

Abstract
Naturally occurring compounds represent a vast pool of pharmacologically active entities. One of such compounds is andrographolide, which is endowed with many beneficial properties, including the activity against severe acute respiratory syndrome coronavirus type 2 (SARS‐CoV‐2). To initiate a drug repurposing or hit optimization campaign, it is imperative to unravel the primary mechanism(s) of the antiviral action of andrographolide. Here, we showed by means of a reporter gene assay that andrographolide exerts its anti‐SARS‐CoV‐2 effects by inhibiting the interaction between Kelch‐like ECH‐associated protein 1 (KEAP1) and nuclear factor erythroid 2‐related factor 2 (NRF2) causing NRF2 upregulation. Moreover, we demonstrated that subtle structural modifications of andrographolide could lead to derivatives with stronger on‐target activities and improved physicochemical properties. Our results indicate that further optimization of this structural class is warranted to develop novel COVID‐19 therapies.
Keywords: SARS-CoV-2, andrographolide, KEAP1/NRF2, natural products, medicinal chemistry
Synthetic andrographolide derivatives were synthesized and found to be potent activators of the KEAP1/NRF2 pathway and inhibitors of SARS‐CoV‐2 replication. Our Medicinal Chemistry efforts yielded highly selective NRF2 activators that are also accessible for the synthesis of tailored semisynthetic drugs or functional molecules.

Introduction
Andrographolide, a diterpenoid lactone with labdane skeleton (Scheme 1), is a well‐studied phytochemical drug that shows a variety of pharmacological effects. [1] In Ayurvedic medicine, herbal extracts from Andrographis paniculata (AP) containing andrographolide are used as a treatment for the common cold due to their anti‐inflammatory and anti‐infective properties. [2] Because of these particular features, andrographolide has also been studied for its antiviral activity against human immunodeficiency virus, influenza A virus, hepatitis B virus, and other viridae. [3] Recent studies revealed that andrographolide exerts most of its cellular effects by modulating the Kelch‐like ECH‐associated protein 1 – nuclear factor erythroid 2‐related factor 2 – antioxidant response element (KEAP1‐NRF2‐ARE) axis.[ 4 , 5 , 6 ] The E3 ligase KEAP1 regulates oxidative stress and detoxification reactions by controlling the levels of the NRF2. The transcription factor NRF2 itself is one of the main orchestrators of the cellular xenobiotic and oxidative stress response. KEAP1 belongs to the Cullin‐RING ligase family and contains an N‐terminal bric‐a‐brac/tram‐track/broad (BTB) domain for CUL3 binding and a C‐terminal Kelch domain for substrate recruitment. The BTB domain is typically homodimeric and therefore associated with two copies of CUL3 (Figure 1).[ 7 , 8 ] Activators of the KEAP1‐NRF2‐ARE axis covalently interact with specific cysteine residues of KEAP1 and thus mimic naturally occurring electrophiles. A modification of Cys151 is considered to alter the KEAP1‐CUL3 interaction, leading to accumulated levels of NRF2.[ 9 , 10 ] A variety of potent electrophilic NRF2 activators are depicted in Scheme 1.
Scheme 1.

Chemical structures of the NRF2 activators andrographolide (1), 14‐deoxy‐14,15‐didehydroandrographolide (2), 14‐deoxy‐11,12‐didehydro‐andrographolide (3), bardoxolone‐methyl (CDDO−Me, 4), sulforaphane (5), and dimethyl fumarate (6). Partial carbon numbering is assigned for andrographolide.
Figure 1.
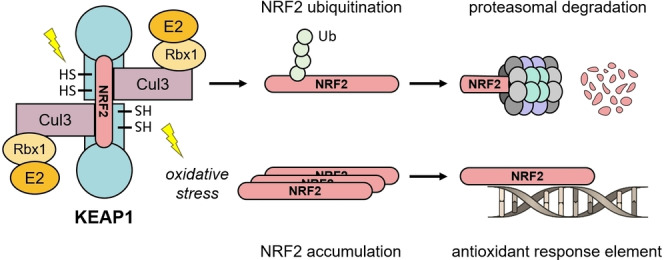
Schematic representation of the KEAP1/NRF2 complex. Under normal conditions, NRF2 undergoes ubiquitination by the CUL3 E3 ligase complex. Polyubiquitination marks the protein for degradation via the proteasome. In case of oxidative stress or chemical modification of specific cysteine residues in the BTB domain of KEAP1, NRF2 ubiquitination is prevented. NRF2 accumulation leads to induced binding to antioxidant response elements (ARE) and subsequent gene transcription.
These comprise synthetic compounds such as the cyanoenone derivative bardoxolone‐methyl (CDDO−Me) and naturally occurring cysteine‐reactive compounds such as andrographolide, disulfiram, carnosol, sulforaphane, and dimethyl fumarate.[ 11 , 12 , 13 , 14 ]
In 2019, the outbreak of the severe acute respiratory syndrome coronavirus type 2 (SARS‐CoV‐2) became a life‐threatening global health issue. Even though the development of potent vaccines has massively reduced the health risks to people, there is an unmet medical need for therapeutics, especially for patients with associated comorbidities and immunodeficiencies.[ 15 , 16 ] Furthermore, tool compounds are needed to investigate the pathophysiology of COVID‐19 infections. Early drug discovery and repurposing campaigns targeting COVID‐19 included the phytoconstituents of AP.[ 3 , 17 ] Bioactive compounds isolated from AP, such as its main ingredient andrographolide, showed potent anti‐SARS‐CoV‐2 activities in plaque reduction assays.[ 18 , 19 ] However, the mechanism of action in this context remains unclear. Andrographolide was found to be a powerful NRF2 activator, but evidence for the correlation of this activity with the observed antiviral effect is still lacking. The present study contributes to developing new antiviral drugs based on the andrographolide scaffold and provides a novel molecular mechanism of action for the treatment of SARS‐CoV‐2 infections, showing that the blockade of the KEAP1‐NRF2 pathway constitutes a new therapeutic option for the fight against COVID‐19.
Results and Discussion
Various andrographolide derivatives have previously been synthesized and investigated for anti‐cancer, anti‐inflammatory, anti‐bacterial, or antiviral properties.[ 20 , 21 , 22 ] However, most compounds probably do not bind to KEAP1 due to steric clashes or inactivation of the critical Michael acceptor group. We generated a focused andrographolide library and paid particular attention to modulating physicochemical properties such as lipophilicity and solubility while maintaining the proposed binding mode into the BTB domain of KEAP1. With tailored structural modifications in the vicinity of activity‐determining groups, structure‐activity relationships could be examined providing valuable information.
Andrographolide (1) was obtained from Soxhlet extraction of powdered Andrographis paniculata leaves with MeOH or by dissolution of 1 from standardized extracts. Subsequently, an isopropylidene protecting group was installed at the two adjacent alcoholic groups of 1 to give building block 7 (Scheme 2). [23] 14ß‐Andrographolide (9) with inverted stereochemistry at C‐14 was obtained via Mitsunobu reaction of 7 and acetic acid, accompanied by a Walden inversion and the subsequent acidic deprotection of 8. Regioselective reduction of the Δ12,13 double bond of 7 with nickel chloride/sodium borohydride, [24] followed by deprotection of the acetal group in 10, gave the reduced compound 11, which is presumably unreactive towards Cys151 of KEAP1. The tert‐butyldimethylsilyl (TBDMS) protection of the remaining OH group at C‐14 yielded 12 after transacetalization with MeOH. As carbon C‐19 was expected to be a suitable position for late‐stage functionalization, 12 was subjected to a mild two‐step oxidation sequence via the intermediate aldehyde 13. Final removal of the silyl ether protecting group with acetyl chloride in dry MeOH gave 15 (Andro‐CO2H) as a common building block for corresponding carboxamides. Its structure was unambiguously confirmed employing NMR spectroscopy and X‐ray crystallography. [25]
Scheme 2.

