

Abstract
Humans have coevolved with the trillions of resident microbes that populate every nook and cranny of the body. At each site, the resident microbiota creates a unique ecosystem specialized to its environment, benefiting the development and maintenance of human physiology through harmonious symbiotic relationships with the host. However, when the resident microbiota is perturbed, significant complications may arise with disastrous consequences that affect the local and distant ecosystems. In this context, periodontal disease results in inflammation beyond the oral cavity, such as in the gastrointestinal tract. Accumulating evidence indicates that potentially harmful oral resident bacteria (referred to as pathobionts) and pathogenic immune cells in the oral mucosa can migrate to the lower gastrointestinal tract and contribute to intestinal inflammation. We will review the most recent advances concerning the periodontal connection with intestinal inflammation from microbiological and immunological perspectives. Potential therapeutic approaches that target the connection between the mouth and the gut to treat gastrointestinal diseases, such as inflammatory bowel disease, will be examined. Deciphering the complex interplay between microbes and immunity along the mouth–gut axis will provide a better understanding of the pathogenesis of both oral and gut pathologies and present therapeutic opportunities.
Keywords: inflammatory bowel disease, oral bacteria, oral‐gut axis, periodontitis, systemic interactions
1. INTRODUCTION
The surface of our body is covered by numerous commensal microorganisms, including bacteria, fungi, and viruses. The oral cavity has the second largest commensal bacterial community, harboring over 770 species of bacteria that live in different habitats, including the lips, teeth, tongue, cheeks, and palate. 1 Oral bacteria are primarily members of the phyla Firmicutes, Fusobacteria, Proteobacteria, and Actinobacteria, creating complex ecosystems by adapting to each unique environment. 2 Although the role of the commensal oral bacteria in oral health is yet to be fully understood, the colonization of the bacteria in the oral cavity after birth appears to be essential for the development of the oral mucosal immune system and terminal maturation of the stratified oral epithelium, which is crucial to the establishment of oral mucosal homeostasis. 3 Also, certain types of commensal oral bacteria serve as the first‐line of defense against the colonization of exogenous pathogens by inhibiting the adhesion of pathogens and the production of bactericidal products (eg, bacteriocins, hydrogen peroxide). 4
Like the oral compartment, unique environments in the human gut (eg, nutrient and anaerobic conditions) shape a complex gut microbiota, consisting of the collection of trillions of microbial cells with thousands of bacterial species. It is the largest bacterial community in the human body and plays an essential role in host physiological homeostasis, including the education of the host immune system, nutrient digestion, and defense against colonization by pathogenic microorganisms. 5 , 6 , 7 , 8 Because of its fundamental role in controlling intestinal physiology, disturbance of the gut microbiota, often referred to as gut dysbiosis, has been demonstrated to underlie multiple intestinal pathologies, including irritable bowel syndrome, inflammatory bowel disease (IBD), and colorectal cancer (CRC). The advances in sequencing technologies have revealed an abnormal enrichment of typical oral resident bacteria in the luminal contents and the mucosal tissues of the gut in patients with gut pathologies. 9 Given the studies depicting the pathological impact of certain oral resident bacteria (eg, Porphyromonas gingivalis and Fusobacterium nucleatum) on gut homeostasis, it is conceivable that the oral cavity serves as a reservoir of oral pathobionts whose ectopic gut colonization contributes to the intestinal pathologies. Studies have clearly shown that patients with gut inflammation, such as IBD, exhibit a significant enrichment of oral bacteria in the gut, including pathogens associated with the oral inflammatory disease periodontitis. 10 , 11 This notion is supported by studies showing the distinct oral microbiota 12 and increased prevalence of periodontitis in IBD patients when compared with healthy individuals. 13 These observations may be indicative of the link between periodontal and gut inflammation established through microbial communications.
2. POTENTIAL ROUTES OF GUT TRANSLOCATION OF ORAL BACTERIA
The translocation of oral bacteria from the oral cavity to the gut mucosa is poorly defined. Two potential routes have been proposed.
2.1. Hematogenous dissemination
Oral resident bacteria can disseminate systemically by the hematogenous route originating in the oral cavity. In this regard, mechanical injuries in the oral cavity can lead to the spread of oral bacteria into the systemic circulation. 14 , 15 Moreover, oral bacteria such as P gingivalis are found in the blood collected from patients with periodontal diseases, including periodontitis. 16 Consistently, ligature‐induced murine periodontitis leads to oral bacterial dissemination to the liver and spleen, indicating that the hematogenous spread of oral bacteria can be determined by oral disease status. 17 Furthermore, it has been shown that hematogenously inoculated Fusobacteria strains are more successful in tumor colonization in the gut than gavaged strains, suggesting the importance of the circulatory system as a route of oral bacteria dissemination. 18 Oral bacteria are also known to invade and survive inside dendritic cells and macrophages, implying the hijacking of host immune cells to serve as Trojan horses for the dissemination of bacteria from the oral to the gut compartment. 19
2.2. Enteral dissemination
People swallow about 600 times a day and produce ~1.5 L of saliva containing 1.5 × 1012 oral bacteria. 20 , 21 Although more than half of the oral resident bacterial species (eg, Streptococcus spp. Veillonella spp.) are detectable in the gut, implying oral–gut translocation of oral bacteria even in healthy individuals, 22 oral bacteria are generally poor colonizers in a healthy gut. This is due to the segregation of mouth and gut bacterial communities through the multiple barriers conferred by the gastrointestinal tract. 9 The first barrier against the oral bacterial translocation to the gut is gastric acidity. 23 , 24 It is estimated that over 99.9% of swallowed bacteria of oral origin cannot survive in the stomach due to its acidic antimicrobial environment, which reduces bacterial numbers by 5‐6 orders of magnitude. 21 , 25 In line with this notion, a significant elevation of gut colonization by oral bacteria (eg, Streptococcus spp., Veillonella spp., Haemophilus spp.) occurs in patients who have gastric achlorhydria caused by the long‐term use of proton pump inhibitors (PPIs). Consistently, patients with gastroesophageal reflux disease treated with long‐term PPI therapy also exhibit a higher oral bacterial accumulation in the gut compared to healthy individuals. 26 Further, individuals who have gastritis after gastric surgery (eg, gastric bypass or removal) exhibit an altered gut microbial composition, accompanied by the accumulation of resident oral bacteria in the gut (eg, Streptococcus spp., Veillonella spp., and Enterobacteriaceae). 27 , 28 Of note, the attenuated gastric acidity is observed in patients with IBD, indicating the potential contribution of a “leaky stomach” in facilitating a profound colonization of oral bacteria in the gut. 29 Importantly, certain types of oral pathogens, such as P gingivalis, can tolerate the acidic environment in the stomach and pass through the stomach barrier. 30 Consequently, although possibly less effective for those bacteria that can tolerate the acidic environment, the prevention of the enteral transmission of oral bacteria by gastric acids is considered as the primary defense mechanism. Secondary, given the colonization resistance conferred by the gut resident microbiota, 31 preservation of the harmonious microbial structure in the gut is also important for preventing ectopic colonization by ingested oral bacteria. This notion is supported by the intestinal expansion of oral bacteria in patients who take certain types of antibiotics (eg, vancomycin), as the antibiotic treatment provokes gut dysbiosis, which generates the niche for ingested oral bacteria. 9 In addition to antibiotics, multiple factors that cause gut dysbiosis, such as gut inflammation, diets, artificial sweeteners, may also contribute to the opportunistic gut colonization by oral bacteria. 9
3. MICROBIAL PATHWAY (VIA DIRECT GUT COLONIZATION OF ORAL PATHOBIONTS)
Disordered gut microbial distribution and discordant immune responses underlie the development of gut inflammation. Once oral pathobionts colonize the gut, they may be the causative agents, responsible for inducing abnormal immune responses in the gut, thereby leading to intestinal inflammation (Figure 1). Multiple oral resident bacteria are reported to be potential oral pathobionts that are conducive to gut inflammation.
FIGURE 1.
