

SUMMARY
The nature of the anti-tumor immune response changes as primary tumors progress and metastasize. We investigated the role of resident memory (Trm) and circulating memory (Tcirm) cells in anti-tumor responses at metastatic locations using a mouse model of melanoma-associated vitiligo. We found that the transcriptional characteristics of tumor-specific CD8+ T cells were defined by the tissue of occupancy. Parabiosis revealed that tumor-specific Trm and Tcirm compartments persisted throughout visceral organs, but Trm cells dominated lymph nodes (LNs). Single cell RNA sequencing profiles of LN Trm and skin Trm cells in LN and skin were distinct, but shared clonotypes that occupied both tissues were preferentially overwhelmingly maintained as Trm in LNs. Whereas Tcirm cells prevented melanoma growth in the lungs, Trm afforded long-lived protection against melanoma seeding in LNs. Expanded Trm populations were also present in melanoma-involved LNs from patients, and their transcriptional signature predicted better survival. Thus tumor-specific Trm cells persist in LNs, restricting metastatic cancer.
eTOC Blurb
Lymph nodes (LNs) are frequent sites of metastasis. Molodtsov, Khatwani et. al. find that melanoma-specific CD8+ T cells persist as functional resident memory (Trm) populations within skin-draining lymph nodes, and these Trm cells prevent melanoma growth upon tumor seeding in LNs. A LN Trm transcriptional signature predicts survival in melanoma patients, highlighting the relevance of LN Trm function in human disease.
Graphical Abstract
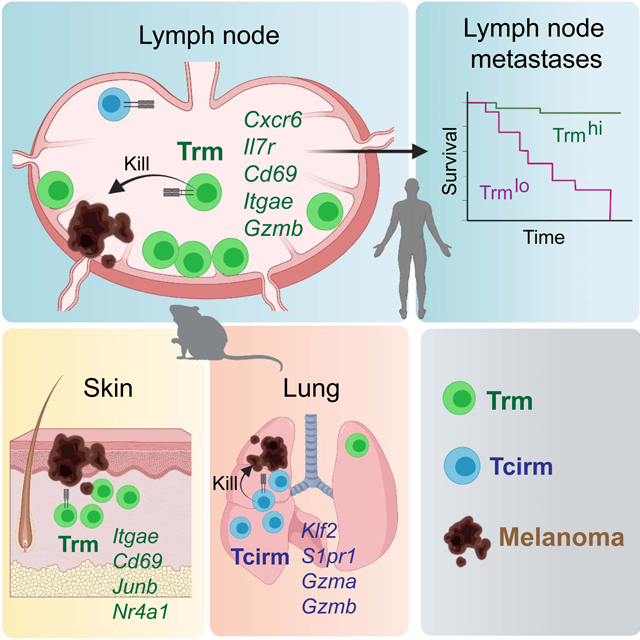
INTRODUCTION
Metastasis is the primary cause of cancer fatality, although immune responses at metastatic locations are not well understood (Blomberg et al., 2018; Gonzalez et al., 2018). CD8 T cells act as guardians against early cancer spreading in mice (Eyles et al., 2010; Lengagne et al., 2008), and T cell-activating immune checkpoint inhibitor drugs can be effective in patients with widely metastatic disease (Riley et al., 2019). Memory T cells, in particular, are critical for durable immunity to cancer (Reading et al., 2018), and are associated with lower rates of metastasis in patients (Pages et al., 2005).
Despite serving as requisite organs for T cell priming, lymph nodes (LNs) are paradoxically the most common sites of early metastasis. Tumor-draining LNs become highly immune suppressed (Jones et al., 2018; Munn and Mellor, 2006; Nunez et al., 2020), and LN vasculature is uniquely capable of disseminating tumor cells to distant organs (Brown et al., 2018; Pereira et al., 2018). This designation of LNs as gateways to metastasis underscores a vital need to understand LN-localized immunity to cancer. The presence of such immunity is inferred by the findings that some metastases never spread beyond the subcapsular regions of lymph nodes (Dewar et al., 2004) and that total regional LN removal lacks therapeutic benefit in patients with metastatic melanoma (Faries et al., 2017).
Following the priming of CD8+ T cells in LNs, clonally expanded populations differentiate into circulating and resident memory subsets. Circulating memory (Tcirm) cells—including central (Tcm), effector (Tem), and stem cell memory (Tscm) subsets— recirculate through the blood and lymphoid organs, whereas resident memory (Trm) populations take up stable residence in tissues (Martin and Badovinac, 2018). As opposed to Tcirm cells, Trm cells express tissue-retention markers, lack the expression of egress markers, and are functionally defined by their inability to recirculate in the steady state (Gebhardt et al., 2018; Jameson and Masopust, 2018). Trm cells are found throughout tissues including the skin, lungs, liver, brain, intestine, and reproductive tract, where they provide a barrier against peripheral re-infections and cancer (Gebhardt et al., 2018; Molodtsov and Turk, 2018). Moreover, CD8+ Trm cells can also occupy regional lymph nodes (Beura et al., 2018; Schenkel et al., 2014). In patients with human immunodeficiency viral (HIV) infection, HIV-specific CD8+ Trm cells are enriched in lymph nodes of elite controllers (Buggert et al., 2018), suggesting a role for LN Trm cells in opposing infections.
The study of Tcirm and Trm cell-mediated immunity to cancer has focused largely on primary tumors, whereas less is known about memory requirements at metastatic sites. Our prior work revealed a key role for skin Trm cells in long-lived protection against dermal melanoma in mice (Malik et al., 2017). Others have shown a requirement for Trm cells against tumors at mucosal sites (Nizard et al., 2017), although the interplay between Trm and Tcm cells may be critical in other settings (Enamorado et al., 2017). Intratumoral CD103+ Trm-like cells are associated with improved prognosis and heightened immunotherapy response (Molodtsov and Turk, 2018). However, circulating memory T cells are also associated with improved outcomes (Huang et al., 2017; Tietze et al., 2017). Tcirm and skin Trm populations together underlie durable immunotherapy responses in melanoma survivors with vitiligo (Han et al., 2021).
Here we examined the contribution of Trm cells to melanoma immunity at metastatic locations using an established model of long-lived immunity to melanoma with associated autoimmunity. Both Trm and Tcirm compartments were generated throughout lymphoid and visceral sites of metastasis, but in contrast to the lung, which was efficiently protected by Tcirm cells, tumor protection in regional lymph nodes was primarily afforded by a transcriptionally distinct population of LN Trm cells. These studies reveal that tumor location defines the optimal features of a protective memory response, and show that Trm cells are critical mediators of immunity to cancer within lymph nodes.
RESULTS
Tissue location dictates the transcriptional characteristics of tumor-specific memory T cells
To investigate CD8+ T cell responses against tumors in metastatic locations, we utilized an established mouse model of postsurgical melanoma-associated vitiligo (MAV) (Byrne et al., 2011; Cote et al., 2011; Malik et al., 2017; Zhang et al., 2007). In response to B16 dermal tumor growth and treatment with anti-CD4 to deplete Treg cells, mice break tolerance to melanocyte differentiation antigens gp100 and TRP-2 (Turk et al., 2004). Following surgical excision of the primary tumor, these mice develop autoimmune vitiligo (Zhang et al., 2007), which is required to sustain memory CD8+ T cell responses against gp100 and TRP-2 in lymph nodes (Byrne et al., 2011) and in the skin (Malik et al., 2017). In mice with MAV, melanoma protection is sustained in the dermis and against i.v. rechallenge in the lungs (Byrne et al., 2011; Zhang et al., 2007). Dermal tumor protection is afforded by Trm cells in the skin (Malik et al., 2017). To identify tumor antigen-specific CD8+ T cells in other tissues, a tracer population of 104 naïve pmel cells (reactive against the shared melanoma/melanocyte-antigen, gp100; (Overwijk et al., 2003)) was introduced prior to MAV induction (Figure 1A). These sentinel pmel cells mirror endogenous CD8+ T cell responses in mice with MAV (Byrne et al., 2011; Malik et al., 2017).
Figure 1. Mice with melanoma-associate vitiligo (MAV) sustain transcriptionally distinct melanoma-specific CD8+ T cell populations throughout lymphoid and peripheral tissues.

(A) Schematic diagram depicting the generation of MAV with sentinel pmel T cell populations. (B-C) proportion of CD44hi pmel cells (gated on total CD8+) in each tissue on the day of tumor excision (priming) or 28–45 days later (memory). (D) Phenotype of pmel cells 150 days post surgery (gated on CD8+Thy1.1+). Symbols represent individual mice; horizontal lines depict means; significance was determined by unpaired (B-C) or paired t test (D), with n.s. (non-significant) denoting p > 0.05. Flow plots depict representative mice. Data were either pooled from 2 independent experiments (B) or taken from a single representative experiment (C-D). Each experiment was conducted twice with similar results. (E) Schematic diagram depicting workflow for bulk RNAseq. (F-G) RNAseq on memory pmel T cells, relative to pmel cells from naïve spleens (Naïve), d12 tumor-draining LNs (D12 LN), or from B16 tumors (TIL); heatmaps show log-transformed transcript per million (TPM) expression of (F) most differentially expressed genes in each tissue, or (G) representative Trm, Activation/Exhaustion, and Tcirm genes. (F-G) Each RNA-seq sample contained sorted pmel cells pooled from a single tissue of 5 mice, and a total of n=4 such pooled samples were analyzed for each group.
Assessing priming on the day of tumor excision, antigen experienced (CD44hi) pmel cells were already distributed throughout tissues (Figure 1B and C, Suppl Figure 1). As expected, highest pmel proportions were detected in the skin. However, pmel cells also accumulated substantively in lungs, liver, and LNs (Figure 1B and C). Forty-five days later, memory populations were still apparent throughout tissues. In perfused lungs and liver, in vivo mAb staining revealed the presence of both parenchymal and vascular-associated pmel memory populations (Suppl. Figure 2A). Relative to priming, responses at memory contracted in the spleen, lungs, and liver, although they remained constant in LNs, and became inflated in autoimmune skin (Figure 1B and C). Thus, in the presence of skin-localized autoimmunity, robust tumor-specific T cell memory developed throughout host lymphoid tissues and visceral organs.
We then assessed phenotypes of tissue localized pmel cells at memory using CD103+CD69+CD62Llo skin Trm cells as a reference population (Malik et al., 2017). While CD103 and CD69 were most strongly expressed on pmel cells in the skin, they were also highly expressed in LNs (Figure 1D). Pmel cells in spleen, lungs, and liver also expressed CD103 and CD69, but at lower levels than in skin or LNs (Figure 1D). CD62L was largely absent from pmel cells in all tissues (Figure 1D), confirming the absence of a robust Tcm compartment in this model (Byrne et al., 2011; Zhang et al., 2007). The CD103+CD69+CD62Llo Trm phenotype was unique to pmel cells in the setting of MAV, as it was distinct from naïve pmel cells prior to adoptive transfer (Suppl Fig. 2B), and distinct from pmel memory populations generated in the context of gp100-expressing herpes simplex viral infection (Suppl Fig. 2C).Thus tumor-specific memory T cells expressed Trm markers, although at varying levels, in all tissue locations.
To identify differences between tumor-specific T cells in different locations, pmel cells were sorted from different tissues and analyzed by bulk RNA sequencing (Figure 1E). Analysis of the most differentially expressed transcripts between tissues revealed tissue-specific transcriptional signatures. Pmel cells in the skin expressed the highest levels of Atf3, Zfp36 and Fabp4/5 (Figure 1F and Suppl. Figure 3), the latter of which is essential for Trm metabolism in the skin (Pan et al., 2017). On the other hand, pmel cells from lungs expressed Gzma, Itgax (CD11c), and Fabp3 (Figure 1F and Suppl Fig. 3). This was in contrast to liver, where cells expressed Fabp1 and Fabp7 (Figure 1F and Suppl Fig. 3), the former of which promotes Trm cell maintenance in the liver (Frizzell et al., 2020). pmel cells from LNs expressed transcripts associated with IL-7 signaling (e.g. Il7r, Stat5a, and Bcl2), and Type I IFN signaling (e.g. Stat1 and Irf3) (Figure 1F and Suppl Fig. 3). This signature was not observed in effector (Teff) pmel cells taken from LNs at a priming/effector timepoint, indicating that it was unique to memory (Figure 1F). Thus, clonally identical memory T cell populations adopted unique gene expression profiles based on their tissues of occupancy.
