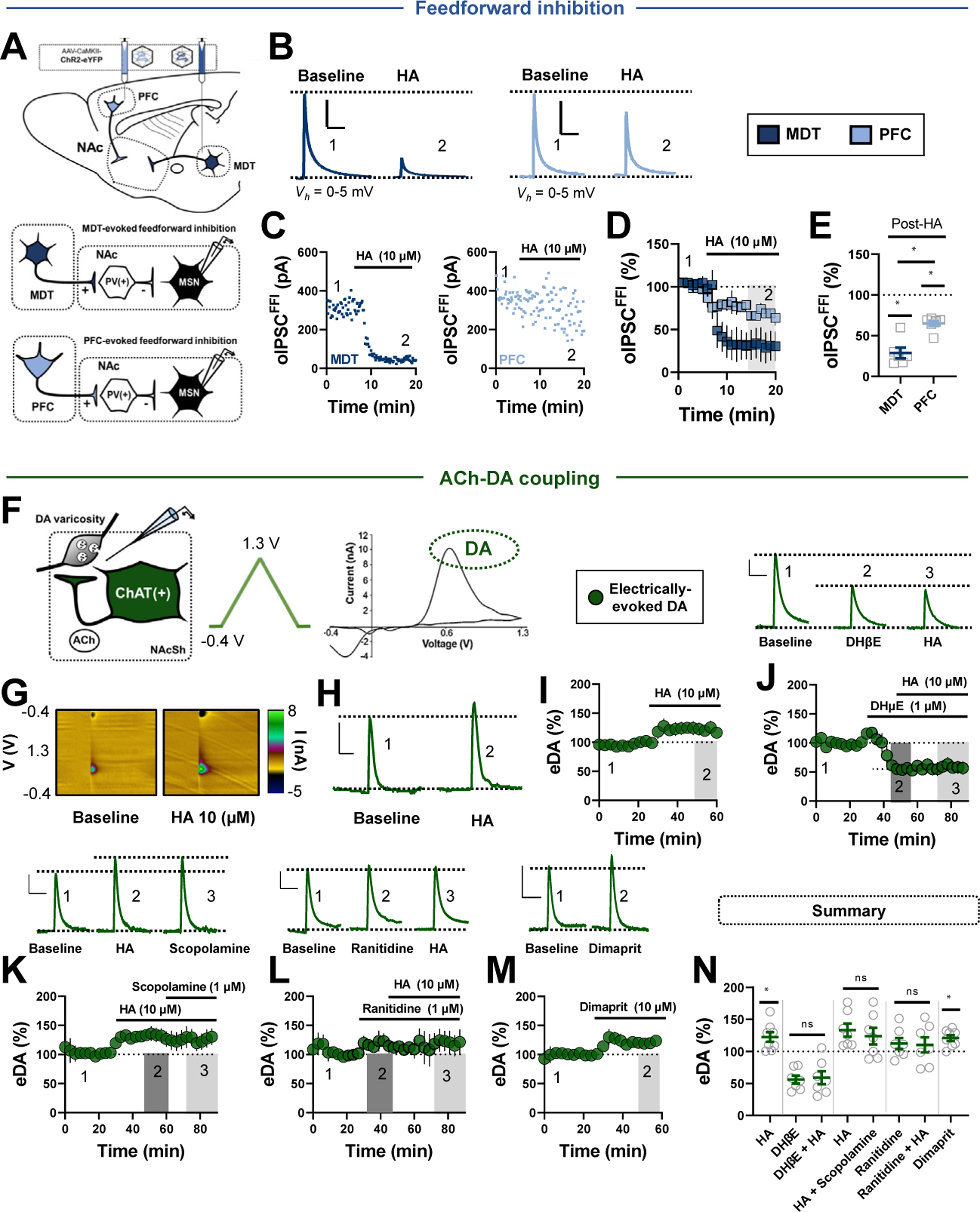
HA divergently modulates afferent-evoked FFI and cholinergic interneuron-dependent DA release. (A) Schematic depicting stereotaxic delivery of channelrhodopsin-2 into the PFC or MDT of wild-type mice (top) and FFI recording strategy in MSNs (bottom). (B) Representative traces of MDT-evoked (dark blue) or PFC-evoked (light blue) oIPSCsFFI in MSNs before and after HA (scale bars = 150 pA/50 ms). (C) Representative experiments depicting the effects of HA on MDT- or PFC-evoked oIPSCsFFI in MSNs. (D, E) Normalized time-course summary and quantification at t(gray) of PFC- and MDT-evoked oIPSCsFFI following HA (MDT HA: 28.92% ± 6.58%, n = 6, p < .0001; PFC HA: 65.27% ± 3.13%, n = 7, p = .0002; MDT vs. PFC, p = .0003). (F) Schematic depicting ACh-DA coupling and the fast-scan cyclic voltammetry recording strategy of DA in NAcSh slices (left). Triangular waveform was applied to the carbon-fiber tip and DA was detected at its oxidation potential (middle). A representative current-voltage plot (right). (G) Representative color plots showing DA (in green) before and after HA application. (H) Representative current vs. time plots showing DA evoked by single-pulse stimulation before and after HA. (I) Normalized time-course summary depicting the effects of HA on DA release (HA: 122.40% ± 7.51%, n = 8, p = .021). (J) Representative DA signals (top) and normalized time-course summary (bottom) depicting the effects of DHβE and DHβE+HA on peak DA amplitude (DHβE alone: 55.98% ± 6.27%, n = 7, p < .0001; HA+DHβE: 58.97% ± 10.12%, n = 7, DHβE vs. HA+DHβE, p = .806). (K) Representative DA traces (top) and normalized time-course summary (bottom) depicting the effects of HA and HA+scopolamine on peak DA amplitude (HA: 133.2% ± 10.33%, HA+scopolamine: 123.8% ± 12.95%, n = 7, HA vs. HA+scopolamine, P = 0.224). (L) Representative DA transients (top) and normalized time-course summary (bottom) depicting the effects of ranitidine and ranitidine+HA on peak DA amplitude (ranitidine: 112.2% ± 8.55%, n = 7, p = .203; HA+ranitidine: 110.2% ± 11.84%, n = 7, ranitidine vs. ranitidine+HA, p = .748). (M) Representative DA transients (top) and normalized time-course summary (bottom) depicting the effects of dimaprit on DA amplitude (dimaprit: 121.7% ± 4.63%, n = 8, p = .003). (N) Average DA amplitude following each pharmacological manipulation relative to baseline. All fast-scan cyclic voltammetry scale bars = 5 nA/3 second. Error bars indicate SEM. *p < .05. ACh, acetylcholine; ANOVA, analysis of variance; ChAT, choline acetyltransferase; DA, dopamine; eDA, electrically evoked DA; FFI, feedforward inhibition; HA, histamine; MDT, mediodorsal thalamus; MSN, medium spiny neuron; NAc, nucleus accumbens; NAcSh, NAc shell; ns, not significant; oIPSC, optically evoked inhibitory postsynaptic current; PFC, prefrontal cortex; PV, parvalbumin.
We next asked whether HA-induced excitation of CINs mounts a shift in mesoaccumbens DA release by measuring electrically evoked DA (eDA) release in the NAcSh using fastscan cyclic voltammetry (Figure 6F). HA significantly increased peak eDA amplitude relative to baseline (Figure 6G–I, N). If exclusively mediated by an increase in ACh release from CINs, AChR antagonists should abolish the HA-induced increase in DA release (34,35). Congruent with studies showing that CINs augment terminal eDA release via nAChRs, superfusion of the β2-selective nAChR antagonist DHβE (dihydro-β-erythroidine) (1 μM) significantly decreased peak eDA amplitude (Figure 6J, N). Further, application of HA in the presence of DHβE failed to evoke a subsequent change in eDA, suggesting that HA increases DA via β2-nAChRs (Figure 6J, N). Accordingly, the pan-mAChR blocker scopolamine (1 μM) failed to reverse the HA-induced increase in DA release (Figure 6K, N). Given our findings that HA potentiates CIN output predominately via H2Rs, we tested the contribution of H2Rs to the HA-induced increase in DA release. Ranitidine did not evoke a change in eDA, pointing to a lack of tonic H2R activity regulating DA release (Figure 6L, N). However, the HA-induced increase in eDA was blocked in ranitidine-containing ACSF (Figure 6L, N). Moreover, the H2R agonist dimaprit recapitulated the effects of HA, mirroring the excitatory actions of H2Rs on CIN activity (Figure 6M, N). Thus, HA recruits H2Rs on CINs to increase NAcSh DA release via β2-containing nAChRs.
