

SUMMARY
Amyotrophic lateral sclerosis (ALS) is a complex disease leading to motor neuron death. Despite heritability estimates of 52%, genome-wide association studies (GWAS) have discovered relatively few loci. We developed a machine learning approach called RefMap which integrates functional genomics with GWAS summary statistics for gene discovery. With transcriptomic and epigenetic profiling of motor neurons derived from induced pluripotent stem cells (iPSCs), RefMap identified 690 ALS-associated genes which represents a 5-fold increase in recovered heritability. Extensive conservation, transcriptome, network, and rare variant analyses demonstrated the functional significance of candidate genes in healthy and diseased motor neurons and brain tissues. Genetic convergence between common and rare variation highlighted KANK1 as a new ALS gene. Reproducing KANK1 patient mutations in human neurons led to neurotoxicity and demonstrated that TDP-43 mislocalization, a hallmark pathology of ALS, is downstream of axonal dysfunction. RefMap can be readily applied to other complex diseases.
eTOC Blurb
Zhang et al. develop a new machine learning method which integrates epigenetic profiling with GWAS summary statistics for gene discovery. Application to ALS identifies 690 risk genes with 5-fold increase in recovered heritability. Leading candidate KANK1 is reproduced in human neurons leading to TDP-43 mislocalization, a hallmark pathology of ALS.
INTRODUCTION
ALS is a fatal and relatively common neurodegenerative disease. The hallmark of the disease is motor neuron loss (Hardiman et al., 2017). 10% of ALS is autosomal dominant, but even for sporadic ALS heritability is estimated to be ~50% (Ryan et al., 2019; Trabjerg et al., 2020). Genome-wide association studies (GWAS) in ALS (van Rheenen et al., 2016, 2021; Nicolas et al., 2018) have identified several risk loci. However, these changes occur in <10% of ALS patients, and thus there are likely to be a large number of ALS risk genes yet to be discovered.
ALS GWAS to date have lost power by considering genetic variants in isolation, whereas in reality, a biological system is the product of a large number of interacting partners (Wang et al., 2011; Li et al., 2019). Moreover, noncoding regulatory regions of the genome have been relatively neglected in efforts to pinpoint the genetic basis of ALS, despite their functional synergy with the coding sequence (Wang et al., 2018; Cooper-Knock et al., 2020). ALS GWAS have demonstrated that missing heritability is distributed throughout noncoding chromosomal regions (van Rheenen et al., 2016; Nicolas et al., 2018). The function of noncoding DNA is often cell-type specific (Heinz et al., 2015). Developments in understanding of cell-type-specific (dys)function in neurological diseases (Lopategui Cabezas, Herrera Batista and Pentón Rol, 2014; Bryois et al., 2020) has created an opportunity to dramatically reduce the search space and so boost power for genetic discovery, by focusing on genomic regions that are functional in the cell type of interest.
Here, we present RefMap (Regional Fine-mapping), a machine learning method for analysis of GWAS summary statistics. RefMap is a hierarchical Bayesian network which performs genome-wide identification of disease-associated genetic variation within active genomic regions, the majority of which are noncoding. RefMap utilizes epigenetic profiling to determine the prior probability of disease-association for each region. ALS is notable for the selective vulnerability of a single cell type, motor neurons (MNs) (Cooper-Knock, Jenkins and Shaw, 2013), which makes it ideally suited to this approach. MNs are difficult to study in post-mortem tissues (Corces et al., 2020) because of their relative sparsity, and so a different approach is needed. We performed exhaustive transcriptomic and epigenetic profiling, including RNA-seq, ATAC-seq, histone ChIP-seq, and Hi-C, of MNs derived from neurologically normal controls via induced pluripotent stem cells (iPSCs). Applying RefMap to ALS GWAS data and molecular profiling of iPSC-derived MNs (Figure 1A) identified 690 ALS-associated genes (RefMap ALS genes), including previous GWAS loci as well as known ALS genes not previously detected by GWAS. This represents a 5-fold increase in recovered SNP-based heritability compared to traditional methods. We replicated RefMap ALS genes in a recent ALS GWAS (van Rheenen et al., 2021). We explored the functional significance of RefMap ALS genes based on a series of orthogonal analyses. Ultimately we determined that RefMap genes suggest that ALS pathogenesis is initiated in the distal axon of diseased MNs. Convergence between ALS-associated common and rare genetic variation highlighted a new ALS gene -- KANK1. Reproducing ALS-associated mutations within KANK1 in human neurons led to toxicity, functional impairment of the distal axon, and TDP-43 mislocalization which is the hallmark pathology of ALS (Neumann et al., 2006).
Figure 1. RefMap identifies ALS risk genes by integrating ALS GWAS data with the molecular profiling of motor neurons.

(A) Schematic of the study design. (1 and 2) We sequenced the transcriptome and epigenome of the iPSC-derived MNs. By integrating (3) ALS GWAS data with functional genomics of MNs, (4) a machine learning model called RefMap was developed to fine-map ALS-associated regions. (5) After linking those identified regions to their regulatory targets, 690 ALS-associated genes were pinpointed. (6) Transcriptome analysis based on iPSC-derived MNs, human tissues, and mouse models, as well as (7) network analysis were performed to demonstrate the functional significance of RefMap ALS genes. (8) CRISPR/Cas9 reproduction of identified ALS-associated mutations experimentally verified the proposed link to neuronal toxicity. The LD heatmap matrix in (4) is visualized in both R2 (red) and D’ (blue) using LDmatrix (https://ldlink.nci.nih.gov/?tab=ldmatrix). cCRE, candidate cis-regulatory element; GO, gene ontology. (B) A region (chr12:112,036,001–112,038,000) around ATXN2 precisely pinpointed by RefMap because of elevated SNP Z-scores as well as enriched epigenetic peaks (ATAC-seq, H3K27ac and H3K4me3 histone ChIP-seq). The output of RefMap is labeled as Q-score. ATAC-seq and ChIP-seq signals are shown in fold change (FC) based on one replicate from sample CS14. See also Figure S1D and Supplemental Note.
RESULTS
Transcriptomic and epigenetic profiling of iPSC-derived motor neurons
To identify genomic regions key to motor neuron function, we performed transcriptomic and epigenetic profiling of iPSC-derived MNs from neurologically normal individuals (Figure S1A; STAR Methods). Consistent with successful differentiation, profiled cells exhibited homogenous expression of lower motor neuron markers (Figure S1B). We prepared RNA-seq (Wang, Gerstein and Snyder, 2009), ATAC-seq (Buenrostro et al., 2015), H3K27ac, H3K4me1, and H3K4me3 ChIP-seq (Creyghton et al., 2010), as well as Hi-C (van Berkum et al., 2010) libraries using two technical replicates and three biological replicates per assay (STAR Methods). Sequencing data were processed and quality control (QC) was performed according to the ENCODE 4 standards (ENCODE Project Consortium et al., 2020) (Table S1; STAR Methods).
MN ATAC-seq peak regions covered only 4.9% of the genome, thereby reducing the search space for ALS-associated genetic variation by >90%. To measure the consistency between distinct motor neuron profiles, we used our RNA-seq dataset to identify promoter regions for highly (>90th centile) and lowly (<10th centile) expressed transcripts. We compared enrichment of ATAC-seq and histone ChIP-seq peak regions, and Hi-C loops in highly versus lowly expressed promoters. Significant enrichment within highly expressed promoters was confirmed for ATAC-seq (P=1.1e-182, odds ratio (OR)=1.9, Fisher’s exact test), H3K27ac ChIP-seq (P=2.0e-57, OR=2.2, Fisher’s exact test), H3K4me1 ChIP-seq (P=8.5e-57, OR=1.9, Fisher’s exact test), H3K4me3 ChIP-seq (P=4.8e-196, OR=2.6, Fisher’s exact test), and Hi-C loops (P=4.0e-14, OR=1.3, Fisher’s exact test) (Figure S1C). Similarly, epigenetic peak regions are enriched in MN Hi-C loops: ATAC-seq (P<1.0e-198, OR=1.9, Fisher’s exact test), H3K27ac ChIP-seq (P<1.0e-198, OR=2.0, Fisher’s exact test), H3K4me1 ChIP-seq (P<1.0e-198, OR=2.0, Fisher’s exact test), and H3K4me3 ChIP-seq (P<1.0e-198, OR=1.7, Fisher’s exact test). These observations confirm that our epigenetic profiling has captured functionally significant regions, and that our epigenetic profiles are internally consistent.
RefMap identifies ALS risk genes
Mismatch between the relatively small number of characterized ALS risk genes and estimates indicating high heritability suggests that a new approach is required to discover ALS-associated genetic variation. We present a machine learning model named RefMap which exploits epigenetic profiling of MNs to reduce the search space and so optimize statistical power for discovery of ALS-associated loci (Figures 1A and S1D, Supplemental Note; STAR Methods). The genome was first divided into linkage disequilibrium (LD) blocks and smaller 1kb regions, and each region was assigned with a disease-association score based on the SNPs within it and their associated GWAS Z-scores. Next, the disease-association score was weighted by an epigenetic score for each region, which was calculated from a linear combination of the relevant MN ATAC-seq and histone ChIP-seq peaks. The final weighted score assigned to every 1kb region is known as a Q-score (STAR Methods). In our study, the Z-scores were calculated based on an ALS GWAS (van Rheenen et al., 2016), including genotyping of 12,577 sporadic ALS patients and 23,475 controls.
Next, we linked ALS-associated regions identified by RefMap to expressed transcripts in MNs (transcript per million (TPM)>1, RNA-seq; STAR Methods). This resulted in 690 ALS-associated genes (Table S2). Among this list, we discovered well-known ALS genes, including C9orf72 (DeJesus-Hernandez et al., 2011) and ATXN2 (Elden et al., 2010) (Figure 1B). Indeed, RefMap genes are enriched with an independently curated list of ALS genes (Eitan et al., 2021) (Table S2) including previous GWAS loci (P=5.2e-03, OR=2.07, Fisher’s exact test), and also with clinically reportable ALS genes (ClinVar) (Landrum et al., 2018) (P=0.03, OR=3.06, Fisher’s exact test). Certain ALS genes, such as UNC13A (Daoud et al., 2010; Diekstra et al., 2012), are missing from the RefMap gene list. We hypothesized that even if there is not an exact overlap between RefMap genes and certain known ALS genes, there might be a functional overlap quantifiable by shared protein-protein interactions (PPIs). To examine this, we mapped RefMap genes (excluding any known ALS genes to produce a gene set hereafter referred to as novel RefMap ALS genes) onto a PPI network (Szklarczyk et al., 2019) (STAR Methods). We discovered that the average path distance between novel RefMap genes and known ALS genes is shorter than expected by chance (curated ALS genes: P=0.015, ClinVar ALS genes: P=0.043, permutation test using 1,000 randomly-selected gene sets of equivalent size; Figure S1E). This demonstrates the functional similarity between genes we identified and known ALS genes.
As a negative control, we randomly shuffled SNP Z-scores and re-derived RefMap genes; in this case there is no overlap between RefMap outputs and known ALS genes. Additional shuffling of epigenetic features disrupted the signal further such that RefMap failed to identify any significant regions. As an additional negative control, we tested for enrichment within a gene list of equivalent size (n=690, hereafter referred to as GWAS-derived genes) derived by taking the nearest gene to GWAS SNPs ranked by P-value. This control list is not significantly enriched with the independently curated ALS gene list (P=0.12, Fisher’s exact test). We also derived a negative control for MN-specific gene expression to check that RefMap genes are not overly dependent on our epigenetic signals. We constructed a gene list of equivalent size (n=690; STAR Methods) associated with expression quantitative trait loci (eQTLs) within human spinal cord (GTEx v7) (Consortium and GTEx Consortium, 2017). Genes associated with spinal cord eQTLs (hereafter referred to as eQTL-derived genes) are not significantly enriched with the independently curated ALS gene list (P=0.73, Fisher’s exact test). Notably, applying RefMap to a recent ALS GWAS dataset including 29,612 ALS patients and 122,656 controls (van Rheenen et al., 2021) replicated the vast majority of RefMap ALS genes (n=585, 84.8%).
As benchmarking, we applied three established approaches for genetic discovery based on GWAS summary statistics, including MAGMA (de Leeuw et al., 2015), Pascal(Lamparter et al., 2016) and PAINTOR(Kichaev et al., 2014), to the same GWAS dataset (STAR Methods). After multiple testing correction, MAGMA and Pascal identified 10 and 5 genes, respectively, both including C9orf72 (P<2.76e-06; Figure S2A, Table S3). Unlike MAGMA and Pascal, PAINTOR includes the capacity to integrate epigenetic annotations. Despite this, PAINTOR identified only two genes: MOB3B and LOC105376001 (Figure S2A, Table S3). In contrast to RefMap ALS genes, genes identified by these three methods do not consist of a significant proportion of the curated ALS genes or of the ClinVar ALS genes (Figure S2A).
We finally used linkage disequilibrium score regression (LDSC) (Finucane et al., 2015) to examine the SNP-based heritability partitioned within RefMap ALS genes (STAR Methods). We discovered that RefMap ALS genes contain 36% of heritability, compared to 6% for genome-wide significant loci, representing a 5-fold increase in the recovered heritability. This is also significantly higher than the 6% of heritability contained within eQTL-derived genes.
Conservation analysis demonstrates the functional importance of RefMap genes
We performed conservation analysis to examine the functional significance of RefMap ALS genes. Compared to all protein-coding genes, RefMap genes are significantly haploinsufficient based on their haploinsufficiency (HI) scores (Huang et al., 2010) (P=2.59e-19, one-sided Wilcoxon rank-sum test; Figure 2A). RefMap genes are intolerant to loss-of-function (LoF) mutations as measured by the LoFtool score (Fadista et al., 2017) (P=2.28e-04, one-sided Wilcoxon rank-sum test; Figure 2B), and to other mutation types as measured by the RVIS score (Petrovski et al., 2013) (P=8.08e-13, one-sided Wilcoxon rank-sum test; Figure 2C); these tests were performed within the ExAC dataset (Lek et al., 2016). Within the larger gnomAD dataset (v2.1), RefMap genes are intolerant to LoF mutations as measured by the o/e score (Karczewski et al., 2020) (P=4.08e-10, one-sided Wilcoxon rank-sum test; Figure 2D). Taken together, these results support the functional importance of RefMap ALS genes and suggest that genetic variation identified by RefMap is likely to be toxic if it leads to altered expression/function of these genes.
Figure 2. RefMap genes are haploinsufficient and intolerant to loss of function.
