

Abstract
Background:
Multiple system atrophy (MSA) is a rare neurodegenerative disease characterized by the aggregation of α-synuclein in glia and neurons. Sirolimus (rapamycin) is an mTOR inhibitor that promotes α-synuclein autophagy and reduces its associated neurotoxicity in preclinical models.
Objective:
To investigate the efficacy and safety of sirolimus in patients with MSA using a futility design. We also analysed 1-year biomarker trajectories in the trial participants.
Methods:
Randomized, double-blind, parallel group, placebo-controlled clinical trial at the New York University of patients with probable MSA randomly assigned (3:1) to sirolimus (2–6 mg daily) for 48-weeks or placebo. Primary endpoint was change in the Unified MSA Rating Scale (UMSARS) total score from baseline to 48-weeks. (ClinicalTrials.gov NCT03589976).
Results:
The trial was stopped after a pre-planned interim analysis met futility criteria. Between Aug 15, 2018, and Nov 15, 2020, 54 participants were screened, and 47 enrolled and randomly assigned (35 sirolimus, 12 placebo). Of those randomized, 34 were included in the intention-to-treat analysis. There was no difference in change from baseline to week-48 between the sirolimus and placebo in UMSARS total score (mean difference: 2.66; 95% CI −7.35 to 6.91; p=0.648). There was no difference in UMSARS-1 and UMSARS-2 scores either. UMSARS scores changes were similar to those reported in natural history studies. Neuroimaging and blood biomarker results were not different. Adverse events were more frequent with sirolimus. Analysis of 1-year biomarker trajectories in all participants showed that increases in blood NfL and reductions in whole brain volume correlated best with UMSARS progression.
Conclusions:
48-weeks of sirolimus was futile to slow the progression of MSA and had no effect on biomarkers compared to placebo. 1-year change in blood NfL and whole brain atrophy are promising biomarkers of disease progression for future clinical trials.
INTRODUCTION
Multiple system atrophy (MSA) is an adult-onset, rare, fatal neurodegenerative disease characterized by the intracellular accumulation of the misfolded protein α-synuclein (αSyn) in glia, as glial cytoplasmic inclusions (GCI), and neurons.1 MSA usually presents during the sixth decade of life and leads to death 8 to 10 years from symptom onset. Despite promising results in multiple preclinical models, no drugs have shown efficacy in placebo-controlled human trials.2–4 There is an urgent unmet medical need for therapies that can halt or slow the progression of this devastating disease.
Synucleinopathies such as MSA are characterized by defects in lysosome-mediated clearance of misfolded αSyn aggregates. Autophagy is the major cellular process that removes damaged macromolecules, organelles and abnormally misfolded proteins.5 Impaired autophagy results in reduced αSyn clearance, contributing to misfolded αSyn aggregation and spread.6, 7
Under normal conditions, autophagy is regulated by the protein mammalian target of rapamycin (mTOR).8, 9 Two mTOR complexes have been described, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Inhibition of mTORC1 results in autophagy activation.
Sirolimus (rapamycin) is an orally active immunosuppressant macrolide that induces autophagy by inhibiting mTORC1.8, 9 It was first isolated from the bacterium Streptomyces hygroscopicus found in soil samples from Easter Island (Rapa Nui in the local language) in a quest to find new antibiotics.10 Sirolimus is lipophilic, and some human evidence suggests it crosses the brain-blood barrier and inhibits mTOR activity in the CNS.11, 12 In preclinical models of synucleinopathies, autophagy activation with mTOR inhibition by sirolimus reduces αSyn accumulation suggesting that treatment with sirolimus could prevent αSyn-induced neurodegeneration.9, 13–17 mTOR inhibition also has neuroprotective effects in preclinical models of other proteinopathies, such as Huntington disease and Alzheimer disease.18–21 Despite compelling preclinical evidence, no clinical trials have yet tested mTOR inhibitors in patients with neurodegenerative disorders.
The safety profile of sirolimus and its analogues is well described, as they have been approved by the U.S. Food and Drug Administration (FDA) since 1999 for the prevention of organ transplant rejection, the treatment of lymphangioleiomyomatosis, and tuberous sclerosis-related angiofibromas.
Owing to its promising effects in animal models of synucleinopathy and its well-known safety profile, we investigated the preliminary efficacy and safety of sirolimus in patients with MSA using a futility design. This design minimizes the number of subjects required for a trial and provides an indication of whether or not to pursue larger trials.22 We also explored the effect of sirolimus on brain and retinal imaging, and blood biomarkers. Finally, we analyzed the combined pool of biomarker information from all participants to calculate 1-year biomarker trajectories and explore what measurement would perform best as outcome measure in future MSA trials.
METHODS
Study design and participants
We performed a 48-week randomized, double-blind, placebo-controlled, parallel-group single-site futility trial at the New York University Grossman School of Medicine. A futility design is appropriate for MSA given: a) the low prevalence of the disease; b) the availability of historical control data from large natural history studies;23, 24 and c) the availability of a validated clinical outcome assessment (Unified MSA Rating Scale, UMSARS) with a defined minimally important clinical decline.25, 26 In a futility design, the null hypothesis assumes that sirolimus will slow the progression of the disease, whereas the alternative hypothesis assumes no benefit of sirolimus. If the null hypothesis is rejected (i.e., sirolimus is futile to slow the progression of MSA), further studies will be discouraged, whereas non-futility will offer strong support for a larger trial to confirm efficacy. Our trial used a 3:1 (sirolimus:placebo) randomization ratio. The placebo group was not used for comparison purposes of the primary endpoint but to facilitate investigators’ and participants’ blinding.
Clinical oversight was provided by an independent data and safety monitoring board (DSMB), composed of an independent medical monitor (IMM) and an independent pharmacist that were unblinded to the allocation and sirolimus blood trough levels.
Eligible participants were 30–80 years old; had probable parkinsonian-predominant or cerebellar-predominant MSA according to current diagnostic criteria;27 were less than 4 years from the time of documented MSA diagnosis; able to walk with or without assistance; with an anticipated survival of at least 3 years in the opinion of the investigator; and able to swallow the study medication capsules. Female patients of childbearing potential were required to use adequate contraceptive methods.
Exclusion criteria included evidence of neurological diseases other than MSA; positive interferon-gamma release-assay indicating past or latent tuberculosis infection; history of active, acute or chronic, or latent hepatitis B or C; human immunodeficiency virus infection, or any other causes of acquired or congenital immunosuppression; uncontrolled diabetes; renal failure; history of malignant cancer within the last 5 years (except for non-metastatic, non-melanoma skin cancers); and use of oral or intravenous glucocorticoids, methotrexate, rituximab or other immunosuppressants in the 3 months prior to baseline; or use of potent inhibitors, inducers or drugs metabolized by CYP3A4. Participants with any condition that, per the investigators’ judgment, might have put them at risk, interfered with their participation in this trial, or confounded the trial results, were excluded. The Supplementary Methods include a full list of the inclusion and exclusion criteria.
The New York University Institutional Review Board (IRB) approved this study and all participants provided written informed consent. A description of the study protocol has been published (NCT03589976).
