

Abstract
Most cancer deaths result from progression of therapy resistant disease, yet our understanding of this phenotype is limited. Cancer therapies generate stress signals that act upon mitochondria to initiate apoptosis. Mitochondria isolated from neuroblastoma cells were exposed to tBid or Bim, death effectors activated by therapeutic stress. Multidrug‐resistant tumor cells obtained from children at relapse had markedly attenuated Bak and Bax oligomerization and cytochrome c release (surrogates for apoptotic commitment) in comparison with patient‐matched tumor cells obtained at diagnosis. Electron microscopy identified reduced ER–mitochondria‐associated membranes (MAMs; ER–mitochondria contacts, ERMCs) in therapy‐resistant cells, and genetically or biochemically reducing MAMs in therapy‐sensitive tumors phenocopied resistance. MAMs serve as platforms to transfer Ca2+ and bioactive lipids to mitochondria. Reduced Ca2+ transfer was found in some but not all resistant cells, and inhibiting transfer did not attenuate apoptotic signaling. In contrast, reduced ceramide synthesis and transfer was common to resistant cells and its inhibition induced stress resistance. We identify ER–mitochondria‐associated membranes as physiologic regulators of apoptosis via ceramide transfer and uncover a previously unrecognized mechanism for cancer multidrug resistance.
Keywords: ceramides, inter‐organelle contacts, mitochondria‐associated membranes, multidrug resistance, sphingolipids
Subject Categories: Cancer, Membranes & Trafficking, Organelles
Impaired ceramide biosynthesis at ER–mitochondria‐associated membranes and lipid transfer into mitochondria decreases apoptotic signaling in relapsed tumor cells, contributing to cancer therapy resistance.
Introduction
Over 600,000 people die of cancer each year in the USA, most with progression of disease that is resistant to available treatments. Our understanding of the mechanisms underlying broad resistance to diverse drug classes and therapeutic modalities includes altered drug transport into or out of the cancer cell, such as from increased activity of ATP‐binding cassette transporters (Holohan et al, 2013), and mutations in genotoxic response genes like TP53 (Olivier et al, 2010). The former have not been shown to contribute to resistance in vivo (Cripe et al, 2010) and does not explain resistance to drugs that are not substrates for such transporters, and the latter does not explain therapy resistance in tumors with retained p53 activity. More recently, the focus has shifted to studying resistance to inhibitors of oncogenic kinases, with secondary mutations in the drug target, activation of bypass signals, and cellular plasticity identified as causal, yet even here a large proportion of acquired resistance remains unexplained (Katayama et al, 2012; Chong & Janne, 2013; Lito et al, 2013; Lord & Ashworth, 2013; Hugo et al, 2015; Wilson et al, 2015). Our incomplete understanding of the contributors to therapy resistance remains a principal barrier to improving cancer outcomes.
We used the highly lethal childhood tumor, neuroblastoma, as a model system to investigate therapy resistance. Children with high‐risk neuroblastoma are treated with intensive chemoradiotherapy, stem cell rescue, surgery, and immunotherapy (Pinto et al, 2015). Many patients have metastatic disease at diagnosis, yet the tumors are chemosensitive and respond to treatment, including complete responses. Still, half of all patients subsequently relapse with lethal therapy‐resistant disease (Keshelava et al, 1997, 1998; Matthay et al, 2009). To study emergent therapy resistance, we established tumor cell lines from the same patients at diagnosis prior to treatment, and again at the time of relapse during or after treatment, providing near‐isogenic tumor models in which post‐relapse tumors demonstrate therapy resistance acquired in situ in response to multimodal therapy (Keshelava et al, 1998). Since mitochondria serve as platforms for integrating cellular stress and survival signals in real time, largely governed by Bcl2 family interactions, we hypothesized they harbor information related to therapeutic stress sensitivity. We used an unbiased assay in which cancer mitochondria are exposed to tBid protein or BimBH3 peptides, death effectors activated downstream of most therapeutic stress (Tan et al, 2005; Kim et al, 2006; Ren et al, 2010; Sarosiek et al, 2013a), to define their sensitivity for activating mitochondrial outer membrane permeabilization (MOMP) as a surrogate for apoptotic commitment. We discovered that mitochondria from therapy‐resistant tumors have markedly attenuated MOMP responses in comparison with patient‐matched therapy‐sensitive tumors, demonstrating that mitochondrial apoptotic signaling dysfunction arises during the course of clinical therapy and contributes to emergent multidrug resistance.
Many mitochondrial functions, including sensitivity to apoptosis, are regulated by signals derived from endoplasmic reticulum (ER) at contact sites with mitochondria [reviewed in (Rowland & Voeltz, 2012; Csordas et al, 2018)]. ER–mitochondria contact sites (ERMCs) consist of a multitude of ER–mitochondria protein bridges that tether the organelles and facilitate inter‐organelle communication. Cell fractionation can isolate these specialized ER domains as mitochondria‐associated ER membranes (MAMs). For simplicity, we use MAM to refer to both ERMCs and MAMs herein. How cells regulate these contacts is incompletely understood, however, the importance of MAMs in tuning the cross‐talk between these dynamic organelle networks is proposed to contribute to many pathophysiologic states, such as diabetes, neurodegeneration, and cancer (Area‐Gomez et al, 2012; Cali et al, 2013; Hedskog et al, 2013; Arruda et al, 2014). Here, we show that post‐relapse tumor cell mitochondria have reduced MAMs (altered numbers, lengths, and gap distances) that directly contribute to their stress resistance, and lead to the multidrug resistance phenotype seen clinically. Depleted MAMs contribute to resistance to chemotherapy, radiotherapy, and molecularly targeted drugs. It acts at a terminal signaling node to imbue cancer cells with resistance to diverse therapy‐induced stressors yet is not exclusive to other resistance mechanisms operative upstream. Ca2+ transfer that occurs at MAMs is reduced in a subset of multidrug‐resistant neuroblastomas but not all, and genetically or biochemically inhibiting this does not attenuate apoptotic signaling. However, synthesis and transfer of the bioactive sphingolipid, ceramide that is enriched at MAMs, is reduced in all multidrug‐resistant neuroblastomas studied and chemical inhibition of this transfer leads to attenuated MOMP. This new framework for understanding therapy resistance may provide opportunities to enhance cancer care, including the measurement of relative resistance by characterizing MAM abundance and proximity, and enabling interventions to restore ER–mitochondrial communication or sphingolipid homeostasis in resistant cancers.
Results
Mitochondria from drug‐resistant tumor cells have attenuated apoptotic signal transduction
Optimizing approaches developed by Letai (Deng et al, 2007), we isolated mitochondria from paired neuroblastoma cell lines derived from the same patients at diagnosis (DX) before therapy, and at relapse (REL) during or after completion of therapy (Fig 1A). Mitochondria‐enriched heavy‐membrane fractions from tumor cells were incubated with recombinant truncated Bid or the death‐activating BH3 domain peptide of Bim (tBid and BimBH3, respectively) across a range of concentrations. Bid and Bim proteins are direct activators of intrinsic apoptosis liberated by diverse cell stressors to either engage Bak or Bax to induce MOMP and cell death, or be sequestered and neutralized by pro‐survival Bcl2 family proteins (Gavathiotis et al, 2008; Sarosiek et al, 2013a). The sensitivity for release of cytochrome c in response to tBid or BimBH3 reflects a cell’s proximity to its apoptotic threshold (Tan et al, 2005; Goldsmith et al, 2012). By delivering terminal death effectors directly to mitochondria, the assay bypasses the contributions of drug transport, metabolism, target engagement, and transcriptional response. Instead, mitochondrial responses reflect the state of the Bcl2 family and related apoptosis‐regulating processes present in the tumor cell at the time of testing [reviewed in (Sarosiek et al, 2013b)]. A Bid BH3 domain with substitution of two highly conserved residues served as a negative control and induced release of < 10% of available cytochrome c in all experiments.
Figure 1. Mitochondria from REL neuroblastomas have attenuated apoptotic responses.
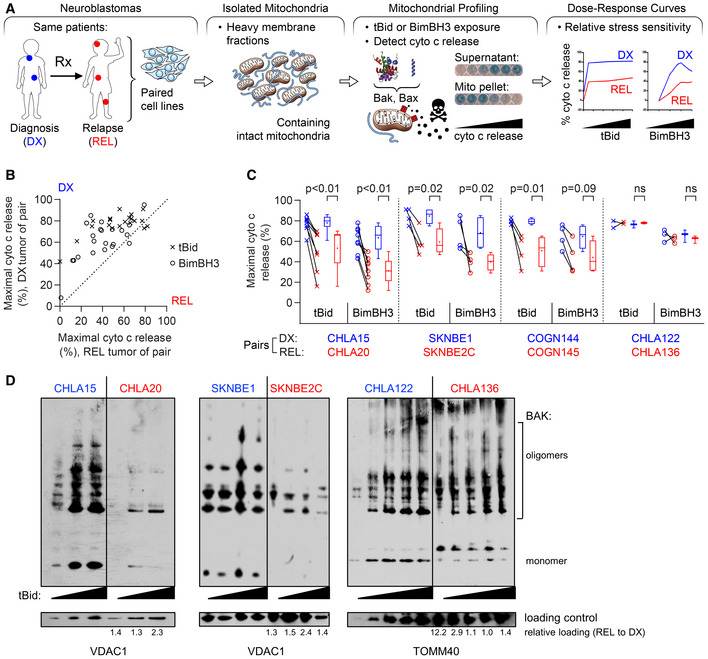
- Tumor models were derived at DX and REL following treatment. Mitochondria‐rich fractions were exposed to tBid or BimBH3 peptide and cytochrome c release measured as a surrogate for apoptotic commitment.
- Maximal cytochrome c release in response to tBid or BimBH3 peptide for each replicate of a DX/REL tumor pair (1–9 biological replicates per DX/REL pair; n = 44 total).
- Maximal cytochrome c release for all DX/REL pairs with ≥ 3 biological replicates; box‐whisker plots summarize data (box 25–75%; belt = median; dot = mean; and whiskers = minimum and maximum values).
- Bak oligomerization in response to escalating tBid concentration for DX/REL pairs. Relative mitochondrial protein loading per lane is assessed by densitometry, showing ration of loading control in REL lane with patient‐matched DX lane at same tBid exposure (no REL lane is underloaded compared to DX lane).
Data information: Statistical analyses in C were performed using an unpaired two‐sided Student’s t‐test, with significance P < 0.05 (trend P < 0.10). Cyto c, cytochrome c; mito, mitochondria.
Seven DX/REL matched cell line pairs were studied, each pair tested concurrently under identical culture conditions. Six of seven pairs showed attenuated cytochrome c release from tumor cells derived at relapse (Figs 1B and C, and EV1A). For five of the seven pairs, REL mitochondria had reduced cytochrome c release in response to both tBid and BimBH3 in every biological replicate, with both reduced sensitivity for release and reduced maximal release. One pair demonstrated reduced cytochrome c release in response to BimBH3 peptide but not tBid; and one pair showed similar release (CHLA122/CHLA136; maximal release differed < 10% in all but one replicate). Overall, in 41 of 44 (93%) assays with tBid or BimBH3, the mitochondria from post‐relapse tumor cells released less cytochrome c than their patient‐matched counterparts from the time of diagnosis. Recombinant tBid was more potent at inducing cytochrome c release than BimBH3 peptide (all experiments), and maximal tBid‐induced release exceeded that of BimBH3 in 39 of 42 (93%) experiments, despite BimBH3 peptide being used to > 1 log higher concentrations.
Figure EV1. Mitochondrial cytochrome c release curves for neuroblastoma cell lines.
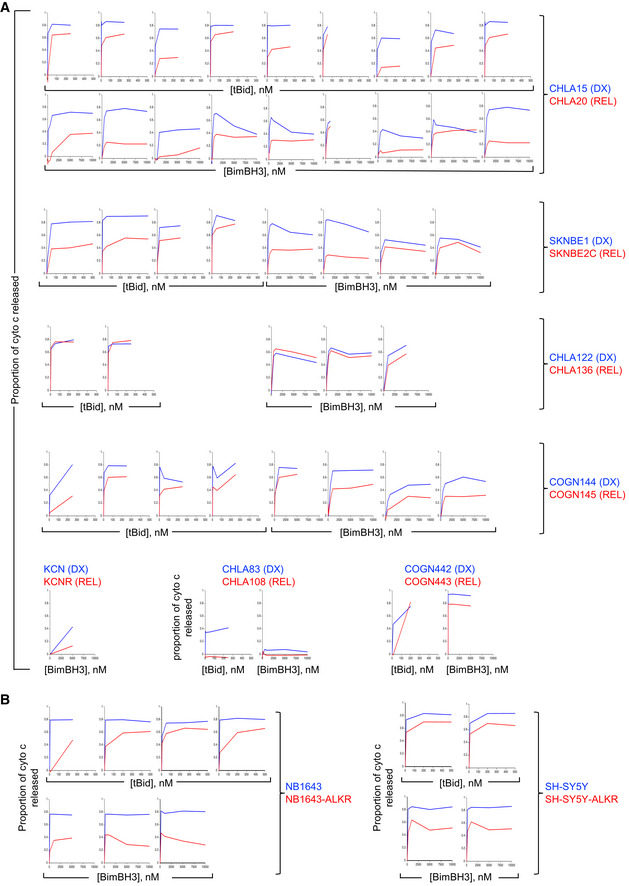
- All mitochondrial profiling data using heavy‐membrane mitochondria‐enriched fractions from DX cells (blue curves) and patient‐matched REL cells (red curves) showing robust qualitative reproducibility.
- All mitochondrial profiling using heavy‐membrane mitochondria‐enriched fractions obtained from ALK mutant neuroblastoma cells selected for resistance to the Alk inhibitor, crizotinib. Cells with crizotinib resistance are designated by “‐ALKR” and are shown relative to parental cells.
Data information: Each plot represents a biological replicate experiment in which cytochrome c release represents the mean of two technical replicates (SD < 0.05 at all points) and all biological replicates are shown.
