

Abstract
Metastasis is the major cause of mortality in patients with breast cancer. Many signaling pathways have been linked to cancer invasiveness, but blockade of few protein components has succeeded in reducing metastasis. Thus, identification of proteins contributing to invasion that are manipulable by small molecules may be valuable in inhibiting spread of the disease. The protein kinase with no lysine (K) 1 (WNK1) has been suggested to induce migration of cells representing a range of cancer types. Analyses of mouse models and patient data have implicated WNK1 as one of a handful of genes uniquely linked to invasive breast cancer. Here, we present evidence that inhibition of WNK1 slows breast cancer metastasis. We show that depletion or inhibition of WNK1 reduces migration of several breast cancer cell lines in wound healing assays and decreases invasion in collagen matrices. Furthermore, WNK1 depletion suppresses expression of AXL, a tyrosine kinase implicated in metastasis. Finally, we demonstrate that WNK inhibition in mice attenuates tumor progression and metastatic burden. These data showing reduced migration, invasion, and metastasis upon WNK1 depletion in multiple breast cancer models suggest that WNK1 contributes to the metastatic phenotype, and that WNK1 inhibition may offer a therapeutic avenue for attenuating progression of invasive breast cancers.
Introduction
The protein kinase with no lysine (K) 1 (WNK1) is the most widely expressed of a family of four related enzymes notable for their unique catalytic lysine location, which distinguishes them from all other members of the eukaryotic protein kinase superfamily (1). Positional cloning and subsequent physiological studies demonstrated that mutations in WNK1 and WNK4 genes can cause a rare form of hypertension resulting from their increased expression (2–5). Among the best described WNK targets are the closely related protein kinases OSR1 (OXSR1, oxidative stress responsive 1) and SPAK (STK39, STE20/SPS-1-related proline-alanine-rich kinase; refs. 6–8). These kinases form complexes with and are activated upon phosphorylation by WNKs and regulate the activities of cation chloride cotransporters that support ion homeostasis throughout the body (9–11). WNKs themselves are sensitive to changes in osmotic stress and intracellular chloride concentrations (1, 12, 13).
WNK1 is an essential gene in mouse. Knockout of the mouse WNK1 gene prevents development of a functional blood vessel architecture resulting in embryonic lethality by embryonic day 12 (14–17). Depletion of WNK1 also prevents migration and angiogenesis in assays with cultured primary human endothelial cells (18). A similar loss of endothelial cell migration is observed upon depletion of OSR1. Although a detailed mechanistic understanding of the effects of WNK1 on endothelial migration is lacking, decreased expression of WNK1 in cultured endothelial cells caused reduced expression of a number of factors that promote angiogenesis including Slug (SNAI2), vascular endothelial growth factor A (VEGF-A), and matrix metalloproteinases (18). These and other findings indicate an effect of the WNK1 cascade on induction of a mesenchymal phenotype essential for endothelial wound healing (18, 19).
Aside from its important functions in vascular biology, WNK1 has been implicated in migration in multiple cancer types including glioblastoma, prostate cancer, non—small cell lung cancer, and breast cancer (20–24). In several of these studies, as well, the actions of WNK1 were associated with a shift towards a mesenchymal phenotype. We chose to focus on examining the impact of WNK1 on breast cancer migration because of an unbiased transposon insertional mutagenesis study in mice designed to identify breast cancer susceptibility genes. Using survival prediction analysis of breast cancer patient progression data, this study categorized WNK1 as one of a handful of driver genes in high-risk, invasive breast cancer (25). A phosphoproteomic analysis also implicated WNK1 in aggressive breast cancers (26). Here we show that in breast cancer model systems depleting or inhibiting WNK1 decreases migration and invasion in vitro. We find that WNK1 promotes invasion through a previously identified network involving Slug and the tyrosine kinase AXL (27). Finally, we show that a WNK inhibitor reduces tumor burden in a mouse xenograft model of metastatic breast cancer.
Materials and Methods
Reagents and consumables
Reagents were obtained from the following sources: WNK463 and BGB324 (Selleck Chemicals, S8358 and S2841), anti-AXL antibody C89E7 (Cell Signaling Technology, 8661S), anti-Vinculin (Sigma Aldrich, B9131), anti-pOSR1 (pT325; EMD Millipore, 07–2273), anti-OSR1 (Cell Signaling Technology, 3729S), anti-WNK1 (Cell Signaling Technology, 4979S), anti-p63 (Cell Signaling Technology, 4892), anti-pan-actin (Cell Signaling Technology, 4968S), anti-N-cadherin (BD Biosciences, 6 10 181), anti-vimentin (Thermo Fisher Scientific, PIMA511883), anti-ERK1/2 (Cell Signaling Technology, 4696S), Optimem (Invitrogen, 51985–034), 2-hydroxypropyl-β-cyclodextrin (Sigma Aldrich, H107-5G), Pluronic F68 (BASF corporation), cytochalasin D (Millipore Sigma, C2618), bumetanide (Sigma Aldrich, B3023), 96-well plates (Corning, 3904 or Greiner, 655090); Alexa Fluor 647 and DAPI Fluoromount-G (00-4959-52) from Thermo Fisher Scientific; phalloidin (Invitrogen, A22283 or Life Technologies, A22287); Hoechst (Invitrogen, H1399); α-tubulin, Developmental Studies Hybridoma Bank 12G10.
siRNAs
Oligonucleotides encoding siRNA for human WNK1 (siWNK1: 5′-CAGACAGUGCAGUAUUCACTT-3′) and control siRNA (#4427037) as in ref. 28 and OSR1 siRNA (s19303 Silencer Select) were purchased from Thermo Fisher Scientific. Transfections of siRNA were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific, #13778150) according to the manufacturer’s protocol. All siRNAs were used at a final concentration of 20 nmol/L, except in MCF-MCF-ductal carcinoma in situ (DCIS) as noted.
Cell culture and treatment
MCF-DCIS cells were as described (27). MDA-MB-231 cells (from ATCC or the Whitehurst lab, UT Southwestern) were maintained in DMEM (Sigma-Aldrich, D5796) with 10% FBS (Sigma-Aldrich, F0926) and 1% penicillin/streptomycin (Thermo Fisher Scientific, SV30010). SUM159 (Asterand) cells were cultured in Ham’s F-12 medium (Sigma-Aldrich, N6658) with 5% FBS, 5 μg/mL insulin (Sigma-Aldrich, I0516), and 1 μg/mL hydrocortisone (Sigma-Aldrich, H0888). BT-549, BT-20, HCC1569, and HCC1419 lines were obtained from the Minna laboratory, UTSW, and grown in RPMI1640 (Sigma-Aldrich, R8758) with 5% FBS and 1% penicillin-streptomycin. Bone-derived metastatic (BOM) MDA-MB-231 cells were obtained from parental MDA-MB-231 as described (29). Cell lines were either fingerprinted (PowerPlex 1.2 Kit, Promega) or independently authenticated annually (ATCC, IDEXX Bioresearch) and mycoplasma-free (e-Myco Kit, Boca Scientific or Universal Mycoplasma Detection Kit 30-1012K, ATCC). All cells were maintained at 37°C and 5% CO2. For immunoblotting, cells were grown to 60% confluency and treated with the indicated siRNA (OSR1 or WNK1) overnight and lysed before cells attained 75% confluency for immunoblotting. Cells were treated with inhibitors WNK463 or BGB324 overnight prior to lysing and immunoblotting.