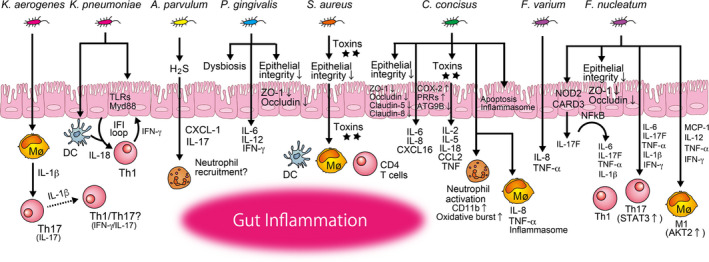
Possible mechanisms of gut inflammation caused by direct colonization by oral pathobionts (microbial pathway). Once oral pathobionts reach the intestine, they first cross the intestinal epithelium. Certain oral pathobionts can adhere to and invade the epithelial cells. The host responses are variable, such as cytoskeletal rearrangement, expression of pattern recognition receptors such as toll‐like receptors (TLRs), inflammasome assembly, cell death, and the release of proinflammatory cytokines. Some oral pathobionts produce cytotoxic substances (eg, hydrogen sulfide [H2S], toxins), leading to disruption of the intestinal integrity. A compromised intestinal epithelium allows oral pathobionts, as well as other commensal microorganisms and their metabolites, to move from the lumen to the lamina propria. Oral pathobionts interact with immune cells including macrophages (Mø), DCs, neutrophils, and T cells in the lamina propria, thereby instigating the development of gut inflammation through the activation of multiple inflammatory cascades, including the induction of proinflammatory cytokines and chemokines and the development of pathogenic T cells
3.1. Fusobacteria spp.
Certain members of the family Fusobacteriaceae, such as F varium and F nucleatum, are enriched in the gut of patients suffering from IBD, and their abundance is significantly elevated when the disease is active, rather than in remission. 9 , 32 , 33 As genetically identical strains of F nucleatum are detectable in both the saliva and colonic tumors of patients with CRC, 34 Fusobacterium strains found in the gut of the IBD patient likely originate from the oral cavity. In addition, considering the inflammatory capacity of Fusobacteria spp. in the oral cavity, 35 , 36 , 37 the involvement of oral‐derived Fusobacteria spp. in the exacerbation of gut inflammation is plausible. F varium can invade the intestinal epithelium and evoke the production of proinflammatory cytokines, such as interleukin (IL)‐8 and TNF‐α, from the intestinal epithelial cells. 38 Similarly, F nucleatum is also highly invasive to intestinal epithelial cells and induces TNF‐α and IL‐1β expression. 39 Moreover, F nucleatum facilitates dextran sulfate sodium (DSS)–induced colitis by disrupting the integrity of the epithelial barrier; reducing tight junction proteins such as ZO‐1 and occludin. 32 , 33 , 40 Activation of the caspase activation and recruitment domain 3 (CARD3)/IL‐17F/nuclear factor‐kappa B (NF‐κB) cascade in the epithelial cells on the colonization of F nucleatum also fuels intestinal inflammation through the secretion of proinflammatory cytokines, such as IL‐6, IL‐17F, IL‐1β, and TNF‐α. 32 , 41 Further, F nucleatum aggravates the progression of DSS‐induced colitis by promoting M1 macrophage polarization through the activation of the AKT2 pathway. 40 F nucleatum also promotes the secretion of proinflammatory cytokines (TNF‐α, IFN‐γ, IL‐1β, IL‐6, and IL‐17) and activates the signal transducer and activator of transcription 3 (STAT3) signaling pathway, thereby inducing the expansion of Th1 and Th17 cells in the DSS‐induced colitis model. 33 However, the administration of F nucleatum to colitis‐associated mouse models (eg, BALB/c IL‐10−/− and BALB/c T‐bet−/− × Rag2−/−) neither accelerates gut inflammation nor increases the number of colorectal adenomas. 42 Although F nucleatum is a well‐recognized oral resident bacterium abundant in colonic tumors, and a known contributor to tumorigenesis, 18 , 43 its role and the mechanisms involved in the development of gut inflammation remain open to debate.
3.2. Porphyromonas gingivalis
P gingivalis is a major periodontopathic bacterium with a wide variety of proinflammatory capacities in the pathogenesis of periodontal diseases, such as periodontitis. 44 , 45 Multiple studies have revealed that the orogastric administration of P gingivalis to mice may impair epithelial integrity in the gut. For instance, continuous administration of P gingivalis (ie, twice a week, for 5 weeks) to C57BL/6N mice causes endotoxemia, accompanied by the decrease of gene expression of tight junction protein ZO‐1 and increase of proinflammatory cytokines IL‐6, IL‐12, and IFN‐γ in the gut. 46 Similarly, administration of a single oral dose of P gingivalis to C57BL/6N mice results in the reduced expression of intestinal tight junction proteins ZO‐1 and occludin in the gut, and the systemic dissemination of enterobacteria to the liver, indicating the disruption of the intestinal barrier function. 47 Interestingly, the gut microbial composition of mice treated with P gingivalis was clearly distinct from that of sham‐treated mice, with the expansion of unclassified Muribaculaceae and Prevotella spp., which are similar to the IgA‐coated colitogenic pathobionts in the gut. 48 This indicates that P gingivalis itself can be colitogenic, yet gut dysbiosis driven by the colonization of P gingivalis may also play a role in the induction or exacerbation of colitis. In a clinical setting, patients with IBD are known to have an increased prevalence of periodontitis compared to individuals who do not have IBD. 13 Given that large quantities of oral bacteria are constantly swallowed and reach the gut, it is plausible that numerous P gingivalis, ranging between 106‐108 cells per mL in subgingival and salivary samples (corresponding to 109‐1011 copies daily), are swallowed by patients with chronic periodontitis. 49 Although the precise impact of gut colonization of P gingivalis on intestinal inflammation remains unexplored, its proinflammatory potential suggests that it may exacerbate the inflammation. On the other hand, it is also reported that monocolonization of P gingivalis in the gut promotes beneficial changes in the gut immune system, including the elevation of genes related to tight junction proteins and the antiinflammatory cytokine IL‐10. 50 Further studies would clarify the impact of gut colonization of P gingivalis on the pathogenesis of intestinal inflammation.
3.3. Atopobium parvulum
A parvulum is frequently isolated from the human oral cavity and found to be associated with oral malodor (halitosis). Research has revealed that patients with IBD, similar to patients with colon cancer, exhibit an accumulation of A parvulum in the gut. 9 Certain oral bacteria (eg, Atopobium spp., Veillonella spp., Prevotella spp., Streptococcus spp., and Aggregatibacter spp.) are known to liberate hydrogen sulfide (H2S), an inflammatory mediator, from sulfur‐containing amino acids. 9 Investigators identified impaired mitochondrial H2S detoxification and the bloom of H2S‐producing pathobionts along with the depletion of butyrate‐producing bacteria in the gut of patients with Crohn's disease (CD) by using system biology approaches that combine metagenomic and proteomic data sets. 51 About one‐quarter of the operational taxonomic units (eg, Atopobium, Fusobacterium, Veillonella, Prevotella, Streptoccocus, and Leptotrichia) that correlate positively with the severity of intestinal disease are known to metabolize sulfur‐containing amino acids into H2S. Importantly, A parvulum is defined as the key pathobiont, serving the central hub of the H2S network. Furthermore, this study demonstrated the colitogenic capacity of A parvulum in an Il10 −/− colitis model, with the increased expression of the chemokine (C‐X‐C motif) ligand 1 (Cxcl1) and Il17 in the gut, compared with controls, which was mitigated by the administration of the H2S scavenger bismuth. 51 In contrast, A parvulum monocolonized germ‐free (GF) Il10 −/− mice did not develop significant colitis, suggesting that other microbes, or their metabolites, are required for A parvulum–driven colitis. Given the ability of H2S to induce proinflammatory molecules (eg, cyclooxygenase (COX)‐2, IL‐8, and CCAAT enhancer binding protein beta [CEBPB]) 52 in epithelial cells and to promote T cell activation, 53 it is conceivable that A parvulum creates niches favorable for the growth of colitogenic pathobionts by inducing H2S. At high concentration, H2S is a strong inhibitor of cytochrome c oxidase, and hence, mitochondrial oxygen (O2) consumption, with deleterious consequences for the epithelial integrity. Furthermore, given that colonocytes obtain more than 70% of their energy from the oxidation of gut bacteria‐derived butyrate, 54 , 55 along with the ability of H2S to inhibit butyrate oxidation, A parvulum may play a role in the epithelial energy deficiency associated with the prevalence of IBD. 56 , 57
3.4. Campylobacter concisus
C concisus is an oral resident bacteria found in the gut of patients with IBD. 58 , 59 , 60 , 61 Genomic comparison of oral and enteric C concisus strains implies that the enteric strains originate from the oral C concisus strains. 62 , 63 Although the mechanistic features of the flagellum of C concisus are not fully understood, C concisus flagellum‐mediated attachment to and invasion of the colonic epithelial cell line Caco‐2 have been documented. 64 Research has also shown that dense bacterial biofilm formation is common in IBD patients and contributes to the disease pathogenesis through the induction of dysbiosis and resistance to treatment, such as antibiotics. 65 In this regard, the flagellum of C concisus enables it to form biofilm and hence survive in the gut. 66 In vitro intestinal epithelial cell culture models (eg, Caco‐2, HT‐29/B6 cells) also suggest that C concisus can increase intestinal permeability through the dislocation (or downregulation) of ZO‐1, occludin, and claudin‐5, together with apoptotic leaks. 64 , 67 Moreover, C concisus impairs sodium (Na+) absorption in HT‐29/B6 cells through the dysfunction of the epithelial Na+ channels. 68 This is dependent on IL‐32–regulated extracellular signal‑regulated protein kinase (ERK)1/2, as well as claudin‐8–dependent barrier dysfunction, both of which contribute to Na+ malabsorption and diarrhea. 68 C concisus also increases the production of proinflammatory molecules such as IL‐8 and COX‐2, which is an enzyme responsible for generating prostaglandins as well as other inflammatory mediators in the intestinal epithelial cells. 69 In parallel, infected HT‐29 epithelial cells express elevated levels of pattern‐recognition receptors (eg, Toll‐like receptor [TLR] 4, but not TLR2 or TLR5), implicating the role of C concisus in modulating the intestinal epithelial responses to bacterial components such as lipopolysaccharide. 69 In response to C concisus colonization of Caco‐2 cells, autophagy‐related genes, such as ATG9B, are significantly reduced, implying the importance of escape from autophagy as a bacterial survival strategy within the intracellular compartment. 70 Interestingly, global gene expression changes in Caco‐2 caused by the exposure to the toxigenic C concisus strain AToCC that expresses zonula occludens toxin were distinct from the changes induced by the nontoxigenic strain AICC. The AToCC strain, compared to AICC, induces a more robust expression of genes related to inflammatory responses (eg, IL‐2, IL‐5, IL‐18, CCL2, and TNF signaling) and the pattern recognition receptors involved in sensing intracellular nucleic acids (eg, TLR3), as well as the assembly of the IFI16 inflammasome. 70