Relative cytochrome c release from tumor cell mitochondria was reproducible (Figs 1C and EV1). Mitochondria from CHLA15 (DX) and CHLA20 (REL) were profiled in 9 biological replicates with both tBid and BimBH3, and maximal cytochrome c release for CHLA15 was greater than CHLA20 in all 18 experiments. The SKNBE1 (DX)/SKNBE2 (REL) and COGN144 (DX)/COGN145 (REL) pairs were profiled in 4 biological replicates each, and DX tumor mitochondria released more cytochrome c to both tBid and BimBH3 in all 16 experiments. In contrast, CHLA122 (DX) and CHLA136 (REL) showed nearly equivalent cytochrome c release in four of five experiments (in one, DX cells released > 10% more cytochrome c than REL cells in response to BimBH3). In all, maximal cytochrome c release in response to tBid or BimBH3 peptide was significantly higher for DX tumor cells compared with patient‐matched REL tumor cells for the CHLA15/CHLA20, SKNBE1/SKNBE2C, and COGN144/COGN145 pairs, consistent with apoptosis resistance at relapse (Table 1). Overall, DX neuroblastomas released > 50% of available cytochrome c in 38 of 43 of experiments (88%) using either tBid or BimBH3 as a stimulus, whereas REL neuroblastomas released > 50% in just 19 of 44 of experiments (43%), further reflecting their attenuated response to stress (P < 0.01). Only the CHLA122/CHLA136 pair demonstrated no difference in cytochrome c release in response to tBid or BimBH3.
Table 1.
Summary of paired diagnostic (DX) and relapse (REL) neuroblastoma models.
| CHLA15 | CHLA20 | SKNBE1 | SKNBE2C | CHLA122 | CHLA136 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mitochondrial response | |||||||||
| Max % cyto c release, tBid (mean ± SD) | 78 ± 8 | 53 ± 19 | P < 0.01 | 85 ± 7 | 59 ± 11 | P < 0.02 | 77 ± 5 | 78 ± 1 | P = ns |
| Max % cyto c release, BimBH3 (mean ± SD) | 63 ± 13 | 31 ± 12 | P < 0.01 | 68 ± 14 | 40 ± 7 | P < 0.02 | 66 ± 6 | 62 ± 4 | P = ns |
| Tumor cell response (fold‐resistance, IC50) | |||||||||
| Mafosfamide | 3.9 | 12.9 | 1.2 | ||||||
| Doxorubicin | 33.0 | 28.6 | 1.7 | ||||||
| Cisplatin | 1.6 | 4.2 | 2.6 | ||||||
| Etoposide | 3.5 | 122.0 | 2.1 | ||||||
| Ionizing Radiation | 2.1 | 2.0 | n.d. | ||||||
| Crizotinib | 5.0 | n.d. | n.d. | ||||||
| ABT737 | 44.0 | n.d. | 1.8 | ||||||
| Mitochondria | |||||||||
| Number analyzed | 196 | 241 | 137 | 160 | 206 | 143 | |||
| Perimeter, mean (nm) | 2,207 | 1,725 | P < 0.01 | 1,653 | 1,810 | P = 0.06 | 1,626 | 1,454 | P = ns |
| Perimeter, median (nm) | 2,055 | 1,675 | 1,503 | 1,725 | 1,404 | 1,330 | |||
| Circularity, median | 0.902 | 0.920 | P < 0.01 | 0.910 | 0.898 | P = ns | 0.913 | 0.905 | P = ns |
| Roundness, median | 0.996 | 0.745 | P = ns | 0.714 | 0.692 | P = ns | 0.676 | 0.655 | P = ns |
| % with 0 MAMs | 18% | 32% | P < 0.01 | 18% | 40% | P < 0.01 | 17% | 8% | P < 0.01 (REL fewer) |
| % with 1 MAMs | 39% | 39% | 37% | 35% | 33% | 34% | |||
| % with 2–3 MAMs | 41% | 25% | P < 0.01 | 38% | 22% | P < 0.01 | 42% | 43% | P = ns |
| % with ≥ 4 MAMs | 3% | 3% | 6% | 4% | 7% | 15% | |||
| %OMM with MAM gap width ≤ 10 nm | 0.7% | 0.4% | (↓43%) | 2.3% | 1.1% | (↓52%) | 2.1% | 2.7% | (↑29%) |
| %OMM with MAM gap width ≤ 25 nm | 2.5% | 2.1% | (↓16%) | 6.0% | 3.5% | (↓42%) | 6.1% | 6.8% | (↑11%) |
| %OMM with MAM gap width ≤ 50 nm | 6.8% | 6.3% | (↓7%) | 12.4% | 7.6% | (↓39%) | 12.8% | 14.2% | (↑13%) |
| %OMM with MAM gap width ≤ 100 nm | 16.9% | 15.5% | (↓8%) | 24.4% | 15.2% | (↓38%) | 24.0% | 25.5% | (↑6%) |
| MAM | |||||||||
| Number analyzed | 267 | 266 | 203 | 154 | 334 | 386 | |||
| MAMs per mitochondria | 1.36 | 1.10 | P < 0.01 | 1.48 | 0.96 | P < 0.01 | 1.62 | 2.00 | P = 0.01 (REL more) |
| MAM frequency (per nm perimeter) | 1,651 | 1,556 | 1,116 | 1,880 | 710 | 1,134 | |||
| %MAM with gap width ≤ 10 nm | 18% | 12% | P = 0.05 | 30% | 21% | P < 0.01 | 36% | 39% | P = ns |
| %MAM with gap width ≤ 25 nm | 40% | 36% | P = ns | 57% | 46% | P < 0.01 | 65% | 69% | P = ns |
| %MAM with gap width ≤ 50 nm | 70% | 63% | 80% | 73% | 86% | 93% | |||
| %MAM with gap width ≤ 100 nm | 100% | 100% | 100% | 100% | 100% | 100% | |||
To confirm that MOMP was being induced by Bak and/or Bax oligomerization in our cytochrome c release assays, we compared their sensitivity to form homo‐oligomers in response to increasing concentrations of tBid. Bak sensitivity to tBid, its preferential target (Sarosiek et al, 2013a), was reduced in CHLA20 and BE2C mitochondria compared with matched DX cells. Bax oligomerization was also reduced in CHLA20, while CHLA136 did not have differential oligomerization of Bak or Bax in response to tBid (Figs 1D and EV2A).
Figure EV2. Bak and Bax analyses.
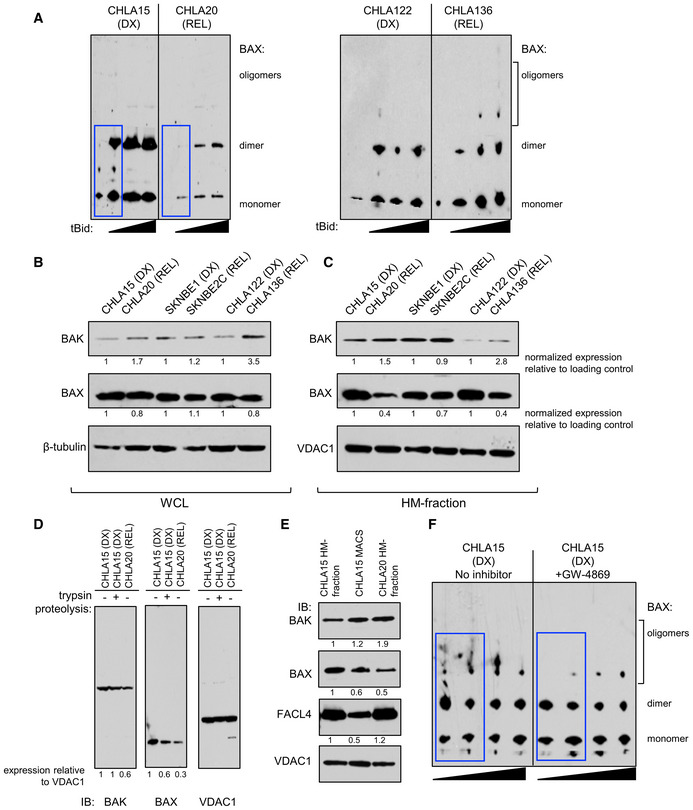
-
ABax oligomerization in response to escalating tBid concentration by immunoblot for DX/REL pairs. Blue boxes highlight key oligomerization differences.
-
B, CDX/REL pair whole‐cell lysates and heavy‐membrane (HM) fractions immunoblotted for Bak and Bax with relevant loading controls.
-
DImmunoblot of CHLA15 HM fraction, with and without tryptic digestion to deplete MAMs, compared with CHLA20 HM fraction, for Bak, Bax, and Vdac1 (loading control).
-
ESimilar to D, but with immunomagnetic separation (MACS) to deplete MAMs, immunoblotted for Bak, Bax, Facl4 (MAM marker), and Vdac1 (loading control).
-
FBax oligomerization in response to escalating tBid concentration by immunoblot for CHLA15 mitochondria pre‐incubated for 30 min with or without the neutral sphingomyelinase inhibitor, GW4869. Blue boxes highlight key oligomerization differences.
Data information: For B and C, relative fold expression (ratio of target gene band to control band normalized to the DX expression for each patient‐matched cell line pair) is shown. For D and E, relative fold expression for each condition is shown, normalized to the first lane for each.
Cytochrome c release from isolated mitochondria correlates with tumor cell sensitivity to diverse therapeutic stressors
Attenuated mitochondrial responses to tBid and Bim, the stress sentinels activated by chemotherapy (Tan et al, 2005; Kim et al, 2006; Ren et al, 2010; Sarosiek et al, 2013a), predicts for reduced cytotoxicity in response to such drugs. We compared sensitivity to chemotherapeutics of different drug classes used to treat neuroblastoma, including cisplatin, mafosfamide, etoposide, and doxorubicin. Etoposide and doxorubicin are substrates for the multidrug resistance protein, P‐glycoprotein‐1, whereas cisplatin and mafosfamide are not. Both the CHLA15/CHLA20 and SKNBE1/SKNBE2C pairs that had attenuated release of cytochrome c from REL mitochondria, showed relative chemoresistance in REL tumor cells to both P‐glycoprotein‐1 substrates and non‐substrates (Fig 2A; Appendix Fig S1A; Table 1). In contrast, the CHLA122/CHLA136 pair without attenuated mitochondrial release of cytochrome c did not show differential chemosensitivity. Comparing IC50 values for DX/REL pairs showed 1.6‐fold to 33‐fold resistance for CHLA15/CHLA20 (three of four drugs with > 3‐fold difference); 4.2‐fold to 122‐fold for SKNBE1/SKNBE2C (all four drugs with > 3‐fold difference); and 1.2‐fold to 2.6‐fold for CHLA122/CHLA136 (no drugs with > 3‐fold difference). We next assessed sensitivity to ionizing radiation as a treatment modality to which there is less cross‐resistance than chemotherapy. Radiation bypasses drug transport and metabolism contributions to deliver genotoxic stress that can be quantified as γ‐H2AX foci. SKNBE2C (REL) cells had a larger induction of γ‐H2AX foci post‐radiation (~10‐fold compared with ~5‐fold for SKNBE1; P = 0.04), yet they were 2‐fold more radiation resistant (Fig EV3). However, SKNBE2C cells harbor an acquired TP53 mutation (C135F) contributing to their resistance, and fail to induce p53, p21, or noxa. CHLA20 (REL) cells, however, are TP53 wild type and were also > 2‐fold radiation resistant in comparison with CHLA15 (DX) cells despite both inducing p53 response genes and deriving equivalent DNA damage, confirming attenuated apoptotic signaling downstream of radiation‐induced genotoxic damage (Fig 2B and C).
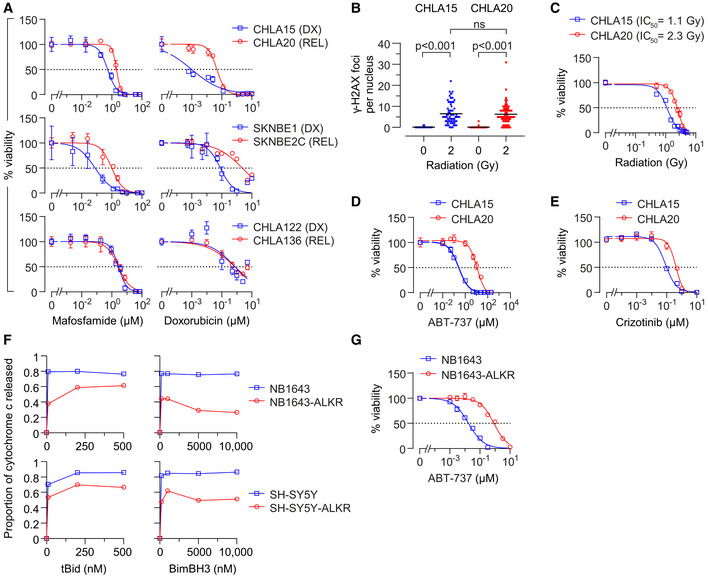
Figure 2. REL neuroblastomas are resistant to multimodal cancer therapeutics.
-
AIn vitro viability curves after 72 h exposure to mafosfamide or doxorubicin. Results are shown for three DX/REL neuroblastoma cell line pairs, two with attenuated cytochrome c release (CHLA15/CHLA20 and SKNBE1/SKNBE2C), and one without (CHLA122/CHLA136).
-
BDNA damage induced by 2 Gy ionizing radiation as measured by γ‐H2AX foci at 1 h (n = 53–77 cell nuclei/condition scored; mean shown); statistical analyses performed using two‐tailed Mann–Whitney U test.
-
C–EIn vitro viability for CHLA15 (DX) and CHLA20 (REL) following ionizing radiation at 7 days, 48 h exposure to the Bcl2/Bclx inhibitor, ABT‐737, or 120 h exposure to the Alk inhibitor, crizotinib.
-
FCytochrome c release from mitochondria after exposure to tBid or BimBH3 peptide for parental NB1643 and SY5Y cells, in comparison to cells cultured in escalating concentrations of crizotinib until resistant (NB1643‐ALKR and SY5Y‐ALKR).
-
GIn vitro viability of NB1643 and NB1643‐ALKR cells following 72 h exposure to ABT‐737.