Randomization and masking
Eligible participants were enrolled and randomly assigned (3:1) to receive sirolimus or matching placebo. A central randomization scheme was used according to a randomization list generated by the IMM, which assigned the participant to receive study drug or placebo as the participant entered with stratification for MSA phenotype (cerebellar or parkinsonian). Study drug consisted of 2-mg sirolimus capsules and matching placebo capsules. Pfizer donated sirolimus (Rapamune®) 2-mg tablets for this study. In a compounding pharmacy, sirolimus 2-mg tablets were powdered and encapsulated in an orange capsule. Matching placebo, composed of powdered lactose, was encapsulated in an orange capsule. Sirolimus and placebo capsules looked identical. The study drug was supplied to each participant according to their assigned treatment in identical bottles.
All participants as well as the investigators and the personnel at the study site conducting the study or handling data were blinded to treatment assignment, with the exception of: i) the personnel labelling and packaging the study drug capsules, and ii) the data and safety monitoring board (DSMB) members.
The randomization list was kept in a secure location until the end of the study. Individual treatment assignment for each treatment kit was available to the study investigators and the IRB in case of an emergency.
Sirolimus dose selection and monitoring
The dosage of oral sirolimus during the study varied between 2 mg (one 2-mg capsule), 4 mg (two 2-mg capsules) or 6 mg (three 2-mg capsules) taken once a day. The number of capsules was adjusted based on i) sirolimus blood levels measured throughout the study; and, ii) the emergence of adverse events related to the study drug. The sirolimus trough blood levels were available only to the unblinded independent pharmacist, who made dosing recommendations to maintain sirolimus trough levels in the therapeutic range of 5 and 20 ng per millilitre in patients allocated to sirolimus, and sham recommendations on dose adjustments for participants allocated to placebo, to maintain the investigator and participant blinding.28 The principal investigator made the final decision to adjust the dosage based on the unblinded pharmacist’s recommendations and the presence and severity of adverse events. The study dose was increased and decreased when necessary during the trial, to maintain trough levels between 5 and 20 ng/ml and a tolerable adverse event profile. The sirolimus therapeutic range was selected based on the FDA-recommended therapeutic levels of sirolimus for the prophylaxis of renal transplant rejection. This daily dosage is within the used clinically and tolerable.29 Evidence in humans suggests that sirolimus at these dosages crosses the blood-brain barrier.11, 12 Blood sirolimus levels above 20 ng/ml are associated with poor tolerability.30
Procedures
Our trial had a 42-day screening period; a 48-week double-blind parallel group treatment period; a follow-up phone call 14±3 days following the last dose of study medication; and a follow-up safety blood and urine analysis 30±5 days following the last dose of study medication, for a total study duration of up to 58 weeks (Supplementary Figure 1).
At screening, after signing the informed consent, each participant underwent physical and neurological examinations, assessments of cognition, blood and urine sampling for clinical laboratory tests and ECG; women of childbearing potential also had a pregnancy test.
After confirmation of eligibility, subsequent visits were held at baseline (week 0) and week 12, week 24, week 36, and week 48. On day −1, after completion of baseline assessments, participants were randomized. Participants received sirolimus 2 mg/day (one 2-mg capsule/day) or matching placebo. During the first month of treatment, participants underwent weekly local blood and urine sample for safety and sirolimus levels (i.e., week 1, week 2, week 3 and week 4). The results were evaluated by the unblinded independent pharmacist who made dosing recommendation, e.g., maintaining, increasing or decreasing the dose of sirolimus or matching placebo. After the first month, patients underwent monthly (i.e., week 8, week 16, week 20, week 28, week 32, week 40, week 44) local safety blood and urine sample for safety and sirolimus levels monitoring.
Electronic case report forms were used to capture data. Adverse events were grouped according to Medical Dictionary for Regulatory Activities (MedDRA) organ class. Serious adverse events were defined as adverse events that led to study discontinuation, hospitalization, or death. Medically necessary changes in concurrent medications were allowed throughout the trial to minimize drop outs. Study drug accountability was performed and compliance discussed with the participant at study site visits during treatment. All used medication bottles, including any unused tablets were reconciled before dispensing new medication bottles and diary cards.
Outcomes
Clinical outcome measures were assessed at screening, baseline and weeks 12, 24, 36 and 48.
The primary outcome was the change from baseline to week 48 in the Unified MSA Rating Scale (UMSARS) total score.26 Higher UMSARS scores denote poorer health. The UMSARS were always assessed by the same highly experienced investigator (J.A.P) at each study visit to ensure consistency. This investigator was blinded to the previous UMSARS of the patient, treatment group assignment, safety laboratory results, and sirolimus blood levels.
Secondary efficacy outcomes were the change from baseline to week 48 in the UMSARS-1 (activities of daily living) and UMSARS-2 (motor examination) subscores;26 as well as brain MRI (putaminal mean diffusivity and putaminal volume, as well as other measures of global and regional atrophy, as well as exploratory measures of kurtosis, fractional anisotropy and axonal water fraction, as assessed by high-resolution isotropic T1-weighted, three-dimensional magnetization-prepared rapid gradient-echo MRI sequences obtained with a 1.5-T MRI machine [Siemens]), and retinal nerve fiber layer (RNFL) and macular ganglion cell complex (GCC) thicknesses assessed by optical coherence tomography (OCT, Cirrus 4000; Carl Zeiss, Dublin, CA).31(Supplementary Results).
Exploratory outcomes were changes from baseline to week 24, and from baseline to week 48, in αSyn-containing neuronal- and oligodendroglia-derived exosomes in plasma, measured as previously described,32 and neurofilament light chain (NfL) levels in plasma using the Simoa platform (Quanterix, Lexington, MA).33
Safety and tolerability outcomes included the number of participants who discontinued the study due to adverse events; the incidence of specific adverse events (i.e., treatment-emergent adverse events); changes in safety laboratory values (basic clinical chemistry); vital signs (blood pressure, heart rate, temperature); physical and neurological examinations; and mortality during the study period. The DSMB monitored unblinded safety data on an ongoing basis to ensure the continued safety of subjects enrolled in the study. The DSMB met at least every 6 months.
Statistical analyses
This was a randomized, double-blind, placebo-controlled futility trial in which the primary outcome measure value (UMSARS total score) in a cohort of patients with MSA taking active agent was compared to a known value from historical controls (i.e., untreated MSA patients) published in the literature.23, 24
The primary statistical analysis involved a comparison of the mean absolute difference in the active sirolimus group with a pre-specified fixed value, constituting a 4-point reduction in the expected mean increase in the UMSARS total score over 48 weeks without treatment. The expected mean increase in total UMSARS outcomes was obtained from the U.S. Natural History Study of MSA and from the placebo group of the phase-3 controlled trial with rifampicin for MSA, also performed in the U.S.2, 23 The pooled UMSARS total score at 48-weeks from these two MSA populations disclosed a mean increase of 8.6 (SD: 6) points from baseline.
The trial was designed to yield a two-sided α of 0.05 at 80% power for a treatment effect of 4 points on the 48-week progression of the UMSARS total score, i.e., 4.6 points in the sirolimus group vs 8.6 points (SD 6) in the historical control group. This difference is above the level for a minimal clinically important difference within 48 weeks of 3.5 points, below which effects cannot be reliably detected.25 Hence, we designed this trial to establish if treatment with sirolimus could lead to meaningful effects on disease progression in patients with MSA. The target sample size was 38 completers in the sirolimus group. Anticipating a 10% drop-out rate, this resulted in 42 completers in the sirolimus group and 14 completers in the placebo arm.