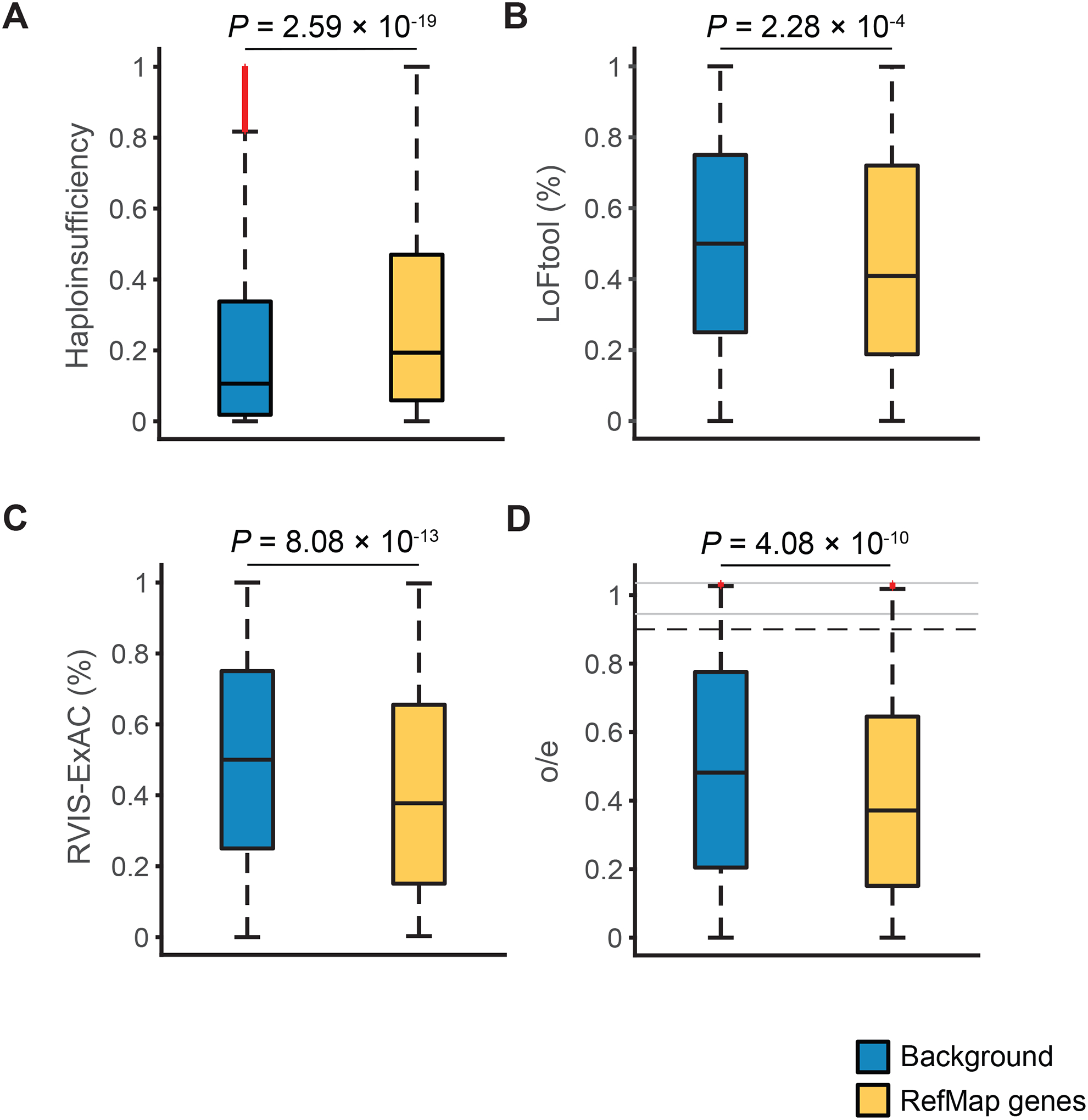
(A-D) Comparison of haploinsufficiency score (A), LoFtool percentile (B), RVIS-ExAC percentile (C), and o/e score (D) between RefMap genes and all protein-coding genes in the background transcriptome. Comparison was performed using the one-sided Wilcoxon rank-sum test. The bottom and top of the boxes indicate the first and third quartiles, respectively, where the black line between indicates the median. Whiskers denote the minimal value within 1.5 interquartile range (IQR) of the lower quartile and the maximum value within 1.5 IQR of the upper quartile. Red symbols denote outliers. In D, black dashed lines indicate the lower and upper limits of the regions with regular scale. Outliers beyond the black dashed lines are visualized with a compressed scale in the regions denoted by gray lines. See also Figures S2B and S2C.
To examine the contribution of transcriptomic and epigenetic profiling in identifying relatively conserved genes, we performed additional control analysis to compare measures of haploinsufficiency and conservation between RefMap ALS genes and genes identified by transcriptomic or epigenetic profiling in isolation. Specifically, we identified all genes within active chromatin defined by MN ATAC-seq peaks, and all genes expressed in MNs (TPM>1); these two gene lists were substituted for the background set in the conservation analysis. Despite this change, we observed that RefMap genes are still more likely to be haploinsufficient and more intolerant to genetic variation compared to both gene lists (P<0.05, one-sided Wilcoxon rank-sum test; Figures S2B and S2C). However, if the contribution of epigenetic profiling is removed entirely then the resulting genes are not conserved: GWAS-derived genes are not intolerant of LoF mutations for all scores including LoFtool, RVIS and o/e (P>0.05, Wilcoxon rank-sum test).
Transcriptome analysis supports functional significance of RefMap genes in MNs and in ALS
We hypothesized that the ALS-associated genetic variation in regulatory regions of MNs identified by RefMap is likely to be pathogenic through reduced expression of the 690 linked genes. To explore this possibility we examined whether change in expression of RefMap genes is associated with ALS, using transcriptomic data from central nervous system (CNS) tissues, patient-derived MNs, and an ALS animal model.
First, we examined the expression of RefMap genes within our iPSC-derived MNs from neurologically normal individuals. RefMap genes are higher expressed (P=3.07e-17, one-sided Wilcoxon rank-sum test; Figure 3A) compared to the overall transcriptome (TPM>1). No differential expression was observed for genes derived from RefMap using randomly shuffled Z-scores.
Figure 3. Transcriptomics supports the functional importance of RefMap genes in motor neurons and in ALS.

(A) Comparative gene expression analysis of RefMap genes in iPSC-derived MNs from neurologically normal individuals (n=3). For a fair comparison, we only considered those genes with evidence for MN expression (TPM>1). (B) Comparative gene expression analysis of RefMap genes in post-mortem CNS tissues from C9orf72-ALS (n=8) and sporadic ALS (n=10) patients versus neurologically normal controls (n=17). FC, frontal cortex; CB, cerebellum. (C) Comparative gene expression analysis of RefMap genes in iPSC-derived MNs from ALS patients (n=55) versus neurologically normal controls (n=15). All comparisons in A-C were performed using the one-sided Wilcoxon rank-sum test, and the Benjamini-Hochberg (BH) correction was carried out in B. In A-C, the bottom and top of the boxes indicate the first and third quartiles, respectively, where the black line in between indicates the median. Whiskers denote the minimal value within 1.5 IQR of the lower quartile and the maximum value within 1.5 IQR of the upper quartile. Red symbols denote outliers. In B and C, black dashed lines indicate the lower and upper limits of the regions with regular scale. Outliers beyond the black dashed lines are visualized with a compressed scale in the regions denoted by gray lines. (D) Heatmap showing hierarchical clustering of expression changes of RefMap genes during disease progression based on the SOD1-G93A mouse model. RefMap genes were mapped to their mouse homologs (n=510). Gene expression levels were estimated using the β scores calculated in (Maniatis et al., 2019), and were averaged across different sections of spinal cords at each time point. Time points p30, p70, p100, and p120 represent presymptomatic, onset, symptomatic, and end-stage, respectively. Difference of gene expression levels between SOD1-G93A and SOD1-WT mice at each time point was quantified by the difference in β (Δβ). Before clustering, Δβ were standardized across genes, and one minus correlation was used as the clustering distance. (E) Two distinct expression patterns (C1: 286 genes; C2: 224 genes) of RefMap genes were identified after clustering. The larger cluster C1 was progressively downregulated during ALS progression. Solid plot represents the mean of expression levels within each cluster, and the standard error is shown as shading. (F) Gene ontology analysis of C1, showing that C1 is enriched with functions related to the MN distal axon and synapse. GO, gene ontology; GOBP, gene ontology biological process; GOCC, gene ontology cellular compartment. Black vertical line represents P=0.05. See also Table S4.
Next, we examined the expression of RefMap ALS genes in CNS tissues derived from ALS patients (n=18) and controls (n=17) (Prudencio et al., 2015). A significant decrease in the expression of RefMap genes was observed in both frontal cortex (C9orf72-ALS (cALS): false discovery rate (FDR)=0.002, one-sided Wilcoxon rank-sum test) and cerebellum (cALS: FDR=0.002, sporadic ALS (sALS): FDR=0.005, one-sided Wilcoxon rank-sum test) of ALS patients compared to the overall transcriptome (Figure 3B; STAR Methods). As an independent validation, we analyzed gene expression in iPSC-derived MNs from ALS patients (n=55, https://www.answerals.org/), and confirmed that RefMap genes are downregulated (P=3.85e-04, one-sided Wilcoxon rank-sum test; Figure 3C) compared to neurologically normal controls (n=15).
Finally, we used longitudinal data to infer whether change in the expression of RefMap ALS genes occurs upstream or downstream in the development of neuronal toxicity. We re-analyzed the transcriptomic data from a spatio-temporal transcriptomics study on the SOD1-G93A-ALS mouse model (Gurney et al., 1994; Maniatis et al., 2019). Longitudinal gene expression averaged across spinal cord sections from SOD1-G93A (n=32) and SOD1-WT (n=24) mice revealed two distinct expression patterns for RefMap homologs (Figures 3D and 3E, Table S4). Interestingly, the largest group (C1, 286/510) of RefMap homologs were progressively downregulated through consecutive disease stages, consistent with our observations in human data. Functional enrichment analysis of C1 genes pointed to distal axon function (Figure 3F), which is consistent with known ALS biology (Cooper-Knock, Jenkins and Shaw, 2013) and with the positioning of ALS as a distal axonopathy (Frey et al., 2000; Moloney, de Winter and Verhaagen, 2014). Functional enrichment of C2 genes did not reach statistical significance (data not shown).
Systems analysis identifies ALS-associated modules
We next examined biological functions associated with RefMap ALS genes by first mapping RefMap genes to the global protein-protein interaction (PPI) network and then inspecting the functional enrichment of ALS-associated network modules (Figure S3A, Table S5; STAR Methods). Two modules were found to be significantly enriched with RefMap genes: M421 (721 genes; FDR<0.1, hypergeometric test; Figure 4A, Table S5) and M604 (308 genes; FDR<0.1, hypergeometric test; Figure 4B, Table S5).
Figure 4. Network analysis associates RefMap genes with distal axonopathy in motor neurons.

(A and B) PPI network analysis revealed two modules that are significantly (FDR<0.1) enriched with RefMap genes: M421 (721 genes) (A) and M604 (308 genes) (B). Hypergeometric test was performed to quantify the enrichment followed by BH correction. Module nodes are colored to demonstrate the enrichment, where RefMap genes are in blue and other module genes are yellow. Edge thickness is proportional to STRING confidence score (>700). (C and D) RefMap modules, including M421 (C) and M604 (D), are enriched for MN functions localized within the distal axon. GOBP, gene ontology biological process; GOCC, gene ontology cellular compartment. Black vertical line represents P=0.05. (E) Representation of pathways enriched in each module (C and D) in MNs. (F) Comparative gene expression analysis of RefMap module genes in control MNs. All comparisons were performed using the one-sided Wilcoxon rank-sum test. The bottom and top of the boxes indicate the first and third quartiles, respectively, where the black line in between indicates the median. Whiskers denote the minimal value within 1.5 IQR of the lower quartile and the maximum value within 1.5 IQR of the upper quartile. Red symbols denote outliers. Black dashed lines indicate the lower and upper limits of the regions with regular scale. Outliers beyond the black dashed lines are visualized with a compressed scale in the regions denoted by gray lines. See also Figures S3A, S3C, S3D and Table S5.
Functionally, both M421 and M604 are enriched with GO/KEGG terms related to the distal axon and neuromuscular junction (NMJ) (Figures 4C, 4D and 4E). This is consistent with previous literature suggesting that ALS pathogenesis is initiated in the distal axon (Frey et al., 2000; Moloney, de Winter and Verhaagen, 2014). Finally, both M421 and M604 are overexpressed in control iPSC-derived MNs (P<1e-06, one-sided Wilcoxon rank-sum test; Figure 4F), in a similar manner to the total set of RefMap genes. Interestingly, many functions ascribed to M421 and M604 overlap with the functions of the C1 cluster from our analysis of the SOD1-G93A mouse model (Figure 3F).
Rare variant analysis validates RefMap genes
Our transcriptome analysis suggests that reduced function of RefMap ALS genes is associated with MN toxicity. On this basis, we hypothesized that rare mutations that disrupt the transcription/translation of RefMap genes would also modify ALS severity. We first screened RefMap genes for rare LoF mutations, including nonsense mutations, high-effect splice-site mutations (Jaganathan et al., 2019), and mutations changing transcription initiation within the 5’UTR (Zhang et al., 2020) (STAR Methods). By analyzing whole-genome sequencing (WGS) data from 5,594 sporadic ALS patients and 2,238 controls (Project MinE ALS Sequencing Consortium, 2018), we observed that 53% of ALS patients carry an ALS-associated rare LoF mutation within a RefMap gene. Notably, patients with a higher burden of rare LoF mutations within RefMap ALS genes suffered earlier age of disease onset (P=7e-03, logrank test; Figure 5A). This is consistent with previous literature in which ALS genetic risk has been associated with the age of disease onset (Shepheard et al., 2021). As a control test, we again examined the two matched gene lists of equivalent size: GWAS-derived genes and eQTL-derived genes. In neither of the control gene lists was the burden of rare LoF mutations associated with the age of disease onset (P>0.05, logrank test).
Figure 5. Rare variant analysis demonstrates the association of RefMap genes with ALS severity.

(A) Survival curves showing the number of rare LoF variants within RefMap ALS genes carried by an ALS patient is inversely correlated with the age of disease onset. Plot shows age of onset for ALS patients grouped by the number of rare LoF variants affecting one or more RefMap ALS genes. P-value by the logrank test. (B-E) Correlation analysis of the expression of ADAMTSL1 (B), BNC2 (C), KANK1 (D), and VAV2 (E) in iPSC-derived MNs obtained from ALS patients (n=55) versus the age of ALS onset. Gene expression level (x-axis) is plotted against the age of onset (y-axis). Lines (blue) of best fit are shown with 95% confidence interval (CI, grey area). The BH method was used for multiple testing correction. See also Figure S3B and Table S6.
As an additional rare variant validation of the total set of RefMap ALS genes, we examined rare deleterious variation within enhancer regions. Enhancer regions were defined as previously described (Fishilevich et al., 2017; Cooper-Knock et al., 2020). Significant variants were identified based on evolutionary conservation, functional annotations, and population frequency (Ritchie et al., 2014; Huang, Gulko and Siepel, 2017; Rentzsch et al., 2019; Karczewski et al., 2020) (STAR Methods). We performed gene-level association testing for differences in rare variant burden between ALS patients and controls (STAR Methods) for all genes expressed in iPSC-derived MNs (n=19,519, TPM>1). The resultant P-values for RefMap ALS genes are significantly smaller than for the total set of genes (P=0.01, Wilcoxon rank-sum test). As a final rare variant validation we analyzed rare (minor allele frequency (MAF)<0.001%) missense variants in an independent exome sequencing cohort (n=3,864 ALS patients and n=7,839 controls) (Farhan et al., 2019). The median P-value for ALS-associated genetic burden in RefMap genes is lower than expected by chance (λ=2.06 versus λ=~1 for all genes; Figure S3B, Table S6; STAR Methods).
To identify candidate genes for further experimental validation, we first ranked RefMap genes by the number of linked regions. Taking top scores over 3 standard deviations from the mean nominated 15 genes (Table S6). We were particularly interested in top RefMap genes with genetic convergence between common and rare ALS-associated variation. To examine this, we tested whether these 15 common-variant-derived genes are enriched with rare LoF variants within coding and noncoding regions. As before, we filtered for rare deleterious variants based on evolutionary conservation, functional annotations, and population frequency but we extended our analysis to enhancer, promoter, and coding regions (STAR Methods). Gene-level association testing for differences in rare variant burden between ALS patients and controls identified significant enrichment in one or more regions for four of the 15 genes, including ADAMTSL1, BNC2, KANK1, and VAV2 (P<0.05, SKAT-O; Table S6), none of which have been linked to ALS previously. In ADAMTSL1, BNC2, and KANK1, ALS patients are enriched with rare deleterious variants which are relatively absent from controls, whereas in VAV2 the direction of association is reversed, suggesting that the loss of VAV2 function is protective against MN toxicity. These four genes were replicated when RefMap was applied to the new ALS GWAS (van Rheenen et al., 2021).
We then examined expression levels of ADAMTSL1, BNC2, KANK1, and VAV2 in iPSC-derived MNs from ALS patients (n=55; https://www.answerals.org/). Expression of all four genes is significantly correlated with the age of ALS onset (FDR<0.05, Pearson correlation; Figures 5B, 5C, 5D and 5E). For ADAMTSL1, BNC2, and KANK1, lower expression is associated with earlier age of ALS onset (Figures 5B, 5C and 5D), which is consistent with a toxic LoF model. For VAV2, the association is in the opposite direction (Figure 5E), which is consistent with our rare variant analysis where LoF mutations are significantly absent from ALS patients. Indeed, VAV2 has been associated with neuroprotection mediated via TREM2 signaling (Painter et al., 2015). To ensure that our analysis was not confounded, we tested for a similar association with age of onset for random selections of four genes, but our findings are highly significant (P<1e-04, permutation test; STAR Methods).