Wound healing assays
Depletion and drug treatment experiments in MCF-DCIS cells were performed as described in ref. 27. Briefly for depletion, 10,000 cells per well were reverse transfected with 50 nmol/L siRNA in 96-well plates and grown to confluence for 72 hours prior to wounding using a 96-pin wounding tool (V&P Scientific, FP6-WP). For inhibitor experiments, cells were grown and wounded as above. In all assays, MCF-DCIS cells were fixed and stained with phalloidin and Hoechst. Montage images (54×) of cells were acquired on a BD Pathway 855 microscope using a 10× objective (Olympus, UplanSApo 10×/0.40). The empty space in the well was defined with Pipeline Pilot software using a custom analysis protocol that determines empty space via a threshold of pixel intensity. Wound closure rate is inversely proportional to empty space. For depletion experiments in MDA-MB-231 cells, 80% confluent cells were treated with siRNA overnight. Wounds were made after 24 to 48 hours, once the cells attained 100% confluence. The cells were imaged with a Zeiss Axio Zoom V16 microscope at time 0 and 24 to 36 hours after treatments until the wound area in the DMSO condition was >90% closed. For inhibitor assays, the medium containing inhibitors or vehicle were replenished every 24 hours. For high-throughput wound healing assays (Fig. 1), cell lines were seeded on 96-well plates. The following day, wounds were made using the pin tool above. Cell debris was removed with an EL406 plate washer (BioTek) and fresh medium was added. Compounds were previously dissolved in anhydrous DMSO. After wound creation, an Echo555 (Labcyte) acoustic liquid dispenser was used to add compound to the wounds at a final DMSO concentration of 0.1%. All plates included replicate wells with 0.1% DMSO as a neutral control. Plates were returned to a Cytomat 2C automated incubator (Thermo Fisher Scientific) immediately after addition. Plates were imaged using an IN Cell Analyzer 6000 automated microscope (GE Healthcare) with a heated stage using a 10× objective and transmitted light. Twelve overlapping fields centered around the monolayer wound were captured for each well. The 12 fields were then stitched together into a single image post-acquisition using the GE Developer Toolbox (v1.9.3; GE Healthcare). All plates were imaged at 8-hour intervals for a total of 7 time points, with the first time point acquired immediately after compound addition. The wound area was measured using ImageJ software (30). The remaining open area was then normalized to that in the DMSO control at 0 time. The lower the ratio (less than 1) means the wound area was closing. Cytochalasin D was used as a positive control.
Figure 1.
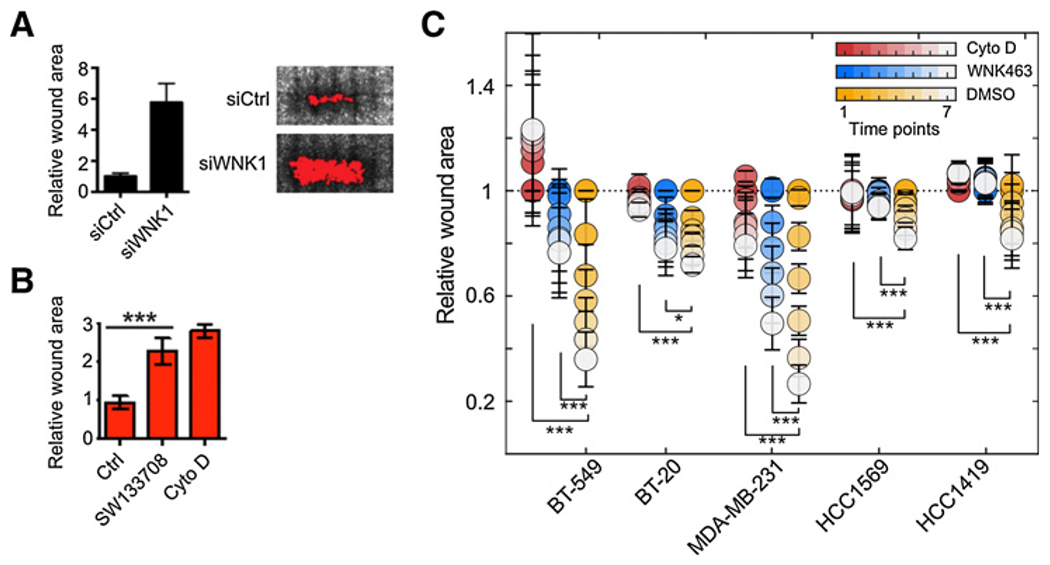
Inhibition of WNK1 decreases breast cancer cell migration. A, Wound closure of MCF-DCIS cells transfected with control or WNK1 siRNA and grown to confluence. Representative images 24 hours after wounding are shown. Graph shows quantification of relative wound area (mean ± SD, n = 6 wounds). B, Wound closure of MCF-DCIS cells grown to confluence and treated with DMSO or SW133708 (10 μmol/L) or cytochalasin D immediately after wounding. Representative images 24 hours after wounding are shown. Graph shows quantitation of wound area (mean ± SD, n = 6 wounds). C, BT-549, BT-20, HCC1569, and HCC1419 cells were grown to confluence, treated with DMSO, WNK463 (1 μmol/L), or cytochalasin D (1 μmol/L) and imaged at time 0 and over 6 time points ending at 48 hours. Results are from n = 8 and are shown as mean ± SE. The last two time points from each treatment were used to determine statistical significance analyzed by Student t test (*, P < 0.05; ***, P < 0.0001).
In vitro kinase assays
Cloning, expression, and purification of pWNK1 (WNK1 residues 194–483, phosphorylated on S382, confirmed by mass spectrometry) and GST-OSR1 (residues 314–344) are generally as described (31). Kinase assays contained 5 nmol/L pWNK1, 10 μmol/L GST-OSR1 fragment, 50 μmol/L ATP, and varying concentrations of inhibitors. 32P incorporation into the OSR1 fragment was quantified by liquid scintillation counting.
Three-dimensional collagen invasion assays
Invasion assays were adapted from Jung and colleagues (32). Briefly, collagen type I (Corning, #354249) was diluted to 1.5 or 2.5 mg/mL in DMEM medium. Thirty microliters of the unsolidified collagen mixture was placed on the bottom of 96-well plates and solidified for 45 minutes at 37°C. siCtrl- or siWNK1-treated MDA-MB-231 cells were seeded at a density of 1 to 3 × 104 cells per well in 0.1 mL of medium, and the three-dimensional (3D) collagen gel culture was incubated at 37° C. After 48 hours, cells were fixed in 3% glutaraldehyde and stained with 0.1% toluidine blue. For inhibitor studies, cells were treated with 1 μmol/L WNK463 or DMSO as the vehicle control at 24 hours after seeding. The following day, cells were fixed and stained as described above. Invasion density was quantified by counting cells below the plane of the monolayer in five random microscopic fields per well under a light microscope (SMZ645, Nikon) at 20× magnification. Representative 3D images were obtained using a Zeiss LSM880 inverted confocal microscope (Carl Zeiss).