A pre-planned interim analysis to detect early evidence of futility was to be performed when 22 patients completed the 48-week assessments. The pre-specified futility criterion was a mean increase in the UMSARS total score from baseline to week-48 of 7 points or more in patients randomized to sirolimus. In such case, the trial was to be stopped. For the primary (UMSARS total score) and secondary (UMSARS-1 and UMSARS-2) outcomes, we used a modified intention-to-treat analysis, which included all participants who completed the 12-week follow-up visit. Study analyses for primary and secondary UMSARS endpoints were performed comparing the sirolimus vs the historical UMSARS data available from natural history studies. The primary outcome endpoint (UMSARS total score) was analyzed comparing the mean decline in UMSARS total score in the sirolimus group (UMSARS total score at week 48 minus UMSARS total score at baseline) with the pre-specified fixed value of 8.6 (SD: 6) using a one-sample t-test. There were no missing individual UMSARS items.
Study analyses for the brain MRI, OCT, and exploratory biomarker endpoints were performed comparing the sirolimus vs the placebo groups, including all participants who had baseline and, at least, one follow-up endpoint acquisition.
Safety was analyzed in the full analysis set, i.e., all randomized participants.
Continuous demographic variables were summarized using descriptive statistics. Categorical demographic variables (e.g., sex, ethnicity) and disease characteristics were summarized in frequency tables (frequency and proportion). Individual subject demographic and baseline disease characteristics were listed. The incidence and severity of treatment-emergent adverse events were summarized by treatment group. Statistical analysis of continuous variables was performed testing change from baseline.
For the longitudinal biomarker analysis, we calculated Pearson’s r for correlation analysis, and the P-value was corrected for multiple comparisons using the Holm method.34 Power calculations were performed with R version 3.5.1. Standardized effect sizes were calculated dividing the annual mean change by the standard deviation of the annual mean change in each of the parameters studied. Power calculations were based on a 2-sided significance level (alpha) of 5%, and a power (1-beta) of 80%, assuming hypothetical 30% and 50% reductions in the progression of each parameter, respectively.
Data were entered independently in an electronic case report database (TrialMaster). Before database lock, the data for all the patients were checked for completeness and quality. All analyses were performed using R (version 3.5.1) unless otherwise specified.
RESULTS
During a 22-month period between August 15, 2018, and November 15, 2020, 54 people were assessed for eligibility and 47 were enrolled and randomly assigned, 35 to the sirolimus group and 12 to the placebo group (Figure 1). All participants fulfilled criteria for probable MSA. Post-mortem histopathological analysis confirmed the diagnosis of MSA in two participants whose brain had been donated. The clinical and demographic characteristics of patients assigned to sirolimus and placebo were similar at baseline (Table 1). At baseline, the median age was 59 years (56–63.5), 20 (43%) participants were women, and the median UMSARS-1 and UMSARS-2 were 21 (17–24) and 23 (19–26.5) respectively.
Figure 1 –
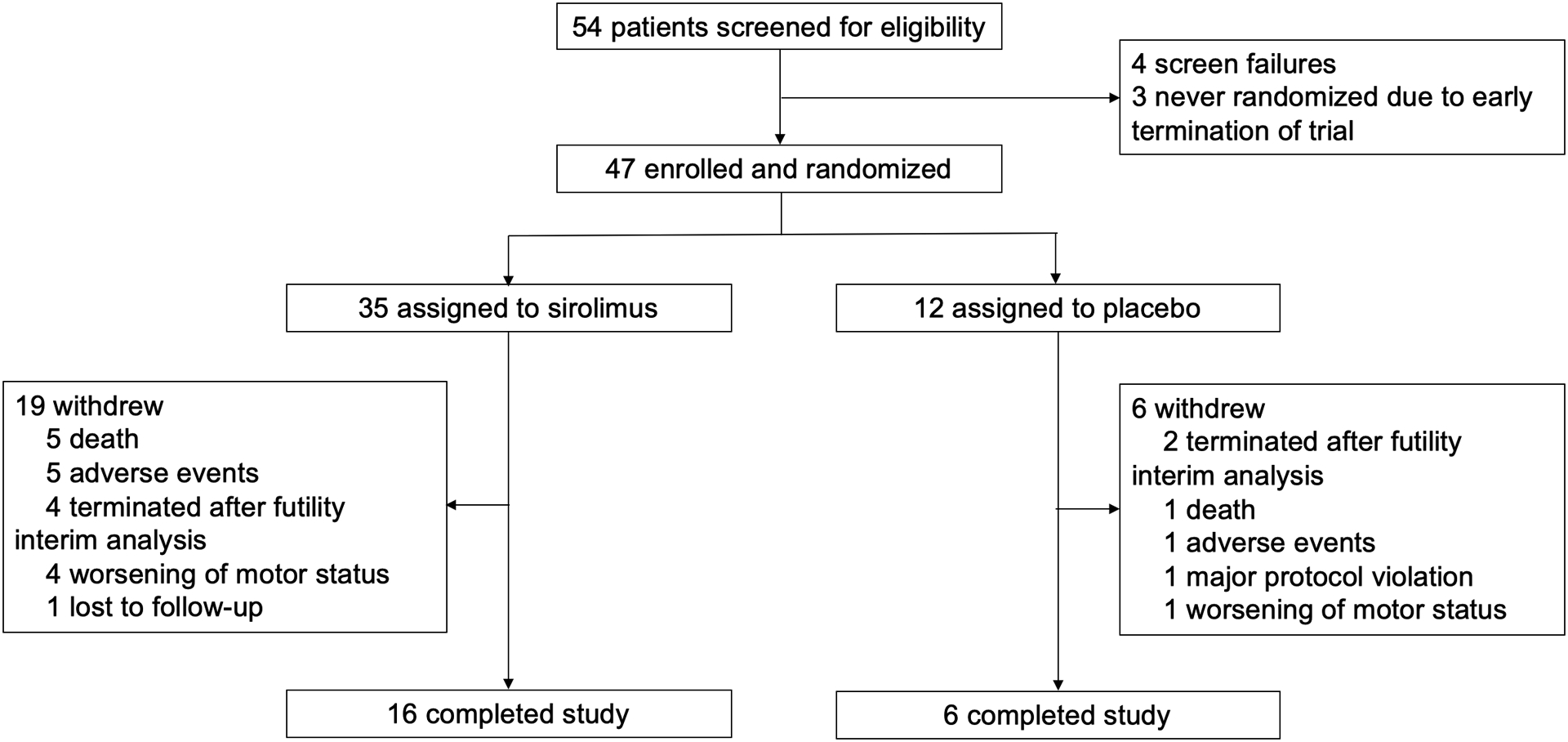
Patient flowchart
Table 1.
