

Abstract
Very little knowledge exists on virus-specific host cell intrinsic mechanisms that prevent hyperproliferation of primary herpes simplex virus type-2 (HSV-2) genital infections. Here we provide evidence that the Nemo-related protein, Optineurin (OPTN), plays a key role in restricting HSV-2 infection both in vitro and in vivo. Contrary to previous reports regarding the proviral role of OPTN during Sendai virus infection, we demonstrate that lack of OPTN in cells causes enhanced virus production. OPTN deficiency negatively affects the host autophagy response and results in a marked reduction of CCL5 induction. OPTN −/− mice display exacerbated genital disease and dysregulated T-cell frequencies in infected tissues and lymph nodes. A human transcriptomic profile dataset provides further credence that a strong positive correlation exists between CCL5 upregulation and OPTN expression during HSV-2 genital infection. Our findings underscore a previously unknown OPTN/CCL5 nexus that restricts hyperproliferative spread of primary HSV-2 infection, which may constitute an intrinsic host defense mechanism against herpesviruses in general.
Introduction
Herpes simplex virus type-2 (HSV-2) is a member of the α-herpesvirus subfamily and has a highly variable global prevalence. Asian countries have reported 10–30% seropositivity, while some regions of sub-Saharan Africa have reached between 30–80% seropositivity (1, 2). In the United States, approximately 22% of the population or 45 million people were seropositive for HSV-2 (3). Transmission typically occurs via sexual contact (4, 5). HSV-2 has been shown to infect the genital, anal, and orofacial regions of the body (6). Neonatal HSV-2 infection can cause eye lesions. Symptomatic infections often result in the formation of blisters, macules, papules, and ulcers (7). Roughly 36% of women and 13% of men with primary HSV-2 genital infections develop meningitis, and a subset of these individuals develop recurrent meningitis after the initial episode (4, 8). Treatments are limited to nucleoside analogs such as acyclovir which relieve symptoms of infection, and there are no vaccines to prevent HSV-2 infections to date (9).
After primary infection, the virus migrates to the sacral ganglia where it establishes latency. Periodic episodes of reactivation can promote symptomatic infections, especially in immunocompromised individuals. However, most individuals (75–90%) remain asymptomatic despite being seropositive for HSV-2 (3, 5). Host factors play essential roles in mediating protection from HSV by regulating the intrinsic, innate, and adaptive immune responses (10–12). Understanding the functions of these host factors may reveal novel strategies to bolster host immunity and inhibit infection.
HSV-1 and HSV-2 require tight regulation of host autophagy to create an optimal environment for replication (13–15). Nemo-related protein, Optineurin (OPTN) is a host autophagy adaptor protein that participates in the clearance of cytosolic bacteria from host cells during infection (16–18). OPTN has also been implicated in the activation of antimicrobial mechanisms and immune response during infection (19–21). Evidence suggest that it negatively impacts IFN-β induction in response to RNA virus infection (22). So far only one study has examined OPTN in context with HSV-1 infection and found that OPTN is down-regulated during infection in vitro (23).This study shows that unlike wild type HSV-1 infection, ICP0 mutant virus produce higher immune response in OPTN depleted condition. However, neither the in vitro nor in vivo roles of OPTN have been studied during HSV-2 infection.
In this study, we investigated the role of OPTN during HSV-2 infection both in vitro and in vivo. We show that the lack of OPTN results in the hyperproliferation of HSV-2 in vitro possibly due to defective autophagic response. We observed similar results in vivo in a murine model of genital HSV-2 infection. We show that loss of OPTN perturbs T cell infiltration in the infected tissue and blocks CCL5 induction, which then dysregulates the host immune response and exacerbates inflammation during HSV-2 pathogenesis. We also establish that a direct correlation exists between CCL5 upregulation and OPTN expression during HSV-2 genital infection in humans.
Materials and Methods
Cells and viruses
OPTN +/+ and −/− HeLa cells were obtained from Dr. Richard Youle (National Institutes of Health) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO) and 1% penicillin-streptomycin (P/S; Life Technologies). African green monkey kidney (Vero) cells were provided by P. Spear (Northwestern University) and passaged in DMEM. Cells were maintained at 37°C and 5% CO2. HSV-2 333-GFP was also provided by P. Spear. Viral stocks were made using Vero cells and stored at −80°C.
Visualization of HSV-2 333-GFP
70–80% confluent HeLa cells were infected with HSV-2 333-GFP at 0.1 MOI. After 2 hours post infection (hpi), the media was changed to DMEM, and a drop of NucBlue Fixed Cell ReadyProbes Reagent (Thermo Fisher Scientific) was added to each well to stain the nuclei of the cells. At various time points post infection (every 4 h from 8–28 hpi), the cells were imaged using a Lionheart LX automated microscope (BioTek Instruments LLX). DAPI and GFP channels were merged to create images with the cellular nuclei (blue) and virus (green) overlaid in the same frame. The images were quantified using the BioTek Gen5 Microplate Reader and Imaging software.
Quantitative polymerase chain reaction assay
Total RNA was extracted from the HeLa cells using TRIzol (Life Technologies) as per the manufacturer’s instructions. Vaginal tissues were homogenized, dissolved in TRIzol, and sonicated prior to the RNA extraction. The RNA was quantified using the NanoDrop (Thermo Fisher Scientific, USA). All samples were equilibrated to 1000 ng before reverse transcription into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, USA). The cDNA was then analyzed with real-time quantitative polymerase chain reaction (qRT-PCR) with the Fast SYBR Green Master Mix (Life Technologies) on the QuantStudio 7 Flex System (Applied Biosystems) and ΔΔCt model was used to calculate the data. Primers used in this are listed as supplementary table1.
Western blot analysis
Cells were collected in Hank’s Balanced Salt Solution (Life Technologies) which was removed and replaced with a protease phosphatase inhibitor cocktail (Thermo Fisher Scientific) in radioimmunoprecipitation assay buffer (Millipore Sigma). The solution was incubated for 30 min at 4°C and centrifuged at 15,000 g for 20 min. The supernatants were transferred to new tubes and mixed with a solution of NuPAGE LDS Sample Buffer (Invitrogen, NP00007) and β-mercaptoethanol. The samples were heated to 90°C for 9 min and allowed to cool. The denatured proteins were loaded onto 4–12% SDS-polyacrylamide gels which ran at 70 V for 3 h. The proteins were transferred onto a nitrocellulose membrane (IB23001, Invitrogen) using the iBlot2 transfer machine (Thermo Fisher Scientific). The membrane was blocked using 5% skim milk in tris-buffered saline (TBS; Thermo Fisher Scientific) and 0.1% Tween 20 (Sigma-Aldrich) (TBST) for 1 h at room temperature. After blocking, each membrane was incubated with a primary antibody (1:1000 in 5% skim milk) overnight at 4°C. The following day, the membranes were washed three times with TBST with 5 min breaks between washes. After the third wash, the membranes were incubated with horseradish peroxidase-conjugated secondary IgG antibody (1:10000 in 5% skim milk) for 1 h at room temperature. After three more washes, SuperSignal West Pico Maximum Sensitivity Substrate (Pierce, 34080) was added to the blots, and the protein bands were then visualized using an ImageQuant LAS 4000 imager. Band densitometry was performed using ImageQuant TL image analysis software (version 7) with GAPDH as the loading control. The following primary antibodies were used in this study: HSV-2 gB mouse monoclonal antibody (Abcam, ab6505), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Proteintech, 10494), LC3I-II (Novus), and OPTN (C-Term) (Cayman Chemical).
The following antibodies were used for immunoblot: Mouse monoclonal [7B] anti-GAPDH (Santa Cruz), Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody.
Plaque Assay
HSV-2 infected HeLa cells were collected in HBSS before resuspension in 1 mL Opti-MEM (Life Technologies). The samples were sonicated using a probe sonication system for 30 s at 70% amplitude. The sonication probe was surface cleaned with 70% ethanol between each sample. Either the cell lysates or vaginal swabs were serially diluted in Opti-MEM before addition to a 90–100% confluent plate of Vero cells. After the cells were incubated with the diluted virus for 2 h at 37°C, the media was changed to 0.5% methylcellulose in DMEM. The plates were incubated for 72 h at 37°C. Afterwards, the 300 μL of 100% methanol was added to each well in the plates for 20 min to fix the cells. The methanol was then removed and replaced with crystal violet to visualize plaque formation.
Confocal microscopy imaging
HeLa cells were plated onto glass-bottom dishes (MatTek Corporation). After they reached 50% confluency, they were transfected with 1 μg of a mCherry-GFP-LC3 plasmid (Addgene, 22418) using Lipofectamine 2000 (Invitrogen, Life Technologies) in Opti-MEM. The media was changed to DMEM after 4 h, and the cells were infected with 0.1 MOI of HSV-2 333 after 24 hours post transfection. The GFP-transfection efficiency was checked in control set under the microscope and approximately 70% cells found GFP positive. The dishes were then washed with PBS before fixation with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA). After two washes with PBS post fixation, the dishes were filled with 1 mL of PBS with one drop of NucBlue Fixed Cell ReadyProbes Reagent. Fluorescence confocal imaging was performed using an LSM 710 confocal microscope under a 63x objective.
For CCL5 expression studies, OPTN+/+ and OPTN −/− HeLa cells were plated onto glass-bottom dishes. After 90% confluency, cells were serum starved for 4 hrs then infected with 5 MOI of HSV-2 333 for 2h then treated with Brefeldin A (2.5uM) to arrest the release of cytokine and kept for another 4h then cells were processed for confocal imaging as explained in above paragraph.