To understand memory T cell subset differentiation within tissues better, we focused on transcripts associated with TRM, activated/effector, and Tcirm subsets (Kumar et al., 2017; Mackay et al., 2016; Milner et al., 2017). For Trm markers, either naïve pmel cells or tumor-infiltrating lymphocytes (TIL) provided a reference for low or absent transcript (Figure 1G). As expected, pmel cells from the skin demonstrated pronounced expression of Trm transcripts (Mackay et al., 2016; Pan et al., 2017), and accordingly lacked most Tcirm transcripts which were highly expressed by control naïve (CD44lo) pmel cells (Figure 1G). Skin pmel cells also expressed high levels of activation/exhaustion transcripts, which were highly expressed in TIL (Figure 1G). This differed from pmel cells from the spleen which expressed lower levels of most Trm and activation/exhaustion transcripts, while expressing high levels of several Tcirm transcripts (Figure 1G). On the other hand, pmel cells in the lung, liver, and LNs were less well defined by known signatures. Whereas each of these populations expressed high levels of multiple Trm markers, they simultaneously expressed intermediate levels of activation/exhaustion markers, and intermediate or high levels of several Tcirm transcripts (Figure 1G). Therefore heterogeneous populations of tumor-specific memory T cells existed within a clonally uniform population even in a single tissue location.
Tumor-specific CD8+ T cells differentiate into Trm populations within LNs and lungs
To conclusively define the presence of Trm and Tcirm subsets in individual tissues, parabiosis studies were conducted (Gebhardt et al., 2018). MAV donor mice containing memory pmel tracer populations were parabiotically joined to congenically marked MAV recipients for 14 days (Figure 2A). This was sufficient to establish vascular anastomoses, which did not further increase by day 30 (Suppl Figure 4). Considering total CD8+ T cells, equilibration of both naïve (CD44lo) and memory (CD44hi) compartments was apparent throughout spleens, lymph nodes and lungs of conjoined mice (Figure 2B). Liver also equilibrated despite its large endogenous Trm populations (Pallett et al., 2020). Skin CD8+ T cells failed to equilibrate (Figure 3B), consistent with the presence of Trm cells in vitiligo-affected skin (Malik et al., 2017).
Figure 2. Melanoma/melanocyte Ag-specific memory T cells durably reside in lungs and lymph nodes.

(A) Schematic diagram depicting parabiosis experiments involving mice with MAV (at least 30 days after tumor excision); CD8+ T cell populations were assessed in different tissues of donor vs. recipient mice either 14 days (B-D and F) or 30 days (E) after parabiotic joining. (B) Distribution of total CD45.1+ cells (representative mice gated on CD8+; left). Right: populations are further divided based on high/low expression of CD44. (C) Distribution of Thy1.1+ pmel cells (gated on CD8+ cells) across tissues. (D) Expression of CD103 and CD62L (gated on CD8+Thy1.1+ pmel cells. (E) Distribution of Thy1.1+ pmel cells (gated on CD8+ cells) after 30 days parabiotic joining. (F) Distribution of Thy1.1+ pmel cells in individual lymph nodes (gated on CD8+ cells). Symbols represent individual mice with lines joining parabiotic partners. Data in each panel are pooled from two independent experiments with the exception of panel (D), which depicts a single experiment. Each experiment was conducted twice with similar results. Significance was determined by paired t test (or by Wilcoxon matched pairs test for data that were not normally distributed); n.s. (non-significant) indicates p > 0.05.
Figure 3. Heterogenous populations of tumor-specific CD8+ T cells with Trm and Tcirm transcriptional features are present throughout lymphoid and peripheral tissues.

Pmel T cells were harvested and FACS sorted from skin, lungs, liver, and lymph nodes of mice with MAV (see Figure 1E). Gene expression was determined by single cell (sc) RNA sequencing using the 10X Genomics platform. Each sample consisted of pmel cells from 5 mice, pooled. Each dot in a tSNE plot corresponds to a single cell. t-SNE plots displaying (A) 5727 total memory pmel cells isolated from all tissues, colored by tissue of origin; (B) feature plots depict representative expression of selected genes. (C) 1266 cells from skin (top) and 575 cells from liver (bottom), re-clustered to identify further heterogeneity. Expression plots (right) depict average Z-transformed normalized expression of representative genes in each cluster. (D-E) From clustering in panel A, further analysis of (D) 2322 pmel cells from lungs and (E) 1625 cells from LNs; Expression plots at right depict average Z-transformed normalized expression of representative genes in each cluster with skin as a reference population. (F) Reconstruction of a trajectory from pseudotemporal analysis (Monocle) of individual pmel cells from all tissues, colored by Seurat-defined clusters. (Left) The trajectory contains two branch points, and (Right) individual clusters are depicted.
Focusing on Ag-specific pmel populations, as expected, no equilibration was observed in the skin (Figure 3C). However, pmel cells in the spleen fully equilibrated, indicating their identity as Tcirm (Figure 2C), which was consistent with their expression of tissue egress molecules by bulk RNA-seq (Figure 1). Lung pmel cells, by comparison, only partially equilibrated, revealing that they were comprised of both Tcirm and Trm subsets (Figure 2B). Moreover, the phenotype of recirculating lung populations was predominantly CD103loCD62Llo (Figure 2D), consistent with these cells being Tem (Sallusto et al., 1999). Pmel populations in livers of recipient mice were smaller than in donors (Figure 2C), indicating that Tcirm cells were prevalent in livers, but not ruling out the presence of a Trm subpopulation. Taken together, these data showed that a combination of tumor specific Trm and Tcirm cells occupy visceral organs of mice with MAV.
Unexpectedly, parabiosis studies further revealed the presence of a large tumor specific Trm population in LNs. A majority of pmel cells failed to equilibrate in inguinal LNs, and were retained in donors with a CD103hiCD62Llow phenotype (Figure 2 C-D). Only a small recirculating fraction was detected in LNs, characterized by a CD103loCD62Lhi/lo phenotype (Figure 2D). LN Trm populations were stable over time, as indicated by minimal equilibration even after thirty days of parabiosis (Figure 2E). Parabiosis alternately conducted using naïve recipients, resulted in similar outcomes, indicating that vitiligo in recipient mice neither prevented nor facilitated pmel T cell residence or recirculation (Suppl. Figure 4). All skin draining LNs contained pmel Trm populations (Figure 2F) whereas mesenteric LNs lacked pmel cells altogether suggesting that proximity to skin could be important for Trm establishment in LNs. Largest populations were observed in regional LNs (RLNs) draining the site of prior tumor excision. Here a majority of pmel cells were TRM, but a small recirculating population was also apparent (Figure 2F). These studies revealed the presence of a previously unappreciated population of tumor specific Trm cells that became durably resident in LNs.
Transcriptional single cell analysis reveals diverse memory T cell populations throughout tissues
To better characterize the heterogeneity of Trm and Tcirm populations, pmel T cells sorted from different tissues were analyzed by single cell RNA sequencing (scRNAseq) using the 10X platform. Pmel cells clearly clustered based on their tissue of origin (Figure 3A, Suppl Fig. 5), except one small mixed cluster that contained cells from all tissues. In general, marker expression by scRNAseq agreed with flow cytometry and bulk RNAseq (Suppl. Table 1). As expected, cells from skin expressed Trm markers (e.g. Itgae,Cd69, CXCR6 and NR4A1), and lacked Tcirm markers (e.g. Klf2 and S1pr1; Figure 3B, Suppl Fig. 5 and 6A). The mixed cluster contained proliferating cells as inferred by their expression of genes such as Stmn1, Mki67, and Mcm3 (Figure 3B, and Suppl Fig. 5). Cells from the skin and liver each formed single homogeneous clusters (Figure 3A). Separate re-clustering of pmel cells from the skin revealed little heterogeneity, with all cells sharing Trm characteristics (Figure 3C, top), consistent with their behavior by parabiosis. However, re-clustering for the liver revealed two clusters that were differentiated by the expression of Klf2, suggesting the presence of both Tcirm and Trm subsets in liver (Figure 3C, bottom).
Pmel T cells in lungs and LNs were heterogeneous even when clustered in the larger dataset (Figure 3A). Lung contained three populations, with Lung_1 and Lung_2 both expressing Tcirm-related transcripts S1pr1 and Klf2, together with high levels of Gzma and Gzmb (Figure 3D and Suppl. Fig. 5). The smaller Lung_3 cluster expressed Bcl2, Bbc3, Pim2 and Ddit3 (Figure 3D and Suppl. Fig. 5), similar to a recently reported lung airway Trm signature (Hayward et al., 2020). Thus, consistent with parabiosis data, lung harbored transcriptionally distinct Trm and Tcirm populations. Notably, lung and liver Tcirm-like populations were transcriptionally distinct (Figure 3A), indicating that they did not represent contaminants from the blood, but rather that Tcirm cells interact with tissues to an extent that alters their transcriptional profiles.
LNs also contained three clusters. LN_1, the smallest, expressed Cd28, Klf2, Slamf6, Tcf7, and Tox but lacked Itgae (Figure 3E, Suppl Fig. 5), consistent with circulating TSCM-like or progenitor exhausted cells (Chen et al., 2019; Hudson et al., 2019; Jansen et al., 2019). By contrast, the larger LN_2 and LN_3 clusters had high Itgae and Cxcr6, and low Klf2 and S1pr1 (Figure 3E, Suppl Fig. 5 and 6A), aligning them with the LN Trm population. Unlike skin Trm cells, LN Trm cells lacked Nr4a1, Fos, and Tnf (Suppl. Fig. 5) and instead expressed high Il7r, Lta, and Cxcr3 (Figure 3A and E). Interestingly, LN_2 was demarcated from LN_3 only by expression of transcripts related to type 1 IFN signaling (Figure 3E and Suppl Fig. 5), which was consistent with bulk RNAseq data (Figure 1 and Suppl Fig. 3) and suggested that LN_2 is a Trm sub-population that is exposed to type-1 IFNs. Thus, there was a high level of transcriptional heterogeneity within this clonal population of tumor-specific memory T cells in lymph nodes.
To identify transcriptional similarities across memory populations in tissues, pseudotemporal ordering was performed using Monocle. Populations formed three distinct branches, with lung and liver largely overlapping and distinct from skin or LN (Figure 3F). Based on distance and trajectories, the main LN Trm cluster (LN_3) appeared most similar to skin Trm cells, with the LN Tcirm cluster being more distinct (Figure 3F). However, no apparent differentiation relationship existed between skin and LN Trm populations based on RNA velocity analysis (Suppl Fig. 6B). Thus, whereas LN and skin populations shared multiple features, LN Trm cells were a transcriptionally distinct memory population.
LN Trm cells are functional, prevalent, and present in the endogenous T cell compartment
The localization and phenotypic characteristic of tumor-specific T cells were more closely examined within RLNs draining sites of prior tumor excision. Immunofluorescence microscopy of RLNs revealed pmel T cell localization within T cell zones (Figure 4A), similar to naïve and Tcm cells (Sung et al., 2012). However, cells were also observed in clusters near CD169-expressing macrophages in the subcapsular sinus (Figure 4A and 4B), consistent with LN Trm localization in a viral infection model (Schenkel et al., 2014). Pmel populations within both the T-cell zones and subcapsular sinus expressed CD103 (Figure 4B) indicating distribution of Trm populations throughout the LN.
Figure 4. LN Trm cells distribute throughout T cell zones and paracortical regions of LNs, and express IFN-γ.

