

Abstract
Herpes simplex virus type 1 (HSV-1) causes recurrent herpes labialis (RHL), a common disease afflicting up to 40% of adults worldwide. Mathematical models are used to analyze the effect of antiviral treatment on the transmission of, and the prevalence of drug resistance in, HSV-1 in the United States. Three scenarios are analyzed: no antiviral use, the current level of use, and a substantial increase in nucleoside analogue use, such as might occur if topical penciclovir were available over-the-counter for the treatment of RHL. A basic model predicts that present level of nucleoside analogue use has a negligible effect on HSV-1 transmission and that even if use of topical penciclovir for (RHL) increased substantially, the overall prevalence of infectious HSV-1 is unlikely to be reduced by more than 5%. An expanded model, which allows for acquired resistance and includes immunocompromised hosts and other more realistic features, predicts that current antiviral use is unlikely to lead to any noticeable increase in resistance. If antiviral use increases, the resulting rise in resistance in the population will depend primarily on the probability that immunocompetent hosts will acquire permanent resistance upon treatment. This probability is known to be small, but its exact value remains uncertain. If acquired resistance occurs less than once per 2,500 treated episodes, then in the community at large, the frequency of HSV-1 resistance is predicted to increase slowly, if at all (remaining below 0.5% for >50 years), even with extensive nucleoside analogue use. If acquired resistance emerges in 1 of 625 treated episodes (the maximum of an approximate 95% confidence interval derived from the results of several studies of resistance in treated hosts), then the prevalence of infection with resistant HSV-1 could rise from about 0.2% to 1.5 to 3% within 50 years. The limitations of existing data on acquired resistance and the potential impact of acquired resistance if it occurs are discussed, and strategies are suggested for enhancing information on acquired resistance. The predictions of this model contrast with the more rapid increases in antimicrobial resistance anticipated by models and observed for other pathogenic bacteria and viruses. The reasons for these contrasting predictions are discussed.
Recurrent herpes labialis (RHL), caused by herpes simplex virus type 1 (HSV-1) affects 15 to 40% of adults in countries around the world (23). Primary oral-facial infection with HSV-1 usually occurs in childhood and may either be asymptomatic or result in oral lesions. Following primary infection, the virus establishes latent infection in the sensory ganglia of the trigeminal nerve. Subsequently, latent HSV-1 may reactivate and spread back to the periphery to initiate a recurrent episode of disease (cold sore). In immunocompetent individuals, HSV-1 replication is self-limited and the cold sore disappears within about 10 days or less (50).
The nucleoside analogues acyclovir (ACV) and penciclovir (PCV) and their respective oral prodrugs, valaciclovir and famciclovir, are used to treat infections caused by HSV-1 or HSV-2 (generally associated with genital herpes). There are two approaches to the therapy of RHL: episodic treatment of a single symptomatic outbreak, which reduces the duration of symptoms and viral shedding (2, 50, 52, 53), and long-term suppressive therapy, which reduces the frequency of recurrences (44, 51). Although neither ACV nor PCV is approved for long-term suppression of RHL, topical PCV has been approved in the United States for treatment of RHL, and in other countries topical ACV and topical PCV are available. The same agents are also effective for the treatment of genital herpes, (35, 42, 45, 58). For both genital and labial herpes, treatment fails to eradicate latent virus and episodes continue to recur periodically after treatment is discontinued (50).
Resistance to ACV is readily selected in vitro and usually results from mutation in the thymidine kinase gene, leading to an absence or reduced expression of thymidine kinase and failure to activate ACV (28). Less commonly, resistance is attributable to a mutation in the viral DNA polymerase (17). ACV-resistant HSV is usually cross-resistant to PCV (11). Based on surveys among immunocompetent individuals, generally with genital herpes, ACV-resistant HSV is rare, appearing in about 0.3% of patients as measured by a plaque reduction assay (PRA) (15; R. Sarisky, K. Esser, R. Saltzman, L. Locke, R. Boon, T. Bacon, and J. Leary, Abstr. 38th Intersci. Conf. Antimicrob. Agents Chemother., abstr. H-10, p. 317, 1998). Because of the self-limiting nature of HSV reactivations in these patients, resistance has minor clinical consequences and resistant virus may occur only transiently (22). In severely immunocompromised people, however, resistance is more common. HSV can cause severe disease in these individuals, and resistance can have more serious clinical consequences (41).
This paper describes the results of mathematical modeling designed to assess the effects of antiviral treatment of RHL on the transmission dynamics of drug-sensitive and drug-resistant HSV-1 infections. Related models have been used recently to assess the impact of antiviral drug use on transmission of and resistance in genital herpes (7) and influenza (56). The models are used to answer two specific questions.
First, to what degree does antiviral treatment reduce the transmission of drug-sensitive HSV-1? This question is addressed, using a basic model, by considering three different scenarios of antiviral use. As a baseline, we consider a hypothetical case in which no antivirals were used to treat HSV-1. This baseline is then compared to the effects of current usage, as measured by recent antiviral prescription data from the United States. We then consider the effects of a large increase in antiviral usage, with specific reference to a substantial increase in topical PCV usage for the treatment of RHL, such as would be anticipated if this treatment were available over the counter (OTC).
Second, to what degree does antiviral treatment promote the development and spread of drug resistance in the community of hosts infected with HSV-1? To address the additional complexities of transmission of resistance, we use an expanded model, which adds several features to those of the basic model, most importantly, age structure, the existence of an immunocompromised class, and the possibility that treatment of an RHL episode can result in acquired resistance in a treated host.
(This work was presented in part at the 39th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, Calif., 26 to 29 September 1999.)
MATERIALS AND METHODS
Basic model.
The structure of the basic mathematical model is shown in Fig. 1, and the equations are given below. This compartmental model considers individuals in one of three states: susceptible (never infected with HSV-1), infected with sensitive virus, and infected with resistant virus. The number of individuals in each category is denoted by S, IS, and IR, respectively. Individuals are born into the susceptible class at rate b per day and live for an average of 1/u days. The overall framework is reflected in the following equations: dS/dt = b − (u + βSwSIS + βRwRIR)S; dIS/dt = βSwSISS − uIS; and dIR/dt = βRwRIRS − uIR.
FIG. 1.

Structure of a basic, compartmental model of HSV-1 transmission. (a) Overall framework, in which individuals are either susceptible (S), infected with sensitive virus (IS), or infected with resistant virus (IR). (b) Proportions of individuals infected with sensitive virus who are asymptomatic (φSA), symptomatic and untreated (φSU), or symptomatic and treated (φST). Infectiousness may differ among individuals in these three states. Untreated individuals remain infectious for longer than untreated individuals. (c) Once infected with resistant virus, individuals may be asymptomatic (a proportion, φRA), or symptomatic (a proportion, φRS); treatment of these persons has no effect.
An HSV-1-infected person may be either asymptomatic or symptomatic; the latter corresponds to an episode of RHL. Antiviral treatment may be applied during the symptomatic phase, and it is assumed that this will affect virus shedding by individuals infected with sensitive virus but have no effect on individuals infected with resistant virus. Individuals in the IS compartment fall into one of three states: asymptomatic (a proportion, φSA), symptomatic and treated (φST), or symptomatic and untreated (φSU), as shown in Fig. 1b. Individuals infected with sensitive virus will be asymptomatic most of the time but will become symptomatic at rate f per day. A proportion p of RHL episodes will be treated; the remainder (1 − p) are untreated. Untreated episodes will last, on average, for 1/dSU days, and treated episodes will last for a shorter period, 1/dST days, according to the equations dφST/dt = fpφSA − dSTφST; dφSU/dt = f(1 − p)φSA − dSUφSU; and φSA = 1 − φST − φSU. Transitions between the symptomatic and asymptomatic states are much faster than the dynamics of host death and new infection, so we make a quasi-steady-state assumption for the processes within the compartment (21, 37). Setting the three dφ/dt to 0, we obtain the solutions φ̂ST = fpdSU/[fpdSU + f(1 − p)dST + dSUdST]; φSU = f(1 − p)dST/[fpdSU + f(1 − p)dST + dSUdST]; and φ̂SA = dSUdST/[fpdSU + f(1 − p)dST + dSUdST]. We make a similar assumption for the IR compartment, where there are only two states, asymptomatic (φRA) and symptomatic (φRS) (Fig. 1c). New episodes occur at a rate of fR per day, with an average duration of 1/dR, according to the equations dφRS/dt = fRφRA − dRφRS and φRA = 1 − φRS. At quasi-steady state, φ̂RA = dR/(dR + fR) and φ̂RS = fR/(dR + fR).