Synthesis of andrographolide derivatives. Reagents and conditions: (a) 2,2‐Dimethoxypropane, p‐TsOH, toluene, DMSO, rt, 6 h; (b) PPh3, DIAD, AcOH, THF, rt, 18 h; (c) p‐TsOH, MeOH, H2O, 40 °C, 4 h; (d) NiCl2, NaBH4, MeOH, 0 °C, 10 min; (e) (i) TBDMSCl, imidazole, DMF, rt, 18 h; (ii) AcOH, H2O, rt, 2 h; (f) TEMPO, TBAI, NCS, CH2Cl2, K2CO3/NaHCO3 buffer pH 8.6, rt, 18 h; (g) 2‐methyl‐2‐butene, NaClO2, NaH2PO4, tBuOH, THF, H2O, rt, 18 h; (h) AcCl, MeOH, rt, 1 h; (j) R‐NH2, HATU, DIPEA, DMF, rt, 18 h.
Recent pharmaceutical studies were aimed at improving the poor bioavailability and solubility of andrographolide via appropriate delivery systems.[ 26 , 27 ] However, appropriate chemical modifications of andrographolide could be an even better option to overcome such issues. Therefore, three different amide derivatives (16 a–16 c) were synthesized from linear aliphatic or aromatic amines. The attachment of a short lipophilic chain in 16 a and 16 b was thought to increase membrane permeability, whereas the polyethylene glycol (PEG) unit was expected to improve solubility of the resulting derivative 16 c.
Next, physicochemical properties of the test compounds were determined to assess their drug‐like characteristics. Table 1 summarizes the experimentally determined lipophilicity index logD7.4 and plasma protein binding data as well as the calculated topological polar surface area. Modification at the labdane scaffold by introduction of the amide residues in 16 influenced the molecular properties. Both compounds 16 a and 16 b displayed increased lipophilicity compared to 1. The presence of the hydrophilic PEG chain in 16 c decreased the logD value. The polar surface area of andrographolide derivatives spans from 67 to 133 Å2. Prior studies revealed andrographolide to be primarily bound to plasma proteins, limiting the amount of drug entering the cells. [28] Interestingly, the 2‐phenylethyl amide derivative 16 b was significantly less bound to human serum albumin than andrographolide, demonstrating that this property can be modulated through such a structural modification.
Table 1.
Physicochemical properties and biological data of andrographolide and derivatives.
|
Compd |
logD7.4 [a] |
TPSA[b] [Å2] |
%PPB[c] |
MM‐GBSA ΔG Bind[d] [kcal/mol] |
ARE specificity ratio, SR[e] |
Vero‐E6 plaque reduction assay NT50 [f] [μM] |
|---|---|---|---|---|---|---|
|
1 |
1.4 |
87 |
88 |
−26.2 |
19.4 |
1.4 |
|
2 |
2.4 |
67 |
93 |
−29.4 |
5.7 |
>10 |
|
3 |
2.4 |
67 |
93 |
−31.8 |
n.d.[g] |
>10 |
|
4 |
n.d. |
n.d. |
n.d. |
−45.8 |
n.d. |
n.d. |
|
9 |
1.4 |
87 |
88 |
−29.4 |
211 |
2.1 |
|
11 |
n.d. |
87 |
n.d. |
n.a.[h] |
n.d. |
>10 |
|
16 a |
1.9 |
96 |
83 |
−27.3 |
35.6 |
3.7 |
|
16 b |
2.5 |
96 |
38 |
−36.4 |
10.8 |
8.1 |
|
16 c |
1.3 |
133 |
92 |
n.a. |
13.3 |
>10 |
[a] Experimental distribution coefficient at pH 7.4. [b] Topological polar surface area. [c] Plasma protein binding values were estimated by an HPLC‐based method. [d] An estimate for the binding affinity derived from computational docking. The estimate does not include contributions from the covalent interaction. A more negative value indicates stronger binding. [e] SR≤1, not specific; 1≤SR≤10, moderately specific; 10≤SR≤100, specific; 100≤SR, highly specific. See also equation 4 in the Supporting Information. [f] Analyte titers at which 50 % neutralization (NT50) was recorded. [g] Not determined. [h] Not available. The binding affinity cannot be reliably estimated: 11 is a non‐electrophilic compound; 16 c has a long and flexible solvent‐exposed linker.
To illustrate the potential of 1 and semi‐synthetic derivatives to bind to the BTB domain of KEAP1, covalent docking simulations were performed. Particular attention was paid to possible interactions with Cys151, which displays the highest reactivity towards electrophiles. Cys151‐targeting compounds such as CDDO−Me and andrographolide may lead to less pronounced NRF2 ubiquitination through a conformational change in the KEAP1‐CUL3 complex.[ 6 , 9 , 12 , 29 ] Binding site alignments of the BTB domain of KEAP1 co‐crystallized with bardoxolone (PDB: 4CXT) [30] and of the apo form of KEAP1 (PDB: 4CXI) show the shallow binding site with some flexible residues (e. g., Arg135, Figure S1) and only limited options for interactions. The covalent docking approach was validated by re‐docking of bardoxolone (CDDO) and CDDO−Me to the BTB domain and comparing the docked and original (PDB: 4CXI) ligand poses (Figure S2). Subsequently, docking with andrographolide (1) and 14‐deoxy‐14,15‐didehydroandrographolide (2) was processed, leading to a pose in which the furan ring is incorporated into a hydrogen‐bond network between Gly148 and the carbonyl group (Figure 2A and 2B). A constraint for a hydrogen bond with Gly148 was applied when docking other compounds. Interestingly, the C‐14 hydroxyl group of 1 was not observed to be involved in interactions with the BTB domain of KEAP1 (Figure S3).
Figure 2.

Covalent docking into the BTB domain of KEAP1 (PDB: 4CXT). Cys151 is represented with yellow surface, Gly148 with blue surface and a dashed yellow line represents a hydrogen bond. (A) Andrographolide (1, cyan). (B) 14‐Deoxy‐14,15‐didehydroandrographolide (2, magenta). (C) The orientation of the furan ring of enantiomer 9 (yellow) deviates from 1 (cyan) but is close to 2 (magenta). (D) Overlay of docking poses of 1 (cyan), 16 a (blue) and 16 b (green).
However, mechanistic considerations highlighted the essentiality of this group in a Michael‐addition/water elimination reaction cascade, forming a covalent adduct of 1 with the target protein. [1] Inversion of the stereocenter in 9 led to a docking pose similar to that of 14‐deoxy‐14,15‐didehydroandrographolide (Figure 2C). Modifications at C‐19 did not significantly influence the docking pose of 16 a and 16 b (Figure 2D), demonstrating that this exit vector might be suitable for the design of BTB‐targeting probes and inhibitors. To estimate the relative binding affinity of the andrographolide derivatives, MM‐GBSA scoring was performed after covalent docking (Table 1). [31] Of all AP phytoconstituents and semi‐synthetic andrographolide derivatives tested, the free‐energy gain by 16 b scored best but did not reach the high value of CDDO.
For the quantitative assessment of the NRF2‐ARE pathway activation, we utilized the AREc32 cell line, derived from the MCF7 cell line, in which ARE is linked to a luciferase reporter gene. [32] The induction of NRF2 and subsequent stimulation of ARE is proportional to the amount of luciferase produced by the cells. The assay was performed as described previously, [33] but with modifications for high‐throughput screening, [34] and cytotoxicity was assessed via cell confluency by an imaging method. [35] The capability of a compound to cause NRF2 activation is expressed as the induction ratio (IR), defined as the ratio of the sample signal divided by the average luminescence of the unexposed cells. For all test compounds, IR and percentage of cell viability were determined at concentrations ranging from 1×10−15 M to 1×10−2 M and measured IRs were plotted against the concentrations (Figure S4). The ECIR1.5 (effective concentration causing an IR of 1.5, i. e., 50 % over the control IR of 1, see equation 2 in the Supporting Information) was derived by a linear concentration‐response model (Table S1). Tert‐butylhydroquinone, an established inducer of oxidative stress, was used as a positive control and its ECIR1.5 of 3.4±0.2 μM was consistent with previous studies. [33] The inhibitory concentration inducing 10 % reduction of cell confluency (IC10, see equation 1 in the Supporting Information) was used as measure of cytotoxicity. [35] By comparison of the experimental IC10 with the IC10,baseline predicted with a baseline toxicity QSAR (see equation 3 in the Supporting Information), [36] it was shown that none of the compounds (except for 16 c) was toxic, because their values approached baseline toxicity (toxicity ratio, TR <10, see equation 5 in the Supporting Information). Finally, the specificity ratios (SR, see equation 4 in the Supporting Information) were calculated, which describe the selectivity of the oxidative stress response relative to the compound's cytotoxicity. Low TR and high SR values are most beneficial for a compound. While andrographolide (1) had a moderate SR value of 19.4 (Table 1), the semi‐synthetic 14ß‐andrographolide (9) had the highest SR measured, in comparison with 332 bioactive chemicals of a total of 7214 tested compounds from the 10 K library of Tox21 that have been screened in the ARE‐BLA assay (Figure 3). [37] Amide‐functionalized compounds 16 a–16 c were able to intrinsically induce ARE activation (SR ∼11 to 36), but, as expected, 2 and 11 lacked this specific mode of action.
