

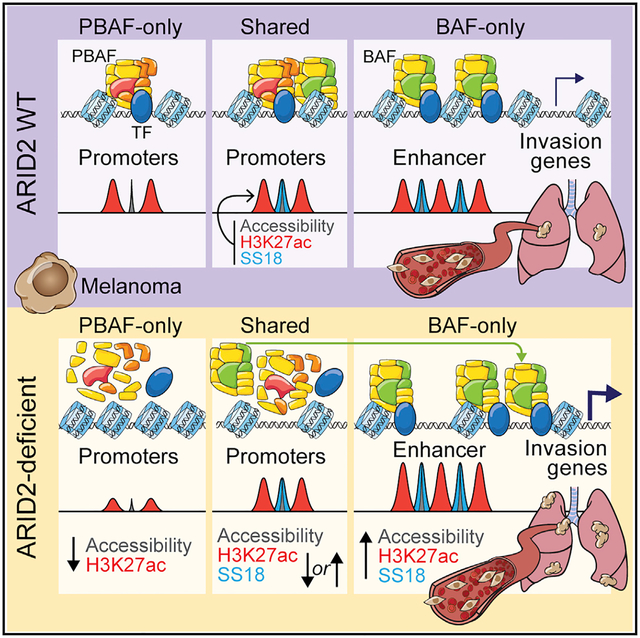
SUMMARY
ARID2 is the most recurrently mutated SWI/SNF complex member in melanoma; however, its tumor-suppressive mechanisms in the context of the chromatin landscape remain to be elucidated. Here, we model ARID2 deficiency in melanoma cells, which results in defective PBAF complex assembly with a concomitant genomic redistribution of the BAF complex. Upon ARID2 depletion, a subset of PBAF and shared BAF-PBAF-occupied regions displays diminished chromatin accessibility and associated gene expression, while BAF-occupied enhancers gain chromatin accessibility and expression of genes linked to the process of invasion. As a function of altered accessibility, the genomic occupancy of melanoma-relevant transcription factors is affected and significantly correlates with the observed transcriptional changes. We further demonstrate that ARID2-deficient cells acquire the ability to colonize distal organs in multiple animal models. Taken together, our results reveal a role for ARID2 in mediating BAF and PBAF subcomplex chromatin dynamics with consequences for melanoma metastasis.
Graphical abstract
In brief
The tumor-suppressive functions of the SWI/SNF subunit ARID2 remain ill-defined in the context of melanoma. Carcamo et al. demonstrate that, upon ARID2 depletion, the PBAF complex fails to assemble, altering BAF genomic occupancy with consequences on chromatin accessibility, transcription factor binding, and transcriptional changes that promote metastasis.
INTRODUCTION
SWI/SNF (switch/sucrose non-fermentable) complex members are mutated in ~20% of all human cancers (Kadoch et al., 2013; Shain and Pollack, 2013), and our understanding of the cellular and physiological consequences of these mutations is emerging (Hodges et al., 2016; Kadoch and Crabtree, 2015). To facilitate access to the nucleosomal DNA, SWI/SNF chromatin remodeling complexes act primarily by sliding nucleosomes to reorganize their positioning, or they may eject or insert histone octamers (Bruno et al., 2003; Dechassa et al., 2010; Saha et al., 2006). These multi-subunit complexes contain a catalytic ATPase (either BRG1 or BRM) and additional non-catalytic subunits that are essential for complex function, stability, and specificity (Khavari et al., 1993; Mashtalir et al., 2018; Wang et al., 1996). The latter often contain DNA-binding and/or chromatin reader domains, which modulate complex targeting and activity. While some SWI/SNF subcomplexes are developmentally regulated or tissue specific (Hargreaves and Crabtree, 2011; Kadoch and Crabtree, 2015), the two most prevalent are the BAF (BRG1-associated factor) and PBAF (polybromo-associated BAF) complexes (Mashtalir et al., 2018). The BAF complex is characterized by the incorporation of ARID1A or ARID1B; DPF1, DPF2, or DPF3; and SS18, while PBAF is distinguished by the presence of ARID2, PBRM1, PHF10, and BRD7 (Hargreaves and Crabtree, 2011; Kadoch and Crabtree, 2015). Pertinent to this study, the members of the ARID family of proteins contain an AT-rich interaction domain that mediates non-sequence-specific DNA interactions (Patsialou et al., 2005). A smaller complex, GBAF (GLTSCR1/like-containing BAF), lacks several core subunits and an ARID protein, yet contains unique subunits, including BRD9 and GLTSCR1/GLTSCRL1 (Alpsoy and Dykhuizen, 2018; Mashtalir et al., 2018; Wang et al., 2019).
Remarkably, specific SWI/SNF subunits are distinctively mutated in different cancers. For example, the BAF and PBAF shared core subunit SNF5 is inactivated in almost all pediatric rhabdoid tumors (Biegel et al., 2000; Roberts et al., 2000; Versteege et al., 1998), and its loss is associated with altered binding of SWI/SNF complexes to enhancers and increased Polycomb repression (Erkek et al., 2018; Nakayama et al., 2017; Wilson et al., 2010). The BAF-specific subunit ARID1A is the most highly mutated in cancer, including bladder, liver, and ovarian clear cell carcinoma, and is thought to act as a tumor suppressor (Kadoch et al., 2013). The PBAF-specific subunits PBRM1 and ARID2 are highly mutated in renal cell carcinoma and melanoma, respectively (Akbani et al., 2015; Hodis et al., 2012; Varela et al., 2011).
Melanoma is a lethal form of skin cancer with rising incidence (Atkins et al., 2021). Recent advances in targeted and immunotherapy have markedly changed the landscape of melanoma treatment (Dummer et al., 2020; Samstein et al., 2019); however, a growing body of evidence suggests that epigenetic regulation plays a key role in melanoma development, progression, and response to therapies (Fontanals-Cirera et al., 2017; Hakimi et al., 2020; Hanniford et al., 2020; Kapoor et al., 2010; Kato et al., 2020; Strub et al., 2018; Vardabasso et al., 2015; Verfaillie et al., 2015). Moreover, melanoma cells can exist in multiple phenotypic states independent of genetic subtype, with distinct transcriptional programs and phenotypes (Rambow et al., 2019; Verfaillie et al., 2015; Widmer et al., 2012). A proliferative state has been linked to high expression levels of the melanocyte lineage-specific TF (transcription factor) MITF, while an invasive state is linked to high AP-1 and TEAD, coupled to low MITF expression (Verfaillie et al., 2015; Widmer et al., 2012).
ARID2 is the most recurrently mutated SWI/SNF subunit in melanoma, with rates similar to the NF1-mutated genetic subtype. Mutations are largely missense or nonsense with few hotspots (Akbani et al., 2015; Hodis et al., 2012), suggesting a tumor-suppressive role for ARID2. Functionally, depletion of ARID2, PBRM1, or BRD7 from mouse melanoma tumors correlates with increased sensitivity to immune checkpoint inhibition and increased T cell killing (Fukumoto et al., 2020; Pan et al., 2018). While intriguing, our molecular understanding of ARID2’s tumor-suppressive function in melanoma remains limited. We herein demonstrate that loss of ARID2 in melanoma, and in turn the PBAF complex, results in BAF redistribution to sites associated with open chromatin and increased TF binding, with consequences for the transcriptional programs that control melanoma metastasis.
RESULTS
Generation of an isogenic ARID2-deficient melanoma cell model
By examining TCGA (The Cancer Genome Atlas) for mutations of SWI/SNF components in melanoma (Akbani et al., 2015), we found that the ARID2 subunit of the PBAF complex is the most commonly mutated SWI/SNF subunit, with a frequency rate of ~13% across 345 melanoma patients (Figures 1A and S1A). Some patients harbor multi-allelic alterations in ARID2, increasing the rate to ~18% (Figure 1A). This was corroborated in a larger study of >1,000 patients (including TCGA) and extended by the finding that ARID2 is a significantly mutated gene in NRAS mutant melanoma (Conway et al., 2020).
Figure 1. Effects of ARID2 depletion on SWI/SNF complexes in melanoma.
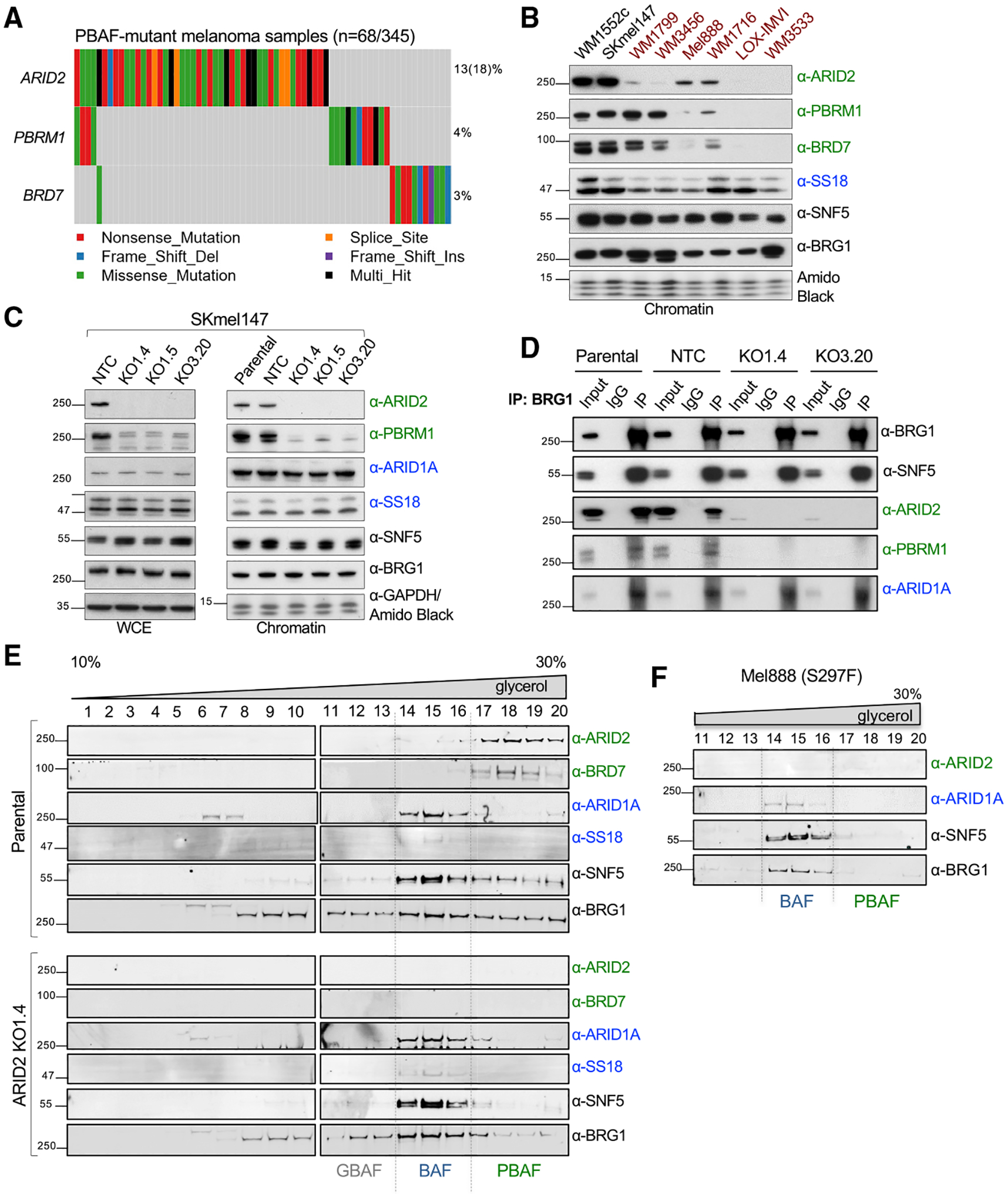
(A) Oncoplot of TCGA skin cutaneous melanoma mutational data highlighting mutation rates of PBAF-specific subunits ARID2, PBRM1, and BRD7. Each column represents one sample.
(B) Chromatin fraction immunoblots of SWI/SNF subunits, including PBAF-specific ARID2, PBRM1, and BRD7 (green); BAF-specific SS18 (blue); and core subunits SNF5 and BRG1 (black), across ARID2 WT and mutant (indicated in red) melanoma cell lines. Amido black staining was used as a loading control (n = 3).
(C) Immunoblot of WCE and chromatin fractions of SWI/SNF subunits in SKmel147 ARID2 WT (parental and NTC) and KO clones. GAPDH was used as a loading control for WCE, amido black for chromatin fractions (n = 3).
(D) Immunoblots of BRG1, SNF5, ARID2, PBRM1, and ARID1A in SKmel147 ARID2 WT and KO cells following immunoprecipitation of BRG1. IgG was used as negative control (n = 2).
(E) Immunoblots of glycerol gradient sedimentation from SKmel147 ARID2 WT and KO1.4 showing sedimentation of SWI/SNF subcomplexes (ARID1A and SS18 in the BAF fractions, ARID2 and BRD7 in the PBAF fractions) in ARID2 WT cells, which is altered in ARID2 KO cells (n = 2).
(F) Immunoblots of ARID2, ARID1A, SNF5, and BRG1 of glycerol gradient sedimentation fractions from Mel888 (n = 1). See also Figure S1 and Table S1.
To probe the effects of ARID2 mutation in melanoma, we first screened a panel of melanoma cell lines by immunoblot, including two ARID2 WT (wild type) and six ARID2 mutant cell lines (Figure 1B and Table S1). We observed reduced levels of ARID2 protein across the ARID2 mutant cell lines. Many of these mutations are annotated as heterozygous (Table S1), and the remaining ARID2 protein in some mutant cell lines can be attributed to the WT allele. In contrast, two cell lines (LOX-IMVI and WM3533) displayed a complete loss of ARID2, as well as BRD7 and PBRM1, without considerable changes in the BAF-specific subunit SS18 or the shared core subunits BRG1 and SNF5 (Figure 1B). WM3533 is annotated as having two ARID2 truncating mutations, an occurrence observed in ARID2 mutant melanomas (Figure 1A) (Akbani et al., 2015; Conway et al., 2020), while LOX-IMVI is annotated as a heterozygous frameshift but displays loss of heterozygosity (Table S1). Therefore, some melanomas exhibit a complete loss of ARID2 and concomitant reduced protein levels of PBAF-specific members, which has been attributed to a deficiency in PBAF subcomplex assembly upon ARID2 loss (Mashtalir et al., 2018; Schick et al., 2019; Yan et al., 2005).
To model ARID2 deficiency, we created an isogenic system using CRISPR-Cas9 genomic editing in the metastatic melanoma cell line SKmel147 (NRASQ61R; MITF low) for which we have extensive epigenomic data (Vardabasso et al., 2015; Fontanals-Cirera et al., 2017). Consistent with the cell line data above, complete loss of ARID2 resulted in depletion of PBAF-specific members in both WCE (whole-cell extract) and chromatin fractions, while BAF-specific and core subunit levels remained intact (Figures 1C and S1B). Importantly, we did not observe significant changes in mRNA levels of the majority of SWI/SNF complex members in ARID2 KO (knockout) clones (Figure S1C), suggesting that loss of the PBAF-specific subunits occurs post-transcriptionally.
ARID2 loss results in deficient PBAF complex assembly
To examine the composition of the BAF and PBAF complexes in the absence of ARID2, we performed co-immunoprecipitation studies of the core subunit BRG1 in ARID2 WT and KO clones. We pulled down all the immunoblotted SWI/SNF subunits with BRG1 in the parental and NTC (non-targeting control) lines (SNF5, ARID1A, ARID2, and PBRM1); however, in the ARID2 KO clones, PBRM1 was no longer associated with BRG1, while SNF5 and ARID1A were still present (Figure 1D). We also performed reverse co-immunoprecipitation of ARID2 and an additional PBAF-specific subunit, PHF10. We found that, while ARID2 and PHF10 pulled down BRG1 and SNF5 in the parental and NTC lines, there was no association with these core subunits, or PBAF-specific subunits, in the ARID2 KO lines (Figures S1D and S1E).