Susceptible individuals acquire new infections with sensitive and resistant viruses at the per capita rates λS and λR, respectively. For sensitive infections, the force of infection is given by λS = ISβSwS, which is the product of the number of individuals currently infected with sensitive HSV-1, a transmission rate constant, βS, and a weighting factor, wS, that represents the sum of the fractions of asymptomatic, symptomatic, and treated individuals, with weights (α with the appropriate subscript) that reflect their levels of infectiousness relative to that of a symptomatic, untreated individual. Thus, wS = φ̂SU + αSTφ̂ST + αSAφ̂SA, where the carets represent the quasi-steady-state values for the fractions in each state, and their values are above.
Likewise, for resistant infections, the force of infection is given by λR = IRβRwR, where the weighting factor (wR) is φ̂RS + αRAφ̂RA. As stated above, IR represents the number of individuals infected with resistant virus. The transmission rate constant βR may be less than that for sensitive infections. This allows for the possibility that resistant infections are less transmissible than sensitive ones (most studies have shown reduced pathogenicity of resistant variants, but direct data on transmissibility are difficult to obtain [16, 27, 40]). The parameter c (which may be 0) is the fractional reduction in transmissibility of resistant infections compared to that of sensitive infections; thus, βR = (1 − c)βS.
In this model, treatment can reduce the transmission of sensitive HSV-1 in two ways. First, it reduces the duration of virus shedding during an episode of RHL, thereby reducing the period of time during which transmission is possible. Second, treatment may reduce the probability of transmission by reducing viral titers. Good data are available for the effect of topical PCV treatment on the duration of shedding in patients with RHL (52), but quantitative virus shedding data are not available. Nonetheless, in case treatment reduces the titer shed, we include this possibility in the model.
This basic model reflects a number of simplifying assumptions about the natural history and epidemiology of HSV-1. Specifically, it assumes (i) that the population is homogeneous and well mixed (for example, no particularly susceptible subpopulation, such as immunocompromised persons, is considered); (ii) that dual infection does not occur (55); and (iii) that treatment of individuals infected with sensitive virus does not result in acquired resistance (41; R. Sarisky et al., 38th ICAAC). These assumptions are relaxed in the expanded model introduced below.
Expanded model.
The expanded model is a generalization of the basic model but incorporates four major changes.
(i) Addition of an immunocompromised class.
In effect, the basic model of Fig. 1 has been duplicated to include variables for a class of immunocompromised individuals that correspond to variables for each class of immunocompetent individuals. Individuals enter an immunocompromised class from the corresponding immunocompetent class at a per capita rate of h per day. Immunocompromised individuals differ from immunocompetent individuals in three ways. First, they are assumed to be more susceptible to new infections by a factor, ς. Second, they may contribute more or less to transmission to other individuals by a factor, κ, than an equivalent immunocompetent person (they may be more infectious because of greater viral shedding; on the other hand, their condition may make them more isolated and therefore less likely to transmit their infection). Finally, their life spans are assumed to be shorter than those of immunocompetent people.
(ii) Dual infection is possible.
In this expanded model, it is assumed that an individual already infected with sensitive HSV-1 can be newly infected with resistant HSV-1 and vice versa. The number of individuals dually infected (with both kinds of virus) is indicated by the variable ID. Dual infection is known to be rare but possible in HSV-2 (12, 46); data for HSV-1 are not available, and we consider both zero and low rates in the model. Dual infection, like primary infection, occurs at a rate proportional to the rates of transmission (or force of infection) of sensitive and resistant viruses, but the model makes the assumption that individuals in the immunocompetent class acquire second infections at a rate δ times the rate at which they would acquire the infection if they were not already infected with the other strain. Because it is thought that infection (seropositivity) with an HSV-1 strain offers considerable protection against infection with another HSV-1 strain, δ is much smaller than 1. The corresponding parameter for the immunocompromised class, δ′, is assumed to be greater than δ but still smaller than 1.
(iii) Age structure.
HSV-1 may be acquired relatively early in life, and infected individuals may continue to be infectious throughout their life spans. Because the duration of infectiousness and the duration of life are comparable for this infection, we felt it was necessary to model host demography with type 2 survivorship, which is more realistic than the exponential (type 1) survivorship used for mathematical convenience in the basic model (3). The expanded model assumes that all immunocompetent individuals live for 70 years and then die. For immunocompromised individuals, type 1 survivorship is maintained. Here, an average life span of 10 years in the immunocompromised state is assumed. This assumption is made to be conservative, as longer life spans for immunocompromised persons increase their ability to spread resistant virus.
(iv) Acquired resistance.
Drug-sensitive infections in patients treating a recurrence with topical PCV may convert to drug-resistant infections. We assume that this occurs with probability m in each treated episode. Acquired resistance is thought to occur at an elevated rate in immunocompromised individuals (15, 24, 25), and this assumption is also reflected in the model.
Figure 2 shows the structure of the expanded model. Within each infected compartment, transitions among asymptomatic, symptomatic treated, and symptomatic untreated individuals are as in the basic model (Fig. 1b to c). All quantities referring to immunocompromised persons are marked with a prime.
FIG. 2.

Structure of an expanded model with consideration of age structure, immunocompromised class, and dual infection. As with the basic model, S denotes the number of individuals susceptible to HSV-1, IS denotes the number infected with sensitive virus only, IR denotes the number infected with resistant virus only, and ID denotes the number dually infected with both resistant and sensitive viruses. Within each infected compartment, transitions among asymptomatic, symptomatic treated, and symptomatic untreated are as described for the basic model (Fig. 1b to c). All quantities referring to immunocompromised persons are marked with a prime. ∗, type 2 survivorship; all deaths are at age 70.
The equations for immunocompetent individuals are (∂/∂a + ∂/∂t)S = b − u(a)S − λSS − λRS − hS; (∂/∂a + ∂/∂t)IS = λSS − [h + u(a) + δλR]IS − mφ̂STIS; (∂/∂a + ∂/∂t)IR = λRS − [h + u(a) + δλS]IR + mφ̂STIS; and (∂/∂a + ∂/∂t)ID = δ(λSIR + λRIS) − [h + u(a)]ID, where t is time and a is age.
The equations for immunocompromised individuals are (∂/∂a + ∂/∂t)S′ = hS − u′S′ − λ′SS′ − λ′RS′; (∂/∂a + ∂/∂t)I′S = hIS + λ′SS′ − (u′ + δ′λ′R)I′S − x′I′S; (∂/∂a + ∂/∂t)I′R = hIR + λ′RS′ − (u′ + δ′λ′S)I′R + x′I′S; and (∂/∂a + ∂/∂t)I′D = hID + δ′(λ′SI′R + λR′I′S) − u′I′D.
The equations for forces of infection are λS = β(IS + ID)wS + kβ(I′S + I′D)w′S; λR = βR(IR + ID)wR + kβR′(I′R + I′D)w′R; λ′S = ςλS; and λ′R = ςλR.
Parameter estimates.
The parameters of the basic model (Table 1) were estimated from published data. The precision of these estimates varies, depending on the quantity and reliability of data available. For each parameter, we have made a best estimate based on a consensus of the data, as well as a range of plausible values where appropriate. Where uncertainty exists, the range has been chosen to encompass values that are reasonable in light of available evidence.
TABLE 1.
Fixed parameters of the basic model that remain constant for all three scenarios
| Parameter | Meaning | Standard value (range) | Comments and/or reference(s) |
|---|---|---|---|
| u | Death rate of hosts (1/life span) | 4 × 10−5 day−1 = 1 / 70 yr | |
| b | Birth rate of hosts | 104 day−1 | 61 |
| αSA | Relative transmission potential (per unit of time) of an asymptomatic host compared to that of an untreated symptomatic host | 3% (0–10%) | Less than or equal to the prevalence of asymptomatic shedding among seropositives (63), which might be an underestimate by ∼50% (60) |
| αST | Relative transmission potential (per unit of time) of a treated symptomatic host compared to that of an untreated symptomatic host | 50% (10–100%) | Depends on the reduction in transmissibility during treatment for RHL but while the individual is still shedding; few data available (54) |
| βNeq | Transmission rate per unit of time of a symptomatic, untreated host carrying sensitive virus times the equilibrium population size | 2.55 × 10−3 (0.76 × 10−3 to 7.6 × 10−3) day−1 | Basic reproductive number for HSV-1 estimated to be in the range of 2–5, based on age-seroprevalence data (48, 63); value of βNeq calculated from equation 1, with Neq = b/u |
| c | Transmissibility cost of resistance | ? (0–50%) | Few data; in the pessimistic case, there may be little or no cost (26, 27) |
| fR | Avg rate at which individuals infected with resistant virus experience new RHL episodes | 0.004 day−1 | Assumed to be equal to f (Table 2) |
| αRA | Relative transmission potential (per unit of time) of an asymptomatic host compared to that of an untreated symptomatic host carrying resistant virus | 3% (0–10%) | Assumed to be equal to αSA (see above) |
| dR | Rate at which persons with resistant RHL episodes cease shedding | 0.17 day−1 | Assumed to be equal to dSU (Table 2) |
The final column of Table 1 gives a brief description of how the parameters were estimated. Briefly, the parameters in Table 1 are estimated from U.S. demographic data and from published data on the natural history of untreated RHL. Parameters for resistant infections are also included, but these estimates are more uncertain, because so few resistant infections have been observed in immunocompetent persons. In this case, a wide range of parameter values is considered to account for the possible variation.