Baseline clinical and demographic characteristics of randomized patients
| Sirolimus group (n=35) |
Placebo group (n=12) |
|
|---|---|---|
| Age, years | 59 (56–63) | 58 (56–63) |
| Sex | ||
| Women | 14 (40%) | 6 (50%) |
| Men | 21 (60%) | 6 (50%) |
| Ethnicity | ||
| Caucasian including Hispanic | 30 (86%) | 12 (100%) |
| Asian | 3 (9%) | 0 |
| Afro-American | 2 (5%) | 0 |
| Time since diagnosis, years | 0.8 (0.5–1.5) | 1.6 (1.1–2.1) |
| Predominant motor phenotype | ||
| Parkinsonian | 18 (51%) | 7 (58%) |
| Cerebellar | 17 (49%) | 5 (42%) |
| Urinary catheterization | ||
| No | 24 (69%) | 5 (42%) |
| Intermittent | 7 (20%) | 5 (42%) |
| Indwelling or suprapubic catheter | 4 (11%) | 2 (16%) |
| Diagnostic certainty | ||
| Probable | 35 (100%) | 12 (100%) |
| UMSARS | ||
| Activities of daily living (UMSARS-1) | 21 (18–24) | 20 (15–24) |
| Motor examination (UMSARS-2) | 23 (20–26) | 22 (18–26) |
| Global disability score (UMSARS-4) | 2 (2–4) | 2 (2–4) |
Data are median (IQR) or n (%). UMSARS=Unified Multiple System Atrophy Rating Scale.
Of the 47 patients who were randomized, 34 (25 in the sirolimus group and 9 in the placebo group) completed, at least, the 12-week study visit and were included in the intention-to-treat analysis set.
When 22 participants -including 16 (45%) of the 35 randomized participants in the sirolimus group, and 6 (50%) of the 12 randomized participants in the placebo group- had completed the 48-week assessment, an interim analysis was performed (November 2020). The completers set included the data of these 22 patients. The interim analysis criterion for futility was met. After discussion among the principal investigators, the DSMB, and the IRB, the trial was stopped on January 2, 2021. At that time, 9 patients who were still active (6 randomized, and 3 screened but not yet randomized) were instructed to stop the study drug (Figure 1).
The median sirolimus dose was 2 capsules (4 mg) a day (Supplementary Figure 2). The median trough sirolimus levels in blood were 8.7 (5.6–12.5) ng/ml. Participants in the sirolimus and placebo groups were compliant with 89% of their scheduled study drug doses.
Unified MSA Rating Scales
There were no UMSARS missing data. In the intention-to-treat analysis set, the change from baseline to week 48 or last available visit in the UMSARS-1 (difference between sirolimus and placebo: 0.32; 95% CI −4.22 to 2.91; p=0.707) and the UMSARS-2 (difference between sirolimus and placebo: 0.08; 95% CI −4.06 to 4.08; p=0.996), and the UMSARS total score (difference between sirolimus and placebo: 2.66; 95% CI −7.35 to 6.91; p=0.648) did not differ between sirolimus and placebo, and was similar to previously reported historical untreated MSA patients enrolled in natural history studies (Table 2; Figure 2). We found no differences in the analysis of the completers set either (data not shown).
Table 2.
Changes in UMSARS scores in the intention-to-treat analysis set
| Sirolimus group (n=25) | Placebo group (n=9) | Difference | P value | |
|---|---|---|---|---|
| Change from baseline to treatment termination in UMSARS-1 | 5.65 (5.91) | 5.33 (3.81) | 0.32 [1.72 (−422, 2.91)] |
0.707 |
| Change from baseline to treatment termination in UMSARS-2 | 6.58 (5.26) | 6.50 (4.46) | 0.08 [1.9 (−4.06, 4.08] |
0.996 |
| Change from baseline to treatment termination in total UMSARS | 13.92 (8.32) | 11.30 (8.00) | 2.66 [3.28 (−7.35, 6.91)] |
0.648 |
Data are mean (SE) or mean (SE; 95% CI). UMSARS=Unified Multiple System Atrophy Rating Scale.
Figure 2 –
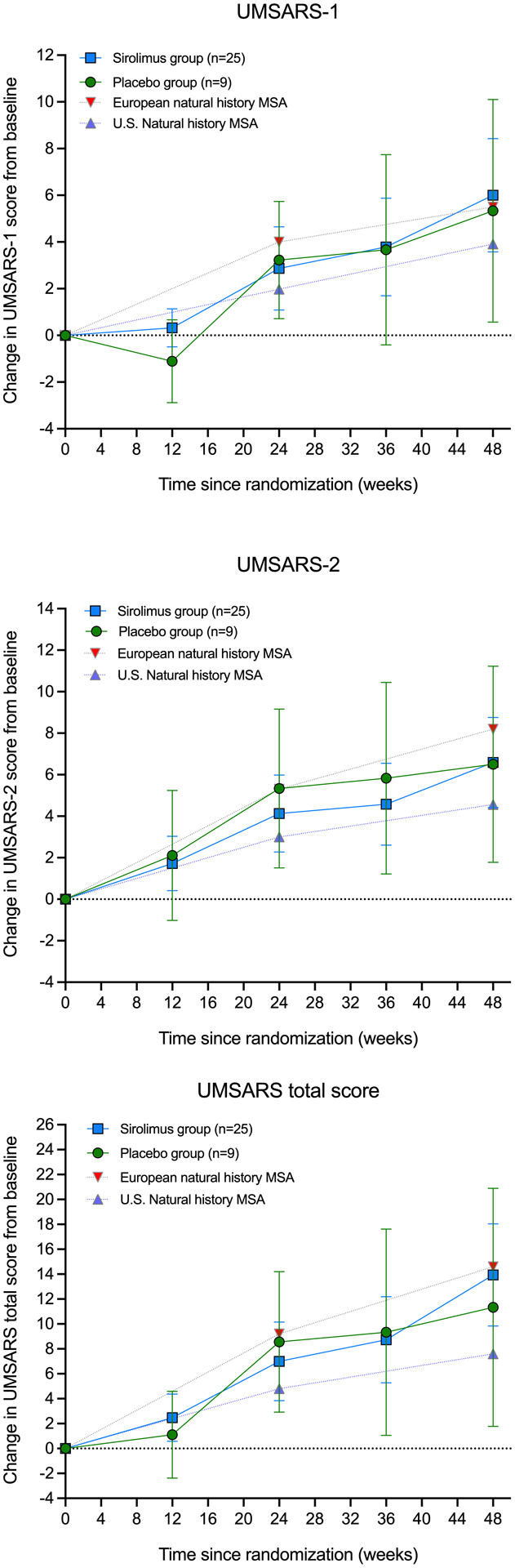
Change from baseline in UMSARS-1, UMSARS-2 and UMSARS total score in the intention-to-treat analysis set. Error bars show 95% CIs. The graphs include UMSARS data from previously reported 1-year European and U.S. natural history studies of MSA.
Adverse events
Adverse events occurred in greater number in the sirolimus group. The most reported adverse events were infection (urinary tract infection, upper respiratory infection, skin infection or sinusitis), oral and labial pathology (aphthae, gingivitis, and herpes-like vesicles), diarrhea, edema in lower limbs, and benign skin pathology (acne, petechiae, rash). All these were significantly more frequent in the sirolimus compared to the placebo group. Other commonly reported (> 5%) adverse events were falls, nausea, worsening of movement disorder, and fatigue, although these were reported with similar frequency in the sirolimus and placebo groups, except for fatigue, which was more frequent in the placebo group. Most adverse events were mild to moderate (Table 3).