DISCUSSION
We report that HA potentiates CIN output by targeting H2Rs and H1Rs, with the primary effect mediated by H2R-dependent actions on SK channels. CIN output is further potentiated by an increase in E/I synaptic balance onto CINs via presynaptically expressed H3Rs at GABAergic synapses. While HA does not alter PV-IN excitability, it triggers a decrease in feedforward glutamatergic drive onto PV-INs via presynaptic H3Rs coupled to AC-cAMP-PKA signaling. This effect is differentially expressed at thalamocortical synapses onto PV-INs with MDT synapses, exerting greater basal control over PV-IN output, undergoing robust H3R-dependent HA-LTD relative to PFC synapses. The HA-induced effects on PV-INs and CINs translate into near-complete escape from MDT-evoked FFI of MSNs and a CIN-dependent increase in DA release. Alongside recent reports of HA-induced plasticity in the NAc, our results highlight circuit specializations mediating the neuromodulatory properties of HA in the NAcSh.
Histamine Increases the Intrinsic Excitability of CINs but Not PV-INs
The NAc, comprising complex interneuron microcircuitry, responds to salient environmental stimuli to guide goal-directed motivated behaviors, including those that become dysregulated in addiction, depression, anhedonia, and chronic pain. Extensive research has described how reward-related monoamine signaling in the NAc contributes to these disease states, yet HA, a noncanonical, wake-active monoamine, remains comparatively unexplored. Data from CINs revealed that HA acts primarily via H2Rs to promote CIN output, with a small but appreciable contribution from H1Rs and synaptic H3Rs. Although synergistic actions of colocalizing H1Rs and H2Rs are difficult to separate ex vivo, this finding diverges from accounts in the dorsal striatum, where HA acts primarily via Gαq-coupled H1Rs to increase CIN output (36). HA receptor subtype notwithstanding, it is clear that HA excites CINs throughout the striatal network, aligning with early reports that HA microinfusion into the NAc increases ACh content (37). Unlike CINs, PV-IN excitability and membrane properties were unchanged by HA or receptor-selective agonists, indicating that HA exerts differential effects on CIN and PV-IN excitability in the NAcSh.
We found that HA potentiates CIN output in parallel by engaging presynaptically expressed H3Rs at local GABAergic, but not glutamatergic, synapses. A GABA-specific synaptic mechanism is unsurprising given that CINs receive less extensive glutamatergic input and are intrinsically coupled to GABAergic microcircuits (38). While the hodological profile of inhibitory synapses onto CINs in the NAcSh have not been resolved, prior studies indicate that CINs are innervated by MSNs and ventral tegmental area-derived GABAergic inputs (13,39,40). Thus, HA may increase E/I synaptic balance onto CINs by targeting presynaptic H3Rs at MSN-to-CIN synapses. Alternatively, HA may engage H3Rs at other GABAergic afferents to the NAcSh, such as the recently described closed-loop return from the ventral pallidum (41). Given that the excitatory actions of HA inversely correlate with basal sAP firing rate, it is also possible that the picrotoxin/SCH 50911-induced increase in basal sAPs reduced the magnitude with which HA increased CIN output. However, convergent observations that HA increased E/I balance by engaging a GABAergic synapse-specific mechanism makes this less likely.