Table 2 contains estimates of parameters that are dependent on the amounts and kinds of antiviral drugs in use. Because we are interested in how the transmission dynamics of HSV-1 are affected, both by current antiviral use and by a hypothetical increase in use of topical PCV, such as would occur in the United States if PCV was to be available OTC for RHL, we consider three scenarios and present parameter estimates for each. Parameters are first calculated for a baseline of no antiviral use. These are readily estimated from data on the natural history of HSV-1 infection in untreated individuals. The parameters are then reestimated for two scenarios of antiviral use. Scenario 1 considers antiviral use at the current level in the United States. To obtain these parameters, audited prescription data on antiherpesvirus drugs are combined with clinical trial data on the effects of treatment on virus shedding (see below). Scenario 2 assumes a substantial increase in the use of topical PCV. Clinical trial data are used to obtain estimates of parameters governing the effect of treatment on viral shedding and transmission.
TABLE 2.
Parameters of the basic model that vary by scenario
| Name | Meaning | Standard value (range)
|
Derivation (reference[s]) | ||
|---|---|---|---|---|---|
| Baseline (no antiviral use) | Scenario 1 (current antiviral use) | Scenario 2 (increased antiviral use) | |||
| dSU | Rate at which persons with untreated, sensitive RHL episodes cease shedding | 0.17 day−1 | 0.17 day−1 | 0.17 day−1 | Calculated from duration of shedding in clinical trial controls (52) (rate = 1/duration); may be a slight overestimate as shedding may begin before symptoms |
| dST | Rate at which persons with treated, sensitive RHL episodes cease shedding | NAa | 0.4 (0.26–0.85) day−1 | 0.23 day−1 | From duration of shedding in trials of oral ACV and famciclovir (2, 52, 53) (scenario 1) and topical PCV (52) (scenario 2); presymptomatic shedding may reduce this estimate modestly, as described above |
| f | Avg rate at which individuals infected with sensitive virus experience new RHL episodes | 0.004 day−1 | 0.004 day−1 | 0.004 day−1 | Baseline calculated from the prevalence of HSV-1 seropositivity (48, 63) and the prevalence and frequency of RHL (23, 31, 47); effect of current usage would be negligible in suppressing RHL at the population level, although it would be effective for individuals (44, 51, 57), because a small proportion of the HSV-1-positive population uses antivirals currently; topical PCV (scenario 2) would not affect recurrence rate |
| p | Proportion of individuals infected with sensitive virus who treat their RHL episodes with topical PCV | 0 | 0.15–2.5% | 20% (10–30%) | Marketing projections from SmithKline Beecham, based on experience with OTC topical ACV cream in the United Kingdom (P. Johnston, personal communication) |
NA, not applicable.
The estimates of parameters for scenario 1 (current antiviral use) were calculated as follows. Prescription data (obtained from the Scott-Levin Source Prescription Audit) for all oral (branded and generic) forms of ACV, valaciclovir, and famciclovir indicate that 339 million units were prescribed from September 1996 to August 1997 in the United States. This was converted to 108 million daily doses (=297,000 patient years) using standard dosing regimens (6). The effect of this use on the duration of shedding (used as a surrogate for transmission) of HSV-1 from RHL depends on how much of this total use was to treat episodes of RHL and how much was for treatment of other conditions, such as genital herpes or non-HSV conditions. Of 2.8 million prescriptions for the ACV family of drugs in the period September 1996 to August 1997, 52,000 (2%) were classified as being for herpetic gingivostomatitis, roughly 1.8 million (64%) were for conditions other than RHL, and 986,000 (34%) were for herpes simplex without complications (anatomical site not specified) (Scott-Levin Source Prescription Audit data). Therefore, 2 to 36% of nucleoside analogue usage may have been for oral or labial HSV-1; if one can extrapolate from the prescriptions that did specify the site of infection, then the actual percentage used for RHL is near the low end of this interval. In calculating the figures in scenario 1 for Table 2, we used data on the reduction of HSV-1 shedding in treated patients from efficacy trials to calculate the effect of the fraction of treatment that is directed against herpes labialis or gingivostomatitis (2, 52, 53). We assumed that the use of these compounds to treat other conditions would affect HSV-1 shedding as if the recipients were taking the drug to suppress RHL (44, 51, 57). Because the number of individuals taking nucleoside analogues for other conditions at any given time is a small fraction of the total HSV-1-seropositive population, the effect on the overall average rate (f) or duration (dSU) of recurrences in the HSV-1-seropositive population is negligible (Table 2).
Table 3 contains the parameter estimates for the expanded model. Where possible, the parameters of the expanded model have been estimated from published data. Because the expanded model includes factors for which data are rare or unavailable, such as the possibility of dual infection and the potentially different transmission dynamics for HSV-1 to and from immunocompromised hosts, greater uncertainties are inherent in the parameters for this model. Consequently, broad ranges of values are considered for some parameters. Brief comments on the derivation of these parameters and relevant references are given in the second column of Table 3.
TABLE 3.
Biological interpretations, names, and ranges of the parameters used in the expanded model
| Parameters | Meaning of the parameter and/or Derivation (reference[s]) | Immunocompetent hosts
|
Immunocompromised hosts
|
||
|---|---|---|---|---|---|
| Symbol | Value (range) | Symbol | Value (range) | ||
| Host demographic parameters | Birth rate of new hosts | b | 104 day−1 | ||
| Death rate (life span) of hosts | u | Life span of 70 yr; type 2 survivorship | u′ | 3 × 10−4 day−1 | |
| Rate at which hosts become immunocompromised; assumed to be equal to the AIDS incidence rate (14). Bone marrow transplants, the other major source of immunosuppression that increases susceptibility to HSV infections (41), are much less frequent (30) and the length of immunosuppression is shorter (36), so this source makes a negligible contribution compared to AIDS. | h | 5 × 10−7 day−1 | |||
| General parameters of the infection process | Susceptibility of an immunocompromised host to new infection, relative to that of an immunocompetent host. No data available; wide range considered | ς | 3 (1–10) | ||
| Proportion of individuals infected with sensitive virus who treat their RHL episodes with topical PCV; parameter is as in basic model. Topical PCV is not indicated for RHL in the immunocompromised; a 10% maximum is considered to account for possible misuse. Systemic antiviral treatment is currently widely used in this population for treatment of herpes infections. | p | 20% (10–30%) | p′ | 10% (0–10%) | |
| Probability that a seropositive individual will become dually infected with the type of virus (sensitive or resistant) not already carried, compared to the probability of infection for the same individual if seronegative; value of this parameter is unknown; thought to be low but possibly higher in immunocompromised individuals | δ | 0.1% (0–1%) | δ′ | 10% (1–100%) | |
| Probability that permanent acquired resistance occurs in a treated episode in an immunocompetent host (explanation in text) | m | (0, 1/625) | |||
| Rate of conversion of sensitive infections to resistant ones in immunocompromised hosts; value that gives approximately 6% prevalence of resistance in the immunocompromised prior to increasing treatment (15) | x′ | 1.7 × 10−5 day−1 | |||
| Parameters relating to drug-sensitive virus infections | Avg rate at which individuals infected with drug-sensitive virus experience new RHL episodes; parameter is as in basic model | f | 0.004 day−1 | f′ | 0.004 day−1 |
| Rate at which untreated, drug-sensitive RHL episodes cease shedding; parameter is as in basic model | dSU | 0.17 day−1 | d′SU | 0.17 day−1 | |
| Rate at which treated, drug-sensitive RHL episodes cease shedding; parameter is as in basic model | dST | 0.23 day−1 | d′ST | 0.23 day−1 | |
| Transmission rate per unit of time of a symptomatic, untreated host carrying sensitive virus; parameter for immunocompetent individuals is as in basic model; no data for compromised individuals; wide range considered | βS | 1 (0.3–3) × 10−11 host−1 day−1 | βS′ = kβS | κ = 1 (0.1–10) | |
| Transmission potential (per unit of time) of an asymptomatic host relative to that of an untreated symptomatic host; parameter is as in basic model | αSA | 1% (0–5%) | α′SA | α′SA = αSA | |
| Transmission potential (per unit of time) of a treated symptomatic host relative to that of an untreated symptomatic host; parameter is as in basic model | αST | 50% (10–100%) | α′ST | α′ST = αST | |
| Parameters relating to drug-resistant virus infections | Avg rate at which individuals infected with resistant virus experience new RHL episodes; parameter is as in basic model | fR | fR = f | f′R | f′R = f′ |
| Rate at which resistant RHL episodes cease shedding; parameter is as in basic model | dR | dR = dSU | d′R | d′R = dR | |
| Transmission rate per unit of time of a symptomatic host carrying resistant virus; parameter is as in basic model | βR = (1 − c)βS | c = 0.1 (0–0.9) | βR′ = κβR (1 − c)β′S | κ = 1 (0.1–10) | |
| Transmission potential (per unit of time) of an asymptomatic host carrying resistant virus relative to that of a symptomatic host carrying resistant virus; parameter is as in basic model | αRA | αRA = αSA | α′RA | α′RA = αRA | |
Acquired resistance.