Another C concisus virulence factor—membrane‐bound hemolytic phospholipase A2 (PLA2)—exhibits cytolytic effects on Chinese hamster ovary cells in tissue culture, indicating the possible mechanism of cell destruction by C concisus during intestinal inflammation. 71 After passing through the epithelial barrier, C concisus can activate immune cells including macrophages and neutrophils in the lamina propria and elicit inflammatory responses. For instance, C concisus enhances the production of IL‐8 and TNF‐α by THP‐1 macrophages. 64 Like the epithelial response against C conscisus, genes associated with the host recognition of C concisus (eg, those encoding TLRs), as well as inflammasome‐related genes (eg, IFI16, ASC), are significantly upregulated after C concisus infection of THP‐1 macrophages. 72 Also, global gene regulation in macrophages on infection with C concisus includes the activation of key inflammatory pathways involving CREB1, NF‐κB, STAT, and interferon regulatory factor signaling. 72 Further, C concisus activates the innate immune system by stimulating CD11b expression in neutrophils, which promotes neutrophil adhesion to the vascular endothelium and an oxidative burst response. 73 To date, published animal studies with C concisus infection are few. The first study, which was conducted in BALB/c mice, showed that the infected mice had marginal gut inflammation with poor colonization. 74 Another study used antibiotic‐treated IL‐10−/− mice (on the C57BL/6J genetic background) and showed that oral administration of C concisus neither induces significant inflammation nor impairs epithelial barrier function in the colon, whereas C concisus colonization can cause dysfunction of the epithelial Na+ channel associated with watery diarrhea. 68 , 75 Despite ample evidence of the colitogenic capacity of C concisus, comprehensive animal studies are required to determine the precise impact of gut colonization of C concisus on intestinal inflammation.
3.5. Staphylococcus aureus
S aureus is a gram‐positive, spherical member of the phylum Firmicutes, and a constituent of the human oral microbiota. 76 , 77 Although this bacterium is well characterized by food poisoning through staphylococcal enterotoxin (SE)–mediated mechanisms, 77 , 78 patients with CD are also known to have higher levels of S aureus in inflamed subgingival sites compared with healthy individuals, even with similar clinical periodontal parameters. 79 Notably, the increased colonization by this bacterium is also reported in the gut of IBD patients compared with non‐IBD controls. 9 , 26 S aureus is reported to adhere to intestinal epithelial cells. 80 It has also been shown that oral administration of S aureus strain RN8098, which produces staphylococcal enterotoxin B (SEB), into antibiotic‐pretreated C57BL/6J mice causes epithelial damage in the small, but not the large intestine, whereas no overt inflammation was observed in mice colonized by a SEB mutant strain. 80 Interestingly, despite the capability of SEs to dampen adherens junction protein expression, 81 disruption of the adherens junction proteins E‐cadherin and β‐catenin in the small intestine of mice with S aureus was detected in both wild‐type and SEB mutant strains. This indicates the possible involvement of virulence factors other than SEB in S aureus–induced epithelial damage in the gut. 82 Furthermore, SEs are known to function as superantigens by binding to the outside of the antigenic peptide binding groove of major histocompatibility complex (MHC)–II on antigen‐presenting cells (eg, macrophages and dendritic cells), as well as to T cell receptors expressing certain Vβ elements. 78 Thus, the massive proliferation of CD4+ T cells with the production of proinflammatory cytokines induced by those interactions may also contribute to the pathogenesis of IBD.
3.6. Klebsiella spp. and Enterobacter spp.
Enterobacteriaceae is a large family of gram‐negative bacteria, including Klebsiella spp. and Enterobacter spp. Most Enterobacteriaceae are part of the gut commensal microbiota. However, investigators have shown that colonization of oral‐derived Klebsiella spp. (eg, K pneumoniae, K aeromobilis) isolated from the saliva of patients with CD results in potent Th1 cell differentiation in the gut of gnotobiotic animals. 26 Importantly, this study showed that oral Klebsiella spp. can facilitate the development of Th1‐skewed IBD‐like colitis in IL‐10−/− mice, whereas no overt inflammation was detected in immune‐competent wild‐type B6 mice despite Th1 induction in the gut. Mechanistically, TLR and IL‐18 signaling are required for the Klebsiella‐mediated Th1 cell induction through the antigen‐presenting CD11b+CD103+ dendritic cells. Also, it was shown that upregulation of IFN‐inducible (IFI) genes, such as those encoding guanylate‐binding proteins, CXCL9, MHC‐related molecules, and dual oxidase 2 (Duox2), may facilitate the gut colonization by K pneumoniae, as well as the development and recruitment of Th1 cells. Further, the investigators observed that mice lacking IFN receptor 1 failed to respond to the K pneumoniae colonization. These results imply that Th1 responses triggered by K pneumoniae are sustained via an IFI‐mediated feed‐forward loop. 26 Of note, these oral‐derived Klebsiella isolates are resistant to multiple antibiotics, indicating the potential risk of antibiotic use in a clinical setting, as such a regimen may allow the bacteria to colonize the gut and induce colitis in IBD‐susceptible hosts.
Ample evidence of the clinical association between periodontitis and IBD 13 prompted us to assess the impact of periodontitis on intestinal inflammation. Our recent study revealed the deleterious contribution of periodontitis, associated with the expansion of oral pathobionts belonging to the Enterobacteriaceae family in the oral cavity, to the development of distant intestinal inflammation. 83 In this study, by combining ligature‐induced murine periodontitis and DSS‐induced colitis models, we revealed that oral inflammation fosters blooms of Enterobacteriaceae including Klebsiella spp. and Enterobacter spp. and enforces colonization of these oral pathobionts in the gut of genetically susceptible IL10−/− mice (but not wild‐type B6 mice), resulting in exacerbation of intestinal inflammation. Further, we showed that direct gut colonization of these oral pathobionts strongly induces colonic IL‐1β production by activating the inflammasome pathway in intestinal macrophages in the inflamed gut, thereby aggravating the intestinal pathology. 83 Importantly, an overt increase of oral pathobionts did not occur in the healthy gut, even in the mice with periodontitis, implying that at least two hits (ie, prerequisites) to the microbiotas in the mouth and gut are essential for the development of oral pathobiont‐driven intestinal inflammation. The first prerequisite is oral dysbiosis, which drastically increases the number of oral pathobionts in the oral cavity, and thus increases the chance of successful transmission to the intestine. As discussed, the physiological barrier functions of the gastrointestinal tract, particularly in the stomach, deter the successful transmission of ingested bacteria. Given the bactericidal effect of gastric acids, the expansion of oral pathobionts in the dysbiotic oral microbiota must be achieved to increase the chance of bacterial survival in the stomach, followed by the successful translocation to the intestine. Attenuation of gastric acidity in patients with IBD or the inhibition of acid secretion may explain why amassed oral bacteria are often found in the gut of IBD patients. 10 , 11 , 84 , 85 The second prerequisite involves the disruption of gut colonization resistance conferred by gut dysbiosis, which may be required to enable oral pathobionts (that successfully passed through the gastric barrier) to colonize the gut. In our study, gut inflammation dampened colonization resistance provided by the gut commensals and allowed ingested oral pathobionts (eg, Klebsiella spp.) to successfully colonize the gut. 83 Given that the inflammatory milieu favors the growth of members of the Enterobacteriaceae family, 86 , 87 , 88 intestinal inflammation may also be a potent driving factor that instigates the ectopic colonization of certain types of oral pathobionts such as Klebsiella spp. that can gain growth benefits in the inflamed gut. Consistent with the previous report of IBD patient‐derived oral Klebsiella spp., 26 the Klebsiella strains that we isolated (eg, K aerogenes) from periodontitis mice also have antibiotic resistance (data not shown), indicating the potential risk of antibiotic use in the development of gut inflammation, which is mediated by ectopically colonized oral pathobionts in the dysbiotic gut environment.