Figure 3.

Comparison of the specificity of ARE activation, expressed as the specificity ratio (SR=IC10/ECIR1.5), by andrographolide derivatives and chemicals from the Tox21 dataset, evaluated with a similar assay (ARE‐BLA). [37]
To exclude that andrographolide induced upregulation of NRF2 via modulation of GPCR‐mediated signaling, [38] we studied its interaction with human adenosine A2A receptors. Of note, several terpene‐derived natural products have previously been described to modulate adenosine receptor binding.[ 39 , 40 ] Andrographolide was tested at different concentrations ranging from 10 nM to 100 μM in radioligand binding assays at human adenosine A2A receptors recombinantly expressed in Chinese hamster ovary (CHO−S) cells. Cell membrane preparations and the A2A‐selective agonist radioligand [3H]CGS‐21680 were employed. Only at the highest concentration of 100 μM, andrographolide showed moderate affinity for the receptor. The K i value was estimated to be well above 100 μM (33 % inhibition at 100 μM, 7 % inhibition at 10 μM, n=3).
Andrographolide is known for its polypharmacological effects. [1] In the context of compound promiscuity analysis, the Δ12,13 double bond of 1 might potentially trigger unspecific binding events. To refute a possible multi‐target Michael acceptor mechanism for 1, we performed several in vitro cysteine reactivity assays with andrographolide (1) and 14‐deoxy‐14,15‐didehydroandrographolide (2). Firstly, the spectrophotometric Ellman's assay was employed that uses 2‐nitro‐5‐thiobenzoate dianion (TNB2−) as a cysteine thiol surrogate (Figure S5). [41] However, under these conditions, i. e. 100 μM compound, 50 μM TNB2−, 37 °C, 12 h, compounds 1 and 2 did not react with TNB2−.
Secondly, potential adduct formation with N‐acetyl‐cysteine (NAC) or glutathione was investigated. Monitoring the reactions by HPLC/MS analyses did not reveal the formation of such adducts (Table S2). Additionally, an NMR method was utilized to identify possible thiol additions. [42] Whereas 1, evidently, did not react with NAC even at elevated temperatures (Figure S6), CDDO−Me underwent temperature‐dependent addition of NAC, as reported previously. [43] The results from our cysteine‐reactivity assays demonstrated that andrographolide is not reactive under conditions typically used to discover (hyper)reactive electrophilic species. Hence, we postulate that andrographolide‘s high specificity for the KEAP1‐NRF2 axis might be due to a covalent trapping reaction that occurs only after tight binding into the pocket of the BTB domain and is therefore highly specific. It has been reported that Cys151 attacks the Δ12,13 double bond of 1, and the intermediate adduct becomes irreversibly bound after elimination of water. [22]
Finally, the anti‐SARS‐CoV‐2 activities of andrographolide and semi‐synthetic derivatives were evaluated in a plaque reduction assay. Vero‐E6 cells were inoculated with SARS‐CoV‐2 (B.3 strain), and the formation of plaques in the cell lawn under methylcellulose overlay was monitored in the presence and absence of test compounds. Andrographolide (1), 14ß‐andrographolide (9), and 16 a showed dose‐dependent inhibition of SARS‐CoV‐2 replication with NT50 values in the low micromolar range (Figure 4), which is in agreement with previous results on 1.[ 18 , 19 ]
Figure 4.

Assessment of SARS‐CoV‐2 inhibition and cytotoxicity of andrographolide derivatives. Andrographolide (A), 14ß‐andrographolide (B), and 16 a (C). All compounds were titrated in triplicates between 0.5 and 10 μM. A representative well is shown for each condition. As vehicle control, DMSO (D) was used. The depicted graphs show the infectivity of the same PFU of virus relative to the untreated control wells according to the decrease in plaques. At 10 μM, 1 and 9 inhibited cell proliferation, as visible in less intense crystal violet staining of these wells. Both compounds did not show toxicity on the attached Vero‐E6 cells at any concentration and no detached cells were observed. Infectivity data represent mean ±SD of biological triplicates. Cell viability was assessed in duplicates.
Importantly, none of the compounds showed cytotoxicity at the concentrations tested (CC50 >10 μM). These three KEAP1 binders were also among the best activators of the KEAP1‐NRF2 axis (Table 1). In contrast, inhibitors lacking NRF2 activation properties did not possess anti‐SARS‐CoV‐2 activities (i. e., 2, 3, and 11). In previous studies, experimental evidence was provided that andrographolide and related derivatives inhibit the main protease (Mpro) of SARS‐CoV and SARS‐CoV‐2 in vitro.[ 44 , 45 ] Accordingly, we investigated whether our newly synthesized analogs mediate their anti‐SARS‐CoV‐2 activity through inhibiting Mpro. Compounds were subjected to our in vitro assay and the cleavage of a fluorogenic SARS‐CoV‐2 Mpro substrate was monitored. [46] Surprisingly, neither the primary herbal constituents of AP nor semi‐synthetic derivatives inhibited this protease at a high concentration of 20 μM (Figure S7).
Conclusion
In conclusion, in the present study, a focused library of andrographolide derivatives was generated and systematically investigated for their capability to activate the KEAP1‐NRF2 pathway and to inhibit replication of SARS‐CoV‐2 in virus‐infected cells. We confirmed previous reports on the anti‐SARS‐CoV‐2 activities of AP phytoconstituents,[ 18 , 19 ] and provided the first evidence that this activity may be correlated to NRF2 activation. NRF2 directly or indirectly (via ARE) interacts with several essential proteins for host cell entry, replication, or defense of SARS‐CoV‐2. These comprise the receptor protein angiotensin‐converting enzyme 2, heme oxygenase 1, and interferon regulatory factor 3. [47] NRF2 downregulates interferon production and may attenuate inflammatory cell damage. As NRF2 gene expression was suppressed in COVID‐19 patients, NRF2 activation with small‐molecule inhibitors is an attractive antiviral approach while limiting overflowing inflammatory host responses. [48] Furthermore, the production of biological antioxidants via ARE could help to balance symptoms induced by the cytokine storm after viral entry. [49] With respect to clinical applications, encouraging results on the use of AP extracts to treat SARS‐CoV‐2 infections were recently published.[ 50 , 51 ] Our medicinal chemistry efforts revealed functional sites of the andrographolide scaffold that can be fine‐tuned to increase NRF2 activation or to modulate physicochemical properties. Notably, andrographolide derivatives displayed negligible cytotoxicity in AREc32 and Vero‐E6 cells. Our work paves the way for the discovery of novel COVID‐19 therapies. Compound 14ß‐andrographolide (9) was identified to be an exceptionally potent inhibitor of the KEAP1/NRF2 interaction, and this ligand might also be of general interest in developing chemical probes covalently interacting with KEAP1. As E3 ligase binders, andrographolide and its derivatives might also be utilized for targeted protein degradation, an emerging area in drug development. [52] Protein degraders are on the rise in developing novel therapies for the treatment of microbial and viral diseases,[ 53 , 54 ] and we assume that KEAP1 degradation could represent a promising strategy to combat SARS‐CoV‐2 infections.