Data information: For A, C–E, and G, data points are mean and SD from triplicate wells, experiments are representative of at least three biological replicates, dotted line represents 50% viability. For F, data points are mean of duplicate wells (SD < 0.05 at all points) in a representative experiment from at least two biological replicates.
Figure EV3. REL neuroblastoma cells are resistant to the cytotoxic effects of ionizing radiation despite incurring an equivalent increase in DNA damage.
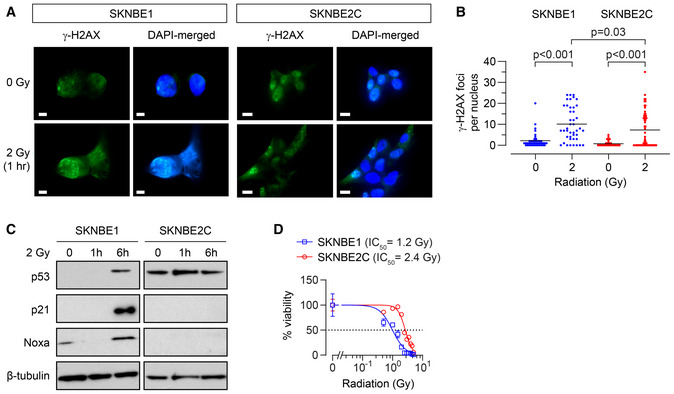
- DNA damage induced by 2 Gy ionizing radiation as measured by γ‐H2AX foci for SKNBE1 (DX, TP53 wild type) and SKNBE2C (REL, TP53 mutant). Fluorescence detection of γ‐H2AX foci and DAPI nuclear counterstain assessed, with representative images shown; SKNBE1 scale bar = 5 µm; SKNBE2C scale bar = 10 µm.
- Foci frequency plotted with mean shown for each condition in A (n = 42–58 nuclei/condition).
- Cell lysates from cells treated with 2 Gy of ionizing irradiation were immunoblotted for p53 and p53 target genes with β‐tubulin as a protein loading control.
- In vitro viability following ionizing radiation at 7 days; dotted line represents 50% viability. Data points are mean and SD error bars from triplicate wells, experiments are representative of at least three biological replicates.
Data information: Statistical analyses in B were performed using the two‐tailed Mann–Whitney U test, with significance P < 0.05.
To determine whether attenuated mitochondrial responses also contribute to resistance to molecularly targeted drugs, we tested Bcl2 inhibitors since their mechanism of activity is localized at mitochondria. We previously showed that CHLA15 (DX) cells are sensitive to Bcl2 inhibitors as they use Bcl2 to sequester Bim and prevent its activation of Bak or Bax (Goldsmith et al, 2012). Both CHLA15 (DX) and CHLA20 (REL) cells have Bim sequestered by Bcl2 and the Bcl2/Bclx inhibitor ABT‐737 displaces Bim with equal potency, yet CHLA20 cells are > 40‐fold more resistant despite the patient the cell line derived from never having been treated with a Bcl2 inhibitor (IC50: CHLA15 = 260 nM, CHLA20 = 11.4 μM; Fig 2D). In contrast, the CHLA122/CHLA136 pair also have Bim sequestered by Bcl2, but these cells have similar cytochrome c release and Bcl2 inhibitor responses (~1.8‐fold IC50 difference). We did not test SKNBE1/SKNBE2C cells with ABT‐737 as they use Mcl1, rather than Bcl2, to neutralize Bim (Goldsmith et al, 2012). We next studied anaplastic lymphoma kinase (Alk) inhibitors since mutations in the ALK gene are found in 10–14% of neuroblastomas, and Alk inhibition has anti‐tumor activity (Bresler et al, 2014). Both CHLA15 (DX) and CHLA20 (REL) cells harbor an ALK R1275Q mutation (variant allele frequency of 0.46 and 0.49, respectively). The patient this tumor pair was obtained from had not been treated with an Alk inhibitor, yet REL cells were > 3‐fold more resistant to the Alk inhibitors crizotinib, ceritinib, and lorlatinib, without having a secondary resistance‐encoding ALK mutation (e.g., crizotinib IC50: CHLA15 = 96 nM and CHLA20 = 349 nM; Fig 2E). SKNBE1/SKNBE2C and CHLA122/CHLA136 have wild‐type ALK and are resistant to Alk inhibitors (Appendix Fig S1B). Therefore, attenuated mitochondrial signaling that arises in response to multimodal therapy in situ confers resistance to chemotherapy, radiotherapy, and molecularly targeted drugs (the latter in the absence of prior selective pressure). We next assessed the reverse: whether selection for resistance to a molecularly targeted agent can induce this mitochondrial phenotype. We exposed SH‐SY5Y (ALK F1174L) and NB1643 (ALK R1275Q) cells to escalating concentrations of crizotinib to generate crizotinib‐resistant clones, and both demonstrated attenuated MOMP responses to tBid and BimBH3 in the absence of secondary ALK mutations, phenocopying REL cells (Fig 2F). In addition, such Alk inhibitor‐resistant cells were more resistant to chemotherapy drugs such as etoposide (> 20‐fold) and molecularly targeted drugs like Bcl2 inhibitors (> 40‐fold; Fig 2G).
Attenuated mitochondrial apoptotic responses are accompanied by reductions in MAMs
We confirmed that loss of Bak or Bax, or upregulation of pro‐survival Bcl2‐family proteins, were not drivers of attenuated MOMP [Fig EV2B and C and (Goldsmith et al, 2012)]. We next compared mitochondrial mass, size, shape, and mitochondrial DNA (mtDNA) from DX and REL pairs, as these have been correlated with apoptotic signaling in other systems. Mitochondrial mass assessed by citrate synthase activity was unchanged in CHLA15 and CHLA20, and reduced in SKNBE2C (relative to SKNBE1) and CHLA136 (relative to CHLA122; Fig EV4A). Mitochondrial DNA content was measured using quantitative PCR for two mitochondrial genes (MT‐CO1 and MT‐ATP6), each normalized to two nuclear genes (CFAP410 and MTTP) that are disomic in > 85% of neuroblastomas. TP53 mutant SKNBE2C cells had markedly reduced mtDNA content compared with TP53 wild‐type SKNBE1 cells, consistent with p53 mutation effects on mtDNA content (Park et al, 2016). CHLA15/CHLA20 and CHLA122/CHLA136 had less divergent mtDNA quantity with modest reductions in REL cells at a subset of loci (Fig EV4B). We next used transmission electron micrographs of each cell line to assess mitochondrial size (circumference) and shape (roundness and circularity; Fig 3A–C). These did not differ for the SKNBE1/SKNBE2C and CHLA122/CHLA136 pairs, although there was a trend toward larger mitochondria in SKNBE2C (P = 0.06). The CHLA15 (DX) and CHLA20 (REL) pair did differ, with REL cells having smaller more circular mitochondria (P < 0.01). No feature correlated with mitochondrial cytochrome c release sensitivity across all pairs.
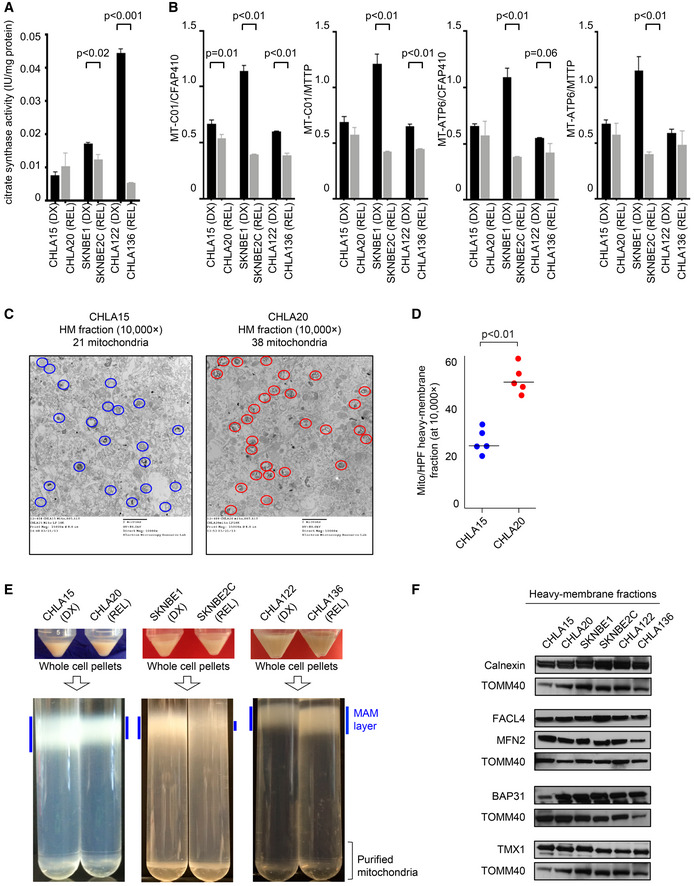
Figure EV4. Only reduced MAM content in neuroblastoma DX/REL pairs is correlated with attenuated mitochondrial responses to stress and chemotherapy resistance.
-
AMitochondrial biomass assessed by citrate synthase activity across DX/REL pairs; in triplicate from three biological replicates; mean with SD error bar shown.
-
BMitochondrial DNA content as quantified by qPCR using the mtDNA/nucDNA ratio for each of the two mitochondrial (MT‐C01 and MT‐ATPase6) and two nuclear (CFAP410 and MTTP) genes; in triplicate from three biological replicates; mean with SD error bar shown.
-
C, DElectron micrograph images of heavy‐membrane mitochondria‐enriched fractions from CHLA15 and CHLA20 cells, identifying relative mitochondria abundance (identified within colored circles); quantification shown in D.
-
EHeavy‐membrane fractions obtained from equal cell input were separated by Percoll gradient ultracentrifugation into MAM fractions (blue bars) and purified mitochondria for DX/REL pairs.
-
FImmunoblot detection of MAM‐ and ER‐localized proteins in heavy‐membrane fractions from DX/REL pairs, with TOMM40 as a loading control for mitochondrial protein input.
Data information: Statistical analyses were performed using an unpaired two‐sided Student’s t‐test.
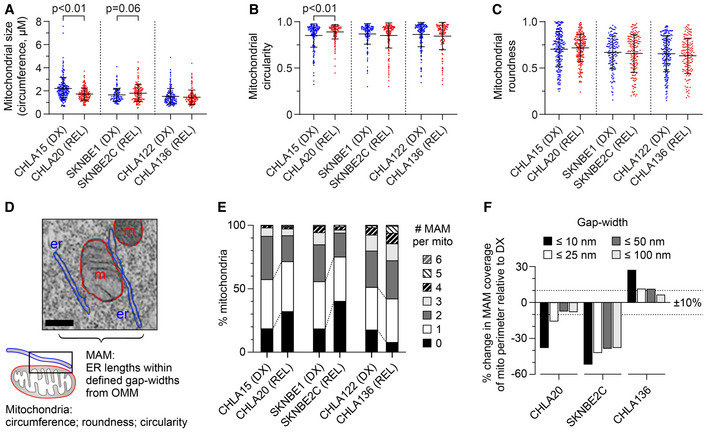
Figure 3. REL neuroblastomas have reduced mitochondria‐associated membranes compared with patient‐matched tumors from DX.
-
A–CTransmission electron microscopy image analysis was used to quantify mitochondrial size, circularity, and roundness in DX/REL neuroblastoma pairs (mean ± SD shown).
-
DElectron micrograph illustrating organelle masking of MAM interfaces; scale bar = 500 nm.
-
EProportion of mitochondria with 0–6 or more MAMs are shown for each cell line, as DX/REL pairs.
-
FPercentage of mitochondria perimeter with an apposed ER within defined gap widths for DX/REL neuroblastoma cell line pairs; dotted lines denote +10% and −10% change.
Data information: For A–C and E, F, n = 137–241 mitochondria per cell line; er, endoplasmic reticulum; m, mitochondria. For A‐C, statistical analyses were performed using a two‐tailed Mann–Whitney U test, with significance P < 0.05 (trend P < 0.10).
We applied electron microscopy to visualize the mitochondria‐enriched heavy‐membrane fractions tested in our mitochondrial profiling assays. Non‐mitochondria organellar membranes were abundant yet there was an increase in the proportion of mitochondria in fractions from REL cells, despite their reduced cytochrome c release when stimulated (Fig EV4C and D). ER, including MAM, is a major contributor to heavy‐membrane fractions. We postulated that fewer ER–mitochondria contacts cause reduced MAM content to be pulled down in heavy‐membrane fractions. We further fractionated these to resolve purified mitochondria from MAMs (Annunziata et al, 2013). With equal cellular input, more MAMs were present from DX cells than REL cells for the CHLA15/CHLA20 and SKNBE1/SKNBE2C pairs, while the CHLA122/CHLA136 pair that lacks attenuated mitochondrial signaling had slightly more MAM from REL cells (Fig EV4E). We also assessed ER‐specific proteins in heavy‐membrane fractions from DX and REL cells but were unable to detect differential expression (Fig EV4F).
We directly visualized MAM interfaces using transmission electron micrographs of DX and REL tumor cells (all MAM interface data can be found in Dataset EV1). We defined MAMs as regions of ER within 100 nm of the outer mitochondrial membrane (OMM), and characterized their number, length, and gap width from the OMM, binned as ≤10 nm, >10–25 nm, >25–50 nm, and >50–100 nm (Fig 3D). CHLA20 (REL) cells had reduced MAM content compared with CHLA15 (DX) cells (Table 1). The number of MAMs per mitochondria was reduced (P < 0.01), the frequency of mitochondria with ≥ 2 MAMs was reduced while those with absent MAMs were increased (both P < 0.01). Because CHLA20 mitochondrial had a mean circumference 22% smaller (P < 0.01), MAMs occurred at equal frequency in DX/REL cells: every 1,651 nm in CHLA15 and every 1,556 nm in CHLA20. However, there was a marked reduction in closely apposed MAMs and MAM lengths in REL cells. The proportion of MAM that came within 10 nm of the OMM was reduced (P = 0.05), as was the proportion of the OMM perimeter in contact with a MAM at < 10 nm. In contrast, the proportion approximated by a MAM at larger gap distances was less reduced (Fig 3E and F). Similarly, SKNBE2C (REL) cells were reduced in the number of MAMs per mitochondria compared to SKNBE1 (DX) cells (P < 0.01), and the frequency of mitochondria with ≥ 2 MAMs was reduced, while those absent MAMs were increased > 2‐fold (both P < 0.01). MAMs occurred on average every 1,116 nm along the OMM of SKNBE1 but only every 1,880 nm in SKNBE2C. In addition to reduced frequency, the total length and proximity of MAMs were reduced in SKNBE2C cells, adjacent to 24% of the mitochondrial perimeter in SKNBE1 but only 15% in SKNBE2C cells, and MAMs were markedly reduced across all gap widths (Fig 3E and F).