CCL5 siRNA transfection
A Dicer-Substrate Short Interfering RNAs (DsiRNAs) TriFECTa® Kit (IDT) with predesigned CCL5 specific siRNA molecules was used for transfections in this study. Cells were plated in 6 well plates and grown to 40–45% confluency. Cells were then transfected as per manufacturer’s protocol using RNAiMAX at 1 μl/mL in OptiMEM (ThermoFisher). Appropriate controls were prepared and tested for tranfection and knockdown efficacy as per the manufacturer. Multiple concentrations for premade CCL5 siRNA molecules were tested and it was determined that siRNA 13.3 at 1 nM produced effective knockdown with minimal cell death after 60 hours of transfection.
In vivo Vaginal Infection
The mice involved in this study were infected as per the approved ACC protocol by the University. Briefly, 8-week-old C57BL/6 female OPTN +/+ and −/− mice were injected subcutaneously (via scruff hold) with 2 mg of medroxyprogesterone (Depo-Provera). On day 5 after injection, mice were given intravaginal dose of 1X106 PFU HSV-2 333 strain. Vaginal swabs were collected on day 1, 3 and 5 post infection using calcium alginate-tipped sterile applicators (Calgiswab; Puritan) to assess the amount of viral shedding in the vaginal epithelium via a plaque assay. Animals were monitored for any change in weight loss and temperature change during this period. We did not see signs of severe distress during the study period as measured by infection score. On 9 and 14 dpi, separate set of animals were euthanized, and their genital tissue, iliac lymph nodes and spleen tissue were collected for the downstream analysis.
Flow cytometry
Mice tissues (Vagina, spleen and iliac lymph node) were collected at 9 and 14dpi and dissociated in collagenase D and Dnase I (Roche, Germany, 2 mg/mL of PBS) for 4 h at 37°C with vortexing intermittently. Resulting sample was thoroughly mixed with pipetting up and down to dissociate the remaining tissue clots. FACS buffer (5% FBS in PBS) was added and the mixture was passed through 70 μm cell strainer. Resulting cells were distributed in round bottom 96 well plate and blocked using TruStain FcX PLUS (156603, Biolegend) according to the manufacturer’s protocol. Appropriate flow cytometry controls including tissue specific non-stained, fluorescence minus one and single color control were prepared for setting the compensation matrix. Cells were then stained for 1 h on ice with the separate mix of following fluor conjugated anti-mouse primary antibodies from BioLegend or Tonbo Biosciences: CD45 (103155), CD3 (100216), CD4 (50–0042-U100), CD8a (100714), Cd11b (101206) CD11c (117310). Cells were washed twice with FACS buffer, and fixed with 4% paraformaldehyde and stored at 4 C until analysis done a BD CytoFlex flow cytometer. 50,000 singlet non-debris events were collected for each sample, and FlowJo X was used to process and analyze the data. A simple gating strategy was used to gate all cells based on SSC and FSC areas, then single cells based on FSC height and FSC area, then single cells were analyzed for respective color channel.
Mouse Tissue Histology and Staining
Mouse vaginal tissue was collected at 10 dpi and embedded in Tissue-Plus O.C.T. (Fisher HealthCare) then frozen on dry ice and kept at −80°C until processing. Tissue sections of 10 μm size were stained with a Hematoxylin and eosin stain following sequential steps. Briefly, 5 minutes ice cold acetone treatment, 2 min water wash, 30 seconds Hematoxylin treatment, 2 min 70% ethanol wash, 2 min 100% ethanol wash, 30 seconds eosin treatment, 2 min 70% ethanol wash, 2 min 100% ethanol wash and 2 min xylene treatment. Finally, stained tissue sections were air dried and stored with coverslip and permount mounting solution. Sections were observed and imaged under the Zeiss Axioskop 2 plus microscope.
HSV-2(333)gJ− replication and spread
Viral infection in the vaginal area was assessed by a non-replicating HSV-2(333)gJ− strain as reported earlier (Antoine et.al., 2016). Briefly, Animals were given Depo 5 days before the virus infection. Vaginal area of wild type and OPTN KO animals was inoculated with 1×107 virus titre. The vaginas were removed at day 1 post infection. Vaginal tissue was then fixed in 2% paraformaldehyde 0.02% Nonidet P-40 in PBS for 6 h. After fixation the organs were stained overnight with 1 mg/ml of X-gal (6-bromo-4-chloro-3-indolyl-b-d-galactopyranisude, V394A, Promega) solution in DPBS. The staining of the vaginal tissue was scored on a scale from 0 to 5: 0 = no blue staining; 1 = 10–20% of surface covered in blue lesions; 2 = 30% of the surface covered in blue lesions; 3 = 40–50% of surface covered in blue lesions; 4 = 60–70% of surface covered in blue lesions; 5 = 80–100% covered in blue lesions. All examiners who have done scoring were masked regarding the treatment condition of each mouse.
Mouse Inflammation Antibody Array
The vaginal tissue from OPTN+/+ and OPTN−/− mice was removed 1day post 1X106 PFU infection with HSV-2 333 strain. The tissues were processed following manufacturer protocol (RayBio® C-Series Mouse Inflammation Antibody Array kit). Briefly, each tissue was homogenized (Bullet Blender Storm Pro, Troy, NY, USA) in the cell lysis buffer with Protease Inhibitor Cocktail (Sigma) and centrifugation (10,000 × g for 15 min at 4°C). Then, the protein concentration was evaluated with a BCA Protein Assay Kit (Thermo), and each sample was diluted (1× Blocking Buffer) to a final concentration of 100 μg/mL. The activated inflammation antibody array membranes were incubated with 1 ml of sample (overnight, 4°C). Next day, the membranes were washed sequentially with wash buffer I and II (RayBio). Each membrane then covered with 1 ml of diluted Biotin-Conjugated Anti-Cytokine primary antibodies (RayBio) and incubated for 2h at room temperature. After, the membranes were washed and incubated (2 h, room temperature) with 2 ml of 1,000-fold diluted HRP-conjugated streptavidin (RayBio). The membranes were washed and detected using Detection Buffer (RayBio) and visualized using an ImageQuant LAS 4000 fluorescent imaging system. The relative levels of immunoreactivity were quantified by densitometry using data calculation template available at RayBiotech.
Bioinformatics analyses
Transcriptional profiling datasets of genital HSV-2 infection in human patients (GSE172423) was obtained from NCBI Gene Expression Omnibus (GEO) repository. Peng T, (2021) recently performed microarray experiments investigating gene expression over the period in human genital skin biopsies at lesion, 2- and 8-weeks post healing and contra-lateral control genital skin biopsies during recurrent HSV-2 infection. Data accessible at NCBI GEO database (24), with accession number GSE172423. GSE172423 dataset contains the analysis of host response at the gene expression level during lesion and post healing periods of recurrent HSV-2 infection as compared to those in control skin. Differential expression analysis was performed based on author-normalized expression values (log2 scale) using the GEO2R platform (https://www.ncbi.nlm.nih.gov/geo/geo2r/). We compared the target gene expression analysis on control skin dataset (n=27) and lesion skin dataset (n=25). The control and lesion groups were further pooled to GEO2R platform for the differential expression analysis with a cutoff of p <0.01. Further we performed correlation analyses between CCL5 and OPTN gene in control and lesion skin condition.
Statistical analysis
Differences between Two groups were analyzed using a parametric, unpaired Student t test (α = 0.05). One-way ANOVA test was used to analyze data from experiments with multiple groups by GraphPad Prism version 9.0.2 (GraphPad Software, la Jolla, CA).Pearson’s correlation coefficient values were determined by imageJ *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001, ns, not significant.
Results
OPTN restricts HSV-2 replication in vitro
To examine the role of OPTN during HSV-2 infection, we infected WT (+/+) or OPTN knockout (−/−) HeLa cells with the 333-GFP strain of HSV-2. We found that OPTN −/− cells fluoresce significantly more GFP than WT HeLa cells, as measured with live cell imaging, from 8–28 hours post infection (hpi) which is indicative of more virus production (Fig. 1A–B). We confirmed these results by observing the elevated transcript levels of the late viral gene, glycoprotein B (gB), using qRT-PCR (Fig. 1C). We further infected the cells with different multiplicity of infection (MOI) to check the expression levels of HSV-2 gB protein. The MOI dependent increase in protein expression was elevated in the OPTN −/− cells at 1, 2 and 5 MOI by 24 hpi (Fig. 1D). We used a host housekeeping gene, GAPDH as a control to compare our observations and ensure similar levels of cell replication in OPTN −/− cells and OPTN +/+ cells. Additionally, total virus produced in infected OPTN +/+ or −/− cells was measured by plaque assay. HSV-2 production was enhanced nearly five-fold in the absence of OPTN (Fig. 1E).
Figure 1. Presence of Optineurin limits HSV-2 infection.