Experimental Section
Chemical synthesis
General remarks
Column chromatography was performed using Merck silica gel 60 or an automated flash chromatography system puriFlash XS 520Plus. Melting points were determined on a Büchi 510 oil bath apparatus and were uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 500 MHz NMR spectrometer or on a Bruker Avance III 600 MHz NMR spectrometer, respectively. NMR spectra were processed and analysed in MestReNova. Chemical shifts are given in parts per million (ppm), coupling constants J are given in Hertz, and spin multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet) or m (multiplet). All multiplets related with J(C,F) couplings in 13C NMR spectra are centred. In the case of overlapping extraneous solvent peaks, multiplet analyses in 1H NMR spectra were performed using qGSD (quantitative Global Spectral Deconvolution). HRMS was recorded on a micrOTOF−Q mass spectrometer (Bruker) with ESI‐source coupled with an HPLC Dionex UltiMate 3000 (Thermo Scientific). The purity and identity of the compounds were determined by HPLC‐UV obtained on an LC–MS instrument (Applied Biosystems API 2000 LC/MS/MS, HPLC Agilent 1100). The purity of all the final compounds was confirmed to be ≥95 %. Reference substances were obtained from Selleckchem, MedChemExpress, or Sigma Aldrich.
Andrographolide (1)
This compound was isolated from plant material as described in the Supporting Information. Yield: 9 % (m/m); m.p. 230–232 °C; lit. m.p. 230–231 °C; Rf =0.44 (10 % MeOH/CH2Cl2); MS (ESI) m/z [M+H]+ calcd for C20H30O5, 351.22; found, 351.0; HPLC purity 99.7 %.
14‐Deoxy‐14,15‐didehydroandrographolide (2)
This compound was isolated from plant material as described in the Supporting Information. Yield: 1.6 % (m/m); m.p. 196–200 °C; lit. m.p. 205–208 °C; Rf =0.53 (10 % MeOH/CH2Cl2); MS (ESI) m/z [M+H]+ calcd for C20H28O4, 333.2 found, 333.0; HPLC purity 99.9 %.
(3E,4S)‐3‐[2‐[(4aR,6aS,7R,10bR)‐3,3,6 a,10 b‐Tetramethyl‐8‐methylene‐1,4 a,5,6,7,9,10,10 a‐octahydronaphtho[2,1‐d][1,3]dioxin‐7‐yl]ethylidene]‐4‐hydroxy‐tetrahydrofuran‐2‐one (7)
Andrographolide (1) (9.58 g, 27.33 mmol) was dissolved in toluene (190.5 mL) and DMSO (27 mL), p‐TsOH (22 mg, cat.) and 2,2‐dimethoxypropane (13.4 mL, 109 mmol) were added. The mixture was stirred at rt for 6 h. Subsequently, Et3N (6 mL) was added and it was diluted with toluene (125 mL). It was extracted with H2O (3×200 mL), and the combined organic layers were washed with brine (100 mL), dried over Na2SO4, filtered, and evaporated. The residue was suspended in Et2O (2×25 mL) and filtered. The filter cake was dried to obtain the desired product as a light yellow solid. Yield (6.83 g, 64 %); m.p. 192–194 °C; lit. m.p. 191–192 °C; Rf =0.56 (7 % MeOH/CH2Cl2); MS (ESI) m/z [M+H]+ calcd for C23H34O5, 391.24; found, 391.2.
[(3R,4E)‐4‐[2‐[(4aR,6aS,7R,10bR)‐3,3,6 a,10 b‐Tetramethyl‐8‐methylene‐1,4 a,5,6,7,9,10,10 a‐octahydronaphtho[2,1‐d][1,3]dioxin‐7‐yl]ethylidene]‐5‐oxo‐tetrahydrofuran‐3‐yl] acetate (8)
Compound 7 (1.56 g, 4.0 mmol) was dissolved in dry THF (40 mL), and triphenylphosphine (1.57 g, 6.0 mmol), anhydrous acetic acid (0.36 g, 0.34 mL, 6.0 mmol), and diisopropyl azodicarboxylate (1.21 g, 1.18 mL, 6.0 mmol) were added at 0 °C. The combined mixture was allowed to warm to rt and it was stirred for 18 h. The solution was diluted with EtOAc (50 mL), and it was washed with brine (5×25 mL), dried over Na2SO4, filtered, and evaporated. The product was purified by flash chromatography (gradient from 0 to 4 % MeOH in CH2Cl2) with detection at 235 nm to give a colorless solid. Yield (0.37 g, 21 %); m.p. 136–138 °C; lit. m.p. 140–142 °C; Rf =0.29 (3 % MeOH in CH2Cl2); MS (ESI) m/z [M+H2O–H]– calcd for C25H36O6, 449.25; found, 449.3
14β‐Andrographolide (9)
Compound 8 (0.30 g, 0.70 mmol) was dissolved in a 4 : 1 mixture of MeOH and H2O (7 mL), and p‐toluenesulfonic acid monohydrate (14 mg, 0.07 mmol) was added. The mixture was stirred at 40 °C for 4 h. It was diluted with EtOAc (50 mL) and washed with saturated NaHCO3 solution (50 mL). The aqueous layer was again extracted with EtOAc (50 mL), and the combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered, and evaporated. The product was purified by flash chromatography (gradient from 0 to 10 % MeOH in CH2Cl2) with detection at 235 nm to give a colorless solid. Yield (0.21 g, 85 %); m.p. 196–198 °C; lit. m.p. 200–202 °C; Rf =0.50 (10 % MeOH in CH2Cl2); MS (ESI) m/z [M+NH4]+ calcd for C20H30O5, 368.24; found, 368.3; HPLC purity 99.9 %.
(4S)‐3‐[2‐[(4aR,6aS,7R,10bR)‐3,3,6 a,10 b‐Tetramethyl‐8‐methylene‐1,4 a,5,6,7,9,10,10 a‐octahydronaphtho[2,1‐d][1,3]dioxin‐7‐yl]ethyl]‐4‐hydroxy‐tetrahydrofuran‐2‐one (10)
Compound 7 (0.78 g, 2.0 mmol) was dissolved in a 1 : 1 mixture of MeOH and 1,2‐dimethoxyethane (30 mL) and cooled to 0 °C. Nickel(II) chloride hexahydrate (95 mg, 0.40 mmol) was added, followed by the portionwise addition of sodium borohydride (0.38 g, 10 mmol). A black precipitate appeared. After 10 min, H2O (10 mL) was added, and the mixture was transferred into a conical centrifuge tube. It was centrifuged at 10000 × g for 5 min at rt. The supernatant was filtered, further diluted with H2O (50 mL), and extracted with EtOAc (3×50 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered, and evaporated. The product was purified by flash chromatography (gradient from 50 to 100 % MTBE in n‐hexanes) with detection at 210 nm. The product fractions were collected and evaporated, and the crude product was recrystallized from n‐hexanes/EtOAc to give a colorless solid. Yield (0.26 g, 33 %); m.p. 196–198 °C; Rf =0.64 (MTBE); HRMS (ESI) m/z [M+H]+ calcd for C23H36O5, 393.2636; found, 393.2618.
(4S)‐3‐[2‐[(1R,5R,6R,8aS)‐6‐Hydroxy‐5‐(hydroxymethyl)‐5,8 a‐dimethyl‐2‐methylene‐decalin‐1‐yl]ethyl]‐4‐hydroxy‐tetrahydrofuran‐2‐one (11)
Compound 10 (100 mg, 255 μmol) was dissolved in a 4 : 1 mixture of MeOH and H2O (10 mL), and p‐toluenesulfonic acid monohydrate (5 mg, 25 μmol) was added. The mixture was stirred at 40 °C for 4 h. It was diluted with EtOAc (50 mL) and washed with saturated NaHCO3 solution (50 mL). The aqueous layer was again extracted with EtOAc (50 mL), and the combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered, and evaporated. The product was purified by flash chromatography (gradient from 0 to 7 % MeOH in CH2Cl2) and all fractions were collected. Product fractions were identified via TLC staining with phosphomolybdic acid, pooled, and evaporated to give a colorless solid. Yield (75 mg, 83 %); m.p. 216–218 °C; lit. m.p. 219–220 °C; Rf =0.26 (7 % MeOH in CH2Cl2); MS (ESI) m/z [2 M+H]+ calcd for C20H32O5, 705.46; found, 705.6; HPLC purity 96.0 %.