To further assess the impact of ARID2 loss on SWI/SNF subcomplex dynamics, we utilized glycerol gradient sedimentation (Pan et al., 2019; Alpsoy and Dykhuizen, 2018). In the parental WT setting, the PBAF subcomplex migrates toward the heavier fractions, as observed by the presence of ARID2, PHF10, and BRD7, while BAF migrates to lighter fractions as detected by the presence of ARID1A and SS18 (Figures 1E and S1F) (Alpsoy and Dykhuizen, 2018; Mashtalir et al., 2018). SNF5 was enriched in the BAF and PBAF fractions, and BRM was predominant in the BAF fractions, while BRG1 was present in all the subcomplex fractions, including the GBAF subcomplex estimated by GLTSCR1 blotting (Alpsoy and Dykhuizen, 2018; Gatchalian et al., 2018; Michel et al., 2018; Wang et al., 2019) (Figures 1E and S1F). ARID2 KO cells displayed depletion of ARID2, PHF10, and BRD7, as expected, but this was also accompanied by a reduction in the core subunits BRG1 and SNF5 in the corresponding PBAF fractions (Figures 1E and S1F). We further validated these findings in Mel888 (confirmed to harbor a S297F mutation), which displays a similar depletion of the core subunits in the PBAF fractions (Figures 1F, S1G, and Table S1) (Akbani et al., 2015; Gao et al., 2013). By comparing ARID2 WT versus KO fractions quantitatively, we observed a modest increased signal of the core subunits in the BAF-containing fractions in the absence of ARID2 (Figures S1H and S1I). This may reflect a redistribution of PBAF-containing core subunits as opposed to partially dissociated PBAF complexes running in the BAF fractions, as the loss of the PBAF-specific subunits would result in a complex of much smaller molecular weight. Similar findings were observed by quantitative mass spectrometry of BRG1 in the absence of ARID2, where PBAF-specific subunits were significantly depleted and BRG1 showed increased association with BAF-specific subunits (Schick et al., 2019). Collectively, these observations suggest that depletion of ARID2 results in a complete loss of the PBAF complex with potential for redistribution of the residual SWI/SNF core subunits to the BAF complex.
PBAF and BAF have distinct yet overlapping genomic occupancy in melanoma
To further dissect the role of SWI/SNF complexes in melanoma, we performed ChIP-seq (chromatin immunoprecipitation with high-throughput sequencing) of ARID2, SS18 (representing a BAF subunit), and BRG1 in the parental SKmel147 cell line (Figures 2A and 2B). We found that, while ARID2 is enriched at promoters, SS18 is found predominantly at distal and intragenic locations (Figure S2A). Importantly, ChIP-seq of ARID2 in SKmel147 ARID2 KO cells yielded <1% of the ARID2 peaks found in WT cells (Figure S2B), likely reflecting non-specific binding (Figure S2C), and these peaks were removed from subsequent analysis. Next, we defined three genomic clusters based on the differential and shared occupancy of ARID2 and SS18: 5% of peaks at PBAF-enriched regions (defined by ARID2 occupancy and low SS18), 71% at BAF-enriched regions (SS18 occupied and low ARID2), and 24% at BAF-PBAF “shared” regions (ARID2- and SS18-bound) (Figures 2A, 2B, and Table S2). All three clusters displayed BRG1 binding, although less signal was detected at the PBAF regions (Figure 2B), as previously observed (Pan et al., 2019). Through genomic annotation analysis, we found that PBAF and shared regions were preferentially enriched at promoters (~55%), whereas the BAF regions were preferentially enriched at distal and intragenic locations (~90%) (Figure 2C).
Figure 2. PBAF and BAF genomic localization in melanoma cells.
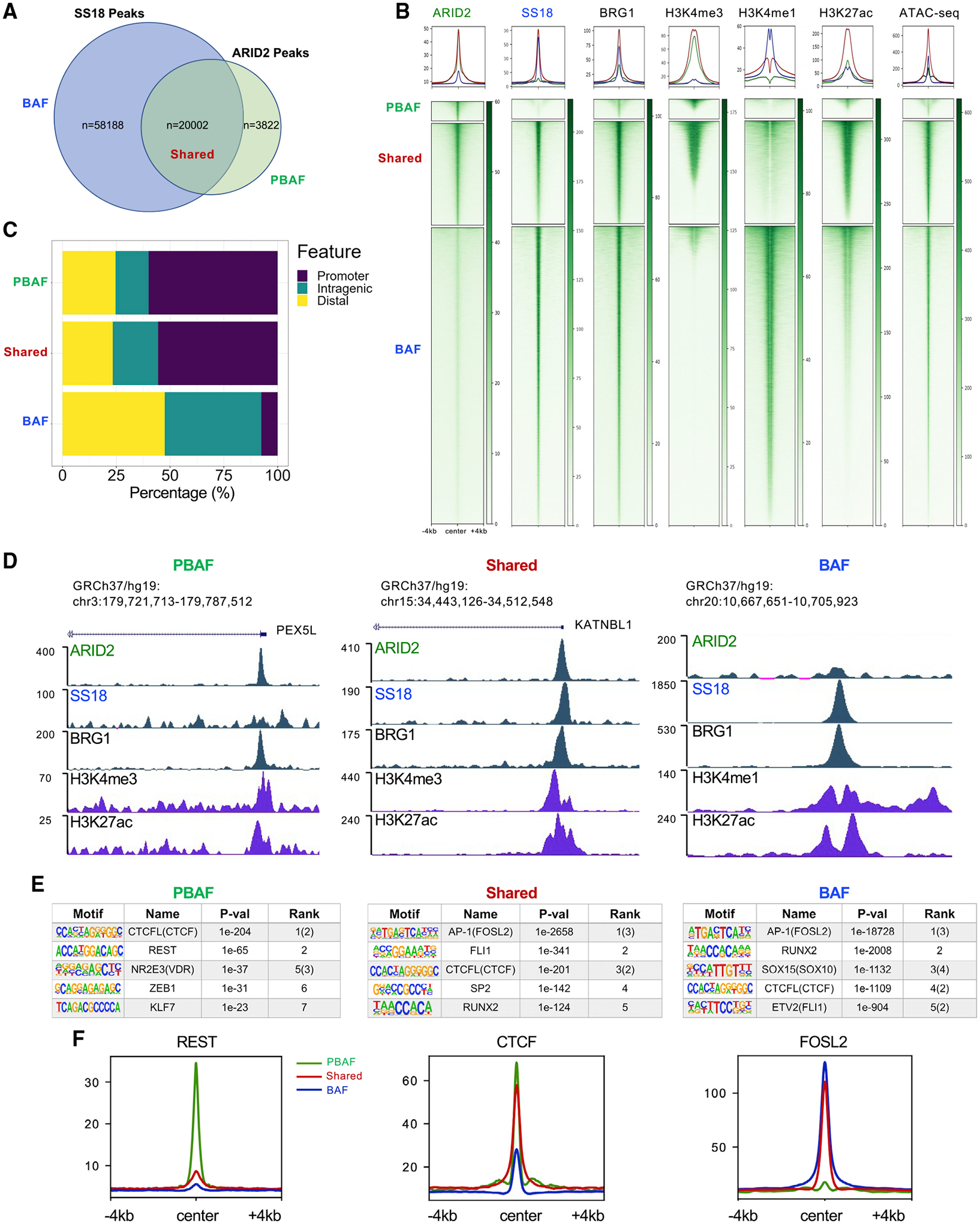
(A) Intersection of significant peaks between ARID2 and SS18 ChIP-seq in SKmel147 used to defined PBAF, BAF, and shared regions.
(B) Heatmaps of ChIP-seq enrichment for ARID2, SS18, BRG1, H3K4me3, H3K4me1, H3K27ac, and ATAC-seq in SKmel147 were centered on PBAF, shared, and BAF regions as defined in (A).
(C) Bar plots displaying the percentage of PBAF, shared, and BAF regions occupying promoter, distal, and intragenic regions.
(D) UCSC genome browser (hg19) snapshots displaying examples of PBAF, shared, and BAF regions.
(E) TF motif analysis of the PBAF, shared, and BAF regions.
(F) Metagene profiles of REST, CTCF, and FOSL2 ChIP-seq in SKmel147 at PBAF, shared, and BAF regions as defined in (A). See also Figure S2 and Tables S1, S2, S4, and S6.
Next, we performed ChIP-seq for H3K27ac, utilized our ChIP-seq data for H3K4me1 and H3K4me3 (Fontanals-Cirera et al., 2017), and performed ATAC-seq (assay for transposase-accessible chromatin with high-throughput sequencing) in SKmel147. Using these data, we found that PBAF-enriched and shared regions are largely associated with promoters marked by H3K4me3, and BAF-enriched regions are associated with enhancers marked by H3K4me1, consistent with the genomic annotation analysis (Figure 2B). Examples of such genomic regions are shown in Figure 2D. Finally, H3K27ac and open chromatin as demonstrated by ATAC-seq were associated with all three clusters (Figure 2B).
We validated these results using a second melanoma cell line, 113/6-4L (herein referred to as 4L), a BRAFV600E mutant, MITF-high line. Briefly, we identified PBAF, shared, and BAF genomic regions in 4L as described above utilizing the differential and shared occupancy of ARID2 and SS18 (Figure S2D and Table S2). Consistent with SKmel147, PBAF regions were preferentially enriched at promoters (~70%) and BAF regions at distal and intragenic locations (~90%), while shared sites were more evenly distributed across these regions than in SKmel147 (Figure S2E). All three clusters displayed BRG1 binding (similar to SKmel147, less signal was detected at PBAF regions) and were associated with H3K27ac and open chromatin as indicated by ATAC-seq (Figure S2F).
To address the functionality of these clusters, we divided all expressed genes in SKmel147 (Table S3) into four categories based on their promoter occupancy: PBAF (n = 1,721), BAF (n = 3,790), shared (n = 8,833), or devoid of either complex, “None” (n = 3,646). Analysis of their gene expression levels revealed that genes occupied by BAF and/or PBAF complexes are associated with significantly higher expression than genes without either complex (None) (Figure S2G). Furthermore, higher gene expression was associated with genes whose promoters were occupied by both complexes, compared with PBAF- or BAF-only occupied promoters (Figure S2G), suggesting cooperation of these complexes when co-bound. These findings were also corroborated in 4L (Figure S2G).
We next examined the TF motifs enriched in each of the SKmel147 clusters to identify putative co-regulators of PBAF and BAF complexes (Figure 2E and Table S4). In the PBAF cluster, we identified various motifs that have been associated with SWI/SNF, including REST, a repressor of neuronal gene expression (Barisic et al., 2019), and VDR, a nuclear receptor of the vitamin D ligand (Wei et al., 2018). The CTCF motif was enriched in all clusters, while the AP-1 motif was most enriched in the shared and BAF clusters, concordant with previous studies associating this family of TFs with BAF (Vierbuchen et al., 2017). AP-1 also emerged as a top motif when all ARID2 and SS18 peaks were examined (Figure S2H and Table S4). Similarly, in 4L, TF motif analysis identified REST as enriched in the PBAF cluster and AP-1 in the shared and BAF clusters (e.g., FOSL1 and ATF3, respectively) (Figure S2I and Table S4). To validate these motif predictions, we performed ChIP-seq for REST, CTCF, and FOSL2 (as a representative AP-1 factor) in SKmel147. Indeed, we observed that REST was most highly enriched in PBAF-enriched regions (although bound at a small subset of sites) and CTCF was enriched in all clusters, while FOSL2 was most enriched in shared and BAF-enriched clusters (Figures 2F and S2J). Taken together, our results indicate that there exists differential and shared enrichment of BAF and PBAF subcomplexes at distinct genomic regions, localizing at unique and common TF-binding sites.
PBAF deficiency results in chromatin accessibility alterations and retargeting of BAF
To understand the changes in chromatin accessibility upon loss of a functional PBAF complex, we performed ATAC-seq in SKmel147 WT (parental and NTC) and ARID2 KO clones (KO1.4 and KO3.20) in duplicate (Figures S3A and S3B). Upon calling significantly differentially accessible regions (adjusted p < 0.05), we identified 6,542 regions with decreased chromatin accessibility and 3,105 regions with increased accessibility in the ARID2 KO samples (Figure S3C and Table S2) with an average 2-fold change at both ATAC increased and ATAC decreased significant regions (Figure 3A). Upon intersection of these regions with the three defined clusters (Figure 2A), we observed that loss of accessibility occurred largely at ARID2-bound regions, with high ARID2 enrichment at baseline (Figures 3B and S3D), whereas increased chromatin accessibility was found predominantly at BAF-enriched regions, which displayed low ARID2 enrichment at baseline (Figures 3B and S3D). This suggests that PBAF primarily maintains open chromatin at a subset of ARID2-bound sites in both PBAF and shared regions, which is not compensated for by the BAF complex, while a fraction of BAF-enriched regions gain chromatin accessibility in the absence of PBAF. We also identified changes in chromatin accessibility in regions lacking ARID2 or SS18 significant peaks (Figure 3B, None), which may be below the threshold of significant peak calling, yet have functional SWI/SNF binding, or may be indirectly affected.
Figure 3. ARID2 depletion results in chromatin accessibility changes and BAF redistribution.
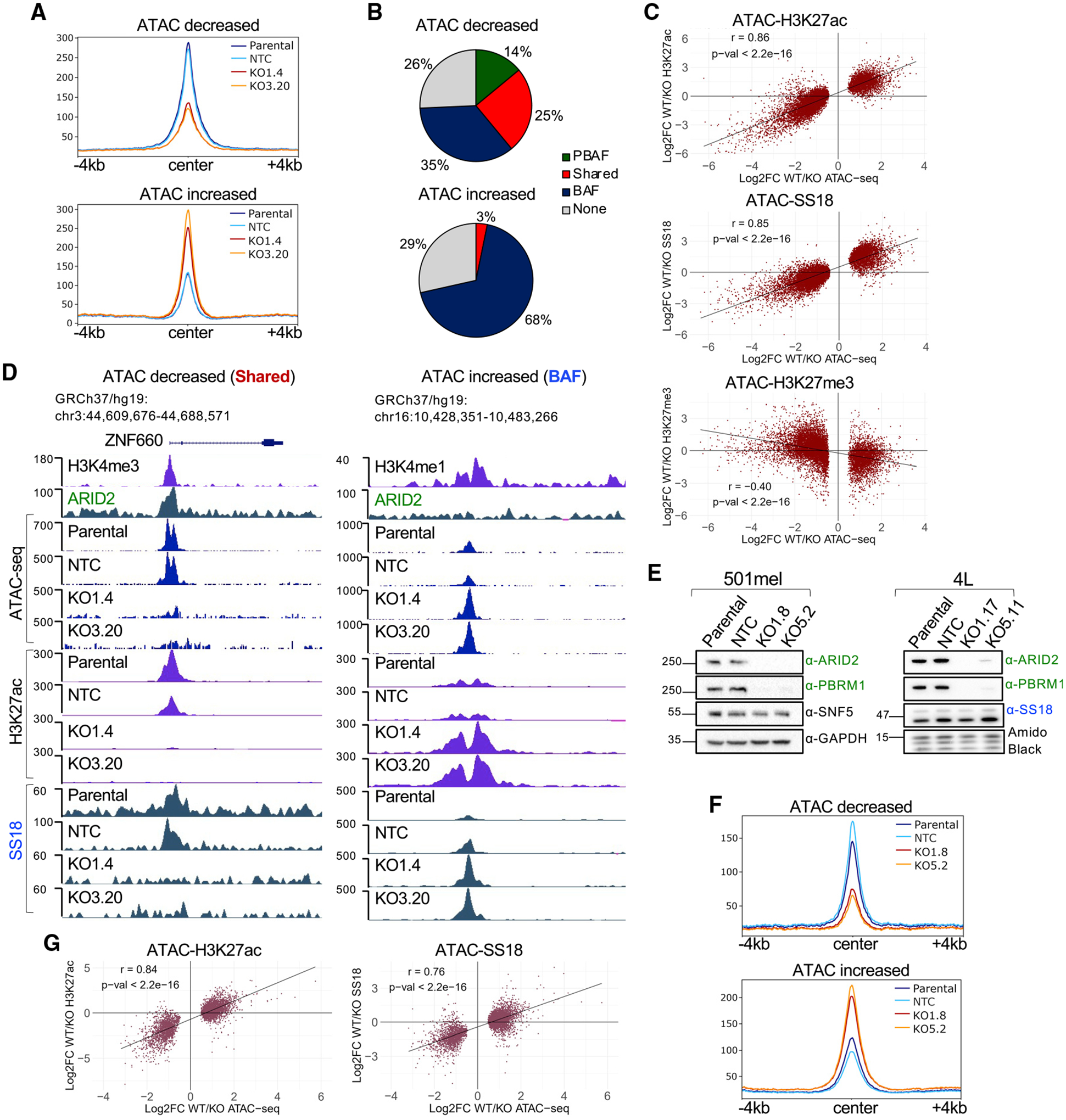
(A) Metagene profiles of ATAC-seq significantly deregulated regions in SKmel147 ARID2 WT (parental and NTC) and KO (KO1.4 and KO3.20) lines. ATAC decreased (n = 6,542) and increased (n = 3,105) regions called with adjusted p < 0.05.