3.7. Other oral bacteria
Like Atopobium spp., certain oral bacteria (eg, Veillonella spp.) enriched in the gut of IBD patients have been identified as major producers of H2S, implicating their proinflammatory potential in the gut. 10 , 89 Also, other indigenous oral bacteria (eg, Streptococcus spp. and Neisseria spp.) can produce acetaldehyde by catabolizing ethanol and glucose. 90 Given the proinflammatory capacity of acetaldehyde through disruption of the epithelial barrier function, 91 , 92 , 93 it is possible that ectopic colonization of the gut by these oral bacteria could instigate gut inflammation. Furthermore, besides the enteral colonization described in Figure 1, certain types of oral pathobionts, such as Streptococcus mutans, may impact the intestinal pathology through hematogenous spread from the oral cavity. S mutans has virulence factors associated with the etiology and pathogenesis of dental caries. 94 , 95 Also, a higher prevalence of dental caries and higher salivary counts of S mutans are reported in CD patients compared to the control group. 96 Several S mutans strains isolated from the oral cavity of patients with ulcerative colitis (UC) caused aggravation of murine DSS‐induced colitis, suggesting the potential involvement of highly virulent S mutans in the occurrence of UC. 97 In this study, the investigators found that intravenous administration of TW295, a serotype k strain of S mutans expressing collagen‐binding protein, can specifically colonize the liver, rather than the intestine, and induce IFN‐γ production (presumably from the hepatocytes), thereby increasing the susceptibility to DSS‐colitis. As oral administration of TW295 did not produce colitis aggravation, it is conceivable that certain oral pathobionts, such as S mutans, coming from the circulating blood, but not from the mucosa surrounding the lumen of the gastrointestinal tract, are involved in the aggravation of colitis. Consistently, it is reported that S mutans can disseminate to the systemic circulation in individuals who have had dental procedures (eg, orthodontics, tooth extraction) or oral disease (eg, oral cancer). 9
4. IMMUNOLOGICAL PATHWAY (VIA TRANSLOCATION OF ORALLY PRIMED IMMUNE CELLS TO THE GUT)
Ample evidence demonstrates that immune cells can move from the gut to other organs (eg, liver, kidney, joints) and contribute to the disease pathogenesis at distant sites. 98 , 99 , 100 The immune cell trafficking between the gut and other organs seems to be bidirectional. It is reported that leukocytes in the oral draining lymph nodes, particularly the cervical lymph nodes (cLNs), can travel to the gut even under steady‐state conditions, 101 indicating the potential role of systemic immune cell circulation in human health and disease. In this context, we unveiled the mechanistic link between the mouth and gut during the development of gut inflammation from an immunological point of view 83 (Figure 2).
FIGURE 2.
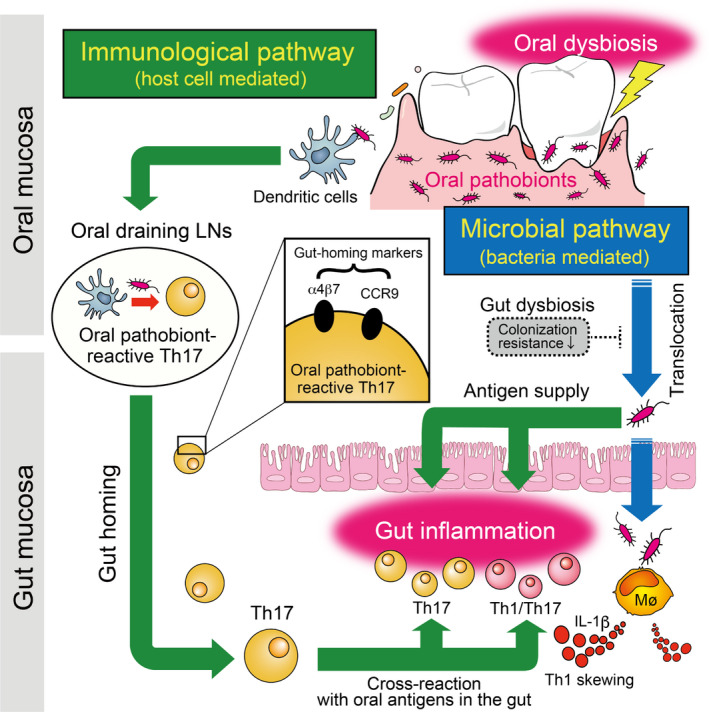
Possible mechanisms of gut inflammation mediated by transmigration of orally primed T cells to the gut (immunological pathway). In parallel with the direct translocation of oral pathobionts to the gut provoked by concurrent oral and gut dysbiosis (Figure 1, microbial pathway), the transmigration of oral immune cells to the gut also plays a key role in the mouth–gut axis during the pathogenesis of oral pathobiont‐driven colitis (ie, the immunological pathway). Mechanistically, during periodontal inflammation, orally primed Th17 cells that recognize oral pathobionts (eg, K aerogenes) are generated in the oral draining lymph nodes (LNs). Oral pathobiont‐reactive Th17 cells express gut‐homing molecules such as CCR9 and α4β7. When Th17 cells of oral origin reach the gut, they can be activated by translocated oral pathobionts and promote the development of colitis. Given the phenotypic changes of oral Th17 cells toward Th1, such as Th17 cells in the gut of mice with periodontitis and the concurrent presence of Th1 skewing factor IL‐1β (produced by intestinal macrophages exposed to oral K aerogenes, as evident in the microbial pathway, Figure 1), it is likely that the microbial and immunological pathways synergistically aggravate the intestinal pathology during the oral pathobiont‐driven gut inflammation
As mentioned above, ligature‐induced murine periodontitis increases the susceptibility to acute DSS‐induced colitis through the direct gut colonization by oral pathobionts 83 (Figure 1). Interestingly, even though the acute DSS‐induced colitis model may lack sufficient time to develop T cell immunity in the gut, we observed a prominent increase of Th17 and Th1 cells in the colonic mucosa of ligature–DSS mice compared with DSS colitis only mice. Given the known cellular trafficking between the oral cavity and the gastrointestinal tract 101 and the role of Th17 in periodontal inflammation, 102 we hypothesized that the pathogenic T cells that accumulate in the gut of ligature–DSS mice originate from the oral cavity. To this end, we first characterized the immune responses provoked by periodontitis in the oral cavity. Then, we showed that CD3+CD4+CD44hiCD62Llo effector memory T (TEM) cells are enriched in the cLNs of mice that developed periodontitis. In accordance with a previous report, 103 we observed that TEM cells accumulated in periodontitis mice display the IL‐17A–producing RORγt+ Th17 phenotype. By coculturing oral antigen‐pulsed dendritic cells (DCs) and isolated orally primed Th17 cells, we discovered that oral Th17 TEM cells were reactive to oral pathobionts, including Klebsiella spp. and Enterobacter spp., all of which expanded in the inflamed, but not the healthy, oral mucosa. These results suggested that oral pathobiont‐reactive Th17 cells are generated during periodontitis, raising the question of whether oral Th17 cells can travel to the gut. Further analysis showed the cell surface expression of gut‐homing markers α4β7 integrin and CCR9 on these oral Th17 cells, indicating their gut tropism. To obtain direct evidence of the transmigration of oral Th17 cells to the gut, we used in vivo photoconversion of cells in the cLNs of transgenic mice expressing the Kaede protein 104 and monitored the ability of these cells to migrate to the gut. In this trafficking system, all cells in Kaede mice constitutively express the photoconvertible Kaede green fluorescent protein. When the photoconvertible protein is exposed to violet light, the cell color changes from Kaede green to Kaede red. 104 As previously reported, 101 we detected Kaede red CD4+ T cells in cLNs in the steady‐state gut, providing concrete evidence of the transmigration of orally primed Th17 cells to the gut mucosa. Interestingly, the influx of oral Th17 cells to the gut was significantly increased in mice with DSS‐induced colitis. Although the precise mechanisms of this transmigration remain unclear, the upregulation of mucosal addressin cell adhesion molecule 1 (MadCAM‐1), a ligand for α4β7 integrin expressed in vessels in the colonic lamina propria of patients with IBD and experimental animal models including DSS‐induced colitis models, 105 , 106 , 107 suggest that an enhanced interaction between α4β7 integrin and MadCAM‐1 plays a role in accelerating the influx of oral Th17 cells into the inflamed gut (Figure 2). To validate the colitogenic capacity of oral Th17 cells in the gut, we conducted multiple immune cell experiments, including adoptive transfer colitis. We found that isolated oral Th17 cells (ie, Kaede red cells isolated from the gut of ligatured mice) induced