Immunofluorescence staining
MDA-MB-231 cells were transfected with control siRNA or WNK1 siRNA. After 36 hours, cells were seeded in 8-well chamber slides (Ibidi, #80827) at densities of 2 × 10+ cells/well and incubated for an additional 36 hours. Cells were fixed with 4% paraformaldehyde and permeabilized for 20 minutes with 0.3% Triton X-100 in PBS and blocked for 30 minutes at room temperature with blocking solution (5% normal goat serum/0.1% Triton X-100/1 × PBS). Cells were incubated with the primary AXL antibody diluted in 1× PBS with 5% BSA overnight at 4°C. Subsequently, cells were incubated with an Alexa Fluor-488 conjugated goat-anti-rabbit secondary antibody (Invitrogen, A-11008) and Alexa Fluor-647 Phalloidin for 1 hour at room temperature, and the slides were mounted with DAPI Fluoromount-G. Immunofluorescence images were acquired using a Zeiss LSM880 inverted confocal microscope and the fluorescence intensity of each cell was analyzed using ImageJ (NIH).
Cell viability assay
A total of 50,000 MDA-MB-231 cells were seeded in a 96-well plate. After cells reached 70% confluence, cells were treated overnight with either 2 or 5 μmol/L BGB324 or 1 μmol/L WNK463 or DMSO control. Cell viability was measured after one day using CellTiter-Blue Cell Viability Kit as per manufacturer’s instructions (Promega, G8080).
Mouse studies
All animal studies were performed according to UTSW Institutional Animal Care and Use Committee guidelines (Animal protocol no. #2017–102099). Four- to five-week-old NSG mice were purchased from the UTSW animal core. For orthotropic implantation assays, 2.5 × 106 MDA-MB-231 BOM cells were resuspended in 1 mL PBS and Matrigel (1:1). Mice were anesthetized by controlled isoflurane administration through a nose cone in a sterile hood. An incision was made between the fourth and fifth nipple of the mouse to expose the mammary fat pad, and 0.1 mL cell suspension was injected using a 28-gauge insulin syringe. Mice were weighed (~25 g) and orally gavaged daily with 0.25 mL containing 25 μg of WNK463 at 1 or 1.5 mg/kg, as indicated, formulated as a suspension in 0.5:0.5:99 (w:w:w) 2-hydroxypropyl β-cyclodextrin:Pluronic F68:purified water. Tumor progression was monitored by measuring the volume of the tumor with Vernier calipers every week. Tumor volume was calculated by using the formula V = 0.5 × a × b2, where “a” and “b” indicate major and minor diameter. At the end point, surgically resected tumors were weighed.
For metastatic colonization assays, 1.0 × 105 MDA-MB-231 BOM cells resuspended in 0.1 mL PBS were intracardially injected into the right ventricle of mice with a 26-gauge tuberculin syringe. Animals were treated daily with WNK463 as above. Tumor progression and metastatic incidence was tracked weekly by bioluminescent imaging (BLI) using an IVIS (In Vivo Imaging System) Spectrum (Perkin-Elmer).
Inclusion and exclusion criteria
All animals were included in the data analysis; outliers were not excluded. Attrition: Animal attrition over time due to metastasis is documented in Fig. 4D. No attrition was observed in mice bearing orthotopic tumors. Sex as a biological variable: Because this is a breast cancer study, we used only female mice for experimental analysis. Randomization: For inhibitor treatment, mice were randomized post-injection based on bioluminescent (BLI) signal. Blinding: not applicable. Power analysis: On the basis of previous experience with these models, animal cohorts of 5 to 10 mice per experimental condition are sufficient to detect differences between groups with 90% power and a 5% type I error rate.
Figure 4.
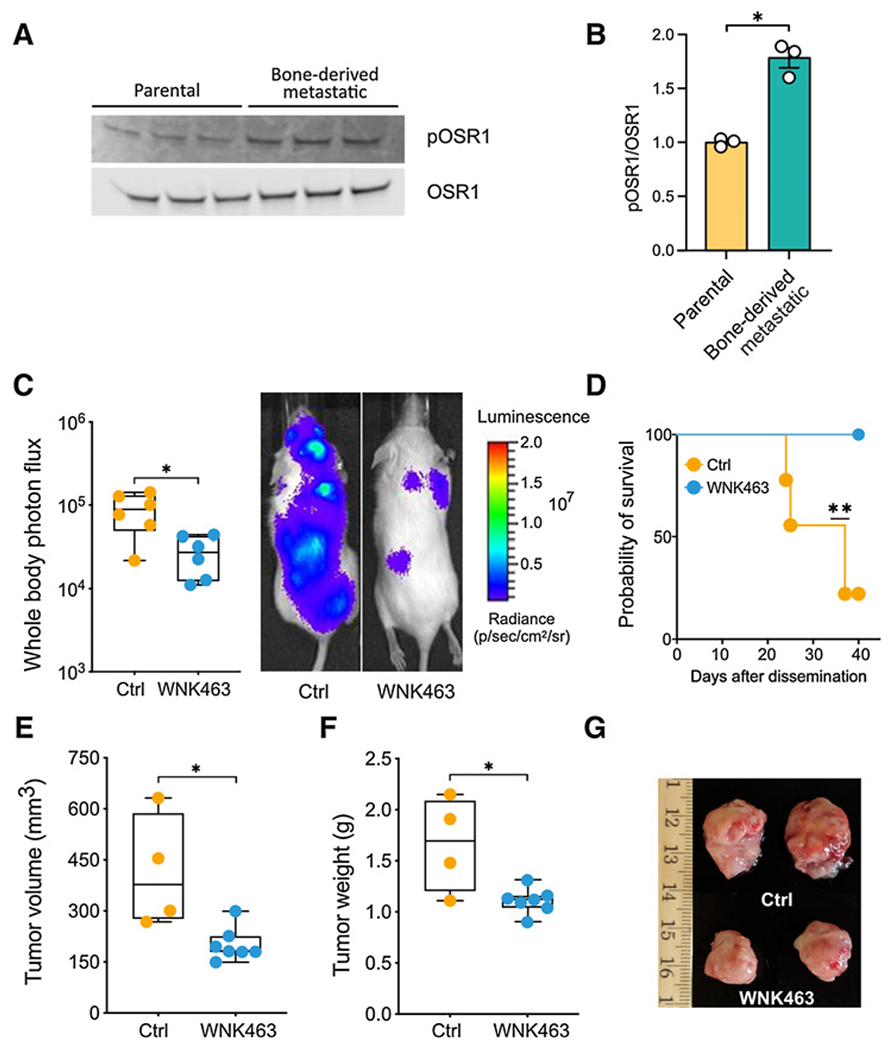
The WNK inhibitor WNK463 decreases tumor burden in mice. A, Lysates from MDA-MB-231 parental cells and bone metastatic cells were immunoblotted with OSR1 and pOSR1 (pT325) antibodies. B, Quantitation of blots in A (*, P < 0.05). C, 105 MDA-MB-231 cells transduced with GFP-luciferase (MDA231-BoM) were injected intracardially into 5- to 6-week-old NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice (The Jackson Laboratory). WNK463 or vehicle was administered by oral gavage every other day, 3 days after injection, at 1 mg/kg for week 1, and at 1.5 mg/kg for weeks 2 to 4. Mice were injected with D-luciferin (150 mg/kg) retro-orbitally and imaged noninvasively using an IVIS spectrum instrument (Perkin-Elmer) weekly. Whole-body photon flux is plotted on the left (*, P < 0.05). Representative mice receiving vehicle or WNK463 are shown on the right. D, Survival is plotted (**, P = 0.01). E, 105 MDA-MB-231 bone metastatic cells transduced with GFP-luciferase as in C were mixed with Matrigel and injected orthotopically in 5- to 6-week-old NSG mice. Two weeks after injection, mice with palpable tumors were treated with either control or WNK463 by oral gavage every other day. During week 1 the dose was 1 mg/kg, and increased from weeks 2–4 to 1.5 mg/kg. Tumor volume for control and WNK463-treated mice was estimated from caliper measurements (*, P = 0.05). Student t test (unpaired). F, Weights of tumors removed from control and WNK463-treated mice (*, P = 0.05). G, Representative tumors for each treatment are shown.