(B) Pie charts displaying the percentage of ATAC decreased and increased regions at PBAF, shared, and BAF sites or None (no SWI/SNF binding).
(C) Scatterplots displaying the log2 fold changes (log2FC) in SKmel147 ARID2 WT versus KO of H3K27ac, SS18, or H3K27me3 (y axis) versus the log2FC of ATAC-seq altered regions (x axis). Pearson correlations are shown.
(D) UCSC genome browser (hg19) snapshots displaying ARID2, H3K4me3/H3K4me1, ATAC, H3K27ac, and SS18 in ARID2 WT and KO SKmel147 lines at ATAC decreased and increased regions.
(E) WCE immunoblots of 501mel ARID2 WT and KO clones probed for ARID2, PBRM1, and SNF5. GAPDH was used as a loading control (n = 2). Chromatin extract immunoblots of 4L ARID2 WT and KO clones probed for ARID2, PBRM1, and SS18. Amido black was used as a loading control (n = 2).
(F) Metagene profiles of 501mel ARID2 WT (parental and NTC) and KO (KO1.8 and KO5.2) lines. ATAC decreased (n = 1,480) and increased (n = 3,762) deregulated regions called with adjusted p < 0.05.
(G) Scatterplots displaying the log2FC in 501mel ARID2 WT versus KO cell lines of H3K27ac or SS18 versus the log2FC of ATAC-seq altered regions. Pearson correlations are shown. See also Figure S3 and Tables S1, S2, S4, and S6.
To probe these changes further, we performed ChIP-seq of SS18 and H3K27ac in ARID2 WT and KO cell lines. We observed that regions with decreased chromatin accessibility displayed concomitant loss of H3K27ac and SS18 (at shared regions), while an increased enrichment of BAF was demonstrated by increased SS18 signal at the ATAC increased regions, along with increased H3K27ac (Figures 3C and S3E–S3G and Table S2). These shifts are further highlighted by the strong positive correlation between ATAC-SS18 (Pearson correlation = 0.86) and ATAC-H3K27ac (Pearson correlation = 0.85) (Figure 3C). Examples of such changes are highlighted in Figure 3D at ATAC decreased and increased regions. Given the reported antagonistic relationship between SWI/SNF and Polycomb repressive complexes (Kia et al., 2008; Kim et al., 2015; Stanton et al., 2017; Wilson et al., 2010), we also performed ChIP-seq of H3K27me3 in ARID2 WT and KO cells and observed a moderate anti-correlation between H3K27me3 and the differential ATAC regions (Pearson correlation = −0.40) (Figure 3C and Table S2). This suggests that PBAF-bound regions may be silenced by different and/or additional mechanisms in the absence of ARID2.
Next, to validate our findings in cell lines with different driver mutations and phenotypic states, we utilized the BRAFV600E-mutant/MITF-high melanoma cell lines 501mel and 4L. In brief, we generated ARID2 KO clones, which, as expected, displayed loss of PBRM1 (Figure 3E), and performed ATAC-seq analysis as described above (Figures S3H and S3I). In 501mel, we identified 1,480 regions with decreased chromatin accessibility and 3,762 regions with increased chromatin accessibility in the ARID2 KO cells (Figure S3J and Table S2). Metagene analysis revealed an ~2-fold change at both ATAC increased and ATAC decreased significant regions (Figure 3F) with concomitant changes of H3K27ac (Pearson correlation = 0.84) and SS18 (Pearson correlation = 0.76) (Figures 3G and S3K–S3N and Table S2), consistent with our results for SKmel147. For reasons that remain unclear, we were unable to ChIP ARID2 in 501mel to further intersect differentially accessible regions with SWI/SNF clusters. Using 4L, however, upon differential ATAC-seq analysis (4L ARID2 WT versus KO) (Figure S3O) and intersection with SWI/SNF clusters (Figure S2F), we confirmed that regions with increased chromatin accessibility occurred primarily at BAF sites, and regions with decreased accessibility occurred mostly at ARID2-bound regions with high ARID2 enrichment at baseline (Figures S3P and S3Q). These data indicate a conserved mechanism of BAF occupancy changes in the absence of a functional PBAF in melanoma cell lines of distinct genetic and phenotypic backgrounds.
Redistribution of SWI/SNF complexes disrupts the melanoma TF network
To investigate TF enrichment at differentially accessible regions in the context of ARID2 KO in SKmel147, we performed de novo motif analysis. The AP-1 motif was the most highly enriched at both ATAC increased and ATAC decreased regions (Figure 4A and Table S4), consistent with this motif being enriched in the shared and BAF clusters (Figure 2E and Table S4). ETS1 and TEAD motifs were highly enriched, and the REST motif less so, at ATAC decreased regions, while the CTCF motif was highly enriched at ATAC increased regions. To investigate whether binding of these TFs was in fact altered at these differentially accessible sites, we performed ChIP-seq of FOSL2, TEAD4, and CTCF in ARID2 WT and KO SKmel147 cells as in Figure 2F. As AP-1/TEAD TFs are master regulators of the melanoma invasive state, and we showed that FOSL2 and TEAD4 regulate the melanoma enhancer landscape (Fontanals-Cirera et al., 2017; Verfaillie et al., 2015), we used these TFs as representative members of their respective families. In addition, FOSL2 represents one of the most highly expressed AP-1 factors in SKmel147 (Figure S4A).
Figure 4. Chromatin accessibility changes in the absence of ARID2 correlate with TF occupancy.
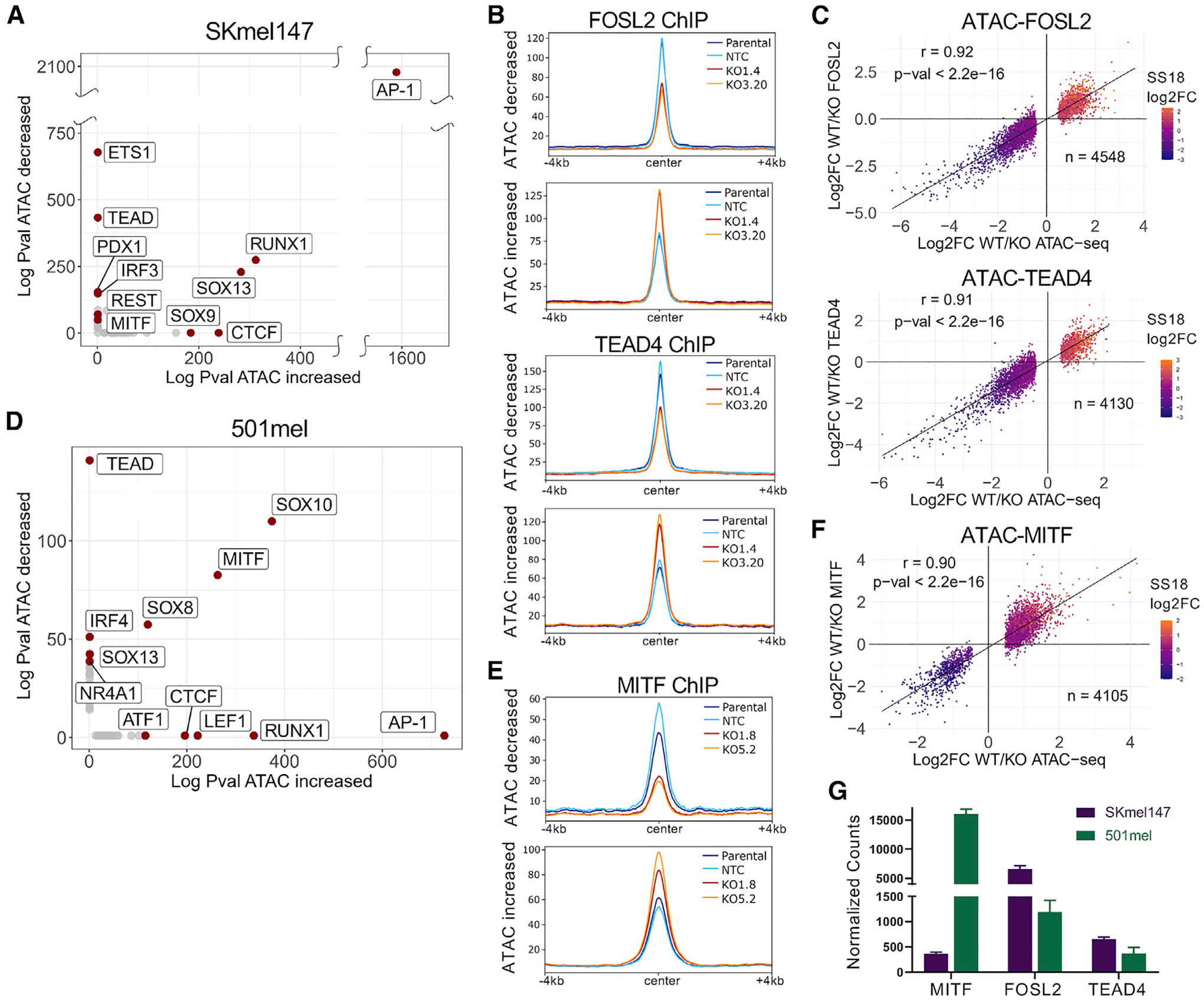
(A) Scatterplot displaying the absolute log p values of the TF motifs (de novo) enriched at SKmel147 ARID2 WT versus KO ATAC-altered regions.
(B) Metagene profiles of SKmel147 ARID2 WT (parental and NTC) and KO (KO1.4 and KO3.20) enrichment of FOSL2 and TEAD4 ChIP-seq centered at ATAC decreased and increased regions that overlap with a TF peak (FOSL2 n = 4,548, TEAD4 n = 4,130).
(C) Scatterplots of SKmel147 ARID2 WT versus KO log2FC of FOSL2 and TEAD4 with ATAC log2FC at ATAC increased and decreased regions that overlap with a TF peak, as in (B). SS18 log2FC was plotted as a color variable and Pearson correlations are shown.
(D) Scatterplot displaying the absolute log p values of TF motifs (de novo) enriched at 501mel ARID2 WT versus KO ATAC-altered regions.
(E) Metagene profiles of 501mel ARID2 WT (parental and NTC) and KO (KO1.8 and KO5.2) enrichment of MITF ChIP-seq intersected with ATAC decreased and increased regions (n = 4,105).
(F) Scatterplot of 501mel ARID2 WT versus KO log2FC of MITF with ATAC log2FC at ATAC increased and decreased regions. SS18 is plotted as in (C).
(G) RNA-sequencing normalized counts of MITF, FOLS2, and TEAD4 in SKmel147 and 501mel. See also Figure S4 and Tables S1, S2, and S6.
By intersecting the TF ChIP-seq with the differential ATAC regions, we found that almost half of the differential ATAC peaks were bound by either FOSL2 or TEAD4, many of which were bound by both (Figures S4B and S4C). Furthermore, we found increased binding of FOSL2 and TEAD4 at ATAC increased regions, and decreased binding of FOSL2 and TEAD4 at ATAC decreased regions (Figure 4B and Table S2). Along with the significant correlation between these changing ATAC-FOSL2 (Pearson correlation = 0.92) and ATAC-TEAD4 (Pearson correlation = 0.91) regions, SS18 enrichment changed in the same direction (Figure 4C). This suggests that the chromatin regions that close lose TF and BAF binding (assessed by SS18 enrichment), while regions that open gain TF and BAF occupancy. Examples are highlighted in Figure S4D. Finally, the intersection between differentially accessible regions and CTCF was not as robust, with many fewer regions affected by ARID2 KO (Figures S4E and S4F). Consistent with previous studies (Barisic et al., 2019), this suggests that CTCF may have limited dependency on SWI/SNF complexes to bind to chromatin.
We also performed motif analysis in 501mel ARID2 WT and KO clones. Unlike SKmel147, an MITF-low cell line, we found that the motifs for the lineage-specific TFs SOX10 and MITF were enriched at both ATAC increased and ATAC decreased regions (Figure 4D and Table S4). MITF ChIP-seq in WT and KO 501mel cells revealed a robust intersection between MITF binding and differentially accessible regions (89% of ATAC increased peaks, 49% of ATAC decreased peaks) (Figure S4G). Similar to FOSL2 and TEAD4 in SKmel147, MITF binding was altered at ATAC differential regions (Figure 4E and Table S2), and we identified a strong correlation between changing ATAC-MITF regions (Pearson correlation = 0.90), along with concordant SS18 differential enrichment (Figure 4F and S4H).
These findings highlight a conserved mechanism of PBAF function across melanoma cells of distinct genetic backgrounds and phenotypic states; however, the TFs affected differ. This appears to correlate with TF expression levels, as in SKmel147, FOSL2 mRNA levels are ~5× higher than in 501mel, and in 501mel, MITF levels are ~15× higher than in SKmel147 (Figure 4G). MITF levels were further validated by immunoblot (Figure S4I). Thus, our findings suggest a dynamic relationship between SWI/SNF complexes and the TFs that utilize these complexes to access chromatin.
Altered transcriptome and TF binding in ARID2-deficient melanoma cells
To identify the biological changes associated with PBAF depletion, we performed RNA sequencing in SKmel147 ARID2 WT (parental and NTC) and ARID2 KO clones (KO1.4, KO1.5 and KO3.20) (Figure S5A). We identified 955 upregulated genes and 1,072 downregulated genes in ARID2 KO samples (Figure S5B and Table S3). Because we identified BAF redistribution in the absence of PBAF, and BAF enrichment at enhancers, we performed Hi-C (chromosome conformation capture assay) in parental SKmel147. This allowed us to impute enhancer-promoter interactions more confidently and to call significant TADs (topologically associated domains) (Table S2). We next integrated the transcriptomic changes with the alterations in chromatin accessibility and TF binding utilizing the significant TADs to delimit the associations. We associated the ATAC increased and decreased regions with positively correlated DEGs (differentially expressed genes) within the same TAD and ± 500 kb of the TSS (transcription start site), and identified 56% of the upregulated genes associated with ATAC increased regions and 80% of the downregulated genes associated with ATAC decreased regions (Table S3). Of the DEGs associated with ATAC-altered regions, ~80% were associated with altered TEAD4 and/or FOSL2 binding (Figure 5A; Table S3). By performing the same analysis in 501mel, we identified 2,457 upregulated genes and 2,355 downregulated genes. As TADs are highly conserved across species and cell types (Dixon et al., 2012; Rao et al., 2014), we utilized the SKmel147 TADs to delineate enhancer-promoter associations in 501mel as described above. We found that chromatin accessibility changes were associated with transcriptional changes (66% of the upregulated genes and 34% of the downregulated genes; Figures S5C–S5E and Table S3). Of the DEGs associated with ATAC-altered regions in 501mel, ~80% were associated with altered MITF binding (Table S3). Together, these data suggest a coordinated control of gene expression via TF access to chromatin through SWI/SNF function.
Figure 5. ARID2 deficiency leads to altered gene expression programs linked to invasion.