A key parameter of the expanded model is m, the probability that treatment of one RHL episode in an immunocompetent individual results in permanent acquired resistance in that individual. Because of the importance of this parameter, we describe the estimation of its possible values in detail. At present, it is unclear whether resistance can be acquired when a symptomatic episode of sensitive HSV-1 in an immunocompetent person is treated with a nucleoside analogue. There are four reports in which treatment of an immunocompetent host may have resulted in the acquisition of resistance in HSV-1 or HSV-2 (22, 33, 38, 58, 59). In none of these is it certain whether treatment itself was responsible for the appearance of resistant virus. More than 1,900 immunocompetent patients have been monitored in clinical trials of various nucleoside analogues for symptomatic treatment or suppressive therapy of HSV-1 and HSV-2, and there has not been a significant increase in antiviral resistance in treated patients compared to that in untreated patients (or compared to that in matched pretreatment isolates) (1, 15, 18–20, 24, 29, 34, 35, 38; R. Sarisky et al., 38th ICAAC). If one assumes that acquired resistance occurred in 0 of 1,900 episodes (a conservative estimate of the total number, taking into account the fact that some clinical trial data seem to be described in more than one published paper and ignoring the possibility that more than one episode may have occurred in some patients) treated with nucleoside analogues, then the 95% confidence interval (CI) for m, the probability of acquired resistance per treated episode, using the binomial assumption is 0 to 0.0016. We use this interval as the possible interval for m in our model, with the following two caveats.
First, it is inevitably difficult to estimate the frequency of rare events from such a small sample size (13, 32). Second, the estimate acquires additional uncertainty from the limitations of the biological assay used to determine the prevalence of resistant HSV. The PRA is the standard assay for measuring the susceptibility of HSV to an antiviral agent; the endpoint of the assay is the concentration of drug that is required to inhibit plaque formation by half of the virus inoculum, known as the 50% inhibitory concentration. Such an assay is unlikely to detect the appearance of highly resistant viruses, if these viruses represent a small percentage (e.g., 5%) of the overall virus population in each sample tested (39, 49) (J. Leary and R. Sarisky, unpublished data). If acquired resistance does occur in treated patients, it may occur by a series of progressive “enrichments” of the resistant population during successive treated episodes, which would not be detected by the PRA in most cases. If this is the case, then the low probability of acquired resistance measured by the PRA in clinical trials may substantially underestimate the true probability. We emphasize these uncertainties in the acquired resistance parameter because of its importance in determining the results (see below).
Evaluation of the model.
The basic model was evaluated analytically for particular parameter values to determine the effect of treatment on the prevalence of HSV-1 infection. Uncertainty about key parameters was quantified by using the range of parameter values described below. The expanded model was evaluated numerically using a FORTRAN program, which treated the partial differential equations as a set of coupled ordinary differential equations in which each 1-year age class was a separate compartment and integrated the differential equations by the Euler method. Again, different parameter values were used to quantify the impact of different assumptions about acquired resistance and transmission of resistance.
In exploratory runs of the expanded model, we found that the most important single factor determining the rate at which resistance spreads was m, the probability of acquired resistance per treated RHL episode in an immunocompetent host. Other parameters influencing this rate were those that determined the selective pressure in favor of resistant virus, including the level of antiviral use, the level of transmission to and from immunocompromised persons (who are more likely to be infected with resistant HSV-1), and the extent to which treatment reduces transmission of sensitive virus. We therefore performed our simulations and present the results for four values of the parameter m and for three sets of values for the other parameters, collectively referred to as the “selective pressure.”
RESULTS
Effect of antiviral treatment on transmission and prevalence of drug-sensitive HSV-1.
The basic model was used to estimate the effect of treatment on the transmission and prevalence of drug-sensitive HSV-1. The transmissibility of HSV-1 can be measured by the basic reproductive number, R0. The basic reproductive number of an infection is defined as the average number of secondary cases that would be generated by a single infected individual placed into a completely uninfected population at equilibrium and is given for this model (for sensitive virus) by the following equation:
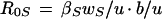 |
1 |
For an infection like HSV-1, the basic reproductive number can be estimated (3) from the equation
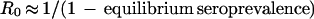 |
2 |
or from the equation
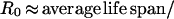 |
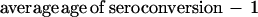 |
3 |
Based on available age-seroprevalence data (48, 63), these equations yield an approximate value for R0 in the range of 2 to 4.5.
Treatment of some individuals with antiviral drugs reduces the transmission of sensitive HSV-1. The extent of this reduction can be measured as the percentage reduction in R0 when antiviral drugs are used at a particular level, compared to the value of R0 when no antiviral drugs are used. Another measure of the effect of treatment is the change in the equilibrium prevalence of infection, which is calculated from the basic reproductive number by equation 2.
Scenario 1: antiviral usage at current levels.
The model predicts that current levels of antiviral usage have a small impact on the transmission and prevalence of sensitive HSV-1. For any given reduction in the basic reproductive number R0, there is a corresponding reduction in the equilibrium seroprevalence of HSV-1, given by equation 2 above. In all cases, the reduction in seroprevalence is smaller than the reduction in R0. Figure 3a shows the estimated reductions in R0 (left-hand scale) and equilibrium seroprevalence (right-hand scale; dashed lines) that result from current antiviral usage. Depending on the assumptions, current antiviral use reduces transmission by between 0 and 2.5%, which translates into a reduction in prevalence of less than 1%.
FIG. 3.

Predicted effect of antiviral treatment on transmission of drug-sensitive HSV-1. In both panels a and b, reduced transmission is shown as the percentages of reduction in the basic reproductive number, R0 (left-hand scale), and in equilibrium seroprevalence (right-hand scale; dashed lines). (a) Current use. On the x axis are the percentages of current use that are directed against RHL episodes, rather than other conditions. The three lines reflect different assumptions about the values of parameters of the model. In the upper line (greatest reduction in transmission), αST = 0.1, αSA = 0, and dST = 0.85; in the middle line, αST = 0.5, αSA = 0.02, and dST = 0.4; and in the lower line (smallest reduction in transmission), αST = 1, αSA = 0.05, and dST = 0.26). Other parameters are as shown in Table 1, scenario 1. (b) Increased use. Reductions are shown for the use of topical PCV to treat up to 30% of all RHL episodes (x axis), with different assumptions being made about the transmissibility of HSV-1 from treated symptomatic and asymptomatic hosts relative to that from untreated symptomatic hosts. In the upper line (greatest reduction in transmission), αST = 0.1 and αSA = 0; in the middle line, αST = 0.5 and αSA = 0.02; and in the lower line (smallest reduction in transmission), αST = 1 and αSA = 0.05). Other parameters are as in Table 1, scenario 2.