(A-B) Immunofluorescence confocal microscopy of pmel cells in RLN of mice with MAV. (A) Colors indicate staining for anti-CD8 (red), anti-B220 (blue), anti-CD169 (green), and anti-Thy1.1+ (pmel cells; white). Inset at right depicts pmel cells (yellow arrows) in proximity to CD169+ cells in the subcapsular sinus. (B) Staining was performed as in panel A, but including anti-CD103 (magenta) instead of B220, to identify CD103-expressing Trm cells. Yellow arrows indicate Thy1.1+ pmel cells (middle), most of which co-express CD103 (right), both in the T cell zone and subcapsular sinus regions. Microscopic images are each representative of at least two sections taken from multiple RLNs, with scale bars indicated. (C) Phenotypic analysis of pmel cells (gated on CD8+Thy1.1+ cells) taken from inguinal LNs of mice with MAV, depicting differences in phenotypes of CD103+ versus CD103− populations. (D) Mice were treated to induce MAV as in Figure 1A, however pmel tracer populations expressed an IFNγ-IRES-YFP reporter. Flow cytometry was conducted on cells taken from RLNs to detect expression of YFP in CD103+ Trm population (direct ex vivo, without peptide restimulation). (C-D) Histograms/dot plots depict representative samples; data shown in each panel are either pooled from two independent experiments (CD127 and CXCR6 and YFP) or from a single experiment (CXCR3). Experiments were conducted twice with similar results. Points represent values from individual mice with lines joining populations within a mouse. Significance was determined by paired t test; n.s. (not significant) denotes p > 0.05.
Based on sequencing and parabiosis data, LN Trm cells were differentiated from their LN Tcirm counterparts by CD103 expression. To define additional phenotypic features, we analyzed surface expression of markers that characterized the LN Trm cluster from scRNA-seq data (Figure 4C). Significantly higher surface expression of IL-7R (CD127), CXCR6, and CXCR3 distinguished CD103+ Trm cells from their CD103− Tcirm counterparts in LNs (Figure 4C). To address potential differences in functionality between these subsets, mice were adoptively transferred with pmel cells expressing an IRES-eYFP reporter for IFN-γ (Reinhardt et al., 2009) prior to MAV induction. Strikingly, rapid ex-vivo analysis of non-restimulated cells, revealed that IFN-γ expression was uniformly high in CD103+ Trm cells in LNs, although clearly absent from the CD103− Tcirm subset (Figure 4D). These data highlight LN Trm cells as a phenotypically and functionally distinct population of tumor-specific T cells, poised for effector function within LNs.
Whereas the above studies assessed transgenic pmel cell populations it was critical to understand whether Trm populations also comprise the endogenous anti-tumor response in lymph nodes. First, we analyzed endogenous TRP-2180–188 specific T cells, which are present in LNs of mice with MAV (Byrne 2011). As these populations are small (Byrne 2011), TRP-2 tetramer enrichment was performed on RLNs prior to phenotypic analysis. Indeed, similar to populations in the skin, most TRP-2 tetramer binding cells in LNs exhibited a CD103+CD69+CD62L− Trm phenotype (Figure 5A). To identify the presence of tumor specific Trm cells in RLNs more broadly, scRNAseq was performed. Parallel scTCRseq enabled matching of clones in LNs to those in vitiligo-affected skin as a reference tissue that is infiltrated with melanocyte/melanoma Ag-specific T cells (Malik 2011). Transcriptional clustering revealed seven clusters in LNs, with one (LN_6) expressing Trm cell markers including Itgae, Cd69, and Cxcr6, but lacking Tcirm markers (Figure 5B and C). Of the twenty most expanded clonotypes identified in vitiligo-affected skin, eight matched to lymph nodes (Figure 5D), and lymph node clonotypes that had matches in skin mapped almost exclusively to the LN Trm cluster (Figure 5D). Together these results reveal that endogenous tumor-specific CD8+ T cells formed Trm responses in tumor-draining LNs.
Figure 5. Lymph node Trm populations are comprised of endogenous tumor-specific T cells.

Endogenous CD8+ T cells from regional LNs of mice with MAV (induced as in Figure 1A, but without pmel cell transfer) were analyzed by either (A) tetramer staining or (B-C) scRNAseq and paired TCRseq, with skin as a reference population. (A) H-2Kb TRP-2180–188 tetramer staining, gated on CD8+CD44+ cells from skin (top row), or draining LN tetramer-enriched and - depleted fractions (middle and bottom rows). Phenotype of each gated population is shown at right; Trm phenotype is highlighted in red. Data are representative of two experiments. (B) tSNE clustering of 3266 FACS-sorted CD8+CD44+ T cells from regional LNs pooled from 15 mice with MAV, analyzed by scRNAseq, revealing seven distinct transcriptional clusters (left) with representative Trm and Tcirm gene expression depicted across clusters (right); red circles highlight the LN_6 cluster. (C) Plots depict average Z-transformed normalized expression of representative Trm and Tcirm genes in LN_6 cluster relative to all other LN clusters. (D) Frequency of twenty most expanded clonotypes in skin of these mice, indicating eight clones that were matched to cells from regional LNs. (E) tSNE projection of LN clusters highlighting (in red) those T cells that were clonally matched to skin. Each dot corresponds to a single cell.
LN TRM cells mediate long-lived immunity to melanoma in regional lymph nodes.
Vitiligo is strongly associated with durable immunotherapy responses and favorable prognosis in metastatic melanoma patients (Hua et al., 2016; Teulings et al., 2015). Accordingly, mice with MAV sustain long-lived protective immunity against melanoma rechallenge in the dermis and the lungs (Byrne et al., 2011; Zhang et al., 2007). To determine the full extent of protective memory in this model, B16 melanoma rechallenge was conducted through a variety of routes, at least 30 days after primary tumor excision. To assess only host-endogenous immunity, pmel cells were not used in these rechallenge studies. In addition to exhibiting strong CD8+ T cell-mediated protection against tail vein rechallenge, which produces metastatic-like disease in the lungs (Figure 6A-B), mice with MAV were also significantly protected against rechallenge through the portal vein route, which seeds melanoma in the liver (Figure 6C). Moreover, protection was afforded against melanoma cells directly instilled into RLNs at the site of prior tumor growth (Figure 6D). As in the lungs, tumor protection in LNs was mediated by CD8+ T cells (Figure 6E) and was observed following inoculation of up to 50,000 B16 cells (Suppl. Figure 4). Therefore, mice with MAV sustained widespread protection against melanoma throughout visceral organs, and within RLNs.
Figure 6. Trm cells provide protection against melanoma in regional lymph nodes.

(A-G) MAV was induced as in Figure 1A, but without transfer of tracer pmel cell populations. Mice were rechallenged 45d post tumor-excision through various routes: (A-B) 1×105 B16 cells were injected into the tail vein, and surface lung metastases were counted 21d later; (B) mice recieved anti-CD8 depleting mAb on days −4, −2, +2, +9 and +16 relative to tumor rechallenge. (C) 0.25 × 105 B16-luciferase cells were injected into the portal vein, and liver metastases were imaged 21d later, or (D-E) 0.25 × 105 B16-luciferace cells were directly injected into regional lymph nodes (RLN) draining the prior site of tumor excision, and LN tumor burden was imaged 7d later. (E) mice received anti-CD8 depleting mAb on days −4, −2, and +2 relative to tumor inoculation. (F) Schematic diagram depicting parabiosis tumor rechallenge experiments. Mice were separated after 14 days and rechallenged as described above to produce tumor growth in lungs (G) or (H) lymph node, with tumor growth assessed as in panels A and D. Naïve controls were C57BL-6 mice that received concurrent tumor inoculation via the indicated route. Symbols represent individual mice, (A-E) horizontal lines depict means, and (G-H) lines join parabiotic pairs. Significance was determined by Mann Whitney test (A, B, and D), unpaired t test (C and E), or Wilcoxon matched pairs test (F and G); n.s. (non-significant) denotes P > 0.05. Data in each panel are pooled from two independent experiments.
Considering our finding that lungs and LNs both contained heterogeneous populations of Trm and Tcirm cells, it remained unclear which of these populations mediated long-lived protective immunity in each location. To address this, mice with MAV were parabiosed to naïve recipients to enable equilibration of Tcirm for 14 days. Mice were then surgically separated and individually rechallenged, on the same day, with B16 cells either through the tail vein, or directly into RLNs (Figure 6F). Lungs of parabiosed donor and recipient mice demonstrated similar resistance to i.v. rechallenge, illustrating that Tcirm cells were sufficient for protective immunity against metastatic-like disease in the lungs (Figure 6G). This also illustrated that protective Tcirm populations could be efficiently transferred to recipients lacking vitiligo. Regardless, protection against LN rechallenge was absent in parabiosed recipient mice, revealing that Trm cells were required for tumor protection in RLNs (Figure 6H). Thus, Tcirm and Trm cells collectively provided protection against melanoma at metastatic sites, with LN Trm cells affording a barrier to melanoma growth within regional LNs.
To extend these findings to humans, regional lymph nodes from melanoma patients were analyzed for the presence of TRM-like cells using microscopic and transcriptional profiling. CD8+CD69+ T cells were observed within human melanoma-infiltrated sentinel LNs (SLNs), within the lymph node parenchyma and the metastatic tumor mass itself but concentrated at the border between the metastasis and its surrounding LN (Figure 7A). These cells were observed in four out of four sentinel lymph nodes with metastatic foci >2mm in diameter (Suppl. Table 2), suggesting the natural generation of Trm responses in melanoma patient SLNs. Immunofluorescence staining further confirmed abundant CD8+CD103+CD69+ TRM-like populations within melanoma-involved LNs (Figure 7B).
Figure 7. LN Trm cells are found in melanoma-involved lymph nodes in humans, where their transcriptional signature portends improved survival.

(A) Immunohistochemistry on patient melanoma-involved sentinel lymph nodes, stained with anti-CD8 (green) and anti-CD69 (red), with co-stained cells (black; arrows) detected throughout regions. Image of LN is from a single patient, but with similar cells identified in a total of n=4 patients. (B) Immunofluorescence of metastatic melanoma patient tumor-involved LN, with CD8+CD103+CD69+ triple-stained cells appearing white. Image is representative of specimens from n=2 patients analyzed. (C-F) ScRNAseq and paired TCR seq of 501 CD8+ T cells FACS sorted from a tumor-invaded LN of a melanoma patient with oligometastatic disease. (C) tSNE clustering, revealing four distinct transcriptional clusters. (D) Representative expression of TRM-associated genes, red circle highlights the Hu_LN1 cluster; each dot corresponds to a single cell. (E) Average Z-transformed normalized expression of representative Trm and Tcirm genes across the clusters. (F) tSNE projection of clusters indicating (in color) individual cells comprising the five most highly expanded TCR clonotypes. (G) Schematic diagram depicting workflow for generating memory T cell subset transcriptional signatures from mouse scRNAseq dataset, and applying signatures to melanoma patient specimen data from The Cancer Genome Atlas. (C) Kaplan-Meier plots indicating the prognostic value of enrichment of the indicated single cell-derived binary memory T cell signatures (skin Trm, lung Tcirm, and LN Trm) in all TCGA melanoma specimens (top, n=431), or regional lymph node metastases only (bottom; n=213); patients were stratified into high and low groups based on the mean. (I) Forest plot showing the hazard ratios estimated based on multivariate Cox proportional hazards regression analysis of TCGA metastatic melanoma specimens (n=345).
To establish the presence of LN Trm populations in humans definitively, scRNAseq and paired scTCR seq were performed on T cells from a melanoma-involved LN surgically harvested from a patient with metastatic disease. Analysis of sorted human CD8+ T cells revealed four transcriptional clusters, one of which (HuLN_1) exhibited pronounced features of Trm cells (Figure 7C-E). Moreover, the top five most-expanded clonotypes in this specimen were enriched in the Trm cluster (Figure 7F), indicating that these human LN Trm cells had undergone clonal expansion. These results collectively confirmed the presence of LN Trm populations in patients.