Notably, of top-ranked RefMap genes only KANK1 (accounting for 3% of heritability) is enriched with ALS-associated rare mutations across both coding and noncoding regions in the Project MinE dataset. Indeed, KANK1 promoter and enhancer regions are independently enriched with ALS-associated rare variants (P<0.05, SKAT-O; Table S6). Nonsense coding variants within KANK1 are absent from controls and present in a small number (n=4) of ALS patients; an additional 8 ALS patients carry a deleterious variant within the KANK1 5’UTR. Combining coding and noncoding associations in a single test revealed a significant enrichment of rare deleterious variants within KANK1 in ALS patients compared to controls (P=5.6e-03, Stouffer’s method(Whitlock, 2005); FDR<0.1 after considering tests applied to all 15 genes). Furthermore, in the independent exome sequencing cohort (Farhan et al., 2019), KANK1 is significantly enriched with ALS-associated rare missense variants after Bonferroni multiple testing correction (P=4.5e-05, OR=1.36, Fisher’s exact test; Figure S3B, Table S6), but not with rare synonymous variants (P=0.11). KANK1 is located within a distinct module (M826, 687 genes; Figure S3C, Table S5) in our network analysis, and this module is enriched with RefMap genes (P=5.6e-03, hypergeometric test), while not after multiple testing correction. Functionally, the KANK1-module is highly expressed in normal MNs (P<1e-06, one-sided Wilcoxon rank-sum test; Figure 4F), and is enriched for biological functions centered on the distal axon and synapse (Figure S3D), which is consistent with other RefMap modules.
Experimental evaluation of BNC2 and KANK1 function in human neurons
We sought to experimentally investigate the effect of LoF of top-ranked RefMap genes on neuronal health (Figure 6A). We tested BNC2 and KANK1; ADAMTSL1 was also tested but we were unable to achieve a knockdown by CRISPR due to the absence of appropriate protospacer adjacent motif (PAM) sites (data not shown). gRNAs were designed to target PAM sites within BNC2 exon 2 and KANK1 exon 2, so as to introduce a series of indels by CRISPR/SpCas9 editing; edited exons coincide with the location of ALS-associated nonsense mutations in our rare variant analysis. BNC2 and KANK1 nonsense mutations were edited into SH-SY5Y neurons (Figure 6A; STAR Methods). Sanger sequencing and waveform decomposition analysis (Hsiau et al., 2019) in undifferentiated SH-SY5Y cells confirmed editing efficiency (Figures S4A, S4B, S4C and S4D).
Figure 6. Loss of function of BNC2 or KANK1 produces neurotoxicity.
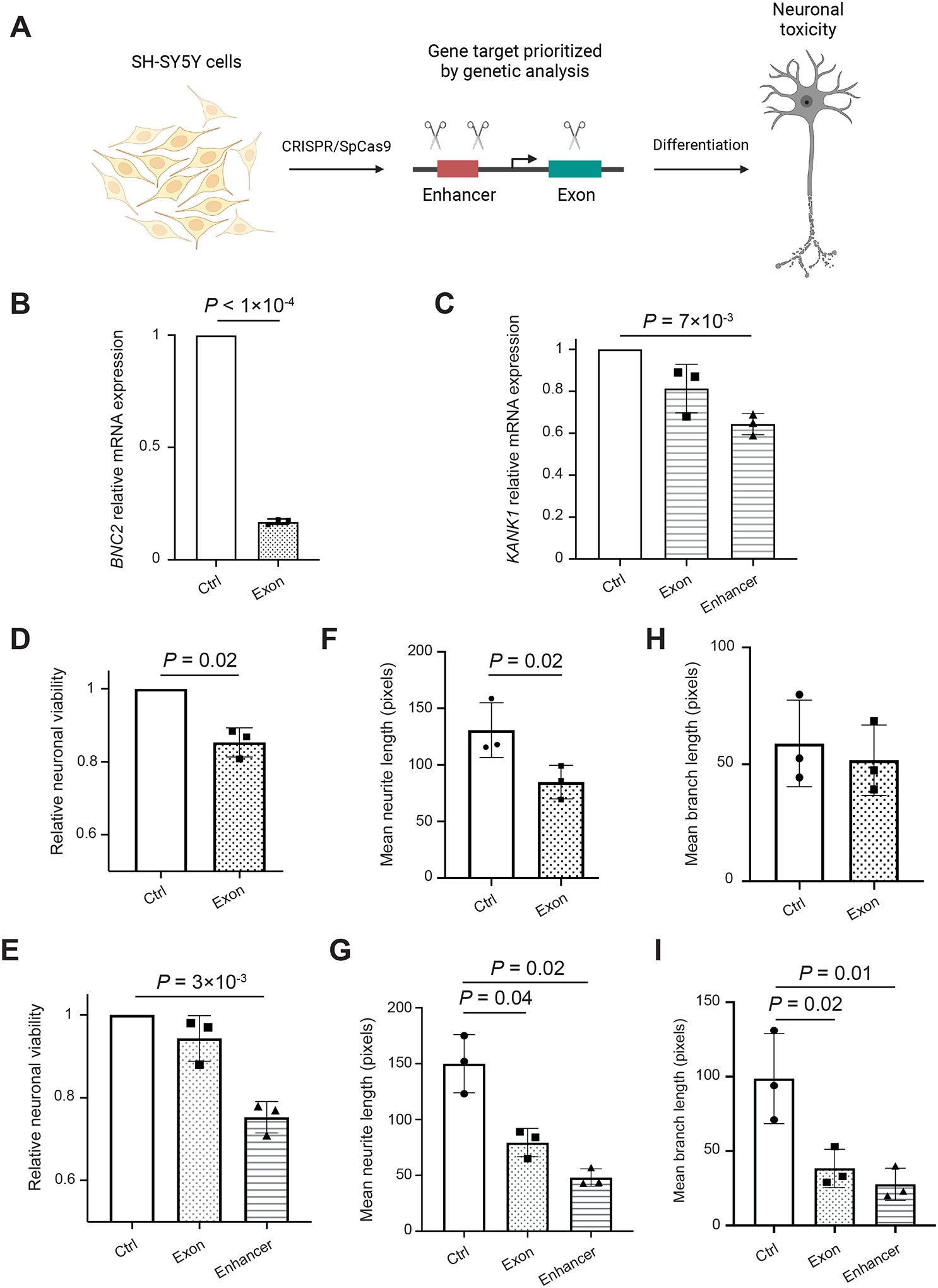
(A) Study design of experimental evaluation of BNC2 and KANK1 function in human neurons. We performed CRISPR/SpCas9 perturbation proximate to patient mutations in coding and enhancer regions of RefMap genes in SH-SY5Y neurons, and then investigated gene expression change and neuronal health. (B and C) Comparison of expression levels of BNC2 (B) and KANK1 (C) in corresponding edited neurons versus in control cells. (D and E) Comparison of neuronal viability by MTT assay between BNC2-edited (D), KANK1-edited neurons (E) and control cells. (F and G) Comparison of axonal length between BNC2-edited (F), KANK1-edited neurons (G) and control cells. (H and I) Comparison of axonal-branch length between BNC2-edited (H), KANK1-edited neurons (I) and control cells. Data are mean ± standard deviation. All comparisons were performed using the paired Student’s t-test. P-values smaller than 0.05 are annotated. See also Figures S4, S5A and S5B.
Among ALS-associated active regions identified by RefMap, chr9:663,001–664,000 has the highest concentration of risk SNPs (22 SNPs). We hypothesized that ALS-associated genetic variation within chr9:663,001–664,000 would reduce the expression of the associated gene, KANK1, leading to MN toxicity. To replicate disruption of this sequence, we designed gRNAs to target PAM sites up- and downstream of chr9:663,001–664,000 so as to delete the entire region (Zheng et al., 2014) in SH-SY5Y cells (Figure 6A; STAR Methods). Successful deletion of the enhancer region was confirmed by RT-PCR in undifferentiated SH-SY5Y cells (Figure S4E).
For experimental evaluation, a commercially available control gRNA targeting HPRT served as a negative control. Edited SH-SY5Y cells were differentiated to a neuronal phenotype (Forster et al., 2016) (Figures S5A and S5B). Differentiated cells were harvested and RNA was extracted for quantitative PCR (qPCR). We confirmed reduced expression of BNC2 and KANK1 mRNA in edited neurons, including BNC2-exon-edited cells, KANK1-exon-edited cells, and KANK1-enhancer-edited cells (BNC2 exon: 83% reduction, P<1e-04; KANK1 exon: 19% reduction, P=0.1; KANK1 enhancer: 36% reduction, P=7e-03, paired Student’s t-test; Figures 6B and 6C).
Reduction in BNC2 and KANK1 expression was associated with reduced neuronal viability in neuronally differentiated cells (MTT assay; BNC2 exon: P=0.02, KANK1 enhancer: P=3e-03, paired Student’s t-test; Figures 6D and 6E; STAR Methods). Edited neurons also exhibited shorter neurites (BNC2 exon: P=0.02, KANK1 exon: P=0.04, KANK1 enhancer: P=0.02, paired Student’s t-test; Figures 6F and 6G) and reduced branch length (KANK1 exon: P=0.02, KANK1 enhancer: P=0.01, paired Student’s t-test; Figures 6H and 6I). When comparing KANK1-exon-edited and KANK1-enhancer-edited neurons, it is notable that in all instances, the measures of neuronal toxicity were correlated with KANK1 expression (Figure 6C).
These experimental observations collectively demonstrate the neuronal toxicity focused on the axon caused by the loss of BNC2 and KANK1 function, which further supports both BNC2 and KANK1 as new ALS risk genes. All of the phenotypes we measured have previously been observed in cell models of ALS (Watanabe et al., 2020; Mehta et al., 2021). In particular, MTT defects reflect bioenergetic dysfunction which has been linked specifically to axonal shortening in C9orf72-ALS models (Mehta et al., 2021), and reduced length of dendrites have been found in C21orf2-ALS cell models (Watanabe et al., 2020).
Loss of KANK1 function in iPSC-derived motor neurons places axonal dysfunction upstream of key ALS molecular phenotypes
In our analysis, we have identified both common and rare ALS-associated mutations which reduce the expression of KANK1, disrupt neuronal function, and modify disease severity. Finally, we sought to investigate the effect of loss of KANK1 function in iPSC-derived MNs, which is the gold standard cell model for ALS (Sances et al., 2016; Fujimori et al., 2018). In particular, we used CRISPR/SpCas9 editing of iPSCs derived from an aged healthy control (STAR Methods) to recapitulate ALS LoF mutations before differentiating cells into mature MNs. MNs were evaluated for evidence of toxicity, electrophysiological dysfunction, and pathology (Figure 7A).
Figure 7. Loss of function of KANK1 in iPSC-derived motor neurons leads to neuronal toxicity, distal axon dysfunction, and TDP-43 mislocalization.
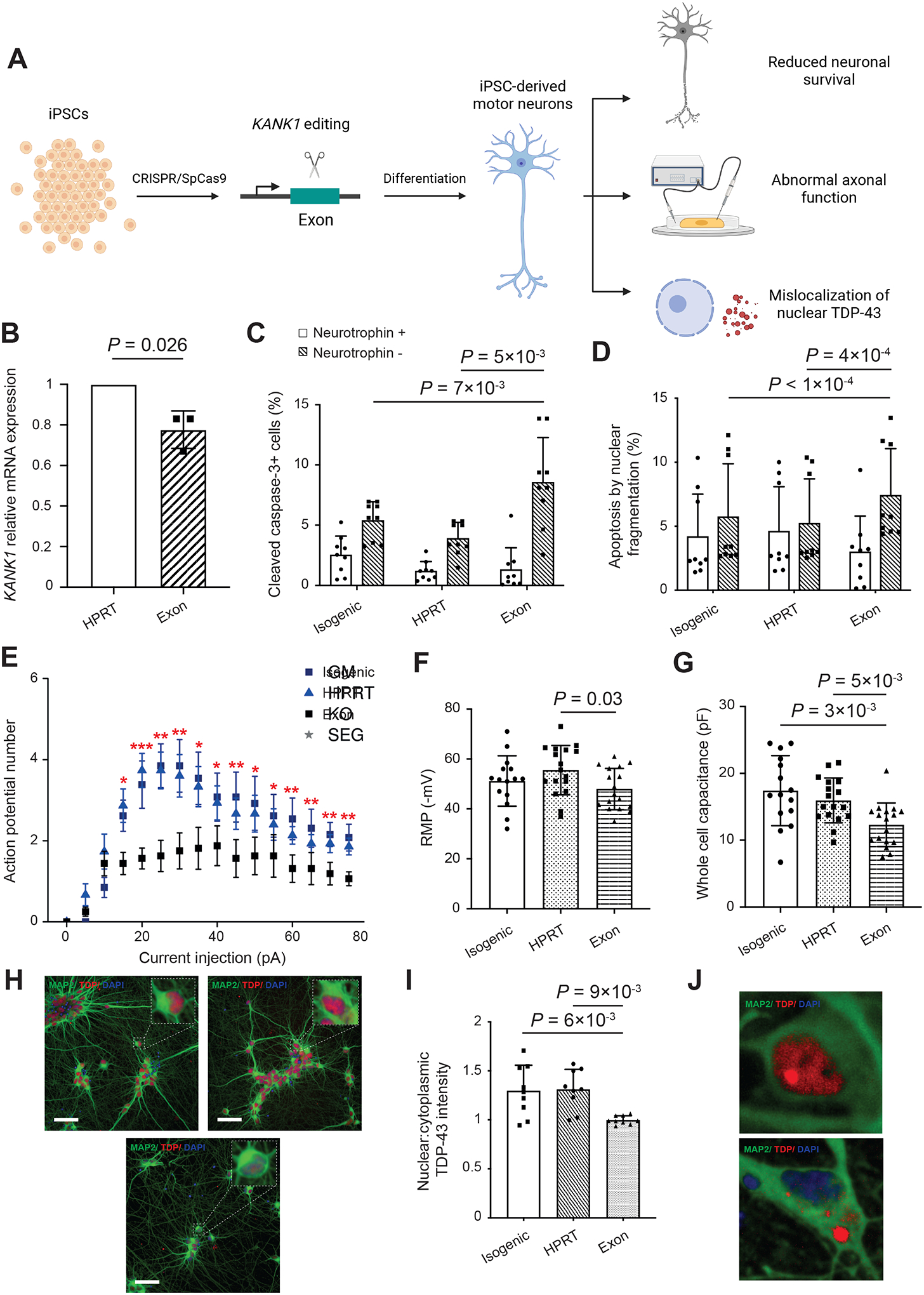
(A) Schematic of experimental study design. To experimentally evaluate the effect of loss of function of KANK1, we performed CRISPR/SpCas9 perturbation proximate to patient KANK1 exonic mutations in iPSCs, which were then differentiated into mature MNs. MNs were evaluated for evidence of toxicity, deficient electrophysiological function, and for molecular phenotypes associated with ALS, including cytoplasmic displacement of TDP-43 with formation of cytoplasmic inclusions. (B) Comparison of KANK1 expression in KANK1-edited versus HPRT-edited cells. (C) Comparison of the proportion of cleaved caspase-3-positive cells between KANK1-edited iPSC-derived motor neurons and controls. (D) Comparison of the proportion of nuclear fragmentation between KANK1 edited motor neurons and controls. Comparisons in B-D were performed using the paired Student’s t-test. (E) Comparison of action potential firing between KANK1-edited motor neurons and controls. *, P<0.05; **, P<0.01; ***, P<0.001. (F) Comparison of resting membrane potential (RMP) between KANK1-edited motor neurons and controls. (G) Comparison of whole cell capacitance between KANK1-edited motor neurons and controls. Comparisons in E-G were performed using the Mann-Whitney U-test. (H) Immunocytochemistry reveals loss of nuclear TDP-43 in KANK1-edited motor neurons. (I) Comparison of the ratio of nuclear to cytoplasmic TDP-43 intensity between KANK1-edited motor neurons and controls. Comparison was performed using the one-way ANOVA. (J) Immunocytochemistry reveals cytoplasmic TDP-43-positive protein aggregates in KANK1-edited motor neurons. Data are mean ± standard deviation. P-values smaller than 0.05 are annotated. See also Figures S5C, S5D, S6 and S7.