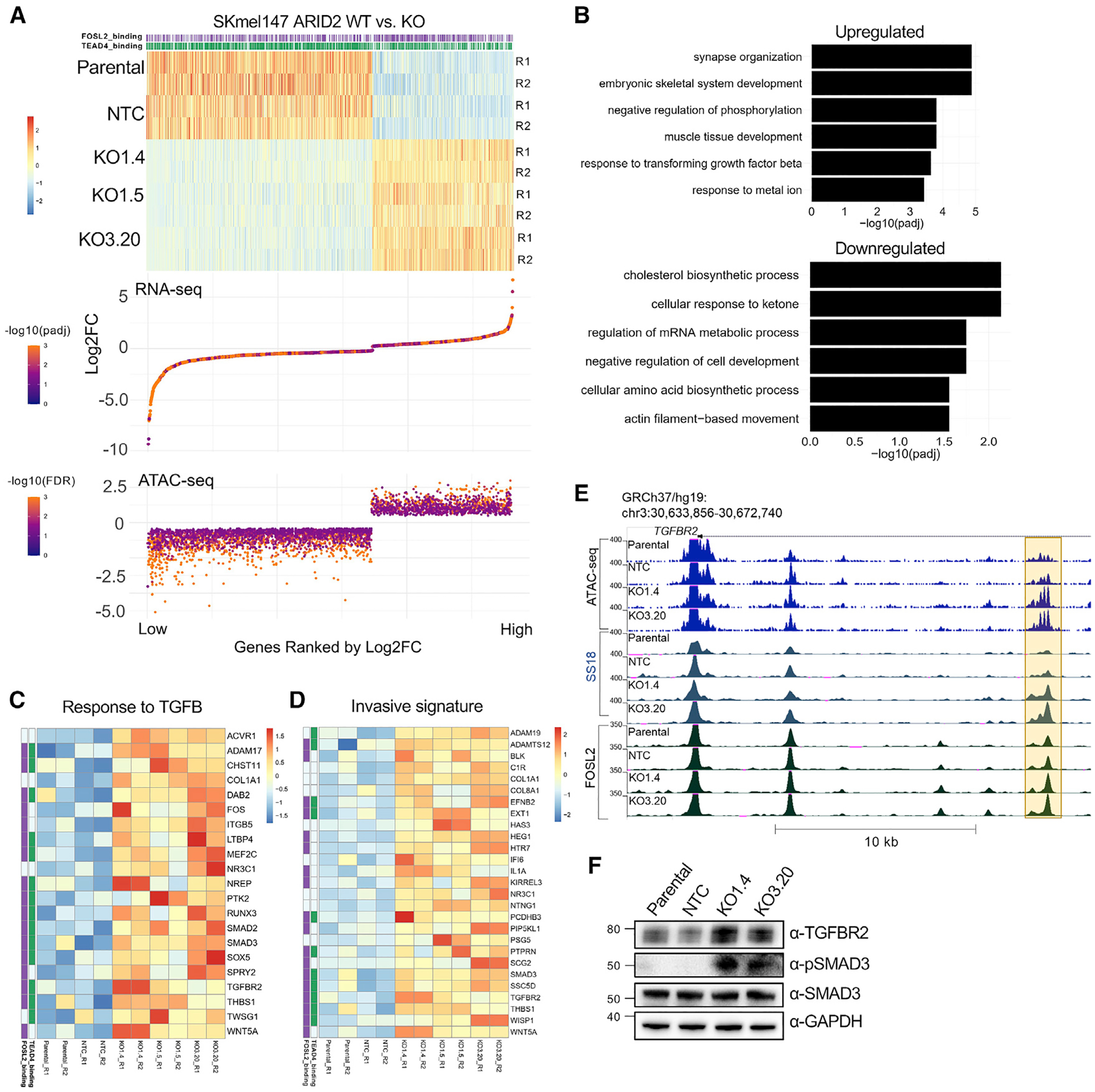
(A) Association of ranked DEGs (heatmap) in SKmel147 ARID2 KO versus WT cells with their associated ATAC increased or decreased peaks (bottom), within a TAD and within ± 500 kb of the TSS. Replicates are shown (R1, R2). TEAD4/FOSL2 status is shown above and called if at least one of the gene-associated ATAC changing peaks is bound by FOSL2 and/or TEAD4.
(B) Biological process gene ontology of genes in (A).
(C) Heatmap of significantly upregulated genes (Z scores) from (B) of the pathway “response to TGFB.” TEAD4/FOSL2 status is shown on the left.
(D) Heatmap of upregulated genes (Z scores) associated with regions with increased chromatin accessibility within a TAD overlapped with the “invasive melanoma signature” from Verfaillie et al. (2015).
(E) UCSC genome browser (hg19) snapshot displaying a putative enhancer (highlighted in yellow) with increased accessibility and SS18/FOSL2 binding in ARID2 KO lines with the promoter of the associated upregulated gene TGFBR2 shown.
(F) WCE immunoblots of SKmel147 ARID2 WT (parental and NTC) and KO (KO1.4 and KO3.20) lines probed for TGFBR2, phospho-SMAD3, and SMAD3. GAPDH was used as a loading control (n = 2). See also Figure S5 and Tables S1, S2, S3, S5, and S6.
ARID2-deficient cells display upregulation of invasive gene signatures
Next, we performed pathway analysis of the DEGs in ARID2 KO versus WT SKmel147 cells that corresponded to differential chromatin accessibility sites within a TAD. Upon GO (gene ontology) analysis, we observed upregulated pathways, including “response to TGFB,” “synapse organization,” and “regulation of protein kinase activity,” among others (Figures 5B and Table S3). Among the downregulated pathways, we found “cholesterol biosynthesis” (Figure S5F), “mRNA metabolic process,” and “cell development” (Figure 5B and Table S3). To extend our results to human melanoma patients, we performed DEG analysis on melanoma samples from the TCGA cohort (n = 334), comparing ARID2 mutant (n = 50) with WT samples (Figure S5G). We found significant intersections between SKmel147 and TCGA deregulated genes (Figure S5H and Table S5). We found pathways related to “synapse” in the upregulated pathways and “metabolic processes” in the downregulated pathways (Figure S5I). Interestingly, in 501mel, the altered pathways were distinct (Figure S5J), highlighting how SWI/SNF complexes regulate different gene expression programs linked to highly expressed TFs that depend on SWI/SNF remodeling activity.
Of interest, we noted key genes involved in melanoma invasion in the upregulated pathway “response to TGFB” in SKmel147 cells (Figure 5C). This includes WNT5A (Weeraratna et al., 2002) and collagen- and integrin-encoding genes (Lu et al., 2012), as well as genes involved in TGFB-induced EMT (epithelial-to-mesenchymal transition), such as TGFBR2, SMAD2, and SMAD3 (Fuxe et al., 2010; Medici et al., 2008), the first and last of which were confirmed by immunoblot (Figure 5F). This led us to intersect the upregulated genes associated with increased accessibility in ARID2 KO cells with published melanoma invasive signatures (Verfaillie et al., 2015). Here we observed overlap with several genes involved in melanoma invasion, which were also associated with altered FOSL2 and TEAD4 binding (Figure 5D), such as the TGFBR2 locus (Figure 5E). TGFBR2 expression is associated with both the TGFB response pathway and the invasive signature (Figures 5B and 5C) and, consistent with our studies, Verfaillie et al. showed that AP-1/TEAD are master regulators of the melanoma invasive gene network (Verfaillie et al., 2015).
Our studies herein modeled ARID2 loss utilizing KO clonal cell lines. As a complementary approach, we also performed shRNA-mediated KD (knockdown) of ARID2 in SKmel147 cells. As expected, we observed loss of PBRM1 and BRD7 upon ARID2 KD (Figure S5K). By examining gene expression changes, we found a significant correlation between the KD and the KO lines (Figure S5L). Importantly, the invasive genes we identified in the KO setting were significantly upregulated upon ARID2 KD (e.g., SMAD3, TGFBR2, and WNT5A) (Figures S5L and S5M).
PBAF loss increases frequency of melanoma metastasis in vivo
Based on the gene expression signatures identified, we examined the phenotypic properties of the ARID2 WT and KO cells using in vitro assays, including proliferation, adhesion, and matrix degradation, but did not identify significant differences between ARID2 WT and KO cells (Figures S6A–S6C). Given the TGFB pathway upregulation in SKmel147 cells, which implicated the potential for invasion and metastasis, we examined the phenotypic differences between ARID2 WT and KO in vivo. We first utilized the chicken CAM (chorioallantoic membrane) model, which allows for examination of primary tumor growth and dissemination into the chicken embryo (Fluegen et al., 2017; Ossowski, 1988) (Figure 6A). While no significant difference in primary tumor growth was observed (Figures 6B and S6D), ARID2 KO cells displayed a striking increase in dissemination to the lower CAM and the liver (Figures 6B, 6D, S6E, and S6F). Next, we employed a tail vein injection model of experimental metastasis. To this end, we quantitated foci of fluorescently labeled cells in the lung to assess metastatic burden in both SKmel147 (MITF low) and 4L (MITF high) at 3 and 5 weeks post-tail-vein injection, respectively. We observed that ARID2 KO significantly increased the number of metastatic colonies compared with ARID2 WT cells as detected by both fluorescence and H&E quantification (Figures 6D–6F and S6G–S6I). While we noted macrometastases in both ARID2 WT and KO conditions, we observed many more micrometastases in the ARID2 KO setting (Figures S6J and S6K). Taken together, this phenotype is consistent with the altered gene expression program identified in ARID2 KO cells, namely, the mesenchymal-like invasive signature, and suggests that chromatin reorganization following ARID2 loss favors melanoma metastasis.
Figure 6. ARID2 depletion leads to melanoma metastasis in vivo.
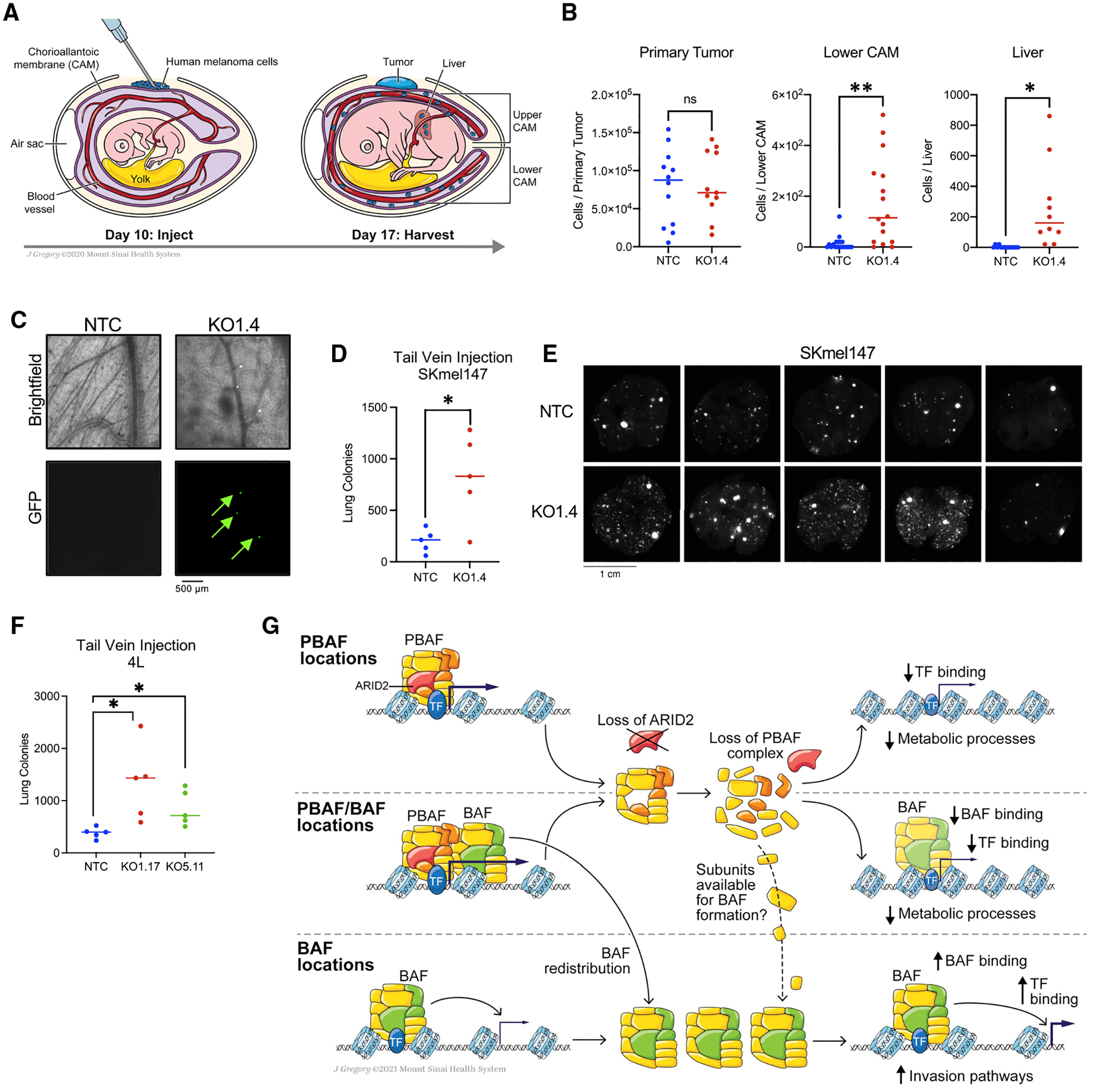
(A) Schematic of the chicken CAM xenograft assay and regions of the chicken embryo where cells disseminate to, including the lower CAM and the liver.
(B) FACS (fluorescence-activated cell sorting) quantification of SKmel147 ARID2 WT (NTC) and KO (KO1.4) cells of the primary tumor (n = 12 and n = 11, respectively), lower CAM (n = 19 and n = 16, respectively), and liver (n = 14 and n = 10, respectively) of the CAM xenografts 7 days post-injection. Nineteen independent replicates are aggregated, line represents the median. Welch’s t test. Primary tumor, p = 0.8580; lower CAM, p = 0.0019; liver, p = 0.0156. *p < 0.05; **p < 0.01; ns, p > 0.05.
(C) Representative images of SKmel147 ARID2 WT (NTC) and KO (KO1.4) GFP+ cells that disseminated to the lower CAM.
(D) Quantification of SKmel147 ARID2 WT (NTC, n = 5) and KO (KO1.4, n = 5) metastatic colonies in the lung 3 weeks post-tail-vein injection detected by fluorescence imaging. Line represents the median. Welch’s t test. p = 0.029. *p < 0.05.
(E) Fluorescence images of lungs from mice with SKmel147 NTC and KO1.4 cells injected into the lateral tail vein.
(F) Quantification of 4L ARID2 WT (NTC, n = 5) and KO (KO1.17, n = 5; KO5.11, n = 5) metastatic colonies in the lung 5 weeks post-tail-vein injection detected by fluorescence imaging. Line represents the median. Welch’s t test. NTC versus KO1.17, p = 0.0421; NTC versus KO.511, p = 0.0334. *p < 0.05.
(G) Model of the mechanisms by which ARID2 loss regulates chromatin dynamics across the three types of SWI/SNF-bound regions, and gene expression pathways affected by PBAF loss. See also Figure S6.
DISCUSSION
Our results shed light onto the mechanisms by which ARID2 loss, and subsequent PBAF depletion, leads to changes in chromatin structure and transcriptional output in melanoma. We found that the PBAF complex predominantly maintains open chromatin at PBAF and PBAF-BAF shared regions, which consequently lose chromatin accessibility upon ARID2 depletion. The majority of chromatin accessibility losses occurred at the promoters of active genes, while gain of chromatin accessibility was almost exclusively found at a subset of BAF-enriched enhancers. Taken together, our biochemical and genomic data implicate both a modest increase in available core subunits upon PBAF loss into the BAF complex and the genomic redistribution of BAF complexes accompanied by increased chromatin accessibility and TF binding (Figure 6G).
Importantly, the mechanistic findings presented here are conserved between melanoma cells lines. The major difference was revealed when we probed the TF motifs of the differentially accessible chromatin regions. We identified that the deregulated transcriptional targets upon PBAF loss were influenced by the dominant TFs in each cell line, resulting in distinct effects on transcriptional programs. Therefore, the transcriptional consequences of ARID2 mutation and/or loss in melanoma tumors might be dictated by the stage of melanoma progression or the state of the cell in which the mutation occurs (e.g., proliferative versus invasive). Nevertheless, we observed increased metastatic potential in cell lines with high and low MITF levels, as well as distinct driver mutations, indicating conservation of the phenotype. Moreover, as melanoma comprises transcriptionally distinct, heterogeneous cell populations (Tsoi et al., 2018), we envision future studies utilizing single-cell methodologies to better understand the nuanced effects of ARID2 loss within subpopulations of cells in human melanoma tumors.