Investigating the potential prognostic significance of LN Trm cells in patients with metastatic melanoma, a transcriptional signature was generated based on the most differentially expressed genes in pmel LN Trm clusters relative to other populations (Figure 7G). Skin-Trm, and lung-TCIRCM signatures were generated for comparison (Figure 7G and Suppl. Table 3). These signatures were then used to infer memory T cell enrichment in melanoma specimens from The Cancer Genome Atlas (TCGA). Assessing all melanoma specimens in TCGA, all three signatures had significant prognostic value (Figure 7H). However, when only analyzing regional LN metastatic melanoma specimens, enrichment of the LN-Trm signature gave the strongest prognostic benefit, with LN-Trmhi patients having a median overall survival of at least 670 days longer than cohorts with other signatures (Figure 7H). This correlation was confirmed in two independent melanoma RLN metastasis datasets and was unchanged even when excluding CD8+T cell-specific genes (Cd8a, Cd8b1, Cd3g, Cd3e, and Trav7–4) from the signature (Suppl Fig. 7). Moreover, by Cox multivariate analysis, enrichment of the LN Trm signature was the only significant protective feature in metastatic melanoma specimens (Figure 7I). Thus, Trm populations were present in melanoma infiltrated LNs, and their transcriptional signature uniquely predicted better outcomes for patients.
DISCUSSION
The generation of durable host-wide anti-cancer immunity is a foundational goal in the field. Recent immunotherapy successes in patients have demonstrated that such outcomes are possible (Eggermont et al., 2016; Larkin et al., 2019; Weber et al., 2015), but cancers metastasize to diverse tissue locations, and T cells must be capable of accessing, occupying, and functioning in these environments. The present study provides new insights into the T cell diversity underpinning such functions. By producing circulating and tissue-resident memory subsets, clonally identical populations of CD8+ T cells form a vast network that confers protective immunity throughout the host. Illuminating the poorly understood phenomenon of tumor rejection within draining LNs, we show that Trm cells are a dominant mediator of tumor immunity at this crucial site of early metastasis.
Our studies exemplify the critical value in combining phenotypic analyses, transcriptomics, and in vivo functional assays, to reveal the identity of discrete memory T cell subpopulations. Only through a combination of parabiosis, flow cytometry, and single cell RNAseq were we able to discern properties of memory T cell subsets with confidence and validity. Whereas it was previously recognized that Trm cells adopt unique characteristics based on tissue location (Kumar et al., 2017; Mackay et al., 2016; Mackay et al., 2013), our studies reveal these precise changes and show that they can arise from clonal populations of tumor-specific T cells. Single cell transcriptomics revealed heterogeneity in the host-wide anti-tumor response. Tumor-specific Trm cells formed a discrete memory population in LNs, with unique transcriptional properties. These properties were prognostically meaningful, as exemplified by their superior ability to predict survival in melanoma patients with RLN metastases. As prior studies of LN Trm responses are limited to settings of viral infection (Beura et al., 2018; Buggert et al., 2018; Schenkel et al., 2014), our work here shows important aspects of this phenomenon in tumor immunity and autoimmunity. In addition to their CD69+CD62Llo phenotype, tumor-specific LN Trm cells are similar to viral-specific LN Trm in their expression of high Itga1 and Xcl1, and low Sell, S1pr1, Klf2, and Nr4a1 (Beura et al., 2018). The LN Trm population was also defined by high Itgae and Cxcr6 expression, which was not observed in Beura et. al. but is prominent in CD69+ LN Trm from HIV patients (Buggert et al., 2018).
The relationship between clonally identical Trm cells in skin and skin-draining LNs remains to be discerned. While sharing many features with skin Trm cells, LN Trm cells lacked the expression of AP-1 family, Nr4a family, and Fabp4/5 transcripts. Instead they expressed high levels of IL-7R, which is known to support memory T cell maintenance (Carrette and Surh, 2012), and markers of type-1 IFN signaling, which can promote tissue residency through the increased expression of CD69 (Kohlmeier et al., 2010; Mackay et al., 2015; Shiow et al., 2006). Our prior studies suggest that distinct properties govern memory T cell maintenance in LNs and skin, as CD103 expression is required for the latter, but not the former (Byrne et al., 2011). Regardless, the present studies do not rule out potential communication between LN and skin compartments by retrograde migration, which is known to sustain LN Trm populations in viral infection models (Beura et al., 2018; Pallett et al., 2020). While we did not observe equilibration of LN Trm cells after 30 days of parabiotic conjoining, more gradual equilibration through the lymphatics could take place over several months in the course of autoimmunity. Moreover, LN Trm cells co-existed with small populations of CD103- Tcirm cells in LNs, which did not make IFNγ, and exhibited a more stem-like or progenitor exhausted-like state (Chen et al., 2019; Hudson et al., 2019; Jansen et al., 2019). A better understanding of the relationships between these tumor-specific compartments in LNs and skin should better inform our understanding of regional anti-tumor immunity.
Viewing the present studies in light of prior work, that nature of the tumor antigen is likely an important factor in LN Trm generation. We previously showed that MAV is required for the formation of memory against shared melanoma/melanocyte antigens in lymph nodes (Byrne et al., 2011). While prior work did not differentiate LN populations based on Trm markers, we now recognize these vitiligo-dependent populations to be Trm cells. In contrast, T cells against B16-expressed OVA (a model tumor-specific antigen) developed into conventional CD62Lhi memory T cells in LNs (Byrne et al., 2011). Melanocyte antigen is persistently sensed by T cells in LNs of mice with MAV (Byrne et al., 2014) and could sustain LN Trm populations. LN Trm populations might, in turn, perpetuate autoimmune vitiligo, although this remains to be explored. Despite chronic antigen exposure, CD8+ T cells can resist exhaustion in autoimmunity (McKinney et al., 2015). One might speculate that antigen guides the positioning of Trm cells within LNs, as pmel Trm cells cluster in the SCS, where antigen is cross-presented by APCs (Grabowska et al., 2018; Louie and Liao, 2019). Notably, tumor antigen cross-presentation and early metastatic invasion both occur within the SCS (Asano et al., 2011; Jones et al., 2018). Studies involving spontaneously metastasizing mouse models will be required to demonstrate that Trm cells prevent true metastasis into the SCS. However, our finding that Trm cells reside in this space, suggests that they are strategically positioned to do so.
Our identification of durable melanoma protection in organs distal to the site of initial antigen encounter refines and extends notions of tumor-specific memory T cell responses as being tissue-restricted (Mullins et al., 2003). In contrast to LNs, tumor protection in lungs was mediated by the population of Tcirm cells in that tissue. A requirement for Tcirm cells in lung melanoma protection may reflect the mechanics of tumor cell introduction into this organ, which occurred through the intravenous route where Tcirm cells are more prevalent. As lung Trm cells typically occupy airways (Hayward et al., 2020), they may be better suited for immunity against primary lung cancers. Technical challenges of parabiosis combined with portal vein injection surgery precluded our investigation of this phenomenon in the liver, although we can speculate that Tcirm populations broadly mediate immunity against melanoma metastasis to visceral organs.
In conclusion, the present studies reveal the tissue-wide diversity of anti-tumor memory T cells, while highlighting a novel population of protective Trm cells within LNs. This work adds to a growing body of literature supporting a role for Trm cells in cancer immunity while filling a long-standing void in our understanding of tumor rejection within lymph nodes. We propose that the installation of memory T cells across lymphoid and peripheral tissues will be a critical path to achieving curative cancer immunity.
Limitations of Study
These studies relied on a mouse model of melanoma-associated vitiligo induced by CD4 depletion, which is not a standard treatment for cancer patients. Thus, it will be important to investigate if LN Trm responses can be generated using anti-PD-1 and anti-CTLA-4 therapies. Ongoing development of humanized anti-CD4 depletion therapy for cancer patients (Shitara et al., 2019) may enable clinical extension of our model in the future. Our studies were limited to a small number of melanoma patient lymph node specimens, all of which were tumor-involved. While we showed that that LN-localized Trm populations can be generated in humans, future studies will be needed to explore potential differences in patient tumor-involved vs. uninvolved lymph nodes, and with varying responses to checkpoint blockade immunotherapy. This investigation was limited to T cells that recognize melanoma-expressed self-antigens. While T cells with such specificities appear important in melanoma patients responding to anti-PD-1 (Lo et al., 2021) and in long-term melanoma survivors with vitiligo (Han et al., 2021), further investigation of LN Trm generation against tumor-expressed neoantigens is clearly warranted.
STAR METHODS
Lead Contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by the lead contact, Mary Jo Turk (Mary.Jo.Turk@dartmouth.edu).
Materials Availability
All reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and Code Availability
Mouse Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. De-identified human scRNAseq data have been deposited at the dbGAP. They are available upon request with IRB approval. To request access, please contact the NIH dbGAP administrator. All database accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies/Tetramer/Beads | ||
| PE/Cy7 anti-mouse CD69 | Biolegend | Cat# 104512; RRID: AB_493564 |
| BV510 anti-mouse CD62L | Biolegend | Cat# 104441; RRID: AB_2561537 |
| FITC anti-mouse/human CD44 | Biolegend | Cat# 103005; RRID: AB_312956 |
| BV421 anti-mouse CD103 | Biolegend | Cat# 121422; RRID: AB_2562901 |
| APC/Cy7 anti-rat CD90/mouse CD90.1 (Thy-1.1) | Biolegend | Cat# 202519; RRID: AB_2201418 |
| PE/Dazzle 594 anti-mouse CD8a | Biolegend | Cat# 100762; RRID: AB_2564027 |
| APC anti-mouse CD45.1 | eBioscience | Cat# 17-0453-82; RRID: AB_469398 |
| PE anti-mouse/human CD44 | Biolegend | Cat# 103008; RRID: AB_312959 |
| BV711 anti-mouse CD186 (CXCR6) | Biolegend | Cat# 151111; RRID: AB_2721558 |
| APC anti-mouse CD183 (CXCR3) | Biolegend | Cat# 126512; RRID: AB_1088993 |
| FITC anti-mose CD127 (Il7ra) | eBioscience | Cat# 11-1271-82; RRID: AB_465195 |
| BV421 anti-mouse CD169 (Siglec-1) | Biolegend | Cat# 142421; RRID: AB_2734202 |
| BV421 anti-mouse/human CD45R/B220 | Biolegend | Cat# 151111; RRID: AB_2721558 |
| AF488 anti-mouse CD103 Antibody | Biolegend | Cat# 121408; RRID: AB_535950 |
| AF647 anti-rat CD90/mouse CD90.1 (Thy-1.1) | Biolegend | Cat# 202508; RRID: AB_492884 |
| PE TRP-2 tetramer(iTAg H-2Kb TRP-2 Tetramer-SVYDFFVWL-P) | MBL International | Cat# TB-5004-1 |
| Anti-PE beads | MACS Miltenyi | Cat# 130-048-801 |
| Purified anti-mouse CD8a | Biolegend | Cat# 100702; RRID: AB_312741 |
| Depleting anti-CD4 (GK1.5) | Bioxcell | Cat# BE0003-1; RRID: AB_1107636 |
| Depleting anti-CD8 (2.43) | Bioxcell | Cat# BE0061; RRID: AB_1125541 |
| Human TruStain FcX | Biolegend | Cat# 422301 |
| BV711 anti-human CD45 | Biolegend | Cat# 304050; RRID: AB_2563466 |