colitis when transferred intravenously into Rag1 −/− mice colonized by the oral pathobiont K aerogenes in the gut associated with an increase of Th17 cells (RORγt+) and Th1/Th17 cells (RORγt+ T‐bet+); in contrast, Kaede green cells isolated from the gut of ligatured mice failed to cause colitis. Interestingly, administration of IL‐1 receptor antagonist (anakinra) ameliorated the severity of colitis in the Kaede red cell–transferred mice. Considering the known role of IL‐1β in skewing Th17 cells toward Th1 phenotypes, intestinal IL‐1β induced by the gut colonization of oral pathobionts (eg, K aerogenes, Figure 1) not only induces proinflammatory innate lymphoid cells and Th17 cells, 108 but also acts as a Th1 skewing factor for generating Th1/Th17 cells, which also accumulate in the gut of individuals with IBD 109 , 110 , 111 , 112 , 113 (Figure 2). In our study, in accordance with the current understanding of a key role of Th17 cells in the commensal‐driven oral inflammation, 103 , 114 , 115 , 116 , 117 we observed the prominent increase of oral pathobiont‐reactive Th17 cells in the oral cavity in response to the ligature‐induced periodontitis. 83 In this context, despite the evidence that commensal‐reactive Th17 cells generated in the gut are not pathogenic, 118 IFN‐γ‐secreting Th1‐like exTh17 cells that arise from Th17 cells under certain circumstances in the gut can induce severe intestinal inflammation. 109 , 119 Interestingly, while the oral commensal pathobiont‐reactive Th17 cells that arise during periodontitis exhibit a Th17 phenotype (RORγt+ T‐bet−) associated with IL‐17A but not IFN‐γ production, when these oral Th17 cells reach the gut mucosal compartment they seem to acquire a Th1‐like Th17 phenotype (RORγt+ T‐bet+) associated with IFN‐γ production (Figure 2). 83 Considering the clinical importance of Th1/Th17 cells, this functional conversion of orally generated Th17 cells into pathogenic Th1/Th17 cells in the gut microenvironment may be an important area for future research.
5. PERSPECTIVES
Over the past decade, the research field of oral microorganisms and intestinal inflammation has been dramatically expanded by studies that primarily focus on the impact of direct colonization of oral pathobionts in the gut (Figure 1, microbial pathway). Furthermore, the use of murine models has revealed the novel aspects of the complex intermucosal connection between the mouth and the gut. Orally primed pathogenic T cells can transmigrate to the gut, where they are reactivated by ingested oral pathobionts, and thus, exacerbate intestinal inflammation (Figure 2, immunological pathway). Yet, despite advances, major knowledge gaps still exist. For example, the considerable microbial dissimilarity between humans and mice 120 challenges the extent to which our findings in the realm of murine studies are readily translatable to humans. In this regard, the colitogenic murine oral pathobionts (eg, K aerogenes) that we identified are genetically very similar to K aeromobilis, which is a strong Th1‐inducing colitogenic oral pathobiont isolated from the saliva of IBD patients. 83 , 121 Although the detailed mechanisms remain unexplored, the genetic similarity of these species, and their functional similarity, considering the induction of Th1‐biased immunity during gut inflammation, suggest that the immunological interaction mediated by oral pathobiont‐reactive immune cells contributes to the pathogenesis of intestinal inflammation in human IBD. At present, neither the class of drugs, nor specific drugs, that target the oral–gut axis are available to treat patients with intestinal inflammation. Future investigations of the oral cavity will lead to a better understanding of the essential steps in the development of novel biomarkers and therapeutics for intestinal inflammation (Figure 3, Oral cavity). For instance, early detection of certain oral pathobionts may help to identify individuals at high risk of the development or relapse of IBD. Also, optimal oral hygiene to reduce the supply of oral pathobionts may attenuate ongoing disease progression in the gut, as well as prevent the development of IBD. A focus on the mode of transmission of oral pathobionts and oral immune cells to the gut could inspire the development of another potential intervention (Figure 3, GI ducts and vessels). For the microbial pathway, this could be achieved by reducing the chance of gut colonization by oral pathobionts through the proper use of PPIs or antibiotics to preserve the physiological barrier functions in the stomach and gut against the invasion of extraintestinal bacteria. In fact, PPI exposure has been associated with adverse clinical consequences (eg, IBD‐related hospitalization or surgery) in patients with both UC and CD. 122 , 123 Further, IBD patients treated with PPIs have been reported to be less likely to achieve remission while taking infliximab. 124 For the immunological pathway, intervention could be achieved by blocking the transmission of orally primed immune cells (eg, pathogenic oral Th17 cells) by inhibitors or biologics specific to the molecules that guide the oral‐derived immune cells to gut. In this context, anti‐α4β7 integrin therapy has been shown to be effective in moderate‐to‐severe CD. 125 , 126 Given the expression of α4β7 on orally primed T cells, the improvement of disease outcomes may be due, in part, to the inhibition of the transmigration of pathogenic orally primed T cells to the gut. Consequently, there remains an unmet need to reliably predict the efficacy of anti‐integrin therapy to maximize the cost‐effectiveness by determining responders and nonresponders. Thus, a better understanding of the immunological link between the mouth and gut of IBD patients may influence clinical decision‐making regarding treatment choices. Furthermore, it would be useful to elucidate the precise mechanisms by which oral‐derived pathobionts and immune cells exacerbate gut inflammation. This may pave the way to develop novel clinical options for IBD (Figure 3, Intestinal tract). Clearly, further research of the complex inflammatory machinery driven by oral pathobionts in the gut (eg, identification of virulence genes, regulatory mechanisms, and downstream immune activations) will become a basis for the future development of novel therapy for IBD.
FIGURE 3.
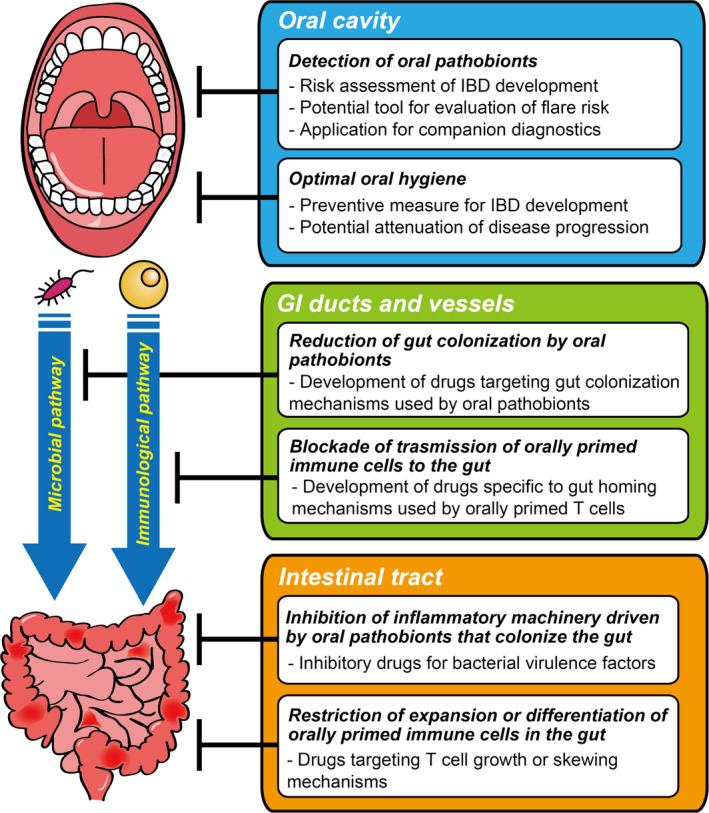
Potential approaches to the development of IBD interventions by targeting the oral–gut axis. The oral–gut axis can be divided into at least three targetable interfaces: (1) the oral cavity where oral pathobionts and potentially pathogenic immune cells are generated, (2) the gastrointestinal (GI) ducts and vessels that are used for the trafficking of oral–derived pathogenic agents to the gut, and (3) the intestinal tract where oral‐derived pathogenic agents can be virulent. Each interface holds potential for the development of clinical interventions in the treatment of IBD
The microbial and immunological connection between the mouth and the gut in the development of intestinal inflammation continues to be an area of intense study. From the clinical standpoint, larger cohorts and longitudinal studies are required to evaluate the importance of the oral–gut axis during the development of intestinal inflammation. In parallel, from the perspectives of basic and translational science, further characterization of the microbial and immune profiles of both sites and the factors affecting the gut colonization by oral pathobionts may present opportunities to develop unique and effective therapies for IBD.