We also observed that TF binding depends on SWI/SNF remodeling activity and the dynamics between the BAF and the PBAF complexes. This is consistent with studies demonstrating that depletion or inhibition of chromatin remodeling subunits plays a critical a role in the genomic occupancy of TFs (Barisic et al., 2019; Iurlaro et al., 2021). On the other hand, it has been shown that pioneer TFs, in cooperation with AP-1 TFs, can recruit SWI/SNF complexes to poised regions to promote accessibility (Vierbuchen et al., 2017). Therefore, such chromatin remodeler-TF interactions appear to be context dependent and might vary between different chromatin remodeling complexes and types of TFs. Moreover, our study does not exclude the possibility that redistribution of TFs upon ARID2 depletion may in fact be recruiting BAF complexes to sites that gain accessibility.
Upon integration of chromatin changes with transcriptional deregulation upon ARID2 depletion, we found that many of the transcriptional changes positively correlated with changes in chromatin structure within a TAD. Moreover, because PBAF is a chromatin remodeling complex, we focused primarily on the gene expression changes that were associated with changes in chromatin accessibility. However, by examining all gene expression changes independent of chromatin structural changes (Table S3), we found additional altered pathways, including the upregulation of antigen processing and presentation genes. This is consistent with previous reports demonstrating that depletion of PBAF members from mouse B16 melanoma tumors correlated with increased sensitivity to immune checkpoint inhibition (Fukumoto et al., 2020; Pan et al., 2018).
Finally, we revealed an unappreciated role for PBAF loss in melanoma metastasis. ARID2 mutations have been detected in early melanoma lesions (Shain et al., 2018), which we speculate may promote early dissemination, a phenomenon that has been clinically observed in melanoma patients (Werner-Klein et al., 2018). Intriguingly, recent studies of human melanoma have reported an enrichment of SWI/SNF mutations, including ARID2 mutations in metastasis, particularly brain metastasis (In et al., 2020; Váraljai et al., 2021). While this remains to be explored, we found prominent enrichment of upregulated synaptic pathways in both melanoma cells and the TCGA, raising the possibility that ARID2 loss might prime melanoma cells for metastasis or adaptation to the brain. Importantly, activation of synaptic activity in cancer cells has been shown to promote cell migration and survival in brain niches (Venkataramani et al., 2019; Venkatesh et al., 2019; Zeng et al., 2019). We look forward to future studies that investigate the role of the PBAF complex, including PBRM1 and BRD7, which are predicted to be melanoma drivers (Alkallas et al., 2020; Martínez-Jiménez et al., 2020), in such critical melanoma processes in order to better tailor treatments for melanoma patients.
Limitations of the study
By creating isogenic ARID2 KO melanoma models, we investigated the direct molecular and cellular consequences of ARID2 loss; however, we recognize that not all ARID2 mutations are complete loss-of-function mutations. Some ARID2 mutations may have distinct consequences on PBAF assembly, possibly leading to the formation of residual aberrant complexes. In addition, while we validated TGFB pathway upregulation in ARID2 KO cells in vitro, activation of this pathway in vivo and its role in metastasis were not explored. We anticipate that studies investigating the transcriptional and signaling changes upon ARID2 depletion during melanoma progression in vivo will be highly informative.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Emily Bernstein (emily.bernstein@mssm.edu).
Materials availability
Reagents used in this study are publicly available or available from the lead contact upon request.
Data and code availability
Raw data from RNA-seq, ATAC-seq, Hi-C and ChIP-seq experiments have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Processed data from the RNA-seq, ATAC-seq, Hi-C and ChIP-seq experiments are available in the Supplemental Tables.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ARID2 | Santa Cruz | Cat# sc-166117; RRID: AB_2060382 |
| ARID2 | Cell Signaling | Cat# 82342; RRID:AB_2799992 |
| PBRM1 | In house | N/A |
| BRD7 | Santa Cruz | Cat# sc-376180; RRID:AB_10989389 |
| SS18 | Cell Signaling | Cat# 21792; RRID:AB_2728667 |
| SNF5 | Santa Cruz | Cat# sc-166165; RRID:AB_2270651 |
| SNF5 | Bethyl | Cat# A301-087A; RRID:AB_2191714 |
| BRG1 | Abcam | Cat# ab110641; RRID:AB_10861578 |
| BRG1 | Santa Cruz | Cat# sc-17796; RRID:AB_626762 |
| ARID1A | Bethyl | Cat# A301-041A; RRID:AB_2060365 |
| ARID1A | Santa Cruz | Cat# sc-32761; RRID:AB_673396 |
| GAPDH | Santa Cruz | Cat# sc-32233; RRID:AB_627679 |
| H3K27ac | Abcam | Cat# ab4729; RRID:AB_2118291 |
| H3K27me3 | Epicypher | Cat# 13-0030; RRID: N/A |
| H3K9me3 | Abcam | Cat# ab176916; RRID:AB_2797591 |
| FOSL2 | Santa Cruz | Cat# sc-604; RRID:AB_2107084 |
| TEAD4 | Santa Cruz | Cat# sc-101184; RRID:AB_2203086 |
| MITF | Sigma A. | Cat# HPA003259; RRID:AB_1079381 |
| MITF | Active Motif | Cat# 91201; RRID:AB_2793801 |
| CTCF | Millipore | Cat# 07-729; RRID:AB_441965 |
| REST | Millipore | Cat# 07-579; RRID:AB_11211936 |
| PHF10 | ThermoFisher | Cat# PA5-30678; RRID:AB_2548152 |
| BRM | Santa Cruz | Cat# sc-166579; RRID:AB_2302397 |
| GLTSCR1 | Santa Cruz | Cat# sc-515086; RRID: N/A |
| CHD5 | Abcam | Cat# ab124736; RRID:AB_10974578 |
| TGBR2 | Abcam | Cat# ab184948; RRID:AB_2818975 |
| Phospho-SMAD3 | Cell Signaling | Cat# 9520; RRID:AB_2193207 |
| SMAD3 | Cell Signaling | Cat# 9523; RRID:AB_2193182 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| DSG | Pierce (ThermoFisher) | Cat# 20593 |
| Protein A + G magnetic beads | Millipore | Cat# LSKMAGAG10 |
| Dynabeads Protein A magnetic beads | Invitrogen | Cat# 10001D |
| KAPA HiFi DNA Polymerase | Roche | Cat# 07958838001 |
| Digitonin | Promega | Cat# G9441 |
| Illumina Tagment DNA Enzyme and Buffer Small Kit | Illumina | Cat# 20034197 |
| AMPure XP beads | NEB | Cat# A63880 |
| T4 PNK | NEB | Cat# M0201S |
| 5U/uL Klenow Fragment (3′–5′ exo-) | NEB | Cat# M0212S |
| Quick T4 DNA ligase | NEB | Cat# M2200S |
| T4 DNA Polymerase | NEB | Cat# M0203S |
| DNA Polymerase Klenow Fragments | NEB | Cat# M0210S |
| dNTPs | Roche | Cat# 11969064001 |
| dATPs | Roche | Cat# 11889516103 |
| Critical commercial assays | ||
| Transit-VirusGEN Transfection Kit | Mirus Bio | Cat# MIR6700 |
| NEXTFLEX Poly(A) Enrichment Beads | PerkinElmer | Cat# NOVA-512992 |
| NEXTFLEX Rapid Directional RNA-seq Kit 2.0 | PerkinElmer | Cat# N0VA-5198-01 |
| RNeasy extraction kit | Qiagen | Cat# 74104 |
| Arima Hi-C Kit | Arima | Cat# A410030 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28106 |
| Kapa HyperPrep Kit | Kapa Biosystems | Cat# KK8500 |
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28704 |
| Deposited data | ||
| Raw RNA sequencing, ATAC-sequencing, ChIP-sequencing, and Hi-C data | GEO | GSE172383 |
| Analyzed RNA sequencing, ATAC-sequencing, ChIP-sequencing, and Hi-C data, See Tables S2, S3, S6 | This paper | N/A |
| The Cancer Genome Atlas Skin Cutaneous Melanoma | Akbani et al. (2015) | https://gdc.cancer.gov/about-data/publications/skcm_2015 |
| Experimental models: Cell lines | ||
| SKmel147 | Eva Hernando Laboratory (NYU) | RRID:CVCL_3876 |
| 501mel | Eva Hernando Laboratory (NYU) | RRID:CVCL_4633 |
| 113/6-4L | Robert Kerbel Laboratory (Sunnybrook Health Sciences Center) | Cruz-Munoz et al., 2008 |
| WM1552c | Rockland | WM1552C-01-0001, RRID: CVCL_6472 |
| WM1799 | Rockland | WM1799-01-0005, RRID: CVCL_A341 |
| WM3456 | Rockland | WM3456-01-0001, RRID: CVCL_AP91 |
| Mel888 | Stuart Aaronson Laboratory (ISMMS) | RRID: CVCL_4632 |
| WM1716 | Rockland | WM1716-01-0001, RRID: CVCL_AP82 |
| LOX-IMVI | Jessie Villanueva Laboratory (Wistar Institute) | RRID: CVCL_1381 |
| WM3533 | Rockland | RRID: CVCL_0B73 |
| Experimental models: Organisms/strains | ||
| Premium specific pathogen-free (SPF) embryonated chicken eggs | Charles River Laboratories | Cat# 10100326 |
| Mouse: female, 6 to 8 weeks old, N0D/SCID/IL2γR−/− | Jackson Laboratories | Cat# 005557 |
| Oligonucleotides | ||
| Non-targeting sgRNA: CACCGTTCCGCGTTACATAACTTAG | This paper | N/A |
| ARID2.1 sgRNA: CACCGAATGGCAAACTCGACGGGGA | This paper | N/A |
| ARID2.3 sgRNA: CACCGATGATGATGAGGTACCACC | This paper | N/A |
| ARID2.5 sgRNA: CACCGTTTACTACTTGCTAATGCCG | This paper | N/A |
| ARID2 shRNA: CGTACCTGTCTTCGTTTCCTA | Sigma-Aldrich | TRCN0000166264 |
| Recombinant DNA | ||
| lentiCas9-Blast | Addgene | Cat# 52962 |
| pLKO.1 eGFP/mCherry | Brian Brown Laboratory (ISMMS) | N/A |
| pLenti-puro | Addgene | Cat# 39481 |
| pLK0.1 eGFP NTsg | This paper | N/A |
| pLKO.1 mCherry ARID2 sg1 | This paper | N/A |
| pLKO.1 mCherry ARID2 sg3 | This paper | N/A |
| lentiCRISPR v2 | Addgene | Cat# 52961 |
| FG12-GFP | Bravo-Cordero Laboratory (ISMMS) | N/A |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| pLKO.1 shARID2 | Sigma-Aldrich | TRCN0000166264 |
| pLL-CMV-rFLuc-T2A-GFP | System Biosciences | Cat# LL300PA-1 |
| Software and algorithms | ||
| Incucyte Life-Cell Imaging System | Essen Bioscience | https://www.sartorius.com/en/products/live-cell-imaging-analysis/live-cell-analysis-software/incucyte-base-software |
| FCS Express | De Novo Software | https://denovosoftware.com/ |
| STAR | Version 2.6.0c, Dobin, 2013 | https://github.com/alexdobin/STAR |
| Rsubread | Version 2.4.3, Liao et al., 2014 | http://subread.sourceforge.net |
| DEseq2 | Version 1.30.1, Love et al. (2014) | https://github.com/mikelove/DESeq2 |
| Bowtie | Version 1.1.12, Langmead et al. (2009) | https://github.com/BenLangmead/bowtie |
| MACS2 | Version 2.1.0, Zhang et al. (2008) | https://pypi.org/project/MACS2/ |
| fastQC | Version 0.11.7, Andrews, 2019 | https://github.com/s-andrews/FastQC |
| PICARD | Version 2.2.4 | https://github.com/broadinstitute/picard/releases |
| Samtools | Version 1.9, Li et al. (2009) | http://www.htslib.org/ |
| SICER | Version 2.0, Zang et al. (2009) | https://zanglab.github.io/SICER2/ |
| deepTools | Version 3.2.1, Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| Bowtie2 | Version 2.1.0, Langmead et al., 2009 | https://github.com/BenLangmead/bowtie2 |
| Topdom | Version 0.0.1, Shin et al., 2015 | https://github.com/HenrikBengtsson/TopDom |
| Hi-C bench | Lazaris et al. (2017) | https://github.com/NYU-BFX/hic-bench |
| BedTools | Version 2.29.2, Quinlan and Hall (2010) | https://bedtools.readthedocs.io/ |
| ChIPSeeker | Version 1.26.2, Yu et al. (2015) | https://github.com/YuLab-SMU/ChIPseeker |
| HOMER | Version 4.11, Heinz et al. (2010) | https://github.com/IGBIllinois/HOMER |
| clusterProfiler | Version 3.18.1, Yu et al., 2012 | https://github.com/YuLab-SMU/clusterProfiler |
| Prism | Version 9 | https://www.graphpad.com |
| ImageJ | Version 2.1.0/1.53c | https://imagej.nih.gov/ij/index.html |
| RStudio | Version 1.1.46 | https://rstudio.com |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell lines
Melanoma cell lines SKmel147, 501mel, 113/6-4L, Mel888 and LOX-IMVI were cultured in DMEM supplemented with 10% FBS, 100 IU of penicillin and 100 μg/mL of streptomycin. Melanoma cell lines WM1552c, WM1799, WM3456, WM1716 and WM3533 were cultured in Tumor 2% media (80% MCDB 153 media, 20% Leibovitz’s L-15 media, 2% FBS, 5 μg/mL bovine insulin, 1.68 mM CaCl2, and 100 IU of penicillin and 100 μg/mL of streptomycin). HEK293T cells were used for virus production and were maintained in DMEM supplemented with 10% FBS, 100 IU of penicillin and 100 μg/mL of streptomycin.
Chicken chorioallantoic membrane (CAM)
For the CAM xenograft assay, specific pathogen-free, fertile, 10-day-incubated embryonated chicken eggs were supplied by Charles River Laboratories.
Mice
For tail vein injection assays, 6–8-week-old NOD/SCID/IL2yR−/− female mice were supplied by Jackson Labs (Cat #005557).
METHOD DETAILS
Chromatin fractionation
Chromatin fractionation was performed as described (Méndez and Stillman, 2000). Protein lysates were resuspended in buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M Sucrose, 10% glycerol,100 nM PMSF, supplemented with protease inhibitors) and protein concentration was determined using Bicinchoninic Acid (BCA) (Pierce) following manufacturer’s guidelines. Samples were mixed with 2× Laemmli loading buffer and boiled prior to immunoblotting.
Whole cell protein extraction
Cells were washed with PBS and lysed on ice for 10 minutes in Nonidet-40 (NP-40) lysis buffer (50 mM Tris pH 7.5, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 10% glycerol) supplemented with protease and phosphatase inhibitors (Roche). Lysates were centrifuged at 20,000 g for 15 minutes and the protein concentration was quantified using BCA (Pierce). Lysates were mixed with 2× Laemmli loading buffer with subsequent boiling prior to immunoblotting.
Immunoprecipitation (IP) of nuclear extracts
Nuclear lysates were prepared as describe (Dignam et al., 1983). Buffers were supplemented with protease and phosphatase inhibitors (Roche). 40–50 million cells were swollen with hypotonic buffer (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2), nuclei were passed through a 27G needle 6x for homogenization, followed by centrifugation, and extraction for 30 min at 4°C with high salt buffer (20 mM HEPES, pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 0.1% NP-40). Extracts were pelleted and the supernatant was used for IP. Before IP, nuclear extract supernatants were diluted with 1.8 volumes of hypotonic buffer with 0.1% NP-40 to reach a final NaCl concentration of 150 mM. Diluted extracts were incubated with antibodies overnight, followed by a 2-h incubation with 50 μL Pierce Protein A/G magnetic beads (ThermoFisher), subsequent washing and elution.