| PE-Dazzle 594 anti-human CD8 | Biolegend | Cat# 344744; RRID: AB_2566515 |
| DAPI | Biolgend | Cat# 422801 |
| anti-human CD8 | Leica Biosystem | Cat# NCL-CD8 (4b11); RRID: AB_563637 |
| anti-human CD69 | Abcam | Cat# ab233396 |
| AF555 anti-human CD8 | Biolgend | Cat# 344702; RRID: AB_1877104 |
| BV421 anti-human CD69 | Biolgend | Cat# 310929; RRID: AB_2561909 |
| Primary anti-human CD103 | ThermoFisher | Cat# 14-1038-82; RRID: AB_467412 |
| AF488 anti-mouse Ig-g | Jackson Immuno Research | Cat# 115-547-003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase Type IV | Worthington Biochemical | Cat# CLSS-4 |
| Collagenase Type II | Sigma | Cat# C6885 |
| Dnase I | Sigma | Cat# 10104159001 |
| BSA | Sigma-Aldrich | Cat# 12659-100GM |
| Liberase | Roche | Cat# 5401020001 |
| Percoll | GE | Cat# 17089101 |
| D-Luciferin, Potassium Salt | GoldBio | Cat# LUCK-3G |
| RPMI 1640 | Corning | Cat# 10-040-CV |
| HBSS | VWR | Cat# VWRL0121-0500 |
| Live Dead Kit | ThrermoFisher | Cat# L34962 |
| Bond Dewax Solution | Leica Biosystem | Cat# AR9222 |
| Bond Epitope Retrieval | Leica Biosystem | Cat# AR9590 |
| Tissue Tek Glas | Sakura | Cat# 6419 |
| Critical Commercial Assays | ||
| AF555 Antibody Labeling Kit | ThermoFisher | Cat# A20187 |
| ChromPlex Kit | Leica Biosystem | Cat# DS9665 |
| Vina Green HRP Chromogen Kit | Biocare | Cat# BRR807 |
| Anti-PE microbeads | MACS Miltenyi | Cat# 130-048-801 |
| Anti-CD8a microbeads | MACS Miltenyi | Cat# 130-117-044 |
| RNA extraction Qiagen RNeasy plus micro kit | Quiagen | Cat# 74034 |
| SMART-Seq v4 Ultra Low Input RNA Kit | ClonTech | Cat# 638490 |
| Nextera DNA flex Library prep kit | Illumina | Cat# 20018704 |
| 10× Single cell 3’ V2 chemistry | 10× Genomics | Cat# PN-120237 |
| Deposited Data | ||
| Bulk Rna-Seq (Fig. 1) | This Paper | GEO GSE169237 |
| scRNA-Seq pmel (Fig. 3) | This Paper | GEO GSE171161 |
| scRNA-Seq endogenous (Fig. 5) | This Paper | GEO GSE169238 |
| scRNA-seq human (Fig. 7) | This Paper | dbGap PHS002309.V1.P1 |
| Experimental Models: Cell Lines | ||
| B16-F10 | Isaiah Fidler, MD Anderson Cancer Center | N/a |
| B16-25K | This paper | N/a |
| B16-Luciferase | Dr. Yina Huang, Dartmouth College | N/a |
| Experimental Models: Organisms/Strains | ||
| C57BL/6NCrl | Charles River Laboratories | Cat# CRL27, RRID: IMSR_CRL:27 |
| B6 CD45.1 | Charles River Laboratories | Cat# CRL:564, RRID: IMSR_CRL:564 |
| B6 Thy1.1 | The Jackson Laboratory | Cat# JAX:000406, RRID: IMSR_JAX:000406 |
| B6 Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J | The Jackson Laboratory | Cat# JAX:005023, RRID: IMSR_JAX:005023 |
| C.129S4(B6)-Ifngtm3.1Lky/J | The Jackson Laboratory | Cat# JAX:017580, RRID: IMSR_JAX:017580 |
| Software and Algorithms | ||
| FlowJo v10 | Treestar Inc | RRID: SCR_008520 |
| Prism 8 | Graphpad Inc | RRID: SCR_002798 |
| Seurat v3.0 | https://satijalab.org/seurat/get_started.html | RRID: SCR_016341 |
| Cell Ranger | 10X Genomics | RRID: SCR_017344 |
| Morpheus | Broad Institute | RRID: SCR_017386 |
| RSEM | Bo Li and Colin Dewey | RRID:SCR_013027 |
| ImageJ | Schneider et al. 2012 | RRID: SCR_018407 |
| RStudio | N/A | https://www.rstudio.com/ |
| Monocle 2.0 | Trapnell Lab | http://cole-trapnell-lab.github.io/monocle-release/ |
| velocyto.R (RNA Velocity) | Kharchenlo Lab | https://github.com/velocyto-team/velocyto.R |
Experimental model and subject details
In vivo animal studies.
Eight to ten-week oldC57BL/6 mice, on CD45.1 and CD45.2congenic backgrounds, were purchased from Charles River Laboratories, and enrolled in experiments between eight and twelve weeks of age. Pmel-1 (pmel) mice (Overwijk et al., 2003) on a Thy1.1 congenic background were bred in house (originally a gift from N. Restifo; The National Cancer Institute) and used by ten weeks of age. IFNγ-IRES-eYFP “Interferon-gamma reporter with endogenous polyA transcript” (GREAT) mice were purchased from the Jackson Laboratory and bred in-house to pmel mice. All animals were housed within the specific pathogen-free section of the Comparative Medicine and Research facility at Dartmouth College. Mice were co-housed in isolator caging units, in a temperature-regulated environment, with standard light and dark cycles. Studies were performed in accordance with Dartmouth’s Institutional Animal Care and Use Committee Guidelines.
Female mice were used for all experiments, as necessitated for successful co-housing in parabiosis studies. Littermates were randomly assigned to experimental groups. Sample sizes and endpoints were based on prior published studies to detect similar biological effects and were selected prior to each experiment. All assessed samples, including outliers, were included in analyses. Detailed descriptions of experimental parameters and data analysis can be found in figure legends and the following paragraphs.
Human studies.
Sentinel lymph nodes from patients undergoing standard of care surgery for melanoma were collected for immunohistological evaluation. Shaofeng Yan, MD, PhD, reviewed IHC slides, and identified four SLNs positive for metastatic melanoma, with intranodal lesions larger than 2mm (Suppl Table 3). Oligometastatic tumor-draining lymph nodes with gross melanoma involvement (Suppl Table 3) were surgically harvested with IRB-approved written informed consent. These studies were performed in accordance with the principles of the Declaration of Helsinki and approved by the committee for the protection of human subjects of Dartmouth-Hitchcock Medical Center (study numbers 00023978 and 00029821).
Cell lines and antibodies.
The B16-F10 mouse melanoma cell line was originally obtained from Isaiah Fidler (MD Anderson Cancer Center) and had previously been passaged intradermally (i.d.) nine times in C57BL/6 mice to ensure reproducible growth in the skin (Malik et al., 2017). This line was used to generate all intradermal primary tumors and was retrovirally transduced with luciferase to produce a B16-luciferase subline that was used for intranodal and portal vein rechallenge studies. Another subline of B16 (“B16–25K”) that had been separately passaged for reproducible growth in the lungs, was administered via tail vein injection for the generation of surface lung metastases; this line did not contain luciferase. All tumor growth experiments involved inoculation of B16 cells at low (less than three) in vitro passages. B16 lines were certified to be free of rodent pathogens by IDEXX BioAnalytics (Columbia, MO).
Depleting anti-CD4 (mAb clone GK1.5) and anti-CD8 (mAb clone 2.43) were produced as bioreactor supernatants from a hybridoma cell lines (American Type Culture Collection) and administered at 200 μg/dose, i.p. Flurorescent antibodies for flow cytometry and microscopy were obtained from Biolegend, unless otherwise indicated.
Method Details
Induction of melanoma-associated vitiligo (MAV).
As previously described (Turk et al., 2004; Zhang et al., 2007), mice were inoculated intradermally on the right flank with 1.75 × 105 B16 cells on day 0, and treated with anti-CD4 mAb i.p. on d4 and 10, to eliminate Treg cells. Tumors were surgically excised on d12. Spontaneous metastases were not observed with this B16 subline, and mice with recurrent primary tumors after surgery (<5%) were removed from the study. Mice were monitored for the development of vitiligo starting 3 weeks post-surgery, and only mice that developed vitiligo (~70%, as previously shown (Byrne et al., 2011)) were used for subsequent studies. Mice with both localized (at the surgical site) and disseminated vitiligo (beyond the surgical site) were used interchangeably for experiments, as previously described (Byrne et al., 2011; Malik et al., 2017). Mice with vitiligo were subsequently enrolled in experiments as described. When more than one group was being compared, initial group assignment was done with the intent of matching the overall level of vitiligo between the groups. Experiments were not blinded.
Pmel cell adoptive transfer.
Naïve CD8+Thy1.1+ pmel cells were isolated from 8–10 week old pmel T cell receptor transgenic mice. Cells were magnetically sorted by anti-CD44:PE negative selection followed by CD8 positive selection using magnetic beads (Miltenyi). T cells were transferred retro-orbitally, at 104 cells per mouse, 1 day prior to intradermal tumor inoculation for the induction of MAV, as described above.
Mouse tissue harvesting and digest.
Tissues were collected from euthanized mice following whole body perfusion through the cardiac ventricle with PBS. To ensure memory formation, all mice with MAV were sacrificed at least 28 days following primary tumor excision. Lymph nodes and spleens were mechanically dissociated. Lungs were minced and then digested in serum-free RPMI containing 16.7 mg/mL of Liberase (Roche) and 0.2 mg/mL of DNase I (Sigma-Aldrich), at 37°C for 30 minutes. Livers were smashed through a mesh filter and then digested in serum-free RPMI containing 0.6 mg/mL Collagenase Type IV (Worthington Biochemical Corporation) and 0.1 mg/mL of DNase I, at 37C for 40 minutes. For both lungs and livers, digests were washed, and buffy coats were isolated by percoll gradient centrifugation. Skin was minced and digested in HBSS containing 3 mg/mL collagenase Type IV and 0.2 mg/mL DNase I at 37 °C for 45 minutes. Unless otherwise indicated, a section of depigmented skin from the right flank (primary tumor excision site) was used for analyses.
Analysis of T cells by flow cytometry.
Samples prepared as described above were blocked with anti-mouse FC blocking Ab (Bioxcell) for 15 min on ice. Cells were then stained for surface markers using the following antibodies; BV421-conjugated anti-mouse CD103 (2E7), BV510-conjugated anti-mouse CD62L (MEL-14), FITC-conjugated anti-mouse/human CD44 (IM7), PE-Dazzle 594-conjugated anti-mouse CD8a (53–6.7), PE-Cy7-conjugated anti-mouse CD69 (H1.2F3), APC-Cy7-conjugated anti-rat CD90.1 (Ox-7), BV711 anti-mouse CXCR6 (SA051D1), APC anti-mouse CXCR3 (Cxcr3–173), FITC anti-mouse CD127 (A7R34) and APC-conjugated anti-mouse CD45.1 (A20; eBioscience). After 30 min incubation at 4°C, cells were washed twice with Flow buffer (1×PBS with 0.2% BSA) and were fixed with 1% Formaldehyde overnight at 4°C. Flow cytometry was performed on a MACSQuant Analyzer 10 (Miltenyi Biotec) or a ZE5 Cell Analyzer (Bio-Rad), and data were analyzed using Flowjo v10 software (Treestar Inc). Phenotypes for pmel cells are only reported for gated populations larger than 50 events.
Intravascular pmel T cell stain.
Mice with MAV that had received pmel adoptive transfer were placed under a heat lamp for 5 minutes, and then injected with 3 ug of biotin-conjugated anti-CD45 anti-mouse antibody (30-F11, Biolegend), followed three minutes later by euthanasia and whole-body PBS perfusion through the cardiac ventricle. APC-conjugated streptavidin (Biolegend) was used to detect anti-CD45 labeled Thy1.1+ pmel cells, as evidence of their presence in the vascular space, in conjunction with Ab staining as described above.
Gp100-expressing herpes simplex viral (HSV) infection.
Recombinant HSV-1 KOS expressing the human gp10025–33 Db epitope KVPRNQDWL under the CMV enhancer/promoter from the UL49-UL50 region, was generated with the assistance of Paul Kinchington (U Pittsburgh) and David Leib (Dartmouth). Live virus was administered i.p. at a dose of 4×105 PFU, in mice that had been transferred with 104 naive Thy1.1+ pmel cells 1 day prior, as described above. The pmel memory response was phenotyped in inguinal LNs 30 days later.
Fluorescence Activated Cell Sorting (FACS).
Tissues from mice with MAV were digested as described above; tissue specimens were each pooled from n=5 mice. Single cell suspensions were prepared for each tissue and stained with anti-CD8 (53–6.7), anti-Thy1.1 (OX-7), and anti-CD44 (IM7). For RNAseq, CD8+Thy1.1+ pmel cell populations were sorted to >90% purity using an ARIA-II cell sorter (BD Biosciences). In mice that did not receive pmel cells, endogenous CD8+CD44hi T cells were sorted to >90% purity using an SH800 cell sorter (SONY).