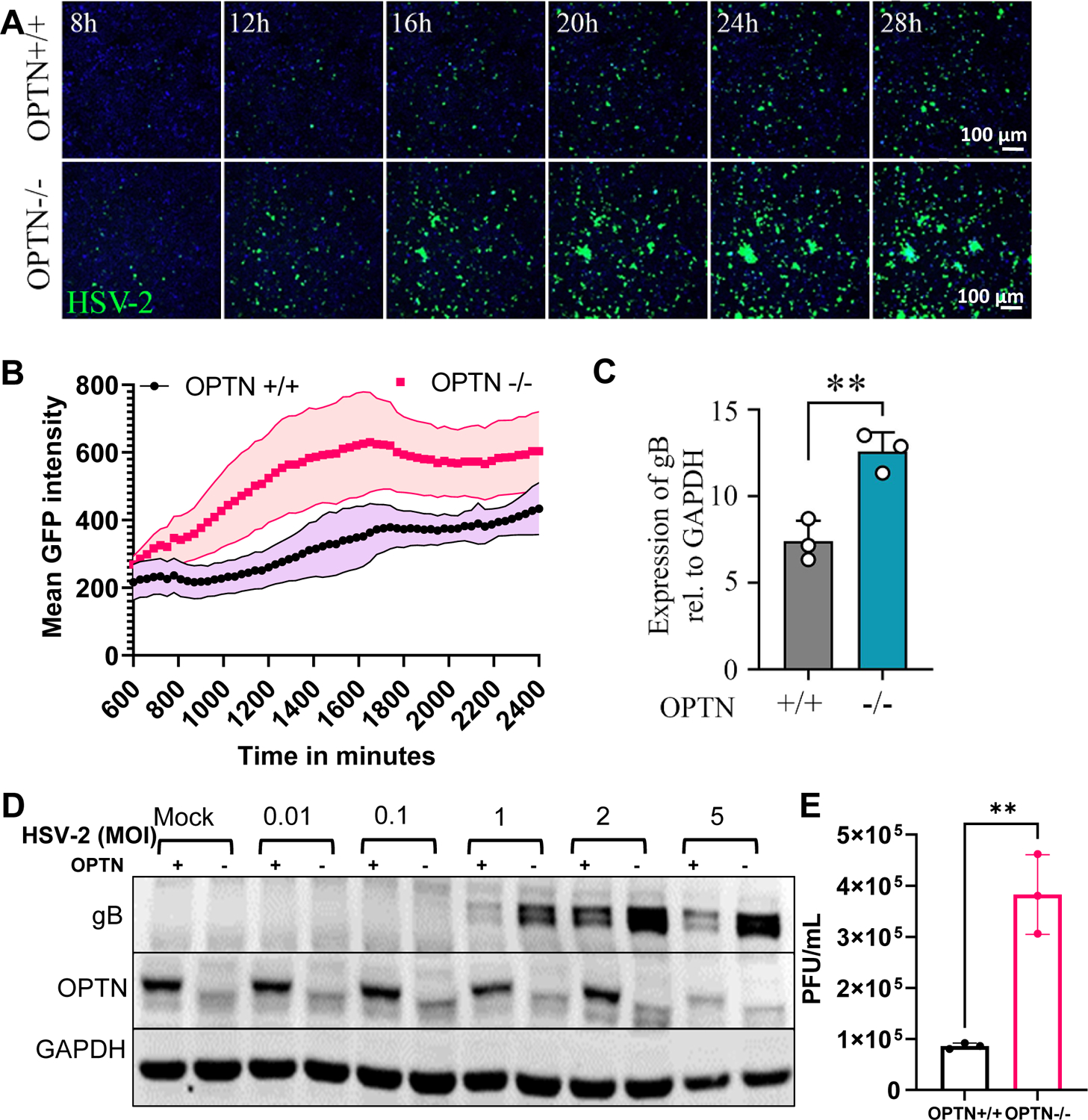
(A)Time lapse imaging of OPTN+/+ and −/− HeLa cells infected with GFP-HSV-2 333-strain at 1 MOI. Scale bar = 100 μm. (B) Quantification of mean green fluorescence intensity over time with error bands. (C) Real-time qPCR analysis of OPTN+/+ and −/− cells infected with HSV-2 333-strain at 1 MOI for 24 hrs mRNA levels of viral late gene are presented. D) Western blot analysis of OPTN+/+ and −/− cells infected with HSV-2 333-strain at different MOI for 24 hpi. (E) Viral plaques in OPTN+/+ and −/− cells infected with HSV-2 333 at 1 MOI for 24 hrs. Data in 1A shows representative time-lapse images from experiments done in triplicate. Data in Fig. 1 B–E represent three independent experiments, n=3 samples/group/genotype/condition. Student’s t-test was performed for statistical analysis (α = 0.05). Error bar in 1C and 1E represent means with SD, *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001, ns, not significant.
Loss of OPTN inhibits autophagic flux during infection
OPTN canonically functions as an autophagy adaptor protein, and perturbations in the regulation of autophagy have been reported to affect HSV-2 replication (13). To identify the mechanism by which OPTN conferred protection against HSV-2 infection, we investigated the changes in microtubule-associated protein light chain 3 (LC3)-I/II OPTN +/+ and −/− cells during infection. LC3-I/II is a well-studied indicator of alterations in autophagic flux (19, 25). We observed that LC3-I to LC3-II conversion was enhanced during HSV-2 infection of OPTN +/+ HeLa cells (Figure 2A–B); however, the conversion was comparatively diminished in the OPTN −/− cells which indicates a decrease in autophagic flux (Figure 2A–B). To confirm these results, we transfected a monomeric mCherry-GFP-LC3 plasmid into the HeLa cells followed by an HSV-2 infection. In the OPTN +/+ cells, we observed the quenching of the GFP signal while the mCherry signal remained prominent, which corresponds to the localization of LC3 to the autolysosome (Fig. 2C–D). However, in the infected OPTN −/− cells, there were yellow puncta corresponding to the colocalization of the GFP and mCherry fluorescence (Fig. 2C–D). This indicates that the tagged LC3 protein is not present in the autolysosome and that autophagic flux is inhibited (26).
Figure 2. Loss of Optineurin interrupts the normal autophagy flux during HSV-2 infection.
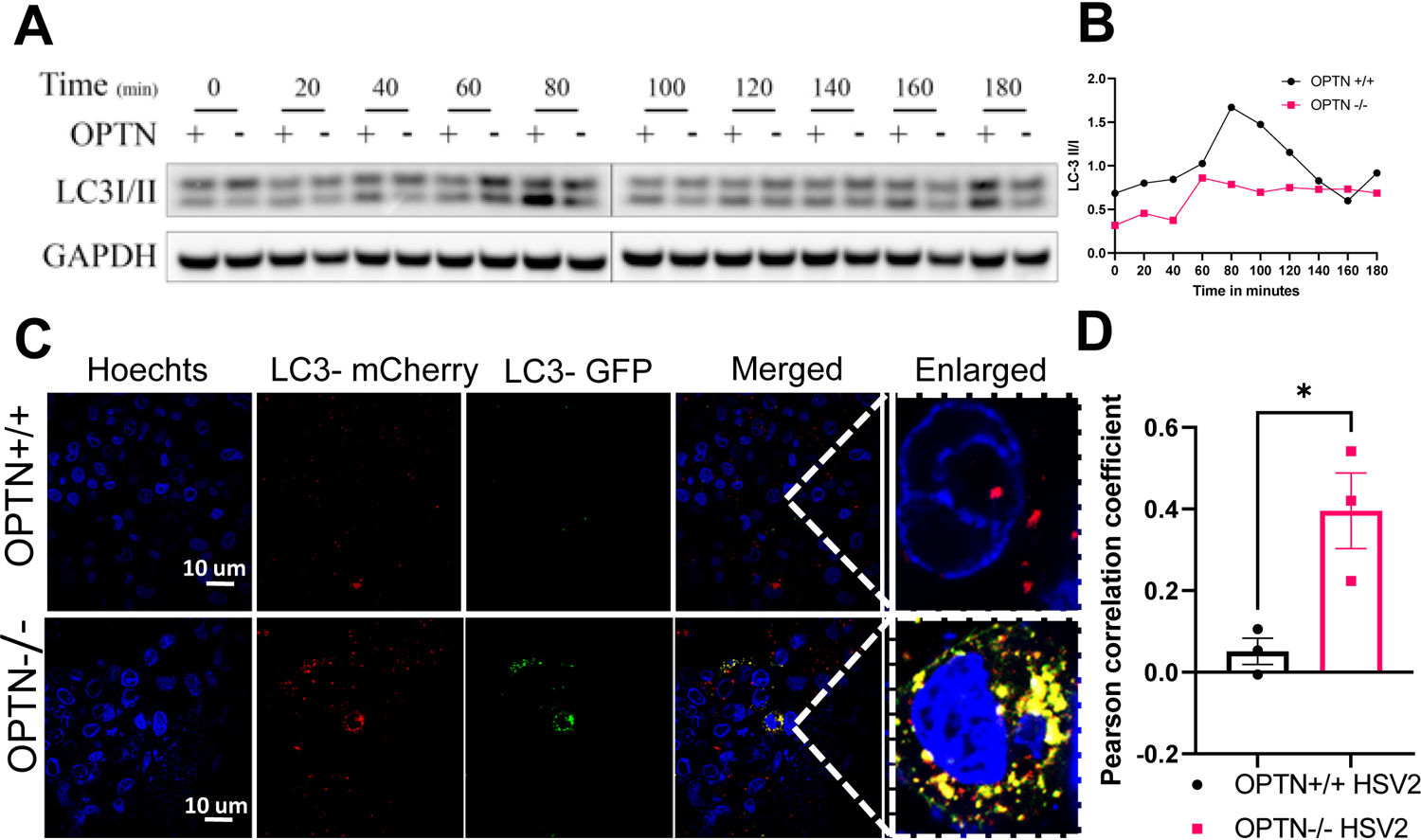
(A) OPTN +/+ or OPTN−/− HeLa cells were infected or mock infected with 2 MOI of HSV-2 333 strain for various time points were analyzed by western blot hybridization using anti-LC3-I/II and anti-GAPDH antibodies. (B) After western blot analysis, the signals from the corresponding bands were analyzed by quantitative densitometry and the ratio of LC3-II over LC3-I was determined after normalization to GAPDH. (C) Autophagy flux was investigated by the transient overexpression of a LC3-GFP-mCherry construct combined, or not, with HSV-2 333 (1MOI) for 24 hours. Representative fluorescent microscopy photographs of each condition are shown (n=3, scale bar = 10 μm). (D) Person’s correlation coefficient representing the co-localization proportion of LC3-mCherry and LC3-GFP. Three images from each group were analyzed for co-localization. Data represent two independent experiments, n=3 samples/group/genotype/condition. Error bar represent means with SEM. Student’s t-test was performed for statistical analysis (α = 0.05). *p < 0.05
Viral pathogenesis is exacerbated and causes severe vaginal tissue infection in OPTN −/− mice