Replication
Representative Supplementary Figure showing one of two animal experiments.
IHC
Orthotopic tumors dissected from NSG mice were fixed in 4% paraformaldehyde and sectioned. Epitope retrieval was performed at pH 9 to unmask antigens. Slides were incubated with a 1:200 dilution of primary pOSR1 antibody or a 1:300 dilution of AXL antibody. Slides were counter-stained with DAPI, mounted and imaged on an EVOS M5000 microscope (Invitrogen).
Statistical analysis
Results are expressed as mean ±SEM. For comparison between only two groups Student t test was used. Single intergroup comparisons between two groups were performed with one-way ANOVA. Two-way ANOVA was used to determine differences between means in two treatments and groups. P ≤ 0.05 was considered statistically significant.
Results
Migration of breast cancer cells is blocked by WNK inhibition
Migration was first examined in MCF-DCIS cells, using a wound healing assay as described (27). Following depletion of WNK1, migration of MCF-DCIS cells was reduced (Fig. 1A). A similar extent of inhibition was also observed in cells exposed to a selective WNK inhibitor, SW133708 (Fig. 1B), identified in a screen in the UT Southwestern High Throughput Screening Core (Supplemental Information; Supplementary Fig. S1). Migration was decreased to nearly the same extent as that caused by cytochalasin D, an inhibitor of actin polymerization. We evaluated the requirement for WNK in migration of several breast cancer cell lines, among them three triple negative (TNBC) lines: BT-549, BT-20, and MDA-MB-231, and two estrogen and progesterone receptor negative, HER2 positive lines: HCC1569 and HCC1419, using the WNK inhibitor WNK463 (Supplementary Fig. S1A; ref. 33). WNK1 protein amount was similar in these cell lines except for BT549 in which WNK1 expression was significantly lower compared with other breast cancer cell lines (Supplementary Figs. S2A and S2B). WNK463 was shown to have high selectivity for WNKs, with remarkably little detectable inhibition of a panel of over 400 other protein kinases (34). WNK463 significantly reduced wound closure of these five migrating lines; it was nearly as effective as cytochalasin D for four of the five lines tested here (Fig. 1C; Supplementary Fig. S2C). These results indicate that interfering with WNK1, either by depletion or via two structurally distinct inhibitors, reduces migration of invasive types of breast cancer cells in two-dimensional culture.
Collagen invasion by breast cancer cells is blocked by WNK inhibition
To determine if WNK1 had an effect on the ability of cells to invade into the extracellular matrix, we examined the ability of MDA-MB-231 cells to migrate into collagen (Fig. 2). Invasion was quantified from the number of cells migrated per field in 10 μm increments in 48 hours using phase contrast microscopy (32, 35). Depleting WNK1 expression resulted in close to a 50% reduction in the number of invading cells per field (Fig. 2A). WNK463 also affected the morphology of the cells and depletion of WNK1 resulted in decreased solidity, as a proxy for less round and more spiky, which are characteristics of nonmalignant cells. In addition, WNK463 decreased the EMT-associated factor N-cadherin (Supplementary Figs. S2D–S2H). We then tested the effect of the WNK inhibitor WNK463. For these experiments, 3 × 104 cells were seeded on two different collagen concentrations for 24 hours prior to the addition of WNK463. Cells invading collagen were then quantified after an additional 24 hours following exposure to the inhibitor or vehicle control. With this protocol, the number of cells invading either collagen concentration was reduced by WNK463 (Fig. 2B). At the higher collagen concentration, the number of cells invading in the presence of DMSO was smaller than at the lower collagen concentration and the reduction in invading cells due to the inhibitor was also close to 50%. At the lower collagen concentration, the inhibitor-induced decrease in invading cells was close to 20%. One factor contributing to the smaller decrease in invasion at the lower collagen concentration compared with WNK1 depletion is the 24 hours of pre-incubation on collagen prior to the addition of the inhibitor, during which time cells in both groups could invade unimpeded by the inhibitor. As was the case for migration in two dimensions, these findings suggest that WNK1 is required for invasion in a 3D matrix.
Figure 2.

Inhibition of WNK1 decreases collagen invasion of MDA-MB-231. A, Equal numbers (1 × 104) of control siRNA (siCtrl)- or WNK1 siRNA (siWNK1)-treated MDA-MB-231 cells were seeded on top of the collagen matrices (1.5 mg/mL) and allowed to invade for 48 hours at 37°C. Cells were fixed and stained, and invading cells were counted from each replicate sample (n = 6). The left panel shows a 3D projection reconstructed from the z-stacks of phalloidin/DAPI stained cells (grayscale images) using a confocal microscope at 20× magnification. The bar graph shows quantification of average numbers of invading cells. B, Twenty-four hours after seeding 3 × 104 cells into 3D collagen gels (1.5 or 2.5 mg/mL), cells were treated with DMSO or WNK463 (1 μmol/L) for 24 hours at 37°C. 3D projection image of phalloidin/DAPI stained cells (grayscale images) taken from a confocal microscope at 40× magnification. Graphs represent average numbers of invading cells (n = 3). Dotted red lines indicate cell monolayer. Data are represented as mean ± SD (**, P < 0.01; ***, P < 0.0001, two-tailed unpaired Student t tests).