CONFLICT OF INTEREST
The authors declare no competing interests.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health grants DK108901, DK119219, AI142047, DK125087 (to NK), the Office of the Assistant Secretary of Defense for Health Affairs endorsed by the Department of Defense through the Peer Reviewed Cancer Research Program under Award No. W81XWH2010547 (to SK), the University of Michigan Clinical and Translational Science Awards Program UL1TR002240, the Prevent Cancer Foundation, and the University of Michigan Center for Gastrointestinal Research Pilot Feasibility Project P30 DK034933 (to SK and NK).
Kitamoto S, Kamada N. Periodontal connection with intestinal inflammation: Microbiological and immunological mechanisms. Periodontol 2000. 2022;98:142–153. doi: 10.1111/prd.12424
Contributor Information
Sho Kitamoto, Email: kitamoto@umich.edu.
Nobuhiko Kamada, Email: nkamada@umich.edu.
REFERENCES
- 1. Escapa IF, Chen T, Huang Y, Gajare P, Dewhirst FE, Lemon KP. New insights into human nostril microbiome from the expanded human oral microbiome database (eHOMD): a resource for the microbiome of the human aerodigestive tract. mSystems. 2018;3(6):00187–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Costalonga M, Herzberg MC. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol Lett. 2014;162(2 Pt A):22‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koren N, Zubeidat K, Saba Y, et al. Maturation of the neonatal oral mucosa involves unique epithelium‐microbiota interactions. Cell Host Microbe. 2021;29(2):197‐209 e5. [DOI] [PubMed] [Google Scholar]
- 4. Zhu L, Kreth J. The role of hydrogen peroxide in environmental adaptation of oral microbial communities. Oxid Med Cell Longev. 2012;2012:717843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. 2018;24(4):392‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guo Y, Kitamoto S, Kamada N. Microbial adaptation to the healthy and inflamed gut environments. Gut Microbes. 2020;12(1):1857505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kitamoto S, Nagao‐Kitamoto H, Kuffa P, Kamada N. Regulation of virulence: the rise and fall of gastrointestinal pathogens. J Gastroenterol. 2016;51(3):195‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13(5):321‐335. [DOI] [PubMed] [Google Scholar]
- 9. Kitamoto S, Nagao‐Kitamoto H, Hein R, Schmidt TM, Kamada N. The bacterial connection between the oral cavity and the gut diseases. J Dent Res. 2020;99(9):1021‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gevers D, Kugathasan S, Denson L, et al. The treatment‐naive microbiome in new‐onset Crohn's disease. Cell Host Microbe. 2014;15(3):382‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dinakaran V, Mandape SN, Shuba K, et al. Identification of specific oral and gut pathogens in full thickness colon of colitis patients: implications for colon motility. Front Microbiol. 2018;9:3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Said HS, Suda W, Nakagome S, et al. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 2014;21(1):15‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. She Y‐Y, Kong X‐B, Ge Y‐P, et al. Periodontitis and inflammatory bowel disease: a meta‐analysis. BMC Oral Health. 2020;20(1):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lockhart PB, Brennan MT, Sasser HC, Fox PC, Paster BJ, Bahrani‐Mougeot FK. Bacteremia associated with toothbrushing and dental extraction. Circulation. 2008;117(24):3118‐3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parahitiyawa NB, Jin LJ, Leung WK, Yam WC, Samaranayake LP. Microbiology of odontogenic bacteremia: beyond endocarditis. Clin Microbiol Rev. 2009;22(1):46‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horliana ACRT, Chambrone L, Foz AM, et al. Dissemination of periodontal pathogens in the bloodstream after periodontal procedures: a systematic review. PLoS One. 2014;9(5):e98271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsukasaki M, Komatsu N, Nagashima K, et al. Host defense against oral microbiota by bone‐damaging T cells. Nat Commun. 2018;9(1):701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abed J, Maalouf N, Manson AL, et al. Colon cancer‐associated fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front Cell Infect Microbiol. 2020;10:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 2021;21:426‐440. doi: 10.1038/s41577-020-00488-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Humphrey SP, Williamson RT. A review of saliva: normal composition, flow, and function. J Prosthet Dent. 2001;85(2):162‐169. [DOI] [PubMed] [Google Scholar]
- 21. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmidt TSB, Hayward MR, Coelho LP, et al. Extensive transmission of microbes along the gastrointestinal tract. Elife. 2019;8:e42693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martinsen TC, Bergh K, Waldum HL. Gastric juice: a barrier against infectious diseases. Basic Clin Pharmacol Toxicol. 2005;96(2):94‐102. [DOI] [PubMed] [Google Scholar]
- 24. Howden CW, Hunt RH. Relationship between gastric secretion and infection. Gut. 1987;28(1):96‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giannella RA, Broitman SA, Zamcheck N. Gastric acid barrier to ingested microorganisms in man: studies in vivo and in vitro. Gut. 1972;13(4):251‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Atarashi K, Suda W, Luo C, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science. 2017;358(6361):359‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Castaner O, Goday A, Park Y‐M, et al. The gut microbiome profile in obesity: a systematic review. Int J Endocrinol. 2018;2018:4095789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paganelli FL, Luyer M, Hazelbag CM, et al. Roux‐Y Gastric Bypass and Sleeve Gastrectomy directly change gut microbiota composition independent of surgery type. Sci Rep. 2019;9(1):10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Press AG, Hauptmann IA, Hauptmann L, et al. Gastrointestinal pH profiles in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 1998;12(7):673‐678. [DOI] [PubMed] [Google Scholar]
- 30. Walker MY, Pratap S, Southerland JH, Farmer‐Dixon CM, Lakshmyya K, Gangula PR. Role of oral and gut microbiome in nitric oxide‐mediated colon motility. Nitric Oxide. 2018;73:81‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamada N, Chen GY, Inohara N, Nunez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14(7):685‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Y, Chen Y, Cao P, Su W, Zhan N, Dong W Fusobacterium nucleatum facilitates ulcerative colitis through activating IL‐17F signaling to NF‐kappaB via the upregulation of CARD3 expression. J Pathol. 2020;250(2):170‐182. [DOI] [PubMed] [Google Scholar]
- 33. Liu H, Hong XL, Sun TT, Huang XW, Wang JL, Xiong H Fusobacterium nucleatum exacerbates colitis by damaging epithelial barriers and inducing aberrant inflammation. J Dig Dis. 2020;21(7):385‐398. [DOI] [PubMed] [Google Scholar]
- 34. Komiya Y, Shimomura Y, Higurashi T, et al. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut. 2019;68(7):1335‐1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krisanaprakornkit S, Kimball JR, Weinberg A, Darveau RP, Bainbridge BW, Dale BA. Inducible expression of human beta‐defensin 2 by Fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect Immun. 2000;68(5):2907‐2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ahn SH, Chun S, Park C, Lee JH, Lee SW, Lee TH. Transcriptome profiling analysis of senescent gingival fibroblasts in response to Fusobacterium nucleatum infection. PLoS One. 2017;12(11):e0188755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhattacharyya S, Ghosh SK, Shokeen B, et al. FAD‐I, a Fusobacterium nucleatum cell wall‐associated diacylated lipoprotein that mediates human beta defensin 2 induction through toll‐like receptor‐1/2 (TLR‐1/2) and TLR‐2/6. Infect Immun. 2016;84(5):1446‐1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ohkusa T, Yoshida T, Sato N, Watanabe S, Tajiri H, Okayasu I. Commensal bacteria can enter colonic epithelial cells and induce proinflammatory cytokine secretion: a possible pathogenic mechanism of ulcerative colitis. J Med Microbiol. 2009;58(Pt 5):535‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dharmani P, Strauss J, Ambrose C, Allen‐Vercoe E, Chadee K Fusobacterium nucleatum infection of colonic cells stimulates MUC2 mucin and tumor necrosis factor alpha. Infect Immun. 2011;79(7):2597‐2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu LE, Liang L, Liang H, et al. Fusobacterium nucleatum aggravates the progression of colitis by regulating M1 macrophage polarization via AKT2 pathway. Front Immunol. 2019;10:1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cao P, Chen Y, Guo X, et al. Fusobacterium nucleatum activates endoplasmic reticulum stress to promote Crohn's disease development via the upregulation of CARD3 expression. Front Pharmacol. 