The uncertainty in the size of the predicted effect results from uncertainty in several parameters: the effect of treatment in reducing the duration of an episode, the effect of treatment in reducing an individual's infectiousness (per unit of time) during an episode, the contribution of asymptomatic individuals to transmission of the virus, and, most importantly, the fraction of current antiviral usage that is directed against RHL, rather than against other conditions (e.g., genital herpes). Available prescription data show that this fraction lies between 2 and 34%, as described in Materials and Methods. As Fig. 3a shows, the size of the reduction in transmission is approximately a linear function of the percentage of current antiviral use that is actually used to treat RHL. The three lines show the effects of different assumptions about the other parameters.
Scenario 2: increased antiviral usage.
If antiviral use for RHL were increased substantially by introduction of topical PCV OTC, the model predicts that the effect on transmission and prevalence would be larger than the effect of current treatment but that it would remain modest in absolute terms. Figure 3b shows the effects on transmission of HSV-1, assuming that topical PCV is used to treat up to 30% of RHL episodes. As before, the left-hand scale shows the percentage reduction in R0 while the right-hand scale shows the corresponding reduction in equilibrium seroprevalence. Taking the intermediate set of parameters, the reduction in seroprevalence of HSV-1 is expected to be less than 5%, even if 30% of RHL episodes are treated.
As in scenario 1, the size of the effect depends on several parameters. The reduction in transmission grows approximately linearly with the level of antiviral use (percentage of RHL episodes treated), which is shown on the x axis. The three lines in Fig. 3b reflect different assumptions about two other parameters that determine the size of the effect: whether individuals who are treated, but still symptomatic, are less infectious than those who are untreated and symptomatic and how much asymptomatic shedders contribute to transmission, relative to the contribution of symptomatic patients.
Effects of antiviral use on the spread of resistant infections.
The expanded model was used to analyze the effect of antiviral use on the spread of resistance.
Effects of current antiviral use.
This model predicts that at current levels of antiviral use, the prevalence of resistance will remain low. In the absence of antiviral use, the basic reproductive number of sensitive infections is expected to be greater than that of resistant infections, as long as resistant infections have even a small (1 to 2%) disadvantage in their rate of transmission. As mentioned in Materials and Methods (see also Tables 1 and 4), the magnitude of this cost of resistance is unknown but appears to be considerable for many resistant isolates. As the amount of antiviral usage increases, the basic reproductive number of sensitive infections (R0S) declines while the corresponding number for resistant infections (R0R) stays constant. The reduction in R0S as a result of treatment can be seen as a burden imposed by treatment on the fitness of the sensitive virus. This burden can be directly compared to the cost (c), the proportional reduction in transmission from hosts infected with resistant virus compared to that from hosts infected with sensitive, untreated virus. To a good approximation, if the burden of treatment on the fitness of sensitive infections is greater than the cost of resistance, then resistant infections will be able to spread in the population. If the cost of resistance is greater, then resistant infections will remain at low levels or decline.
TABLE 4.
Pessimistic, moderate, and optimistic sets of parameter values for selective pressure
| Parameter | Name(s) | Pessimistic value | Moderate value | Optimistic value |
|---|---|---|---|---|
| Basic reproductive no. (prior to treatment) | R0 | 4.4 | 3.3 | 2.3 |
| Cost of resistance | c | 0 | 10% | 50% |
| Proportion of RHL episodes treated in immunocompetent | p | 30% | 20% | 10% |
| Proportion of RHL episodes treated in immunocompromised | p′ | 10% | 0 | 0 |
| Relative rate of secondary HSV-1 infection in immunocompetent | δ | 0.01 | 0.001 | 0 |
| Relative rate of secondary HSV-1 infection in immunocompromised | δ′ | 1 | 0.1 | 0.01 |
| Relative ability of an immunocompromised individual to transmit | κ | 3 | 1 | 0.3 |
| Relative susceptibility of an immunocompromised individual to infection | ς | 10 | 3 | 1 |
| Relative rate of transmission per unit of time from an asymptomatic individual | αSA, α′SA, αRA, α′RA | 0 | 0.03 | 0.1 |
| Relative rate of transmission per unit of time from a treated individual | αST, α′ST | 0.1 | 0.5 | 1 |
As seen above, using the basic model, the burden imposed by current antiviral treatment on the transmission of sensitive virus is very low. As a result, even a small cost of resistance (26, 27) is enough to outweigh the burden imposed by current treatment and resistant infections remain rare.
Effects of increased antiviral use.
We first performed exploratory simulations using the parameter ranges described in Table 3. From these simulations, it was clear that the probability of acquired resistance per treated RHL episode, m, is the single most important factor determining whether and how fast resistance increases in the population following an increase in the rate of topical PCV use. As described in Materials and Methods, the 95% CI for m estimated from a combination of studies is (0, 0.0016). Therefore, we present the simulation results separately for four different values of this key parameter: 0, 0.0016 (1 in 6,250 treated episodes or 1/10 of the maximum of the 95% CI), 0.0004 (1 in 2,500 treated episodes or 1/4 of the maximum of the 95% CI), and 0.0016 (1 in 625 treated episodes or the maximum of the 95% CI). These results are shown in Fig. 4.
FIG. 4.

Changes in the prevalence of infection with resistant HSV-1 following an increase in topical PCV usage. (a to d) Increasing probabilities of acquired resistance per treated episode in an immunocompetent host (m), within the rough 95% CI estimated in the text, with m = 0, (a), m = 1/6,250, (b), m = 1/2,500, (c), and m = 1/625 (d). Curves (from top to bottom within each panel) represent pessimistic, moderate, and optimistic assumptions concerning the selective pressure (antiviral use, effect of use in reducing transmissibility, etc.). In each scenario, it is assumed that 0.3% of HSV-1-infected persons are infected with resistant virus at time zero but that the starting levels of prevalence of resistant infection are different under different scenarios (and less than 0.3%) because the figure shows the prevalence of resistant infection (prevalence of HSV-1 infection times the proportion of individuals who are resistant) in the whole population, and the levels of prevalence of HSV-1 infection are different in different scenarios.
The other key parameters of the model, which determined the strength of selective pressure for resistant virus, were varied together to create three parameter sets. The “optimistic” parameter set uses those values, chosen from the plausible range of each parameter (Table 3), which result in the slowest increase in resistance, for example, a low total transmissibility (R0), high cost of resistance, and low levels of transmission to and from the immunocompromised class. The “pessimistic” parameter set, on the contrary, uses those values for each parameter that result in the most rapid ascent in the frequency of resistant infection, for example, high R0, no cost of resistance, and high levels of transmission of virus to and from the immunocompromised class. The “moderate” set takes what we judged to be the most plausible values, intermediate between these extremes, for each of the changeable parameters. The values of these parameters for each parameter set are given in Table 4, and each panel of Fig. 4 contains three curves corresponding to the three parameter sets.
If treatment of RHL episodes in immunocompetent persons does not produce acquired resistance (m = 0) (Fig. 4a), or if it does so at a sufficiently low rate (m = 0.00016 or 1 of 6,250 treated episodes) (Fig. 4b), then the prevalence of resistant HSV-1 will remain relatively low, less than 0.6% over 50 years, regardless of the degree of selective pressure. If the probability of acquired resistance takes on an intermediate value (m = 1 of 2,500 treated episodes) (Fig. 4c), then the rise of resistance will depend strongly on the selective pressure; if this is high, then resistant infections could reach 1% of the population within 50 years; however, if it is moderate or low, the prevalence of resistance will remain below 0.5%. Finally, if acquired resistance is very common (m = 0.0016 or 1 of 625 treated episodes), then the increase can be faster, as long as there is a moderate or large (pessimistic) degree of selective pressure. In these circumstances, the prevalence of infection in the population with resistant HSV-1 could reach 1.5 to 3% after 50 years, an increase of 7- to 12-fold over its starting value of about 0.2%, depending on the selective pressure.
DISCUSSION
We have used a mathematical model to analyze the effect of antiviral treatment of RHL on the transmission dynamics of HSV-1. Two basic questions were addressed: (i) to what degree does antiviral treatment reduce the transmission of the virus, thereby providing a public health benefit in the form of reduced prevalence of HSV-1 infection, and (ii) does the selective pressure imposed by antiviral treatment cause the spread of antiviral-resistant HSV-1 infections, and if so, how fast will these infections spread? We first modeled the effects of current antiviral use and then used the model to predict the effects on transmission and resistance in HSV-1 if there were a substantial increase in the use of topical PCV to treat recurrent herpes labialis, such as might be expected if PCV was approved for OTC sales.