WNK1 is in a regulatory cascade with AXL and OSR1
The Tyro3-AXL-Mer (TAM) receptor tyrosine kinase AXL was shown to form a network that promotes migration and a partial epithelial—mesenchymal transition (EMT) in MCF-DCIS cells (27, 36, 37). Numerous studies have suggested the importance of AXL as a target in breast and other cancers (38–46). AXL causes feed-forward transcriptional activation of Slug (SNAI2) through autocrine TGFβ signaling as well as more directly through SMAD3 (47, 48). The p53 family transcription factor p63 also regulates the expression of AXL in TGFβ-dependent EMT (49). Because we previously found interactions of WNK1 with SMAD3 function and we showed that WNK1 influences Slug expression in endothelial cells, we asked whether inhibiting WNK1 changed expression of AXL in MCF-DCIS cells (Fig. 3; refs. 18, 50). Silencing WNK1 decreased expression of AXL and Slug in this invasive MCF-10 model, as detected by immunoblotting, similar to the known AXL regulator p63 (Fig. 3A). A significant reduction of AXL expression upon WNK1 depletion was observed in MDA-MB-231 cells (Fig. 3B, E, and E) and a smaller but clear reduction was noted in the triple-negative breast cancer line SUM159 (Fig. 3C). In contrast, WNK1 depletion did not impair expression of its substrate kinase OSR1, although as much as half of WNK1 and OSR1 are colocalized in a number of cell types (Fig. 3F; ref. 51). Similar to WNK1 siRNA, 1 μmol/L WNK463, which caused minimal cytotoxicity (Supplementary Fig. S3A), decreased expression of AXL in MDA-MB-231 cells (Fig. 3D). Confirmation of the effect of WNK1 depletion on AXL expression was further demonstrated by immunofluorescence in MDA-MB-231 cells (Fig. 3G and H).
Figure 3.
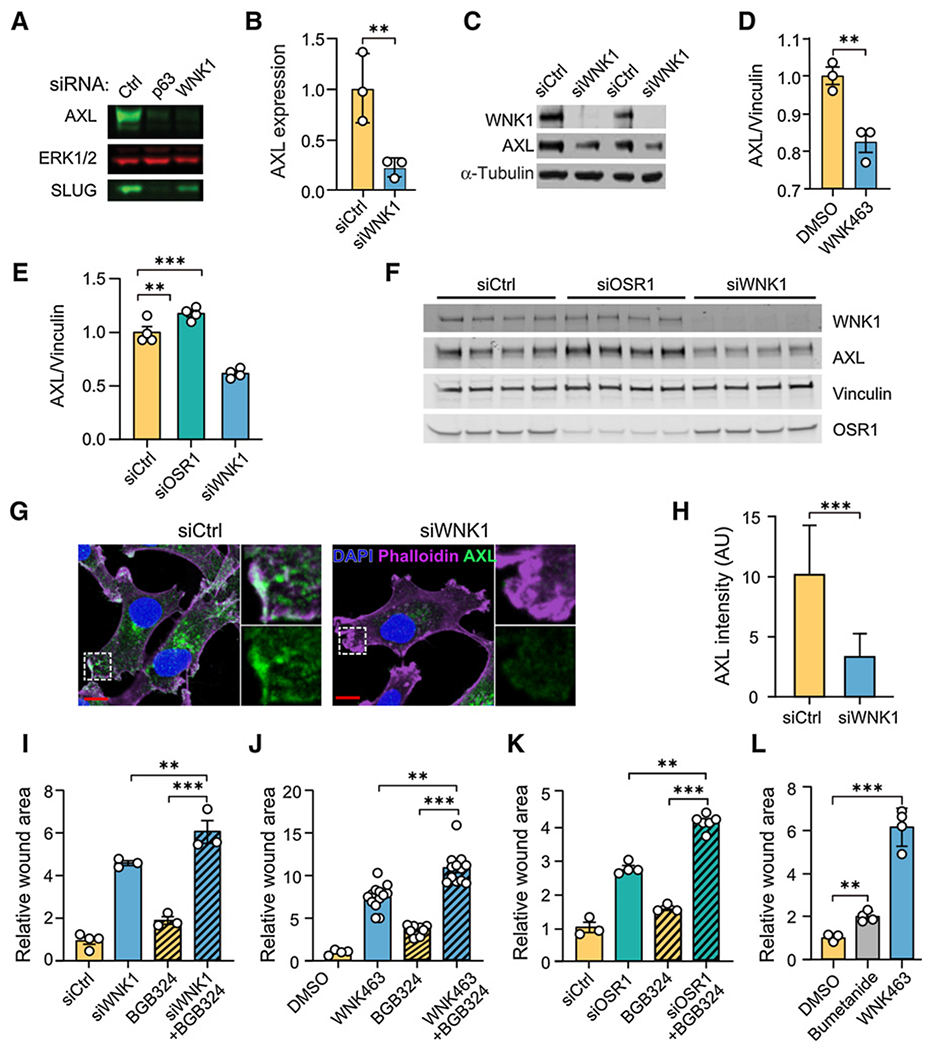
Inhibition of WNK1 reduces AXL expression. A, MCF-DCIS cells (70% confluent) were treated with either control, p63, or WNK1 siRNA for 72 hours. Cells were lysed and immunoblotted for AXL, and Slug, ERK as the loading control. B, Quantification of normalized AXL expression from A; n = 3. C, SUM159 cells treated with siCtrl or siWNK1 were harvested 72 hours after transfection, and lysates were analyzed by immunoblotting with the indicated antibodies. α-Tubulin is the loading control. D, Quantification of AXL protein expression from MDA-MB-231 cells treated with WNK463 (1 μmol/L) overnight (n = 3). E, Quantification of AXL protein expression from MDA-MB-231 cells treated with control, OSR1, or WNK1 siRNA overnight (n = 4). F, Immunoblots from E. G, MDA-MB-231 cells were treated with WNK1 or control siRNAs for 72 hours. Cells were stained for AXL (green), F-actin (phalloidin, purple), and nuclei (DAPI, blue). Representative images of AXL immunofluorescence staining. Areas marked with white squares are amplified and shown in insets. Scale bars, 10 μm. H, Quantification of mean AXL immunofluorescence intensity as shown in G. An unpaired t test was performed (***, P < 0.0001; N = 60). AU, arbitrary units. I-L, Migration of MDA-MB-231 cells treated as indicated with wound area normalized to that at time 0. I, Control siRNA (n = 4), WNK1 siRNA (n = 3), BGB324 (2 μmol/L; n = 3), and WNK1 siRNA+BGB324 (n = 3) for 24 hours. J, DMSO (n = 4), WNK463 (1 μmol/L; n = 13), BGB324 (2 μmol/L; n = 10), and WNK463+BGB324 (n = 12) for 36 hours. K, Control siRNA (n = 3), OSR1 siRNA (n = 4), BGB324 (2 μmol/L; n = 3), and OSR1 siRNA+BGB324 (n = 6) for 24 hours. L, DMSO, bumetanide (20 μmol/L) and WNK463 (1 μmol/L) overnight (n = 4 for each). Data in I-L are mean ± SE; analyzed by one-way ANOVA (**P < 0.01; ***P < 0.0001). Higher concentrations of BGB324 inhibited migration more but also decreased cell viability (Supplementary Figs. S3B and S3C).
Disruption of the OSR1 gene in endothelial cells in mouse recapitulated the WNK1 gene disruption phenotype (16). As a result, we asked if OSR1 depletion also reduced AXL expression in breast cancer cells. In contrast to WNK1 siRNA, OSR1 siRNA did not decrease AXL protein (Fig. 3E and F); in fact, a small increase in AXL was detected in OSR1-depleted cells. We then tested effects of the AXL inhibitor BGB324 (52). This inhibitor has an IC50 14 nmol/L and blocks tumor spread and prolongs survival in models of metastatic breast cancer. The AXL inhibitor alone reduced migration of MDA-MB-231 cells, as reported previously (52), but its effects were less pronounced than depletion of WNK1 (Fig. 3I), the inhibitor WNK463 (Fig. 3J), or depletion of OSR1 (Fig. 3K). Nevertheless, modest but significant effects of BGB324 could be detected even in cells in which WNK1 or OSR1 had been depleted as well as in WNK463-treated cells, suggesting that their effects may be additive. From these findings, it appears that the control of AXL expression by WNK1 occurs through a mechanism independent of its effector kinase OSR1. The results also suggest that control of AXL expression alone is not sufficient to account for the actions of WNK1 on migration.