In contrast, CHLA136 (REL) cells do not have attenuated mitochondrial responses, multidrug resistance, reduced MAM content by fractionation, nor reduced MAM interfaces in comparison with CHLA122 (DX) cells. In fact, CHLA136 (REL) cells had a trend toward more mitochondria with ≥ 2 MAMs (P = 0.10) and fewer orphan mitochondria (P < 0.01), more MAMs per mitochondria overall (P = 0.01), and a slightly higher proportion of MAMs along the OMM across all gap widths. The characteristics of individual MAMs by their length and relative proximity to the mitochondrial outer membrane were otherwise similar (Fig 3E and F; Table 1). In all, reduced ER–mitochondria connectivity (in particular, a reduction in the proportion of MAMs in close proximity to the OMM) was recurrently present in tumor cells with multidrug resistance and attenuated mitochondrial responses to death stimuli, nominating this feature as a contributor to therapy resistance.
The role of MAMs in mitochondrial apoptotic signaling can be functionally demonstrated
Murine liver and HeLa cell mitochondria separated from MAMs using limiting proteolysis have reduced cytochrome c release in response to Bid and BimBH3 domains (Chipuk et al, 2012). We tested limited proteolysis and immunomagnetic bead separation to mechanically isolate mitochondria from ER in DX neuroblastoma cells. Both induced non‐specific cytochrome c release, dampening the dynamic range of the assay. Neither process reduced the integrity or abundance of Bak (the preferential target of tBid), although the latter led to reduced MAM markers such as Facl4 protein (Fig EV2D and E). Immunomagnetic bead separation induced less membrane disruption, and MOMP sensitivity in response to BimBH3 was greater for CHLA15 heavy‐membrane fractions (intact MAMs) compared with purified mitochondria (reduced MAMs; Fig 4A; SKNBE1 could not be reliably assessed due to high non‐specific release with both methods).
Figure 4. Chemically or genetically reducing MAMs leads to MOMP resistance.
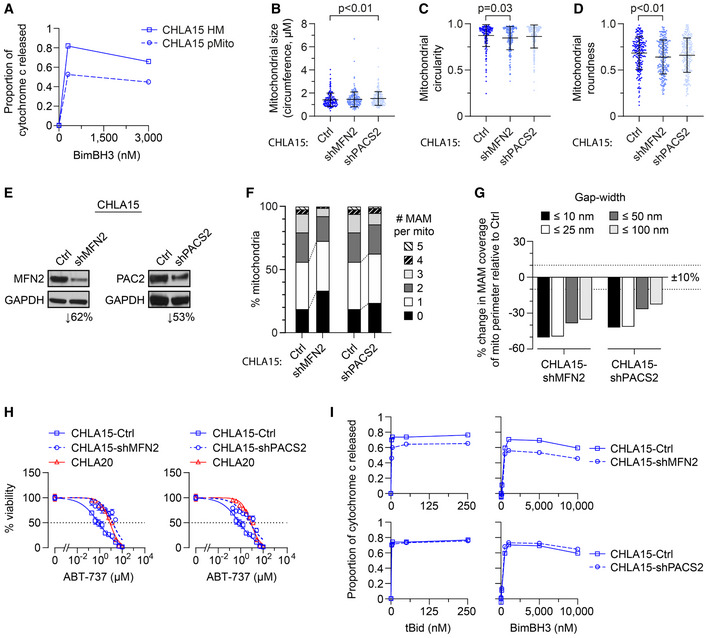
-
AHeavy‐membrane (HM) fractions of CHLA15 were rendered MAM depleted by immunomagnetic separation to derive purified mitochondria (pMito), and cytochrome c release measured in response to BimBH3 peptide.
-
B–DTEM analyses were used to quantify mitochondrial size, circularity, and roundness in CHLA15 cells transfected with a sh‐control (Ctrl), shMFN2 or shPACS2 constructs (mean ± SD shown).
-
EImmunoblot assessment of Mfn2 and Pacs2 protein knockdown.
-
FProportion of mitochondria with 0–5 MAMs are shown for each.
-
GPercentage of mitochondria perimeter with an apposed ER within defined gap widths; dotted lines denote +10% and −10% change.
-
HIn vitro viability of CHLA15‐ctrl, CHLA15‐shMFN2, CHLA15‐shPACS2, and CHLA20 cells following 72 h exposure to ABT‐737; dotted line represents 50% viability.
-
IMitochondrial cytochrome c release in response to tBid and BimBH3 peptide in CHLA15‐Ctrl, CHLA15‐shMFN2, and CHLA15‐shPACS2 cells.
Data information: For B–D and F, G, n = 214–246 mitochondria per cell line. For A and I, data points are mean of duplicate wells (SD < 0.05 at all points) in a representative experiment from at least two biological replicates; for H, data points are mean and SD from triplicate wells, experiments, are representative of at least two biological replicates. For B–D, statistical analyses were performed using a two‐tailed Mann–Whitney U test, with significance P < 0.05.
We next knocked‐down expression of phosphofurin acid cluster sorting protein‐2 (PACS2) and mitofusin‐2 (MFN2) as these have been shown to reduce MAMs (Simmen et al, 2005; de Brito & Scorrano, 2008; Moulis et al, 2019). Mitochondria in CHLA15‐shPACS2 cells with Pacs2 expression reduced > 50% were larger than CHLA15‐shCtrl cells but similar in roundness and circularity (Fig 4B–E). There was a trend toward reduced MAM per mitochondria, although the distribution of MAMs to mitochondria was preserved (Fig 4F; Table 2). MAM length and proximity were reduced, with Pacs2 knockdown cells having a reduced MAM per mitochondrial perimeter across all gap widths (Fig 4F and G; Table 2). The proportion of MAM that came within 10 or 25 nm of the OMM was reduced (P = 0.02 and P < 0.05, respectively), as was the proportion of the OMM perimeter in contact with a MAM at all gap widths (Fig 4G). By comparison, mitochondria in CHLA15‐shMFN2 cells with Mfn2 reduced ~60% had reduced circularity and roundness (P = 0.03 and P < 0.01, respectively) but size was maintained (Fig 4B–E). There were fewer MAMs per mitochondria (P < 0.01), and the frequency of mitochondria with ≥ 2 MAMs was reduced while those absent MAMs were increased (both P < 0.01; Fig 4F). Moreover, MAM lengths were reduced across all gap widths (Fig 4G). We tested the sensitivity of these cells to the Bcl2/Bclx‐inhibitor, ABT‐737, a pharmacological enhancer of MOMP in CHLA15 cells, and knockdown of Mfn2 or Pacs2 phenocopied the ABT‐737 resistance seen in CHLA20 cells, shifting the IC50 > 18‐fold and 8‐fold, respectively (Fig 4H). In addition, CHLA15‐shMFN2 cells, which had more substantial MAM alterations than did CHLA15‐shPACS2 cells, showed a resistance to tBid and BimBH3 in mitochondrial profiles that phenocopied multidrug‐resistant neuroblastomas (Fig 4I).
Table 2.
Summary of CHLA15‐Ctrl (DX) and CHLA15‐shMFN2 and ‐shPACS2 models.
| CHLA15‐ctrl | CHLA15‐shMFN2 | CHLA15‐shPACS2 | |||
|---|---|---|---|---|---|
| Mitochondria | |||||
| Number analyzed | 214 | 234 | 246 | ||
| Perimeter, mean (nm) | 1,416 | 1,468 | P = ns | 1,530 | P < 0.01 |
| Perimeter, median (nm) | 1,319 | 1,381 | 1,453 | ||
| Circularity, median | 0.918 | 0.892 | P = 0.03 | 0.914 | P = ns |
| Roundness, median | 0.715 | 0.655 | P < 0.01 | 0.686 | P = ns |
| % with 0 MAMs | 19% | 33% | P < 0.01 | 24% | P = ns |
| % with 1 MAMs | 37% | 39% | 39% | ||
| % with 2–3 MAMs | 37% | 26% | P < 0.01 | 22% | P = ns |
| % with ≥ 4 MAMs | 6% | 1% | 5% | ||
| %OMM with MAM gap width ≤ 10 nm | 1.7% | 0.9% | (↓47%) | 1.0% | (↓41%) |
| %OMM with MAM gap width ≤ 25 nm | 5.9% | 3.0% | (↓49%) | 3.5% | (↓41%) |
| %OMM with MAM gap width ≤ 50 nm | 12.6% | 7.8% | (↓38%) | 9.3% | (↓26%) |
| %OMM with MAM gap width ≤ 10 nm | 27.4% | 17.8% | (↓35%) | 21.2% | (↓23%) |
| MAM | |||||
| Number analyzed | 330 | 241 | 331 | ||
| MAMs per mitochondria | 1.54 | 1.03 | P < 0.01 | 1.35 | P = 0.07 |
| MAM frequency (per nm perimeter) | 918 | 1,425 | 1,137 | ||
| %MAM with gap width ≤ 10 nm | 22% | 17% | P = 0.09 | 15% | P = 0.02 |
| %MAM with gap width ≤ 25 nm | 47% | 43% | P = ns | 40% | P < 0.05 |
| %MAM with gap width ≤ 50 nm | 68% | 73% | P = ns | 71% | P = ns |
| %MAM with gap width ≤ 100 nm | 100% | 100% | 100% | ||
Reduced transfer of Ca2+ from MAMs to mitochondria is not required for attenuated apoptotic signaling
Mitochondria‐associated ER membranes support mitochondria as platforms to integrate stress signals. ER provides the major intracellular reservoir for Ca2+, and MAMs are enriched with Ca2+ release constituents to create localized microdomains that enable calcium transfer into mitochondria through the low‐affinity mitochondrial calcium uniporter [mtCU; (Rizzuto & Pozzan, 2006)], which has been linked to Bcl2 family function and apoptotic sensitivity (Hajnoczky et al, 2000; Rizzuto & Pozzan, 2006). Indeed, synthetically decreasing ER–mitochondria tethering in rat liver cells and basophils reduces Ca2+ transfer and apoptotic sensitivity, while augmenting tethering increases both (Csordas et al, 2006).
We posited that the reduction in MAMs in REL tumors reduces ER‐to‐mitochondria Ca2+ transfer to attenuate apoptotic signaling. We measured Ca2+ transfer in two DX/REL pairs with differential MAM content. Mitochondrial calcium concentration ([Ca2+]m) was detected with a fluorescent protein‐based Ca2+ sensor targeted to the mitochondrial matrix, GCamp6f. Cytoplasmic calcium concentration ([Ca2+]c) was monitored with fura2 loaded to the cells as fura2AM. Calcium responses evoked by the IP3R‐linked stimulus, carbachol, were recorded and the time courses for the corresponding fluorescence signals calculated. CHLA20 (REL) cells have attenuated MOMP responses, multidrug‐resistant phenotype, and reduced MAM content in comparison with CHLA15 cells (DX), yet both have similar [Ca2+]c and [Ca2+]m signals and coupling time (< 2 s), the time difference between [Ca2+]c and [Ca2+]m achieving 50% of maximum (Fig 5A; Appendix Fig S2E). Like CHLA20, SKNBE2C (REL) cells also have attenuated MOMP responses, multidrug‐resistant phenotype, and reduced MAM content in comparison with SNKBE1 (DX) cells. However, unlike CHLA20, SKNBE2C cells have markedly reduced Ca2+ transfer compared with SKNBE1 (DX) cells, with a > 3‐fold increase in coupling time (P < 0.01; Fig 5B). We genetically augmented MAMs in SKNBE2C cells using a monomeric linker composed of a fluorescent protein extended with ER and OMM membrane anchor domains (Csordas et al, 2006). Linker‐induced augmentation of MAMs normalized the Ca2+ transfer coupling time to ~1.5 s, compared with a control OMM‐only linker, confirming the reduction was a consequence of reduced MAMs (Fig 5C). In both the CHLA15/CHLA20 and SKNBE1/SKNBE2C pairs, the REL cells showed similar proportional reductions in MAM coverage at ≤10 nm ER–mitochondrial gap width (Table 1). However, CHLA20 cells had preserved MAM coverage at larger gap widths capable of accommodating the Ca2+ transfer machinery, while SKNBE2C cells were reduced at all gap widths, limiting Ca2+ transfer.
Figure 5. Reduced Ca2+ transfer at MAMs is not required for attenuated mitochondrial responses to stress.
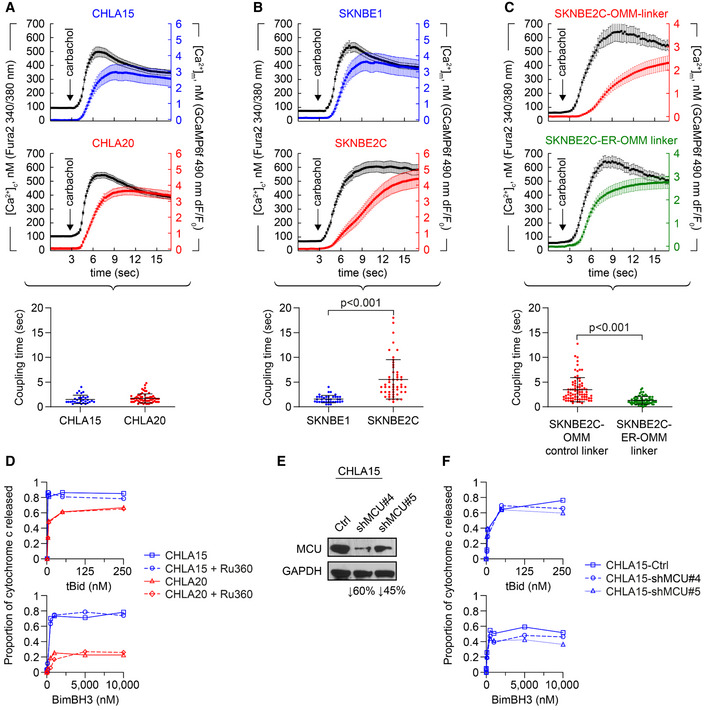
-
A, BCytosolic (black tracing) and mitochondrial (colored tracing) Ca2+ concentration measured using fluorescent reporters in the CHLA15 (n = 29 cells)/CHLA20 (n = 65 cells) and SKNBE1 (n = 37 cells)/SKNBE2C (n = 52 cells) pairs. ER Ca2+ release was induced by 100 μM carbachol, an IP3R‐agonist; time added indicated by arrow; tracings are mean ± SD error bars. Coupling time (time between achieving 50% of maximal cytosolic and mitochondrial concentrations) is an index of ER–mitochondrial proximity and transfer efficiency; plotted below.