Glycerol gradient sedimentation
Glycerol gradient sedimentations were performed as described (Alpsoy and Dykhuizen, 2018). Briefly, cells (40–50 million) were collected, lysed in buffer A (20 mM HEPES pH 7.9, 25 mM KCl, 10% glycerol, 5 mM MgCl2, 0.05 mM EDTA, 0.1% NP-40, 100 nM PMSF, supplemented with protease inhibitors) and washed once with buffer A without NP-40. Next, the nuclei were resuspended in buffer C (10 mM HEPES pH 7.6, 3 mM MgCl2, 100 mM KCl, 0.1 mM EDTA, 10% glycerol). Ammonium sulfate powder was added to (300 mg/mL), and the supernatant was incubated on ice for 20 min. Proteins were precipitated by ultracentrifugation at 150,000 g for 30 min. The pelleted protein was resuspended with 100 μL of HEG1000 buffer (25 mM HEPES pH 7.6, 0.1 mM EDTA, 12.5 mM MgCl2, 100 mM KCl, supplemented with protease inhibitors). A glycerol gradient (10–30%) was prepared using HEG1000 buffer without glycerol and with 30% glycerol. Following, the protein lysates were layered on top of the 10–30% glycerol gradients (10 mL) and fractionated by centrifugation at 40,000 rpm for 16 h using the SW32Ti rotor (Beckman Coulter). Fractions (20x) of 500 μL were collected sequentially from the top of the gradient and used for immunoblotting after dilution with 4× Laemmli buffer with subsequent boiling.
Immunoblotting
Protein lysates (10–20 ug of total protein) were run on a 4–12% Bis-Tris gradient gels (Life Technologies). After wet transfer onto a PVDF membrane for 2 h at 110 volts, membranes were incubated with blocking buffer (5% milk in Tris-Buffered Saline, 0.1% Tween (TBS-T)) followed by an overnight incubation with the respective primary antibodies (Table S1). Next, membranes were incubated with the respective secondary antibodies, washed with TBS-T, and developed.
Plasmids, lentiviral vector production, and cell transfection
The lentiCas9-Blast plasmid (Addgene, Cat# 52962) was used to generate SKmel147 cells stably expressing Cas9. The sgRNAs were cloned into a pLKO.1 lentiviral vector (kind gift from Brown Laboratory, ISMMS), using the restriction enzyme BbsI downstream of the U6 promoter which also contained eGFP or mCherry downstream of the human PGK promoter. Viral particles were generated as previously described (Fontanals-Cirera et al., 2017). SKmel147 was first infected with the lentiCas9-Blast plasmid lentiviral particles and selected with blasticidin (10 μg/mL for 8 days) and then infected with Plenti-GFP/mCherry ARID2 sgRNAs (Table S1). To generate ARID2 KO 501mel and 4L clonal cell lines, the ARID2 sgRNAs (Table S1) were cloned into the Cas9 containing lentiCRISPR V2 vector following the established protocol (Shalem et al., 2014). Upon infection, cells were selected with puromycin (2 μg/mL for 2 days). To track SKmel147 ARID2 WT (NTC) and ARID2 KO (KO1.4) cells in the CAM model and in the tail vein assay, the lentiviral FG12-GFP vector (kind gift from the Bravo-Cordero laboratory, ISMMS) was used for transduction. To track 4L ARID2 WT (NTC) and ARID2 KO (KO1.17 and KO5.11) cells in the tail vein assay, the lentiviral pLL-CMV-rFLuc-T2A-GFP vector (System Biosciences) was used for transduction. To produce lentiviral particles, HEK293T cells were seeded in 10 cm tissue culture dishes and incubated at 37°C and 5% CO2. When cells reached 80% confluency, they were co-transfected with 5 μg of lentiviral expression constructs, 3.75 μg of psPAX2 and 1.25 μg pMD2.G vectors using the Transit-VirusGen (Mirus) transfection reagent following manufacturer’s recommendations. At 48 h post-transfection, supernatants were collected and passed through a 0.45 μm PVDF filter to remove cells and debris.
Single cell clonal expansion
For clonal isolation, serial dilutions of cells were grown in 10 cm plates to seed cells at a very low density. Individual colonies were isolated by pipetting, and further expanded for approximately three weeks prior to protein extraction for immunoblotting screening. ARID2 KO clones are named and identified by two numbers: the sgRNA used followed by the clone number (e.g., KO3.20).
Cell proliferation
Equal number of cells (SKmel147: 1 × 104 cells; 501mel: 2 × 104; 4L: 6 × 104) were plated into 12- or 24-well plates in triplicate. Cells were imaged on brightfield, and confluence of each well was quantified using the Incucyte Life-Cell Imaging System every 24 h for five to six days.
Adhesion assays
96-well plates were coated with the respective matrix (fibronectin or collagen) and incubated 1 h at 37°C. 2 × 105 cells were plated per well and allowed to adhere for 2.5 h. Cells were washed and fixed, followed by crystal violet staining. To quantify, crystal violet was solubilized with 2% SDS and absorbance of the staining was measured at 550nm.
Matrix degradation assay
Assay was performed as described by (Di Martino et al., 2017). Briefly, coverslips were coated with Alexa Flour 488-gelatin. 3.5 × 104 cells per coverslip were plated on the gelatin-coated coverslips for 16 h before fixation. Cells were fixed and stained with DAPI and Alexa Flour 647-phalloidin to visualize the nucleus and cytoplasmic area, respectively. Gelatin degradation was analyzed by quantifying the average area of degraded gel (pixels) per field divided by the number of nuclei in those fields. 10–15 fields were randomly selected per condition and quantified.
CAM xenografts
Cells were washed and harvested with 2 mM EDTA-PBS and resuspended at a concentration of 16 million cells/ml in PBS without calcium and magnesium. To prepare the chicken embryos for inoculation, a small opening in the shell was created followed by the removal of air from the eggs to allow detachment of the developing CAM. 8 × 105 human melanoma cells were inoculated on top of the CAM in a volume of 50 μL. The opening was sealed with transparent adhesive tape and the eggs were incubated for seven days. After incubation, chicken embryos were sacrificed, the primary tumor, the lower CAM and the liver were collected for further processing. All tissues were washed and minced on ice upon collection. Tissue was incubated with collagenase and DNase for 30–35 min at 37°C, followed by neutralization of collagenase with DMEM (10% FBS). Samples were passed through 70 μm cell strainers, washed with FACS buffer (PBS, 2 mM EDTA, 0.5% FBS), and resuspended with FACS buffer in a final volume of 0.5–2 mL. Samples were acquired on an Attune NxT flow cytometer (Thermo Scientific) and analyzed using FCS Express (De Novo Software). Debris and doublets were eliminated based on scatter properties and disseminated tumor cells were identified by GFP and mCherry fluorescence read in FITC and PE-Texas Red channels, respectively. 10% of the primary tumor and lower CAM cell suspension and 5% of the liver cell suspension were analyzed by volume-based acquisition, and results were extrapolated to the entire tissue.
Tail vein injections
The experiments were conducted following protocols approved by ISMMS Institutional Animal Care and Use Committee (IACUC) (protocol number LA11-00122). 6–8-week-old NOD/SCID/IL2yR−/− female mice (Jackson Labs, Cat# 005557) were used. Cells were harvested with 2mM EDTA-PBS, resuspended in 100 μL HBSS (SKmel147: 1.25 × 105 cells; 4L: 5 × 104 cells), and injected into one of the lateral tail veins with a 27-gauge needle. At the experimental endpoint (SKmel147: 3 weeks; 4L: 5 weeks), mice were euthanized by cervical dislocation, lungs were cleared of blood by right ventricle PBS perfusion, and imaged ex vivo for GFP and mCherry fluorescence on a Cytation 7 instrument (BioTek). Quantification of lung colonies was performed using Gen 5 software (BioTek). Harvested lungs were fixed in neutral buffered formalin for 24 h, paraffin embedded and sectioned at 5 μM thickness. Sections were obtained from three levels spaced 100 μM, stained with H&E and acquired at 20× magnification on a NanoZoomer S210 slide scanner (Hamamatsu). Metastatic colonies were identified morphologically and counted using NDP.view 2 software (Hamamatsu). Lung area on each section was calculated by NDP.view 2 software by outlining the edges of the tissue. Slides were blinded to the reviewer (C.N.) prior to analysis.
RNA-sequencing
Total RNA was extracted from cell lines using the RNAeasy Mini Kit (Qiagen) according to manufacturer’s protocol, and libraries were prepared from 1–2 μg of total RNA. Poly-A RNA was isolated using NETFLEX Poly(A) beads, and sequencing libraries were prepared using the NETFLEX Rapid Directional RNA-seq Kit 2.0 following manufacturer’s protocols (PerkinElmer). Single-end 75-bp reads were sequenced on the NextSeq550 according to the manufacturer’s guidelines (Illumina).
ChIP-sequencing
ChIP samples were processed as previously described (Fontanals-Cirera et al., 2017), with several modifications. For histone modification ChIP (H3K27ac and H3K27me3), ~10–15 million cells per sample were cross-linked with 1% Formaldehyde for 10 min at room temperature. For chromatin remodeler subunits and transcription factors (ARID2, SS18, BRG1, FOSL2, TEAD4, MITF, CTCF, and REST) ~10–40 million cells per sample were double cross-linked with 0.25 M disuccinimidyl glutarate (DSG) for 45 min, followed with 1% formaldehyde for 10 min. Single and double cross-linked cells were quenched with 0.125 M glycine for 5 min at room temperature, followed by pelleting at 400 g for 3 min at 4°C. Cells were washed once with ice-cold PBS and resuspended at ~10–20 million cells in 500 μL of cell lysis buffer (10 mM Tris pH 8, 10 mM NaCl, 0.2% NP-40, 100 nM PMSF, supplemented with protease inhibitors), followed by 15 min incubation on ice. Next, cells were centrifuged at 400 g for 5 min and resuspended in 500 μL cold nuclear lysis buffer (50 mM Tris pH 8, 10 mM EDTA, 1% SDS, 100 nM PMSF, supplemented with protease inhibitors) and incubated on ice for 10 min. Cells were sonicated for 15–20 cycles, 30 s on, 30 s off, at low intensity in a Bioruptor sonicator (Diagenode). After sonication, samples were centrifuged at 13,000 g for 10 min at 4°C, and the supernatant-containing chromatin was diluted 1:4 with IP Dilution buffer (20 mM Tris pH 8, 2 mM EDTA, 150 mM NaCl, 1% Triton X-, 0.01% SDS, 100 nM PMSF, supplemented with protease inhibitors). Drosophila spike-in chromatin was added to the diluted chromatin followed by pre-clearing with protein A (Invitrogen) or A + G (Millipore) magnetic beads pre-conjugated with rabbit or mouse IgG respectively, for 2 h at 4°C. After pre-clearing, 50 μL of chromatin were saved for input control. Pre-conjugated antibodies (50–100μL slurry and 5–10μg antibody per ~10–20 million cells for H3K27ac, H3K27me3, ARID2, BRG1, SS18, TEAD4, FOSL2, MITF, CTCF, REST), were added to the pre-cleared chromatin and rotated overnight at 4°C. Following overnight incubation, beads were washed once with cold IP Wash I buffer (20 mM Tris pH 8, 2 mM EDTA, 50 mM NaCl, 1% Triton X-, 0.1% SDS, 100 nM PMSF, supplemented with protease inhibitors), twice with cold High Salt buffer (20 mM Tris pH 8, 2 mM EDTA, 500 mM NaCl, 1% Triton X-, 0.01% SDS, 100 nM PMSF, supplemented with protease inhibitors), once with cold IP Wash II buffer (10 mM Tris pH 8, 1 mM EDTA, 0.25 LiCl, 1% NP-40, 1% deoxycholic acid, 100 nM PMSF supplemented with protease inhibitors) and twice with cold TE buffer (5 mM Tris pH 7.4, 1 mM EDTA). DNA was eluted twice in 100 μL elution buffer (1% SDS, 100 mM NaHCO3) at 65°C for 30 min and shaking. For input, 130 μL TE buffer, 12 μL 5 M NaCl, 20 μL 10% SDS and 2 μL of RNase A (10 mg/mL) were added followed by overnight incubation at 65°C. For the ChIP fractions, 12 μL of 5 M NaCl, 2 μL of RNase A (10 mg/mL) were added, followed by overnight incubation at 65°C. Following, 4 μL of Proteinase K (20 mg/mL) were added and samples were incubated for 2 h at 42°C. DNA was purified using QIAquick PCR Purification Kit (Qiagen) following manufacturer’s protocol. Libraries were generated and barcoded for multiplexed sequencing according to Illumina’s recommendations. Briefly, 1–8 ng of DNA was end-repaired and Poly-A tailed with deoxyadenosine. Truseq adaptors (Illumina) were ligated, libraries were size-selected on a 2% agarose gel (350–650bp), and amplified using KAPA HiFi DNA Polymerase (12–15 cycles). NEB enzymes were used to carry out all the steps of the library preparation. Libraries were diluted to 5–10ng/μL and run on an Agilent 2100 Bioanalyzer High Sensitivity DNA chip. Libraries were sequenced on Illumina’s NextSeq550 at 75-bp single- end reads.
ATAC-sequencing
To generate ATAC libraries (Corces et al., 2017), 5 × 104 cells were harvested, treated with digitonin and tagmented with ~2.5 μL Tn5 Transposase for 30 min. DNA was amplified up to 10 cycles and purified using MinElute PCR purification columns (Qiagen). Following, libraries were size-selected on a 2% agarose gel (150–700bp), followed by purification using AMPure XP beads (NEB). Libraries were diluted to 5–10 ng/μL and run on an Agilent 2100 Bioanalyzer High Sensitivity DNA chip. Libraries were sequenced on Illumina NextSeq550 at 40 bp paired-end reads.
Hi-C
SKmel147 cells were grown in exponential growth phase in duplicates, washed twice with PBS, and 5–10 × 106 cells were fixed in 4% paraformaldehyde/1x PBS for 15 min, quenched by the addition 0.125 M glycine final concentration for 5 min at room temperature. Cells were then washed with PBS, harvested and dried pellets were snap-frozen and stored at −80°C until use. Thereafter, in situ Hi-C was performed using Arima Hi-C kit (#A410030) according to the manufacturers’ recommendations. DNA input quantities ranged from approx. 0.9 – 3μg per replicate. Both proximity ligated DNA Hi-C samples passed QC1 checkpoint with a biotinylation efficiency >36% (high quality Arima-QC1 values are expected to be >15%). Next, output proximity ligated samples where mechanically fragmented by Covaris Ultrasonicator LE220, size selected to 200–600 bp (average fragment size of 400 bp) using AMPure XP Beads and monitored for correct size range by Agilent TapeStation 2200 analysis. After final biotin enrichment, processed Hi-C samples were converted to sequencing libraries with Kapa HyperPrep Kit (Kapa Biosystems #KK8500) and Illumina TruSeq sequencing adapters according to the manufacturers’ guidelines with 7 cycles for the final PCR amplification step (yielding Arima QC2 values of 0.45% and 1.94%; >0.2% are expected as high quality samples) and sequenced with Illumina NovaSeq 6000 using 50 cycles paired- end mode.
TCGA SKCM oncoplots
Mutational calls from the TCGA Skin Cutaneous melanoma (SKCM) cohort (primary and metastatic samples) were downloaded from the Broad Institute GDAC portal (http://gdac.broadinstitute.org/). The R package maftools 2.6.05 (Mayakonda et al., 2018) was used to generate oncoplots.
RNA-sequencing
Single-end 75-bp reads were aligned to the human reference genome (hg19/GRCh37.p13) with STAR v2.6.0.c (Dobin, 2013) using the parameters –sjdbOverhang 100 –outFilterMultimapNmax 10 –outFilterMismatchNmax 10 –outFilterType BySJout –outFilterIntronMotifs RemoveNoncanonicalUnannotated (Table S6). Following, featureCounts from the Rsubread 2.4.3 R package (Liao et al., 2014) was used to assign reads to coding genes. Assigned reads were then normalized and DEGs were identified using the R package DEseq2 1.30.1 (Love et al., 2014). Genes were considered expressed if the sum of raw counts across all samples was >200 for any given gene. Differentially expressed genes were called using an adjusted p value < 0.05 (using the Benjamini and Hochberg procedure). Principal component analysis (PCA) was generated using regularized log-transformed reads with the DEseq2 package. Volcano plots were generated with the ggplot2 3.3.3 package using DEseq2 results statistics. Heatmaps were generated with the pheatmap 1.0.12 package, using DEseq2 median-ratio normalized counts.