To reproduce ALS-associated variants in iPSCs, we re-used the CRISPR gRNAs utilized in the SH-SY5Y experiment targeting KANK1 exon 2. We confirmed the editing efficiency at the iNPC and iPSC stages (Figures S5C and S5D; STAR Methods). Reduction in KANK1 expression was confirmed in mature MNs (23% reduction, P=0.026, paired Student’s t-test; Figure 7B) compared to a separate line edited with a commercially available control gRNA targeting HPRT. We also utilized isogenic unedited iPSCs as an additional control. We confirmed successful differentiation of mature motor neurons for all lines at day 45 post-differentiation (Figure S6; STAR Methods). All molecular phenotypes were confirmed in a minimum of three technical replicates and >100 MNs per condition (STAR Methods).
We observed increased apoptosis (STAR Methods) in KANK1-edited MNs as evidenced by staining for cleaved caspase-3 (versus isogenic control cells: 59% increase in the mean number of cleaved caspase-3-positive cells, P=7e-03; versus HPRT-edited cells: 120% increase in the mean number of cleaved caspase-3-positive cells, P=5e-03, paired Student’s t-test; Figures 7C and S7A) and nuclear fragmentation (versus isogenic control cells: 29% increase in the mean number of cells with nuclear fragmentation, P<1e-04; versus HPRT-edited cells: 42% increase in the mean number of cells with nuclear fragmentation, P=4e-04, paired Student’s t-test; Figure 7D). Excessive cell death was observed with withdrawal of neurotrophic factors from the culture media, suggesting that KANK1 may have a downstream role in neurotrophic signaling.
At day 40 post-differentiation motor neurons were electrophysiologically functional. However, patch-clamp electrophysiology (STAR Methods) demonstrates that KANK1-edited lines were hypoexcitable relative to isogenic control cells (up to 53% reduction in number of action potentials, P=4e-03, Mann-Whitney U-Test; Figures 7E and S7B) as well as to HPRT-edited cells (up to 54% reduction in number of action potentials, P=8e-04, Mann-Whitney U-Test; Figures 7E and S7B). This is reflected in a moderate increase in input resistance (versus isogenic control cells: P=0.09, versus HPRT-edited cells: P=0.05, Mann-Whitney U-test; Figure S7C), reduced resting membrane potential (RMP) (versus HPRT-edited cells: P=0.03, Mann-Whitney U-test; Figure 7F), and reduced whole cell capacitance (versus isogenic control cells: P=3e-03, versus HPRT-edited cells: P=5e-03, Mann-Whitney U-test; Figure 7G). These observations together demonstrate electrophysiological dysfunction in the distal axon of motor neurons with loss of KANK1 function.
The pathological hallmark of ALS is TDP-43 mislocalization, including nuclear loss and the formation of cytoplasmic protein aggregates (Neumann et al., 2006). Specifically, loss of nuclear TDP-43 has been linked to splicing changes with a role in downstream pathogenesis (Melamed et al., 2019; Green et al., 2021; Ule et al., 2021). At day 45, post-differentiation iPSC-derived motor neurons were fixed and stained for TDP-43 (STAR Methods). In the absence of any exogenous stressor, KANK1-edited neurons exhibited dramatic loss of nuclear TDP-43, which was not present in either isogenic control cells (30% reduction, P=6e-03, one-way ANOVA; Figures 7H and 7I) or in HPRT-edited cells (31% reduction, P=9e-03, one-way ANOVA; Figures 7H and 7I). KANK1-edited neurons also displayed evidence of cytoplasmic TDP-43-positive protein aggregates (Figure 7J).
Overall, our experimental data indicates that the loss of KANK1 function produces neuronal toxicity, disrupts distal axon function, and reproduces key phenotypes associated with ALS, including TDP-43 mislocalization (Neumann et al., 2006), hypoexcitability (Sareen et al., 2014; Devlin et al., 2015; Naujock et al., 2016; Martínez-Silva et al., 2018), and failure of neurotrophic signaling (Lamas et al., 2014; Sances et al., 2016; Shi et al., 2018).
DISCUSSION
Study of the genetic architecture of complex diseases has been advanced by GWAS. However, previous studies have not considered cell-type-specific aspects of genomic function, which is particularly relevant for noncoding regulatory sequence (Heinz et al., 2015). This may explain why complex diseases such as ALS have been linked to relatively few risk genes despite substantial estimates of heritability (Ryan et al., 2019; Trabjerg et al., 2020). Fine-mapping methods have been proposed to disentangle causal SNPs from genetic associations (Hormozdiari et al., 2014; Kichaev et al., 2014; Pickrell, 2014; Benner et al., 2016; Chen et al., 2016; Schaid, Chen and Larson, 2018), but these approaches are not integrated with cell-type-specific biology (Hormozdiari et al., 2014; Benner et al., 2016), or assume a fixed number of causal SNPs per locus (Kichaev et al., 2014; Pickrell, 2014; Chen et al., 2016), limiting their power for gene discovery. We have characterized epigenetic features of MNs, which are the key cell type for ALS pathogenesis. Integrating MN epigenetic features with ALS GWAS data in our RefMap model revealed 690 ALS risk genes and 36% of SNP-based heritability, which represents a 5-fold increase in recovered SNP-based heritability compared to conventional methods.
Others have performed more limited epigenetic profiling of motor neurons (Song et al., 2019), but our data are unique with respect to the depth and number of assessments.
RefMap ALS genes were identified through analysis of common genetic variation; LD between common and rare variants is minimal (Pritchard and Cox, 2002) and therefore analysis of rare variants provides an orthogonal test of underlying disease biology. Rare deleterious genetic variation within the total set of RefMap genes is associated with ALS. Moreover, rare LoF mutations within RefMap genes modify ALS clinical severity. Finally, we have shown experimentally that LoF of top-ranked RefMap genes produces key molecular phenotypes associated with ALS, including TDP-43 mislocalization (Neumann et al., 2006), hypoexcitability (Sareen et al., 2014; Devlin et al., 2015; Naujock et al., 2016; Martínez-Silva et al., 2018), and disruption of neurotrophic signaling (Lamas et al., 2014; Sances et al., 2016; Shi et al., 2018). Altogether, our data strongly support the utility of the RefMap framework for gene discovery.
In addition to rare genetic variation within the total set of RefMap genes, we also identified a significant enrichment of ALS-associated rare variants in a number of top-ranked RefMap genes including ADAMTSL1, BNC2, KANK1, and VAV2. For KANK1 in particular, rare genetic variation is ALS associated even after stringent multiple testing correction across multiple independent cohorts. Expression of these genes in patient-derived MNs is correlated with ALS severity. We were able to use CRISPR editing to replicate ALS-associated mutations in two of these genes -- KANK1 and BNC2 -- in human neurons.
BNC2 is not well characterized in the context of human neurons. We discovered that reduced expression of BNC2 is toxic and leads to an axonopathy in human neurons. Moreover, the expression of BNC2 in patient-derived motor neurons is directly correlated with disease severity. BNC2 is localized to nuclear speckles and has been associated with regulation of RNA splicing (Vanhoutteghem and Djian, 2006). It is interesting that sequestration of SC35/SRSF2, the major marker of nuclear speckles, has been previously associated with ALS (Lee et al., 2013; Cooper-Knock et al., 2014). Further work is needed to understand how LoF of BNC2 leads to axonal dysfunction and neuronal death.
KANK1 is functionally related to a number of known ALS genes which are important for cytoskeletal function, including PFN1, KIF5A, and TUBA4A. In particular, PFN1, like KANK1, is implicated in actin polymerization (Chandra Roy, Kakinuma and Kiyama, 2009; Kakinuma et al., 2009; Boopathy et al., 2015). Disruption of actin polymerization has been associated with alterations in synaptic organization (Dillon and Goda, 2005), including the NMJ (Mallik and Kumar, 2018), but also with nucleocytoplasmic transport defects (Giampetruzzi et al., 2019). We have experimentally verified the link between variants identified by our analysis and KANK1 expression. Moreover, we have demonstrated that the reduced expression of KANK1 in iPSC-derived MNs is toxic and reproduces key pathological hallmarks of ALS, including TDP-43 pathology (Neumann et al., 2006). It is plausible that KANK1 upregulation could be a therapeutic target for ALS patients carrying mutations that disrupt KANK1 function, and possibly more broadly. Of note, we showed that expression of KANK1 in patient-derived motor neurons is directly correlated with disease severity, without selecting for patients carrying KANK1 mutations.
Consistent with previous literature, RefMap ALS genes are functionally associated with the distal axon (Frey et al., 2000; Moloney, de Winter and Verhaagen, 2014). Several known ALS risk genes are related to axonal function and axonal transport in particular (De Vos and Hafezparast, 2017). Unlike previous literature, our work is based on a comprehensive genome-wide screening and not on a small number of rare variants. As a result, our data suggest that the distal axon may be the site of disease initiation in the majority of ALS patients, and should be a major focus of future translational research. It has previously been proposed that axonal dysfunction is secondary to TDP-43 dysfunction (Herzog et al., 2017; Briese et al., 2020), but importantly our work suggests that the opposite is true. KANK1 is a key protein for distal axon function (Chandra Roy, Kakinuma and Kiyama, 2009; Kakinuma et al., 2009), and ALS-associated mutations within KANK1 disrupt distal axon function, but also lead to TDP-43 mislocalization from the nucleus in iPSC-derived MNs. We conclude that axonal dysfunction precedes TDP-43 pathology in the cascade of pathogenesis.
Our study has certain limitations. First, 84.8% of RefMap ALS genes were re-discovered when we replicated the RefMap analysis in a new GWAS dataset with a more heterogeneous population structure (van Rheenen et al., 2021), but some genes were not replicated. This may be a result of mismatch in population structure between the two GWAS datasets. Second, in order to make our method more portable we rely on out-sample estimation of LD structure using the 1000 Genomes dataset, which is suboptimal in modeling common variants (Benner et al., 2017) compared to in-sample estimation which is often not widely available. Third, we present evidence for the functional significance of the total set of RefMap genes including enrichment with known ALS genes, transcriptomics, conservation and systems analysis, and rare variant burden within exons, promoters and enhancers. However, our experimental validation is limited to top-ranked RefMap genes and we reproduce ALS-associated genetic variants in iPSC-derived motor neurons only for KANK1. We await future efforts to perform similar study of other RefMap genes.
In summary, our study generates significant new resources, including transcriptomic and epigenetic profiling of MNs for future study of motor neuron diseases such as ALS, and we also provide a general framework which can be applied for the identification of risk genes involved in a large number of complex diseases and traits.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Micheal P. Snyder (mpsnyder@stanford.edu).
Consortia
Project MinE ALS Sequencing Consortium includes Ian Blair, Naomi Wray, Matthew Kiernan, Miguel Mitne Neto, Adriano Chio, Ruben Cauchi, Wim Robberecht, Philip van Damme, Phillippe Corcia, Phillipe Couratier, Orla Hardiman, Russel McLaughlin, Marc Gotkine, Vivan Drory, Nicola Ticozzi, Vincenzo Silani, Jan Veldink, Leonard van den Berg, Mamede de Carvalho, Jesus Mora Pardina, Monica Povedano, Peter Andersen, Markus Wber, Nazli Başak, Ammar Al-Chalabi, Christopher Shaw, Pamela Shaw, Karen Morrison, John Landers, and Jonathan Glass.
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Data files for ATAC-seq, Hi-C and histone ChIP-seq are available at encodeproject.org with the following link or accessions numbers mentioned below: https://www.encodeproject.org/publications/de19555b-a49f-471c-bfbc-be3b628fe9bf/.
The source code of RefMap can be accessed at https://github.com/szhang1112/refmap.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study cohorts
iPSC-cells were derived from fibroblasts obtained from three neurologically normal controls of different ages: 55-year old male, a 52-year old female and a 6-year old male (Figure S1A). Human SH-SY5Y neuroblastoma cells were utilised within the range of 7–32 passages.
GWAS summary statistics were previously published (van Rheenen et al., 2016). The 5,594 patients and 2,238 controls subject to WGS and included in this study were recruited at specialized neuromuscular centers in the UK, Belgium, Germany, Ireland, Italy, Spain, Turkey, the United States and the Netherlands (Project MinE ALS Sequencing Consortium, 2018). Patients were diagnosed with possible, probable or definite ALS according to the 1994 El-Escorial criteria (Brooks, 1994). All controls were free of neuromuscular diseases and matched for age, sex and geographical location.
The study was approved by the South Sheffield Research Ethics Committee. Also, this study followed study protocols approved by Medical Ethical Committees for each of the participating institutions. Written informed consent was obtained from all participating individuals. All methods were performed in accordance with relevant national and international guidelines and regulations.
METHOD DETAILS
Cell culture
Human SH-SY5Y neuroblastoma cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Lonza) supplemented with 10% (v/v) foetal bovine serum (FBS) (Thermo-Fisher Scientific), 50 units/mL of penicillin and 50 μg/mL of streptomycin. Cell lines were maintained at 5% CO2 in a 37°C incubator and split every 3–4 days.
Human induced pluripotent stem cells iPSCs were maintained in Matrigel-coated plates (Corning) according to the manufacturer’s recommendations in complete mTeSR-Plus Medium (StemCell Technologies). The culture medium was replaced daily and confirmed mycoplasma free. Cells were passaged every four to six days as clumps using ReLeSR, an enzyme-free reagent for dissociation (StemCell Technologies) according to the manufacturer’s recommendations. For all the experiments in this study, iPSCs were between passage 20 and 32.
iPSC-derived motor neuron differentiation
iPSCs derived from unaffected controls were differentiated to motor neurons using the modified version of the dual SMAD inhibition protocol (Du et al., 2015). Briefly, iPCS cells were transferred for Matrigel-coated plate (Corning). On the day after plating (day 1), after the cells had reached ~100% confluence, the cells were washed once with PBS and then the medium was replaced with neural medium (50% of KnockOut DMEM/F-12, 50% of Neurobasal), 0.5× N2 supplement (ThermoFisher), 1x Gibco GlutaMAX Supplement (ThermoFisher), 0.5x B-27 (ThermoFisher), 50 U ml−1 penicillin and 50 mg ml−1 streptomycin, supplemented with SMAD inhibitors (DMH-1 2 μM; SB431542 10 μM; and CHIR99021 3 μM). This medium was changed every day for 6 days, on day 7, the medium was replaced for neural medium supplemented with DMH-1 2 μM, SB431542–10 μM and CHIR 1 μM, All-Trans Retinoic Acid 0.1 μM (RA), and Purmorphamine 0.5 μM (PMN), the cells were kept in this medium until day 12 when it was possible to observe a uniform neuroepithelial sheet. At this point the cells were split 1:6 with Accutase (Gibco), onto matrigel substrate in the presence of 10 μM of ROCK inhibitor (Y-27632 dihydrochloride, Tocris), giving rise to a sheet of neural progenitor cells (NPC). After 24 hours of incubation the medium was changed for neural medium supplemented with RA 0.5 μM and PMN 0.1 μM, the medium was changed every day for 6 more days. On day 19 motor neuron progenitors were split with accutase onto to matrigel-coated plates and the medium was replaced with neural medium supplemented with RA 0.5 μM, PMN 0.1 μM, compound E 0.1 μM (Cpd E, Tocris), BDNF 10ng/mL, CNTF 10ng/mL and IGF 10ng/mL until day 28. On day 29, the media was replaced with neuronal media (neurobasal media supplemented with 1% of B27, BDNF 10ng/mL, CNTF 10ng/mL and IGF 10ng/mL). The cells were then fed alternate days with neuronal medium until day 40.
ATAC-seq
50,000 viable motor neurons were spun down at 500 RCF at 4°C for 5 min. Supernatant was discarded. 50 μl cold ATAC Resuspension Buffer (RSB) (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, sterile H2O) containing 0.1% NP40, 0.1% Tween-20, and 0.01% Digitonin was added and carefully mixed. Tubes were incubated on ice for 3 min. 1 ml of cold ATAC-RSB containing 0.1% Tween-20 was added and the tubes were inverted three times. Nuclei were spun down at 500 RCF for 10 min at 4°C. Supernatant was aspirated. Cell pellet was resuspended in 50 μl of transposition mix (25 μl 2x TD buffer, 2.5μl transposase (100 nM final), 16.5 μl PBS, 0.5 μl 1% digitonin, 0.5 μl 10% Tween-20, 5 μl H2O) by pipetting up and down 6 times. TD buffer consists of 20 mM Tris-HCl pH 7.6, 10 mM MgCl2, 20% DMF, sterile H2O. pH was adjusted with acetic acid before adding DMF.