Our principal findings were as follows. First, current antiviral use has at most a small effect on the transmission and prevalence of HSV-1. Consequently, it also exerts a small selective effect in favor of antiviral resistance, so the model predicts that current antiviral use is unlikely to increase the prevalence of antiviral resistance. This prediction is consistent with recent surveillance data largely from patients with genital herpes (15, 43).
Second, a substantial increase in antiviral treatment of RHL would produce only a modest reduction in the transmission and prevalence of HSV-1. Estimating this effect precisely is difficult, primarily because adequate data on the relationship between symptomatic and asymptomatic viral shedding and also on transmission of HSV-1 infection are not available (9, 65). Nonetheless, using the best available estimates (the middle line of Fig. 3b), the model predicts that the prevalence of HSV-1 infection would decline by less than 5%, even if 30% of all recurrences were treated with topical PCV.
Third were the effects of increased antiviral use on resistance. The most important parameter determining the rate at which the prevalence of resistant HSV-1 rises following an increase in antiviral usage is m, the probability that a treated RHL episode will result in acquired resistance. Unfortunately, this parameter is also the most uncertain; with approximately 1,900 patients studied for emergence of resistance during nucleoside analogue treatment of HSV infection, the 95% CI for m is (0, 0.0016), corresponding to the emergence of acquired resistance in between 0 and 1 in 625 treated episodes. If the true probability of acquired resistance is less than or equal to about 1 in 6,250 treated episodes (10% of the maximum value), then the increase in resistant infections will be very slow, remaining below 1% prevalence after 50 years even if the selective pressure in favor of resistance is very strong. If the true probability of acquired resistance takes a value intermediate between these figures, approximately 1 in 2,500 patients, then rapid increases in the prevalence of resistant infection will occur only if the selective pressure for resistance is very high. If the true probability of acquired resistance were indeed 1 in 625 treated episodes, then the spread of resistance could be faster; in the extreme, the prevalence of resistant HSV-1 could exceed 1% within less than 20 years. However, we note that even under these assumptions, which are pessimistic with respect to both acquired resistance and selective pressure, the predicted rate of increase is still considerably slower than that observed (4, 5, 8) or predicted (10, 56) for many other viral and bacterial pathogens.
In determining the optimistic and pessimistic values for the parameters that underlie the selective pressure, we used values consistent with available data that would produce the slowest and fastest increases in the prevalence of resistant infection, respectively; the moderate values were intermediates between these extremes. In some cases, the choice of optimistic and pessimistic values was counterintuitive. For example, in the case of αSA, the relative contribution of asymptomatic individuals to transmission, one might expect that high values would result in a faster rise in resistance. In fact, the fastest rise in resistance occurs when asymptomatic individuals contribute least to transmission. Topical PCV is applied only to symptomatic recurrences of RHL, so when symptomatic persons are the main sources of transmission, treatment exerts the maximum selective pressure against sensitive virus. Similarly, the greatest selective pressure (and therefore the fastest rise in resistance) occurs when treatment is highly effective in reducing transmission of sensitive virus.
The predictions of the model are consistent with the observation that the prevalence of resistance in HSV-1 has remained relatively flat, despite almost 20 years of nucleoside analogue use. The modeling framework used here demonstrates, perhaps counter to intuition, that although current usage of nucleoside analogues looks large in absolute terms (see “Parameter estimates” above), the selection exerted by present usage in favor of resistance (in immunocompetent hosts) is rather small. Another prediction of the model that was surprising, at least to its authors, is the sensitive dependence of its predictions on the probability of emergence of resistance within treated, immunocompetent hosts. The continued validity of the model's predictions will be tested over time as levels of nucleoside analogue use for and resistance in HSV-1 are monitored.
Our predictions may be compared to the predictions of other, recently published models of virus transmission and antiviral treatment. Blower et al. (7) study oral ACV treatment of genital herpes caused by HSV-2. In contrast to our predictions for labial herpes, they find that widespread use of ACV for treatment of genital herpes might substantially reduce the prevalence and incidence of the infection. Several biological differences between HSV-1 and HSV-2 account for this divergent prediction, including route of transmission, seroprevalence, and frequency of recurrences. In addition, Blower et al. consider higher levels of antiviral treatment (up to 50%) than those considered here; another model of ACV treatment of HSV-2 found that treatment would have to be widespread and continued for a long period in order to reduce HSV-2 transmission substantially (62). On the question of drug resistance, our conclusions are in general accord with those of Blower et al. in predicting that drug resistance will remain rare (less than 5% of infections over 50 years), even under pessimistic assumptions.
In contrast to these models of HSV infection, models of amantadine or rimantadine treatment in an influenza epidemic predict extremely rapid increases in resistance, reaching 10% within weeks of the onset of treatment (56).
There are at least four key reasons why the emergence of resistance is expected to be slower for HSV-1 or HSV-2 than for influenza virus. (i) Acquired resistance is less common. Despite the uncertainty about just how rare acquired resistance is, it is clear that it is less common in treated HSV-1 patients than the 20% assumed for amantadine and rimantadine treatment of influenza (56). As we have seen, the probability of acquired resistance is crucial to the rate of ascent of resistance in the population. (ii) HSV infection, and presumably infectiousness, is life-long (64). The time scale on which resistance increases in a population is proportional to the duration of the infectiousness (10), so long-lived infections like HSV-1 have much slower dynamics than acute infections. (iii) Many resistant HSV-1 mutants may be less transmissible (less infectious and/or less likely to reactivate from latency) than sensitive wild-type virus, as shown by reduced virulence in animal models (16, 27). (iv) Treatment of sensitive infections causes a relatively modest reduction in shedding and (presumably) transmission. Topical PCV reduces the duration of shedding by 25% in patients with RHL (52), which means that the selective pressure in favor of resistance is relatively weak. This finding is compounded by the fact that treatment does not preclude future episodes of shedding, so that the impact of a single treatment episode on the total transmission from an infected individual is very small.
To address the uncertainty surrounding the predictions of the rate of the ascent of resistance in the population, it is crucial that ongoing surveillance for resistance be targeted to long-term monitoring of virus isolates from individuals who repeatedly treat recurrences with antiviral drugs. Such studies would be most likely to yield maximal information about acquired resistance per unit effort. Furthermore, methodologies should be developed and standardized to measure subpopulations of resistant viruses within a heterogeneous virus sample; such methods would be valuable for testing whether there are gradual increases in the proportion of resistant viruses during successive rounds of treatment. One such method, the plating efficiency assay (39), is currently being evaluated alongside the PRA (J. Leary and R. Sarisky, unpublished data).
ACKNOWLEDGMENTS
S. L. Spruance, D. M. Coen, M. Levin, M. Reyes, J. Copeland, P. Johnston, G. Westerbeck, and R. Boon are thanked for valuable comments on previous versions of this work and for helpful discussions.
This work was supported by an educational grant from SmithKline Beecham to Emory University.