ChIP-seq alignment and peak sets
For each individual sample, reads were aligned to the human reference genome (GRCh37/hg19) using Bowtie v1.1.2 (Langmead et al., 2009) with parameters –l 65 –n 2 –best –k 1 –m 1, and reads quality was assessed using fastQC (Simon, 2019). Alignment statistics for each sample are provided in Table S6. Duplicate reads were removed with PICARD v2.2.4 (Broad Institute Picard Toolkit, nd). Binary alignment map (BAM) files were generated with samtools v1.9 (Li et al., 2009), and were used in downstream analysis. For ChIP-seq used for differential binding analysis, including H3K27ac, SS18, H3K27me3, TEAD4, FOSL2, CTCF and MITF, the bam files of 2 WT and 2 KO samples (For SKmel147: Parental and NTC, and KO1.4 and KO3.20, and for 501mel: Parental and NTC, and KO1.8 and KO5.2) were concatenated using samtools merge to generate ‘master’ bam files. Significant peaks were called on ‘master’ bam files and matching input controls using MACS2 v2.1.0 (Zhang et al., 2008) for narrow peaks, and SICER 2.0 (Zang et al., 2009) for broad peaks. Significance q-value cut-offs were determined post-hoc, testing several q-values based on signal to background ratio (for SKmel147 ChIP-seqs: ARID2, BRG1 q-value < 1e-3, SS18 q-value < 1e-7, FOSL2 and TEAD4 q-value 1e-13, CTCF q-value < 1e-5, H3K27ac q-value < 1e-7, and H3K27me3 q-value < 1e-2. For 501mel ChIP-seqs: SS18 q-value < 1e-2, H3K27ac and MITF q-value < 1e-3). Peaks in ENCODE blacklisted regions were removed. Quantification of reads in significant peak for all samples was performed using BedTools multicov 2.29.2 (Quinlan and Hall, 2010). Differential peak analysis was performed using DEseq2 1.30.1 (adjusted p value < 0.05 (using the Benjamini and Hochberg procedure)). Coverage tracks were generated from BAM files for the master bam files and the individual replicates and conditions using deepTools 3.2.1 (Ramírez et al., 2016) bamCoverage with parameters –normalizeUsingRPKM –binsize 10 and normalized with DEseq2 scaling factors. PCA plots were generated using DEseq2. ChIP-seq enrichment plots were visualized on the UCSC genome browser.
ATAC-seq alignment and peak sets
For each individual sample, paired-end 40-bp reads were aligned to the human reference genome (GRCh37/hg19) using Bowtie2 2.1.0 with parameters –q –X 2000 (Table S6), and read quality was assessed using fastQC. Duplicate reads were removed with PICARD v2.2.4, and BAM files were generated with samtools v1.9 (removing mitochondrial alignments and only keeping reads with MAPQ ≥30) for downstream analysis. WT and KO replicates for each cell line were then concatenated using samtools merge. MACS2 was used to identify significant peaks (master bam files) using parameters –nomodel –nolambada –keepdup all –slocal 10000. Quantification of reads in significant peak for all samples was performed using BedTools multicov 2.29.2 (Quinlan and Hall, 2010). Differential peak analysis was performed using DEseq2 1.30.1 (adjusted p value < 0.05 (using the Benjamini and Hochberg procedure)). Coverage tracks (Bigwig files) were generated from BAM files for the master bam files and the individual replicates and conditions using deepTools 3.2.1 (Ramírez et al., 2016) bamCoverage with parameters –normalizeUsingRPKM –binsize 10 and normalized with DEseq2 scaling factors. PCA plots were generated using DEseq2. ATAC-seq enrichment plots were visualized on the UCSC genome browser.
ATAC-seq correlation matrices
DeepTools 3.2.1. was used to generate the correlation plots at ATAC deregulated regions. First, multiBigwigsummary files were generated to average scores for each sample at ATAC deregulated regions. Next, the plotCorrelation command was used with multiBigwigsummary files to calculate the spearman correlation between the samples and generate the plots.
ATAC-gene associations
Differentially enriched distal/intragenic ATAC regions were associated to the positively correlated promoters of differentially expressed genes within a genomic range of ± 500 kb. Next the significant TADs from two TAD callers (Topdom and Hi-C bench) (Lazaris et al., 2017; Shin et al., 2015) were merged, and only those within the same TAD were kept.
ATAC-seq correlation scatterplots
Log2fold changes generated with DEseq2 over the regions of interest were used to generate the scatterplots. Correlation values were calculated with the R 4.0.3 function cor.test using Pearson’s correlation test. Plots were generated with ggplot2 3.3.3.
Upset intersection plots
Bed files of FOSL2/TEAD4 significant peaks and ATAC deregulated regions were used to generated Upset plots. Common intersected regions (FOSL2/TEAD4/ATAC) are displayed. BedTools 2.29.2 was used to generate intersections and Upset plots were generated with the R package UpSetR 1.4.0 (Conway et al., 2017).
Genomic annotation analysis
Promoters +2 kb relative to the TSS were defined according to the human GRCh37/hg19 Gencode v19 genome annotation. The CHIPSeeker 1.26.2 package (Yu et al., 2015) was modified and used to determined feature distribution for peak sets.
TF motif analysis
Transcription Factor (TF) motif enrichments analysis of the different SWI/SNF clusters was performed on the intersection of all bound regions within a cluster (i.e., PBAF, shared and BAF clusters), and ATAC-seq summits. De novo motifs were identified within a +200bp window around ATAC peak summit using the HOMER v.4.11 suite (Heinz et al., 2010) with parameters; findMotifsGenome.pl hg19 -size 200 with the default Homer generated background regions. Only significant motifs (p value < 1e-12) with corresponding expressed TFs were reported for each cell line. For differentially enriched ARID2 WT/KO ATAC regions, de novo motifs were identified within a +200bp window around the ATAC peak summit with same parameters as above. Differential motif enrichment scatterplot was generated with ggplot2.
Metagenes and heatmaps
Metagene and heatmaps of genomic regions were generated with deepTools 3.2.1. The command computeMatrix was used to calculate scores at genomic regions and generate a matrix file to use with plotHeamap or plotProfile, to generate heatmaps or metagene profile plots, respectively. Bigwigs used to generate matrix files were first scaled with normalization values produced by DEseq2 1.30.1.
Gene ontology
Gene ontology (GO) enrichment (Ashburner et al., 2000) was performed with the R package clusterProfiler 3.18.1 (Yu et al., 2012) using the GO over representation test function. Significant pathways were called with an adjusted p value < 0.05 (using the Benjamini and Hochberg procedure).
Hi-C
Hi-C data from SKMel147 was analyzed using the HiC-bench platform. Data was aligned to the human reference genome (GRCh37/hg19) with bowtie2 2.1.0. Aligned reads were filtered with the GenomicTools 0.2.9.7 (Tsirigos et al., 2012) tools-hic filter command and only the accepted intra-chromosomal read-pairs were used for downstream analysis (~200 million reads). Interaction matrices were generated using the HiC-bench 0.1 (Akdemir and Chin, 2015; Imakaev et al., 2012; Lazaris et al., 2017) platform at 40kb resolution. Filtered read counts were normalized using iterative correction and eigenvector decomposition (ICE). Distance normalization was performed to account for variances in read counts for more distant loci. Topologically associating domains (TADs) were called using HiC-bench’s hicratio algorithm as well as topdom 0.10.1 (Shin et al., 2015).
TCGA data analysis
From the Broad Institute GDAC portal (http://gdac.broadinstitute.org/), the SKCM log2 normalized RNA-seq reads, clinical annotations, mutational calls, and copy number annotation datasets were downloaded. When annotating ARID2 mutant samples, mutations of unknow significance and synonymous mutations were removed from the analysis. Using R 4.0.3, a linear model with the covariates: sex, age, tumor type (primary vs. metastatic), copy number variation, and ARID2 mutation status, was used to call significant differences between ARID2 mutant samples and ARID2 WT samples. Given the high biological heterogeneity within patient samples, and the relatively low number of ARID2 mutant samples (n = 50), we utilized a cut-off of p value < 0.05 to call significant changes.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests were performed using RStudio and GraphPad Prism 9 (GraphPad Software). The statistical analysis details for each experiment can be found in figure legends and in the method details section.
Data deposition
The GEO accession number for the raw and processed sequencing data reported in this paper is GSE172383.
Supplementary Material
Highlights.
ARID2 loss results in impaired PBAF complex assembly and BAF genomic redistribution
Altered SWI/SNF dynamics results in chromatin accessibility and TF binding changes
PBAF loss drives an invasive gene expression signature and phenotype in melanoma
ACKNOWLEDGMENTS
The authors thank the laboratories of David Fisher, Brian Brown, Javier Bravo-Cordero, Eva Hernando, Robert Kerbel, and Poulikos Poulikakos for reagents and advice; Ana Hahn, Kent Madrid, and Kevin Mohammed for NGS support; Daniel Munguia for experimental assistance; the Bernstein lab for discussions and feedback; Jill Gregory for illustrations; NYU Genome Technology Center (GTC), and both the Applied Bioinformatics Laboratories (ABL) at NYU and the Bioinformatics for Next Generation Sequencing (BiNGS) core at the Icahn School of Medicine at Mount Sinai (ISMMS) for bioinformatic support. This study was supported by NIH/NCI F30 CA253988-02 to C.B.N.; Philippe Foundation to T.C.M.; American-Italian Cancer Foundation and National Cancer Center to E.G.; American Skin Association to D.F.; NIH/NCI P01CA229086, R01CA242020, and R01CA228135 to I.A.; NIH/NCI P01CA229086 and NIH/NCI R01CA252239 to A.T.; NIH/NCI U01CA207532 to E.C.D.; NCI P30 CA196521 to R.P.; NIH/NCI CA109182, CA216248, CA218024, and CA196521 to J.A.A.-G.; and NIH/NCI R01CA154683 and CA218024 to E.B. J.A.A.-G. is a Samuel Waxman Cancer Research Foundation Investigator. This work was supported in part through the Oncological Sciences Sequencing Core supported by Tisch Cancer Institute of the ISMMS Cancer Center, support grant P30CA196521; Scientific Computing supported by the Office of Research Infrastructure of the NIH under award no. S10OD026880 to ISMMS; and the ISMMS Genomics Technology Facility, NYU GTC and ABL and shared resources partially supported by the Cancer Center support grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. This work utilized computing resources at the NYU High Performance Computing Facility.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110637.
DECLARATION OF INTERESTS
J.A.A.-G. is a scientific co-founder, scientific advisory board member, and equity owner of HiberCell and receives financial compensation as a consultant.
REFERENCES
- Andrews S FastQC, 2019. GitHub. https://github.com/s-andrews/FastQC [Google Scholar]
- Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, et al. (2015). Genomic classification of cutaneous melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akdemir KC, and Chin L (2015). HiCPlotter integrates genomic data with interaction matrices. Genome Biol. 16, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkallas R, Lajoie M, Moldoveanu D, Hoang KV, Lefrançois P, Lingrand M, Ahanfeshar-Adams M, Watters K, Spatz A, Zippin JH, et al. (2020). Multi-omic analysis reveals significantly mutated genes and DDX3X as a sex-specific tumor suppressor in cutaneous melanoma. Nat. Cancer. 16, 635–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpsoy A, and Dykhuizen EC (2018). Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem 293, 3892–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins MB, Curiel-Lewandrowski C, Fisher DE, Swetter SM, Tsao H, Aguirre-Ghiso JA, Soengas MS, Weeraratna AT, Flaherty KT, Herlyn M, et al. (2021). The state of melanoma: emergent challenges and opportunities. Clin. Cancer Res 27, 2678–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barisic D, Stadler MB, Iurlaro M, and Schübeler D (2019). Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 569, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegel JA, Fogelgren B, Wainwright LM, Zhou JY, Bevan H, and Rorke LB (2000). Germline INII mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosom. Cancer 28, 31–37. [DOI] [PubMed] [Google Scholar]
- Broad Institute Picard Toolkit. Broad Institute Picard Toolkit. http://broadinstitute.github.io/picard/.
- Bruno M, Flaus A, Stockdale C, Rencurel C, Ferreira H, and Owen-Hughes T (2003). Histone H2A/H2B dimer exchange by ATP-dependent chromatin remodeling activities. Mol. Cell 12, 1599–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Lex A, and Gehlenborg N (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Dietlein F, Taylor-Weiner A, AlDubayan S, Vokes N, Keenan T, Reardon B, He MX, Margolis CA, Weirather JL, et al. (2020). Integrated molecular drivers coordinate biological and clinical states in melanoma. Nat. Genet 52, 1373–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Munoz W, Man S, Xu P, and Kerbel RS (2008). Development of a preclinical model of spontaneous human melanoma central nervous system metastasis. Cancer Res 68, 4500–4505. [DOI] [PubMed] [Google Scholar]
- Dechassa ML, Sabri A, Pondugula S, Kassabov SR, Chatterjee N, Kladde MP, and Bartholomew B (2010). SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol. Cell 38, 590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, and Roeder RG (1983). Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A Spliced Transcripts Alignment to a Reference, 2013. GitHub; https://www.ncbi.nlm.nih.gov/pubmed/23104886 [Google Scholar]
- Dummer R, Ascierto PA, Nathan P, Robert C, and Schadendorf D (2020). Rationale for immune checkpoint inhibitors plus targeted therapy in metastatic melanoma: a review. JAMA Oncol. 6, 1957–1966. [DOI] [PubMed] [Google Scholar]
- Erkek S, Johann PD, Finetti MA, Drosos Y, Chou H-C, Zapatka M, Sturm D, Jones DTW, Korshunov A, Rhyzova M, et al. (2018). Comprehensive analysis of chromatin states in atypical teratoid/rhabdoid tumor identifies diverging roles for SWI/SNF and polycomb in gene regulation. Cancer Cell 35, 95–110.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, Calvo V, Cheung JF, Bravo-Cordero JJ, Entenberg D, et al. (2017). Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol 19, 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanals-Cirera B, Hasson D, Vardabasso C, Di Micco R, Agrawal P, Chowdhury A, Gantz M, de Pablos-Aragoneses A, Morgenstern A, Wu P, et al. (2017). Harnessing BET inhibitor sensitivity reveals AMIGO2 as a melanoma survival gene. Mol. Cell 68, 731–744.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto T, Lin J, Fatkhutdinov N, Liu P, Somasundaram R, Herlyn M, Zhang R, and Nishigori C (2020). ARID2 deficiency correlates with the response to immune checkpoint blockade in melanoma. J. Invest. Dermatol 141, 1564–1572.e4. [DOI] [PubMed] [Google Scholar]
- Fuxe J, Vincent T, and De Herreros AG (2010). Transcriptional crosstalk between TGFβ and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle 9, 2363–2374. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchalian J, Malik S, Ho J, Lee D-S, Kelso TWR, Shokhirev MN, Dixon JR, and Hargreaves DC (2018). A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun 9, 5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi AA, Attalla K, DiNatale RG, Ostrovnaya I, Flynn J, Blum KA, Ged Y, Hoen D, Kendall SM, Reznik E, et al. (2020). A pan-cancer analysis of PBAF complex mutations and their association with immunotherapy response. Nat. Commun 11, 4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanniford D, Ulloa-Morales A, Karz A, Berzoti-Coelho MG, Moubarak RS, Sánchez-Sendra B, Kloetgen A, Davalos V, Imig J, Wu P, et al. (2020). Epigenetic silencing of CDR1as drives IGF2BP3-mediated melanoma invasion and metastasis. Cancer Cell 37, 55–70.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, and Crabtree GR (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21, 396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges C, Kirkland JG, and Crabtree GR (2016). The many roles of BAF (mSWI/SNF) and PBAF complexes in cancer. Cold Spring Harb. Perspect. Med 6, a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In GK, Poorman K, Saul M, O’Day S, Farma JM, Olszanski AJ, Gordon MS, Thomas JS, Eisenberg B, Flaherty L, et al. (2020). Molecular profiling of melanoma brain metastases compared to primary cutaneous melanoma and to extracranial metastases. Oncotarget 11, 3118–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iurlaro M, Stadler MB, Masoni F, Jagani Z, Galli GG, and Schübeler D (2021). Mammalian SWI/SNF continuously restores local accessibility to chromatin. Nat. Genet 53, 279–287. [DOI] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci. Adv 1, e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, and Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet 45, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, LeRoy G, Vidal CI, et al. (2010). The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Weng QY, Insco ML, Chen KY, Muralidhar S, Pozniak J, Diaz JMS, Drier Y, Nguyen N, Lo JA, et al. (2020). Gain-of-function genetic alterations of g9a drive oncogenesis. Cancer Discov 10, 980–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavari PA, Peterson CL, Tamkun JW, Mendel DB, and Crabtree GR (1993). BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature 366, 170–174. [DOI] [PubMed] [Google Scholar]
- Kia SK, Gorski MM, Giannakopoulos S, and Verrijzer CP (2008). SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol 28, 3457–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, et al. (2015). SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med 21, 1491–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaris C, Kelly S, Ntziachristos P, Aifantis I, and Tsirigos A (2017). HiC-bench: comprehensive and reproducible Hi-C data analysis designed for parameter exploration and benchmarking. BMC Genomics 18, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Weaver VM, and Werb Z (2012). The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol 196, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, Mularoni L, Pich O, Bonet J, Kranas H, et al. (2020). A compendium of mutational cancer driver genes. Nat. Rev. Cancer 20, 555–572. [DOI] [PubMed] [Google Scholar]
- Di Martino J, Henriet E, Ezzoukhry Z, Mondal C, Bravo-Cordero JJ, Moreau V, and Saltel F (2017). 2D and 3D matrices to study linear invadosome formation and activity. J. Vis. Exp 124, e54911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, Zullow HJ, McKenzie ZM, Kubiak RL, St. Pierre R, et al. (2018). Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 175, 1272–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayakonda A, Lin DC, Assenov Y, Plass C, and Koeffler HP (2018). Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28, 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D, Hay ED, and Olsen BR (2008). Snail and slug promote epithelial-mesenchymal transition through β-catenin-T-cell factor-4-dependent expression of transforming growth factor-β3. Mol. Biol. Cell 19, 4875–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez J, and Stillman B (2000). Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol 20, 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel BC, D’Avino AR, Cassel SH, Mashtalir N, McKenzie ZM, McBride MJ, Valencia AM, Zhou Q, Bocker M, Soares LMM, et al. (2018). A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol 20, 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, Ku WL, Teng M, Cui K, Williams RT, et al. (2017). SMARCB1 is required for widespread BAF complex–mediated activation of enhancers and bivalent promoters. Nat. Genet 49, 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski L (1988). Plasminogen activator dependent pathways inthe dissemination of human tumor cells in the chick embryo. Cell 52, 321–328. [DOI] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, De Andrade LF, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, McKenzie ZM, D’Avino AR, Mashtalir N, Lareau CA, St. Pierre R, Wang L, Shilatifard A, and Kadoch C (2019). The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity–independent genomic targeting. Nat. Genet 51, 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsialou A, Wilsker D, and Moran E (2005). DNA-binding properties of ARID family proteins. Nucleic Acids Res 33, 66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Marine JC, and Goding CR (2019). Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev 33, 1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CWM, Galusha SA, McMenamin ME, Fletcher CDM, and Orkin SH (2000). Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. U S A 97, 13796–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Wittmeyer J, and Cairns BR (2006). Chromatin remodelling: the industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol 7, 437–447. [DOI] [PubMed] [Google Scholar]
- Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. (2019). Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet 51, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick S, Rendeiro AF, Runggatscher K, Ringler A, Boidol B, Hinkel M, Májek P, Vulliard L, Penz T, Parapatics K, et al. (2019). Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat. Genet 51, 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, and Pollack JR (2013). The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One 8, e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, Ruben B, North J, Pincus L, Yeh I, et al. (2018). Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell 34, 45–55.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Shi Y, Dai C, Tjong H, Gong K, Alber F, and Zhou XJ (2015). TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res 44, e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A (2019). Babraham Bioinformatics - FastQC A Quality Control Tool for High Throughput Sequence Data.