-
CMAM proximity was enforced in SKNBE2C cells using an ER‐OMM linker construct, with Ca2+ transfer coupling time (n = 57 cells) compared with cells expressing an OMM‐only control linker (n = 70 cells); tracings are mean ± SEM error bars.
-
DMitochondrial cytochrome c release for CHLA15 and CHLA20 cells following exposure to tBid or BimBH3 peptide, with or without Ru360 treatment.
-
ESilencing of the mitochondrial calcium uniporter (MCU) was achieved in two CHLA15‐shMCU clones.
-
FCytochrome c release in response to tBid and BimBH3 assessed.
Data information: For A–C, data are derived from at least three separate cell transfections (biological replicates). For D and F, data points are mean of duplicate wells (SD < 0.05 at all points) in a representative experiment from at least two biological replicates. For A–C, statistical analyses were performed using an unpaired two‐sided Student’s t‐test, with significance P < 0.05.
We used biochemical and genetic approaches to further evaluate Ca2+ transfer and stress sensitivity. We treated CHLA15 heavy‐membrane fractions with Ru360 that inhibits the mtCU, before measuring cytochrome c release in response to tBid or BimBH3. No reduction in release was seen (Fig 5D). Similarly, CHLA20 (REL) cells did not have further attenuated cytochrome c release following exposure to Ru360. We next used siRNA to reduce expression of MCU, the pore‐forming component of the mtCU (Baughman et al, 2011; De Stefani et al, 2011). MCU was not differentially expressed in DX/REL pairs (Appendix Fig S2A). MCU knockdown of ~45% and 60% was achieved in two CHLA15 subclones and MOMP sensitivity was only modestly reduced (Fig 5E and F). We also tested MCU knockdown and Ru360 exposure in SKNBE1 cells and obtained similar results (Appendix Fig S2B–D). Collectively, these data support that reduced Ca2+ transfer from ER to mitochondria is not required for the attenuated MOMP phenotype.
MAM‐reduced tumor cells have diminished ceramide pools, and repressing ceramide generation at MAMs induces relative MOMP resistance
Mitochondria are dependent on the transfer of lipids from MAMs to maintain their OMM composition, including sphingolipids that are implicated in regulating Bak and Bax sensitivity to stress signals (Chipuk et al, 2012). ER is the primary organelle for bulk lipid synthesis and distribution, and regulatory enzymes are highly compartmentalized within ER subdomains, including MAMs. Neutral sphingomyelinases are enriched at MAMs where they hydrolyze sphingomyelin to ceramide and facilitate its transfer to the OMM through incompletely understood mechanisms (Morad & Cabot, 2013). Ceramide in the OMM facilitates the oligomerization of Bak and/or Bax (Ganesan et al, 2010).
We quantified bioactive sphingolipids in DX and REL cell pairs with altered MAM content, focusing on ceramide (Cer) and sphingomyelin (SM) species. SKNBE2C cells demonstrated the most altered sphingolipid profile in comparison with matched DX SKNBE1 cells, consistent with their markedly disrupted MAM content. This included reductions in multiple bioactive Cer species (chain length C32‐C36) and total Cers, elevation in SMs, and an elevated SM:Cer ratio. CHLA20 cells also had reductions in bioactive Cers, although no alterations in SMs or SM:Cer ratio. Finally, CHLA136 cells did not have alterations in Cers, SMs, or their ratio, consistent with their intact MAM content at DX and REL (Fig 6A–F and Fig EV5). To determine whether reduced MAM‐synthesized ceramides specifically were relevant in regulating MOMP in our models, we inhibited neutral sphingomyelinase in isolated mitochondria by pre‐incubating them with the inhibitor, GW4869. Prolonged incubation of isolated mitochondria with GW4869 led to mitochondrial dysfunction and failure to elicit specific cytochrome c release, but drug incubation for 30 min was tolerated, albeit with a reduced dynamic range. Thirty minutes of sphingomyelinase inhibition reduced tBid‐induced cytochrome c release in DX therapy‐sensitive CHLA15 (median 35% inhibition; P = 0.09) and SKNBE1 (median 34%; P < 0.01; Fig 6G and H). CHLA15 cells were most inhibited at lower tBid concentrations and Bax oligomerization was also inhibited at these concentrations (Fig EV2F). To exclude direct effects of GW4869 on cytochrome c release, as opposed to lipid‐mediated mechanisms, we pre‐incubated mitochondria without the inhibitor, and added it at the time of tBid, and no inhibition of cytochrome c release was observed (Fig 6G).
Figure 6. Reduced ceramides are present in REL cells with depleted MAMs.
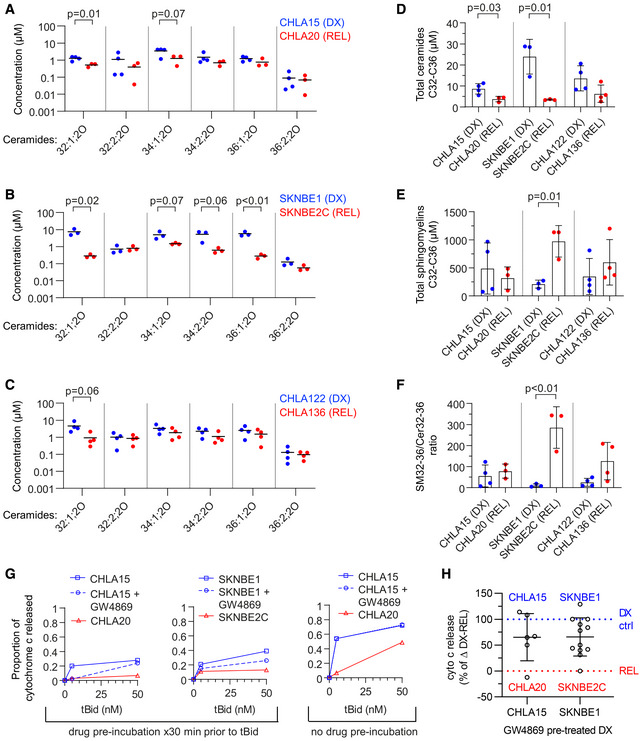
-
A–CConcentration of ceramide species as measured by LC/MS from DX and REL pair whole‐cell pellets.
-
D, ECumulative ceramides and sphingomyelins (of C32–C36 chain length) for DX/REL pairs.
-
FRatio of total C32‐C36 sphingomyelins:ceramides for DX/REL pairs.
-
GCytochrome c release for CHLA15 and SKNBE1 mitochondria following exposure to tBid, pre‐incubated for 30 min with or without GW4869, and CHLA20 and SKNBE2C pre‐incubated without GW4869 (left panel); same experiment but with GW4869 added with tBid, after the 30 min pre‐incubation (right panel).
-
HSummary data for CHLA15/CHLA20 and SKNBE1/SKNBE2C showing relative cytochrome c release when pre‐incubated with GW4869 to inhibit ceramide generation, compared with untreated cells.
Data information: A–F show mean of three to four biological replicates plotted; three technical replicates each; D–F error bars are ± SD. For G, data points are mean of duplicate wells (SD < 0.05 at all points) in a representative experiment from at least two biological replicates. All data points from two (CHLA15) or three (SKNBE1) biological replicates are depicted in H. Statistical analyses were performed using an unpaired two‐sided Student’s t‐test, with significance P < 0.05 (trend, P < 0.10).
Figure EV5. Sphingomyelin concentrations in DX/REL neuroblastoma cells.
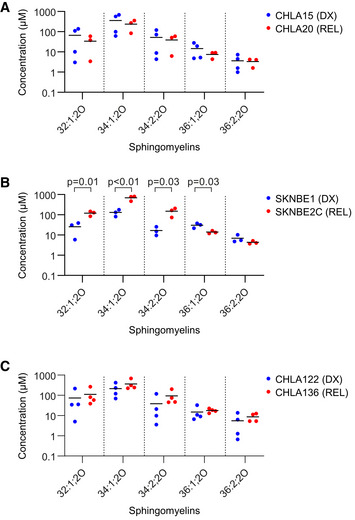
-
A–CConcentration of sphingomyelin species as measured by LC/MS from DX and REL pair whole‐cell pellets. Data information: three to four biological replicates plotted per cell line; three technical replicates each; statistical analyses were performed using an unpaired two‐sided Student’s t‐test, with significance P < 0.05.
Discussion
Most cancer deaths result from progression of disease that is resistant to available therapies, yet its principal contributors remain incompletely understood. The diversity of targets and stress signals engaged by cancer treatments, and the myriad adaptations cancer cells use to subvert them, pose fundamental challenges. Cancer therapies activate stress signals that converge on mitochondria (Danial & Korsmeyer, 2004; Deng et al, 2007; Ni Chonghaile et al, 2011; Vo et al, 2012; Sarosiek et al, 2013b; Montero et al, 2015), whose ability to undergo MOMP is regulated by MAMs, essential contacts for the transfer of metabolites. Here, we identify reduced MAMs as a novel convergence‐based mechanism of resistance arising from attenuation of stress‐induced MOMP. Since mitochondria act at a terminal node integrating stress signals, this attenuated responsiveness contributes to multidrug resistance. Importantly, this mechanism is not exclusive to other mechanisms of resistance operative upstream of mitochondria.
We identified the MOMP‐attenuated phenotype measuring mitochondrial responses to the Bak and Bax activators, tBid and Bim. Mitochondrial profiling was developed by Letai (Deng et al, 2007) and used to define Bcl2 family dependencies and predict responses to chemotherapy (Ni Chonghaile et al, 2011; Vo et al, 2012; Montero et al, 2015) and molecularly targeted drugs (Hata et al, 2015; Montero et al, 2015). We studied neuroblastoma, a tumor that often responds to chemoradiotherapy with tumor regression (Uccini et al, 2006) followed by lethal relapse (Matthay et al, 2009). Limited clinical samples had constrained such investigations, so we derived patient‐matched tumor models at diagnosis and relapse, the latter frequently manifesting multidrug resistance (Keshelava et al, 1998). We show that in six of seven pairs, mitochondria from relapsed neuroblastomas have reduced MOMP responses to tBid and Bim compared with their at‐diagnosis counterparts, providing evidence that attenuated mitochondrial signaling arises during multimodal therapy. Attenuated MOMP was identified in response to either death activator, although tBid was more potent. Indeed, BimBH3 is an intrinsically unstructured peptide (Hinds et al, 2007) that engages Bak or Bax at micromolar exposures, while recombinant tBid binds lipid membranes to activate Bak or Bax at nanomolar exposures, highlighting the importance of lipid membranes in facilitating MOMP (Walensky et al, 2006; Sarosiek et al, 2013a). Quantitative variability among biological replicates reflects myriad events impinging on Bak/Bax‐mediated pore formation, including temperature, pH, detergents, and metabolites (Kale et al, 2018), but working with patient‐matched pairs in parallel enabled reproducible qualitative differences to be identified. A mitochondrial basis has been proposed for the selective killing of tumor cells by chemotherapy (Vo et al, 2012; Sarosiek et al, 2013b), and here we expand this notion to demonstrate a mitochondrial basis for multidrug resistance in response to therapeutic pressure during multimodal therapy.
Neuroblastoma cell lines represent oligoclonal outgrowths from tumors. If MOMP sensitivity were highly heterogeneous within a tumor, we predict some diagnosis–relapse pairs to have attenuated mitochondrial responses present at diagnosis from chance alone. Instead, our data support that contributions from intra‐tumoral heterogeneity are minor compared with the effect of therapeutic selective pressure. We confirmed that attenuated mitochondrial responses correlated with resistance to chemotherapy and ionizing radiation. For the latter, differential cell death follows equivalent genotoxic stress, emphasizing this as a post‐target resistance mechanism. Resistance is also conferred to molecularly targeted drugs such as Bcl2 and Alk inhibitors in the absence of prior exposure, since these operate upstream of mitochondria. More surprising is evidence that selecting for resistance to targeted agents in vitro can induce the attenuated MOMP phenotype and multidrug resistance. This provides a potential mechanism for the significant proportion of patients with emergent resistance to therapeutic kinase inhibitors that have no on‐pathway mechanism identified (Katayama et al, 2012; Chong & Janne, 2013; Lito et al, 2013; Hugo et al, 2015; Wilson et al, 2015).
We identified reductions in MAM content from tumors with attenuated MOMP when visualized by fractionation or quantified by EM morphometry. ER–mitochondria contacts at MAMs regulate fission/fusion dynamics to optimize mitochondrial shape (Hoppins & Nunnari, 2012), which impacts Bax‐induced MOMP in murine hepatocytes and embryonic fibroblasts (MEFs) by altering cooperation among Bcl2 family members. Smaller mitochondria with increased membrane curvature are more resistant to stress‐induced MOMP, possibly contributing to chemoresistance (Renault et al, 2015). Consistent with this, the CHLA15/CHLA20 pair had smaller more circular mitochondria in the post‐relapse MOMP‐resistant tumor cells, however, the reverse was true for the SKNBE1/SKNBE2C pair. While mitochondrial size, shape, mass, and mtDNA content differed, only the reduction in MAMs was present across all models with attenuated MOMP, while absent in the single pair without this feature.