Our analyses implicate medium afterhyperpolarization-regulating SK channels in mediating the actions of H2Rs on CINs. Whereas manipulating the AC-cAMP-PKA pathway had negligible impact on the dimaprit-induced increase in sAP firing, dimaprit decreased depolarization-evoked ITail and was occluded by the selective SK channel blocker apamin. A key observation supporting an SK channel-dependent effector mechanism is that CINs exhibiting a high basal sAP firing rate transition to burst discharge when exposed to dimaprit. Although several electrochemical mechanisms may underlie this switch, we postulated that H2Rs drive tonic CIN sAP firing to a physiological maximum, beyond which CINs undergo a gradual shift to burst firing. This phenomenon may explain why the excitatory effect of HA inversely correlated with basal CIN sAP firing rate. The present findings do not exclude the possibility that H2Rs engage molecular intermediates that gate SK channel function, such as SK channel-coupled N-type VGCCs during the AP upstroke (28). Nevertheless, our data indicate that HA signaling via H2Rs ultimately reduces SK channel function to potentiate CIN output in the NAcSh.
HA Dampens Feedforward Glutamatergic Drive Onto PV-INs
We found that HA recruits presynaptic H3Rs to reduce feedforward glutamatergic transmission onto PV-INs, while PV-IN excitability was unaffected by HA signaling. At local (i.e., electrically stimulated) glutamatergic synapses onto PV-INs, multiple pharmacological assays suggest, but do not prove, that H3R effector function requires AC-cAMP-PKA activity but not N- or P/Q-type VGCCs (30). Additionally, a qualitatively notable reduction in eEPSCs by HA persisted following cAMP-/PKA-specific manipulations, suggesting that other presynaptic signaling partners are recruited in parallel or this mechanism is input specific.
As PV-IN AP firing relies on time-contingent shifts in glutamatergic drive, inhibitory H3R activity at this synaptic locus is similarly efficacious in modulating PV-IN output (24). Our input-specific optogenetic analysis encourages this notion, as MDT-to-PV-IN synapses, imposing greater basal control over PV-IN output than PFC synapses, underwent robust H3R-dependent HA-LTD that virtually abolished MDT-evoked FFI of MSNs. Conversely, HA elicited only a modest reduction in synaptic strength at PFC-to-PV-IN synapses with a slight but discernible attenuation in PFC-evoked FFI. Interestingly, we recently showed the opposite finding at MSN synapses in which glutamatergic afferents from the MDT were minimally regulated by HA relative to those from the PFC (8). Therefore, a working model emerges in which HA differentially regulates thalamocortical drive onto PV-INs and MSNs in the NAcSh, ultimately leading to increased MSN responsiveness to information encoded by the MDT.
Histaminergic Effects on PV-IN- and CIN-Regulated Microcircuit Motifs
As discussed above, HA application resulted in near-complete escape from MDT-evoked FFI, consistent with the robust expression of HA-LTD at MDT-to-PV-IN synapses. In contrast to PV-IN-mediated FFI, a direct readout of CIN microcircuit activity is complicated by the fact that ACh exerts a vast array of nAChR- and mAChR-dependent effects on NAc and striatal circuits (21). CINs extend diffuse axonal arbors from which background ACh tone can modulate assorted circuit elements (42–46). We focused our analysis on ACh-DA coupling, a process by which CIN activation can drive and/or enhance DA release from ventral tegmental area projections to the NAc (33). Consistent with the secretagogue actions of CINs on NAcSh DA release, the β2-containing nAChR antagonist DHβE decreased DA by approximately 50%, mirroring observations that ACh signaling via nAChRs drives evoked and spontaneous DA release in the NAc, with complex, activity-dependent effects mediated by multiple mAChR and nAChR subtypes (22,47). Accordingly, chemical or experiential alterations of CIN sAP firing in the NAc often result in altered DA release dynamics through nAChR- or mAChR-dependent pathways (48–50). Our data extend these findings by showing that HA increases mesoaccumbens DA release through a H2R-dependent mechanism reliant on β2-containing nAChRs targeted by CINs. Indeed, this similarly corroborates early studies showing that microinfusion of HA into the NAc increases ACh and DA release (37). As the neuromodulatory actions of HA within the mesolimbic network continue to be unveiled, it is our hope that a renewed emphasis will be placed on HA and ascending tuberomammillary nucleus pathways for the treatment of neuropsychiatric disease states.