To study the relationship between OPTN and viral infection in vivo, we infected OPTN +/+ and −/− mice with 1×106 plaque forming units of HSV-2 333 and measured viral titers in vaginal swabs at 1, 3, and 5 days post infection (dpi) (Fig. 3A). At 1 and 3 dpi, viral shedding was significantly enhanced in the absence of OPTN (Fig. 3B–C). By 5 dpi, both groups exhibited similar levels of virus shedding (Fig.S1A). We also wanted to know whether the genital tissues from OPTN −/− animals retain higher HSV-2 infection. To check this, we infected the OPTN +/+ and −/− mice with HSV-2(333)gJ− strain at 1×107 titer. The amount of β-galactosidase production directly correlates to the infective HSV-2(333)gJ− particles. With the x-gal β-galactosidase detection assay, we found that 1dpi, OPTN−/− vaginal tissue stained significantly higher than the OPTN+/+ mice (Fig.3D). Both groups of mice displayed a reduction in body temperature with infection, but the effect was more pronounced in OPTN −/− mice at 7 dpi (Fig.S1B). Additionally, animals lacking OPTN showed increased vaginal inflammation as the infection score was approximately two-fold higher in the knockout animals (Fig. 3E–F). H&E staining of the vaginal tissue post infection revealed a modest loss of epithelial integrity in the OPTN +/+ mice compared to more severe epithelial damage and swelling in the OPTN −/− mice (Fig. 3G). Furthermore, the vaginal epithelium in infected tissues of OPTN −/− mice were approximately two-fold thicker than the +/+ tissues at 9 dpi (Fig. 3H) indicating that lack of OPTN increased leukocyte infiltration into the infected vaginal tissue leading to severe inflammation (Fig. 3F). Collectively, the OPTN −/− mice showed worsened viral pathogenesis and tissue damage compared to the +/+ mice. Furthermore, iliac lymph nodes from OPTN −/− mice weighed significantly more at both 9 and 14 dpi than the OPTN+/+ mice (Fig. 3I). However, the weight of the spleens were comparable between the two groups (Fig. 3J).
Figure 3. Loss of Optineurin leads to severe HSV-2 genital infection.
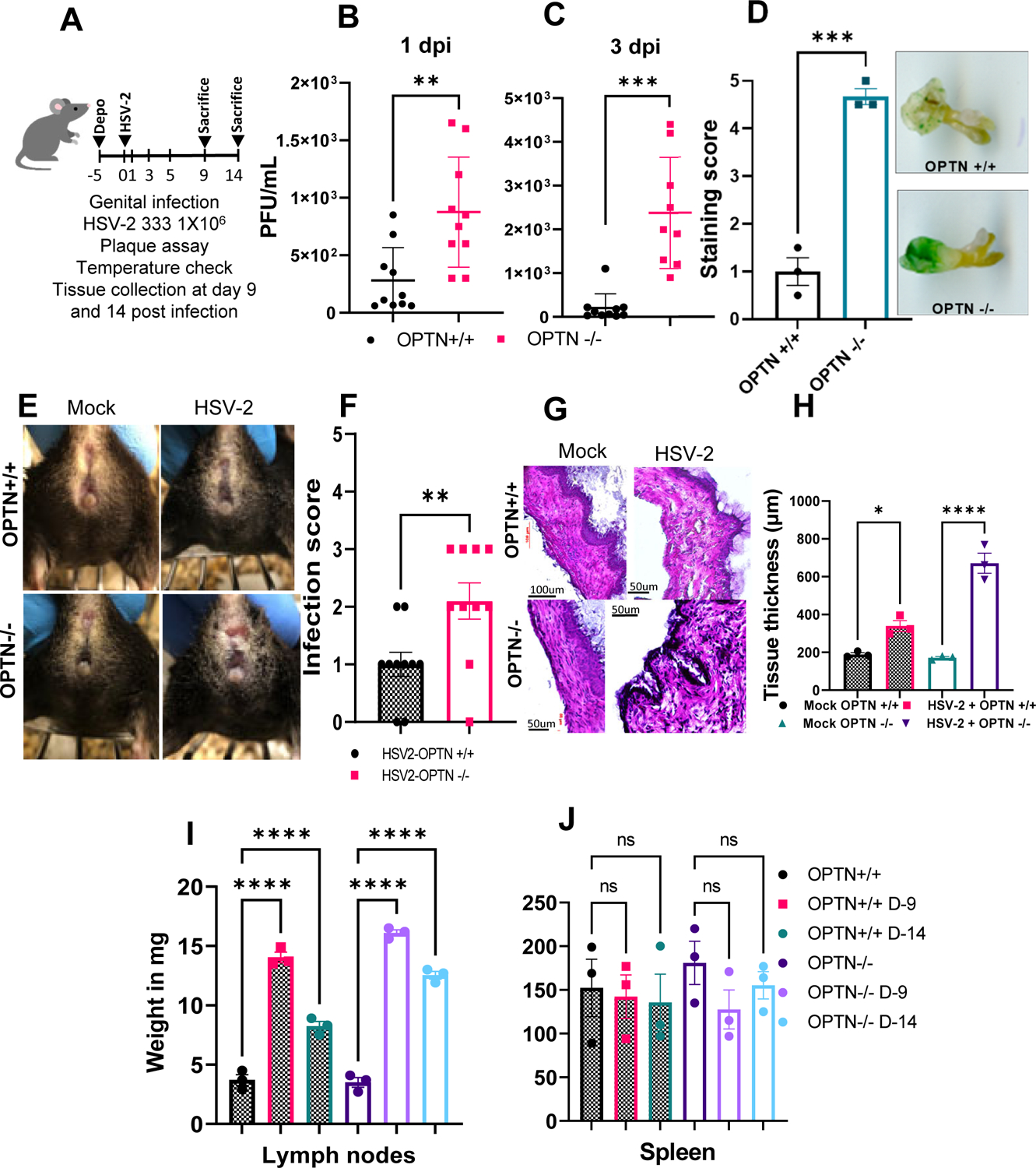
(A) Schematic showing the overall animal experiment. Mice were infected with 1×106 PFU of HSV-2 333 strain and monitored over 14 days (B-C) Genital swabs were taken from HSV-2 333 infected OPTN +/+ and OPTN −/− animals at different time interval post infection. (D)Vaginal area of OPTN +/+ and OPTN−/− animals was inoculated with 1×107 virus titer of a non-replicating HSV-2(333)gJ− strain for 24h. Tissue were stained with x-gal and staining score was determined on the scale of 0–5. (E) Representative photographs of vaginal area from each group were taken. (F) Genital disease infection score (0–5, 5 being severe) in mice. (G) Representative images of hematoxylin and eosin stained vaginal tissue of mock and infected mice at 9 dpi. Images at 100um scale and observed under 45X magnification. (H) Quantitation of vaginal tissue thickness with or without HSV-2 infection. (I-J) Inflammation in the iliac lymph nodes and spleen tissues represented total weight of collected tissue at day14 post infection. Data represent three independent experiments with similar results, n=3–5 samples/group/genotype/condition. Error bar (B-C) represent means ± SD. Error bar (D,F,H,I,J) represent means ± SEM. Significance was determined by one way ANOVA with Šídák’s multiple comparisons test. Student’s t-test was performed for statistical analysis (α = 0.05). *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001, ns, not significant.
CCL5 expression is not induced in the absence of OPTN during HSV-2 vaginal infection.
While OPTN participates in selective autophagy, it also functions to recruit ubiquitinated tank-binding kinase 1 (TBK1) to the Golgi apparatus where it is auto-phosphorylated and activated. Activated TBK1 phosphorylates IRF3 which goes on to stimulate the production of antiviral type I interferons (27). Given the role of OPTN in stimulating the interferon response, we profiled cytokine transcripts in OPTN +/+ and −/− HeLa cells during HSV-2 infection. Interestingly, loss of OPTN did not restrict the production of the type I interferons IFN-α and IFN-β (Fig. 4A–B) or the cytokines IL-6 and TNF-α (Fig. 4C–D). However, we observed that the chemokine CCL5 was not induced in OPTN −/− cells during infection (Fig. 4E).
Figure 4. Loss of OPTN impacts CCL5 induction during HSV-2 infection.