Table 3.
Adverse events by system, outcome, grade and attribution in the full analysis set.
| Sirolimus group (n=180 events / 35 participants) |
Placebo group (n=34 events / 12 participants) |
|
|---|---|---|
| By event | ||
| Infection (UTI, upper respiratory, skin, sinusitis) | 29 (16%) / 21 (60%) | 6 (18%) / 4 (33%) |
| Oral and labial benign pathology (aphthae, gingivitis, and oral herpes-like vesicles) | 21 (12%) /15 (43%) | 2 (6%) / 2 (17%) |
| Diarrhea | 17 (9%) / 16 (46%) | 2 (6%) / 2 (17%) |
| Edema in lower limbs | 17 (9%) / 17 (49%) | 1 (3%) / 1 (8%) |
| Skin benign pathology (acne, petechiae, rash) | 15 (8%) / 13 (37%) | 2 (6%) / 2 (17%) |
| Fall | 10 (5%) / 6 (17%) | 6 (18%) / 4 (33%) |
| Nausea | 5 (3%) / 3 (9%) | 3 (9%) / 1 (8%) |
| Worsening of movement disorder | 4 (2%) / 4 (11%) | 1 (3%) / 1 (8%) |
| Fatigue | 1 (0.5%) / 1 (3%) | 7 (21%) / 7 (58%) |
| Other | 56 (31%) / 25 (71%) | 10 (29%) / 4 (33%) |
| By outcome | ||
| Fatal | 3 (2%) / 3 (8%) | 1 (4%) / 1 (8%) |
| Not resolved | 23 (15%) / 14 (40%) | 1 (4%) / 1 (8%) |
| Resolved | 133 (84%) / 29 (83%) | 26 (93%) / 9 (75%) |
| By grade | ||
| Mild | 128 (72%) / 28 (80%) | 29 (85%) / 9 (75%) |
| Moderate | 43 (24%) / 22 (63%) | 3 (9%) / 2 (17%) |
| Severe | 7 (4%) / 5 (14%) | 2 (6%) / 2 (17%) |
| By attribution to study treatment | ||
| Definitely not related | 52 (30%) / 23 (66%) | 14 (45%) / 6 (50%) |
| Probably not related | 21 (12%) / 13 (37%) | 5 (16%) / 4 (33%) |
| Possibly related | 25 (15%) / 15 (43%) | 4 (13%) / 2 (17%) |
| Probably related | 30 (17%) / 16 (46%) | 5 (16%) / 2 (17%) |
| Definitely related | 45 (26%) / 20 (57%) | 3 (10%) / 2 (17%) |
| By seriousness | ||
| Non-serious | 160 (93%) / 31 (86%) | 29 (88%) / 9 (75%) |
| Serious | 12 (7%) / 9 (26%) | 4 (12%) / 2 (17%) |
Data are number of events (%) / number of participants (%)
Five patients died in the sirolimus group and one in the placebo group. In all cases, deaths were considered not related to the study drug. Five patients in the sirolimus group and one in the placebo group discontinued the study because of adverse events. Of the five patients in the sirolimus group who discontinued, the adverse event in four of them (sepsis of urinary origin, severe skin rash, overwhelming fatigue, and recurrent urinary tract infections) was considered probably or possibly related to sirolimus. Seven serious adverse events (in 5 patients) were noted in the sirolimus group and two (in two patients) in the placebo group. Of the 7 serious adverse events in the sirolimus group, two were considered probably related.
Brain magnetic resonance imaging
All randomized participants (35 to the sirolimus and 12 to the placebo group) had a brain MRI at baseline. Of those 47, 15 participants (11 in the sirolimus group and 4 in the placebo group) had 48-week brain MRI, and were considered for the analysis. The change from baseline to week-48 in putaminal mean diffusivity (difference between sirolimus and placebo in right putaminal mean diffusivity: −0.072; 95% CI –0.16 to 0.3; p=0.403; and in left putaminal mean diffusivity: 0.063; 95% CI –0.19 to 0.06; p=0.238) and putaminal volume (difference between sirolimus and placebo in right putaminal volume: −116.8 ml; 95% CI –405 to 619; p=0.595; and in left putaminal volume: 29.8 ml; 95% CI −1092 to 1032; p=0.938) did not differ between sirolimus and placebo (Figure 3). The change from baseline to week-48 was not different between sirolimus and placebo either in any of the other multiple studied parameters and brain regions (Supplementary Results).
Figure 3 –
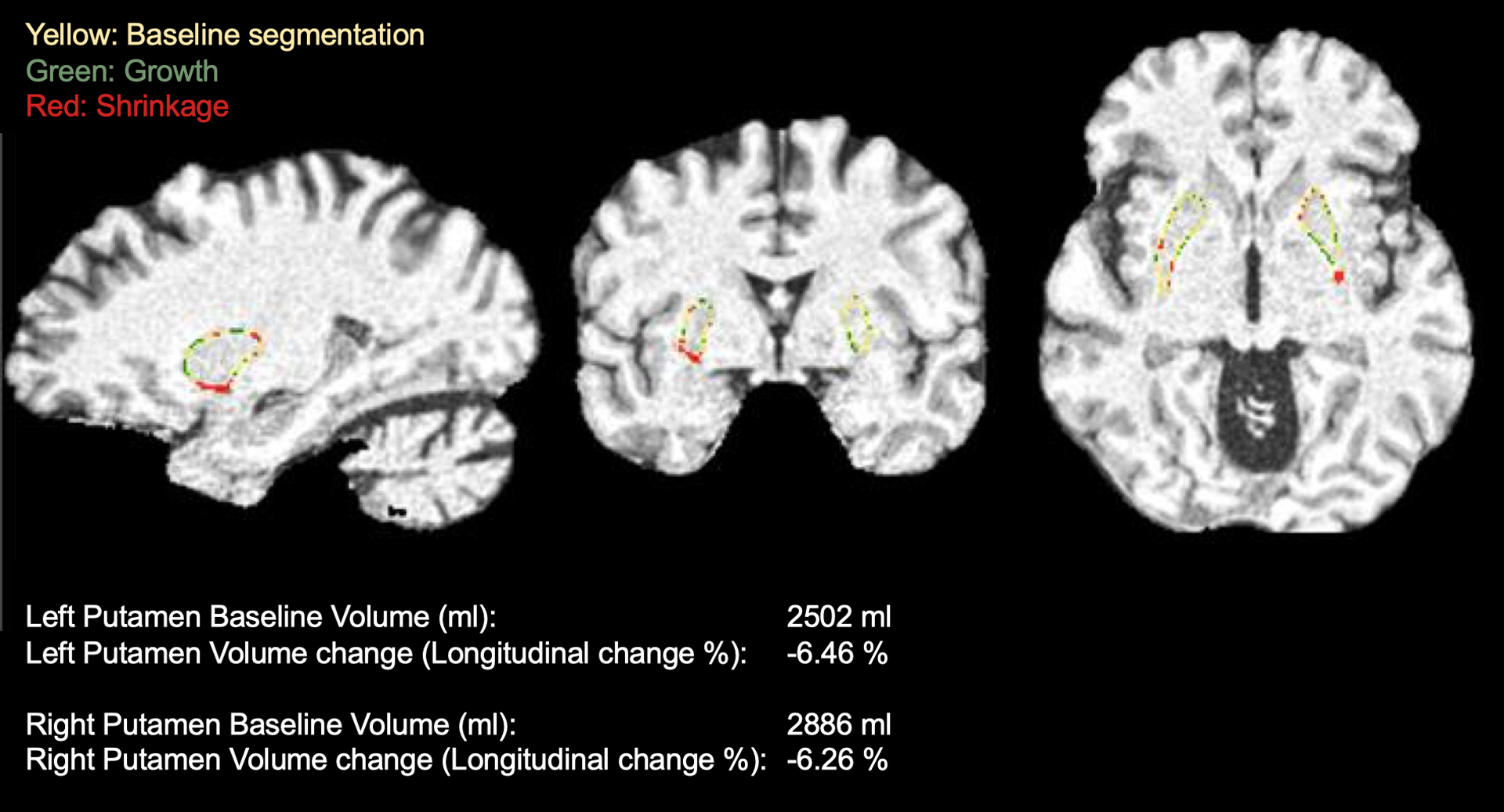
Example of putaminal volume assessment in one of the study participants. T1-weighted brain MRI sequences automated putaminal segmentation at baseline and at 48-month visit, showing a significant longitudinal volume reduction.