Supplementary Material
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Chemical Compound, Drug | Histamine dihydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 5818 | |
| Chemical Compound, Drug | Scopolamine hydrobromide | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 6603108 | |
| Chemical Compound, Drug | Cetirizine dihydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 55182 | |
| Chemical Compound, Drug | Ranitidine hydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 657344 | |
| Chemical Compound, Drug | (R)-(-)-α-methylhistamine dihydrobromide | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 45037031 | |
| Chemical Compound, Drug | Dimaprit dihyrdrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 90045 | |
| Chemical Compound, Drug | Mecamylamine | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 4032 | |
| Chemical Compound, Drug | SCH 50911 | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 5311429 | |
| Chemical Compound, Drug | ZD7288 | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 123983 | |
| Chemical Compound, Drug | db-cAMP | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 9934146 | |
| Chemical Compound, Drug | Apamin | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 91808947 | |
| Chemical Compound, Drug | ω-conotoxin | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 16133838 | |
| Chemical Compound, Drug | ω-agatoxin-IVA | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 56841669 | |
| Chemical Compound, Drug | Dihydro-β-erythroidine hydrobromide (DHβE) | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 11957537 | |
| Chemical Compound, Drug | 2-Pyridylethylamine dihydrochloride (2-PyEA) | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 201148 | |
| Chemical Compound, Drug | Forskolin | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 47936 | |
| Chemical Compound, Drug | NBQX disodium salt | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 3272523 | |
| Chemical Compound, Drug | D-(-)-2-Amino-5-Phosphonopentanoic Acid (APV) | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 135342 | |
| Chemical Compound, Drug | Picrotoxin | Sigma-Aldrich, Stl Louis, MO | PubChem ID: 6473767 | |
| Chemical Compound, Drug | H89 dihydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 5702541 | |
| Chemical Compound, Drug | BAPTA-AM | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 2293 | |
| Chemical Compound, Drug | 4-Aminopyridine (4-AP) | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 1727 | |
| Chemical Compound, Drug | Tetrodotoxin (TTX) | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 11174599 | |
| Organism, Strain | PVCre, Pvalbtm1(cre)Arbr/J | The Jackson Laboratory, Sacramento, CA | Stock No: 008069 | |
| Organism, Strain | Ai9, Gt(ROSA)26Sortm9(CAG-tdTomato)Hze | The Jackson Laboratory, Sacramento, CA | Stock No: 007909 | |
| Organism, Strain | ChAT-IRES-Cre-Chattm2(cre)Lowl/J | The Jackson Laboratory, Sacramento, CA | Stock No: 018957 | |
| Organism, Strain | Mouse: C57BL/6J | The Jackson Laboratory, Sacramento, CA | Stock No: 000664 | |
| Bacterial or viral strain | Virus: pAAV5-CaMKIIα-hChR2(H134R)-EYFP | Addgene, Watertown, MA | Plasmid No: 26969 | |
| Software, Algorithm | DEMON voltammetry | Wake Forest Innovations, Winston Salem, NC | http://www.wfubmc.edu/OTAM/Technologies/Computer-Software-and-copyright.htm#Demon. | |
| Software, Algorithm | pClamp 10 | Molecular Devices, Sunnyvale, CA | www.moleculardevices.com | |
| Software, Algorithm | GraphPad Prism | GraphPad Software, Inc., La Jolla, CA | www.graphpad.com/scientific-software/prism/ |
ACKNOWLEDGMENTS AND DISCLOSURES
This work was supported by the National Institute on Drug Abuse (Grant No. R01 DA040630 [to BAG]), National Institute of General Medical Sciences (Grant No. T32 GM108554 [to KMM]), National Institutes of Health (Grant Nos. DA042111 and DA048931 [to ESC] and Grant No. DA052641 [to LJB]), Academic Pathways Program at Vanderbilt University (to LJB), Brain and Behavior Research Foundation (to ESC), Whitehall Foundation (to ESC), and Edward Mallinckrodt, Jr. Foundation (to ESC).