Our image analyses were done in 2D, so to ensure representative findings we analyzed > 2,400 MAMs from > 1,700 mitochondria across nine tumor models. In both the CHLA15/CHLA20 and SKNBE1/SKNBE2C pairs, MAMs were markedly reduced in the therapy‐resistant post‐relapse model, as was the proportion of mitochondrial surface with an apposed MAM. In contrast, the only tumor pair without attenuated MOMP responses or chemoresistance, CHLA122/CHLA136, did not have such alterations. That MAMs are reduced in cancer cells was originally posited by Howatson and Ham from EM studies of mouse liver tumors (Howatson & Ham, 1955). A role in mediating apoptotic sensitivity was demonstrated using MEF heavy‐membrane fractions, as MOMP sensitivity to Bid was reduced when mitochondria were purified away from their associated MAMs (Chipuk et al, 2012). Similarly, we could phenocopy attenuated MOMP responses in DX tumors by reducing MAMs from mitochondria with chemical proteolysis or genetic perturbation. In the latter, we used EM morphometry to confirm the MAM‐reduced phenotype.
These data support that an essential factor for mitochondrial sensitization to MOMP is provided by MAMs. Korsmeyer’s group showed that many intrinsic cell death signals require both functional Bak or Bax and ER‐released Ca2+ (Scorrano et al, 2003), and deficiencies in MAM‐derived Ca2+ transfer can confer apoptosis resistance in mesothelioma cells (Patergnani et al, 2015). We found a > 3‐fold increase in Ca2+ transfer coupling time (reflecting reduced transfer) in therapy‐resistant SKNBE2C cells compared with DX cells, which was normalized using a linker that tightens MAM contacts. However, we found no change in coupling time for the CHLA15/CHLA20 pair. CHLA20 have reduced MAMs with tight gap widths with relative preservation at larger gap widths that enable Ca2+ transfer (Csordas et al, 2006, 2010). While altered Ca2+ transfer may contribute to MOMP regulation, it is not indispensable for therapy resistance in neuroblastoma and disrupted Ca2+ transfer did not phenocopy MOMP attenuation. It is postulated that Ca2+ transfer at MAMs regulates the mitochondrial permeability transition pore and sphingolipid transfer regulates MOMP (Lemasters et al, 2009; Bauer & Murphy, 2020).
Narrow gap width MAMs are unable to accommodate the Ca2+ transfer machinery but are critical for lipid transport, and tight contacts (< 10 nm) have been referred to as lipid‐ERMCs or lipid‐MAMs (Schauder et al, 2014; Martino Adami et al, 2019). We show that resistant neuroblastoma cells have reduced ceramide levels reflecting reduced MAM activities and inhibiting the hydrolysis of sphingomyelin that occurs at MAMs for only 30 min partially phenocopies the attenuated MOMP phenotype. Ceramides are synthesized by neutral sphingomyelinases enriched at MAMs, and transferred to the OMM where they synergize with Bak and Bax to induce pore formation (Ganesan et al, 2010). Multiple ceramide regulators (including sphingomyelinase) were identified as determinants of multidrug resistance in an siRNA screen across diverse carcinoma cell lines. That work was agnostic to consideration of MAM content yet knockdown of six of six ceramide‐metabolizing enzymes led to drug sensitivity changes (increased or decreased) predicted by a ceramide–OMM–MOMP model (Swanton et al, 2007). Chipuk and Green demonstrated that the reduced MOMP sensitivity of mitochondria purified away from MAMs results from loss of neutral sphingomyelinase activity. Sensitivity was restored by the addition of microsomes containing MAM domains, active sphingomyelinase and sphingomyelin, or ceramide and/or ceramide‐derived lipids (Chipuk et al, 2012). Walensky’s group identified a covalent lipid‐binding site on Bax regulating its oligomerization potential (Cohen et al, 2020), and crystal structures have revealed Bak sites preferentially bound by lipids and necessary to support its oligomerization (Cowan et al, 2020). Targeting neutral sphingomyelinases to various organelles demonstrated that ceramide’s effect on apoptosis induction requires its presence at mitochondria (Birbes et al, 2001) where it creates ceramide‐rich macrodomains that enhance Bak or Bax oligomerization (Lee et al, 2011). Finally, mistargeting of the ceramide transporter, CERT, to mitochondria enhances Bax‐dependent MOMP and this requires its ceramide transfer domain (Jain et al, 2017). Here, we propose that MAMs serve this “targeting” role to facilitate ceramide transfer to the OMM, and this is frequently disrupted in REL tumor cells.
Since MAMs serve as physiological regulators of apoptosis by maintaining an OMM lipid milieu that supports Bak and/or Bax oligomerization (Renault & Chipuk, 2014), MAM reductions may comprise an adaptive response providing a novel multidrug resistance mechanism (Fig 7). Additional work defining the lipid requirements downstream of ceramide that support MOMP, and how they are disrupted in cancer, is warranted. Alterations in MAMs have increasingly been linked to diverse pathological states, including cardiomyocyte recovery after reperfusion injury (Paillard et al, 2013), neurodegeneration (Zampese et al, 2011; Area‐Gomez et al, 2012; Cali et al, 2013; Hedskog et al, 2013; Ottolini et al, 2013), obesity‐related diabetes (Rieusset, 2011; Arruda et al, 2014), and systemic lipid homeostasis (Anastasia et al, 2021). Importantly, our attenuated MOMP model proposes that multidrug resistance does not reflect an absence of death signals but their insufficiency to trigger MOMP. Investigational cancer drugs are often tested in patients with multidrug resistance. For drugs that do not induce a tumor response, we cannot differentiate those that engaged their target to liberate death‐activating signals that remain subthreshold for MOMP versus those that did not. This may lead to drugs that might have clinical efficacy in other settings being abandoned. Finally, recognition of this phenotype can facilitate the development of tools to measure ER–mitochondria interactions for clinical use in predicting therapy resistance and provide a novel framework for testing interventions to prevent emergent resistance or restore mitochondrial competence to resistant cancers.
Figure 7. MAM‐mediated mitochondrial resistance model.
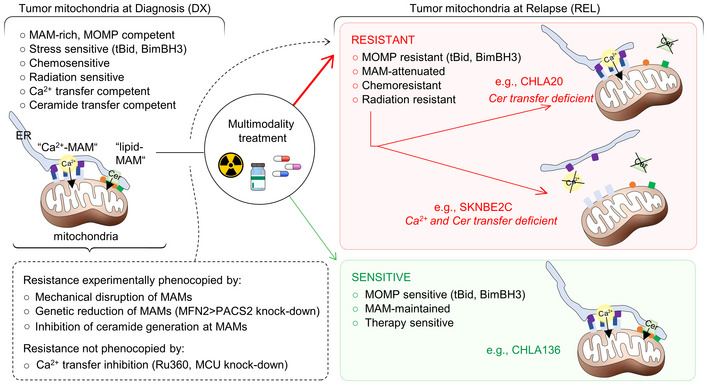
DX neuroblastoma cells have MAM‐replete mitochondria that are MOMP competent and sensitive to therapeutic stress. REL tumor cells have reduced MAMs (by number and/or proximity) and are relatively multidrug resistant due to attenuated MOMP (red box). Rare tumors at relapse retain MOMP competence and relative therapy sensitivity (green box). Ca2+ transfer may be reduced but is not essential for the multidrug‐resistant phenotype. Reduced transfer of sphingolipids such as ceramides (Cer) at MAMs is common in REL cells and contributes to attenuated MOMP via altered Bak/Bax oligomerization in response to stress.
Materials and Methods
Cell lines
Cell lines were obtained from the COG/ALSF Childhood Cancer Repository (www.CCcells.org) and grown in IMDM Media 12440 (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal bovine serum, 2 mM l‐glutamine, 1% ITS, 100 U/ml of penicillin, and 100 μg/ml gentamycin. Those obtained at diagnosis (DX) were kept physically separate from those at relapse (REL) to avoid cross‐contamination. All were subjected to STR identity and pathogen testing every 4–6 months. Culture conditions were 37°C in a humidified atmosphere of 5% CO2. Cell lines were interrogated for cancer gene mutations using the FoundationOne CDx assay (Foundation Medicine) to confirm TP53, ALK, and additional cancer gene mutation status, including variant allele frequencies.
Isolation of heavy‐membrane, purified mitochondria, and MAM fractions
Heavy‐membrane fractions were obtained by rupturing cells in isolation buffer A (250 mM Sucrose, 20 mM HEPES, 1 mM DTT, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, and 1.5 mM MgCl2) with protease inhibitor cocktail by 20 strokes in a 2‐ml glass Dounce homogenizer, followed by removal of debris and nuclei by centrifugation at 800 g for 10 min and then 1,050 g for 10 min at 4°C. Mitochondria‐enriched fractions were collected by centrifuging the supernatant of the second spin at 12,000 g for 10 min at 4°C. To visualize MAM content, heavy‐membrane fractions were resuspended in 2 ml Isolation Medium (250 mM Mannitol, 5 mM HEPES pH 7.4, 0.5 mM EGTA, and 0.1% BSA) with freshly added protease inhibitors and slowly layered onto 8 ml of 30% Percoll gradient prepared with Gradient Buffer (225 mM Mannitol, 25 mM HEPES pH 7.5, 1 mM EGTA, and 0.1% BSA) in an Ultra‐Clear Beckman Centrifuge Tube, and spun at 95,000 g for 60 min to resolve MAM from more pure mitochondria, as described (Annunziata et al, 2013).
To prepare purified mitochondria, we used Magnetic Activated Cell Sorting (MACS) according to the MidiMACS™ Starting Kit (LS) protocol (130‐042‐301, Mitenyi Biotec, Germany). Cells were collected in cold PBS and resuspended in cold Buffer A with protease inhibitor at 10 million cells/ml. After 20–25 strokes of homogenization using a 7 ml Dounce homogenizer, 1 ml of cell lysate was mixed with 9 ml of ice‐cold 1× Separation Buffer (130‐091‐221, Mitenyi Biotec) and incubated with 50 μl anti‐TOM22 microbeads at 4°C on a shaker for 1 h. The mixture was passed stepwise through an LS column (130‐042‐401, Mitenyi Biotec), pre‐rinsed with 3 ml of 1× Separation Buffer, and installed in the magnetic field of the MidiMACS Separator (130‐042‐302, Mitenyi Biotec). After washing three times with 3 ml of 1× Separation Buffer, the column was removed and magnetically labeled mitochondria were flushed into a collection tube using 1.5 ml of 1× Separation Buffer. Purified mitochondria were used immediately for analysis or centrifuged at 13,000 g for 2 min at 4°C and stored in 100 μl storage buffer on ice.
Mitochondrial profiling
Heavy‐membrane or purified mitochondria fractions were resuspended in buffer C [10 mM Tris‐MOPS (pH 7.4), 1 mM KH2PO4, 10 mcM Tris‐EGTA, 5 mM glutamate, 2 mM malate, and 125 mM KCl]. Mitochondria (50 μl of 1 μg/μl protein) were treated with caspase‐8‐cleaved tBid from 5 to 500 nM (882‐B8‐050, R&D Systems, Minneapolis, MN), Bim‐BH3 from 10 to 10,000 nM (Ac‐DMRPEIWIAQELRRIGDEFNAYYARR‐amide; New England Peptide, Gardner, MA), or 300 μM BidAltBH3 (Ac‐EDIIRNIARHAAQVGASMDR‐amide; New England Peptide, Inc.). Reactions were incubated at 37°C for 30 mins and spun at 12,000 g for 10 min at 4°C: 10 μl of the supernatant (SN) and the 10 μl pellet (mitochondria; MITO) fractions were resuspended in 50 μl of 0.1% Triton X in PBS in duplicate in Quantikine, Human Cyto C Immunoassay 96‐well plates followed by ELISA detection (R&D Systems, Minneapolis, MN). The fractional release of mitochondrial cytochrome c was calculated for each condition as [mean intensitySN/(mean intensityMITO + mean intensitySN)]. We report percent cytochrome c release normalized to the non‐specific release induced by DMSO as [(fractional releaseRX ‐ fractional releaseDMSO)/(1‐fractional releaseDMSO)]*100. To test GW4869 (D1692, Sigma Aldrich), the drug (0–250 nM) was pre‐incubated with mitochondria for 30 min at 30°C prior to tBid (pre‐incubation), or added with tBid after 30 min pre‐incubation (control). Experiments with < 30% cytochrome c release to DMSO and BidAltBH3 peptide were analyzed.
Bak and Bax oligomerization
Mitochondria (1 μg/μl) suspended in buffer A/C were treated with tBid (0–250 nM) or DMSO for 30 min at 37°C followed by a 30 min incubation with 0.9 mM of 1,6‐bismaleimidohexane (Pierce, #22330) at RT to crosslink oligomers, and centrifuged at 12,000 g (4°C) to pellet mitochondria. Supernatant was removed and the pellet dissolved in NuPage loading buffer (Invitrogen) and immunoblotted for Bak and Bax.
Cytotoxicity
Cells were seeded into Corning® 96‐well Flat Clear Bottom White Polystyrene TC‐treated luminescent microplates (3610, Corning, Corning, NY) in triplicate at a density of 10,000 cells/well and allowed a 24‐h recovery period, then treated with 100 μl of control Gibco™ IMDM full culture media (12440‐053, Gibco by Life Technologies, Carlsbad, CA). ABT‐737 was tested from 1 nM to 200 µM, added to IMDM culture media for 48 h. Chemotherapy exposure was 72 h, concentrations tested were as follows: etoposide (341205‐25MG, EMD Millipore, Billerica, MA) 10 nM to 10 µM, cisplatin (63323‐103‐51, Fresenius Kabi, Lake Zurich, IL) 0.5 nM to 75 µM, carboplatin (216100‐25MG, EMD Millipore) 6 nM to 10 µM, doxorubicin (5927S, Cell Signaling Technology, Danvers, MA) 0.5 nM to 10 µM, and mafosfamide (M110300 Toronto Research Chemicals, Inc., Toronto, Canada) 2 nM to 75 µM. ALK inhibitors [crizotinib (C‐7900, LC Laboratories, Woburn, MA), ceritinib (C‐2086, LC Laboratories), and lorlatinib (5640, Tocris Bioscience, Minneapolis, MN)] were tested from 0.01 nM to 10 µM and assessed after 5 days. Cells were irradiated on a Cs‐137 Gammacell 40 irradiator S/N 186 (Nordion Ltd, Kanata, Ontario, Canada) at a dose rate of 1.3 cGy/s from 0.5 to 5 Gy and assessed after 7 days. Viability was assessed using CellTiter‐Glo® Assay according to the manufacturer’s instructions (Promega, Madison, WI; #G7571). A Synergy™ 2 microplate reader with Gen5™ software (BioTek Instruments, Winooski, VT) was used to measure luminescence. For each plate, mean relative luminescence was calculated from at least three technical replicates, normalized to control samples. Non‐linear regression algorithms in Prism software (GraphPad8) were used to calculate IC50 values.