Retinal optical coherence tomography
All randomized participants (35 to the sirolimus and 12 to the placebo group) had an OCT at baseline. Only 10 participants (8 in the sirolimus group and 2 in the placebo group) had 48-week OCT, and were considered for analysis. The change from baseline to week 48 in the average retinal nerve fibre layer (RNFL) thickness as measured by OCT (difference between sirolimus and placebo in the right eye: −0.357; 95% CI −5.18 to 5.89; p=0.882; left eye: 1.333; 95% CI −9.26 to 6.6; p=0.693) and the average macular ganglion cell complex (GCC) thickness (difference between sirolimus and placebo in the right eye: 2.375; 95% CI −17 to 12.3; p=0.345; left eye: 1.833; 95% CI −6.98 to 3.31; p=0.384) did not differ between sirolimus and placebo. There were no differences in specific RNFL and GCC quadrants either (Supplementary Results).
Neurofilament light chain (NfL) and αSyn-containing CNS-originated exosomes in plasma
46 randomized participants (12 placebo and 34 sirolimus) underwent plasma NfL measurements at baseline, 27 (10 placebo and 17 sirolimus) at the 24-week visit, and 12 (4 placebo and 8 sirolimus) at the 48-week visit. The change from baseline to week-48 in NfL concentrations in plasma did not differ between sirolimus and placebo. Forty-six randomized participants (12 placebo and 34 sirolimus) underwent measurements of αSyn putative neuronal and oligodendroglial exosomes at baseline, 27 (8 placebo and 19 sirolimus) at the 24-week visit, and 14 (3 placebo and 11 sirolimus) at the 48-week visit. The change from baseline to week-48 in any of the studied exosomal parameters did not differ between sirolimus and placebo (Supplementary Figure 3).
Biomarker longitudinal changes
Given the lack of differences between sirolimus and placebo in the clinical, neuroimaging, and biomarker endpoints studied suggesting that sirolimus has no biological effect on MSA, we sought to describe longitudinal changes in the mentioned biomarkers in all participants, as a single group, with the goal of identifying the ones with the highest standardized effect sizes that could be used in future clinical trials. Table 4 shows the values at baseline and 48-week follow-up, the mean differences between baseline and follow-up, and the standardized effect sizes. The last two columns display the required numbers of patients per group in a future hypothetical clinical trial with an expected treatment effect of 30% and 50% respectively, i.e., when the annual change expected by the natural history of the disease is reduced by those percentages. The measurements with the highest standardized effect size, i.e., left and right putaminal volumes, required a 60% smaller number of patients than the clinical rating scale, with the highest effect size (UMSARS total score) to detect a treatment effect of 50%.
Table 4.
Longitudinal biomarker analysis
| Measurement | Baseline (SD) | 48-week follow-up (SD) | Mean annual change (SD) | Standardized effect size | Power calculation for clinical trials (subjects per group) | |
|---|---|---|---|---|---|---|
| 30% reduction | 50% reduction | |||||
| UMSARS | Points | |||||
| UMSARS-1 (n=22) | 19.9 (4.8) | 26.0 (6.4) | 6.1 (4.5) | 1.30 | 118 | 43 |
| UMSARS-2 (n=22) | 21.4 (5.4) | 27.9 (7.7) | 6.5 (4.6) | 1.40 | 101 | 37 |
| UMSARS-total (n=22) | 41.2 (9.5) | 53.9 (13.2) | 12.6 (8.8) | 1.40 | 100 | 36 |
| Blood biomarkers | pg./ml | |||||
| NfL (N=12) | 23.0 (10.8) | 42.9 (20.6) | 19.9 (15.3) | 1.30 | 104 | 38 |
| αSyn neuronal exosomes (n=14) | 351.4 (299.7) | 69.7 (120.2) | −281.7 (331.5) | −0.8 | 241 | 140 |
| αSyn glial exosomes (n=14) | 1030 (920.9) | 67.1 (80.4) | −962.9 (919.9) | −1.1 | 159 | 58 |
| Retinal OCT | μm | |||||
| RNFL OD (n=8 eyes) | 91.1 (9.5) | 92.9 (11.6) | 1.3 (5.5) | 0.2 | 3123 | 1125 |
| RNFL OS (n=9 eyes) | 90.6 (9.7) | 91.8 (10.5) | 1.1 (5.7) | 0.2 | 4685 | 1687 |
| GCC OD (n=8 eyes) | 78.4 (6) | 79.5 (6.3) | 0.4 (1.8) | 0.2 | 3534 | 1272 |
| GCC OS (n=9 eyes) | 78.3 (5.4) | 78.6 (7.0) | 0.2 (2.7) | 0.1 | >10,000 | >10,000 |
| Volumetric brain MRI | % | |||||
| Whole brain (n=12) | 1353.36 (933) | 1337.9 (913) | −1.11 (0.60) | −1.85 | 49 | 19 |
| Left putamen (n=12) | 2.72 (0.83) | 2.50 (0.80) | −8.26 (2.91) | −2.83 | 22 | 8 |
| Right putamen (n=12) | 3.44 (0.78) | 3.21 (0.64) | −6.38 (2.59) | −2.46 | 29 | 11 |
| Lateral ventricles (n=12) | 26.13 (11.6) | 28.42 (32.2) | 10.14 (9.94) | 1.03 | 168 | 61 |
| Cerebellum (n=12) | 138.2 (231.2) | 136.7 (210.1) | −1.17 (0.73) | −1.60 | 68 | 25 |
| Midbrain (n=12) | 9.4 (0.6) | 9.3 (0.4) | −1.60 (1.44) | −1.11 | 84 | 31 |
| Pons (n=12) | 14.7 (5) | 14 (4) | −4.69 (2.26) | −2.07 | 34 | 13 |
| Medulla (n=12) | 4.2 (0.6) | 4 (0.4) | −3.95 (1.83) | −2.16 | 38 | 14 |
UMSARS: Unified MSA Rating Scale; OCT: Optical Coherence Tomography; OD: Right Eye; OS: Left Eye; SD: Standard Deviation;
Correlation analysis
We correlated the annual change in plasma NfL, αSyn in neuronal and oligodendroglial exosomes isolated from plasma, and volumetric MRI with the annual change in the UMSARS-1, UMSARS-2 and UMSARS total score (Table 5). Increases in UMSARS scores were significantly associated with reductions in whole brain volume and increases in plasma NfL.