Footnotes
The authors report no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online at https://doi.org/10.1016/j.biopsych.2021.10.004.
REFERENCES
- 1.Takagi H, Morishima Y, Matsuyama T, Hayashi H, Watanabe T, Wada H (1986): Histaminergic axons in the neostriatum and cerebral cortex of the rat: A correlated light and electron microscopic immunocytochemical study using histidine decarboxylase as a marker. Brain Res 364:114–123. [DOI] [PubMed] [Google Scholar]
- 2.Watanabe T, Taguchi Y, Shiosaka S, Tanaka J, Kubota H, Terano Y, et al. (1984): Distribution of the histaminergic neuron system in the central nervous system of rats; a fluorescent immunohistochemical analysis with histidine decarboxylase as a marker. Brain Res 295:13–25. [DOI] [PubMed] [Google Scholar]
- 3.Panula P, Nuutinen S (2013): The histaminergic network in the brain: 22. Basic organization and role in disease [no. 7]. Nat Rev Neurosci 14:472–487. [DOI] [PubMed] [Google Scholar]
- 4.Passani MB, Blandina P (2011): Histamine receptors in the CNS as 23. targets for therapeutic intervention. Trends Pharmacol Sci 32:242249. [DOI] [PubMed] [Google Scholar]
- 5.Brabant C, Alleva L, Quertemont E, Tirelli E (2010): Involvement of the 24. brain histaminergic system in addiction and addiction-related behaviors: A comprehensive review with emphasis on the potential therapeutic use of histaminergic compounds in drug dependence. Prog Neurobiol 92:421–441.. [DOI] [PubMed] [Google Scholar]
- 6.Taylor KM, Snyder SH (1971): Brain histamine: Rapid apparent turnover altered by restraint and cold stress. Science 172:1037–1039. [DOI] [PubMed] [Google Scholar]
- 7.Zhang XY, Peng SY, Shen LP, Zhuang QX, Li B, Xie ST, et al. (2020): Targeting presynaptic H3 heteroreceptor in nucleus accumbens to improve anxiety and obsessive-compulsive-like behaviors. Proc Natl Acad Sci U S A 117:32155–32164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manz KM, Becker JC, Grueter CA, Grueter BA (2021): Histamine H3 27. receptor function biases excitatory gain in the nucleus accumbens. Biol Psychiatry 89:588–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolam JP, Wainer BH, Smith AD (1984): Characterization ofcholinergic 28. neurons in the rat neostriatum. A combination of choline acetyl-transferase immunocytochemistry, Golgi-impregnation and electron microscopy. Neuroscience 12:711–718. [DOI] [PubMed] [Google Scholar]
- 10.Castro DC, Bruchas MR (2019): A motivational and neuropeptidergic 29. hub: Anatomical and functional diversity within the nucleus accumbens shell. Neuron 102:529–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morse AK, Leung BK, Heath E, Bertran-Gonzalez J, Pepin E, Chieng BC, et al. (2020): Basolateral amygdala drives a GPCR-mediated striatal memory necessary for predictive learning to influence choice. Neuron 106:855–869.e8. [DOI] [PubMed] [Google Scholar]
- 12.Schall TA, Wright WJ, Dong Y(2021): Nucleus accumbens fast-spiking 31. interneurons in motivational and addictive behaviors. Mol Psychiatry 26:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown MTC, Tan KR, O’Connor EC, Nikonenko I, Muller D, Lüscher C (2012): Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature 492:452–456.. [DOI] [PubMed] [Google Scholar]
- 14.Collins AL, Aitken TJ, Huang IW, Shieh C, Greenfield VY, Monbouquette HG, et al. (2019): Nucleus accumbens cholinergic interneurons oppose cue-motivated behavior. Biol Psychiatry 86:388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JH, Ribeiro EA, Kim J, Ko B, Kronman H, Jeong YH, et al. (2020): Dopaminergic regulation of nucleus accumbens cholinergic interneurons demarcates susceptibility to cocaine addiction. Biol Psychiatry 88:746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Gallegos DA, Pogorelov VM, O’Hare JK, Calakos N, Wetsel WC, West AE (2018): Parvalbumin interneurons of the mouse nucleus accumbens are required