- Stanton BZ, Hodges C, Calarco JP, Braun SMG, Ku WL, Kadoch C, Zhao K, and Crabtree GR (2017). Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet 49, 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub T, Ghiraldini FG, Carcamo S, Li M, Wroblewska A, Singh R, Goldberg MS, Hasson D, Wang Z, Gallagher SJ, et al. (2018). SIRT6 haploinsufficiency induces BRAFV600E melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat. Commun 9, 3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirigos A, Haiminen N, Bilal E, and Utro F (2012). GenomicTools: a computational platform for developing high-throughput analytics in genomics. Bioinformatics 28, 282–283. [DOI] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 33, 890–904.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Váraljai R, Horn S, Sucker A, Piercianek D, Schmitt V, Carpinteiro A, Becker KA, Reifenberger J, Roesch A, Felsberg J, et al. (2021). Integrative genomic analyses of patient-matched intracranial and extracranial metastases reveal a novel brain-specific landscape of genetic variants in driver genes of malignant melanoma. Cancers (Basel). 13, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardabasso C, Gaspar-Maia A, Hasson D, Pünzeler S, Valle-Garcia D, Straub T, Keilhauer EC, Strub T, Dong J, Panda T, et al. (2015). Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol. Cell 59, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al. (2011). Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, Körber C, Kardorff M, Ratliff M, Xie R, et al. (2019). Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538. [DOI] [PubMed] [Google Scholar]
- Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, Tam LT, Espenel C, Ponnuswami A, Ni L, et al. (2019). Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van Den Mooter L, et al. (2015). Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun 6, 6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, and Delattre O (1998). Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394, 203–206. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell 68, 1067–1082.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, and Crabtree GR (1996). Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev 10, 2117–2130. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang S, Troisi EC, Howard TP, Haswell JR, Wolf BK, Hawk WH, Ramos P, Oberlick EM, Tzvetkov EP, et al. (2019). BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, and Trent JM (2002). Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 1, 279–288. [DOI] [PubMed] [Google Scholar]
- Wei Z, Yoshihara E, He N, Hah N, Fan W, Pinto AFM, Huddy T, Wang Y, Ross B, Estepa G, et al. (2018). Vitamin D switches BAF complexes to protect β cells. Cell 173, 1135–1149.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner-Klein M, Scheitler S, Hoffmann M, Hodak I, Dietz K, Lehnert P, Naimer V, Polzer B, Treitschke S, Werno C, et al. (2018). Genetic alterations driving metastatic colony formation are acquired outside of the primary tumour in melanoma. Nat. Commun 9, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer DS, Cheng PF, Eichhoff OM, Belloni BC, Zipser MC, Schlegel NC, Javelaud D, Mauviel A, Dummer R, and Hoek KS (2012). Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment Cell Melanoma Res 25, 343–353. [DOI] [PubMed] [Google Scholar]
- Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, and Roberts CWM (2010). Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, Parsons R, Zhao K, and Wang W (2005). PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev 19, 1662–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). ClusterProfiler: an R package for comparing biological themes among gene clusters. Omi. A. J. Integr. Biol 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIP seeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, and Peng W (2009). A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, McCabe BD, Galván JA, Robinson HPC, Zlobec I, et al. (2019). Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data from RNA-seq, ATAC-seq, Hi-C and ChIP-seq experiments have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Processed data from the RNA-seq, ATAC-seq, Hi-C and ChIP-seq experiments are available in the Supplemental Tables.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ARID2 | Santa Cruz | Cat# sc-166117; RRID: AB_2060382 |
| ARID2 | Cell Signaling | Cat# 82342; RRID:AB_2799992 |
| PBRM1 | In house | N/A |
| BRD7 | Santa Cruz | Cat# sc-376180; RRID:AB_10989389 |
| SS18 | Cell Signaling | Cat# 21792; RRID:AB_2728667 |
| SNF5 | Santa Cruz | Cat# sc-166165; RRID:AB_2270651 |
| SNF5 | Bethyl | Cat# A301-087A; RRID:AB_2191714 |
| BRG1 | Abcam | Cat# ab110641; RRID:AB_10861578 |
| BRG1 | Santa Cruz | Cat# sc-17796; RRID:AB_626762 |
| ARID1A | Bethyl | Cat# A301-041A; RRID:AB_2060365 |
| ARID1A | Santa Cruz | Cat# sc-32761; RRID:AB_673396 |
| GAPDH | Santa Cruz | Cat# sc-32233; RRID:AB_627679 |
| H3K27ac | Abcam | Cat# ab4729; RRID:AB_2118291 |
| H3K27me3 | Epicypher | Cat# 13-0030; RRID: N/A |
| H3K9me3 | Abcam | Cat# ab176916; RRID:AB_2797591 |
| FOSL2 | Santa Cruz | Cat# sc-604; RRID:AB_2107084 |
| TEAD4 | Santa Cruz | Cat# sc-101184; RRID:AB_2203086 |
| MITF | Sigma A. | Cat# HPA003259; RRID:AB_1079381 |
| MITF | Active Motif | Cat# 91201; RRID:AB_2793801 |
| CTCF | Millipore | Cat# 07-729; RRID:AB_441965 |
| REST | Millipore | Cat# 07-579; RRID:AB_11211936 |
| PHF10 | ThermoFisher | Cat# PA5-30678; RRID:AB_2548152 |
| BRM | Santa Cruz | Cat# sc-166579; RRID:AB_2302397 |
| GLTSCR1 | Santa Cruz | Cat# sc-515086; RRID: N/A |
| CHD5 | Abcam | Cat# ab124736; RRID:AB_10974578 |
| TGBR2 | Abcam | Cat# ab184948; RRID:AB_2818975 |
| Phospho-SMAD3 | Cell Signaling | Cat# 9520; RRID:AB_2193207 |
| SMAD3 | Cell Signaling | Cat# 9523; RRID:AB_2193182 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| DSG | Pierce (ThermoFisher) | Cat# 20593 |
| Protein A + G magnetic beads | Millipore | Cat# LSKMAGAG10 |
| Dynabeads Protein A magnetic beads | Invitrogen | Cat# 10001D |
| KAPA HiFi DNA Polymerase | Roche | Cat# 07958838001 |
| Digitonin | Promega | Cat# G9441 |
| Illumina Tagment DNA Enzyme and Buffer Small Kit | Illumina | Cat# 20034197 |
| AMPure XP beads | NEB | Cat# A63880 |
| T4 PNK | NEB | Cat# M0201S |
| 5U/uL Klenow Fragment (3′–5′ exo-) | NEB | Cat# M0212S |
| Quick T4 DNA ligase | NEB | Cat# M2200S |
| T4 DNA Polymerase | NEB | Cat# M0203S |
| DNA Polymerase Klenow Fragments | NEB | Cat# M0210S |
| dNTPs | Roche | Cat# 11969064001 |
| dATPs | Roche | Cat# 11889516103 |
| Critical commercial assays | ||
| Transit-VirusGEN Transfection Kit | Mirus Bio | Cat# MIR6700 |
| NEXTFLEX Poly(A) Enrichment Beads | PerkinElmer | Cat# NOVA-512992 |
| NEXTFLEX Rapid Directional RNA-seq Kit 2.0 | PerkinElmer | Cat# N0VA-5198-01 |
| RNeasy extraction kit | Qiagen | Cat# 74104 |
| Arima Hi-C Kit | Arima | Cat# A410030 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28106 |
| Kapa HyperPrep Kit | Kapa Biosystems | Cat# KK8500 |
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28704 |
| Deposited data | ||
| Raw RNA sequencing, ATAC-sequencing, ChIP-sequencing, and Hi-C data | GEO | GSE172383 |
| Analyzed RNA sequencing, ATAC-sequencing, ChIP-sequencing, and Hi-C data, See Tables S2, S3, S6 | This paper | N/A |
| The Cancer Genome Atlas Skin Cutaneous Melanoma | Akbani et al. (2015) | https://gdc.cancer.gov/about-data/publications/skcm_2015 |
| Experimental models: Cell lines | ||
| SKmel147 | Eva Hernando Laboratory (NYU) | RRID:CVCL_3876 |
| 501mel | Eva Hernando Laboratory (NYU) | RRID:CVCL_4633 |
| 113/6-4L | Robert Kerbel Laboratory (Sunnybrook Health Sciences Center) | Cruz-Munoz et al., 2008 |
| WM1552c | Rockland | WM1552C-01-0001, RRID: CVCL_6472 |
| WM1799 | Rockland | WM1799-01-0005, RRID: CVCL_A341 |
| WM3456 | Rockland | WM3456-01-0001, RRID: CVCL_AP91 |
| Mel888 | Stuart Aaronson Laboratory (ISMMS) | RRID: CVCL_4632 |
| WM1716 | Rockland | WM1716-01-0001, RRID: CVCL_AP82 |
| LOX-IMVI | Jessie Villanueva Laboratory (Wistar Institute) | RRID: CVCL_1381 |
| WM3533 | Rockland | RRID: CVCL_0B73 |
| Experimental models: Organisms/strains | ||
| Premium specific pathogen-free (SPF) embryonated chicken eggs | Charles River Laboratories | Cat# 10100326 |
| Mouse: female, 6 to 8 weeks old, N0D/SCID/IL2γR−/− | Jackson Laboratories | Cat# 005557 |
| Oligonucleotides | ||
| Non-targeting sgRNA: CACCGTTCCGCGTTACATAACTTAG | This paper | N/A |
| ARID2.1 sgRNA: CACCGAATGGCAAACTCGACGGGGA | This paper | N/A |
| ARID2.3 sgRNA: CACCGATGATGATGAGGTACCACC | This paper | N/A |
| ARID2.5 sgRNA: CACCGTTTACTACTTGCTAATGCCG | This paper | N/A |
| ARID2 shRNA: CGTACCTGTCTTCGTTTCCTA | Sigma-Aldrich | TRCN0000166264 |
| Recombinant DNA | ||
| lentiCas9-Blast | Addgene | Cat# 52962 |
| pLKO.1 eGFP/mCherry | Brian Brown Laboratory (ISMMS) | N/A |
| pLenti-puro | Addgene | Cat# 39481 |
| pLK0.1 eGFP NTsg | This paper | N/A |
| pLKO.1 mCherry ARID2 sg1 | This paper | N/A |
| pLKO.1 mCherry ARID2 sg3 | This paper | N/A |
| lentiCRISPR v2 | Addgene | Cat# 52961 |
| FG12-GFP | Bravo-Cordero Laboratory (ISMMS) | N/A |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| pLKO.1 shARID2 | Sigma-Aldrich | TRCN0000166264 |
| pLL-CMV-rFLuc-T2A-GFP | System Biosciences | Cat# LL300PA-1 |
| Software and algorithms | ||
| Incucyte Life-Cell Imaging System | Essen Bioscience | https://www.sartorius.com/en/products/live-cell-imaging-analysis/live-cell-analysis-software/incucyte-base-software |
| FCS Express | De Novo Software | https://denovosoftware.com/ |
| STAR | Version 2.6.0c, Dobin, 2013 | https://github.com/alexdobin/STAR |
| Rsubread | Version 2.4.3, Liao et al., 2014 | http://subread.sourceforge.net |
| DEseq2 | Version 1.30.1, Love et al. (2014) | https://github.com/mikelove/DESeq2 |
| Bowtie | Version 1.1.12, Langmead et al. (2009) | https://github.com/BenLangmead/bowtie |
| MACS2 | Version 2.1.0, Zhang et al. (2008) | https://pypi.org/project/MACS2/ |
| fastQC | Version 0.11.7, Andrews, 2019 | https://github.com/s-andrews/FastQC |
| PICARD | Version 2.2.4 | https://github.com/broadinstitute/picard/releases |
| Samtools | Version 1.9, Li et al. (2009) | http://www.htslib.org/ |
| SICER | Version 2.0, Zang et al. (2009) | https://zanglab.github.io/SICER2/ |
| deepTools | Version 3.2.1, Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| Bowtie2 | Version 2.1.0, Langmead et al., 2009 | https://github.com/BenLangmead/bowtie2 |
| Topdom | Version 0.0.1, Shin et al., 2015 | https://github.com/HenrikBengtsson/TopDom |
| Hi-C bench | Lazaris et al. (2017) | https://github.com/NYU-BFX/hic-bench |
| BedTools | Version 2.29.2, Quinlan and Hall (2010) | https://bedtools.readthedocs.io/ |
| ChIPSeeker | Version 1.26.2, Yu et al. (2015) | https://github.com/YuLab-SMU/ChIPseeker |
| HOMER | Version 4.11, Heinz et al. (2010) | https://github.com/IGBIllinois/HOMER |
| clusterProfiler | Version 3.18.1, Yu et al., 2012 | https://github.com/YuLab-SMU/clusterProfiler |
| Prism | Version 9 | https://www.graphpad.com |
| ImageJ | Version 2.1.0/1.53c | https://imagej.nih.gov/ij/index.html |
| RStudio | Version 1.1.46 | https://rstudio.com |