The reaction was incubated at 37°C for 30 minutes in a thermomixer while shaking at 1000 RPM. Reaction was cleaned up with a Qiagen MiElute kit. DNA was eluted in 20 μL elution buffer. DNA was amplified using the NEBNext 2xMasterMix. Cycling conditions: 5 min at 72°C, 30 sec at 98°C, followed by 5 cycles of 10 sec at 98°C, 30 sec at 63°C and 1 min at 72°C, hold at 4°C. 5μl (10% of the pre-amplified mixture) were used for qPCR to determine the number of additional cycles needed (3.76 μL H2O, 0.5 μL 25 μM Primer1, 0.5 μL 25 μM Primer2, 0.24 μL 25x SYBR Green, 5 μL NEBNext MasterMix). Cycling conditions: 30 sec at 98°C, followed by 20 cycles of 10 sec at 98°C, 30 sec at 63°C and 1 min at 72°C, hold at 4°C. Amplification profiles were assessed as previously described (Buenrostro et al., 2015). The remainder of the pre-amplified DNA (45μL) was used to run the required number of additional cycles. The final PCR reaction was cleaned up using Qiagen MinElute kit and eluted in 20 μl H2O. Libraries were quantified with the KAPA Library Quantification kit (Roche) and sequenced on a NovaSeq 6000 system (Illumina). Raw data were processed with the ENCODE 4 pipeline for ATAC-seq according to ENCODE 4 standards (https://www.encodeproject.org/atac-seq/). All samples exceeded ENCODE 4 standards for % mapped reads, enrichment of transcription start sites, the fraction of reads that fall within peak regions (FRiP), and reproducibility between technical replicates (Table S1).
Files are available at encodeproject.org with the following accession numbers: ENCSR065CER, ENCSR410DWV, ENCSR812ZKP, ENCSR634WYX, ENCSR459PVP, ENCSR913OWV, ENCSR704VZY, ENCSR131HOY, ENCSR516YAD, ENCSR709QRD.
Histone ChIP-seq
5 million motor neurons were crosslinked and resuspended in 10 mL of cold L1 buffer (50mM Hepes KOH, pH 7.5, 140mM NaCl, 1mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100, dH2O, 1 protease inhibitor tablet (Roche) per 50ml buffer). Cells were incubated on a rocking platform at 4°C for 10 minutes and spun down at 3000 rpm at 4°C for 10 minutes. Pellets were resuspended in 10 mL of L2 buffer (200mM NaCl 1mM EDTA pH 8 0.5mM EGTA 10mM Tris, pH 8, dH2O, 1 protease inhibitor tablet (Roche) per 50ml buffer, room temperature). Tubes were incubated at room temperature for 10 minutes and spun down at 3000 rpm for 10 minutes at 4°C. Nuclei were resuspended in 3 mL 1X RIPA buffer and incubated on ice for 30 minutes. Samples were sonicated with Branson 250 Sonifier to shear the chromatin. 3 mL of sheared chromatin lysate were transferred to two 2 mL tubes and spun down at 14,000 rpm at 4°C for 15 minutes. 50 μL were saved from each replicate and pooled as input (no antibody added, kept at −20°C). 2 μL histone modification antibody was added to each 3 mL lysates and incubated at 4°C on a neutator for 12–16 hours. The following antibodies were used: H3K4me1 (Cell Signaling Technologies), H3K4me3 (Cell Signaling Technologies), H3K27ac (ActiveMotif). 80 μL of Protein A/G-agarose for each sample were washed twice with 1 mL of ice cold 1X RIPA buffer, spun down at 5000 rpm for 1 minute at 4°C and resuspended in 80μL in 1x RIPA buffer. Beads were added to tubes containing Ag-Ab complex (80 μ L 1X RIPA to wash out the beads) and incubated for 1 hour at 4°C with neutator rocking. Tubes were spun down at 1500 rpm for 3 minutes, beads were washed 3 times 15 minutes each with 10 mL of fresh, ice cold 1x RIPA buffer supplemented per 50 mL with 1 protease inhibitor tablet, 250 μL of 100 mM PMSF, 50 μL of 1M DTT, 2 ml of phosphatase inhibitor (sodium pyrophosphate 1mM, sodium orthovanadate 2mM, sodium fluoride 10mM). Afterwards, beads were washed once with ice cold 1 × PBS for 15 minutes. Beads were resuspended in 1200 μL ice cold 1x PBS, transferred to an 1.5mL Eppendorf tube and spun down at 5000 rpm for 1 minute. PBS was removed and 100 μL of Elute 1 solution (1% SDS, 1x TE, dH2O) was added to resuspend beads and tubes were incubated at 65°C for 10 minutes with gentle mixing every 2 minutes. Beads were spun down at 5000 rpm for 1 minute at room temperature and the supernatant was kept as Elute 1. 150 μL of Elute 2 solution (0.67% SDS, 1x TE) was added to the bead pellets and incubated at 65°C for 10 minutes with gentle vortexing. After spinning down for 1 minute at 5000 rpm, the second elute was combined with the first. Input DNA was thawed and 150 μL of Elute 1 solution was added. All samples incubated at 65°C overnight to reverse cross-linking. 250 μL 1X TE containing 100 μg RNase was added to each sample and incubated for 30 minutes at 37°C. 5 μL of 20 mg/mL Proteinase K was added to each sample and incubated at 45°C for 30 minutes. After transferring samples to 15 mL tubes, DNA was purified (Qiaquick PCR purification kit, Qiagen). DNA was eluted in elution buffer (50μL for input, 35μL for ChIP sample).
The following components were combined and mixed in a microfuge tube: ChIP DNA to be end-repaired (25ng) 34 μL, 5 μL 10X End-Repair Buffer, 5 μL 2.5 mM dNTP Mix, 5 μL10 mM ATP, 1 μL End-Repair Enzyme Mix. The mixture was incubated at room temperature for 45 minutes. DNA was purified (MinElute PCR purification kit, Quiagen) and eluted in 19 μL EB. Adapter ligated DNA was run on a 2% EX-Gel and excised in the range of 450–650 bp with a clean scalpel. DNA was purified (Gel extraction kit, Quiagen) and eluted in 20 μL EB. The following components were mixed in a PCR tube: 20 μL of purified DNA, 25 μL KAPA HiFi HotSTARt ReadyMix (2X), 5 μL KAPA Library Amplification Primer Mix (10X). DNA was amplified with the following conditions: 45 sec at 98°C, 15x [15 sec at 98°C, 30 sec at 60°C, 30 sec at 72°C], 60 sec at 72°C, hold at 4°C. The PCR product was purified (MinElute PCR purification kit, Quiagen) and eluted in 19 μL EB. DNA was run on a 2% EX-Gel and excised in the range of 300–450 bp (or brightest smear) with a clean scalpel. DNA was purified (Qiaquick Gel extraction kit, Quiagen) and eluted in 12 μL EB. Library concentration was measured using Qubit and each library was run on the Bioanalyzer. Equal concentrations of different barcoded libraries were pooled and sequenced on a NovaSeq 6000 system (Illumina). Raw data were processed with the ENCODE 4 pipeline for Histone ChIP-seq according to ENCODE 4 standards (https://www.encodeproject.org/chip-seq/histone/). All samples exceeded ENCODE standards for % mapped reads, the fraction of reads that fall within peak regions (FRiP), and reproducibility between technical replicates (Table S1).
Files are available at encodeproject.org with the following accession numbers: ENCSR754DRC, ENCSR672RKZ, ENCSR571HAY, ENCSR503HWR, ENCSR207VLY, ENCSR962OTG, ENCSR745TRI, ENCSR595HWK, ENCSR312HLG, ENCSR682BFG, ENCSR680IWU, ENCSR564EFE, ENCSR358AOC, ENCSR698HPK, ENCSR778FKK, ENCSR425FUS, ENCSR489LNU, ENCSR540KQC.
Hi-C
We generated Hi-C libraries following the protocol previously described (Rao et al., 2014, 2017). In brief, 2–5 million cells were crosslinked with formaldehyde. Nuclei were permeabilized and DNA was digested with 100U of MboI. DNA fragments were labelled with biotinylated nucleotides. Ligated DNA was purified and sheared to a length of 300–500 bp after reverse cross-linking. Ligation junctions were pulled-down with magnetic streptavidin beads. Libraries were amplified by PCR and purified. Library concentrations were measured (Qubit). Hi-C libraries were paired-end sequenced on a NovaSeq 6000 system (Illumina). Raw data were processed with the ENCODE 4 pipeline for Hi-C according to ENCODE 4 standards (https://www.encodeproject.org/documents/75926e4b-77aa-4959-8ca7-87efcba39d79/). Files are available at encodeproject.org with the following accession numbers: ENCSR305RTT, ENCSR866FWQ, ENCSR550JLK, ENCSR094EIC, ENCSR350NJV, ENCSR379CII, ENCSR228TUX, ENCSR794RDS, ENCSR444BAR.
RNA-seq
RNA libraries were prepared by first depleting ribosomal RNA using the Illumina Ribo-Zero rRNA depletion kit. Strand-specific libraries were then prepared using NEBext Ultra RNA prep kit. RNAseq libraries were paired-end sequences on a NovaSeq 6000 system (Illumina). A minimum of 80 million reads were obtained per sample. Raw Fastq files were trimmed for the presence of Illumina adapter sequences using Cutadapt v1.2.1 (Martin, 2011). The reads were further trimmed using Sickle v1.200 with a minimum window quality score of 20. Reads shorter than 15 bp after trimming were removed. Reads were aligned to hg19 transcripts (n=180,253) using Kallisto v0.46.0 (Bray et al., 2016).
Model design and inference of RefMap
In this study, allele Z-scores were calculated as Z=b/se, where b and se are effect size and standard error, respectively, and they were estimated from the mixed linear model implemented in an ALS GWAS (van Rheenen et al., 2016). Given allele Z-scores and the epigenetic profiling of iPSC-derived motor neurons, we are interested in predicting causal associations of individual genomic regions with ALS risk. Suppose we have K 1Mb LD blocks with non-zero alleles, whose approximate between-block independence has been verified in previous literature (Loh et al., 2015). Also suppose each LD block contains Jk (k=1, …, K) 1kb regions and each region harbors Ij,k (j=1, …, Jk, Ij,k>0) SNPs. We further denote the Z-score for the i-th SNP in the j-th region of the k-th block as zi,j,k (i=1, …, Ij,k). Under a linearity hypothesis, we can prove that zk follows a multivariate normal distribution (Pasaniuc and Price, no date; Han, Kang and Eskin, 2009; Kichaev et al., 2014; Bulik-Sullivan et al., 2015, no date; Finucane et al., 2015; Joo et al., 2016) (Supplemental Note), i.e.,
| (1) |
in which uk are the effect sizes of individual SNPs which can be expressed as
| (2) |
Moreover, in Eq. (1) represents the in-sample LD matrix comprising of the pairwise Pearson correlation coefficients between SNPs within the k-th block, where Ik is the total number of SNPs given by . Here, since we have no access to the individual genotypes, we used European (EUR) samples from the 1000 Genomes Project phase 3 to estimate ∑k (i.e., out-sample LD matrix).
Here, the latent variables uk can be treated as the disentangled Z-scores from LD confounding, leaving the right place for independence assumption and facilitating downstream modeling. Indeed, we assume ui,j,k (i=1, …, Ij,k) are independent and identically distributed (i.i.d.), following a normal distribution given by
| (3) |
where the precision follows λj,k a Gamma distribution, i.e.,
| (4) |
Moreover, to characterize the shift of expectation in Eq. (3) from the background due to its functional effect, we model mj,k by a three-component Gaussian mixture model, i.e.,
| (5) |
where the precisions follow
| (6) |
and v−1 and v+1 are non-negative variables quantifying the absolute values of effect size shifts for the negative and positive components, respectively.
To impose non-negativity over v−1 and v+1, here we employ the rectification nonlinearity technique proposed previously (Harva and Kabán, 2007). In particular, we assume v−1 and v+1 follow
| (7) |
| (8) |
in which the rectified Gaussian distribution is defined via a dumb variable. Specifically, we first define v−1 and v+1 by
| (9) |
| (10) |
which guarantee that v−1 and v+1 are non-negative. The dump variable r−1 and r+1 follow Gaussian distributions given by
| (11) |
| (12) |
where m± and λ± follow the Gaussian-Gamma distributions, i.e.,
| (13) |
| (14) |
The indicator variables in Eq. (5) denote whether that region is ALS-associated or not. Indeed, we define the region to be disease-associated if or , and to be non-associated otherwise. To simplify the analysis, we put a symmetry over and , and define the distribution by
| (15) |
Furthermore, the probability parameter πj,k in Eq. (15) is given by
| (16) |
where σ(·) is the sigmoid function, sj,k is the vector of epigenetic features for the j-th region in the k-th LD block, and the weight vector w follows a multivariate normal distribution, i.e.,
| (17) |
and Λ follows
| (18) |
In our study, the epigenetic features sj,k were calculated as the overlapping ratios of that region with the narrow peaks of ATAC-seq and histone ChIP-seq, respectively.
Since our modeling is fully Bayesian, all hyperparameters were set to be non-informative, i.e., a0=1×10−6, b0=1×10−6, μ0=0, β0=1,W0=I5, and ν0=5. Based on Eqs. (1) to (18), we are interested in calculating the posterior probability p(T | Z, S) wherein the calculation of integrals is intractable. Here we seek for approximate inference based on the mean-field variational inference (MFVI) (Blei, Kucukelbir and McAuliffe, 2017). To control the false positive rate, we set a hard threshold for with respect to the ATAC-seq signal, where we set if the corresponding region overlaps no ATAC-seq peak. This was motivated by our particular interest in active regions. More technical details, including a coordinate ascent-based inference algorithm, were provided in Supplemental Note.
In this study, we ran the inference algorithm per chromosome to accelerate the computation. The Q+- and Q−-scores were defined as q(t(+1) = 1) and q(t(−1) = 1), respectively, and we also defined the Q-score as Q=Q++Q−. To prioritize RefMap-scored regions, we set a cutoff of 0.95 and defined those regions with either Q+- or Q−-score larger than the cutoff as significant regions (i.e., ALS-associated regions).
Mapping target genes
After identifying ALS-associated genomic regions using RefMap, we linked those regions to their target genes. Mapping to target genes was performed based on two principles: (i) a region was mapped to a particular gene if the region overlaps the gene or an area +/− 10kb either side of the gene body; (ii) a region was mapped to a particular gene if the region overlapped a loop anchor harboring the transcription start site (TSS) of that gene. Loops were identified from Hi-C data profiling of iPSC-derived MNs. Only expressed transcripts/genes (TPM>1) were taken forward for downstream analysis.
Benchmarking details
MAGMA (v1.08) (de Leeuw et al., 2015) and Pascal (Lamparter et al., 2016) were applied using default settings. Input consisted of summary statistics for all SNPs as measured in our ALS GWAS (van Rheenen et al., 2016). We employed PAINTOR (v3.0) following the guidance provided in (Kichaev et al., 2014) and https://github.com/gkichaev/PAINTOR_V3.0/. The genome was annotated based on the epigenetic features (ATAC-seq, H3K27ac, H3K4me1 and H3K4me3 ChIP-seq peaks) in MNs. We ran the algorithm in MCMC mode and specified the number of casuals to be 3. All other parameters in PAINTOR were left to be default. In all cases, we estimated the LD structure using EUR samples from the 1000 Genomes Project phase 3.
Heritability analysis
Partitioned heritability analysis was carried out using LD score regression as previously described (Finucane et al., 2015). Heritability was quantified within RefMap ALS genes and significant GWAS loci, respectively. As a control, we calculated the heritability linked to genes associated with significant eQTLs within spinal cord tissue (GTEx v7). GTEx eQTLs were first ranked by P-values along with their associated genes, and then the top 690 unique genes were retrieved to match the number of RefMap genes.
Transcriptome analysis
All transcriptomic data after QC were downloaded from original studies (Prudencio et al., 2015; Maniatis et al., 2019). Fold change was calculated as the ratio of gene expression levels in ALS cases compared to controls.