(3E,4S)‐3‐[2‐[(1R,5R,6R,8aS)‐6‐Hydroxy‐5‐(hydroxymethyl)‐5,8 a‐dimethyl‐2‐methylene‐decalin‐1‐yl]ethylidene]‐4‐[tert‐butyl(dimethyl)silyl]oxy‐tetrahydrofuran‐2‐one (12)
Compound 7 (2.07 g, 5.3 mmol) and imidazole (0.69 g, 10.07 mmol) were dissolved in dry DMF (20 mL), and TBDSCl (1.44 g, 9.54 mmol) was added at 0 °C. The mixture was stirred at rt for 18 h. After evaporation of the solvent, it was diluted with EtOAc (100 mL) and washed with H2O (100 mL). The aqueous layer was again extracted with EtOAc (100 mL). The combined organic layers were evaporated and suspended in H2O (3.5 mL). AcOH (8.1 mL) was added and it was stirred at rt for 2 h. Subsequently, it was diluted with CH2Cl2 (50 mL) and slowly saturated NaHCO3 solution (50 mL) was added. The two phases were separated and the organic layer was washed with brine (50 mL), dried over Na2SO4, filtered, and evaporated. The product was purified by flash chromatography (gradient from 10 to 95 % EtOAc in cyclohexane) to give a colorless solid. Yield (1.23 g, 50 %); m.p. 106–110 °C; lit. m.p. 98–99 °C; Rf =0.32 (50 % EtOAc/petroleum ether); MS (ESI) m/z [M+H]+ calcd for C26H44O5Si, 465.29; found, 465.5.
(1S,2R,4aS,5R)‐5‐[(2E)‐2‐[(4S)‐4‐[tert‐Butyl(dimethyl)silyl]oxy‐2‐oxo‐tetrahydrofuran‐3‐ylidene]ethyl]‐2‐hydroxy‐1,4 a‐dimethyl‐6‐methylene‐decalin‐1‐carbaldehyde (13)
This compound was prepared analogous to a previously reported method on the selective oxidation of primary alcohols to aldehydes. [55] Compound 12 (4.12 g, 8.68 mmol) was dissolved in CH2Cl2 (100 mL) and K2CO3/NaHCO3 Buffer (100 mL, pH 8.6). TBAI (0.32 g, 0.87 mmol) and TEMPO (0.14 g, 0.87 mmol) were added at 0 °C and N‐chlorosuccinimide (2.32 g, 17.36 mmol) was added in portions. The orange mixture was stirred at rt for 18 h. Subsequently, the two phases were separated and the aqueous layer was extracted with CH2Cl2 (100 mL). The combined organic phases were washed with brine (100 mL), dried over Na2SO4, filtered, and evaporated. The product was purified by column chromatography (gradient of EtOAc/cyclohexane 2 : 1 to 1 : 1) to give a light yellow solid. Yield (3.94 g, 98 %); m.p. 100–102 °C; lit. m.p. 101–102 °C; Rf =0.33 (50 % EtOAc/petroleum ether); MS (ESI) m/z [M+H]+ calcd for C26H42O5Si, 463.28; found, 463.3.
(1S,2R,4aS,5R)‐5‐[(2E)‐2‐[(4S)‐4‐[tert‐Butyl(dimethyl)silyl]oxy‐2‐oxo‐tetrahydrofuran‐3‐ylidene]ethyl]‐2‐hydroxy‐1,4 a‐dimethyl‐6‐methylene‐decalin‐1‐carboxylic acid (14)
Compound 13 (3.93 g, 8.49 mmol) was dissolved in tBuOH (120 mL) and THF (60 mL) at 0 °C. 2‐Methyl‐2‐butene (18 mL, 170 mmol) and a freshly produced solution of NaClO2 (4.61 g, 51 mmol) and NaH2PO4 (7.95 g, 66 mmol) in H2O (40 mL) were added dropwise. The yellow mixture was stirred at rt for 18 h. Afterwards it was diluted with EtOAc (200 mL) and washed with brine (100 mL). The aqueous layer was extracted with EtOAc (200 mL) and the combined organic layers were dried over Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography (gradient of 0 % to 100 % EtOAc/cyclohexane) to give a colorless solid. Yield (3.25 g, 80 %); m.p. 102–104 °C; lit. m.p. 104–106 °C; Rf =0.24 (50 % EtOAc/petroleum ether); MS (ESI) m/z [M+H]+ calcd for C26H42O6Si, 479.28; found, 479.3.
(1S,2R,4aS,5R)‐2‐Hydroxy‐5‐[(2E)‐2‐[(4S)‐4‐hydroxy‐2‐oxo‐tetrahydrofuran‐3‐ylidene]ethyl]‐1,4 a‐dimethyl‐6‐methylene‐decalin‐1‐carboxylic acid (15)
Several techniques for the deprotection of the TBDMS protecting group were tested. However, only acetyl chloride in dry MeOH achieved good conversions without dehydration of the 14‐OH group. [56] Compound 14 (1.63 g, 3.40 mmol) was dissolved in dry MeOH (30 mL), AcCl (10 drops) were added and the mixture was stirred at 0 °C for 30 min and at rt for 1 h. Subsequently, it was diluted with CH2Cl2 (80 mL), washed with H2O (2×60 mL) and brine (20 mL), dried over Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography (gradient of 0 % to 10 % MeOH/CH2Cl2) to give the desired product as a colorless solid. Yield (0.68 g, 55 %); m.p. 178–182 °C; Rf =0.34 (10 % MeOH/CH2Cl2); HRMS (ESI) m/z [M+H]+ calcd for C20H28O6, 363.1813; found, 363.1815.
General Procedure I for the synthesis of andrographolide‐amide derivatives
Andro‐COOH (15, 55 mg, 0.15 mmol), the corresponding amine (0.30 mmol), and DIPEA (39 mg, 52 μL, 0.30 mmol) were dissolved in dry DMF (4 mL). Subsequently, HATU (63 mg, 0.165 mmol) was added and the mixture was stirred at rt for 18 h. It was diluted with EtOAc (50 mL), washed with half‐saturated NH4Cl solution, and extracted again with EtOAc (50 mL). The combined organic layers were washed with 5 % LiCl solution and brine (each 50 mL), dried over Na2SO4, filtered, and concentrated in vacuo.
(1S,2R,4aS,5R)‐2‐Hydroxy‐5‐[(2E)‐2‐[(4S)‐4‐hydroxy‐2‐oxo‐tetrahydrofuran‐3‐ylidene]ethyl]‐1,4 a‐dimethyl‐6‐methylene‐N‐propyl‐decalin‐1‐carboxamide (16 a)
This compound was prepared using the General Procedure I and n‐propylamine (44 mg, 62 μL, 0.75 mmol). The crude product was purified by flash chromatography (gradient of 60 % to 100 % EtOAc/cyclohexane) to give the title compound as a colorless solid. Yield: 58 %; m.p. 144–146 °C; Rf =0.48 (EtOAc); HRMS (ESI) m/z [M+H]+ calcd for C23H35NO5, 406.2588; found, 406.2570; HPLC purity 99.8 %.
(1S,2R,4aS,5R)‐2‐Hydroxy‐5‐[(2E)‐2‐[(4S)‐4‐hydroxy‐2‐oxo‐tetrahydrofuran‐3‐ylidene]ethyl]‐1,4 a‐dimethyl‐6‐methylene‐N‐(2‐phenylethyl)decalin‐1‐carboxamide (16 b)
This compound was prepared using the General Procedure I and phenethylamine (36 mg, 38 μL). The crude product was purified by flash chromatography (gradient of 13 % to 100 % EtOAc/cyclohexane) to give the desired product as a colorless semi‐solid. Yield: 8 %; Rf =0.47 (EtOAc); HRMS (ESI) m/z [M+H]+ calcd for C28H37NO5, 468.2744; found, 468.2734; HPLC purity 98.6 %.
(1S,2R,4aS,5R)‐2‐Hydroxy‐5‐[(2E)‐2‐[(4S)‐4‐hydroxy‐2‐oxo‐tetrahydrofuran‐3‐ylidene]ethyl]‐N‐[2‐[2‐[2‐(2‐methoxy‐ethoxy)ethoxy]ethoxy]ethyl]‐1,4 a‐dimethyl‐6‐methylene‐decalin‐1‐carboxamide (16 c)
This compound was prepared using the General Procedure I and m‐dPEG®4‐amine (62 mg). The crude product was purified by flash chromatography (gradient of 13 % to 100 % EtOAc/cyclohexane) to give the title compound as a colorless oil. Yield: 41 %; Rf =0.39 (EtOAc); HRMS (ESI) m/z [M+H]+ calcd for C29H47NO9, 554.3324; found, 554.3308; HPLC purity 99.5 %.