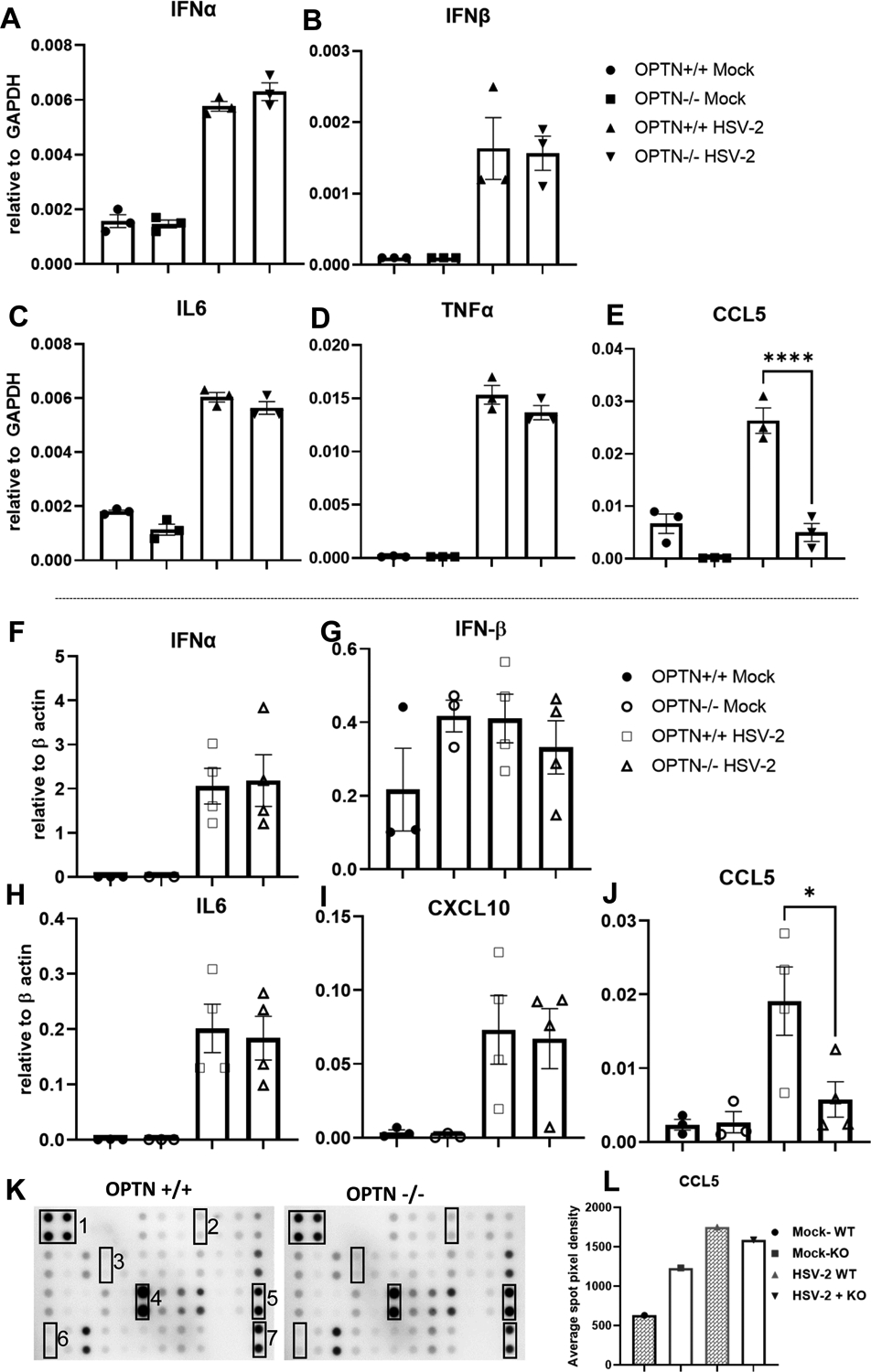
(A-E) Real-time qPCR analysis of OPTN+/+ and −/− cells infected with HSV-2 333-strain at 1 MOI for 24 hrs mRNA levels of Viral late gene are presented. (F-J) Mock and HSV-2 infected vaginal tissues were subjected to cytokine expression analysis at 9dpi by qRT-PCR. Copy numbers relative to β-actin are shown. (K) Inflammation antibody array blots of mouse inflammatory proteins in vaginal tissue of OPTN +/+ and OPTN−/− animals infected with 1X106 PFU HSV-2 333 strain for 24h. Numbers indicate the position of controls and selective cytokines on array; 1and 7 - Positive control, 2-Eotaxin, 3-IL-1-β, 4-Lix, 5-MIP-1-gamma, 6-CCL5, each cytokine expression was determined in duplicate. (L) Relative expression of CCL5 in mock and HSV-2 infected vaginal tissue lysate. Bar graph showing average spot pixel density of CCL5 run in duplicate. Data (A-J) represent one of the three independent experiments with similar results n=3–4 sample/group/genotype/condition. Error bar (A-J) represent means ± SEM of at least triplicates. Experiment (K-I) was performed once and the average spot pixel density of CCL5 spot in duplicate is presented. Significance was determined by one way ANOVA with Šídák’s multiple comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001, ns, not significant.
Given the differences in immune infiltration between the OPTN +/+ and −/− mice, we also examined the cytokine levels in their vaginal tissues using qRT-PCR. Consistent with our in vitro findings, we observed no differences in the levels of IFN-α, IFN-β, IL-6 and CXCL10 between the infected OPTN +/+ and −/− animals (Fig. 4H–I). However, we found that the induction of CCL5 with infection was again inhibited in the absence of OPTN (Fig. 4J). With the mouse inflammation antibody array, we detected the semi-quantitative levels of 40 mouse proteins including CCL5 in the vaginal tissue lysate of OPTN +/+ and −/− mouse after 1dpi (Fig.4K, Fig.S2). The relative expression of CCL5 increased nearly 3.5 fold in OPTN +/+ samples after infection compared to non-infected controls while infected OPTN −/− samples had a very minimal change in CCL5 expression compared to non-infected OPTN−/− tissue (Fig.4L).
Loss of OPTN results in differential T-cell infiltration during HSV-2 infection
Since CCL5 functions as a chemoattractant, we wanted to identify the differential T- cell populations present in the vaginal tissue, iliac lymph nodes (which neighbors the site of HSV-2 infection) and spleen in each genotype during infection. We gated total singlet leukocyte (CD45+CD11b-CD11c-CD3+) to selectively measure the CD4+ and CD8+ T cell population (Fig. 5A). The frequency of helper T cells (CD4+) were significantly elevated over the period of infection in the OPTN +/+ in vagina, lymph node and spleen tissue compared to non-infected tissue (Fig. 5B–D). Whereas infected OPTN −/− tissue showed dysregulated pattern of CD4+ cells among the tissues (Fig 5B,E). The frequency of CD4+ cells in OPTN −/− spleen remained comparable between infected and non-infected mice (Fig 5D).
Figure 5. Loss of OPTN shows differential expression of T cells.
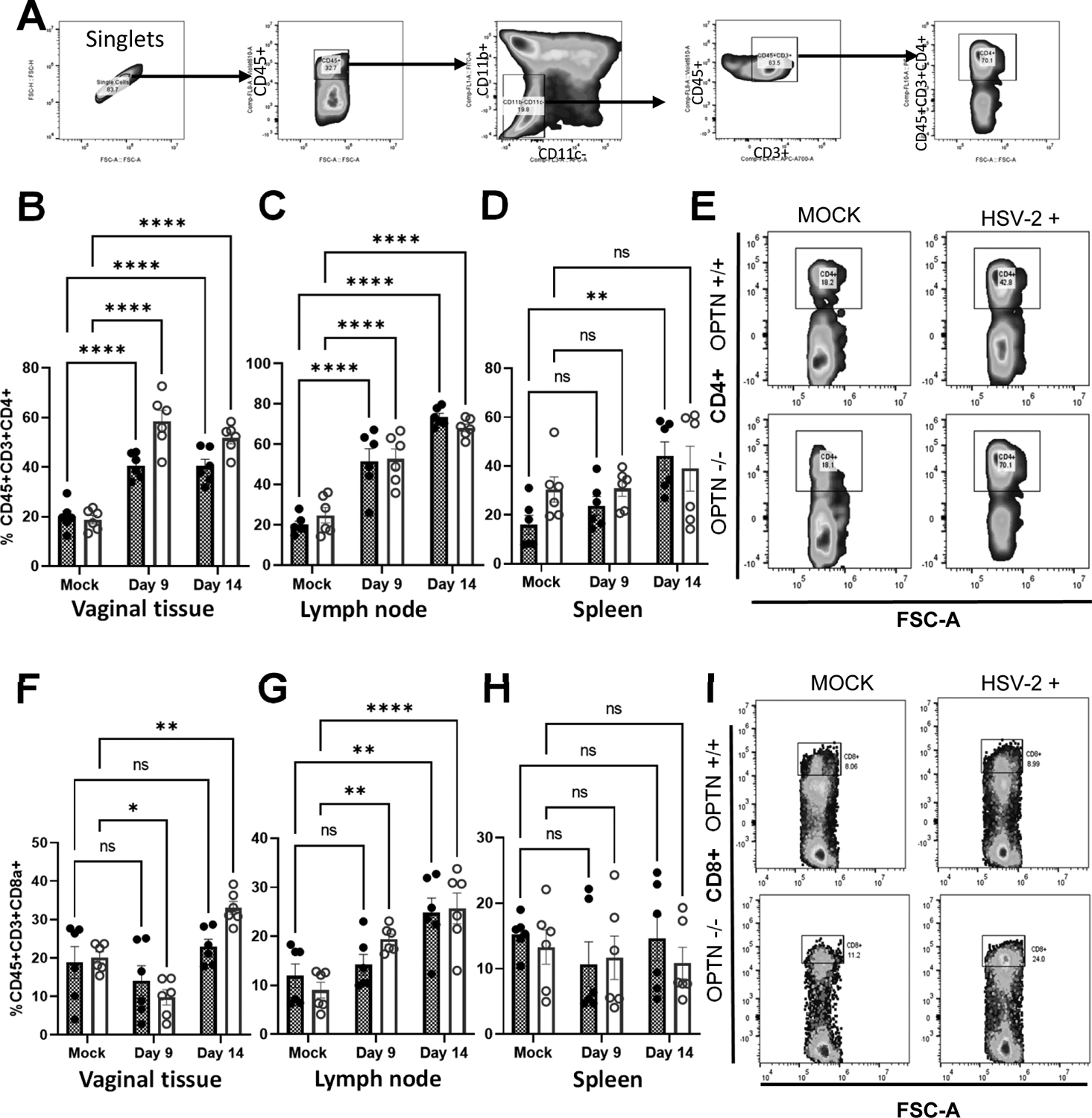
Mice vaginal tissue collected at 9 and 14 dpi were subjected to flow cytometry analysis of immune cell populations A) The dot plot shows selected windows and gating strategy applied to identify T cell population in mice tissues. From left to right directional arrows indicate gating for singlets, CD45+ leucocytes, then CD45+CD11b-CD11c- cells were selected as T cell population. Which further gated for CD3+CD45+ population. Finally, T helper (CD4+) or T cytotoxic (CD8+) cells were identified from CD45+CD3+ population. (B-D) Individual percent population of CD4+ positive immune cells from vaginal tissue, lymph node and spleen collected at 9 and 14 dpi from OPTN +/+ and −/− mock or infected mice. (E) Representative flow cytometry plot showing differential CD4+ expression in vaginal tissue. (F-H) Individual percent population of CD8+ positive immune cells from vaginal tissue, lymph node and spleen collected at 9 and 14 dpi from OPTN +/+ and −/− mock or infected mice. (I) Representative flow cytometry plot showing differential CD8+ expression in lymph node tissue. Data represent results generated from two independent experiment using a total of n=6 mice/group/genotype/condition. The results are presented as means ± SEM.Statistical significance was measured by two way ANOVA Šídák’s multiple comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001, ns, not significant.