OSR1 knockdown in cell culture prevented endothelial cell migration (18). As noted above, depletion of OSR1 also attenuated migration of MDA-MB-231 cells (Fig. 3K), consistent with its actions in endothelial cells. Among best validated targets of OSR1 are the sodium, potassium, two chloride cotransporters, NKCC1 and NKCC2, that regulate ion balance and cell volume (9–11). NKCC1 is broadly expressed in tissues and has been implicated in the invasiveness of certain cancers such as glioblastoma (20, 53). NKCCs are inhibited by furosamide-related diuretics such as bumetanide. We compared effects of bumetanide to WNK463 and observed that bumetanide inhibited migration of MDA-MB-231 cells, but not nearly as effectively as WNK463 (Fig. 3L). These findings suggest that OSR1 is a significant mediator of the migratory ability of WNK1; however, NKCC1 is only one of multiple inputs that contribute to WNK1-dependent migration downstream of OSR1 (Supplementary Fig. S4).
WNK1 activity is elevated in cells isolated from breast cancer metastases
A study by Kang and colleagues (29) examined features of breast cancer cells that colonized bone by injecting MDA-MB-231 cells into mice, and then re-isolating and analyzing properties of the cells with a propensity to metastasize to this site. We used MDA-MB-231 parental cells and bone metastatic derivatives (MDA-BOM) isolated as previously described to evaluate any differences in WNK activity in these cells (29). Because OSR1 is phosphorylated and activated by WNK, OSR1 phosphorylation, detected with phospho-antibodies to activating phosphorylation sites, is a useful readout of WNK activity. To estimate relative WNK activity in the parental cells and cells derived from bone metastases, lysates of these cells were immunoblotted for phospho-OSR1. The amount of phospho-OSR1 relative to total OSR1 in MDA-MB-231 BOM cells was nearly twice that in the parental MDA-MB-231 cells (Fig. 4A and B), but there was no detectable difference in WNK1 (Supplementary Figs. S3E and S3F).
WNK463 treatment attenuates tumor growth and metastatic burden in mice injected with metastatic breast cancer cells
On the basis of the evidence above that WNK1 inhibition reduces migration and invasion of breast cancer cells, we determined the effectiveness of WNK463 in blocking tumor progression and metastasis in mice using MDA-MB-231 xenograft models. Because increased phospho-OSR1 suggested higher WNK activity in MDA-MB-231 BOM cells, we injected these cells intracardially into 5- to 6-week-old immune compromised (NSG) mice to examine effects of WNK inhibition on metastasis. WNK463 was shown to reduce hypertension in mouse models, but was associated with toxicity at the most effective concentrations reported (33). Because of these concerns for in vivo use, we chose relatively low doses (1–1.5 mg/kg) of drug rather than higher doses tested for hypertension (up to 10 mg/kg; ref. 33). At these lower doses of WNK463, no effect on body weight was observed after 2 weeks (Supplementary Fig. S3G), but there was a substantial decrease in overall metastatic burden, as assessed by bioluminescence (Fig. 4C). In addition, treatment with WNK463 was associated with improved survival compared with the vehicle-treated animals (Fig. 4D). To test the potential efficacy of WNK463 on the growth of orthotopic tumors, MDA-MB-231 cells were injected into mammary fat pads. WNK463 or DMSO was administered for 4 weeks after tumors were palpable (Fig. 4E–G). Using this protocol, tumors in the inhibitor-treated animals were smaller by volume and weight than those in the control animals. In addition, IHC indicated a significant decrease in AXL and pOSR1 staining in tumors from WNK463-treated animals (Supplementary Figs. S3H–S3K). These results provide evidence that WNK1 inhibition attenuates tumor progression and metastasis. These findings indicate that WNK1 may be a worthwhile target to be considered for therapy of invasive breast cancer.
Discussion
Invasive breast cancer is the second leading causing of mortality in women (54). Poor survival is associated with metastasis to distant sites including bone, brain, liver, and lung (55). The majority of therapeutic approaches for breast and other cancers have focused on cell proliferation, whereas success in interfering with the migratory and invasive events that reduce survival of patients with breast cancer has been more limited (37). This problem motivated our study to evaluate WNK1 as an initiator of invasion.
WNK1 was recognized early as a regulator of blood pressure (2). Studies in mice revealed that, in addition to well-documented actions on ion transport, WNK1 is required for the elaboration of the circulatory system through its essential function in endothelial angiogenesis (14–17). Paralleling these findings in endothelial cells, WNK1 has been implicated in migration with EMT features through knockdown studies in other cancer types, as well as a contributor to stem-like properties in metastatic breast cancers (22–24). Mechanistic information has suggested links to expression of EMT factors Slug and Snail, microRNA networks, changes in expression of cell surface proteins, altered vesicle trafficking, and effects on actin polymerization (22, 23, 56). Genetic analysis integrating data from insertional mutagenesis and human patients has further supported the idea that WNK1 may be a relevant sensitivity in invasive breast cancer (25). In testing this concept, we find that interfering with WNK1 reduced the migratory behavior of breast cancer cells in 2D- and 3D in vitro, reduced the dispersion of metastatic cells in mice, and reduced orthotopic tumor volume. In addition to elevated phospho-OSR1 in bone metastatic cells, we also found that lung-derived metastatic MDA-MB-231 had higher phospho-OSR1 (Supplementary Fig. S3D), suggesting that increased WNK activity may be a feature of breast cancers that can metastasize to multiple sites.
Previous findings on WNK1 in endothelial migration indicated a large number of factors that may contribute to WNK1-dependent migration, some or all of which may be relevant in breast cancers (18). Top among these is OSR1, which in contrast to many other WNK1 partners, is a direct WNK1 substrate. Disruption of the OSR1 gene in endothelial cells in mouse recapitulated the impaired angiogenesis caused by WNK1 gene disruption (16). Here we show that depletion of OSR1 also reduced migration of breast cancer cells. Inhibition of the OSR1 substrate NKCC has been shown to reduce migration in glioma (20,53); thus, we examined its contribution to breast cancer migration and found that its inhibition was not as effective as OSR1 depletion in slowing migration of breast cells. As a result, we expect that other events downstream of OSR1, in addition to this cotransporter, also contribute to the breast cancer migratory phenotype.