Computational docking
For each compound, a 3D structure of one stereoisomer was generated before docking using LigPrep (Schrödinger Suite 2020–2, Schrödinger, LLC, New York, NY, 2020). The BTB domain of KEAP1 in complex with CDDO (PDB: 4CXT29) was prepared using Protein Preparation Wizard. [57] The covalently bound ligand CDDO was extracted, hydrogen atoms were added, residues were protonated at pH 7.0, the hydrogen bonding network was refined, missing side chains (missing atoms for Lys131) were filled in using Prime, [58] and restrained minimization was performed. The receptor‘s grid box was centered on the co‐crystallized ligand CDDO. Covalent docking was performed with the program CovDock, [59] and using the pose prediction mode with the default setup and Cys151 selected as the reactive residue. The reaction type was set to Michael addition, and an additional hydrogen bond constraint with Gly148 (NH) was applied. MM‐GBSA scoring was performed after docking, and the five highest scoring poses were manually inspected to select one that best aligned with the co‐crystallized ligand CDDO. The co‐crystallized ligand was redocked in the active site to validate the docking protocol. Structural visualizations were created in PyMOL 2.4.0.
AREc32 assay
The AREc32 assay was performed as reported,[ 34 , 60 ] and the cell confluency served as surrogate for cell viability as previously described. [35] Briefly, 2650 cells in 30 μL medium were plated in each well of a white 384‐well polystyrene microtiter plate with clear bottom (cat# 3765, Corning, Maine, USA) and incubated for 24 h at 37 °C, 5 % CO2 to allow the cells to attach. All medium components were purchased from Gibco. The medium for the for AREc32 cells was composed of 90 % DMEM+GlutaMAX plus 10 % FBS and 100 U/mL penicillin and 100 μg/mL streptomycin. During the initial 24 h, the cell number did not increase visibly, but cells attached. [35] Plated cell numbers were adjusted to 2650 cells per well that the confluency was around 30 to 50 % prior to dosing and no more than 80 % after 24 h of exposure. [35] Cells were treated with 10 μL/well of the dosing medium containing the test compound and incubated for 24 h.
The detection of activation in the luciferase reporter gene was performed as previously described.[ 33 , 34 , 60 ] Briefly, after 24 h of exposure and measurement of cell confluency, AREc32 cells were washed twice with PBS. Subsequently 10 μL of lysis buffer per well was added (25 mM Tris (AppliChem, A1379,0500), 1 % Triton‐X 100 (Geyer Chemsolute, 8059), 2 mM EDTA (AppliChem, A1104,0500), 2 mM DTT (Sigma‐Aldrich, D0632), 10 % glycerol (AppliChem, A1123,1000)) followed by a 15–20 min incubation at room temperature and shaking at 1500 rpm to allow complete lysis of cells. Afterwards 40 μL of luciferase substrate buffer (20 mM Tricine (Sigma‐Aldrich, T0377), 2.67 mM MgSO4 (AppliChem, 131404.1210), 33.3 mM DTT (Sigma‐Aldrich, D0632), 0.1 mM EDTA (AppliChem, A1104,0500), 0.261 mM coenzyme A (Sigma‐Aldrich, C3144), 0.53 mM ATP (Sigma‐Aldrich, A2383), 0.235 mM D‐luciferin (AREc32) and 0.059 mM D‐luciferin (AhR) (AAT Bioquest, ABD‐12506) was added and luminescence was read with a Tecan Infinite M1000 plate reader. The effect concentration causing an induction ratio (IR) of 1.5 (ECIR1.5) was derived from the linear regression of the IR against the concentration (IR=1+slope×concentration for IR<4). [61]
Cytotoxicity screenings
AREc32: Before dosing and after additional 24 h of exposure the cell confluency was measured with an IncuCyte S3 live cell imaging system (Essen BioScience, Ann Arbor, Michigan, USA). Cytotoxicity was expressed as % inhibition of cell viability as compared to unexposed cells (ratio of confluency of exposed to confluency of unexposed cells). The inhibitory concentration for 10 % cytotoxicity (IC10) was determined from the linear range of the concentration‐cytotoxicity curves (% cytotoxicity=slope × concentration) as described previously. [35] Tert‐butyl‐hydroquinone (tBQH) was used as reference chemical.
Vero‐E6: 1.25×105 Vero‐E6 cells (ATCC, CRL‐1586™) per well were plated in tissue culture‐treated 24‐well plates and incubated over night at 37 °C and 5 % CO2. Cells were either infected with 80 PFU in vitro propagated SARS‐CoV‐2 (variant B.3) in OptiPRO™ serum‐free media for 1 h or left uninfected. Then, virus or media were aspirated and replaced with 1 mL/well of MEM +0.75 % (w/v) carboxymethylcellulose containing andrographolide, its derivatives, or DMSO, and incubated for 72 h at 37 °C and 5 % CO2. Afterwards, the uninfected wells were inspected via light microscope for detached cells, washed with DPBS, detached from the plate by a 3 min 0.25 % (v/v) trypsin‐EDTA treatment, and resuspended in DMEM +10 % FBS. The cells were then pelleted, washed in DPBS, and resuspended in 100 μL Zombie Aqua™ Fixable Viability Dye (BioLegend, 1 : 100 dilution). Staining was performed for 15 min at room temperature in the dark, after which 2 mL DBPS +2 % (v/v) FBS were added, the cells were pelleted, and fixed with 4 % (v/v) formaldehyde in DPBS for 30 min in the dark (room temperature). Finally, cells were washed twice with 2 mL DBPS +2 % (v/v) FBS. As a positive control, one well of uninfected and untreated cells was fixed and permeabilized before viability staining for 30 min with BD Cytofix/Cytoperm™, followed by one wash with 2 mL BD Perm/Wash™.
Plaque reduction assay [62]
Andrographolide and its derivatives were tested for their capacity to inhibit the formation of SARS‐CoV‐2‐induced plaques in confluent Vero‐E6 cells. Vero‐E6 were plated as described above, infected for 1 h with 80 PFU SARS‐CoV‐2/well, and then overlaid with MEM +0.75 % (w/v) carboxymethylcellulose containing andrographolide, its derivatives, or DMSO, and incubated for 72 h at 37 °C and 5 % CO2. The overlay media was then aspirated and the 24‐well plates submerged in 6 % (v/v) formaldehyde for 30 min. After fixation each well was rinsed repeatedly with H2O, the liquid shaken out, and the cell proteins stained with 1 % (w/v) crystal violet in 50 % (v/v) EtOH for 30 min. Excess staining solution was drained and rinsed off repeatedly with H2O. After airdrying the plates plaques were counted.
Thiol reactivity assays
Spectrophotometric Ellman's assay: The assay was performed as previously described. [41] Briefly, 100 μM of compound was incubated at 37 °C with a mixture of 100 μM TCEP and 25 μM DTNB (generating 50 μM TNB2− in situ) or in a parallel experiment with 50 μM TNB2− (without TCEP). All reagent solutions were freshly prepared before performing the experiments. Reactions were performed in buffer (20 mM sodium phosphate, 150 mM NaCl, pH 7.4) with 5 % final DMSO concentration. Absorbance at 412 nm was measured every 5 min for 12 h (Synergy H4, BioTek Instruments, Inc., USA) to monitor TNB2− depletion. Compound background absorbance was subtracted from each measurement. 2‐Chloro‐N‐(3‐chlorophenyl)acetamide (NSC8368) was used as a control.
HPLC‐MS assay: The reaction mixture consisting of 2.5 mM compound, 5 mM NAC or glutathione, 25 % DMSO in buffer (50 mM HEPES, pH 7.4) was incubated overnight at 37 °C in a shaker. Next, the reaction mixture was 5‐fold diluted with MeCN/H2O 4 : 1 (v/v). The final mixture was analyzed using the Agilent 1260 Infinity II LC System equipped with Advion Expression L CMS detector (Advion Inc., Ithaca, NY, USA). An XBridge C18 column (4.6 mm ×150 mm, 3.5 μm) was thermostated at 40 °C, with a flow rate of 1.5 mL/min, an injection volume of 10 μL, and an eluent system of A, 0.1 % aqueous HCOOH with 1 % MeCN; B, MeCN. The following gradients were used: 0–6 min, 25 % to 98 % B; 6–6.5 min, 98 % B; 6.5–7 min, 98 % to 25 % B; 7–10 min, 25 % B. Adducts were detected using a photodiode array detector (220 and 254 nm) and an ESI‐MS analyser in the range of 100–900 m/z. 2‐Chloro‐N‐(3‐chlorophenyl)acetamide (NSC8368) was used as a control.