2020;11:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kostic A, Chun E, Robertson L, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor‐immune microenvironment. Cell Host Microbe. 2013;14(2):207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu J, Li Q, Fu X Fusobacterium nucleatum contributes to the carcinogenesis of colorectal cancer by inducing inflammation and suppressing host immunity. Transl Oncol. 2019;12(6):846‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rafiei M, Kiani F, Sayehmiri F, Sayehmiri K, Sheikhi A, Zamanian AM. Study of Porphyromonas gingivalis in periodontal diseases: a systematic review and meta‐analysis. Med J Islam Repub Iran. 2017;31:355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol. 2016;7:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arimatsu K, Yamada H, Miyazawa H, et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. 2014;4:4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nakajima M, Arimatsu K, Kato T, et al. Oral administration of P gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS One. 2015;10(7):e0134234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Palm N, de Zoete M, Cullen T, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158(5):1000‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saygun I, Nizam N, Keskiner I, et al. Salivary infectious agents and periodontal disease status. J Periodontal Res. 2011;46(2):235‐239. [DOI] [PubMed] [Google Scholar]
- 50. Sato K, Yokoji M, Yamada M, Nakajima T, Yamazaki K. An orally administered oral pathobiont and commensal have comparable and innocuous systemic effects in germ‐free mice. J Periodontal Res. 2018;53(6):950‐960. [DOI] [PubMed] [Google Scholar]
- 51. Mottawea W, Chiang C‐K, Mühlbauer M, et al. Altered intestinal microbiota‐host mitochondria crosstalk in new onset Crohn's disease. Nat Commun. 2016;7:13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Attene‐Ramos MS, Nava GM, Muellner MG, Wagner ED, Plewa MJ, Gaskins HR. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ Mol Mutagen. 2010;51(4):304‐314. [DOI] [PubMed] [Google Scholar]
- 53. Miller TW, Wang EA, Gould S, et al. Hydrogen sulfide is an endogenous potentiator of T cell activation. J Biol Chem. 2012;287(6):4211‐4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Riviere A, Selak M, Lantin D, Leroy F, De Vuyst L. Bifidobacteria and butyrate‐producing colon bacteria: importance and strategies for their stimulation in the human gut. Front Microbiol. 2016;7:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. 1980;21(9):793‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roediger WE, Moore J, Babidge W. Colonic sulfide in pathogenesis and treatment of ulcerative colitis. Dig Dis Sci. 1997;42(8):1571‐1579. [DOI] [PubMed] [Google Scholar]
- 57. Roediger WE. The colonic epithelium in ulcerative colitis: an energy‐deficiency disease? Lancet. 1980;2(8197):712‐715. [DOI] [PubMed] [Google Scholar]
- 58. Zhang L, Budiman V, Day AS, et al. Isolation and detection of Campylobacter concisus from saliva of healthy individuals and patients with inflammatory bowel disease. J Clin Microbiol. 2010;48(8):2965‐2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang L, Man SM, Day AS, et al. Detection and isolation of Campylobacter species other than C. jejuni from children with Crohn's disease. J Clin Microbiol. 2009;47(2):453‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Man SM, Zhang L, Day AS, Leach ST, Lemberg DA, Mitchell H Campylobacter concisus and other Campylobacter species in children with newly diagnosed Crohn's disease. Inflamm Bowel Dis. 2010;16(6):1008‐1016. [DOI] [PubMed] [Google Scholar]
- 61. Mukhopadhya I, Thomson JM, Hansen R, Berry SH, El‐Omar EM, Hold GL. Detection of Campylobacter concisus and other Campylobacter species in colonic biopsies from adults with ulcerative colitis. PLoS One. 2011;6(6):e21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ismail Y, Mahendran V, Octavia S, et al. Investigation of the enteric pathogenic potential of oral Campylobacter concisus strains isolated from patients with inflammatory bowel disease. PLoS One. 2012;7(5):e38217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chung HKL, Tay A, Octavia S, et al. Genome analysis of Campylobacter concisus strains from patients with inflammatory bowel disease and gastroenteritis provides new insights into pathogenicity. Sci Rep. 2016;6:38442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Man SM, Kaakoush NO, Leach ST, et al. Host attachment, invasion, and stimulation of proinflammatory cytokines by Campylobacter concisus and other non‐Campylobacter jejuni Campylobacter species. J Infect Dis. 2010;202(12):1855‐1865. [DOI] [PubMed] [Google Scholar]
- 65. Swidsinski A, Weber J, Loening‐Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol. 2005;43(7):3380‐3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lavrencic P, Kaakoush NO, Huinao KD, Kain N, Mitchell HM. Investigation of motility and biofilm formation by intestinal Campylobacter concisus strains. Gut Pathog. 2012;4(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nielsen HL, Nielsen H, Ejlertsen T, et al. Oral and fecal Campylobacter concisus strains perturb barrier function by apoptosis induction in HT‐29/B6 intestinal epithelial cells. PLoS One. 2011;6(8):e23858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nattramilarasu PK, Bücker R, Lobo de Sá FD, et al. Campylobacter concisus impairs sodium absorption in colonic epithelium via ENaC dysfunction and claudin‐8 disruption. Int J Mol Sci. 2020;21(2):373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ismail Y, Lee H, Riordan SM, Grimm MC, Zhang L. The effects of oral and enteric Campylobacter concisus strains on expression of TLR4, MD‐2, TLR2, TLR5 and COX‐2 in HT‐29 cells. PLoS One. 2013;8(2):e56888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Deshpande NP, Wilkins MR, Castaño‐Rodríguez N, et al. Campylobacter concisus pathotypes induce distinct global responses in intestinal epithelial cells. Sci Rep. 2016;6:34288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Istivan TS, Coloe PJ, Fry BN, Ward P, Smith SC. Characterization of a haemolytic phospholipase A(2) activity in clinical isolates of Campylobacter concisus . J Med Microbiol. 2004;53(Pt 6):483‐493. [DOI] [PubMed] [Google Scholar]
- 72. Kaakoush NO, Deshpande NP, Man SM, et al. Transcriptomic and proteomic analyses reveal key innate immune signatures in the host response to the gastrointestinal pathogen Campylobacter concisus . Infect Immun. 2015;83(2):832‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sorensen NB, Nielsen HL, Varming K, Nielsen H. Neutrophil activation by Campylobacter concisus . Gut Pathog. 2013;5(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Aabenhus R, Stenram U, Andersen LP, Permin H, Ljungh A. First attempt to produce experimental Campylobacter concisus infection in mice. World J Gastroenterol. 2008;14(45):6954‐6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Barmeyer C, Erko I, Awad K, et al. Epithelial barrier dysfunction in lymphocytic colitis through cytokine‐dependent internalization of claudin‐5 and ‐8. J Gastroenterol. 2017;52(10):1090‐1100. [DOI] [PubMed] [Google Scholar]
- 76. Jackson MS, Bagg J, Gupta MN, Sturrock RD. Oral carriage of staphylococci in patients with rheumatoid arthritis. Rheumatology. 1999;38(6):572‐575. [DOI] [PubMed] [Google Scholar]
- 77. Ohara‐Nemoto Y, Haraga H, Kimura S, Nemoto TK. Occurrence of staphylococci in the oral cavities of healthy adults and nasal oral trafficking of the bacteria. J Med Microbiol. 2008;57(Pt 1):95‐99. [DOI] [PubMed] [Google Scholar]
- 78. Pinchuk IV, Beswick EJ, Reyes VE. Staphylococcal enterotoxins. Toxins. 2010;2(8):2177‐2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Brito F, Zaltman C, Carvalho ATP, et al. Subgingival microflora in inflammatory bowel disease patients with untreated periodontitis. Eur J Gastroenterol Hepatol. 2013;25(2):239‐245. [DOI] [PubMed] [Google Scholar]
- 80. Misawa Y, Kelley KA, Wang X, et al. Staphylococcus aureus colonization of the mouse gastrointestinal tract is modulated by wall teichoic acid, capsule, and surface proteins. PLoS Pathog. 2015;11(7):e1005061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Perez‐Bosque A, Moreto M. A rat model of mild intestinal inflammation induced by Staphylococcus aureus enterotoxin B. Proc Nutr Soc. 2010;69(3):447‐453. [DOI] [PubMed] [Google Scholar]