For AnswerALS data, gene expression profiling of iPSC-derived MNs and phenotype data were obtained for 55 ALS patients and 15 controls (https://www.answerals.org/). Gene expression was normalized for gene length and then sequencing depth to produce transcripts per kilobase million (TPM). Age of onset and disease status were available for all individuals and these parameters were used to check for the correlation between expression of top-ranked RefMap ALS genes and age at disease onset. For each of ADAMTSL1, BNC2, KANK1, and VAV2, we performed a Pearson correlation analysis to determine whether gene expression within MNs was significantly associated with age of disease onset. As a control, we selected 10,000 random sets of four expressed genes in MNs and compared the product of P-values to the equivalent value for ADAMTSL1, BNC2, KANK1, and VAV2.
For the SOD1-G93A-ALS mouse model data (Maniatis et al., 2019), four time points were sampled, including presymptomatic (p30), onset (p70), symptomatic (p100), and end-stage (p120). The model-estimated expression levels (β) from the original paper were adopted to quantify the gene expression difference (Δβ) between diseased and control mice at different time points. To determine the expression changes of RefMap genes over the course of ALS pathogenesis, we first mapped RefMap genes to their mouse homologs (n=510), and then performed unsupervised clustering based on gene expression levels over time.
Network analysis
We downloaded the human PPIs from STRING v11.0 (Szklarczyk et al., 2019), including 19,567 proteins and 11,759,455 protein interactions. We extracted high-confidence (combined score >700) PPIs for all downstream analysis, including 17,161 proteins and 839,522 protein interactions. To eliminate the bias caused by hub proteins (Krishnan et al., 2016), we first carried out the random walk with restart algorithm (Wang et al., 2015) over the PPI network, wherein the restart probability was set to 0.5, resulting in a smoothed network after preserving the top 5% predicted edges. To decompose the network into different subnetworks/modules, we performed the widely-used Louvain algorithm (Blondel et al., 2008), a classic community detection algorithm which searches for densely connected modules by optimizing the modularity. After the algorithm converged, we obtained 912 modules with an average size of 18.39 nodes (Table S5). Two modules (M421 and M604) are significantly enriched (FDR<0.1) with our RefMap genes based on the hypergeometric test followed by the BH correction.
As a negative control, we constructed 100 shuffled networks by randomly rewiring the PPI network while keeping the same number of neighbors (Milo et al., 2002). None of the randomized networks achieved the same modularity of our smoothed network after clustering, demonstrating the significance of our derived gene modules (P<1e-6; Figure S3A).
Identification of rare deleterious variants and rare variant association testing
For analysis of WGS data from 5,594 sporadic ALS patients and 2,238 controls (Project MinE ALS Sequencing Consortium, 2018), variants within promoter, enhancer and coding regions were determined to be rare if the minor allele frequency (MAF) within the Genome Aggregation Database (gnomAD) is <1/100 control alleles (Lek et al., 2016). Additional filtering varied reflecting differences in function between promoter, enhancer and coding sequences. In promoter regions, we utilized two independent scores for functionality and pathogenicity: variants were included in association testing if their CADD (Rentzsch et al., 2019) score >25 and GWAVA (Ritchie et al., 2014) score >0.5. In enhancer regions, variants were included only if evolutionary conserved based on a LINSIGHT score >0.8 (Huang, Gulko and Siepel, 2017). We also utilized an independently compiled score for ALS-associated regulatory variation (Chen, Jin and Qin, 2016): variants were excluded with a DIVAN score <0.5. In coding regions, we annotated variants using VEP (McLaren et al., 2016); LoF variants were defined as nonsense mutations, high-effect splice-site mutations (Jaganathan et al., 2019), or 5’UTR variants involving a gain/loss of a start/stop codon (Zhang et al., 2020). The optimal unified test (SKAT-O) was used to perform rare variant association testing in promoter and enhancer regions because it is optimized for large numbers of samples and for regions where a significant number of variants may not be causal (Lee et al., 2012). SKAT-O tests upweight significance of rare variants according to a beta density function of MAF, i.e., wj = Beta(pj, a1, a2), where pj is the estimated MAF for SNPj using all cases and controls, parameters a1 and a2 are prespecified. Here a2=2500 was chosen for all statistical tests. In coding regions where all LoF variants were proposed to be significant, we applied Firth logistic regression because SKAT-O can lose power when variants are expected to have equivalent functional impact (Basu and Pan, 2011). To adjust for confounders including population structure we used the first ten eigenvectors generated by principal components analysis of common variants as covariates. Sequencing platform and sex were also included as covariates.
Analysis of the exome sequencing data was performed as previously described (Farhan et al., 2019). Briefly, rare variants were defined as MAF<0.001% in the exome datasets of DiscovEHR (Dewey et al., 2016) and ExAC (Lek et al., 2016). Burden testing for each gene was performed using a Fisher’s exact test. Burden testing was performed separately for missense and synonymous variants within each gene.
CRISPR/Cas9 editing of SH-SY5Y and iPS cells
Guide RNAs (gRNAs) were designed using the Crispor tool (http://crispor.tefor.net/) to target KANK1 regulatory and coding regions and BNC2 coding regions. Design was guided by proximity to patient enhancer mutation sites, available protospacer adjacent motifs (PAM), and predicted on- and off-target efficiencies.
gRNAs targeting within 30bp either side of the KANK1-enhancer containing ALS-associated mutations (chr9:663,001–664,000, hg19) were considered and screened for editing efficiency. One pair of guide sequences (5’ -UCAUGGGAACUCUUCAAAUA-3’ and 5’-UCAUGGGAACUCUUCAAAUA-3’) was most efficient and chosen for subsequent experimentation. Validated, commercially available CRISPR control targeting HPRT (IDT), BNC2-exon targeting (IDT, 5’-GTTCGGAACCAGAACGACTA) and KANK1 exon-targeting (IDT, 5’-GUCUAGUUGAUAACCAUAGG-3’) gRNAs were also obtained. gRNA duplexes were assembled from tracrRNA and crRNA in a thermocycler according to manufacturer’s instructions under RNAse-free conditions. Cells were cultured to ensure 70–90% confluency on the day of transfection. iPSCs were pre-treated with 2μM ROCK inhibitor for 2 hours prior to electroporation. 24-well plates containing either 500uL antibiotic-free DMEM (Lonza) (SH-SY5Y) or 500μL complete mTeSR-Plus Medium (StemCell Technologies) (iPSC) were incubated at 37°C. CRISPR/Cas9 Ribonucleoproteins were formed by complexing 240ng gRNA duplex with 1250ng Alt-R V3 Cas9 Protein (IDT) in 10μL buffer R (from 10μL Neon transfection kit, ThermoFisher Scientific) - a 1:1 molar ratio - for 10 minutes. 100,000 viable cells were aliquoted per transfection and centrifuged at either 400 × g for 4 minutes (SH-SY5Y) or 200 × g for 3 minutes (iPSC). Cells were washed in calcium- and magnesium-free Dulbecco’s Phosphate Buffered Saline (Sigma) and centrifuged at either 400 × g for 4 minutes (SH-SY5Y) or 200 × g for 3 minutes (iPSC). Cell pellets were resuspended in 10μL buffer R containing Cas9 protein and gRNA duplexes. 2μL of 10.8μM electroporation enhancer (IDT) was added and the solution mixed thoroughly to ensure a suspension of single cells. 10μL of this mixture was loaded into a Neon transfection system (ThermoFisher Scientific) and electroporated according to manufacturer’s instructions (1200V, 3 pulse, 20s pulse width for SH-SY5Y cells; 1400V, 3 pulse, 5ms pulse width for iPSCs). Cells were transferred to pre-warmed media in 24-well plates for expansion. For iPSCs, media was replaced with fresh mTeSR1 without ROCK inhibitor 24 hours post electroporation. For SH-SY5Ys, media was replaced with fresh antibiotic-free DMEM 48 hours post electroporation.
Determining CRISPR editing efficiency
Genomic DNA was isolated from edited and control cells using a GenElute Mammalian DNA Miniprep Kit (Sigma) according to manufacturer’s instructions. A ~400bp region around the expected cas9 cut site was amplified by polymerase chain reaction using VeriFi mix (PCRbio). Expected amplification was confirmed using gel electrophoresis, and the products were Sanger-sequenced. Sequencing trace files were uploaded to both TIDE (Brinkman et al., 2014) and ICE (https://ice.synthego.com), and an indel efficiency calculated.
Quantitative PCR (RT-PCR)
Cells were cultured until at least 70% confluent, lysed on ice using an appropriate volume of Tri Reagent (Sigma) for 5 minutes and transferred to 1.5ml RNAse-free tubes. Total RNA was extracted using a Direct-zol RNA Miniprep Kit (Zymo) according to manufacturer’s instructions, and RNA concentration confirmed using a NanoDrop spectrophotometer (ThermoFisher Scientific). 2μg of total RNA was then converted to cDNA by adding 1μL 10mM dNTPs, 1μL 40μM random hexamer primer (ThermoFisher Scientific), and DNAse/RNAse-free water to a total volume of 14μL. This mixture was heated for 5 minutes at 70°C then placed on ice for 5 minutes. 4μL of 5x FS buffer, 2μL 0.1M DTT, and 1μL M-MLV reverse transcriptase (200U/μL) (ThermoFisher Scientific) were then added and cDNA conversion performed in a PCR thermocycler (37°C for 60 minutes, 85°C for 10 minutes). cDNA was amplified using RT-PCR with Brilliant III SYBR Green (Agilent) as per manufacturer’s instructions. Ct analysis was performed using CFX Maestro software (BioRad). GAPDH was chosen as a reference gene because expression is relatively stable in SH-SY5Y cells (Hoerndli et al., 2004). GAPDH and β-actin were both tested as endogenous controls in iPSC-derived motor neurons, with no significant differences observed in their expression levels (data not shown); expression data from iPSC-derived cells in this manuscript were normalised to GAPDH.
SH-SY5Y neuronal differentiation
Human SH-SY5Y neuroblastoma cells were seeded at densities of either 5×104 cells per well of a 6-well culture plate, or 2×103 cells per well of a 96-well culture plate in DMEM (Lonza) supplemented with 10% (v/v) FBS, 50 units/mL penicillin and 50 μg/mL of streptomycin. 24 hours after seeding the media was changed to DMEM supplemented with 5% (v/v) FBS, 50 units/mL penicillin, 50 μg/mL of streptomycin, 4mM l-glutamine and 10μM retinoic acid. After 72 hours, the medium was switched to neurobasal media (ThermoFisher Scientific) containing 1% (v/v) N-2 supplement 100x, 50 units/mL penicillin, 50 μg/mL of streptomycin, 1% l-glutamine and 50ng/mL human BDNF. Cells were cultured for an additional 3 days until fully differentiated.
Immunocytochemistry for SH-SY5Y cells
SH-SY5Y cells were fixed with 4% paraformaldehyde for 15 minutes and washed 3x with PBS. Cells were blocked in 5% normal horse serum containing 0.1% Triton X-100 for 1 hour at RT. All primary antibodies were diluted in blocking solution (α-tubulin, 1:2000; anti-Pax6, 1:200). Cells were incubated in the primary antibody for 2 hours at RT and washed 3x in PBS before incubation in the appropriate secondary antibody (1:1000 in PBS) for 1 hour at RT. Nuclear counterstain (Hoechst 33342) was applied for 10 minutes followed by a 3x wash in PBS. Cells were imaged using an Opera Phenix High Content Screening System (PerkinElmer).
Immunocytochemistry for iPS-derived cells
For immunostaining, neural progenitor cells (NPC) and motor neurons (MN) were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 10 min at room temperature. After fixation samples were washed three times with PBS and permeabilized with 0.3% Triton X-100 diluted in PBS for 5 min. The cells were subsequently blocked in 5% Donkey serum for 1h at room temperature. After blocking, cell cultures were incubated with the appropriate primary antibodies diluted in PBS containing 5% of DS overnight. Cells were then washed with PBS three times. Fluorescent secondary antibodies (Alexa Fluor 488, 555, 594 or 647, diluted 1:400 with DS) were subsequently added to the cells and incubated for 1h. The samples were washed with PBS three more times and incubated with Hoechst 33342 for nuclear staining for 5 minutes. All experiments included cultures where the primary antibodies were not added, non-specific staining was not observed in these negative controls. Images were obtained from the Opera Phenix™ High Content Screening System at × 40 magnification using the Harmony™ Image analysis system. We used 405, 488 and 594 nm and 647 lasers, along with the appropriate excitation and emission filters. These settings were kept consistent while taking images from all cultures.
High-content image screening (HCS)
To investigate whether introduced KANK1 mutations recapitulate MN death observed in ALS patients, MN were kept in medium with and without neurotrophic factors (IGF, BDNF and CNTF) and then were stained for active caspase 3, a classical apoptotic marker. MN cells were plated on matrigel-coated 96-well plates. On day 40, MNs were fixed and stained for active caspase 3 and MAP2, which was used as a marker that defines the boundary of cells and DAPI for nuclear staining. Quantitative imaging analysis of the MN was conducted through the Opera Phenix™ High Content Screening System at × 40 magnification using the Harmony™ Image analysis system. The following morphological features were assessed for all the groups (Isogenic control, HPRT, Exon-edited cells): percentage Caspase 3 positive cells and the number of fragmented nuclei. At least 25 fields were randomly selected and scanned per well of a 96-well plate in triplicate. To identify and remove any false readings generated by the system, three random treated and untreated wells were selected and counted manually (blind to group).
To investigate whether the introduced KANK1 mutations recapitulate the nuclear loss of TDP-43 observed in ALS patients MN were stained for TDP-43. On day 40, MN were fixed and stained for TDP-43, MAP2 which was used as a marker to define the cytoplasmic boundary of cells, and DAPI for nuclear staining. A quantitative imaging analysis of the MN was conducted through the Opera Phenix™ High Content Screening System at × 40 magnification using the Harmony™ Image analysis system. The following morphological features were assessed for all the groups (Isogenic control, HPRT, Exon-edited cells): Nuclear TDP-43 intensity (Arbitrary Fluorescence Unit) and ratio of Nuclear/Cytoplasmic intensity. At least 25 fields were randomly selected and scanned per well of a 96-well plate in triplicate.
MTT assays
A colorimetric assay using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) dye was used to assess neuronally differentiated SH-SY5Y cellular metabolic activity and hence neuronal viability. 55 μL of 5mg/mL of MTT reagent in PBS was added per well of a 24-well culture plate and incubated at 37°C for 1 hour. 550 μL of unprecipitated 20% SDS in 50% di-methyl formamide (DMF) + dH2O (pH 7.4) was added per well and mixed thoroughly to lyse the cells. Cells were incubated in a dark environment on an orbital shaker for 1 hour. The colorimetric change was measured using a PHERAstar FS spectrophotometer (BMG Biotech), and absorbance readings taken at 590nm were normalized to media-only wells. Mean absorbance readings were calculated for each biological repeat and expressed as a percentage of controls.
Patch-clamp electrophysiology for iPSC-derived motor neurons
Whole-cell patch-clamp recordings were performed in the current-clamp configuration and were performed as described (Bilican et al., 2014; Perkins et al., 2021) using electrodes filled with (in mM): 155 K-gluconate, 2 MgCl2, 10 Na-HEPES, 10 Na-PiCreatine, 2 Mg2-ATP, and 0.3 Na3-GTP, pH 7.3, 300 mOsm. Cells were typically bathed in an extracellular recording solution comprising (in mM): 152 NaCl, 2.8 KCl, 10 HEPES, 2 CaCl2, 1.5 MgCl2, 10 glucose, pH 7.3, 320–330 mOsm and supplemented with picrotoxin (50 μM) CNQX (5 μM) and D-APV (50 μM) to block synaptic activity.
Recordings were performed at room temperature (20–23 °C). Measurements were typically low-pass filtered online at 2 kHz, digitized at 10 kHz and recorded to computer using the WinEDR V2 7.6 Electrophysiology Data Recorder (J. Dempster, Department of Physiology and Pharmacology, University of Strathclyde, UK; www.strath.ac.uk/Departments/PhysPharm/). Recordings were omitted from analysis if the series resistance changed by more than 20% during the experiment, or if the resistance exceeded 20 MΩ.