Table 5.
Correlation between 1-year changes in UMSARS and 1-year plasma NfL and volumetric brain MRI changes
| UMSARS-1 | UMSARS-2 | UMSARS total | |
|---|---|---|---|
| Plasma NfL |
r=0.564 p=0.05 |
r=0.795
P=0.002* |
r=0.739
P=0.006* |
| aSyn neuronal exosomes | r=0.196 p=0.50 |
r=0.173 p=0.55 |
r=0.200 p=0.49 |
| aSyn glial exosomes | r=0.148 p=0.61 |
r=0.139 p=0.63 |
r=0.155 p=0.59 |
| Whole brain |
r=−0.554
P=0.049* |
r=−0.591
P=0.033* |
r=−0.618
P=0.024* |
| Cerebellum |
r=0.238 P=0.58 |
r=0.269 P=0.42 |
r=0.297 P=0.31 |
| Right putamen |
r=−0.227 P=0.45 |
r=−0.145 P=0.63 |
r=−0.20 P=0.51 |
| Left putamen |
r=0.08 P=0.77 |
r=−0.316 P=0.29 |
r=−0.123 P=0.68 |
| Midbrain |
r=−0.140 P=0.64 |
r=0.267 P=0.37 |
r=0.067 P=0.82 |
| Pons |
r=−0.063 P=0.83 |
r=0.265 P=0.38 |
r=0.108 P=0.72 |
| Medulla oblongata |
r=−0.114 P=0.71 |
r=0.234 P=0.44 |
r=0.064 P=0.83 |
Pearson’s r was calculated using the 12 patients who had complete UMSARS, NfL and MRI data. Shown P values are corrected for multiple comparisons using the Bonferroni-Holm’s method. UMSARS: Unified MSA Rating Scale.
DISCUSSION
In this randomized, double-blind, placebo-controlled trial, there was no evidence that sirolimus modified disease progression in patients with MSA, as measured by the change in the UMSARS score at 48 weeks. Sirolimus was relatively well tolerated, with frequent mild and moderate adverse events compared to placebo. The adverse events were comparable to the ones already described in the literature in patients receiving sirolimus to prevent organ transplant rejection.30 There was no evidence that sirolimus could modify the progression of brain MR imaging markers, retinal abnormalities, or exploratory blood biomarkers such as plasma NfL and αSyn in putative CNS-originating exosomes in patients with MSA. Overall, the results of this clinical trial do not support further clinical development of sirolimus in patients with MSA.
Basic science data provide a solid rational for the use of autophagy activators, and in particular sirolimus, as a potential therapeutic strategy for neurodegenerative proteinopathies.8, 9, 35 Thus, the rationale for repurposing sirolimus as potential disease-modifying agent in MSA and other neurodegenerative disorders is well established preclinically. Ours has been the first clinical trial using an mTOR inhibitor with the goal of slowing neurodegeneration. A clinical trial testing sirolimus for ALS is ongoing.36
Despite its limitations,37 the UMSARS remains the most widely used clinical outcome assessment and has been used as primary endpoint in most clinical trials investigating disease modification in MSA.2, 4, 38 We chose a 48-week observation period, which was sufficient in previous trials to investigate the disease progression of UMSARS. The placebo-controlled futility design allows a relatively rapid low-cost hypothesis testing, based on the availability of high-quality data from natural history studies, used as a comparison group. The placebo arm was to facilitate blinding and to define if placebo-treated patients in trials have a similar evolution to natural history controls. Indeed, the UMSARS progression in the sirolimus and placebo groups was within the rage of the described UMSARS progression in natural history studies,23, 24 confirming the feasibility of our approach.
Trial recruitment was stopped after the declaration of futility by the pre-planned interim analysis. We were able to enroll 47 patients within a 22-month period at our single site. However, the frequency of dropouts in our trial was higher than in previous trials, perhaps reflecting the inclusion of patients with slightly more advanced disease in our trial. Baseline UMSARS-2 scores in our trial (~22 points) were similar than those in the PROMESA (~21 points)4, but higher than the rasagiline (~17 points)3, and rifampicin (~12 points)2 trials. In spite of the high dropout rate, our criteria for overwhelming futility were met, indicating that the study was adequately powered to detect futility.
The main finding of our trial is that, at 48-weeks, sirolimus was not associated with disease modification in patients with MSA compared with previously reported natural history controls. We also found no evidence that sirolimus had an effect on secondary clinical endpoints (i.e., UMSARS-1 and UMSARS-2). This study also included exploratory neuroimaging, retinal, and blood biomarkers. There were no differences in any of the studied parameters between sirolimus and placebo.
Given the lack of differences between sirolimus and placebo, we considered that sirolimus had no biological effect on any of the biomarkers studied and, therefore, combined the sirolimus and placebo datasets to perform a 1-year biomarker trajectory analysis. We found that the annual changes in plasma NfL and whole brain volume in brain MRI correlated best with the changes in UMSARS, and provided higher standardized effect sizes than the UMSARS, suggesting that these biomarkers have clinical relevance to track disease progression. This is, to our knowledge, the most comprehensive MSA longitudinal biomarker study published in MSA so far, and the conclusions should aid in the design of future clinical trials.
Our study has limitations. The dropout was significantly higher than expected, which should inform planning for future clinical trials of MSA. We found no evidence that oral sirolimus has any clinical or biomarker effect in patients with MSA. Potential explanations for this include that the mTOR-autophagy pathway is not essential in synucleinopathies, that the burden of synucleinopathy in patients with probable MSA is too high to be impacted by autophagy inhibitors, and that CNS biodistribution and target engagement were poor. Unfortunately, our study did not include CSF sirolimus levels or putative autophagy-related biomarkers, which could have provided information on the CNS activity of sirolimus. Whether higher dosages of sirolimus could have been neuroprotective is unknown, however this is limited by the emergence of systemic tolerability side effects. It is tempting to speculate that high-doses of CNS-selective mTOR inhibitors, or non-mTOR CNS-selective autophagy activators, without systemic adverse effects, may have neuroprotective effects.
Supplementary Material
Acknowledgements
This study was funded by the NINDS (R01NS107596). Sirolimus (Rapamune®) 2-mg tablets were donated by Pfizer (New York, NY), who provided no funding for this study and had no input in the design, analysis or result reporting. The sirolimus capsules and matching placebo were prepared by Mr. Joseph Navarra at Town Total Compounding Center Pharmacy (Woodbury, NY). We thank the Multiple System Atrophy (MSA) Coalition for their support with recruitment; the patients and families for their participation; Dr. Codrin Lungu (NINDS) for strategic advice; Amir S. Steinberg, and Sharon Simon for their role as data safety and monitoring committee members; Zenith Khan, DNP, Kaia Dalamo DNP, and Erin Barnes, NP for their assistance with the study; IXICO for support with volumetric brain MRI analysis; Biogen Inc. for logistic support with the NfL biomarker analysis. This trial was designed, in part, during the NINDS Clinical Trials Methodology Course, supported by R25NS088248.