REFERENCES
- 1.Al-Hasani A M, Barton I G, Al-Omer L S, Kinghorn G R, Potter C W. Susceptibility of HSV strains from patients with genital herpes treated with various formulations of acyclovir. J Antimicrob Chemother. 1986;18(Suppl. B):113–119. doi: 10.1093/jac/18.supplement_b.113. [DOI] [PubMed] [Google Scholar]
- 2.Amir J, Harel L, Smetana Z, Varsano I. Treatment of herpes simplex gingivostomatitis with aciclovir in children: a randomised double blind placebo controlled study. Br Med. 1997;314:1800–1803. doi: 10.1136/bmj.314.7097.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson R M, May R M. Infectious diseases of humans: dynamics and control. Oxford, United Kingdom: Oxford University Press; 1991. [Google Scholar]
- 4.Baquero F. Evolving resistance patterns of Streptococcus pneumoniae: a link with long-acting macrolide consumption? J Chemother. 1999;11(Suppl. 1):35–43. doi: 10.1179/joc.1999.11.Supplement-2.35. [DOI] [PubMed] [Google Scholar]
- 5.Baquero F. Trends in antibiotic resistance of respiratory pathogens: an analysis and commentary on a collaborative surveillance study. J Antimicrob Chemother. 1996;38(Suppl. A):117–132. doi: 10.1093/jac/38.suppl_a.117. [DOI] [PubMed] [Google Scholar]
- 6.Bartlett J G. Pocket book of infectious disease therapy. Baltimore, Md: Williams & Wilkins; 1997. [Google Scholar]
- 7.Blower S M, Porco T C, Darby G. Predicting and preventing the emergence of antiviral drug resistance in HSV-2. Nat Med. 1998;4:673–678. doi: 10.1038/nm0698-673. [DOI] [PubMed] [Google Scholar]
- 8.Boden D, Hurley A, Zhang L, Cao Y, Guo Y, Jones E, Tsay J, Ip J, Farthing C, Limoli K, Parkin N, Markowitz M. HIV-1 drug resistance in newly infected individuals. JAMA. 1999;282:1135–1141. doi: 10.1001/jama.282.12.1135. [DOI] [PubMed] [Google Scholar]
- 9.Bodurtha J, Adler S P, Nance W E. Seroepidemiology of cytomegalovirus and herpes simplex virus in twins and their families. Am J Epidemiol. 1988;128:268–276. doi: 10.1093/oxfordjournals.aje.a114967. [DOI] [PubMed] [Google Scholar]
- 10.Bonhoeffer S, Lipsitch M, Levin B R. Evaluating treatment protocols to prevent antibiotic resistance. Proc Natl Acad Sci USA. 1997;94:12106–12111. doi: 10.1073/pnas.94.22.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyd M R, Safrin S, Kern E R. Penciclovir: a review of its spectrum of activity, selectivity, and cross-resistance pattern. Antivir Chem Chemother. 1993;4(Suppl. 1):3–11. [Google Scholar]
- 12.Buchman T G, Roizman B, Nahmias A J. Demonstration of exogenous genital reinfection with herpes simplex virus type 2 by restriction endonuclease fingerprinting of viral DNA. J Infect Dis. 1979;140:295–304. doi: 10.1093/infdis/140.3.295. [DOI] [PubMed] [Google Scholar]
- 13.Carson J L, Strom B L, Maislin G. Screening for unknown effects of newly marketed drugs. In: Strom B L, editor. Pharmacoepidemiology. Chichester, United Kingdom: John Wiley & Sons; 1994. pp. 431–448. [Google Scholar]
- 14.Centers for Disease Control and Prevention. HIV/AIDS surveillance report 9 (no. 1). Atlanta, Ga: Centers for Disease Control and Prevention; 1997. [Google Scholar]
- 15.Christophers J, Clayton J, Craske J, Ward R, Collins P, Trowbridge M, Darby G. Survey of resistance of herpes simplex virus to acyclovir in northwest England. Antimicrob Agents Chemother. 1998;42:868–872. doi: 10.1128/aac.42.4.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coen D M. Antiviral drug resistance in herpes simplex virus. Adv Exp Med Biol. 1996;394:49–57. doi: 10.1007/978-1-4757-9209-6_7. [DOI] [PubMed] [Google Scholar]
- 17.Coen D M. The implications of resistance to antiviral agents for herpesvirus drug targets and drug therapy. Antivir Res. 1991;15:287–300. doi: 10.1016/0166-3542(91)90010-o. [DOI] [PubMed] [Google Scholar]
- 18.Collins P, Nixon Ellis M. Sensitivity monitoring of clinical isolates of herpes simplex virus to acyclovir. J Med Virol. 1993;1993(Suppl. 1):58–66. doi: 10.1002/jmv.1890410512. [DOI] [PubMed] [Google Scholar]
- 19.Collins P, Oliver N M. Sensitivity monitoring of herpes simplex virus isolates from patients receiving acyclovir. J Antimicrob Chemother. 1986;18(Suppl. B):103–112. doi: 10.1093/jac/18.supplement_b.103. [DOI] [PubMed] [Google Scholar]
- 20.Dekker C, Ellis M N, McLaren C, Hunter G, Rogers J, Barry D W. Virus resistance in clinical practice. J Antimicrob Chemother. 1983;12(Suppl. B):137–152. doi: 10.1093/jac/12.suppl_b.137. [DOI] [PubMed] [Google Scholar]
- 21.Edelstein-Keshet L. Mathematical models in biology. New York, N.Y: Random House; 1988. [Google Scholar]
- 22.Ellis M N, Keller P M, Fyfe J A, Martin J L, Rooney J F, Straus S E, Lehrman S N, Barry D W. Clinical isolate of herpes simplex virus type 2 that induces a thymidine kinase with altered substrate specificity. Antimicrob Agents Chemother. 1987;31:1117–1125. doi: 10.1128/aac.31.7.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Embil J A, Stephens R G, Manuel F R. Prevalence of recurrent herpes labialis and aphthous ulcers among young adults on six continents. Can Med Assoc J. 1975;113:627–630. [PMC free article] [PubMed] [Google Scholar]
- 24.Englund J A, Zimmerman M E, Swierkosz E M, Goodman J L, Scholl D R, Balfour H H., Jr Herpes simplex virus resistant to acyclovir. A study in a tertiary care center. Ann Intern Med. 1990;112:416–422. doi: 10.7326/0003-4819-76-3-112-6-416. [DOI] [PubMed] [Google Scholar]
- 25.Erlich K S, Mills J, Chatis P, Mertz G J, Busch D F, Follansbee S E, Grant R M, Crumpacker C S. Acyclovir-resistant herpes simplex virus infections in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1989;320:293–296. doi: 10.1056/NEJM198902023200506. [DOI] [PubMed] [Google Scholar]
- 26.Field H J. Development of clinical resistance to acyclovir in herpes simplex virus-infected mice receiving oral therapy. Antimicrob Agents Chemother. 1982;21:744–752. doi: 10.1128/aac.21.5.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Field H J, Goldthorpe S E. The pathogenicity of drug-resistant variants of herpes simplex virus. Res Virol. 1992;143:120–124. doi: 10.1016/s0923-2516(06)80092-0. [DOI] [PubMed] [Google Scholar]
- 28.Field H J, Larder B A, Darby G. Isolation and characterization of acyclovir-resistant strains of herpes simplex virus. Am J Med. 1982;73(1A):369–371. doi: 10.1016/0002-9343(82)90124-3. [DOI] [PubMed] [Google Scholar]
- 29.Fife K H, Crumpacker C S, Mertz G J, Hill E L, Boone G S. Recurrence and resistance patterns of herpes simplex virus following cessation of > or = 6 years of chronic suppression with acyclovir. Acyclovir Study Group. J Infect Dis. 1994;169:1338–1341. doi: 10.1093/infdis/169.6.1338. [DOI] [PubMed] [Google Scholar]
- 30.Graves E J, Kozak L J. Detailed diagnoses and procedures. National Hospital Discharge Survey 1996. Vital health statistics no. 138. Atlanta, Ga: National Center for Health Statistics; 1998. [PubMed] [Google Scholar]
- 31.Grout P, Barber V E. Cold sores—an epidemiologic survey. J R Coll Gen Pract. 1976;26:428–434. [PMC free article] [PubMed] [Google Scholar]
- 32.Hanley J A, Lippman-Hand A. If nothing goes wrong, is everything all right? Interpreting zero numerators. JAMA. 1983;249:1743–1745. [PubMed] [Google Scholar]
- 33.Kost R G, Hill E L, Tigges M, Straus S E. Brief report: recurrent acyclovir-resistant genital herpes in an immunocompetent patient. N Engl J Med. 1993;329:1777–1782. doi: 10.1056/NEJM199312093292405. [DOI] [PubMed] [Google Scholar]
- 34.McLaren C, Corey L, Dekket C, Barry D W. In vitro sensitivity to acyclovir in genital herpes simplex viruses from acyclovir-treated patients. J Infect Dis. 1983;148:868–875. doi: 10.1093/infdis/148.5.868. [DOI] [PubMed] [Google Scholar]