QUANTIFICATION AND STATISTICAL ANALYSIS
Morphological assessment of differentiated SH-SY5Y cells and iPSC-derived motor neurons
To confirm neuronal differentiation and to assess for changes consistent with axonopathy, semi-automated analysis of neurite length was performed using the SimpleNeuriteTracer plugin for FIJI (Longair, Baker and Armstrong, 2011). 2D images were converted to 8-bit grayscale and successive points along the midline of a neural process were selected. The software automatically identified the path between the two points. Tracing accuracy was improved using Hessian-based analysis of image curvatures. The AnalyzeSkeleton plugin (Arganda-Carreras et al., 2010) was used to quantify the morphology of the traces including the length of neurites. In the case of joined neurites, the shorter path length was assigned to ‘branches’. To determine whether observed changes in neurite length are significant, three fields of view were analyzed and differences were assessed by a Student’s t-test, where a one-tailed test was chosen based on the hypothesis that ALS-associated mutations would reduce neurite length.
Quantitative PCR and MTT assays
Relative mRNA expression values were calculated using the 2−ΔΔCT method (Schmittgen and Livak, 2008). Statistical analysis was conducted in GraphPad Prism 7 (La Jolla, CA) and R (v4.0.2). All bar plots show the mean ± standard deviation. To identify statistical differences between treatment groups the Student’s t-test was utilized.
Supplementary Material
Table S1 Quality control measures for RNA-seq, ATAC-seq, histone ChIP-seq and Hi-C of iPSC-derived motor neurons, Related to Figure 1.
Table S2 ALS-associated regions and genes identified by RefMap, and manually curated ALS gene list with evidence for association, including references, Related to Figure 1.
Table S3 ALS genes discovered by established methods, Related to Figure 1.
Table S4 Clustering of RefMap homologs using transcriptomic data from SOD1-G93A ALS mouse model, Related to Figure 3.
Table S5 Gene modules detected by the Louvain algorithm, including modules enriched with RefMap ALS genes, Related to Figure 4.
Table S6 Rare variant association testing for RefMap genes in Project MinE and the ALS exome cohorts, Related to Figure 5.
Supplemental Note Additional method details, Related to STAR Methods.
KEY RESOURCES TABLE
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Anti-H3K4me1 | Cell Signaling Technologies | #5326S, lot 3 |
| Anti-H3K4me3 | Cell Signaling Technologies | #9751S, lot 10 |
| Anti-H3K27ac |
ActiveMotif | #93133, lot 28518012 |
| Nestin | Biolegend | #841901 |
| Pax6 | Millipore | #MAB5552 |
| Anti-Beta III Tubulin | Millipore | #AB9354 |
| Pax6 | Abcam | #AB5790 |
| Anti-Alpha Tubulin | Sigma | #T9206 |
| Islet 1/2 | Abcam | #AB109517 |
| NeuN | Millipore | #MAB377 |
| Chat | Millipore | #AB144P |
| SMI32 | Biolegend | #801701 |
| phospho TDP-43 (Ser409) | Affinity Biosciences | #AF7365 |
| TDP-43 | Proteintech | #12892-1-AP |
| Anti-MAP-2 | Synaptic systems | #188004 |
| Caspase 3 | Millipore | #AB3623 |
| Donkey anti-Mouse IgG Alexa Fluor 488 | Thermofisher | #A-21202 |
| Donkey anti-Rabbit IgG Alexa Fluor 568 | Thermofisher | #A-10042 |
| Donkey anti-Goat IgG Alexa Fluor 555 | Thermofisher | #A-21432 |
| Goat anti-Guinea Pig IgG Alexa Fluor 647 | Thermofisher | #A-21450 |
| Donkey anti-Mouse IgG Alexa Fluor 594 | Thermofisher | #A-32744 |
| Donkey anti-Rabbit IgG, Alexa Fluor 488 | Thermofisher | #A-21206 |
| Donkey anti-Mouse IgG Alexa Fluor 568 | Thermofisher | #A-10037 |
| Goat anti-Chicken IgY (H+L) Alexa Fluor Plus 488 | Thermofisher | #A-32931 |
| Chemicals, Peptides and Recombinant Proteins | ||
| Thiazolyl Blue Tetrazolium Bromide (MTT) | Sigma-Aldrich | #M2128 |
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA technologies | #1081059 |
| Alt-R Cas9 Electroporation Enhancer | Integrated DNA technologies | #1075915 |
| Dulbecco’s Modified Eagle medium | Lonza | #12-604F |
| KnockOut DMEM/F-12 | ThermoFisher Scientific | #12660012 |
| Neurobasal medium | ThermoFisher Scientific | #12348017 |
| Penicillin-Streptomycin | Sigma | #P4333 |
| Fibronectin | Merck | #FC010 |
| 10x Trypsin | Sigma | #59427C |
| Foetal bovine serum | ThermoFisher Scientific | #10270106 |
| Matrigel | Corning | #356230 |
| mTeSR-Plus Medium | StemCell Technologies | #05825 |
| ReLeSR | StemCell Technologies | #05872 |
| Ethidium bromide solution | Sigma | #E1510 |
| VeriFi mix red | PCRBio | #PB10.42-01 |
| Tri reagent | Sigma | #93289-100ML |
| M-MLV reverse transcriptase | ThermoFisher Scientific | #28025-013 |
| 5x First Strand buffer | ThermoFisher Scientific | #18057-018 |
| 0.1M Dithiothreitol | ThermoFisher Scientific | #707265ML |
| dNTP Mix | ThermoFisher Scientific | #10534823 |
| SYBR Green Brilliant III master mix | Agilent | #600882 |
| Random hexamer primer | ThermoFisher Scientific | #SO142 |
| Purmorphamine | Tocris Bioscience | #4551 |
| StemPro Accutase Cell Dissociation Reagent | Gibco | #A1110501 |
| ROCK inhibitor (Y-27632 dihydrochloride) | Tocris Bioscience | #1254 |
| Compound E | Tocris Bioscience | #6476 |
| NEBNext 2xMasterMix | New England Biolabs | M0541 |
| EDTA | Sigma | #E5134 |
| HEPES | Sigma | #H3375 |
| PMSF protease inhibitor | ThermoFisher Scientific | #36978 |
| Protease inhibitor tablet | Roche | #1697498 |
| Gibco GlutaMAX Supplement | ThermoFisher Scientific | #35050061 |
| TracrRNA | Integrated DNA technologies | #1072533 |
| TE Buffer, RNAse-free pH 8 | ThermoFisher Scientific | #AM9849 |
| Dulbecco’s Phosphate Buffered Saline | Sigma | #D8537-500ML |
| Triton X-100 | Sigma-Aldrich | #T8787 |
| Normal horse serum | Vector | #S-2000-20 |
| Hoechst 33342 | ThermoFisher Scientific | #62249 |
| All-trans retinoic acid | Sigma | #R2625 |
| BDNF | PeproTech | #450-02 |
| IGF | ThermoFisher Scientific | #PHG0078 |
| CNTF | ThermoFisher Scientific | #PHC7015 |
| N-2 supplement | ThermoFisher Scientific | #17502048 |
| B-27 supplement | ThermoFisher Scientific | #17504001 |
| DMH-1 | Tocris Bioscience | #4126 |
| SB431542 | Tocris Bioscience | #1614 |
| CHIR99021 | Tocris Bioscience | #4423 |
| Critical Commercial Assays | ||
| Pierce BCA Assay Protein Assay Kit | ThermoFisher Scientific | #23225 |
| GenElute Mammalian Genomic DNA Miniprep Kit | Sigma | #G1N350 |
| Direct-zol RNA Miniprep Kit | Zymo Research | #R2050 |
| Neon Transfection System 10 μL Kit | ThermoFisher Scientific | #MPK1096 |
| Alt-R CRISPR-Cas9 Control Kit, Human, 2 nmol | Integrated DNA technologies | #1072554 |
| QIAquick PCR Purification kit | Qiagen | #28104 |
| MiElute kit. | Qiagen | #28004 |
| KAPA Library Quantification kit | Roche | #07960140001 |
| KAPA HiFi HotSTARt ReadyMix | Roche | #07958927001 |
| KAPA Library Amplification Primer Mix | Roche | #07958978001 |
| QIAquick Gel Extraction Kit | Qiagen | #28506 |
| Ribo-Zero rRNA depletion kit | Illumina | #20040526 |
| NEBext Ultra RNA prep kit | New England Biolabs | #E7530 |
| Experimental Models: Cell Lines | ||
| SH-SY5Y | ATCC | Cat.#CRL-2266 |
| GM23338 (iPSC line derived from healthy volunteer) | Coriell Institute | #CVCL_F182 |
| Epigenetic profiling see Figure S1A | ||
| Software and Algorithms | ||
| Sickle v1.200 | https://github.com/najoshi/sickle | |
| Cutadapt v1.2.1 | https://pypi.org/project/cutadapt/1.2.1/ | |
| Kallisto v0.46.0 | https://pachterlab.github.io/kallisto/ | |
| SKAT-O | https://cran.r-project.org/web/packages/SKAT/index.html | |
| R v4.0.1 | https://cran.r-project.org/mirrors.html | |
| snpStats | https://www.bioconductor.org/packages/release/bioc/html/snpStats.html | |
| VariantAnnotation | https://www.bioconductor.org/packages/release/bioc/html/VariantAnnotation.html | |
| VAutils | https://github.com/oyhel/vautils/ | |
| PLINK V1.90 | http://zzz.bwh.harvard.edu/plink/download.shtml | |
| PRISM 7 | GraphPad | |
| ICE CRISPR analysis tool | https://ice.synthego.com/#/ | |
| CRISPOR guide RNA design tool | http://crispor.tefor.net/ | |
| CFX Maestro | Bio-Rad | |
| Harmony Imaging Analysis Software | PerkinElmer | |
| FIJI (FIJI Is Just ImageJ) | NIH | |
| IGV v2.4.16 | https://software.broadinstitute.org/software/igv/ | |
| MATLAB R2018b | MathWorks | |
| MAGMA v1.08 | https://ctg.cncr.nl/software/magma | |
| Pascal | https://www2.unil.ch/cbg/index.php?title=Pascal | |
| PAINTOR v3.0 | https://github.com/gkichaev/PAINTOR_V3.0 | |
| LD Score Regression | https://github.com/bulik/ldsc | |
| RefMap | https://github.com/szhang1112/refmap | DOI: 10.5281/zenodo.5774249 |
Highlights:
Machine learning method identifies risk genes by integrating GWAS and epigenetic data
Discovered ALS risk genes lead to a 5-fold increase in recovered heritability
Genetic and experimental support for initiation of ALS pathogenesis in the distal axon
Convergent genetic and experimental data establish KANK1 as a new ALS gene
ACKNOWLEDGEMENTS
We thank the anonymous reviewers for all the constructive comments. This work used the Genome Sequencing Service Center by Stanford Center for Genomics and Personalized Medicine Sequencing Center, supported by the grant award NIH S10OD025212, and NIH/NIDDK P30DK116074. We acknowledge the Stanford Genetics Bioinformatics Service Center for providing computational infrastructure for this study, including the UV-300 supercomputer supported by NIH 1S10OD023452-01. We thank J. Adrian for the help to initiate the project. We also thank J. Zhai and X. Yang for the help with histone ChIP-seq assays, and I. Gabdank and M. Kagda for running the Hi-C pipeline. This work was supported by the National Institutes of Health (CEGS 5P50HG00773504, 1P50HL083800, 1R01HL101388, 1R01-HL122939, S10OD025212, P30DK116074, and UM1HG009442 to M.P.S.), the Wellcome Trust (216596/Z/19/Z to J.C.-K.), and NIHR (NF-SI-0617-10077 to P.J.S.). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement n° 772376 - EScORIAL). The collaboration project is co-funded by the PPP Allowance made available by Health~Holland, Top Sector Life Sciences & Health, to stimulate public-private partnerships. This study was also supported by the ALS Foundation Netherlands, by research grants from IWT (n° 140935), the ALS Liga België, the National Lottery of Belgium, the KU Leuven Opening the Future Fund. We also acknowledge support from a Kingsland fellowship (T.M.), the My Name’5 Doddie Foundation (J.F.), and the NIHR Sheffield Biomedical Research Centre for Translational Neuroscience. Biosample collection was supported by the MND Association and the Wellcome Trust (P.J.S.). We are very grateful to the ALS patients and control subjects who generously donated biosamples. We acknowledge transcriptomic data provided by the AnswerALS Consortium. Figures 1A, 4E, 6A and 7A were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.P.S is a co-founder and member of the scientific advisory board of Personalis, Qbio, January, SensOmics, Protos, Mirvie, NiMo, Onza and Oralome. He is also on the scientific advisory board of Danaher, Genapsys and Jupiter. J.L. is a cofounder of SensOmics. No other authors have competing interests.