Surprisingly, we found that inhibiting WNK1 decreased expression of the tyrosine kinase AXL. Preliminary results suggest that this is not due to a change in AXL mRNA. This effect of WNK1 was independent of OSR1. AXL expression is associated with metastasis and poor prognosis in a variety of tumor types including breast cancer (45, 52, 57, 58). BGB324 is a first-in-class AXL inhibitor, currently in phase II clinical trials, exhibiting promising therapeutic characteristics. It displays AXL-selective antitumor and antimetastatic activity in murine models of breast cancer (52). The connection revealed here between AXL and WNK1 raises the possibility that WNK1 may be a therapeutic option in other AXL-dependent tumor types as well. AXL inhibition in tumor cells decreases the secretion of pro-angiogenic factors such as endothelin and VEGF-A and impairs functional properties of endothelial cells in vivo, suggesting its important role in the initiation of tumor angiogenesis (58). Interestingly, we previously reported that mRNAs encoding these factors were also decreased upon knockdown of WNK1 in endothelial cells (18). Perhaps WNK1 knockdown diminishes these mRNAs in part via suppression of AXL (59). We also found that inhibition of WNK1 had a more pronounced effect than the AXL inhibitor to attenuate migration in MDA-MB-231 cells. Yet, the combination of AXL and WNK inhibitors was more effective at reducing migration than inhibition of either alone. This observation warrants future studies examining the combination of AXL and WNK inhibitors on tumor progression and metastasis in animal models, which could potentially inform future clinical trials.
WNK463 is the first reported highly selective WNK inhibitor. It displays 100% oral bioavailability in C57BL/6 mice. At a dose of 10 mg/kg, WNK463 decreased blood pressure and produced multiple other clinical signs, consistent with the broad physiological actions of WNKs (33). Given the high WNK selectivity of the molecule, it remains to be determined how many of the observed clinical changes were the result of on-target actions of the inhibitor or instead may have been due to off-target effects on molecules outside the kinase family. However, in this study, not only did the inhibitor decrease tumor size and metastatic burden at the lower doses we used (1–1.5 mg/kg), no obvious untoward effects of the inhibitor were observed. No drug-treated animals died over the 4-week time course. These results offer promise that there will be a therapeutic window for WNK inhibitors to treat metastatic breast cancer.
Supplementary Material
Acknowledgments
The authors thank Mike Kalwat (Indiana Biomedical Research Institute) and members of contributing labs for valuable suggestions, Matt Esparza for assistance with MCF-DCIS cells, and Dionne Ware for administrative assistance. We would like to thank UT Southwestern colleagues Dr. Yan Peng for consultation on tumor immunohistochemical staining, Dr. James Collins for use of the Zeiss Axio Zoom V16 microscope, and Dr. John Minna for breast cancer cell lines. These studies were supported by NIH R01 HL147661, Mary Kay Foundation grant 18-18, and Welch Foundation grant I1243 to M.H. Cobb; Cancer Prevention and Research Institute of Texas (CPRIT) grant RP190421, NIH R01 DK110538, and Welch Foundation grant I1128 to E.J. Goldsmith; NCI R01 CA155241 and R01 CA218670 to G.W. Pearson; CPRIT RP170003 to S. Malladi; American Heart Association postdoctoral fellowship 18POST34030438 to A.B. Jaykumar; CPRIT training grant RP160157 for support of P.K. Parida and early support of A.B. Jaykumar; and the Simmons Comprehensive Cancer Center NCI grant (P30 CA142543) for assistance from the UTSW High Throughput Screening (HTS), Small Animal Imaging, Tissue Management, and Microscopy Shared Resources. High throughput wound healing assays (Fig. 1) were performed in collaboration with Hanspeter Niederstrasser, PhD, in the High Throughput Screening Core. This work was also in part supported by an NIH-sponsored S10 grant (1S10OD018005-01 to Bruce A. Posner) for the IN Cell Analyzer 6000.
Authors’ Disclosures
T.T. Dang reports grants from NIH during the conduct of the study. G.W. Pearson reports grants from NIH during the conduct of the study. No disclosures were reported by the other authors.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Supplementary Material Access the most recent supplemental material at: (http://mct.aacrjournals.org/content/suppl/2021/06/29/1535-7163.MCT-21-0174.DC1).
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
References
- 1.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem 2000;275:16795–801. [DOI] [PubMed] [Google Scholar]
- 2.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, et al. Human hypertension caused by mutations in WNK kinases. Science 2001;293:1107–12. [DOI] [PubMed] [Google Scholar]
- 3.Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, et al. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J 2013;451:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep 2013;3:858–68. [DOI] [PubMed] [Google Scholar]
- 5.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci U S A 2013;110:7838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, et al. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem 2005;280:42685–93. [DOI] [PubMed] [Google Scholar]
- 7.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 2005;391:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anselmo AN, Earnest S, Chen W, Juang YC, Kim SC, Zhao Y, et al. WNK1 and OSR1 regulate the Na+, K+, 2Cl-cotransporter in HeLa cells. Proc Natl Acad Sci U S A 2006;103:10883–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 2002;277:50812–9. [DOI] [PubMed] [Google Scholar]
- 10.Dowd BF, Forbush B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1). J Biol Chem 2003;278:27347–53. [DOI] [PubMed] [Google Scholar]
- 11.Markadieu N, Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch 2014;466:91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zagorska A, Pozo-Guisado E, Boudeau J, Vitari AC, Rafiqi FH, Thastrup J, et al. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol 2007;176:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 2014;7:ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zambrowicz BP, Abuin A, Ramirez-Solis R, Richter LJ, Piggott J, BeltrandelRio H, et al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci U S A 2003;100:14109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie J, Wu T, Xu K, Huang IK, Cleaver O, Huang CL. Endothelial-specific expression of WNK1 kinase is essential for angiogenesis and heart development in mice. Am J Pathol 2009;175:1315–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie J, Yoon J, Yang SS, Lin SH, Huang CL. WNK1 protein kinase regulates embryonic cardiovascular development through the OSR1 signaling cascade. J Biol Chem 2013;288:8566–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai JG, Tsai SM, Tu HC, Chen WC, Kou FJ, Lu JW, et al. Zebrafish WNK lysine deficient protein kinase 1 (wnk1) affects angiogenesis associated with VEGF signaling. PLoS One 2014;9:e106129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dbouk HA, Weil LM, Perera GKS, Dellinger MT, Brekken RA, Cobb MH. Actions of the protein kinase WNK1 on endothelial cells are differentially mediated by its substrate kinases OSR1 and SPAK. Proc Natl Acad Sci U S A 2014;111:15999–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasan SS, Siekmann AF. The same but different: signaling pathways in control of endothelial cell migration. Curr Opin Cell Biol 2015;36:86–92. [DOI] [PubMed] [Google Scholar]