Financial disclosures:
JAP: Research funding from the NIH, Michael J. Fox Foundation, MSA Coalition, Familial Dysautonomia Foundation, FDA; advisory board member for Takeda, Astellas, and Dr. Reddy’s Laboratories; managing editor of Clinical Autonomic Research; principal investigator in studies funded by Biohaven Pharmaceuticals, Theravance Biopharma, and Biogen. Salary from Novartis. JM: Research funding from the NIH, Michael J. Fox Foundation, MSA Coalition, Familial Dysautonomia Foundation, FDA. PMV: research funding from the Familial Dysautonomia Foundation and Theravance Biopharma. Principal investigator in studies funded by Biohaven Pharmaceuticals, Theravance Biopharma. MAP: research funding from the Familial Dysautonomia Foundation. HK: research funding from the NIH, Michael J. Fox Foundation, MSA Coalition, Familial Dysautonomia Foundation, FDA, and Biogen; is an advisory Board Member for Lundbeck, Biogen, Biohaven, Theravance, PTC Therapeutics, ONO, Takeda, Vaxxinity, and Lilly; is Editor-in-Chief of Clinical Autonomic Research. TMS: research funding from the NIH UJK: research funding from the NIH. Advisory board member of Amprion. GB, JZ, YQ, SD, KNM, IS, and BAA report no conflicts
Funding:
National Institutes of Neurological Diseases and Stroke (R01NS107596 and R25NS088248)
REFERENCES
- 1.Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med 2015;372(3):249–263. [DOI] [PubMed] [Google Scholar]
- 2.Low PA, Robertson D, Gilman S, et al. Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13(3):268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poewe W, Seppi K, Fitzer-Attas CJ, et al. Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. Lancet Neurol 2015;14(2):145–152. [DOI] [PubMed] [Google Scholar]
- 4.Levin J, Maass S, Schuberth M, et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2019;18(8):724–735. [DOI] [PubMed] [Google Scholar]
- 5.Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 2004;24(8):1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004;305(5688):1292–1295. [DOI] [PubMed] [Google Scholar]
- 7.Lan AP, Chen J, Zhao Y, Chai Z, Hu Y. mTOR Signaling in Parkinson’s Disease. Neuromolecular Med 2017;19(1):1–10. [DOI] [PubMed] [Google Scholar]
- 8.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015;125(1):25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nature reviews Neuroscience 2011;12(8):437–452. [DOI] [PubMed] [Google Scholar]
- 10.Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975;28(10):721–726. [DOI] [PubMed] [Google Scholar]
- 11.Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med 2008;5(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parker WE, Orlova KA, Parker WH, et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med 2013;5(182):182ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proceedings of the National Academy of Sciences of the United States of America 2013;110(19):E1817–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu K, Shi N, Sun Y, Zhang T, Sun X. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem Res 2013;38(1):201–207. [DOI] [PubMed] [Google Scholar]
- 15.Bai X, Wey MC, Fernandez E, et al. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol Aging Age Relat Dis 2015;5:28743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Cuina M, Guerin P, Bezard E, Meissner W, Fernagut PO. Rapamycin for treating MSA: A preclinical proof of concept study. Movement Disord 2018;33:S441–S441. [Google Scholar]
- 17.Gao J, Perera G, Bhadbhade M, Halliday GM, Dzamko N. Autophagy activation promotes clearance of alpha-synuclein inclusions in fibril-seeded human neural cells. J Biol Chem 2019;294(39):14241–14256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004;36(6):585–595. [DOI] [PubMed] [Google Scholar]
- 19.Siman R, Cocca R, Dong Y. The mTOR Inhibitor Rapamycin Mitigates Perforant Pathway Neurodegeneration and Synapse Loss in a Mouse Model of Early-Stage Alzheimer-Type Tauopathy. PloS one 2015;10(11):e0142340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caccamo A, Magri A, Medina DX, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013;12(3):370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frederick C, Ando K, Leroy K, et al. Rapamycin ester analog CCI-779/Temsirolimus alleviates tau pathology and improves motor deficit in mutant tau transgenic mice. J Alzheimers Dis 2015;44(4):1145–1156. [DOI] [PubMed] [Google Scholar]
- 22.Koch MW, Korngut L, Patry DG, et al. The promise of futility trials in neurological diseases. Nat Rev Neurol 2015;11(5):300–305. [DOI] [PubMed] [Google Scholar]
- 23.Low PA, Reich SG, Jankovic J, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 2015;14(7):710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013;12(3):264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krismer F, Seppi K, Wenning GK, Abler V, Papapetropoulos S, Poewe W. Minimally clinically important decline in the parkinsonian variant of multiple system atrophy. Mov Disord 2016;31(10):1577–1581. [DOI] [PubMed] [Google Scholar]
- 26.Wenning GK, Tison F, Seppi K, et al. Development and validation of the Unified Multiple System Atrophy Rating Scale (UMSARS). Mov Disord 2004;19(12):1391–1402. [DOI] [PubMed] [Google Scholar]
- 27.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71(9):670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesosky M, Joska J, Decloedt E. Simulating therapeutic drug monitoring results for dose individualisation to maintain investigator blinding in a randomised controlled trial. Trials 2017;18(1):261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kasiske BL, Nashan B, Del Carmen Rial M, et al. A prospective, multinational pharmacoepidemiological study of clinical conversion to sirolimus immunosuppression after renal transplantation. J Transplant 2012;2012:107180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen LS, Vautier M, Allenbach Y, et al. Sirolimus and mTOR Inhibitors: A Review of Side Effects and Specific Management in Solid Organ Transplantation. Drug Saf 2019;42(7):813–825. [DOI] [PubMed] [Google Scholar]
- 31.Mendoza-Santiesteban CE, Gabilondo I, Palma JA, Norcliffe-Kaufmann L, Kaufmann H. The Retina in Multiple System Atrophy: Systematic Review and Meta-Analysis. Front Neurol 2017;8:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutta S, Hornung S, Kruayatidee A, et al. alpha-Synuclein in blood exosomes immunoprecipitated using neuronal and oligodendroglial markers distinguishes Parkinson’s disease from multiple system atrophy. Acta Neuropathol 2021;142(3):495–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin CH, Li CH, Yang KC, et al. Blood NfL: A biomarker for disease severity and progression in Parkinson disease. Neurology 2019;93(11):e1104–e1111. [DOI] [PubMed] [Google Scholar]
- 34.Holm S A simple sequentially rejective multiple test procedure. Scandinavian journal of statistics 1979:65–70. [Google Scholar]
- 35.Scrivo A, Bourdenx M, Pampliega O, Cuervo AM. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol 2018;17(9):802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandrioli J, D’Amico R, Zucchi E, et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine (Baltimore) 2018;97(24):e11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palma JA, Vernetti PM, Perez MA, et al. Limitations of the Unified Multiple System Atrophy Rating Scale as outcome measure for clinical trials and a roadmap for improvement. Clin Auton Res 2021;31(2):157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singer W, Dietz AB, Zeller AD, et al. Intrathecal administration of autologous mesenchymal stem cells in multiple system atrophy. Neurology 2019;93(1):e77–e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.