REFERENCES
- Arganda-Carreras I et al. (2010) ‘3D reconstruction of histological sections: Application to mammary gland tissue’, Microscopy Research and Technique, pp. 1019–1029. doi: 10.1002/jemt.20829. [DOI] [PubMed] [Google Scholar]
- Basu S and Pan W (2011) ‘Comparison of statistical tests for disease association with rare variants’, Genetic epidemiology, 35(7), pp. 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C et al. (2016) ‘FINEMAP: efficient variable selection using summary data from genome-wide association studies’, Bioinformatics, 32(10), pp. 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C et al. (2017) ‘Prospects of Fine-Mapping Trait-Associated Genomic Regions by Using Summary Statistics from Genome-wide Association Studies’, American journal of human genetics, 101(4), pp. 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkum NL et al. (2010) ‘Hi-C: a method to study the three-dimensional architecture of genomes’, Journal of visualized experiments: JoVE, (39). doi: 10.3791/1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilican B et al. (2014) ‘Physiological normoxia and absence of EGF is required for the long-term propagation of anterior neural precursors from human pluripotent cells’, PloS one, 9(1), p. e85932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blei DM, Kucukelbir A and McAuliffe JD (2017) ‘Variational Inference: A Review for Statisticians’, Journal of the American Statistical Association, pp. 859–877. doi: 10.1080/01621459.2017.1285773. [DOI] [Google Scholar]
- Blondel VD et al. (2008) ‘Fast unfolding of communities in large networks’, Journal of statistical mechanics, 2008(10), p. P10008. [Google Scholar]
- Boopathy S et al. (2015) ‘Structural basis for mutation-induced destabilization of profilin 1 in ALS’, Proceedings of the National Academy of Sciences of the United States of America, 112(26), pp. 7984–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL et al. (2016) ‘Near-optimal probabilistic RNA-seq quantification’, Nature biotechnology, 34(5), pp. 525–527. [DOI] [PubMed] [Google Scholar]
- Briese M et al. (2020) ‘Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function’, Acta neuropathologica communications, 8(1), p. 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK et al. (2014) ‘Easy quantitative assessment of genome editing by sequence trace decomposition’, Nucleic acids research, 42(22), p. e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR (1994) ‘El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors’, Journal of the neurological sciences, 124 Suppl, pp. 96–107. [DOI] [PubMed] [Google Scholar]
- Bryois J et al. (2020) ‘Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease’, Nature genetics, 52(5), pp. 482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD et al. (2015) ‘ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide’, Current protocols in molecular biology / edited by Frederick M. Ausubel … [et al.], 109, pp. 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan B et al. (2015) ‘An atlas of genetic correlations across human diseases and traits’, Nature genetics, 47(11), pp. 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan BK et al. (no date) ‘LD Score Regression Distinguishes Confounding from Polygenicity in Genome-Wide Association Studies’. doi: 10.1101/002931. [DOI] [PMC free article] [PubMed]
- Chandra Roy B, Kakinuma N and Kiyama R (2009) ‘Kank attenuates actin remodeling by preventing interaction between IRSp53 and Rac1’, The Journal of cell biology, 184(2), pp. 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Jin P and Qin ZS (2016) ‘DIVAN: accurate identification of non-coding disease-specific risk variants using multi-omics profiles’, Genome biology, 17(1), p. 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W et al. (2016) ‘Incorporating Functional Annotations for Fine-Mapping Causal Variants in a Bayesian Framework Using Summary Statistics’, Genetics, pp. 933–958. doi: 10.1534/genetics.116.188953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, G. and GTEx Consortium (2017) ‘Genetic effects on gene expression across human tissues’, Nature, pp. 204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J et al. (2014) ‘Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions’, Brain: a journal of neurology, 137(Pt 7), pp. 2040–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J et al. (2020) ‘Rare Variant Burden Analysis within Enhancers Identifies CAV1 as a New ALS Risk Gene’. doi: 10.2139/ssrn.3606796. [DOI] [PMC free article] [PubMed]
- Cooper-Knock J, Jenkins T and Shaw PJ (2013) Clinical and Molecular Aspects of Motor Neuron Disease. Biota Publishing. [Google Scholar]
- Corces MR et al. (2020) ‘Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases’, Nature genetics, 52(11), pp. 1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP et al. (2010) ‘Histone H3K27ac separates active from poised enhancers and predicts developmental state’, Proceedings of the National Academy of Sciences of the United States of America, 107(50), pp. 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoud H et al. (2010) ‘Analysis of the UNC13A gene as a risk factor for sporadic amyotrophic lateral sclerosis’, Archives of neurology, 67(4), pp. 516–517. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M et al. (2011) ‘Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS’, Neuron, 72(2), pp. 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin A-C et al. (2015) ‘Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability’, Nature communications, 6, p. 5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ and Hafezparast M (2017) ‘Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research?’, Neurobiology of disease, 105, pp. 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey FE et al. (2016) ‘Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study’, Science, 354(6319). doi: 10.1126/science.aaf6814. [DOI] [PubMed] [Google Scholar]
- Diekstra FP et al. (2012) ‘UNC13A is a modifier of survival in amyotrophic lateral sclerosis‘, Neurobiology of aging, 33(3), pp. 630.e3–8. [DOI] [PubMed] [Google Scholar]
- Dillon C and Goda Y (2005) ‘The actin cytoskeleton: integrating form and function at the synapse’, Annual review of neuroscience, 28, pp. 25–55. [DOI] [PubMed] [Google Scholar]
- Du Z-W et al. (2015) ‘Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells’, Nature communications, 6, p. 6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitan C et al. (2021) ‘Non-Coding Genetic Analysis Implicates Interleukin 18 Receptor Accessory Protein 3’UTR in Amyotrophic Lateral Sclerosis’, bioRxiv. Available at: https://www.biorxiv.org/content/10.1101/2021.06.03.446863v1.abstract. [Google Scholar]
- Elden AC et al. (2010) ‘Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS’, Nature, 466(7310), pp. 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium et al. (2020) ‘Expanded encyclopaedias of DNA elements in the human and mouse genomes’, Nature, 583(7818), pp. 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadista J et al. (2017) ‘LoFtool: a gene intolerance score based on loss-of-function variants in 60 706 individuals’, Bioinformatics, 33(4), pp. 471–474. [DOI] [PubMed] [Google Scholar]
- Farhan SMK et al. (2019) ‘Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein’, Nature neuroscience, 22(12), pp. 1966–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK et al. (2015) ‘Partitioning heritability by functional annotation using genome-wide association summary statistics’, Nature genetics, 47(11), pp. 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishilevich S et al. (2017) ‘GeneHancer: genome-wide integration of enhancers and target genes in GeneCards’, Database: the journal of biological databases and curation, 2017. doi: 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster JI et al. (2016) ‘Characterization of Differentiated SH-SY5Y as Neuronal Screening Model Reveals Increased Oxidative Vulnerability’, Journal of biomolecular screening, 21(5), pp. 496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey D et al. (2000) ‘Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases’, The Journal of neuroscience: the official journal of the Society for Neuroscience, 20(7), pp. 2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori K et al. (2018) ‘Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent’, Nature medicine, 24(10), pp. 1579–1589. [DOI] [PubMed] [Google Scholar]
- Giampetruzzi A et al. (2019) ‘Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis’, Nature communications, 10(1), p. 3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green EM et al. (2021) ‘TDP-43 represses cryptic exon inclusion in FTD/ALS gene UNC13A’, bioRxiv. Available at: https://www.biorxiv.org/content/10.1101/2021.04.02.438213v1.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME et al. (1994) ‘Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation’, Science, 264(5166), pp. 1772–1775. [DOI] [PubMed] [Google Scholar]
- Han B, Kang HM and Eskin E (2009) ‘Rapid and accurate multiple testing correction and power estimation for millions of correlated markers’, PLoS genetics, 5(4), p. e1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardiman O et al. (2017) ‘Amyotrophic lateral sclerosis’, Nature reviews. Disease primers, 3, p. 17071. [DOI] [PubMed] [Google Scholar]
- Harva M and Kabán A (2007) ‘Variational learning for rectified factor analysis’, Signal Processing, pp. 509–527. doi: 10.1016/j.sigpro.2006.06.006. [DOI] [Google Scholar]
- Heinz S et al. (2015) ‘The selection and function of cell type-specific enhancers’, Nature reviews. Molecular cell biology, 16(3), pp. 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog JJ et al. (2017) ‘TDP-43 misexpression causes defects in dendritic growth’, Scientific reports, 7(1), p. 15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoerndli FJ et al. (2004) ‘Reference genes identified in SH-SY5Y cells using custom-made gene arrays with validation by quantitative polymerase chain reaction’, Analytical biochemistry, 335(1), pp. 30–41. [DOI] [PubMed] [Google Scholar]
- Hormozdiari F et al. (2014) ‘Identifying causal variants at loci with multiple signals of association’, Genetics, 198(2), pp. 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiau T et al. (2019) ‘Inference of CRISPR Edits from Sanger Trace Data. bioRxiv, 251082’. [DOI] [PubMed] [Google Scholar]
- Huang N et al. (2010) ‘Characterising and predicting haploinsufficiency in the human genome’, PLoS genetics, 6(10), p. e1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-F, Gulko B and Siepel A (2017) ‘Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data’, Nature genetics, 49(4), pp. 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan K et al. (2019) ‘Predicting Splicing from Primary Sequence with Deep Learning’, Cell, 176(3), pp. 535–548.e24. [DOI] [PubMed] [Google Scholar]
- Joo JWJ et al. (2016) ‘Multiple testing correction in linear mixed models’, Genome biology, 17, p. 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma N et al. (2009) ‘Kank proteins: structure, functions and diseases’, Cellular and molecular life sciences: CMLS, 66(16), pp. 2651–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ et al. (2020) ‘The mutational constraint spectrum quantified from variation in 141,456 humans’, bioRxiv. doi: 10.1101/531210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichaev G et al. (2014) ‘Integrating functional data to prioritize causal variants in statistical fine-mapping studies’, PLoS genetics, 10(10), p. e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A et al. (2016) ‘Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder’, Nature neuroscience, 19(11), pp. 1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamas NJ et al. (2014) ‘Neurotrophic requirements of human motor neurons defined using amplified and purified stem cell-derived cultures’, PloS one, 9(10), p. e110324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamparter D et al. (2016) ‘Fast and Rigorous Computation of Gene and Pathway Scores from SNP-Based Summary Statistics’, PLoS computational biology, 12(1), p. e1004714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ et al. (2018) ‘ClinVar: improving access to variant interpretations and supporting evidence’, Nucleic acids research, 46(D1), pp. D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S et al. (2012) ‘Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies’, American journal of human genetics, 91(2), pp. 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw CA et al. (2015) ‘MAGMA: generalized gene-set analysis of GWAS data’, PLoS computational biology, 11(4), p. e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-B et al. (2013) ‘Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic’, Cell reports, 5(5), pp. 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M et al. (2016) ‘Analysis of protein-coding genetic variation in 60,706 humans’, Nature, 536(7616), pp. 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J et al. (2019) ‘Gene-Environment Interaction in the Era of Precision Medicine’, Cell, 177(1), pp. 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R et al. (2015) ‘Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis’, Nature genetics, 47(12), pp. 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longair MH, Baker DA and Armstrong JD (2011) ‘Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes’, Bioinformatics, 27(17), pp. 2453–2454. [DOI] [PubMed] [Google Scholar]
- Lopategui Cabezas I, Herrera Batista A and Pentón Rol G (2014) ‘The role of glial cells in Alzheimer disease: potential therapeutic implications’, Neurologia, 29(5), pp. 305–309. [DOI] [PubMed] [Google Scholar]
- Mallik B and Kumar V (2018) ‘Regulation of actin-Spectrin cytoskeleton by ICA69 at the Drosophila neuromuscular junction’, Communicative & Integrative Biology, 11(1), p. e1381806. [Google Scholar]
- Maniatis S et al. (2019) ‘Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis’, Science, 364(6435), pp. 89–93. [DOI] [PubMed] [Google Scholar]
- Martínez-Silva M. de L. et al. (2018) ‘Hypoexcitability precedes denervation in the large fast-ccontracting motor units in two unrelated mouse models of ALS’, eLife, 7. doi: 10.7554/eLife.30955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) ‘Cutadapt removes adapter sequences from high-throughput sequencing reads’, EMBnet.journal, 17(1), pp. 10–12. [Google Scholar]
- McLaren W et al. (2016) ‘The Ensembl Variant Effect Predictor’, Genome biology, 17(1), p. 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AR et al. (2021) ‘Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis’, Acta neuropathologica, 141(2), pp. 257–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed Z et al. (2019) ‘Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration’, Nature neuroscience, 22(2), pp. 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R et al. (2002) ‘Network motifs: simple building blocks of complex networks’, Science, 298(5594), pp. 824–827. [DOI] [PubMed] [Google Scholar]
- Moloney EB, de Winter F and Verhaagen J (2014) ‘ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease’, Frontiers in neuroscience, 8, p. 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujock M et al. (2016) ‘4-Aminopyridine induced activity rescues hypoexcitable motor neurons from amyotrophic lateral sclerosis patient-derived induced pluripotent stem cells’, Stem cells, 34(6), pp. 1563–1575. [DOI] [PubMed] [Google Scholar]
- Neumann M et al. (2006) ‘Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis’, Science, 314(5796), pp. 130–133. [DOI] [PubMed] [Google Scholar]
- Nicolas A et al. (2018) ‘Genome-wide Analyses Identify KIF5A as a Novel ALS Gene’, Neuron, 97(6), pp. 1268–1283.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MM et al. (2015) ‘TREM2 in CNS homeostasis and neurodegenerative disease’, Molecular neurodegeneration, 10, p. 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasaniuc B and Price AL (no date) ‘Dissecting the genetics of complex traits using summary association statistics’. doi: 10.1101/072934. [DOI] [PMC free article] [PubMed]
- Perkins EM et al. (2021) ‘Altered network properties in C9ORF72 repeat expansion cortical neurons are due to synaptic dysfunction’, Molecular neurodegeneration, 16(1), p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S et al. (2013) ‘Genic intolerance to functional variation and the interpretation of personal genomes’, PLoS genetics, 9(8), p. e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK (2014) ‘Joint analysis of functional genomic data and genome-wide association studies of 18 human traits’, American journal of human genetics, 94(4), pp. 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK and Cox NJ (2002) ‘The allelic architecture of human disease genes: common disease–common variant… or not?’, Human molecular genetics, 11(20), pp. 2417–2423. [DOI] [PubMed] [Google Scholar]
- Project MinE ALS Sequencing Consortium (2018) ‘Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis’, European journal of human genetics: EJHG, 26(10), pp. 1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M et al. (2015) ‘Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS’, Nature neuroscience, 18(8), pp. 1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP et al. (2014) ‘A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping’, Cell, 159(7), pp. 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP et al. (2017) ‘Cohesin Loss Eliminates All Loop Domains’, Cell, 171(2), pp. 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P et al. (2019) ‘CADD: predicting the deleteriousness of variants throughout the human genome’, Nucleic acids research, 47(D1), pp. D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen W et al. (2016) ‘Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis’, Nature genetics, 48(9), pp. 1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen W et al. (2021) ‘Common and rare variant association analyses in Amyotrophic Lateral Sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology’, medRxiv, p. 2021.03.12.21253159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie GR et al. (2014) ‘Functional annotation of noncoding sequence variants’, Nature methods, 11(3), pp. 294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M et al. (2019) ‘Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis’, JAMA neurology. doi: 10.1001/jamaneurol.2019.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sances S et al. (2016) ‘Modeling ALS with motor neurons derived from human induced pluripotent stem cells’, Nature neuroscience, 19(4), pp. 542–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D et al. (2014) ‘Human induced pluripotent stem cells are a novel source of neural progenitor cells (iNPCs) that migrate and integrate in the rodent spinal cord’, The Journal of comparative neurology, 522(12), pp. 2707–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ, Chen W and Larson NB (2018) ‘From genome-wide associations to candidate causal variants by statistical fine-mapping’, Nature reviews. Genetics, 19(8), pp. 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD and Livak KJ (2008) ‘Analyzing real-time PCR data by the comparative C(T) method’, Nature protocols, 3(6), pp. 1101–1108. [DOI] [PubMed] [Google Scholar]
- Shepheard SR et al. (2021) ‘Value of systematic genetic screening of patients with amyotrophic lateral sclerosis’, Journal of neurology, neurosurgery, and psychiatry. doi: 10.1136/jnnp-2020-325014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y et al. (2018) ‘Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons’, Nature medicine, 24(3), pp. 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M et al. (2019) ‘Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes’, Nature genetics, 51(8), pp. 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D et al. (2019) ‘STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets’, Nucleic acids research, 47(D1), pp. D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabjerg BB et al. (2020) ‘ALS in Danish Registries: Heritability and links to psychiatric and cardiovascular disorders’, Neurology. Genetics, 6(2), p. e398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J et al. (2021) ‘Common ALS/FTD risk variants in UNC13A exacerbate its cryptic splicing and loss upon TDP-43 mislocalization’, bioRxiv. Available at: https://www.biorxiv.org/content/10.1101/2021.04.02.438170v1.abstract. [Google Scholar]
- Vanhoutteghem A and Djian P (2006) ‘Basonuclins 1 and 2, whose genes share a common origin, are proteins with widely different properties and functions’, Proceedings of the National Academy of Sciences of the United States of America, 103(33), pp. 12423–12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D et al. (2018) ‘Comprehensive functional genomic resource and integrative model for the human brain’, Science, 362(6420). doi: 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L et al. (2011) ‘Gene set analysis of genome-wide association studies: methodological issues and perspectives’, Genomics, 98(1), pp. 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S et al. (2015) ‘Exploiting ontology graph for predicting sparsely annotated gene function’, Bioinformatics, 31(12), pp. i357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M and Snyder M (2009) ‘RNA-Seq: a revolutionary tool for transcriptomics’, Nature reviews. Genetics, 10(1), pp. 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y et al. (2020) ‘An Amyotrophic Lateral Sclerosis-Associated Mutant of C21ORF2 Is Stabilized by NEK1-Mediated Hyperphosphorylation and the Inability to Bind FBXO3’, iScience, 23(9), p. 101491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock MC (2005) ‘Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach’, Journal of evolutionary biology, 18(5), pp. 1368–1373. [DOI] [PubMed] [Google Scholar]
- Zhang X et al. (2020) ‘Annotating high-impact 5’untranslated region variants with the UTRannotator’, Bioinformatics. doi: 10.1093/bioinformatics/btaa783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q et al. (2014) ‘Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells’, BioTechniques, 57(3), pp. 115–124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Quality control measures for RNA-seq, ATAC-seq, histone ChIP-seq and Hi-C of iPSC-derived motor neurons, Related to Figure 1.
Table S2 ALS-associated regions and genes identified by RefMap, and manually curated ALS gene list with evidence for association, including references, Related to Figure 1.
Table S3 ALS genes discovered by established methods, Related to Figure 1.
Table S4 Clustering of RefMap homologs using transcriptomic data from SOD1-G93A ALS mouse model, Related to Figure 3.
Table S5 Gene modules detected by the Louvain algorithm, including modules enriched with RefMap ALS genes, Related to Figure 4.
Table S6 Rare variant association testing for RefMap genes in Project MinE and the ALS exome cohorts, Related to Figure 5.
Supplemental Note Additional method details, Related to STAR Methods.
Data Availability Statement
Data files for ATAC-seq, Hi-C and histone ChIP-seq are available at encodeproject.org with the following link or accessions numbers mentioned below: https://www.encodeproject.org/publications/de19555b-a49f-471c-bfbc-be3b628fe9bf/.
The source code of RefMap can be accessed at https://github.com/szhang1112/refmap.