Real‐time qPCR detection of mtDNA
mtDNA content was quantified by qPCR to define the mtDNA/nucDNA ratio using each of the two mitochondrial genome (MT‐C01 and MT‐ATPase6) and two nuclear genome (CFAP410 and MTTP) genes. Assays were run in triplicate using Taqman Gene Expression Mastermix (4369016, Applied Biosystems) and the following primer/probes from Invitrogen: MT‐CO1 (Hs02596864_g1), MT‐ATPase6 (Hs02596862_g1), CFAP410 (Hs00223770_cn), and MTTP (HS0405900_cn). mtDNA/nucDNA ratio was determined as the mean and SD of all biological replicates.
Citrate synthase measurements
Mitochondrial mass was assessed by measuring citrate synthase activity from 4 million whole cells normalized to protein input (U/mg) in triplicate from three biological replicates using the Citrate Synthase Assay kit (CS0720, Sigma‐Aldrich, St, Louis, MO).
Electron microscopy
Intact cells for 2D electron microscopic morphometry were fixed with 2% glutaraldehyde with 1% tannic acid in 0.1 M sodium cacodylate buffer, pH 7.4, overnight at 4°C. After buffer washes, the samples were post‐fixed in 2.0% osmium tetroxide with 1.5% K3Fe(CN)6 for 1 h at room temperature, and rinsed in ddH2O prior to en bloc staining with 2% uranyl acetate. After dehydration through a graded ethanol series, the tissue was infiltrated and embedded in EMbed‐812 (Electron Microscopy Sciences, Fort Washington, PA). Thin 70 nm sections were stained with uranyl acetate and SATO lead and examined with a JEOL 1010 electron microscope (JEOL, Peabody, MA) at 70 kV fitted with a Hamamatsu digital camera and AMT Advantage NanoSprint500 software. MAM and mitochondria characteristics were obtained from TEM images at 50,000× magnification analyzed blinded in Image J after hand masking the mitochondria perimeter and ER interface. Analysis of the ER–mitochondria interfaces were extracted with a custom macro (Bartok et al, 2019) available at: http://sites.imagej.net/MitoCare/ that quantifies ER interface metrics binned within a 10, 25, 50, or 100 nm gap distance from the mitochondria (Csordas et al, 2006).
Calcium transfer
Mitochondria‐targeted calcium reporter 4mtGCamp6f was electroporated into cells with Amaxa Nucleofactor (Lonza, Basel, Switzerland). Cells recovered in full IMDM media for 24 h and were plated on poly‐D lysine covered slides, washed three times with Ca2+‐free HBSS medium (Life Technologies Inc., Grand Island, NY), and loaded with 1 μM Fura 2‐AM (Life Technologies Inc., Eugene, OR) cytoplasmic calcium indicator in the presence of 0.03% Pluronic F127 and 100 μM sulfinpyrazone at 35°C for 30 min. Fura‐2AM‐loaded cells were washed with Ca2+‐free HBSS medium and mounted into microscope loading chambers on a thermostat‐controlled (37°C) stage of Olympus IX70 inverted microscope with LambdaDG4 wavelength‐switch xenon light source (Sutter, Novato, CA) optimized by custom Spectralyzer software. Fura2 fluorescence was recorded with 340/11 nm and 380/11 nm excitation while 485/15 nm excitation was used for the mitochondrial matrix‐targeted GCaMP6f using dual‐band dichroic and emission filters (Chroma, customized set 59022). Images were collected on a high quantum efficiency cooled CCD camera every 0.25 s. [Ca2+]c measurements with fura2 were calibrated in vitro by adding 1 mM CaCl2, followed by 10 mM EGTA/Tris, pH 8.5. Basal fluorescent levels were measured for 120 s, after which calcium release was evoked by addition of 100 μM carbachol. To determine [Ca2+], the following formula was used:
where K d is the Ca2+ dissociation constant (0.224 µM), R is the ratio of the fluorescence intensities at 340/380 nm excitation, R min and R max are the fluorescence ratios in Ca2+‐free and Ca2+‐saturated conditions, respectively, and Sf2/Sb2 is the ratio of fluorescence intensities measured at 380 nm excitation in Ca2+‐free (f)/Ca2+‐saturated (Ca2+ bound, b) conditions (Csordas & Hajnoczky, 2001). [Ca2+] is measured over regions of interest (cells) and cells with cytosolic distributed GCaMP6f were excluded from analyses. To determine coupling time, the delay between ½ max [Ca2+]c and ½ max [Ca2+]m was calculated for each cell.
Lipidomics
Modified Bligh and Dyer lipid extraction was used. Cell pellets were added to 900 μl ice‐chilled chloroform/methanol (1:2; v/v) and vortexed 15 s, then incubated in 4°C on a mixer (300 rpm) for 1 h. After agitation, 300 μl of ice‐chilled chloroform and 250 μl of ice‐chilled MQ water were added sequentially, followed by vortex for 15 s and centrifugation at 9,000 rpm for 2 min to separate the phase. Bottom organic phases were collected, and the aqueous phases were re‐extracted with 500 μl of chilled chloroform. Collected organic phases were dried in a vacuum concentrator and stored lyophilized at −80°C. Samples were solubilized in 100 μl chloroform/methanol (1:1; v/v) and analyzed using a Waters Acquity UPLC I class coupled with a Waters Synapt G2‐Si (Waters Corp, Milford, MA). Lipids were separated in reverse phase using an Acquity UPLC HSS T3 1.8 μm column with the following conditions: mobile phase A (water:acetronitrile, 40:60, with 10 μM ammonium acetate and 0.1% acetic acid), B (water:acetonitrile:isopropanol:acetic acid, 5:10:85:0.1, with 10 μM ammonium acetate and 0.1% acetic acid); flow rate of 300 μl/min; injection volume of 5 μl; column temperature at 55°C; 20% B for 1.5 min; linear change to 100% B over next 16.5min; and maintained at 100% B for 3 min. Then, the gradient was reverted back to initial state 20% B for 1 min then held for next 1 min at 20% B. QC samples were injected prior to, and after every 5 samples, to monitor the stability of the instrument. Samples were run under untargeted positive and negative electrospray ionization (ESI) modes in a data‐independent manner (MSE mode). The following ESI conditions were used: for positive, capillary at 2 kV, sampling cone at 35 V, source temperature at 100°C, desolvation gas at 500 l/h, and nebulizer at 6.5 bar; and for negative, capillary at 2.2 kV, sampling cone at 40 V, source temperature at 80°C, desolvation gas at 500 l/h, and nebulizer at 6.5 bar. For lock mass correction, leucine enkephain was used at 1 ng/ml in acetonitrile/water (1:1, v/v) with 0.1% formic acid and at a flow rate of 10 μl/min. The low collision energy was set to 4 eV and high collision energy was set between 25 and 40 eV for both positive and negative modes. Raw data were converted into ABF format using Reifycs Analysis Base File Converter, then used in MS‐Dial (ver. 4.60) for peak picking and retention time alignment using default settings. Lipid species were manually verified and named using Lipid Maps abbreviations. The intensities were normalized with total ionic current. These species were exported into R (ver. 4.0.3) to calculate the concentration using the known concentration of spiked internal standards.
Plasmids, retroviral constructs, and reagents
A synthetic mitochondria–ER linker was used to recouple ER and mitochondria, as described (Csordas et al, 2006). Lentiviral shRNAs to MFN2 (RHS5086‐EG9927‐GIPZ MFN2), PACS2 (RHS5086‐EG23241‐GIPZ PACS2) and GIPZ non‐silencing lentiviral shRNA control were purchased from GE Health Dharmacon, Inc., Lafayette, CO. 4mtGCamp6f was provided by Dr. Diego De Stefani. Primary antibodies were as follows: Bak (3814, Cell Signaling Technologies, Danvers, MA), Bax (2774, Cell Signaling), Bap31 (ab109304, Abcam Inc., Cambridge, MA), β‐tubulin (T8328, Sigma‐Aldrich, St. Louis, MO), calnexin (C7617, Sigma‐Aldrich), FACL4 (22401‐1‐AP, Proteintech, Rosemond, IL), Gapdh (2118, Cell Signaling), γ‐H2AX (NB100‐384, Novus, Littleton, CO), Mfn2 (ab56889, Abcam Inc.), Mcu (HPA016480, Sigma‐Aldrich), noxa (OP180, Calbiochem/EMD Chemicals, San Diego, CA), PACS2 (ab129402, Abcam Inc.), p53 (sc‐126, Santa Cruz Biotechnology, Dallas, TX), p21 (sc‐6246, Santa Cruz Biotechnology), Tmx1 (256‐270, SAB1105403, Sigma‐Aldrich), Tomm40 (18409‐1‐AP, Proteintech), and Vdac1 (D73D12, Cell Signaling). To quantify protein abundance, densitometry was performed using Image J. The intensity of each band was normalized to its respective loading control for comparison.
DNA damage detection
A total of 10,000 cells were plated onto poly‐L lysine‐coated microscope slides, allowed to adhere 24 h, and irradiated at 2 Gy for 1 h on a Cs‐137 Gammacell 40 irradiator S/N 186 (Nordion Ltd, Kanata, Ontario, Canada) at a dose rate of 1.3 cGy/s. Cells were washed once with warm PBS and fixed with pre‐warmed paraformaldehyde for 10 min, permeabilized with 2% Triton‐PBS for 5 min at 4°C, washed twice with PBS‐5% triton, and blocked for 10 min in Duolink blocking solution. Cells were then incubated for 1 h at 37°C with γ‐H2AX primary antibody (NB100‐384, Novus Biologicals, Centennial, CO) at 1:1,000 dilution, washed twice with PBS‐T, and incubated at 37°C with AlexaFluor goat anti‐rabbit 488 (ab150077; Abcam Inc., Cambridge, MA) at 1:500 dilution for 45 min. After washing four times with PBS‐T, they were stained with VectaShield DAPI staining, covered with a microscope coverslip, and analyzed using a Leica DMR fluorescent microscope at 40× magnification, and the number of γ‐H2AX foci/cell quantified.
Statistical analyses
Statistical comparisons for mitochondrial size, roundness, circularity, MAM content per mitochondria, and γ‐H2AX foci were performed with the Mann–Whitney U test, two tailed; comparisons for cytochrome c release, Ca2+ transfer coupling time, lipid content, mtDNA content, and mass were performed with the Student’s t‐test, independent values, two tailed; and comparisons for the proportion of mitochondria with diverse tether numbers were performed using the Chi‐Square test. For all, significance was defined as P < 0.05, trend P < 0.10, and non‐significant as P ≥ 0.10.
Author contributions
Jorida Çoku: Conceptualization; Data curation; Formal analysis; Validation; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review and editing. David M Booth: Software; Validation; Investigation; Visualization; Methodology; Writing—review and editing. Jan Skoda: Formal analysis; Validation; Investigation; Visualization; Writing—review and editing. Madison C Pedrotty: Data curation; Formal analysis; Investigation. Jennifer Vogel: Data curation; Formal analysis; Investigation; Methodology; Writing—review and editing. Kangning Liu: Conceptualization; Data curation; Formal analysis; Supervision; Validation; Investigation; Methodology; Writing—review and editing. Annette Vu: Data curation; Formal analysis; Investigation; Methodology; Writing—review and editing. Erica L Carpenter: Data curation; Formal analysis; Investigation; Visualization; Methodology; Writing—review and editing. Jamie C Ye: Data curation; Formal analysis; Investigation; Methodology. Michelle A Chen: Data curation; Formal analysis; Investigation; Methodology. Peter Dunbar: Data curation; Formal analysis; Validation; Investigation; Visualization; Methodology. Elizabeth Scadden: Data curation; Formal analysis; Investigation; Methodology; Writing—review and editing. Taekyung D Yun: Validation; Investigation; Visualization; Methodology; Writing—review and editing. Eiko Nakamaru‐Ogiso: Data curation; Formal analysis; Supervision; Validation; Investigation; Visualization; Methodology; Writing—review and editing. Estela Area‐Gomez: Conceptualization; Funding acquisition; Investigation; Methodology; Project administration; Writing—review and editing. Yimei Li: Data curation; Software; Formal analysis; Validation; Writing—review and editing. Kelly C Goldsmith: Data curation; Formal analysis; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—review and editing. C Patrick Reynolds: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review and editing. Gyorgy Hajnoczky: Conceptualization; Data curation; Formal analysis; Funding acquisition; Validation; Investigation; Visualization; Methodology; Project administration; Writing—review and editing. Michael D Hogarty: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
JC, DMB, JV, CPR, JS, GH, EA‐G, and MDH designed the experiments; CPR provided tumor models and model validation; JC, DMB, KCG, MCP, JV, KL, JCY, MAC, ES, PD, EN‐O, ELC, JS, and AV conducted the experiments and acquired data related to ER–mitochondria connectivity, signaling, and stress response; JC, DMB, and GH conducted the experiments and acquired data related to calcium transfer; TDY and EA‐G conducted lipidomic experiments; YL performed statistical analyses; JC, DMB, MCP, JV, KL, JCY, ELC, ES, AV, CPR, JS, GH, TDY, EA‐G, and MDH analyzed the data; and JC, JS, DMB, CPR, KCG, GH, EA‐G, and MDH wrote and edited the manuscript.