for amphetamine-induced locomotor sensitization and conditioned place preference. Neuropsychopharmacology 43:953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, et al. (2012): Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: Setting the tone for reward processing. Cell Rep 2:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manz KM, Ghose D, Turner BD, Taylor A, Becker J, Grueter CA, Grueter BA (2020): Calcium-permeable AMPA receptors promote endocannabinoid signaling at parvalbumin interneuron synapses in the 38. nucleus accumbens core. Cell Rep 32:107971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mateo Y, Johnson KA, Covey DP, Atwood BK, Wang HL, Zhang S, et al. (2017): Endocannabinoid actions on cortical terminals orchestrate local modulation of dopamine release in the nucleus accumbens. Neuron 96:1112–1126.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Threlfell S, Cragg SJ (2011): Dopamine signaling in dorsal versus 40. ventral striatum: the dynamic role of cholinergic interneurons. Front Syst Neurosci 5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mamaligas AA, Ford CP (2016): Spontaneous synaptic activation of muscarinic receptors by striatal cholinergic neuron firing. Neuron 91:574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yorgason JT, Zeppenfeld DM, Williams JT (2017): Cholinergic interneurons underlie spontaneous dopamine release in nucleus accumbens. J Neurosci 37:2086–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawaguchi Y (1993): Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci 13:4908–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu J, Yan Y, Li KL, Wang Y, Huang YH, Urban NN, et al. (2017): Nucleus accumbens feedforward inhibition circuit promotes cocaine self-administration. Proc Natl Acad Sci U S A 114:E8750-E8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji MJ, Zhang XY, Peng XC, Zhang YX, Chen Z, Yu L, et al. (2018): Histamine excites rat GABAergic ventral pallidum neurons via co-activation of H1 and H2 receptors. Neurosci Bull 34:1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorst MC, Tokarska A, Zhou M, Lee K, Stagkourakis S, Broberger C, et al. (2020): Polysynaptic inhibition between striatal cholinergic interneurons shapes their network activity patterns in a dopamine-dependent manner. Nat Commun 11:5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng J, Umschweif G, Leung J, Sagi Y, Greengard P (2019): HCN2 channels in cholinergic interneurons of nucleus accumbens shell regulate depressive behaviors. Neuron 101:662–672.e5. [DOI] [PubMed] [Google Scholar]
- 28.Goldberg JA, Wilson CJ (2005): Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J Neurosci 25:10230–10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hopf FW, Bowers MS, Chang SJ, Chen BT, Martin M, Seif T, et al. (2010): Reduced nucleus accumbens SK channel activity enhances alcohol seeking during abstinence. Neuron 65:682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeshita Y, Watanabe T, Sakata T, Munakata M, Ishibashi H, Akaike N (1998): Histamine modulates high-voltage-activated calcium channels in neurons dissociated from the rat tuberomammillary nucleus. Neuroscience 87:797–805. [DOI] [PubMed] [Google Scholar]
- 31.Vázquez-Vázquez H, Gonzalez-Sandoval MDC, Vega AV, Arias-Montaño JA, Barral J (2020): Histamine H3 receptor activation modulates glutamate release in the corticostriatal synapse by acting at CaV2.1 (P/Q-type) calcium channels and GIRK (KIR3) potassium channels [published online ahead of print Oct 17]. Cell Mol Neurobiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drutel G, Peitsaro N, Karlstedt K, Wieland K, Smit MJ, Timmerman H, et al. (2001): Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Mol Pharmacol 59:1–8. [PubMed] [Google Scholar]