Higher CD8+ T-cell frequency in infected OPTN−/− mice vaginal tissues at 14dpi compared to OPTN+/+ mice also points towards the prolonged inflammatory consequences in the absence of OPTN (Fig. 5F, I). Lymph node tissue also had higher frequency of CD8+ T-cells in infected OPTN−/− mice than the OPTN+/+ mice both at 9dpi and 14dpi. Interestingly, the baseline levels of CD8+ T-cells in both OPTN +/+ and −/− spleen tissue remained comparable over the course of infection (Fig.5H). The differences in immune populations were variable between the two groups.
HSV-2 infection correlates with OPTN and CCL5 expression
CCL5 was the only analyzed cytokine which transcriptionally differed with the loss of OPTN during HSV-2 infection in vitro and in vivo. CCL5 is a chemoattractant known to inhibit respiratory syncytial virus and CCR5-mediated HIV infection in vitro alongside protecting against Mycobacterium tuberculosis infections in vivo (28–31). As CCL5 induction was impeded in the OPTN −/− HeLa cells during infection, we examined whether it made up a second mechanism by which OPTN protects the host against HSV-2 infection. We transfected HeLa cells with an siRNA targeting CCL5 and observed the effect on viral replication. Knockdown of CCL5 increased viral encoded protein synthesis as measured by fluorescence imaging, plaque assay, qRT-PCR and western blotting (Fig. 6A–F). We confirmed that CCL5 induction was inhibited in OPTN +/+ HeLa by the siRNA-mediated knockdown (Fig. 6D) and found that loss of CCL5 significantly increased HSV-2 gB transcript production in OPTN +/+ HeLa cells (Fig. 6E–F). Interestingly, gB transcripts in the siCCL5 OPTN +/+ cells were approximately equivalent to their levels in OPTN −/− HeLa cells (Fig. 6E). Much of the increase in viral production in the absence of OPTN may be due to the lack of CCL5 induction during infection. With the immunofluorescence imaging, we also checked whether HSV-2 infection induces CCL5 protein expression in the presence of OPTN. OPTN +/+ HeLa cells showed higher CCL5 expression while OPTN −/− cells barely showed signs of CCL5 expression (Fig.6G). On the other hand, externally supplied rhCCL5 showed an increase in the phosphorylated form of OPTN in serum starved HeLa cells over a short period of time (Fig.6H) which suggests a yet unknown interaction between OPTN and CCL5 with a possible connection to autophagy. With these observations in mind we were curious to determine whether a similar relationship exists between OPTN and CCL5 in human subjects with HSV-2 genital infection. We therefore analyzed a publically available genital skin biopsy microarray profile dataset from HSV-2 infected patients (GSE172423). As predicted, we observed significantly upregulated CCL5 gene expression in HSV-2 infected genital lesions than the non-infected biopsy samples (Fig.6I). Next, we assessed the correlation between OPTN and CCL5, with and without HSV-2 infection. The control group did not correlate significantly (Fig.6J). In contrast, HSV-2 infected group showed strong positive correlation (Fig.6K), suggesting that the upregulation of CCL5 is accompanied by a simultaneous increase OPTN expression which corroborates with our in vitro experiment data (Fig. 6H).
Figure 6. HSV-2 infection correlates with OPTN and CCL5 expression.
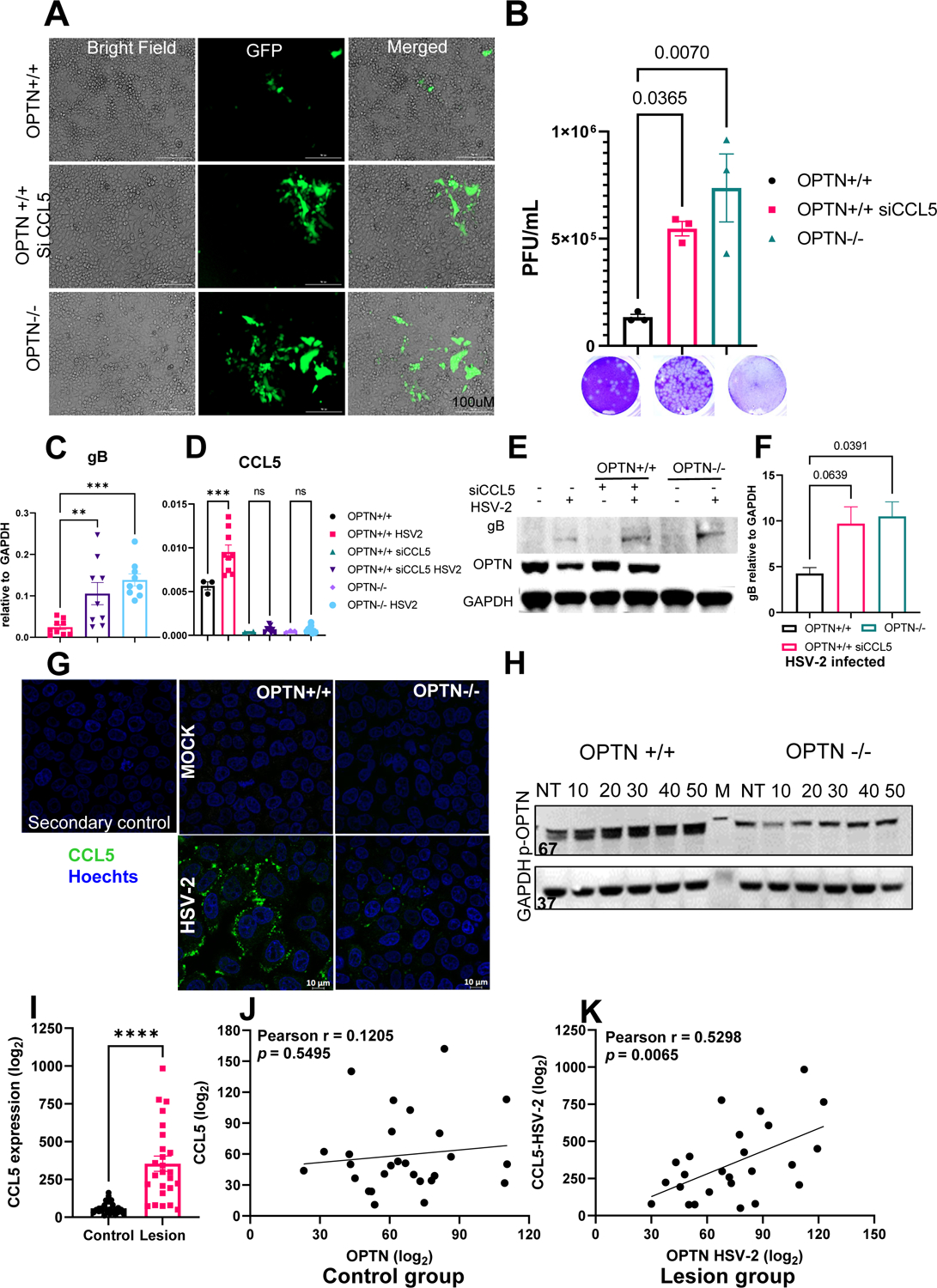
OPTN+/+, siCCL5 treated OPTN+/+ or OPTN −/− HeLa cells infected with GFP-HSV-2 333-strain at 1 MOI for 24h. (A) Representative images showing the infected cells emitting green fluorescence. (B) Viral titer from infected cells was determined by plaque assay. Representative plaque images are presented. (C-D) gB and CCL5 expression analysis by qRT-PCR. Copy numbers relative to GAPDH are shown. (E) Mock, infected or siCCL5 transfected cells were subjected to western blot analysis for viral protein relative to GAPDH. Immunoblot showing gB and OPTN. (F) Quantitative expression of gB in OPTN +/+, −/−infected or siCCL5 infected samples relative to GAPDH. (G) Immunofluorescence imaging of CCL5 expression in OPTN+/+ and OPTN−/− HeLa cells. Cells were infected with 5 MOI of HSV-2 for 2h then treated with Brefeldin A to arrest the release of cytokine and kept for another 4h then cells were processed for imaging. Green color represent the CCL5, blue denotes nuclear stain with Hoechts stain. (H) Optineurin phosphorylation with the external addition of rhCCL5 (50ng/mL) was monitored over 50 minutes by western blot. (I) Relative expression analysis of CCL5 gene in genital tissue with or without HSV-2 infection obtained from the public human transcriptomic dataset (GSE172423). (J-K) Correlation analysis on GSE172423 dataset between OPTN and CCL5 expression with (n=25) or without (n=27) HSV-2 infection. Each experiment (A-H) was performed 3 or more times (n=3/group/genotype/condition). Statistical analysis was performed using unpaired t test (I) or One-way ANOVA Šídák’s multiple comparison test (B,C,D,F) or Pearson correlation test (J-K). Error bar represent means ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001, ns, not significant
Discussion
HSV-2 is the leading cause of genital ulcers in the world (32). Given its relatively high recurrence rate and its ability to cause severe neurological disorders, HSV-2 is a pathogen of great public interest to combat (8). A lack of vaccine candidates or diverse treatment modalities compound the severity of the issue (33). A thorough understanding of host factors responsible for enhancing or inhibiting viral pathogenesis would allow for more effective therapies to be developed (34, 35). To our knowledge, this study is the first report on the protective role of OPTN during HSV-2 infection. OPTN −/− HeLa cells are more susceptible to infection, likely through their defects in selective autophagy and inducing CCL5 expression. The loss of CCL5 alone increases viral replication as demonstrated in the study (Fig. 6). Certain mutations in the Optn gene are associated with glaucoma and amyotrophic lateral sclerosis (16, 36–38). If these defects produce loss-of-function mutations, they may make individuals more vulnerable to HSV-2 infections and the associated manifestations.