Supporting information
Appendix
Dataset EV1
Acknowledgements
This study is dedicated to the memory of our dear friend and co‐author Madison Pedrotty. We thank Renata Sano (Children’s Hospital of Philadelphia) for stimulating discussions related to this work; Ocean Malka, Rohan Vemu, and Kiera Patton for technical assistance; the Childhood Cancer Repository powered by Alex’s Lemonade Stand Foundation (www.cccells.org) for tumor models; Diego De Stefani for provision of the GCamp6f plasmid; John Maris for cancer gene sequencing data; and the children and families providing tumor samples for research via the Children’s Oncology Group (COG). This study was supported by NIH CA198430, St. Baldrick’s Foundation, and CURE Childhood Cancer Foundation (to MDH); Alex’s Lemonade Stand Foundation (to EAG and MDH); NIH CA216254 (to GH); and the Czech Science Foundation (No. GJ20‐00987Y) to JS. The Metabolomics Workbench repository for archiving the lipidomic dataset is supported by NIH grant U2C‐DK119886.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
The EMBO Journal (2022) 41: e108272.
Data availability
This study includes lipidomics data that are available for public access at the NIH Common Fund's National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench (https://www.metabolomicsworkbench.org), where it has been assigned Study ID ST002054 (Sur et al, 2016).
References
- Anastasia I, Ilacqua N, Raimondi A, Lemieux P, Ghandehari‐Alavijeh R, Faure G, Mekhedov SL, Williams KJ, Caicci F, Valle G et al (2021) Mitochondria‐rough‐ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep 34: 108873 [DOI] [PubMed] [Google Scholar]
- Annunziata I, Patterson A, d'Azzo A (2013) Mitochondria‐associated ER membranes (MAMs) and glycosphingolipid enriched microdomains (GEMs): isolation from mouse brain. J vis Exp 73: e50215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area‐Gomez E, del Carmen Lara Castillo M, Tambini MD, Guardia‐Laguarta C, de Groof AJC, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL et al (2012) Upregulated function of mitochondria‐associated ER membranes in Alzheimer disease. EMBO J 31: 4106–4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda AP, Pers BM, Parlakgul G, Guney E, Inouye K, Hotamisligil GS (2014) Chronic enrichment of hepatic endoplasmic reticulum‐mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 20: 1427–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartok A, Weaver D, Golenár T, Nichtova Z, Katona M, Bánsághi S, Alzayady KJ, Thomas VK, Ando H, Mikoshiba K et al (2019) IP3 receptor isoforms differently regulate ER‐mitochondrial contacts and local calcium transfer. Nat Commun 10: 3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer TM, Murphy E (2020) Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ Res 126: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL et al (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbes H, El Bawab S, Hannun YA, Obeid LM (2001) Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. FASEB J 15: 2669–2679 [DOI] [PubMed] [Google Scholar]
- Bresler S, Weiser D, Huwe P, Park J, Krytska K, Ryles H, Laudenslager M, Rappaport E, Wood A, McGrady P et al (2014) ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26: 682–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- Cali T, Ottolini D, Brini M (2013) Calcium and endoplasmic reticulum‐mitochondria tethering in neurodegeneration. DNA Cell Biol 32: 140–146 [DOI] [PubMed] [Google Scholar]
- Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148: 988–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CR, Janne PA (2013) The quest to overcome resistance to EGFR‐targeted therapies in cancer. Nat Med 19: 1389–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonghaile TN, Sarosiek KA, Vo T‐T, Ryan JA, Tammareddi A, Moore VDG, Deng J, Anderson KC, Richardson P, Tai Y‐T et al (2011) Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334: 1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DT, Wales TE, McHenry MW, Engen JR, Walensky LD (2020) Site‐dependent cysteine lipidation potentiates the activation of proapoptotic BAX. Cell Rep 30: 3229–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan AD, Smith NA, Sandow JJ, Kapp EA, Rustam YH, Murphy JM, Brouwer JM, Bernardini JP, Roy MJ, Wardak AZ et al (2020) BAK core dimers bind lipids and can be bridged by them. Nat Struct Mol Biol 27: 1024–1031 [DOI] [PubMed] [Google Scholar]
- Cripe LD, Uno H, Paietta EM, Litzow MR, Ketterling RP, Bennett JM, Rowe JM, Lazarus HM, Luger S, Tallman MS (2010) Zosuquidar, a novel modulator of P‐glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo‐controlled trial of the Eastern Cooperative Oncology Group 3999. Blood 116: 4077–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Hajnoczky G (2001) Sorting of calcium signals at the junctions of endoplasmic reticulum and mitochondria. Cell Calcium 29: 249–262 [DOI] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174: 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T, Hajnoczky G (2010) Imaging interorganelle contacts and local calcium dynamics at the ER‐mitochondrial interface. Mol Cell 39: 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Weaver D, Hajnoczky G (2018) Endoplasmic reticulum‐mitochondrial contactology: structure and signaling functions. Trends Cell Biol 28: 523–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219 [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A (2007) BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT‐737 and conventional chemotherapeutic agents. Cancer Cell 12: 171–185 [DOI] [PubMed] [Google Scholar]
- Ganesan V, Perera MN, Colombini D, Datskovskiy D, Chadha K, Colombini M (2010) Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 15: 553–562 [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu H‐C, Kim H, Cheng E‐Y, Tjandra N et al (2008) BAX activation is initiated at a novel interaction site. Nature 455: 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith KC, Gross M, Peirce S, Luyindula D, Liu X, Vu A, Sliozberg M, Guo R, Zhao H, Reynolds CP et al (2012) Mitochondrial Bcl‐2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res 72: 2565–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Madesh M, Pacher P (2000) Control of apoptosis by IP(3) and ryanodine receptor driven calcium signals. Cell Calcium 28: 349–363 [DOI] [PubMed] [Google Scholar]
- Hata AN, Engelman JA, Faber AC (2015) The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov 5: 475–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedskog L, Pinho CM, Filadi R, Ronnback A, Hertwig L, Wiehager B, Larssen P, Gellhaar S, Sandebring A, Westerlund M et al (2013) Modulation of the endoplasmic reticulum‐mitochondria interface in Alzheimer's disease and related models. Proc Natl Acad Sci USA 110: 7916–7921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds MG, Smits C, Fredericks‐Short R, Risk JM, Bailey M, Huang DC, Day CL (2007) Bim, Bad and Bmf: intrinsically unstructured BH3‐only proteins that undergo a localized conformational change upon binding to prosurvival Bcl‐2 targets. Cell Death Differ 14: 128–136 [DOI] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13: 714–726 [DOI] [PubMed] [Google Scholar]
- Hoppins S, Nunnari J (2012) Cell Biology. Mitochondrial dynamics and apoptosis–the ER connection. Science 337: 1052–1054 [DOI] [PubMed] [Google Scholar]
- Howatson AF, Ham AW (1955) Electron microscope study of sections of two rat liver tumors. Cancer Res 15: 62–69 [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun LU, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman K, Johnson D et al (2015) Non‐genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 162: 1271–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Beutel O, Ebell K, Korneev S, Holthuis JC (2017) Diverting CERT‐mediated ceramide transport to mitochondria triggers Bax‐dependent apoptosis. J Cell Sci 130: 360–371 [DOI] [PubMed] [Google Scholar]
- Kale J, Osterlund EJ, Andrews DW (2018) BCL‐2 family proteins: changing partners in the dance towards death. Cell Death Differ 25: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R, Shaw AT, Khan TM, Mino‐Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C et al (2012) Mechanisms of acquired crizotinib resistance in ALK‐rearranged lung Cancers. Sci Transl Med 4: 120ra117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshelava N, Seeger RC, Reynolds CP (1997) Drug resistance in human neuroblastoma cell lines correlates with clinical therapy. Eur J Cancer 33: 2002–2006 [DOI] [PubMed] [Google Scholar]
- Keshelava N, Seeger RC, Groshen S, Reynolds CP (1998) Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res 58: 5396–5405 [PubMed] [Google Scholar]
- Kim H, Rafiuddin‐Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2006) Hierarchical regulation of mitochondrion‐dependent apoptosis by BCL‐2 subfamilies. Nat Cell Biol 8: 1348–1358 [DOI] [PubMed] [Google Scholar]
- Lee H, Rotolo JA, Mesicek J, Penate‐Medina T, Rimner A, Liao W‐C, Yin X, Ragupathi G, Ehleiter D, Gulbins E et al (2011) Mitochondrial ceramide‐rich macrodomains functionalize Bax upon irradiation. PLoS One 6: e19783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL (2009) Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 1787: 1395–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Rosen N, Solit DB (2013) Tumor adaptation and resistance to RAF inhibitors. Nat Med 19: 1401–1409 [DOI] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A (2013) Mechanisms of resistance to therapies targeting BRCA‐mutant cancers. Nat Med 19: 1381–1388 [DOI] [PubMed] [Google Scholar]
- Martino Adami PV, Nichtova Z, Weaver DB, Bartok A, Wisniewski T, Jones DR, Do Carmo S, Castano EM, Cuello AC, Hajnoczky G et al (2019) Perturbed mitochondria‐ER contacts in live neurons that model the amyloid pathology of Alzheimer's disease. J Cell Sci 132: jcs229906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas‐Kogan D, Gerbing RB, London WB, Villablanca JG (2009) Long‐term results for children with high‐risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13‐cis‐retinoic acid: a children's oncology group study. J Clin Oncol 27: 1007–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero J, Sarosiek K, DeAngelo J, Maertens O, Ryan J, Ercan D, Piao H, Horowitz N, Berkowitz R, Matulonis U et al (2015) Drug‐induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 160: 977–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morad SA, Cabot MC (2013) Ceramide‐orchestrated signalling in cancer cells. Nat Rev Cancer 13: 51–65 [DOI] [PubMed] [Google Scholar]
- Moulis M, Grousset E, Faccini J, Richetin K, Thomas G, Vindis C (2019) The multifunctional sorting protein PACS‐2 controls mitophagosome formation in human vascular smooth muscle cells through mitochondria‐ER contact sites. Cells 8: 638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2: a001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolini D, Cali T, Negro A, Brini M (2013) The Parkinson disease‐related protein DJ‐1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum‐mitochondria tethering. Hum Mol Genet 22: 2152–2168 [DOI] [PubMed] [Google Scholar]
- Paillard M, Tubbs E, Thiebaut P‐A, Gomez L, Fauconnier J, Crola Da Silva C, Teixeira G, Mewton N, Belaidi E, Durand A et al (2013) Depressing mitochondria‐reticulum interactions protects cardiomyocytes from lethal hypoxia‐reoxygenation injury. Circulation 128: 1555–1565 [DOI] [PubMed] [Google Scholar]
- Park JH, Zhuang J, Li J, Hwang PM (2016) p53 as guardian of the mitochondrial genome. FEBS Lett 590: 924–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patergnani S, Giorgi C, Maniero S, Missiroli S, Maniscalco P, Bononi I, Martini F, Cavallesco G, Tognon M, Pinton P (2015) The endoplasmic reticulum mitochondrial calcium cross talk is downregulated in malignant pleural mesothelioma cells and plays a critical role in apoptosis inhibition. Oncotarget 6: 23427–23444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, Nakagawara A, Berthold F, Schleiermacher G, Park JR et al (2015) Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol 33: 3008–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2010) BID, BIM, and PUMA are essential for activation of the BAX‐ and BAK‐dependent cell death program. Science 330: 1390–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault TT, Chipuk JE (2014) Death upon a kiss: mitochondrial outer membrane composition and organelle communication govern sensitivity to BAK/BAX‐dependent apoptosis. Chem Biol 21: 114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault T, Floros K, Elkholi R, Corrigan K‐A, Kushnareva Y, Wieder S, Lindtner C, Serasinghe M, Asciolla J, Buettner C et al (2015) Mitochondrial shape governs BAX‐induced membrane permeabilization and apoptosis. Mol Cell 57: 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieusset J (2011) Mitochondria and endoplasmic reticulum: mitochondria‐endoplasmic reticulum interplay in type 2 diabetes pathophysiology. Int J Biochem Cell Biol 43: 1257–1262 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86: 369–408 [DOI] [PubMed] [Google Scholar]
- Rowland AA, Voeltz GK (2012) Endoplasmic reticulum‐mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13: 607–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, Letai A (2013a) BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 51: 751–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarosiek KA, Ni Chonghaile T, Letai A (2013b) Mitochondria: gatekeepers of response to chemotherapy. Trends Cell Biol 23: 612–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauder CM, Wu X, Saheki Y, Narayanaswamy P, Torta F, Wenk MR, De Camilli P, Reinisch KM (2014) Structure of a lipid‐bound extended synaptotagmin indicates a role in lipid transfer. Nature 510: 552–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139 [DOI] [PubMed] [Google Scholar]
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G (2005) PACS‐2 controls endoplasmic reticulum‐mitochondria communication and Bid‐mediated apoptosis. EMBO J 24: 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Sreekumaran Nair K et al (2016) Metabolomics workbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nuc Acids Res 44: D463–D470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA et al (2007) Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 11: 498–512 [DOI] [PubMed] [Google Scholar]
- Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves‐Neira W, Bouillet P, Villunger A, Adams JM, White E (2005) Key roles of BIM‐driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7: 227–238 [DOI] [PubMed] [Google Scholar]
- Uccini S, Colarossi C, Scarpino S, Boldrini R, Natali PG, Nicotra MR, Perla FM, Mannarino O, Altavista P, Boglino C et al (2006) Morphological and molecular assessment of apoptotic mechanisms in peripheral neuroblastic tumours. Br J Cancer 95: 49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, Letai A (2012) Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151: 344–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ (2006) A stapled BID BH3 helix directly binds and activates BAX. Mol Cell 24: 199–210 [DOI] [PubMed] [Google Scholar]
- Wilson F, Johannessen C, Piccioni F, Tamayo P, Kim J, Van Allen E, Corsello S, Capelletti M, Calles A, Butaney M et al (2015) A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell 27: 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P (2011) Presenilin 2 modulates endoplasmic reticulum (ER)‐mitochondria interactions and Ca2+ cross‐talk. Proc Natl Acad Sci USA 108: 2777–2782 [DOI] [PMC free article] [PubMed] [Google Scholar]