- 33.Nolan SO, Zachry JE, Johnson AR, Brady LJ, Siciliano CA, Calipari ES (2020): Direct dopamine terminal regulation by local striatal microcircuitry. J Neurochem 155:475–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brimblecombe KR, Threlfell S, Dautan D, Kosillo P, Mena-Segovia J, Cragg SJ (2018): Targeted activation of cholinergic interneurons accounts for the modulation of dopamine by striatal nicotinic receptors. eNeuro 5.. ENEURO.0397–17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kosillo P, Zhang YF, Threlfell S, Cragg SJ (2016): Cortical control of striatal dopamine transmission via striatal cholinergic interneurons. Cereb Cortex 26:4160–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bell MI, Richardson PJ, Lee K (2000): Histamine depolarizes cholinergic interneurones in the rat striatum via a H1-receptor mediated action. Br J Pharmacol 131:1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galosi R, Lenard L, Knoche A, Haas H, Huston JP, Schwarting RK (2001): Dopaminergic effects of histamine administration in the nucleus accumbens and the impact of H1-receptor blockade. Neuropharmacology 40:624–633. [DOI] [PubMed] [Google Scholar]
- 38.Choi SJ, Ma TC, Ding Y, Cheung T, Joshi N, Sulzer D, et al. (2020): Alterations in the intrinsic properties of striatal cholinergic interneurons after dopamine lesion and chronic L-DOPA. Elife 9:e56920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Francis TC, Yano H, Demarest TG, Shen H, Bonci A (2019): High-frequency activation of nucleus accumbens D1-MSNs drives excitatory potentiation on D2-MSNs. Neuron 103:432–444.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki E, Momiyama T (2021): M1 muscarinic acetylcholine receptor-mediated inhibition of GABA release from striatal medium spiny neurons onto cholinergic interneurons. Eur J Neurosci 53:796–813. [DOI] [PubMed] [Google Scholar]
- 41.Vachez YM, Tooley JR, Abiraman K, Matikainen-Ankney B, Casey E, Earnest T, et al. (2021): Ventral arkypallidal neurons inhibit accumbal firing to promote reward consumption. Nat Neurosci 24:379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Assous M, Kaminer J, Shah F, Garg A, Koós T, Tepper JM (2017): Differential processing of thalamic information via distinct striatal interneuron circuits. Nat Commun 8:15860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elghaba R, Vautrelle N, Bracci E (2016): Mutual control of cholinergic and low-threshold spike interneurons in the striatum. Front Cell Neurosci 10:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faust TW, Assous M, Tepper JM, Koós T (2016): Neostriatal GABAergic interneurons mediate cholinergic inhibition of spiny projection neurons. J Neurosci 36:9505–9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melendez-Zaidi AE, Lakshminarasimhah H, Surmeier DJ (2019): Cholinergic modulation of striatal nitric oxide-producing interneurons. Eur J Neurosci 50:3713–3731. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, et al. (2006): Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50:443–452. [DOI] [PubMed] [Google Scholar]
- 47.Shin JH, Adrover MF, Wess J, Alvarez VA (2015): Muscarinic regulation of dopamine and glutamate transmission in the nucleus accumbens. Proc Natl Acad Sci U S A 112:8124–8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adrover MF, Shin JH, Quiroz C, Ferré S, Lemos JC, Alvarez VA (2020): Prefrontal cortex-driven dopamine signals in the striatum show unique spatial and pharmacological properties. J Neurosci 40:7510–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lemos JC, Shin JH, Alvarez VA(2019): Striatal cholinergic interneurons are a novel target of corticotropin releasing factor. J Neurosci 39:5647–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stouffer MA, Woods CA, Patel JC, Lee CR, Witkovsky P, Bao L, et al. (2015): Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat Commun 6:8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.