There exists some debate on whether OPTN supports or detracts from host immunity. Evidence from bacterial studies suggests that OPTN assists in the clearance of intracellular pathogens via autophagy. Phosphorylation of OPTN has been shown to restrict Salmonella enterica infection through its enhanced LC3-binding affinity and subsequent autophagic clearance of ubiquitinated bacteria (18, 39). OPTN −/− mice were more susceptible to Salmonella infection as well (40). Loss of OPTN reduces the autophagic clearance of Mycobacterium marinum in a zebrafish model of infection, and overexpression of OPTN produces the opposite effect (19). Therefore, bacterial studies describing OPTN’s enhancement of intrinsic immunity are consistent with our results.
However, studies on the role of OPTN during viral infection are limited and primarily in vitro. OPTN has been shown to inhibit the production of IFN-β during Sendai virus infection, an ssRNA virus (22). Additionally, the silencing of OPTN using siRNAs promoted resistance to Sendai virus. (22). The apparent differences between our study and these previous reports may be virus-specific as OPTN may differentially interact with the evolutionarily distinct ssRNA Sendai virus and the dsDNA HSV-2 during infection. Our ongoing in vitro and in vivo studies with HSV-1 suggests the similar type of viral restriction in presence of OPTN. A recent study using HSV-1 found that the viral protein ICP0 down-regulated OPTN during infection, and depletion of OPTN did not impact production of HSV-1 virions. Our results differ from those of this study, possibly due to cell type and viral differences in the experimental designs. Our study was performed using HSV-2 infections in HeLa cells, while their study utilized HSV-1 infections in immortalized human embryonic lung fibroblasts. Furthermore, our in vitro findings of increased viral production and decreased CCL5 induction in the absence of OPTN are consistent with our in vivo data.
In vivo studies assessing viral infection OPTN-deficient mice are limited but suggest OPTN enhances antiviral responses to infection. One study reported that OPTN −/− mice displayed slightly higher mortality in response to a mouse-adapted influenza A infection (40). While there were no mortality differences between OPTN +/+ and −/− mice in response to HSV-2 infection, the knockout mice exhibited significantly more vaginal inflammation and tissue damage, viral shedding, and a reduction in body temperature during infection (Fig. 3). Other studies have found that loss of OPTN in vivo negatively regulates IFN-β upon pattern-recognition receptor stimulation (20, 41). While we did not observe differences in IFN-β production in the infected vaginal tissue, we did note that the antiviral chemokine CCL5 was not up-regulated in the OPTN −/− mice during HSV-2 infection. Thus, our findings are generally consistent with other in vivo OPTN-deficient mice studies indicating the impaired cytokine secretion and chronic inflammatory response upon bacterial infection (42).
As CCL5 is a potent chemokine, one would expect a dramatic shift in immune cell activation and recruitment in its absence. CCL5 expression is crucial and known for maintaining survival, migration and differentiation of T cells (43–45). CCL5−/− mice showed diminished cytotoxic ability of CD8+ T cells that led to develop chronic LCMV infection (46). In this study, we observed variable changes in T cell expression within the vaginal tissue with more CD4+ and CD8+ T cells in the OPTN −/− mice (Fig. 5). CD8+ T cells were similar in the lymph nodes tissue between the two infected genotypes (Fig. 5). Given the hyper-inflammation observed in the OPTN −/− vaginal tissue, it is possible that the immune cells present in abundance there over the period of time (CD4+, CD8+) may exacerbate the inflammatory response to viral infection in a similar manner to macrophage activation syndrome (47, 48). Also a delayed viral clearance and excessive airway inflammation due to influenza virus induced apoptotic death in macrophages was observed in mice lacking CCL5 (49). However, additional studies would be needed to confirm if this mechanism is the causative agent for hyper-inflammation observed during genital HSV-2 infection in the absence of OPTN. We noted here an interesting correlation between autophagy, Optineurin and CCL5. CCL5 has been reported to induce autophagy and regulate the cell migration through AMPK pathway (50). Although we did not explore the precise mechanism of CCL5 and OPTN correlation, our findings still indicate a high degree interaction between CCL5 with OPTN during the virus infection. This important correlation, as we found, also exists during the genital disease in humans (Fig. 6).
In conclusion, this study highlights OPTN as a host restriction factor against a potentially hyperproliferative spread of HSV-2 primary infection. This role is demonstrated in both in vitro and in vivo models of infection. We add HSV and possibly all herpesviruses under the antimicrobial roles of OPTN and provide an interesting link between autophagy adapter protein OPTN and the induction of CCL5 during infection in animals and humans alike. We also corroborate our in vitro findings with in vivo studies demonstrating enhanced viral pathogenesis and differential T-cell populations in vaginal and lymph node tissues in the absence of OPTN. Future studies investigating whether the differential immune responses account for the inflammation and tissue damage in the OPTN −/− mice will be useful in uncovering the relationship between OPTN and host immunity. Likewise, understanding polymorphic changes including loss-of-function OPTN mutations is likely to shed light on why certain human populations are more susceptible to acquiring HSV-2 infections or developing severe complications.
Supplementary Material
Key points:
OPTN mediates protection from HSV-2 by regulating the intrinsic immune response.
OPTN loss results in hyperproliferation of HSV-2 and reduced CCL5 induction.
OPTN/CCL5 nexus restricts hyper proliferative spread of primary HSV-2 infection.
Acknowledgements
We acknowledge Ruth Zhelka for help with imaging. This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
This work was supported by RO1 grants from the NIH (EY029426 and EY024710) to D.S. and a core grant (P30 EY001792)
This work was supported by NIH grants R01EY024710 (DS), R01AI1397686 (DS), and a departmental core grant P30EY001792 (DS).