Analysis of mouse T cells by RNA-sequencing (RNA-seq).
For bulk RNA-seq and single cell (sc) RNA-seq experiments, CD8+Thy1.1+ pmel cells were isolated from MAV mice 45 days following primary tumor excision. In scRNA-seq a small contamination population of B-cell and myeloid transcript expressing cells were identified and excluded from subsequent analysis.
Bulk RNA-seq.
Cells were sorted directly into 200ul Qiagen RLT buffer and snap frozen on dry ice, after which RNA extraction was performed using Qiagen RNeasy plus micro kit following the manufacturer’s instructions. cDNA was made from the extracted RNA using the SMART-Seq v4 Ultra Low Input Kit (Takara) and 10 cycles of cDNA amplification. Libraries were generated from 10ng cDNA using the Nextera DNA Flex library prep kit (Illumina). Libraries underwent quality control by Fragment Analyzer (Agilent) and Qubit (ThermoFisher) to determine the size distribution and the quantity of the libraries, respectively. Libraries were sequenced on a NextSeq 500 (Illumina) to generate and average of 40 million singe-end 75bp per sample. Fastq files were aligned to the mm10 genome using bowtie2 (8) and normalized to obtain Transcripts Per Kilobase Million (TPM) for each RNA-seq sample using the software RSEM (9, 10).
Mouse Single-cell RNAseq and TCRseq.
For each sample, cells were FACS sorted into a single well of a 96 well plate to minimize sample loss. In order to obtain the maximum number of cells from limited material, the entire sorted volume (up to 33.8ul) was loaded onto a single lane of a Single Cell A chip and processed on a Chromium instrument (10X Genomics). Libraries were prepared using the 10X Single cell 3’ V2 chemistry according to the manufacturer’s protocol. Libraries underwent quality control by Fragment Analyzer and Qubit (ThermoFisher) to determine size distribution and the quantity of the libraries prepared. Amplified cDNA and TCR-specific libraries were prepared following the standard 10× procedure to generate libraries for Illumina sequencing. Samples were uniquely barcoded, pooled and sequenced across multiple Illumina NextSeq500High Output runs to generate 50,000 and 5000 reads/cell for gene expression and TCR libraries, respectively. Paired end sequencing was performed using 26 cycles for read 1 to decode the 16bp cell barcode and 10bp UMI sequences, and 98 cycles for read 2 corresponding to the transcript sequence. Raw sequencing data was processed through the Cell Ranger v3.0 pipeline (10× Genomics) using the mouse reference genome mm10 to generate gene expression matrices for single cell 5’ RNA-seq data and, in some instances, fully reconstructed, paired TRA/TRB sequences.
Bulk RNA-seq Data Analysis.
To identify tissue specific gene expression, TPM values from individual replicates were log10 transformed and z-transformed followed by the collapse of the cell replicates by taking the mean z-score of each replicate. Genes with a mean z-score >0 were considered upregulated, and genes with a z-score<0 downregulated, in each sample. Only the upregulated profiles were used to generate tissue specific signatures by first converting the upregulated z-score into -log10(p value). The transformed profiles where then rescaled from 0 to 1 scale. This rescaled data was named as a weight matrix in which a weight of 0 represents no expression difference across cell population, and values >0 represent upregulation of gene expression relative to other profiles in the dataset (Varn et al., 2016). Genes with a weight of 0.3 and higher were determined to be tissue-specific. Subsequently these genes with their corresponding TPM were used to generate heatmaps using the browser-based software, Morpheus (https://software.broadinstitute.org/morpheus).
Mouse single-cell RNAseq and TCRseq analysis.
As previously published (Butler et al., 2018; Stuart et al., 2019), the “Seurat v3” R package was applied to filter out low-quality cells, normalize gene expression profiles, and cluster cells. Cells expressing >5% mitochondrial gene counts or expressing less than 500 genes were discarded using the FilterCells function. Then, the NormalizeData function was applied to normalize and log transformed the raw counts for each cell based on the library size. To cluster cells based on their gene expression, the K-nearest neighbor (KNN) algorithm was used with parameter dimensions set to 20 in the FindNeighbors function, and a resolution parameter set to 0.4 in the FindClusters function. Cells isolated from skin and liver were also each analyzed separately using the same parameters indicated above, but without lymph node or liver samples within the clustering analysis. Pseudotemporal analysis was done using the “Monocle 2” (Qiu et al., 2017a; Qiu et al., 2017b; Trapnell et al., 2014), and RNA Velocity (La Manno et al., 2018) R packages. Cells were predefined and subsequently colored according to “Seurat” defined clusters. For TCR analysis, the10× Cellranger VdJ pipeline was used to determine each TCR α-chain and β-chain CDR3 sequence for a corresponding cell. Only productive TCRs were used to identify TCR clonotypes. Identical CDR3 sequences were required for cells to be identified as a matched clonotype.
Parabiosis studies.
When assessing T cell populations by parabiosis, donor mice were first transferred with naïve pmel tracer populations prior to induction of MAV, as described above. Forty-five days following tumor excision in both groups, pairs of individual donors and recipients were cohoused for 14 days in preparation for parabiosis. Donors and recipients were then surgically joined; donors were joined on their left sides, which was distal to tumor growth/excision sites. The parabiosis surgical technique was performed according to a published protocol (Kamran et al., 2013), which was modified to provide buphenorphen SR (ZooPharm) for pain, and 5% dexterose in saline during recovery. Mice remained surgically joined for either 14 days or 30 days. CD8+ T cell equilibration in the spleen was confirmed by the assessing presence of CD45.1/CD45.2 congenic markers. After either 14 or 30 days, pairs were separated, perfused with PBS, and euthanized. Tissues were processed and analyzed by flow cytometry for the presence of Thy1.1+ pmel cells, as described above. Pmel cell equilibration was only reported for conjoined pairs in which donors had detectable pmel populations (at least 0.03% of total CD8+ T cells across all tissues).
When assessing tumor protection by parabiosis, mice with MAV did not contain pmel cells, and naïve, age-matched C57BL/6 mice were used as recipients. As stated above, forty-five days following tumor excision, pairs of individual donors and recipients were cohoused for 14 days, then surgically joined. Donors were joined on their left sides, which was distal to tumor growth/excision sites. After 14 days, parabionts were surgically separated and rechallenged with B16 cells either in the tail vein or directly into the regional lymph node (draining the site of prior tumor excision, but distal to the site of parabiosis), as described below.
TRP-2 tetramer enrichment and staining.
Pooled skin-draining lymph nodes were collected from mice with MAV (at least 30 days post-surgery), but lacking pmel cells. Single cell suspensions were stained with iTAg H-2Kb TRP-2 Tetramer-SVYDFFVWL-P (MBL International) and enriched for tetramer bound cells using a Miltenyi LS magnetic column and anti-PE beads. Tetramer-enriched and depleted (flow-through) fractions were stained with additional antibodies and flow cytometry was performed, as described above. Single cell suspensions from the skin were tetramer stained, but not enriched.
Tumor rechallenge studies.
Forty-five to ninety days following excision of primary tumors, mice with MAV were rechallenged with B16 cells through various routes. In each experiment, age and sex-matched naïve mice were simultaneously rechallenged as controls; these experiments were not blinded. All tumor rechallenge experiments were done in mice lacking pmel cells to assess endogenous anti-tumor immunity.
Regional lymph node rechallenge was accomplished based on an adaptation of a published protocol for lymph node injection (Johansen and Kundig, 2014). Briefly, a small skin incision (<5mm) was used to expose inguinal lymph nodes. Using forceps to stabilize the lymph node, B16-luciferase cells (either 2.5×104 or 5×104 live cells, as noted) were injected directly into the_lymph node through a 28 gauge needle, in a volume of 5 μl. Steel wound clips were used to close incisions, and B16-luciferase tumor growth was imaged 7 days later after removal of clips from healed wound sites. Lung rechallenge was accomplished by injection of 1×105 B16–25K cells directly into the tail vein. Mice were euthanized 21d later, and pigmented surface lung metastases were counted. Where indicated, a separate group of mice with MAV were depleted of CD8+ T cells, by i.p. injection with anti-CD8 mAb, on days −2, −4 and 3 relative to lymph node or lung rechallenge.
Liver rechallenge was based on an adaptation of a published protocol for portal vein injection (Goddard et al., 2016). Following surgical exposure of the portal vein, B16-luciferase cells (2.5×104 live cells) were injected directly into the portal vein through a 31 gauge needle, in a volume of 25 μl. Bleeding was controlled by applying hemostatic gauze and pressure; wounds were closed by internal and external suturing. B16-luciferase tumor growth was imaged 21 days later.
Tumor luminescence imaging.
To assess tumor burden in lymph nodes and livers, mice were given luciferin at concentration of 150 mg/kg i.p. and then visualized using a Xenogen IVIS 200 system with exposures set at 30 seconds for each tissue. The luminescence scale from 100 to 5000 was used for all analyses, with ROI area set constant for all images. Background was subtracted using a mouse injected with luciferin but containing no B16-Luciferase cells.
Immunohistochemistry (IHC) and confocal microscopy of mouse LN.
Mice received 104 naïve pmel cells, and MAV was induced as described above. At least 60 days post tumor excision, mice were euthanized, and regional lymph nodes were harvested and incubated in 4% paraformeldehyde for 15 minutes at 4°C followed by a 1-hour incubation in 30% sucrose in PBS. Lymph nodes were then embedded in optimum cutting temperature (OCT; Tissue Tek, Sakura) and ten micrometer sections were cut using a cryostat, air-dried, fixed in cold methanol, and then rehydrated in PBS. Before staining, sections were blocked with 5% BSA, 1% goat serum, 1% rat serum and 1% donkey serum in PBS for 1 hour at room temperature. Sections were then stained overnight at 4°C with combinations of Anti-mouse CD8-AF555 (53–6.7), Thy1.1-AF647 (OX7), CD169-AF488 (3D6.112), CD103-AF488 (2E7) and B220-BV421 (RA3–6B2) antibodies. Anti-CD8 clone was labeled with Alexa Fluor 555 antibody labeling kit (ThermoFisher). Sections were washed with PBS and analyzed with a Zeiss LSM 800 confocal microscope fitted with a Hamamatsu Orca Flash 4 camera, using a 20× Pan-App/0.8 NA. Images were taken with ZEN Pro 2012 (Zeiss) software and processed with Fiji software.
IHC and Immunofluorescence (IF) of human lymph nodes.
IHC slides were cut from paraffin embedded human melanoma sentinel lymph nodes, at 4 microns. Slide preparation was performed on a Leica Bond Rx Automated Stainer using the automated protocol that included baking slides at 60C, incubation in Bond Dewax solution, and rinse in alcohol and Bond Wash Buffer. Antigen retrieval was done using the Bond Epitope Retrieval Kit at pH 9.0, for 20 min at 100C. Antibody cocktail containing primary anti-human CD8 (clone 4b11, Leica Biosystems; dilution 1:80) and anti-human CD69 (Abcam; dilution 1:1000) was applied for 30 min, followed by Poly-HRP IgG mouse antibody, for 8 min and Poly AP IgG rabbit antibody, for 20 min. Staining was developed using the Leica Bond Dual Detection kit containing via red for anti-CD69 (ChroPlex kit) or vina green HRP chromogen (Biocare) for anti-CD8. Slides were rinsed, mounted, and examined for dual-staining cells under a Leica light microscope, by a pathologist.
For immunofluorescence, tumor-involved lymph node tissue was embedded in optimum cutting temperature (OCT; Tissue Tek) and flash frozen. Ten micrometer sections were cut using a cryostat, air-dried, fixed in cold methanol, and rehydrated in PBS. Before staining, sections were blocked with 5% BSA and 1% goat serum in PBS for 1 hour at room temperature. Sections were stained overnight at 4°C with directly conjugated antibodies targeting CD8-Alexa Flour 555 (Biolegend) and CD69-BV421 (Biolegend) and primary antibody against CD103 (Thermo; Cat# 14–1038-82). The next day, slides were incubated with a secondary antibody Anti-Mouse IgG-Alexa Flour 488 (Jackson Immuno Research) for 1 hour at RT in the dark. Slides were washed with PBS, and then imaged as described for mouse lymph nodes (above).