- 35.Mertz G J, Loveless M O, Levin M J, Kraus S J, Fowler S L, Goade D, Tyring S K. Oral famciclovir for suppression of recurrent genital herpes simplex virus infection in women. A multicenter, double-blind, placebo-controlled trial. Collaborative Famciclovir Genital Herpes Research Group. Arch Intern Med. 1997;157:343–349. [PubMed] [Google Scholar]
- 36.Momin F, Chandrasekar P H. Antimicrobial prophylaxis in bone marrow transplantation. Ann Intern Med. 1995;123:205–215. doi: 10.7326/0003-4819-123-3-199508010-00008. [DOI] [PubMed] [Google Scholar]
- 37.Murray J D. Mathematical biology, 2nd, corrected ed. Berlin, Germany: Springer Verlag; 1993. [Google Scholar]
- 38.Nugier F, Colin J N, Aymard M, Langlois M. Occurrence and characterization of acyclovir-resistant herpes simplex virus isolates: report on a two-year sensitivity screening survey. J Med Virol. 1992;36:1–12. doi: 10.1002/jmv.1890360102. [DOI] [PubMed] [Google Scholar]
- 39.Parris D S, Harrington J E. Herpes simplex virus variants resistant to high concentrations of acyclovir exist in clinical isolates. Antimicrob Agents Chemother. 1982;22:71–77. doi: 10.1128/aac.22.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelosi E, Mulamba G B, Coen D M. Penciclovir and pathogenesis phenotypes of drug-resistant herpes simplex virus mutants. Antivir Res. 1998;37:17–28. doi: 10.1016/s0166-3542(97)00054-5. [DOI] [PubMed] [Google Scholar]
- 41.Pottage J C, Kessler H A. Herpes simplex virus resistance to acyclovir: clinical relevance. Infect Agents Dis. 1995;4:115–124. [PubMed] [Google Scholar]
- 42.Reichman R C, Badger G J, Mertz G J, Corey L, Richman D D, Connor J D, Redfield D, Savoia M C, Oxman M N, Bryson Y. Treatment of recurrent genital herpes simplex infections with oral acyclovir. A controlled trial. JAMA. 1984;251:2103–2107. [PubMed] [Google Scholar]
- 43.Reyes M, Graber J M, Weatherall N, Hodges-Savola C, Reeves W C. Acyclovir-resistant herpes simplex virus: preliminary results from a national surveillance system. Antivir Res. 1998;37:A44. [Google Scholar]
- 44.Rooney J F, Straus S E, Mannix M L, Wohlenberg C R, Alling D W, Dumois J A, Notkins A L. Oral acyclovir to suppress frequently recurrent herpes labialis. A double-blind, placebo-controlled trial. Ann Intern Med. 1993;118:268–272. doi: 10.7326/0003-4819-118-4-199302150-00004. [DOI] [PubMed] [Google Scholar]
- 45.Sacks S L, Aoki F Y, Diaz-Mitoma F, Sellors J, Shafran S D. Patient-initiated, twice-daily oral famciclovir for early recurrent genital herpes. A randomized, double-blind multicenter trial. Canadian Famciclovir Study Group. JAMA. 1996;276:44–49. [PubMed] [Google Scholar]
- 46.Schmidt O W, Fife K H, Corey L. Reinfection is an uncommon occurrence in patients with symptomatic recurrent genital herpes. J Infect Dis. 1984;149:645–646. doi: 10.1093/infdis/149.4.645. [DOI] [PubMed] [Google Scholar]
- 47.Ship I I, Morris A L, Durocher R T, Burket L W. Recurrent aphthous ulcerations and recurrent herpes labialis in a professional school student population. 1. Experience. Oral Med Oral Surg Oral Pathol. 1960;13:1191–1202. [Google Scholar]
- 48.Siegel D, Golden E, Washington E, Morse S A, Fullilove M T, Catania J A, Marin B, Hulley S B. Prevalence and correlates of herpes simplex infections: the population-based AIDS in Multiethnic Neighborhoods Study. JAMA. 1992;268:1702–1708. [PubMed] [Google Scholar]
- 49.Smith K O, Kennell W L, Poirier R H, Lynd F T. In vitro and in vivo resistance of herpes simplex virus to 9-(2-hydroxyethoxymethyl)guanine (acycloguanosine) Antimicrob Agents Chemother. 1980;17:144–150. doi: 10.1128/aac.17.2.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spruance S L. Herpes simplex labialis. In: Sacks S L, Straus S E, Whitley R J, Griffiths P D, editors. Clinical management of herpes viruses. Amsterdam, The Netherlands: IOS Press; 1995. pp. 3–42. [Google Scholar]
- 51.Spruance S L. Prophylactic chemotherapy with acyclovir for recurrent herpes simplex labialis. J Med Virol. 1993;1993(Suppl. 1):27–32. doi: 10.1002/jmv.1890410507. [DOI] [PubMed] [Google Scholar]
- 52.Spruance S L, Rea T L, Thoming C, Tucker R, Saltzman R, Boon R. Penciclovir cream for the treatment of herpes simplex labialis: a randomized, multicenter, double-blind, placebo-controlled trial. JAMA. 1997;277:1374–1379. [PubMed] [Google Scholar]
- 53.Spruance S L, Rowe N H, Raborn G W, Thibodeau E A, D'Ambrosio J A, Bernstein D A. Peroral famciclovir in the treatment of experimental ultraviolet radiation-induced herpes simplex labialis: a double-blind, dose-ranging, placebo-controlled, multicenter trial. J Infect Dis. 1999;179:303–310. doi: 10.1086/314605. [DOI] [PubMed] [Google Scholar]
- 54.Spruance S L, Schnipper L E, Overall J C, Kern E R, Wester B, Modlin J, Wenerstrom G, Burton C, Arndt K A, Chiu G L, Crumpacker C L. Treatment of herpes simplex labialis with topical acyclovir in polyethylene glycol. J Infect Dis. 1982;146:85–90. doi: 10.1093/infdis/146.1.85. [DOI] [PubMed] [Google Scholar]
- 55.Stanberry L R, Jorgensen D M, Nahmias A J. Herpes simplex viruses 1 and 2. In: Evans A S, Kaslow R A, editors. Viral infections of humans: epidemiology and control. 4th ed. New York, N.Y: Plenum; 1997. pp. 419–454. [Google Scholar]
- 56.Stilianakis N I, Perelson A S, Hayden F G. Emergence of drug resistance during an influenza epidemic: insights from a mathematical model. J Infect Dis. 1998;177:863–873. doi: 10.1086/515246. [DOI] [PubMed] [Google Scholar]
- 57.Straus S E, Seidlin M, Takiff H E, Rooney J F, Felser J M, Smith H A, Roane P, Johnson F, Hallahan C, Ostrove J M, Nusinoff-Lehrman S. Effect of oral acyclovir treatment on symptomatic and asymptomatic virus shedding in recurrent genital herpes. Sex Transm Dis. 1989;16:107–113. doi: 10.1097/00007435-198904000-00013. [DOI] [PubMed] [Google Scholar]
- 58.Straus S E, Takiff H E, Seidlin M, Bachrach S, Lininger L, DiGiovanna J J, Western K A, Smith H A, Lehrman S N, Creagh-Kirk T. Suppression of frequently recurring genital herpes. A placebo-controlled double-blind trial of oral acyclovir. N Engl J Med. 1984;310:1545–1550. doi: 10.1056/NEJM198406143102401. [DOI] [PubMed] [Google Scholar]
- 59.Swetter S M, Hill E L, Kern E R, Koelle D M, Posavad C M, Lawrence W, Safrin S. Chronic vulvular ulceration in an immunocompetent woman due to acyclovir-resistant, thymidine kinase-deficient herpes simplex virus. J Infect Dis. 1998;177:543–550. doi: 10.1086/514229. [DOI] [PubMed] [Google Scholar]
- 60.Tateishi K, Toh Y, Minagawa H, Tashiro H. Detection of herpes simplex virus (HSV) in the saliva from 1,000 oral surgery outpatients by the polymerase chain reaction (PCR) and virus isolation. J Oral Pathol Med. 1994;23:80–84. doi: 10.1111/j.1600-0714.1994.tb00261.x. [DOI] [PubMed] [Google Scholar]
- 61.Ventura S J, Martin J A, Curtin S C, Mathews T J. Report of final natality statistics, 1995. Mon Vital Stat Rep. 1997;45(Suppl. 11):1–84. [Google Scholar]
- 62.White P J, Garnett G P. Use of antiviral treatment and prophylaxis is unlikely to have a major impact on the prevalence of herpes simplex virus type 2. Sex Transm Infect. 1999;75:49–54. doi: 10.1136/sti.75.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whitley R J, Gnann J W. The epidemiology and clinical manifestations of herpes simplex virus infections. In: Roizman B, Whitley R J, Lopez C, editors. The human herpesviruses. New York, N.Y: Raven Press, Ltd.; 1993. pp. 69–105. [Google Scholar]
- 64.Wildy P, Field H J, Nash A A. Classical herpes latency revisited. In: Mahy M W J, Minson A C, Darby G K, editors. Virus persistence: 33rd Symposium of the Society for General Microbiology. Cambridge, United Kingdom: Cambridge University Press; 1982. [Google Scholar]
- 65.Young T B, Rimm E B, D'Alessio D J. Cross-sectional study of recurrent herpes labialis. Prevalence and risk factors. Am J Epidemiol. 1988;127:612–625. doi: 10.1093/oxfordjournals.aje.a114837. [DOI] [PubMed] [Google Scholar]