References
- 1.Weiss H 2004. Epidemiology of herpes simplex virus type 2 infection in the developing world. Herpes 11 Suppl 1: 24A–35A. [PubMed] [Google Scholar]
- 2.Weiss HA, Buvé A, Robinson NJ, Van Dyck E, Kahindo M, Anagonou S, Musonda R, Zekeng L, Morison L, Caraël M, Laga M, and Hayes RJ. 2001. The epidemiology of HSV-2 infection and its association with HIV infection in four urban African populations. Aids 15 Suppl 4: 97. [DOI] [PubMed] [Google Scholar]
- 3.Fleming DT, McQuillan GM, Johnson RE, Nahmias AJ, Aral SO, Lee FK, and St Louis ME. 1997. Herpes simplex virus type 2 in the United States, 1976 to 1994. N Engl J Med 337: 1105–1111. [DOI] [PubMed] [Google Scholar]
- 4.Corey L, Adams HG, Brown ZA, and Holmes KK. 1983. Genital herpes simplex virus infections: clinical manifestations, course, and complications. Ann. Intern. Med 98: 958–972. [DOI] [PubMed] [Google Scholar]
- 5.Cowan FM, Johnson AM, Ashley R, Corey L, and Mindel A. 1994. Antibody to herpes simplex virus type 2 as serological marker of sexual lifestyle in populations. Bmj 309: 1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corey L and Holmes KK. 1983. Genital herpes simplex virus infections: current concepts in diagnosis, therapy, and prevention. Ann. Intern. Med 98: 973–983. [DOI] [PubMed] [Google Scholar]
- 7.Whitley RJ and Roizman B. 2001. Herpes simplex virus infections. Lancet 357: 1513–1518. [DOI] [PubMed] [Google Scholar]
- 8.Berger JR and Houff S. 2008. Neurological complications of herpes simplex virus type 2 infection. Arch Neurol 65: 596–600. [DOI] [PubMed] [Google Scholar]
- 9.Koganti R, Yadavalli T, and Shukla D. 2019. Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections. Microorganisms 7: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffiths SJ, Koegl M, Boutell C, Zenner HL, Crump CM, Pica F, Gonzalez O, Friedel CC, Barry G, Martin K, Craigon MH, Chen R, Kaza LN, Fossum E, Fazakerley JK, Efstathiou S, Volpi A, Zimmer R, Ghazal P, and Haas J. 2013. A systematic analysis of host factors reveals a Med23-interferon-λ regulatory axis against herpes simplex virus type 1 replication. PLoS Pathog 9: e1003514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moraru M, Cisneros E, Gómez-Lozano N, de Pablo R, Portero F, Cañizares M, Vaquero M, Roustán G, Millán I, López-Botet M, and Vilches C. 2012. Host genetic factors in susceptibility to herpes simplex type 1 virus infection: contribution of polymorphic genes at the interface of innate and adaptive immunity. J Immunol 188: 4412–4420. [DOI] [PubMed] [Google Scholar]
- 12.Tallóczy Z, Virgin HW, and Levine B. 2006. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2: 24–29. [DOI] [PubMed] [Google Scholar]
- 13.Yakoub AM and Shukla D. 2015. Basal Autophagy Is Required for Herpes simplex Virus-2 Infection. Sci. Rep 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yakoub AM and Shukla D. 2015. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci. Rep 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yakoub AM and Shukla D. 2015. Herpes simplex virus-1 fine-tunes host’s autophagic response to infection: A comprehensive analysis in productive infection models. PLoS ONE 10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong YC and Holzbaur ELF. 2014. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U. S. A 111: 4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ying H and Yue Beatrice Y. J. T. 2012. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol 294: 223–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, and Dikic I. 2011. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang R, Varela M, Vallentgoed W, Forn-Cuni G, van der Vaart M, and Meijer AH. 2019. The selective autophagy receptors Optineurin and p62 are both required for zebrafish host resistance to mycobacterial infection. PLoS Pathog 15: e1007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Outlioua A, Pourcelot M, and Arnoult D. 2018. The Role of Optineurin in Antiviral Type I Interferon Production. Front Immunol 9: 853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Génin P, Cuvelier F, Lambin S, Filipe JC, Autrusseau E, Laurent C, Laplantine E, and Weil R. 2015. Optineurin Regulates the Interferon Response in a Cell Cycle-Dependent Manner. PLOS Pathogens 11: e1004877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mankouri J, Fragkoudis R, Richards KH, Wetherill LF, Harris M, Kohl A, Elliott RM, and Macdonald A. 2010. Optineurin Negatively Regulates the Induction of IFNβ in Response to RNA Virus Infection. PLoS Pathog 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waisner H and Kalamvoki M. 2019. The ICP0 Protein of Herpes Simplex Virus 1 (HSV-1) Downregulates Major Autophagy Adaptor Proteins Sequestosome 1 and Optineurin during the Early Stages of HSV-1 Infection. J. Virol 93: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar R, Domrachev M, and Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, and Dikic I. 2016. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U. S. A 113: 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, Choi S, and Kang D. 2020. Quantitative and qualitative analysis of autophagy flux using imaging. BMB Reports 53: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Outlioua A, Pourcelot M, and Arnoult D. 2018. The Role of Optineurin in Antiviral Type I Interferon Production. Front Immunol 9: 853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vesosky B, Rottinghaus EK, Stromberg P, Turner J, and Beamer G. 2010. CCL5 participates in early protection against Mycobacterium tuberculosis. J Leukoc Biol 87: 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katsounas A, Schlaak JF, and Lempicki RA. 2011. CCL5: a double-edged sword in host defense against the hepatitis C virus. Int Rev Immunol 30: 366–378. [DOI] [PubMed] [Google Scholar]
- 30.Elliott MB, Tebbey PW, Pryharski KS, Scheuer CA, Laughlin TS, and Hancock GE. 2004. Inhibition of respiratory syncytial virus infection with the CC chemokine RANTES (CCL5). J Med Virol 73: 300–308. [DOI] [PubMed] [Google Scholar]
- 31.Coffey MJ, Woffendin C, Phare SM, Strieter RM, and Markovitz DM. 1997. RANTES inhibits HIV-1 replication in human peripheral blood monocytes and alveolar macrophages. Am J Physiol 272: 1025. [DOI] [PubMed] [Google Scholar]
- 32.Roett MA 2020. Genital Ulcers: Differential Diagnosis and Management. Am Fam Physician 101: 355–361. [PubMed] [Google Scholar]
- 33.Koujah L, Suryawanshi RK, and Shukla D. 2019. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cellular and Molecular Life Sciences 76: 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yadavalli T, Suryawanshi R, Koganti R, Hopkins J, Ames J, Koujah L, Iqbal A, Madavaraju K, Agelidis A, and Shukla D. 2020. Standalone or combinatorial phenylbutyrate therapy shows excellent antiviral activity and mimics CREB3 silencing. Sci. Adv 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadigal SR, Agelidis AM, Karasneh GA, Antoine TE, Yakoub AM, Ramani VC, Djalilian AR, Sanderson RD, and Shukla D. 2015. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat. Commun 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chalasani ML, Swarup G, and Balasubramanian D. 2009. Optineurin and its mutants: molecules associated with some forms of glaucoma. Ophthalmic Res 42: 176–184. [DOI] [PubMed] [Google Scholar]
- 37.Swarup G and Sayyad Z. 2018. Altered Functions and Interactions of Glaucoma-Associated Mutants of Optineurin. Front Immunol 9: 1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bansal M, Swarup G, and Balasubramanian D. 2015. Functional analysis of optineurin and some of its disease-associated mutants. IUBMB Life 67: 120–128. [DOI] [PubMed] [Google Scholar]
- 39.van Wijk Sjoerd J. L., Fricke F, Herhaus L, Gupta J, Hötte K, Pampaloni F, Grumati P, Kaulich M, Sou Y, Komatsu M, Greten FR, Fulda S, Heilemann M, and Dikic I. 2017. Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-κB and restricts bacterial proliferation. Nat Microbiol 2: 17066. [DOI] [PubMed] [Google Scholar]
- 40.Slowicka K, Vereecke L, Mc Guire C, Sze M, Maelfait J, Kolpe A, Saelens X, Beyaert R, and van Loo G. 2016. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-κB signaling. Eur J Immunol 46: 971–980. [DOI] [PubMed] [Google Scholar]
- 41.Meena NP, Zhu G, Mittelstadt PR, Giardino Torchia ML, Pourcelot M, Arnoult D, Ashwell JD, and Munitic I. 2016. The TBK1-binding domain of optineurin promotes type I interferon responses. FEBS Lett 590: 1498–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chew TS, O’Shea NR, Sewell GW, Oehlers SH, Mulvey CM, Crosier PS, Godovac-Zimmermann J, Bloom SL, Smith AM, and Segal AW. 2015. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Disease Models & Mechanisms 8: 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang L, Lin Y, Mahalingam J, Huang C, Chen T, Kang C, Peng H, Chu Y, Chiang J, Dutta A, Day Y, Chen T, Yeh C, and Lin C. 2012. Tumor-derived chemokine CCL5 enhances TGF-β-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res 72: 1092–1102. [DOI] [PubMed] [Google Scholar]
- 44.Shadidi KR, Aarvak T, Henriksen JE, Natvig JB, and Thompson KM. 2003. The chemokines CCL5, CCL2 and CXCL12 play significant roles in the migration of Th1 cells into rheumatoid synovial tissue. Scand J Immunol 57: 192–198. [DOI] [PubMed] [Google Scholar]
- 45.Luther SA and Cyster JG. 2001. Chemokines as regulators of T cell differentiation. Nat Immunol 2: 102–107. [DOI] [PubMed] [Google Scholar]
- 46.Crawford A, Angelosanto JM, Nadwodny KL, Blackburn SD, and Wherry EJ. 2011. A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog 7: e1002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crayne CB, Albeituni S, Nichols KE, and Cron RQ. 2019. The Immunology of Macrophage Activation Syndrome. Frontiers in Immunology 10: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ombrello MJ and Schulert GS. 2021. COVID-19 and cytokine storm syndrome: are there lessons from macrophage activation syndrome? Transl Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tyner JW, Uchida O, Kajiwara N, Kim EY, Patel AC, O’Sullivan MP, Walter MJ, Schwendener RA, Cook DN, Danoff TM, and Holtzman MJ. 2005. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med 11: 1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao H, Chen D, Cao R, Wang S, Yu D, Liu Y, Jiang Y, Xu M, Luo J, and Wang S. 2018. Alcohol consumption promotes colorectal carcinoma metastasis via a CCL5-induced and AMPK-pathway-mediated activation of autophagy. Sci Rep 8: 8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.