NMR assay: The thiol reactivity studies were performed as reported previously. [43] Compounds (10 mg) were dissolved in DMSO‐d 6 (0.75 mL) and combined with one equivalent N‐acetylcysteine (NAC) at 25 °C. NMR measurements were performed on a Bruker Advance 500 MHz spectrometer. To demonstrate thermal lability, NMR tubes were heated to 37 or 50 °C prior to measurement of NMR signals.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank Prof. A.C. Filippou, R. Pacher, M. Schneider, C. Vielmuth, L.P. Vu (University of Bonn, Bonn, Germany), R. Schlichting (Helmholtz Centre for Environmental Research‐UFZ, Leipzig, Germany), and M. Frelih (University of Ljubljana, Ljubljana, Slovenia), for their support and technical assistance. This project was funded by the Volkswagen Foundation (project 9 A894, COOP). Open Access funding enabled and organized by Projekt DEAL.
B. Schulte, M. König, B. I. Escher, S. Wittenburg, M. Proj, V. Wolf, C. Lemke, G. Schnakenburg, I. Sosič, H. Streeck, C. E. Müller, M. Gütschow, C. Steinebach, ChemMedChem 2022, 17, e202100732.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Tran Q. T. N., Tan W. S. D., Wong W. S. F., Chai C. L. L., Nat. Prod. Rep. 2020. [DOI] [PubMed] [Google Scholar]
- 2. Okhuarobo A., Ehizogie Falodun J., Erharuyi O., Imieje V., Falodun A., Langer P., Asian Pac. J. Trop. Dis. 2014, 4, 213–222. [Google Scholar]
- 3. Jadhav A. K., Karuppayil S. M., Phytother. Res. 2021, 35, 5365–5373. [DOI] [PubMed] [Google Scholar]
- 4. Li B., Jiang T., Liu H., Miao Z., Fang D., Zheng L., Zhao J., J. Cell. Physiol. 2019, 234, 561–571. [DOI] [PubMed] [Google Scholar]
- 5. Wong S. Y., Tan M. G. K., Wong P. T. H., Herr D. R., Lai M. K. P., J. Neuroinflammation 2016, 13, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wong D. P. W., Ng M. Y., Leung J. Y., Boh B. K., Lim E. C., Tan S. H., Lim S., Seah W. H., Hu C. Z., Ho B. C., Ng D. H. P., Hagen T., PLoS One 2018, 13, e0204853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Canning P., Cooper C. D. O., Krojer T., Murray J. W., Pike A. C. W., Chaikuad A., Keates T., Thangaratnarajah C., Hojzan V., Marsden B. D., Gileadi O., Knapp S., von Delft F., Bullock A. N., J. Biol. Chem. 2013, 288, 7803–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Canning P., Sorrell F. J., Bullock A. N., Free Radical Biol. Med. 2015, 88, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horie Y., Suzuki T., Inoue J., Iso T., Wells G., Moore T. W., Mizushima T., Dinkova-Kostova A. T., Kasai T., Kamei T., Koshiba S., Yamamoto M., Commun. Biol. 2021, 4, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taguchi K., Yamamoto M., Cancers 2020, 13, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu K., McDonald P., Liu J., Klaassen C., Planta Med. 2013, 80, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Naidu S. D., Muramatsu A., Saito R., Asami S., Honda T., Hosoya T., Itoh K., Yamamoto M., Suzuki T., Dinkova-Kostova A. T., Sci. Rep. 2018, 8, 8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rojo de la Vega M., Dodson M., Chapman E., Zhang D. D., Curr. Opin. Toxicol. 2016, 1, 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao S., Huang T., Song L., Xu S., Cheng Y., Cherukupalli S., Kang D., Zhao T., Sun L., Zhang J., Zhan P., Liu X., Acta Pharm. Sin. B 2021, doi: 10.1016/j.apsb.2021.08.027. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang X., Yu Y., Xu J., Shu H., Xia J., Liu H., Wu Y., Zhang L., Yu Z., Fang M., Yu T., Wang Y., Pan S., Zou X., Yuan S., Shang Y., Lancet Respir. Med. 2020, 8, 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiu S., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S., Zhan P., Liu X., J. Med. Chem. 2020, 63, 12256–12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vellingiri B., Jayaramayya K., Iyer M., Narayanasamy A., Govindasamy V., Giridharan B., Ganesan S., Venugopal A., Venkatesan D., Ganesan H., Rajagopalan K., Rahman P. K. S. M., Cho S.-G., Kumar N. S., Subramaniam M. D., Sci. Total Environ. 2020, 725, 138277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanjanasirirat P., Suksatu A., Manopwisedjaroen S., Munyoo B., Tuchinda P., Jearawuttanakul K., Seemakhan S., Charoensutthivarakul S., Wongtrakoongate P., Rangkasenee N., Pitiporn S., Waranuch N., Chabang N., Khemawoot P., Sa-ngiamsuntorn K., Pewkliang Y., Thongsri P., Chutipongtanate S., Hongeng S., Borwornpinyo S., Thitithanyanont A., Sci. Rep. 2020, 10, 19963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sa-ngiamsuntorn K., Suksatu A., Pewkliang Y., Thongsri P., Kanjanasirirat P., Manopwisedjaroen S., Charoensutthivarakul S., Wongtrakoongate P., Pitiporn S., Chaopreecha J., Kongsomros S., Jearawuttanakul K., Wannalo W., Khemawoot P., Chutipongtanate S., Borwornpinyo S., Thitithanyanont A., Hongeng S., J. Nat. Prod. 2021, 84, 1261–1270. [DOI] [PubMed] [Google Scholar]
- 20. Kumar G., Singh D., Tali J. A., Dheer D., Shankar R., Bioorg. Chem. 2020, 95, 103511. [DOI] [PubMed] [Google Scholar]
- 21. Zhang H., Li S., Si Y., Xu H., Eur. J. Med. Chem. 2021, 113710. [DOI] [PubMed] [Google Scholar]
- 22. Tran Q. T. N., Tan D. W. S., Wong W. S. F., Chai C. L. L., Eur. J. Med. Chem. 2020, 204, 112481. [DOI] [PubMed] [Google Scholar]
- 23. Chen D., Song Y., Lu Y., Xue X., Bioorg. Med. Chem. Lett. 2013, 23, 3166–3169. [DOI] [PubMed] [Google Scholar]
- 24. Dai G.-F., Xu H.-W., Wang J.-F., Liu F.-W., Liu H.-M., Bioorg. Med. Chem. Lett. 2006, 16, 2710–2713. [DOI] [PubMed] [Google Scholar]
- 25.The X-ray crystallographic data collection of 15 was performed on a Bruker D8 Venture diffractometer at 165(2) K. The diffractometer was equipped with a low-temperature device (Cryostream 800er series) and used Cu−Kα radiation (Λ=1.54178 Å). Intensities were measured by fine-slicing ϕ- and ω-scans and corrected for background, polarization and lorentz effects. A semi-empirical absorption correction was applied for the data sets by using Bruker's SADABS program. The structures were solved by direct methods and refined anisotropically by the least-squares procedure implemented in the ShelX-2014/7 program system. H atoms were included isotropically using a riding model on the bound C atoms. CCDC 2104432 contains the supplementary crystallographic data. These data are provided free of charge by the CCDC.