- 20.Garzon-Muvdi T, Schiapparelli P, Ap Rhys C, Guerrero-Cazares H, Smith C, Kim DH, et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol 2012;10:e1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fulford L, Milewski D, Ustiyan V, Ravishankar N, Cai Y, Le T, et al. The transcription factor FOXF1 promotes prostate cancer by stimulating the mitogen-activated protein kinase ERK5. Sci Signal 2016;9:ra48. [DOI] [PubMed] [Google Scholar]
- 22.Hung JY, Yen MC, Jian SF, Wu CY, Chang WA, Liu KT, et al. Secreted protein acidic and rich in cysteine (SPARC) induces cell migration and epithelial mesenchymal transition through WNK1/snail in non-small cell lung cancer. Oncotarget 2017;8:63691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shyamasundar S, Lim JP, Bay BH. miR-93 inhibits the invasive potential of triple-negative breast cancer cells in vitro via protein kinase WNK1. Int J Oncol 2016;49:2629–36. [DOI] [PubMed] [Google Scholar]
- 24.Pio GM, Xia Y, Piaseczny MM, Chu JE, Allan AL. Soluble bone-derived osteopontin promotes migration and stem-like behavior of breast cancer cells. PLoS One 2017;12:e0177640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Jenjaroenpun P, Pillai AM, Ivshina AV, Ow GS, Efthimios M, et al. Transposon insertional mutagenesis in mice identifies human breast cancer susceptibility genes and signatures for stratification. Proc Natl Acad Sci U S A 2017;114:E2215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang KL, Wu Y, Primeau T, Wang YT, Gao Y, McMichael JF, et al. Regulated phosphosignaling associated with breast cancer subtypes and druggability. Mol Cell Proteomics 2019;18:1630–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang TT, Esparza MA, Maine EA, Westcott JM, Pearson GW. DeltaNp63alpha promotes breast cancer cell motility through the selective activation of components of the epithelial-to-mesenchymal transition program. Cancer Res 2015;75:3925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sengupta S, Lorente-Rodriguez A, Earnest S, Stippec S, Guo X, Trudgian DC, et al. Regulation of OSR1 and the sodium, potassium, two chloride cotransporter by convergent signals. Proc Natl Acad Sci U S A 2013;110:18826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003;3:537–49. [DOI] [PubMed] [Google Scholar]
- 30.Schneider CA, Rasband WS, Eliceiri KW. NIH image to Image J: 25 years of image analysis. Nat Methods 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min X, Lee BH, Cobb MH, Goldsmith EJ. Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure 2004;12:1303–11. [DOI] [PubMed] [Google Scholar]
- 32.Jung JU, Ravi S, Lee DW, McFadden K, Kamradt ML, Toussaint LG, et al. NIK/MAP3K14 regulates mitochondrial dynamics and trafficking to promote cell invasion. Curr Biol 2016;26:3288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada K, Park HM, Rigel DF, DiPetrillo K, Whalen EJ, Anisowicz A, et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat Chem Biol 2016;12:896–8. [DOI] [PubMed] [Google Scholar]
- 34.Yamada K, Zhang JH, Xie X, Reinhardt J, Xie AQ, LaSala D, et al. Discovery and characterization of allosteric WNK kinase inhibitors. ACS Chem Biol 2016;11:3338–46. [DOI] [PubMed] [Google Scholar]
- 35.Bayless KJ, Kwak HI, Su SC. Investigating endothelial invasion and sprouting behavior in three-dimensional collagen matrices. Nat Protoc 2009;4:1888–98. [DOI] [PubMed] [Google Scholar]
- 36.Dang TT, Westcott JM, Maine EA, Kanchwala M, Xing C, Pearson GW. DeltaNp63alpha induces the expression of FAT2 and Slug to promote tumor invasion. Oncotarget 2016;7:28592–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearson GW. Control of invasion by epithelial-to-mesenchymal transition programs during metastasis. J Clin Med 2019;8:646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T, et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A 2010;107:1124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J, et al. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009;28:3442–55. [DOI] [PubMed] [Google Scholar]
- 40.Holland SJ, Powell MJ, Franci C, Chan EW, Friera AM, Atchison RE, et al. Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res 2005;65:9294–303. [DOI] [PubMed] [Google Scholar]
- 41.Ruan GX, Kazlauskas A. VEGF-A engages at least three tyrosine kinases to activate PI3K/Akt. Cell Cycle 2012;11:2047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leconet W, Chentouf M, du Manoir S, Chevalier C, Sirvent A, Aït-Arsa I, et al. Therapeutic activity of anti-AXL antibody against triple-negative breast cancer patient-derived xenografts and metastasis. Clin Cancer Res 2017;23:2806–16. [DOI] [PubMed] [Google Scholar]
- 43.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal 2013;6:ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goyette MA, Duhamel S, Aubert L, Pelletier A, Savage P, Thibault MP, et al. The receptor tyrosine kinase AXL is required at multiple steps of the metastatic cascade during HER2-positive breast cancer progression. Cell Rep 2018;23:1476–90. [DOI] [PubMed] [Google Scholar]
- 45.Engelsen AST, Wnuk-Lipinska K, Bougnaud S, Pelissier Vatter FA, Tiron C, Villadsen R, et al. AXL Is a driver of stemness in normal mammary gland and breast cancer. iScience 2020;23:101649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cruz VH, Arner EN, Du W, Bremauntz AE, Brekken RA. Axl-mediated activation of TBK1 drives epithelial plasticity in pancreatic cancer. JCI Insight 2019;5:e126117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HJ, Jeng YM, Chen YL, Chung L, Yuan RH. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis 2014;35:769–75. [DOI] [PubMed] [Google Scholar]
- 48.Reichl P, Dengler M, van ZF, Huber H, Fuhrlinger G, Reichel C, et al. Axl activates autocrine transforming growth factor-beta signaling in hepatocellular carcinoma. Hepatology 2015;61:930–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oh JE, Kim RH, Shin KH, Park NH, Kang MK. DeltaNp63alpha protein triggers epithelial-mesenchymal transition and confers stem cell properties in normal human keratinocytes. J Biol Chem 2011;286:38757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee BH, Chen W, Stippec S, Cobb MH. Biological cross-talk between WNK1 and the transforming growth factor beta-Smad signaling pathway. J Biol Chem 2007;282:17985–96. [DOI] [PubMed] [Google Scholar]
- 51.Tu S, Bugde A, Luby-Phelps K, Cobb MH. WNK1 is required for mitosis and abscission. Proc Natl Acad Sci U S A 2011;108:1385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res 2010;70:1544–54. [DOI] [PubMed] [Google Scholar]
- 53.Zhu W, Begum G, Pointer K, Clark PA, Yang SS, Lin SH, et al. WNK1-OSR1 kinase-mediated phospho-activation of Na+−K+−2Cl− cotransporter facilitates glioma migration. Mol Cancer 2014;13:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang Y, Zhang H, Song X, Yang Q. Metastatic heterogeneity of breast cancer: molecular mechanism and potential therapeutic targets. Semin Cancer Biol 2020;60:14–27. [DOI] [PubMed] [Google Scholar]
- 55.Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov 2019;9:176–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright PK, May FE, Darby S, Saif R, Lennard TW, Westley BR. Estrogen regulates vesicle trafficking gene expression in EFF-3, EFM-19 and MCF-7 breast cancer cells. Int J Clin Exp Pathol 2009;2:463–75. [PMC free article] [PubMed] [Google Scholar]
- 57.Levin PA, Brekken RA, Byers LA, Heymach JV, Gerber DE. Axl receptor axis: a new therapeutic target in lung cancer. J Thorac Oncol 2016;11:1357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ludwig KF, Du W, Sorrelle NB, Wnuk-Lipinska K, Topalovski M, Toombs JE, et al. Small-molecule inhibition of Axl targets tumor immune suppression and enhances chemotherapy in pancreatic cancer. Cancer Res 2018;78:246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanaka M, Siemann DW. Axl signaling is an important mediator of tumor angiogenesis. Oncotarget 2019;10:2887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.