- 82. Larcombe S, Jiang JH, Hutton ML, Abud HE, Peleg AY, Lyras D. A mouse model of Staphylococcus aureus small intestinal infection. J Med Microbiol. 2020;69(2):290‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kitamoto S, Nagao‐Kitamoto H, Jiao Y, et al. The intermucosal connection between the mouth and gut in commensal pathobiont‐driven colitis. Cell. 2020;182(2):447‐62 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Imhann F, Bonder MJ, Vich Vila A, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016;65(5):740‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jackson MA, Goodrich JK, Maxan M‐E, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut. 2016;65(5):749‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kitamoto S, Alteri CJ, Rodrigues M, et al. Dietary L‐serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat Microbiol. 2020;5(1):116‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Winter SE, Winter MG, Xavier MN, et al. Host‐derived nitrate boosts growth of E coli in the inflamed gut. Science. 2013;339(6120):708‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhu W, Winter MG, Byndloss MX, et al. Precision editing of the gut microbiota ameliorates colitis. Nature. 2018;553(7687):208‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Washio J, Shimada Y, Yamada M, Sakamaki R, Takahashi N. Effects of pH and lactate on hydrogen sulfide production by oral Veillonella spp. Appl Environ Microbiol. 2014;80(14):4184‐4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tagaino R, Washio J, Abiko Y, Tanda N, Sasaki K, Takahashi N. Metabolic property of acetaldehyde production from ethanol and glucose by oral Streptococcus and Neisseria. Sci Rep. 2019;9(1):10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Alvarez R, Stork CA, Sayoc‐Becerra A, Marchelletta RR, Prisk GK, McCole DF. A simulated microgravity environment causes a sustained defect in epithelial barrier function. Sci Rep. 2019;9(1):17531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dunagan M, Chaudhry K, Samak G, Rao RK. Acetaldehyde disrupts tight junctions in Caco‐2 cell monolayers by a protein phosphatase 2A‐dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2012;303(12):G1356‐G1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rao RK. Acetaldehyde‐induced barrier disruption and paracellular permeability in Caco‐2 cell monolayer. Methods Mol Biol. 2008;447:171‐183. [DOI] [PubMed] [Google Scholar]
- 94. Lemos JA, Palmer SR, Zeng L, et al. The biology of Streptococcus mutans. Microbiol Spectr. 2019;7(1). doi: 10.1128/microbiolspec.GPP3-0051-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chen X, Daliri EB, Kim N, Kim JR, Yoo D, Oh DH. Microbial etiology and prevention of dental caries: exploiting natural products to inhibit cariogenic biofilms. Pathogens. 2020;9(7):569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Szymanska S, Lordal M, Rathnayake N, Gustafsson A, Johannsen A. Dental caries, prevalence and risk factors in patients with Crohn's disease. PLoS One. 2014;9(3):e91059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kojima A, Nakano K, Wada K, et al. Infection of specific strains of Streptococcus mutans, oral bacteria, confers a risk of ulcerative colitis. Sci Rep. 2012;2:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Krebs C, Paust H‐J, Krohn S, et al. Autoimmune renal disease is exacerbated by S1P‐receptor‐1‐dependent intestinal Th17 cell migration to the kidney. Immunity. 2016;45(5):1078‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tajik N, Frech M, Schulz O, et al. Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis. Nat Commun. 2020;11(1):1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lee K‐C, Chen P, Maricic I, et al. Intestinal iNKT cells migrate to liver and contribute to hepatocyte apoptosis during alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2019;316(5):G585‐G597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D. Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proc Natl Acad Sci USA. 2014;111(18):6696‐6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Dutzan N, Abusleme L. T helper 17 cells as pathogenic drivers of periodontitis. Adv Exp Med Biol. 2019;1197:107‐117. [DOI] [PubMed] [Google Scholar]
- 103. Dutzan N, Kajikawa T, Abusleme L, et al. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. 2018;10(463):eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tomura M, Yoshida N, Tanaka J, et al. Monitoring cellular movement in vivo with photoconvertible fluorescence protein "Kaede" transgenic mice. Proc Natl Acad Sci USA. 2008;105(31):10871‐10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Berlin C, Berg EL, Briskin MJ, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM‐1. Cell. 1993;74(1):185‐195. [DOI] [PubMed] [Google Scholar]
- 106. Briskin M, Winsor‐Hines D, Shyjan A, et al. Human mucosal addressin cell adhesion molecule‐1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am J Pathol. 1997;151(1):97‐110. [PMC free article] [PubMed] [Google Scholar]
- 107. Kato S, Hokari R, Matsuzaki K, et al. Amelioration of murine experimental colitis by inhibition of mucosal addressin cell adhesion molecule‐1. J Pharmacol Exp Ther. 2000;295(1):183‐189. [PubMed] [Google Scholar]
- 108. Coccia M, Harrison OJ, Schiering C, et al. IL‐1beta mediates chronic intestinal inflammation by promoting the accumulation of IL‐17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med. 2012;209(9):1595‐1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci USA. 2015;112(22):7061‐7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bsat M, Chapuy L, Rubio M, et al. Differential pathogenic Th17 profile in mesenteric lymph nodes of Crohn's disease and ulcerative colitis patients. Front Immunol. 2019;10:1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204(8):1849‐1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Calderón‐Gómez E, Bassolas‐Molina H, Mora‐Buch R, et al. Commensal‐specific CD4(+) cells from patients with Crohn's disease have a T‐helper 17 inflammatory profile. Gastroenterology. 2016;151(3):489‐500 e3. [DOI] [PubMed] [Google Scholar]
- 113. Hegazy AN, West NR, Stubbington MJT, et al. Circulating and tissue‐resident CD4(+) T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology. 2017;153(5):1320‐37 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Beklen A, Ainola M, Hukkanen M, Gurgan C, Sorsa T, Konttinen YT. MMPs, IL‐1, and TNF are regulated by IL‐17 in periodontitis. J Dent Res. 2007;86(4):347‐351. [DOI] [PubMed] [Google Scholar]
- 115. Cheng WC, van Asten SD, Burns LA, et al. Periodontitis‐associated pathogens P gingivalis and A actinomycetemcomitans activate human CD14(+) monocytes leading to enhanced Th17/IL‐17 responses. Eur J Immunol. 2016;46(9):2211‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Eskan MA, Jotwani R, Abe T, et al. The leukocyte integrin antagonist Del‐1 inhibits IL‐17‐mediated inflammatory bone loss. Nat Immunol. 2012;13(5):465‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Moutsopoulos NM, Konkel J, Sarmadi M, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL‐17‐driven inflammatory bone loss. Sci Transl Med. 2014;6(229):229ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ono Y, Kanai T, Sujino T, et al. T‐helper 17 and interleukin‐17‐producing lymphoid tissue inducer‐like cells make different contributions to colitis in mice. Gastroenterology. 2012;143(5):1288‐1297. [DOI] [PubMed] [Google Scholar]
- 119. Ahern PP, Schiering C, Buonocore S, et al. Interleukin‐23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33(2):279‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hugenholtz F, de Vos WM. Mouse models for human intestinal microbiota research: a critical evaluation. Cell Mol Life Sci. 2018;75(1):149‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Diene SM, Merhej V, Henry M, et al. The rhizome of the multidrug‐resistant Enterobacter aerogenes genome reveals how new "killer bugs" are created because of a sympatric lifestyle. Mol Biol Evol. 2013;30(2):369‐383. [DOI] [PubMed] [Google Scholar]
- 122. Juillerat P, Schneeweiss S, Cook EF, Ananthakrishnan AN, Mogun H, Korzenik JR. Drugs that inhibit gastric acid secretion may alter the course of inflammatory bowel disease. Aliment Pharmacol Ther. 2012;36(3):239‐247. [DOI] [PubMed] [Google Scholar]
- 123. Shah R, Richardson P, Yu H, Kramer J, Hou JK. Gastric acid suppression is associated with an increased risk of adverse outcomes in inflammatory bowel disease. Digestion. 2017;95(3):188‐193. [DOI] [PubMed] [Google Scholar]
- 124. Lu TX, Dapas M, Lin E, Peters T, Sakuraba A. The influence of proton pump inhibitor therapy on the outcome of infliximab therapy in inflammatory bowel disease: a patient‐level meta‐analysis of randomised controlled studies. Gut. 2021;70:2076‐2084 [DOI] [PubMed] [Google Scholar]
- 125. Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. 2013;369(8):711‐721. [DOI] [PubMed] [Google Scholar]
- 126. Dulai PS, Singh S, Jiang X, et al. The real‐world effectiveness and safety of vedolizumab for moderate‐severe Crohn's disease: results from the US VICTORY Consortium. Am J Gastroenterol. 2016;111(8):1147‐1155. [DOI] [PubMed] [Google Scholar]