- 26. Casamonti M., Risaliti L., Vanti G., Piazzini V., Bergonzi M. C., Bilia A. R., Engineering 2019, 5, 69–75. [DOI] [PubMed] [Google Scholar]
- 27. Oseni B. A., Azubuike C. P., Okubanjo O. O., Igwilo C. I., Panyam J., Front. Bioeng. Biotechnol. 2021, 9, 639409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panossian A., Hovhannisyan A., Mamikonyan G., Abrahamian H., Hambardzumyan E., Gabrielian E., Goukasova G., Wikman G., Wagner H., Phytomedicine 2000, 7, 351–364. [DOI] [PubMed] [Google Scholar]
- 29. Seo J. Y., Pyo E., An J.-P., Kim J., Sung S. H., Oh W. K., Mediators Inflammation 2017, 2017, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cleasby A., Yon J., Day P. J., Richardson C., Tickle I. J., Williams P. A., Callahan J. F., Carr R., Concha N., Kerns J. K., Qi H., Sweitzer T., Ward P., Davies T. G., PLoS One 2014, 9, e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Genheden S., Ryde U., Expert Opin. Drug Discovery 2015, 10, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang X. J., Hayes J. D., Wolf C. R., Cancer Res. 2006, 66, 10983–10994. [DOI] [PubMed] [Google Scholar]
- 33. Escher B. I., Dutt M., Maylin E., Tang J. Y. M., Toze S., Wolf C. R., Lang M., J. Environ. Monit. 2012, 14, 2877–2885. [DOI] [PubMed] [Google Scholar]
- 34. Neale P. A., Altenburger R., Aït-Aïssa S., Brion F., Busch W., de Aragão Umbuzeiro G., Denison M. S., Du Pasquier D., Hilscherová K., Hollert H., Morales D. A., Novák J., Schlichting R., Seiler T.-B., Serra H., Shao Y., Tindall A. J., Tollefsen K. E., Williams T. D., Escher B. I., Water Res. 2017, 123, 734–750. [DOI] [PubMed] [Google Scholar]
- 35. Escher B. I., Glauch L., König M., Mayer P., Schlichting R., Chem. Res. Toxicol. 2019, 32, 1646–1655. [DOI] [PubMed] [Google Scholar]
- 36. Escher B. I., Henneberger L., König M., Schlichting R., Fischer F. C., Environ. Health Perspect. 2020, 128, 077007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee J., Braun G., Henneberger L., König M., Schlichting R., Scholz S., Escher B. I., Chem. Res. Toxicol. 2021, 34, 2100–2109. [DOI] [PubMed] [Google Scholar]
- 38. Mittal S. P. K., Khole S., Jagadish N., Ghosh D., Gadgil V., Sinkar V., Ghaskadbi S. S., Biochim. Biophys. Acta. 2016, 1860, 2377–2390. [DOI] [PubMed] [Google Scholar]
- 39. Lacher S. K., Mayer R., Sichardt K., Nieber K., Müller C. E., Biochem. Pharmacol. 2007, 73, 248–258. [DOI] [PubMed] [Google Scholar]
- 40. Saitoh O., Saitoh Y., Nakata H., NeuroReport 1994, 5, 1317–1320. [PubMed] [Google Scholar]
- 41. Kollár L., Gobec M., Szilágyi B., Proj M., Knez D., Ábrányi-Balogh P., Petri L., Imre T., Bajusz D., Ferenczy G. G., Gobec S., Keserű G. M., Sosič I., Eur. J. Med. Chem. 2021, 219, 113455. [DOI] [PubMed] [Google Scholar]
- 42. Avonto C., Taglialatela-Scafati O., Pollastro F., Minassi A., Di Marzo V., De Petrocellis L., Appendino G., Angew. Chem. Int. Ed. 2011, 50, 467–471; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 487–491. [Google Scholar]
- 43. Wong M. H. L., Bryan H. K., Copple I. M., Jenkins R. E., Chiu P. H., Bibby J., Berry N. G., Kitteringham N. R., Goldring C. E., O'Neill P. M., Park B. K., J. Med. Chem. 2016, 59, 2396–2409. [DOI] [PubMed] [Google Scholar]
- 44. Shi T.-H., Huang Y.-L., Chen C.-C., Pi W.-C., Hsu Y.-L., Lo L.-C., Chen W.-Y., Fu S.-L., Lin C.-H., Biochem. Biophys. Res. 2020, 533, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Enmozhi S. K., Raja K., Sebastine I., Joseph J., J. Biomol. Struct. Dyn. 2020, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Breidenbach J., Lemke C., Pillaiyar T., Schäkel L., Al Hamwi G., Diett M., Gedschold R., Geiger N., Lopez V., Mirza S., Namasivayam V., Schiedel A. C., Sylvester K., Thimm D., Vielmuth C., Phuong Vu L., Zyulina M., Bodem J., Gütschow M., Müller C. E., Angew. Chem. Int. Ed. 2021, 60, 10423–10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cuadrado A., Pajares M., Benito C., Jiménez-Villegas J., Escoll M., Fernández-Ginés R., Garcia Yagüe A. J., Lastra D., Manda G., Rojo A. I., Dinkova-Kostova A. T., Trends Pharmacol. Sci. 2020, 41, 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olagnier D., Farahani E., Thyrsted J., Blay-Cadanet J., Herengt A., Idorn M., Hait A., Hernaez B., Knudsen A., Iversen M. B., Schilling M., Jørgensen S. E., Thomsen M., Reinert L. S., Lappe M., Hoang H.-D., Gilchrist V. H., Hansen A. L., Ottosen R., Nielsen C. G., Møller C., van der Horst D., Peri S., Balachandran S., Huang J., Jakobsen M., Svenningsen E. B., Poulsen T. B., Bartsch L., Thielke A. L., Luo Y., Alain T., Rehwinkel J., Alcamí A., Hiscott J., Mogensen T. H., Paludan S. R., Holm C. K., Nat. Commun. 2020, 11, 4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Singh E., Matada G. S. P., Abbas N., Dhiwar P. S., Ghara A., Das A., Inflammopharmacology 2021, 29, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang X., Lv L., Zhou Y., Xie L., Xu Q., Zou X., Ding Y., Tian J., Fan J., Fan H., Yang Y., Ye X., Phytother. Res. 2021, ptr.7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Narimanyan M., Jamalyan K., Balyan A., Barth A., Palm S., Wikman G., Panossian A., J. Tradit. Complement. Med. 2021, S2225411021000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gabizon R., London N., Curr. Opin. Chem. Biol. 2021, 62, 24–33. [DOI] [PubMed] [Google Scholar]
- 53. Powell M., Blaskovich M. A. T., Hansford K. A., ACS Infect. Dis. 2021, 7, 2050–2067. [DOI] [PubMed] [Google Scholar]
- 54. Ma Y., Frutos-Beltrán E., Kang D., Pannecouque C., De Clercq E., Menéndez-Arias L., Liu X., Zhan P., Chem. Soc. Rev. 2021, 50, 4514–4540. [DOI] [PubMed] [Google Scholar]
- 55. Einhorn J., Einhorn C., Ratajczak F., Pierre J.-L., J. Org. Chem. 1996, 61, 7452–7454. [DOI] [PubMed] [Google Scholar]
- 56. Khan A. T., Mondal E., Synlett 2003, 5, 694–698. [Google Scholar]
- 57. Sastry G. M., Adzhigirey M., Day T., Annabhimoju R., Sherman W., J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 58. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J. F., Honig B., Shaw D. E., Friesner R. A., Proteins 2004, 55, 351–367. [DOI] [PubMed] [Google Scholar]
- 59. Zhu K., Borrelli K. W., Greenwood J. R., Day T., Abel R., Farid R. S., Harder E., J. Chem. Inf. Model. 2014, 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- 60. König M., Escher B. I., Neale P. A., Krauss M., Hilscherová K., Novák J., Teodorović I., Schulze T., Seidensticker S., Kamal Hashmi M. A., Ahlheim J., Brack W., Environ. Pollut. 2017, 220, 1220–1230. [DOI] [PubMed] [Google Scholar]
- 61. Escher B. I., Neale P. A., Villeneuve D. L., Environ. Toxicol. Chem. 2018, 37, 2273–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Streeck H., Schulte B., Kümmerer B. M., Richter E., Höller T., Fuhrmann C., Bartok E., Dolscheid-Pommerich R., Berger M., Wessendorf L., Eschbach-Bludau M., Kellings A., Schwaiger A., Coenen M., Hoffmann P., Stoffel-Wagner B., Nöthen M. M., Eis-Hübinger A. M., Exner M., Schmithausen R. M., Schmid M., Hartmann G., Nat. Commun. 2020, 11, 5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.