Human lymph node tissue processing and scRNASeq/TCRseq.
A fresh surgical lymph node tumor specimen was minced and digested in 5 mg/mL of collagenase type II and 10 mg/mL of DNaseI in RPMI-1640 at 37 °C for 1 hour. Cold PBS+0.2% BSA was added to stop the digest. The sample was blocked with TruStain FcX 198 (Biolegend) and stained with BV711-conjugated anti-human CD45 (HI30;), PE-Dazzle 594-conjugated anti-human CD8 (SK1), and DAPI viability stain, followed by sorting to isolate live CD8+CD45+ T cells on a SONY SH800 Cell Sorter. The entire sorted volume was loaded onto a Single Cell A chip and processed on a Chromium instrument (10X Genomics). The RNA-seq library and V(D)J library for TCR-seq were generated and data were processed as described above for mouse scRNAseq above, however the human reference genome GRCh38 was used to generate gene expression matrices.
Preparation of TCGA melanoma patient gene expression datasets.
The RNA-seq expression dataset from skin cutaneous melanoma (SKCM) patients, generated by The Cancer Genome Atlas (TCGA) project, was downloaded from FireHose (http://gdac.broadinstitute.org/) as level 3 processed RNA-seq data. The total SKCM dataset consisted of 431 primary and metastatic melanoma specimens with gene expression determined by reads per kilobase per million mapped reads (RPKM). Of the 431 specimens, a subset of 345 specimens were identified as metastatic, and a smaller subset of 213 specimens was defined based upon their identity as regional lymph node metastases.
In addition to the SKCM TCGA dataset we used one-channel microarray datasets of regional lymph node metastases from the publicly available Gene Expression Omnibus (GEO) database datasets with accession IDs GSE53118 (n = 79) and GSE65904 (n = 195) were used. These datasets were generated from one-channel microarray platforms, and were downloaded as a matrix containing the expression levels of all probesets.
Gene Signature Generation and Survival Analyses.
To examine the role of individual tissue-localized memory T cell populations as markers for patient prognosis, we derived population specific gene signatures from scRNA seq data. For cluster-defining gene signatures we selected upregulated genes defined by the “Seurat” build-in function FindMarkers. Upregulated genes with expression having a log fold change higher than 0.25, and being detected in more than 25% of cells in a cluster, were considered marker genes for that cluster. In total, we identified 195 genes specific to the skin cluster, 171 genes to the lung cluster (Lung_1), and 94 genes to the lymph node cluster (LN_3). Because each of these populations were comprised of pure CD8+ T cells, the resulting tissue-specific gene lists were further integrated with or without CD8+ T cell specific genes (Cd8a, Cd8b1, Cd3g, Cd3e, and Trav7–4) to generate the final signatures for each subpopulation.
To estimate the relative enrichment of memory T cell subpopulations in patient melanoma specimens, we assed enrichment of subpopulation signatures through the Binding Association with Sorted Expression (BASE) algorithm, as previously described (Varn et al., 2016; Varn et al., 2017). The BASE algorithm allows the generation of cell subpopulation enrichment scores, in which a higher score is indicative of higher enrichment for a given cell signature. We applied this approach to the above TCGA and GEO melanoma datasets with associated patient survival information, to infer survival based on enrichment scores. Patients were stratified based on having high or low enrichment scores, defined as being above or below the median, respectively. Survival differences between patients with high versus low enrichment scores were assessed by Kaplan-Meier, using the survfit function from the R “survival” package. Multivariable Cox regression analysis was performed using the coxph function from the R “survival” package comparing the LN, lung and skin transcriptional signatures and using CD8A and CD3G gene expression as confounding variables. Data were visualized using the gforest function from the R “survminer” package.
Statistical analyses.
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software Inc). Data were tested for Gaussian distribution using the D’Agostino & Pearson test and the Shapiro-Wilk test. When normally distributed according to one of these tests, statistical differences were analyzed with unpaired t-test for comparing two groups from separate mice, or paired t-test for comparing two values from the same mouse or from parabiotic partners. When not normally distributed, or when sample size was too small to determine the distribution, the Mann-Whitney test was used to compare two unrelated groups, and the Wilcoxon matched pairs test was used to compare two values from the same mouse or from parabiotic partners. t tests were two-sided, and a p value of < 0.05 was considered significant.
Supplementary Material
HIGHLIGHTS.
Tissue localization defines the transcriptional profile of tumor specific CD8 T cells
Melanoma-specific Trm populations develop in skin draining LNs
Trm cells protect against melanoma tumor seeding in LNs
A LN Trm transcriptional signature uniquely predicts survival in melanoma patients
Acknowledgments:
Keep on jammin MCVC. We thank David Findley, Kayla Fay, Fatima Haider, and Brian Highhouse for their help in acquiring patient specimens. Funding: This work was supported by NIH R01 CA225028 to MJT, NIH R01 CA205965 to MJT and TJC, NIH R21 CA227996 to CC, a Cancer Prevention Research Institute of Texas (CPRIT RR180061) to CC (CC is a CPRIT Scholar in Cancer Research), NIH F31CA232554 to AKM, The Knights of the York Cross of Honour and Munck-Pfefferkorn Research Funds to MJT, NIH R01 CA054174, CA231325 and an Owens Foundation Grant to TJC, and support from 5P30 CA023108-40 (Immune Monitoring Shared Resource). Graphical abstract was created with BioRender.com.
Footnotes
Declaration of Interests: Authors declare no competing interests.
Inclusion and diversity: We worked to ensure gender balance in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science, and one or more authors received support from a program designed to increase minority representation in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, Kanagawa O, Fujii S, and Tanaka M. (2011). CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity 34, 85–95. [DOI] [PubMed] [Google Scholar]
- Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, Schenkel JM, Mitchell JS, Vezys V, Fife BT, et al. (2018). T Cells in Nonlymphoid Tissues Give Rise to Lymph-Node-Resident Memory T Cells. Immunity 48, 327–338 e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg OS, Spagnuolo L, and de Visser KE (2018). Immune regulation of metastasis: mechanistic insights and therapeutic opportunities. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M, Assen FP, Leithner A, Abe J, Schachner H, Asfour G, Bago-Horvath Z, Stein JV, Uhrin P, Sixt M, and Kerjaschki D. (2018). Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 359, 1408–1411. [DOI] [PubMed] [Google Scholar]
- Buggert M, Nguyen S, Salgado-Montes de Oca G, Bengsch B, Darko S, Ransier A, Roberts ER, Del Alcazar D, Brody IB, Vella LA, et al. (2018). Identification and characterization of HIV-specific resident memory CD8(+) T cells in human lymphoid tissue. Sci Immunol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne KT, Cote AL, Zhang P, Steinberg SM, Guo Y, Allie R, Zhang W, Ernstoff MS, Usherwood EJ, and Turk MJ (2011). Autoimmune melanocyte destruction is required for robust CD8+ memory T cell responses to mouse melanoma. J Clin Invest 121, 1797–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne KT, Zhang P, Steinberg SM, and Turk MJ (2014). Autoimmune vitiligo does not require the ongoing priming of naive CD8 T cells for disease progression or associated protection against melanoma. J Immunol 192, 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrette F, and Surh CD (2012). IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin Immunol 24, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, et al. (2019). TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 51, 840–855 e845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote AL, Byrne KT, Steinberg SM, Zhang P, and Turk MJ (2011). Protective CD8 memory T cell responses to mouse melanoma are generated in the absence of CD4 T cell help. PLoS One 6, e26491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar DJ, Newell B, Green MA, Topping AP, Powell BW, and Cook MG (2004). The microanatomic location of metastatic melanoma in sentinel lymph nodes predicts nonsentinel lymph node involvement. J Clin Oncol 22, 3345–3349. [DOI] [PubMed] [Google Scholar]
- Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, et al. (2016). Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N Engl J Med 375, 1845–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martinez-Cano S, Mejias-Perez E, Esteban M, Melero I, Hidalgo A, and Sancho D. (2017). Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun 8, 16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y, et al. (2010). Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 120, 2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faries MB, Thompson JF, Cochran AJ, Andtbacka RH, Mozzillo N, Zager JS, Jahkola T, Bowles TL, Testori A, Beitsch PD, et al. (2017). Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N Engl J Med 376, 2211–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizzell H, Fonseca R, Christo SN, Evrard M, Cruz-Gomez S, Zanluqui NG, von Scheidt B, Freestone D, Park SL, McWilliam HEG, et al. (2020). Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci Immunol 5. [DOI] [PubMed] [Google Scholar]
- Gebhardt T, Palendira U, Tscharke DC, and Bedoui S. (2018). Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev 283, 54–76. [DOI] [PubMed] [Google Scholar]
- Goddard ET, Fischer J, and Schedin P. (2016). A Portal Vein Injection Model to Study Liver Metastasis of Breast Cancer. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez H, Hagerling C, and Werb Z. (2018). Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 32, 1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowska J, Lopez-Venegas MA, Affandi AJ, and den Haan JMM (2018). CD169(+) Macrophages Capture and Dendritic Cells Instruct: The Interplay of the Gatekeeper and the General of the Immune System. Front Immunol 9, 2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Zhao Y, Shirai K, Molodtsov A, Kolling FW, Fisher JL, Zhang P, Yan S, Searles TG, Bader JM, et al. (2021). Resident and circulating memory T cells persist for years in melanoma patients with durable responses to immunotherapy. Nature Cancer 2, 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward SL, Scharer CD, Cartwright EK, Takamura S, Li ZT, Boss JM, and Kohlmeier JE (2020). Environmental cues regulate epigenetic reprogramming of airway-resident memory CD8(+) T cells. Nat Immunol 21, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua C, Boussemart L, Mateus C, Routier E, Boutros C, Cazenave H, Viollet R, Thomas M, Roy S, Benannoune N, et al. (2016). Association of Vitiligo With Tumor Response in Patients With Metastatic Melanoma Treated With Pembrolizumab. JAMA Dermatol 152, 45–51. [DOI] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin JX, Konieczny BT, Im SJ, Freeman GJ, et al. (2019). Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity 51, 1043–1058 e1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson SC, and Masopust D. (2018). Understanding Subset Diversity in T Cell Memory. Immunity 48, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, et al. (2019). An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen P, and Kundig TM (2014). Intralymphatic immunotherapy and vaccination in mice. J Vis Exp, e51031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D, Pereira ER, and Padera TP (2018). Growth and Immune Evasion of Lymph Node Metastasis. Front Oncol 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamran P, Sereti KI, Zhao P, Ali SR, Weissman IL, and Ardehali R. (2013). Parabiosis in mice: a detailed protocol. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, and Woodland DL (2010). Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity 33, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, et al. (2017). Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep 20, 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. (2019). Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 381, 1535–1546. [DOI] [PubMed] [Google Scholar]
- Lengagne R, Graff-Dubois S, Garcette M, Renia L, Kato M, Guillet JG, Engelhard VH, Avril MF, Abastado JP, and Prevost-Blondel A. (2008). Distinct role for CD8 T cells toward cutaneous tumors and visceral metastases. J Immunol 180, 130–137. [DOI] [PubMed] [Google Scholar]
- Lo JA, Kawakubo M, Juneja VR, Su MY, Erlich TH, LaFleur MW, Kemeny LV, Rashid M, Malehmir M, Rabi SA, et al. (2021). Epitope spreading toward wild-type melanocyte-lineage antigens rescues suboptimal immune checkpoint blockade responses. Sci Transl Med 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie DAP, and Liao S. (2019). Lymph Node Subcapsular Sinus Macrophages as the Frontline of Lymphatic Immune Defense. Front Immunol 10, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, Carbone FR, and Gebhardt T. (2015). Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol 194, 2059–2063. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, et al. (2016). Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. (2013). The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14, 1294–1301. [DOI] [PubMed] [Google Scholar]
- Malik BT, Byrne KT, Vella JL, Zhang P, Shabaneh TB, Steinberg SM, Molodtsov AK, Bowers JS, Angeles CV, Paulos CM, et al. (2017). Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MD, and Badovinac VP (2018). Defining Memory CD8 T Cell. Front Immunol 9, 2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney EF, Lee JC, Jayne DR, Lyons PA, and Smith KG (2015). T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 523, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. (2017). Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molodtsov A, and Turk MJ (2018). Tissue Resident CD8 Memory T Cell Responses in Cancer and Autoimmunity. Front Immunol 9, 2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins DW, Sheasley SL, Ream RM, Bullock TN, Fu YX, and Engelhard VH (2003). Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J Exp Med 198, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, and Mellor AL (2006). The tumor-draining lymph node as an immune-privileged site. Immunol Rev 213, 146–158. [DOI] [PubMed] [Google Scholar]
- Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, et al. (2017). Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun 8, 15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez NG, Tosello Boari J, Ramos RN, Richer W, Cagnard N, Anderfuhren CD, Niborski LL, Bigot J, Meseure D, De La Rochere P, et al. (2020). Tumor invasion in draining lymph nodes is associated with Treg accumulation in breast cancer patients. Nat Commun 11, 3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. (2003). Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med 198, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, et al. (2005). Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 353, 2654–2666. [DOI] [PubMed] [Google Scholar]
- Pallett LJ, Burton AR, Amin OE, Rodriguez-Tajes S, Patel AA, Zakeri N, Jeffery-Smith A, Swadling L, Schmidt NM, Baiges A, et al. (2020). Longevity and replenishment of human liver-resident memory T cells and mononuclear phagocytes. J Exp Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, et al. (2017). Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira ER, Kedrin D, Seano G, Gautier O, Meijer EFJ, Jones D, Chin SM, Kitahara S, Bouta EM, Chang J, et al. (2018). Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science 359, 1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma YA, and Trapnell C. (2017a). Single-cell mRNA quantification and differential analysis with Census. Nat Methods 14, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C. (2017b). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading JL, Galvez-Cancino F, Swanton C, Lladser A, Peggs KS, and Quezada SA (2018). The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol Rev 283, 194–212. [DOI] [PubMed] [Google Scholar]
- Reinhardt RL, Liang HE, and Locksley RM (2009). Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 10, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RS, June CH, Langer R, and Mitchell MJ (2019). Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov 18, 175–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Forster R, Lipp M, and Lanzavecchia A. (1999). Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712. [DOI] [PubMed] [Google Scholar]
- Schenkel JM, Fraser KA, and Masopust D. (2014). Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J Immunol 192, 2961–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, and Matloubian M. (2006). CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440, 540–544. [DOI] [PubMed] [Google Scholar]
- Shitara K, Ueha S, Shichino S, Aoki H, Ogiwara H, Nakatsura T, Suzuki T, Shimomura M, Yoshikawa T, Shoda K, et al. (2019). First-in-human phase 1 study of IT1208, a defucosylated humanized anti-CD4 depleting antibody, in patients with advanced solid tumors. J Immunother Cancer 7, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R. (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JH, Zhang H, Moseman EA, Alvarez D, Iannacone M, Henrickson SE, de la Torre JC, Groom JR, Luster AD, and von Andrian UH (2012). Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 150, 1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teulings HE, Limpens J, Jansen SN, Zwinderman AH, Reitsma JB, Spuls PI, and Luiten RM (2015). Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol 33, 773–781. [DOI] [PubMed] [Google Scholar]
- Tietze JK, Angelova D, Heppt MV, Reinholz M, Murphy WJ, Spannagl M, Ruzicka T, and Berking C. (2017). The proportion of circulating CD45RO(+)CD8(+) memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. Eur J Cancer 75, 268–279. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, and Houghton AN (2004). Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med 200, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varn FS, Andrews EH, Mullins DW, and Cheng C. (2016). Integrative analysis of breast cancer reveals prognostic haematopoietic activity and patient-specific immune response profiles. Nat Commun 7, 10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varn FS, Wang Y, Mullins DW, Fiering S, and Cheng C. (2017). Systematic Pan-Cancer Analysis Reveals Immune Cell Interactions in the Tumor Microenvironment. Cancer Res 77, 1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr., Lao CD, et al. (2015). Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16, 375–384. [DOI] [PubMed] [Google Scholar]
- Zhang P, Cote AL, de Vries VC, Usherwood EJ, and Turk MJ (2007). Induction of postsurgical tumor immunity and T-cell memory by a poorly immunogenic tumor. Cancer Res 67, 6468–6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mouse Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. De-identified human scRNAseq data have been deposited at the dbGAP. They are available upon request with IRB approval. To request access, please contact the NIH dbGAP administrator. All database accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies/Tetramer/Beads | ||
| PE/Cy7 anti-mouse CD69 | Biolegend | Cat# 104512; RRID: AB_493564 |
| BV510 anti-mouse CD62L | Biolegend | Cat# 104441; RRID: AB_2561537 |
| FITC anti-mouse/human CD44 | Biolegend | Cat# 103005; RRID: AB_312956 |
| BV421 anti-mouse CD103 | Biolegend | Cat# 121422; RRID: AB_2562901 |
| APC/Cy7 anti-rat CD90/mouse CD90.1 (Thy-1.1) | Biolegend | Cat# 202519; RRID: AB_2201418 |
| PE/Dazzle 594 anti-mouse CD8a | Biolegend | Cat# 100762; RRID: AB_2564027 |
| APC anti-mouse CD45.1 | eBioscience | Cat# 17-0453-82; RRID: AB_469398 |
| PE anti-mouse/human CD44 | Biolegend | Cat# 103008; RRID: AB_312959 |
| BV711 anti-mouse CD186 (CXCR6) | Biolegend | Cat# 151111; RRID: AB_2721558 |
| APC anti-mouse CD183 (CXCR3) | Biolegend | Cat# 126512; RRID: AB_1088993 |
| FITC anti-mose CD127 (Il7ra) | eBioscience | Cat# 11-1271-82; RRID: AB_465195 |
| BV421 anti-mouse CD169 (Siglec-1) | Biolegend | Cat# 142421; RRID: AB_2734202 |
| BV421 anti-mouse/human CD45R/B220 | Biolegend | Cat# 151111; RRID: AB_2721558 |
| AF488 anti-mouse CD103 Antibody | Biolegend | Cat# 121408; RRID: AB_535950 |
| AF647 anti-rat CD90/mouse CD90.1 (Thy-1.1) | Biolegend | Cat# 202508; RRID: AB_492884 |
| PE TRP-2 tetramer(iTAg H-2Kb TRP-2 Tetramer-SVYDFFVWL-P) | MBL International | Cat# TB-5004-1 |
| Anti-PE beads | MACS Miltenyi | Cat# 130-048-801 |
| Purified anti-mouse CD8a | Biolegend | Cat# 100702; RRID: AB_312741 |
| Depleting anti-CD4 (GK1.5) | Bioxcell | Cat# BE0003-1; RRID: AB_1107636 |
| Depleting anti-CD8 (2.43) | Bioxcell | Cat# BE0061; RRID: AB_1125541 |
| Human TruStain FcX | Biolegend | Cat# 422301 |
| BV711 anti-human CD45 | Biolegend | Cat# 304050; RRID: AB_2563466 |
| PE-Dazzle 594 anti-human CD8 | Biolegend | Cat# 344744; RRID: AB_2566515 |
| DAPI | Biolgend | Cat# 422801 |
| anti-human CD8 | Leica Biosystem | Cat# NCL-CD8 (4b11); RRID: AB_563637 |
| anti-human CD69 | Abcam | Cat# ab233396 |
| AF555 anti-human CD8 | Biolgend | Cat# 344702; RRID: AB_1877104 |
| BV421 anti-human CD69 | Biolgend | Cat# 310929; RRID: AB_2561909 |
| Primary anti-human CD103 | ThermoFisher | Cat# 14-1038-82; RRID: AB_467412 |
| AF488 anti-mouse Ig-g | Jackson Immuno Research | Cat# 115-547-003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase Type IV | Worthington Biochemical | Cat# CLSS-4 |
| Collagenase Type II | Sigma | Cat# C6885 |
| Dnase I | Sigma | Cat# 10104159001 |
| BSA | Sigma-Aldrich | Cat# 12659-100GM |
| Liberase | Roche | Cat# 5401020001 |
| Percoll | GE | Cat# 17089101 |
| D-Luciferin, Potassium Salt | GoldBio | Cat# LUCK-3G |
| RPMI 1640 | Corning | Cat# 10-040-CV |
| HBSS | VWR | Cat# VWRL0121-0500 |
| Live Dead Kit | ThrermoFisher | Cat# L34962 |
| Bond Dewax Solution | Leica Biosystem | Cat# AR9222 |
| Bond Epitope Retrieval | Leica Biosystem | Cat# AR9590 |
| Tissue Tek Glas | Sakura | Cat# 6419 |
| Critical Commercial Assays | ||
| AF555 Antibody Labeling Kit | ThermoFisher | Cat# A20187 |
| ChromPlex Kit | Leica Biosystem | Cat# DS9665 |
| Vina Green HRP Chromogen Kit | Biocare | Cat# BRR807 |
| Anti-PE microbeads | MACS Miltenyi | Cat# 130-048-801 |
| Anti-CD8a microbeads | MACS Miltenyi | Cat# 130-117-044 |
| RNA extraction Qiagen RNeasy plus micro kit | Quiagen | Cat# 74034 |
| SMART-Seq v4 Ultra Low Input RNA Kit | ClonTech | Cat# 638490 |
| Nextera DNA flex Library prep kit | Illumina | Cat# 20018704 |
| 10× Single cell 3’ V2 chemistry | 10× Genomics | Cat# PN-120237 |
| Deposited Data | ||
| Bulk Rna-Seq (Fig. 1) | This Paper | GEO GSE169237 |
| scRNA-Seq pmel (Fig. 3) | This Paper | GEO GSE171161 |
| scRNA-Seq endogenous (Fig. 5) | This Paper | GEO GSE169238 |
| scRNA-seq human (Fig. 7) | This Paper | dbGap PHS002309.V1.P1 |
| Experimental Models: Cell Lines | ||
| B16-F10 | Isaiah Fidler, MD Anderson Cancer Center | N/a |
| B16-25K | This paper | N/a |
| B16-Luciferase | Dr. Yina Huang, Dartmouth College | N/a |
| Experimental Models: Organisms/Strains | ||
| C57BL/6NCrl | Charles River Laboratories | Cat# CRL27, RRID: IMSR_CRL:27 |
| B6 CD45.1 | Charles River Laboratories | Cat# CRL:564, RRID: IMSR_CRL:564 |
| B6 Thy1.1 | The Jackson Laboratory | Cat# JAX:000406, RRID: IMSR_JAX:000406 |
| B6 Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J | The Jackson Laboratory | Cat# JAX:005023, RRID: IMSR_JAX:005023 |
| C.129S4(B6)-Ifngtm3.1Lky/J | The Jackson Laboratory | Cat# JAX:017580, RRID: IMSR_JAX:017580 |
| Software and Algorithms | ||
| FlowJo v10 | Treestar Inc | RRID: SCR_008520 |
| Prism 8 | Graphpad Inc | RRID: SCR_002798 |
| Seurat v3.0 | https://satijalab.org/seurat/get_started.html | RRID: SCR_016341 |
| Cell Ranger | 10X Genomics | RRID: SCR_017344 |
| Morpheus | Broad Institute | RRID: SCR_017386 |
| RSEM | Bo Li and Colin Dewey | RRID:SCR_013027 |
| ImageJ | Schneider et al. 2012 | RRID: SCR_018407 |
| RStudio | N/A | https://www.rstudio.com/ |
| Monocle 2.0 | Trapnell Lab | http://cole-trapnell-lab.github.io/monocle-release/ |
| velocyto.R (RNA Velocity) | Kharchenlo Lab | https://github.com/velocyto-team/velocyto.R |